
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergistic effect between carbon-confined bismuth nanoparticles and K+-ether co-intercalation enables high-rate potassium storage at −50 °C
Jiawei
Gu†
a,
Jiali
Lin†
a,
Ling
Huang†
a,
Miao
Liu
*a,
Zhefei
Sun
b,
Xing
Chen
a,
Jiaqi
Zhang
a,
Mingxuan
Luo
a,
Qiaobao
Zhang
 ab and
Li
Zhang
*ac
ab and
Li
Zhang
*ac
aCollege of Chemistry and Chemical Engineering, State Key Laboratory of Physical Chemistry of Solid Surfaces, Xiamen 361005, Fujian, China. E-mail: zhangli81@xmu.edu.cn; liumiao11@xmu.edu.cn
bCollege of Materials, Fujian Key Laboratory of Surface and Interface Engineering for High Performance Materials, Xiamen Key Laboratory of High Performance Metals and Materials, Xiamen University, Xiamen, 361005, Fujian, China
cTan Kah Kee Innovation Laboratory, Xiamen University, Collaborative Innovation Center of Chemistry for Energy Materials, Xiamen University, Xiamen 361005, Fujian, China
First published on 6th January 2026
Abstract
The sluggish desolvation kinetics of K+ cations is widely recognized as a major bottleneck limiting the electrochemical performance of potassium ion batteries (PIBs) at low temperatures (LTs). Recent studies have identified the Li+/Na+-solvent co-intercalation mechanism is an effective strategy to lower desolvation energy and enhance low-temperature electrochemistry, yet its implementation remains rarely unexplored in PIBs, particularly below −40 °C. Meanwhile, the rational design of electrode structures is also crucial for achieving effective K+-solvent co-intercalation. Herein, we propose a rationally engineered architecture in which bismuth nanoparticles (∼19 nm) are uniformly confined within a conductive carbon framework (Bi@CFs). Experimental and theoretical analyses collectively reveal that the Bi@CFs configuration facilitates K+-ether co-intercalation, thereby lowering the desolvation energy barrier and achieving high-capacity and high-rate potassium storage at −50 °C. Accordingly, the Bi@CFs half-cell stably cycles over 400 cycles at −50 °C and 1 A g−1, maintaining an ultrahigh reversible capacity of 345.61 mAh g−1 with negligible degradation. Paired with an activated carbon (AC) cathode, the Bi@CFs/AC full cell presents an impressive energy density of 121.66 Wh kg−1 and power density of 9658.28 W kg−1 at −50 °C, along with an ultra-long lifespan exceeding 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles. This work lays the foundation for high-performance PIBs capable of stable operation at extremely low temperatures.
000 cycles. This work lays the foundation for high-performance PIBs capable of stable operation at extremely low temperatures.
Broader contextAdvancing rechargeable batteries toward higher energy density and wider operating temperature ranges, particularly at ultra-low temperatures, is essential for applications in high-latitude regions. Recently, potassium-ion batteries (PIBs) have attracted considerable attention owing to their low redox potential and intrinsic adaptability to low-temperature environments. However, PIBs face a major scientific challenge: the sluggish desolvation kinetics of K+ ions at the solid electrolyte interphase on the anode side during cycling. This issue becomes particularly severe below −40 °C, greatly limiting the electrochemical performance of PIBs. Herein, we successfully achieved the direct intercalation of solvated or partially solvated K+ ions, fundamentally resolving the challenge of sluggish K+ desolvation. Theoretical and experimental analyses reveal that the anchoring of ultrafine Bi nanoparticles within a conductive carbon network, along with the selection of 1,2-dimethoxyethane as the solvent, jointly contributed to this breakthrough. Therefore, we unlocked state-of-the-art high-capacity and high-rate K-ion storage at −50 °C. This work will have a broad impact on two levels: First, the proposed approach also offers a valuable solution for reducing interfacial kinetic barriers of Li+ and Na+ ions at extremely low temperatures. Second, our innovative strategy is expected to greatly accelerate the large scale application of high-energy-density batteries in cold regions. |
1. Introduction
Lithium-ion batteries (LIBs) have emerged as the prevailing power sources for electric vehicles and portable electronic devices, primarily owing to their high energy density and excellent cycling stability.1,2 However, their large-scale deployment in harsh cold environments remains fundamentally challenged by the escalating cost and geographically limited reserves of lithium, compounded by their intrinsically poor electrochemical performance at low-temperatures (LTs).3,4 As a promising alternative to LIBs, potassium-ion batteries (PIBs) have attracted considerable attention owing to the earth-abundant potassium resources,5,6 a lower redox potential (−2.93 V vs. SHE),7,8 and, most importantly, their intrinsic adaptability to low-temperature environments.9,10 This favorable LT performance is primarily attributed to the small Stokes radius of K+ and its relatively weak ion-solvent interaction energy compared to Li+, Na+, and Mg2+, which together facilitate enhanced ionic conductivity and rapid K+ diffusion within electrode materials, thereby enabling efficient charge transport even at LTs.11 However, a key limitation hindering the LT performance of PIBs is the high energy barrier associated with K+ desolvation at the solid electrolyte interphase (SEI).12,13Recent findings have revealed that the K+-solvent co-intercalation mechanism14 is regarded as one of the most effective strategies for graphene-based electrodes to reduce the high desolvation energy barrier, thereby enhancing interfacial ion transport kinetics and ultimately enabling superior K+ storage performance at −20 °C. Consequently, the rational design of high-capacity anodes similar to graphene-based electrodes (∼0.335 nm) with enlarged interlayer spacing is crucial to facilitate K+-solvent co-intercalation, thereby ensuring the reliable performance of PIBs at LTs, particularly below −40 °C.
Among the various anodes for PIBs, bismuth (Bi), owing to its enlarged interlayer spacing (∼0.395 nm), high theoretical capacity of 385 mAh g−1 and a relatively low voltage plateau,15 has emerged as a particularly promising candidate for achieving high-capacity K+ storage. However, commercial Bi electrodes face significant challenges, including pronounced volume expansion during cycling and the formation of unstable SEI films,16 making it difficult to achieve K+-solvent co-intercalation below −40 °C (Scheme 1A). What is particularly inspiring is that recent studies13 have demonstrated that the rational Bi@C architecture can facilitate Na+-solvent co-intercalation, thus enabling excellent Na+ storage performance at −40 °C. This highlights the potential of employing similar architectural designs is beneficial to drive K+-ether-solvent co-intercalation behavior, while enhancing the low-temperature performance of Bi-based anodes for PIBs operating below −40 °C.
