
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnomalous pseudocapacitance of vanadium carbide MXene in water-in-salt electrolytes
Rituprava
Dash†
,
Suman
Yadav†
and
Narendra
Kurra
 *
*
Department of Chemistry, Indian Institute of Technology Hyderabad, Kandi, Sangareddy, Telangana State-502284, India. E-mail: narendra@chy.iith.ac.in
First published on 26th March 2026
Abstract
MXenes are known for intercalation-induced capacitive charge storage in dilute electrolytes, resulting in a low specific capacity with a limited anodic potential window of operation (<0.2 V vs Ag/AgCl). In this work, we report the anomalous pseudocapacitance of V2CTx MXene beyond 0.2 V vs Ag/AgCl by the deployment of mono- (Li+ and Na+) and divalent (Ca2+ and Zn2+) metal-ion-based concentrated electrolytes. Typically, a four-fold enhancement in the reversible sodiation capacity (131 mAh g−1) and a 0.9 V enlargement of the anodic potential window of V2CTx are observed compared to the dilute counterpart. Three-dimensional Bode analysis confirms the pseudocapacitive charge storage characteristics of V2CTx in concentrated electrolytes at anodic potentials. Tafel analysis further confirms the sluggish oxygen evolution and suppressed electrode irreversible anodic oxidation at the V2CTx electrode–concentrated electrolyte interface, as evidenced by an order of magnitude lower exchange current density (0.1 mA cm−2) relative to the dilute electrolyte interface (1.2 mA cm−2). Thus, suppressed water activity not only expands the anodic potential window of operation of V2CTx MXene but also enhances the charge storage capacity and electrochemical stability in concentrated electrolytes. This study opens new doors for exploring MXenes in concentrated electrolytes leading towards the design of high-performance MXene-based aqueous electrochemical energy storage devices.
Broader contextPseudocapacitive charge storage bridges the gap between non-Faradaic and bulk Faradaic processes by leveraging rapid intercalation- and surface-induced redox reactions. Transition metal oxides such as RuO2, MnO2, WO3, and Nb2O5 have demonstrated cation-induced pseudocapacitance in specific electrolytes. MXenes – an emerging and versatile class of two-dimensional transition metal carbides and nitrides – are known to exhibit proton-induced pseudocapacitance in protic electrolytes. However, MXenes have a tendency to undergo irreversible oxidation at anodic potentials, thus limiting their stable operational window to 0.2 V vs Ag/AgCl in aqueous electrolytes. Fundamentally, the reaction of water molecules with defective vanadium sites is the dominant mechanism driving this irreversible oxidation, preventing access to the V3+/V4+ redox chemistry. In the current work, the unusual pseudocapacitance of vanadium carbide MXene within this forbidden anodic potential window is demonstrated through the deployment of concentrated aqueous electrolytes. Water-in-salt imide electrolytes (Li-, Na-, Ca-, and Zn-based) suppress water activity, allowing for the activation of redox capacitance in vanadium carbide MXene. This study opens new doors for exploring MXenes in concentrated electrolytes toward the design of high-performance and safe aqueous electrochemical energy-storage devices. |
Introduction
The origin of pseudocapacitance in charge storage materials is based on diffusion-less redox reactions without phase transformations, in addition to providing long-term cycling stability.1 Most importantly, pseudocapacitive materials bridge the gap between electrical double-layer capacitors and metal-ion batteries with simultaneous achievement of high charge storage capacities and high rates.2 Traditionally, RuO2·0.5H2O is known for its surface pseudocapacitance in acidic electrolytes, where the insertion of protons, facilitated by structural water, leads to a continuous change in the oxidation state of Ru across the applied potential range.3,4 A similar kind of surface pseudocapacitance has been observed in a variety of transition metal oxides, including, but not limited to, MnO2 and WO3.5,6 Interestingly, intercalation pseudocapacitance has been reported for Nb2O5 in non-aqueous Li-ion electrolytes.7 A versatile class of pseudocapacitive materials includes two-dimensional (2D) functional transition metal carbides (MXenes), which are well known for their proton-induced pseudocapacitive response.8,9 Rapid ion intercalation in layered host structures may give rise to capacitive-like kinetics while exhibiting redox-type charge storage characteristics.10–12Owing to their rare combination of redox activity and metallic conductivity, MXenes are the candidate materials for high-rate pseudocapacitive energy storage. Specifically, top-down wet chemical etching of MAX phases results in the formation of MXenes along with the inherent introduction of polar functional groups such as ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, –OH, –F, and –Cl.13 However, MXenes exhibit limited charge storage capacities in neutral aqueous electrolytes due to the formation of electrical double layers by the hydrated cations across the interlayer spaces.14 Moreover, MXenes are prone to irreversible anodic oxidation beyond 0.2 V (vs Ag/AgCl), limiting the operational voltage window of symmetric MXene devices below 1 V.15,16 Despite the capability of vanadium to exhibit multiple oxidation states, the electrochemistry of vanadium carbide (V2CTx) MXene has been restricted to negative potential windows (−1.2 V to 0.2 V vs Ag/AgCl) so far. This is possibly due to poor chemical stability and limited anodic potential stability in salt-in-water electrolytes.17,18 Notably, Ti3C2Tx MXene exhibited desolvation-free Li-ion insertion in the case of a 19.8 M lithium chloride (LiCl) water-in-salt electrolyte (WiSE), achieving an anodic stability window of 0.8 V (vs Ag wire).19
O, –OH, –F, and –Cl.13 However, MXenes exhibit limited charge storage capacities in neutral aqueous electrolytes due to the formation of electrical double layers by the hydrated cations across the interlayer spaces.14 Moreover, MXenes are prone to irreversible anodic oxidation beyond 0.2 V (vs Ag/AgCl), limiting the operational voltage window of symmetric MXene devices below 1 V.15,16 Despite the capability of vanadium to exhibit multiple oxidation states, the electrochemistry of vanadium carbide (V2CTx) MXene has been restricted to negative potential windows (−1.2 V to 0.2 V vs Ag/AgCl) so far. This is possibly due to poor chemical stability and limited anodic potential stability in salt-in-water electrolytes.17,18 Notably, Ti3C2Tx MXene exhibited desolvation-free Li-ion insertion in the case of a 19.8 M lithium chloride (LiCl) water-in-salt electrolyte (WiSE), achieving an anodic stability window of 0.8 V (vs Ag wire).19
Water-in-salt (WiS) electrolytes are known to widen the electrochemical stable potential window of operation by suppressing water activity while increasing the overpotential for hydrogen and oxygen evolution reactions.20 For instance, Suo et al. demonstrated an operational potential window of 3.0 V by employing a WiSE with a concentration of 21 m lithium bis(trifluoromethanesulfonyl)imide (LiTFSI), where the typical salt-to-water molar ratio is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.6.21–26 Such a reduction in water activity was achieved through the formation of a dense solid electrolyte interphase (SEI) on the anode, composed of fluoride-based products, which expands the operational potential window to approximately 3.0 V.27,28 However, an expanded potential window of operation has also been observed for electrodes in WiSE without SEI formation.29,30 While Ti3C2Tx is known to exhibit relatively better electrochemical stability than V2CTx in aqueous media, the reaction of water with defective vanadium sites is the major cause of degradation. Thus, both the chemical instability and the restricted anodic potential window of operation have hindered the exploration of the electrochemical behavior of V2CTx MXene. By choosing WiSE, the reaction of water with defective vanadium sites can be minimized through ion–water interactions in concentrated electrolytes. Remarkably, the anodic potential window of operation of V2CTx in WiSE was extended by 0.6 to 0.8 V compared to that in dilute electrolytes.
:2.6.21–26 Such a reduction in water activity was achieved through the formation of a dense solid electrolyte interphase (SEI) on the anode, composed of fluoride-based products, which expands the operational potential window to approximately 3.0 V.27,28 However, an expanded potential window of operation has also been observed for electrodes in WiSE without SEI formation.29,30 While Ti3C2Tx is known to exhibit relatively better electrochemical stability than V2CTx in aqueous media, the reaction of water with defective vanadium sites is the major cause of degradation. Thus, both the chemical instability and the restricted anodic potential window of operation have hindered the exploration of the electrochemical behavior of V2CTx MXene. By choosing WiSE, the reaction of water with defective vanadium sites can be minimized through ion–water interactions in concentrated electrolytes. Remarkably, the anodic potential window of operation of V2CTx in WiSE was extended by 0.6 to 0.8 V compared to that in dilute electrolytes.
In this work, WiSEs of Li, Na, Zn, and Ca were employed to investigate the reversible redox chemistry of V2CTx MXene in the anodic potential window of operation. Unlike the pseudocapacitance of V2CTx in traditional electrolytes at cathodic potentials, concentration-induced pseudocapacitance was observed at anodic potentials beyond 0.2 V vs Ag/AgCl. Such forbidden redox chemistry of V2CTx MXene was unlocked through the employment of bis(trifluoromethanesulfonyl)imide (TFSI) anion-based concentrated electrolytes, irrespective of the nature of the cations (mono- or divalent). Furthermore, the electrochemical stability of V2CTx is enhanced due to the engagement of water molecules in coordination with a high ratio of salt ions, and a reduced amount of free water is available to react with the defective vanadium metal sites.
Experimental section/methods
Chemicals
The V2AlC (particle size < 32 µm) MAX precursor was procured from Y-Carbon, Ukraine Ltd. Hydrofluoric acid (HF, 40 wt%) and hydrochloric acid (HCl, 12 M) were acquired from AVRA and Fisher Chemicals respectively. All the salts were purchased from Tokyo Chemical Industry (TCI) Co. Ltd. All chemicals and salts were used as received without further purification.Synthesis of V2CTx MXene
V2CTx MXene was synthesized using a top-down wet chemical etching method from its MAX precursor, V2AlC, as reported in the literature.31 The synthesis process involves the selective etching of the ‘Al’ atomic layer from its MAX precursor through a mixed-acid etching method. The etchant solution, containing 12 mL of hydrofluoric acid (HF, 40 wt%) and 8 mL of hydrochloric acid (HCl, 12 M), was mixed in a volumetric ratio of 6:4 in a polypropylene bottle. During the synthesis, 1 g of V2AlC MAX phase was gradually added to 20 mL of the etchant solution with continuous stirring under an ice bath maintained at 0 °C to prevent local heat generation at the beginning of the reaction, which is exothermic in nature. By maintaining a low temperature, plausible dissolution or oxidation of the V2AlC MAX phase can be avoided. After bubble formation ceases during the initial stages of the etching reaction, the polypropylene bottle containing the MAX phase and the etchant solution was transferred to an oil bath maintained at a constant temperature of 50 °C. Now, the etching reaction is allowed to proceed for 80 h under continuous stirring at 400 rpm. Furthermore, the obtained acidic multilayered V2CTx was washed several times with deionized water via centrifugation at 3500 rpm for 5 minutes each time. Thus, the top acidic supernatant obtained was discarded each time, and washing was continued until the pH reached ∼6. The wet multilayered V2CTx MXene was filtered and dried under vacuum to obtain the multilayered V2CTx powder.
Processing of V2CTx MXene electrodes
The V2CTx electrodes were prepared by mixing the active material (V2CTx), carbon black as a conductive additive (Super P C45, Timical), and polyvinylidene fluoride (PVDF, Sigma Aldrich) as a binder in a weight ratio of 8:1:1. A homogeneous, thick slurry was prepared using a mortar and pestle with a minimal amount of N-methyl-2-pyrrolidone (NMP) solvent. The obtained slurry was cast onto a graphite foil current collector, followed by vacuum drying for 12 h at a temperature of 80 °C. The dried electrodes were calendared to have better contact between the active material and the current collector. The areal mass loading of the electrodes was typically 1.5–2 mg cm−2.
Preparation of activated carbon self-standing films
Activated carbon films were prepared by blending 95 wt% activated carbon (YP50F, Kuraray, Japan) with 5 wt% polytetrafluoroethylene (PTFE, 60 wt% in water, Sigma Aldrich) as a binder, using an appropriate amount of ethanol in a mortar and pestle. The obtained wet slurry was rolled into an activated carbon (AC) thin film (thickness of ∼100 µm). The AC thin films were dried at 70 °C for 12 h in a vacuum oven to remove trapped water and ethanol molecules in the porous structure.Material characterization
The X-ray diffraction (XRD) patterns of V2AlC MAX and V2CTx MXene were recorded using a powder X-ray diffractometer (PANalytical, Netherlands) equipped with a Cu Kα cathode (λ = 1.5406 Å) at a step size of 0.012° with a dwell time of 0.13 seconds. The morphology of the V2AlC MAX precursor and V2CTx MXene was imaged using field-emission scanning electron microscopy (FESEM) (JEOL JIB-4700F). The electronic absorption spectra of V2CTx dispersions were recorded using a JASCO model V-770 UV-Vis-NIR spectrophotometer in a screw-capped quartz cell (optical path length = 10 mm). Fourier transform infrared (FTIR) spectra were recorded using the JASCO model FTIR-4600 instrument. The attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectra of the fresh and cycled electrolytes were recorded using an Alpha instrument (Bruker) equipped with a diamond crystal. Ex situ X-ray diffraction (XRD) of the V2CTx electrode (both in its pristine state and after irreversible anodic oxidation) was performed to investigate the structural changes induced by anodic oxidation. Ex situ FESEM analysis of the cycled electrode was conducted to investigate the morphological changes of the electrode after 10000 cycles.
Electrochemical measurements
Electrochemical measurements, including cyclic voltammetry (CV), galvanostatic charge–discharge (GCD), electrochemical impedance spectroscopy (EIS), 3D Bode analysis, chronoamperometry, cycling stability tests and linear sweep voltammetry (LSV) for Tafel analysis, were performed at room temperature (25 °C) using an electrochemical workstation (Biologic VSP-3e, France). The electrochemical behavior of V2CTx MXene was investigated in various monovalent and divalent dilute and saturated electrolytes such as lithium chloride (LiCl), lithium bis(trifluorosulfonyl)imide (LiTFSI), sodium bis(trifluorosulfonyl)imide (NaTFSI), calcium bis(trifluorosulfonyl)imide [Ca(TFSI)2], and zinc bis(trifluorosulfonyl)imide [Zn(TFSI)2], using a perfluoroalkyl alkane (PFA) Swagelok cell. In dilute electrolyte solutions, molality and molarity differ only slightly because the weight of solvent (density of water is 0.99 g cm−3) and the volume of solution are approximately equal. However, the concentration of water-in-salt electrolytes are reported in molarity (M) for the ease of comparison with dilute electrolytes. The cell consists of V2CTx MXene as the working electrode, activated carbon as the counter electrode, and Ag/AgCl (3 M KCl) as the reference electrode. The Tafel slope and exchange current density values were estimated from linear sweep voltammetry (LSV) measurements conducted in a three-electrode Swagelok cell at a scan rate of 2 mV s−1. CV curves were recorded at various scan rates ranging from 1 to 50 mV s−1. Galvanostatic charge–discharge profiles were recorded at various current densities ranging from 0.2 to 10 A g−1. Chronoamperometry measurements were performed to monitor the leakage current at two extreme potentials.The differential capacity 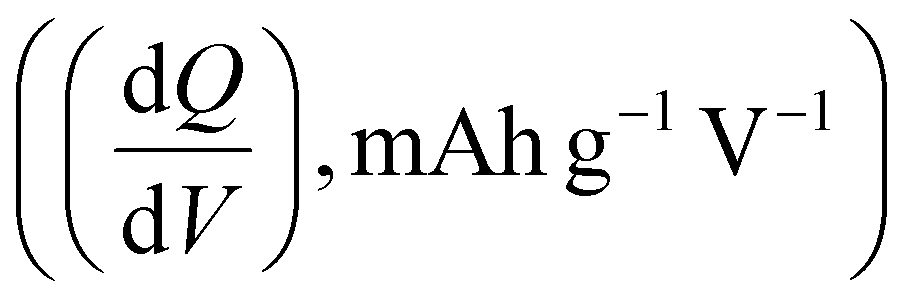 was calculated by integrating the discharge current from the CV curves, dividing the operational potential window (V), followed by normalizing the mass of the electrode and the corresponding scan rates (eqn (1)):
was calculated by integrating the discharge current from the CV curves, dividing the operational potential window (V), followed by normalizing the mass of the electrode and the corresponding scan rates (eqn (1)):
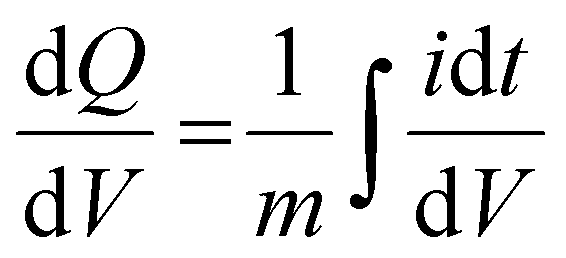 | (1) |
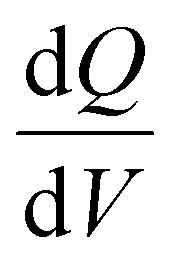 is the differential capacity, i is the current (mA), V is the operational potential window (V), and m is the active mass of the electrode (mg) (here, Q is the interfacial charge per unit mass).
is the differential capacity, i is the current (mA), V is the operational potential window (V), and m is the active mass of the electrode (mg) (here, Q is the interfacial charge per unit mass).
