
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Predicting the formation of mud cracks in Li-ion battery electrodes during the drying process with in situ X-ray computed tomography
Will J.
Dawson
 abc,
Andrew R. T.
Morrison
*bc,
Simon M.
Tonge
cg,
Matthew P.
Jones
b,
Kofi
Coke
bcl,
Isabel C.
Antony
bcdef,
Kaz
Wanelik
d,
Vyacheslav
Kachkanov
d,
Partha P.
Paul
ghj,
Bratislav
Lukić
ghi,
Robert Scott
Young
b,
Zifa
Zuhair
bd,
James
Parker
ck,
Inez
Kesuma
bf,
Gargi
Giri
bcf,
Liam
Bird
bcf,
Alexander J. E.
Rettie
ab,
Rhodri
Jervis
abc,
James B.
Robinson
ac,
Denis
Cumming
ck,
Thomas S.
Miller
abc and
Paul R.
Shearing
*cf
abc,
Andrew R. T.
Morrison
*bc,
Simon M.
Tonge
cg,
Matthew P.
Jones
b,
Kofi
Coke
bcl,
Isabel C.
Antony
bcdef,
Kaz
Wanelik
d,
Vyacheslav
Kachkanov
d,
Partha P.
Paul
ghj,
Bratislav
Lukić
ghi,
Robert Scott
Young
b,
Zifa
Zuhair
bd,
James
Parker
ck,
Inez
Kesuma
bf,
Gargi
Giri
bcf,
Liam
Bird
bcf,
Alexander J. E.
Rettie
ab,
Rhodri
Jervis
abc,
James B.
Robinson
ac,
Denis
Cumming
ck,
Thomas S.
Miller
abc and
Paul R.
Shearing
*cf
aAdvanced Propulsion Lab, UCL East, University College London, London, E20 2AE, UK
bElectrochemical Innovation Lab, University College London, London, WC1E 7JE, UK. E-mail: andrewrt.morrison@ucl.ac.uk
cThe Faraday Institution, Didcot, OX11 0RA, UK. E-mail: paul.shearing@eng.ox.ac.uk
dDiamond Light Source, Harwell Science and Innovation Campus, Didcot, Oxfordshire OX11 0DE, UK
eISIS Neutron and Muon Source, Rutherford Appleton Laboratory, OX11 0QX Harwell, UK
fThe ZERO Institute University of Oxford, Holywell House, Osney Mead, Oxford, OX2 0ES, UK
gESRF – The European Synchrotron Radiation Facility, 71 Av. des Martyrs, 38000 Grenoble, France
hHenry Royce Institute, Department of Materials, University of Manchester, Manchester, M13 9PL, United Kingdom
iInstitut Max von Laue - Paul Langevin, ILL, 71 Av. des Martyrs, 38000 Grenoble, France
jSLAC National Accelerator Laboratory, Menlo Park, CA 94025-7015, USA
kSchool of Chemical, Materials and Biological Engineering, The University of Sheffield, Sheffield, S1 3JD, UK
lSchool of Chemistry and Chemical Engineering, University of Southampton, Southampton, SO17 1BJ, UK
First published on 12th February 2026
Abstract
Drying of slurry cast electrodes is amongst the most energy intensive unit operations in battery manufacture. In spite of this, the optimisation of drying processes has been highly empirical, and there remains limited understanding of the interplay between drying dynamics and resulting microstructure. In this work, we used synchrotron X-ray computed microtomography in order to study the formation of mud cracks during the drying process, and evaluate their impact on the electrode microstructure. This was achieved by applying a reduced drying rate, which proved to be an effective means of studying the drying mechanism with a greater resolution and image contrast than otherwise possible. The rate of crack growth is measured, and the differing crack morphology resultant from changes in thickness (between 300 and 800 µm doctor blade gaps) and the presence of air bubbles in the slurry is demonstrated. Digital volume correlation is utilised to identify the specific location of crack formation before these cracks were visible, suggesting image correlation methods as an appropriate tool for process feedback in order to control or eliminate mud cracking. This new approach which enables direct quantification of the evolving microstructure during dynamic drying, in 3D, is therefore transformative in our understanding of the underlying physical processes and will guide rational optimisation of this industrially significant process.
Broader contextSolvent-based slurry processing is the means by which the majority of battery electrodes of all chemistries are presently manufactured, typically with a slot die coater head, as part of a roll-to-roll electrode manufacturing line. These approaches are also increasingly applied in the manufacture of perovskite solar cells and other functional coatings. The slurry drying process can have a major impact on the structure and performance of coatings. One such impact is the formation of mud cracks, which result from capillary stress which builds as solvent evaporates. Here we apply in situ X-ray computed tomography in a time-resolved study of the electrode drying process, developing key insights necessary to better understand, control or prevent mud cracking. |
1 Introduction
Roll-to-roll fabrication of battery electrodes includes mixing, coating, drying and calendering steps, after which the electrodes are slit and wound into cells. The drying process of Li-ion battery electrodes, including solvent recovery, has been estimated to account for 15% of total cell cost, and contributes profoundly to the mechanical and electrochemical performance of electrodes.1–10 Understanding, predicting and controlling this process is thus crucial to progressing knowledge of battery manufacturing. The drying process begins with a coated electrode slurry where active particles and conductive carbon are suspended in a solution of binder and solvent – typically polyvinylidene fluoride (PVDF) and N-methyl pyrrolidone (NMP), respectively – in conventional Li-ion positive electrodes. As solvent evaporates, the solid components consolidate such that inter-particle distances are shorter until all particles are compacted. In granular coatings, such as Li-ion battery electrodes, at the point the solvent level begins to drop below the upper layer of particles, concurrently with air intrusion into the porous structure, a capillary action of solvent towards the evaporating surface results.11–15 This capillary action causes capillary stress, σcap, which can be approximated by the relationship: | (1) |
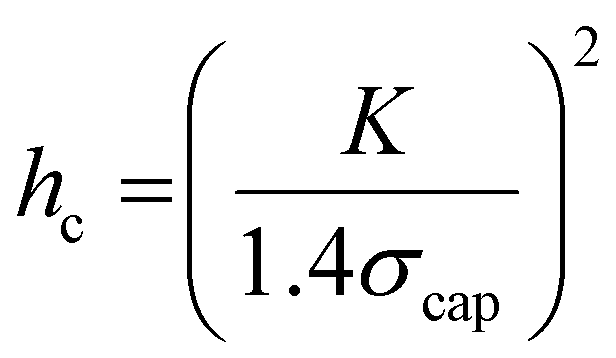 | (2) |
In Li-ion battery electrodes, the capillary action process has also been shown to cause the migration of binder and conductive carbon towards the upper region of the electrode.2–8,19,20 This effect, which is influenced by drying conditions such as drying temperature and time, can negatively affect adhesion to the current collector and resistivity of the electrode.2–10 In addition to binder migration, Jaiser et al. showed using cryo-SEM with EDS mapping (in work following on from studies in silica coatings by Price et al.) that the formation of ‘pendular rings’ of solvent between particles in the final stage of the drying process causes the binder to precipitate largely between closely-packed active particles, whilst binder is not present in larger pore spaces.16,21 The conditions of the drying process are therefore a major influence on electrode performance.
