
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aging of commercial sodium-ion batteries with layered oxides: how to measure and analyze it?
Mathias
Rehm
 *a,
Julius
Bahrke
a,
Johannes
Natterer
ab,
Leon
Milutinovic
a,
Franz
Roehrer
a and
Andreas
Jossen
a
*a,
Julius
Bahrke
a,
Johannes
Natterer
ab,
Leon
Milutinovic
a,
Franz
Roehrer
a and
Andreas
Jossen
a
aTechnical University of Munich (TUM), TUM School of Engineering and Design; Department of Energy and Process Engineering, Chair of Electrical Energy Storage Technology (EES), Arcisstr. 21, 80333 Munich, Germany. E-mail: mathias.rehm@tum.de
bInfineon Technologies AG, Am Campeon 1-15, 85579 Neubiberg, Germany
First published on 22nd January 2026
Abstract
Improving the understanding of the aging of sodium-ion batteries (SIBs) is vital for their subsequent improvement and commercialization. Nevertheless, the electrode-resolved aging of SIBs has not been thoroughly investigated to date. Here, we use commercial SIBs that were cyclically aged for almost two years and show how to use three-electrode cells to measure the changes of both kinetics and balancing of the electrodes over the aging. The area-normalized comparison between the three-electrode cells and the 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 650 cells shows that both state-of-charge resolved pulse resistance and capacity are transferable. Using a modified setup with a tin-wire reference electrode, we demonstrate how to use the three-electrode cells to measure the electrode-resolved impedance. In addition, we use the open-circuit potentials from three-electrode cells to perform degradation mode analysis on the 18650 cells. The results of the degradation mode analysis are validated both by the low difference between measured and reconstructed full-cell voltage over the aging and by post-mortem analysis. Furthermore, we verify that the fundamental assumption of the degradation mode analysis, i.e., that the shapes of the electrode open-circuit potentials do not change during aging, is valid for the investigated SIBs.
650 cells shows that both state-of-charge resolved pulse resistance and capacity are transferable. Using a modified setup with a tin-wire reference electrode, we demonstrate how to use the three-electrode cells to measure the electrode-resolved impedance. In addition, we use the open-circuit potentials from three-electrode cells to perform degradation mode analysis on the 18650 cells. The results of the degradation mode analysis are validated both by the low difference between measured and reconstructed full-cell voltage over the aging and by post-mortem analysis. Furthermore, we verify that the fundamental assumption of the degradation mode analysis, i.e., that the shapes of the electrode open-circuit potentials do not change during aging, is valid for the investigated SIBs.
Broader contextSodium-ion batteries are a promising alternative to lithium-ion batteries. This is particularly relevant for Europe, where materials such as hard carbon can help mitigate supply chain risks. Among the possible cathode materials, layered oxides, including nickel–manganese–iron compounds, are especially promising. Nonetheless, systematic aging analyses of commercial sodium-ion batteries remain rare. Here, we analyze one fresh cell and three end-of-life cells post mortem. From these cylindrical cells, we build three-electrode cells to quantify electrode-resolved contributions to resistance increase and capacity loss. We show that, despite the few features in the half-cell potentials, the early-life half-cell curves still enable degradation mode analysis to identify capacity losses at all aging states. This analysis reveals that cathode capacity can limit the overall aging of these cells, which contrasts with previous knowledge from lithium-ion batteries. Overall, this study shows how to quantify limiting aging mechanisms in sodium-ion batteries, which is crucial for operation and future cell design. |
1 Introduction
The annual demand for batteries is expected to increase three to four times between 2025 and 2030 in Europe, probably surpassing 1 TWh per year.1 Despite its huge demand, Europe contributes to only 2% of the global battery raw-material supply2,3 compared to a share of 25% of the global cell demand.4 This imbalance leads to geopolitical dependencies.5Here, sodium-ion batteries (SIBs) are a promising alternative to lithium-ion batteries (LIBs) to reduce the risk of supply chain disruptions2,5 as they can be produced with similar production processes and working principles, while requiring more abundant materials such as sodium and manganese.6
Despite their lower energy density, SIBs with nickel–manganese–iron (NMF) cathodes and hard carbon anodes are less prone to material price fluctuations and might be more cost-effective than LIBs with nickel–manganese–cobalt (NMC) or lithium–iron phosphate (LFP) cathodes.7 Furthermore, SIBs can be even more economically competitive if they exhibit less aging.8,9
Analogous to LIBs, the aging of SIBs can be organized hierarchically into aging effects, degradation modes (DMs), and underlying mechanisms. Aging effects include capacity loss and, induced by increased resistances, power fade. They can be directly quantified and are thus widely used to assess the aging of LIBs and SIBs.10–12 A more detailed understanding of the aging effects can be achieved by distinguishing between different DMs, namely loss of active material at the positive electrode (LAMca), loss of active material at the negative electrode (LAMan), and loss of charge carrier inventory (LI).13–15 Reasonably, we use LI instead of, e.g., loss of lithium inventory (LLI), to avoid a distinction in the nomenclature between SIBs and LIBs. Although DMs provide higher information density, they are not directly measurable, and their estimation is non-trivial.13,16 At the mechanism level, component aging results from the superposition of processes such as ongoing solid electrolyte interphase (SEI) formation driven by an unstable SEI at the anode,17–19 cathode electrolyte interphase (CEI) growth at the cathode,20,21 and particle cracking at both electrodes,21,22 which can accelerate SEI and CEI growth.20
Regarding SIBs with layered oxides, only a few publications systematically address their aging behavior.11,12,23–25 Among these, Streck et al.24 investigated the calendar aging of a commercial 18650 SIB with an NMF cathode and hard carbon anode, finding less than 2% capacity loss over two months of storage, including four reference performance tests (RPTs). Significant reversible side reactions reduced coulombic efficiency, not solely due to LI. For the same cell type, Laufen et al.11 showed that cycling could be continued for at least 1000 cycles before the current interrupt device (CID) was triggered, using current rates of up to 5C and 8C for charge and discharge, respectively. Later, Klick et al.25 revealed that reducing the depth of discharge (DOD) to 80% reduced aging, while low temperatures of −10 °C or high temperatures of 40 °C and 50 °C accelerated capacity fade.
A precise determination of the DMs is imperative in order to achieve a comprehensive understanding of the aging process of the SIBs and to ensure the accurate modeling of their aging behavior, as previously outlined by Li et al.26 Nonetheless, to the best of our knowledge, any non-destructive quantitative analysis of the DMs relies either on a clear assignment of specific features in the full-cell open-circuit voltage (OCV) or its derivatives to specific electrodes, or on high quality half-cell data.13,16,27–29 Voltage or its derivatives are used as the input signals in most cases, with only a few exceptions.30,31
However, previously published half-cell open-circuit potentials (OCPs) of commercial SIBs suffered from high hysteresis between the sodiation and desodiation in the case of the hard carbon anode OCP and low coulombic efficiency in the case of the layered oxide cathode OCP.11,25,32 For the sake of clarity, we note that within the discussed work, we always refer OCP to half-cell potentials, while OCV refers to full-cell potential. We expect that within these coin-cells, the metallic sodium is not stable in the electrolyte.33,34 As demonstrated by Fitzpatrick et al.,35 the SEI for hard carbon in a half-cell configuration with a metallic sodium working electrode exhibits a greater increase in the relative concentration of organic species, such as alkyl carbonates and carboxylates, when compared to a full-cell setup. Additionally, the SEI thickness increases more significantly in the half-cell configuration. As a result, the resistance is strongly increased36 and thus the voltage difference between charge and discharge during pseudo-open-circuit voltage (pOCV) measurements is also increased.32 In addition, recent publications including our own work have shown a low coulombic efficiency for NMF cathodes in half-cell setups against metallic sodium.11,25,32,37 While for the anode, the additional hysteresis of the coin-cell might be mitigated by calculating the mean of the sodiation and desodiation curve, the low coulombic efficiency of the cathode leads to different shapes of the OCP in charge and discharge direction.11,25,32 In addition, it is hard to distinguish whether the charge or discharge OCP is closer to the true OCP in the full-cell setup. As a result, the few studies who have determined the DMs for SIBs have only performed a qualitative analysis of the DMs by tracking changes in the electrode balancing via specific peaks in the differential voltage (DV),23 or used synthetic data to compare to the measured data.25
Within this work, we solve this problem by using three-electrode cells, i.e. Swagelok T-cells. Instead of measuring against metallic sodium, we measure against the hard carbon anode harvested from the 18650 cell. Neither sodium metal nor, in the case of the modified setup for electrode-resolved electrochemical impedance spectroscopy (EIS) measurements, the tin-wire reference electrode (TWRE) serves as the working electrode. Thus, for both setups, a potential increase of resistance at the reference electrode interface does not affect the measurements.38
As the T-cells allow for measuring the half-cell OCPs of both electrodes, we can use those OCPs for the so-called degradation mode analysis (DMA) to determine the DMs over aging. This method has been widely used for LIBs,13,16,27,28,39,40 but to the best of our knowledge, has not yet been applied to SIBs with layered oxide cathodes. Here, the full-cell OCV or its derivatives are reconstructed by shifting and scaling of half-cell OCPs at the beginning of test (BOT) of the cell.13,16,27,28 The DMA was successfully shown to extract the DMs of LIBs based on the OCV, whether in the full range13,41,42 or in a limited range of the state-of-charge (SOC).16,43 Both the pOCV13,16,41,42 and the relaxed-OCV43 can be used as input data. To the current state DMA for SIBs has been performed solely on synthetic aging data44 or using such synthetic data to compare to measured data25 as described above.
To apply the DMA, the OCP curves need to be invariant over aging.13,41,42,45 While this assumption generally holds for LIBs, many authors have reported exceptions. For blended anodes or cathodes, the ratio of those materials determines the shape of the OCP.46,47 Thus, the stronger degradation of one of the blended materials, for example silicon in silicon-graphite blend anodes, can change the OCP shape over aging.28,48,49 Changes in the OCP have also been reported for cathodes for certain cathode chemistries, for example: minor changes in relaxed-OCP of LFP cathodes due to increased hysteresis50,51 and in pOCVs of NMC in a full-cell33 have been reported. For cobalt oxide cathodes, changes in the pOCVs of cobalt oxide both in a full-cell52 and in a half cell setup at high potentials53 were shown. Furthermore, small changes in the OCP for Ni-rich cathodes at high potentials both in half-cell54 and full-cell55 setups have been reported. Here, Friedrich et al.56 showed that a resistive surface layer in Ni-rich NMC cathodes can lead to a strong increase in the overpotentials during pOCV measurements. Furthermore, lithium- and manganese-rich (LMR) materials are known to change their shape over aging.57–59 Interestingly, a change of OCP has also been observed for layered oxide cathodes for SIBs.60,61 Thus, one may conclude that the assumption of an invariant OCP curve over aging for NMFs is not unconditionally true, which would complicate the application of DMA for SIBs with layered oxide cathodes.
Besides the change of the electrode balancing, the changes of the electrode kinetics play an important role for aging determination. These changes are quantified using both pulse and EIS measurements at electrode level. While such data on the electrode-level are scarce for SIBs with layered oxide cathodes, this knowledge is crucial for understanding their aging. Several publications investigated the dependence of pulse resistance and impedance on temperature11,23,32,62,63 or SOC11,32. Among these, only few resolved the resistances on the electrode level. The rare exceptions are either for non-commercial materials,64 which limits the transferability to commercial cells, or they do not resolve over the SOC.65
Therefore, we cycled four commercial cells in the SOC range from 20% to 80% and built 19 three-electrode cells (T-cells). T-cells were built to show that they are suited to measure and analyze the electrode-resolved pulse resistance and impedance, and that half-cell data from T-cells can be used for the DMA over the aging trajectory. For the latter, we conducted the following investigations: (a) we show how to obtain stable half-cell OCPs, (b) we investigate the stability assumption of the OCP for SIBs with layered oxide cathodes, which is a crucial factor for the validity of the DMA, (c) we show how the DMA can be applied to SIBs with layered oxide cathodes, (d) we methodologically validate the conducted DMA with measurements from early-life and end of test (EOT) cells, and (e) we discuss the results of the DMA and what they imply about the aging of SIBs with layered oxide cathodes.
2 Experimental
2.1 Investigated cells
To investigate the aging behavior of commercially available SIBs, we used cells in the 18650 format from Shenzhen Mushang Electronics Co., Ltd (CY-18650-1250 mAh). These cells have been extensively characterized in previous studies, both regarding their electrochemical performance on the full-cell level25,32,62,63,66 as well as on the material level.11,32 The cell has been designed with an NMF cathode and a hard carbon anode.11,32 The nominal capacity (Cnom) of 1.25 Ah is used as the reference for all current rates. The cells allow maximum charge and discharge currents of 5C and 8C, respectively. For more details, we refer to our previous publication.32
2.2 Cyclic aging study
Cycling of the cells was performed in a climate chamber at 25 °C using a CTS battery test system from BaSyTec. All cells were contacted using 4-wire sensing, where both connections for power and for voltage were connected separately at both terminals of the cells with two solder joints per connection. A negative-temperature-coefficient thermistor sensor was placed as temperature sensor on the negative terminal and thermally insulated from the environment with a 10 mm thick piece of insulation material (ArmaFlex, Armacell, Germany).The investigated cells were constant current constant voltage (CCCV) charged and discharged at 1C between 3.475 V and 2.358 V. The constant voltage (CV) cut-off current was 0.05C, while the CV phase and the subsequent break combined lasted 720 s. The lower voltage limit is the voltage at 20% SOC of the discharge pOCVs, while the upper limit is the voltage at 80% SOC of the charge pOCVs. For both charge and discharge voltage limits, the mean values of the pOCVs of 20 cells were used.
2.3 Reference performance tests
RPT measurements were conducted at BOT and after every 333 cycles, which refers to 200 equivalent full cycle (EFC), as detailed in Fig. S1, SI. Each RPT included two charge–discharge cycles using CCCV at 0.5C with a cut-off current of 0.05C. In the first RPT, we performed 10 cycles instead of 2 to mitigate possible break-in effects.67 Moreover, we inserted pauses of 2 h before the RPT and 0.5 h between the charging and discharging steps. Each RPT included pOCV and relaxed-OCV measurements, both conducted in charge and discharge directions. We measured pOCV at 0.05C and performed relaxed-OCV using a current rate of 1C in steps of 5% SOC. The relaxed-OCV measurements are only used to determine the resistance values and thus short relaxation pauses of 300 s were used. The discharge pulses of the relaxed-OCV at 50% after 10 s were also used to calculate the resistance of the cells.2.4 Preparation of three-electrode cells
In this study, a total of 19 Swagelok T-cells were produced, of which 16 were built from cyclic aged cells, exhibiting divergent aging trajectories, and three were built from early-life cells. For harvested-material analysis, cells were CCCV-discharged to end of discharge (EOD), opened, the jelly roll unrolled, the separator removed, and one side of the coated sheets delaminated with ethylene carbonate under argon; for positive electrodes, a scalpel was additionally used to remove the coating. Subsequently, circular samples with a diameter of 10.95 mm were punched from both the anode and cathode sheets with a punching machine manufactured by NOGAMI to ensure reproducible results, clean edges, and closely comparable sample capacity. All samples were taken from the mid-plane of the electrode sheets (approximately 3 cm above and below the sheet's centerline in the jelly-roll's axial direction, please refer to ref. 32 for electrode sheet dimensions). Along the roll length two radial positions were sampled, (i) an outer region, located about 10 cm inward from the roll's outer edge, and (ii) an inner region, located about 18 cm outward from the innermost winding. The inner region was chosen as compromise between being close to the core and stable electrode coatings after unwinding the jelly-roll both at BOT and EOT. In every case the sample's face corresponded to the electrochemically active surface that points toward the core of the jelly-roll.It is worth noting that we deliberately did not wash the harvested electrodes to prevent any alteration of the electrode surface chemistry or passivation layers, likely being crucial for the assessment of the degradation state and kinetics of the electrodes. Furthermore, we avoided areas with visible deposits of metallic sodium on the anode samples. We note that within this work, we did not perform any structural analysis such as transmission electron microscopy or chemical analysis of the harvested electrodes, but rely on electrochemical analysis and visually inspecting the electrodes for metallic sodium deposits.
The obtained samples were then measured for their thickness with a digital caliper (Hoffmann Group, Germany) and weighed with a high-precision scale (Sartorius, Germany), before being assembled into a three-electrode configuration in laboratory Swagelok T-cell setup (Swagelok, USA). All required parts of this setup were dried overnight with at least 80 °C in dynamic vacuum in a glass oven (Büchi, Switzerland) before being transferred to a glove box with argon atmosphere (H2O, O2 < 0.1 ppm, MBraun, Germany). For a detailed view of all components used, please refer to Friedrich et al.68 For assembly, first the anode sample was inserted into an high-density polyethylene (HDPE) cylinder, which is held in place by a pin and nut with HDPE sealing in the housing, followed by two glass fiber separators of 260 μm thickness and 11 mm diameter (691, VWR, USA). The second separator was equipped with a small extension, orthogonal to the stack, which eventually ensures the ionic connection to the reference. After applying 60 μL of electrolyte (1.0 M of NaPF6 in EC:EMC 3:7 by weight) onto the separators, the cathode sample was placed on top, always making sure that the separators extension was well-aligned with the HDPE spacer's opening. After insertion of the countering stamp, pin, spring, HDPE sealing and nut, the homogeneous compression of the stack was ensured using an in-house apparatus with a micro head screw (Mitutoyo, Japan), allowing replicable tensioning of the inserted spring, which in turn ensures uniform pressure on the stack of 0.20 MPa.
Next, a separator with a diameter of 10 mm was placed into the third opening of the housing. This separator rests on the orthogonal extension of the separator within the stack and, together with an additional 40 μL of electrolyte, ensures reliable ionic contact with the metallic sodium reference (99.7 purity sodium metal with thickness of ≈0.45 mm and coated on aluminum; bought from Xiamen AOT Electronics Technology Co., Ltd). Finally, a pin was inserted to secure the reference electrode, and an HDPE ring was added to seal the assembly.
To measure the electrode-resolved impedance, cells using TWRE were built in a modified process. The orthogonal separator extension was omitted, and the prepared TWRE was inserted through the reference opening in the housing and HDPE tube before adding the second separator and cathode to the stack. The tip of the TWRE was positioned as centrally as possible within the circular active area, placing it between two separator layers for electrical isolation. As no third separator was used, only 60 μL of electrolyte were added directly to the stack. Using TWREs instead of metallic sodium required extensive preparation, which is detailed in ref. 69, but should be briefly outlined herein. The tinned copper wire (diameter = 50 μm, Eurowire, Great Britain) was initially uncoated, which prevented direct measurement of the reference potential. To prepare the wire, it was first cleaned with isopropanol and then coated with a polyurethane spray (Urethan 71, Kontakt Chemie, Germany). After coating, the wire was cut to a length of about 10 mm using a scalpel. The end facing the reference pin was then scraped to remove the coating, ensuring a reliable electrical clamp connection between the reference pin and the wire, which was formed using a small screw.
2.5 Measurements with three-electrode cells
All three-electrode cells, in our case T-cells, were tested at 25 °C in a climate chamber using a BioLogic VMP-300. After 10 h post-assembly rest, T-cells without TWRE were cycled between 1.5 V and 3.8 V using different current rates. Between each charge and discharge step a 30 min pause was used. First, the cells were cycled with 0.05C and then 0.02C. Then, the cells were charged to 100% SOC with 0.1C and another 0.05C cycle was performed. Subsequently, the cells were discharged at 0.05C in 10% SOC steps, each with 30 min relaxation. At each SOC level from 90% to 10%, 11 s discharge and charge pulses at 0.05C, 0.1C, and 0.5C were applied, with a 10 min relaxation between pulses. Finally, the cells were cycled again at 0.05C and 0.02C to assure they did not lose capacity during the measurement.For T-cells with TWRE, first the reference electrode was sodiated with a current of 50 nA for ≈30 h as outlined in ref. 69. We note that less time for sodiation led to a drift in the potential after less than 24 h, even if the potential was stable during sodiation within less than 1 mV for several hours. The amount of charge used for sodiation of the TWRE corresponds to ≈0.1% of the total capacity of the cathode and can therefore be deemed negligible. After sodiation of the TWRE, a shortened version of the protocol for T-cells without TWRE was used, as we expect TWRE T-cells to be less stable over time. The measurement started with a charge, discharge and another charge with 0.05C. Then, the cells were discharged in 10% SOC steps with 0.05C. Following a 60 min relaxation period, galvanostatic electrochemical impedance spectroscopy (GEIS) measurements were recorded. After an additional 10 min rest, potentiostatic electrochemical impedance spectroscopy (PEIS) measurements were recorded at each SOC level. Both GEIS and PEIS measurements were performed in a frequency range from 200 kHz to 25 mHz with 8 points per decade and 9 average measures per frequency. The amplitude was 120 μA for GEIS and 10 mV for PEIS. Within this work only the PEIS data are discussed, as the GEIS data have shown artifacts at high frequencies and are therefore omitted. After the PEIS measurement and another pause of 10 min, pulses of 11 s with 0.05C, 0.1C and 0.5C were applied. For each current rate first the discharge pulse, then the charge pulse was applied. Between each pulse, a relaxation time of 10 min was used.
3 Implementation of the DMA
For DMA, we used the pOCVs from the RPTs in charge direction as input data. We note that using the pOCVs in discharge direction led to very similar results, but was slightly less robust in our case. The core MATLAB (R2024b, MathWorks Inc.) implementation of the DMA tool has been further developed starting from our previous framework.16 Parts of the improvements were adapted from ref. 40. The framework is described in detail in a separate publication70 and is available as open-source code on GitHub (https://github.com/tum-ees/DegradationModeAnalysis).71 The algorithm minimizes the residual between the measured and the estimated OCV curves by shifting and scaling the OCP curves of pristine electrodes.13,16 As the OCP shapes are assumed invariant during aging, which will be shown within this work for the investigated cells in section 4.5.1, aging effects can be captured by shifting and scaling both electrodes toward each other.27,28,41,42,45 A concise synopsis of the implementation process is hereby presented. Readers who seek a more comprehensive examination of the subject are advised to refer to the following publication.16,28,40To determine the DMs, both the residual between the measured and reconstructed OCV as well as between the measured and reconstructed DV are minimized. The DV is defined as
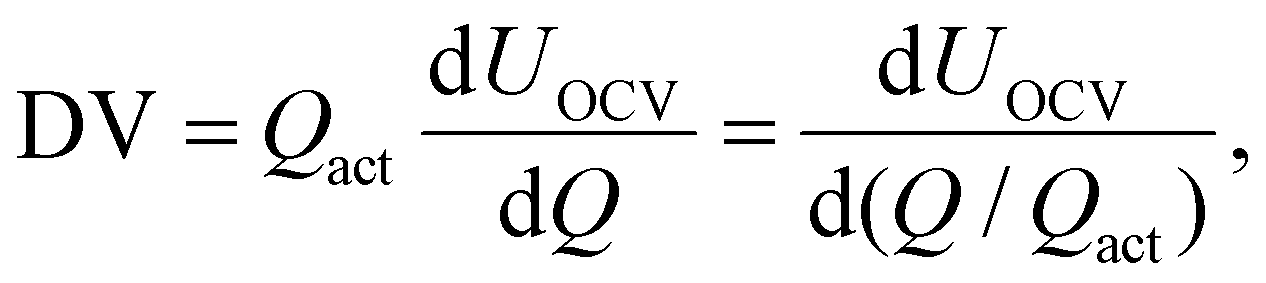 | (1) |
The DMA uses the following cost function:
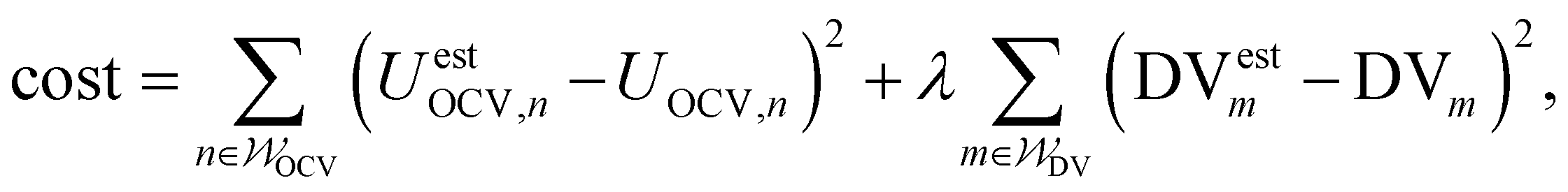 | (2) |
 and
and 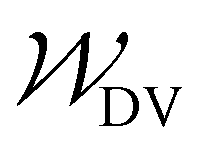 specify the individual fitting windows for the pseudo-OCV and DV, respectively, which may differ in location and length along the capacity axis. The weighting factor λ allows us to weight the residuals of the fitting of the DV region against the OCV region. It is set to 50 based on empirical evaluation whether features in both OCV and DV are well represented in the fitting result. For the sake of clarity, we note that the weighting factor is not normalized and thus a weighting factor of 50 means that both parts of the cost function have a significant influence on the fitting result.
specify the individual fitting windows for the pseudo-OCV and DV, respectively, which may differ in location and length along the capacity axis. The weighting factor λ allows us to weight the residuals of the fitting of the DV region against the OCV region. It is set to 50 based on empirical evaluation whether features in both OCV and DV are well represented in the fitting result. For the sake of clarity, we note that the weighting factor is not normalized and thus a weighting factor of 50 means that both parts of the cost function have a significant influence on the fitting result.
In the proposed DMA framework, we use OCP curves from T-cells harvested from an early-life cell as input for the fitting procedure to overcome the limitations of half-cell OCP measurements against metallic sodium.11,25,32,37 Thus, our half-cell OCPs are only measured within the full-cell potential window. We note that in principle, measuring outside of the voltage limits of the full-cell would be possible. However, cycling 150 mV above the upper voltage limit did subsequently decrease the reproducibility of the OCP measurements and hinder subsequent measurements in the T-cells. Thus, to ensure that our fitting results are not constrained by the OCPs of the early-life electrodes, the highest and lowest SOC regions below 5% and above 95% are excluded from the fitting procedure.
To minimize the cost function, we tested several algorithms previously used in the literature for determination of DMs, including lsqnonlin,28 fmincon,16,45 GlobalSearch,75 patternsearch,43 particleswarm,76 and genetic algorithm (ga).43,77 We obtained the best fitting results with the ga algorithm. In addition, we increased population size to 500 compared to the default value of 50 for 4 variables, which further improved both the robustness and the best fitting results. While it remains unclear to us why the ga outperforms the particleswarm algorithm (with population size 500), we expect that the high randomness and high computational effort of the ga allows to escape local minima more easily than e.g. fmincon, lsqnonlin or GlobalSearch. To adapt for the random nature of the ga algorithm, the fitting was repeated at least 20 times. Additional runs up to a maximum of 50 total runs were performed if the fitting error was above 8 mV between measured and reconstructed OCV curve. The best fitting result was selected based on the lowest root mean square error (RMSE) between measured and reconstructed OCV curve in the investigated SOC range. These settings were used for maximum quality of the results. We note that the number of maximum repitions can be reduced from 20 to 5 or even lower for future degradation mode estimations without substantial loss of quality.
3.1 Loss of charge carrier inventory
The amount of charge carrier inventory Cinv is directly dependent on the cyclable sodium residing in the electrodes when the full-cell is at an arbitrary SOC within the utilization window. Following Schmitt et al.,28 we obtain| Cinv = (αcat − βan + βcat)Cfull | (3) |
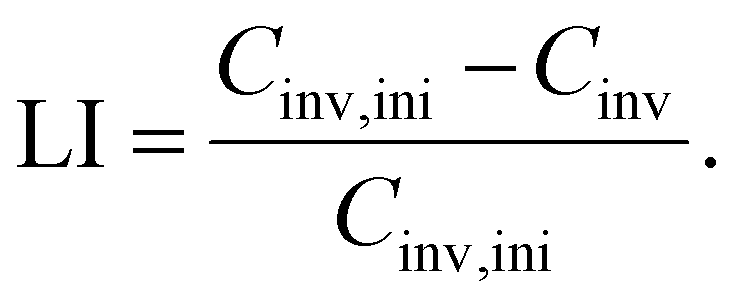 | (4) |
Please refer to Fig. 9 for an illustration of the four alignment parameters and their shifts over aging.
3.2. Loss of active material
According to Schmitt et al.,28 the loss of active material (LAM) at both electrodes is tracked in terms of the capacity fade relative to their initial capacity with the following equations:| Can = αan·Cfull | (5) |
| Ccat = αcat·Cfull. | (6) |
Thus, the LAM for anode and cathode can be expressed as
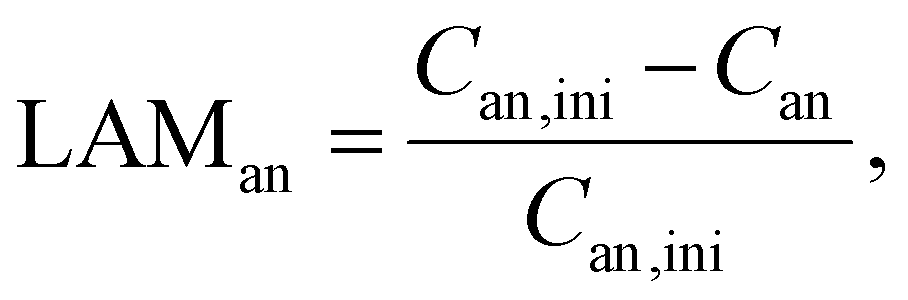 | (7) |
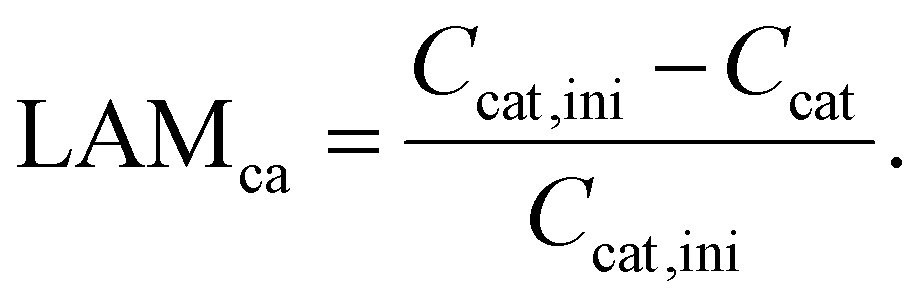 | (8) |
4 Results and discussion
This section reports cyclic-aging results for SIBs, aging-related kinetic changes, electrode-resolved impedance spectra, and the change of balancing over aging. The latter includes both electrode-resolved OCP curves and the results of the DMA.4.1 Cyclic aging results
Fig. 1 visualizes the resistance (a) and capacity (b) evolution over the cycling process. All cells were cycled at exactly the same conditions. However, as shown in Fig. S2, SI (cell ② in panel b and cell ④ in panel c), both the anode of cell ② and cell ④ show edge plating, i.e., stripes of deposits of metallic sodium at the upper and lower edges of the anode, which can be induced by the anode overhang.24,78,79 Notably, while in cell ② the plating is only visible at the edge, in cell ④ the edge plating is much stronger, and, at some points, the plating even reaches the center of the anode. | ||
| Fig. 1 Resistance (a) and capacity (b) evolution of four cells cycled between 20% and 80% SOC at 1C in both charge and discharge direction. Both the resistance and the capacity values are normalized to the initial values of the first cycle. The resistance is evaluated after 10 s of a 1C pulse at 50% SOC in discharge direction, while the capacity is measured during discharge with 0.5C with constant current. | ||
A possible reason for the strong variance in aging behavior between the four cells are inhomogeneities within the electrodes during the production process. Specifically, Laufen et al.11 showed an uneven binder distribution at the anode and derived poor overall production quality from computer tomography scans of the cell. We thus expect that this low production quality, for instance due to an uneven binder distribution and subsequent local delamination of active material from the current collector, can lead to locally higher current densities during cycling, which eventually causes deposition of metallic sodium on the anode.80,81 Such local deposits are known to be a self-reinforcing process for lithium-based cells.82 However, further investigations explicitly examining the distribution of the binder in the electrodes are necessary to prove this hypothesis. The high variance in edge plating between the cells could be related to the uneven coating width of the electrodes of this cell type,11 which leads to different anode overhangs and thus different current density distributions at the edges of the electrodes.78,79 We note that the cells, that showed higher aging, also exhibited reduced coulombic efficiency after the initial pause in the RPT, which supports the theory of plating, and, connected to this, internal short circuits within the cells that age faster.24 The reduced coulombic efficiency was less pronounced or absent during the cycling procedure, which suggests that the effect, which is likely related to plating, gets more pronounced after rest periods.
While the observed variance in aging behavior complicates the quantitative analysis of the impact of the cyclic conditions, we deem this set of cells to be ideal for showcasing how to analyze and quantify the aging behavior of SIBs with layered oxide cathodes. Despite high aging variance, all cells achieved at least 2800 EFC with less than 30% capacity reduction; excluding cell ④, the others remained even above 90% of their initial capacity after 4000 EFC.
4.2 Change of kinetics over aging
To investigate the pulse resistance resolved over the SOC of the cells at electrode level, we built multiple T-cells in a three-electrode setup of the harvested electrodes of cells ①, ②, and ④ at EOT as described in section 2.4. Furthermore, we built several cells from the harvested electrodes of cell ① equipped with a TWRE for enhanced impedance analysis.650 cell is compared with those of two T-cells, all normalized to the cathode area.32 Due to the thicker separator in the T-cells, the absolute resistance values of the T-cells are expected to be higher than those of the cylindrical cell.83 Nonetheless, we expect that the pressure in the T-cells is higher than in the cylindrical cell due to the spring used for compression of the electrodes in the T-cell setup. Thus, we expect that the contact between the electrodes and separator is improved for the T-cell.84 This, together with the high porosity of more than 90% of this separator85 and the decrease in separator thickness due to the applied pressure,68 may be reasons why the T-cells do not show higher normalized resistance values than the 18650 cell.
 | ||
| Fig. 2
R
DC of an early-life 18650 cell compared to corresponding T-cells (a) and electrode-level resolved RDC over SOC of these T-cells (b). The resistance values are normalized to the area of the electrodes (T-cells) and the area of the cathode measured in ref. 32, respectively. The resistance is calculated from a 0.1C pulse in discharge direction of the full-cell for both the 18650 cell and the T-cells. Error bars represent the minimum and maximum values of two T-cells. | ||
As a result, the resistance of the T-cells is very similar to that of the early-life 18650 cell. We thus expect that the results of the T-cells are comparable to the cylindrical cells.
At EOT, the T-cells from cell ① show similar area-normalized resistance values compared to their corresponding 18650 cell (see Fig. S3(b), SI). However, similar to the T-cells of the early-life cell, at low SOC values of maximum 20%, the T-cell shows a higher resistance than the 18650 cell. The exact reasons for this are unclear. Nevertheless, the time constants of the 18650 cell and the T-cell are very similar over the whole SOC range. Thus, we expect that the cathode/anode resistance ratio and the electrode time constants remain consistent with the 18650 cell, whereas the absolute resistance values are only transferable at medium and high SOC values.
In addition to the RDC at full-cell level, we investigated the electrode-resolved resistances of two T-cells both from the outer and inner part of the jelly roll as explained in section 2.4. The cathode generally shows lower resistance than the anode. Starting from 40% SOC, the anode shows nearly identical resistance values at 100 ms and 10000 ms, while the cathodic resistance increases with time constant. This pattern suggests the anode is governed by processes with time constants below 100 ms in the plateau region, and is likely not limited by diffusion of sodium ions in the solid phase, which is in accordance with findings that quasi-metallic sodium is formed during the so-called pore-filling process.86–88 However this could not be observed for T-cells built from aged cells as shown in Fig. 3 and thus requires further investigation. In contrast, the cathode's strong time constant dependence indicates limitation by diffusion of sodium in the solid phase.89
 | ||
| Fig. 3 Electrode-resolved resistances from T-cells harvested from cells ① (a), ② (b), ④ (c) at EOT. The resistances are calculated from discharge pulses with a current rate of 0.1C. The error bars represent the maximum and minimum values of at least three T-cells. | ||
In Fig. 2, we can see that, in accordance with the results for the 18650 full-cells, at EOT the T-cells from cells ② and ④ show higher full-cell resistance values compared to the T-cells from cell ①. Here, for all cells the anodic resistance has increased compared to the early-life cell, while the cathodic resistance has remained relatively stable. The formation of a thicker SEI layer on the hard carbon anode during cycling could explain the increase of the anodic resistance.90–92 Such growth of the SEI would consume cyclable sodium and thus lead to a significant LI.19 For the T-cells of cell ①, no significant LI could be observed as explained in section 4.5.2, which is counterintuitive to the assumption of a thicker SEI layer on the anode. Possible other explanations for the increase in resistance at the anode are a more disordered structure of the SEI over the aging93 or changes in the particle structure of the hard carbon.94 To prove these hypotheses, further investigations explicitly focusing on the SEI at the anode of such commercial SIBs would be beneficial.
The cathodic resistance of the T-cells from cell ② has strongly increased compared to both the early-life cell and the other two aged cells. This increase is significant for all three investigated T-cells from cell ②, independent of whether they were built from the inner or outer part of the jelly roll. Thus, we further investigate the reason for this strong increase of the cathodic resistance in Fig. S4, SI. Here, we show the RDC at EOT of the T-cells built from cells ① (a), ② (b), and ④ (c) together with the resistances of the corresponding 18650 cells. Similar to the early-life cell shown in Fig. 2(a), the T-cells from cell ① and cell ④ at EOT show similar resistance values as their corresponding 18650 cells. In contrast, the T-cells from cell ② show substantially higher area-normalized resistance values than the corresponding 18650 cell. Thus, we except the high cathodic resistance of the T-cells from cell ② to be an artifact of the T-cell building process rather than a property of the cathode itself. As the procedure for the harvesting of all electrodes was identical, we cannot pinpoint the exact reason for this artifact.
4.3 Electrode-resolved impedance after aging
In addition to the pulse resistance, we also measured the electrode-resolved impedance spectra. The regular Swagelok T-cell setup with metallic sodium as reference used for the pulse resistance measurements is not suitable for impedance measurements.95,96 Thus, we built T-cells with TWRE which can be assumed to allow artifact-free electrode-resolved impedance measurements for SIBs.69,97 In Fig. 4, the PEIS of a T-cell with material harvested from cell ① is shown for the full-cell and each electrode. The impedance spectra support the previous findings of the pulse resistance measurements, where the cathode shows a lower resistance than the anode. Furthermore, the area-normalized impedance of both cathode and anode increases at low SOCs, whereas the increase is more pronounced for the anode. Specifically, at 10% SOC, the real part of the impedance at a frequency of 100 mHz is more than twice as high compared to the real part of the impedance at 20% SOC, which agrees with previous findings of drastically increased impedance for hard carbon at low SOCs and low frequencies, while high-frequency impedance is rarely affected.97 | ||
| Fig. 4 PEIS over SOC of a T-cell harvested from cell ① at EOT. Data of full-cell (a), cathode (b), and anode (c) are shown. PEIS was measured in the frequency range from 200 kHz to 25 mHz with an amplitude of 10 mV. For each tenfold decrease of the frequency, starting from 100 kHz, a cross represents real and imaginary part of the impedance at the respective frequency. | ||
We point out that the TWRE T-cells are indeed well-suited and reproducible, if the impedance spectra of SIBs are needed. Nevertheless, their higher complexity in the building procedure increases the risk of non-functioning T-cells. In addition, we do not recommend using TWRE cells for measurements of OCPs, as, despite the long time of sodiation of the tin-wire of ≈30 h, a drift in potential cannot be completely ruled out during the long time spans needed for the OCP measurements.