The specific capacity (mAh g−1) was calculated by integrating the current vs time plot:
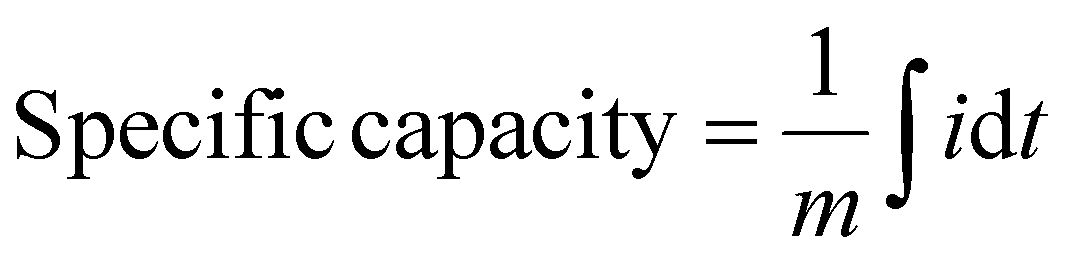 | (2) |
The contributions from diffusion-controlled and surface-controlled processes can be analysed using the power law relationship, as shown in eqn (3):
| i = aνb | (3) |
| logi = loga + blogν | (4) |
Staircase potentio electrochemical impedance spectroscopy (SPEIS) was performed over a frequency range from 100 kHz to 10 mHz by varying the applied DC potential using a step size of 50 mV. The V2CTx electrode was held at various DC potentials along with a sinusoidal voltage with an amplitude of 10 mV, and electrochemical impedance spectra were recorded at intervals of 50 mV DC applied potentials. The real part of the differential areal capacity 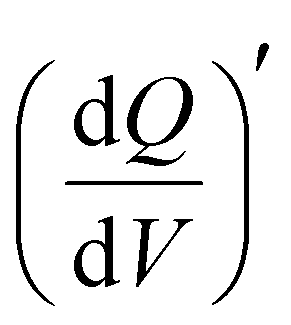 (mAs cm−2 V−1) and the imaginary differential capacity
(mAs cm−2 V−1) and the imaginary differential capacity 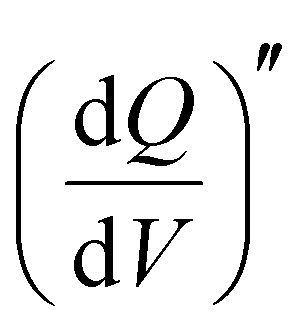 (mAs cm−2 V−1) were estimated within the corresponding frequency range using eqn (5) and (6), respectively (where Q is the interfacial charge per unit area):
(mAs cm−2 V−1) were estimated within the corresponding frequency range using eqn (5) and (6), respectively (where Q is the interfacial charge per unit area):
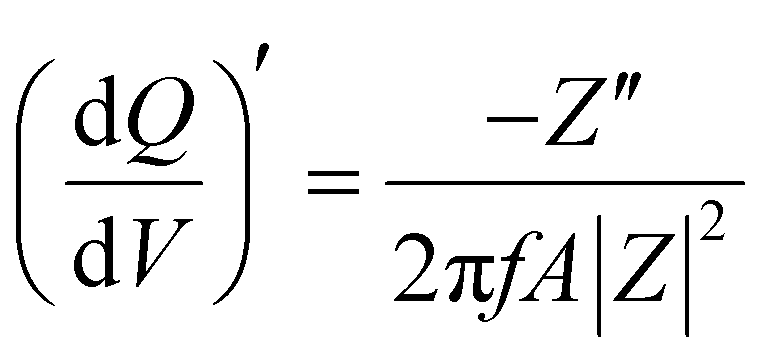 | (5) |
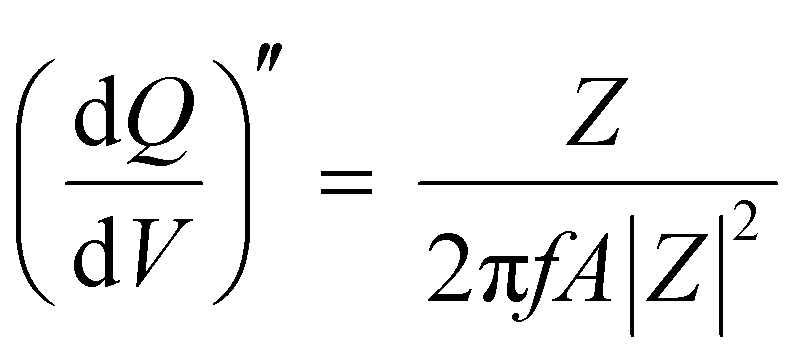 | (6) |
Results and discussion
Vanadium carbide (V2CTx) MXene was synthesized by employing a mixed-acid (HF/HCl) etching method, followed by electrode processing through the doctor-blade coating technique. Presumably, the by-products such as AlFx and VFx generated during the etching reaction may undergo dissolution in the presence of supporting acid (HCl), which are eventually washed away during centrifugation. The advantage of mixed acid over HF etching was demonstrated in the preparation of high-quality Ti3C2Tx MXene.32 The multilayer V2CTx MXene was dispersed in deionized water through bath sonication to record the ultraviolet-visible-near infrared (UV-Vis-NIR) spectrum (Fig. S1a, SI). The absorption peak in the low UV region at 283 nm is characteristic of V2CTx, which is in agreement with the reported literature.31 Furthermore, Fourier transform infrared (FTIR) spectroscopy was employed to investigate the confined water region (4000–1400 cm−1) and the fingerprint region (1400–450 cm−1) of the V2CTx MXene (Fig. S1b, SI). The intense band in the 3200–3600 cm−1 region corresponds to O–H stretching, and the peak at 1380 cm−1 is due to the surface-terminated O–H bending vibration. The peaks at 1685, 1624, and 989 cm−1 correspond to CO, C–O, and C–F stretching vibrations, respectively. Apparently, the attachment of functional groups to the core carbon layer is due to the presence of defective vanadium sites created during the etching reaction. The intense peak in the fingerprint region at 490 cm−1 is due to the V–O bending mode, which matches well with previously reported literature.33
The structural evolution of V2CTx MXene from its V2AlC MAX precursor was characterized by X-ray diffraction (XRD) (Fig. S1c, SI). V2AlC showed the (002) peak at 2θ ∼ 13.40°, which corresponds to a d-spacing of 6.6 Å and was downshifted to 7.40° with an enlarged d-spacing of 11.9 Å in V2CTx MXene. The enlargement of the d-spacing is attributed to the selective extraction of Al atomic layers, with the simultaneous incorporation of surface functional groups, such as O, –OH, –F, and –Cl, onto the V2C MXene during the etching process.31 The coating of V2CTx MXene was found to be uniform (a typical thickness of ∼25 μm) over a graphite foil current collector (Fig. S1d, SI).
It is well-known that V2CTx MXene exhibits pseudocapacitive charge storage in acidic and basic electrolytes.34 Typically, the potential window of operation of V2CTx was found to be in the range of −1.2 to 0.2 V (vs Ag/AgCl). Owing to the negative surface functionalities and negative operating potentials, the spontaneous intercalation of hydrated cations is trivial across the interlayer spaces of MXenes. In the case of neutral aqueous electrolytes, a usual capacitive type of electrochemical response is observed in V2CTx MXene due to the formation of electrical double layers (EDLs) by the hydrated ions in the interlayer spaces.14 To investigate the electrochemistry of V2CTx MXene in dilute and concentrated electrolytes, three-electrode electrochemical measurements were performed by employing various monovalent and divalent aqueous electrolytes. Fig. 1a depicts the atomistic view of a typical local environment in the interlayer spaces of V2CTx MXene at dilute and concentrated regimes of an electrolyte. In the case of dilute electrolytes, there are strong ion–dipole interactions between ions and surrounding solvent molecules, which hinder the direct electrostatic attraction between oppositely charged ions. Therefore, hydrated ions diffuse into the interlayer spaces of V2CTx MXene due to partial screening of the charge on the ions by the hydration shell. In contrast, concentrated electrolytes tend to form contact ion pairs and aggregated structures due to the lower weight-to-volume ratio of water molecules in comparison with the salt ions. Thus, fewer water molecules are associated with the ions, resulting in suppressed water activity in the case of concentrated electrolytes.35
 | ||
| Fig. 1 (a) Atomistic view of a typical ion–water environment across the V2CTx MXene interlayer space when employing dilute and concentrated aqueous electrolytes. (b) Cyclic voltammograms (CVs) of V2CTx MXene in 1 M LiCl aqueous electrolyte and an abrupt rise in the current beyond 0.1 V (vs Ag/AgCl), subjected to anodic oxidation (AO) at a scan rate of 2 mV s−1. (c) CVs of V2CTx MXene in 1 M LiCl electrolyte before and after anodic oxidation at a scan rate of 2 mV s−1. (d) CVs of V2CTx MXene in 19.8 M LiCl electrolyte with anodic potential cut-offs up to 0.3 and 0.6 V (vs Ag/AgCl) at a scan rate of 2 mV s−1. (e) CVs of V2CTx MXene in 19.8 M LiCl before and after anodic oxidation at a scan rate of 2 mV s−1. | ||