As discussed above, the drying process can also result in mud cracking of Li-ion battery electrodes above a critical thickness. Mud cracking refers to the formation of cracks in the electrode during the drying process as a consequence of drying stress.22 Mud crack formation in Li-ion battery electrodes has been shown to be promoted by high drying temperatures, as well as increased electrode thickness, both of which are a hindrance to the manufacture of thick electrodes for higher energy density batteries.19,22–25 The inverse relationship between critical cracking thickness and solvent surface tension is also a barrier to a move towards aqueous processing and fluorine-free binders, since water has a higher surface tension than alternative organic solvents.14,22,23,26,27 Similarly, LFP and LMFP particles are commonly processed as primary particles hundreds of nanometres in diameter, rather than micron-scale secondary particles, so are likely to have a lower critical cracking thickness and so be even less amenable to thick slurry coating.28,29
In contrast, studies by Bryntesen et al., and our own work, have demonstrated a relationship between the presence of cracking and improved electrochemical performance of electrodes.25,30 It is believed that this improvement is due in part to enhanced ion transport in cracked electrodes, since cracks provide a directional pore channel which facilitates ion transport through the electrode thickness.25,31 In this respect mud cracks are similar to various pore structure engineering methodologies, such as laser ablation,32–36 templating,37–41 mechanical pore punching42 and acoustic patterning.43 These approaches aim to provide enhanced ion transport through directional porosity, enabling improved rate performance in thick electrodes, which are otherwise hindered by poor ion transport.44 Controlled mud cracking offers the potential to achieve this effect without additional electrode processing.45 The motivation to understand crack formation is therefore two-fold: to prevent excessive cracking which can result in coating delamination, electronic disconnection or other electrode failure mode; and to identify and facilitate an optimal degree of cracking which enhances ion transport without harming performance.
In order to understand the mechanism of the drying process, several imaging approaches have been applied to observe structural changes. Jaiser et al. applied time-resolved cryo-SEM of electrode cross sections to investigate solvent binder distribution during the drying process.21 They also proposed heterogeneous pore emptying at the drying electrode surface with in situ optical fluorescence microscopy.5 However, the inherently three dimensional nature of the drying process limited the extent of their study. Higa et al. used high speed X-ray radiography to monitor the changing density of drying aqueous SiOx slurries, and paired this in situ analysis with three dimensional X-ray computed tomography (X-ray CT) to show that particles settled in a ring around a drying drop of coating.9 A second study on NMC-based cathodes drying showed poor image contrast of active material, and in both studies in situ X-ray CT was not attempted, since the drying times of several minutes were too short to carry out tomographic imaging.46 Yang et al. resolved this issue using a high-speed dynamic tomography mode which achieved 0.5 s per tomograph, and applied digital volume correlation (DVC) to resulting reconstructions of a drying slurry of NMC 622 in NMP with a PVDF binder, in order to capture particle motion.47 Whilst the rough position of active particles could be resolved, the full electrode microstructure was not visible, again due to the short drying time of under 10 minutes.
In previous work, we have studied the three dimensional structure of mud cracks with ex situ X-ray CT, showing the importance of three dimensional crack structure in influencing ion transport and electrode performance, but without clear resolution of the full electrode microstructure.25 In this study we extend that work, to analyse the electrode drying and mud cracking processes in situ, with full resolution of the electrode microstructure (Fig. 1). The relationship between mud crack formation and the local microstructural reorganisation is probed, and we demonstrate using DVC that the position of mud cracks can be predicted minutes before they are visible in tomograms.
 | ||
| Fig. 1 (a) Schematic of the slurry coating procedure and imaging apparatus; (b) a volume rendering of the imaged coated slurry with cross-sectional cut-outs in the vertical and horizontal planes; and (c) a schematic showing the three tested doctor blade gaps coated on steel pins. | ||
2 Experimental
2.1 Preparation of Li-ion positive electrode slurry imaging samples
Positive electrode slurries were prepared using the active material LiNi0.6Mn0.2Co0.2 (BASF), conductive additive carbon black (C-Nergy Super C65, Timcal) and polyvinylidene fluoride binder (PVDF, Solef, Solvay Specialty Polymers) dispersed in N-methyl pyrrolidone solvent (NMP, anhydrous 99.5%, Sigma Aldrich). The ratio of components is described in Table 1 below, where the increased C65 content and PVDF, compared with typical commercial formulations, were used in order to encourage cracking and enhance the flow behaviour of the slurry, respectively, as in our previous work.25 PVDF was added to stirred NMP to prevent agglomeration and the mixture was stirred until dissolved to give an 8 wt% solution. C65 was added to a portion of this binder solution, and mixed in a planetary mixer (Thinky ARE-250, Intertronics) for 1 minute at 500 rpm and 5 minutes at 2000 rpm. NMC622 was then added alongside the additional NMP required to give the desired solids content and mixed for 10 minutes at 2000 rpm with a subsequent 3 minutes degassing at 2200 rpm in the planetary mixer. Prepared slurries were kept for a maximum of 12 hours before being replaced with a new batch to limit the effects of slurry degradation over time. To prepare samples for dynamic imaging experiments, slurries were coated with doctor blade gaps of 300, 500 and 800 µm onto a Perspex sheet with a small perforation, into which a cylindrical steel pillar of 4 mm diameter and a machined flat surface was placed. Slurry casting used a draw down coater and a calibrated doctor blade (3600/4, Elcometer). The coated pillar was then removed from the perforated sheet by pushing it upwards from below, and transferred to the sample imaging stage (Fig. 1a). Samples were then dried at ambient temperature in the X-ray beam. Temperature measurements using a thermocouple embedded in the electrode slurry indicated that the beam induced an increase of 1–3 °C during exposure, which returned to baseline when the sample was removed from the beam. Beam induced heating affects are therefore assumed to be negligible.| Mass of solid components/% | Total solid content/% | ||
|---|---|---|---|