 | ||
| Scheme 1 Schematic illustration of two distinct mechanisms for K+ transport from the bulk electrolyte to the anode through the SEI at low temperatures. (A) Classical K+ desolvation process at the anode-electrolyte interface. (B) Partial K+-solvent co-intercalation into the designed anode. | ||
In this study, we propose a rationally designed architecture, in which bismuth nanoparticles are uniformly embedded into a conductive carbon framework (denoted as Bi@CFs), to effectively promote partial K+-ether-solvent co-intercalation (Scheme 1B). This synergistic strategy not only mitigates the volume changes associated with K+ insertion but also reduces the desolvation energy barrier during the alloying/dealloying processes. Simultaneously, it promotes the formation of a compact bilayer SEI with high ionic conductivity, thus collectively enabling rapid K+ storage at 50 °C. Both experimental results and density functional theory (DFT) calculations demonstrate that Bi@CFs architecture facilitates the K+-solvent co-intercalation, thereby lowering reaction energy barrier and boosting ionic transport kinetics within the Bi@CFs electrode at −50 °C. As expected, the Bi@CFs electrode delivers a remarkable K+ ion storage capability across a wide low-temperature range from −20 to −50 °C, notably achieving a high reversible capacity of 345.61 mAh g−1 at 1 A g−1 over 400 cycles at −50 °C, significantly outperforming previously reported values in the literature at LTs. More impressively, when paired with an activated carbon (AC) cathode to assemble a full cell, the Bi@CFs/AC device demonstrates an outstanding low-temperature durability, maintaining an ultra-long lifespan exceeding 10000 cycles at −50 °C. This work opens a new pathway for efficient K-ion storage under ultra-low temperature conditions.
2. Results and discussion
2.1. Preparation and characterizations
As illustrated in Fig. 1A, Bi@CFs is synthesized via a facile annealing approach using commercially available bismuth subgallate hydrate (C7H5BiO6). A combined analysis of infrared spectroscopy (IR, Fig. S1A) and X-ray diffraction (XRD, Fig. S1B) patterns reveals the thermal decomposition behavior of C7H5BiO6. Specifically, the disappearance of vibrational peaks at 3500 cm−1 and 1800 cm−1 above 150 °C and 250–280 °C, respectively, indicates the removal of -OH and –COOH groups. Meanwhile, the gradual weakening of the Bi–O vibrational peak at 800 cm−1 within the 280–320 °C range suggests the breakdown of Bi–O coordination.17 Furthermore, the disappearance of the characteristic benzene ring vibrational peak (1421.52 cm−1, 1475.52 cm−1 and 1554.6 cm−1) between 350–450 °C confirms the decomposition of the organic ligand structure.17 Ultimately, the formation of Bi@CFs is observed at temperatures above 450 °C. | ||
| Fig. 1 Design concept and simulations for the stress distribution of Bi@CFs and pure Bi. (A) The possible decomposition mechanism of basic bismuth gallate. (B and C) SEM image (B) and TEM image (C) of Bi@CFs. (D and E) Schematic illustration of the potassiation and depotassiation processes of Bi@CFs (D) and pure Bi (E) electrodes, respectively. (F and G) COMSOL Multiphysics simulations showing the stress distribution in Bi@CFs (F) and pure Bi (G) electrodes at different potassiation states. | ||
The morphology and structure of the Bi@CFs composites were systematically investigated using scanning electron microscopy (SEM) and transmission electron microscopy (TEM). As shown in Fig. 1B and C, the composite exhibits a highly uniform structure, wherein nano-Bi particles (ca. 19 nm, Fig. S2) are tightly embedded within a carbon framework that presents a cross-linked architecture, effectively mitigating the stress associated with volume fluctuations during cycling and offering efficient electron transport pathways.18 High-resolution TEM (HRTEM) image further reveals that the Bi nanoparticles are uniformly encapsulated by a carbon layer with a thickness below 5 nm (Fig. S3), where well-resolved lattice fringes with an interplanar spacing of 0.326 nm are observed, corresponding to the (012) plane of metallic Bi (JCPDS No. 85-1329).12 Moreover, the successful formation of crystalline metallic Bi within the composite is corroborated by selected area electron diffraction (SAED, Fig. S4) and XRD (Fig. S5) analyses, both of which reveal characteristic diffraction features of metallic Bi. Furthermore, elemental mapping confirms the homogeneous distribution of Bi and C throughout the composite (Fig. S6), corroborating the uniform embedding of Bi within the carbon matrix. Additionally, thermogravimetric analysis (TGA, Fig. S7) further quantifies the Bi content in the composite to be 73.12%. Raman spectrum (Fig. S8) verifies the presence of a graphitic carbon coating, as evidenced by the prominent D and G bands at ∼1350 and 1584 cm−1, respectively, with an ID/IG ratio of 0.82, indicative of a defect-rich, partially disordered structure that provides abundant electroactive sites and facilitates favorable K+ ion diffusion.19
As illustrated in Fig. 1D, the Bi@CFs electrode, leveraging the synergistic integration of a cross-linked carbon framework and nano-sized Bi domains, effectively mitigates the mechanical stress arising from volume fluctuations during cycling, while the interconnected conductive network ensures sustained electron transport,20 thereby enabling superior K-ion storage. In contrast, the pure Bi electrode, which lacks the structural confinement and buffering effect provided by the carbon framework, coupled with the bulk Bi to accommodate large volume variations (Fig. 1E), inevitably suffers from severe pulverization and exhibits markedly poor electrochemical performance. Especially, finite element simulations were conducted to investigate the mechanical strain distribution in Bi@CFs (Fig. 1F) and pure Bi (Fig. 1G) anodes during the potassiation process, wherein a stark contrast in internal stress distribution is observed. Notably, the pure Bi electrode exhibits significantly higher mechanical strain than the Bi@CFs counterpart beyond 60% state of potassiation, as evidenced by a pronounced color transition from blue to red, indicating intensified internal stress accumulation. In contrast, the Bi@CFs electrode demonstrates a more uniform strain distribution throughout the potassiation process, which can be attributed to the synergistic effect of the nano-sized Bi and the cross-linked carbon framework, whose dual structural protection effectively buffers volume expansion and mitigates stress concentration.
2.2. Investigation of potassium storage mechanisms
The potassium storage mechanism of the Bi@CFs electrode during cycling was investigated using in situ XRD. As presented in Fig. 2A and Fig. S9, no obvious change in the diffraction peaks of Bi is detected within the potential range of 1.5–0.8 V, suggesting that the initial potassiation process primarily occurs at the surface of carbon frameworks. As the voltage decreases to 0.4 V, two new peaks gradually emerge at 30.8° and 32.2°, corresponding to the formation of cubic KBi2 (PDF#03-0698).21 Upon further discharge, the KBi2 peaks vanish, while three new diffraction peaks appear at 18.2°, 28.6°, and 29.3°, which are attributed to the formation of hexagonal K3Bi (PDF#04-0642).21 During the subsequent charging process, when the voltage reaches 0.69 V, the characteristic peak of K3Bi disappear, while two weak peaks emerge at 29.8° and 31.1°, associated with the intermediate phase K3Bi2. With further increase in voltage, the reappearance of distinct peaks at 30.8° and 32.2° confirms the reversible formation of KBi2, whereas the emergence of a peak at 26.8° is attributed to the reformation of metallic Bi. Beginning with the second cycle, the potassiation and depotassiation processes proceed through a well-defined three-step phase transition during discharge and charge (Fig. S10), respectively, involving the sequential transformations of Bi → KBi2 → K3Bi2 → K3Bi and K3Bi → K3Bi2 → KBi2 → Bi, which reflects a highly reversible alloying/dealloying mechanism. This potassium storage mechanism is further corroborated by ex situ HRTEM analysis (Fig. 2C–E), where distinct lattice fringes corresponding to the (311) plane of KBi2, the (103) plane of K3Bi2, and the (110) plane of K3Bi are observed at discharge voltages of 0.88 V, 0.35 V, and 0.30 V, respectively. Furthermore, ex situ FTIR measurements were performed to investigate the Bi@CFs electrodes at various potassiation states (Fig. 2B), where characteristic peaks at 557.3 cm−1, 1027.8 cm−1, 983.5 cm−1, 1101.2 cm−1, 1199.5 cm−1, 1363.4 cm−1 were observed after discharging to 0.01 V,12,13 representing the occurrence of partial K+-ether-solvent co-intercalation during the potassiation process. However, the disappearance of these peaks after charging to 1.5 V, consistent with the spectra of pristine Bi@CFs electrode, indicates that the fast potassium storage kinetics at LTs are primarily driven by the K+-ether-solvent co-intercalation mechanism. | ||
| Fig. 2 Potassium-ion storage mechanism analysis of the Bi@CFs anode in PIBs. (A) In situ XRD patterns of the Bi@CFs electrode during the initial discharge and charge processes. (B) Ex situ FTIR spectra of the Bi@CFs electrode at various discharge and charge stages. (C–E) TEM images of the Bi@CFs electrode after discharge to 0.88 V, 0.35 V and 0.30 V. (F and G) HRTEM images of Bi@CFs (F) and the pure Bi (G) electrodes after 20 cycles in 1,2-dimethoxyethane (DME)-based electrolytes. (H–K) High-resolution C 1 and F 1s XPS spectra of the Bi@CFs (H and I) and pure Bi (J and K) electrodes after cycling. | ||
Given that the desolvation step is widely regarded as a major energy barrier in the diffusion process, the emergence of this co-intercalation pathway significantly reduces the activation energy, thereby facilitating accelerated ion transport.13 Notably, this phenomenon has not been observed in carbonate-based electrolytes such as EC and DEC (Fig. S11A), nor has any evidence of such a mechanism been detected for pure Bi electrodes in ether-based electrolyte (Fig. S11B). Consequently, the engineered Bi@CFs nanostructure facilitates the partial K+-solvent co-intercalation, which synergistically mitigates volume expansion during cycling and reduces the desolvation energy barrier, thereby enabling superior potassium storage performance at LTs. In addition, the chemical composition of the SEI formed on both electrodes was systematically analyzed. As shown in Fig. 2F, the SEI formed on the Bi@CFs electrode remains uniformly distributed and relatively thin. In contrast, the SEI on the pure Bi electrode becomes markedly uneven and significantly thicker after 20 cycles (Fig. 2G). Furthermore, ex situ X-ray photoelectron spectroscopy (XPS) with Ar+ sputtering was conducted to resolve the depth-dependent composition of both SEI layers. As illustrated in the C 1s spectrum (Fig. 2H), characteristic binding energies corresponding to –C–C–, –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and O–CO moieties can be identified at 284.8 eV, 287 eV and 288.8 eV, respectively.18 Notably, signals corresponding to –C–C– and –C–O– bonds, typically originating from solvent decomposition, drops sharply beyond 5 nm sputtering depth. This rapid attenuation, followed by a more gradual decrease upon further sputtering, implies that these organic constituents are predominantly localized within a thin surface layer. Concurrently, the peak at 683.5 eV in the F 1s region (Fig. 2I), attributed to inorganic K–F species,12 becomes increasingly prominent with progressive sputtering, suggesting an accumulation of fluorinated inorganic compounds beneath the organic-rich interface. Collectively, the results indicate a bilayer SEI structure on the Bi@CFs electrode, comprising an organic-rich outer layer and an inorganic-rich inner layer. In comparison, the SEI formed on the pure Bi electrode surface exhibits a greater abundance of both organic (Fig. 2J) and inorganic (Fig. 2K) components. Furthermore, the K–F signal exhibits an initial attenuation followed by progressive intensification with sputtering depth, indicative of a mixed organic–inorganic SEI structure.
O, and O–CO moieties can be identified at 284.8 eV, 287 eV and 288.8 eV, respectively.18 Notably, signals corresponding to –C–C– and –C–O– bonds, typically originating from solvent decomposition, drops sharply beyond 5 nm sputtering depth. This rapid attenuation, followed by a more gradual decrease upon further sputtering, implies that these organic constituents are predominantly localized within a thin surface layer. Concurrently, the peak at 683.5 eV in the F 1s region (Fig. 2I), attributed to inorganic K–F species,12 becomes increasingly prominent with progressive sputtering, suggesting an accumulation of fluorinated inorganic compounds beneath the organic-rich interface. Collectively, the results indicate a bilayer SEI structure on the Bi@CFs electrode, comprising an organic-rich outer layer and an inorganic-rich inner layer. In comparison, the SEI formed on the pure Bi electrode surface exhibits a greater abundance of both organic (Fig. 2J) and inorganic (Fig. 2K) components. Furthermore, the K–F signal exhibits an initial attenuation followed by progressive intensification with sputtering depth, indicative of a mixed organic–inorganic SEI structure.
2.3. Electrochemical performance of Bi@CFs half-cells
Benefiting from the rationally engineered Bi@CFs architecture that facilitates partial K+-ether-solvent co-intercalation, the Bi@CFs half-cell demonstrates an exceptional potassium storage performance in PIBs, even at −50 °C. As demonstrated in Fig. S12, the CV curves were recorded at a scan rate of 0.2 mV s−1 within a voltage range of 0.1–1.5 V (V vs. K+/K). During the initial cathodic scan, an irreversible peak appears at approximately 0.5 V, which can be attributed to the reduction of surface oxides as well as the irreversible insertion of K+ ions into defect sites within the carbon framework.12 In addition, two small shoulder peaks at 0.31 V and 0.27 V are assigned to the alloying reactions between K+ and metallic bismuth. In subsequent cycles, three cathodic peaks, which emerge at 0.88 V, 0.35 V, and 0.30 V, are corresponding to the three–step alloy reactions involving the progressive formation of K–Bi alloys, whereas the anodic peaks at 0.51 V, 0.62 V, and 1.20 V, which are associated with the corresponding dealloying steps. What is particularly noteworthy is that these pairs of cathodic and anodic peaks, which become increasingly overlapped in the subsequent cycles, provide compelling evidence for the excellent reversibility and structural stability of the Bi@CFs anode at low temperature. Similar results were confirmed by the initial galvanostatic charge–discharge (GCD) profiles, as displayed in Fig. 3A, where all curves exhibit distinct voltage plateaus and demonstrate good consistency, except for the first cycle. | ||
| Fig. 3 Electrochemical performance of the Bi@CFs anode in PIBs at low temperatures. (A) Charge/discharge profiles at 0.5 A g−1 at −20 °C. (B) Long-term cycling performance at 1 A g−1 at −50 °C. (C) Cycle performance at 1 A g−1 from −20 °C to −50 °C. (D) Rate performance at current densities from 0.5 to 2 A g−1. (E) The comparison of electrochemical performance between the Bi@CFs anode and previously reported anodes for SIBs and PIBs at various temperatures. (F) Cycle performance of the Bi@CFs/K half-cell pouch battery at 0.1 A g−1 and −50 °C. | ||
Furthermore, the Bi@CFs anode receives a reversible capacity of 345.61 mAh g−1 at 1 A g−1 (Fig. 3B), while maintaining nearly 100% capacity retention over 400 cycles at −50 °C, which includes an initial activation process conducted for 20 cycles at −20 °C. Fig. S13 displays the charge/discharge profiles of the Bi@CFs at 1 A g−1 over several cycles, where the well-overlapped curves and distinct plateaus collectively indicate highly reversible electrochemical behavior and excellent cycling stability at −50 °C. For comparison, the pure Bi electrode exhibits an inferior electrochemical performance, evidenced by a rapid capacity decline to 253.21 mAh g−1 and pronounced overcharging behavior after 23 cycles (Fig. S14A). Meanwhile, the Bi@CFs electrode displays poor K+ storage behavior in carbonate-based electrolyte (Fig. S15). In this sense, the enhanced electrochemical performance of the Bi@CFs electrode is ascribed to the tailored Bi@CFs architecture accelerates partial K+-ether-solvent co-intercalation mechanism, which simultaneously mitigate volume fluctuation and reduce activation energy, thereby facilitating efficient K+ storage at LTs. Additionally, the rational selection of the DME-based electrolyte, which exhibits a low freezing point of −55.5 °C (ref. 12) and sufficient ionic conductivity, enables the Bi@CFs electrode to operate stably at −50 °C (Fig. S16). Moreover, the Bi@CFs anode exhibits remarkable K+ ion storage capability across a wide low-temperature range (Fig. 3C), as evidenced by the average capacities of 365.32 mAh g−1, 363.23 mAh g−1, 358 mAh g−1 and 347.77 mAh g−1 at −20 °C, −30 °C, −40 °C and −50 °C, respectively, while exhibiting minimal polarization in the GCD curves and negligible capacity degradation (Fig. S17). Upon returning to −20 °C, a high capacity of 375.86 mAh g−1 is recovered, with a capacity retention exceeding 100%, highlighting the excellent electrochemical reversibility and structural stability of the Bi@CFs anode under fluctuating low temperature. Moreover, the average rate capabilities of Bi@CFs are 363.13, 344.46, 331.22, 307.12, and 245.79 mAh g−1 at current densities of 0.5, 0.8, 1, 1.5 and 2 A g−1 (Fig. 3D), respectively, which is significantly higher than that of the pure Bi electrode (Fig. S14B). When switching back to 0.5 A g−1, a high average capacity of 367.06 mAh g−1 is recovered, and the charge/discharge curves retain similar shapes with clearly defined alloying/dealloying plateaus even at high rates (Fig. S18), thereby demonstrating the favorable reversibility and outstanding rate retention capability of the Bi@CFs anode at −50 °C. It is worth noting that the reversible capacity of the Bi@CFs anode significantly surpasses that of most previously reported anode materials for LIBs, SIBs, and PIBs at low temperature (Fig. 3E and Table S1),10,13,14,22–31 with particularly few studies demonstrating stable performance at −50 °C. More importantly, a half-cell pouch battery with mass loading of 15.87 mg was conducted at 0.1 A g−1 at −50 °C (Fig. 3F), displaying a stable K-ion storage and delivering a favorable reversible capacity of 345.69 mAh g−1 over 200 cycles without any noticeable capacity degradation, which strongly indicates the excellent electrochemical stability and practical applicability of the Bi@CFs electrode at low temperature.
2.4. Dynamic analysis and theoretical simulations
COMSOL Multiphysics simulation was carried out to evaluate the K+ concentration in the Bi@CFs and pure Bi electrodes at −50 °C during the potassiation process. As shown in Fig. 4A, the K+ concentration in the bottom region of the Bi@CFs electrode exceeds 50% of that in the top region within 30 min, whereas the same level of K+ diffusion in the pure Bi anode requires approximately 50 min, indicating an enhanced K+ diffusion kinetics in the Bi@CFs electrode.15 Moreover, the ratio of K+ concentration between the bottom and top regions in the Bi@CFs electrode reaches nearly 100% after 50 min, significantly outperforming the corresponding ratio of only ∼30% observed in the pure Bi anode (Fig. 4B). This again indicates that the Bi@CFs structure can effectively promote more uniform ion diffusion at low temperatures. Furthermore, DFT calculations were performed to simulate the K+ adsorption on the pure Bi and Bi@CFs electrodes at −50 °C, in which a graphene monolayer was employed as a simplified representation of the carbon layer to approximate the complexity of the actual carbon frameworks coating the Bi nanospheres.18 As presented in Fig. 4C and D, the migration pathways of K+ on the Bi/C and Bi surfaces for the Bi@CFs electrode are simulated from top-view perspective and side-view paths, while pathways on the Bi surface of the pure Bi are illustrated in Fig. S19. Based on the simulated migration pathways, the migration energy barriers associated with pure Bi and Bi@CFs electrodes are calculated (Fig. 4E), in which Bi@CFs electrode consistently exhibits lower energy barriers compared to the pure Bi electrode, thereby indicating more favorable K+ ions diffusion kinetics within the Bi@CFs electrode. In situ electrochemical impedance spectroscopy (EIS) measurements were applied to investigate the reaction kinetics of the Bi@CFs electrode at −50 °C, as shown in Fig. 4F and Fig. S20, where the charge transfer resistance (Rct) in the high frequency region and the Warburg impedance in the low frequency region were derived from the equivalent circuit model (inset of Fig. S20).32 What is noteworthy is that the Bi@CFs electrode delivers the Rct of less than 30 Ω during the potassiation/depotassiation processes at −50 °C, which is markedly lower than the values of the pure Bi electrode (Fig. S21) and the reported in previous studies (60–500 Ω) at low temperatures (Table S2),33–36 and this result should be responsible for the accelerated K+ ion storage kinetics at such low temperatures. | ||
| Fig. 4 Potassium ion transport behavior in Bi@CFs and pure Bi electrodes. (A and B) COMSOL Multiphysics simulations of K+ transport kinetics in Bi@CFs (A) and pure Bi (B) anodes at different potassiation states at −50 °C. (C and D) Top-view (C) and side-view (D) illustration of K+ migration pathways on Bi@CFs structures. (E) The migration energy barriers in the Bi@CFs and pure Bi. (F) Charge transfer resistances (Rct) of the Bi@CFs electrode during discharge/charge processes at −50 °C. | ||
In addition, the contact angle measurements (Fig. S22) reveal the superior wettability of DME on the Bi@CFs surface, in contrast to that of the pure Bi electrode in DME-based electrolyte and the Bi@CFs electrode in carbonate-based electrolyte, which contributes to a lower resistance and consequently improves the reaction kinetics.12,37 Moreover, galvanostatic intermittent titration technique (GITT) measurements further confirmed that the Bi@CFs electrode exhibits high K+ diffusion coefficients at −50 °C (Fig. S23), which not only facilitates rapid ion transport during the potassiation/depotassiation process, but also contributes to the excellent K-ion storage performance that is maintained at −50 °C. Meanwhile, the electrode gradually develops a uniformly porous nanostructure (Fig. S24) after prolonged cycling, whose nanoscale porosity shortens K+ diffusion pathways and mitigates structural strain and volume changes, thereby promoting excellent capacity retention and cycle stability.
2.5. Electrochemical performance of K-ion hybrid capacitors
Driven by the engineered designed Bi@CFs architecture and K+-ether-solvent co-intercalation behavior, a full-cell K-ion hybrid capacitor (PIHC), with Bi@CFs as anode and AC as the cathode, was constructed and referred to as Bi@CFs/AC. During the charging process, the PF6− ions from the electrolyte are absorbed onto the AC surface, while K+ ions are intercalated into the Bi@CFs anode (Fig. 5A).38 The discharge process occurs in the reverse order. As disclosed in Fig. 5B and Fig. S25, the mass ratio of anode to cathode has been precisely optimized to 1:2. Under this condition, the Bi@CFs/AC device, for the first time, achieves a record-high energy density (121.66 Wh kg−1) and power density (9658.28 W kg−1) at −50 °C, ranging from 0.1 A g−1 to 2 A g−1. These results clearly demonstrate the exceptional rate capability of the Bi@CFs/AC PIHC even under low-temperature conditions. Fig. 5C displays the GCD profiles of Bi@CFs/AC PIHC with the 1:2 mass ratio of anode to cathode, revealing a deviated-linear sloping fit with redox peaks, primarily due to the hybrid energy storage mechanism involving the Bi@CFs anode and the AC cathode.32 Remarkably, for the first time, the Bi@CFs/AC PIHC delivers up to 10000 cycles with the 75% capacity retention at 1 A g−1 (Fig. 5D) at −50 °C, with the GCD profiles maintaining stable electrochemical behavior without noticeable deformation. Note that the electrochemical performance of the Bi@CFs/AC device at −50 °C surpasses that of previously reported lithium-ion hybrid capacitors (LIHCs) and sodium-ion hybrid capacitors (SIHCs) at −20 °C and −40 °C, respectively (Fig. 5E).39–42 More importantly, a Bi@CFs/AC pouch cell was assembled to validate its commercial viability for large-scale applications at −50 °C (Fig. 5F). The cell featured a mass loading of 31.65 mg and an electrode area of 26.1 cm2 for both the anode and cathode (Fig. 5G), delivering a high energy density of 73.65 W kg−1 at 0.02 A g−1.
 | ||
| Fig. 5 Electrochemical performance of the Bi@CFs/AC PIHC at −50 °C. (A) Schematic illustration of the Bi@CFs/AC PIHC device. (B) Rate performance of the Bi@CFs/AC PIHC with varying anode-to-cathode mass ratios (1:1, 1:2, and 1:3). (C) GCD profiles of the Bi@CFs/AC PIHC with a 1:2 mass ratio of anode to cathode at various current densities. (D) Long-term cycling performance of the Bi@CFs/AC PIHC with a 1:2 mass ratio of anode to cathode at 1 A g−1, with GCD curves from 2500 to 2512 cycles (left), 5200 to 5212 cycles (middle) and 7900 to 7912 cycles (right) (inset of D). (E) Ragone plots in comparison with previously reported PIHCs, SIHCs and LIHCs. (F) Digital photograph of the assembled Bi@CFs/AC pouch cell. (G) Rate performance of the Bi@CFs/AC pouch cell at the current densities from 0.02 A g−1 to 0.2 A g−1 with a high mass loading of 31.65 mg at −50 °C. | ||
3. Conclusion
In conclusion, we have successfully developed a bismuth-based anode capable of high-performance K-ion storage at extreme low temperatures. The specially designed Bi@CFs architecture, with uniformly distributed bismuth nanoparticles confined in a conductive carbon matrix, facilitates partial K+-ether-solvent co-intercalation during cycling, thereby synergistically mitigating structural strain and overcoming desolvation energy barriers to enable efficient low-temperature K+ storage at −50 °C. DFT calculations confirm the favorable reaction free energy of the Bi@CFs electrode throughout the entire potassiation process, facilitating rapid K+ transport at −50 °C. Moreover, COMSOL simulations further demonstrate that the engineered architecture not only improves mechanical robustness under repeated cycling but also ensures long-term structural integrity. In combination with an ether-based electrolyte, this design promotes the formation of a dense bilayer SEI, composed of an inorganic-rich inner layer and an organic-rich outer layer, thereby ensuring a high ionic conductivity and stable interfaces. Benefiting from the combined advantages of its robust structure and K+-ether-solvent co-intercalation mechanism, the Bi@CFs anode exhibits an outstanding electrochemical performance at −50 °C, delivering a high reversible capacity of 345.61 mAh g−1 at 1 A g−1 over 400 cycles. More impressively, the as-derived Bi@CFs/AC hybrid capacitor delivers a record-high energy density of 121.66 Wh kg−1 and power density of 9658.28 W kg−1 at −50 °C, while maintaining exceptional durability over 10000 cycles. Our work marks the beginning of high-capacity and high-rate K-ion storage under ultra-low temperature conditions.
4. Experimental
4.1. Material synthesis
Commercial bismuth powder and bismuth subgallate hydrate powder were purchased from Shanghai Maclin Co., Ltd, and were used as received without further purification. Initially, the bismuth subgallate hydrate powder was manually ground for 30 min to ensure uniformity, followed by sintering in a tube furnace at 750 °C for 2 h under an argon atmosphere with a heating rate of 3 °C min−1. After natural cooling, the obtained product was collected and denoted as bismuth carbon frameworks (Bi@CFs). For comparison, commercial bismuth powder was referred to as Bi powder.4.2. Materials characterization
The morphological and structural characteristics of both samples were examined using field-emission scanning electron microscopy (FESEM, Hitachi-SU8010, Zeiss Merlin Gemini) operated at an accelerating voltage of 5–15 kV. TEM analyses, including SAED and energy-dispersive X-ray spectroscopy (EDX), were conducted on an FEI Tecnai F20 microscope. For the ex situ HRTEM analyses, cycled electrodes were taken from coin cells at designated discharge voltages (0.88, 0.35, and 0.30 V) inside an Ar-filled glovebox (O2/H2O < 0.1 ppm). The electrodes were rinsed with pure DME to remove residual salts, after which the active material was scraped off, ultrasonically dispersed in DME, and dropped onto a lacey carbon TEM grid. FTIR spectra were acquired using a Bruker Vertex 70 V spectrometer. TGA was performed using a thermogravimetric analyzer (STA449F5), and the content of Bi in the Bi@CFs can be calculated by the eqn (1): | (1) |
The elemental composition and chemical states were characterized by XPS (Thermo Fisher Scientific, ESCALAB 250Xi+). Phase identification was conducted using XRD (D8 Advance, Bruker) with Cu-Kα radiation, while Raman spectra were recorded using a Raman spectrometer equipped with a 532 nm laser source. For in situ XRD measurement, slurries containing Bi@CFs powder, super-P and polyvinylidene fluoride (PVDF) binder in a mass ratio of 8:1:1 were homogeneously dispersed in N-methyl-2-pyrrolidone (NMP) and subsequently cast onto Cu meshes (200 mesh), followed by drying at 100 °C under vacuum for 12 h. In situ XRD measurements were carried out on a Bruker D8 Advance diffractometer (Cu Kα), equipped with a custom-designed in situ chamber (Beijing Scistar Technology Co., Ltd) featuring a Be window for X-ray transmission. Data were collected over a 2θ range of 15–45° with a step size of 0.02°, during galvanostatic charge–discharge cycling at a current density of 0.3 A g−1 within a voltage window of 0.1–1.5 V vs. K/K+. The contact angle of electrode was measured by a JC-2000C1 tester.
4.3. Battery assembly and electrochemistry
To prepare the working electrodes, a homogeneous slurry was obtained by mixing the active material, Super-P, and PVDF binder in a weight ratio of 8:1:1 using NMP as the solvent, which was then uniformly coated onto Cu foil and dried at 120 °C under vacuum for 12 h. The mass loading of active materials was approximately 1 mg cm−2 with a diameter of 11 mm and a thickness of 100 μm. The cathodes were prepared by mixing AC, PVDF, and Super-P with a mass ratio of 8:1:1 using NMP as the solvent, which was coated onto a carbon-coated Al foil and vacuum-dried at 100 °C for 12 h. The mass loading of AC active material was approximately 2 mg cm−2 with a diameter of 11 mm and a thickness of 23 μm. And the pouch cell was constructed with electrodes sized at 45 mm × 58 mm (geometric area of 26.1 cm2). All batteries were assembled with coin-type CR2032 configurations, in which K metal foil severed as the counter electrode, Whatman GF/D glass fiber as the separator, and 1 M KPF6 in DME as the electrolyte, within a glovebox where O2 and H2O levels were maintained below 0.1 ppm. Prior to assembling PIHCs, the Bi@CFs anode was pre-activated for five cycles at a current density of 0.5 A g−1 in a half-cell, then held at the discharged state. The anode was then disassembled under an argon-filled glovebox atmosphere. The PIHC device was constructed using Bi@CFs as the anode and AC as the cathode with a mass ratio of 1:2. Galvanostatic charge/discharge profiles were recorded at various current densities with a voltage range of 0.1–1.5 V for half-cells and 0.3–3.8 V for PIHCs. The energy density (E, Wh kg−1) and power density (P, W kg−1) of PIHCs were determined through numerical integration of the galvanostatic discharge curves, using eqn (2) and (3):43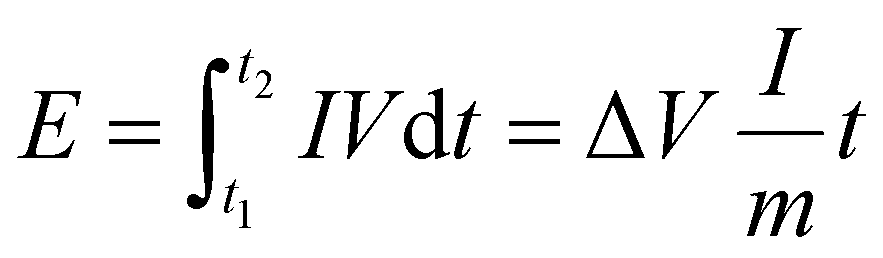 | (2) |
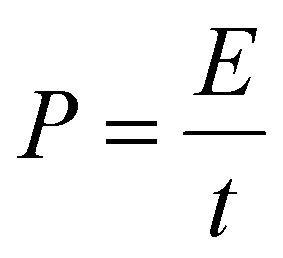 | (3) |
GITT measurement was employed with a pulse current of 50 mA g−1 for 10 min, followed by a 40 min relaxation period within the voltage range of 0.1–1.5 V. Cyclic voltammetry measurements were conducted using an electrochemical workstation (CHI700e, Shanghai) at varying scan rates. EIS was performed using an IVIUM-VERTEX.C workstation (Netherlands) with an amplitude of 10 mV over a frequency range of 0.01 Hz to 100 kHz.
4.4. Stress distribution simulation
The finite element simulations were performed using COMSOL Multiphysics® version 6.3 to investigate the volume expansion behavior of pure Bi and Bi@CFs models during potassium storage. The model employed a direct multiphysics coupling framework to solve the relevant partial differential equations, incorporating both thermal expansion and ion-intercalation-induced volumetric changes.15,39 The solid mechanics module and heat transfer in solids module were used to capture the mechanical stress distribution and temperature effects, respectively. All simulations were conducted under the assumption of material isotropy.Specifically, a simplified case study was constructed with nominal parameters for demonstration purposes. The initial volume of pure bismuth was set to 1.000 cm3. The linear thermal expansion coefficient of bismuth was assumed to be α (room temperature), noting that this value may vary significantly at sub-zero temperatures such as −50 °C. The temperature rise (ΔT) induced by the exothermic potassium storage reaction was introduced as a user-defined parameter, which could be estimated based on the reaction enthalpy and thermal conditions. The volumetric expansion due to temperature variation was calculated using the relation:44
| ΔVT = 3αV0ΔT | (4) |
| Vfinal = V0 (1 + 3αΔT)(1 + β) | (5) |
Based on this simplified analysis, the final volume was estimated to be approximately 1.200486 cm3, indicating a significant expansion relative to the initial state. This result provides guidance for defining boundary conditions and inputting deformation-dependent material properties in subsequent structural mechanics simulations. It is emphasized that, in practical modelling, experimentally validated parameters and precise thermodynamic data should be incorporated to ensure simulation fidelity.
4.5. Simulation of K-ion transport in a rectangular channel
To investigate K-ion transport behavior under electro-diffusive control, a two-dimensional rectangular channel (length 0.1 m, width 0.02 m) filled with stationary electrolyte was constructed using COMSOL Multiphysics® 6.3.45 The simulation considered only diffusion and migration under an applied electric field, while convection was neglected. Potassium ions (z = +1) were defined as the mobile species, with a diffusion coefficient of D = 1.96 × 10−9 m2 s−1. The electrolyte conductivity was set to σ = 0.1 S m−1, and the temperature was maintained at 298 K.Two coupled physics interfaces – “Transport of Diluted Species” and “Electrostatics” – were used. The Nernst–Planck equation governed ion transport, and electrostatic potential was applied along the x-direction to induce ionic migration. Boundary conditions were defined as follows: a fixed potassium ion concentration of 0.1 mol m−3 at the inlet (x = 0), zero concentration gradient at the outlet (x = 0.1 m), and no-flux conditions at the upper and lower channel boundaries (y = 0 and 0.02 m).46 Electric potential was specified at the inlet and outlet to form a uniform electric field along the flow direction.
A free triangular mesh was applied, with refined boundary elements and a maximum element size of 0.005 m. The steady-state solver was used to compute the results. Postprocessing included contour and vector plots of concentration and flux distributions, as well as line profiles extracted along the channel centerline to evaluate the spatial evolution of potassium ion concentration under the combined influence of diffusion and migration.
4.6. DFT simulation
All first-principal calculations were carried out using Density Functional Theory (DFT) as implemented in the Vienna Ab initio Simulation Package.47–49 The calculations utilized projector augmented wave pseudo potentials combined with the Perdew–Burke–Enrnzerhof exchange–correlation functional. The Brillouin zone was sampled at the gamma point using a 3 × 3 × 1 Monkhorst–Pack k-point grid. The energy cut-off was set to 400 eV, with convergence criteria for total energy at 10−5 eV and force at 0.02 eV per Å per atom for structural optimization and self-consistent calculations. The Gibbs free energy was calculated as ΔG = ΔE + ΔZPE − TΔS, where ΔE represents the electron energy; ΔZPE, the zero-point correction energy; T, the reaction temperature; and ΔS, the entropy change, determined via standard vibrational correction for entropy.Author contributions
J. G., J. L. and L. H. contributed equally to this work. L. Z. and M. L. designed and supervised this work. J. G., J. L. and L. H. carried out the synthesis and the electrochemical experiments. J. G. and L. H. participate in DFT and COMSOL calculations. J. G, J. L. and M. L. performed in XRD, Raman, IR, XPS, FESEM characterizations. Z. S. carried out TEM measurement. X. C. performed in contact angle characterization. M. L., and J. G. wrote the manuscript. L. Z., Q. Z. appraised and revised the paper. All the authors participated in the analysis of experimental data and the discussion of results, as well as the preparation of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Data availability
The data that support the findings of this study are available on request from the corresponding authors, upon reasonable request.The Supplementary Information (SI) includes cycling performance, rate performance, capacity-voltage curves for PIBs, EIS, GITT, and material characterization data (SEM, XRD, in-situ XRD, TEM, Raman, TGA, FTIR, viscosity and ionic conductivity, and contact angle measurements). The Supplementary Information is available at DOI: https://doi.org/10.1039/d5eb00151j.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 92372101, 52122211, 92472203, 92472104), the Fundamental Research Funds for the Central Universities (20720220010), the National Key Research and Development Program of China (2021YFA1201502). M. Liu acknowledges the support of China Postdoctoral Science Foundation (2024M751764). L. Zhang and Q. B. Zhang acknowledge the support of Nanqiang Young Top-notch Talent Fellowship in Xiamen University. J. W. Gu, J. L. Lin and L. H. contribute equally to this work.References
- A. Mahmood, Z. Bai, T. Wang, Y. Lei, S. Wang, B. Sun, H. Khan, K. Khan, K. Sun and G. Wang, Chem. Soc. Rev., 2025, 54, 2369–2435 RSC.
- S. Yuan, S. Cao, X. Chen, J. Wei, Z. Lv, H. Xia, L. Chen, R. B. F. Ng, F. L. Tan, H. Li, X. J. Loh, S. Li, X. Feng and X. Chen, J. Am. Chem. Soc., 2025, 147, 4089–4099 CrossRef PubMed.
- J. Chen, H. Lan, S. Wang, X. Liu, Q. Zhu, X. Zhang, M. Tang, S. Dong, J. Yang, M. Kurbanov, L. Guo and H. Wang, J. Am. Chem. Soc., 2025, 147, 2393–2402 CrossRef.
- Y. Niu, J. Yang, F. Meng, Z. Sun, C. Jiang, Y. Liu, H. Xu, M. Wang, H. Yang, Y. Zhu, G. Wu and W. Chen, Angew. Chem., Int. Ed., 2025, 64, e202416720 Search PubMed.
- S. Dhir, S. Wheeler, I. Capone and M. Pasta, Chem, 2020, 6, 2442–2460 Search PubMed.
- H. Wang, D. Zhai and F. Kang, Energy Environ. Sci., 2020, 13, 4583–4608 RSC.
- K. Lei, Z. Zhu, Z. Yin, P. Yan, F. Li and J. Chen, Chem, 2019, 5, 3220–3231 Search PubMed.
- L. Fan, H. Xie, Y. Hu, Z. Caixiang, A. M. Rao, J. Zhou and B. Lu, Energy Environ. Sci., 2023, 16, 305–315 RSC.
- L. Que, J. Wu, Z. Lan, Y. Xie, F. Yu, Z. Wang, J. Meng and X. Zhang, Adv. Mater., 2023, 35, 2307592 Search PubMed.
- J. Chen, D. An, S. Wang, H. Wang, Y. Wang, Q. Zhu, D. Yu, M. Tang, L. Guo and H. Wang, Angew. Chem., Int. Ed., 2023, 62, e202307122 CrossRef PubMed.
- M. Okoshi, Y. Yamada, S. Komaba, A. Yamada and H. Nakai, J. Electrochem. Soc., 2016, 164, A54–A60 Search PubMed.
- G. Chen, B. Feng, G. Xu, Q. Gong, L. Yan, C. Zhou, J. Jiang, L. Yang, Q. Wu, X. Wang and Z. Hu, ACS Energy Lett., 2025, 10, 1821–1828 CrossRef.
- L. Liu, S. Li, L. Hu, X. Liang, W. Yang, X. Yang, K. Hu, C. Hou, Y. Han and S. Chou, Carbon Energy, 2024, 6, e531 Search PubMed.
- L. Cheng, H. Lan, Y. Gao, S. Dong, Y. Wang, M. Tang, X. Sun, W. Huang and H. Wang, Angew. Chem., Int. Ed., 2024, 63, e202315624 Search PubMed.
- X. Liu, Z. Sun, Y. Sun, H. Lin, Z. Chen, X. Chen, L. Niu, Q. Zhang and H. Li, Adv. Funct. Mater., 2023, 33, 2307205 Search PubMed.
- W.-C. Lin, Y.-C. Yang and H.-Y. Tuan, Energy Storage Mater., 2022, 51, 38–53 Search PubMed.
- P. Xiong, P. Bai, A. Li, B. Li, M. Cheng, Y. Chen, S. Huang, Q. Jiang, X. H. Bu and Y. Xu, Adv. Mater., 2019, 31, 1904771 CrossRef.
- Y. Liang, N. Song, Z. Zhang, W. Chen, J. Feng, B. Xi and S. Xiong, Adv. Mater., 2022, 34, 2202673 Search PubMed.
- Z. Sun, Y. Liu, W. Ye, J. Zhang, Y. Wang, Y. Lin, L. Hou, M. S. Wang and C. Yuan, Angew. Chem., Int. Ed., 2021, 60, 7180–7187 CrossRef PubMed.
- C. Wang, L. Wang, F. Li, F. Cheng and J. Chen, Adv. Mater., 2017, 29, 1702212 Search PubMed.
- Y. Feng, Y. Lv, H. Fu, M. Parekh, A. M. Rao, H. Wang, X. Tai, X. Yi, Y. Lin, J. Zhou and B. Lu, Natl. Sci. Rev., 2023, 10, nwad118 CrossRef.
- D. Wang, J. Zhang, X. Li, L. Liu, M. Yuan, B. Cao, A. Li, X. Chen, R. Yang and H. Song, Chem. Eng. J., 2022, 429, 132272 CrossRef.
- K. Lee, M. J. Lee, J. Lim, K. Ryu, M. Li, S. Noda, S. J. Kwon and S. W. Lee, Adv. Funct. Mater., 2022, 33, 2209775 CrossRef.
- D.-R. Deng, X.-Y. Cui, Q.-H. Wu, M.-S. Zheng and Q.-F. Dong, J. Alloys Compd., 2020, 835, 155413 CrossRef.
- Q. S. Lai, J. J. Mu, Z. M. Liu, L. K. Zhao, X. W. Gao, D. R. Yang, H. Chen and W. B. Luo, Batteries Supercaps, 2023, 6, e202200549 CrossRef.
- Q. Li, K. Jiang, X. Li, Y. Qiao, X. Zhang, P. He, S. Guo and H. Zhou, Adv. Energy Mater., 2018, 8, 1801162 CrossRef.
- L. Wang, B. Wang, G. Liu, T. Liu, T. Gao and D. Wang, RSC Adv., 2016, 6, 70277–70283 Search PubMed.
- X. Zhang, M. Zeng, Y. She, X. Lin, D. Yang, Y. Qin and X. Rui, J. Power Sources, 2020, 477, 228735 CrossRef CAS.
- B. Wang, H. Yang, Y. Feng, S. Zeng, L. Tan, R. Xu, L. Wang, R. Hu and Y. Yu, Mater. Today Energy, 2021, 20, 100627 Search PubMed.
- Y. Tian, J. Lu, H. Tang, X. Wang, L. Zhang, P. Hu, L. Zhou, Y. Wang, Y. Guo, R. Khatoon, Q. Zhang, Q. He, Y. He, M. Qiu, Y. Hou and Z. Ye, Chem. Eng. J., 2021, 422, 130054 CrossRef CAS.
- J. Chen, Y. Peng, Y. Yin, Z. Fang, Y. Cao, Y. Wang, X. Dong and Y. Xia, Angew. Chem., Int. Ed., 2021, 60, 23858–23862 CrossRef CAS PubMed.
- M. Liu, Y. Xing, J. Wang, D. Wang, L. Huang, X. Wu, Z. Liu and Y. Wu, Adv. Funct. Mater., 2021, 31, 2101868 CrossRef CAS.
- Y. Yu, M. I. Levine, Z. Yang, S. Jeon, E. A. Stach and J. Xie, ACS Appl. Energy Mater., 2023, 6, 12371–12378 Search PubMed.
- Z. Pu, H. Li, Z. Yang, Y. Zhang, Y. Liu, G. Dong and Y. Li, Mater. Today Chem., 2022, 26, 101145 Search PubMed.
- B. Lu, J.-X. Song, D.-R. Deng, G.-F. Li, Y. Zeng, S.-L. Cai, J.-C. Weng, X.-H. Fan, Y. Li and Q.-H. Wu, J. Energy Storage, 2024, 102, 114056 CrossRef.
- D.-J. Yoo, Q. Liu, O. Cohen, M. Kim, K. A. Persson and Z. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 11910–11918 Search PubMed.
- L.-K. Zhao, X.-W. Gao, Q. Gu, X. Ge, Z. Ding, Z. Liu and W.-B. Luo, eScience, 2024, 4, 100201 CrossRef.
- L. Yu, X. He, B. Peng, W. Wang, G. Wan, X. Ma, S. Zeng and G. Zhang, Matter, 2023, 6, 1604–1621 CrossRef CAS.
- J. Ruan, S. Luo, S. Wang, J. Hu, F. Fang, F. Wang, M. Chen, S. Zheng, D. Sun and Y. Song, Adv. Energy Mater., 2023, 13, 2301509 CrossRef CAS.
- J. Zhang, J. Wang, Z. Shi and Z. Xu, J. Electroanal. Chem., 2018, 817, 195–201 Search PubMed.
- M. L. Divya, S. Natarajan, Y.-S. Lee and V. Aravindan, J. Mater. Chem. A, 2020, 8, 4950–4959 Search PubMed.
- M. Li, C. Li, C. Zuo, J. Hu, C. Li, W. Luo, S. Luo, A. Duan, J. Wang, X. Wang, W. Sun and L. Mai, Adv. Mater., 2024, 36, 2407233 Search PubMed.
- D. Sui, M. Wu, Y. Liu, Y. Yang, H. Zhang, Y. Ma, L. Zhang and Y. Chen, Nanotechnology, 2021, 32, 015403 CrossRef CAS PubMed.
- M. Hadipeykani, F. Aghadavoudi and D. Toghraie, Physica A, 2020, 546, 123995 Search PubMed.
- D. Wang, H. Peng, S. Zhang, H. Liu, N. Wang and J. Yang, Angew. Chem., Int. Ed., 2023, 62, e202315834 Search PubMed.
- G. Cheng, W. Zhang, W. Wang, H. Wang, Y. Wang, J. Shi, J. Chen, S. Liu, M. Huang and D. Mitlin, Carbon Energy, 2022, 4, 986–1001 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 Search PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2026 |