To examine the electrochemical stability window (ESW) of the electrolytes, linear sweep voltammetry (LSV) was performed (Fig. S2, SI). To investigate the effect of concentration, the V2CTx MXene electrode was tested in 1 M lithium chloride (LiCl) (dilute) and 19.8 M LiCl WiSE. As shown in Fig. 1b, the V2CTx MXene exhibited a rectangular-type cyclic voltammogram (at a scan rate of 2 mV s−1) in 1 M LiCl electrolyte, signifying a capacitive response within the potential window of −0.4 to 0.15 V (vs Ag/AgCl). The narrow potential window is possibly due to irreversible anodic oxidation (IAOR) and irreversible cathodic reduction reactions (ICRRs) (Fig. S3, SI). When the V2CTx electrode was polarized to an anodic potential of 0.3 V (vs Ag/AgCl), an abrupt rise in the current was observed. A subsequent CV scan in the stable potential window (−0.4 to 0.15 V vs Ag/AgCl) was observed with a diminished current response. Such an electrochemical response is possibly due to irreversible anodic oxidation of V2CTx, which is responsible for diminishing the overall charge storage capacity (Fig. 1c). In the case of 19.8 M LiCl WiS electrolyte, the anodic potential limit of the V2CTx MXene electrode is 0.3 V (vs Ag/AgCl), increased by 150 mV in comparison with 1 M LiCl (Fig. 1d). Interestingly, the V2CTx MXene exhibited a pair of redox peaks at potentials ∼−0.35/−0.45 V (vs Ag/AgCl) in 19.8 M LiCl. Such an appearance of redox peaks at negative potentials could be due to V2+/V3+ redox chemistry (Fig. S4, SI).36 When the electrode was subjected to polarization at a potential of 0.6 V (vs Ag/AgCl), irreversible anodic oxidation was observed with a reduction in the electrochemical performance of the V2CTx MXene electrode (Fig. 1e). Such irreversible anodic oxidation was also observed in the case of Ti3C2Tx electrodes in aqueous electrolytes.37
To examine the effect of the anion on the electrochemistry of V2CTx MXene, the electrode was tested in 1 and 21 M LiTFSI electrolytes. Unlike the spherical nature of the chloride anion, TFSI is asymmetric in nature with a delocalized charge distribution. Such imide-based anions are known to exhibit weak coordination with the counter cations with inherent hydrophobicity influencing the water association significantly.38 To analyze the ion–solvent interaction, hydrogen bonding, and solvation structure, attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectra were recorded for 1 M and 21 M LiTFSI electrolytes. At high salt concentrations, it was observed that the absorption intensity of the O–H stretching vibration reduced while TFSI− anion absorption intensity increased, signifying the unavailability of free water molecules. Such a scenario leads to suppressed water activity in the WiSE (Fig. S5a, SI). Fig. 2a shows the comparative CVs of V2CTx MXene electrodes in dilute (1 M) and WiS (21 M) LiTFSI electrolytes at a scan rate of 1 mV s−1. The V2CTx electrode exhibited an operational potential window of −0.5 to 0.1 V (vs Ag/AgCl) in 1 M LiTFSI electrolyte with an electrical double-layer response due to the intercalation of hydrated Li+ ions across interlayer spaces (Fig. S5b, SI). Unusually, the V2CTx electrode exhibited a wide operational potential window from −0.8 to 0.6 V (vs Ag/AgCl) with two pairs of reversible redox peaks at potentials of −0.37/−0.43 and −0.05/0.04 V (vs Ag/AgCl) in 21 M LiTFSI electrolyte, which could be possibly due to the reversible redox chemistry from the V2+/V3+ and V3+/V4+ redox couples, respectively (SI).39,40 The anodic potential stability of V2CTx was extended by 0.5 V (vs Ag/AgCl), accompanied by the emergence of broad redox signatures (Fig. 2a and S5d, SI). Apparently, in the case of WiSE, the asymmetric and hydrophobic nature of the TFSI− anion forms an extended ion network that enables fast cation transport, whereas chloride ions remain hydrated even at high saturation.41
 | ||
Fig. 2 Electrochemical performance of V2CTx MXene in 1 and 21 M LiTFSI electrolytes. (a) Comparative CVs and (b) galvanostatic charge–discharge (GCD) profiles of V2CTx MXene in 1 and 21 M LiTFSI electrolytes. (c) Chronoamperometry profiles at different anodic and cathodic potentials in 1 M and 21 M LiTFSI electrolytes. (d) Kinetic parameter, b-value, obtained from the slope of log (current) vs log (scan rate) plots at anodic potentials of −0.02 V and 0.03 V of 1 M and 21 M LiTFSI electrolytes, respectively. 3D Bode maps of the 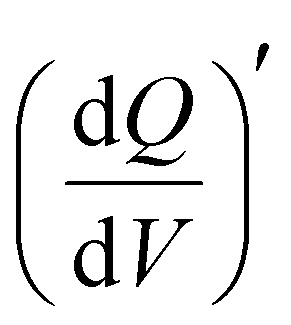 as a function of frequency (Hz) and applied potential V (vs Ag/AgCl) for V2CTx MXene in (e) 1 M and (f) 21 M LiTFSI electrolytes. as a function of frequency (Hz) and applied potential V (vs Ag/AgCl) for V2CTx MXene in (e) 1 M and (f) 21 M LiTFSI electrolytes. | ||
Fig. 2b shows the comparative galvanostatic charge–discharge (GCD) profiles of V2CTx MXene in 1 M and 21 M LiTFSI electrolytes at a current density of 0.2 A g−1. V2CTx showed a specific capacity of only 15 mAh g−1 at a current density of 0.2 A g−1 in 1 M LiTFSI (Fig. S5c, SI). In comparison, the V2CTx MXene exhibited a specific capacity of 90 mAh g−1 in 21 M LiTFSI electrolyte at a current density of 0.2 A g−1 (Fig. S5e SI). Notably, the potential window of operation of V2CTx almost doubled when changing the electrolyte concentration from 1 M (ΔV ∼ 0.6 V vs Ag/AgCl) to 21 M LiTFSI (ΔV ∼ 1.4 V vs Ag/AgCl) in addition to a four-fold enhancement in the specific capacity. To confirm the stable cut-off potentials, chronoamperometry measurements were performed. The chronoamperograms (CAs) showed a steady-state leakage current of <0.1 A g−1 in the extreme potential ranges of −0.5 V to 0.1 V (vs Ag/AgCl) in 1 M and −0.8 to 0.6 V (vs Ag/AgCl) in 21 M LiTFSI electrolytes (Fig. 2c). Thus, the extended potential window of V2CTx in 21 M LiTFSI electrolyte is attributed to the reversible electrochemical phenomenon without parasitic reactions associated with the electrolyte.
To understand the charge transfer kinetics of the V2CTx electrode in both dilute (1 M) and WiS (21 M) LiTFSI electrolytes, electrochemical impedance spectroscopy was performed from the high (100 kHz) to the low (10 mHz) frequency region under open circuit potential (OCP) conditions (Fig. S5f, SI). In the high-frequency regime, the equivalent series resistance (ESR) corresponding to the x-axis intercept was found to be 6.6 and 2.4 Ω for 1 and 21 M LiTFSI electrolytes, respectively. The lower ESR value in the highly concentrated Li+ electrolyte is possibly due to facile ion diffusion within the electrodes and reduced electrostatic interactions between the oppositely charged species.42 In the case of 21 M LiTFSI electrolyte, a 45° line is prevalent in the low-frequency region, unlike that in the case of 1 M electrolyte with a predominant capacitive response.
Furthermore, to understand the electrochemical kinetics of the V2CTx MXene electrode in Li-ion electrolytes, the kinetic parameter (b-value) was calculated from the slope of the logarithm of current (logi) vs the logarithm of scan rate (logν) using eqn (3) and (4) (see the Experimental section). A slope (b-value) of 0.5 corresponds to diffusion-controlled processes, whereas a b-value of 1 corresponds to surface-controlled processes.43 The b-values estimated at potentials of −0.02 V and −0.03 V (vs Ag/AgCl) were found to be 0.9 and 0.8 for 1 and 21 M LiTFSI, respectively (Fig. 2d). Thus, in the case of 1 M LiTFSI electrolyte, V2CTx exhibited predominant surface-controlled processes due to the formation of an EDL by hydrated Li+ ions at the electrode/electrolyte interface. However, in the case of 21 M LiTFSI electrolyte, the relative increase in diffusion-controlled current is attributed to the concentration factors.
Furthermore, 3D Bode maps were constructed to better visualize the charge storage dynamics across V2CTx MXene in both dilute (1 M) and WiS (21 M) LiTFSI electrolytes. The 3D Bode maps were generated by plotting the 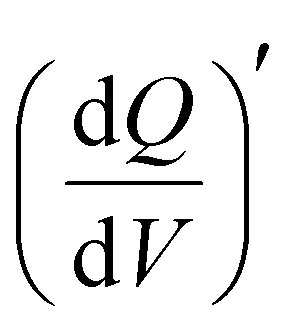 in the low-frequency regime (10 Hz to 10 mHz) along with a third independent variable, i.e., the applied DC potential.34 V2CTx MXene exhibits capacitive-type behaviour in dilute 1 M LiTFSI electrolyte, where the electrode showed a rectangular 3D waterfall shape throughout the potential window of −0.5 to 0.1 V (vs Ag/AgCl) (Fig. 2e). Possibly, the hydration shell surrounding lithium ions prevents orbital coupling between bare Li+ ions and V2CTx surface atoms, resulting in capacitive behaviour at the V2CTx – 1 M LiTFSI electrochemical interface.44 Similarly,
in the low-frequency regime (10 Hz to 10 mHz) along with a third independent variable, i.e., the applied DC potential.34 V2CTx MXene exhibits capacitive-type behaviour in dilute 1 M LiTFSI electrolyte, where the electrode showed a rectangular 3D waterfall shape throughout the potential window of −0.5 to 0.1 V (vs Ag/AgCl) (Fig. 2e). Possibly, the hydration shell surrounding lithium ions prevents orbital coupling between bare Li+ ions and V2CTx surface atoms, resulting in capacitive behaviour at the V2CTx – 1 M LiTFSI electrochemical interface.44 Similarly, 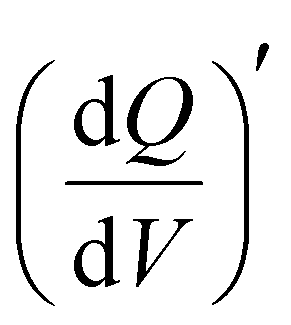 of the V2CTx MXene in (21 M LiTFSI) WiSE at 0.05 V (vs Ag/AgCl) was calculated using eqn (5), and the maximum
of the V2CTx MXene in (21 M LiTFSI) WiSE at 0.05 V (vs Ag/AgCl) was calculated using eqn (5), and the maximum  normalized with the area of the electrode was found to be 0.2 mAs cm−2 V−1 in the low frequency (10 mHz) regime which is 2.5 times higher than that of the dilute electrolyte (0.08 mAs cm−2 V−1). In the case of 21 M WiSE, the 3D Bode map showed a mountain-shaped hump, indicating redox-type charge storage in V2CTx MXene, which corroborates well with the voltammetric analysis (Fig. 2f).
normalized with the area of the electrode was found to be 0.2 mAs cm−2 V−1 in the low frequency (10 mHz) regime which is 2.5 times higher than that of the dilute electrolyte (0.08 mAs cm−2 V−1). In the case of 21 M WiSE, the 3D Bode map showed a mountain-shaped hump, indicating redox-type charge storage in V2CTx MXene, which corroborates well with the voltammetric analysis (Fig. 2f).
To examine the effect of the cations, V2CTx MXene electrodes were tested in imide-based concentrated electrolytes, including 9 M NaTFSI, 5 M Ca(TFSI)2, and 3 M Zn(TFSI)2. The V2CTx electrode showed a reversible and stable extended anodic potential window in all concentrated imide-based electrolytes. Due to the maximum enlarged potential window and high charge storage capacity shown by V2CTx MXene in 9 M NaTFSI WiSE, detailed investigations were further carried out (Fig. 3a and S6, SI). V2CTx MXene showed an enlarged anodic potential window of operation (−0.6 to 0.7 V vs Ag/AgCl) when the concentration of Ca(TFSI)2 electrolyte changed from 1 to 5 M.45 Similarly, the V2CTx MXene showed a stable anodic potential window of −0.7 to 0.6 V (vs Ag/AgCl) in 3 M Zn(TFSI)2 (Fig. S7a and c, SI). Therefore, concentration plays a crucial role in governing the stable operational potential window at both anodic and cathodic potentials. When moving from monovalent-based (Li+, Na+) to divalent-based (Zn2+, Ca2+) electrolytes, the local coordination environment around vanadium sites in V2CTx MXene may be altered. Even though the V3+/V4+ redox couple is the major contributor to the observed redox chemistry, differences in charge density of ions could lead to observable shifts in the potential values. Based on galvanostatic charge–discharge profiles, it was shown that V2CTx exhibited the highest specific capacity of 131 mAh g−1 in 9 M NaTFSI WiSE at a current density of 0.2 A g−1. The electrochemical performance of V2CTx MXene in 9 M NaTFSI electrolyte was compared with that in the reported literature for mono- and divalent metal-ion WiSE (Table S2, SI).
 | ||
Fig. 3 Electrochemistry of V2CTx MXene in concentrated metal-ion electrolytes. (a) Comparative CVs of V2CTx MXene in monovalent (9 M NaTFSI) and divalent [5 M Ca(TFSI)2 and 3 M Zn(TFSI)2] electrolytes at a scan rate of 1 mV s−1. (b) Galvanostatic charge–discharge (GCD) profiles of V2CTx MXene in 9 M NaTFSI, 5 M Ca(TFSI)2 and 3 M Zn(TFSI)2 electrolytes at a current density of 0.2 A g−1. (c) Rate performances of V2CTx MXene in 9 M NaTFSI, 5 M Ca(TFSI)2 and 3 M Zn(TFSI)2 electrolytes at different current densities from 0.2 to 19 A g−1. 3D Bode maps portraying  frequency (Hz), and potential (V vs Ag/AgCl) for V2CTx MXene in different concentrated electrolytes, such as (d) 9 M NaTFSI, (e) 5 M Ca(TFSI)2, and (f) 3 M Zn(TFSI)2. (g) Long-term cycling performance of V2CTx MXene in 9 M NaTFSI electrolyte for 10000 cycles at a scan rate of 20 mV s−1 with 100% coulombic efficiency; the inset shows the CVs for cycle numbers 1 and 10000. frequency (Hz), and potential (V vs Ag/AgCl) for V2CTx MXene in different concentrated electrolytes, such as (d) 9 M NaTFSI, (e) 5 M Ca(TFSI)2, and (f) 3 M Zn(TFSI)2. (g) Long-term cycling performance of V2CTx MXene in 9 M NaTFSI electrolyte for 10000 cycles at a scan rate of 20 mV s−1 with 100% coulombic efficiency; the inset shows the CVs for cycle numbers 1 and 10000. | ||
In comparison, V2CTx MXene showed specific capacity values of 70 and 90 mAh g−1 at a current density of 0.2 A g−1 in 5 M Ca(TFSI)2 and 3 M Zn(TFSI)2 electrolytes, respectively (Fig. 3b, S7b and S7d, SI). The rate performance of V2CTx MXene in all concentrated electrolytes showed a high degree of rate retention with 100% coulombic efficiency (Fig. 3c). Furthermore, to understand the charge transfer kinetics between the V2CTx MXene and various concentrated electrolytes, electrochemical impedance spectroscopy was recorded from the high-frequency region (100 kHz) to the low-frequency (10 mHz) region under open-circuit potential (OCP) conditions. In the high-frequency region, the equivalent series resistance (ESR), corresponding to the x-axis intercept, was found to be 4.4, 7.8, and 8.5 Ω for 9 M NaTFSI, 5 M Ca(TFSI)2, and 3 M Zn(TFSI)2, respectively. This indicates rapid Na+ ion diffusion into the 2D galleries of V2CTx MXene (Fig. S8, SI).
Furthermore, 3D Bode maps were constructed by plotting 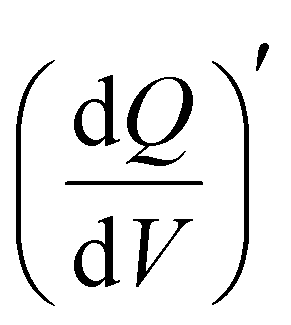 (mAs cm−2 V−1), which was represented against the low-frequency range (10 Hz to 10 mHz) along the third independent variable, i.e., applied DC potential (V). Ko et al. employed 3D Bode analysis to distinguish between double-layer, pseudocapacitive, and battery-like mechanisms in activated carbon, Nb2O5, and LiFePO4 in a non-aqueous (1 M LiPF6 in 1:1 EC:DEC) electrolyte.46 In this study, we have used this analysis to evaluate the redox charge storage dynamics of V2CTx MXene in various concentrated TFSI-based electrolytes (Fig. 3d–f and Fig. S9, SI). In all three electrolytes, the electrode showed a mountain-shaped hump in the corresponding potential region of >0 V vs Ag/AgCl, confirming the pseudocapacitive characteristics of V2CTx MXene, which is well corroborated by their respective cyclic voltammograms (CVs).
(mAs cm−2 V−1), which was represented against the low-frequency range (10 Hz to 10 mHz) along the third independent variable, i.e., applied DC potential (V). Ko et al. employed 3D Bode analysis to distinguish between double-layer, pseudocapacitive, and battery-like mechanisms in activated carbon, Nb2O5, and LiFePO4 in a non-aqueous (1 M LiPF6 in 1:1 EC:DEC) electrolyte.46 In this study, we have used this analysis to evaluate the redox charge storage dynamics of V2CTx MXene in various concentrated TFSI-based electrolytes (Fig. 3d–f and Fig. S9, SI). In all three electrolytes, the electrode showed a mountain-shaped hump in the corresponding potential region of >0 V vs Ag/AgCl, confirming the pseudocapacitive characteristics of V2CTx MXene, which is well corroborated by their respective cyclic voltammograms (CVs).