| NMC 622 | C65 | PVDF | |
| 84 | 9 | 7 | 45 |
2.2 Synchrotron X-ray computed tomography
Series of 3D tomograms of electrode samples of 4 mm diameter were taken at ID19 beamline for X-ray computed microtomography at ESRF, using a polychromatic beam with a peak at 70 keV. The sample was rotated through 360° with 5000 exposures of 0.015 s each, and 21 dark field and 40 flat field images. An sCMOS detector (PCOedge HS, 2560 × 2160 pixels) was used with a 5x optical magnification, giving a voxel size of 1.3 µm. The ‘half acquisition’ imaging protocol, where the sample rotation axis is offset from the detector center line and rotated over 360° instead of 180°, was used to increase the FOV to 5.15 mm × 2.6 mm (horizontal × vertical) to ensure the full 4 mm samples could be reconstructed by off-setting the axis of rotation from the centre of the field of view. Additionally, an array of 8 compound refractive X-ray lenses was used to increase X-ray flux at the sample to allow for lower exposure times and faster tomography. Tomograms were reconstructed using a standard filtered back projection algorithm using the ESRF in-house software NABU.48Tomographic series at Diamond Light Source (DLS) beamline i13-2 were taken using a pink beam with a peak at 30 keV and an sCMOS detector (pco.edge 5.5, 2560 × 2160 pixels) with 2x optical magnification to give an FOV of 4.5 × 3.2 mm (horizontal × vertical) and an effective pixel size of 1.625 µm, allowing for the imaging of the full sample within the field of view. Samples were therefore rotated through 180° with a step size of 0.18° and an exposure time of 0.05 s. For each tomogram 20 dark field and 20 flat field images were taken. Tomograms were reconstructed using a standard filtered back projection algorithm available in Savu.49
2.3 Image analysis
 | ||
| Fig. 2 Analysis of electrode consolidation and densification prior to crack formation. (a) Plot showing the change in coating height over time for 2 coating thicknesses, where solid lines indicate the solvent level above the coating and the shaded area indicates standard deviation. (b) the average porosity over time, during electrode consolidation, for the 300 µm coating, where the shaded area represents the standard deviation from 3 measurements; (c) the relative porosity as a function of distance from the base of the electrode coating, with a range of drying times shown; (d) 2D slices in the x–z plane from the centre of the reconstructed X-ray CT volume for the 300 µm coating showing initial and consolidated electrode thickness, with an additional cut out showing the solvent above the solid electrode material at the beginning of drying, indicated by a white dashed line; (e) indicative volumes showing the reduction in coating thickness and densification of solid material during this consolidation stage of the drying process, before cracking, where the solvent still occupies the space between active particles. | ||
The fine cracks present in the electrode after 49 minutes of drying were segmented in ilastik, preserving voxel size such that the segmented image could be used as a mask to separate cracked and uncracked regions from localised particle number and volume maps. The greyscale histogram, plotted as a distribution, was used to compare particle packing density in cracked and uncracked regions.
3 Results and discussion
For X-ray CT imaging of the electrode drying process, electrode slurries were coated using a typical draw down coater and doctor blade with gaps of 300 µm, 500 µm, and 800 µm onto steel pins of 4 mm diameter as shown in Fig. 1a and c. Samples were dried at ambient temperature over the course of 1–3 hours whilst imaging was carried out continuously, with interruptions only for dark and flat field images. In general, where cracks form they are clearly interconnected with the pore network, but are distinct from pores in size and morphology.25,31 In this work, cracks were defined as roughly vertical channels (perpendicular to the plane of the coating) which opened during drying, without solid material impeding their connection to the upper electrode surface. Film thickness and crack formation were studied in all three electrodes, whilst further detailed analysis of the electrode microstructure during drying and cracking, as well as DVC analysis, was carried out for the 300 µm electrode.3.1 Initial electrode drying process
Before cracks initiate, the electrode undergoes an initial phase of drying where the coating reduces in thickness and active particles move closer together, until the active particles have fully consolidated but are still immersed in solvent. This process is represented in Fig. 2e.20Fig. 2a and Fig. S2 in the SI show the reduction in height of the three drying coating thicknesses during this period. In images of the drying 300 µm electrode, two levels of material were visible – a solid layer of electrode active material, and a solvent meniscus above the electrode, which are represented as solid lines in Fig. 2a. In the 300 µm electrode, this excess solvent layer was 30 µm thick and began reducing thickness immediately after coating. On meeting the upper solid material in the electrode, the solvent level could no longer be resolved by X-ray CT due to the highly X-ray attenuating nature of the NMC622 active material. Concurrently, the solid phase thickness reduced linearly to a final thickness of 129 µm after 37 minutes.The solvent level was also clearly visible above the solid electrode material in the 800 µm coatings, which is shown in X-ray CT cross sections in Fig. 2d. The much larger gap between solvent and solid layers (of 180 µm) in the 800 µm coating likely resulted from a combination of the tendency of the solvent to form a curved meniscus on 4 mm pins, causing separation between solvent and solid material, a behaviour more marked at higher thickness, and possibly a degree of slurry degradation over time which caused poorer slurry phase suspension (electrode slurries were retained for 12 hours during the experiment before being replaced). In the 800 µm electrode, the solid layer did not change in thickness until 24 minutes, at which point the excess solvent had reduced from 180 µm to 50 µm above the solid layer. The solid layer then reduced in a linear fashion until the coating thickness stopped reducing (in other words, the electrode was consolidated) after 72 minutes and at 298 µm thick. The 300 µm electrode, which began with an excess solvent layer only 30 µm thick, began reducing thickness linearly in the solid phase immediately after coating, to a final thickness of 129 µm after 37 minutes. Imaging of the 500 µm failed during the first 35 minutes of the drying process, due to an error in the centre of rotation, and the coating had largely consolidated once imaging was successful. The coating height for this sample is shown in Fig. S2b. The experiments reveal a complex interplay between slurry age, coated thickness and solvent height.
In addition to evaluating solvent height and macro-crack behaviour, the experimental approach employed in this study enables the analysis of the porosity within the electrode microstructure. Fig. 2b and c represent the porous phase of the composite electrode (where porosity is defined here for simplicity as the non-active particle volume of the coating, shown in Fig. 2e) of the 300 µm electrode during the initial 37 minutes of the drying process. Fig. 2b shows overall porosity and Fig. 2c shows the porosity as a function of the percentage distance from the base of the electrode coating, relative to the porosity at the base of the electrode. Porosity data was extracted from segmented tomograms at each time step. In Fig. 2b the porosities given are an average of those determined from three machine learning-based segmentations of each volume, in order to adapt to the challenge of segmenting a large series of images where the materials are in motion. The total porosity of the coating reduced from 79% to 73% over the course of this initial drying period. This relatively high porosity reflects the large proportion (9%) of low-density porous conductive carbon binder domain (CBD) in the slurry formulation (Table 1), which cannot be resolved directly by X-ray CT, and is therefore included within the pore phase. Porosity calculations continued up to 37 minutes, since beyond this time cracking occurred, so porosity changes no longer related to vertical electrode consolidation. Further porosity changes are analysed in Fig. 6.
In order to achieve comparison despite changing electrode thickness and variation in image characteristics between time steps, the x and y axes in Fig. 2c are normalised. The bottom of the electrode was chosen as the normalised 0 point, since theoretically its porosity shouldn't change over the drying time, as active particles were resting on the bottom surface at the time of coating. This principle can been seen in the cut-outs in Fig. 2e. At the start of the drying process the porosity was greater with increased distance from the base of the electrode, from which observation it can be inferred that the active particles had a greater degree of compaction at the bottom of the coating, and were suspended more sparsely towards the upper surface. As drying progressed this trend reduced rapidly towards a roughly even distribution of porosity through the thickness, as can be seen between 8 and 10 minutes. Between 10 and 37 minutes the decrease in porosity in upper portions of the electrode continued, such that up to 50% of the full distance from the base of the electrode, the porosity remained roughly constant, but closer to the upper surface porosity reduced sharply, reversing the original trend. This difference was stark, with a minimum porosity at the top of electrode 4% lower than that at the base of the electrode after 37 minutes of drying. A reduced porosity at the upper surface would clearly have negative impacts on ion transport, and this effect may have been a consequence of the meniscus of the coating having lower initial density of active material causing greater compaction at the highest point as it dried. This further highlights the importance of consistent coating thickness. The steep increase in porosity in the top 5–10% of the electrode coating was due to the tendency of porosity towards 100% above the upper surface.
3.2 Growth of mud cracks
After particle consolidation, which occurs in the initial drying stage described above, mud-cracks may initiate in the electrode, particularly in thick electrode coatings. Experimental results, shown in Fig. 3, showed that crack initiation occurred after the drying electrode thickness ceased reducing (39 and 74 minutes for the 300 µm and 800 µm electrodes respectively). In the 500 µm electrode, crack initiation occurred at 45 minutes, which event is shown in Fig. S3h. This is in agreement with studies by Moorhead and Price et al., who studied ceramic and silica particulate coatings respectively, showing that crack initiation is expected to occur at the point where solvent drops below the upper layer of active particles, when there is a sharp increase in stress acting on the coating.16,18 | ||
| Fig. 3 3D volume renderings of (a) the 300 µm electrode and (b) the 800 µm electrode, each with two 2D slices from the reconstructed volumes showing initiation and growth of cracks. The scale bars represent 0.5 mm. | ||
Following crack initiation, shown in Fig. 3, in the case of the 300 µm and 500 µm electrodes, crack propagation was immediate, with cracks progressing from near the electrode surface downwards towards the substrate. Crack volume increased linearly in both cases until 75 minutes, as shown in Fig. 4g, in which the 3D cracking intensity factor (3D CIF; defined in previous work as the crack volume fraction25) is plotted against drying time.25 In both electrodes, the crack volume increased at approximately the same rate, peaking at a 3D CIF of 18.7% and 19.0% for 300 µm and 500 µm electrodes respectively. Though similar in volume, the crack morphology showed clear structural differences, with a greater degree of crack branching and inter-connectedness in the 300 µm electrode. In the 500 µm electrodes cracks developed which were larger with a lesser degree of cross-linking, as has been shown previously in Li-ion battery electrodes and a number of other particulate coating systems, where thicker coatings have larger crack spacings.12,25,52,53 The 300 µm electrode also formed a number of fine radial cracks around the outer edge of the electrode, not present in thicker coatings. These radial cracks are considered to be stochastic, rather than a consequence of lateral solvent transport towards the edges during drying, as has been noted previously in drying droplets, since this mechanism would result in crack initiation at the droplet edge, which is evidently not the case in this study.53,54
 | ||
| Fig. 4 Volume renderings of crack growth for electrode coatings of 300 µm, 500 µm and 800 µm doctor blade gap, where (a, c and e) show crack initiation and (b, d and f) show final cracking extent, respectively. For each volume, a vertical cut out showing a single slice of the reconstructed X-ray CT image. The scale bars and axes represent 1 mm. Plot (g) shows the increase of 3D CIF over time for each thickness, whilst (h–j) show the 2DCIF as a function of distance from the electrode base for coatings of 300 µm, 500 µm and 800 µm doctor blade gap respectively. | ||
The air bubble present in the 800 μm electrode (Fig. 3b) which occurred due to incomplete degassing of the sample, resulted in a different pattern of crack initiation. In the first scan at t = 0 (Fig. S3o and q) the bubble had a maximum height and width of 84 µm and 229 µm respectively, which increased to 125 µm and 405 µm after 19 minutes. A second smaller bubble with a maximum height and width of 63 µm and 109 µm respectively was visible from the beginning of drying, and did not change in size during the initial phase of the drying process described. The larger bubble retained consistent geometry between 19 and 55 minutes, at which point air was seen to escape from the bubble towards the upper surface, which caused permanent deformation of the highly viscous, partially-dried coating. This feature underwent no further growth until 74 minutes, during which the electrode coating continued to consolidate and contained an excess of solvent (Fig. 2a). As in the other electrodes, once consolidation had completed, crack propagation occurred, forming a connection between the upper surface and the bubble, and extending across the electrode.
As a consequence, cracking in the 800 µm electrode was starkly morphologically different, and much greater in volume (the final 3D CIF was 41.5%), than the two thinner coatings, though the rate of increase of the 3D CIF was similar. The air bubble acted as the nucleation point of a crack when the drying capillary stress increased, concentrating stress in a single region. A single large crack subsequently formed through the centre of the electrode, with branching at either end. Two further examples of crack initiation at bubbles are show in Fig. S4, where both electrodes were coated with a 500 µm doctor blade gap. The first (Fig. S4a) had a single small bubble and the other (Fig. S4b) contained seven bubbles of varying sizes. In both cases cracks nucleated exclusively from bubbles, and the largest four bubbles in Fig. S4b had similar protrusions connecting them to the electrode surface, seven minutes before crack initiation. The final crack morphology differed from the electrode of the same blade gap without a bubble shown in Fig. 4d, with fewer, less connected cracks, concentrated around the position of the bubbles. This suggests that the morphology of the cracking in the 800 µm electrode (Fig. 4f and Fig. S3o–v), where only a single crack was observed, likely resulted due to the bubble, rather than as a consequence of the trend towards fewer, larger cracks with increasing coating thickness. Although bubbles were not the target of this study, they are known to arise in manufacturing and their presence in this study has provided valuable mechanistic insight.
As shown in Fig. 3a, cracks propagate downwards from the upper electrode surface, and it was believed that they may initiate and propagate where there were less closely-packed particles since, as noted above, the binder forms between particles which are close together at the end of the drying process.21 The temporal resolution (two minutes per image) of the tomographic imaging was insufficient to capture the precise position of crack initiation, so in order to investigate the principle that local binder formation may determine crack nucleation position, an analysis of the volume and number of neighbouring particles for each voxel in cracked and uncracked regions, as a proxy for inter-particle distance, was carried out. This analysis, which is shown in Fig. S5, found no clear correlation between crack position and the density of particles in a region. Whilst cracks may initiate at such structural features, they may be too small in comparison to the full volume to resolve by this method.
3.3 Delamination from the current collector
Alongside cracking, delamination from the substrate, in this case a steel pin, was observed. As highlighted in Fig. 5, this delamination began where cracks had grown downwards to reach the substrate, and the junctions of cracks with the substrate acted as nucleation points for delamination, which in essence is an interfacial cracking between the coating and current collector. Delamination is therefore a related but distinct process from the formation of vertical mud cracks, and the 3D CIF does not capture their different contributions to overall crack volume. Further examples of delamination are shown in Fig. S3(e, l and u). | ||
| Fig. 5 3D volume rendering of the 500 μm electrode 93 minutes after drying (left) and three segments from 2D radiographs of a region of interest showing the progression of delamination of the electrode coating from the pin at three time steps. The scale bar for the 3D volume represents 1 mm, whilst those for the 2D cut-outs show 0.25 mm. | ||
Fig. 4h–j show the 2D CIF, which represents the cracking area fraction for each horizontal slice through the coating thickness, enabling the disentanglement of the magnitudes of mud cracking and delamination. Since the coating thickness did not change measurably in the period during which cracking occurred, distance from the electrode base can be given in absolute terms. In the 300 µm electrode, between crack initiation at 39 minutes and 56 minutes, the 2D CIF remained fairly even through the thickness, with a slight increase towards the upper surface, in line with ex situ observations in previous work, indicating an absence of delamination.25 Between 63 and 78 minutes a small increase in the 2D CIF at the current collector was seen, indicating a delamination distinct from the reduction of contact resultant between electrode coating and current collector resulting from vertical cracking. Delamination events are seen clearly in the 2D image slices from 51 and 82 minutes (Fig. S3e and g). Concurrent with this delamination, far larger increases in the 2D CIF close to the electrode surface were observed, as 2D CIF increased at the surface increased from 11% to 20.5% at the upper surface between 56 and 63 minutes. This trend developed through 70 and 78 minutes of drying, with the appearance of a region of near constant 2D CIF between 15 µm and 60 µm from the electrode base where no substantial increase was seen over time, and large increases in 2D CIF at the electrode surface, suggesting the vertical mud cracking had ceased, but drying stresses acting in parallel to vertical cracks continued to cause delamination.
A similar pattern could be seen in the 500 µm and 800 µm electrodes (Fig. 4i and j respectively), with three distinct regions within the 2D CIF profile: delamination at the base; a plateau in the middle; and greater separation at the electrode surface. Delamination occurred sooner and with greater severity in thicker electrodes; whilst for the 300 µm electrode delamination in excess of the 2D CIF plateau was notable after 20 minutes of cracking, it occurred within 10 and 9 minutes for the 500 µm and 800 µm electrodes respectively. The maximum 2D CIF values at the electrode base were 16%, 19% and 33% for electrodes in ascending order of thickness. Peak 2D CIF values at the electrode surface were 34%, 37% and 30% for 300 µm, 500 µm and 800 µm electrodes respectively. The trend, showing higher 2D CIF at the surface than the base, was reversed at the surface of the 800 µm electrode after 89 minutes of drying, with delamination making an increased contribution to the overall 3D CIF. At some points the curves overlap, showing decreasing 2D CIF over time, which is likely the result of the degree of uncertainty in segmentation. These uncertainties are greater at the electrode surface and where cracks are wider, owing to challenges differentiating between cracks and the empty space above the electrode. In Fig. 4j the two small peaks seen from 70–74 minutes, centred at 40 µm and 255 µm from the current collector, correspond to the two air bubbles present in the slurry (Fig. S3o and p). As cracking progressed at 79 minutes these signals were no longer apparent in the data, instead forming part of the baseline 2D CIF.
The higher degree of delamination in thicker electrodes indicates a greater stress resulting from the drying process, which even where cracking does not occur is likely to cause crimping around the edge of coatings on a conventional metal foil current collector. The increase in 2D CIF close to the upper electrode surface towards the end of the drying process likely has two components. In part, a separation between opposite crack walls is driven by the delamination at the base of the electrode, and this in turn increases 2D CIF at the upper surface. However increased 2D CIFs near the upper electrode surface were also observed in previous work in the absence of delamination, so it can be concluded that additional expansion occurs towards the upper region of the electrode.25 This through thickness analysis also suggests that crack growth does not continue below the surface when it has stopped at the surface. Surface optical imaging can therefore be used reliably to determine the period during which cracks grow, which may enable in-line surface characterisation to predict 3-dimensional features.
3.4 Impact of cracking on local microstructure
Fig. 6 shows an analysis of the impact of cracking on the porosity of the remainder of the electrode. The porosity of a 300 × 300 µm square of the 300 µm electrode between a number of cracks, cropped between 30–110 µm of the electrode thickness to exclude the top and bottom of the electrode (Fig. 5a), was analysed during crack propagation. As was expected, the total porosity of the cropped region reduced as cracking progressed, at a decreasing rate between 39 and 78 minutes (Fig. 6b), which corresponds well with the plateauing of the overall increase of 3D CIF shown in Fig. 4g. Peaks and troughs, which can be seen in Fig. 6c, which occur consistently at each time step, appear to shift towards the current collector as crack growth progresses. This indicates a compaction of the electrode which could result from additional contraction due to drying, but is more probably caused by greater crack opening at the upper surface (Fig. 4h), which resulted in a downward compression of solid material. The anomalous characteristics of data points at 61 and 66 minutes resulted from more rapid movements of the electrode which appeared to occur during this period, resulting in less clear imaging of active particles, reducing segmentation accuracy and increasing the calculated overall porosity. The compaction of the pore phase adjacent to cracks identified here is likely to limit ion transport in the bulk electrode which, despite the enhanced performance in cracked electrodes noted previously may cause increased inhomogeneity in ion transport and active a particle utilisation. | ||
| Fig. 6 Plots describing the change in porosity over time for a drying electrode coating of 300 µm thickness, where (a) the region analysed at 39 and 78 minutes, with scale bars indicating 0.5 mm; (b) is an indicative diagrams demonstrating a single crack opening and compressing adjacent particles closer together; (c) shows porosity as a function of distance from the base of the electrode over time; and (d) shows total porosity as a function of time. | ||
3.5 Predicting cracking with DVC
This compaction of particles suggests that volume correlation may yield insights into the movement of particles during mud cracking. Fig. 7 shows the combined displacement maps generated by DVC analysis of the 300 µm electrode during the scan prior to measured crack opening (37 minutes) and the subsequent two scans (39–42 minutes), and maps of displacement separated into its x-, y- and z-direction components (x and y are in plane of the coating direction, and z is the electrode height) are shown in Fig. S6. DVC displacement maps show the local displacement, given in µm, within the electrode by comparing a reference image (an initial X-ray CT scan) with a deformed image (the subsequent scan, where movement has occurred). In this case, for instance, the displacement map for 37 minutes was generated by comparison of local voxel intensities at 37 minutes (the deformed image) with those of the preceding scan at 34 minutes (the reference image). Three cracks are identified within white ellipses in Fig. 7, which show that displacement was observable by DVC prior to cracks becoming visible within the raw image. In displacement maps at 34 (Fig. S6) and 37 minutes, there was a y-direction displacement of approx. 5 µm across the full slice, due to slight fluctuations in image intensity causing the artefact during DVC computation. At 37 minutes, therefore, the combined displacement from the first crack was 1–2 µm above the background level, with a largely x–y character and very little z-component. This far smaller displacement than is likely to be observed by visual comparison highlights the value of DVC for early identification of crack opening. | ||
| Fig. 7 DVC heat maps of total displacement for the 300 µm electrode between 37–42 minutes from a representative central slice of the volume, compared with the unprocessed slice from the reconstructed X-ray volume. Three regions indicated within white ellipses show that displacement was visible by DVC prior to the crack in that region being evident in the raw CT data. | ||
At 39 and 42 minutes, the background displacement observed outside cracking regions was 0 µm, so displacement magnitude could be determined directly from the map values. In these maps, larger cracks became visible, again showing that displacement could be identified before each crack itself was clearly visible in the raw image. Longer, wider cracks showed larger displacement magnitudes, and a more rapid growth rate. Whilst the first, smallest crack had a maximum displacement of 7 µm, the largest crack (approximately 3 mm in total length) had a maximum displacement of 10 µm at 39 minutes. Additionally, hot spot regions of high displacement were identified, with a maximum combined value of 13 µm at 42 minutes. The largest displacement values tended to be focused at the centre of the cracks’ length, rather than at the opening tip of the cracks, where the perpendicular driving force of cracking acts (Fig. S1). At all time-steps the z-direction displacement was lesser in magnitude, with a maximum of 5 µm, and was entirely negative, indicating the downwards movement of material towards the electrode substrate. This corroborates the observation in Fig. 6c, which suggested that cracking resulted in the forcing of some electrode material downwards. It is not believed that this effect is a continuation of the settling of particles during drying, since the coating thickness reached a plateau prior to crack initiation.
The displacement fields around cracks were observed are over a much longer length scale and encompass regions that had not yet cracked. DVC displacement could therefore be observed earlier and more clearly than cracks themselves. Given previous work correlating 3D crack structure with surface cracking, this work suggests digital image correlation, the 2D equivalent of DVC, may be a viable tool to rapidly identify crack formation before crack growth has caused critical damage to the electrode.25 Coupled with an understanding of processing parameters which influence cracking, such a coating thickness and drying temperature, this approach may enable the rapid feedback and control to halt cracking, particularly during the initial period of process optimisation during battery manufacturing. An in situ analysis of crack distribution and rate of opening may also enable the responsive tailoring of drying conditions to control, rather than totally eliminate, cracking.
4 Conclusions
In this work the first three dimensional time-resolved imaging of the microstructure and the mud cracking process of a Li-ion electrode during the drying process was reported using synchrotron X-ray computed tomography. This was achieved by drying at ambient temperature over 1–3 hours, coupled with the high X-ray flux of synchrotron sources, which was shown to be a highly effective approach to image changes to the electrode microstructure in detail. As in previous reports, cracking was shown to initiate at the time that the solid material had consolidated and the solvent level dropped below the upper layer of active particles. Cracking was confirmed to increase in intensity with increased coating thickness, and the cracks formed in thicker coatings were fewer in number and greater in volume. The presence of air bubbles was shown to act as a nucleating point for cracks, and concentrated the crack volume in a smaller region, which highlights the importance of thorough degassing of electrode slurries prior to drying. Furthermore, cracks which reached the electrode substrate were shown to cause the nucleation of delamination processes, which resulted in additional damage to the electrode structure. The growth of cracking was shown to compress the surrounding active material, reducing the porosity of these regions, and the combination of open pore channels (cracks) and otherwise constricted porosity may result in increased inhomogeneity of active material utilisation in cracked electrodes. Finally, digital volume correlation was demonstrated as an effective analytical tool for the identification of crack position prior to major crack growth, and the potential of image correlation approaches as tools for process feedback and control was discussed.These experiments provide new mechanistic insight into the drying process, which remains one of the most stubbornly energy intensive operations in battery manufacture. Whilst the experimental conditions studied here demand the use of model samples, we have corroborated our findings with studies of more conventional drying setups to provide generalisable insight. Moreover, we propose future directions for integration of advanced metrology within the manufacturing processes, which can be validated against these laboratory measurements. Collectively, this provides a pathway to optimise the manufacturing process, and the resultant electrode microstructure towards high performance and sustainable battery manufacture.
Author contributions
WJD and ARTM conceptualised and led the experiments, in which they were supported by ICA, VK, KW, PPP, BL, RSY, ZZ, JP, IK and GG. Writing and data analysis was carried out primarily WJD, with additional analysis and interpretation by ST, MPJ, ARTM, LB, and KC aided in data visualisation. AJER, JBR, DC, TSM and PRS supervised research, acquired funding and supported in project administration. The work was reviewed and edited by ARTM, KW, JBR, RJ, TSM, PPP, BL and PRS.Conflicts of interest
There are no conflicts of interest to declare.Data availability
The authors affirm that the data supporting the discussion and conclusions are fully available within the article and in the supplementary information (SI). Raw X-ray CT data is available from the Diamond Light Source data gateway under experiment number MG33315-1, and from the ESRF data repository through https://doi.esrf.fr/10.15151/ESRF-ES-1308732998. Supplementary information is available. See DOI: https://doi.org/10.1039/d5eb00201j.Acknowledgements
The authors acknowledge Beamline ID19 at The European Synchrotron Radiation Facility (ESRF) for experiment ME-1661 (https://doi.org/10.15151/ESRF-ES-1308732998) and Diamond Light Source Beamline I13-2 for experiment MG33315-1, as well as the Faraday Institution through the NEXTRODE (Grant FIRG015, FIRG066), LiSTAR program (FIRG083, EP/S003053/1), and Characterisation and Manufacturing of Advanced LFP Batteries (FIRG081). P. R. S., P. P. P. and B. L. thank EPSRC for the International Centre to Centre Collaboration with the ESRF grant reference EP/W003333/1, and the Henry Royce Institute established through EPSRC grants EP/R00661X/1, EP/P025498/1 and EP/P025021/1. P. R. S. was supported by the Department of Science, Innovation and Technology (DSIT) and the Royal Academy of Engineering under the Chair in Emerging Technologies program (CiET1718/59).References
- Y. Liu, R. Zhang, J. Wang and Y. Wang, iScience, 2021, 24, 102332 CrossRef CAS PubMed.
- B. Westphal, H. Bockholt, T. Günther, W. Haselrieder and A. Kwade, ECS Trans., 2015, 64, 57–68 CrossRef CAS.
- B. G. Westphal and A. Kwade, J. Energy Storage, 2018, 18, 509–517 CrossRef.
- M. Baunach, S. Jaiser, S. Schmelzle, H. Nirschl, P. Scharfer and W. Schabel, Drying Technol., 2016, 34, 462–473 CrossRef CAS.
- S. Jaiser, L. Funk, M. Baunach, P. Scharfer and W. Schabel, J. Colloid Interface Sci., 2017, 494, 22–31 CrossRef CAS PubMed.
- S. Jaiser, M. Müller, M. Baunach, W. Bauer, P. Scharfer and W. Schabel, J. Power Sources, 2016, 318, 210–219 CrossRef CAS.
- M. Müller, L. Pfaffmann, S. Jaiser, M. Baunach, V. Trouillet, F. Scheiba, P. Scharfer, W. Schabel and W. Bauer, J. Power Sources, 2017, 340, 1–5 CrossRef.
- A. Kukay, G. Polizos, E. Bott, A. Ielvev, R. Tao, J. Sharma and J. Li, Batteries Supercaps, 2024, 7, e202400113 CrossRef CAS.
- K. Higa, H. Zhao, D. Y. Parkinson, H. Barnard, M. Ling, G. Liu and V. Srinivasan, J. Electrochem. Soc., 2017, 164, A380–A388 CrossRef CAS.
- L. Pfaffmann, S. Jaiser, M. Müller, P. Scharfer, W. Schabel, W. Bauer, F. Scheiba and H. Ehrenberg, J. Power Sources, 2017, 363, 460–469 CrossRef CAS.
- R. C. Chiu and M. J. Cima, J. Am. Ceram. Soc., 1993, 76, 2769–2777 CrossRef CAS.
- W. P. Lee and A. F. Routh, Langmuir, 2004, 20, 9885–9888 CrossRef CAS PubMed.
- R. C. Chiu, T. J. Garino and M. J. Cima, J. Am. Ceram. Soc., 1993, 76, 2257–2264 CrossRef CAS.
- R. Sahore, D. L. Wood, A. Kukay, K. M. Grady, J. Li and I. Belharouak, ACS Sustainable Chem. Eng., 2020, 8, 3162–3169 CrossRef CAS.
- K. B. Singh and M. S. Tirumkudulu, Phys. Rev. Lett., 2007, 98, 218302 CrossRef PubMed.
- K. K. Price, Y. Wu, A. V. McCormick and L. F. Francis, J. Am. Ceram. Soc., 2015, 98, 2214–2222 CrossRef CAS.
- S. K. Maiti, in Fracture Mechanics, Cambridge University Press, 2015, pp. 6–64 Search PubMed.
- A. Moorhead and L. F. Francis, J. Am. Ceram. Soc., 2024, 107, 2837–2848 CrossRef CAS.
- J. Kumberg, M. Müller, R. Diehm, S. Spiegel, C. Wachsmann, W. Bauer, P. Scharfer and W. Schabel, Energy Technol., 2019, 7, 1900722 CrossRef CAS.
- Y. S. Zhang, N. E. Courtier, Z. Zhang, K. Liu, J. J. Bailey, A. M. Boyce, G. Richardson, P. R. Shearing, E. Kendrick and D. J. L. Brett, Adv. Energy Mater., 2022, 12, 2102233 CrossRef CAS.
- S. Jaiser, J. Kumberg, J. Klaver, J. L. Urai, W. Schabel, J. Schmatz and P. Scharfer, J. Power Sources, 2017, 345, 97–107 CrossRef CAS.
- K. Rollag, D. Juarez-Robles, Z. Du, D. L. Wood and P. P. Mukherjee, ACS Appl. Energy Mater., 2019, 2, 4464–4476 CrossRef CAS.
- Z. Du, K. M. Rollag, J. Li, S. J. An, M. Wood, Y. Sheng, P. P. Mukherjee, C. Daniel and D. L. Wood, J. Power Sources, 2017, 354, 200–206 CrossRef CAS.
- S. Yari, L. Bird, S. Rahimisheikh, A. C. Reis, M. Mohammad, J. Hadermann, J. Robinson, P. R. Shearing and M. Safari, Adv. Energy Mater., 2024, 14(46), 2402163 CrossRef CAS.
- W. J. Dawson, A. R. Morrison, F. Iacoviello, A. M. Boyce, G. Giri, J. Li, T. S. Miller and P. Shearing, Batteries Supercaps, 2024, 7, e202400260 CrossRef CAS.
- S. C. Mun, Y. H. Jeon and J. H. Won, Prog. Nat. Sci.:Mater. Int., 2024, 34, 194–206 Search PubMed.
- D. Bresser, D. Buchholz, A. Moretti, A. Varzi and S. Passerini, Energy Environ. Sci., 2018, 11, 3096–3127 RSC.
- L. Chen, Z. Chen, S. Liu, B. Gao and J. Wang, J. Electrochem. Energy Convers. Storage, 2018, 15(4), 041011 CrossRef.
- E. Lyle, R. Vaeli, M. Cormier and M. Metzger, J. Electrochem. Soc., 2022, 169, 060527 Search PubMed.
- S. N. Bryntesen, A. Kahrom, J. J. Lamb, I. Tolstorebrov and O. S. Burheim, Batteries, 2023, 9, 96 Search PubMed.
- B.-S. Lee, Z. Wu, V. Petrova, X. Xing, H.-D. Lim, H. Liu and P. Liu, J. Electrochem. Soc., 2018, 165, A525 CrossRef CAS.
- N. A. Dunlap, D. Kern, F. Usseglio-Viretta, D. P. Finegan and B. J. Tremolet de Villers, in ECS Meeting Abstracts, The Electrochemical Society, 2021 Search PubMed.
- P. Zhu, A. Boyce, S. R. Daemi, B. Dong, Y. Chen, S. Guan, M. Crozier, Y.-L. Chiu, A. J. Davenport, R. Jervis, P. Shearing, R. N. Esfahani, P. R. Slater and E. Kendrick, Energy Storage Mater., 2024, 69, 103373 CrossRef.
- B. J. Tremolet de Villers and D. P. Finegan, in AMMTO and EDO Joint Peer Review, Washington, D.C., 2023 Search PubMed.
- K.-H. Chen, M. J. Namkoong, V. Goel, C. Yang, S. Kazemiabnavi, S. M. Mortuza, E. Kazyak, J. Mazumder, K. Thornton, J. Sakamoto and N. P. Dasgupta, J. Power Sources, 2020, 471, 228475 CrossRef CAS.
- J. B. Habedank, J. Kriegler and M. F. Zaeh, J. Electrochem. Soc., 2019, 166, A3940–A3949 CrossRef CAS.
- J. S. Sander, R. M. Erb, L. Li, A. Gurijala and Y.-M. Chiang, Nat. Energy, 2016, 1, 16099 CrossRef CAS.
- J. Billaud, F. Bouville, T. Magrini, C. Villevieille and A. R. Studart, Nat. Energy, 2016, 1, 16097 CrossRef CAS.
- C. Huang, M. Dontigny, K. Zaghib and P. S. Grant, J. Mater. Chem. A, 2019, 7, 21421–21431 RSC.
- C. Huang and P. S. Grant, J. Mater. Chem. A, 2018, 6, 14689–14699 RSC.
- F. Jiao, K. M. Shaju and P. G. Bruce, Angew. Chem., Int. Ed., 2005, 44, 6550–6553 CrossRef CAS PubMed.
- S. N. Bryntesen, P. H. Finne, A. M. Svensson, P. R. Shearing, N. Tolstik, I. T. Sorokina, J. Vinje, J. J. Lamb and O. S. Burheim, J. Mater. Chem. A, 2023, 11, 6483–6502 RSC.
- Y. Zhang, M. Shahriar and S. Hu, J. Mater. Chem. A, 2023, 11, 11849–11858 RSC.
- A. M. Boyce, X. Lu, D. J. L. Brett and P. R. Shearing, J. Power Sources, 2022, 542, 231779 CrossRef CAS.
- R. Seghir and S. Arscott, Sci. Rep., 2015, 5, 14787 CrossRef CAS PubMed.
- K. Higa, B. Zhang, D. K. Chandrasiri, D. Tan, D. Collins-Wildman, P. Bloemhard, E. Lizotte, G. Martin-Nyenhuis, D. Y. Parkinson, R. Prasher and V. S. Battaglia, ACS Appl. Energy Mater., 2024, 7, 2989–3008 CrossRef CAS PubMed.
- Y. Yang, Z. Xu, J. D. Steiner, Y. Liu, F. Lin and X. Xiao, Appl. Phys. Lett., 2020, 116, 081904 CrossRef CAS.
- P. Paleo, A. Mirone, C. Nemoz and N. R. Viganò, Nabu, 2019, https://gitlab.esrf.fr/tomotools/nabu.
- D. Kazantsev, N. Wadeson and M. Basham, SoftwareX 202219101157 Search PubMed.
- S. Berg, D. Kutra, T. Kroeger, C. N. Straehle, B. X. Kausler, C. Haubold, M. Schiegg, J. Ales, T. Beier, M. Rudy, K. Eren, J. I. Cervantes, B. Xu, F. Beuttenmueller, A. Wolny, C. Zhang, U. Koethe, F. A. Hamprecht and A. Kreshuk, Nat. Methods, 2019, 16, 1226–1232 CrossRef CAS PubMed.
- S. M. Tonge, C. A. Simpson, C. Reinhard, T. Connolley, A. H. Sherry, T. J. Marrow and M. Mostafavi, J. Mech. Phys. Solids, 2020, 141, 103956 CrossRef.
- M. I. Smith and J. S. Sharp, Langmuir, 2011, 27, 8009–8017 CrossRef CAS PubMed.
- P. Lilin, M. Ibrahim and I. Bischofberger, Sci. Adv., 2024, 10(34) DOI:10.1126/sciadv.adp3746.
- H. M. van der Kooij, G. T. van de Kerkhof and J. Sprakel, Soft Matter, 2016, 12, 2858–2867 RSC.
| This journal is © The Royal Society of Chemistry 2026 |