
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxidation of organic films at the mineral–water interface by aqueous-phase ozone affects aerosol light scattering
Edward J. Stuckey ab,
Rebecca J. L. Welbournbc,
Tobias W. D. Robsonad,
Philipp Gutfreunde,
Katherine C. Thompsonf,
Adrian R. Rennieg and
Martin D. King*a
ab,
Rebecca J. L. Welbournbc,
Tobias W. D. Robsonad,
Philipp Gutfreunde,
Katherine C. Thompsonf,
Adrian R. Rennieg and
Martin D. King*a
aDepartment of Earth Sciences, Royal Holloway University of London, Egham, Surrey TW20 0EX, UK. E-mail: m.king@rhul.ac.uk
bISIS Neutron & Muon Source, Rutherford Appleton Laboratory, Didcot, Oxfordshire OX11 0QX, UK
cOak Ridge National Laboratory, 1 Bethel Valley Road, Oak Ridge, TN 37830, USA
dForestry Research, Alice Holt Lodge, Wrecclesham, Farnham, GU10 4LH, UK
eInstitut Laue-Langevin, 71 Avenue des Martyrs, CS 20156, 38042 Grenoble Cedex 9, France
fSchool of Natural Sciences, Birkbeck, University of London, Malet Street, London WC1E 7HX, UK
gDepartment of Chemistry, Ångström Laboratory, Uppsala University, Lägerhyddsvägen 1, Box 538, 751 21 Uppsala, Sweden
First published on 1st May 2026
Abstract
Interfacial organic films on atmospheric aerosol particulates can modify their light scattering properties, potentially impacting Earth's radiative balance. Here, a proxy for organic films at the aerosol mineral–water interface is measured using neutron reflectometry, and the results are contextualized in a single particle light scattering model, N-Mie. Model lipid-bilayers, composed of DPPC (no double bonds), POPC (1 carbon–carbon double bond), and DOPC (2 carbon–carbon double bonds), were deposited on a silica–water interface and oxidized with aqueous ozone to simulate the oxidation of organic films on aqueous mineral aerosol particles. Neutron reflectometry measurements showed that these bilayers initially formed films approximately 4 nm thick, and oxidation by aqueous ozone led to thickness reductions proportional to the number of carbon–carbon double bonds. Post-oxidation thicknesses were 4 nm (DPPC), 1.6 nm (POPC), and 0.4 nm (DOPC). Light scattering modelling of particulates with a mineral core and water film revealed that a 1 nm lipid-like organic film at the mineral–water interface had similar effects on the single scattering albedo (ω) asymmetry parameter (g), and forcing efficiency  of an aerosol particulate as at the air–water interface. The Mie modelling also revealed that an increase in the thickness of the organic film at the mineral–water interface of up to 10 nm results in an increase in forward scattering of light (Δg = 0.025), a decrease in the amount of light scattered (Δω = −0.016) and an aerosol forcing efficiency
of an aerosol particulate as at the air–water interface. The Mie modelling also revealed that an increase in the thickness of the organic film at the mineral–water interface of up to 10 nm results in an increase in forward scattering of light (Δg = 0.025), a decrease in the amount of light scattered (Δω = −0.016) and an aerosol forcing efficiency 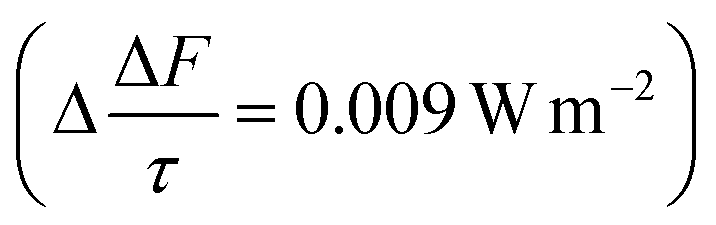 . The findings presented here indicate that organic films with experimentally measured thicknesses at the mineral–water interface can significantly alter aerosol forcing efficiency.
. The findings presented here indicate that organic films with experimentally measured thicknesses at the mineral–water interface can significantly alter aerosol forcing efficiency.
Environmental significanceAtmospheric aerosols influence Earth's climate by scattering and absorbing sunlight, yet the role of organic films at aerosol interfaces remains poorly constrained. Understanding how these interfacial layers modify light scattering is essential for accurately representing aerosol–radiation interactions in climate models. The work presented here combines neutron reflectometry with light-scattering simulations to quantify how ozone oxidation alters the thickness and optical effects of lipid-like organic films on aqueous mineral surfaces. The results show that film oxidation markedly influence scattering properties and aerosol forcing efficiency. These findings demonstrate that nanometer scale interfacial organic layers, commonly present on atmospheric particles, can meaningfully affect radiative forcing, emphasizing the need to include interfacial chemistry in climate-relevant aerosol models. |
1 Introduction
Organic thin films can form at the air–water interface of aqueous atmospheric aerosol and hydrometeors and have been shown to have significant atmospheric impacts,1–6 altering the light scattering and cloud-water nucleating potential of atmospheric aerosol.1,2,4,7–9 In prior studies, the authors of the work presented here have shown that there are significant and similar light scattering effects on atmospheric aerosol owing to the presence of thin films of the same thicknesses at the air–water10,11 and mineral–air interface.12 However, the importance of organic thin films at the alternative, but linked, mineral–water interface of the aerosol remains relatively unknown. A simplistic representation of atmospheric aerosol with films at the “air–water” and “mineral–water” interfaces are provided in Fig. 1. Here, the authors undertake a combination of experimental measurements and single particle Mie scattering modelling to better understand the atmospheric importance of organic thin films at the aerosol mineral–water interface. | ||
| Fig. 1 A schematic of mineral aerosol particles encapsulated by water with an organic film at the mineral–water (top-left) and air–water (top-right) interfaces, and a schematic representation of the core–shell model of the aerosol used in the N-Mie program to calculate the change in single scattering albedo and asymmetry parameter of the aerosol owing to the presence of the organic film discussed later in Sections 2.4, 3.3 and 4.2. The grey/light component represents the mineral core, the blue/dark component represents the water around the particle, and the orange striped layer represents the organic film. | ||
A significant proportion of atmospheric aerosol is comprised of mineral material, with mineral dust contributions to global aerosol emissions reaching an estimated 1000–3000 Mt per year;13 constituting more than half of the total global aerosol burden.14–16 Mineral aerosols can affect the atmosphere by scattering and absorbing light,17–20 by serving as cloud condensation nuclei,20–23 and by providing a solid surface for chemistry in the atmosphere.24–27 The atmospheric impact of mineral aerosol is determined in part by their chemical and morphological properties,14,20,28,29 which may vary spatially, and temporally.8,30–35 The size distribution of these particles is typically characterized by a multi-modal log-normal distribution by particle radius; while the largest number densities are concentrated in the sub-micron “accumulation mode” (Dp < 1 µm), the vast majority of the mass density is sequestered within the coarse mode, often exceeding 10 µm in diameter near source regions.36 Beyond its physical presence, dust serves as a critical heterogeneous reaction site for atmospheric chemistry, facilitating the uptake and partitioning of trace gases.27 For laboratory-based kinetic and optical studies, silica (SiO2) is frequently employed as a robust proxy for natural dust due to its dominance as a primary mineralogical constituent (averaging approximately 60% by mass in crustal samples) and its well-defined surface hydroxyl groups which mimic the reactive behaviour of authentic desert aerosols.36–38 Owing to the substantial amount of mineral aerosol particulates in the atmosphere, quantifying how they may change over their lifetime is important for refining climate, aerosol, and cloud processing models. One way in which a mineral aerosol may change with time is through the accumulation of organic material at the interfaces of the host particulate as a thin film.1,39,40 Interfacial organic thin films have been shown to be susceptible to oxidation.9,41–46 The oxidation of interfacial thin films can alter the optical and hygroscopic properties of the host aerosol particulate, leading to subsequent effects on the weather and climate.9–12,47,48
One prevalent oxidant in the atmosphere is ozone. Many molecules in the atmosphere are susceptible to chemical transformation by ozone through reaction pathways such as the addition of oxygen to the carbon–carbon double bond of alkenes49–52 and triple bonds of alkynes,52,53 the addition to aromatic rings,52,54,55 and the oxidation of nitrogen oxides52,56,57 and sulfides.52,55 Furthermore, ozone is readily soluble in water,58,59 which allows aqueous-phase reactions to occur in bulk water.60,61 Ozone resides in the atmosphere at approximate background concentrations of 30–40 ppb in the gas-phase,62 and approximately 30 ppb in the aqueous phase, dissolved in atmospheric water,63 with some spatial and temporal variation in the atmospheric concentrations of ozone across the Earth.64 Owing to the relatively large concentrations of ozone in the atmosphere, the numerous reaction pathways, and its aqueous solubility, ozone is particularly relevant for the study of thin-film oxidation reactions at atmospheric aerosol interfaces.
Some of the methods of characterizing the chemical and physical properties of films at aerosol and aerosol proxy interfaces include spectrometry,65–72 spectroscopy,73–77 chromatography,78–80 surface tensiometry,81–83 neutron and X-ray scattering,10,11,42,48,84–92 and microscopy.93–95 Despite extensive research into the aerosol air–liquid, liquid–liquid, and solid–air interfaces,2,3,10,11,41,42,48,73,84–86,88,91,92,96–104 little research has been done on the role of atmospheric organics at the solid–liquid interface, in part due to the complexity of measuring “buried” interfaces. Neutron reflectometry is one technique that enables the observation of buried interfaces105–107 owing to the ability for neutrons to penetrate certain materials, and has been successfully used elsewhere to investigate organics at the solid–liquid interface and an overview of the recent and historic use of neutron reflectometry to measure the solid–liquid interface can be found in the reviews by Welbourn and Clarke105 and Fragneto-Cusani108 respectively. While neutron reflectometry is well adopted in fields such as biochemistry and soft matter, it remains a novel measurement technique for atmospheric sciences. Neutron reflectometry is the chosen measurement technique for the work presented here owing to its non-destructive, penetrative surface measurement capabilities, and, so far as the authors are aware, the work herein is the first application of neutron reflectometry to consider the atmospheric implications of organic films at the aerosol mineral–water interface.