4.4 Change of balancing over aging
Similar to the resistance measurements, the change of the OCPs of the electrodes was investigated.650 cell, we show the full-cell pOCVs of both a T-cell built from an early-life cell and the corresponding 18650 cell with respect to the cathode area32 in Fig. 5(a). Here, both the pOCVs and the corresponding DVs show good agreement, while the difference in area-normalized capacity between the T-cell and the 18650 cell is less than 1%. We thus expect that both the pOCV and the OCPs of the T-cell allow insights for the 18650 cell.
 | ||
| Fig. 5 The pOCVs of an early-life 18650 cell compared to the corresponding full-cell T-cell (a) and pOCVs and OCPs of the respective T-cell (b). The capacities are normalized to the area of the electrodes (T-cell) and the area of the cathode of the 18650 cell measured in ref. 32, respectively. The T-cell was tested at a current of 0.05C, and the 18650 cell at a current of 0.1C. For both, pOCVs and OCPs, the charge and discharge direction are shown in solid and dashed lines, respectively. In addition, the differential voltage in discharge direction is shown. | ||
At EOT, the deviation between the area-normalized capacity of the T-cell harvested from cell ① and the corresponding 18650 cell is below 2% as shown in Fig. S3(a), SI, while the discharge pOCV and the corresponding DV of the 18650 cell are shifted towards higher capacity values compared to the T-cell. Furthermore, a characteristic peak in the DV of the 18650 cell at 0.35 mAh cm−2 is not visible in the T-cell. This additional feature as well as the deviation in area-normalized capacity might be explained, at least to some extent, by the fact that the T-cells do not have any anode overhang, while the 18650 cell has an geometrical anode overhang of 1.5 mm if upper and lower edge are combined.32
In Fig. 6, we show the OCPs and pOCVs of T-cells harvested from three different cells at EOT. Here, the cathode voltage is lowest for the T-cell from ①, and increases for ② and even further for ④, while simultaneously the anode shows the opposite trend. In addition, a characteristic peak in the DV reveals that the cathode is less utilized at low SOCs. Altogether, this shift indicates that the usable capacity of cell ② and especially cell ④ is dominated by LI.13 Apart from that, especially for cell ④ the electrode capacities seem to be strongly reduced compared to the early-life cell shown in Fig. 5(a), as distances between characteristic features in both anode and cathode are reduced. The quantitative analysis of this capacity loss is shown in section 4.5.2.
 | ||
| Fig. 6 The pOCVs and OCPs from T-cells harvested from cells ① (a), ② (b), and ④ (c). The T-cells were tested at a current of 0.05C and their capacities are normalized to the area of the electrodes (T-cell) and the area of the cathode of the 18650 cell measured in ref. 32, respectively. For both pOCVs and OCPs, the charge and discharge direction are shown in solid and dashed lines, respectively. In addition, the differential voltage in discharge direction is shown. | ||
4.5 Degradation mode analysis
We quantitatively determined the DMs of the cells using the DMA as described in section 3. To validate the numerically fitted DMs, we compare the reconstructed half-cell OCPs with the measured half-cell OCPs at EOT. Then, we show the results of the DMA for all investigated cells and explain the implications of the results of the DMA. | ||
| Fig. 7 The OCPs obtained from cell ① at EOT measured in a T-cell in comparison with the fitted OCPs including their shifting and scaling. The RMSE of the voltage difference between reconstructed and measured OCP is 5.91 mV for the cathode and 14.18 mV for the anode in the whole range of the reconstructed half-cell OCPs. | ||
A common metric for evaluating fitting quality is the RMSE between the measured and reconstructed pOCV curves.16,28,42 Nevertheless, a low voltage difference between those curves does not necessarily guarantee that the electrode OCPs are reconstructed correctly.40 For instance, a low voltage difference can also be achieved by shifting the anode toward lower and the cathode toward higher SOCs. Furthermore, a drift of the reference potentials during the measurement of the half-cell OCPs at BOT could occur.34,99 Such deviations could also be compensated during the fitting procedure by shifting the opposite electrode. Nevertheless, the physical meaning of the DMA relies on correct reconstruction of the scaling and shifting of the electrodes, and not the reconstruction of the full-cell OCV.
We therefore also compare the reconstructed OCP curves with the measured half-cell OCP curves at EOT. Our three-electrode setup (see section 2.4) ensures that the fitting captures not only the shape of the OCPs but also the stoichiometric drift between the two electrodes.40,100
The reconstructed and measured curves agree closely: the cathode exhibits an RMSE of 5.91 mV, while the anode shows 14.18 mV. The small residual deviation for the anode arises mainly from a slightly higher scaling of the reconstructed curve. This behavior may stem in part from minor fitting inaccuracies, but we expect that inhomogeneous anode aging, which is possibly related to anode edge plating (see Fig. S2, SI), also contributes to this deviation.101 Furthermore, as shown in Fig. S3(a), SI, the T-cell from cell ① already shows a slightly different pOCV compared to the 18650 cell. Thus, even a perfect reconstruction of the electrode potentials within the cylindrical cell would not lead to a perfect match between measured and reconstructed half-cell OCPs of the T-cell.
The low RMSE values between measured and reconstructed OCP prove the accuracy of the DM fitting procedure. Besides reproducing the full-cell OCV (in this case with an RMSE of 5.1 mV between measured and reconstructed OCV in the SOC region from 5% to 95% SOC), the method accurately captures the scaling and shifting of the half-cell OCPs, which demonstrates the capability of the DMA to quantify the LI and LAM over aging for SIBs.40
The results of the DMA are shown in Fig. 8. The fitting accuracy is shown in Fig. 8(d) as the RMSE of the voltage difference between the measured and reconstructed pOCV curves in the SOC range from 5% to 95%. For cells ①, ② and ③, the RMSE remains below 7.5 mV across all RPTs and cells. Lower fitting errors are possible by increasing the weighting of the accuracy of the OCV reconstruction in comparison to the correct fit of the DV in the fitting process (see eqn (2)). Despite the lower RMSE values, the accuracy of reconstructing the half-cells, and as a consequence also the DMs, is worse if the DV is not fitted correctly.72,73 The minor jumps over the aging (brief capacity drop and immediate recovery) in both anode and cathode capacity occur because the fitting did not explicitly forbid negative capacity losses, while it remains unclear whether these jumps are caused by fitting inaccuracies or real capacity recovery effects.
 | ||
| Fig. 8 DMs of the cells investigated in the study. Capacity loss of cathode and anode, as well as the loss of sodium inventory is shown with respect to the initial RPT for each cell. Fitting quality is measured by the RMSE of the voltage difference between the reconstructed and the measured OCVs from 5% to 95% SOC of the measured OCV. | ||
Interestingly, despite the high variance in capacity decline, cells ① and ② show similar LAMca as shown in Fig. 8(a). Cell ③ shows a similar trend, but exhibits a more pronounced increase in LAMca compared to the other cells. In contrast, the LAMan of these three cells varies strongly, whereas cell ③ shows more than twice the LAMan of cell ①. Despite this high variance in LAMan, the LAMca of all cells is considerably higher than the LAMan at EOT. This behavior is in contrast to LIBs, where the anode usually shows a higher LAM than the cathode for both anodes using pure graphite102,103 and silicon containing anodes16,40,41 under most test conditions.
The loss of sodium inventory differs strongly among the cells: for cells ② and ③, LI is dominating or at least comparable to LAMca. Here, the LI is matching the loss of capacity of the full-cell (see Fig. 1(b)) very well including both the shape of the capacity decay and the absolute values. More specifically, cell ② and ③ show capacity losses of 7.5% and 9.0% at EOT, respectively, which is in good agreement with values for LI of 8.0% and 9.4% at EOT. In contrast, for cell ①, LI reaches only 0.3% at EOT, whereas the capacity loss of the full-cell is 3.1%.
While the observed variance in LI is unexpected, it explains the pronounced capacity-loss differences in Fig. 1(b). We attribute the higher LI in cells ② and ③ not to a thicker SEI, but mainly to severe sodium edge plating confirmed in Fig. S2, SI and reported elsewhere.24 Roth et al.78 and Oehler et al.79 showed that local current peaks at jelly-roll ends due to anode overhang can lead to plating. The low potentials of the anode, reaching low values of approximately 50 mV versus metallic sodium at full-cell SOC of 100% as shown in Fig. 5(b), might further increase the risk of plating in comparison to LIB under inhomogeneous conditions. We expect that inhomogeneities in, for instance, anode geometry or bent ends at the anode layers even at BOT11 might strongly increase the risk of sodium plating. Furthermore, uneven distribution of binder material in the anode11 might increase the risk of inhomogeneous pressure distribution in the cell, which can itself lead to inhomogeneous aging82,101,104 and thus lead to plating even under moderate cycling conditions.82,105 The sodium plating, which is at least to some extent irreversible, also passivates regions of the hard carbon anode. As a consequence, also the opposite cathode regions may be partially deactivated. Thus, less plating can also explain the lower LAMca of cell ① compared to cells ②, ③ and ④.
Based on the low LI of cell ①, we hypothesize that the SEI formation in commercial SIBs with hard carbon anodes is, as proposed by Fitzpatrick et al.,35 less severe than expected from half-cell measurements against metallic sodium.17–19 More specifically, the LI related to continuous SEI growth might be even lower than in LIBs with graphite anodes.41,106 Here, further investigations using pristine cells could allow to explicitly quantify the LI due to initial SEI growth and continuous SEI growth separately. The combination with post-mortem analysis of the SEI could allow to show whether the assumption of a very stable SEI on hard carbon anodes in full-cell setups holds true.
In contrast to cells ①, ②, and ③, the DMs of cell ④ are hard to determine, which is represented in the strongly increased RMSE values between measured and reconstructed OCV. Furthermore, the 18650 cell shows a strongly increased hysteresis between charge and discharge pOCV at low SOCs, which is only visible for the cylindrical cell but not for the T-cells from the same cell as shown in Fig. S5, SI. The T-cells are built from the parts of the electrodes which are not affected by plating. Thus, we expect that the plated areas lead to an increase in overpotentials even at low current rates. Possible explanations for this behavior are higher local resistances of the plated areas, higher local current densities in the surrounding areas, and longer diffusion paths into the cathodic regions opposite to the plated anodic regions.107 In principle, such inhomogeneities can also be quantified with the DMA. Nonetheless, such inhomogeneities are usually assumed to be either equidistant108 or Gaussian distributed.45 Notably, this assumption may not hold true in the context of such pronounced plating. Furthermore, we expect that due to the few features in the OCPs and their derivatives of both electrodes, the fitted results are not reliable in case of such strong inhomogeneities.
The dominant degradation mode is the LAMca with approximately 5.7% at EOT for cell ①, where no extensive plating occurred. NMF cathodes are known to show strong LAM during cycling at high end of charge (EOC) such as 4.0 V or even 4.2 V due to structural degradation.109,110 Within this study, even during the RPTs the EOC voltage did not exceed 3.8 V. Thus, the observed LAMca being higher than LI and LAMan despite the moderate cycling conditions is unexpected.111 While the root cause cannot be isolated from the present data, cathode doping, which was not done for this cell type,32 is a plausible lever to enhance stability and potentially reduce the observed LAMca.23,112
Given the low values of LI and LAMan, the NMF cathode is limiting the capacity of the full-cell at EOT. Interestingly, despite limiting the capacity of the full-cell, LAMca is significantly higher than the loss of capacity of the full-cell, which is only 3.1% at EOT. This discrepancy is possible as the degree of cathode utilization is strongly increased over the lifetime of the cell, as shown in Fig. 9 as shift of βcathode from BOT (a) to EOT (b). In addition, the value of αcathode is substantially diminished at EOT compared to BOT. This indicates that a more extensive range of the cathode's SOC is utilized.
 | ||
| Fig. 9 Balancing of both electrodes of cell ① at BOT and EOT obtained via applying the DMA. The gray lines represent the measured charge pOCVs of the 18650 cells at BOT (a) and EOT (b). The positive values for βcat and βan indicate that the OCPs of the early-life cell are not sufficient to fully reconstruct the OCV at low SOCs, which is compensated by performing the fitting only in the SOC range from 5% to 95%. | ||
It is noteworthy that the decay of full-cell capacity exhibits a slight increase, even though the degressive trend of the LAMca is evident throughout the cell's lifespan. This indicates that at the beginning of the cycling, the cathode aging is hidden. As shown in Fig. 1, starting at ≈2000 EFC, the cathode is gradually starting to limit the full-cell capacity at both low and high full-cell SOCs. Such behavior, where suddenly either LI or LAMca becomes the limiting factor of the full-cell, is described for LIBs as knee-points.113,114 However, despite the theoretical case of LAMca starting to limit the full-cell capacity at a certain point in the lifetime of the cell,27,114 the shapes of anode and cathode lead to a continuous increase of usage of the cathode at low full-cell SOCs over the lifetime of the cell instead of a sudden limitation of the full-cell capacity by the cathode (see Fig. 9). We further note that here, despite LIBs which usually limit capacity either by negative or positive electrode at low or high full-cell SOC, respectively,13,27 the positive electrode limits the capacity of the full-cell both at low and high full-cell SOC at EOT. While more research is needed to show the generality of these findings for SIBs, this implies that (i) electrode-resolved aging estimation for SIBs can increase the prediction of the aging of SIBs at an early stage of aging and (ii) a capacitive overdimensioned cathode might further increase the lifetime of SIBs. Conversely, in the latter scenario, a gradual increase of the EOC voltage over the cell's lifespan might prove advantageous in preventing overcharging of the anode due to the oversized cathode.
5 Conclusion
In the present study, we showed, how to measure and analyze the change of kinetics and the balancing of commercial SIBs over aging, by using four commercial 18650 SIBs cyclically aged for at least 3400 EFC as examples. We built 19 three-electrode T-cells to measure the electrode-resolved resistances and OCPs from an early-life cell as well as three aged cells showing different aging trajectories.
We have shown how three-electrode setups, in our case Swagelok T-cells, can be used to differentiate between pulse resistance contributions of anode and cathode over SOC. A comparison of the resistance of the 18650 cell and T-cells reveals that the findings of the three-electrode setup are transferable to cylindrical cells. In addition, we built T-cells with TWRE to measure the PEIS at electrode level. These results show that the resistance is dominated by the anode over the whole SOC range for both T-cells from an early-life cell as well as for the three different cells at EOT. The strong increase of pulse resistance and impedance at low SOC is dominated by an increase of the anode resistance.
After validating the transferability of the pOCV between T-cell and 18650 cell, we used the OCPs of an early-life cell to apply the so-called DMA to quantitatively determine the DMs of the investigated cells. We used a limited SOC range of the measured OCPs for the DMA, as the T-cell was only measured within the recommended full-cell voltage limits of the 18650 cell. The comparison between the reconstructed and measured half-cell OCPs at EOT allows us to show that despite the limited features in both electrodes, both the anode and cathode OCP can be accurately captured over the aging process. This is reflected in the RMSE between the reconstructed and measured OCP being as low as 5.91 mV for the cathode and 14.18 mV for the anode. The high accuracy of the fitting is not only reflected in the low RMSE values of the difference between measured and reconstructed half-cell OCPs at EOT, but also in the low RMSE of the difference between measured and reconstructed full-cell OCV staying below 7 mV for all cells and RPTs except one outlier cell. The direct comparison of the OCPs from T-cells from the early-life cell and from aged cells at EOT allows us to prove that the invariance of the shape of the OCPs over aging, is valid for the investigated cells.
The results of the DMA reveal that for all investigated cells, the loss of capacity at the cathode is significantly higher than at the anode. The aging of those cells showing the fastest capacity decay was dominated by the loss of charge carrier inventory, which we connect with severe plating visible at the anode in the post mortem analysis (PMA). We expect that this plating might be a self-reinforcing process, which will be discussed in more detail in our upcoming publications. The cell which was not affected by plating showed almost negligible LI and low loss of anode capacity. This cell clearly indicates that the aging of these SIBs (if they reach high production quality and do not suffer from severe plating) is dominated by the cathode. However, this loss of capacity at the cathode is partially counterbalanced by the increased utilization of the cathode in the early phase of aging.
Author contributions
Mathias Rehm: conceptualization, methodology, software, data curation, visualization, writing – original draft, writing – review & editing. Julius Bahrke: conceptualization, methodology, data curation, writing – review & editing. Johannes Natterer: methodology, data curation, writing – original draft, writing – review & editing. Leon Milutinovic: data curation, writing – review & editing. Franz Roehrer: conceptualization, writing – review & editing. Andreas Jossen: supervision, writing – review & editing.Conflicts of interest
All authors declare that they have no conflicts of interest.Data availability
Data for this article is available at https://doi.org/10.14459/2025mp1837893.Supplementary information (SI): data includes (i) the pOCVs in both charge and discharge direction and the 0.5C discharge as well as the resistance over SOC for all four cells at all aging steps, (ii) the time series data of the T-cells with sodium reference including the pulse data, (iii) PEIS and GEIS data of T-cells with TWRE, and (iv) the results of the DMA. See DOI: https://doi.org/10.1039/d5eb00221d.
Acknowledgements
This research was financially supported by the Federal Ministry for Economic Affairs and Energy in the project ProMoBiS (03ETE046A) and by the Technical University of Munich. The responsibility for this publication rests with the authors. The support in building TWRE T-cells by Rebecca Wilhelm and fruitful discussions with Philipp Rapp are gratefully acknowledged. Moreover, we would like to thank Igor Brakus for his help in conducting the aging study, Can Korkmaz and Yannis Riedel for their help in the implementation and visualization of the DMA and Lysander Sirach for proofreading the manuscript.Declaration of AI and AI-assisted technologies in the writing process: during the preparation of this work the authors used Grammarly and ChatGPT for individual sections to improve language and readability. After using these tools, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
References
- S. Link, L. Schneider, A. Stephan, L. Weymann and P. Plötz, Nat. Energy, 2025, 10, 526–534 CrossRef.
- S. Bobba, C. Latunussa, F. M. Manni and F. Mathieux, Deep dive on critical raw materials for batteries in the EU, 2025 Search PubMed.
- S. Carrara, S. Bobba, D. Blagoeva, P. Alves Dias, K. Georgitzikis, M. Grohol, A. Itul, T. Kuzov, C. E. L. Latunussa, L. Lyons, G. Malano, T. Maury, Á. Prior, J. Somers, T. Telsnig, C. Veeh, D. M. A. G. Wittmer, C. Black, D. W. Pennington and M. Christou, Supply chain analysis and material demand forecast in strategic technologies and sectors in the EU – A foresight study, Publications Office of the European Union, 2023 Search PubMed.
- T. Wicke, L. Weymann, C. Neef and J. Tübke, Batteries, 2025, 11, 76 CrossRef CAS.
- A. Stephan, T. Hettesheimer, C. Neef, T. Schmaltz, S. Link, M. Stephan, J. L. Heizmann and A. Thielmann, Alternative Battery Technologies Roadmap 2030+, Fraunhofer Institute for Systems and Innovation Research ISI, 2023 Search PubMed.
- N. Tapia-Ruiz, A. R. Armstrong, H. Alptekin, M. A. Amores, H. Au, J. Barker, R. Boston, W. R. Brant, J. M. Brittain, Y. Chen, M. Chhowalla, Y.-S. Choi, S. I. R. Costa, M. Crespo-Ribadeneyra, S. A. Cussen, E. J. Cussen, W. I. F. David, A. V. Desai, S. A. M. Dickson, E. I. Eweka, J. D. Forero-Saboya, C. P. Grey, J. M. Griffin, P. Gross, X. Hua, J. T. S. Irvine, P. Johansson, M. O. Jones, M. Karlsmo, E. Kendrick, E. Kim, O. V. Kolosov, Z. Li, S. F. L. Mertens, R. Mogensen, L. Monconduit, R. E. Morris, A. J. Naylor, S. Nikman, C. A. O’Keefe, D. M. C. Ould, R. G. Palgrave, P. Poizot, A. Ponrouch, S. Renault, E. M. Reynolds, A. Rudola, R. Sayers, D. O. Scanlon, S. Sen, V. R. Seymour, B. Silván, M. T. Sougrati, L. Stievano, G. S. Stone, C. I. Thomas, M.-M. Titirici, J. Tong, T. J. Wood, D. S. Wright and R. Younesi, J. Phys.: Energy, 2021, 3, 031503 CAS.
- J. Ruppert, P. Voß, L. Ihlbrock, J. Palm, S. Lux and J. Leker, J. Power Sources Adv., 2025, 36, 100190 CrossRef CAS.
- A. Yao, S. M. Benson and W. C. Chueh, Nat. Energy, 2025, 10, 404–416 CrossRef.
- X. Cai, Y. Yue, Z. Yi, J. Liu, Y. Sheng and Y. Lu, Nano Energy, 2024, 129, 110052 CrossRef CAS.
- M. Naumann, M. Schimpe, P. Keil, H. C. Hesse and A. Jossen, J. Energy Storage, 2018, 17, 153–169 CrossRef.
- H. Laufen, S. Klick, H. Ditler, K. L. Quade, A. Mikitisin, A. Blömeke, M. Schütte, D. Wasylowski, M. Sonnet, L. Henrich, A. Schwedt, G. Stahl, F. Ringbeck, J. Mayer and D. U. Sauer, Cell Rep. Phys. Sci., 2024, 5, 101945 CrossRef CAS.
- E. Irujo, A. Berrueta, P. Sanchis and A. Ursúa, 2024 International Conference on Renewable Energies and Smart Technologies (REST), 2024, pp. 1–5.
- C. R. Birkl, M. R. Roberts, E. McTurk, P. G. Bruce and D. A. Howey, J. Power Sources, 2017, 341, 373–386 CrossRef CAS.
- J. S. Edge, S. O'Kane, R. Prosser, N. D. Kirkaldy, A. N. Patel, A. Hales, A. Ghosh, W. Ai, J. Chen, J. Yang, S. Li, M.-C. Pang, L. Bravo Diaz, A. Tomaszewska, M. W. Marzook, K. N. Radhakrishnan, H. Wang, Y. Patel, B. Wu and G. J. Offer, Phys. Chem. Chem. Phys., 2021, 23, 8200–8221 RSC.
- W. Vermeer, G. R. Chandra Mouli and P. Bauer, IEEE Trans. Transp. Electrif., 2022, 8, 2205–2232 Search PubMed.
- J. Schmitt, M. Rehm, A. Karger and A. Jossen, J. Energy Storage, 2023, 59, 106517 CrossRef.
- J. B. Robinson, D. P. Finegan, T. M. M. Heenan, K. Smith, E. Kendrick, D. J. L. Brett and P. R. Shearing, J. Electrochem. Energy Convers. Storage, 2018, 15, 1–9 Search PubMed.
- A. Le Ma, A. J. Naylor, L. Nyholm and R. Younesi, Angew. Chem., Int. Ed., 2021, 60, 4855–4863 CrossRef PubMed.
- A. Le Ma, A. Buckel, A. Hofmann, L. Nyholm and R. Younesi, Adv. Sci., 2024, 11, e2306771 Search PubMed.
- J. Weaving, J. Robinson, D. Ledwoch, G. He, E. Kendrick, P. Shearing and D. Brett, Sodium–Ion Batteries, Wiley, 2022, pp. 501–530 Search PubMed.
- T. Zhang, Y. Li, Z. Song, Y. Huang, F. Li, S. Cheng and F. Li, J. Energy Chem., 2025, 103, 294–315 CrossRef CAS.
- M. Sarkar, R. Hossain and V. Sahajwalla, J. Power Sources, 2023, 584, 233577 Search PubMed.
- F. A. Dorau, A. Sommer, J. Koloch, R. Röß-Ohlenroth, M. Schreiber, M. Neuner, K. Abo Gamra, Y. Lin, J. Schöberl, P. Bilfinger, S. Grabmann, B. Stumper, L. Katzenmeier, M. Lienkamp and R. Daub, J. Electrochem. Soc., 2024, 171, 090521 CrossRef CAS.
- L. Streck, T. Roth, H. Bosch, C. Kirst, M. Rehm, P. Keil and A. Jossen, J. Electrochem. Soc., 2024, 171, 080531 Search PubMed.
- S. Klick, H. Laufen, M. Schütte, B. Qian, K. L. Quade, C. Rahe, M. Dubarry and D. U. Sauer, Batteries Supercaps, 2025, 8, 1–14 Search PubMed.
- R. Li, N. D. Kirkaldy, F. F. Oehler, M. Marinescu, G. J. Offer and S. E. J. O'Kane, Nat. Commun., 2025, 16, 2776 CrossRef CAS PubMed.
- M. Dubarry, C. Truchot and B. Y. Liaw, J. Power Sources, 2012, 219, 204–216 Search PubMed.
- J. Schmitt, M. Schindler, A. Oberbauer and A. Jossen, J. Power Sources, 2022, 532, 231296 CrossRef CAS.
- S. Friedrich, V. Savvin, R. Wilhelm and A. Jossen, J. Electrochem. Soc., 2025, 172, 010521 CrossRef CAS.
- R. Prosser, G. Offer and Y. Patel, J. Electrochem. Soc., 2021, 168, 030532 CrossRef CAS.
- A. Kunz, C. Kirst, A. Durdel, J. P. Singer and A. Jossen, J. Power Sources, 2025, 627, 235732 Search PubMed.
- M. Rehm, M. Fischer, M. R. Gomez, M. Schütte, D. U. Sauer and A. Jossen, J. Power Sources, 2025, 633, 236290 CrossRef CAS.
- S. Lee, J. B. Siegel, A. G. Stefanopoulou, J.-W. Lee and T.-K. Lee, J. Electrochem. Soc., 2020, 167, 090531 CrossRef CAS.
- K. Pfeifer, S. Arnold, J. Becherer, C. Das, J. Maibach, H. Ehrenberg and S. Dsoke, ChemSusChem, 2019, 12, 3312–3319 CrossRef CAS PubMed.
- J. R. Fitzpatrick, B. E. Murdock, P. K. Thakur, T.-L. Lee, S. Fearn, A. J. Naylor, D. Biswas and N. Tapia-Ruiz, Adv. Sci., 2025, e04717 Search PubMed.
- J. Conder and C. Villevieille, Chem. Commun., 2019, 55, 1275–1278 RSC.
- M. Schütte, H. Laufen, D. Luder, H. Ditler, J. Kern, S. Klick, M. Junker, G. Stahl, F. Frie and D. U. Sauer, J. Energy Storage, 2025, 107, 114931 CrossRef.
- Z. Li, Z. Jian, X. Wang, I. A. Rodríguez-Pérez, C. Bommier and X. Ji, Chem. Commun., 2017, 53, 2610–2613 RSC.
- I. Bloom, J. Christophersen and K. Gering, J. Power Sources, 2005, 139, 304–313 CrossRef CAS.
- S. Friedrich, F. Dengler, M. Bock, M. Stuckenberger and A. Jossen, J. Electrochem. Soc., 2025, 070516 CrossRef CAS.
- L. Wildfeuer, A. Karger, D. Aygül, N. Wassiliadis, A. Jossen and M. Lienkamp, J. Power Sources, 2023, 560, 232498 CrossRef CAS.
- M. Rogge and A. Jossen, Batteries Supercaps, 2024, 7, 1–12 Search PubMed.
- T. Hofmann, J. Li, J. Hamar, S. Erhard and J. P. Schmidt, J. Power Sources, 2024, 596, 234107 CrossRef CAS.
- M. Dubarry and D. Beck, J. Electrochem. Soc., 2024, 171, 080541 CrossRef CAS.
- C. Kirst, A. Karger, J. P. Singer and A. Jossen, J. Power Sources, 2024, 602, 234341 CrossRef CAS.
- J. P. Schmidt, H. Y. Tran, J. Richter, E. Ivers-Tiffée and M. Wohlfahrt-Mehrens, J. Power Sources, 2013, 239, 696–704 CrossRef CAS.
- S. Jung, J. Power Sources, 2014, 264, 184–194 CrossRef CAS.
- J. Schmitt, M. Schindler and A. Jossen, J. Power Sources, 2021, 506, 230240 CrossRef CAS.
- N. Kirkaldy, M. A. Samieian, G. J. Offer, M. Marinescu and Y. Patel, ACS Appl. Energy Mater., 2022, 5, 13367–13376 CrossRef CAS PubMed.
- A. Farmann and D. U. Sauer, J. Power Sources, 2017, 347, 1–13 CrossRef CAS.
- J. Hodakovska, L. Britala, A. Mezulis, L. Grinberga, G. Bajars and G. Kucinskis, J. Solid State Electrochem., 2025, 29, 4187–4198 CrossRef CAS.
- H. Chen, S. Yang, W. Zhang, C. Zhang, B. Sun and D. Chen, J. Power Sources, 2024, 614, 234921 CrossRef CAS.
- Y. Zou, Y. Cheng, J. Lin, Y. Xiao, F. Ren, K. Zhou, M.-S. Wang, D.-Y. Wu, Y. Yang and J. Zheng, J. Power Sources, 2022, 532, 231372 CrossRef CAS.
- A. Saleem, L. L. Shaw, R. Iqbal, A. Hussain, A. R. Akbar, B. Jabar, S. Rauf and M. K. Majeed, Energy Storage Mater., 2024, 69, 103440 CrossRef.
- M.-T. F. Rodrigues, J. Gim, A. Tornheim, O. Kahvecioglu, M. Luo, A. Y. R. Prado, S. E. Trask, J. Croy and D. P. Abraham, J. Electrochem. Soc., 2022, 169, 040539 CrossRef CAS.
- F. Friedrich, B. Strehle, A. T. S. Freiberg, K. Kleiner, S. J. Day, C. Erk, M. Piana and H. A. Gasteiger, J. Electrochem. Soc., 2019, 166, A3760–A3774 CrossRef CAS.
- H. Koga, L. Croguennec, M. Ménétrier, K. Douhil, S. Belin, L. Bourgeois, E. Suard, F. Weill and C. Delmas, J. Electrochem. Soc., 2013, 160, A786–A792 CrossRef CAS.
- P. M. Csernica, S. S. Kalirai, W. E. Gent, K. Lim, Y.-S. Yu, Y. Liu, S.-J. Ahn, E. Kaeli, X. Xu, K. H. Stone, A. F. Marshall, R. Sinclair, D. A. Shapiro, M. F. Toney and W. C. Chueh, Nat. Energy, 2021, 6, 642–652 CrossRef CAS.
- L. Zeng, H. Liang, B. Qiu, Z. Shi, S. Cheng, K. Shi, Q. Liu and Z. Liu, Adv. Funct. Mater., 2023, 33, 1–34 Search PubMed.
- W. Zhao, S. Luo, L. Qian, R. Huang, G. Wang, H. Zhang, G. Hao and S. Yan, J. Mater. Chem. C, 2025, 13, 4938–4948 RSC.
- P.-F. Wang, Y. You, Y.-X. Yin and Y.-G. Guo, Adv. Energy Mater., 2018, 8, 1–23 Search PubMed.
- P. Jocher, F. Roehrer, M. Rehm, T. Idrizi, A. Himmelreich and A. Jossen, J. Energy Storage, 2024, 98, 112931 CrossRef.
- M. R. Gomez, S. Ludwig, P. Jocher, A. Frank, Y. Fedoryshyna and A. Jossen, J. Energy Storage, 2025, 123, 116640 CrossRef.
- L. A. Middlemiss, A. J. R. Rennie, R. Sayers and A. R. West, J. Electrochem. Soc., 2024, 171, 010528 CrossRef CAS.
- V. Marangon, K. Bischof, A. A. Regalado, M. Keppeler, M. Pogosova, M. Wan, J. Choi, S. Fleischmann, T. Diemant, M. Wohlfahrt-Mehrens, M. Hölzle, T. Waldmann and D. Bresser, J. Power Sources, 2025, 634, 236496 CrossRef CAS.
- F. Roehrer, P. Jocher, M. Rehm, A. Graule and A. Jossen, J. Power Sources, 2025, 640, 236573 CrossRef CAS.
- K. Smith, P. Gasper, A. M. Colclasure, Y. Shimonishi and S. Yoshida, J. Electrochem. Soc., 2021, 168, 100530 CrossRef CAS.
- S. Friedrich, S. Stojecevic, P. Rapp, S. Helmer, M. Bock, A. Durdel, H. A. Gasteiger and A. Jossen, J. Electrochem. Soc., 2024, 171, 050540 CrossRef CAS.
- F. Linsenmann, D. Pritzl and H. A. Gasteiger, J. Electrochem. Soc., 2019, 166, A3668–A3674 CrossRef CAS.
- M. Rehm, J. Natterer, J. Eizenhammer, M. Guenthner, S. Kuecher, F. Roehrer, C. Korkmaz and A. Jossen, How to determine the degradation modes of lithium-ion batteries with silicon–graphite blend electrodes, J. Power Sources, 2026, 1–14 Search PubMed.
- M. Rehm, J. Eizenhammer and M. Günthner, tum-ees/DegradationModeAnalysis: v1.1.1 (paper release), Zenodo, 2025 DOI:10.5281/zenodo.17979026.
- M. Lewerenz, A. Marongiu, A. Warnecke and D. U. Sauer, J. Power Sources, 2017, 368, 57–67 CrossRef CAS.
- H. Kato, Y. Kobayashi and H. Miyashiro, J. Power Sources, 2018, 398, 49–54 Search PubMed.
- S. L. Hahn, M. Storch, R. Swaminathan, B. Obry, J. Bandlow and K. P. Birke, J. Power Sources, 2018, 400, 402–414 CrossRef CAS.
- I. Bin-Mat-Arishad, B. Wimarshana and A. Fly, J. Energy Storage, 2023, 70, 107884 CrossRef.
- A. Singh, P. Nakhanivej, Y. Monaghan, M. J. Loveridge and A. Barai, J. Power Sources Adv., 2025, 33, 100175 CrossRef.
- J. Chen, M. N. Marlow, Q. Jiang and B. Wu, J. Energy Storage, 2022, 45, 103669 CrossRef.
- T. Roth, A. Frank, F. Oehler, A. Graule, S. Kücher and A. Jossen, J. Electrochem. Soc., 2024, 171, 050547 CrossRef CAS.
- F. F. Oehler, T. Roth, A. Frank, A. Graule, S. Kücher and A. Jossen, J. Electrochem. Soc., 2025, 172, 010505 CrossRef CAS.
- H. Hijazi, Z. Ye, L. Zhang, J. Deshmukh, M. B. Johnson, J. R. Dahn and M. Metzger, J. Electrochem. Soc., 2023, 170, 070512 Search PubMed.
- K. Bischof, M. Feinauer, A. Aracil Regalado, M. Wohlfahrt-Mehrens, M. Hölzle and T. Waldmann, J. Electrochem. Soc., 2025, 172, 090520 CrossRef CAS.
- A. J. Smith, Y. Fang, A. Mikheenkova, H. Ekström, P. Svens, I. Ahmed, M. J. Lacey, G. Lindbergh, I. Furó and R. W. Lindström, J. Power Sources, 2023, 573, 233118 CrossRef CAS.
- D. V. Horváth, R. Tian, C. Gabbett, V. Nicolosi and J. N. Coleman, J. Electrochem. Soc., 2022, 169, 030503 CrossRef.
- A. Schommer, M. O. Corzo, P. Henshall, D. Morrey and G. Collier, J. Power Sources, 2025, 629, 236019 CrossRef CAS.
- S. Kücher, T. Schabenberger, E. Zonta and A. Jossen, J. Electrochem. Soc., 2025, 172, 070512 CrossRef.
- Z. Tang, R. Zhang, H. Wang, S. Zhou, Z. Pan, Y. Huang, D. Sun, Y. Tang, X. Ji, K. Amine and M. Shao, Nat. Commun., 2023, 14, 6024 Search PubMed.
- H. Au, H. Alptekin, A. C. S. Jensen, E. Olsson, C. A. O'Keefe, T. Smith, M. Crespo-Ribadeneyra, T. F. Headen, C. P. Grey, Q. Cai, A. J. Drew and M.-M. Titirici, Energy Environ. Sci., 2020, 13, 3469–3479 RSC.
- L. Kitsu Iglesias, S. D. Marks, N. Rampal, E. N. Antonio, R. de Ferreira Menezes, L. Zhang, D. Olds, S. E. Weitzner, K. G. Sprenger, L. F. Wan and M. F. Toney, Small, 2025, e2505561 Search PubMed.
- Y.-J. Guo, R.-X. Jin, M. Fan, W.-P. Wang, S. Xin, L.-J. Wan and Y.-G. Guo, Chem. Soc. Rev., 2024, 53, 7828–7874 RSC.
- W. Li, H. Xie, S. Lin, Y. Qin, J. Zeng, P. Zhang and J. Zhao, J. Power Sources, 2025, 639, 236635 CrossRef CAS.
- S. Daboss, N. Franke, B. Fraboni, C. Kranz and T. Cramer, J. Microsc., 2025 DOI:10.1111/jmi.13417.
- K. Schutjajew, T. Tichter, J. Schneider, M. Antonietti, C. Roth and M. Oschatz, Phys. Chem. Chem. Phys., 2021, 23, 11488–11500 RSC.
- J. Meng, G. Jia, H. Yang and M. Wang, Front. Chem., 2022, 10, 986541 Search PubMed.
- Y. Zhang, S.-W. Zhang, Y. Chu, J. Zhang, H. Xue, Y. Jia, T. Cao, D. Qiu, X. Zou, D.-W. Wang, Y. Tao, G. Zhong, Z. Peng, F. Kang, W. Lv and Q.-H. Yang, Nat. Commun., 2025, 16, 3634 Search PubMed.
- M. Ender, A. Weber and I.-T. Ellen, J. Electrochem. Soc., 2011, 159, A128–A136 CrossRef.
- S. Solchenbach, D. Pritzl, E. J. Y. Kong, J. Landesfeind and H. A. Gasteiger, J. Electrochem. Soc., 2016, 163, A2265–A2272 CrossRef CAS.
- F. Linsenmann, D. Pritzl and H. A. Gasteiger, J. Electrochem. Soc., 2021, 168, 010506 Search PubMed.
- P. Rodríguez-Iturriaga, S. Thatipamula and S. Onori, Future Batteries, 2025, 6, 100056 CrossRef.
- S. E. Lee and M. H. Tang, J. Electrochem. Soc., 2019, 166, A3260–A3264 Search PubMed.
- W. Diao, J. Kim, M. H. Azarian and M. Pecht, Electrochim. Acta, 2022, 431, 141143 CrossRef CAS.
- H. Wang and J. F. Whitacre, J. Energy Storage, 2021, 35, 102244 CrossRef.
- N. Kim, N. Shamim, A. Crawford, V. V. Viswanathan, B. M. Sivakumar, Q. Huang, D. Reed, V. Sprenkle and D. Choi, J. Power Sources, 2022, 546, 231949 CrossRef CAS.
- W. Xie, R. He, X. Gao, X. Li, H. Wang, X. Liu, X. Yan and S. Yang, Electrochim. Acta, 2021, 392, 138979 CrossRef CAS.
- P. Blazek, P. Westenberger, S. Erker, A. Brinek, T. Zikmund, D. Rettenwander, N. P. Wagner, J. Keckes, J. Kaiser, T. Kazda, P. Vyroubal, M. Macak and J. Todt, J. Energy Storage, 2022, 52, 104563 Search PubMed.
- Y. Xie, S. Wang, R. Li, D. Ren, M. Yi, C. Xu, X. Han, L. Lu, B. Friess, G. Offer and M. Ouyang, J. Power Sources, 2022, 542, 231753 CrossRef CAS.
- A. Karger, S. E. J. O'Kane, M. Rogge, C. Kirst, J. P. Singer, M. Marinescu, G. J. Offer and A. Jossen, J. Electrochem. Soc., 2024, 171, 090512 CrossRef CAS.
- S. Paarmann, K. Schuld and T. Wetzel, Energy Technol., 2022, 10, 1–13 Search PubMed.
- J. P. Fath, D. Dragicevic, L. Bittel, A. Nuhic, J. Sieg, S. Hahn, L. Alsheimer, B. Spier and T. Wetzel, J. Energy Storage, 2019, 25, 100813 CrossRef.
- S. Lin, W. Li, H. Yang, M. Chen, H. Xie, Y. Qin, J. Zeng, P. Zhang and J. Zhao, EES Batteries, 2025, 1, 935–946 Search PubMed.
- X. Zhao, L. Hou, Q. Liu, Y. Zhao, D. Mu, Z. Zhao, L. Li, R. Chen and F. Wu, J. Mater. Chem. A, 2024, 12, 12443–12451 RSC.
- L. F. Pfeiffer, Y. Li, M. Mundszinger, J. Geisler, C. Pfeifer, D. Mikhailova, A. Omar, V. Baran, J. Biskupek, U. Kaiser, P. Adelhelm, M. Wohlfahrt-Mehrens, S. Passerini and P. Axmann, Chem. Mater., 2023, 35, 8065–8080 CrossRef CAS.
- G. Chen, Y. Luo, J. Liu, X. Gao, Y. Pu, P. Niu, W. Zhao, W. Tong, T. Zeng, X. Wang, L. Cao, J. Zheng, Z. Ma, N. Zhang, W. Ji, Z. Tan, P. Miao, J. Zhang, J. Wang, R. Wang and Y. Xiao, J. Energy Chem., 2025, 109, 839–849 CrossRef CAS.
- P. M. Attia, A. Bills, F. Brosa Planella, P. Dechent, G. dos Reis, M. Dubarry, P. Gasper, R. Gilchrist, S. Greenbank, D. Howey, O. Liu, E. Khoo, Y. Preger, A. Soni, S. Sripad, A. G. Stefanopoulou and V. Sulzer, J. Electrochem. Soc., 2022, 169, 060517 CrossRef CAS.
- K. Smith, A. Saxon, M. Keyser, B. Lundstrom, Z. Cao and A. Roc, 2017 American Control Conference (ACC), 2017, pp. 4062–4068.
| This journal is © The Royal Society of Chemistry 2026 |