Furthermore, to investigate the intrinsic redox behaviour of V2CTx MXene in the extended potential window, the electrode was swept towards a high anodic potential up to 1.5 V (vs Ag/AgCl) at a scan rate of 1 mV s−1 for ∼10 cycles in 9 M NaTFSI WiSE (Fig. S10, SI). The onset potential for irreversible anodic oxidation was observed above 0.9 V (vs Ag/AgCl). A subsequent CV was recorded in the stable potential window exhibiting a diminished capacity value for V2CTx, possibly due to the oxidation of electrochemically redox-active vanadium sites. To examine long-term electrochemical cycling stability, the V2CTx electrode was swept at a scan rate of 20 mV s−1 for 10000 cycles. The V2CTx electrode showed a capacity retention of 64% after 10000 cycles with 100% coulombic efficiency in 9 M NaTFSI WiSE (Fig. 3g). The subsequent decrease in the capacity of V2CTx MXene over the cycling range is possibly due to reactions of co-intercalated water molecules along with the metal cations. The inserted water molecules undergo parasitic reactions at extreme potentials, which degrade the 2D sheets of MXene. Additionally, the inserted water molecules occupy the V-deficient sites and facilitate the anodic oxidation of the MXene framework and convert redox-active sites into inactive ones.
Moreover, the electrodes at varying mass loadings were tested in 9 M NaTFSI WiSE to understand how the increase in mass loading influences the redox characteristics and specific capacity of V2CTx MXene (Fig. S11, SI). Furthermore, ex situ SEM images were recorded for the cycled V2CTx electrode and it was found that the morphology of V2CTx is maintained, but glass microfibers from the separator were deposited over the surface of the electrode (Fig. S12, SI).
Water activity plays a critical role in influencing the electrochemical stability window of aqueous electrolytes based on the interplay between intermolecular association and ion–water interactions. Thermodynamically, the potential difference between the oxygen and hydrogen evolution reaction is 1.23 V, irrespective of the pH of the aqueous medium.47 However, kinetic factors play an important role in affecting the reaction rates which significantly enlarge the thermodynamic stability window of water beyond 1.23 V. In the context of V2CTx–aqueous electrolyte interfaces, the kinetics of irreversible anodic oxidation of V2CTx are dominant over the oxygen evolution reaction at anodic potentials. At high salt concentrations, the ESW of the electrolyte can become enlarged by 25 mV based on thermodynamic factors. Thus, kinetic factors play a significant role in expanding the potential window by 0.9 V in WiSE. Such extended potential stability is either due to the formation of a solid electrolyte interphase (SEI) or reduced water activity. However, when switching from concentrated to dilute media, an expansion of the potential window was observed, but the capacity fading of V2CTx MXene is not rapid, even after operation in the anodic potential window. This is possibly due to surface coverage of salt on V2CTx MXene in highly concentrated media, rather than the permanent formation of a stable SEI layer (Fig. S13, SI).
To further understand the electrochemical kinetics of the irreversible anodic oxidation reaction at the electrode–electrolyte interface, LSV and Tafel analyses were performed in both dilute and concentrated electrolytes. The stability window was determined by recording the linear sweep voltammograms over a potential range of −1.4 V to 0.2 V (vs Ag/AgCl) in 1 M NaTFSI and −1.4 V to 1.1 V (vs Ag/AgCl) in 9 M NaTFSI electrolyte, as shown in Fig. 4a. The potential window operation of V2CTx MXene in 9 M NaTFSI electrolyte extends to 2.5 V (vs Ag/AgCl), a characteristic of WiSE.21 At overpotentials, η > 120 mV, the contribution from the cathodic reaction current becomes insignificant, and thus, the reaction kinetics is solely governed by the rate of anodic processes.
 | ||
| Fig. 4 (a) Linear sweep voltammograms (LSVs) of the V2CTx electrode in 1 M and 9 M NaTFSI aqueous electrolytes at a scan rate of 2 mV s−1. (b) Significant anodic potential window enlargement by 0.9 V in 9 M NaTFSI electrolyte. Tafel analysis of the V2CTx electrode in (c) dilute [1 M NaTFSI, 1 M LiTFSI, 1 M Zn(TFSI)2 and 1 M Ca(TFSI)2] electrolytes and (d) concentrated [9 M NaTFSI, 21 M LiTFSI, 3 M Zn(TFSI)2 and 5 M Ca(TFSI)2] electrolytes performed at a current density of ∼10 mA cm−2. | ||
The Tafel equation describes the irreversible nature of the reaction, and two key parameters such as exchange current density and Tafel slope can be determined from the following equation:48
| η = a + blogi | (7) |
The Tafel constant (a = (RT/αnF)lnio, where R is the universal gas constant, T is the absolute temperature, α is the transfer coefficient, n is the number of electrons transferred, and F is the Faraday constant = 96485 C mol−1), used to extract the exchange current (io), which is a characteristic parameter that describes the rate of an electrochemical process at equilibrium. It depends on the nature of the electrolyte, electrode–electrolyte interface, and temperature. The Tafel slope (b = RT/αnF) signifies the sensitivity of the reaction rate to overpotential and determines the overpotential necessary for increasing the current (and hence the reaction rate) by an order of magnitude. At high salt concentrations, enhanced anodic stability arises from prominent interionic interactions relative to solvent–ion interactions, imparting distinct physicochemical properties.49 The irreversible anodic oxidation reaction (IAOR) exhibits a substantial increase in overpotential when transitioning from dilute to concentrated electrolytes (Fig. 4b and S14, SI). The difference in the onset potential between 9 and 1 M NaTFSI is 0.9 V at a current density of 1 mA cm−2, while the reduction reaction remains unaffected. The corresponding Tafel plots exhibit a Tafel slope value of 47 mV dec−1 at an exchange current density (j0) of 1.2 mA cm−2 for 1 M and 89 mV dec−1 at an exchange current density j0 of 0.1 mA cm−2 for 9 M NaTFSI electrolyte, respectively (Fig. 4c and d). The j0 value decreased by 12-fold with an increase in the salt concentration from 1 to 9 M NaTFSI. Thus, the kinetics of irreversible oxidation of V2CTx MXene is suppressed significantly at high salt concentrations. Recently, Vazquez et al. observed a similar kind of decrease in the exchange current density (j0) value in the case of a non-SEI-forming 17 m NaClO4 electrolyte.50 The mechanism of sluggish water transport within the double-layer structure limits hydrogen evolution kinetics, thus extending the potential window of operation.
Similarly, in other concentrated [21 M LiTFSI, 3 M Zn(TFSI)2, and 5 M Ca(TFSI)2] electrolytes, their corresponding Tafel slopes (61, 40, and 35 mV dec−1) are larger than their counterparts of dilute electrolytes [1 M LiTFSI, 1 M Zn(TFSI)2, and 1 M Ca(TFSI)2], which signifies sluggish irreversible anodic oxidation (IAOR) in concentrated electrolytes. A lower Tafel slope and a higher exchange current density (j0) result in faster reaction kinetics.20 On the other hand, a higher Tafel slope value with a small j0 leads to sluggish reaction kinetics at higher concentrations. Therefore, a larger overpotential is required to achieve the current values associated with the irreversible anodic oxidation process. Among all four concentrated electrolytes (Li+, Na+, Zn2+, and Ca2+), Ca2+ showed the lowest j0 value of 4 × 10−4 mA cm−2 due to its high enthalpy of hydration and high charge density, whereas Na+ showed a comparatively higher j0 (0.1 mA cm−2) due to its larger cation size and lower enthalpy of hydration (Fig. S15, SI).51,52 These results confirmed that, highly concentrated electrolytes effectively inhibit parasitic anodic oxidation reactions of V2CTx, thereby enabling an expanded anodic potential window of operation and, consequently, improving electrochemical performance. The comparative electrochemical kinetics of dilute as well as concentrated regimes with their corresponding exchange current densities are tabulated in Table S3 (SI).
MXene-based symmetric devices operate below 1 V due to irreversible anodic oxidation of the positive MXene electrode.15 Given the extended anodic stability of V2CTx in WiSE, further demonstration of a voltage window of operation of a symmetric V2CTx//V2CTx device provides evidence for the practical high voltage window of operation beyond 1.23 V. The V2CTx//V2CTx symmetric device was fabricated by using V2CTx MXene as both positive and negative electrodes in 1 and 9 M NaTFSI electrolytes (Fig. 5a). The symmetric device was operable in a voltage window of 0.01 to 0.6 V in the case of 1 M dilute NaTFSI electrolyte, which was also evident from the three-electrode configurations, where the potential window of operation is −0.5 to 0.1 V (vs Ag/AgCl) as shown in Fig. 5b. Similarly, the V2CTx electrode showed a stable wide potential window of −0.9 to 0.9 V (vs Ag/AgCl) in 9 M NaTFSI WiSE. Therefore, it was expected to have a voltage window exceeding 1 V for the symmetric device. A V2CTx//V2CTx symmetric cell showed a stable voltage window of 1.4 V at a scan rate of 5 mV s−1 (Fig. 5c). As the thermodynamic water decomposition potential is 1.23 V, the extended voltage window was attributed to the kinetic factors contributing to the observed overpotentials of the irreversible anodic oxidation reaction (IAOR).
 | ||
| Fig. 5 Electrochemical performance of the V2CTx MXene-based symmetric device in 1 and 9 M NaTFSI electrolytes. (a) Schematic representation of the V2CTx//V2CTx symmetric device in 1 M (dilute) and 9 M NaTFSI (WiSE). Comparative cyclic voltammograms of the V2CTx//V2CTx symmetric device in (b) 1 M NaTFSI and (c) 9 M NaTFSI electrolytes. (d) Cycling stability of the V2CTx//V2CTx symmetric device in 9 M WiSE over 10000 cycles at a scan rate of 20 mV s−1. (e) Attenuated total reflectance Fourier transform infrared (ATR–FTIR) spectra of pristine and cycled V2CTx electrodes over 10000 cycles in 9 M NaTFSI electrolyte. | ||
Furthermore, to investigate the long-term cycling stability of the symmetric device in 9 M WiSE, a cyclic voltammogram was recorded at a scan rate of 20 mV s−1 over 10000 cycles. During cycling, the capacity increased until 2000 cycles, then decreased until 5500 cycles, and finally stabilized after 6000 cycles. This could be due to the inaccessibility of active sites in V2CTx MXene in the initial cycles, resulting from the restacking of MXene layers. During continuous charge–discharge cycling, the electrode achieves a redox-active accessible surface area due to the intercalation of ions into MXene interlayer spaces. After the interlayer activation of the MXene surface area, the device showed a capacity retention of 62% with 99% coulombic efficiency throughout 10000 cycles (Fig. 5d). Wei et al. observed a similar trend in CoSe2/ZnSe composites nano-confined in Ti3C2Tx MXene 2D sheets (CoSe2/ZnSe@MX), where the capacity was increased up to 300 charge/discharge cycles at 1 A g−1. In contrast, the electrodes without MXene confinement exhibited inferior long-term cycling stability, improved ion accessibility of MXene over continuous charge/discharge cycles.53 Recent MXene-based studies have improved cycling performance mainly through binder-free three-dimensional thick electrode designs (71.5% retention over 1000 cycles), interfacial hydrogen-bond engineering of Ti3C2Tx MXene with carbon felt (81.8% after 5000 cycles), or artificial redox activation via low-valence Zn injection into Ti3C2Tx. Unlike these structurally or chemically engineered systems, the present V2CTx//V2CTx symmetric device achieves long-term stability based on its intrinsic material properties.54–56
To examine the modifications of the electrolyte after cycling over 10000 cycles, attenuated total reflectance Fourier transform infrared (ATR–FTIR) spectra were recorded before and after the cycling test. The characteristic O–H stretching and fingerprint regions remain unaffected, which clearly indicates that there is hardly any modification in the WiS (9 M) NaTFSI electrolyte (Fig. S16, SI). Additionally, ATR-FTIR spectra were recorded to examine any modification in the electrode after 10000 cycles. The electrode was washed and dried properly after the long-term cycling stability test. The spectra were recorded to examine the confined water region (4000–1400 cm−1) and the fingerprint region (1400–450 cm−1) for pristine and cycled electrodes. As this technique is sensitive to surface functional groups, there is a slight shift in the confined water region of the cycled electrode compared to the pristine electrode. Similarly, surface functional group bonds such as C–O, O–H, and C–F in the fingerprint regions shifted from 1554, 1393, and 1007 cm−1 to 1550, 1382, and 1003 cm−1, indicating negligible changes in bond strength. The relative intensity of absorption peaks in the cycled electrode was possibly reduced due to the deposition of glass microfibers from the separator.57
There are a few key aspects of the current work that are worth mentioning. MXenes are known to exhibit proton-induced pseudocapacitive behavior but suffer from irreversible anodic oxidation at anodic potentials. Unlike the previously reported literature, the current study demonstrated the hidden pseudocapacitance of vanadium carbide MXene in an otherwise forbidden anodic potential window. Thanks to suppressed water activity at high salt concentrations, not only expanded the anodic potential window, but also the redox chemistry of the V3+/V4+ couple was unlocked. Such an observation is demonstrated in various monovalent and divalent metal-ion-based concentrated electrolytes. This study opens new avenues for the exploration of MXene-concentrated electrolytes to unravel fundamental charge storage processes for enabling the design of safe and high-performance aqueous energy storage devices.
Conclusions
In summary, the anomalous pseudocapacitance of V2CTx MXene was unlocked through the deployment of water-in-salt (WiS) electrolytes. Traditionally, V2CTx MXene exhibits limited charge storage capacity and narrow anodic potential window of operation, besides its poor electrochemical stability in dilute aqueous electrolytes. Thanks to suppressed water activity at high concentrations, which not only unlocked the forbidden V3+/V4+ redox chemistry, but also enlarged the anodic potential window of V2CTx MXene up to a maximum of 0.9 V in both monovalent-based and divalent-based concentrated electrolytes. 3D Bode analysis further confirmed the distinct charge storage dynamics at V2CTx MXene-concentrated electrolyte interfaces, unlike the traditional double-layer characteristics of their dilute electrolyte counterparts. The key electrochemical kinetic parameter, the exchange current density, was found to be at least an order of magnitude lower for the concentrated electrolyte interfaces in comparison with dilute electrolyte interfaces, which suppressed the rate of irreversible oxidation of V2CTx MXene through strong ion–water interactions. This study opens new avenues for the exploration of MXene-concentrated aqueous electrolytes for the design of high-voltage, safe, and environmentally benign energy storage devices.Author contributions
The manuscript was written with contributions from all three authors. All authors have approved the final version of the manuscript. Rituprava Dash and Suman Yadav contributed equally.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Material characterization of V2CTx MXene, including, X-ray diffraction (XRD), scanning electron micrscope (SEM), ultraviolet-visible-near-infrared (UV-Vis-NIR) spectroscopy and fourier transform infrared (FTIR) spectroscopy were provided. Along with that electrochemical characterizations including, cyclic voltammetry (CV), galvanostatic charge-discharge (GCD), electrochemical impedance spectroscopy (EIS) and linear sweep voltammetry (LSV) analysis were provided. See DOI: https://doi.org/10.1039/d6eb00012f.Acknowledgements
R. D. acknowledges the scholarship support from the Ministry of Education (MOE), India. S. Y. acknowledges the scholarship support from the PMRF, India. N. K. acknowledges financial support from the Indian Institute of Technology Hyderabad, India, the Council of Scientific and Industrial Research (CSIR/HRDG/EMR-II/01-3103-23), India, and the Anusandhan National Research Foundation (ANRF/MAHA/2024/000126/EVB), India.References
- S. Fleischmann, J. B. Mitchell, R. Wang, C. Zhan, D.-e. Jiang, V. Presser and V. Augustyn, Chem. Rev., 2020, 120, 6738–6782 CrossRef CAS.
- Y. Gogotsi and R. M. Penner, ACS Nano, 2018, 12, 2081–2083 CrossRef CAS.
- V. Augustyn, P. Simon and B. Dunn, Energy Environ. Sci., 2014, 7, 1597–1614 RSC.
- J. P. Zheng, P. J. Cygan and T. R. Jow, J. Electrochem. Soc., 1995, 142, 2699 CrossRef CAS.
- J. B. Mitchell, W. C. Lo, A. Genc, J. LeBeau and V. Augustyn, Chem. Mater., 2017, 29, 3928–3937 CrossRef CAS.
- H. Y. Lee and J. B. Goodenough, J. Solid State Chem., 1999, 144, 220–223 CrossRef CAS.
- X. Han, Q. Meng, X. Wan, B. Sun, Y. Zhang, B. Shen, J. Gao, Y. Ma, P. Zuo, S. Lou and G. Yin, Nano Energy, 2021, 81, 105635 CrossRef CAS.
- C. Zhan, M. Naguib, M. Lukatskaya, P. R. C. Kent, Y. Gogotsi and D.-e. Jiang, J. Phys. Chem. Lett., 2018, 9, 1223–1228 CrossRef CAS.
- M. B. Cabré, D. Spurling, P. Martinuz, M. Longhi, C. Schröder, H. Nolan, V. Nicolosi, P. E. Colavita and K. McKelvey, Nat. Commun., 2023, 14, 374 CrossRef.
- G. Nagaraju, S. Tagliaferri, A. Panagiotopoulos, M. Och, R. Quintin-Baxendale and C. Mattevi, J. Mater. Chem. A, 2022, 10, 15665–15676 RSC.
- G. Nagaraju, S. C. Sekhar, B. Ramulu and J. S. Yu, Energy Storage Mater., 2021, 35, 750–760 CrossRef.
- G. Nagaraju, S. C. Sekhar, B. Ramulu and J. S. Yu, Small, 2019, 15, 1805418 CrossRef PubMed.
- R. Ibragimova, P. Erhart, P. Rinke and H.-P. Komsa, J. Phys. Chem. Lett., 2021, 12, 2377–2384 CrossRef CAS PubMed.
- S. Yadav and N. Kurra, Energy Storage Mater., 2024, 65, 103094 CrossRef.
- J. Tang, T. S. Mathis, N. Kurra, A. Sarycheva, X. Xiao, M. N. Hedhili, Q. Jiang, H. N. Alshareef, B. Xu and F. Pan, Angew. Chem., 2019, 131, 18013–18019 CrossRef.
- Q. Gao, W. Sun, P. Ilani-Kashkouli, A. Tselev, P. R. C. Kent, N. Kabengi, M. Naguib, M. Alhabeb, W.-Y. Tsai, A. P. Baddorf, J. Huang, S. Jesse, Y. Gogotsi and N. Balke, Energy Environ. Sci., 2020, 13, 2549–2558 RSC.
- Y. Liu, Q. Liu, C. Zhao, L. Liu, Z. Liu, A. Ying, Z. Pang, X. Sun, P. Chen and G. Chen, Adv. Funct. Mater., 2024, 34, 2407497 CrossRef CAS.
- F. Cao, Y. Zhang, H. Wang, K. Khan, A. K. Tareen, W. Qian, H. Zhang and H. Ågren, Adv. Mater., 2022, 34, 2107554 CrossRef CAS PubMed.
- X. Wang, T. S. Mathis, Y. Sun, W.-Y. Tsai, N. Shpigel, H. Shao, D. Zhang, K. Hantanasirisakul, F. Malchik, N. Balke, D.-e. Jiang, P. Simon and Y. Gogotsi, ACS Nano, 2021, 15, 15274–15284 CrossRef CAS PubMed.
- Y. Zhigalenok, S. Abdimomyn, M. Levi, N. Shpigel, M. Ryabicheva, M. Lepikhin, A. Galeyeva and F. Malchik, J. Mater. Chem. A, 2024, 12, 33855–33869 RSC.
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef CAS PubMed.
- S. Ko, Y. Yamada and A. Yamada, Electrochem. Commun., 2020, 116, 106764 CrossRef CAS.
- Q. Nian, X. Zhang, Y. Feng, S. Liu, T. Sun, S. Zheng, X. Ren, Z. Tao, D. Zhang and J. Chen, ACS Energy Lett., 2021, 6, 2174–2180 CrossRef CAS.
- L. Zhang, I. A. Rodríguez-Pérez, H. Jiang, C. Zhang, D. P. Leonard, Q. Guo, W. Wang, S. Han, L. Wang and X. Ji, Adv. Funct. Mater., 2019, 29, 1902653 CrossRef.
- J. Han, H. Zhang, A. Varzi and S. Passerini, ChemSusChem, 2018, 11, 3704–3707 CrossRef CAS PubMed.
- S. Chen, R. Lan, J. Humphreys and S. Tao, App. Mater. Today, 2020, 20, 100728 CrossRef.
- L. Suo, D. Oh, Y. Lin, Z. Zhuo, O. Borodin, T. Gao, F. Wang, A. Kushima, Z. Wang, H.-C. Kim, Y. Qi, W. Yang, F. Pan, J. Li, K. Xu and C. Wang, J. Am. Chem. Soc., 2017, 139, 18670–18680 CrossRef CAS PubMed.
- H.-G. Steinrück, C. Cao, M. R. Lukatskaya, C. J. Takacs, G. Wan, D. G. Mackanic, Y. Tsao, J. Zhao, B. A. Helms, K. Xu, O. Borodin, J. F. Wishart and M. F. Toney, Angew. Chem., Int. Ed., 2020, 59, 23180–23187 CrossRef PubMed.
- K. W. Leong, W. Pan, Y. Wang, S. Luo, X. Zhao and D. Y. C. Leung, ACS Energy Lett., 2022, 7, 2657–2666 CrossRef CAS.
- D. Reber, R. Grissa, M. Becker, R.-S. Kühnel and C. Battaglia, Adv. Energy Mater., 2021, 11, 2002913 CrossRef CAS.
- K. Matthews, T. Zhang, C. E. Shuck, A. VahidMohammadi and Y. Gogotsi, Chem. Mater., 2021, 34, 499–509 CrossRef.
- M. Anayee, N. Kurra, M. Alhabeb, M. Seredych, M. N. Hedhili, A.-H. Emwas, H. N. Alshareef, B. Anasori and Y. Gogotsi, Chem. Commun., 2020, 56, 6090–6093 RSC.
- T. Parker, D. Zhang, D. Bugallo, K. Shevchuk, M. Downes, G. Valurouthu, A. Inman, B. Chacon, T. Zhang and C. E. Shuck, Chem. Mater., 2024, 36, 8437–8446 CrossRef CAS PubMed.
- N. Kurra, S. Uzun, G. Valurouthu and Y. Gogotsi, Energy Storage Mater., 2021, 39, 347–353 CrossRef.
- H. Zhang, X. Liu, H. Li, I. Hasa and S. Passerini, Angew. Chem., Int. Ed., 2021, 60, 598–616 CrossRef CAS PubMed.
- R. Huang, S. Liu, Z. He, W. Zhu, G. Ye, Y. Su, W. Deng and J. Wang, Adv. Funct. Mater., 2022, 32, 2111661 CrossRef CAS.
- J. Tang, T. S. Mathis, N. Kurra, A. Sarycheva, X. Xiao, M. N. Hedhili, Q. Jiang, H. N. Alshareef, B. Xu, F. Pan and Y. Gogotsi, Angew. Chem., Int. Ed., 2019, 58, 17849–17855 CrossRef CAS PubMed.
- J.-P. Hoffknecht, A. Wettstein, J. Atik, C. Krause, J. Thienenkamp, G. Brunklaus, M. Winter, D. Diddens, A. Heuer and E. Paillard, Adv. Energy Mater., 2023, 13, 2202789 CrossRef CAS.
- H. Agarwal, J. Florian, B. R. Goldsmith and N. Singh, ACS Energy Lett., 2019, 4, 2368–2377 CrossRef CAS.
- P. Leuaa, D. Priyadarshani, A. K. Tripathi and M. Neergat, J. Electroanal. Chem., 2020, 878, 114590 CrossRef CAS.
- Y. Zhang and E. J. Maginn, J. Phys. Chem. B., 2021, 125, 13246–13254 CrossRef CAS PubMed.
- D. Inoue and Y. Moritomo, RSC Adv., 2024, 14, 6292–6297 RSC.
- T. S. Mathis, N. Kurra, X. Wang, D. Pinto, P. Simon and Y. Gogotsi, Adv. Energy Mater., 2019, 9, 1902007 CrossRef CAS.
- N. Kurra, S. Uzun, G. Valurouthu and Y. Gogotsi, Energy Storage Mater., 2021, 39, 347–353 CrossRef.
- S. Yadav and N. Kurra, Small, 2025, 21, 2503657 CrossRef CAS PubMed.
- J. S. Ko, C.-H. Lai, J. W. Long, D. R. Rolison, B. Dunn and J. N. Weker, ACS Appl. Mater. Interfaces, 2020, 12, 14071–14078 CrossRef CAS PubMed.
- H. Kim, J. Hong, K.-Y. Park, H. Kim, S.-W. Kim and K. Kang, Chem. Rev., 2014, 114, 11788–11827 CrossRef CAS PubMed.
- A. J. Bard, L. R. Faulkner and H. S. White, Electrochemical methods: fundamentals and applications, John Wiley & Sons, 2022 Search PubMed.
- L. Suo, Y.-S. Hu, H. Li, M. Armand and L. Chen, Nat. Commun., 2013, 4, 1481 CrossRef PubMed.
- D. G. Vazquez, J. Ingenmey, K. Trapp, D. Ciliak, M. Salanne and M. R. Lukatskaya, J. Am. Chem. Soc., 2025, 147, 35953 CrossRef PubMed.
- T. J. Webb, J. Am. Chem. Soc., 1926, 48, 2589–2603 CrossRef CAS.
- S. E. Rodriguez-Cruz, R. A. Jockusch and E. R. Williams, J. Am. Chem. Soc., 1999, 121, 8898–8906 CrossRef CAS PubMed.
- F. Wei, F. Liang, Y. Zhao, Z. Ji, T. Yan, R. Li, H. Liu, Y. Kong, H. He, W. Huang, C. Cao, W. Zhang, B. Fei and M. Ge, J. Mater. Chem. A, 2025, 13, 13070–13080 RSC.
- S. M. M. Alonzo and B. P. Bastakoti, Mater. Today Nano, 2025, 32, 100704 CrossRef CAS.
- Z. Cao, Y. Wu, L. Jin, X. Li, D. Qian and H. Hu, Adv. Funct. Mater., 2025, e26006 Search PubMed.
- Z. Cao, G. Liang, D. Ho, C. Zhi and H. Hu, Adv. Funct. Mater., 2023, 33, 2303060 CrossRef CAS.
- T. Parker, Y. Zhang, K. Shevchuk, T. Zhang, V. Khokhar, Y.-H. Kim, G. Kadagishvili, D. Bugallo, M. Tanwar, B. Davis, J. Kim, Z. Fakhraai, Y.-J. Hu, D.-e. Jiang, D. V. Talapin and Y. Gogotsi, ACS Nano, 2025, 19, 22228–22239 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2026 |