
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Atmospheric fates and global warming potential of HFO-1234ze(E) and its degradation product trifluoroacetaldehyde (CF3CHO)
Beth Killen *a,
Jenny A. Fisher*bc,
Christopher S. Hansena,
Paul B. Krummeld,
Martin K. Vollmere and
Scott H. Kablea
*a,
Jenny A. Fisher*bc,
Christopher S. Hansena,
Paul B. Krummeld,
Martin K. Vollmere and
Scott H. Kablea
aSchool of Chemistry, UNSW Sydney, NSW 2052, Australia. E-mail: b.killen@unsw.edu.au
bCollege of Science and Engineering, James Cook University, Douglas, QLD 4811, Australia. E-mail: jenny.fisher@jcu.edu.au
cEnvironmental Futures, University of Wollongong, Wollongong, NSW 2522, Australia
dCSIRO Environment, Aspendale, VIC 3195, Australia
eLaboratory for Air Pollution/Environmental Technology, Empa, Swiss Federal Laboratories for Materials Science and Technology, 8600 Dübendorf, Switzerland
First published on 21st May 2026
Abstract
Hydrofluoroolefins (HFOs) are replacing high-GWP hydrofluorocarbons (HFCs) across multiple applications including foam blowing, refrigeration, and aerosols, but their atmospheric degradation and climate consequences remain uncertain. We use the GEOS-Chem 3-D chemical transport model, supported by AtChem2 box-model simulations, to develop a complete representation of the atmospheric chemistry and fate of HFO-1234ze(E) and its key intermediate product, trifluoroacetaldehyde (CF3CHO). We focus on HFO-1234ze(E) as it is the dominant isomer in commercial use. The model includes newly measured CF3CHO photolysis quantum yields to form fluoroform (HFC-23), the recently identified chemical pathways of HFO-1234ze(E) ozonolysis and CF3CHO reversible reaction with HO2, and explicit wet and dry deposition parameterisations. Using observationally constrained global HFO-1234ze(E) emissions of 15 Gg year−1, simulated HFO-1234ze(E) surface mixing ratios agree well with 2020–2024 observations at 8 Advanced Global Atmospheric Gases Experiment (AGAGE) network sites. We find that 99.6% of HFO-1234ze(E) is removed by reaction with OH, with the remaining 0.4% lost to ozonolysis. Sensitivity tests for effective Henry's law constants  spanning 10–106 M atm−1 show sensitivity of CF3CHO fate to
spanning 10–106 M atm−1 show sensitivity of CF3CHO fate to  up to 104 M atm−1 and saturation at higher
up to 104 M atm−1 and saturation at higher  . Using an upper bound of 105 M atm−1, deposition accounts for ≈51% of total CF3CHO loss in GEOS-Chem (20% dry, 31% wet), with photolysis contributing ≈33% and OH reaction ≈15%. The reversible reaction with HO2 contributes around 1% to net CF3CHO loss due to rapid conversion of the reaction products back to reactants. We calculate a total (direct + indirect) GWP100 for HFO-1234ze(E) of 11.4+3.1−1.9, with CF3CHO photolysis to HFC-23 contributing 8.2+3.1−1.9. We also estimate a maximum potential formation of 4.5 Gg year−1 of trifluoroacetic acid (TFA) under current emissions assuming complete conversion of wet-deposited CF3CHO from HFO-1234ze(E), suggesting a potential unrecognised TFA source from all CF3CHO sources.
. Using an upper bound of 105 M atm−1, deposition accounts for ≈51% of total CF3CHO loss in GEOS-Chem (20% dry, 31% wet), with photolysis contributing ≈33% and OH reaction ≈15%. The reversible reaction with HO2 contributes around 1% to net CF3CHO loss due to rapid conversion of the reaction products back to reactants. We calculate a total (direct + indirect) GWP100 for HFO-1234ze(E) of 11.4+3.1−1.9, with CF3CHO photolysis to HFC-23 contributing 8.2+3.1−1.9. We also estimate a maximum potential formation of 4.5 Gg year−1 of trifluoroacetic acid (TFA) under current emissions assuming complete conversion of wet-deposited CF3CHO from HFO-1234ze(E), suggesting a potential unrecognised TFA source from all CF3CHO sources.
Environmental significanceHydrofluoroolefins (HFOs) are replacing climate-warming hydrofluorocarbons (HFCs), but their environmental impacts remain uncertain. Using 3-D atmospheric modelling incorporating recent experimental findings, we provide a comprehensive assessment of global HFO-1234ze(E) degradation, including photochemical and deposition processes. Whilst HFO-1234ze(E) photochemistry produces the potent greenhouse gas HFC-23, the combined direct and indirect global warming potential of HFO-1234ze(E) (∼11) remains far below regulatory thresholds and two orders of magnitude lower than the HFCs it replaces. We identify wet deposition of the intermediate trifluoroacetaldehyde (CF3CHO) as a potentially significant source of trifluoroacetic acid (TFA), a persistent environmental contaminant. These findings confirm that HFO-1234ze(E) offers substantial climate benefits over traditional refrigerants whilst highlighting the need for TFA monitoring as HFO use increases. |
1 Introduction
Fluorine-containing compounds have been a part of modern life for almost a century. Over recent decades, however, they have received significant attention in scientific and popular media for their environmental persistence and unintended consequences. Industrial fluorine-containing gases have been used for a multitude of purposes, including air conditioning, heat pumps, refrigeration, foam-blowing agents and insulation materials. In 1987, the Montreal Protocol initiated the phase-out of first- and second-generation refrigerants (chlorofluorocarbons, CFCs, and hydrochlorofluorocarbons, HCFCs)1 after these substances were found to significantly deplete the ozone layer, increasing exposure to ultraviolet radiation at the Earth's surface. In 2016, the Kigali Amendment to the Montreal Protocol stipulated the phase out of the third-generation refrigerants (hydrofluorocarbons, HFCs),2 which do not deplete the ozone layer but are potent greenhouse gases with high Global Warming Potential (GWP).The search for alternatives to high-GWP gases led to the development of hydrofluoroolefins (HFOs). HFOs are similar to HFCs, except that they possess a carbon–carbon double bond, making them prone to oxidation by hydroxyl (OH) radicals in the troposphere. This reactivity results in shorter atmospheric lifetimes and is the reason HFOs are generally considered to have low GWP, making them appealing as climate-friendly refrigerants.3 However, the environmental impact of HFOs depends on the fate of their atmospheric degradation products. Several HFOs, including HFO-1234yf, HFO-1225ye(Z) and HFO-1225ye(E) degrade to trifluoroacetic acid (TFA), a persistent compound in aquatic environments.4 HFO-1234ze(E), HFO-1336mzz and HCFO-1233zd degrade to trifluoroacetaldehyde (CF3CHO).5 CF3CHO is of particular concern because it can photolyse to form fluoroform (CHF3, widely known as HFC-23), a potent greenhouse gas.
We focus in this work on HFO-1234ze(E) (1,3,3,3-tetrafluoropropene), a widely used HFC replacement compound that produces CF3CHO via its dominant atmospheric removal pathway, reaction with OH,6 and also directly produces HFC-23 via ozonolysis.7 HFO-1234ze(E) is already in widespread use, with its use set to increase as HFCs are rapidly phased down.8 HFO-1234ze(E) was originally developed for foam blowing applications (particularly in extruded polystyrene and polyurethane foams) but is also used in refrigeration, air conditioning (including chillers and heat pumps), and as an aerosol propellant.
A summary of the currently understood atmospheric chemistry of HFO-1234ze(E) is shown in Fig. 1.
 | ||
| Fig. 1 Proposed atmospheric degradation pathway of HFO-1234ze(E) and CF3CHO to form HFC-23. The Criegee intermediate from ozonolysis reaction (R2) decomposes to form HFC-23 with a yield of 7.9%, along with CO2 and CHFO.20 HFC-23 is also produced via minor channel photolysis of CF3CHO, with yield as discussed in the text. | ||
Reaction with the OH radical leads to the formation of CF3CHO:
| HFO-1234ze(E) + OH → CF3CHO + CHFO | (R1) |
| HFO-1234ze(E) + O3 → HFC-23 + CHFO + CO2 | (R2) |
The end-products of (R2) are CO2 and HFC-23, plus CHFO, which decomposes to HF + CO. While CO2 and HFC-23 are greenhouse gases, HF and CO are not persistent in the atmosphere. (R1) also forms CHFO, along with CF3CHO that undergoes its own series of atmospheric reactions, including reaction with OH and photolysis:
| CF3CHO + OH → CF3CO + H2O | (R3) |
| CF3CHO + hν → CF3 + HCO | (R4) |
| CF3CHO + hν → HFC-23 + CO | (R5) |
The CF3 radical produced from (R4) reacts rapidly with O2 under atmospheric conditions to form CF3O2, which undergoes further oxidation to produce COF2 (carbonyl fluoride) and ultimately CO2 and HF.9–11 The CF3CO radical produced from (R3) either decomposes to give CF3 and CO2, or reacts with O2 to form the CF3CO2 peroxy radical, which subsequently decomposes to yield CF3 and CO2.12 Neither pathway produces HFC-23.
Recently, a computational chemistry study13 proposed that CF3CHO also reacts facilely with the HO2 radical in a reversible reaction:
| CF3CHO + HO2 ⇌ CF3CH(OH)OO | (R6) |
However, that study did not evaluate whether the reverse reaction limits the atmospheric significance of this pathway. If (R6) represents a significant CF3CHO sink, it would reduce photolytic production of HFC-23 via (R5) and affect the total climate impact of HFO-1234ze(E).
HFC-23 is a very strong greenhouse gas with a 100-year GWP100 = 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600,14 and therefore the production of HFC-23 from (R2) and (R5) has significant implications for assessing the climate impact of HFO-1234ze(E). The rate coefficient for (R2) was published recently by McGillen et al.,7 but historical values for the quantum yield of (R5) have varied by orders of magnitude. The earliest results by Dodd et al. used 313 nm radiation and reported quantum yields of ϕ5 = 2.1%.15 Subsequent studies by Pearce et al. found no evidence of ϕ5 at 313 nm.16 More recent results from Sulbaek Andersen and Nielsen17 also did not detect formation of HFC-23 across pressures ranging from 100–700 torr. Two recent papers provide consistent results for wavelength- and pressure-dependent quantum yields for (R4) and (R5). In 2024, Thomson et al.18 reported quantum yields for (R4) and (R5) at 308 nm for pressures from 75 to 750 torr, including ϕ5 (750 torr) = 0.023%. Shortly thereafter, Van Hoomissen et al.19 reported pressure-dependent quantum yields for ϕ4 and ϕ5 at 248, 266, 281 and 308 nm at 100 and 650 torr, including ϕ5 = 0.0302% at 308 nm and 650 torr. The pressure dependence of ϕ5 means that HFC-23 yields will vary with altitude, requiring a 3-D model to quantify the atmospheric implications.
600,14 and therefore the production of HFC-23 from (R2) and (R5) has significant implications for assessing the climate impact of HFO-1234ze(E). The rate coefficient for (R2) was published recently by McGillen et al.,7 but historical values for the quantum yield of (R5) have varied by orders of magnitude. The earliest results by Dodd et al. used 313 nm radiation and reported quantum yields of ϕ5 = 2.1%.15 Subsequent studies by Pearce et al. found no evidence of ϕ5 at 313 nm.16 More recent results from Sulbaek Andersen and Nielsen17 also did not detect formation of HFC-23 across pressures ranging from 100–700 torr. Two recent papers provide consistent results for wavelength- and pressure-dependent quantum yields for (R4) and (R5). In 2024, Thomson et al.18 reported quantum yields for (R4) and (R5) at 308 nm for pressures from 75 to 750 torr, including ϕ5 (750 torr) = 0.023%. Shortly thereafter, Van Hoomissen et al.19 reported pressure-dependent quantum yields for ϕ4 and ϕ5 at 248, 266, 281 and 308 nm at 100 and 650 torr, including ϕ5 = 0.0302% at 308 nm and 650 torr. The pressure dependence of ϕ5 means that HFC-23 yields will vary with altitude, requiring a 3-D model to quantify the atmospheric implications.
In addition to chemical loss mechanisms, summarised as (R1)–(R6), atmospheric species can be physically removed via wet and dry deposition. Wet and dry deposition can be important sinks for water-soluble species, with the efficiency of uptake into cloud droplets and precipitation governed by the effective Henry's Law constant,  . HFO-1234ze(E) has a low water solubility of 0.373 g L−1 at 20 °C, and thus is not considered to undergo deposition, consistent with treatment in previous modelling studies.21–23 Deposition of the CF3CHO intermediate, however, is the subject of ongoing debate.
. HFO-1234ze(E) has a low water solubility of 0.373 g L−1 at 20 °C, and thus is not considered to undergo deposition, consistent with treatment in previous modelling studies.21–23 Deposition of the CF3CHO intermediate, however, is the subject of ongoing debate.
The  for CF3CHO has not been reported. Pérez-Peña et al.5 were the first to incorporate its deposition in model simulations, parameterising combined wet and dry depositional losses through a single loss constant. Their simulations revealed that even with conservative solubility assumptions, deposition could reduce the CF3CHO atmospheric lifetime by 20–40%, thus altering its distribution between atmospheric and surface reservoirs. Their analysis suggested that deposition can play a significant role in CF3CHO loss, although the magnitude of this effect depends on
for CF3CHO has not been reported. Pérez-Peña et al.5 were the first to incorporate its deposition in model simulations, parameterising combined wet and dry depositional losses through a single loss constant. Their simulations revealed that even with conservative solubility assumptions, deposition could reduce the CF3CHO atmospheric lifetime by 20–40%, thus altering its distribution between atmospheric and surface reservoirs. Their analysis suggested that deposition can play a significant role in CF3CHO loss, although the magnitude of this effect depends on  assumptions. The fate of CF3CHO is particularly important because deposition represents a pathway to TFA formation (via hydrolysis of deposited CF3CHO) while simultaneously reducing photolytic HFC-23 production via (R5). Nielsen et al.24 argued that the values of
assumptions. The fate of CF3CHO is particularly important because deposition represents a pathway to TFA formation (via hydrolysis of deposited CF3CHO) while simultaneously reducing photolytic HFC-23 production via (R5). Nielsen et al.24 argued that the values of  chosen by Pérez-Peña et al. were too small and that if the solubility of CF3CHO mirrored its chlorinated analogue, deposition would become the dominant mechanism controlling CF3CHO's atmospheric fate. Using a lifetime-based estimate, Pérez-Peña et al.25 showed that CF3CHO loss to deposition was only moderately sensitive to
chosen by Pérez-Peña et al. were too small and that if the solubility of CF3CHO mirrored its chlorinated analogue, deposition would become the dominant mechanism controlling CF3CHO's atmospheric fate. Using a lifetime-based estimate, Pérez-Peña et al.25 showed that CF3CHO loss to deposition was only moderately sensitive to  , changing by a factor of 2.5 in response to a three-order-of-magnitude increase in
, changing by a factor of 2.5 in response to a three-order-of-magnitude increase in  . Both authors highlighted the need to explore the atmospheric implications of a higher
. Both authors highlighted the need to explore the atmospheric implications of a higher  as essential for understanding the atmospheric fate of CF3CHO.
as essential for understanding the atmospheric fate of CF3CHO.
Modelling of the atmospheric fate of HFO-1234ze(E) has not kept pace with the rapid evolution of new chemistry concerning HFO-1234ze(E), including accurate quantum yields for (R4) and (R5), ozonolysis rate coefficient for (R2), and the recent identification of reversible reaction of CF3CHO with HO2 (R6). This has left a number of unanswered questions, including (i) what is the overall yield of the strong greenhouse gas, HFC-23, when both ozonolysis (R2) and photochemistry (R5) are included, (ii) what is the impact of HO2 chemistry, (R6), which was reported to have a faster rate coefficient than the OH reaction (R1); (iii) what is the sensitivity of the atmospheric fate of CF3CHO to reasonable values of  ; and (iv) considering the above, what is the final distribution of fates for HFO-1234ze(E) and its resultant indirect GWP? Answering those questions requires integrating spatial and temporal variability in emissions, photolytic and chemical reaction rates, and deposition.
; and (iv) considering the above, what is the final distribution of fates for HFO-1234ze(E) and its resultant indirect GWP? Answering those questions requires integrating spatial and temporal variability in emissions, photolytic and chemical reaction rates, and deposition.
In this work, we incorporate all these new advances to develop a comprehensive simulation of the atmospheric chemistry of HFO-1234ze(E) and its main degradation product CF3CHO in a global 3-D chemical transport model (GEOS-Chem). We first use a box model (AtChem2) to evaluate the importance of the reversible reaction between CF3CHO and HO2. We then test the sensitivity of CF3CHO depositional losses to  . Using the results from these initial studies, we implement all relevant reactions and processes in GEOS-Chem, which we use to quantify the fate of HFO-1234ze(E) and its total GWP including indirect impact through HFC-23 production.
. Using the results from these initial studies, we implement all relevant reactions and processes in GEOS-Chem, which we use to quantify the fate of HFO-1234ze(E) and its total GWP including indirect impact through HFC-23 production.
2 Model configuration
We use two models in this work: the AtChem2 0D box model incorporating the Master Chemical Mechanism (AtChem2-MCMv3.3.1) and the GEOS-Chem 3-D global chemical transport model. In Section 2.1, we outline the setup and implementation of the AtChem2-MCMv3.3.1 simulations used to test the reaction between CF3CHO and HO2. In Section 2.2, we describe the GEOS-Chem model configuration, including the new emissions (Section 2.2.1), chemical mechanism (Section 2.2.2), and deposition parameters (Section 2.2.3) required to implement the HFO-1234ze(E) → CF3CHO → HFC-23 chemical cascade.2.1 Implementation of HFO-1234ze(E) chemistry in the AtChem2-MCMv3.3.1 box model
We first test the relative importance of the CF3CHO + HO2 reaction to total CF3CHO loss using the AtChem2 box model implementing the Master Chemical Mechanism (MCM) v3.3.1, a near-explicit chemical mechanism describing detailed gas-phase oxidation in the troposphere. The current version of the MCM includes 17224 reactions and 5832 species. It does not currently contain any HFO species or chemistry. Table 1 summarises the new reactions added to the MCM. Both HFO-1234ze(E) and CF3CHO have well-characterised reactions with the OH radical ((R1) and (R3), respectively). The reaction between CF3CHO and HO2 (R6) yields a hydroxy-peroxy radical (CF3CH(OH)OO). This reaction is reversible, and the reverse reaction (R6′) was also added to the MCM.13 We also added removal of the CF3CH(OH)OO radical by reaction with NO (R7).13 As these simulations were designed solely to determine the relative contribution of the HO2 reaction (R6) to total CF3CHO loss, we did not separate the two photolysis channels but rather used the total photolysis quantum yield from Chiappero et al.28 (R8). For the same reason, we did not include the reaction between HFO-1234ze(E) and O3.
| No. | Reaction | Rate coefficient | Quantum yield |
|---|---|---|---|
| a Antiñolo et al.26b Calvert et al.27c Long et al.13d Chiappero et al.28 | |||
| R1 | HFO-1234ze(E) + OH → CF3CHO + CHFO | 7.06 × 10−13a | |
| R3 | CF3CHO + OH → CF3CO + H2O | 5.80 × 10−13b | |
| R6 | CF3CHO + HO2 → CF3CH(OH)OO | 2.80 × 10−13c | |
| R6′ | CF3CH(OH)OO CF3CHO + HO2 | 9.71 × 102c † | |
| R7 | CF3CH(OH)OO + NO → CF3CH(OH)O + NO2 | 1.5 × 10−11c | |
| R8 | CF3CHO + hν → products | 0.17d | |
We constrain the box model using surface observations from the July–August 2012 ClearfLo (Clean Air for London) measurement campaign to represent a typical urban environment,29 with a fixed temperature of 298 K. We therefore consider this simulation indicative of surface-level conditions in Northern Hemisphere summer. This is a limitation of the box model simulation, as reaction with HO2 may become more significant at higher altitudes, which are characterised by lower temperatures and pressures. More realistic temporal, spatial and meteorological variability are explored in the subsequent simulations using the global model.
2.2 Implementation of HFO-1234ze(E) simulation in GEOS-chem
GEOS-Chem is a widely used global 3D atmospheric chemical transport model30 driven by assimilated meteorology from the NASA Global Modelling and Assimilation Office (GMAO) Goddard Earth Observing System (GEOS). GEOS-Chem does not currently include an HFO simulation capability. Here, we started from GEOS-Chem Classic version 14.3.0 (doi: 10.5281/zenodo.10640536) and modified it to include all known chemical and physical processes associated with HFO-1234ze(E) and CF3CHO. The simulations were driven by Modern-Era Retrospective Analysis for Research and Applications, Version 2 (MERRA-2) meteorology. The native horizontal resolution of MERRA-2 (0.5° × 0.667°) was downgraded to 4° × 5° for computational efficiency, with 72 vertical layers. Standard global full-chemistry simulations were run for a one-year period from 01 January 2019 to 31 December 2019 following a six-month spin-up, which is sufficient to equilibrate the short-lived species in this study (HFO-1234ze(E), ∼16 days; CF3CHO, ∼2 days). These chemical lifetimes are many orders of magnitude longer than typical boundary layer turbulent mixing timescales, ensuring that both species are well-mixed within model grid boxes before significant chemical conversion occurs. This is a standard assumption in global chemical transport models.31,32 In GEOS-Chem, transport is driven by meteorological fields from the NASA GMAO, which includes parameterised boundary layer mixing and convective transport.33 Note that we do not simulate HFC-23 in this work; instead, we calculate HFC-23 production from HFO-1234ze(E), directly and via the CF3CHO intermediate.We added two different estimates of gridded HFO-1234ze(E) emissions into HEMCO: a hypothetical China-only HCFC-141b replacement scenario used previously by Wang et al.8 and Pérez-Peña et al.,5 and a more realistic global emissions scenario developed in this work. We used the hypothetical China-only emissions for Henry's law sensitivity tests (Section 3.2), and the global emissions for all other simulations (Section 3.3). The two emission inventories are described in the following sub-sections.
2.2.1.1 HFO-1234ze(E) as a replacement for HCFC-141b in China. Two previous modelling studies have examined the degradation of HFO-1234ze(E) using the emissions scenario described in Wang et al.5,8 In that scenario, Wang et al. assume complete replacement of HCFC-141b with HFO-1234ze(E), one of its proposed replacements, on a 1:1 mass basis.8 HCFC-141b was developed as an interim compound during the phase down of CFCs, and its production has been gradually phased out since 2003.38
As the emissions inventory developed by Wang et al. is not publicly available, we reproduced a variant for this work. Gridded emissions of HFO-1234ze(E) over China were generated as input for the HEMCO emissions component in GEOS-Chem. A total annual emission of 12.6 Gg year−1 was implemented, matching Wang et al.8 The geographical distribution of emissions within China was modelled using 2015 population density data as a proxy (see Fig. S1 for population density map).39 The resulting China emissions inventory is shown in Fig. 2c.
 | ||
| Fig. 2 HFO-1234ze(E) emission scenarios used in GEOS-Chem (see text for details). (a) Global baseline scenario (0.1° × 0.1°), total = 15 Gg year−1. (b) China subset from panel (a) regridded to 1° × 1°, regional total = 3.9 Gg year−1. (c) Alternative China scenario (1° × 1°), after Wang et al.,8 regional total = 12.6 Gg year−1. | ||
2.2.1.2 Observationally constrained global HFO-1234ze(E) emissions estimate. To better represent the distribution of HFO emissions beyond China, we developed a global emissions inventory through an iterative approach combining literature estimates with observational constraints. We start with an initial constraint implied by a 2024 Montreal Protocol Scientific Assessment Panel (SAP) report that estimates ∼150 tonnes year−1 of HFC-23 would be produced from HFO-1234ze(E) degradation.40 Based on the assumptions detailed in that report (100% conversion of HFO-1234ze(E) to CF3CHO; fractional loss of CF3CHO to photolysis, Yphotolysis = 0.75; HFC-23 yield from CF3CHO photolysis, αphotolysis = 0.003) we estimate approximately 110 Gg HFO-1234ze(E) emitted annually as follows:
where PHFC-23 is the estimated mass of HFC-23 produced from HFO-1234ze(E), MHFC-23 is the molar mass of HFC-23 and MHFO-1234ze(E) is the molar mass of HFO-1234ze(E). The SAP report acknowledges this likely overestimates global emissions as it extrapolates from limited European measurements, and because 0.003 is an upper limit for HFC-23 formation from CF3CHO.40,41

We tested this 110 Gg year−1 estimate in GEOS-Chem and compared modelled surface mixing ratios against preliminary 2020 Advanced Global Atmospheric Gases Experiment (AGAGE) observations from two European monitoring sites42 (Dübendorf and Jungfraujoch, both in Switzerland). The model overestimated observations at these sites by a factor of approximately 7, suggesting true emissions are lower. Assuming linearity between emissions and mixing ratios, we scaled the global emissions inventory by this ratio, resulting in total global emissions of 15 Gg year−1 (approximately 14% of the SAP-derived upper bound). Subsequently released AGAGE data from additional monitoring sites43 provided independent validation of this emission estimate, as detailed in Section 3.3.1. We spatially distributed these emissions using the 0.1° × 0.1° EDGAR 2018 HFC-134a emissions44 as a proxy. We selected 2018 (pre Kigali Amendment implementation) HFC emissions because HFO-1234ze(E) is being adopted as a replacement for HFCs in the same applications and geographic regions. While HFO-1234ze(E) was originally developed primarily for foam blowing applications, it is also used in refrigeration and air conditioning systems. HFC-134a serves these same multiple applications, making it an appropriate spatial proxy from combined end uses of HFO-1234ze(E), in the absence of bottom-up HFO-1234ze(E) emission inventories.
Fig. 2a shows the final global distribution of HFO-1234ze(E) emissions used in the GEOS-Chem simulations. The figure also compares emissions over China in the global inventory (regridded to 1° × 1° for comparison; Fig. 2b) to those in the hypothetical replacement inventory (Section 2.2.1.2; Fig. 2c). In the global scenario, annual emissions over China are 3.9 Gg year−1. This suggests that the 12.6 Gg year−1 derived by Wang et al.8 from full replacement of 2015 HCFC-141b emissions is likely too high. However, without HFO-1234ze(E) observations in China or downwind regions, it is impossible to quantitatively evaluate either estimate.
The new reactions implemented into the model along with their corresponding rate constants are detailed in Table 2. These are largely the same as those used in AtChem-MCM (Table 1), but with the addition of temperature dependence ((R1), (R3) and (R6)), separating the two photolysis channels ((R4) and (R5)), and ozonolysis of HFO-1234ze(E) (R2). These are implemented in GEOS-Chem via the Kinetic PreProcessor (KPP) used to solve chemical kinetics.47
| No. | Reaction | Rate coefficient, k | Quantum yield, ϕ |
|---|---|---|---|
| a Antiñolo et al.26b McGillen et al.7c Baumann et al.48d Long et al.13e JPL Data Evaluation 19–5.46f Van Hoomissen et al.19 | |||
| R1 | HFO-1234ze(E) + OH → CF3CHO + CHFO |  a a |
|
| R2 | HFO-1234ze(E) + O3 → HFC-23 + CHFO + CO2 | 2.44 × 10−21b | |
| R3 | CF3CHO + OH → CF3CO + H2O |  c c |
|
| R4 | CF3CHO + hν → CF3 + HCO | See Table S1e | |
| R5 | CF3CHO + hν → HFC-23 + CO | See Table S1f | |
| R6 | CF3CHO + HO2 → CF3CH(OH)OO |  d d |
|
| R6′ | CF3CH(OH)OO → CF3CHO + HO2 |  d † d † |
|
| R7 | CF3CH(OH)OO + NO → CF3CH(OH)O + NO2 | 1.5 × 10−11d | |
 , which is currently unknown for CF3CHO. Pérez-Peña et al. used a value of 13.17 M atm−1 by analogy to the hydrogenated analogue CH3CHO,5 while Nielsen et al. suggested
, which is currently unknown for CF3CHO. Pérez-Peña et al. used a value of 13.17 M atm−1 by analogy to the hydrogenated analogue CH3CHO,5 while Nielsen et al. suggested  would be closer to the chlorinated equivalent value of 3.44 × 105 M atm−1.24 We therefore performed sensitivity tests using
would be closer to the chlorinated equivalent value of 3.44 × 105 M atm−1.24 We therefore performed sensitivity tests using  values ranging from 10 to 106 M atm−1, spanning the estimates proposed in recent literature to constrain the impact of the uncertainty of
values ranging from 10 to 106 M atm−1, spanning the estimates proposed in recent literature to constrain the impact of the uncertainty of  on the atmospheric chemistry of CF3CHO.
on the atmospheric chemistry of CF3CHO.The dry deposition parameterisation also depends on a reactivity factor, f0, which has not been measured for CF3CHO. Pérez-Peña et al.5 found that there was limited sensitivity to the choice of f0. Following Pérez-Peña et al.,5 we use f0 = 1 for our simulations.
3 Results and discussion
3.1 Box model evaluation of CF3CHO + HO2 reaction
We first use the AtChem-MCM box model to evaluate the relative contribution of reaction with HO2 to total photochemical loss of CF3CHO. Fig. 3 shows the relative importance of each CF3CHO photochemical loss process as simulated by the box model. In Fig. 3a, we show the results when the box model includes the forward reaction ((R6), CF3CHO + HO2) but does not include the reverse reaction (R6′, CF3CH(OH)OO decomposition). In this scenario, reaction with HO2 is the dominant sink for CF3CHO. However, when the reverse reaction is introduced, as shown in Fig. 3b, reaction with HO2 becomes negligible, accounting for 0.1% of total photochemical loss of CF3CHO. CF3CHO loss is dominated by photolysis (≈80%), followed by reaction with OH (≈20%). This result highlights that although almost 80% of CF3CHO is initially removed via reaction with HO2, the resulting radical is unstable and rapidly decomposes back to CF3CHO and HO2 before it can react further, and thus the net effect is almost zero. We note that these simulations include chemical losses only. When deposition is included, the overall contribution of reaction with HO2 to total CF3CHO is reduced further. | ||
| Fig. 3 Percentage contribution of each of the photochemical loss processes to the total photochemical loss of CF3CHO derived from the AtChem2 box model simulations using the modified MCMv3.3.1 with (a) excluding the reverse reaction (R6′) and (b) including the reverse reaction (R6′). | ||
To account for uncertainties, we tested the sensitivity of our results to different conditions, independently increasing the HO2 mixing ratio and the forward reaction rate by a factor of 10 each. We also performed box model simulations representative of conditions at around 5 km altitude where the pressure-dependent reaction is more favorable. The results from these simulations are presented in Fig. S2 in the SI. In all scenarios, CF3CHO loss to reaction with HO2 remained under 4%. Our results indicate that the reaction is unlikely to dominate under tropospheric conditions, contrary to the findings of the original study.13
3.2 Sensitivity of CF3CHO fate to Henry's law constant
Given the uncertainty of (CF3CHO) and its implication for depositional losses, we performed a series of sensitivity tests in GEOS-Chem, using
(CF3CHO) and its implication for depositional losses, we performed a series of sensitivity tests in GEOS-Chem, using  as input to both the dry and wet deposition schemes. These values were tested during a northern hemisphere winter month (January) and a summer month (July). For these simulations, we used the simulation with HFO-1234ze(E) emissions over China only as described in Section 2.2.1.1. This allows us to compare the results with those of Pérez-Peña et al.,5 who used the same emissions data.
as input to both the dry and wet deposition schemes. These values were tested during a northern hemisphere winter month (January) and a summer month (July). For these simulations, we used the simulation with HFO-1234ze(E) emissions over China only as described in Section 2.2.1.1. This allows us to compare the results with those of Pérez-Peña et al.,5 who used the same emissions data.
Fig. 4 shows a linear-log plot of the wet, dry and total deposition fluxes as a function of  in both January (Fig. 4a) and July (Fig. 4b). Wet deposition loss increases approximately linearly with
in both January (Fig. 4a) and July (Fig. 4b). Wet deposition loss increases approximately linearly with  between
between  , evincing a logarithmic relationship between deposition loss and
, evincing a logarithmic relationship between deposition loss and  over this range. For
over this range. For  , the curve flattens, and loss remains constant for
, the curve flattens, and loss remains constant for  .
.
 | ||
Fig. 4 Wet (blue squares), dry (light blue triangles), and total (dark blue circles) deposition (kg m−2 s−1) in (a) January and (b) July as a function of  . . | ||
Previous studies have also shown that wet deposition contributions typically increase with  until around 105 M atm−1, where the efficiency of wet deposition peaks and the process becomes saturated.51 This saturation occurs because at sufficiently high
until around 105 M atm−1, where the efficiency of wet deposition peaks and the process becomes saturated.51 This saturation occurs because at sufficiently high  , the dissolution process becomes so thermodynamically favourable that every collision between a gas phase molecule and an aerosol droplet results in uptake. At this point, the wet deposition rate is limited by the rate of diffusion across droplet surfaces rather than by
, the dissolution process becomes so thermodynamically favourable that every collision between a gas phase molecule and an aerosol droplet results in uptake. At this point, the wet deposition rate is limited by the rate of diffusion across droplet surfaces rather than by  .51 The saturation behaviour is consistent in both January and July. The difference in the magnitude of the wet deposition fluxes between the two months can be attributed to rainfall frequency and intensity, which are typically much higher in July.52
.51 The saturation behaviour is consistent in both January and July. The difference in the magnitude of the wet deposition fluxes between the two months can be attributed to rainfall frequency and intensity, which are typically much higher in July.52
Fig. 4 also shows how the dry deposition flux varies with  in the two months tested.
in the two months tested.  has much less effect on dry deposition, primarily affecting the non-stomatal pathway by affecting leaf cuticle resistance.49 Higher
has much less effect on dry deposition, primarily affecting the non-stomatal pathway by affecting leaf cuticle resistance.49 Higher  values increase the solubility of the species at the leaf surface, enhancing non-stomatal deposition. However, since stomatal deposition remains largely unaffected by changes in
values increase the solubility of the species at the leaf surface, enhancing non-stomatal deposition. However, since stomatal deposition remains largely unaffected by changes in  , dry deposition is much less sensitive to changes in
, dry deposition is much less sensitive to changes in  than wet deposition. Dry deposition flux rates are higher in winter months when temperatures are lower.53 This is primarily due to the influence of temperature on the stomata of plants. Higher temperatures cause the plant stomata to close, thus reducing the surface area available for gas exchange.54 Overall, the total (wet plus dry) depositional loss is relatively invariant between the two months, with more wet deposition in July largely balanced by more dry deposition in January.
than wet deposition. Dry deposition flux rates are higher in winter months when temperatures are lower.53 This is primarily due to the influence of temperature on the stomata of plants. Higher temperatures cause the plant stomata to close, thus reducing the surface area available for gas exchange.54 Overall, the total (wet plus dry) depositional loss is relatively invariant between the two months, with more wet deposition in July largely balanced by more dry deposition in January.
We use the sensitivity tests to assess the impact of  on the fate of CF3CHO. Fig. 5 illustrates the seasonal competition between photochemical and depositional loss processes, showing the fractional contribution of each loss process to total CF3CHO removal. In January (Fig. 5a), photochemical loss dominates up to
on the fate of CF3CHO. Fig. 5 illustrates the seasonal competition between photochemical and depositional loss processes, showing the fractional contribution of each loss process to total CF3CHO removal. In January (Fig. 5a), photochemical loss dominates up to  and deposition dominates at higher
and deposition dominates at higher  . In contrast, in July (Fig. 5b), deposition never exceeds photochemical loss. While summer conditions enhance deposition rates, increased sunlight hours also increase the photolysis rate and OH reactivity. We find that from
. In contrast, in July (Fig. 5b), deposition never exceeds photochemical loss. While summer conditions enhance deposition rates, increased sunlight hours also increase the photolysis rate and OH reactivity. We find that from  , the fractional loss to deposition saturates at ∼60% in January (35% wet, 25% dry) and ∼45% in July (35% wet, 11% dry).
, the fractional loss to deposition saturates at ∼60% in January (35% wet, 25% dry) and ∼45% in July (35% wet, 11% dry).
 | ||
Fig. 5 Relative contributions of dry deposition, wet deposition and photochemical loss to total CF3CHO removal as a function of  in (a) January and (b) July. in (a) January and (b) July. | ||
 for CF3CHO has not been measured experimentally. Previous work has posited a value of 3.3 × 104 M atm−1 or higher, based on scaling of its chlorinated analogue.24 Based on this proposed value combined with our sensitivity analysis showing near-saturation behaviour from 105 M atm−1 (Fig. 4 and 5), we use
for CF3CHO has not been measured experimentally. Previous work has posited a value of 3.3 × 104 M atm−1 or higher, based on scaling of its chlorinated analogue.24 Based on this proposed value combined with our sensitivity analysis showing near-saturation behaviour from 105 M atm−1 (Fig. 4 and 5), we use  in subsequent simulations as indicative of the upper end of the plausible range.
in subsequent simulations as indicative of the upper end of the plausible range.
3.3 Global atmospheric modelling of HFO-1234ze(E) chemistry
We performed a 1-year global simulation using GEOS-Chem to evaluate the atmospheric implications of 15 Gg year−1 of HFO-1234ze(E) emissions. In this section, we first evaluate the model's performance against surface observations of HFO-1234ze(E) to assess how well it captures observed spatial gradients and seasonal cycles, analysing the discrepancies to identify potential limitations in our emission assumptions (Section 3.3.1). We then quantify CF3CHO and HFC-23 production from the model (Sections 3.3.2 and 3.3.3). Finally, we evaluate the contribution of HFC-23 formation to the indirect GWP100 of HFO-1234ze(E) and estimate how this process may contribute to the observed annual increase of 1 ppt of atmospheric HFC-23 (Section 3.3.3). | ||
| Fig. 6 Annual average HFO-1234ze(E) mixing ratios simulated by GEOS-Chem (a) at the surface, with 2022–2024 observed mixing ratios overlaid as filled circles, (b) as a function of longitude and pressure (averaged over latitudes), and (c) as a function of latitude and pressure (averaged over longitudes). Observation sites in (a) are indicated by the first letter of the site name: Zeppelin (Z), Mace Head (M), Jungfraujoch (J), Trinidad Head (T), Gosan (G), Ragged Point (R), Cape Matatula (C), Kennaook/Cape Grim (K). | ||
Fig. 6a shows the annual mean HFO-1234ze(E) mixing ratios at the surface as simulated by GEOS-Chem. The simulated global average surface HFO-1234ze(E) mixing ratio is 0.10 ppt. Mixing ratios peak over major industrial regions in Eastern China (≈2.1 ppt), and the Middle East (≈1.5 ppt), followed by Europe and the US (≈1.0 ppt). Elevated HFO-1234ze(E) mixing ratios remain highly localised near emission sources. Fig. 6b and c show simulated vertical cross sections of HFO-1234ze(E) mixing ratios. The majority of HFO-1234ze(E) reacts in the boundary layer, with limited transport into the mid-troposphere. There is some vertical transport, particularly northward towards the Arctic, driven by large-scale circulation patterns.60 The longitudinal cross section (Fig. 6b) shows elevated HFO-1234ze(E) mixing ratios centred over key emissions regions, while the latitudinal cross section (Fig. 6c) highlights the asymmetry between hemispheres, with elevated mixing ratios between 20 and 45°N where major emission sources are located and very low mixing ratios in the Southern Hemisphere.
Fig. 7 compares the simulated mixing ratios to the 2020–2024 observations at the 8 AGAGE sites. The model captures broad spatial patterns between most sites. For example, the model reproduces the interhemispheric gradient, with higher mixing ratios at Northern Hemisphere sites compared to Southern Hemisphere sites, and seasonal patterns. However, there are notable discrepancies in absolute mixing ratios, with the model overestimating observations by a factor of 5 at Gosan and by a factor of 7 at Kennaook/Cape Grim. Averaged over all 8 sites, the model overestimates observed mixing ratios by roughly 60% (0.188 vs. 0.114 ppt). However, the modelled mean is skewed by the high model bias at Gosan. Excluding this site reduces the model mean to 0.106 ppt. Further comparison statistics can be found in Table S2 in the supplement.
 | ||
| Fig. 7 Seasonal cycles of HFO-1234ze(E) at eight advanced global atmospheric gases experiment (AGAGE) network sites ordered by latitude from north to south. Observations (black circles, solid lines) show the mean seasonal cycle for 2020–2024, with shading indicating ±1 standard deviation of monthly means across the five years. Vertical bars indicate the full range of monthly mean values across those 5 years. Model results (red triangles, dashed lines) show the 2019 monthly means. All data are monthly mean dry air mixing ratios expressed in units of parts per trillion (ppt). Observations are from Vollmer et al.43 | ||
The biases in our simulation primarily reflect limitations in our emissions estimate, which uses 2018 HFC-134a emissions as a proxy in the absence of an existing bottom-up inventory for HFO-1234ze(E). Our results highlight the need for a dedicated inventory, with particular attention to the spatial distribution of emissions. At 4° × 5° resolution, near-source concentration gradients and complex transport pathways are not well resolved, amplifying biases at sites such as Gosan that receive polluted air masses from multiple nearby source regions. Further, HFO-1234ze(E) adoption patterns likely differ across countries and application sectors. For example, uptake rates may vary between China, Japan and South Korea in ways the HFC-134a distribution does not capture, leading to misallocation of regional emissions within East Asia. Gosan receives diverse air masses from all three countries and is particularly sensitive to this misallocation. By contrast, our European emissions total of 1.1 Gg year−1 agrees well with the 0.96 Gg year−1 reported by Vollmer et al.,43 providing confidence in the inventory where observational constraints are available and highlighting the need for improved constraints on East Asian emissions.
Remote background sites are less sensitive to the choice of emissions proxy because mixing ratios are controlled by total hemispheric emissions rather than their regional distribution, explaining the generally better model agreement at these sites. The exception is the high bias at Kennaook/Cape Grim, which likely reflects a mix of uncertainties in Southern Hemisphere mid-latitude emission totals and local oxidation chemistry. Overestimated interhemispheric exchange rates are unlikely to be drivers, as the interhemispheric exchange in GEOS-Chem has been validated in previous studies using long-lived tracers including SF6 and CH3CCl3,61 and HFO-1234ze(E) has a tropospheric lifetime of approximately 16 days, much shorter than the interhemispheric exchange time of approximately 1.4 years.61 In addition, there is no equivalent high bias at the more equatorward Cape Matatula site. Local chemistry, on the other hand, may play a role. Our simulations do not include chlorine (Cl)-initiated oxidation of HFO-1234ze(E),6 as tropospheric Cl concentrations are roughly three orders of magnitude lower than OH concentrations62 and therefore unlikely to be of major consequence at the global scale. However, at coastal sites such as Kennaook/Cape Grim, Cl concentrations can be elevated due to sea salt and halogen activation chemistry,8 and HFO-1234ze(E) loss to Cl oxidation could be higher locally. The regional impacts of this chemistry should be tested in future work; however, it is unlikely to have a significant impact on the overall global outcomes that are the focus of this work.
Despite lingering biases at individual sites, based on the model's ability to simulate the observed inter-hemispheric gradient, seasonal cycles, and order of magnitude of HFO-1234ze(E) mixing ratios, we consider our simulation sufficient for identifying broad global-scale impacts of HFO-1234ze(E) emissions. Two previous modelling studies of HFO-1234ze(E) degradation also provide useful benchmarks for our results. Wang et al.8 used emissions of 12.6 Gg year−1 over China in an alternative GEOS-Chem implementation and reported much higher surface mixing ratios of HFO-1234ze(E), with a global average of 0.55 ppt and 10.47 ppt over China. In comparison, our global emissions inventory assigns 3.9 Gg year−1 to China (Fig. 2). Despite our Chinese emissions being about three times lower than those from Wang et al., our simulated HFO-1234ze(E) mixing ratios over China are approximately 20 times lower. With 15 Gg year−1 of total global emissions distributed across all regions, our simulated global average mixing ratio (≈0.1 ppt) is approximately five times lower than Wang et al.'s reported 0.55 ppt, based on 12.6 Gg year−1 of emissions from China and none elsewhere. Our evaluation against AGAGE observations suggests our mixing ratios are more realistic than those simulated by Wang et al., possibly due to differences in their model setup. Pérez-Peña et al.5 used a box model to simulate the global boundary layer with 12.6 Gg year−1 of emissions and calculated mixing ratios of 0.08 ppt averaged over the global planetary boundary layer, consistent with our results.
 | ||
| Fig. 8 Annual average CF3CHO mixing ratios simulated by GEOS-Chem (a) at the surface, (b) as a function of longitude and pressure (averaged over latitudes), and (c) as a function of latitude and pressure (averaged over longitudes). | ||
The HFO-1234ze(E) differences between our simulation and Wang et al.8 discussed in the previous section propagated to the modelled CF3CHO mixing ratios. Wang et al. reported an average global CF3CHO surface mixing ratio of 0.18 ppt,8 compared to 0.01 ppt from our simulations. In addition to likely overestimating HFO-1234ze(E) in their model (as discussed above), Wang et al. did not include depositional losses for CF3CHO, which we demonstrate below to be a significant sink. Our results are more consistent with those reported by Pérez-Peña et al., who found an average global mixing ratio of 0.02 ppt.5
Fig. 8b and c show the vertical distribution of CF3CHO mixing ratios. Although the highest HFO-1234ze(E) mixing ratios were found at the surface, the CF3CHO mixing ratios peak around 900 hPa (≈2 km). This offset is primarily driven by the altitude dependence of the chemical lifetime of HFO-1234ze(E), shown in Fig. S3 in the SI. HFO-1234ze(E) is lost most rapidly at around 900 hPa, leading to enhanced production of CF3CHO at this altitude. Surface removal processes such as dry deposition further suppress CF3CHO mixing ratios near the ground.
Table 3 displays the global budget of atmospheric CF3CHO. Sources and sinks of CF3CHO are balanced over the year. The dominant sink is deposition, accounting for on average 51% of total CF3CHO loss. Photolysis to CF3 and HCO radicals (R4) represents the next largest CF3CHO sink at 33%. This pathway does not produce HFC-23, which is formed from CF3CHO exclusively via the concerted molecular elimination channel (R5). We tested the sensitivity of the CF3CHO loss branching to uncertainty in the photolysis quantum yield using the AtChem2 box model. Varying the total photolysis quantum yield by ±20%, consistent with uncertainties reported by IUPAC, shifted the relative contributions of photolysis and OH to chemical loss by ±1.5%, indicating limited sensitivity to this parameter. Consistent with our box model results (Section 3.1), we find using GEOS-Chem that the net loss of CF3CHO to reaction with HO2 is small (0.14 Gg year−1, 1.2%).
| Sources/sinks | Absolute (Gg year−1) | Relative (%) |
|---|---|---|
| HFO-1234ze(E) oxidation | 12.5 | 100 |
| Total sources | 12.5 | 100 |
| Photolysis (CF3 + HCO) | 4.1 | 32.5 |
| Wet deposition | 3.9 | 31.0 |
| Dry deposition | 2.5 | 19.8 |
| OH oxidation | 1.9 | 15.4 |
| Reaction with HO2 | 0.1 | 1.2 |
| Photolysis (HFC-23 + CO) | 0.01 | 0.1 |
| Total sinks | 12.5 | 100 |
We added tracers to the chemical mechanism to quantify the extent to which CF3CHO reacts with HO2 (R6) before undergoing the reverse reaction (R6′). Although this reaction is the dominant initial pathway (with 88% of CF3CHO first forming the CF3CHOHOO intermediate), the intermediate rapidly decomposes back to CF3CHO + HO2 under typical tropospheric conditions. As a result there is little net forward reaction. These results support our finding in Section 3.1 that the reaction between CF3CHO and HO2 is of little atmospheric significance. Further comparison between the AtChem2-MCM and GEOS-Chem results can be found in the SI (Section S1, Fig. S4).
Fig. 9 shows the relative contributions of the different loss processes to total CF3CHO loss as a function of altitude. The loss mechanisms exhibit strong altitude dependence. At the surface, dry deposition accounts for almost 92%. Because dry deposition is confined to the surface layer, the sharp initial decrease in depositional loss with altitude is driven by the absence of this pathway above the surface. Photolysis processes become increasingly important in the upper atmosphere, where UV radiation is more intense, quantum yields are higher (lower pressure) and water vapour drops off. The reaction with HO2, shown in purple, exhibits a notable pressure dependence. At the surface, this process contributes less than 0.1% to total CF3CHO loss, consistent with our AtChem2 box model (Section 3.1, Fig. 3b). However, the contribution increases with altitude, reaching 80% in the upper troposphere due to the pressure-dependent forward rate coefficient (R6). Despite this altitude dependence, the global column-integrated contribution remains small (1.2%, Table 3 and Fig. 9b) because the CF3CH(OH)OO intermediate formed in the forward reaction rapidly decomposes back to CF3CHO + HO2 under typical tropospheric conditions. The altitude profile in Fig. 9 reveals that while the forward reaction becomes more favourable at lower pressures, the net atmospheric significance of this pathway remains limited even in the upper troposphere, confirming our box model conclusions that this recently identified reaction5 does not substantially alter the atmospheric fate of CF3CHO. The contribution of the photolysis channel leading to HFC-23 production (shown in dark orange) also increases as pressure decreases, rising from less than 0.01% at the surface to a maximum of 0.4% at 300 hPa.
 | ||
| Fig. 9 Altitude dependence of CF3CHO loss pathways from HFO-1234ze(E) degradation as simulated by GEOS-Chem. (a) Fractional contribution to annual mean total loss at each pressure level. (b) Absolute annual loss rates at each pressure level. Note that dry deposition is a surface-only process. | ||
The CF3CHO lifetime as simulated by GEOS-Chem is shown in Table 4 and Fig. S3 in the SI as a function of season. We find a tropospheric lifetime for CF3CHO of 2.1 days, with individual contributions of 3.7–4.6 days against deposition, 5.3–8.5 days against photolysis, and 9.2–21.4 days against OH oxidation. The lifetimes vary seasonally, with shorter lifetimes in boreal summer for all processes due to faster deposition, OH reaction and photolysis. Our results are in close agreement with the overall 2.2 days tropospheric lifetime estimated in the SAP report40 and substantially lower than the 13 ± 4 days lifetime at 5 km altitude reported by Sulbaek Andersen et al.,41 who did not include depositional losses. For individual processes, our photolysis lifetime is close to the values reported by Chiappero et al.28 and Nielsen et al.,24 who estimated 3–6 days and 7 days, respectively. Our deposition lifetime is consistent with the estimates of 4–8 days by Nielsen et al.24 (wet scavenging only) and 5.5 days by Pérez-Peña et al.25 (wet and dry deposition), both assuming  of CF3CHO of order 104 M atm−1 (vs. 105 M atm−1 here). Our OH oxidation lifetimes exhibit significant seasonal variability, ranging from 9.2 days to 21.4 days, compared to the 20 days lifetime reported by Nielsen et al.24
of CF3CHO of order 104 M atm−1 (vs. 105 M atm−1 here). Our OH oxidation lifetimes exhibit significant seasonal variability, ranging from 9.2 days to 21.4 days, compared to the 20 days lifetime reported by Nielsen et al.24

| Loss process | Lifetime (days) | ||||
|---|---|---|---|---|---|
| Annual | DJF | MAM | JJA | SON | |
| Deposition (dry + wet) | 4.2 | 4.3 | 4.6 | 3.7 | 4.2 |
| Photolysis (CF3 + HCO) | 6.6 | 8.5 | 6.0 | 5.3 | 7.1 |
| Oxidation by OH | 13.9 | 21.4 | 14.1 | 9.2 | 14.3 |
| Overall lifetime | 2.1 | 2.5 | 2.2 | 1.7 | 2.2 |
 | ||
| Fig. 10 Annual average HFC-23 production rates simulated by GEOS-Chem (a) at the surface (b) as a function of longitude and pressure (averaged over latitudes) and (c) as a function of latitude and pressure (averaged over longitudes). | ||
Using the simulated production rates, we calculate total HFC-23 production to be approximately 11 Mg year−1 during our one-year global simulation. This corresponds to a growth rate of less than 0.001 ppt year−1, three orders of magnitude smaller than the observed annual increase of 1 ppt year−1.63 Therefore, our simulations indicate that HFO-1234ze(E) emission makes a negligible contribution to current HFC-23 growth.
We note that our choice of  is at the upper end of the plausible range, and a lower
is at the upper end of the plausible range, and a lower  would reduce CF3CHO loss to deposition, potentially increasing loss to photolysis and associated HFC-23 production. In addition, the substantial growth in HFO emissions projected for some parts of the world8,64 would increase the additional HFC-23 source from HFO-1234ze(E). Regardless of these uncertainties, we expect HFC-23 production from HFO-1234ze(E) to remain small relative to the present-day HFC-23 growth rate.
would reduce CF3CHO loss to deposition, potentially increasing loss to photolysis and associated HFC-23 production. In addition, the substantial growth in HFO emissions projected for some parts of the world8,64 would increase the additional HFC-23 source from HFO-1234ze(E). Regardless of these uncertainties, we expect HFC-23 production from HFO-1234ze(E) to remain small relative to the present-day HFC-23 growth rate.
In our simulations, 99.6% of HFO-1234ze(E) reacts with OH to form CF3CHO, with the remainder undergoing ozonolysis. Since ozonolysis forms HFC-23 from HFO-1234ze(E) rather than via CF3CHO, we treat these pathways differently. We calculate an overall atmospheric molar yield of HFC-23 from CF3CHO of 9.0 × 10−4 mol mol−1. Combining our calculated HFC-23 yield from CF3CHO with the published GWP100 of HFC-23 (14600 (ref. 14)), we calculate an indirect GWP100 of 8.2 for HFO-1234ze(E) from the CF3CHO photolysis pathway. The uncertainty in the indirect GWP100 from the CF3CHO photolysis pathway is dominated by the uncertainty in ϕ5. Van Hoomissen et al. report ϕ5 = (3.02 ± 0.70) × 10−4 at 308 nm and 650 torr, corresponding to a relative uncertainty of approximately 23%.19 Since HFC-23 production scales linearly with ϕ5, this propagates directly to the indirect GWP100 from photolysis, giving 8.2 ± 1.9. Additional uncertainty arises from  , which we have partially characterised through sensitivity tests in Section 3.2. Reducing
, which we have partially characterised through sensitivity tests in Section 3.2. Reducing  from 105 to 103 M atm−1 decreases CF3CHO deposition by 30%, resulting in proportional increases to other loss process and thereby to the indirect GWP100 from photolysis. Increasing
from 105 to 103 M atm−1 decreases CF3CHO deposition by 30%, resulting in proportional increases to other loss process and thereby to the indirect GWP100 from photolysis. Increasing  from 105 M atm−1 has no impact on the fate of CF3CHO, so there is no equivalent decrease in the indirect GWP100 from photolysis. Incorporating both of these uncertainties, our calculated indirect GWP100 from photolysis is 8.2+3.1−1.9.
from 105 M atm−1 has no impact on the fate of CF3CHO, so there is no equivalent decrease in the indirect GWP100 from photolysis. Incorporating both of these uncertainties, our calculated indirect GWP100 from photolysis is 8.2+3.1−1.9.
Recent experimental work provides benchmarks for the molar yield of HFC-23 from CF3CHO. Most recently, Van Hoomissen et al.19 reported a molar product yield for HFC-23 formation from CF3CHO of (1.71 ± 0.70) × 10−3 mol mol−1 at 308 nm, 650 torr. This is in close agreement with Thomson et al.,18 who reported a molar yield of HFC-23 from CF3CHO of (1.17 ± 0.27) × 10−3 mol mol−1 at 308 nm, 1 bar N2. Thomson et al. also estimated an atmospheric molar yield of HFC-23 from HFO-1234ze(E) of 6.4 × 10−4 mol mol−1, assuming 41% depositional loss of CF3CHO (vs. 50% here), from which they estimated an indirect GWP100 of around 6. Our results are consistent with these estimates.
Our simulation also includes HFC-23 production from HFO-1234ze(E) ozonolysis, yielding an indirect GWP100 of 2.2 ± 0.3. We find ozonolysis accounts for 0.36% of total HFO-1234ze(E) loss—substantially lower than the 2.96% initially reported by McGillen et al.7 but consistent with their updated model estimates reported in Garavagno et al.20 Our results are also consistent with theoretical expectations: assuming average mixing ratios of OH (1 × 106 molecules cm−3) and O3 (7.5 × 1011 molecules cm−3)67,68 with rate coefficients at 298 K, we calculate an ozonolysis contribution of ∼0.4%, consistent with our 3-D model results. The kO3/kOH ratio at 298 K (3.46 × 10−9) falls below the 10−8 threshold where ozonolysis typically becomes significant.69 While kOH is temperature-dependent, our 3-D simulations account for this variability across all atmospheric conditions and confirm that ozonolysis remains a minor contributor to both HFO-1234ze(E) loss and HFC-23 formation globally.
The combined indirect GWP100 of HFO-1234ze(E) from HFC-23 formed via ozonolysis (GWP100 = 2.2 ± 0.3) and via photolysis of CF3CHO (GWP100 = 8.2+3.1−1.9) is around GWP100 = 10.4+3.1−1.9. There is also a direct radiative forcing contribution from HFO-1234ze(E) with a GWP100 of approximately 1.3,70 These contributions are shown together in the bar chart in Fig. 11a. Taken together, our results imply a total GWP100 (indirect + direct) for HFO-1234ze(E) of GWP100 = 11.4+3.1−1.9. Note that the uncertainty reported here does not include uncertainties in emissions or other parameters of the global transport model.
 | ||
Fig. 11 (a) Total GWP100 of HFO-1234ze(E), separated into contributions from direct radiative forcing (grey) and indirect effects from HFC-23 formation via HFO-1234ze(E) ozonolysis (yellow) and HFC-23 formation via CF3CHO photolysis (orange). Error bars indicate the combined uncertainty from the CF3CHO photolysis quantum yield (±23%) and Henry's Law constant sensitivity for  . (b) Comparison of the HFO-1234ze(E) GWP100 calculated in this work with common HFCs on a logarithmic scale. Dashed lines indicate regulatory thresholds under EU F-gas regulations (150) and the US AIM Act (700).65,66 . (b) Comparison of the HFO-1234ze(E) GWP100 calculated in this work with common HFCs on a logarithmic scale. Dashed lines indicate regulatory thresholds under EU F-gas regulations (150) and the US AIM Act (700).65,66 | ||
Although an order of magnitude larger than the currently reported total GWP100, Fig. 11b shows that the updated HFO-1234ze(E) GWP100 determined in this work is far below the threshold of concern in current regulatory frameworks, even under the most conservative combination of assumptions (lower bound  and upper bound ϕ5). Under the European Union F-gas regulation, the use of F-gases is prohibited in commercial refrigeration systems if their total GWP100 exceeds 150, and in industrial refrigeration if it exceeds 2500.65 Similar thresholds are being adopted by the US legislation under the American Innovation and Manufacturing (AIM) Act, which will restrict refrigerants to GWP100 values below 700, 300, or 150, depending on the use.66 Against these benchmarks, the total GWP100 we calculate for HFO-1234ze(E) is of no regulatory significance. This value also remains far below the GWP100 of the HFCs that HFO-1234ze(E) is designed to replace, including HFC-134a (GWP100 = 1430), HFC-32 (GWP100 = 675), and HFC-125 (GWP100 = 3500), as shown in Fig. 11b. Even accounting for the uncertainties in the indirect contributions quantified in this work, HFO-1234ze(E) offers a climate benefit of roughly two orders of magnitude compared to these legacy refrigerants.
and upper bound ϕ5). Under the European Union F-gas regulation, the use of F-gases is prohibited in commercial refrigeration systems if their total GWP100 exceeds 150, and in industrial refrigeration if it exceeds 2500.65 Similar thresholds are being adopted by the US legislation under the American Innovation and Manufacturing (AIM) Act, which will restrict refrigerants to GWP100 values below 700, 300, or 150, depending on the use.66 Against these benchmarks, the total GWP100 we calculate for HFO-1234ze(E) is of no regulatory significance. This value also remains far below the GWP100 of the HFCs that HFO-1234ze(E) is designed to replace, including HFC-134a (GWP100 = 1430), HFC-32 (GWP100 = 675), and HFC-125 (GWP100 = 3500), as shown in Fig. 11b. Even accounting for the uncertainties in the indirect contributions quantified in this work, HFO-1234ze(E) offers a climate benefit of roughly two orders of magnitude compared to these legacy refrigerants.
4 Conclusions
Despite its increasing use as a replacement for ozone-destroying and climate-warming CFCs, HCFCs and HFCs, the true climate impact of HFO-1234ze(E) is not well characterised. In this work, we modelled the degradation of HFO-1234ze(E) using the GEOS-Chem 3-D chemical transport model to quantify the fates of HFO-1234ze(E) and its primary oxidation product CF3CHO, the resulting production of the potent greenhouse gas HFC-23, and the total GWP100 of HFO-1234ze(E) accounting for indirect effects. We modified GEOS-Chem to add the relevant chemistry, including newly determined reaction rate constants and photolysis quantum yields. We also used the AtChem2 box model with MCM v3.3.1 to test specific aspects of the chemistry before implementation in GEOS-Chem. We developed two emissions scenarios: a China-only emission scenario assuming complete HCFC-141b replacement (12.6 Gg year−1) following previous work5,8 that we used for Henry's Law sensitivity studies, and a new global emission scenario with 15 Gg year−1 distributed using HFC-134a emissions patterns and scaled to match observational constraints that we used for all other results.We tested the recently proposed CF3CHO + HO2 reaction13 using the AtChem2 box model incorporating the MCMv3.3 and found the net reaction contributes less than 0.1% to CF3CHO removal at the surface because the product of the forward reaction rapidly decomposes back to its reactants. We then assessed the sensitivity of CF3CHO loss to the effective Henry's Law Constant,  , using GEOS-Chem. In the absence of experimental measurements of
, using GEOS-Chem. In the absence of experimental measurements of  (CF3CHO), we tested a range of values from 10–106 M atm−1. We found that wet deposition saturates for
(CF3CHO), we tested a range of values from 10–106 M atm−1. We found that wet deposition saturates for  above 104 M atm−1, with the choice of
above 104 M atm−1, with the choice of 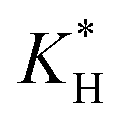 fundamental to the atmospheric fate of CF3CHO. Our results show that at high
fundamental to the atmospheric fate of CF3CHO. Our results show that at high  , deposition accounts for up to 60% of total CF3CHO loss, but this varies considerably with
, deposition accounts for up to 60% of total CF3CHO loss, but this varies considerably with  . Without measurements of
. Without measurements of  (CF3CHO), significant uncertainty remains. Future experimental determination of
(CF3CHO), significant uncertainty remains. Future experimental determination of  is required to reduce these uncertainties.
is required to reduce these uncertainties.
Using an upper bound of  and global HFO-1234ze(E) emissions of 15 Gg year−1, we found good agreement between GEOS-Chem simulated HFO-1234ze(E) and observations at 8 AGAGE sites representing diverse global environments. The model reasonably captures both the magnitude and seasonal variability of HFO-1234ze(E) mixing ratios across Northern and Southern Hemisphere sites. We find that 99.6% of HFO-1234ze(E) is removed by reaction with OH, with the remaining 0.4% undergoing ozonolysis. Our simulations reveal that the atmospheric fate of CF3CHO is dominated by deposition (51%) and photolysis (33%), with reaction with OH playing a more minor role (15%). The choice of Henry's Law constant affects the balance between these loss pathways: at
and global HFO-1234ze(E) emissions of 15 Gg year−1, we found good agreement between GEOS-Chem simulated HFO-1234ze(E) and observations at 8 AGAGE sites representing diverse global environments. The model reasonably captures both the magnitude and seasonal variability of HFO-1234ze(E) mixing ratios across Northern and Southern Hemisphere sites. We find that 99.6% of HFO-1234ze(E) is removed by reaction with OH, with the remaining 0.4% undergoing ozonolysis. Our simulations reveal that the atmospheric fate of CF3CHO is dominated by deposition (51%) and photolysis (33%), with reaction with OH playing a more minor role (15%). The choice of Henry's Law constant affects the balance between these loss pathways: at  , deposition becomes a major sink, reducing the amount of CF3CHO available for photolysis. This has direct implications for HFC-23 formation, as photolysis is the only pathway that produces HFC-23 from CF3CHO. We calculate a global tropospheric CF3CHO lifetime of 2.1 days, consistent with previous estimates.40 The overall atmospheric fate of HFO-1234ze(E) and CF3CHO is summarised in Fig. 12.
, deposition becomes a major sink, reducing the amount of CF3CHO available for photolysis. This has direct implications for HFC-23 formation, as photolysis is the only pathway that produces HFC-23 from CF3CHO. We calculate a global tropospheric CF3CHO lifetime of 2.1 days, consistent with previous estimates.40 The overall atmospheric fate of HFO-1234ze(E) and CF3CHO is summarised in Fig. 12.
 | ||
Fig. 12 Summary of the atmospheric fate of HFO-1234ze(E) and its primary oxidation product CF3CHO as simulated by GEOS-Chem using  . Percentages indicate the fraction of total removal attributed to each pathway. Note that 0.36% of HFO-1234ze(E) reacts with ozone, and this process produces HFC-23 with a yield of 7.9%.20 CF3CHO loss is dominated by deposition (51%), followed by photolysis (33%) and reaction with OH (15%). The CF3CHO + HO2 reaction accounts for a net 1.2% of CF3CHO removal. . Percentages indicate the fraction of total removal attributed to each pathway. Note that 0.36% of HFO-1234ze(E) reacts with ozone, and this process produces HFC-23 with a yield of 7.9%.20 CF3CHO loss is dominated by deposition (51%), followed by photolysis (33%) and reaction with OH (15%). The CF3CHO + HO2 reaction accounts for a net 1.2% of CF3CHO removal. | ||
From our simulations, we estimate total HFC-23 production of approximately 11 Mg year−1, which corresponds to an HFC-23 growth rate of less than 0.001 ppt year−1. This is negligible compared to the current observed annual increase of 1 ppt year−1.63 Our findings result in an indirect GWP100 of 10.4+3.1−1.9 for HFO-1234ze(E), of which 8.2+3.1−1.9 is due to photolysis of the CF3CHO intermediate and the 2.2 ± 0.3 from HFO-1234ze(E) ozonolysis. The indirect GWP100 from CF3CHO photolysis is similar to the recently reported value of 6 estimated from experimental measurements of the quantum yield.18 Combined with the direct GWP100 of approximately 1,3,70 the total GWP100 for HFO-1234ze(E) is 11.4+3.1−1.9, well below the current legislative thresholds everywhere in the world,65,66 and substantially lower than the GWP100 values of the HFCs that HFO-1234ze(E) is replacing (e.g., 1430 for HFC-134a). This total GWP100 estimate represents a lower bound due to our use of the upper bound  , which maximises deposition and therefore minimises HFC-23 formation via photolysis of CF3CHO. If the true
, which maximises deposition and therefore minimises HFC-23 formation via photolysis of CF3CHO. If the true  is lower, e.g. 103 M atm−1, our sensitivity tests (Section 3.2) indicate that deposition would decrease by 30%, increasing the indirect GWP100 contribution proportionally. However, even under this scenario, the total GWP100 remains well below regulatory thresholds,65,66 and represents a substantial improvement over the HFCs it is replacing. Experimental measurement of
is lower, e.g. 103 M atm−1, our sensitivity tests (Section 3.2) indicate that deposition would decrease by 30%, increasing the indirect GWP100 contribution proportionally. However, even under this scenario, the total GWP100 remains well below regulatory thresholds,65,66 and represents a substantial improvement over the HFCs it is replacing. Experimental measurement of  would refine this estimate but would not alter the fundamental conclusions.
would refine this estimate but would not alter the fundamental conclusions.
The dominant role of deposition as a CF3CHO sink has implications for the formation of trifluoroacetic acid (TFA). TFA is resistant to atmospheric degradation and accumulates in the environment, particularly in water bodies.71,72 We find from our simulation using  (an upper bound) that up to 31% of CF3CHO may undergo wet deposition. When CF3CHO comes into contact with water, it hydrates to form the stable gem-diol CF3CH(OH)2,24 which is then almost completely oxidised to TFA.73 Using this upper bound, and assuming complete hydrolysis of all-wet deposited CF3CHO to TFA from HFO-1234ze(E) degradation yields a maximum potential formation of 4.5 Gg year−1. This represents the maximum theoretical TFA formation from this source. Our sensitivity tests (Section 3.2) show that reducing
(an upper bound) that up to 31% of CF3CHO may undergo wet deposition. When CF3CHO comes into contact with water, it hydrates to form the stable gem-diol CF3CH(OH)2,24 which is then almost completely oxidised to TFA.73 Using this upper bound, and assuming complete hydrolysis of all-wet deposited CF3CHO to TFA from HFO-1234ze(E) degradation yields a maximum potential formation of 4.5 Gg year−1. This represents the maximum theoretical TFA formation from this source. Our sensitivity tests (Section 3.2) show that reducing  from 105 to 103 M atm−1 decreases the wet deposition fraction from ∼31% to ∼22%, corresponding to a TFA formation range of approximately 2.8–4.5 Gg year−1. For context, 2022 global TFA deposition from HCFC and HFC sources was estimated as 21.8 Gg year−1.74 These results suggest that wet deposition of CF3CHO formed via HFO-1234ze(E) oxidation may be a previously unrecognised source of TFA accumulation, particularly in regions close to emission sources.
from 105 to 103 M atm−1 decreases the wet deposition fraction from ∼31% to ∼22%, corresponding to a TFA formation range of approximately 2.8–4.5 Gg year−1. For context, 2022 global TFA deposition from HCFC and HFC sources was estimated as 21.8 Gg year−1.74 These results suggest that wet deposition of CF3CHO formed via HFO-1234ze(E) oxidation may be a previously unrecognised source of TFA accumulation, particularly in regions close to emission sources.
Author contributions
BK conducted the GEOS-Chem and AtChem2 simulations, analysed the data, and prepared the manuscript. JAF, CSH and SHK supervised the research and contributed to manuscript preparation. MKV and PBK provided observational data from AGAGE measurements, assisted with model-observation comparisons and data analysis, and contributed to manuscript preparation. All authors reviewed and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
Observational data for HFO-1234ze(E) were obtained from the Advanced Global Atmospheric Gases Experiment (AGAGE) network. AGAGE data are available from https://zenodo.org/records/20020038. GEOS-Chem model configuration files and emissions inventories are available at https://github.com/bethkillen.GEOS-Chem (https://geos-chem.readthedocs.io/en/stable/)) and AtChem2 (https://github.com/AtChem/AtChem2)) are open-source software. The full model output is available on request.
Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d6ea00034g.
Acknowledgements
The authors thank Paolo Sebastianelli and Stephen MacFarlane for their assistance with GEOS-Chem model configuration and development. This research was supported by the Australian Research Council (grants DP220102466 and DP220100891). The work was undertaken with the assistance of resources provided at the NCI National Facility systems at the Australian National University through the National Computational Merit Allocation Scheme supported by the Australian Government (project q90).References
- United Nations Environment Programme Montreal Protocol on Substances that Deplete the Ozone Layer, 1987, https://ozone.unep.org/treaties/montreal-protocol, 26 I.L.M. 1550.
- United Nations Environment Programme Kigali Amendment to the Montreal Protocol on Substances that Deplete the Ozone Layer, 2016, https://ozone.unep.org/treaties/montreal-protocol/amendments/kigali-amendment, C.N.872.2016.TREATIES-XXVII.2.f.
- Inc., H. I. Solstice® ze Refrigerant (HFO-1234ze (E)): The Environmental Alternative to Traditional Refrigerants Ultra-low GWP Hydrofluoroolefins (HFO), Technical Data Sheet, 2018.
- M. A. H. Khan, D. C. Mendes, R. E. T. Holland, M. A. Garavagno, A. J. Orr-Ewing, K. M. Stanley, S. J. O'Doherty, D. Young, M. K. Vollmer, A. J. Antony, F. Karamshahi, T. J. Wallington, C. J. Percival, A. Bacak, R. G. Derwent and D. E. Shallcross, Global modeling of trifluoroacetic acid surface concentration and deposition from the gas-phase oxidation of a wide range of precursor hydrofluoroolefins, Environ. Sci.: Atmos., 2026, 6, 195–212 CAS.
- M. P. Pérez-Peña, J. A. Fisher, C. Hansen and S. H. Kable, Assessing the atmospheric fate of trifluoroacetaldehyde (CF3CHO) and its potential as a new source of fluoroform (HFC-23) using the AtChem2 box model, Environ. Sci.: Atmos., 2023, 3, 1767–1777 Search PubMed.
- M. S. Javadi, R. Søndergaard, O. J. Nielsen, M. D. Hurley and T. J. Wallington, Atmospheric chemistry of trans-CF3CH=CHF: products and mechanisms of hydroxyl radical and chlorine atom initiated oxidation, Atmos. Chem. Phys., 2008, 8, 3141–3147 CrossRef CAS.
- M. R. McGillen, Z. T. P. Fried, A. H. Khan, K. T. Kuwata, C. M. Martin, S. O'Doherty, F. Pecere, D. E. Shallcross, K. M. Stanley and K. Zhang, Ozonolysis can produce long-lived greenhouse gases from commercial refrigerants, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2312714120 CrossRef CAS PubMed.
- Y. Wang, Z. Wang, M. Sun, J. Guo and J. Zhang, Emissions, degradation and impact of HFO-1234ze from China PU foam industry, Sci. Total Environ., 2021, 780, 146631 CrossRef CAS PubMed.
- T. J. Wallington, M. D. Hurley and W. F. Schneider, Kinetic Study of the Reaction CF3O + O3 → CF3O2 + O2, Chem. Phys. Lett., 1993, 213, 442–448 CrossRef CAS.
- J. S. Francisco, On the Mechanism of the Gas-Phase Hydrolysis of Carbonyl Fluoride, J. Am. Chem. Soc., 1993, 115, 6045–6046 CrossRef.
- M. R. Zachariah, P. R. Westmoreland and D. R. Burgess, A Study of the Gas-Phase Reaction of Carbonyl Fluoride with Water, J. Phys. Chem., 1995, 99, 11801–11808 CrossRef.
- T. J. Wallington, M. D. Hurley, O. J. Nielsen and J. Sehested, Atmospheric Chemistry of CF3COx Radicals: Fate of CF3CO Radicals, the UV Absorption Spectrum of CF3C(O)O2 Radicals, and Kinetics of the Reaction CF3C(O)O2 + NO → CF3C(O)O + NO2, J. Phys. Chem., 1994, 98, 5686–5694 CrossRef CAS.
- B. Long, Y. Xia and D. G. Truhlar, Quantitative Kinetics of HO2 Reactions with Aldehydes in the Atmosphere: High-Order Dynamic Correlation, Anharmonicity, and Falloff Effects Are All Important, J. Am. Chem. Soc., 2022, 144, 19910–19920 CrossRef CAS PubMed.
- V. Masson-Delmotte, P. Zhai, A. Pirani, S. L. Connors, C. Péan, S. Berger, N. Caud, Y. Chen, L. Goldfarb, M. I. Gomis, M. Huang, K. Leitzell, E. Lonnoy, J. B. R. Matthews, T. K. Maycock, T. Waterfield, O. Yelekçi, R. Yu and B. Zhou, Climate Change 2021: The Physical Science Basis, Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 2021 Search PubMed.
- R. E. Dodd and J. W. Smith, 282. The photolysis of trifluoroacetaldehyde, J. Chem. Soc., 1957, 1465–1473 RSC.
- C. Pearce and D. A. Whytock, The photolysis of trifluoroacetaldehyde at 313 nm, J. Chem. Soc. D, 1971, 1464–1466 RSC.
- M. P. Sulbaek Andersen and O. J. Nielsen, Tropospheric photolysis of CF3CHO, Atmos. Environ., 2022, 272, 118935 Search PubMed.
- J. D. Thomson, J. S. Campbell, E. B. Edwards, C. Medcraft, K. Nauta, M. P. Pérez-Peña, J. A. Fisher, D. L. Osborn, S. H. Kable and C. S. Hansen, Fluoroform (CHF3) Production from CF3CHO Photolysis and Implications for the Decomposition of Hydrofluoroolefins and Hydrochlorofluoroolefins in the Atmosphere, J. Am. Chem. Soc., 2025, 147, 33–38 Search PubMed.
- D. Van Hoomissen, A. Chattopadhyay, S. A. Montzka and J. B. Burkholder, CHF3 (HFC-23) and CF3CHO Quantum Yields in the Pulsed Laser Photolysis of CF3CHO at 248, 266, 281, and 308 nm, ACS Earth Space Chem., 2025, 9, 589–602 Search PubMed.
- M. A. Garavagno, A. Wenger, R. E. T. Holland, B. R. Fena, S. D. Goldstein, D. E. Hicks, F. Liu, J. B. Madell, S. J. Solomon, K. T. Kuwata, M. R. McGillen, M. A. H. Khan, D. E. Shallcross, K. M. Stanley and A. J. Orr-Ewing, Atmospheric Oxidation of Hydrofluoroolefins and Hydrochlorofluoroolefins by Ozone Produces HFC-23, PFC-14, and CFC-13, Environ. Sci. Technol., 2025, 41317145 Search PubMed.
- Harp International Ltd, Safety Data Sheet: R1234ze (Trans-1,3,3,3-tetrafluoropropene), Safety Data Sheet, 2020 Search PubMed.
- S. G. Tewari, K. Vijayaraghavan, K. Zhao, L. M. David, K. Tuite, F. Kristanovich, Y. Zhuang, B. Yang, C. Hurtado, D. K. Papanastasiou, P. Giffen, H. Kimko, M. Gibbs and S. Platz, Atmospheric and watershed modelling of HFO-1234ze(E) emissions from prospective pressurized metered-dose inhalers usage, Atmos. Chem. Phys., 2025, 25, 15469–15486 CrossRef CAS.
- M. P. Sulbaek Andersen, J. A. Schmidt, A. Volkova and D. J. Wuebbles, A three-dimensional model of the atmospheric chemistry of E and Z-CF3CH − CHCl (HCFO-1233(zd) (E/Z)), Atmos. Environ., 2018, 179, 250–259 Search PubMed.
- O. J. Nielsen, M. P. Sulbaek Andersen and J. Franklin, Comment on “Assessing the atmospheric fate of trifluoroacetaldehyde (CF3CHO) and its potential as a new source of fluoroform (HFC-23) using the AtChem2 box model” by Pérez-Peña et al., Environ. Sci.: Atmos., 2023, 3, 1767–1777, 10.1039/D3EA00120B.
- M. P. Pérez-Peña, J. A. Fisher, C. Hansen and S. H. Kable, Reply to the ‘Comment on “Assessing the atmospheric fate of trifluoroacetaldehyde (CF3CHO) and its potential as a new source of fluoroform (HFC-23) using the AtChem2 box model”’ by O. J. Nielsen, M. P. Sulbaek Andersen and J. Franklin, Environ. Sci.: Atmos., 2025, 5, 535–538, 10.1039/D4EA00123K.
- M. Antiñolo, I. Bravo, E. Jiménez, B. Ballesteros and J. Albaladejo, Atmospheric Chemistry of E- and Z-CF3CH − CHF (HFO-1234ze): OH Reaction Kinetics as a Function of Temperature and UV and IR Absorption Cross Sections, J. Phys. Chem. A, 2017, 121, 8322–8331 CrossRef PubMed.
- J. Calvert, A. Mellouki, J. Orlando, M. Pilling, T. Wallington, Mechanisms of Atmospheric Oxidation of the Oxygenates, Oxford University Press, 2011 Search PubMed.
- M. S. Chiappero, F. E. Malanca, G. A. Argüello, S. T. Wooldridge, M. D. Hurley, J. C. Ball, T. J. Wallington, R. L. Waterland and R. C. Buck, Atmospheric Chemistry of Perfluoroaldehydes (CxF2x+1CHO) and Fluorotelomer Aldehydes (CxF2x+1CH2CHO): Quantification of the Important Role of Photolysis, J. Phys. Chem. A, 2006, 110, 11944–11953 CrossRef CAS PubMed.
- S. I. Bohnenstengel, S. E. Belcher, A. Aiken, G. Allen, A. Bacak, T. J. Bannan, J. F. Barlow, D. C. S. Beddows, W. J. Bloss, A. M. Booth, C. Chemel, O. Coceal, C. F. Di Marco, M. K. Dubey, K. H. Faloon, Z. L. Fleming, M. Furger, J. K. Gietl, R. R. Graves, D. C. Green, C. S. B. Grimmond, C. H. Halios, J. F. Hamilton, R. M. Harrison, M. R. Heal, D. E. Heard, C. Helfter, S. C. Herndon, R. E. Holmes, J. R. Hopkins, A. M. Jones, F. J. Kelly, S. Kotthaus, B. Langford, J. D. Lee, R. J. Leigh, A. C. Lewis, R. T. Lidster, F. D. Lopez-Hilfilker, J. B. McQuaid, C. Mohr, P. S. Monks, E. Nemitz, N. L. Ng, C. J. Percival, A. S. H. Prévôt, H. M. A. Ricketts, R. Sokhi, D. Stone, J. A. Thornton, A. H. Tremper, A. C. Valach, S. Visser, L. K. Whalley, L. R. Williams, L. Xu, D. E. Young and P. Zotter, Meteorology, Air Quality, and Health in London: The ClearfLo Project, Bull. Am. Meteorol. Soc., 2015, 96, 779–804 CrossRef.
- I. Bey, D. J. Jacob, R. M. Yantosca, J. A. Logan, B. D. Field, A. M. Fiore, Q. Li, H. Y. Liu, L. J. Mickley and M. G. Schultz, Global modeling of tropospheric chemistry with assimilated meteorology: Model description and evaluation, J. Geophys. Res.:Atmos., 2001, 106, 23072–23095 Search PubMed.
- D. J. Jacob, Introduction to Atmospheric Chemistry, Princeton University Press, Princeton, 1999 Search PubMed.
- G. P. Brasseur and D. J. Jacob, Modeling of Atmospheric Chemistry, Cambridge University Press, Cambridge, 2017 Search PubMed.
- S. Wu, L. J. Mickley, D. J. Jacob, J. A. Logan, R. M. Yantosca and D. Rind, Why are there large differences between models in global budgets of tropospheric ozone?, J. Geophys. Res.:Atmos., 2007, 112, D05302 Search PubMed.
- H. Lin, D. J. Jacob, E. W. Lundgren, M. P. Sulprizio, C. A. Keller, T. M. Fritz, S. D. Eastham, L. K. Emmons, P. C. Campbell, B. Baker, R. D. Saylor and R. Montuoro, Harmonized Emissions Component (HEMCO) 3.0 as a versatile emissions component for atmospheric models: application in the GEOS-Chem, NASA GEOS, WRF-GC, CESM2, NOAA GEFS-Aerosol, and NOAA UFS models, Geosci. Model Dev., 2021, 14, 5487–5506 CrossRef CAS.
- R. M. Hoesly, S. J. Smith, L. Feng, Z. Klimont, G. Janssens-Maenhout, T. Pitkanen, J. J. Siebert, L. Vu, R. J. Andres, R. M. Bolt, T. C. Bond, L. Dawidowski, N. Kholod, J. Kurokawa, M. Li, L. Liu, Z. Lu, M. C. P. Moura, P. R. O’Rourke and Q. Zhang, Historical (1750–2014) anthropogenic emissions of reactive gases and aerosols from the Community Emissions Data System (CEDS), Geosci. Model Dev., 2018, 11, 369–408 Search PubMed.
- J. T. Randerson; G. R. van der Werf; L. Giglio; G. J. Collatz; P. S. Kasibhatla Global Fire Emissions Database, Version 4.1 (GFEDv4). 2017, DOI:10.3334/ORNLDAAC/1293, Date Accessed: 2025-09-25.
- A. B. Guenther, X. Jiang, C. L. Heald, T. Sakulyanontvittaya, T. Duhl, L. K. Emmons and X. Wang, The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 2012, 5, 1471–1492 Search PubMed.
- G. M. Rusch, The development of environmentally acceptable fluorocarbons, Crit. Rev. Toxicol., 2018, 48, 615–665 Search PubMed.
- Center for International Earth Science Information Network – CIESIN – Columbia University Gridded Population of the World, Version 4 (GPWv4): Population Density, Revision 11. 2018 Search PubMed.
- S. A. Montzka and J. B. Burkholder, Report of the Scientific Assessment Panel in Response to Decision XXXV/7: Emissions of HFC-23; UNEP Report, Prepared for the Meeting of the Parties to the Montreal Protocol, 2024 Search PubMed.
- M. P. Sulbaek Andersen, S. Madronich, J. M. Ohide, M. Frausig and O. J. Nielsen, Photolysis of CF3CHO at 254 nm and potential contribution to the atmospheric abundance of HFC-23, Atmos. Environ., 2023, 314, 120087 Search PubMed.
- M. K. Vollmer, S. Reimann, M. Hill and D. Brunner, Update to: First Observations of the Fourth Generation Synthetic Halocarbons HFC-1234yf, HFC-1234ze(E), and HCFC-1233zd(E) in the Atmosphere, Environ. Sci. Technol., 2015, 49, 2703–2708 Search PubMed.
- M. K. Vollmer, J. R. Pitt, D. Young, S. Henne, B. Mitrevski, J. Mühle, A. Ganesan, A. Arduini, A. J. Manning, T. Wagenhäuser, A. L. Redington, D. B. Melo, B. Murphy, R. Gluckmann, K. Stanley, P. B. Krummel, C. R. Lunder, J. Yun, D. Rust, A. Wenger, M. Guillevic, J. Kim, R. H. J. Wang, T. S. Rhee, T. S. Constantin, A. Frumau, A. Harth, P. K. Salameh, O. Hermansen, M. Rigby, L. M. Western, A. Engel, S. O’Doherty, S. Park, M. Maione, P. J. Fraser, R. G. Prinn, R. F. Weiss and S. Reimann, Global Observations and European Emissions of the Halogenated Olefins HFO-1234yf, HFO-1234ze(E), and HCFO-1233zd(E) from the AGAGE (Advanced Global Atmospheric Gases Experiment) Network, EGUsphere, 2025, 26, 6993–7012 Search PubMed.
- M. Crippa, D. Guizzardi, F. Pagani, M. Banja, M. Muntean, E. Schaaf, F. Monforti-Ferrario, W. Becker, R. Quadrelli, A. Risquez Martin, P. Taghavi-Moharamli, J. Köykkä, G. Grassi, S. Rossi, J. Melo, D. Oom, A. Branco, J. San-Miguel, G. Manca, E. Pisoni, E. Vignati and F. Pekar, EDGAR – Emissions Database for Global Atmospheric Research, release EDGAR 2024 GHG (1970–2023), Pub-lications Office of the European Union, Luxembourg, 2023, DOI:10.2760/4002897, https://edgar.jrc.ec.europa.eu/dataset_ghg2024.
- W. E. van Caspel, D. Simpson, J. E. Jonson, A. M. K. Benedictow, Y. Ge, A. di Sarra, G. Pace, M. Vieno, H. L. Walker and M. R. Heal, Implementation and evaluation of updated photolysis rates in the EMEP MSC-W chemistry-transport model using Cloud-J v7.3e, Geosci. Model Dev., 2023, 16, 7433–7459 Search PubMed.
- J. B. Burkholder, S. P. Sander, J. P. D. Abbatt, J. R. Barker, C. Cappa, J. D. Crounse, T. S. Dibble, R. E. Huie, C. E. Kolb, M. J. Kurylo, V. L. Orkin, J. C. Percival, D. M. Wilmouth and P. H. Wine, Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies: Evaluation Number 19, National Aeronautics and Space Administration, Jet Propulsion Laboratory, California Institute of Technology, Pasadena, California, 2020 Search PubMed.
- V. Damian, A. Sandu, M. Damian, F. Potra and G. R. Carmichael, The kinetic preprocessor KPP-a software environment for solving chemical kinetics, Comput. Chem. Eng., 2002, 26, 1567–1579 Search PubMed.
- F. Baumann, C. Fernholz, J. Lelieveld and J. N. Crowley, Kinetics of the reaction of CF3CHO with OH between 204 K and 361 K, Phys. Chem. Chem. Phys., 2025, 27, 18907–18916 Search PubMed.
- M. L. Wesely, Parameterization of surface resistances to gaseous dry deposition in regional-scale numerical models, Atmos. Environ., 1989, 23, 1293–1304 Search PubMed.
- H. Liu, D. J. Jacob, I. Bey and R. M. Yantosca, Constraints from 210Pb and 7Be on wet deposition and transport in a global three-dimensional chemical tracer model driven by assimilated meteorological fields, J. Geophys. Res.:Atmos., 2001, 106, 12109–12128 Search PubMed.
- C. Bi and G. Isaacman-VanWertz, Estimated timescales for wet deposition of organic compounds as a function of Henry's law constants, Environ. Sci.: Atmos., 2022, 2, 1526–1533 Search PubMed.
- China Meteorological Association China Meteorological Association – Climate Data Center, 2014, https://web.archive.org/web/20140727001008/, https://cdc.cma.gov.cn/cdc_en/home.dd, archived version, accessed: 28-Jan-2025.
- W. C. Porter and C. L. Heald, The mechanisms and meteorological drivers of the summertime ozone–temperature relationship, Atmos. Chem. Phys., 2019, 19, 13367–13381 Search PubMed.
- S. C. Kavassalis and J. G. Murphy, Understanding ozone-meteorology correlations: A role for dry deposition, Geophys. Res. Lett., 2017, 44, 2922–2931 Search PubMed.
- R. G. Prinn, R. F. Weiss, J. Arduini, T. Arnold, H. Langley DeWitt, P. J. Fraser, A. L. Ganesan, J. Gasore, C. M. Harth, O. Hermansen, J. Kim, P. B. Krummel, S. Li, Z. M. Loh, C. R. Lunder, M. Maione, A. J. Manning, B. P. Miller, B. Mitrevski, J. Mühle, S. O’Doherty, S. Park, S. Reimann, M. Rigby, T. Saito, P. K. Salameh, R. Schmidt, P. G. Simmonds, P. Steele, M. K. Vollmer, R. H. Wang, B. Yao, Y. Yokouchi, D. Young and L. Zhou, History of Chemically and Radiatively Important Atmospheric Gases from the Advanced Global Atmospheric Gases Experiment (AGAGE), Earth Syst. Sci. Data, 2018, 10, 985–1018 Search PubMed.
- R. G. Prinn, R. F. Weiss, P. J. Fraser, P. G. Simmonds, D. M. Cunnold, F. N. Alyea, S. O’Doherty, P. Salameh, B. R. Miller, J. Huang, R. H. J. Wang, D. E. Hartley, C. Harth, L. P. Steele, G. Sturrock, P. M. Midgley and A. McCulloch, A history of chemically and radiatively important gases in air deduced from ALE/GAGE/AGAGE, J. Geophys. Res.:Atmos., 2000, 105, 17751–17792 CrossRef CAS.
- S. Reimann, D. Schaub, K. Stemmler, D. Folini, M. Hill, P. Hofer, B. Buchmann, P. G. Simmonds, B. R. Greally and S. O'Doherty, Halogenated greenhouse gases at the Swiss High Alpine Site of Jungfraujoch (3580 m asl): Continuous measurements and their use for regional European source allocation, J. Geophys. Res.:Atmos., 2004, 109, D05307 CrossRef.
- J. Kim, J. Lee, S.-D. Choi, Y. Kim and Y. Ghim, Gaseous and particulate polycyclic aromatic hydrocarbons at the Gosan background site in East Asia, Atmos. Environ., 2012, 49, 311–319 Search PubMed.
- S. M. Platt, Ø. Hov, T. Berg, K. Breivik, S. Eckhardt, K. Eleftheriadis, N. Evangeliou, M. Fiebig, R. Fisher, G. Hansen, H. C. Hansson, J. Heintzenberg, O. Hermansen, D. Heslin-Rees, K. Holmén, S. Hudson, R. Kallenborn, R. Krejci, T. Krognes, S. Larssen, D. Lowry, C. Lund Myhre, E. Nisbet, P. B. Nizzetto, K. T. Park, C. A. Pedersen, K. Aspmo Pfaffhuber, T. Röckmann, N. Schmidbauer, S. Solberg, A. Stohl, J. Ström, T. Svendby, P. Tunved, K. Tørnkist, C. van der Veen, S. Vratolis, Y. J. Yoon, K. E. Yttri, P. Zieger, W. Aas and K. Tørseth, Atmospheric composition in the European Arctic and 30 years of the Zeppelin Observatory, Ny-Ålesund, Atmos. Chem. Phys., 2022, 22, 3321–3369 Search PubMed.
- S. Eckhardt, A. Stohl, S. Beirle, N. Spichtinger, P. James, C. Forster, C. Junker, T. Wagner, U. Platt and S. G. Jennings, The North Atlantic Oscillation controls air pollution transport to the Arctic, Atmos. Chem. Phys., 2003, 3, 1769–1778 Search PubMed.
- P. K. Patra, S. Houweling, P. Krol, D. Bousquet, D. Belikov, D. Bergmann, H. Bian, P. Cameron-Smith, M. P. Chipperfield, K. Corbin, A. Fortems-Cheiney, A. Fraser, E. Gloor, P. Hess, A. Ito, S. R. Kawa, R. M. Law, Z. Loh, S. Maksyutov, L. Meng, P. I. Palmer, R. G. Prinn, M. Rigby, R Saito and C. Wilson, TransCom model simulations of CH4 and related species: linking transport, surface flux and chemical loss with CH4 variability in the troposphere and lower stratosphere, Atmos. Chem. Phys., 2011, 11, 12813–12837 Search PubMed.
- R. Hossaini, M. P. Chipperfield, A. Saiz-Lopez, R. Fernandez, S. Monks, W. Feng, P. Brauer and R. von Glasow, A global model of tropospheric chlorine chemistry: Organic versus inorganic sources and impact on methane oxidation, J. Geophys. Res.:Atmos., 2016, 121, 14271–14297 Search PubMed.
- K. M. Stanley, D. Say, J. Mühle, C. M. Harth, P. B. Krummel, D. Young, S. J. O'Doherty, P. K. Salameh, P. G. Simmonds, R. F. Weiss, R. G. Prinn, P. J. Fraser and M. Rigby, Increase in global emissions of HFC-23 despite near-total expected reductions, Nat. Commun., 2020, 11, 397 Search PubMed.
- Y. Wang, L. Liu, X. Qiao, M. Sun, J. Guo, J. Zhang and B. Zhao, Projections of National-Gridded Emissions of Hydrofluoroolefins (HFOs) in China, Environ. Sci. Technol., 2023, 57, 8650–8659 Search PubMed.
- European Commission Regulation (EU), 2024/573 of the European Parliament and of the Council of 7 February 2024 on fluorinated greenhouse gases, Official Journal of the European Union, L series, 2024, 759, 1–76 Search PubMed ; https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32024R0573.
- United States Congress American Innovation and Manufacturing Act of 2020. Public Law No: 116-260, Division S, Title VI, 2020, https://www.congress.gov/bill/116th-congress/senate-bill/2754 Search PubMed.
- Y. Lu and M. Khalil, Tropospheric OH: model calculations of spatial, temporal, and secular variations, Chemosphere, 1991, 23, 397–444 CrossRef CAS.
- D. A. Malashock, M. N. Delang, J. S. Becker, M. L. Serre, J. J. West, K. Chang, O. R. Cooper and S. C. Anenberg, Estimates of ozone concentrations and attributable mortality in urban, peri-urban and rural areas worldwide in 2019, Environ. Res. Lett., 2022, 17, 958–967 Search PubMed.
- S. M. Saunders, M. E. Jenkin, R. G. Derwent and M. J. Pilling, Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 2003, 3, 161–180 Search PubMed.
- Ø. Hodnebrog, M. Etminan, J. S. Fuglestvedt, G. Marston, G. Myhre, C. J. Nielsen, K. P. Shine and T. J. Wallington, Global warming potentials and radiative efficiencies of halocarbons and related compounds: A comprehensive review, Rev. Geophys., 2013, 51, 300–378 CrossRef.
- M. Scheurer, K. Nödler, F. Freeling, J. J. Janda, O. Happel, M. Riegel, U. Müller, F. Rüdiger Storck, M. Fleig, F. T. Lange, A. Brunsch and H.-J. Brauch, Small, mobile, persistent: Trifluoroacetate in the water cycle – Overlooked sources, pathways, and consequences for drinking water supply, Water Res., 2017, 126, 460–471 CrossRef CAS PubMed.
- F. Freeling and M. K. Björnsdotter, Assessing the environmental occurrence of the anthropogenic contaminant trifluoroacetic acid (TFA), Curr. Opin. Green Sustainable Chem., 2023, 41, 100807 Search PubMed.
- M. P. Sulbaek Andersen, A. Toft, O. J. Nielsen, M. D. Hurley, T. J. Wallington, H. Chishima, K. Tonokura, S. A. Mabury, J. W. Martin and D. A. Ellis, Atmospheric Chemistry of Perfluorinated Aldehyde Hydrates (n-CxF2x+1CH(OH)2, x = 1, 3, 4): Hydration, Dehydration, and Kinetics and Mechanism of Cl Atom and OH Radical Initiated Oxidation, J. Phys. Chem. A, 2006, 110, 9854–9860 Search PubMed.
- L. Hart, R. Hossaini, O. Wild, A. Mazzeo, C. Halsall, X. Hou, Z. Wang, M. P. Chipperfield, J. Arduini, P. B. Krummel, C. R. Lunder, J. Mühle, S. O'Doherty, S. Park, S. Reimann, K. M. Stanley, R. Weiss and D. Young, Growth in Production and Environmental Deposition of Trifluoroacetic Acid Due To Long-Lived CFC Replacements and Anesthetics, Geophys. Res. Lett., 2026, 53, e2025GL119216 Search PubMed.
| This journal is © The Royal Society of Chemistry 2026 |