For this initial investigation into the atmospheric significance of organic thin films at the mineral–water interface, phosphocholine lipids have been used as an atmospheric organic film proxy owing to their ability to self assemble into reproducible organic films at the mineral–water interface.109 The selected atmospheric proxies for this work are DPPC (1,2-dipalmitoylphosphatidylcholine), POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine), and DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine), chosen for their similar molecular structure and varying number of unsaturated carbon–carbon double bonds (0, 1, and 2 respectively). The chemical structure of DPPC, POPC, and DOPC is provided in Fig. 2. The compositional and structural similarities of DPPC, POPC, and DOPC allow for consideration of the significance of the number of unsaturated carbon–carbon double bonds in the extent and duration of organic film oxidation.
 | ||
| Fig. 2 Diagrams of the chemical structure of the three lipids used in this work, 1,2-dipalmitoylphosphatidylcholine (DPPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), and 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC). Diagrams adapted from relevant chemical diagrams at Sigma-Aldrich. | ||
Virkkula et al.110 reported that aerosol samples collected in the Eastern Mediterranean contained three main phospholipid classes: phosphatidylcholines (present in 73% of samples), followed by phosphatidylglycerols (20%) and phosphatidylethanolamines (7%). Griffen et al.111 suggested that dust and soil aerosols provide effective surfaces for biological material, including cellular and fungal debris as well as pollen. The lysis of such biological material has been identified as a potential source of phosphocholine lipids.112,113 Van Acker et al.114 reported the presence of the phosphocholine lipid dipalmitoylphosphatidylcholine (DPPC) in sea spray aerosol in Belgium, with median and maximum air concentrations of 7.1 and 33 pg m−3, respectively. Decesari et al.115 also provided tentative evidence for the presence of phosphocholine lipids in aerosols from the Southern Ocean. More broadly, sea spray aerosol is recognised as a significant source of organic biological material in the atmosphere,116–118 and that some of this material will form films at the air–water interface.10,11,86
The questions this study aims to answer are threefold: what are the thicknesses of films formed by cellular (lipid) material, to what extent are these films removed by exposure to ozone; and, lastly, what are the light scattering impacts of the presence and oxidation of the experimentally measured films on atmospheric aerosol?
2 Methods
The work presented here employs neutron reflectometry, a surface measurement technique, to analyze thin films at the mineral–water interface as a proxy for similar interfaces in atmospheric aerosols. The thickness of thin films before, during, and after oxidation are incorporated into single-particle Mie scattering models to evaluate the atmospheric significance of these films over an atmospheric aerosol's physical lifetime. From the results of the Mie scattering model, the aerosol forcing efficiency is estimated.2.1 Neutron reflectometry
Neutron reflectometry is a surface measurement technique that can derive the thickness, inter-layer roughness, and density of a sample at an interface by measurement of the intensity of a beam of specularly reflected neutrons before, I0, and after, I, interacting with a surface, or “sample”. The reflectivity is then the proportion of the incident beam reflected by the sample, as demonstrated in eqn (1).
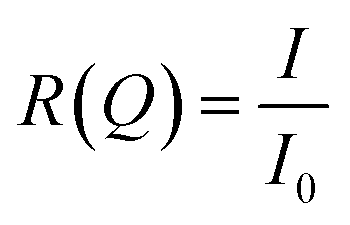 | (1) |
The reflectivity is a function of both neutron wavelength, λn, and angle of incidence, θ, so the momentum transfer, Q, is used to combine these dependencies as defined in eqn (2). A plot of the reflectivity versus momentum transfer is known as the reflectivity profile.
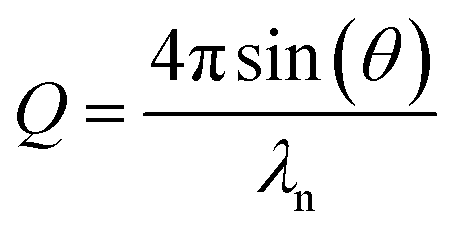 | (2) |
The intensity of the scattering interaction of a neutron with an atom is described by the square of the neutron scattering length, b. For larger materials composed of multiple atoms the average scattering effect per unit volume, known as the scattering length density, ρ, can be calculated as the sum of the scattering lengths of all atoms within the material divided by their collective volume, Vn, as shown in eqn (3).
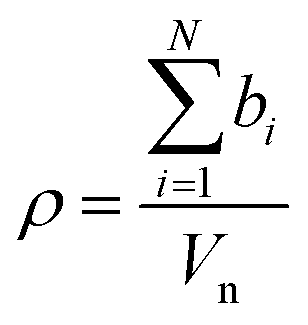 | (3) |
A comparison of the scattering length densities of an uncoated surface and a surface with a lipid bilayer on, and the reflectivities produced by these, is demonstrated in Fig. 3.
 | ||
| Fig. 3 The neutron scattering length density (left) and reflectivity (right) profiles of an uncoated and POPC lipid bilayer coated silica surface. The best fits shown were produced using the Abelès method in Refnx.119 Common parameters are used to describe the silica layer and bulk components. The shaded area, the area between the scattering length density profile of the uncoated surface and the surface with a POPC Bilayer, was used to determine the proportion of material remaining at the interface. | ||
The neutron scattering length of a material is isotopically variable; hence, isotopic substitution can be employed to modify the neutron contrast, effectively masking or enhancing information about certain parts of a sample. In this study, isotopic substitution is utilized to match the neutron scattering length density of water to that of silicon by mixing H2O and D2O, thereby isolating information about the lipid bilayer—this solution is referred to as silicon contrast-matched water (SiMW).
The reflectivity measurements in the work presented here120–124 were made at the ISIS Neutron and Muon Source, UK, using the multi-wavelength instrument INTER,125–127 with preliminary measurements performed at the Institut Laue-Langevin, France. The data were reduced following standard reduction procedures.128,129 The high-flux of INTER125–127 provides a relatively fine time resolution and was used here to monitor the oxidation of organic thin films at the mineral–water interface by aqueous ozone. Throughout the measurements presented here, the neutron footprint was maintained within the sample area of the solid–liquid cell and a constant δq/q resolution of 0.03 was controlled using the incident slits. Two incident angles of 0.7° and 2.3° were used to measure over a momentum transfer range between 0.01–0.3 Å−1.
2.2 Reflectometry data analysis
In order to determine the structure of a film at an interface from neutron reflectometry data, a model is required owing to a phenomena known as “the phase problem”.130 Instead of using the reflectivity directly, a model of the scattering length density profile (scattering length density as a function of the distance normal to the interface) is optimized until it most accurately reproduces a measured reflectivity profile, an example of this is shown in Fig. 3. Models of neutron reflectometry data typically consist of a series of uniform slabs between two infinitely thick bulk materials of given values of scattering length density. Each slab is usually parametrized by a thickness, scattering length density, and interlayer roughness. For solid–liquid experiments, the infinite bulk materials are the liquid subphase and the solid substrate.The work described here has used the Abelès method131 through the fitting package Refnx.119 Model scattering length density profiles were parameterized by a set of five slabs consisting of the native oxide layer on the silicon, and the head and tail regions of both lipid bilayer leaflets, all between bulk scattering length densities of silicon and the relevant aqueous contrast. A schematic diagram of this is shown in Fig. 4. The scattering length density, thickness, solvation, and interfacial roughness of the lipid components, as well as the thickness and roughness of the oxide layer, were variable parameters. The initial values of the pre-fit parameters were determined from existing literature for the lipid components,132 and the values of the scattering length density of the bulk and silica layer were determined from the NIST Scattering Length Density Calculator, with the fitting bounds constrained by reasonable physical limitations. For the measurements of the uncoated interface and the lipid bilayers before oxidation, contrasts of the sample with H2O, D2O, and SiMW were combined in a co-fitting approach, allowing the values of thickness, interlayer roughness, scattering length densities, and solvation to be retained between the models of each contrast while varying the bulk aqueous phase scattering length density, thereby improving confidence in the fit. To fit the data during the ozonolysis reaction, the scattering length density and thickness of the lipid components were allowed to vary with all other model parameters held at the pre-reaction values. In order to account for the possibility of complete removal of the film, the lipid thickness lower bound was set to zero and the lipid scattering length densities were allowed to vary up to the scattering length density of the subphase.
 | ||
| Fig. 4 A simplified representation of a diagram, ‘slab’ model, and simplified scattering length density profile of a mineral–water interface with and without a lipid bilayer, i.e. “coated” and “uncoated”. | ||
2.3 Experimental setup & sample preparation
The lipids DPPC, POPC, and DOPC, obtained from Sigma-Aldrich, were dissolved in ultrapure (≥99.9) chloroform at a concentration of 1 mg mL−1 and stored in amber glass vials at −18 °C in the dark until experimental use. A maximum of 90 minutes prior to measurement, 1 mL of the lipid-chloroform solution was transferred into an amber glass vial, dried gently under nitrogen, and then combined with ultrapure MilliQ water (>18 MΩ cm) and vigorously shaken until a cloudy appearance was achieved. The resulting water–lipid suspension was sonicated using an ultratip sonicator, employing 10-second pulses for 2 minutes to produce lipid vesicles suspended in a water solution (1 mg mL−1). Vesicle formation was evident from the appearance of a transparent solution with a characteristic faint blue tinge when held up to a light source.Single-crystal silicon substrates, oriented along the (001) plane, were obtained from and polished by Crystech to a root-mean-square surface roughness of approximately 2 Å. The surface flatness was <5 µm, and the parallelism was maintained within ∼0.05°. The crystals were thoroughly washed with water, ethanol, and sulfuric acid, then rinsed with water and cleaned in a UV-Ozone Cleaner for 20 minutes, followed by another rinse with water to ensure they were hydrophilic. The blocks were mounted in solid–liquid cells and measured by neutron reflection under three contrasts: H2O, D2O, and SiMW. Lipids were subsequently deposited via vesicle fusion, a widely adopted procedure, where, on contact with the solid interface, the vesicles rupture and self-assemble into bilayers. POPC and DOPC were injected at room temperature, whereas DPPC was heated to 50 °C for deposition.133 The initial state of each bilayer film was measured under D2O, H2O, and SiMW contrasts. The ozonolysis reaction was carried out by flowing silicon contrast-matched water containing known concentrations of aqueous ozone continuously through the cell. All fluid exchanges were performed at a rate of 0.5 mL min−1, regulated with a Jasco HPLC pump with the de-gasser bypassed.
The aqueous solutions containing dissolved ozone were prepared by passing gaseous oxygen through a corona discharge ozonizer at a rate of 0.005 L min−1 to generate gas-phase ozone at mixing ratios exceeding 1000 ppm, which was then bubbled into 200 mL of silicon contrast-matched water whilst stirring. The concentration of ozone in the water did not increase beyond 2 hours, plateauing at a concentration of approximately 42 µmol dm−3, as determined by UV-vis spectroscopy134 using the absorbance at 260 nm and a molar absorption coefficient of 2992 M−1 L cm−1.135 Dilutions of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 v/v (8.3 µmol dm−3) and 1:19 (2.1 µmol dm−3) were prepared and kept in amber bottles in the dark at 4 °C, with a single drop of concentrated sulfuric acid added to each 1 L solution (pH ∼1) to prevent decay.136 Aqueous ozone solutions were prepared a maximum of 2 hours before use.
:4 v/v (8.3 µmol dm−3) and 1:19 (2.1 µmol dm−3) were prepared and kept in amber bottles in the dark at 4 °C, with a single drop of concentrated sulfuric acid added to each 1 L solution (pH ∼1) to prevent decay.136 Aqueous ozone solutions were prepared a maximum of 2 hours before use.
2.4 Mie scattering
Mie scattering is the dominant form of light scattering caused by aerosols in the atmosphere owing to the similarity of their size and the wavelength of sunlight,33,137,138 and is the focus of a wide range of atmospheric33,137,139 and astronomical140–142 research. Despite atmospheric aerosol comprising a variety of complex particle shapes, it is common to approximate the aerosol particles to homogeneous spheres. Typically the aerosol particles are described as one or more concentric spheres with given refractive indices,143 and an equivalent approach is used herein.The atmospheric aerosol considered in the work presented here consist of three components, a mineral core (silica), surrounded by layers of organic (e.g. lipid-like) material and water, and hence are considered as a “multi-layered sphere”, as depicted in Fig. 1. To calculate the optical properties of a multi-layered sphere, the Mie scattering program N-Mie by Voshchinnikov et al.,144 based on the recursive algorithms by Wu and Wang,145 is implemented. N-Mie represents multi-layered spheres as a series of concentric spheres, each with a given radius/thickness and complex refractive index. N-Mie has been used extensively elsewhere, including to determine the effect of thin films on aerosol optical properties.110 The Mie scattering analysis in this work focuses primarily on changes in film thickness rather than chemical composition as the refractive index of the organic material is unlikely to significantly change from chemical reaction with ozone. As an example, the ozonolysis of oleic acid (real component of the refractive index (RI) = 1.47), one of the tail components of the unsaturated lipid, produces products such as nonanoic acid (RI = 1.43) and azelaic acid (RI = 1.43)).146
A single wavelength of light of 500 nm was used in the N-Mie model to be broadly representative of shortwave wavelengths within the lower atmosphere. The optical properties of an atmospheric aerosol were simplified to the single scattering albedo, ω, and the asymmetry parameter (the average of the cosine of the scattering angle of light by the particle), g. The value of ω ranges from 0 (completely absorbed) to 1 (completely scattered), and the value of g ranges from −1 (backward scattering) to 1 (forward scattering). The complex refractive indices of the components of the aerosol model were obtained from experimentally acquired measurements at 500 nm for silica (complex refractive index, n = 1.45 + 0.00i),147 atmospheric organics (n = 1.54 + 0.01i),12,148 and water (n = 1.34 + 0.00i).149 The imaginary components of the complex refractive indices for water and silica were considered negligible. The thickness of the organic film was varied from 0.1 to 10 nm (0.1 nm steps), and the water film from 1 to 100 nm (1 nm steps). The values of ω and g for an aerosol particle with a given organic and water film thickness on a range of mineral core sizes between 10–1000 nm (10 nm steps between 10–100, 100 nm steps between 100–1000) were calculated. The values of ω and g for a “typical” aerosol particle with a given organic and water film thickness were determined by producing a weighted average of the values of ω and g for core sizes in a range of 10 to 1000 nm. The values of ω and g for a given organic and water film thickness were weighted according to a number density distribution of the urban atmospheric aerosol size distribution given by Jaenicke,33 where the weighting was relative to the radius of the core. A summary of the N-Mie modelling parameters used herein is presented in Table 1.
| Layer | Description | δ/nm | Δδ/nm | RI |
|---|---|---|---|---|
| Core | Silica core | 10–1000 | 10/100 | (1.45, 0.00) |
| Inner shell | Organic film | 0.1–10 | 0.1 | (1.54, 0.01) |
| Outer shell | Water film | 1–100 | 1 | (1.34, 0.00) |
To determine the effect of an organic film on the light scattering properties of an aerosol at the mineral–water (and for comparison, air–water) interface, the differences in single scattering albedo Δω and asymmetry parameter Δg were computed, as described in eqn (4) and (5).
| Δω = ωcoated − ωuncoated | (4) |
| Δg = gcoated − guncoated | (5) |
The total size of the coated and uncoated aerosol were kept constant for this comparison, to eliminate any size related changes to ω and g, as shown in Fig. 1.
N-Mie was tested against Bohren & Huffman143 and MiePlot150 to ensure the models were in agreement for single homogeneous sphere and core–shell calculations of ω and g for the size range and optical properties used in the work presented here.
2.5 Aerosol forcing efficiency
The mean of the annual, global effect of an aerosol on the top of atmosphere aerosol forcing, ΔF can be estimated using a given value for aerosol single scattering albedo, ω, backscattering efficiency, b, and optical depth (AOD), τ. In the current work, the values of aerosol single scattering albedo and backscattering efficiency can be calculated from the Mie scattering calculation, allowing the value of aerosol radiative forcing per unit optical depth (aerosol forcing efficiency) as described elsewhere,151,152 presented in eqn (6), to be calculated.
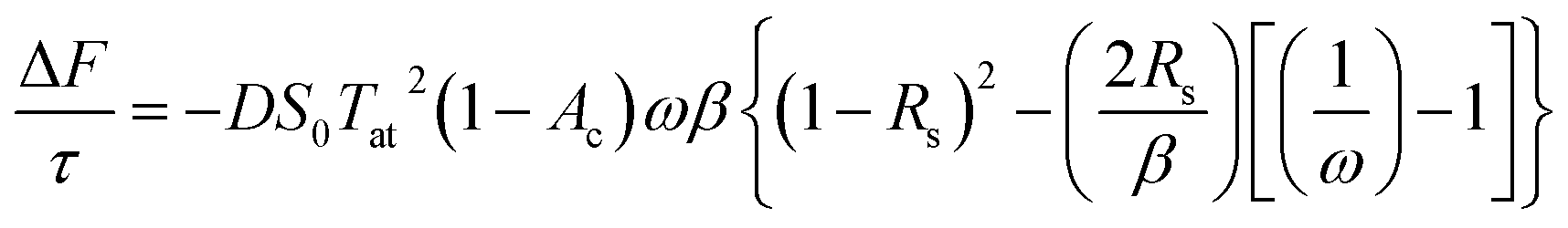 | (6) |
The results calculated using eqn (6) for an aerosol with and without a thin, interfacial film are used to determine the change in aerosol forcing efficiency owing to the presence of a thin film at the aerosol air–water and mineral–water interface 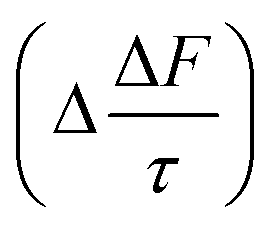 and presented alongside the respective values of Δω and Δg.
and presented alongside the respective values of Δω and Δg.
3 Results
The results presented here combine neutron reflectometry, Mie scattering, and aerosol forcing efficiency calculations. The neutron reflectometry measurements are used to determine the thickness of thin organic films at the mineral–water interface, and investigate how these films change with time (see Fig. 3). The model, N-Mie, is then used to calculate the change in single scattering albedo and asymmetry parameter owing to the presence of thin films of experimentally-measured thicknesses at the air–water versus mineral water interface. Mie scattering calculations are also made for films of varying thicknesses at the mineral–water interface. The results from the N-Mie model were then used to approximate the change in aerosol forcing efficiency owing to the presence of the interfacial organic film.3.1 Initial characterisation of bilayers
Neutron reflectometry measurements confirmed the presence of a deposited film at the mineral–water interface in all samples studied, as evidenced by a noticeable change in the reflectivity profiles before and after vesicle deposition (see Fig. 3). The formation of lipid bilayers at the mineral–water interface was verified by optimizing scattering length density models based on the structure shown in Fig. 4, which reproduced the measured reflectivity data. The optimization of the scattering length density models provided values for the approximate thickness of the overall bilayer used in the Mie Modelling. Note that the surface coverage of lipid bilayers can vary between substrates.3.2 Exposure of films to ozone
Fig. 5 shows an approximation for the relative proportion of the DPPC, POPC and DOPC bilayers remaining at the mineral–water interface as a function of the duration the films were exposed to 42 µmol per dm3 aqueous ozone. The proportion of material in the POPC and DOPC bilayers decreased over the first ∼1000 s, to approximately 40% and 10% of the original film respectively, at which point no more material was lost for the rest of the measurement period. The proportion of material in the DPPC bilayer did not appear to be altered by exposure to 42 µmol dm−3 ozone over ∼8000 s. | ||
| Fig. 5 The proportion of material remaining at the mineral–water interface as a function of time exposed to 42 µmol dm−3 ozone, determined directly from the total neutron counts. | ||
Fig. 6 presents the scattering length density of the POPC and DOPC bilayers as a function of time and distance from the interface, when exposed to aqueous ozone of concentrations of 8.3 and 2.1 µmol dm−3, respectively. As regions of the plot darken with time, this can be interpreted as organic material being replaced with water, highlighting a reduction in overall film thickness but some material remains. The decrease in ozone concentration slowed the rate of loss of material from the interface from ∼1000 s in 42 µmol dm−3 ozone, to ∼5000 s for POPC in 8.3 µmol dm−3, and ∼30000 s for DOPC in 2.1 µmol dm−3. The use of significantly lower concentrations of ozone allowed the acquisition of the more detailed structural information presented in Fig. 6. The method used in Fig. 6 provides unprecedented graphic detail regarding what is happening at the mineral–water interface and allows mechanistic details to be elucidated.
 | ||
| Fig. 6 The scattering length density profiles of POPC and DOPC bilayers at the mineral–water interface as a function of time when exposed to continuously flown aqueous ozone (8.3 µmol dm−3 for POPC, 2.1 µmol dm−3 for DOPC). The brightness intensity of the plot is determined by the scattering length density, allowing clear identification of different film components and changes over time. Sample components with a scattering length density equal to the scattering length density of the solution, i.e., 2.065 × 10−6 Å−2, are shown in black, while lighter shading indicates a lower scattering length density. Thus, a decrease in component brightness is considered representative of the film decaying away and being replaced with silicon contrast-matched water. Aqueous solutions of ozone are added when the exposure time is zero. | ||
3.3 Light scattering and aerosol forcing efficiency
Fig. 7 shows the changes in single scattering albedo, asymmetry parameter and aerosol forcing efficiency of a model mineral aerosol owing to the presence of a 1 nm thick organic film at the mineral–water or air–water interface (as shown in Fig. 1) as a function of the thickness of the water layer, where the water layer thickness varies between 1–100 nm. For the model particle with the organic layer at the air–water interface, the value of Δω varies from −3.5 × 10−3 to −0.8 × 10−3, the values of Δg vary from 3.8 × 10−3 to 0.5 × 10−3, and the values of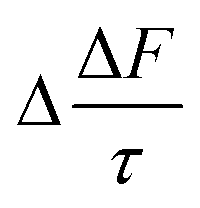 vary from 0.11 to 0.03 W m−2. For the model particle with the organic film at the mineral–water interface, the value of Δω vary from −3.4 × 10−3 to −0.3 × 10−3, Δg varies from 3.6 × 10−3 to −0.6 × 10−3, and the values of
vary from 0.11 to 0.03 W m−2. For the model particle with the organic film at the mineral–water interface, the value of Δω vary from −3.4 × 10−3 to −0.3 × 10−3, Δg varies from 3.6 × 10−3 to −0.6 × 10−3, and the values of 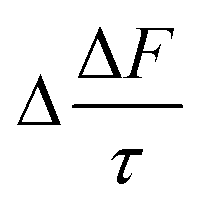 vary from 0.11 to 0.01 W m−2.
vary from 0.11 to 0.01 W m−2.
 | ||
| Fig. 7 Modeled changes in single scattering albedo, asymmetry parameter, and aerosol forcing efficiency for a model water-coated silica particle owing to the presence of a 1 nm thick organic film located at either the mineral–water or air–water interface. The parameters are shown as a function of water layer thickness, varying from 1 to 100 nm. | ||
Fig. 8 shows the values of Δω, Δg, and  as a function of the organic film thickness for a model particle with an organic film present at the mineral–water interface. The thicknesses of the organic film vary between 0.1–10 nm, and the water layer's thickness is 10 nm. The results in Fig. 8 demonstrate that the value of ω of the model particle decreases and the values of g and
as a function of the organic film thickness for a model particle with an organic film present at the mineral–water interface. The thicknesses of the organic film vary between 0.1–10 nm, and the water layer's thickness is 10 nm. The results in Fig. 8 demonstrate that the value of ω of the model particle decreases and the values of g and  increase as a function of the thickness of the organic film. The greatest effect on the model particles considered here was for an organic film at 10 nm, which produced a value of −0.016 for Δω, 0.025 for Δg, and 0.52 W m−2 for
increase as a function of the thickness of the organic film. The greatest effect on the model particles considered here was for an organic film at 10 nm, which produced a value of −0.016 for Δω, 0.025 for Δg, and 0.52 W m−2 for  .
.
 | ||
| Fig. 8 The calculated change in single scattering albedo, asymmetry parameter, and aerosol forcing efficiency of a silica particulate coated in a 10 nm thick water film owing to the presence of an organic film of 0.1–10 nm thickness at the mineral–water interface. The dashed lines provide a guide to contextualise the results of the experimental component of this work. | ||
The Mie scattering calculations presented here were performed for light with a wavelength of 500 nm, chosen to be broadly representative of shortwave solar radiation in the lower atmosphere. At longer wavelengths, the size parameter of a given aerosol particle decreases which tends to increase the ratio of forward scattered light, relative to backward scattered light, thus increasing the value of the asymmetry parameter, g. It could be expected that there would be a decrease light absorption by the particle with wavelength in the visible regime, increasing the value of the single scattering albedo, ω. The inverse is expected at shorter wavelengths. However, it should be noted that such suggestions are tendencies only and a Mie scattering calculation should be performed. Also given the complex interplay of scattering and absorption effects introduced by a thin interfacial film, the values of Δω and Δg for an aerosol particle of a given morphology are wavelength-dependent, and accurate determination would require individual calculation at each wavelength of interest.
4 Discussion
4.1 Film oxidation
The results depicted in Fig. 6 demonstrate that lipid bilayers at the mineral–water interface, containing molecules with unsaturated carbon–carbon double bonds, are susceptible to oxidation by aqueous ozone which can penetrate the bilayer. After the lipid bilayers were exposed to aqueous ozone for extended periods of time (POPC; 42 µmol dm−3 for 6000 s & 8.3 µmol dm−3 for 40000 s, DOPC; 42 µmol dm−3 for 6000 s & 2.1 µmol dm−3 40000 s) a product film appeared to form that persisted at the mineral–water interface and resisted further oxidation by ozone. The proportion of the original organic film removed from the mineral–water interface due to oxidation by aqueous ozone depended on the number of unsaturated double bonds in the film, with higher ozone concentrations increasing the rate of material loss. The results presented here are consistent with the results of both experimental observations88,153,154 and molecular dynamics simulations.155–158 Thompson et al.159 studied the oxidation of a phosphocholine lipid, POPC, at the air–water interface and noted that any reactive intermediate (by inference here a Criegee intermediate) does not react intermolecularly with another lipid molecule or intra-moleculary with the other hydrocarbon ‘tail’ of the lipid but that bond scission occurs with material leaving the interface. The scattering length density profiles with time in Fig. 6 support equivalent chemistry occurring at the silica–water interface as shown in the simplified and stylised chemical mechanisms in Fig. 9 and 10.
 | ||
| Fig. 9 A ‘cartoon’ chemical mechanism for the initial reaction of aqueous ozone with a bilayer of POPC at the air–water interface, demonstrating the ozonolysis and bond scission of the carbon–carbon double bond to produce two carboxylic acid groups and the loss of nonanoic acid to the bulk solution based on the chemistry reported in Thompson et al.159 Note the ozonolysis of the double bond is shown producing carboxylic acid groups but may produce aldehydes, alcohol or peroxy hydroxy groups. The diagram is for the initial reaction and the remaining double bonds in the film will react removing some material from the interface but leaving a slightly thinner film with less material at the interface as consistent with Fig. 6. | ||
 | ||
| Fig. 10 A ‘cartoon’ chemical mechanism for the initial reaction of aqueous ozone with a bilayer of DOPC at the air–water interface, demonstrating the ozonolysis and bond scission of the carbon–carbon double bond to produce two carboxylic acid groups and the loss of two nonanoic acids and remnants of the DOPC lipid to the bulk solution based on the chemistry reported in Thompson et al.159 The diagram is for the initial reaction and the remaining double bonds in the film will react removing the majority of the lipid material from the interface as consistent with Fig. 6. | ||
4.2 Light scattering and aerosol forcing efficiency
The experimental component of this work measured the thicknesses of lipid bilayers at the mineral–water interface, the values of which were used to approximate the thickness of lipid-like films at the same interface on atmospheric aerosol particulates. The lipid films measured before and after oxidation (approximately 4 nm, 1.6 nm, and 0.4 nm based on the proportion of material remaining after oxidation shown in Fig. 5), and thus the light-scattering modelling and aerosol-forcing efficiency calculations based on these values, represent interfacial films that are considerably thinner than those investigated in many core–shell studies such as Hu et al. (2021)160 (core is ∼12% of the total particle mass), Hu et al. (2023)161 (∼2%) and Hartikainen et al. (2025)162 (0.02–10%). Comparisons between the results presented here and studies of core–shell aerosols with substantially thicker films should be made with caution, as differences in organic film or shell thickness may limit the direct comparability of these works.The light scattering effects of thin films on atmospheric aerosol have previously been modelled using experimental measurements performed by the authors of this work, including using complex refractive indexes determined from laser trapping experiments12,148 and film thicknesses obtained through X-ray and neutron reflectometry.10,11 These works10–12,148 conclude that thin, interfacial films at the air–water, and mineral–air, interface have atmospherically significant effects on the light scattering of atmospheric aerosol particles. The results presented here in Fig. 7 demonstrate that organic films of 1 nm thickness broadly have a similar order of magnitude effect on the light scattering of an aerosol particle regardless of whether the film is located at the air–water or mineral–water interface, over a range of water layer thicknesses of 1–100 nm. Interestingly, as the thickness of the water layer increases, both Δω and  follow very similar trends between the model with the organic film at the air–water, and mineral–water interface. However, there is some variation in the changes to Δg as a function of water layer thickness.
follow very similar trends between the model with the organic film at the air–water, and mineral–water interface. However, there is some variation in the changes to Δg as a function of water layer thickness.
Broadly, Fig. 8 demonstrates that for a model aerosol particle with an organic film at the mineral–water interface, both the light absorbed and the light forward scattered increase as a function of the organic film thickness. When considering the experimental observations of the current work with the results of the modelling shown in Fig. 8 (where the dashed lines indicate the thicknesses of pristine and oxidised films), it is evident that the oxidation of lipid-like films containing unsaturated carbon–carbon double bonds at the mineral–water interface would decrease, but not entirely remove, the influence of the organic film on the optical properties of the aerosol particle. For example, the results depicted in Fig. 8 indicate that oxidation of a pristine lipid-like film (approximately 4 nm) by aqueous ozone (to approximately 0.4 nm) would change the values of Δω, Δg, and  from −0.08, 0.011, and 0.26 W m−2 to −0.001, 0.001 and 0.03 W m−2, respectively.
from −0.08, 0.011, and 0.26 W m−2 to −0.001, 0.001 and 0.03 W m−2, respectively.
A similar approach to the current work was taken in Shepherd et al.11 where the authors estimated the changes to ω and g owing to the oxidation of an organic film at the atmospheric aerosol air–water and mineral–air interface by gas-phase hydroxyl radicals, and this oxidation is approximated to be a halving of the film thickness. Note, that Shepherd et al. uses a core–shell (two-layer) system of a mineral core and organic shell, or water core and organic shell. Similarly to the current work, Shepherd et al. suggest that the oxidation of thin films at the air–water and solid–air interface of “urban” aerosol would result in an increase in the ω of the aerosol particulate and a decrease in g. The results in Fig. 9 of Shepherd et al. indicate the oxidation of a 4 nm thick organic thin film will cause an increase in ω of approximately 0.02 and a decrease in g of 0.01. Comparatively, in the work presented here, the oxidation of an organic film of 4 nm thickness is found to cause an increase of 0.004 in ω and a decrease of 0.004 in g. Comparison with Shepherd et al. shows that oxidation of thin organic films induces similar-magnitude changes in aerosol ω and g, regardless of the film's interface.
The results of the work presented here and in Shepherd et al. are applicable to particulates with highly scattering, weakly absorbing cores, however the effect of organic thin films may have different behaviours on more absorbing cores such as black carbon. Hu et al.161 demonstrate experimentally that the development of organic films, with similar absorption to those used here, on the surface of black carbon cores results in an increase in the aerosol's ω as the film thickness increases. While Hu et al. use significantly larger shell to core ratios than here, a comparison of their work and the work presented here demonstrates the variable effects that organic thin films may have on different host particulates.
5 Atmospheric implications
It is demonstrated here that the presence of an organic film at the presence of an organic film at the mineral–water interface of an atmospheric aerosol particle has significant effects on the optical properties of the aerosol, and these effects are of a similar order of magnitude regardless of the interface the film is present at. As shown in Fig. 7, when the thickness of the water component of a model aerosol particle changes, such as during cloud processing, the ω and of each aerosol particle model varies similarly regardless of the interface the organic film is present at, while changes in g are different. The differences in Δg highlight an interface-specific effect that organic thin films may have on aerosol optical properties, suggesting that films of the same thickness can influence the atmospheric impact of aerosols in similar yet distinct ways depending on the interface it is present at. Furthermore, the similarities in magnitude of the values of Δg and Δω in this study and those from Shepherd et al.11 indicate that, similarly to Shepherd's conclusions for thin films at the air–water interface, organic films at the mineral–water interface may also affect the top-of-atmosphere albedo.
of each aerosol particle model varies similarly regardless of the interface the organic film is present at, while changes in g are different. The differences in Δg highlight an interface-specific effect that organic thin films may have on aerosol optical properties, suggesting that films of the same thickness can influence the atmospheric impact of aerosols in similar yet distinct ways depending on the interface it is present at. Furthermore, the similarities in magnitude of the values of Δg and Δω in this study and those from Shepherd et al.11 indicate that, similarly to Shepherd's conclusions for thin films at the air–water interface, organic films at the mineral–water interface may also affect the top-of-atmosphere albedo.
The effect of an organic film at the mineral–water interface has been shown to increase in magnitude as a function of the organic film thickness, depicted in Fig. 8. The increase in  as a function of organic film thickness shown in Fig. 8 effectively demonstrates the warming effect on the atmosphere of organic films at the mineral–water interface of atmospheric particles. The experimental component of this work has demonstrated that the oxidation of films containing proxies for atmospheric organic material would reduce the thickness of these films, ultimately having a small cooling effect. The experimental component of the work presented here also validates the findings of the Mie modelling by demonstrating that atmospherically relevant organic materials can adsorb to the mineral–water interface, replicating one formation pathway of organic thin films at the atmospheric aerosol mineral–water interface. Atmospherically relevant proxy organic films have been shown to remain at the interface despite a continuous flow of water, suggesting that films of similar thickness and material may remain stable at the aerosol mineral–water interface in atmospheric contexts, unless disrupted by photochemical reaction.
as a function of organic film thickness shown in Fig. 8 effectively demonstrates the warming effect on the atmosphere of organic films at the mineral–water interface of atmospheric particles. The experimental component of this work has demonstrated that the oxidation of films containing proxies for atmospheric organic material would reduce the thickness of these films, ultimately having a small cooling effect. The experimental component of the work presented here also validates the findings of the Mie modelling by demonstrating that atmospherically relevant organic materials can adsorb to the mineral–water interface, replicating one formation pathway of organic thin films at the atmospheric aerosol mineral–water interface. Atmospherically relevant proxy organic films have been shown to remain at the interface despite a continuous flow of water, suggesting that films of similar thickness and material may remain stable at the aerosol mineral–water interface in atmospheric contexts, unless disrupted by photochemical reaction.
The work presented here also shows that organic films containing unsaturated carbon–carbon double bonds are partially removed from the mineral–water interface through oxidation by aqueous ozone. The removal of organic films from the mineral–water interface reduces the film thickness in a manner that may influence aerosol optical properties. However, despite oxidation, some organic material remains at the interface regardless of the number of carbon–carbon double bonds in the organic material. The persistence of some material at the mineral–water interface suggests that aqueous ozone may not be sufficient for the complete removal of organic films, allowing these films, and their impacts on ω g, and  to persist throughout an aerosol's atmospheric lifetime, depending on the action of other atmospherically relevant oxidants.
to persist throughout an aerosol's atmospheric lifetime, depending on the action of other atmospherically relevant oxidants.
6 Conclusions
Three lipids with varying numbers of carbon–carbon double bonds, DPPC, POPC, and DOPC, were deposited at the silica–water interface of a silicon block and all appeared to form films of approximately 4 nm in thickness.Exposure of the POPC and DOPC films to aqueous ozone resulted in the films decaying to approximately 40% and 10% of their respective pristine films, where the product films appeared to resist further oxidation by ozone. The DPPC film did not appear to change under exposure to aqueous ozone.
The results of the Mie modelling and aerosol forcing efficiency approximation indicated that a film thickness of up to 10 nm could decrease the aerosol particle's single-scattering albedo (ω) by 0.016, increase the asymmetry parameter (g) by 0.025, and increase the aerosol forcing efficiency  by 0.52 W m−2. It was also shown that organic films of the same thickness have a similar order of magnitude of effects on the light scattering of aerosol particle regardless of whether the film is at the air–water or mineral–water interface. Consideration of the thickness measurements taken using neutron reflectometry demonstrated that the oxidation of organic films at the mineral–water interface of an atmospheric aerosol can significantly alter the optical effects the film has on the aerosol.
by 0.52 W m−2. It was also shown that organic films of the same thickness have a similar order of magnitude of effects on the light scattering of aerosol particle regardless of whether the film is at the air–water or mineral–water interface. Consideration of the thickness measurements taken using neutron reflectometry demonstrated that the oxidation of organic films at the mineral–water interface of an atmospheric aerosol can significantly alter the optical effects the film has on the aerosol.
The results presented here demonstrate the ability for atmospherically relevant organic material to adsorb at the mineral–water interface, forming thin films that can have important optical effects on their host aerosol. Thin films at the mineral–water interface have been shown to be susceptible to oxidation by ozone, relative to the number of carbon–carbon double bonds present within the material, and it has been shown that this oxidation can result in significant changes to the optical effects of the thin film which may be interpreted as a minor cooling effect. Owing to the results presented here, it is evident that organic thin films at the aerosol mineral–water interface should be considered for implementation into larger scale aerosol chemistry models in order to further determine the significance of these thin films in the atmosphere.
Author contributions
Contributions have been defined by the CRediT contributor role taxonomy. Edward J. Stuckey: data curation, formal analysis, investigation, methodology, software, validation, visualization, writing – original draft, writing – review & editing. Rebecca J. L. Welbourn: data curation, formal analysis, funding acquisition, investigation, methodology, project administration, resources, software, supervision, validation, writing – review & editing. Tobias W. D. Robson: formal analysis, investigation, writing – review & editing. Philipp Gutfreund: investigation, resources, writing – review & editing. Katherine C. Thompson: investigation, writing – review & editing. Adrian R. Rennie: investigation, writing – review & editing. Martin D. King: conceptualization, funding acquisition, investigation, methodology, project administration, resources, supervision, validation, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Data availability
The data presented in this article are available as part of the ISIS & ILL open access data policy. See DOI: https://doi.org/10.5286/ISIS.E.RB1820245, https://doi.org/10.5286/ISIS.E.RB2210207-1, https://doi.org/10.5286/ISIS.E.RB2210371-2, https://doi.org/10.5286/ISIS.E.RB2210371-1, https://doi.org/10.5291/ILL-DATA.9-10-1517 and https://doi.org/10.5291/ILL-DATA.9-10-1241. Data for experiment 9-10-1079 is available through the ILL legacy data portal for measurement numbers 082149–082474.Acknowledgements
The authors would like to acknowledge the work by the support network at the ISIS Neutron and Muon Source and the Institut Laue-Langevin for providing the beam time and making this experiment possible by maintaining the facility, providing engineering help for our sample environment and providing constant support throughout the experiments RB1820245, RB2210207, RB2210371(ISIS), and 9-10-1079, 9-10-1517, and 9-10-1241(ILL). Acknowledgements are made to NERC (NE/T00732X/1) for funding this work.References
- P. S. Gill, T. E. Graedel and C. J. Weschler, Rev. Geophys., 1983, 21, 903 CrossRef CAS.
- D. J. Donaldson and V. Vaida, Chem. Rev., 2006, 106, 1445–1461 CrossRef CAS PubMed.
- J. F. Davies, R. E. Miles, A. E. Haddrell and J. P. Reid, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8807–8812 CrossRef CAS PubMed.
- D. B. Collins and V. H. Grassian, in Gas-liquid Interfaces in the Atmosphere: Impacts, Complexity, and Challenges, Elsevier, 2018, pp. 271–313 Search PubMed.
- G. B. Ellison, A. F. Tuck and V. Vaida, J. Geophys. Res. Atmos., 1999, 104, 11633–11641 CrossRef CAS.
- H. Tervahattu, J. Juhanoja, V. Vaida, A. F. Tuck, J. Niemi, K. Kupiainen, M. Kulmala and H. Vehkamäki, J. Geophys. Res. Atmos., 2005, 110, D06207 CrossRef.
- D. A. Lack and C. D. Cappa, Atmos. Chem. Phys., 2010, 10, 4207–4220 CrossRef CAS.
- C. H. Song and G. R. Carmichael, Atmos. Environ., 1999, 33, 2203–2218 CrossRef CAS.
- T. Eliason, J. Gilman and V. Vaida, Atmos. Environ., 2004, 38, 1367–1378 CrossRef CAS.
- R. H. Shepherd, M. D. King, A. R. Rennie, A. D. Ward, M. M. Frey, N. Brough, J. Eveson, S. D. Vento, A. Milsom, C. Pfrang, M. W. A. Skoda and R. J. L. Welbourn, Environ. Sci.: Atmos., 2022, 2, 574–590 CAS.
- R. H. Shepherd, M. D. King, A. D. Ward, E. J. Stuckey, R. J. L. Welbourn, N. Brough, A. Milsom, C. Pfrang and T. Arnold, Atmos. Chem. Phys., 2025, 25, 2569–2588 CrossRef CAS.
- C. R. Barker, M. L. Poole, M. Wilkinson, J. Morison, A. Wilson, G. Little, E. J. Stuckey, R. J. L. Welbourn, A. D. Ward and M. D. King, Environ. Sci.: Atmos., 2023, 3, 1008–1024 CAS.
- C. S. Zender, R. L. L. Miller and I. Tegen, EOS Trans. Am. Geophys. Union, 2004, 85, 509–512 CrossRef.
- P. R. Buseck and M. Pósfai, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3372–3379 Search PubMed.
- M. O. Andreae, R. J. Charlson, F. Bruynseels, H. Storms, R. V. Grieken and W. Maenhaut, Science, 1986, 232, 1620–1623 CrossRef CAS PubMed.
- C. Textor, M. Schulz, S. Guibert, S. Kinne, Y. Balkanski, S. Bauer, T. Berntsen, T. Berglen, O. Boucher, M. Chin, F. Dentener, T. Diehl, R. Easter, H. Feichter, D. Fillmore, S. Ghan, P. Ginoux, S. Gong, A. Grini, J. Hendricks, L. Horowitz, P. Huang, I. Isaksen, I. Iversen, S. Kloster, D. Koch, A. Kirkevåg, J. E. Kristjansson, M. Krol, A. Lauer, J. F. Lamarque, X. Liu, V. Montanaro, G. Myhre, J. Penner, G. Pitari, S. Reddy, O. Seland, P. Stier, T. Takemura and X. Tie, Atmos. Chem. Phys., 2006, 6, 1777–1813 CrossRef CAS.
- R. A. McCormick and J. H. Ludwig, Science, 1967, 156, 1358–1359 CrossRef CAS.
- R. W. Bergstrom, P. Pilewskie, P. B. Russell, J. Redemann, T. C. Bond, P. K. Quinn and B. Sierau, Atmos. Chem. Phys., 2007, 7, 5937–5943 CrossRef CAS.
- J. Li, B. E. Carlson, Y. L. Yung, D. Lv, J. Hansen, J. E. Penner, H. Liao, V. Ramaswamy, R. A. Kahn, P. Zhang, O. Dubovik, A. Ding, A. A. Lacis, L. Zhang and Y. Dong, Nat. Rev. Earth Environ., 2022, 3, 363–379 CrossRef CAS.
- J. F. Kok, T. Storelvmo, V. A. Karydis, A. A. Adebiyi, N. M. Mahowald, A. T. Evan, C. He and D. M. Leung, Nat. Rev. Earth Environ., 2023, 4, 71–86 CrossRef.
- V. A. Karydis, P. Kumar, D. Barahona, I. N. Sokolik and A. Nenes, J. Geophys. Res. Atmos., 2011, 116, D23204 CrossRef.
- P. Kumar, I. N. Sokolik and A. Nenes, Atmos. Chem. Phys., 2011, 11, 3527–3541 CrossRef CAS.
- H. Herich, T. Tritscher, A. Wiacek, M. Gysel, E. Weingartner, U. Lohmann, U. Baltensperger and D. J. Cziczo, Phys. Chem. Chem. Phys., 2009, 11, 7804 RSC.
- S. E. Bauer, Y. Balkanski, M. Schulz, D. A. Hauglustaine and F. Dentener, J. Geophys. Res. Atmos., 2004, 109, D02304 CrossRef.
- F. J. Dentener, G. R. Carmichael, Y. Zhang, J. Lelieveld and P. Crutzen, J. Geophys. Res. Atmos., 1996, 101(22), 869–889 Search PubMed.
- V. H. Grassian, J. Phys. Chem. A, 2002, 106, 860–877 CrossRef CAS.
- C. R. Usher, A. E. Michel and V. H. Grassian, Chem. Rev., 2003, 103, 4883–4940 CrossRef CAS PubMed.
- T. Claquin, M. Schulz, Y. Balkanski and O. Boucher, Tellus B, 1998, 50, 491 CrossRef.
- L. Su and O. B. Toon, Atmos. Chem. Phys., 2011, 11, 3263–3280 CrossRef CAS.
- M. Masmoudi, M. Chaabane, D. Tanré, P. Gouloup, L. Blarel and F. Elleuch, Atmos. Res., 2003, 66, 1–19 Search PubMed.
- N. Mahowald, C. Luo, J. Corral and C. Zender, J. Geophys. Res. Atmos., 2003, 108, 4206 Search PubMed.
- O. Dubovik, B. Holben, T. F. Eck, A. Smirnov, Y. J. Kaufman, M. D. King, D. Tanré and I. Slutsker, J. Atmos. Sci., 2002, 59, 590–608 CrossRef.
- R. Jaenicke, Science, 2005, 308, 73 Search PubMed.
- C. Denjean, F. Cassola, A. Mazzino, S. Triquet, S. Chevaillier, N. Grand, T. Bourrianne, G. Momboisse, K. Sellegri, A. Schwarzenbock, E. Freney, M. Mallet and P. Formenti, Atmos. Chem. Phys., 2016, 16, 1081–1104 CrossRef CAS.
- E. R. Gibson, Geophys. Res. Lett., 2006, 33, L13811 Search PubMed.
- C. L. Ryder, E. J. Highwood, A. Walser, P. Seibert, A. Philipp and B. Weinzierl, Atmos. Chem. Phys., 2019, 19, 15353–15376 Search PubMed.
- M. Tang, X. Huang, K. Lu, M. Ge, Y. Li, P. Cheng, T. Zhu, A. Ding, Y. Zhang, S. Gligorovski, W. Song, X. Ding, X. Bi and X. Wang, Atmos. Chem. Phys., 2017, 17, 11727–11777 Search PubMed.
- P. K. Mogili, K. H. Yang, M. A. Young, P. D. Kleiber and V. H. Grassian, J. Geophys. Res. Atmos., 2007, 112(D21204) CAS.
- S. E. Bauer, M. I. Mishchenko, A. A. Lacis, S. Zhang, J. Perlwitz and S. M. Metzger, J. Geophys. Res. Atmos., 2007, 112, D06307 CrossRef.
- O. Möhler, S. Benz, H. Saathoff, M. Schnaiter, R. Wagner, J. Schneider, S. Walter, V. Ebert and S. Wagner, Environ. Res. Lett., 2008, 3, 025007 CrossRef.
- M. Brüggemann, N. Hayeck, C. Bonnineau, S. Pesce, P. A. Alpert, S. Perrier, C. Zuth, T. Hoffmann, J. Chen and C. George, Faraday Discuss., 2017, 200, 59–74 Search PubMed.
- S. H. Jones, M. D. King, A. R. Rennie, A. D. Ward, R. A. Campbell and A. V. Hughes, J. Phys. Chem. A, 2023, 127, 8922–8934 CrossRef CAS PubMed.
- K. S. Docherty and P. J. Ziemann, J. Phys. Chem. A, 2006, 110, 3567–3577 CrossRef CAS PubMed.
- I. J. George, A. Vlasenko, J. G. Slowik, K. Broekhuizen and J. P. D. Abbatt, Atmos. Chem. Phys., 2007, 7, 4187–4201 CrossRef CAS.
- A. T. Lambe, A. T. Ahern, L. R. Williams, J. G. Slowik, J. P. S. Wong, J. P. D. Abbatt, W. H. Brune, N. L. Ng, J. P. Wright, D. R. Croasdale, D. R. Worsnop, P. Davidovits and T. B. Onasch, Atmos. Meas. Tech., 2011, 4, 445–461 CrossRef CAS.
- C. Y. Lim, E. C. Browne, R. A. Sugrue and J. H. Kroll, Geophys. Res. Lett., 2017, 44, 2949–2957 CrossRef CAS.
- A. T. Lambe, C. D. Cappa, P. Massoli, T. B. Onasch, S. D. Forestieri, A. T. Martin, M. J. Cummings, D. R. Croasdale, W. H. Brune, D. R. Worsnop and P. Davidovits, Environ. Sci. Technol., 2013, 47, 6349–6357 Search PubMed.
- M. D. King, A. R. Rennie, K. C. Thompson, F. N. Fisher, C. C. Dong, R. K. Thomas, C. Pfrang and A. V. Hughes, Phys. Chem. Chem. Phys., 2009, 11, 7699–7707 RSC.
- S. E. Paulson and J. J. Orlando, Geophys. Res. Lett., 1996, 23, 3727–3730 CrossRef CAS.
- A. R. Rickard, D. Johnson, C. D. McGill and G. Marston, J. Phys. Chem. A, 1999, 103, 7656–7664 Search PubMed.
- R. E. Keay and G. A. Hamilton, J. Am. Chem. Soc., 1976, 98, 6578–6582 Search PubMed.
- P. S. Bailey, Chem. Rev., 1958, 58, 925–1010 CrossRef CAS.
- W. B. Demore, Int. J. Chem. Kinet., 1971, 3, 161–173 CrossRef CAS.
- S. N. Chu, S. Sands, M. R. Tomasik, P. S. Lee and V. F. McNeill, J. Am. Chem. Soc., 2010, 132, 15968–15975 Search PubMed.
- Y. Pi, J. Schumacher and M. Jekel, Water Res., 2005, 39, 83–88 CrossRef CAS PubMed.
- R. R. Dickerson, D. H. Stedman and A. C. Delany, J. Geophys. Res., Oceans, 1982, 87, 4933–4946 CrossRef CAS.
- E. Berezina, K. Moiseenko, A. Skorokhod, N. V. Pankratova, I. Belikov, V. Belousov and N. F. Elansky, Atmosphere, 2020, 11, 1262 CrossRef CAS.
- J. A. Roth and D. E. Sullivan, Ind. Eng. Chem. Fundam., 1981, 20, 137–140 Search PubMed.
- J. Sotelo, F. Beltrán, F. Benitez and J. Beltrán-Heredia, Water Res., 1989, 23, 1239–1246 CrossRef CAS.
- C. G. Hewes and R. R. Davison, AIChE J., 1971, 17, 141–147 Search PubMed.
- M. S. Siddiqui, G. L. Amy and B. D. Murphy, Water Res., 1997, 31, 3098–3106 CrossRef CAS.
- R. E. Brandt, J. J. Schwab, P. W. Casson, U. K. Roychowdhury, D. Wolfe, K. L. Demerjian, K. L. Civerolo, O. V. Rattigan and H. D. Felton, Aerosol Air Qual. Res., 2016, 16, 873–884 CrossRef CAS.
- B. Ervens, C. George, J. E. Williams, G. V. Buxton, G. A. Salmon, M. Bydder, F. Wilkinson, F. Dentener, P. Mirabel, R. Wolke and H. Herrmann, J. Geophys. Res. Atmos., 2003, 108, 4426 Search PubMed.
- V. P. Aneja, C. S. Claiborn, Z. Li and A. Murthy, Atmos. Environ., 1994, 28, 1781–1790 Search PubMed.
- X. Zhang, K. M. Barraza and J. L. Beauchamp, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3255–3260 CrossRef CAS PubMed.
- R. M. Kirpes, D. Bonanno, N. W. May, M. Fraund, A. J. Barget, R. C. Moffet, A. P. Ault and K. A. Pratt, ACS Cent. Sci., 2019, 5, 1760–1767 CrossRef CAS.
- J. V. Trueblood, M. R. Alves, D. Power, M. V. Santander, R. E. Cochran, K. A. Prather and V. H. Grassian, ACS Earth Space Chem., 2019, 3, 1614–1623 Search PubMed.
- J. A. Faust and J. P. D. Abbatt, J. Phys. Chem. A, 2019, 123, 2114–2124 CrossRef CAS PubMed.
- H. Yu, W. Li, Y. Zhang, P. Tunved, M. Dall'Osto, X. Shen, J. Sun, X. Zhang, J. Zhang and Z. Shi, Atmos. Chem. Phys., 2019, 19, 10433–10446 Search PubMed.
- M. Passananti, L. Kong, J. Shang, Y. Dupart, S. Perrier, J. Chen, D. J. Donaldson and C. George, Angew. Chem., Int. Ed., 2016, 55, 10336–10339 CrossRef CAS PubMed.
- T. C. Burdette and A. A. Frossard, J. Environ. Sci., 2021, 108, 164–174 Search PubMed.
- N. Hayeck, I. Mussa, S. Perrier and C. George, ACS Earth Space Chem., 2020, 4, 1247–1253 Search PubMed.
- J. Shang, M. Passananti, Y. Dupart, R. Ciuraru, L. Tinel, S. Rossignol, S. Perrier, T. Zhu and C. George, Environ. Sci. Technol. Lett., 2016, 3, 67–72 Search PubMed.
- M. Xu, N. T. Tsona, S. Cheng, J. Li and L. Du, Sci. Total Environ., 2021, 782, 146893 CrossRef CAS PubMed.
- Y. Dubowski, J. Vieceli, D. J. Tobias, A. Gomez, A. Lin, S. A. Nizkorodov, T. M. McIntire and B. J. Finlayson-Pitts, J. Phys. Chem. A, 2004, 108, 10473–10485 CrossRef CAS.
- H. C. Allen, J. M. Laux, R. Vogt, B. J. Finlayson-Pitts and J. C. Hemminger, J. Phys. Chem., 1996, 100, 6371–6375 Search PubMed.
- A. A. Frossard, S. M. Burrows, S. M. Elliott, T. S. Bates and P. K. Quinn, J. Geophys. Res. Atmos., 2014, 119(13), 134–146 Search PubMed.
- M. S. Rana and M. I. Guzman, J. Phys. Chem. A, 2022, 126, 6502–6516 Search PubMed.
- B. M. Connelly and M. A. Tolbert, Environ. Sci. Technol., 2010, 44, 4603–4608 Search PubMed.
- S. Langenberg, V. Proksch and U. Schurath, Atmos. Environ., 1998, 32, 3129–3137 CrossRef CAS.
- B. A. Wellen, E. A. Lach and H. C. Allen, Phys. Chem. Chem. Phys., 2017, 19, 26551–26558 RSC.
- J. R. Lawrence, S. V. Glass and G. M. Nathanson, J. Phys. Chem. A, 2005, 109, 7449–7457 CrossRef CAS PubMed.
- S. Decesari, M. C. Facchini, S. Fuzzi and E. Tagliavini, J. Geophys. Res. Atmos., 2003, 108, 4685 CrossRef.
- M. D. King, A. R. Rennie, C. Pfrang, A. V. Hughes and K. C. Thompson, Atmos. Environ., 2010, 44, 1822–1825 Search PubMed.
- M. D. King, S. H. Jones, C. O. Lucas, K. C. Thompson, A. R. Rennie, A. D. Ward, A. A. Marks, F. N. Fisher, C. Pfrang, A. V. Hughes and R. A. Campbell, Phys. Chem. Chem. Phys., 2020, 22, 28032–28044 RSC.
- S. H. Jones, M. D. King, A. D. Ward, A. R. Rennie, A. C. Jones and T. Arnold, Atmos. Environ., 2017, 161, 274–287 Search PubMed.
- A. Milsom, A. M. Squires, I. Quant, N. J. Terrill, S. Huband, B. Woden, E. R. Cabrera-Martinez and C. Pfrang, J. Phys. Chem. A, 2022, 126, 7331–7341 CrossRef CAS PubMed.
- K. C. Thompson, A. R. Rennie, M. D. King, S. J. O. Hardman, C. O. M. Lucas, C. Pfrang, B. R. Hughes and A. V. Hughes, Langmuir, 2010, 26, 17295–17303 Search PubMed.
- C. Pfrang, K. Rastogi, E. R. Cabrera-Martinez, A. M. Seddon, C. Dicko, A. Labrador, T. S. Plivelic, N. Cowieson and A. M. Squires, Nat. Commun., 2017, 8, 1724 Search PubMed.
- B. Woden, M. W. A. Skoda, A. Milsom, C. Gubb, A. Maestro, J. Tellam and C. Pfrang, Atmos. Chem. Phys., 2021, 21, 1325–1340 CrossRef CAS.
- E. J. Stuckey, R. J. L. Welbourn, S. H. Jones, A. J. Armstrong, M. Wilkinson, J. I. L. Morison and M. D. King, Environ. Sci.: Atmos., 2024, 4, 1309–1321 Search PubMed.
- E. J. Stuckey, R. J. L. Welbourn, T. W. D. Robson, C. R. Barker, M. Wilkinson, J. I. L. Morison and M. D. King, Atmos. Environ., 2026, 365, 121690 Search PubMed.
- P.-M. Wu and K. Okada, Atmos. Environ., 1994, 28, 2053–2060 Search PubMed.
- T. M. McIntire, A. S. Lea, D. J. Gaspar, N. Jaitly, Y. Dubowski, Q. Li and B. J. Finlayson-Pitts, Phys. Chem. Chem. Phys., 2005, 7, 3605 Search PubMed.
- S. Sobanska, J. Barbillat, M. Moreau, N. Nuns, I. D. Waele, D. Petitprez, Y. Tobon and C. Brémard, Phys. Chem. Chem. Phys., 2015, 17, 10963–10977 Search PubMed.
- F. Sebastiani, R. A. Campbell, K. Rastogi and C. Pfrang, Atmos. Chem. Phys., 2018, 18, 3249–3268 CrossRef CAS.
- D. J. Donaldson and K. T. Valsaraj, Environ. Sci. Technol., 2010, 44, 865–873 Search PubMed.
- J. M. Anglada, M. T. Martins-Costa, J. S. Francisco and M. F. Ruiz-López, J. Am. Chem. Soc., 2020, 142, 16140–16155 CrossRef CAS PubMed.
- L. F. Voss, M. F. Bazerbashi, C. P. Beekman, C. M. Hadad and H. C. Allen, J. Geophys. Res. Atmos., 2007, 112, D06209 Search PubMed.
- M. Shiraiwa, C. Pfrang and U. Pöschl, Atmos. Chem. Phys., 2010, 10, 3673–3691 Search PubMed.
- C. Pfrang, M. Shiraiwa and U. Pöschl, Atmos. Chem. Phys., 2010, 10, 4537–4557 CrossRef CAS.
- F. Sebastiani, R. A. Campbell and C. Pfrang, Environ. Sci.: Atmos., 2022, 2, 1324–1337 CAS.
- J. He, H. Zhang, W. Wang, Y. Ma, M. Yang, Y. He, Z. Liu, K. Yu and J. Jiang, Environ. Res., 2022, 212, 113232 CrossRef CAS PubMed.
- J. B. Gilman, H. Tervahattu and V. Vaida, Atmos. Environ., 2006, 40, 6606–6614 CrossRef CAS.
- R. Welbourn and S. Clarke, Curr. Opin. Colloid Interface Sci., 2019, 42, 87–98 CrossRef CAS.
- A. R. Rennie, M. S. Hellsing, E. Lindholm and A. Olsson, Rev. Sci. Instrum., 2015, 86, 016115 CrossRef PubMed.
- J. Penfold and R. K. Thomas, Curr. Opin. Colloid Interface Sci., 2014, 19, 198–206 CrossRef CAS.
- G. Fragneto-Cusani, J. Phys.: Condens. Matter, 2001, 13, 4973–4989 CrossRef CAS.
- R. P. Richter, R. Bérat and A. R. Brisson, Langmuir, 2006, 22, 3497–3505 CrossRef CAS PubMed.
- A. Virkkula, H. Grythe, J. Backman, T. Petäjä, M. Busetto, C. Lanconelli, A. Lupi, S. Becagli, R. Traversi, M. Severi, V. Vitale, P. Sheridan and E. Andrews, Atmos. Chem. Phys., 2022, 22, 5033–5069 CrossRef CAS.
- D. W. Griffin, N. Kubilay, M. Koçak, M. A. Gray, T. C. Borden and E. A. Shinn, Atmos. Environ., 2007, 41, 4050–4062 CrossRef CAS.
- K. Hayakawa, N. Handa, K. Kawanobe and C. S. Wong, Mar. Chem., 1996, 52, 233–244 CrossRef CAS.
- Z. Liu, E. Van Acker, M. De Rijcke, F. Van Nieuwerburgh, C. Janssen and J. Asselman, Environ. Int., 2025, 195, 109255 CrossRef CAS PubMed.
- E. Van Acker, M. De Rijcke, Z. Liu, J. Asselman, K. A. C. De Schamphelaere, L. Vanhaecke and C. R. Janssen, Environ. Sci. Technol., 2021, 55, 15989–16000 CrossRef CAS PubMed.
- S. Decesari, M. Paglione, M. Rinaldi, M. Dall'Osto, R. Simó, N. Zanca, F. Volpi, M. C. Facchini, T. Hoffmann, S. Götz, C. J. Kampf, C. O'Dowd, D. Ceburnis, J. Ovadnevaite and E. Tagliavini, Atmos. Chem. Phys., 2020, 20, 4193–4207 CrossRef CAS.
- T. H. Bertram, R. E. Cochran, V. H. Grassian and E. A. Stone, Chem. Soc. Rev., 2018, 47, 2374–2400 RSC.
- R. E. Cochran, O. Laskina, T. Jayarathne, A. Laskin, J. Laskin, P. Lin, C. Sultana, C. Lee, K. A. Moore, C. D. Cappa, T. H. Bertram, K. A. Prather, V. H. Grassian and E. A. Stone, Environ. Sci. Technol., 2016, 50, 2477–2486 CrossRef CAS PubMed.
- K. A. Prather, T. H. Bertram, V. H. Grassian, G. B. Deane, M. D. Stokes, P. J. DeMott, L. I. Aluwihare, B. P. Palenik, F. Azam, J. H. Seinfeld, R. C. Moffet, M. J. Molina, C. D. Cappa, F. M. Geiger, G. C. Roberts, L. M. Russell, A. P. Ault, J. Baltrusaitis, D. B. Collins, C. E. Corrigan, L. A. Cuadra-Rodriguez, C. J. Ebben, S. D. Forestieri, T. L. Guasco, S. P. Hersey, M. J. Kim, W. F. Lambert, R. L. Modini, W. Mui, B. E. Pedler, M. J. Ruppel, O. S. Ryder, N. G. Schoepp, R. C. Sullivan and D. Zhao, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7550–7555 CrossRef CAS PubMed.
- A. R. J. Nelson and S. W. Prescott, J. Appl. Crystallogr., 2019, 52, 193–200 CrossRef CAS PubMed.
- M. D. King, K. C. Thompson, T. Arnold, A. R. Rennie, T. W. D. Robson and R. J. L. Welbourn, Oxidation of a bilayer of DOPC by aqueous ozone – a chemical, kinetic and mechanistic investigation, 2018, https://data.isis.stfc.ac.uk/doi/STUDY/103198184/.
- E. J. Stuckey, R. J. L. Welbourn and M. D. King, Oxidation of a natural organic matter at air-mineral interface by aqueous ozone – a proof of concept, 2022, https://data.isis.stfc.ac.uk/doi/STUDY/115144602/.
- E. J. Stuckey, R. J. L. Welbourn and M. D. King, Finishing the study of Oxidation of a bilayer of DOPC, POPC and DPPC by aqueous ozone – a chemical, kinetic and mechanistic investigation, 2022, https://data.isis.stfc.ac.uk/doi/STUDY/115144567/.
- M. D. King, S. H. Jones, A. R. Rennie, K. C. Thompson, A. D. Ward and E. Watkins, Bilayer oxidation using labelled POPC to confirm chemical mechanism and structure of decay by ozone oxidation, 2012, DOI:10.5291/ILL-DATA.9-10-1241.
- M. D. King, P. Gutfreund, A. R. Rennie, T. W. D. Robson, A. D. Ward and R. J. L. Welbourn, Oxidation of bilayer of labelled DPPC by aqueous hydroxyl radical; elucidating a mechanism for atmospheric science, 2018, DOI:10.5291/ILL-DATA.9-10-1517.
- J. Webster, S. Holt and R. Dalgliesh, Phys. B, 2006, 385–386, 1164–1166 CrossRef CAS.
- J. R. P. Webster, S. Langridge, R. M. Dalgliesh and T. R. Charlton, Eur. Phys. J. Plus., 2011, 126, 112 CrossRef.
- T. R. Charlton, R. L. S. Coleman, R. M. Dalgliesh, C. J. Kinane, C. Neylon, S. Langridge, J. Plomp, N. G. J. Webb and J. R. P. Webster, Neutron News, 2011, 22, 15–18 CrossRef.
- O. Arnold, J. Bilheux, J. Borreguero, A. Buts, S. Campbell, L. Chapon, M. Doucet, N. Draper, R. Ferraz Leal, M. Gigg, V. Lynch, A. Markvardsen, D. Mikkelson, R. Mikkelson, R. Miller, K. Palmen, P. Parker, G. Passos, T. Perring, P. Peterson, S. Ren, M. Reuter, A. Savici, J. Taylor, R. Taylor, R. Tolchenov, W. Zhou and J. Zikovsky, Nucl. Instrum. Methods Phys. Res., Sect. A, 2014, 764, 156–166 CrossRef CAS.
- P. Gutfreund, T. Saerbeck, M. A. Gonzalez, E. Pellegrini, M. Laver, C. Dewhurst and R. Cubitt, J. Appl. Crystallogr., 2018, 51, 606–615 CrossRef CAS.
- P. S. Pershan, Phys. Rev. E, 1994, 50, 2369–2372 CrossRef CAS PubMed.
- O. S. Heavens, Rep. Prog. Phys., 1960, 23, 1–65 CrossRef.
- S. Chiu, E. Jakobsson, S. Subramaniam and H. Scott, Biophys. J., 1999, 77, 2462–2469 Search PubMed.
- T. K. Lind, M. Cárdenas and H. P. Wacklin, Langmuir, 2014, 30, 7259–7263 Search PubMed.
- H. Bader and J. Hoigné, Water Res., 1981, 15, 449–456 Search PubMed.
- A. V. Levanov, O. Y. Isaikina, A. N. Tyutyunnik, E. E. Antipenko and V. V. Lunin, J. Anal. Chem., 2016, 71, 549–553 Search PubMed.
- J. Hoigné, in The Chemistry of Ozone in Water, Springer US, 1988, pp. 121–141 Search PubMed.
- K. Bullrich, in Scattered Radiation in the Atmosphere and the Natural Aerosol, 1964, pp. 99–260 Search PubMed.
- D. Sinclair and V. K. La Mer, Chem. Rev., 1949, 44, 245–267 Search PubMed.
- H. Moosmüller, R. Chakrabarty and W. Arnott, J. Quant. Spectrosc. Radiat. Transf., 2009, 110, 844–878 Search PubMed.
- D. R. Huffman, Adv. Phys., 1977, 26, 129–230 Search PubMed.
- F. Hoyle and C. Wickramasinghe, Astrophys. Space Sci., 1999, 268, 249–261 Search PubMed.
- M. Matsumura and M. Seki, Astrophys. Space Sci., 1986, 126, 155–165 Search PubMed.
- C. F. Bohren and D. R. Huffman, Absorption and Scattering of Light by Small Particles, Wiley, 1998 Search PubMed.
- N. V. Voshchinnikov and J. S. Mathis, Astrophys. J., 1999, 526, 257–264 Search PubMed.
- Z. S. Wu and Y. P. Wang, Radio Sci., 1991, 26, 1393–1401 Search PubMed.
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen, B. Yu, L. Zaslavsky, J. Zhang and E. E. Bolton, Nucleic Acids Res., 2025, 53, D1516–D1525 Search PubMed.
- I. H. Malitson, J. Opt. Soc. Am., 1965, 55, 1205 Search PubMed.
- R. H. Shepherd, M. D. King, A. A. Marks, N. Brough and A. D. Ward, Atmos. Chem. Phys., 2018, 18, 5235–5252 Search PubMed.
- G. M. Hale, Appl. Opt., 1973, 12, 555–563 Search PubMed.
- P. Laven, MiePlotv4.6, 2024 Search PubMed.
- J. M. Haywood and K. P. Shine, Geophys. Res. Lett., 1995, 22, 603–606 Search PubMed.
- D. J. Delene and J. A. Ogren, J. Atmos. Sci., 2002, 59, 1135–1150 Search PubMed.
- E. Rudolphi-Skórska, M. Filek and M. Zembala, J. Membr. Biol., 2017, 250, 493–505 Search PubMed.
- I. O. Bacellar, M. S. Baptista, H. C. Junqueira, M. Wainwright, F. Thalmann, C. M. Marques and A. P. Schroder, Biochim. Biophys. Acta Biomembr., 2018, 1860, 2366–2373 Search PubMed.
- B. Bagheri, P. Boonnoy, J. Wong-ekkabut and M. Karttunen, Phys. Chem. Chem. Phys., 2023, 25, 18310–18321 Search PubMed.
- H. L. Smith, M. C. Howland, A. W. Szmodis, Q. Li, L. L. Daemen, A. N. Parikh and J. Majewski, J. Am. Chem. Soc., 2009, 131, 3631–3638 Search PubMed.
- L. Cwiklik and P. Jungwirth, Chem. Phys. Lett., 2010, 486, 99–103 Search PubMed.
- M. Lis, A. Wizert, M. Przybylo, M. Langner, J. Swiatek, P. Jungwirth and L. Cwiklik, Phys. Chem. Chem. Phys., 2011, 13, 17555 Search PubMed.
- K. C. Thompson, S. H. Jones, A. R. Rennie, M. D. King, A. D. Ward, B. R. Hughes, C. O. M. Lucas, R. A. Campbell and A. V. Hughes, Langmuir, 2013, 29, 4594–4602 Search PubMed.
- D. Hu, M. R. Alfarra, K. Szpek, J. M. Langridge, M. I. Cotterell, C. Belcher, I. Rule, Z. Liu, C. Yu, Y. Shao, A. Voliotis, M. Du, B. Smith, G. Smallwood, P. Lobo, D. Liu, J. M. Haywood, H. Coe and J. D. Allan, Atmos. Chem. Phys., 2021, 21, 16161–16182 Search PubMed.
- D. Hu, M. R. Alfarra, K. Szpek, J. M. Langridge, M. I. Cotterell, M. J. Flynn, Y. Shao, A. Voliotis, M. Du, D. Liu, B. Johnson, G. McFiggans, J. M. Haywood, H. Coe and J. D. Allan, J. Geophys. Res. Atmos., 2023, 128, e2023JD039178 Search PubMed.
- A. Hartikainen, M. Ihalainen, D. Shukla, M. Rohkamp, A. Mukherjee, Q. He, S. Piel, A. Virkkula, D. Li, T. Kokkola, S. Jeong, H. Koponen, U. Etzien, A. Das, K. Luoma, L. Schwalb, T. Gröger, A. Barth, M. Sklorz, T. Streibel, H. Czech, B. Gündling, M. Kalberer, B. Buchholz, A. Hupfer, T. Adam, T. Hohaus, J. Øvrevik, R. Zimmermann and O. Sippula, Atmos. Chem. Phys., 2025, 25, 9275–9294 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |