
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Benefits and drawbacks of high-temperature formation cycling for lithium-ion batteries
Sebastian Klick *abc,
Philipp Finsterd,
Karl Martin Graffbcf,
Carlos Ziebertd,
Gereon Stahlabc,
Felix Weberbcef,
Egbert Figgemeierbcef and
Dirk Uwe Sauerabce
*abc,
Philipp Finsterd,
Karl Martin Graffbcf,
Carlos Ziebertd,
Gereon Stahlabc,
Felix Weberbcef,
Egbert Figgemeierbcef and
Dirk Uwe Sauerabce
aChair for Electrochemical Energy Conversion and Storage Systems, Institute for Power Electronics and Electrical Drives (ISEA), RWTH Aachen University, Campus-Boulevard 89, 52074 Aachen, Germany. E-mail: batteries@isea.rwth-aachen.de
bJuelich Aachen Research Alliance, JARA-Energy, Germany
cCenter for Ageing, Reliability and Lifetime Prediction of Electrochemical and Power Electronic Systems (CARL), Campus-Boulevard 89, 52074 Aachen, Germany
dKarlsruhe Institute of Technology (KIT), Institute for Applied Materials-Applied Materials Physics (IAM-AWP), Kaiserstraße 12, 76131 Karlsruhe, Germany
eHelmholtz Institute Münster (HIMS), IMD-4, Forschungszentrum, Jülich, Jülich 52425, Germany
fAging Processes and Lifetime Prediction of Batteries, Institute for Power Electronics and Electrical Drives (ISEA), RWTH Aachen University, Campus-Boulevard 89, 52074 Aachen, Germany
First published on 9th April 2026
Abstract
Formation is a key process in battery manufacturing. During this process the solid electrolyte interface (SEI) and cathode electrolyte interface (CEI) are formed influencing the performance and lifetime of the cell. In this work, we investigate the effects of three different formation temperatures (45 °C, 60 °C, and 80 °C) on cell properties after formation and degradation of the battery during cycle life testing. We found that formation at 80 °C causes a higher loss of lithium and active material during formation. Nonetheless, this high-temperature formation reduces capacity fade during cycling. By coupling degradation mode analysis, statistical tools, and analysing the composition of the electrolyte, we conclude that soluble SEI components cause faster capacity fade in cells formed at the two lower temperatures. In addition, transesterification was suppressed by the high temperature formation. This was confirmed by gas chromatography and Thermal Electrolyte Analysis (TEA).
Broader contextLithium ion batteries power a wide range of different applications: from grid storage to small wearables. The long lifetime of lithium ion batteries is enabled by the formation of protective layers during the first cycle(s). During these formation cycles protective layers (SEI and CEI) are formed on the electrodes. These protective layers enable the exceptional lifetimes achieved with modern batteries. However, the links between formation conditions, SEI/CEI composition, cell performance and lifetime are not well understood. The formation process is a slow and costly process during battery manufacturing. An improved understanding of the formation processes enables more efficient battery manufacturing and improved lifetime. Our work contributes to this understanding by comparing three formation temperatures with respect to their effects on electrolyte composition, cell performance and degradation under two different ageing conditions. We identified increased loss of lithium and active material during formation at 80 °C as a major cause of a lower capacity of these cells after formation. Despite the superior initial capacity of cells formed at 45 °C and 60 °C, cells formed at 80 °C exhibited improved stability resulting in significant lifetime improvements. |
Introduction
The formation of lithium ion batteries is an important step during battery production. During this process, which can include several cycles, the SEI is formed.1–5 Battery manufacturers use proprietary processes which are not disclosed to the research community.6 Thus, despite its importance for battery lifetime and performance, little is published about the choice of different factors and their influence on SEI-composition, battery performance and the electrolyte.7 In addition to achieving stable, well-performing batteries, cost reduction is another focus for optimised formation protocols.6Key factors for the design of formation protocols are the current rate applied during cycling,8,9 the number and voltage range of cycles,1,4,10 and the temperature during cycling.9,11–15
Schomburg et al.7 provide an extensive review on recent publications related to the formation process. They identified conflicting evidence with regard to the benefits of increased formation temperature. Higher temperature in the range of up to 50 °C reduces formation time12 and lead to a more homogeneous SEI according to ref. 11 and 14. Cui et al.9 also identified positive effects of higher formation temperatures up to 55 °C on the resistance and the stability of the cells with single crystal NMC532 and artificial graphite as positive and negative electrode active material, respectively. However, German et al.13 identified lower formation temperatures as beneficial for long-term stability. They investigated formation at temperatures between 15 and 45 °C in large format NMC111/graphite cells with an EC-based electrolyte and LiPF6 as conductive salt.13 In addition to attempts of analysing differences in SEI composition by FTIR and XPS they investigated cyclic ageing, showing that the lowest formation temperature in their test leads to the best long-term stability.13
The highest temperature that was investigated in the aforementioned studies does not exceed 60 °C. Rodrigues et al.15 applied an electrolyte system based on ionic liquids and LiTFSI and identified benefits of high temperature formation at 90 °C. Despite a higher impedance, cells formed at this temperature showed improved thermal stability. However, these results obtained from a cell using ionic liquids cannot simply be transferred to commonly used carbonate electrolytes. Hildenbrand et al. also used a high formation temperature of 80 °C in their study on the combination of commonly used additives (vinylene carbonate (VC) or fluorethylene carbonate (FEC)).16 However, they neither investigated the formation procedure nor compared different temperatures during formation. Nonetheless, their results indicate that formation temperatures as high as 80 °C can result in cells lasting over 2000 cycles.
In our previous publication, we compared different additives and a baseline electrolyte without additives and their impact on formation cycling and cell properties also using a formation temperature of 80 °C.17 In that work,17 we found CO2 after formation at 80 °C even in cells without additives known to generate this gas. Strehle et al.18 reported that CO2 prevents the transesterification of ethylmethyl carbonate (EMC) to dimethyl carbonate (DMC) and diethylcarbonate (DEC) during formation. The transesterification usually occurs during formation of cells with EC/EMC electrolytes without additives and is driven by lithium alkoxides.18,19 FEC and VC, often used as additives, suppress transesterification and improve cell performance.16,18
The fact that CO2 is present in cells after formation at 80 °C raises the question whether a high-temperature formation can suppress transesterification of the bulk electrolyte. In addition, the effects of such high formation temperatures on cell properties such as available active material and lithium loss during formation have not been investigated and compared to results obtained from lower temperatures yet.
In this study, we compare three different formation temperatures: 45 °C, 60 °C, and 80 °C. We analyse the impact of formation at these different temperatures on the gasses generated during formation, the electrolyte composition after formation, and the initial performance in terms of resistance and capacity. This is complemented by a comparative analysis of the active material loss (LAM) and loss of lithium inventory (LLI) during formation and subsequent cycling at 1.5C, the maximum charging current according to the manufacturer.
A total of 49 cells (15 formed at 80 °C and 17 each formed at 45 °C and 60 °C) is used for analysing formation an initial performance. 12 cells each (4 per formation temperature) are cycled with 1.5C at a cycling temperature of 25 °C and 45 °C, respectively.
Bulk electrolyte changes during formation are identified using the non-destructive Thermal Electrolyte Analysis (TEA) to test whether high temperature formation can actually prevent transesterification of the electrolyte. Gas chromatography mass spectrometry (GC-MS) and differential scanning calorimetry (DSC)-measurements are used to validate the results obtained from TEA.
As TEA has not yet been discussed in a research paper, it is explained in more detail in the SI. We also discuss the qualitative and quantitative comparison of TEA with DSC results of the electrolyte to validate this method.
Experimental section
This section describes the materials and methods used in this study. We introduce the battery cells used in this work and give an overview of the preparation of the cells. Further sections provide more details on the analytical methods used in this work, and our approach to analysing the data obtained from cycling the cells.Fig. 1 provides an overview of the cell preparation and cycling experiment conducted in this study.
 | ||
| Fig. 1 A schematic overview of the testing procedure. Formation is conducted at three different temperatures and the respective colors will be used consistently in this publication to indicate cells formed at the respective temperature. Cycle life testing was interrupted in irregular intervals (roughly once a month) for an RPT. | ||
Materials and preparation of battery cells
The cells used in this work are purchased dry (without electrolyte) from Li-FUN Technology Corporation Limited. The active material is artificial graphite and nickel manganese cobalt oxide (NMC622) for negative and positive electrode, respectively. The cells are industrially manufactured with the same specifications for all cells and the nominal capacity is 1 Ah. The specifications of the cells can be found in Table S1 in the SI. The electrolyte was purchased from E-Lyte Innovations and consisted of 1 M LiPF6 in ethylene carbonate (EC) and ethyl methyl carbonate (EMC) (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 by weight). Prior to filling, cells were dried under vacuum at 40 °C for about 2.5 days. Cells were filled in a dry room (dew point <−50 °C). After filling, cells were connected to a cycler (Maccor Series 4200), charged to 1.5 V, and kept at that voltage for at least 12 h in an oven (Memmert GmbH + Co. KG) at 25 °C. Subsequently cells were clamped between plates as reported in ref. 17 for the formation and placed in the oven at the respective formation temperature (45 °C, 60 °C and 80 °C). The formation consists of another constant voltage step at 1.5 V for 8 h followed by a C/5 constant current (CC) charge to 4.2 V and a discharge with the same current to 3.9 V. After this partial discharge, cells remained in the oven for at least 8 h before being degassed in the dry room. The protocol is the same as used in our previous publication17 except for the temperatures. After formation gas samples were taken from cells using a syringe for GC analysis. To sample the gas the gasbag of the cells was pierced with the needle of the syringe and approximately 100 μl of gas were drawn from the gasbag. Subsequently, all cells were degassed by cutting the gasbag open and resealing the cell with a vacuum sealer.
:7 by weight). Prior to filling, cells were dried under vacuum at 40 °C for about 2.5 days. Cells were filled in a dry room (dew point <−50 °C). After filling, cells were connected to a cycler (Maccor Series 4200), charged to 1.5 V, and kept at that voltage for at least 12 h in an oven (Memmert GmbH + Co. KG) at 25 °C. Subsequently cells were clamped between plates as reported in ref. 17 for the formation and placed in the oven at the respective formation temperature (45 °C, 60 °C and 80 °C). The formation consists of another constant voltage step at 1.5 V for 8 h followed by a C/5 constant current (CC) charge to 4.2 V and a discharge with the same current to 3.9 V. After this partial discharge, cells remained in the oven for at least 8 h before being degassed in the dry room. The protocol is the same as used in our previous publication17 except for the temperatures. After formation gas samples were taken from cells using a syringe for GC analysis. To sample the gas the gasbag of the cells was pierced with the needle of the syringe and approximately 100 μl of gas were drawn from the gasbag. Subsequently, all cells were degassed by cutting the gasbag open and resealing the cell with a vacuum sealer.
In addition, lint-free swabs were used to obtain samples of the electrolyte from a total of 10 cells (4 × 45 °C and 60 °C each and 2 × 80 °C). More details on the sampling can be found below. We refer to these samples as samples taken after formation as opposed to other samples taken during the tear-downs after the initial reference performance test (RPT). After degassing, cells were connected to another cycler (Neware BTS4000-5V6A) in an oven at 45 °C. Cells were first discharged to the lower cut-off voltage of 3 V with a current of C/5. Subsequently, cells underwent 10 break-in cycles with a current of C/5 and a constant voltage (CV) phase until an end of charge current of C/20 at 4.2 V and a CC discharge to 3 V. At the end of discharge no CV phase was applied. These break-in cycles were used to analyse the initial behaviour of the cells and ensure a stable capacity and impedance prior to the reference performance test conducted at 25 °C. The RPT consists of a C/2 capacity test, a C/20 qOCV, and pulse measurements at 0% and 50% SOC. A more detailed description is provided in the SI.
Thermal electrolyte analysis
The Thermal Electrolyte Analysis (TEA) analyses the electrolyte using its melting properties. It is inspired by the differential thermal analysis (DTA) of Day et al.20 The basic principle consists of measuring the energy required to melt the electrolyte and the melting point.While this can be achieved using differential scanning calorimetry (DSC) as we show in the SI, these devices can be costly or too small to accommodate larger cells (e.g. pouch cells in the range of 1 Ah or more). The DTA, introduced by Day et al.20 provides a qualitative method for investigating electrolyte in lithium-ion batteries. Their approach does not require a calorimeter but a cryostat which provides a linear temperature ramp. This requires a closed-loop control of the temperature. The temperature of a sample cell is compared to a reference cell containing a non-freezing electrolyte.20
DTA has been used to complement traditional ageing tests,21,22 to investigate the tortuosity of different cells,23 and to analyse changes of the electrolyte in commercial cylindrical cells.22 This demonstrates the usefulness of a non-destructive method to identify changes within the electrolyte. However, the fact that this method has not been more widely used, indicates that the setup can be a challenge. The TEA aims to simplify the required instrumentation while still providing comparable qualitative meaning compared to the DTA. Furthermore, quantification of TEA results is possible by using calibration measurements of samples with a known heat-capacity or a liquid with a known melting point and melting enthalpy (preferably in the same temperature range as the melting point of the electrolyte under test).
The experiment consists of two steps: first the battery is submerged in liquid nitrogen, to rapidly freeze the electrolyte. This avoids phase separation during cool down.24 The cell remains in the liquid nitrogen for at least 1 h to ensure enough time for freezing of the electrolyte. This time was used in this work without investigation of shorter cooling times. It has to be adjusted to the cell size. Second, the cell is put in an indentation between two polystyrene foam blocks. The blocks provide good thermal insulation and a low heat capacity. The temperature on the outer surface of the blocks and the cell surface is measured and provides the data required for our analysis. The polystyrene foam blocks can be adjusted to accommodate various cell sizes and formats. One key requirement is that the thermal conductivity from the outside of the polystyrene foam block is small compared to the thermal conductivity across the cell so that a homogeneous temperature of the cell can be assumed.
To visualise the qualitative data we will plot the temperature measured at the cell on the x-axis and 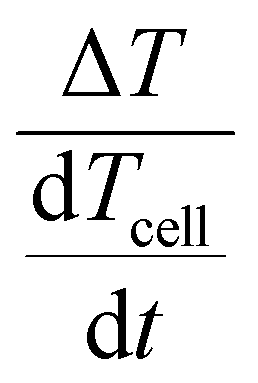 , which we call the TEA-Signal, on the y-axis. Deviation upwards from the baseline indicates endothermic processes, while downwards deviation indicate exothermic processes.
, which we call the TEA-Signal, on the y-axis. Deviation upwards from the baseline indicates endothermic processes, while downwards deviation indicate exothermic processes.
More details including measurements to calibrate the TEA and the derivation of the respective equations can be found in the SI.
Gas analysis
Gas samples were analysed using gaschromatography (PerkinElmer Clarus 690, GC) with a thermal conductivity detector (TCD) and a flame ionisation detector (FID) equipped with a methaniser. The system is the same as described in ref. 17. Argon was used as a carrier gas to enable the quantification of hydrogen in the gas samples via the TCD. The GC was calibrated using the same calibration gas described in our earlier publication.17 Thus, the following gasses can be quantified: hydrogen, nitrogen, ethene, ethane, methane, carbonmonoxide and carbondioxide. Quantification is performed by comparing the peak area of the respective peaks in the TCD-signal with the peak observed during calibration. A linear correlation between peak area and injected volume of the respective gas species is assumed.The share of the respective gas species is normalised the nitrogen content of the sample. The rationale behind this normalisation is that we do not expect any substantial source or sink of nitrogen in the cells. Hence, the nitrogen is left in the cells after sealing and is diluted by the gasses generated during formation. Therefore, normalising the gas-composition to nitrogen gives a rough idea about the relation between the absolute amounts of gas species generated during formation at different temperatures.
Tear down of cells
A total of four cells were disassembled after the initial reference performance test: two cells formed at 80 °C and one each for the two lower temperatures (45 °C and 60 °C). The cells were disassembled inside an argon-filled glovebox. The electrode sheets were scanned using a flatbed scanner and electrolyte samples for electrolyte composition analysis were taken. These samples include a swab and a separator-sample from each cell.Electrolyte composition analysis
The electrolyte composition after formation was analysed using GC (PerkinElmer Clarus 590) coupled with mass spectroscopy (PerkinElmer Clarus SQ 8 S). Samples for these measurements were taken from cells during degassing after formation using lint-free swabs (Cleanroom Swab HUBY-340 CA-008) purchased from Hein Reinraum GmbH, Braunschweig, Germany. The edge of the cell stack was wiped with the tip of a swab, which was then cut off from the stick and placed in a vial. This data is complemented by samples from cells disassembled after the initial RPT, obtained in the same way, and separator samples from the disassembled cells containing some electrolyte.Analysis of electrical data
The capacity is measured under three different conditions after formation and prior to the ageing test. The initial break-in cycles conducted with C/5 and a CC-CV charge and CC discharge at 45 °C are complemented by a reference performance test (RPT) with a capacity test (C/2, CCCV charge, CC discharge), and a qOCV-cycle (C/20, CC-charge, CC-discharge) conducted at 25 °C. For all tests a voltage range between 3 V and 4.2 V was used. Capacity measurement inevitably contain small errors. As the effect size in our experiments is small, the errors can play a substantial role. Therefore, the upper limit of the capacity error has been calculated based on the accuracy provided in the datasheet for cyclers used. The method of calculating the errors is explained in the SI.The capacity error during the RPT is in the range of ±10 mAh for the C/2 capacity and below ±2.8 mAh for the C/20 capacity. The error of the C/2 pulse resistances is below ±10 mΩ.
The initial loss during formation is calculated by subtracting the capacity of the first cycle (part of which is discharged before and after degassing, respectively) and the charge input during the formation (including 8 h of wetting and temperature adjustment but excluding room-temperature wetting). The second part of the discharge process is conducted in an oven set to 45 °C using a Neware BTS4000-5V6A as part of the break-in cycles. Thus, the ambient temperature at the end of discharge is the same for all formation temperatures.
The formation cycle were analysed using incremental capacity analysis (ICA). An adapted version of the LEAN-algorithm described in ref. 25 is used to calculate the incremental capacity analysis. The adaptations can be found in the SI. Fitting half-cell potentials to the full cell voltage provides information about usable active material and lithium trapped in the graphite, SEI, or CEI.26 Data obtained from the break-in cycles and the qOCV of the RPTs is analysed in this way. The fitting procedure is based on minimising a cost function, which is the weighted sum of the absolute error of the fitted cell voltage and the fitted differential voltage. More details can be found in ref. 27. Furthermore, the differential voltage can be affected by transient processes at the start of charge and discharge respectively. To avoid these processes from influencing the fitting result the weight at high and low SOCs (<5% and >95% SOC) is set to zero.
Resistance-related properties are investigated using the voltage relaxation at the end of (dis-)charge of the initial break-in cycles, pulse resistance measurements conducted as part of the RPT and the voltage difference between charge and discharge processes with different current rates. The latter is used to calculate a resistance which we refer to as cycling resistance Rcycling(SOC) as opposed to pulse resistance throughout this publication. This resistance is a function of the SOC. It is calculated according to eqn (1). Where Vdiff(SOC) and Idiff(SOC) represent the difference between the voltage and current at any given SOC, respectively. In this work V1 and I1 refer to the voltage and current from the C/2 cycle while V2 and I2 represent voltage and current during the C/20 cycle. Fig. S28 in the SI provides a graphical illustration. This resistance includes processes with time constants substantially higher than 10 s and thus differs from the pulse resistance.
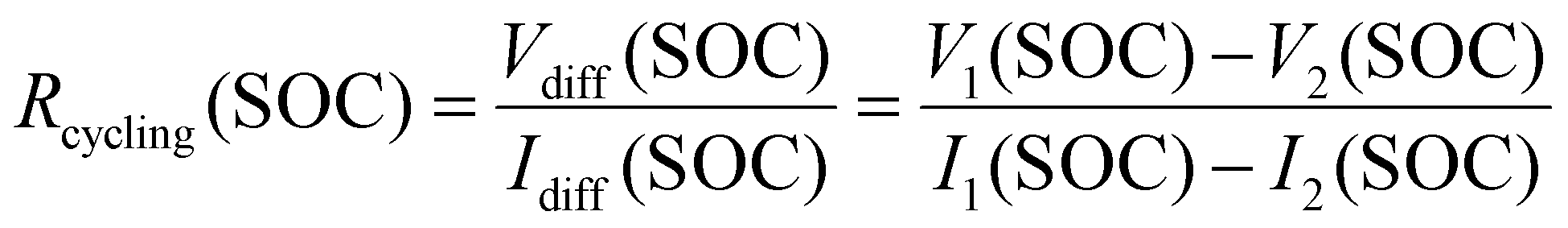 | (1) |
Cyclic ageing protocol
After the initial RPT cells undergo a cyclic ageing test at 25 °C and 45 °C. The C-rate is set to 1.5C for both charge and discharge. This is the maximum charging current according to the datasheet. Cells are charged with a CC-CV protocol with a cut-off current of C/20 and discharged without a CV-phase. The voltage range is 3 V to 4.2 V. Each (dis-)charge step is followed by a rest-period of 1 h. After 100 fast cycles, three C/10 cycles are added. Additional RPTs conducted roughly once a month provide further insights and enable a comparison of cells tested at 25 °C and those tested at 45 °C.Cell properties and electrolyte composition after formation
Here we present the results of our measurements starting with results obtained from the electrochemical data of the formation cycle. Subsequently, we present results obtained from the break-in cycles and the RPT. This is complemented by an analysis of the composition of the gasses generated during formation, the composition of the electrolyte after formation, and TEA-measurements.Formation cycling
The formation cycle is analysed by using incremental capacity analysis. Fig. 2 shows the ICA curves for the different formation temperatures. A higher value of the incremental capacity means more electrons have to be moved from the NMC to the graphite to increase the voltage of the full cell. A peak shift to lower voltages is observed for higher temperatures, which is expected due to improved kinetics at elevated temperatures. However, the first peak between 2.6 V and 3.2 V displayed in Fig. 2b also changes its shape: at 45 °C it is a rather narrow single peak for all cells. At 60 °C the peak is wider and evolves into a double-peak. At 80 °C the peak is a superposition of a narrow peak just below 2.8 V and a wider peak at 2.85 V. | ||
| Fig. 2 Overall formation ICA (a) and magnified sections (b–d), showing the differences between cells formed at 45, 60 and 80 °C, respectively. The small initial peak shifts to lower voltages for higher temperatures (b). The first major peak exhibits a similar shift but is reduced in height for cells formed at 80 °C. Panel (d) shows the upper SOC range of the first charge. A small peak around 3.77 V occurs for cells formed at 45 and 60 °C which is not visible for formation at 80 °C (marked with I). At the end of charge the incremental capacity for cells formed at 80 °C increases while it decreases for the lower temperatures (marked with II) except for two outliers. | ||
The second peak between 3.4 V and 3.55 V which is displayed in Panel c of Fig. 2 shifts to lower voltages at higher temperatures but also decreases in height. A shift to lower voltages at higher temperatures is expected due to faster kinetics and reduced overpotentials at elevated temperatures.
In the high voltage region, a minor peak between 3.75 and 3.8 V is detected for cells with formation temperature of 45 or 60 °C. This is marked with I in Fig. 2d. This peak is not clearly visible during formation at 80 °C. The last feature where a clear difference between the different formation temperatures is visible, is the trend of the incremental capacity at the end of the first charge (Fig. 2d marked with II). For cells formed at 80 °C an increase at the end of charge is visible, while cells formed at 60 and 45 °C show a decreasing trend.
Gas composition
Gas samples were taken from cells during the degassing step after formation. Fig. 3 visualises the N2-normalised gas composition. The results of the quantification prior to the normalisation to the nitrogen content can be found in the SI. Substantially more CO2 and CO was found in cells formed at 80 °C compared to lower temperatures where no or only minor amounts of CO2 were found. Furthermore, a similar amount of hydrogen and methane, respectively was detected in cells regardless of formation temperature. The nitrogen-normalised value of CO was essentially the same for cells formed at 45 °C and 60 °C but substantially higher for cells formed at 80 °C. The nitrogen-normalised share of ethene found after formation increased with increasing temperature. | ||
| Fig. 3 Share of different gases based on the samples analysed via GC. The bars represent the mean values of n measurements. Each gas volume has been divided by the volume of nitrogen of the respective sample. The error bars indicate the respective maximal and minimal measured gas share. | ||
Summarising the results of our gas analysis, we confirmed the fact that CO2 is released during formation at 80 °C reported in our previous publication.17 At lower formation temperatures no or very little CO2 was found. Either not CO2 was generated at lower temperatures, or all CO2 that was generated at lower temperatures was consumed in other reactions.18 As CO2 can prevent transesterification18 and it has been reported for cells formed at 45 °C when no additives were used,20 we expect to observe transesterification in cells after formation at 45 and 60 °C but no transesterification after formation at 80 °C. This alters the melting process and the electrolyte composition as discussed in the next sections.
TEA
By identifying melting and crystallisation processes TEA can support our analysis by providing information on changes of the bulk electrolyte composition. A detailed discussion of TEA including a quantitative comparison with DSC is included in the SI. Two cells for each formation temperature were investigated as described above after the initial RPT. Fig. 4 presents the signal of the TEA for these cells, together with one dry cell, and a cell immediately after filling (labelled Prior to Formation in the legend). This time no baseline correction was performed as the results are analysed qualitatively. The electrolyte prior to formation shows two overlapping peaks labelled a and b in Fig. 4: a broad one in the range of −70 °C and a more narrow peak at −60 °C. In addition, an exothermic crystallisation feature (c) around −95 °C is observed. | ||
| Fig. 4 TEA-signal for 6 cells after formation at different temperatures together with the signal of a cell filled with electrolyte prior to formation immediately after filling and the signal obtained from a dry cell. Three major features are observed for the electrolyte prior to formation: a broad endothermic feature (a), a more narrow endothermic peak with higher amplitude (b), and an exothermic process (c). Peak (b) vanishes after formation at 45 and 60 °C but is preserved after formation at 80 °C. The broader peak (a) shifts to lower temperatures after formation at 45 and 60 °C but is suppressed after formation at 80 °C. The endothermic feature (c) is less pronounced and shifted to higher temperatures after formation at 45 and 60 °C. | ||
After formation at 80 °C, the broad peak vanishes, while both the narrow peak and the exothermic process can still be detected. The narrow peak shifts slightly to higher temperatures, possibly indicating a reduced salt concentration. In contrast to these results, after formation at lower temperatures of 45 and 60 °C the broad peak seems to shift to lower temperatures. The narrow peak disappears completely and the exothermic process is much less pronounced compared to the pristine electrolyte and the electrolyte in cells formed at 80 °C. Concluding the observations of our TEA measurements, we observe very similar changes of the electrolyte after formation at 45 °C and 60 °C. Formation at 80 °C causes fewer changes of the melting process of the electrolyte, suggesting less changes in electrolyte composition than after formation at lower temperatures.
Electrolyte composition after formation, RPT, and cycle ageing
To complement the non-destructive results from the TEA, electrolyte samples taken from cells during degassing and tear downs using swabs are analysed using GC-MS. In addition, samples from the separator are also analysed with GC-MS. Fig. 5 shows the relevant solvent peaks of the chromatogram for samples taken after formation. Samples taken from cells after the initial RPT were also analysed. The results are shown in Fig. S10 in the SI. The intensities in Fig. 5 are normalized to the peak intensity of EMC, based on the assumption that EMC, DEC, and DMC evaporate in comparable proportions during sampling due to the similar vapour pressure. The analysis shows that both after formation and after the RPT, only negligible amounts of DMC and DEC are present in the electrolyte samples from cells formed at 80 °C. Significant amounts of these substances are present in cells after formation at 45 °C and 60 °C. At higher retention times from minute 22 to 24, a small but reproducible pattern of three signals (in some cases up to five), all showing a molecular ion peak at m/z = 281, can be observed at formation temperatures of 45 °C and 60 °C. Based on comparison with the NIST database and the corresponding mass fractions, these signals can be assigned to oligomeric hydrocarbons (OHC). At 80 °C, none of these signals are observed, neither after formation nor after the RPT. In the samples taken after the RPT, these signals shift to slightly shorter retention times compared to the samples taken after formation. One exception is the cell formed at 60 °C, where the samples which were collected from the cell edge using a swab, show no such shift. However, the samples taken from the separator of that cell exhibit the same shift as observed for the cell that underwent formation at 45 °C. | ||
| Fig. 5 GC–MS results of the electrolyte samples from the cells after formation. (a) Shows the signal for DMC, (b) indicates EMC, (c) shows DEC, (d) shows OHCs. Due to the large differences in signal intensities between the various components, the y-axis range is adjusted to the respective peak. Results from samples taken after the first RPT can be found in the SI. | ||
Overvoltages, capacity, and cell balancing during break-in cycles
The ten break-in cycles are analysed in terms of the capacity, and voltage relaxation after charge and discharge, respectively.Fig. 6 shows the evolution of the capacity (top), the maximum voltage after discharge (middle), and the minimum voltage after charge (bottom) for the break-in cycles performed at 45 °C for all cells. The capacity decreases with increasing cycle number for all cells. Cells formed at 45 °C exhibit the largest spread with the minimal capacity being lower and the maximum one higher than for the cells formed at 60 °C. The capacity is lowest for cells formed at 80 °C.
 | ||
| Fig. 6 Capacity (top), voltage at the end of relaxation after discharge (middle) and end of relaxation after charge (bottom) over the 10 break-in cycles. The differences in the OCV after charge between individual cells are in the range of the quantification of the cycler. Hence, curves of several cells overlap. | ||
Cells formed at 80 °C relax to a lower voltage after discharge and a higher voltage after charge compared to both other groups indicating reduced overvoltages at low and high SOCs, respectively. For cells formed at 45 °C and 60 °C a transient behaviour covering the initial 5–6 cycles is visible: overvoltages decrease after both charge and discharge. The relaxed voltage after the charging process in the first cycles is lowest for cells formed at 45 °C, indicating higher overvoltages. The difference between 60 °C and 45 °C formation temperature vanishes after 5 cycles for the end of charge relaxation. For the end of discharge relaxation, the difference is reduced but cells formed at 45 °C still exhibit higher overvoltages than cells formed at 60 °C after 10 cycles.
For a more detailed analysis the cycles were fitted to charge-dependent electrode potentials for graphite and NMC622 respectively. The data for graphite and NMC622 from our previous publication27 was used, as the active material (graphite and NMC622) is the same. The fitting procedure is described in more detail in that publication.
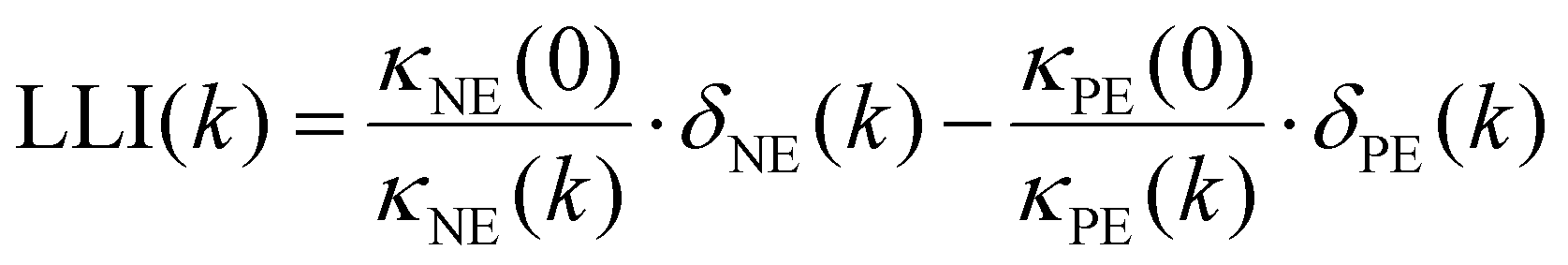 | (2) |
Four parameters are fitted: κPE and κNE represent the available capacity of NMC622 and graphite, respectively, under the same utilisation range as in the coin cells used to obtain the potential curves versus Li–Li+. To compare our results with information given by the cell manufacturer regarding electrode loadings we normalised κPE to a voltage range between 3.2 V and 4.3 V in contrast to our previous work. δNE and δPE shift the electrode potential of the respective half cell along the capacity axis. Thus, these parameters represent the remaining electrochemically active lithium in the graphite and electrochemically active unoccupied lithium sites in the NMC. To obtain the lithium loss, the parameters δPE,k and δNE,k obtained from the fitting have to be scaled with 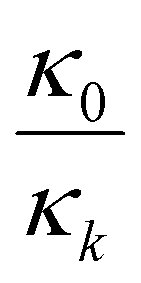 where k indicates the cycle while κ0 is the initial capacity of the respective electrode. The difference between these scaled values (eqn (2)) is the lithium loss, albeit with a shift. This shift is due to remaining lithium in the graphite of the coin cell and slow diffusion in highly lithiated NMC hindering the full lithiation of the NMC622 in the coin cells.28 This kinetic limitation causes some of the observed first cycle losses in NMC28 and effects the end of discharge of NMC622—Lithium coin cells. Nonetheless, the shift of LLI compared to the actual amount of LLI is due to the coin cell measurement which is the same for all cells investigated here. Thus, the difference between the obtained results indicate the difference in LLI between the cells. Nonetheless, it should not be mistaken as the absolute amount of LLI. This also explains why the LLI is negative in the initial cycles.
where k indicates the cycle while κ0 is the initial capacity of the respective electrode. The difference between these scaled values (eqn (2)) is the lithium loss, albeit with a shift. This shift is due to remaining lithium in the graphite of the coin cell and slow diffusion in highly lithiated NMC hindering the full lithiation of the NMC622 in the coin cells.28 This kinetic limitation causes some of the observed first cycle losses in NMC28 and effects the end of discharge of NMC622—Lithium coin cells. Nonetheless, the shift of LLI compared to the actual amount of LLI is due to the coin cell measurement which is the same for all cells investigated here. Thus, the difference between the obtained results indicate the difference in LLI between the cells. Nonetheless, it should not be mistaken as the absolute amount of LLI. This also explains why the LLI is negative in the initial cycles.
Fig. 7 shows the fitting results for κNE and κPE for each of the ten break-in cycles and the LLI calculated based on the fitting results. The graphite capacity κNE remains rather stable during the ten break-in cycles. However, it is slightly lower after formation at 80 °C compared to lower formation temperatures. The NMC capacity κPE is also lowest after formation at 80 °C. However, it is more stable after high temperature formation, evidenced by a small decrease of κPE for cells formed at 45 °C and 60 °C which is not observed after formation at 80 °C. LLI is highest after formation at 80 °C and increases faster during the 10 break-in cycles (C/5, 45 °C). Note that higher values mark more lithium loss. The values are negative due to the aforementioned first cycle losses of NMC622 in the coin cell. The difference is in the range of 10 mAh and increases to values in the range of 25 mAh during the 10 cycles. Based on the datasheet, we can compare our fitting results with the expected values. For the positive electrode a loading of 2.24 mAh cm−2 is given resulting in an expected capacity in the range of 1.16 Ah. The negative electrode loading is given as 2.7 mAh cm−2 resulting in an expected capacity of 1.4 Ah. κPE is roughly in the range of the data provided by the manufacturer. However, κNE is slightly lower, which might be the result of different definitions of the potential window to measure the capacity or loss of active material during the formation cycle.
 | ||
| Fig. 7 Fitting results for four each break-in cycle. κNE (top) and κPE (middle), representing the available graphite and NMC622 capacity, respectively. The graphite capacity remains stable during the break in cycles, while the NMC622 capacity decreases slightly after formation at 45 °C and 60 °C. The loss during formation is highest for TForm = 80 °C LLI (bottom) is negative in this case. This is due first-cycle losses of the NMC622 as explained in the text. Higher values of the LLI still indicate more lithium loss. Thus, lithium loss is highest at 80 °C. Furthermore it increases faster during the low rate cycling at 45 °C. The NMC622 capacity (κNMC622) declines slightly and is lower for cells after formation at 80 °C compared to their counterparts formed at 60 or 45 °C. The graphite capacity exhibit a less pronounced trend with respect to formation temperature and cycle count. Nonetheless, cells formed at 80 °C show a lower available graphite capacity. Higher values of LLI at 80 °C indicate an increased lithium loss during high temperature formation. LLI is negative due to the first cycle loss of NMC622 as explained in the text. | ||
Concluding the results obtained from break-in cycles, we found that formation at 80 °C negatively affects the availability of active material and increases LLI. It also leads to faster loss of lithium during the break-in cycles with C/5 at 45 °C. However, the overvoltage of cells formed at 80 °C is lower than the one of their counterparts formed at lower temperatures. Nonetheless, the capacity after high temperature formation is lower compared to formation at 60 °C and 45 °C.
Initial reference performance test
The reference performance test consists of a capacity test with a current of C/2, a low-current qOCV (C/20), and pulses at 0% and 50% SOC with 1C and 0.5C in charge and discharge direction.Fig. 8 shows a summary of the results. On the left (a) the capacity distribution for both the C/2 and C/20 cycle is shown. The capacity for cells formed at 45 and 60 °C is at a similar level and higher compared to the cells formed at 80 °C. This is true for both the C/20 and the C/2 test but the difference is more pronounced at C/20. This indicates a kinetic limitation after formation at lower temperatures.
 | ||
| Fig. 8 Summary of the RPT results: (a) distribution of the measured capacity of the C/2 and C/20 cycle, (b) 10 s pulse resistance at 0% SOC, (c) 10 s pulse resistance at 50% SOC both measured with a C/2 discharge pulse, (d) cycling resistance for selected SOCs for the charging process (e) cycling resistance for selected SOCs for the discharging process in this case values for an SOC of 0.1 are too high to be displayed here. The median for cells formed at 80 °C at this point is 137 mΩ compared to 226 mΩ and 223 mΩ for cells formed at 45 °C and 60 °C, respectively. | ||
Pulse-resistance measurements shown in Fig. 8b, support a lower resistance for cells formed at 80 °C compared to 45 and 60 °C at low states of charge. At 50% SOC (Fig. 8c) the difference is substantially smaller.
In addition to pulse resistance measurements at 0% and 50% SOC, we analysed the voltage difference between charge and discharge by calculating Rcycling as described above. The results are shown in Fig. 8d. A total of six outliers are shifted to higher resistance values. Two of these cells were formed at 80 °C, three at 60 °C and one at 45 °C. These outliers are included in the dataset but not included in Fig. 8d and e.
We analysed the charge and discharge process separately. Rcycling,cha is calculated based on the C/2 charge and C/20 charge using eqn (1). Rcycling,dch was calculated using the voltages obtained from the C/2 and C/20 discharge using the same equation. Here we focus on the 10% SOC-steps displayed in Fig. 8d and e. Regardless of formation temperature the Rcycling,cha is lowest around 30–40% SOC. Below this SOC it rises and does so faster in cells formed at 45 °C and 60 °C compared to cells formed at 80 °C. A similar trend can be observed in the discharge direction, where the values for 10% SOC are too high to be displayed in the graph. The median for cells formed at 80 °C at this point is 137 mΩ compared to 226 mΩ and 223 mΩ for cells formed at 45 °C and 60 °C, respectively. The differences between formation temperatures in the SOC-range from 60–70% can be attributed to different usages of the graphite: increased loss of lithium during high temperature formation causes a shift of the graphite plateau with respect to the SOC, even though it may remain the same when using the absolute capacity. At high SOCs the resistance is slightly lower for cells formed at 80 °C.
To summarise the results from the RPT, cells formed at high temperature exhibit lower overvoltages at low SOCs. This can explain the smaller difference of the C/2 capacities compared to the C/20 capacities between the three groups: given the CC discharge, an increased resistance after formation at lower temperatures results in an earlier end of discharge at C/2 At medium SOCs this difference vanishes and cells perform very similarly.
Cell tear down
A total of four cells were disassembled after the initial RPT, one each for a formation temperature of 45 °C and 60 °C, and two formed at 80 °C. In cells formed at 80 °C, we found that parts of the separator stick to the positive electrode and parts of the negative electrode coating sticks to the separator. This was observed for both cells opened after high temperature formation. The cells formed at 45 °C and 60 °C, could be easily disassembled without the separator sticking to either electrode. Other than that no difference was observed.In addition, two aged cells were opened (E_45_001 and E_45_002). E_45_002 was opened after an RPT and an additional week in discharged state for relaxation, the other one (E_45_001) within 3 h after rapid cycling.
The scanned electrodes shown in Fig. 9 from both of these cells show traces which might indicate lithium plating as well as considerable inhomogeneities of the negative electrode. Parts of the near overhang in the cell opened immediately after rapid ageing contains considerable amounts of lithium evidenced by a golden hue. This is not the case after an RPT and a resting period of a week in the discharged state.
 | ||
| Fig. 9 Scans from negative electrode sheets from the two cells opened after cyclic ageing. On the left from cell E_45_002, opened after a final RPT and a week of relaxation in the discharged state. On the right, from cell E_45_001, opened after rapid cycling. Both cells were formed at 45 °C. | ||
Discussion of cell properties and electrolyte composition after formation
We will first discuss the results related to the formation cycle including the different charge throughputs, losses, and ICA results. This is complemented by TEA measurements. We then continue by discussing the links between the formation temperature and the cell performance.Formation cycle
The differences observed in the ICA between the three formation temperatures during the initial formation cycle point at different reactions during formation. Most notably the emergence of a second peak in the low voltage range at higher temperatures and the differences at high voltages between formation at 80 °C and lower temperatures indicate different reactions. The different behaviour at high SOC (above 4.1 V in Fig. 2) may indicate additional reactions on the positive electrode especially as the thermal stability of NMC622 decreases with decreasing lithiation.29Another notable difference is the charge input during formation (see Fig. S11 in the SI). It is highest for cells formed at 60 °C, despite cells formed at 80 °C showing a lower overpotential after formation and higher losses. Due to the flat graphite potential in the fully charged cell the charge throughput is essentially limited by the amount of lithium that has to be removed from the NMC622 to reach the corresponding PE potential including the overpotentials. Hence, it should be similar for all cells regardless of formation temperature, when neglecting the minor contribution of the entropy effect (discussed in the SI based on ref. 30) and overvoltages discussed below. This is not the case.
Based on the observed difference in charge throughput in the first cycle, we identified two possible explanations:
1. formation at 80 °C increases the first cycle loss of NMC622
2. formation at 60 °C (and likely also 45 °C) increase the amount of side reactions which transport charge but do not consume lithium
The first cycle loss of layered transitions metal oxides is well known28,31,32 but the temperature dependency has not yet been studied in detail to the best of our knowledge. A large share of the apparent first cycle loss is attributed to kinetic limitations at high lithiation degrees of the NMC caused by poor lithium diffusion.28,31,32 In addition, active material can be lost due to side reactions forming a surface layer.32,33 Furthermore, the first cycle charge capacity can be increased by irreversible electrochemical reactions with surface contaminants such as Li2CO334 or the electrolyte.32 The last case essentially corresponds to the increased amount of charge consumed in side reactions mentioned above. Supplemental coin-cell measurements described in the SI, indicate that the highest loss occurs when NMC is delithiated to a potential of 3.7 V. It corresponds with an increase of the relaxed voltage after lithiation: when NMC is charged to 3.65 V with a current of C/25, the voltage after discharge to 2.8 V, relaxes to about 3 V within 10 min. Once the NMC is delithiated to 3.75 V the voltage after discharge relaxes to about 3.6 V, marking an increased overvoltage. This is in line with kinetic limitations being responsible for the majority of the losses. However, the kinetic limitation of the lithiation of NMC does not affect the initial charge input, as we only consider the delithiation of the NMC622.
Thus, the higher charge throughput observed for formation at 60 °C is either due to increased loss of PE active material at 80 °C, or due to side reactions. Indeed, we observe a lower NMC capacity after formation at 80 °C compared to the other two temperatures. It is well-known that highly delithiated NMC can become unstable and react chemically with the electrolyte.35 However, the lower charge input occurs in the medium SOC range.
Considering the observations during cell tear down, where we found that the separator sticks to the positive electrode active material, the loss of active material could also be related to passivation of NMC and graphite particles by covering of the surfaces by separator material. This may also occur at lower SOCs.
Effects of formation temperature on gassing and electrolyte composition
Gas analysis shows that during formation at 80 °C CO2 is released. Formation at lower temperatures of 60 °C and 45 °C results in no or only minor traces of CO2 after formation. Based on the results of Strehle et al.18 the CO2 may prevent transesterification of the EMC in the electrolyte which would influence the melting behaviour of the electrolyte.36 Based on the GC measurements alone no statement regarding the suppression of transesterification at lower temperatures is possible, as CO2 is consumed when scavenging lithium alkoxides.18 Combining TEA, gas analysis and the analysis of the electrolyte composition, we get a more holostic picture.The results of TEA suggest significant changes of the electrolyte during formation at both lower temperatures amid little changes after high-temperature formation: after formation at 80 °C the main melting feature only shifts slightly to higher temperatures, possibly indicating a decrease in salt concentration. In contrast, after formation at 45 and 60 °C the sharp endothermic peak vanishes and the broader peak shifts to lower temperatures. This shift of the melting process is in line with the expected transesterification of EMC to DEC and DMC and has also been reported by Day et al.20,36 Ding et al.36 reported a solidus feature at −86.6 °C and −74.3 °C for mixtures of DEC with EMC and EC, respectively. Considering melting point suppression due to LiPF6, this is in a similar range of the observed melting feature after formation at 45 °C and 60 °C. In addition, GC-MS confirmed substantial amounts of both DMC and DEC in the samples taken after formation from cells formed at 45 °C and 60 °C, whereas only negligible amounts of these substances were found in cells formed at 80 °C. TEA shows that transesterification does not only occur in specific locations but affects the melting behaviour of the bulk electrolyte.
The suppression of transesterification is most likely due to the presence of CO2.18 This gas makes up roughly 16% (by volume) of the gaseous species sampled from the cells after formation. This is more than the 10% Strehle et al.18 added in the head-space of a cell to reduce the amount of transesterification. Thus, based on our results and the mechnistic insights by Strehle et al.18 CO2 released from side-reactions during high-temperature formation prevents the transesterification reaction while the amount of CO2 produced during formation at lower temperature, if there is any, is too little to effectively prevent transesterification.
Another effect of the formation temperature on the electrolyte composition is the formation or modification of oligomeric hydrocarbons (OHCs). These OHCs are presumably formed through the condensation or polymerization of carbonate decomposition products.37,38 At a formation temperature of 80 °C, OHCs are not detectable neither after the formation process nor after the RPT, suggesting that higher formation temperatures suppress their formation or promote further reactions yielding insoluble, or gaseous products.
Cell properties
During the initial break-in cycles as well as the RPT conducted after a total of 11 cycles, we identified improved kinetics especially at low states of charge after formation at 80 °C. Despite the de-facto increased voltage window of the cells formed at 80 °C we observe a lower capacity due to a higher loss of lithium and some loss of active material in these cells. At least a part of the improved kinetics at low SOCs can be attributed to a shift in cell balancing: formation at 80 °C causes a higher loss of lithium which results in a lower state of lithiation of the NMC host material at the positive electrode at low full-cell SOCs. Thus, the NMC does not reach highly lithiated states with poor kinetics.39 This is in line with the results from the voltage difference analysis which shows a clear difference at low SOCs but a similar resistance at medium SOCs. This also explains the lower differences of the C/2 capacities between the different formation temperatures when compared with the C/20 capacities (Fig. 8) the contribution of the poor kinetic of the highly lithiated NMC to the end of discharge is higher in cells after formation at 45 °C and 60 °C than in cells formed at 80 °C. However, the poor kinetic of highly lithiated NMC fails to explain the differences in relaxation after charge during the break-in cycles: in a fully charged cell the degree of lithiation of the NMC should be similarly low, as the graphite-potential is rather flat in this region. These differences are likely related to differences in the SEI, CEI, and the electrolyte.Formation at 80 °C also reduced the available active material on the positive electrode. This may point at a different CEI which is usually comprised of some material from the electrode as well as decomposition products from solvents.40 The degradation mode analysis adds further evidence to differences in the behaviour of the positive electrode: it is more stable in cells formed at a higher temperature. Interestingly, formation at 60 °C improves the stability of the positive electrode compared to formation at 45 °C, despite no significant difference in the initial capacity of the positive electrode. Further research on the role of the CEI and how formation conditions affect this interphase is required.
Insights from cycle ageing tests
After discussing the effects of different formation temperatures on the initial cell performance, we turn to the consequences of formation at different temperatures on cyclic ageing.Cyclic ageing
High-temperature formation results in a lower initial capacity but also an improved capacity retention during the 1.5C cycles, for both tested cycling temperatures. We first focus on the results from cycling at 25 °C displayed in Fig. 10. A notable difference between the three formation temperatures is the effect of low-rate cycles and the interruption by RPTs: cells with lower formation temperatures (45 °C and 60 °C) exhibit a capacity recovery during low rate cycles and test interruptions. Subsequent cycles with high C-rates cause a faster capacity fade compared to the cycles before the low rate cycles. This results in a “sawtooth”-type ageing trajectory as shown in Fig. 10. In contrast, cells formed at 80 °C exhibit almost no influence of the low rate cycles on the capacity fade trajectory regardless of testing temperature. | ||
| Fig. 10 Capacity evolution during cyclic ageing with 1.5C. The points show the intermittent C/10 cycles. The large spread observed for cells formed at 45 °C and 60 °C is due to the capacity recovery during these cycles. The inset (b) shows a magnification of the first 160 cycles. | ||
During cycling at 45 °C the differences in terms of the ageing-trajectory are similar but the capacity recovery during low rate cycles of cells formed at 45 °C and 60 °C is less pronounced. The results of cycling at 45 °C can be found in Fig. S13 in the SI.
The RPTs conducted at irregular intervals always at 25 °C add more information. Pulse resistance measured in charge direction at 0% and 50% SOC during the RPTs are shown in Fig. 11. The discharge pulse Resistance can be found in Fig. S12 in the SI.
 | ||
| Fig. 11 Pulse resistance obtained from the C/2 charge pulses during the RPT at 25 °C and 0% SOC (top) and 50% SOC (bottom). The left panels show the results from cells cycled at 25 °C. Results from cells cycled at 45 °C are shown on the right. | ||
Initially, cells formed at 80 °C exhibit the lowest pulse resistances at 0% SOC, compared to cells formed at lower temperatures. Over the course of the ageing experiment this changes and cells formed at 80 °C exhibit a higher increase of the pulse resistance at 0% SOC compared to the other cells, resulting in a higher pulse resistance towards the end of the test, despite a lower loss of capacity. However, at 50% SOC all cells follow a roughly linear trajectory of increasing resistance with similar gradients, despite differences in capacity fade. Here cells formed at high temperatures maintain the comparatively lower resistance.
Cycling cells at 45 °C leads to different results: the pulse resistance at 0% SOC increases for all cells. The charge pulse resistance at this low SOC increases fastest for cell formed at 45 °C, followed by cells formed at 60 °C. Cells which underwent high-temperature formation at 80 °C exhibit the lowest resistance increase. At 50% SOC this trend is even more pronounced. In discharge direction (Fig. S12) the differences of the pulse resistance at 0% SOC is lower and cells formed at 80 °C exhibit essentially the same discharge pulse resistance as cells formed at lower tempertures. Nonetheless, at 50%, the discharge pulse resistance remains lowest for cells formed at 80 °C.
Degradation modes are analysed by fitting of the qOCV data obtained from the RPTs. Results are obtained from the charging process. We used the same procedure as described in ref. 27. Both voltage and differential voltage 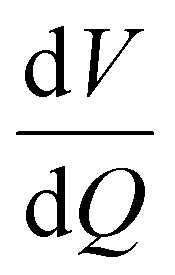 were used for fitting with equal weights. The outermost 5% of the charging process was neglected during fitting. This removes the influence of transient processes at the beginning of charge on the fitting results. The results for κPE, κNE, and LLI evolution for cells cycled at 25 °C are displayed in Fig. 12.
were used for fitting with equal weights. The outermost 5% of the charging process was neglected during fitting. This removes the influence of transient processes at the beginning of charge on the fitting results. The results for κPE, κNE, and LLI evolution for cells cycled at 25 °C are displayed in Fig. 12.
 | ||
| Fig. 12 Degradation Mode Analysis of cells cycled at 25 °C (left, a, b, e and f) and those cycled at 45 °C (right, c, d, g and h). Lower capacity values of the electrodes of (κNE and κPE) after formation at 80 °C are compensated by less LLI during cycling at 25 °C. During cycling at 45 °C LLI increases for cells formed at 80 °C but remains lower than the decreased LLI observed in cells formed at lower temperatures (g). During cycle ageing at 45 °C cells LAMPE increases with decreasing formation temperature (d). | ||
Degradation mode analysis indicates substantially reduced LLI for cells formed at 80 °C compared to those formed at 45 °C and 60 °C for both cycling at 45 °C and 25 °C. However, while cells formed at 80 °C exhibit an increase in LLI during cycling at 45 °C (see Fig. 12) compared to 25 °C no such increase is observed for cells formed at 45 °C and 60 °C. Cells formed at the lower two formation temperatures exhibit roughly the same LLI at both cycling temperatures.
The loss of active material (LAM) during cycling at 25 °C is similar for cells formed at 45 °C and 60 °C. The available active material on the negative electrode (κNE in Fig. 12a) shows an initial increase followed by a plateau between approximately 750 and 2000 cycles followed by a decrease. The positive electrode capacity (κPE) exhibits a slow but steady decrease for all formation temperatures.
During cycling at 45 °C κNE starts to decrease earlier and the decrease in κPE is more pronounced and shows a clear dependency on formation temperature: the decrease is lowest for cells formed at 80 °C, followed by those formed at 60 °C with cells formed at 45 °C exhibiting the highest LAMPE.
This might explain results by Cui et al.,9 who observed that an increased formation temperature delays the knee-point and end of life but has little impact on initial performance or performance after 120 cycles.
Furthermore, Cui et al.9 suggested that shifting positive and negative electrode in a way to avoid the kinetically inhibited highly lithiated operating range of NMC can increase cycle life. On first sight, our data supports this conclusion: cells formed at 80 °C exhibit higher LLI, thus operating mostly outside the highly lithiated domain of the NMC. However, LLI in absolute terms is and remains lower starting with the first RPT after high rate cycling. For cyclic ageing at 25 °C, this results in an increase of the low-SOC resistance to values higher than observed for cells formed at 45 °C and 60 °C.
In summary, a higher formation temperature shows benefits in the long term, even though the initial capacity of these cells is lower: both capacity fade and resistance increase are lower for cells formed at 80 °C. In addition, LLI is reduced by a higher formation temperature. During cycling at 45 °C a higher formation temperature also leads to a more stable positive electrode.
Capacity recovery during slow cycles
Substantially different ageing trajectories have been observed between cells formed at 80 °C and the other cells formed at 45 °C and 60 °C. The latter exhibit a sawtooth-like capacity fade, where interruptions of the 1.5C cycles by RPTs and low C-rate cycles lead to partial recovery of the lost capacity. In contrast, cells formed at 80 °C show a slower, linear capacity fade which is barely affected by the lower C-rate cycles or RPTs.Different mechanisms can explain the capacity recovery observed in cells formed at lower temperatures:
1. Salt inhomogeneity caused by electrolyte motion41
2. Slow lithiation of the overhang during rapid cycling42
3. Slow recovery of plated lithium (similar to the effects reported in ref. 43 for lithium metal batteries)
The recently proposed EMSI-effect mainly affects large format cells and is caused by a change of porosity of the negative electrode during cycling.41 Cells tested in this study were cycled without external pressure which has recently been shown to mitigate the EMSI-effect.44 Furthermore, cells tested here are rather small and use negative electrodes without silicon, resulting in a limited volume change of the active material. Thus, we regard the EMSI-effect as an unlikely explanation for the observed capacity loss and recovery. Furthermore, this EMSI should affect cells after high-temperature formation as well. However, we do not observe the sawtooth-ageing in these cells.
The overhang lithiation is a slow process with time constants possibly in the range of weeks or even months.42,45 It is affected by the mean lithiation of the graphite in the cell. The different formation temperatures affect the operating point of the graphite in the cells. The average charging voltage of the cells formed at lower temperatures and the relaxed voltage at low SOCs is higher compared to cells after high-temperature formation. In addition, the CV-phases take longer. This can affect the usage of the overhang during cycling. This may result in a higher lithiation of the overhang during rapid cycling. Decreasing the overpotential and discharging the cell to lower states of charge during the low-current cycles activates lithium from the overhang.
A third explanation is based on the differences in the SEI. Formation at 45 °C and 60 °C yields oligomeric hydrocarbons (OHCs) which dissolve in the electrolyte as evidenced by the GC-MS measurements. While the exact characterisation of these molecules is beyond the scope of this work, they provide a baseline for this hypothesis to explain the ageing pattern observed in these cells. These components dissolve in the electrolyte. On newly opened surfaces high current densities lead to highly localised plating. Eventually the dissolution of these soluble species is hindered by the increased concentration in the electrolyte. Thus, the capacity fade slows down. Some of the plating is recovered during the low current cycles as the cells are kept at low SOC for a longer time. It has been shown, that rest periods at low SOC can lead to the recovery of lost lithium in lithium metal batteries.43 Tasaki and Harris46 investigated the solubility of species found in the SEI with the result that organic salts such as lithium ethylene dicarbonate or lithium–alkyl carbonates can be dissolved in both EC and DMC. No statement regarding the solubility in EMC has been made in that publication. However, DMC and EC is both present in the cells formed at 45 °C and 60 °C. Thus, the dissolution of SEI-components can affect the cell behaviour. We will refer to this hypothesis as the dynamic SEI hypothesis in the following discussion.
We now turn to the comparison of the overhang and a dynamic SEI as causes of the observed sawtooth ageing. The overhang should be charged primarily during times with a high SOC, where the anode potential is lowest. This includes the constant voltage phase during charging. In addition we consider the average charging voltage as another proxy for time spent at high SOCs. If the reversible loss is primarily caused by the overhang, we would expect a positive correlation between the duration of the accumulated CV-phase in 100 cycles and the losses observed during the set of 100 cycles, or between the average charge voltage and the losses. This correlation might go into saturation at extremely long CV-phases if the overhang is essentially fully charged. The recovered capacity should also follow a similar correlation pattern because the lithiation of the overhang is reversible.42 It might even be a slightly better indicator, as it does not include irreversible losses. However, it will saturate earlier because not all reversible losses are recovered in three C/10 cycles.
In this case, the difference between cells formed at 80 °C and those formed at lower temperatures is caused by differences in the operating point. Thus, cells formed at 80 °C have to be included in this correlation analysis and should show a similar correlation.
To test this, all sets of three C/10 cycles and the corresponding 100 fast cycles before are analysed. We refer to the first of the three C/10 cycle as the nth cycle. We define the recovery as the differences between the discharged capacity of the first fast charging cycle after the C/10 cycles (n + 3) and the discharged capacity of the last fast charging cycle prior to the C/10 cycles (n − 1). The loss is defined as the difference between the discharged capacity 100 cycles before the low current cycles (n − 100) and the discharge capacity of cycle n − 1. The accumulated CV-time is the sum of the CV-time of cycles n − 100 to n − 1.
The correlations are visualised in Fig. S15 in the SI. While both CV-time and average voltage increase due to ageing effects, initially the capacity loss observed in 100 fast charging cycles and the recovery decreases or remain roughly constant, respectively. Both increase later on for heavily aged cells. Cells formed at 80 °C exhibit a markedly different behaviour with both recovery and losses lower compared to cells with the same average charge voltage or accumulated CV-time. Thus, while the overhang might play a role, it is insufficient to explain the observed differences.
The dynamic SEI would result in a linear correlation between capacity fade and electrolyte volume as long as other effects such as dry-out can be neglected. Less electrolyte would result in a reduced loss because a lower amount of SEI components can be dissolved until the electrolyte is saturated or a steady state concentration gradient is reached. As this hypothesis is based on different SEI properties between cells formed at 80 °C and cells formed at lower temperatures, cells formed at 80 °C should not exhibit the same correlation. To test this hypothesis cells were filled with lower electrolyte volumes, formed at 60 °C and cycled at 25 °C. Material, cell preparation, and the testing protocol have been the same for these cells.
Indeed, a linear correlation between capacity fade during the initial 100 cycles and electrolyte mass is observed for cells cycled at 25 °C as shown in Fig. 13b. The correlation is very strong and highly significant (r = 0.967, p < 1.3 × 10−6). The resulting slope is about 17 ± 3.4 mAh gelectrolyte−1. This indicates a significant role of the electrolyte mass in the degradation process.
 | ||
| Fig. 13 Scatter plots showing the capacity loss until the 10th (a) and 100th cycle (b) of the cycling experiment at 25 °C. Panel c shows the slope of the regression for the capacity loss until the respective cycle for 25 °C. The shaded green areas correspond to the 95% confidence interval. The low current cycles (101–103) are marked by the grey rectangle. | ||
Based on the dynamic SEI hypothesis the slope, representing the additional loss of lithium per gram of electrolyte should increase for each cycle until reaching a saturation level because the dissolution will likely require multiple cycles to complete.
Further analysis presented in Fig. 13 confirms a trajectory that resembles such a saturation process when calculating the slope of the linear regression for the loss between the initial cycle and cycle k and plotting the result over the corresponding cycles. After the interruption by the C/10 cycles the trajectory starts again, albeit not being fully reset to the initial values.
If the lost capacity was indeed recovered from lithium plating barely connected to the negative electrode, the capacity should be recovered on the negative electrode by re-intercalation of the plated lithium. This would result in a shift of the discharge endpoint to lower charge values, causing a negative discharge endpoint slippage.47 While such an analysis should be treated prudently given the limited accuracy of the cyclers used in this work, the observed discharge endpoint slippage is much higher than the charge endpoint slippage during the three C/10 cycles. Both are displayed in Fig. 14. This is in line with recovery of lost lithium on the negative electrode which can be explained by the mobilisation of previously plated lithium.
 | ||
| Fig. 14 Comparison of charge endpoint slippage and discharge endpoint slippage between the last 1.5C cycle before and the first 1.5C cycle after the C/10 cycles after the first 100 cycles at 25 °C. Open markers indicate the additional cells with lower electrolyte volume. Note the different scales of the axes. | ||
The consideration of the degradation modes complements this picture: cells formed at 45 °C and 60 °C exhibit a higher LLI compared to cells formed at 80 °C during cyclic ageing at 25 °C. Plating is known to be mitigated by elevated temperatures.48 Thus, capacity fade caused by plating is reduced during cycling at 45 °C. This results in a slightly lower LLI as evidenced by the degradation mode analysis, despite the fact that other side reactions are likely aggravated by the increased temperature.
To conclude the discussion of the mechanisms responsible for the sawtooth-ageing, the combined data from a statistical analysis of the capacity fade and electrolyte mass as well as degradation mode analysis suggests that plating leads to higher losses during high rate cycling at 25 °C if parts of the SEI are soluble. This is the case after formation at 45 °C and 60 °C but not after formation at 80 °C. The more stable SEI obtained after formation at 80 °C limits further LLI.
To the best of our knowledge, the consequences of the dissolution of partial SEI components during non-abusive testing have not been explicitly investigated. However, in the light of our findings, different outcomes of publications discussing the effect of electrolyte volume on capacity retention can be explained. One example is the publication of Lechtenfeld et al.49 where additional electrolyte negatively affected cells without additives and with VC cycled with 1C at 20 °C. This is contrasted by results from our group where more electrolyte had no effect on the initial performance but prevented the occurrence of a knee-point in cells cycled with C/5 at 45 °C.27 An SEI containing more soluble components combined with the higher current at lower temperature might have contributed to highly localised plating similar to our observation in this work. Higher temperatures and lower C-rates combined with a high-temperature formation in ref. 27 possibly yields a less soluble SEI and mitigates the effect of SEI-dissolution.
Limitations of high temperature formation
In this work we investigated the effects of formation temperature on cells with an electrolyte containing widely used solvents (EC and EMC) but not additives. Further research is required to identify whether high formation temperatures are beneficial for other electrolyte systems, especially those that readily release CO2 during formation at lower temperatures such as FEC or VC.18 SEI-forming additives are typically less stable compared to the solvent molecule so that they are reduced at higher potential.40 Increasing the temperature may result in a larger contribution of chemical rather than electrochemical decomposition, altering the properties of SEI and/or CEI. Whether such a shift results in an improved SEI likely depends on the specific additive. We would like to emphasise that a limited thermal stability of film-forming additives may not prevent their use in a high temperature formation, as these sacrificial additives decompose during formation to create the SEI-layer anyway.18,40While a high-temperature formation may not be beneficial in cells using additives, our results demonstrate that high formation temperatures are beneficial for cells using electrolyte without additives. Thus, a comparison of cells formed at moderate temperatures with additive-containing electrolyte and with additive-free electrolyte will underestimate the achievable lifetime of cells without additives. This should be considered when reporting the benefits of film-forming additives.
Furthermore, in battery production additional concerns such as safety, energy consumption, and costs of equipment have to be considered.6 Furthermore, heating cells to the high temperatures reported here, may introduce additional rest times and thus costs, or significant thermal inhomogeneities especially in large-format cells. The authors are not aware of any study investigating the effect of thermal inhomogeneities during the formation process on cell performance and lifetime.
Conclusions
In this study, we investigated the effects of high-temperature formation at 80 °C and compared it with lower temperatures of 60 °C and 45 °C. We reproduced prior results showing that formation at 80 °C releases CO2. Furthermore, we identified changes of the melting behaviour after formation at 45 °C and 60 °C which are in line transesterification of the EMC. This is contrasted by cells formed at 80 °C where the major melting behaviour does not change. Additionally, transesterification of electrolyte during formation at 45 °C and 60 °C is confirmed by GC-MS measurements, which identified DMC and DEC in electrolyte samples collected after formation at lower temperatures but found only negligible amounts of these solvents after formation at 80 °C. Thus, we demonstrated that high-temperature formation can prevent the transesterification of the EMC observed at lower temperatures.High temperature formation shifts the cell balancing, due to a higher LLI during formation. The benefit of this shift is a reduced low-SOC resistance. Furthermore, high-rate cycling shows that LLI during cycling is suppressed by formation at 80 °C for both cycling temperatures (45 °C and 25 °C). In addition, higher formation temperatures reduce LAMPE during cycling at 45 °C. This results in superior stability of cells formed at higher temperatures, despite their initially lower capacity. The increased formation temperature leads to a significant reduction of both reversible and irreversible capacity fade. For lower formation temperatures our results indicate that dissolution of soluble SEI-components and subsequent plating contribute to faster capacity loss during 1.5C cycling at 25 °C. This effect is at least partially reversible: during low rate cycles a fairly large share of the lost capacity is recovered. The exact role, secondary effects and potential benefits of the more soluble SEI obtained at lower formation temperature requires further research.
Our results highlight that increasing the formation temperature can improve long-term capacity retention under high rate cycling even though the initial capacity is lower compared to lower formation temperatures. Further research is on-going to compare degradation at lower C-rates to investigate the results of formation temperature on the lifetime of the cell under less aggressive ageing regimes.
From a development perspective, further investigations will help in identifying whether these results can be transferred to more complex electrolyte systems containing several additives, or whether it is a way to achieve some of the benefits typically assigned to additives without using them. Our results indicate that it is possible to tune battery properties by carefully designing formation protocols. A better understanding of the relationship and mechanisms linking the formation process with cell performance enables formation protocols tuned for specific applications. Thus, we support and underline the call by Schomburg et al. for more research on formation processes to achieve the required understanding for knowledge driven formation process design.7
Author contributions
Sebastian Klick: conceptualization, methodology, software, formal analysis, investigation, data curation, writing – original draft, visualization, validation. Philipp Finster: investigation, writing – original draft, validation, formal analysis. Karl Martin Graff: writing – original draft, investigation, formal analysis, validation. Felix Weber: writing – review & editing, supervision, project administration, conceptualization. Gereon Stahl: writing – review & editing, supervision, project administration. Carlos Ziebert: writing – review & editing, supervision, project administration, funding acquisition. Egbert Figgemeier: supervision, project administration, funding acquisition. Dirk Uwe Sauer: supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Data availability
Data for this article from electrochemical cycling of the cells, including aggregated data and time series data together with basic scripts to load the data written in python is available at RWTH Publications at https://doi.org/10.18154/RWTH-2026-03769.Raw data from TEA measurements, DSC measurements and TEA results, are also available in this repository at the same URL. Further data can be obtained from the authors upon reasonable request. Supplementary information (SI) including a more detailed description of TEA and other methods used in this work is available under the following DOI: https://doi.org/10.1039/d5eb00218d.
Acknowledgements
The authors gratefully acknowledge funding from the German Federal Ministry of Education and Research as part of the Batgasmod project (03XP0311B). S. K. would like to thank Hyunsang Joo, Sascha Berg, and Hendrik Laufen for the productive discussions which contributed to the development of the explanations of the observed ageing.References
- S. J. An, J. Li, Z. Du, C. Daniel and D. L. Wood, J. Power Sources, 2017, 342, 846–852 CrossRef CAS.
- P. M. Attia, S. J. Harris and W. C. Chueh, J. Electrochem. Soc., 2021, 168, 050543 CrossRef CAS.
- D. L. Wood, J. Li and S. J. An, Joule, 2019, 3, 2884–2888 CrossRef.
- C. Mao, S. J. An, H. M. Meyer, J. Li, M. Wood, R. E. Ruther and D. L. Wood, J. Power Sources, 2018, 402, 107–115 CrossRef CAS.
- E. Peled, J. Electrochem. Soc., 1979, 126, 2047 CrossRef CAS.
- D. L. Wood, J. Li and C. Daniel, J. Power Sources, 2015, 275, 234–242 CrossRef CAS.
- F. Schomburg, B. Heidrich, S. Wennemar, R. Drees, T. Roth, M. Kurrat, H. Heimes, A. Jossen, M. Winter, J. Y. Cheong and F. Röder, Energy Environ. Sci., 2024, 17, 2686–2733 RSC.
- M. Leißing, F. Horsthemke, S. Wiemers-Meyer, M. Winter, P. Niehoff and S. Nowak, Batteries Supercaps, 2021, 4, 1344–1350 CrossRef.
- X. Cui, S. D. Kang, S. Wang, J. A. Rose, H. Lian, A. Geslin, S. B. Torrisi, M. Z. Bazant, S. Sun and W. C. Chueh, Joule, 2024, 8, 3072–3087 CrossRef CAS.
- P. M. Attia, S. J. Harris and W. C. Chueh, J. Electrochem. Soc., 2021, 168, 050543 CrossRef CAS.
- S. J. An, J. Li, C. Daniel, D. Mohanty, S. Nagpure and D. L. Wood, Carbon, 2016, 105, 52–76 CrossRef CAS.
- H. H. Heimes, C. Offermanns, A. Mohsseni, H. Laufen, U. Westerhoff, L. Hoffmann, P. Niehoff, M. Kurrat, M. Winter and A. Kampker, Energy Technol., 2020, 8, 1900118 CrossRef CAS.
- F. German, A. Hintennach, A. LaCroix, D. Thiemig, S. Oswald, F. Scheiba, M. J. Hoffmann and H. Ehrenberg, J. Power Sources, 2014, 264, 100–107 CrossRef CAS.
- S. Bhattacharya, A. R. Riahi and A. T. Alpas, Carbon, 2014, 67, 592–606 CrossRef CAS.
- M.-T. F. Rodrigues, F. N. Sayed, H. Gullapalli and P. M. Ajayan, J. Power Sources, 2018, 381, 107–115 CrossRef CAS.
- F. Hildenbrand, F. Aupperle, G. Stahl, E. Figgmeier and D. U. Sauer, Batteries Supercaps, 2022, 5, e202200038 CrossRef CAS.
- S. Klick, K. M. Graff, G. Stahl, E. Figgemeier and D. U. Sauer, Batteries Supercaps, 2024, e202400291 CrossRef CAS.
- B. Strehle, S. Solchenbach, M. Metzger, K. U. Schwenke and H. A. Gasteiger, J. Electrochem. Soc., 2017, 164, A2513 CrossRef CAS.
- R. Petibon, L. Rotermund, K. J. Nelson, A. S. Gozdz, J. Xia and J. R. Dahn, J. Electrochem. Soc., 2014, 161, A1167 CrossRef CAS.
- R. P. Day, J. Xia, R. Petibon, J. Rucska, H. Wang, A. T. B. Wright and J. R. Dahn, J. Electrochem. Soc., 2015, 162, A2577–A2581 CrossRef CAS.
- M. K. G. Bauer, J. Harlow, T. Hynes and J. R. Dahn, J. Electrochem. Soc., 2023, 170, 030543 CrossRef CAS.
- F. M. Maddar, R. Genieser, C. C. Tan and M. J. Loveridge, J. Electrochem. Soc., 2023, 170, 030522 CrossRef CAS.
- M. K. G. Bauer and J. R. Dahn, J. Electrochem. Soc., 2021, 168, 020501 CrossRef CAS.
- M. M. Nolasco, S. N. Pedro, C. Vilela, P. D. Vaz, P. Ribeiro-Claro, S. Rudić, S. F. Parker, C. S. Freire, M. G. Freire and A. J. D. Silvestre, Front. Phys., 2022, 10, 834571 CrossRef.
- X. Feng, Y. Merla, C. Weng, M. Ouyang, X. He, B. Y. Liaw, S. Santhanagopalan, X. Li, P. Liu, L. Lu, X. Han, D. Ren, Y. Wang, R. Li, C. Jin, P. Huang, M. Yi, L. Wang, Y. Zhao, Y. Patel and G. Offer, eTransportation, 2020, 3, 100051 CrossRef.
- M. Dubarry, C. Truchot and B. Y. Liaw, J. Power Sources, 2012, 219, 204–216 CrossRef CAS.
- S. Klick, M. Harmsen, C. Rahe and D. U. Sauer, J. Electrochem. Soc., 2025, 172, 070530 CrossRef CAS.
- J. Kasnatscheew, M. Evertz, B. Streipert, R. Wagner, R. Klöpsch, B. Vortmann, H. Hahn, S. Nowak, M. Amereller, A.-C. Gentschev, P. Lamp and M. Winter, Phys. Chem. Chem. Phys., 2016, 18, 3956–3965 RSC.
- C. Tian, Y. Xu, W. H. Kan, D. Sokaras, D. Nordlund, H. Shen, K. Chen, Y. Liu and M. Doeff, ACS Appl. Mater. Interfaces, 2020, 12, 11643–11656 CrossRef CAS PubMed.
- W. Zhao, M. Rohde, I. U. Mohsin, C. Ziebert and H. J. Seifert, Batteries, 2020, 6, 55 CrossRef CAS.
- M. J. Lain and E. Kendrick, J. Power Sources, 2021, 493, 229690 CrossRef CAS.
- H. Zhou, F. Xin, B. Pei and M. S. Whittingham, ACS Energy Lett., 2019, 4, 1902–1906 CrossRef CAS.
- J. Choi and A. Manthiram, Electrochem. Solid-State Lett., 2005, 8, C102 CrossRef CAS.
- S. E. Renfrew and B. D. McCloskey, ACS Appl. Energy Mater., 2019, 2, 3762–3772 CrossRef CAS.
- R. Jung, P. Strobl, F. Maglia, C. Stinner and H. A. Gasteiger, J. Electrochem. Soc., 2018, 165, A2869 CrossRef CAS.
- M. S. Ding, K. Xu, S. Zhang and T. R. Jow, J. Electrochem. Soc., 2001, 148, A299 CrossRef CAS.
- G. Gachot, S. Grugeon, M. Armand, S. Pilard, P. Guenot, J.-M. Tarascon and S. Laruelle, J. Power Sources, 2008, 178, 409–421 CrossRef CAS.
- J. Henschel, C. Peschel, S. Klein, F. Horsthemke, M. Winter and S. Nowak, Angew. Chem., 2020, 132, 6184–6193 CrossRef.
- A. Weng, P. Mohtat, P. M. Attia, V. Sulzer, S. Lee, G. Less and A. Stefanopoulou, Joule, 2021, 5, 2971–2992 CrossRef CAS.
- K. Xu, Electrolytes, Interfaces and Interphases, The Royal Society of Chemistry, 2023 Search PubMed.
- S. Solchenbach, C. Tacconis, A. Gomez Martin, V. Peters, L. Wallisch, A. Stanke, J. Hofer, D. Renz, B. Lewerich, G. Bauer, M. Wichmann, D. Goldbach, A. Adam, M. Spielbauer, P. Lamp and J. Wandt, Energy Environ. Sci., 2024, 17, 7294–7317 RSC.
- M. Lewerenz, G. Fuchs, L. Becker and D. U. Sauer, J. Energy Storage, 2018, 18, 149–159 CrossRef.
- W. Zhang, P. Sayavong, X. Xiao, S. T. Oyakhire, S. B. Shuchi, R. A. Vilá, D. T. Boyle, S. C. Kim, M. S. Kim, S. E. Holmes, Y. Ye, D. Li, S. F. Bent and Y. Cui, Nature, 2024, 626, 306–312 CrossRef CAS PubMed.
- E. Petursdottir, S. Solchenbach, A. Gomez Martin, V. Peters, A. Stanke, L. Wallisch, M. Kohlhuber, H. Ehrenberg and J. Wandt, J. Electrochem. Soc., 2025, 100504 CrossRef CAS.
- F. Frie, H. Ditler, S. Klick, G. Stahl, C. Rahe, T. Ghaddar and D. U. Sauer, ChemElectroChem, 2024, 11, e202400020 CrossRef CAS.
- K. Tasaki and S. J. Harris, J. Phys. Chem. C, 2010, 114, 8076–8083 CrossRef CAS.
- A. J. Smith, J. C. Burns, D. Xiong and J. R. Dahn, J. Electrochem. Soc., 2011, 158, A1136 CrossRef CAS.
- H. Ditler, T. Tegetmeyer-Kleine, G. Stahl, C. Rahe and D. U. Sauer, J. Energy Storage, 2025, 132, 117706 CrossRef CAS.
- C.-T. Lechtenfeld, J. Buchmann, J. Hagemeister, M. M. Bela, S. van Wickeren, S. Stock, R. Daub, S. Wiemers-Meyer, M. Winter and S. Nowak, Adv. Sci., 2024, 11, 2405897 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |