
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrografting solid polymer electrolytes for separator-less structural sodium batteries
Elvira
Lind
 *a,
Vincent
Nieboer
b,
Martina
Cattaruzza
c,
Mats
Johansson
c,
Karin
Odelius
b,
Dan
Zenkert
d and
Göran
Lindbergh
a
*a,
Vincent
Nieboer
b,
Martina
Cattaruzza
c,
Mats
Johansson
c,
Karin
Odelius
b,
Dan
Zenkert
d and
Göran
Lindbergh
a
aApplied Electrochemistry, Chemical Engineering, KTH Royal Institute of Technology, SE-100 44 Stockholm, Sweden. E-mail: elvirali@kth.se
bPolymer Technology, Fiber and Polymer Technology, KTH Royal Institute of Technology, SE-100 44 Stockholm, Sweden
cCoating Technology, Fiber and Polymer Technology, KTH Royal Institute of Technology, SE-100 44 Stockholm, Sweden
dMaterial and Structural Mechanics, Engineering Mechanics, KTH Royal Institute of Technology, SE-100 44 Stockholm, Sweden
First published on 9th February 2026
Abstract
Sodium ion batteries (SIBs) are emerging as an attractive energy storage technology due to the accessibility, global abundance and low cost of sodium. However, improving their energy density to reduce system weight, particularly for mobile applications, remains a challenge. Structural batteries address this issue by integrating energy storage and mechanical load-bearing functionality into a single material. Here, we present electrografting as a single-step method to uniformly coat individual carbon fibres with a 1.1 μm thick, chemisorbed PEG-acrylate/NaTFSI-based solid polymer electrolyte (SPE). This enables the fabrication of separator-less structural sodium batteries with high energy and power density. The SPE rapidly forms a passivating layer on the carbon fibre surface, exhibiting excellent electrochemical stability, thermal resilience, and low overall resistance. A post-synthesis leaching step is critical to remove unreacted monomer, thereby minimising irreversible first-cycle capacity loss and the SPE resistance. The resulting SPE-coated electrodes deliver high specific capacities (150 mAh g−1) and coulombic efficiencies >99% over 100 cycles. This approach opens new pathways for lightweight, high-performance structural and conventional sodium battery systems.
Broader contextAs the global shift toward clean energy and electrification accelerates, there is a growing need for safe, cost-effective, and sustainable battery technologies. Sodium-ion batteries are an attractive option, as they use abundant, accessible and inexpensive materials. This research focuses on increasing their energy density, which is essential for use in mobile and high-performance systems. The key innovation is the use of electrografting, a single-step, scalable technique to coat a thin, chemisorbed polymer electrolyte directly onto a carbon fibre electrode. This technique creates strong adhesion between the electrolyte and the electrode, works on complex geometries, and produces uniform, ultra-thin coatings. These properties open the door to new battery architectures that can enhance performance or enable novel applications. For example, such solid polymer electrolyte coated electrodes can be used in separator-less battery designs, which reduce inactive materials and allow for improved energy density. The coated carbon fibres can also serve as electrodes in structural batteries that combine mechanical strength with energy storage, offering system-level weight reduction. Overall, this work presents a novel approach to advancing sodium-ion battery technology and supports broader efforts toward scalable, multifunctional, and sustainable energy storage solutions for sectors such as electric transport, aerospace, and portable electronics. |
1. Introduction
The ongoing global transition to a carbon-neutral energy economy has given rise to major technological shifts in the energy and transport sectors. The energy sector is shifting to renewable electric energy sources, such as wind and solar power, which are inherently intermittent in nature. This has prompted the need for stationary, grid-connected energy storage systems – such as batteries – that can store energy, distribute energy, and regulate grid frequency. Beyond stationary applications, the demand for batteries from the transport sector have skyrocketed in recent years as a result of the increasing number of battery electric vehicles (BEVs). Both grid-connected battery energy storage and batteries for BEVs are vastly dominated by LIBs.1 However, there is a growing interest for employing sodium-ion batteries (SIBs) for these applications since sodium is abundant, accessible and cheap. Moreover, the intrinsic properties of sodium enable the use of other inexpensive and abundant materials in the cells. For example, unlike lithium, sodium does not readily alloy with aluminium, allowing the use of aluminium current collectors. Additionally, sodium can be inserted into hard carbon negative electrodes, which offer high capacities and can be derived from renewable feedstocks.2,3SIBs offer strong power performance and are resilient to temperature fluctuations, which is beneficial for both grid storage and mobile applications.4,5 However, the lower energy density of SIBs compared to LIBs has driven research into strategies for enhancing their energy density, aiming to make them more competitive in weight-sensitive applications. One approach is the development of solid-state electrolytes, which could enable the use of high capacity sodium metal negative electrodes (1165 mAh g−1).6 This approach faces challenges such as ensuring good electrode/electrolyte interface compatibility and overcoming the lower ionic conductivity of solid electrolytes compared to liquids.7 Another strategy is to explore novel battery concepts, such as structural batteries. Structural batteries can replace load bearing components, such as the body of a car, thereby integrating multiple functions into a single structure. Substituting monofunctional components with multifunctional ones can significantly reduce the overall system weight.8,9
Carbon fibres (CFs) play a central role in structural batteries due to their excellent mechanical strength, electrical conductivity, and hard carbon-like microstructure.10 This enables them to function as both structural negative electrodes and current collectors in structural sodium batteries. Most CF based structural battery designs use a laminated configuration, where CF electrodes are stacked.11–13 These stacks are typically infused via vacuum-assisted infusion with a hybrid solid/liquid structural battery electrolyte (SBE), developed by Ihrner et al.14 In this system, the solid matrix transfers mechanical loads between CF electrodes, while the liquid phase conducts ions, achieving both high mechanical performance (Young's modulus ∼400 MPa) and high ionic conductivity (1.5–2.9 × 10–4 S cm−1).14–17 However, monofunctional separators with a thickness of 20–200 µm are employed in the electrode stack solely to separate the positive and negative electrodes and prevent short circuits. The use of such monofunctional separators adds inactive material and reduces the CF volume fraction, which lowers both energy density and mechanical performance.18–22 Eliminating monofunctional separators reduces inactive material, enabling higher energy and power densities compared to their separator-based counterparts.
Solid polymer electrolytes (SPEs), a class of solid-state electrolytes, are attractive because they combine the roles of both separator and electrolyte into a single material, allowing the removal of monofunctional separators. Thus, SPEs facilitate separator-less configurations, since no additional separator is required.23 However, their inherently low ionic conductivity and the challenge of achieving effective electrode/electrolyte interfacial contact remain critical issues.24 Poor interfacial contact, often due to inadequate wetting, can lead to high cell resistance and reduced rate performance.25 This can be mitigated by synthesis methods that promote direct contact between the SPE precursor and the electrode surface, such as electrografting.26
Cathodic electrografting is suitable for grafting electrically insulating polymers like SPEs.27,28 It involves applying a specific grafting/passivation potential to the substrate in the presence of a methacrylate- or acrylate-based monomer, a high donor number (DN) solvent, and a supporting electrolyte. This chemisorbs and initiates the polymerisation of the monomer, which then propagates chemically by an anionic mechanism from the CF surface and finally terminates through side reactions with impurities in the environment.26,27 The resulting coating consists of chemisorbed polymer brushes. Since the propagation of the grafted polymer chains is kinetically limited, meaning that the thickness of the SPE can be tailored by changing the monomer concentration.29,30
The DN of the solvent reflects its tendency to adsorb onto the substrate, with higher-DN solvents being less prone to adsorption. As a result, the DN of the solvent relative to that of the monomer governs the availability of monomer for grafting at the substrate surface. When the solvent DN is too low relative to the monomer, strong solvent adsorption can hinder monomer access to the surface and suppress electrografting. In such cases, polymer coatings may still form via polymerisation in solution, followed by precipitation onto the substrate once a critical chain length is reached. However, these precipitated coatings are not chemically bound to the surface and are typically thicker than grafted coatings.27,31,32
As the DN of the solvent increases relative to that of the monomer, solvent adsorption at the substrate is reduced, leading to a higher effective monomer concentration at the surface. Consequently, the solvent–monomer DN relationship strongly influences the density and uniformity of grafted polymer brushes.27,29,31 This, in turn, affects the ionic conductivity of grafted SPEs, as higher densities generally result in lower conductivities. Therefore, careful selection of the solvent–monomer system is required to achieve the desired coating properties.
Electrografting thus presents a way to form homogeneous, chemically bonded SPEs onto any complex geometry of electrodes, including CFs, with excellent electrode/electrolyte interfacial contact. Since electrografting yields very thin coatings, it concurrently addresses the issue of low ionic conductivities of SPEs by shortening the ion transport distance between the electrodes.
Electrografting of lithium-based solid polymer electrolytes (SPEs) onto CFs has been demonstrated by Leijonmarck et al., who compared monofunctional and bifunctional PEG-methacrylate monomers. Dimethylformamide (DMF) served as the solvent, and lithium trifluoromethanesulfonate (Li-triflate) was used as the supporting electrolyte.28 While the bifunctional monomer produced a brittle SPE incompatible with the flexibility of CFs, the monofunctional monomer yielded a flexible, strongly adhering coating. The resulting SPE was approximately 470 nm thick and uniformly coated each filament in a 1.5K CF tow. These coated CFs exhibited high specific capacities for lithium ions of 260 mAh g−1 at C/10.28 However, electrografting sodium-based SPEs onto CFs using sustainable, dual-purpose solvents – also used as liquid battery electrolytes – remains unexplored. In addition, key factors such as the effect of electrografting on the intrinsic properties of the polymer and the first-cycle irreversible capacity loss have not been systematically investigated, despite their relevance not only to structural batteries but also to the broader field of solid-state batteries.
This study demonstrates electrografting to produce sodium-SPE coated CFs using an EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PC solvent mixture and NaTFSI as the supporting electrolyte. The resulting Na-SPE coated CFs are evaluated electrochemically, thermally and by applying spectroscopic techniques to evaluate their suitability in separator-less structural sodium battery applications. The results reveal an electrochemically durable, thermally stable, passivating, low resistance, thin sodium SPE. Being significantly thinner than separators in structural battery laminates, the Na-SPE coated CFs can be used to realise high energy and power density structural batteries. Since electrografting addresses the issue of electrode/electrolyte interfacial contact and yields a low overall internal cell resistance, the findings of this work are applicable beyond structural batteries to the general field of solid-state SIBs. Furthermore, the 3D nature of the coated CFs facilitates high areal energy densities, making them suitable also for novel microbattery applications, where this metric is paramount.
:PC solvent mixture and NaTFSI as the supporting electrolyte. The resulting Na-SPE coated CFs are evaluated electrochemically, thermally and by applying spectroscopic techniques to evaluate their suitability in separator-less structural sodium battery applications. The results reveal an electrochemically durable, thermally stable, passivating, low resistance, thin sodium SPE. Being significantly thinner than separators in structural battery laminates, the Na-SPE coated CFs can be used to realise high energy and power density structural batteries. Since electrografting addresses the issue of electrode/electrolyte interfacial contact and yields a low overall internal cell resistance, the findings of this work are applicable beyond structural batteries to the general field of solid-state SIBs. Furthermore, the 3D nature of the coated CFs facilitates high areal energy densities, making them suitable also for novel microbattery applications, where this metric is paramount.
2. Experimental
2.1 Cathodic electrografting
Poly(ethylene glycol) methyl ether acrylate (Mn = 480) (PEGMEA) (Merck KGaA) mixed with a solvent containing ≥99% ethylene carbonate (EC) (Merck KGaA) and 99.7% anhydrous propylene carbonate (PC) (Merck KGaA) and a sodium trifluoromethanesulfonimide (NaTFSI) (Solvionic) supporting electrolyte was used for the electrografting. The solvent contained an EC:PC ratio of 1:1 (wt/wt) and 0.8 M NaTFSI. This EC:PC solvent mixture was used since it also performs well as a liquid electrolyte for SIBs,33 which minimised the post-processing of the coated CFs prior to the half-cell assembly. The reaction mixture was prepared by mixing a monomer-to-solvent ratio of 1:1 (wt/wt).
A Toho Tenax IMS65 24K fibre tow unsized carbon fibres (Toho Tenax Europe GmbH), was divided manually into tows containing approximately 1.5K fibres. The CF tow was cut with a scalpel into lengths of 8 cm and weighed with a 0.1 mg resolution Sauter Re 1614 balance. These tows were attached to 20 μm thick copper current collectors (Advent Research Materials) by silver conductive paint (Electrolube) and placed in a purpose-built Teflon mould (4 × 7 cm), see Fig. 1. The mould containing the CF tow was dried in a vacuum drier overnight at 60 °C before it was brought into a glove box (H2O and O2 < 1 ppm) and placed in a 3D printed polypropylene reaction bath. A three-electrode setup was arranged in the reaction bath with the CF tow as the working electrode (WE), an aluminium foil as the counter electrode (CE) and a sodium foil prepared from 99.9% pure sodium cubes in mineral oil (Sigma Aldrich), as a reference electrode (RE). All potentials are given versus the RE (Na/Na+). The WE and CE were kept separate by a Whatman glass microfibre GF/A (260 μm) fabric separator (Whatman, GE Healthcare Life Sciences, Chicago, IL, USA).
 | ||
| Fig. 1 The cross-section side view of the electrografting reaction bath containing the three-electrode setup and the reaction mixture constituents, ethylene carbonate, propylene carbonate, sodium trifluoromethanesulfonimide and poly(ethylene glycol) methyl ether acrylate. | ||
The grafting potential was found by linear sweep voltammetry (LSV) from 2 to −2 V vs. Na/Na+ at the sweep rate of 10 mV s−1. To verify that the solvents were not involved in any reaction during the electrografting, the same LSV test containing only the solvent mixture was performed for comparison. A fresh tow of CFs was used for the electrografting, which was performed by holding the WE at the grafting potential for 250 s while measuring the current output using an Ivium XP(20) potentiostat. The duration of the potentiostatic hold was selected to allow full passivation of the CF surface area. To evaluate the passivation efficiency, cyclic voltammograms (CVs) were conducted at a scan rate of 10 mV s−1 across the grafting potential (1.1–0 V vs. Na/Na+).
The coated CFs were subjected to either a rinsing protocol or a leaching protocol before further characterisation. In the rinsing protocol, the coated CFs were flushed with the EC:PC 1:1 (wt/wt) and 0.8 M NaTFSI solvent for approximately one minute. In the leaching protocol, the freshly coated CFs were submerged and left in 10 mL vials, containing the same solvent as was used for the rinsing protocol, for 48 hours. To avoid contact with air, both the rinsing and leaching were conducted inside a glove box.
2.2. Bulk polymer preparation
Two types of bulk polymer films, one with no salt and the other containing salt, were prepared to provide reference values for the coated polymer. Two different resins were prepared inside a glove box (H2O and O2 < 1 ppm), one containing only PEGMEA and 1 wt% of the thermal initiator 2,2′-azobis(2-methylpropionitrile) (AIBN) (Sigma-Aldrich) and the other containing PEGMEA, 0.8 M NaTFSI and 1 wt% of AIBN. The resins were poured into aluminium moulds with the dimensions 30 × 6×0.5 mm3 upon which glass slabs were placed and clamped. To avoid oxygen and moisture contamination, the glass covered moulds were vacuum sealed in a polyethylene terephthalate (PET) coated aluminium pouch, PET/Al/PE (12 μm/9 μm/17 μm) (Skultuna Flexible). The vacuum sealed specimens were subsequently taken out of the glove box and cured in an oven at 90 °C for 45 minutes. The crude polymer was analysed without further purification.2.3 Polymer characterisation
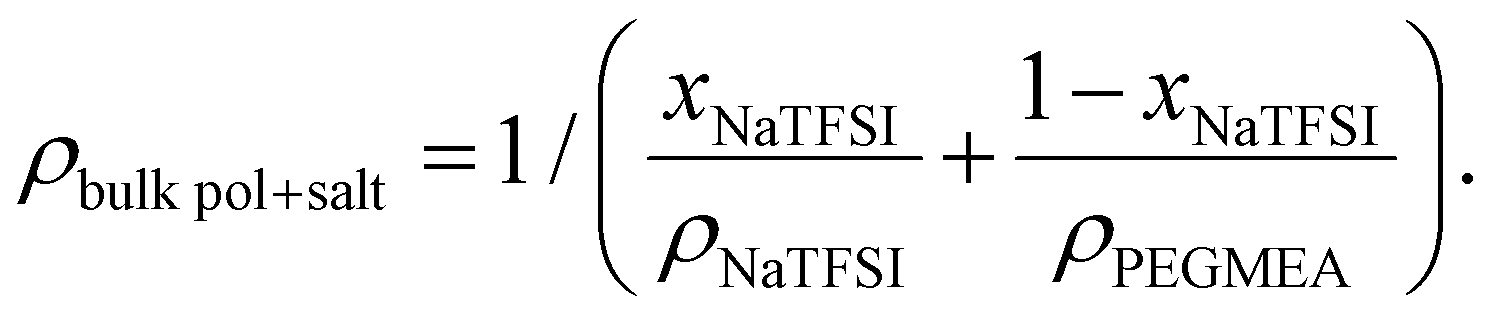 | (1) |
2.4. Electrochemical characterisation
:PC 1:1 (wt/wt) and 0.8 M NaTFSI and a sodium foil as the counter electrode with a nickel foil current collector. The half-cell stack was placed in a PET coated aluminium pouch, PET/Al/PE (12 μm/9 μm/17 μm) (Skultuna Flexible), and vacuum sealed inside an argon filled glove box.
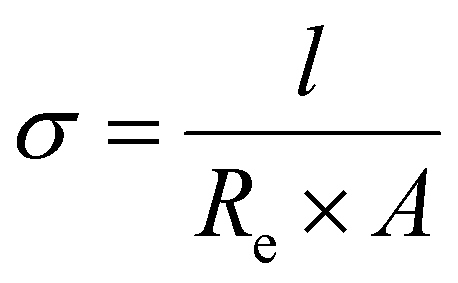 | (2) |
The thickness of the polymer disc, l, was taken to be 400 μm at maximum compression and A was the area of the disc. The activation energy was calculated for the ion movement in the SPE by eqn (3).
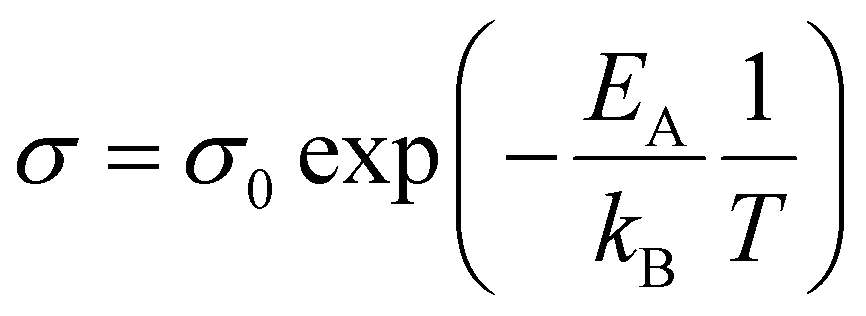 | (3) |
3. Results and discussion
3.1. Electrografting
The characteristic grafting potential for the reaction mixture used in the present study was identified by an LSV between 2 and −2 V vs. Na/Na+. As shown in the potential sweep of the monomer–solvent mixture in Fig. 2(a), a small peak at 0.65 V vs. Na/Na+ is observed, which is characteristic of grafting.27 At lower potentials, the equilibrium shifts toward polymerisation in solution, resulting in an increasing current below the grafting potential. In contrast, no current is observed in the grafting potential region for the pure solvent, indicating solvent stability at this potential.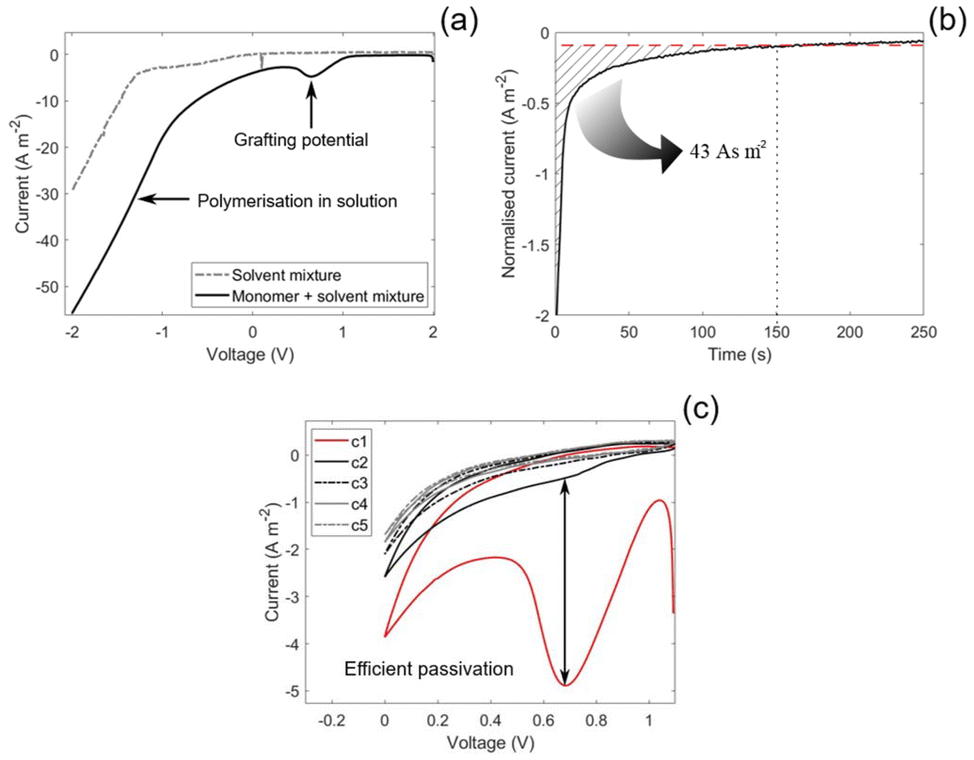 | ||
| Fig. 2 (a) Linear sweep voltammetry for the solvent mixture (ethylene carbonate:propylene carbonate 1:1 (wt/wt) and 0.8 M sodium trifluoromethanesulfonimide) and the solvent mixture with the monomer (poly(ethylene glycol) methyl ether acrylate) with a monomer-to-solvent ratio of 1:1. (b) The measured current in the three electrode electrografting setup as a function of time during the potentiostatic hold (0.6 V vs. Na/Na+). The CFs (working electrode) are submerged in the reaction mixture with aluminium foil as counter electrode and a sodium foil as the reference electrode. (c) The CVs performed under argon atmosphere at a scan rate of 10 mV s−1 across the grafting potential between 1.1 V and 0 V vs. Na/Na+, where c1–c5 refers to cycle 1–5. | ||
The electrografting was conducted by holding the submerged CFs at the 0.65 V vs. Na/Na+ grafting potential for 250 s in the EC:PC 1:1 (wt/wt) and 0.8 M NaTFSI reaction mixture while measuring the current output. The measured current, normalised by the surface area of the CFs, initially measures at −2 A m−2 and decays to −0.1 A m−2 after the initial 150 s. After 150 s, the current density stabilises at a constant background current for the remaining duration of the potentiostatic hold, see Fig. 2(b). A background current was also observed in the study by Leijonmarck et al.,28 which was attributed to side reactions related to the electrolyte and the insertion of Li into the CFs. At the grafting potential for the present study (0.65 V vs. Na/Na+), sodium is expected to insert into the CFs.34 However, since the solvents are stable at this potential for the present system, we propose that the background current is purely caused by sodium insertion.
The passivation efficiency of the electrografting was evaluated by CVs where the voltage was swept across the grafting potential. The efficiency is indicated by the reduction of the grafting peak between cycles, where a larger reduction indicates a more efficient grafting. As can be seen in Fig. 2(c), the current of the grafting peak was reduced from −5 to −0.5 A m−2 by the second cycle and reached a value of 0 A m−2 by the third cycle. This indicates a highly efficient passivation of the CF surface.
An efficient passivation is further elucidated from the low grafting charge of 43 As m−2, which is the integrated area marked in Fig. 2(b), indicating a rapid grafting and polymerisation event.27 By comparison, the grafting charge observed in the present study is an order of magnitude lower than the grafting charge in the study by Leijonmarck et al.28 This is expected since acrylates, as were used herein, are more reactive to electrografting compared to their methacrylate counterparts which were used by Leijonmarck et al., given that the solvent is not interfering with the grafting. Since the solvent is not interfering with the grafting, as indicated by the efficient passivation, and is stable at the grafting potential, the present system appears compatible. This shows that the donor numbers of EC (16.4) and PC (15.1) are sufficiently high and that they do not compete for adsorption sites on the CF surface with the PEGMEA monomer.24,35
3.2. Polymer characteristics
The polymer characteristics of the coated SPE was studied and compared to the bulk polymer. If not otherwise specified, the coated CFs in this section refers to samples that were subjected to the rinsing protocol described in section 2.1.The thickness of the coated SPE was evaluated by studying the cross-section of the coated fibres by SEM-EDS and by TGA measurements. The SEM images reveal a homogeneous layer of SPE covering all individual CFs, see Fig. 3(a), where the brighter SPE covering the CFs longitudinally stands in contrast to the darker CFs seen at the cross section where the cut was made on the epoxy embedded CFs. The agglomerates observed in this image are residues from the epoxy that was used to embed the coated CFs. Fig. 3(b) shows the cross section of a single CF from a cryo-cut sample, which is clearly coated with the brighter SPE. The corresponding EDS line-scan performed across the cross section, which depicts the oxygen levels, is shown in Fig. 3(c). The oxygen level was found to be a useful marker to differentiate between the CFs and the SPE, low oxygen levels indicate CF, and high levels indicate the oxygen rich SPE. Higher oxygen levels are seen across a span of approximately 1.1–1.5 μm at both ends of the line scan and lower oxygen levels span across roughly 5 μm inbetween. This coincides well with the average fibre diameter specifications for IMS65 (5 μm). It should be noted that the CF is slightly tilted in the image, which can cause a minor over or under estimation of the SPE thickness.
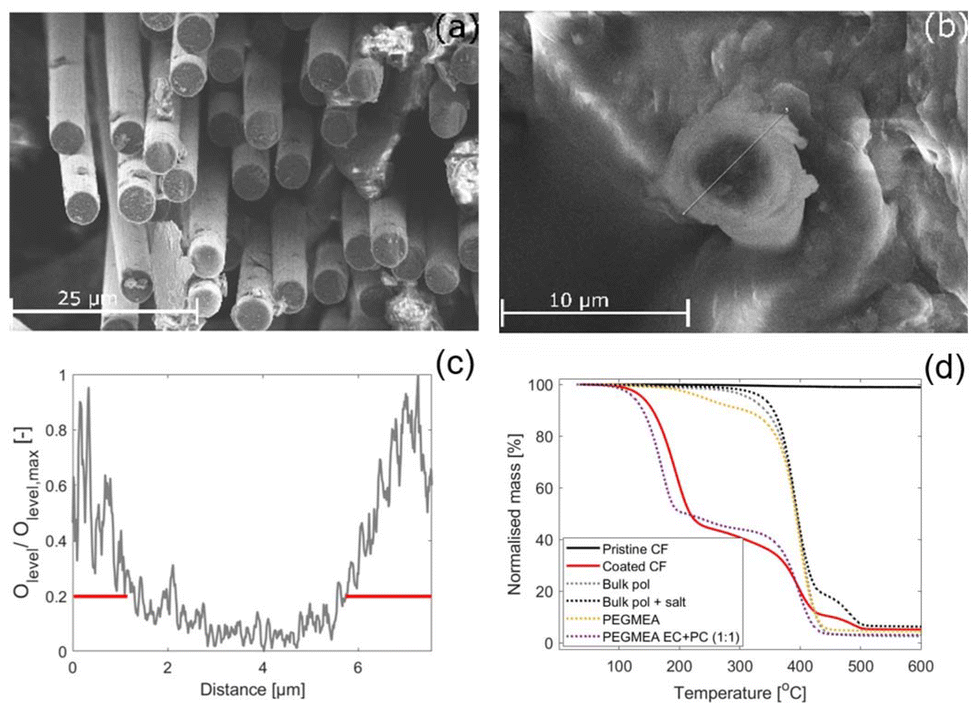 | ||
| Fig. 3 (a) Epoxy embedded SPE coated CFs, (b) the cross section of a single CF in a tow of CFs that were cryo-cut. (c) The oxygen levels obtained from the EDS line-scan performed along the line marked in (b), which are normalised by the maximum oxygen level. (d) The TGA results obtained from heating the sample from 30 to 600 °C at a heating rate of 10 °C min−1 under nitrogen flow (50 mL min−1). | ||
The TGA results show that mass loss in the coated CFs occurs in three distinct steps, see Fig. 3(d). For comparison, the TGA curves of pristine CFs, bulk polymer (with and without salt), the PEGMEA monomer, and a 1:1 (wt/wt) mixture of PEGMEA and the EC:PC (1:1, wt/wt) solvent are superimposed. This comparison helps differentiate mass losses due to decomposition of the polymer, monomer, solvent, or salt. As the pristine CFs exhibit no mass loss across the temperature range (30–600 °C), all observed mass losses in the coated samples can be attributed to the SPE, the PEGMEA monomer, the NaTFSI salt or the solvents.
The first mass loss in the coated CFs begins at 100 °C and is attributed to solvent evaporation or decomposition, as a similar loss is observed in the PEGMEA–solvent mixture. Pure PEGMEA decomposes in two steps at higher temperatures, a minor loss between 180–280 °C and a major loss between 280–450 °C. In the coated CFs, the second and third mass loss steps occur at around 350 and 460 °C, and are assigned to polymer degradation and salt decomposition, respectively. The mass loss at 350 °C may also include some unreacted monomer, as this overlaps with the second decomposition step of the PEGMEA monomer. Based on the total mass loss from the first and second steps and using the estimated density of the salt-containing polymer, the thickness of the SPE coating is calculated to be approximately 1.1 μm, which is consistent with the thickness observed in SEM images.
The ratio between the weight loss step corresponding to the polymer and the salt for the coated CFs (Fig. 3(d)) yields the polymer-to-salt ratio, which can be used as an evaluation of salt enrichment in the polymer during electrografting.28 For reference, the polymer-to-salt ratio for the bulk polymer, which contains the same salt content as the reaction mixture used for electrografting, is estimated to 5.4 from the weight loss steps in Fig. 3(d). By comparison, the polymer-to-salt ratio is 5.5 for the coated CFs, which shows that no salt enrichment occurs in the coated polymer. This stands in contrast to what was observed in28 where the salt concentration had more than doubled in the coated polymer. The average ethylene oxide group to sodium (EO:Na) ratio in the coated SPE for the present system was found to be approximately 30:1. For PEGs, an EO:Na ratio of 10:1–20:1 typically yield a conductivity maxima, according to literature.36 This means that increasing the salt concentration in the reaction mixture in the present study could increase the SPE conductivity. However, increasing the salt concentration can reduce the coating thickness and induce inhomogeneities, as previously observed in a lithium-based system.28 Consequently, higher salt concentrations were not explored using the single-step process employed in the present study. Nevertheless, higher salt concentrations may be attainable by using a higher donor number (DN) solvent, increasing the monomer concentration alongside the salt concentration in the reaction mixture, or through post-polymerisation swelling of the grafted SPE.
The conductivity for the bulk polymer shows Arrhenius-type behaviour and a linear increase with temperature, see Fig. 4. At room temperature, the ionic conductivity is σ = 6 × 10−5 S cm−1 and at the maximum temperature (80 °C) the conductivity is an order of magnitude larger (σ = 6 × 10−4 S cm−1). This agrees with values found in literature.7,36–38 The activation energy for the ion movement in the PEG is calculated from the Arrhenius equation and is found to be EA = 38 kJ mol−1.
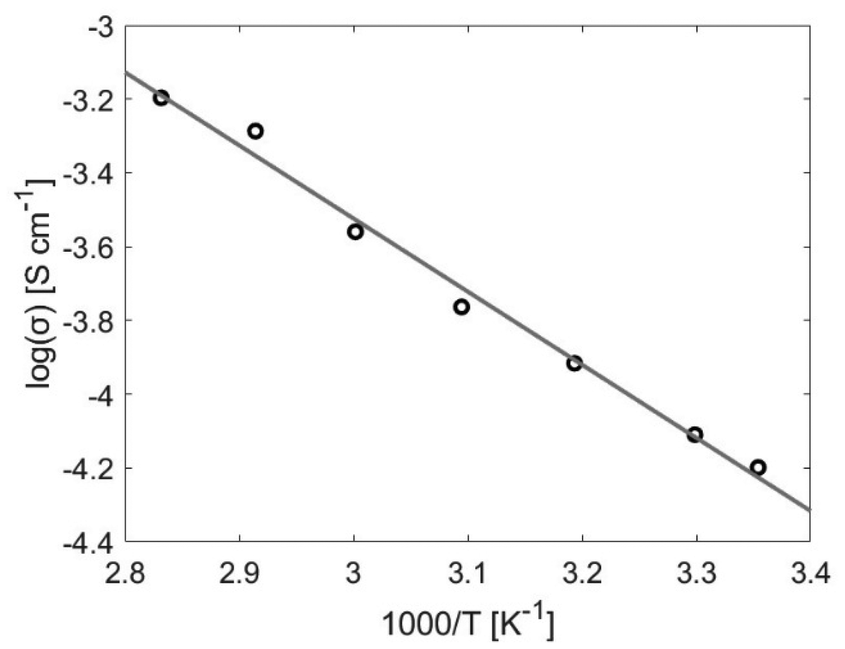 | ||
| Fig. 4 The Arrhenius plot calculated from the average high frequency resistance obtained from three EIS measurements conducted on the bulk polymer in a Swagelock, stainless steel blocking electrode cell at temperatures 25, 30, 40, 50, 60, 70 and 80 °C. | ||
The thermal transitions of the SPE were evaluated by DSC, the results of which is depicted in Fig. 5(a). The bulk polymer containing no salt exhibits a Tg of −60 °C, which agrees with values found in literature for PEGs,39 and a melting point at 3.5 °C. Thus, the bulk polymer containing no salt is in a molten state at room temperature. In comparison, the bulk polymer containing salt has a slightly higher Tg (−53 °C) and no observed melting point. This shows that the presence of the sodium salt, which coordinates to the conducting EO groups, completely supresses the crystallinity of the PEG. The coordination between the EO groups and sodium salt also decreases the mobility of the polymer chains, which explains the higher Tg. For the coated polymer, the Tg is increased further to −49 °C. The additional increase in Tg can be explained by the fact that the polymer chains are anchored to the CFs, which increases the energy required for the polymer chains to move freely. A complete suppression of crystallinity is also observed for the coated CFs, which is beneficial for the SPE conductivity.40 No other thermal events are observed for the salt containing samples during the second heating scan at temperatures higher than their respective Tg values within the studied temperature range.
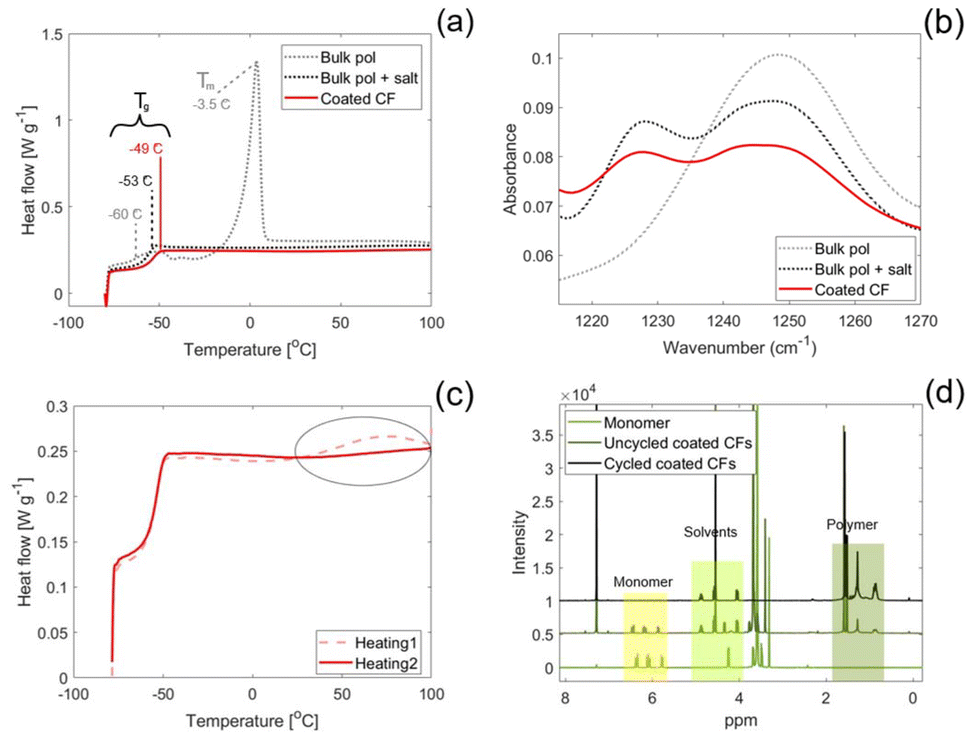 | ||
| Fig. 5 (a) The second DSC heating cycle between −80 and 100 °C for the bulk polymer, bulk polymer containing salt and the coated CFs. (b) The peaks related to uncoordinated (1250 cm−1) and coordinated (1230 cm−1) C–O–C groups found in the FTIR spectra for the bulk polymer, bulk polymer containing salt and the coated CFs. (c) The first and second DSC heating cycles for the coated CFs. (d) The NMR spectra for the PEGMEA monomer, uncycled coated CFs and the cycled coated CFs. | ||
The coordination between the salt and the conducting EO groups is confirmed by FTIR, where the peak for C–O–C stretching found at 1250 cm−1 is split into two peaks for the samples containing salt, see Fig. 5(b). The new peak, found at 1230 cm−1, corresponds to the coordinated conducting groups whereas the peak with reduced intensity found at 1250 cm−1 corresponds to the uncoordinated groups. This behaviour is observed both for the bulk polymer containing salt and the coated polymer.
The DSC measurement showed a thermal event in the first heating cycle that was not present during the second heating cycle for both the coated CFs, Fig. 5(c), and the bulk polymer containing salt. This thermal event can be explained by a heat-initiated polymerisation of unreacted monomer in the SPE, causing the heat flux observed during the first heating cycle. Since the monomer reacts in the first cycle, a similar event is not observed in the second heating cycle. Interestingly, the same behaviour was not observed for the bulk sample without salt, Fig. S1, implying that the salt affects the polymerisation.
To confirm the presence of unreacted monomer in the SPE and to evaluate whether it is initiated electrochemically upon cycling the coated CFs, 1H NMR measurements were performed (Fig. 5(d)). The peaks between 1 and 2 ppm stem from the polymer and are clearly present for both coated CF samples, which shows that unanchored polymer chains exist both prior to and after electrochemical cycling. Such unanchored polymer chains are expected since the polymerisation reaction is an equilibrium between forming grafted polymer chains and polymerising in bulk solution.27 Whereas the equilibrium favours the formation of grafted polymer chains at the grafting potential, bulk polymerisation will also occur in these conditions. Additional peaks are seen for the coated CFs between 4 and 5 ppm which are related to the solvents, EC and PC. Evidently, the peak around 6 ppm related to the vinyl bond in the PEGMEA monomer, is visible for the uncycled coated CFs. However, the cycled coated CFs show no peaks around this chemical shift, confirming the absence of monomer after cycling. Thus, the rinsing protocol is not sufficient in removing unreacted monomer and this is electrochemically relevant since the monomer reacts upon cycling.
3.3. Electrochemical performance
The rinsed and leached SPE coated CFs and the uncoated CFs were assembled in half-cell configurations and cycled against sodium metal to evaluate their electrochemical performance. The rate performance test reveals that all three samples regain their initial capacity by the final five cycles, see Fig. 6(a). The uncoated CFs outperform both types of coated CF samples in terms of capacity for all three specific currents. Of the two types of coated CF samples, the leached sample show a higher specific capacity for all three currents. This suggests that the resistance of the SPE on leached samples is lower than the rinsed samples. As evidenced by the 1H NMR spectra, the rinsed samples contain unreacted monomer that reacts upon electrochemical cycling. Although the CVs indicate complete passivation of the CF surface, Fig. 2(c), we propose that this reaction still occur near the CF/SPE interface during electrochemical cycling. This is because the longer cycling time scale, compared to that of the CVs, allows unreacted monomers to diffuse to the interface. This is supported by the fact that it appears as a loss in capacity, which is attributed to electron transfer to monomers at the CF surface. Formation of chemisorbed anions can subsequently react further with neighbouring monomers, leading to densification of the SPE at the interface and in the bulk of the polymer film, thereby increasing its ionic resistance, as shown in the rate performance tests in Fig. 6(a). Thus, by removing unreacted monomer by the leaching procedure prior to electrochemical cycling, the densification is supressed and thereby also the resistance increase.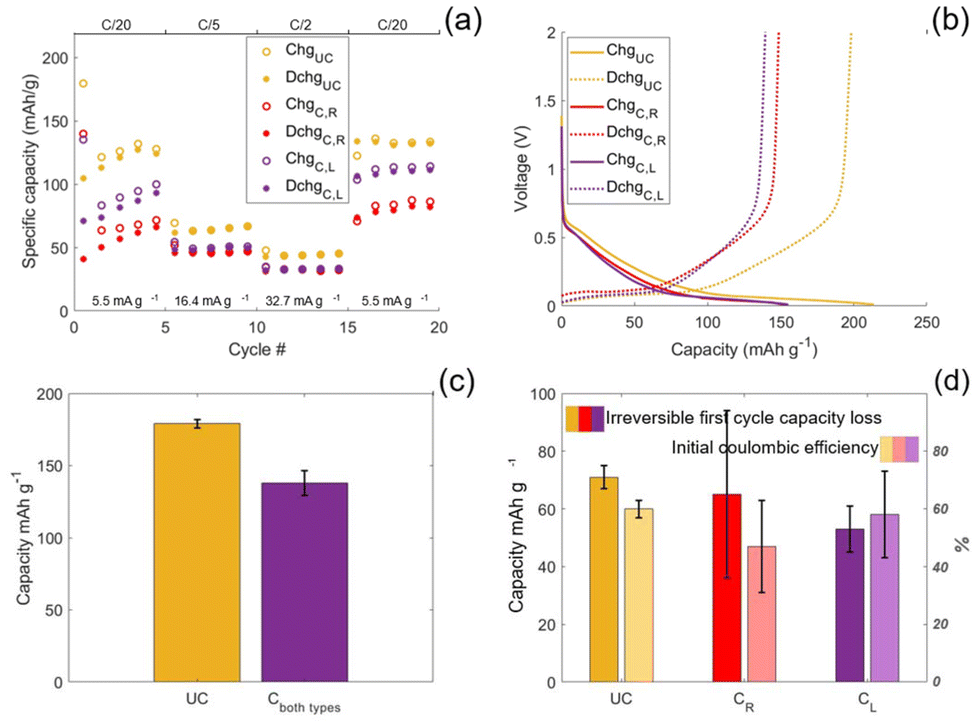 | ||
| Fig. 6 (a) Rate performance tests for half-cells containing coated-rinsed (C,R), coated-leached (C,L) and uncoated (UC) CFs, respectively. The voltage window is 2–0.01 V vs. Na/Na+. The specific current for cycles 1–5 is 5.5 mA g−1, for cycles 6–10 it is 16.4 mA g−1, for cycles 11–15 it is 32.7 mA g−1 and the last five cycles are cycled at the initial current, 5.5 mA g−1. (b) Maximum capacity tests performed at a low specific current of 1.4 mA g−1 for the rinsed and leached coated CFs and the uncoated CFs. (c) The average capacity for both types of coated and uncoated CFs when cycled at 1.4 mA g−1, which equates to ∼C/100. The red bar indicated both the rinsed and leached coated CFs and the error bars show the standard deviation. (d) The average irreversible first-cycle capacity loss and the initial coulombic efficiency (ICE) observed in the rate performance tests for the uncoated, coated and rinsed, and coated and leached samples, respectively. The error bars show the standard deviations. | ||
Cycling at low specific currents show maximum capacities reaching 200 and 150 mAh g−1 for the pristine and coated IMS65 fibres, respectively. The pristine IMS65 outperform the capacity for other polyacrolynitrile (PAN)-based fibre types found in literature, which exhibit a 170 mAh g−1 capacity.41 The charge/discharge profile of the IMS65 carbon fibres exhibits the typical characteristics of hard carbon materials, featuring both a slope and a plateau region, see Fig. 6(b).34 Most of the capacity (∼60%) originates from the plateau region, the flat section of the curve below 0.1 V vs. Na/Na+ close to the lower cut-off voltage. Due to its flat nature and proximity to the cut-off voltage, even small overpotentials can result in a portion of this capacity becoming inaccessible. In contrast, the slope region, which contributes ∼40% of the total capacity and is accessed at higher potentials (>0.1 V vs. Na/Na+) is much less sensitive to overpotentials. Interestingly, the relative capacity difference between coated and uncoated samples is more pronounced at low specific currents, which may seem counterintuitive. However, charge/discharge curves from the rate performance tests reveal that the plateau region is only accessed at the lowest specific current (5.5 mA g−1), while at higher currents (16.4 and 32.7 mA g−1), only the slope region is utilised, see Fig. S2. This implies that although overpotentials increase with current, the low-current condition (5.5 mA g−1) affects capacity more severely due to its reliance on the overpotential-sensitive plateau region. Thus, the impact of the coated SPE on the capacity is largest at low specific currents due to the inherent charge/discharge profile of the carbon fibres.
A capacity difference between the coated and uncoated samples is also observed when eliminating the influence of overpotentials by cycling at a very low specific current (1.4 mA g−1), see Fig. 6(b). The charge/discharge profiles are parallel, indicating no mechanistic differences in the sodiation/desodiation of the samples. Taking the intercept of the charge and discharge curves as the shift between the slope and plateau regions, the capacity is divided by approximately 40:60 for both coated and the uncoated CFs. Since the capacity of the coated samples is lower for both the slope and plateau region by an equal amount, the difference in capacity can be explained by a loss of active mass during electrografting. During manufacturing, some fibres do not establish direct electrical contact with the current collector when the CF tow is attached. This is a direct consequence of manually dividing the as received 24K fibre tow into the ∼1.5K tow sample size used herein. Specifically, the manual division process displaces a fraction of the fibres below the attachment point to the current collector, preventing direct contact. As a result, these fibres are electrically connected to the current collector only indirectly, via contact with neighbouring fibres. This indirect electrical contact is nevertheless sufficient to enable electrochemical grafting and the initiation of a monolayer of monomer on the CF surface, because this initiation step occurs instantaneously.27 Once initiated, propagation proceeds as a purely chemical process and therefore continues even if the indirect electrical contact is subsequently lost. Consequently, SEM images (Fig. 3a and b) show a homogeneous coating across all fibres. However, coating of the SPE removes fibre–fibre contact points, which in turn eliminates the indirect electrical pathways to the current collector. This electrically isolates fibres that previously relied on indirect contact. Because electrically isolated fibres cannot participate in electrochemical cycling, this loss of connectivity directly results in a reduction of the electrochemically active mass. This explains why the specific capacity is lower for the coated samples even at very low specific currents.
On average, the loss of active mass equates to 23% for both the rinsed and leached coated CFs, see Fig. 6(c). Considering this loss of active mass, the influence of the coated SPE on the rate performance test, Fig. 6(a), is exaggerated. Even with this exaggerated influence on capacity, the SPE coated CFs perform similarly to the uncoated CFs. This implies that resistance of the SPE is indeed sufficiently low to allow high power and energy density separator-less structural sodium batteries.
The influence of the coated SPE on irreversible first-cycle capacity loss and initial coulombic efficiency (ICE) is evaluated for uncoated, coated-rinsed, and coated-leached CFs, see Fig. 6(d). Both coated-rinsed and coated-leached CFs exhibit lower irreversible capacity loss compared to the uncoated samples, which is consistent with a reduction in active CF mass during the electrografting process. Between the two, the coated-rinsed CFs display higher capacity loss and lower ICE than the coated-leached CFs, which is attributed to the initiation of unreacted monomer. This is further supported by the large standard deviations observed for the coated-rinsed samples, suggesting variability in monomer content within the SPE. Therefore, to assess the intrinsic effect of the coated SPE without the influence of unreacted monomer, the uncoated CFs are compared to the coated-leached CFs. Interestingly, the reduction in irreversible first-cycle capacity loss (25%) closely matches the reduction in maximum capacity (23%), indicating that the reduction in irreversible loss is proportional to the loss in active CF mass.
The ICE remains comparable for the uncoated (60%) and coated-leached (58%) CFs. Surprisingly, this suggests that the presence of the coated SPE does not significantly impact SEI formation or the irreversible entrapment of sodium ions within the CF microstructure. This observation indicates that, although the SPE film is homogeneous (Fig. 3(a) and (b)) and clearly passivating (Fig. 2(c)), the density of the grafted polymer chains is probably low and therefore may not entirely suppress reactions between the liquid electrolyte and the CF surface that lead to SEI formation. Increasing the solvent DN could enhance the density of grafted polymer brushes and improve the ICE. However, such an increase in grafting density may also adversely affect the ionic conductivity of the SPE.
Stability tests were conducted on both coated-rinsed and uncoated CFs, during which 100 charge/discharge cycles were completed. After initial stabilisation, both samples exhibited high coulombic efficiencies exceeding 99%, as shown in Fig. 7(a). Notably, an increase in both coulombic efficiency and cyclable capacity was observed during the first 40 cycles before reaching a steady state, see Fig. 7(a) and (b). The most significant rise in coulombic efficiency occurred within the initial five cycles for both coated and uncoated CFs, consistent with expected capacity losses due to solid electrolyte interphase (SEI) formation and the irreversible entrapment of sodium ions within the CF structure.10
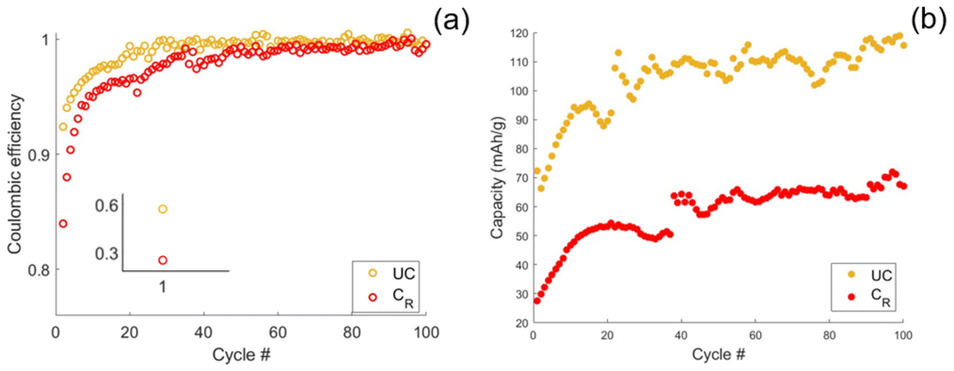 | ||
| Fig. 7 (a) Stability tests cycling at cycles 10.9 mA g−1, corresponding to a C-rate of approximately C/10, over 100 cycles for rinsed coated and uncoated CFs in half-cell configurations showing the coulombic efficiency. (b) The discharge capacity over the 100 cycles for the rinsed coated and uncoated CF half-cells. | ||
Although the absolute capacity increase over the first 40 cycles is similar for both samples, the relative increase is greater for the coated CFs. This difference can be attributed to the initially lower capacity of the rinsed coated CFs, which is attributed to active mass loss during electrografting and increased overpotential induced by the SPE. The comparable absolute increase across both samples suggests that the capacity enhancement originates primarily from the CFs themselves or the sodium counter electrode, rather than from the SPE.
Similar behaviour has previously been observed in other studies of hard carbon materials, where the capacity increases over the initial 10–50 cycles.42,43 This may be caused by voltage drift, which is known to occur for sodium metal in organic solvents,44 effectively leading to cycling over a larger voltage window. As most of the capacity is typically delivered in the low-voltage plateau region, even small changes in the voltage window can lead to a substantial increase in capacity.
4. Conclusions
This study demonstrates an efficient method for coating carbon fibres (CFs) with a thin, fully amorphous PEG-based solid polymer electrolyte that exhibits excellent electrochemical durability, thermal stability, and low ionic resistance. The electrografting process enables uniform SPE deposition over the CF surface within a short time frame (150 seconds). Following electrografting, leaching of the coated CFs to remove unreacted monomer is essential for reducing internal cell resistance and mitigating irreversible capacity loss during the first electrochemical cycle. The resulting SPE layer has a thickness of approximately 1.1 μm, an EO:Na ratio of 30:1, and an ionic conductivity of 6 × 10–5 S cm−1 at 25 °C. Given that PEG/NaTFSI systems typically exhibit maximum conductivity at higher salt concentrations, an increase in the salt content is expected to further enhance ionic conductivity in this system. The thermal stability of the SPE is high, with decomposition occurring at 390 °C, indicating suitability for battery applications. Despite partial loss of active CF mass during electrografting, the SPE-coated CFs exhibit high reversible sodium storage capacities of approximately 150 mAh g−1. The influence of the SPE on rate performance is minor, especially when accounting for the reduction in active material. Interestingly, the sodium storage capacity increases over the initial 40 cycles before stabilising, which could be the results of a voltage drift of the sodium counter electrode. After stabilisation, the coated CFs maintain high coulombic efficiencies exceeding 99% through 100 cycles.
Collectively, these results highlight the significant potential of sodium-SPE coated CFs in the development of sustainable, high energy and power density separator-less structural sodium batteries. This controlled way of creating SPEs could likely be of interest also for conventional sodium-ion batteries.
Author contributions
Conceptualisation, E. L., G. L., D. Z., M. J., K. O., methodology, E. L., investigation, E. L., V. N., M. C., funding acquisition, G. L., D. Z. The original draft was written by E. L. and reviewed by all other co-authors.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5eb00212e.Acknowledgements
The authors would like to sincerely thank Björn Eriksson for his help in acquiring SEM images and EDS scans, Noé Fanjul-Mosteirín for his help in performing the TGA, DSC and FTIR measurements and Alfredo Bici for his support with processing data. This work was supported by the Swedish Energy Agency [project number 50508-1], the Swedish Research Council [project numbers 2021-05276 and 2024-04344], Air Force Office of Scientific Research [grant number FA8655-25-1-7047], the StandUp for Energy initiative.References
- Energy storage, https://www.iea.org/energy-system/electricity/grid-scale-storage, accessed October 3, 2025.
- L. Wang, H. Tian, X. Yao, Y. Cai, Z. Gao and Z. Su, ChemElectroChem, 2024, 11, e202300414 Search PubMed.
- X.-X. He, L. Li, X. Wu and S.-L. Chou, Adv. Mater., 2025, 37, 2506066 CrossRef CAS PubMed.
- T. Liu, Y. Zhang, Z. Jiang, X. Zeng, J. Ji, Z. Li, X. Gao, M. Sun, Z. Lin, M. Ling, J. Zheng and C. Liang, Energy Environ. Sci., 2019, 12, 1512–1533 RSC.
- H. Pan, Y.-S. Hu and L. Chen, Energy Environ. Sci., 2013, 6, 2338–2360 Search PubMed.
- Y. Sun, J.-C. Li, H. Zhou and S. Guo, Energy Environ. Sci., 2023, 16, 4759–4811 RSC.
- Q. Liu, X. Zhao, Q. Yang, L. Hou, D. Mu, G. Tan, L. Li, R. Chen and F. Wu, Adv. Mater. Technol., 2023, 8, 2200822 Search PubMed.
- L. E. Asp, M. Johansson, G. Lindbergh, J. Xu and D. Zenkert, Funct. Compos. Struct., 2019, 1, 042001 Search PubMed.
- S. Kalnaus, L. E. Asp, J. Li, G. M. Veith, J. Nanda, C. Daniel, X. C. Chen, A. Westover and N. J. Dudney, J. Energy Storage, 2021, 40, 102747 CrossRef.
- R. Harnden, K. Peuvot, D. Zenkert and G. Lindbergh, J. Electrochem. Soc., 2018, 165, B616–B622 CrossRef CAS.
- R. Mao, Z. Lei, J. Di, Y. Shang, R. Bai and C. Yan, J. Electrochem. Energy Convers. Storage, 2025, 22(1) DOI:10.1115/1.4065094.
- E. S. Greenhalgh, S. Nguyen, M. Valkova, N. Shirshova, M. S. P. Shaffer and A. R. J. Kucernak, Compos. Sci. Technol., 2023, 235, 109968 CrossRef CAS.
- D. Zenkert, R. Harnden, L. E. Asp, G. Lindbergh and M. Johansson, Composites Part B, 2024, 273, 111240 CrossRef CAS.
- N. Ihrner, W. Johannisson, F. Sieland, D. Zenkert and M. Johansson, J. Mater. Chem. A, 2017, 5, 25652–25659 RSC.
- S. Duan, M. Cattaruzza, V. Tu, R. M. Auenhammer, R. Jänicke, M. K. G. Johansson, F. Liu and L. E. Asp, Commun. Mater., 2023, 4, 49 CrossRef CAS.
- M. Cattaruzza, Y. Fang, I. Furó, G. Lindbergh, F. Liu and M. Johansson, J. Mater. Chem. A, 2023, 11, 7006–7015 RSC.
- R. Tavano, M. Spagnol, N. Al-Ramahi, R. Joffe, J. Xu and L. E. Asp, Polymer, 2024, 312, 127646 CrossRef CAS.
- W. Johannisson, D. Zenkert and G. Lindbergh, Multifunct. Mater., 2019, 2, 035002 CrossRef CAS.
- R. Harnden, D. Carlstedt, D. Zenkert and G. Lindbergh, ACS Appl. Mater. Interfaces, 2022, 14, 33871–33880 CrossRef CAS PubMed.
- L. E. Asp, K. Bouton, D. Carlstedt, S. Duan, R. Harnden, W. Johannisson, M. Johansen, M. K. G. Johansson, G. Lindbergh, F. Liu, K. Peuvot, L. M. Schneider, J. Xu and D. Zenkert, Adv. Energy Sustainability Res., 2021, 2, 2000093 CrossRef.
- K. Bouton, L. Schneider, D. Zenkert and G. Lindbergh, Compos. Sci. Technol., 2024, 256, 110728 CrossRef CAS.
- W. Johannisson, N. Ihrner, D. Zenkert, M. Johansson, D. Carlstedt, L. E. Asp and F. Sieland, Compos. Sci. Technol., 2018, 168, 81–87 CrossRef CAS.
- D. Zhou, D. Shanmukaraj, A. Tkacheva, M. Armand and G. Wang, Chem, 2019, 5, 2326–2352 CAS.
- N. A. Y. Razamin, R. H. Y. Subban and T. Winie, Mater. Today: Proc., 2019, 17, 459–464 CAS.
- X. Chen, W. He, L.-X. Ding, S. Wang and H. Wang, Energy Environ. Sci., 2019, 12, 938–944 RSC.
- V. Vijayakumar, B. Anothumakkool, S. Kurungot, M. Winter and J. Ravi Nair, Energy Environ. Sci., 2021, 14, 2708–2788 RSC.
- S. Gabriel, R. Jérôme and C. Jérôme, Prog. Polym. Sci., 2010, 35, 113–140 CrossRef CAS.
- S. Leijonmarck, T. Carlson, G. Lindbergh, L. E. Asp, H. Maples and A. Bismarck, Compos. Sci. Technol., 2013, 89, 149–157 CrossRef CAS.
- N. Baute, C. Jérôme, L. Martinot, M. Mertens, V. M. Geskin, R. Lazzaroni, J.-L. Brédas and R. Jérôme, Eur. J. Inorg. Chem., 2001, 2001, 1097–1107 CrossRef.
- N. Baute, C. Calberg, P. Dubois, C. Jérǒme, R. Jérdme, L. Martinot, M. Mertens and P. Teyssié, Macromol. Symp., 1998, 134, 157–166 CrossRef CAS.
- N. Baute, P. Teyssié, L. Martinot, M. Mertens, P. Dubois and R. Jérôme, Eur. J. Inorg. Chem., 1998, 1998, 1711–1720 CrossRef.
- N. Baute, C. Jérôme, L. Martinot, M. Mertens, V. M. Geskin, R. Lazzaroni, J.-L. Brédas and R. Jérôme, Eur. J. Inorg. Chem., 2001, 2001, 1097–1107 CrossRef.
- D. Monti, E. Jónsson, A. Boschin, M. Rosa Palacín, A. Ponrouch and P. Johansson, Phys. Chem. Chem. Phys., 2020, 22, 22768–22777 Search PubMed.
- X. Chen, C. Liu, Y. Fang, X. Ai, F. Zhong, H. Yang and Y. Cao, Carbon Energy, 2022, 4, 1133–1150 CrossRef CAS.
- F. Cataldo, Eur. Chem. Bull., 2015, 4(2), 92–97 Search PubMed.
- A. Boschin and P. Johansson, Electrochim. Acta, 2015, 175, 124–133 CrossRef CAS.
- F. Gebert, J. Knott, R. Gorkin, S.-L. Chou and S.-X. Dou, Energy Storage Mater., 2021, 36, 10–30 CrossRef.
- M. M. Doeff, A. Ferry, Y. Ma, L. Ding and L. C. D. Jonghe, J. Electrochem. Soc., 1997, 144, L20 CrossRef CAS.
- Y. An, X. Han, Y. Liu, A. Azhar, J. Na, A. K. Nanjundan, S. Wang, J. Yu and Y. Yamauchi, Small, 2022, 18, 2103617 CrossRef CAS PubMed.
- V. St-Onge, M. Cui, S. Rochon, J.-C. Daigle and J. P. Claverie, Commun. Mater., 2021, 2, 83 CrossRef CAS.
- R. Harnden, K. Peuvot, D. Zenkert and G. Lindbergh, J. Electrochem. Soc., 2018, 165, B616 CrossRef CAS.
- Z. Lu, H. Yang, Y. Guo, H. Lin, P. Shan, S. Wu, P. He, Y. Yang, Q.-H. Yang and H. Zhou, Nat. Commun., 2024, 15, 3497 CrossRef CAS PubMed.
- Y. He, F. Yu, K. Liu, L. Bai, Y. Liu, C. Liu, H. Niu, J. Xu, K. Bu, J. Zhao, Z. Chang and H. Zhou, Adv. Energy Mater., e04760.
- D. S. Tchitchekova, D. Monti, P. Johansson, F. Bardé, A. Randon-Vitanova, M. R. Palacín and A. Ponrouch, J. Electrochem. Soc., 2017, 164, A1384 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |