
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputational investigation of perfluorcarbonates as multi-functional diluents in localized high concentration electrolytes for lithium metal batteries: insights from MD and DFT simulations
Zhuijun Xua,
Tianle Zheng a,
Ying Yub,
Yaozong Guc,
Yinghui Licd,
Jie Gaoc,
Siqi Shid,
Yonggao Xia*ce,
Ya-Jun Cheng*bc and
Peter Müller-Buschbaum*a
a,
Ying Yub,
Yaozong Guc,
Yinghui Licd,
Jie Gaoc,
Siqi Shid,
Yonggao Xia*ce,
Ya-Jun Cheng*bc and
Peter Müller-Buschbaum*a
aTechnical University of Munich, TUM School of Natural Sciences, Department of Physics, Chair for Functional Materials, Garching, 85748, Germany. E-mail: muellerb@ph.tum.de
bCollege of Renewable Energy, Hohai University, 1915 Hohai Ave, Jintan District, Changzhou, Jiangsu 213200, P. R. China. E-mail: xiayg@nimte.ac.cn; chengyajun@hhu.edu.cn
cNingbo Institute of Materials Technology & Engineering, Chinese Academy of Sciences, Ningbo, Zhejiang 315201, P. R. China
dSchool of Materials Science and Engineering, Shanghai University, Shanghai, 200444, P. R. China
eCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Shijingshan District, Beijing 100049, P. R. China. E-mail: xiayg@nimte.ac.cn; chengyajun@hhu.edu.cn; muellerb@ph.tum.de
First published on 24th April 2026
Abstract
Diluents play a vital role in the preparation of localized high concentration electrolytes (LHCEs) for 500 Wh kg−1 lithium metal batteries (LMBs). Although the current 1,1,2,2-tetrafluoroethyl-2,2,3,3-tetrafluoropropylether (TTE)-containing diluent electrolytes offer good performance, they still face challenges (e.g., side reactions with nickel-rich layered materials, gas production, and high volatility). Therefore, it is crucial to develop the next generation of diluents. In this study, a variety of solvents, including 4-fluoro-, 4,4-difluoro-, cis- and trans-4,5-difluoro-, 4,4,5-trifluoro-, and 4,4,5,5-tetrafluoro-1,3-dioxolan-2-one (Tetra-FEC), and TTE, are analyzed using molecular dynamics (MD) simulations and density functional theory (DFT) calculations. Key physical and chemical properties are examined, including the densities, dielectric constant, highest occupied molecular orbital (HOMO), and lowest unoccupied molecular orbital (LUMO) values, binding energies with Li+, as well as adsorption and dissociation capability on the LiNiO2 cathodes. Further MD and DFT calculations on corresponding LHCE systems reveal densities, Li+ concentration, Li+ diffusion coefficients, solvation structures, and HOMO–LUMO levels of solvation clusters, highlighting the potential of Tetra-FEC as a promising diluent. Moreover, this study demonstrates a green research approach by utilizing MD and DFT calculations, eliminating the need for chemical experiments, which also showcases a viable process for future electrolyte research through artificial intelligence.
Broader contextThis study presents a systematic computational investigation of fluorinated vinyl carbonates as advanced diluents for high-energy-density lithium-metal batteries (LMBs). The results clearly identify 4,4,5,5-tetrafluoro-1,3-dioxolan-2-one (Tetra-FEC) as a superior alternative to the conventional diluent 1,1,2,2-tetrafluoroethyl-2,2,3,3-tetrafluoropropylether (TTE) by employing molecular dynamics and density functional theory (MD/DFT), demonstrating enhanced ability to a spectrum of physical properties. This work successfully establishes a green, computation-driven design strategy, which effectively eliminates the need for resource-intensive experimental screening. It thereby provides a rational and efficient pathway for accelerating the development of next-generation electrolyte systems, highlighting the power of theoretical modeling in sustainable battery innovation. |
Introduction
Alkali metals, such as lithium metal and sodium metal, are known for their significant theoretical capacity in combination with a low electrochemical potential. For instance, lithium metal has a specific capacity of 3860 mAh g−1 with the lowest electrochemical potential (−3.04 V vs. SHE).1,2 Therefore, lithium metal batteries (LMBs), which utilize lithium metal as anodes, can offer higher energy densities compared to current lithium-ion batteries. However, uncontrolled dendrite growth, side reactions, electrolyte depletion, and thickening of the SEI layer accelerate capacity fading and pose severe safety risks.3To address the above issues, various approaches have been adopted, including electrolyte additive engineering, construction of artificial SEI/CEI layers, and design of novel electrode current collectors.4–8 Among these, high-concentration electrolytes (HCEs) and localized high-concentration electrolytes (LHCEs) are simple and effective strategies to stabilize Li plating/stripping.9–11 For example, Ren et al. demonstrated that using 1,1,2,2-tetrafluoroethyl-2,2,3,3-tetrafluoropropyl ether (TTE) as a diluent in LiFSI/1,2-DME/TTE (1,2-DME: 1,2-dimethoxyethane and 1,2-DME![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TTE = 1.2:3, molar ratio) significantly improved Li‖NCM811(LiNi0.8Co0.1Mn0.1O2) cycling at 4.4–4.5 V even under limited Li and electrolyte.12 Nevertheless, Ni-rich layered cathodes (Ni > 0.9) operating at high voltages (>4.5 V) promote spontaneous ether decomposition, limiting the oxidative stability (∼4.5 V) of conventional LHCEs and inducing gas evolution, electrode cracking, and shortened pouch cell lifetimes.12–17 Although widely used as a diluent, TTE contributes little to Li+ solvation, SEI/CEI formation, or interfacial stabilization, functioning primarily as a volumetric filler. Its limited oxidative stability under high-voltage conditions further promotes its decomposition, generating gas and raising safety concerns.14,18 In addition, the large quantities of TTE required reduce the overall energy density, and its high volatility further restricts practical large-scale applications.19
:TTE = 1.2:3, molar ratio) significantly improved Li‖NCM811(LiNi0.8Co0.1Mn0.1O2) cycling at 4.4–4.5 V even under limited Li and electrolyte.12 Nevertheless, Ni-rich layered cathodes (Ni > 0.9) operating at high voltages (>4.5 V) promote spontaneous ether decomposition, limiting the oxidative stability (∼4.5 V) of conventional LHCEs and inducing gas evolution, electrode cracking, and shortened pouch cell lifetimes.12–17 Although widely used as a diluent, TTE contributes little to Li+ solvation, SEI/CEI formation, or interfacial stabilization, functioning primarily as a volumetric filler. Its limited oxidative stability under high-voltage conditions further promotes its decomposition, generating gas and raising safety concerns.14,18 In addition, the large quantities of TTE required reduce the overall energy density, and its high volatility further restricts practical large-scale applications.19
Fluorination of low-volatility ester solvents lowers their HOMO/LUMO levels, enhancing the electrolytes’ oxidative stability and promoting reduction ability. At the cathode/anode, these solvents are readily decomposed to form LiF-rich SEI/CEI layers, further mitigating lithium dendrite growth and minimizing electrolyte degradation.20 For example, 4-fluoro-1,3-dioxolan-2-one (FEC) improve cycling stability: partial replacement of TTE with FEC in LiFSI/LiPF6-FEC/1,2-DME/TTE yielded 57.1% capacity retention after 500 cycles at 45 °C for SiOx/C‖NCM90 cells.21 Similarly, trans-4,5-difluoro-1,3-dioxolan-2-one in LHCEs enhanced interfacial compatibility, maintaining >80% capacity over 1000 cycles in Li‖LiCoO2 at 0.3 C with the voltage range of 3.0–4.5 V.22–24 These studies highlight that fluorinated vinyl carbonates and their analogs can serve dual roles as co-solvents and diluents, contributing to the formation of robust interfacial layers (CEI and SEI layers) that enhance battery performance. Moreover, fluorination also increases solvent flash points and reduces flammability; when F/H > 4 and F/C > 1, electrolytes become nonflammable.25–27
The exceptional properties of fluorinated vinyl carbonates and similar compounds motivate the exploration of their potential as multifunctional solvents and diluents for next-generation safe, high-energy-density lithium metal batteries.20,25 Compounds such as 4,4,5,5-tetrafluoro-1,3-dioxolane-2-one (Tetra-FEC) offer promising avenues for performance enhancement; however, current synthetic limitations impede their scalable production, precluding experimental evaluation and highlighting the need for alternative approaches to probe their electrochemical potential.
In this study, we systematically investigated the physical and electrochemical properties of a series of solvents – including FEC, 4,4-difluoro-1,3-dioxolan-2-one (DFEC), cis-4,5-difluoro-1,3-dioxolan-2-one (DFEC1), trans-4,5-difluoro-1,3-dioxolan-2-one (DFEC2), 4,4,5-trifluoro-1,3-dioxolan-2-one (Tri-FEC), Tetra-FEC, 1,2-DME, and TTE – using molecular dynamics (MD) simulations, and further analyzed their HOMO/LUMO levels, Li binding energies, and interactions with LiNiO2 cathodes via density functional theory (DFT) calculation. Tetra-FEC, with the highest fluorination, lowest dielectric constant (∼2), and weakest Li+ binding energy, indicate its potential as a diluent.28 Additionally, the lower HOMO of Tetra-FEC, weak adsorption energy with LiNiO2, and the theoretically absent dissociation process confer stronger antioxidant properties on the cathode side, while lower LUMO levels of Tetra-FEC indicate that it is more easily reduced on the anode and forms a robust SEI layer.
Subsequently, seven localized high-concentration electrolyte (LHCE) systems were constructed by combining LiFSI/1,2-DME with different diluents (FEC, DFEC, DFEC1, DFEC2, Tri-FEC, Tetra-FEC, or TTE) in MD simulations. Essential physical properties, including density, lithium-ion molar concentrations, and lithium-ion diffusion coefficient, are analyzed. Compared to the classical LiFSI/1,2-DME/TTE LHCEs system, LiFSI/1,2-DME/Tetra-FEC LHCEs system exhibits slightly higher electrolyte density (9.6% increase) and lower lithium-ion diffusion coefficient (maintaining the same order of magnitude). Radial distribution function (RDF) curves further revealed that FEC mainly functions as a co-solvent participating in Li+ solvation, TTE is largely inert, while Tetra-FEC partially enters the solvation shell, promoting FSI− coordination and potential LiF generation. Finally, DFT calculations confirmed that LiFSI/1,2-DME/Tetra-FEC clusters possess the lowest HOMO and LUMO levels among all tested systems, demonstrating enhanced oxidative resistance and favorable SEI/CEI formation. Consequently, all the above-mentioned results highlight the potential of Tetra-FEC as a promising diluent for high-energy-density LMBs. Moreover, this study demonstrates the relevant content to clearly frame this work as a foundational computational workflow, and explicitly propose that this approach can be further extended, combined, and integrated with machine learning and artificial intelligence algorithms in future research, so as to provide a feasible research paradigm for AI-driven electrolyte design and optimization.
DFT computational method
DMol3 uses numerical atomic orbital basis sets (e.g., DNP), enabling efficient and accurate all-electron calculations for molecular and cluster systems, making it ideal for molecular properties such as Li+ binding energies, solvation structures, charge distributions, and HOMO/LUMO levels. However, it is inherently limited for periodic surfaces: its localized, finite-cutoff basis sets poorly describe delocalized electrons, d-band states, and long-range interactions at transition metal surfaces, with lower accuracy for slab models, vacuum layers, interlayer interactions, and adsorption hybridization. By contrast, Vienna Ab initio Simulation Package-VASP employs plane-wave basis sets naturally suited for periodic boundary conditions, providing more reliable electronic structure and energetic descriptions of metal surfaces, interfaces, and adsorbates.HOMO and LUMO of all solvents, anions and clusters within seven LHCE systems
For all the selected solvents, including FEC, DFEC, DFEC1, DFEC2, Tri-FEC, Tetra-FEC, and TTE, as well as salts consisting of LiFSI, DFT calculations for geometric optimization and energy are conducted in the Materials Studio with the DMol3 package. The hybrid Becke's three-parameter and Lee–Yang–Parr hybrid functional (B3LYP) density functional method is used to solve the exchange correlation energy when solving the Schrödinger equation, and the solvation effect is not considered.29 The HOMO and LUMO level calculations of all clusters within the selected seven LHCEs system are conducted using the framework of the generalized gradient approximation (GGA) and the Perdew–Burke–Ernzerhof (PBE) functional, and the corresponding solvation effects are taken into consideration (detailed explanations are provided in Table S1 and the accompanying discussion below.).30,31To improve accuracy, vibrational frequency analyses were performed for optimized structures, and zero-point energy (ZPE) corrections were added to the total electronic energies in subsequent binding energy and orbital calculations.
Electrostatic potential (ESP) charges distribution on the oxygen atoms of various solvents
In the calculation step for “HOMO and LUMO of all solvents, anions and clusters within seven LHCE systems”, during the calculation of solvent energies, by selecting Electrostatic potential (ESP) charges distribution in the population analysis, the ESP charge distribution on oxygen atoms of all solvent molecules was obtained.Binding energy (Eb) between Li+ and solvents/anions
The binding energy (Eb) is further ascribed in the Materials Studio with the DMol3 package and the B3LYP density functional method for the electrostatic interactions, specifically ion-dipole interactions, that occur between the solvent and lithium ions (Li+) within the electrolytes. This energy is defined as follows:| Eb = ELi+−x − Ex − ELi+ |
Computational details
In the above three calculations with Materials Studio, to account for van der Waals interactions, the Grimme approach for DFT-D is applied as an empirical correction. The valence electron wave functions are described using the double-numeric with polarization (DNP) basis set version 4.4.29 As for the electronic, the DFT Semi-core Pseudopots (DSPP) is selected in the core treatment. Electrostatic potential (ESP) charges are applied to optimize all molecules.32,33 During geometric optimization and energy calculations, the k-points are set at Gamma (1 × 1 × 1). As for the convergence criteria for energy, force, and displacement are set to 1.0 × 10−5 Ha, 2.0 × 10−3 Ha Å−1, and 5 × 10−3 Å, respectively. Furthermore, to expedite the convergence of the electronic computational steps, a thermodynamic damping factor of 0.005 Ha is introduced.Absorption energy, dissociation energy between solvents and LiNiO2 (10![[1 with combining macron]](https://www.rsc.org/images/entities/h3_char_0031_0304.gif) 4) surface
4) surface
Periodic plane-wave DFT calculations are carried out to investigate the bulk properties of LiNiO2 and the adsorption behavior of various solvents, including FEC, DFEC, DFEC1, DFEC2, Tri-FEC, Tetra-FEC, and TTE-on its oxide surfaces.34 The calculations use the PBE exchange–correlation functional as implemented in the VASP, with the Projector Augmented Wave (PAW) method used to describe core-electron interactions.35,36 A plane-wave cutoff energy of 450 eV is applied. The DFT+U method is utilized to account for strong correlation effects in the transition metal d orbitals, with an effective U value (Ueff = U − J) of 6.4 eV for Ni.37–40
All LiNiO2 structures are modeled in the layered R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group. The Brillouin zone of the primitive bulk unit cell is sampled using a 9 × 9 × 9 k-point mesh. Ferromagnetic ordering is imposed on all oxide structures to ensure consistency and tractability in magnetic configurations. Particles of nickel-rich layered oxide cathode materials are mostly terminated by the (10
m space group. The Brillouin zone of the primitive bulk unit cell is sampled using a 9 × 9 × 9 k-point mesh. Ferromagnetic ordering is imposed on all oxide structures to ensure consistency and tractability in magnetic configurations. Particles of nickel-rich layered oxide cathode materials are mostly terminated by the (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 4) surface under oxygen-rich conditions. The nonpolar surface (104), possessing the lowest surface energy, provides sufficient active adsorption sites, and, therefore, is chosen as the representative surface for adsorption studies.41,42 Surface models consist of five oxide layers with a vacuum spacing of at least 15 Å to eliminate inter-slab interactions, and dipole corrections are applied to suppress artificial electrostatic effects perpendicular to the surface. The bottom three layers are fixed at bulk positions, while the top layers and adsorbed species are fully relaxed. To minimize lateral interactions between adsorbates, a (3 × 2) surface supercell is used.
4) surface under oxygen-rich conditions. The nonpolar surface (104), possessing the lowest surface energy, provides sufficient active adsorption sites, and, therefore, is chosen as the representative surface for adsorption studies.41,42 Surface models consist of five oxide layers with a vacuum spacing of at least 15 Å to eliminate inter-slab interactions, and dipole corrections are applied to suppress artificial electrostatic effects perpendicular to the surface. The bottom three layers are fixed at bulk positions, while the top layers and adsorbed species are fully relaxed. To minimize lateral interactions between adsorbates, a (3 × 2) surface supercell is used.
Initial adsorption geometries of the various solvents on the LiNiO2 (104) surface are constructed using the ADF modeling suite and pre-relaxed using the Universal Force Field (UFF) in molecular mechanics. These configurations are then further relaxed using DFT, allowing full optimization of the top two surface layers and solvent molecules until the forces converged below 0.04 eV Å−2 (0.01 eV Å−2 for bulk and clean surface calculations), while the bottom layers remained fixed at bulk positions. Vibrational frequency calculations were performed for adsorbed configurations to obtain zero-point energy (ZPE) corrections, which were added to the total electronic energies.
For adsorption configurations, the carbonyl oxygen in fluorinated vinyl carbonates and analogous compounds, or the ether oxygen in TTE, interacts with metal sites on the oxide surface. For dissociation configurations, a C–H bond in fluorinated vinyl carbonates and analogous compounds or in TTE undergoes cleavage, accompanied by the formation of surface hydroxyl groups. During this process, no oxygen vacancies are generated in the oxide, and the oxide oxygen atoms deviate from their original positions.
The absorption energy (Eab) or dissociation energy (Ediss) is calculated as follows:
| Eab/diss = ELiNiO2(104)−y − ELiNiO2(104) − Ey |
4)−y is the ZPE-corrected total energy of the various solvents on the (104) plane of LiNiO2 (when calculating the adsorption energy and dissociation energy of a given solvent molecule, ELiNiO2(104)−y corresponds to its different adsorption configuration and dissociation configuration, respectively), and ELiNiO2(104) and Ey are the ZPE-corrected energies of bulk LiNiO2 with (104) surface and corresponding solvents, respectively.
MD calculation
All MD simulations are conducted using the COMPASS force field within Material Studio, facilitated by the Forcite package. The Ewald summation method is used to account for long-range electrostatic interactions, while the van der Waals interactions are handled using an atom-based approach. To determine the densities and the dielectric constant of various molecules, including FEC, DFEC, DFEC1, DFEC2, Tri-FEC, Tetra-FEC, 1,2-DME, and TTE at room temperature (25 °C), 200 instances of each solvent molecule are configured within an amorphous cell. For each cell, a geometry optimization is conducted, followed by a 100 ps pre-equilibrium simulation under the constant number, pressure, and temperature (NPT) ensemble conditions at room temperature with 0.1 MPa. The densities of the various molecules are calculated. Post-equilibration, an equilibrium simulation of 10000 ps (i.e., 10 ns) is executed at the respective temperature under the constant number of particles, volume, and temperature (NVT) ensemble. After NVT conditions, the total dipole moment of the system (M) and the square of it (M2) are both averaged from 5000 ps to the end of the NVT production run. Subsequently, the dielectric constant is calculated according to the following formula:where ε represents the dielectric constant of the solvents, ε0 is the dielectric constant of vacuum, V denotes the volume of the simulation cells, kB is the Boltzmann constant, and T is the absolute temperature.43
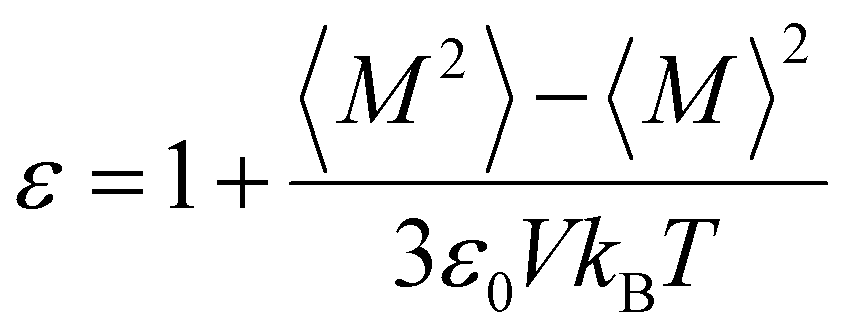
For the bulk electrolyte MD simulations, which encompass seven lithium ion electrolytes, a geometry optimization is initially carried out. This step is followed by a 20 ps pre-equilibrium run under NPT ensemble conditions at room temperature. Post-equilibration, an equilibrium simulation of 200 ps is executed at the respective temperature under the constant number of particles, volume, and temperature (NVT) ensemble.44
Throughout the MD computational process, the Nosé algorithm and Berendsen algorithm methods are utilized to regulate temperature and pressure, respectively.
The specific quantities of species used to simulate the seven types of electrolytes are detailed in Table 1.
| No. | FEC | DFEC | DFEC1 | DFEC2 | Tri-FEC | Tetra-FEC | TTE | 1,2-DME | LiFSI |
|---|---|---|---|---|---|---|---|---|---|
| No. 1–7 are LiFSI/1,2-DME/FEC, LiFSI/1,2-DME/DFEC, LiFSI/1,2-DME/DFEC1, LiFSI/1,2-DME/DFEC2, LiFSI/1,2-DME/Tri-FEC, LiFSI/1,2-DME/Tetra-FEC, LiFSI/1,2-DME/TTE, respectively. | |||||||||
| 1 | 205 | 0 | 0 | 0 | 0 | 0 | 0 | 49 | 20 |
| 2 | 0 | 185 | 0 | 0 | 0 | 0 | 0 | 49 | 20 |
| 3 | 0 | 0 | 196 | 0 | 0 | 0 | 0 | 49 | 20 |
| 4 | 0 | 0 | 0 | 184 | 0 | 0 | 0 | 49 | 20 |
| 5 | 0 | 0 | 0 | 0 | 176 | 0 | 0 | 49 | 20 |
| 6 | 0 | 0 | 0 | 0 | 0 | 162 | 0 | 49 | 20 |
| 7 | 0 | 0 | 0 | 0 | 0 | 0 | 100 | 49 | 20 |
The lithium ion diffusion coefficient (DLi+) is derived by the following formula:
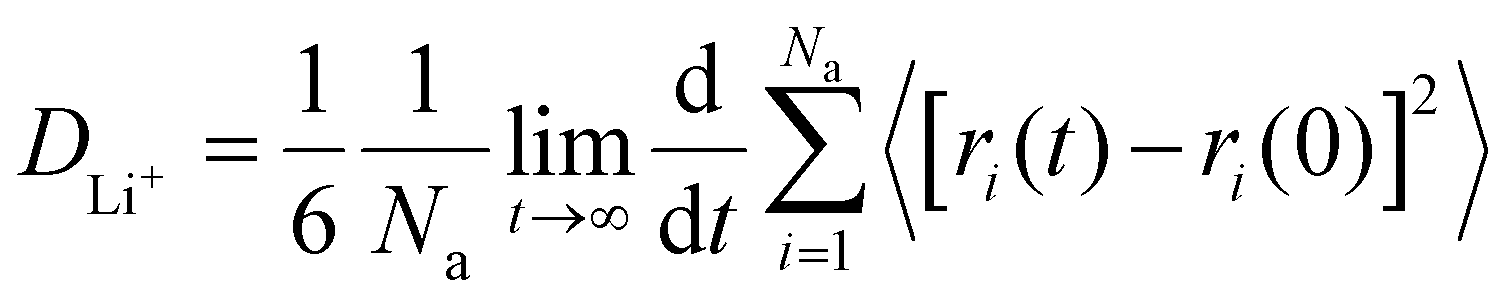
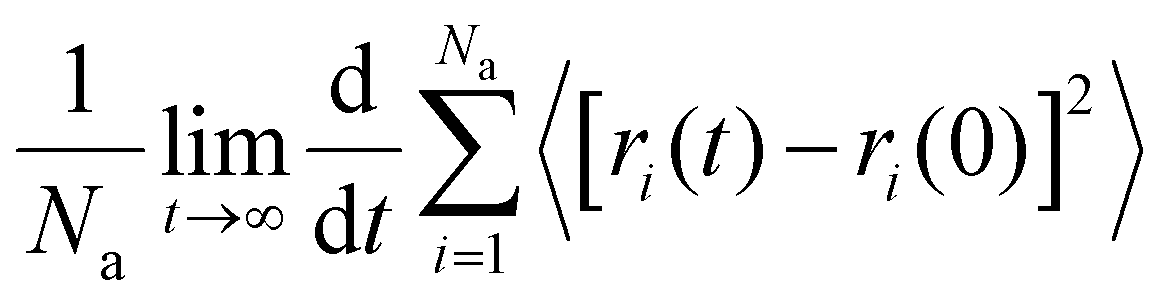 is the slope of the straight-line by fits of the mean square displacement (MSD) curves.45
is the slope of the straight-line by fits of the mean square displacement (MSD) curves.45
The density of various LHCEs is determined, and their corresponding solvation structures are analyzed using the radial distribution function (RDF) curves. The coordination number (CN) is calculated by the following formula:
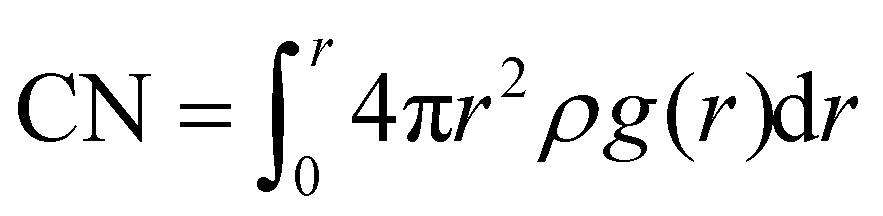
Results and discussion
All solvents consisting of FEC, DFEC, DFEC1, DFEC2, Tri-FEC, Tetra-FEC,1,2-DME, and TTE are shown in Fig. 1a. When the number of fluorine atoms increases to 3, in Tri-FEC, the F/C and F/H ratios are 1.5 and 3, respectively; in Tetra-FEC, the F/C ratio is 2, while the F/H ratio is positive infinity. Both of F/H ratios are larger than that of TTE (F/H ratio = 2). These results mean that these two solvents are not flammable.26 During the NPT process of the MD calculations following geometry optimization, as shown in Fig. 1b, the densities of all solvents increase, and after 60 ps, the density of all solvents stabilizes. The average value of the last 20 ps is taken as the theoretical density of each solvent and is presented in Fig. 1c. The calculated densities of FEC, DFEC, DFEC1, DFEC2, Tri-FEC, Tetra-FEC,1,2-DME, and TTE are 1.55 g cm−3, 1.70 g cm−3, 1.80 g cm−3,1.70 g cm−3,1.86 g cm−3, 1.97 g cm−3, 0.96 g cm−3 and 1.73 g cm−3, respectively. The literature values of the densities of FEC (CAS No. 114435-02-8), DFEC2 (CAS No. 171730-81-7), 1,2-DME (CAS No. 110-71-4), and TTE (CAS No. 16627-68-2) are 1.45 g cm−3, 1.52 g cm−3, 0.84 g cm−3, and 1.53 g cm−3, respectively (Fig. 1c). These actual values are approximately 90% ± 3% of the calculated values. The difference between the actual and calculated values arises from factors such as molecular field, pressure, and other parameters set during the MD simulation. Through the above analysis, we can infer that the density of DFEC, DFEC1, Tri-FEC, and Tetra-FEC is approximately 1.53 ± 0.05 g cm−3, 1.62 ± 0.05 g cm−3, 1.67 ± 0.06 g cm−3, and 1.73 ± 0.06 g cm−3. These densities of all solvents are essential for the bulk electrolyte MD simulations i.e. corresponding LHCEs of LMBs. Next, the dielectric constant of all solvents is calculated by MD simulations. As shown in Fig. 1d, the dielectric constants of FEC, DFEC, DFEC1, DFEC2, Tri-FEC, Tetra-FEC, 1,2-DME, and TTE are 78.81, 28.16, 33.48, 10.18, 8.82, 2.25, 7.90, and 6.26, respectively. The dielectric constants of FEC, 1,2-DME, and TTE are in good agreement with those reported in the literature, confirming the accuracy of our method. | ||
| Fig. 1 (a) Structure of the studied solvents FEC, DFEC, DFEC1, DFEC2, Tri-FEC, Tetra-FEC, TTE and 1,2-DME (red atoms represent oxygen; gray atoms represent carbon; white atoms represent hydrogen; and cyan atoms represent fluorine); (b) densities of all solvents during the NPT process from 0 ps to 100 ps; (c) densities of all solvents determined by MD calculations (solid column on the left) along with the actual densities (half-hollow column on the right if available); (d) dielectric constant of all solvents determined by MD calculations in the NVT ensemble. | ||
As the number of fluorine atoms increases, the dielectric constant decreases among the six perfluorocarbonates. When the number of fluorine atoms reaches four, as in Tetra-FEC, its dielectric constant decreases to 2.37, which is lower than that of TTE, suggesting that Tetra-FEC may serve as a potential diluent.28 Furthermore, the precise dielectric constant offers theoretical support for the accurate calculation of the HOMO and LUMO levels in the solvation structure.
The LUMO and HOMO values for FEC, DFEC, DFEC1, DFEC2, Tri-FEC, Tetra-FEC, FSI−, LiFSI, TTE, and 1,2-DME are illustrated in Fig. 2. The LUMO and HOMO levels of FEC are 0.20 eV and −8.80 eV, respectively, which aligns closely with prior research.2 The results of DFEC2, 1,2-DME, TTE are also consistent with the literatures.24,46 As hydrogen atoms are progressively substituted by fluorine atoms, there is a downward trend in the LUMO energies, from 0.20 eV to −0.96 eV, and a similar reduction in the HOMO energies, from −8.8 eV to −10.00 eV. These findings suggest that molecules with a higher fluorine content exhibit a higher propensity for reduction and a more robust resistance to oxidation.47 In comparison to 1,2-DME and TTE, Tetra-FEC demonstrates the most negative HOMO value of approximately −10.00 eV, indicating its superior antioxidant capacity. Additionally, the HOMO level of LiFSI is relatively high, indicating its limited high-voltage resistance. While constructing a locally high-concentration electrolyte, such as LiFSI/1,2-DME/TTE, and increasing the lithium salt concentration can raise the HOMO and thereby enhance the oxidation voltage, this effect is limited. However, the addition of a small amount of FEC to LiFSI/1,2-DME/TTE significantly improves its antioxidant potential. This improvement is attributed to FEC's participation in the solvation structure, which substantially decreases the HOMO level of the solvation structure. This finding suggests that using a solvent with a lower HOMO value than TTE and FEC, such as Tetra-FEC, and incorporating it into the solvation structure can further enhance the oxidation potential. This aspect will be analyzed in detail later. The above-mentioned characteristics imply that the electrochemical stability window can be significantly broadened when Tetra-FEC is utilized as a multi-functional diluent.
 | ||
| Fig. 2 Lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO) values for a comprehensive range of solvents, lithium salts, and anions, including FEC, DFEC, DFEC1, DFEC2, Tri-FEC, Tetra-FEC, FSI−, LiFSI, TTE, and 1,2-DME, without considering the solvation model. | ||
The LUMO energies of FEC, DFEC, DFEC1, DFEC2, Tri-FEC, and Tetra-FEC are all lower than those of 1,2-DME and TTE, signifying that any of these fluorinated ethylene carbonates are more susceptible to reduction when applied as a solvent for lithium salts. More importantly, it is widely acknowledged that FEC is an exceptionally potent electrolyte additive, a fact attributed to the substantial formation of inorganic LiF on the anode surface during the discharge process. This LiF formation imparts a robust mechanical and chemical stability to the solid electrolyte interphase (SEI) layer.48 Consequently, it can be inferred that as the molecular fluorine content increases (for instance, transitioning from FEC to Tetra-FEC), the likelihood of reduction increases, leading to the formation of a SEI or cathode electrolyte interphase (CEI) layer enriched with more lithium fluoride (LiF).3,48,49 This enrichment enhances the passivation of the anode or cathode surface, thereby improving the overall performance and stability of the electrochemical system.25
As electronegativity is an intrinsic atomic property, fluorine atoms inherently possess a higher electronegativity compared to oxygen atoms. As displayed in the Fig. 3a, among the fluorinated vinyl carbonates and their analogs, there is a progressive transfer of electrons from the carbonyl oxygen to the fluorine atoms. In the cases of DFEC, DFEC1 and DFEC2, where the distances between the two fluorine atoms are 2.193 Å, 2.559 Å, and 3.484 Å, respectively, the proximity of the two fluorine atoms to each other correlates with an increased repulsion, which in turn diminishes the electron transfer to the carbonyl oxygen. Consequently, the ESP charges of the carbonyl oxygen in DFEC2 are the least, followed by DFEC1, with DFEC exhibiting the most negative ESP charges. As more fluorine atoms are incorporated into the molecule, the charge on the carbonyl oxygen progressively decreases, leading to a reduction in ESP charges. Just like in TTE, there are fewer electrons on the oxygen atom originating from the presence of multiple fluorine atoms in TTE. Consequently, the binding energies between Li+ and the fluorinated vinyl carbonates and their analogs increase, as depicted in Fig. 3b. This trend is observed as the binding energies range from −1.932 eV to −1.248 eV, indicating that FEC exhibits the strongest affinity for Li+, while Tetra-FEC demonstrates the weakest binding among the compounds analyzed. However, without taking the surrounding solvent into account, the high electronegativity of these fluorine atoms and the flexibility of the alkyl chains in TTE allow lithium ions to bind to both oxygen and fluorine atoms (Fig. 3b). In contrast, for cyclic fluorocarbonates, the steric hindrance and rigidity of the ring structure, as seen in Tetra-FEC, restrict the lithium-ion binding to the carbonyl oxygen alone, resulting in a lower binding energy compared to Li+-TTE. Although the ESP charges of oxygen in fluorinated vinyl carbonate and its analogues, as depicted in Fig. 3a, is lower than that of 1,2-DME, the binding affinity of Li+ to fluoroethylene carbonate and its analogues is weaker compared to the interaction between lithium ions and 1,2-DME. This trend is attributed to the fact that in the electrolyte, Li+ is coordinated with two oxygen atoms in 1,2-DME, as shown in Fig. S1. For FSI−, due to the presence of sulfur atoms, the oxygen atoms have a higher electronegativity, and more importantly, one lithium ion combines with two oxygen atoms on FSI− (Fig. S1), which leads to a larger binding energy between FSI− and the lithium ion. Similarly, as shown in Fig. 3c and Table S2, as the charge on the oxygen atom in the solvent molecules decreases (Fig. 3a), the adsorption interaction between the solvent molecules and LiNiO2 weakens, along with the elongation of the Ni–O bond (which refers to the carbonyl oxygen in FEC and its derivatives). For example, the adsorption energy of FEC on the LiNiO2 (104) surface is −0.63 eV, with a Ni–O bond length of 2.551 Å, whereas the adsorption energy of tetra-FEC decreases to −0.34 eV, with a Ni–O bond length of 2.864 Å. TTE exhibits the lowest adsorption energy, only −0.28 eV, and the Ni–O bond stretches to 3.466 Å. This lower adsorption energy and longer Ni–O bond can prevent solvent molecules from adsorbing onto the cathode surface and thus being oxidized. More importantly, with the increasing number of fluorine atoms, these fluorine atoms not only withdraw electrons from the oxygen atom but also from nearby hydrogen atoms. This electron withdrawal renders the hydrogen atoms on the solvent more susceptible to bonding with oxygen atoms on the LiNiO2 (104) surface, leading to the formation of OH groups.41 The resulting OH can subsequently combine with Li+ on the cathode surface to form LiOH, which reduces capacity, and it can also further decompose the electrolyte, contributing to gas evolution in the cell.14,18 Even more critically, the departure of hydrogen leaves behind solvent molecules that can further react with transition metals (e.g., Ni) on the LiNiO2 (104) surface, causing metal dissolution and structural degradation, ultimately leading to cathode failure.18 As shown in Fig. 3d, as for FEC and its derivatives, the dissociation energy increases with the number of fluorine atoms. However, in the case of TTE, the number of fluorine atoms (eight) is much larger than in FEC and its derivatives. As a result, the terminal hydrogen atoms in TTE are highly prone to undergoing dissociation processes with LiNiO2. Although the hydrogens of TTE in the chain segment are spatially hindered, which reduces the likelihood of such reactions, the dissociation energy still reaches −1.56 eV. These findings well explain the experimental observation that using TTE as a diluent leads to increased gas evolution in the battery. In contrast, for tetra-FEC, since all hydrogen atoms are replaced by fluorine, dissociation processes are much less likely to occur.
 | ||
| Fig. 3 (a) Electrostatic potential (ESP) charges distribution on the carbonyl oxygen atoms of FEC, DFEC, DFEC1, DFEC2, Tri-FEC, and Tetra-FEC, as well as on the ether oxygen atoms of TTE, without considering the solvation model. (b) Binding energies between various solvents and Li+; (c) adsorption energy for different solvents adsorbed on the LiNiO2 (104) surface; (d) dissociation energy for different solvents adsorbed on the LiNiO2 (104) surface. Red atoms represent oxygen atoms, grey atoms represent carbon atoms, cyan atoms represent fluorine atoms, white atoms represent hydrogen atoms, blue atoms represent nitrogen atoms, and purple atoms represent lithium atoms. | ||
Consequently, it is hypothesized that with the increasing number of fluorine atoms, when fluoroethylene carbonate (FEC) and its analogues are used to replace TTE in 1,2-DME/TTE localized high-concentration electrolyte systems, these fluorinated carbonates tend to function more as diluents rather than co-solvents and exhibit fewer side reactions on the surface of the nickel-rich layered cathode materials.
In the classical LiFSI/1,2-DME/TTE LHCEs system, TTE is replaced with FEC, DFEC, DFEC1, DFEC2, Tri-FEC, and Tetra-FEC, respectively. As shown in the Fig. 4a and Fig. S3, molecular dynamics simulations are executed to reveal the various key physical properties, including the Li+ molar concentrations, densities, and Li+ diffusion coefficients (DLi+), and solvation structures, providing deeper insight into their underlying behavior. As depicted in Fig. 4b, Li+ molar concentrations of all LHCEs systems remain around 1 M. From FEC to Tetra-FEC, as solvent densities increase, the densities of the corresponding LHCEs systems also rise (Fig. 4c), ranging from approximately 1.448 g cm−3 to 1.705 g cm−3. The density of the classic system LiFSI/1,2-DME/TTE is about 1.555 g cm−3. When DFEC, DFEC1, or DFEC2 is used as a diluent, the electrolyte density remains largely unchanged, having minimal impact on the battery energy density. Compared to the LiFSI/1,2-DME/TTE system, FEC can slightly reduce the electrolyte density, thereby potentially enhancing the battery energy density. In contrast, when Tri-FEC or Tetra-FEC is used as the diluent, the electrolyte density increases, but the increase is limited to only about 5.5% and 9.6%, respectively. The diffusion coefficient of lithium ions in the electrolyte is a key factor influencing the rate performance of lithium-ion or lithium-metal batteries. Therefore, the diffusion coefficients of all LHCE systems are determined by fits of the mean square displacement (Fig. S4). Lithium-ion diffusion is influenced by multiple factors, including the solvent density and the binding energy between lithium ions and solvent molecules. A higher solvent density increases the viscosity, which can hinder ion mobility. Conversely, weaker binding interactions facilitate lithium-ion diffusion. Among FEC, DFEC, DFEC1, DFEC2, Tri-FEC, and Tetra-FEC, FEC exhibits the lowest viscosity, while Tetra-FEC has the weakest binding energy with lithium ions. As a result, in the Fig. 4d, the lithium-ion diffusion coefficients in LiFSI/1,2-DME/FEC and LiFSI/1,2-DME/Tetra-FEC are relatively high, measuring 1.23 × 10−10 m2 s−1 and 1.04 × 10−10 m2 s−1, respectively. Although these values are slightly lower than those of LiFSI/1,2-DME/TTE (1.41 × 10−10 m2 s−1), they remain within the same order of magnitude, ensuring comparable ion transport properties.
 | ||
| Fig. 4 (a) Snapshot of the LiFSI/1,2-DME/Tetra-FEC LHCEs system; (b–d) Li+ molar concentrations, densities and Li+ diffusion coefficients (DLi+) of different LHCEs systems including LiFSI/1,2-DME/FEC, LiFSI/1,2-DME/DFEC, LiFSI/1,2-DME/DFEC1, LiFSI/1,2-DME/DFEC2, LiFSI/1,2-DME/Tri-FEC, LiFSI/1,2-DME/Tetra-FEC, LiFSI/1,2-DME/TTE. | ||
Subsequently, the coordination structures of the last frame in the LHCEs systems are depicted through radial distribution function (RDF) curves as presented in Fig. 5 and Fig. S5. For the first solvation shell (2–3 Å), except in the LiFSI/1,2-DME/DFEC2 system, the radial distribution function (RDF) curves of Li+–O1,2-DME nearly overlap.19,50 This finding indicates that 1,2-DME has minimal influence on the lithium-ion coordination across all LHCE systems. Regarding different diluents, the oxygen in TTE exhibits almost no complexation with lithium ions, explaining its effectiveness as a diluent. In contrast, for multifunctional diluents, such as FEC, DFEC, DFEC1, DFEC2, Tri-FEC, and Tetra-FEC, due to the binding energy between carbonyl oxygen and lithium ions gradually decreasing, as shown in Fig. 5b, Tetra-FEC has the lowest probability of appearing in lithium-ion clusters. Conversely, as shown in Fig. 5c, more oxygen atoms from FSI− participate in the solvation structure. Additionally, only trace amounts of fluorine (F) elements from FSI− or the diluents are directly involved in the solvation shell (Fig. S5); therefore, they can be ignored.
 | ||
| Fig. 5 (a–c) RDF curves of Li+–O1,2-DME, Li+–Odiluents (in esters, it should be carbonyl oxygen), Li+–OFSI− of LiFSI/1,2-DME/FEC, LiFSI/1,2-DME/DFEC, LiFSI/1,2-DME/DFEC1, LiFSI/1,2-DME/DFEC2, LiFSI/1,2-DME/Tri-FEC, LiFSI/1,2-DME/Tetra-FEC, LiFSI/1,2-DME/TTE at 298 K; (d–f) corresponding coordination number around Li+ in all the LHCE systems. | ||
Then, by integrating the RDF curves, the average coordination number around the lithium ion is obtained, as displayed in Fig. 5d–f. The details are as follows:
LiFSI/1,2-DME/FEC: Li+–O1,2-DME(1.76)OFSI−(1.14)OFEC(2.67);
LiFSI/1,2-DME/DFEC: Li+–O1,2-DME(1.83)OFSI−(1.25)ODFEC(2.44);
LiFSI/1,2-DME/DFEC1: Li+–O1,2-DME(1.66)OFSI−(1.67)ODFEC1(2.10);
LiFSI/1,2-DME/DFEC2: Li+–O1,2-DME(2.25)OFSI−(1.44)ODFEC2(1.36);
LiFSI/1,2-DME/Tri-FEC: Li+–O1,2-DME(1.97)OFSI−(1.99)OTri–FEC (1.26);
LiFSI/1,2-DME/Tetra-FEC: Li+–O1,2-DME(1.97)OFSI−(2.37)OTetra-FEC(1.00);
LiFSI/1,2-DME/TTE: Li+–O1,2-DME(1.88)OFSI−(2.51)OTTE(0.00).
The binding strength between Li+ and oxygen decreases with the ESP charge on oxygen. Accordingly, among the above solvation structures shown in Fig. 5b, the total number of molecules participating in the solvation shells also gradually decreases for FEC, DFEC, DFEC1, DFEC2, Tri-FEC, and Tetra-FEC, with values of 53, 49, 42, 27, 25, and 20, respectively (values rounded to the nearest integer) and most of them are still solvents; therefore, these solvents can be regarded as diluents. For all LHCE systems, the composition of the solvent is listed in Table S1. Therefore, FEC, DFEC, DFEC1, DFEC2, Tri-FEC, and Tetra-FEC can be regarded as diluents. In LHCE systems where FEC, DFEC, DFEC1, DFEC2, Tri-FEC, and Tetra-FEC serve as diluents, the LiFSI/1,2-DME/Tetra-FEC system exhibits the highest participation of FSI− in the first solvation structure, with an average of 2.37. Additionally, since Tetra-FEC contains four fluorine atoms, all four indirectly contribute to the solvation structure. In comparison, the indirect participation of fluorine atoms in the LiFSI/1,2-DME/FEC, LiFSI/1,2-DME/DFEC, LiFSI/1,2-DME/DFEC1, LiFSI/1,2-DME/DFEC2, LiFSI/1,2-DME/Tri-FEC, and LiFSI/1,2-DME/TTE systems is 2.67, 4.88, 4.20, 2.72, 3.78, and 0, respectively. As a result, the LiFSI/1,2-DME/Tetra-FEC system contains the highest proportion of inorganic components in its lithium-ion solvation structure. These kinds of lithium clusters lead to a significant increase in the inorganic components within the SEI layer when they are reduced on the lithium metal surface, thereby enhancing the electrode cycling stability.
In order to better characterize the solvated structure, the proportions of free FSI− (also named as solvent-separated ion pairs (SSIP)), contact ion pair (CIP), and aggregate (AGG) in a total of 150 frames are counted every 1 ps between 50 ps and 200 ps.10,51 As shown in Fig. 6, in the LiFSI/1,2-DME/FEC, LiFSI/1,2-DME/DFEC, LiFSI/1,2-DME/DFEC1, LiFSI/1,2-DME/DFEC2, and LiFSI/1,2-DME/Tri-FEC LHCE systems, a portion of SSIP remains, which aligns with the Li cluster coordination numbers analyzed in Fig. 5. When the coordination number of Li+–OFSI− is less than 2, the presence of AGG necessitates the existence of some SSIP. Conversely, when the coordination number of Li+–OFSI− exceeds 2, the likelihood of SSIP is significantly reduced. For instance, in the LiFSI/1,2-DME/Tetra-FEC and LiFSI/1,2-DME/TTE systems, the probability of SSIP is only 0.1% and 0.8%, respectively. A lower free FSI− concentration helps enhance the oxidative stability window and mitigate aluminum foil corrosion. More importantly, as the number of fluorine atoms in the ester diluent increases, the proportion of CIP + AGG also rises. When Tetra-FEC is used as a diluent, the AGG fraction reaches 78.6%, even surpassing the AGG percentage observed in the TTE-based system (71.7%). This observation aligns with the findings from the final frame analysis of the lithium-ion cluster solvation structure, confirming that in the LiFSI/1,2-DME/Tetra-FEC system, the solvation structure contains more inorganic components, an essential factor in stabilizing the lithium metal anode. Finally, based on the solvent environment and corresponding dielectric constants listed in Table S1, the HOMO and LUMO energy levels of lithium ion clusters (last frame) and free FSI− anions (if present) in all LHCE systems are calculated to assess the electrolyte's oxidative stability and its reducibility at the anode.52 All results are depicted in Fig. 6b, c, and Fig. S6–S11. Regarding the HOMO value, it is influenced by two factors: the dielectric constant of the solvent and the molecular structures within the clusters. A higher dielectric constant or a larger involvement of fluorine-containing ester molecules in the cluster results in a lower HOMO level, leading to improved antioxidant properties. Compared to LiFSI/1,2-DME/TTE, the highest HOMO level of all lithium-ion clusters in LiFSI/1,2-DME/FEC, LiFSI/1,2-DME/DFEC, LiFSI/1,2-DME/DFEC1, LiFSI/1,2-DME/DFEC2, LiFSI/1,2-DME/Tri-FEC, and LiFSI/1,2-DME/Tetra-FEC are generally lower than that of LiFSI/1,2-DME/TTE. The only exception is a single CIP structure in LiFSI/1,2-DME/DFEC, which has a slightly higher HOMO level than LiFSI/1,2-DME/TTE (Fig. 6b and Fig. S11). This reduction in the HOMO level is primarily attributed to the higher dielectric constants of most ester diluents (except Tetra-FEC) or the increased presence of fluorinated esters in the solvation structure. More importantly, in LiFSI/1,2-DME/TTE, the highest HOMO level of the lithium clusters corresponds to an aggregate (AGG), as shown in Fig. S11, indicating that a significant portion of lithium-ion clusters exhibit poor high-voltage resistance. The lower HOMO of the lithium ion clusters in LiFSI/1,2-DME/Tetra-FEC also indicates its excellent antioxidant performance, which is beneficial to improve the energy density of lithium metal batteries. Concerning the LUMO, compared to LiFSI/1,2-DME/TTE, the lowest LUMO levels of all lithium-ion clusters in LiFSI/1,2-DME/FEC, LiFSI/1,2-DME/DFEC, LiFSI/1,2-DME/Tri-FEC, and LiFSI/1,2-DME/Tetra-FEC are generally lower. This trend can be attributed to the presence of fluorinated esters in the solvation structure, which influence LUMO levels based on the number of fluorine atoms, their molecular symmetry, and steric hindrance. Among all LHCEs, the lithium-ion clusters in LiFSI/1,2-DME/Tetra-FEC exhibit the lowest LUMO, indicating their tendency for reduction. Considering the high proportion of CIP and AGG in LiFSI/1,2-DME/Tetra-FEC, we speculate that using Tetra-FEC as a diluent facilitates the formation of a robust SEI.
 | ||
| Fig. 6 (a) Representative all Li+ cation solvation species (SSIP, CIP, and AGG) in all LHCE systems statistically counted from 50 ps to 200 ps; (b) LUMO and HOMO levels of Li clusters in the last frame in LiFSI/1,2-DME/Tetra-FEC LHCEs system; (c and d) HOMO, LUMO levels of Li clusters in the last frame in all LHCE systems, respectively. | ||
Finally, a basic computational framework is presented, as illustrated in Fig. 7. First, molecular dynamics and DFT calculations are performed to investigate the fundamental properties of solvents, including density (ρ), dielectric constant (ε), binding energy with lithium ions, interaction with the cathode, and HOMO/LUMO energy levels. If the solvent meets the required criteria, electrolyte design proceeds, with a focus on key electrolyte properties such as Li+ molar concentration (cLi+), Li+ diffusion coefficient (DLi+), the population of Li+ ion clusters including SSIP, CIP and AGG in the solvation structure, and the corresponding HOMO/LUMO energy levels. If the electrolyte satisfies the design targets, the design process is completed; otherwise, the procedure returns to the solvent selection step to identify suitable candidates and repeat the cycle. In the future, this framework can be further integrated with artificial intelligence and machine learning to accelerate electrolyte design.
 | ||
| Fig. 7 A basic computational framework enabling future integration with AI and machine learning for electrolyte development. | ||
Conclusion
Firstly, the fundamental physical properties of solvents, including TTE, 1,2-DME, FEC, DFEC, DFEC1, DFEC2, Tri-FEC, and Tetra-FEC, are investigated using MD and DFT calculations. These properties included density, dielectric constant, HOMO, LUMO, their binding energy with lithium ions, as well as their adsorption and dissociation capability on the LiNiO2 cathode material. Subsequently, in the classical LiFSI/1,2-DME/TTE system, TTE is replaced with FEC, DFEC, DFEC1, DFEC2, Tri-FEC, and Tetra-FEC, and MD simulations are applied for the study of LHCEs systems including LiFSI/1,2-DME/FEC, LiFSI/1,2-DME/DFEC, LiFSI/1,2-DME/DFEC1, LiFSI/1,2-DME/DFEC2, LiFSI/1,2-DME/Tri-FEC, LiFSI/1,2-DME/Tetra-FEC, and LiFSI/1,2-DME/TTE. The key physical properties of these systems, such as density, lithium-ion molar concentration, lithium-ion diffusion coefficient, and solvation structure, are determined. Additionally, the percentages of SSIP, CIP, and AGG in each system are statistically analyzed. Finally, the HOMO and LUMO of all clusters in the final frame of the MD simulations are further calculated using DFT. Compared to the classical LiFSI/1,2-DME/TTE LHCEs system, the LiFSI/1,2-DME/Tetra-FEC LHCEs system exhibits a slightly higher electrolyte density and a lower lithium-ion diffusion coefficient. However, its clusters with the lowest HOMO and LUMO levels possess strong antioxidative properties and are more prone to reduction. Besides, these clusters contain the highest proportion of CIP and AGG components. Hence, this LiFSI/1,2-DME/Tetra-FEC system facilitates the construction of stable, high-energy-density lithium-metal batteries, highlighting the potential of Tetra-FEC as a promising diluent. Moreover, this study revises the content to frame this work as a basic computational workflow, and explicitly state that it can be further integrated with AI and machine learning for future electrolyte design.Conflicts of interest
There are no conflicts to declare.Data availability
The data that support the findings of this study are available from the following public repository: https://doi.org/10.14459/2026mp1840938.Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d5eb00209e.
Acknowledgements
P.M-B acknowledges funding from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) via project MU 1487/38-1 (AOBJ: 673749), by the International Research Training Group 2022 Alberta/Technical University of Munich International Graduate School for Environmentally Responsible Functional Hybrid Materials (ATUMS) with the associated grant number IRTG2022, and under Germany's Excellence Strategy EXC 2089/1-390776260 (e-conversion). This research is supported by the National Natural Science Foundation of China (52061135110), Natural Science Foundation of Zhejiang Province (Grant No. LD22E020003), and the Ningbo Science & Technology Innovation 2025 Major Project (2021Z121, 2022Z021). T. Zheng acknowledges the financial support from the China Scholarship Council (CSC).References
- Y. Wu, Q. Hu, H. Liang, A. Wang, H. Xu, L. Wang and X. He, Adv. Energy Mater., 2023, 13, 2300259 CrossRef CAS.
- Q. Wang, Z. Yao, C. Zhao, T. Verhallen, D. P. Tabor, M. Liu, F. Ooms, F. Kang, A. Aspuru-Guzik, Y.-S. Hu, M. Wagemaker and B. Li, Nat. Commun., 2020, 11, 4188 CrossRef PubMed.
- Q. Xu, T. Li, Z. Ju, G. Chen, D. Ye, G. I. N. Waterhouse, Y. Lu, X. Lai, G. Zhou, L. Guo, K. Yan, X. Tao, H. Li and Y. Qiu, Nature, 2025, 637, 339–346 CrossRef CAS PubMed.
- Z. Xu, M. Peng, G. Pan, T. Zheng, Y. Xiao, W. Xie, Y. Li, J. Gao, S. Yin, Q. Ji, B. Wu, Y. Miao, S. Shi, Y.-J. Cheng, Y. Xia and P. Müller-Buschbaum, Energy Storage Mater., 2026, 84, 104779 CrossRef.
- H. Yang, Y. Zhao, T. Qin, H. Pan, S. Feng, Q. Li, X. Wang, X. Yu and H. Li, ACS Energy Lett., 2024, 9, 4475–4484 CrossRef CAS.
- H. Yang, P. Luo, N. Sun, S. Jiao, J. Yue, B. Cao, J. Song, Q. Li, X. Yu, H. Li and X. Huang, Energy Storage Mater., 2025, 83, 104703 CrossRef.
- C. P. Yang, Y. X. Yin, S. F. Zhang, N. W. Li and Y. G. Guo, Nat. Commun., 2015, 6, 8058 CrossRef CAS PubMed.
- Q. Wu, Z. Xu, Y. Yu, M. Peng, J. Gao, L. Nie, Y. J. Cheng, P. Müller-Buschbaum and Y. Xia, Langmuir, 2024, 40, 14863–14871 CrossRef CAS PubMed.
- X. Cao, H. Jia, W. Xu and J.-G. Zhang, J. Electrochem. Soc., 2021, 168, 010522 CrossRef CAS.
- C. M. Efaw, Q. Wu, N. Gao, Y. Zhang, H. Zhu, K. Gering, M. F. Hurley, H. Xiong, E. Hu, X. Cao, W. Xu, J.-G. Zhang, E. J. Dufek, J. Xiao, X.-Q. Yang, J. Liu, Y. Qi and B. Li, Nat. Mater., 2023, 22, 1531–1539 CrossRef CAS PubMed.
- X. Chen and H. Yu, ChemElectroChem, 2024, 11, e202400444 CrossRef CAS.
- X. Ren, L. Zou, X. Cao, M. H. Engelhard, W. Liu, S. D. Burton, H. Lee, C. Niu, B. E. Matthews, Z. Zhu, C. Wang, B. W. Arey, J. Xiao, J. Liu, J.-G. Zhang and W. Xu, Joule, 2019, 3, 1662–1676 CrossRef CAS.
- S. Zhang, R. Li, T. Deng, Q. Ma, X. Hong, H. Zhang, R. Zhang, S. Ding, Y. Wu, H. Zhu, M. Li, H. Zhang, D. Lu, B. Ma, L. Lv, Y. Li, L. Chen, Y. Shen, R. Guo and X. Fan, Nat. Energy, 2024, 9, 1285–1296 CrossRef CAS.
- R. Sim and A. Manthiram, Adv. Energy Mater., 2024, 14, 2300259 Search PubMed.
- W. W. van Ekeren, M. Albuquerque, G. Ek, R. Mogensen, W. R. Brant, L. T. Costa, D. Brandell and R. Younesi, J. Mater. Chem. A, 2023, 11, 4111–4125 RSC.
- J. Kim, H. Lee, H. Cha, M. Yoon, M. Park and J. Cho, Adv. Energy Mater., 2017, 8, 1702028 CrossRef.
- L. Ni, S. Zhang, A. Di, W. Deng, G. Zou, H. Hou and X. Ji, Adv. Energy Mater., 2022, 12, 2201510 CrossRef CAS.
- M. Jiang, D. L. Danilov, R. A. Eichel and P. H. L. Notten, Adv. Energy Mater., 2021, 11, 2103005 CrossRef CAS.
- S. Li, H. Liu, L. Zheng, C. Ma, H. Yu, X. Wu, X. Niu and L. Wang, Nano Energy, 2024, 131, 110234 CrossRef CAS.
- X. Zheng, L. Huang, X. Ye, J. Zhang, F. Min, W. Luo and Y. Huang, Chem, 2021, 7, 2312–2346 CAS.
- G. Liu, M. Xia, J. Gao, Y. Cheng, M. Wang, W. Hong, Y. Yang and J. Zheng, ACS Appl. Mater. Interfaces, 2023, 15, 3586–3598 CrossRef CAS PubMed.
- H. Ren, G. Zheng, Y. Li, S. Chen, X. Wang, M. Zhang, W. Zhao, H. Yi, W. Huang, J. Fang, T. Liu, L. Yang, M. Liu, Q. Zhao and F. Pan, Energy Environ. Sci., 2024, 17, 7944–7957 RSC.
- A. Zhang, Z. Bi, G. Wang, S. Liao, P. Das, H. Lin, M. Li, Y. Yu, X. Feng, X. Bao and Z.-S. Wu, Energy Environ. Sci., 2024, 17, 3021–3031 RSC.
- C. Luo, Q. Liu, X. Wang, Y. Tian, Z. Liu, F. Kang and B. Li, Nano Energy, 2023, 109, 108323 CrossRef CAS.
- X. Fan, L. Chen, O. Borodin, X. Ji, J. Chen, S. Hou, T. Deng, J. Zheng, C. Yang, S.-C. Liou, K. Amine, K. Xu and C. Wang, Nat. Nanotechnol., 2018, 13, 715–722 CrossRef CAS PubMed.
- J. Liu, W. Hao, M. Fang, X. Chen, Y. Dong, Y. Chen, Z. Wang, X. Yue and Z. Liang, Nat. Commun., 2024, 15, 9356 CrossRef CAS PubMed.
- T. H. K.-C. Möller, W. K. Appelb, M. Winter and J. O. Besenhard, J. Power Sources, 2001, 97–98, 595–597 CrossRef.
- Y. Wu, A. Wang, Q. Hu, H. Liang, H. Xu, L. Wang and X. He, ACS Cent. Sci., 2022, 8, 1290–1298 CrossRef CAS PubMed.
- P. Liu and J. A. Rodriguez, J. Am. Chem. Soc., 2005, 127, 14871–14878 CrossRef CAS PubMed.
- A. V. David, J. Henry and I. Yarovsky, J. Phys. Chem. A, 2008, 112, 9835–9844 CrossRef PubMed.
- R. Hasan and D. Datta, Energy Adv., 2025, 4, 1049–1066 RSC.
- T. Zheng, J. Xiong, X. Shi, B. Zhu, Y.-J. Cheng, H. Zhao and Y. Xia, Energy Storage Mater., 2021, 38, 599–608 CrossRef.
- Z. Wang, Y. Sun, Y. Mao, F. Zhang, L. Zheng, D. Fu, Y. Shen, J. Hu, H. Dong, J. Xu and X. Wu, Energy Storage Mater., 2020, 30, 228–237 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- J. F. G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef PubMed.
- V. I. Anisimov, F. Aryasetiawan and A. I. Lichtenstein, J. Condens. Matter Phys., 1997, 9, 767–808 CrossRef CAS.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505 CrossRef CAS.
- L. Wang, T. Maxisch and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 195107 CrossRef.
- Y.-L. Lee, J. Kleis, J. Rossmeisl and D. Morgan, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 224101 CrossRef.
- L. Giordano, P. Karayaylali, Y. Yu, Y. Katayama, F. Maglia, S. Lux and Y. Shao-Horn, J. Phys. Chem. Lett., 2017, 8, 3881–3887 CrossRef CAS PubMed.
- A. Taborosi, H. Shiiba, M. Koyama and N. Zettsu, ACS Omega, 2024, 9, 42116–42126 CrossRef CAS PubMed.
- N. Yao, X. Chen, X. Shen, R. Zhang, Z. H. Fu, X. X. Ma, X. Q. Zhang, B. Q. Li and Q. Zhang, Angew. Chem., Int. Ed., 2021, 60, 21473–21478 CrossRef CAS PubMed.
- X. Q. Zhang, X. Chen, X. B. Cheng, B. Q. Li, X. Shen, C. Yan, J. Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2018, 57, 5301–5305 CrossRef CAS PubMed.
- Z. Li, Z. Chen, N. Sun, D. Wang, X. Yao and Z. Peng, Angew. Chem., Int. Ed., 2024, 63, e202400876 CrossRef CAS PubMed.
- X. Cao, P. Gao, X. Ren, L. Zou, M. H. Engelhard, B. E. Matthews, J. Hu, C. Niu, D. Liu, B. W. Arey, C. Wang, J. Xiao, J. Liu, W. Xu and J. G. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2020357118 CrossRef CAS PubMed.
- E. Quartarone and P. Mustarelli, J. Electrochem. Soc., 2020, 167, 050508 CrossRef CAS.
- A. L. Michan, B. S. Parimalam, M. Leskes, R. N. Kerber, T. Yoon, C. P. Grey and B. L. Lucht, Chem. Mater., 2016, 28, 8149–8159 CrossRef CAS.
- Y. Liu, X. Tao, Y. Wang, C. Jiang, C. Ma, O. Sheng, S. Lu and X. W. Lou, Science, 2022, 375, 739–745 CrossRef CAS PubMed.
- H. Cheng, Q. Sun, L. Li, Y. Zou, Y. Wang, T. Cai, F. Zhao, G. Liu, Z. Ma, W. Wahyudi, Q. Li and J. Ming, ACS Energy Lett., 2022, 7, 490–513 CrossRef CAS.
- Z. Yu, N. P. Balsara, O. Borodin, A. A. Gewirth, N. T. Hahn, E. J. Maginn, K. A. Persson, V. Srinivasan, M. F. Toney, K. Xu, K. R. Zavadil, L. A. Curtiss and L. Cheng, ACS Energy Lett., 2021, 7, 461–470 CrossRef.
- M. Shakourian-Fard, G. Kamath and S. K. Sankaranarayanan, ChemPhysChem, 2016, 17, 2916–2930 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |