DOI:
10.1039/D5EA00138B
(Critical Review)
Environ. Sci.: Atmos., 2026,
6, 248-272
Atmospheric chemistry of halogenated OVOCs: a review of rate coefficients, dielectric strength, and environmental impact
Received
17th October 2025
, Accepted 16th December 2025
First published on 23rd December 2025
Abstract
Anthropogenic activities have increased atmospheric emissions of halogenated compounds. Some of these are potent greenhouse gases; however, their current atmospheric concentrations are low. We assessed the potential risks and hazards of novel chemical substances before their introduction into the environment. Evaluation systems consider both the direct impacts of exposure to new compounds and the indirect effects associated with their deposition in soil and surface waters when evaluating atmospheric hazards and risks. Therefore, in this study, we reviewed previously published rate coefficient values for oxidants such as OH radicals and Cl atoms and the lifetime values of 94 oxygenated halogenated compounds, including halogenated esters, hydrofluoroalcohols, halogenated aldehydes, halogenated carboxylic acids, halogenated ketones, and diketones. We also reviewed previously calculated radiative efficiencies (REs) and global warming potentials (GWPs) for halogenated compounds over 20-, 100-, and 500-year time horizons. We also calculated instantaneous and lifetime-corrected REs and GWPs for 45 halogenated compounds that have not been previously mentioned in the literature. This study also includes global temperature change potential (GTP) values for more than 94 halogenated compounds over 20-, 50-, and 100-year time horizons. Among these, we report GTPs, including instantaneous and lifetime-corrected values, for 59 halogenated compounds for the first time in this study. We also calculated ozone-depleting potential (ODP) values for 14 chlorine-containing halogenated compounds. The calculated ODP values for these 14 halogenated compounds were low. In this study, we also considered the photochemical ozone creation potential (POCP) and acidification potential (AP) of halogenated oxygenated volatile organic compounds (XOVOCs). This review also provides a thorough investigation of the possible replacement of SF6, a gas frequently used in electric insulation because of its high dielectric strength (DS). To identify an alternative, the DS values of all the compounds were determined and are provided in this review.
Environmental significance
Anthropogenic activities are increasing atmospheric levels of halogenated oxygenated volatile organic compounds (XOVOCs). This exhaustive list includes halogenated esters, alcohols, aldehydes, ketones, and diketones. It is essential to consider degradation and environmental impact. This study compiles kinetic and radiative data for 94 such compounds and provides new lifetime-corrected impact metrics. The findings reveal their atmospheric behaviour and climatic effects, likely helping identify low-impact alternatives to potent gases like SF6 and guiding environmental regulations.
|
1. Introduction
Chlorofluorocarbons (CFCs) were a group of substances used for various industrial and household purposes in the 1970s. Their environmental impact became evident ten years later with the discovery of the Antarctic ozone hole, a less severe but widespread depletion of stratospheric ozone.1 In general, CFCs exhibit good chemical stability, low toxicity, low flammability, low gas-phase thermal conductivity, and reasonable cost, and hence were used as cleaning solvents, foam-blowing agents, aerosol propellants, refrigerants, and fire-extinguishing agents.2,3 A ban on ozone-depleting substances was imposed by the Montreal Protocol (1987)4,5 and the Kyoto Protocol (1997)6 to recover stratospheric ozone.
Hydrofluorocarbons (HFCs) and hydrochlorofluorocarbons (HCFCs) were considered replacements for CFCs. HFCs have no ozone depletion potential (ODP), and HCFCs impact stratospheric ozone because they contain chlorine atoms. Both compound classes are often highly potent greenhouse gases with significant global warming potentials (GWPs) and are frequently characterized by long atmospheric lifetimes and strong infrared (IR) absorption.7 Researchers have been motivated to seek innovative alternatives that meet numerous industrial and commercial purposes while having minimal impact on ozone depletion and climate change. Hydrofluorocarbons (HFCs) and perfluorocarbons (PFCs) are considered second-generation alternatives because they do not contain chlorine or bromine atoms that destroy ozone. They have minimal or no tendency to deplete the ozone layer in the atmosphere.8,9 HFCs and PFCs have highly significant global warming potentials (GWPs) and are considered potent greenhouse gases (GHGs) under the Kyoto Protocol.10
Hydrofluoroethers are used as substitutes for chlorofluorocarbons in various industrial applications and undergo atmospheric oxidation to form halogenated esters.11,12 The atmospheric breakdown of halogenated esters results in the formation of trifluoroacetic acid (TFA) and its derivatives, as well as HF and CO2.13 TFA has been found in surface waters (lakes, rivers, and seas) and in snow, fog, and rainwater samples.14–16 TFA is considered an omnipresent accumulating component of the hydrosphere and is not a natural element in freshwater environments.17 TFA accumulation may impact aquatic and agricultural systems.16
Halogenated oxygenated volatile organic compounds (XOVOCs) are a superior choice owing to their increased reactivity brought about by the presence of an O atom. To evaluate their environmental effects, a comprehensive understanding of the atmospheric chemistry of different XOVOCs is required. The widespread use of these chemicals is anticipated to significantly affect air quality and climate. Many classes of XOVOCs have emerged, including hydrofluoroethers, fluorinated esters, hydrofluoroalcohols, fluorinated aldehydes, fluorinated ketones, fluorinated diketones, and perfluorinated carboxylic acids. These chemicals are considered prospective substitutes for CFCs, HCFCs, and HFCs.18
Hydrofluoroalcohols are utilized in various industrial applications, including coatings, paints, adhesives, polymers, waxes, polishes, and the cleaning of electronic components.19 It is well known that the main oxidation products of hydrofluoroalcohols are fluorinated aldehydes.20–23 Fluorinated ketones are used in various industries as reagents in organic synthesis and as fire-extinguishing agents.24 Fluorinated diketones also have several applications, particularly in metallurgy, where they may be utilized as chelating agents, and in the organic synthesis of pharmaceuticals, specialty dyes, and coatings.25–28 A possible source of airborne diketones is metal–organic chemical vapor deposition methods.29 Perfluorinated carboxylic acids are minor products emitted to the atmosphere during the thermolysis of fluoropolymers.30
XOVOCs contain C–F bonds, which make them potent and effective greenhouse gases. They efficiently absorb infrared radiation emitted by the Earth within the “atmospheric transparency window,” that is, between 8 and 12 µm in wavelength. Thus, these molecules may contribute to global warming.31 In the atmosphere below 1400 cm−1, water absorbs infrared (IR) radiation, whereas above 800 cm−1, water and carbon dioxide absorb IR radiation efficiently. Consequently, a significant fraction of the Earth's heat radiation can escape through the spectral region between 800 and 1400 cm−1.32 Different gases have different radiative efficiencies owing to variations in the wavelength positions and intensities of their IR absorption bands.
Radiative efficiency (RE), which quantifies the radiative forcing per unit change in the atmospheric concentration of a gas, is frequently expressed in units of W m−2 ppb−1 for halocarbons and similar chemicals.33 For the assessment of radiative efficiency and GWP, precise knowledge of the atmospheric lifetime and IR absorption spectra is necessary to comprehend the severe dangers posed by a chemical as a potent greenhouse gas. A molecule reacts with different atmospheric oxidants, which determines its lifetime in the atmosphere. The GWP of a molecule is defined as the time-integrated radiative forcing resulting from the instantaneous emission of a kilogram of a trace gas, relative to that of one kilogram of a reference gas, usually CO2.34 Shorter atmospheric lifetimes often translate to lower GWPs, depending on where and how strongly the molecule absorbs infrared light.
XOVOCs in the atmosphere are mostly degraded via reactions with OH radicals during the entire day.7,18,29,31,35–38 Although chlorine atoms can contribute to the removal of XOVOCs, their overall impact is usually negligible due to their much lower atmospheric abundance compared to OH radicals. However, Cl-initiated reactions may still be relevant in some specific environments such as the marine boundary layer, coastal regions, or certain polluted urban areas where elevated chlorine levels have been observed.18 Therefore, including Cl-initiated pathways provides a complete understanding of the possible atmospheric fates of XOVOCs under such region-specific conditions.39 Photolysis and other chemical degradation mechanisms, such as interactions with NO3 and O3, are considered minor atmospheric degradation pathways for XOVOCs. Hydroxyl radicals are anticipated to play a vital role in determining the atmospheric lifetimes of XOVOCs.34
The primary goal of this review is to offer a thorough and consistent study of input variables, such as rate coefficients and lifetimes, needed to determine atmospheric impact parameters, including RE, GWP, GTP, ODP, POCP, and AP values for a large number of XOVOCs. This review addresses the kinetic and mechanistic information currently known for the oxidation of oxygenated organic molecules under tropospheric conditions. The atmospheric lifetime and other crucial input parameters required for calculating GWPs, GTPs, and other metrics are also discussed. Atmospheric lifetime values were obtained from the literature. This review presents a detailed analysis of the instantaneous and lifetime-corrected REs for 105 compounds, including global warming potential, global temperature change potential, photochemical ozone creation potential, and acidification potential for 94 chemical compounds and the ozone-depleting potential for 14 chlorine-containing compounds. This review also examines the dielectric strengths of all halogenated OVOCs to determine their suitability as substitutes for SF6 in electrical insulation applications. The dielectric strength (DS) of each compound was determined.
2. Methodology
There are two approaches for determining the rate coefficients and atmospheric implications of XOVOCs: (1) experimental and (2) theoretical methods. There are two methods for calculating the rate coefficients for reactions of organic compounds with OH radicals or Cl atoms experimentally: the absolute and relative rate methods. Here, we briefly discuss each approach.
2.1 Experimental technique
2.1.1 Kinetics study.
2.1.1.1 Absolute technique.
When the rate constant for OH radicals is determined using an absolute procedure, OH radicals are produced utilizing either flash photolysis (FP), laser photolysis (LP), or discharge flow (DF) techniques.40 Since it is necessary to monitor OH radicals as a function of reaction time, the sensitive pulsed dye laser-induced fluorescence (LIF) method is used to detect absolute rate constants for OH-initiated reactions under tropospheric environmental conditions.41 In the FP approach, a xenon (Xe) flash lamp is used to photolyze water vapor directly. The photolyzing light is directed down the axial line to the reaction cell through a quartz window. Argon (Ar) was used as the carrier gas for the FP technique. Similarly, three approaches for OH radical generation have been studied using LP methods. The first is the production of OH radicals through the reaction O(1D) + H2O → 2OH(˙) (LP-H2O method), in which O(1D) atoms are created by photodissociation of N2O using an ArF excimer laser. The second method for producing OH radicals without the use of water is the O(1D) + CH4 → OH(˙) + CH3 reaction (LP-CH4 method). Third, the LP-H2O2 method, which utilizes a KrF excimer laser to photolyze H2O2 directly, is investigated. Helium (He) is used as the carrier gas in the LIF method. Experimental setup description and protocol for the kinetic measurements can be found elsewhere.42–48
2.1.1.2 Relative rate technique.
Most often, relative rate approaches in smog chamber experiments have been used to evaluate rate coefficients for reactions with OH radicals under tropospheric conditions or at atmospheric pressure (1 atm).41 Various techniques have been used to determine the relative rate constants for the reactions of OH radicals with organic molecules.49–53 The basic concept behind the relative rate approach is that the rate constant for the reaction between a reference chemical and OH radicals must be known. Subsequently, a competitive response mechanism can be used to analyze the simultaneous decay of the compound of interest and reference compounds in the presence of OH radicals to obtain the rate constant for the compound of interest. Furthermore, the reference rate coefficient is crucial for selecting reference compounds to increase the sensitivity. Taking Z (the reference compound) and A (the compounds of interest) as examples, the reactions with OH radicals can be described as follows:| | | Z + OH → products (kZ) | (1) |
| | | A + OH → products (kA) | (2) |
| |  | (3) |
| | 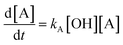 | (4) |
| | 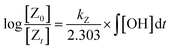 | (5) |
| | 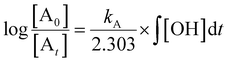 | (6) |
| | 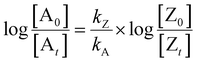 | (7) |
In this case, [Z0] and [A0] represent the initial concentrations of the reference compound and the compound of interest, respectively, before the light is turned on. Similarly, [Zt] and [At] represent the corresponding concentrations after light exposure for time t. The reference compounds and the compounds of interest, along with their respective second-order rate coefficients for reaction with OH radicals, are denoted by kZ and kA.
2.1.2 Atmospheric implication calculation.
2.1.2.1 Absorption cross-section (σi).
Two methods are generally used to compute absorption cross-sections: experimental techniques and theoretical calculations. In the case of experimental techniques, infrared absorption spectra are usually obtained using analytical infrared instruments in the 4000–400 cm−1 mid-infrared region. In general, measurements are obtained using different spectrophotometers, such as a Bomem DA8, a Mattson Sirius, and a Bruker spectrometer, with a stainless-steel cell fitted with KBr windows at room temperature.54–57 Operating at a spectral resolution of 0.01 to 0.001 cm−1, the spectrometer produced interferograms based on 100 to 1000 co-added scans.54 Spectra of the pure vapour or mixtures containing 60–506 hPa (1 hPa = 9.87 × 10−4 atm) of N2 diluent were recorded. The absorption spectrum was measured in two stages: first, a long path-length cell was evacuated to less than 10−3 torr to obtain the background spectrum; second, the cell was filled with the sample gas to a specific pressure (5–165 torr).| | 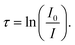 | (8) |
In the evacuated cell, I represents the background intensity, and I0 is the intensity of the sample.where σi is the absorption cross-section (cm2 per molecule), L is the path length (cm), and n is the number density of the absorbing molecule (molecule per cm3).| | 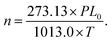 | (10) |
In eqn (10), L0 is the Loschmidt number, which is 2.69 × 1019 molecules per cm3, T is the gas temperature (kelvin), and P is the pressure of the absorber in hectopascal (hPa). Additionally, a comprehensive assessment of atmospheric metrics, including lifetime, radiative efficiency (RE), global warming potential (GWP), global temperature change potential (GTP), ozone depletion potential (ODP), photo ozone creation potential (POCP), and dielectric strength (DS), is provided in the SI.
2.2 Theoretical techniques
2.2.1 Kinetics calculation.
According to previous investigations,58–63 the rate coefficients of organic compounds with oxidizing radicals, such as OH radicals, Cl atoms, and NO3 radicals, were theoretically determined using Gaussian software based on DFT. Gaussian 16 is a software program for calculating electronic energies.64 GaussView software is used for visualizing molecular structures.65 First, using DFT methodologies, such as M06-2X66–68 and BH&HLYP,69 with different basis sets depending on the computational cost as well as accuracy (e.g., cc-pVTZ and aug-cc-pVTZ),70–73 the geometries of stationary points involved in the oxidation of interest (A) organic compounds by oxidants were optimized. The M06-2X approach is reliable for calculating thermochemistry, reaction mechanisms, and rate coefficients for atmospheric reactions involving main-group elements.74,75 Following stationary point optimization and frequency computations using a reaction channel, transition state concepts (one negative frequency) and stable minima on the potential energy surface were confirmed using vibrational frequencies. Each local minimum was described using real frequencies, and the presence of one imaginary frequency verified the existence of transition stages. To confirm that reactants transform into their intended products via transition state crossings, intrinsic reaction coordinate (IRC)76,77 calculations were performed for all the transition states at the same level of theory. Using the optimized geometries, single-point energy calculations were performed using the high-level coupled-cluster with single, double, and perturbative triple excitations (CCSD(T))78 along with a basis set (depending on accuracy and computational cost) to refine the potential energy surface and obtain more accurate energies.
Rate coefficients were calculated using the canonical variational transition state theory (CVT) for the tropospheric temperature range, considering atmospheric significance.79–81 Two methods are used for tunneling corrections: zero-curvature tunneling (ZCT)81–83 and small curvature tunneling (SCT).84,85 Canonical variational theory rate coefficients, kCVT(T), were derived by minimizing along the dividing surface (s) at a given temperature (T). They are represented as follows:
and
| | 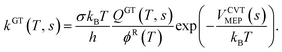 | (12) |
Here,
kGT(
T,
s) is the generalized transition state theory rate coefficient at the dividing surface
s,
σ is the symmetry factor,
kB is the Boltzmann constant,
h is Planck's constant, and
QGT(
T,
s) is the partition function of the generalized transition state at
s, with a local zero of energy at
VCVTMEP(
s).
ϕR(
T) is the partition function of the reactants per unit volume. The CVTST rate coefficient is multiplied by the transmission coefficient,
κT, to account for tunneling effects.
κT is calculated using zero-curvature tunneling (ZCT) and small-curvature tunneling (SCT). The modified Arrhenius equation was fitted to the computed CVT/SCT rate coefficients over the temperature range.
| | 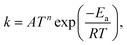 | (13) |
where
T is the temperature,
n is the temperature exponent,
R is the universal gas constant,
Ea is the barrier height, and
A is the Arrhenius prefactor.
2.2.2 Molecular descriptors calculations.
All compounds included in this review underwent geometric optimization and frequency calculations at the B3LYP/6-31G(d,p) level of theory.70,86–99 The Becke 3-parameter, Lee–Yang–Parr (B3LYP) method, a widely used DFT technique, was used to calculate the RE values. The B3LYP functional includes the exchange functional of HF, the empirical correlation functional of Lee, Yang, and Parr, and Becke's empirical exchange functional.100 Analysis of the vibrational frequencies revealed that all frequencies were positive, indicating that the optimized geometries correspond to minima on the potential energy surface at the B3LYP/6-31G(d,p) level of theory. The B3LYP DFT calculations exhibited noticeably greater agreement with experimental data. Bravo et al. compared experimental results with DFT calculations across the range of 700–1400 cm−1; the parameters of the best-fit line showed variations of less than 2% for DFT but over 5% for ab initio calculations.89
Utilizing the optimized geometries and vibrational frequencies of XOVOCs at the B3LYP level with the Pople 6-311+G(d,p) basis set, molecular descriptors such as frontier molecular orbital (FMO), reactivity parameters such as molecular electrostatic potential (MEP), and non-covalent interactions (NCIs) were studied. FMO, MEP, and NCI analyses were performed using Multiwfn software.101 Wavefunction files of the oxygenated organic compounds were obtained using Gaussian 16. Subsequently, these wavefunction files were used as input files for Multiwfn. All isosurface maps were generated using the Visual Molecular Dynamics (VMD) 1.9.3 program based on Multiwfn analysis results.102 The B3LYP/6-311+G(d,p) level of theory was used to determine the dielectric strengths (DS) of all oxygenated organic compounds included in this review. According to earlier research, the B3LYP/6-311+G(d,p) level of theory is the most effective DFT method for estimating the dielectric strength (DS).103–106
3. Results and discussion
3.1 Halogenated esters
3.1.1 Rate coefficient of halogenated esters.
The literature rate coefficients and lifetime data for reactions between halogenated esters and oxidants (OH radicals and Cl atoms) are presented in Tables S2 and S3, respectively. In these SI tables, we reviewed the lifetime values and rate coefficients of various halogenated esters, such as fluoroalkyl formates,12,107–112 fluoroalkyl acetates,113,114 and fluoroalkyl fluoroacetates.115–117 The reactivity of alkyl esters was not significantly affected by the extension of the alkyl chain length, whereas formates, acetates, propionates, and butyrates exhibited a greater chain-length dependency.118–120 Blanco et al.115 previously showed that the reaction rates of halogenated esters are greater than those of the equivalent halocarbons. The ester linkage, –C(O)O–, often activates neighboring C–H bonds. Mechanistic insights show that halogen-atom abstraction plays a modest role in OH-initiated reactions, which mostly occur through hydrogen abstraction, frequently preceded by the formation of hydrogen-bonded complexes.121,122 Furthermore, the combined effects of H-atom abstraction and radical addition across the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond result in significantly greater rate coefficients for unsaturated halogenated esters, such as fluorinated acrylates and methacrylates, compared to their saturated counterparts. In electrophilic addition processes, the –OC(O)R group associated with the double bond activates this bond. Therefore, the electrical density of the π system increases owing to the lone pair electrons on the oxygen atom (Fig. S4). These results demonstrate how the oxidative persistence of halogenated esters in the environment is controlled by their electronic structure and substitution patterns.
C bond result in significantly greater rate coefficients for unsaturated halogenated esters, such as fluorinated acrylates and methacrylates, compared to their saturated counterparts. In electrophilic addition processes, the –OC(O)R group associated with the double bond activates this bond. Therefore, the electrical density of the π system increases owing to the lone pair electrons on the oxygen atom (Fig. S4). These results demonstrate how the oxidative persistence of halogenated esters in the environment is controlled by their electronic structure and substitution patterns.
3.1.2 Atmospheric fate of halogenated esters.
Several halogenated esters have mechanistic data available for hydrogen abstraction, including fluoroalkyl formates,12,107–112 fluoroalkyl acetates,113,114 and fluoroalkyl fluoroacetates.115–117 These compounds have been investigated with oxidants such as OH radicals and Cl atoms. The OH radical-initiated atmospheric degradation of the investigated saturated halogenated esters is predicted to produce halogenated acids, alcohols, aldehydes, and dicarbonyl compounds, which are prone to additional reactions with OH radicals and photolysis. Lower molecular weight acids, which are degradation products of halogenated esters, are highly soluble substances that can be rapidly absorbed into cloud droplets, leading to precipitation.123
The degradation of halogenated esters with oxidants has been previously investigated, and we have reviewed some of the detailed degradation mechanisms of halogenated esters that fall under the category of saturated halogenated esters. The radical products are generated via H atom abstraction, and in the case of fluoroalkyl acetates such as CF3C(O), the OCH2CH3 moiety undergoes an α-ester rearrangement via a five-membered ring intermediate to yield the corresponding fluoroacetic acid. The detailed mechanism is shown in Scheme 1. These radicals may also react directly with O2 to form the corresponding fluoroanhydride.123 When the –CH3 is group is substituted with a –CF3 group in CF3C(O)OCH2CH3, H abstraction occurs at the –CH2 position by OH radicals, producing the alkyl radical product CF3C(O)OCH(˙)CF3. After that, the peroxy radical CF3C(O)OCH(OO˙)CF3 is produced via the reaction of alkyl radicals with ambient oxygen (Scheme 2). Then, the peroxy radical quickly combines with NO to form NO2 and, consequently, CF3C(O)OCH(O˙)CF3 radicals. At 296 K and 1 atm atmospheric pressure, 65% of the CF3C(O)OCH(OO˙)CF3 radicals react with ambient O2 molecule. Subsequently, they produce CF3C(O)OCH(O)CF3, and 18% undergo an α-ester rearrangement to produce CF3C(O)OH and CF3C(O) radicals. The fate of the remaining 17% is still unknown.124 Prior research on the atmospheric degradation of methyl dichloroacetate (CHCl2C(O)OCH3) has concentrated on its reactivity with OH radicals. The methyl group (–CH3) or the α-position next to the carbonyl (–CHCl2) are the two main routes for hydrogen abstraction. CCl2(˙)COOCH3 is the radical intermediate that is produced when abstraction at the α-site is energetically favored. This mechanistic preference highlights the role of halogen substitution in stabilizing radical intermediates and influencing the oxidative routes of halogenated esters in the environment. Following the formation of alkyl radical products (Scheme 3), these radicals react with atmospheric oxygen to produce the peroxy radical CCl2OO(˙)COOCH3, which further reacts with NO to produce NO2 and CCl2(O)C(O)OCH3.125,126 Furthermore, our research group investigated the atmospheric deterioration of CFxH3−xCF2C(O)OCH3 (x = 3 and 2), including reaction kinetics and radical product degradation with OH radicals. After forming alkyl radical products, they reacted with ambient oxygen to generate CFxH3−xCF2C(O)OCH2(OO) radicals. Following their synthesis, CFxH3−xCF2C(O)OCH2(OO) radicals react with other radicals through several paths to produce halo alkoxy radicals (Scheme 4).87
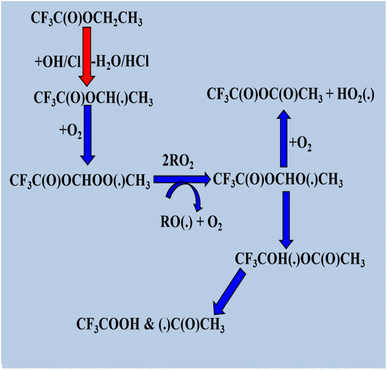 |
| | Scheme 1 Degradation process of CF3C(O)OCH2CH3. | |
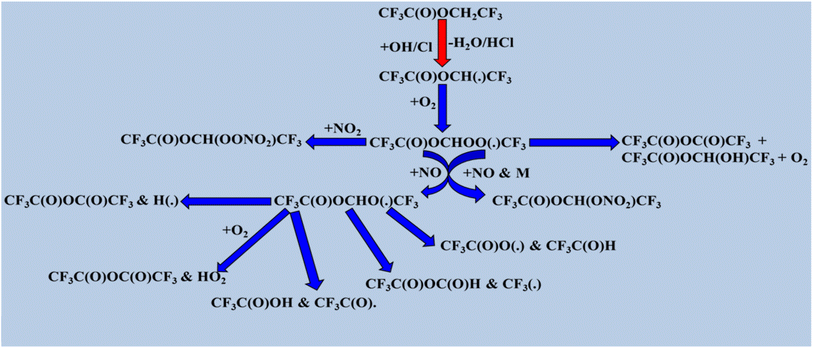 |
| | Scheme 2 Degradation process of CF3C(O)OCH2CF3. | |
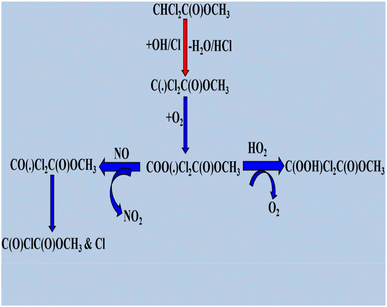 |
| | Scheme 3 Degradation process of CHCl2(O)C(O)OCH3. | |
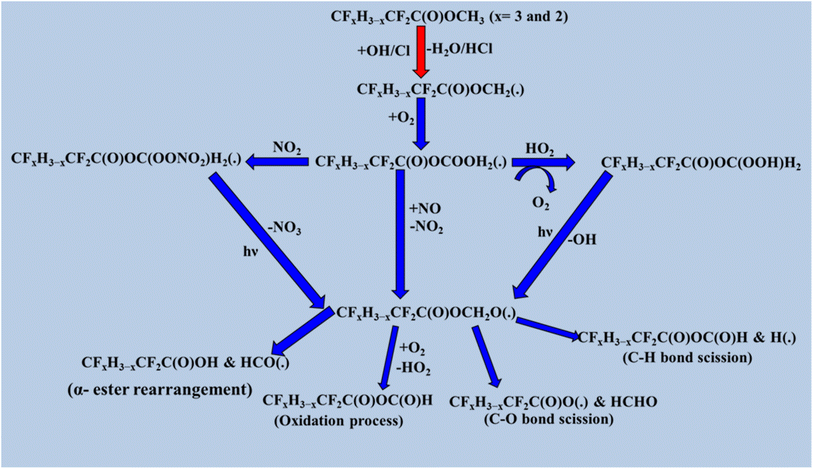 |
| | Scheme 4 Degradation process of CFxH3−xCF2C(O)OCH3. | |
Unsaturated halogenated esters degrade to halogenated glyoxylate and pyruvate, which can further react with OH radicals or Cl to generate halogenated aldehydes and contribute to the generation of ozone and secondary organic aerosols (SOAs). Halogenated carboxylic acids, considered potential environmental contaminants, are believed to be produced via the oxidation of halogenated glyoxylates and pyruvates; their detailed degradation mechanisms have been studied by Rodríguez et al.,127 Teruel et al.114 and Wallington et al.128
3.1.3 Atmospheric implications of halogenated esters.
The atmospheric lifetimes of halogenated esters were calculated by determining their rate coefficients for reactions with OH radicals and Cl atoms. Although OH-initiated oxidation dominates in most environments, reactions with Cl atom play an important role in coastal and marine boundary layer conditions. For compounds with reported Cl rate constants, the total lifetime was calculated by combining the OH- and Cl-initiated pathways; for compounds without such data, lifetimes were estimated based only on OH reactivity. The calculated lifetimes of 41 halogenated esters are presented in Table S2. The atmospheric lifetimes of the considered halogenated esters vary, from 5 hours for CH2C(CH3)C(O)OCH2CF3, as reported by Tovar and Teruel,114 to 3.6 years for HC(O)OCF3 and HC(O)OCF2CF3.34 Unsaturated esters like CF3C(O)OCHCH2, CF3C(O)OCH2CHCH2, CH2C(CH3)C(O)OCH(CF3)2, CH2C(CH3)C(O)OCH2CF3, CH2CHC(O)OCH2CF3, CH2CHC(O)OCH(CF3)2, and CH2CHC(O)O(CH2)2C4F9 have negligible lifetimes because of the presence of unsaturation. Compared to their saturated counterparts, unsaturated halogenated esters exhibit much shorter lifetimes because of the extra OH addition channel across the CC bond, which increases their overall reactivity. In contrast, due to the absence of such addition routes, saturated esters exhibit considerably longer atmospheric persistence.127 These differences are further demonstrated by photochemical ozone creation potential (POCP) measurements. Compared to ethene (POCP = 100), most halogenated esters have low POCP values, suggesting a restricted role in tropospheric ozone generation and minimal risk to biological health or air quality. Nevertheless, the POCP values of unsaturated fluorinated acrylates and methacrylates are significantly greater, indicating their dual reactivity through H abstraction and OH addition processes. These results show that the atmospheric lifetimes and subsequent ozone production potential of halogenated esters are controlled by their structural characteristics, especially the presence of unsaturation.
The optimized geometry, infrared absorption cross-section, molecular electrostatic potential (MEP), and frontier molecular orbital (FMO) surfaces for HCOOCHFCF2CF3 are shown in Fig. 1 for representation, while corresponding data for the other halogenated esters are illustrated in SI Fig. S2–S4. Absorption cross-sections and the resultant radiative efficiencies (REs) for all 52 compounds were determined from calculated spectra based on their infrared frequencies and intensities. This enabled the computation of GWPs over 20-, 100-, and 500-year time horizons for 41 esters, while estimates for the remaining compounds including FC(O)OCFH2, FC(O)OCF2H, FC(O)OCF3, FC(O)OCF2CF3, FC(O)OCH2CF3, FC(O)OCF2CF2CF3, CF3C(O)OCF3, CF3C(O)OCF2CF3, CF3C(O)OCH(CF3)2, and CF3C(O)OPh, are still unavailable due to their expected fast atmospheric elimination via wet deposition processes. These findings highlight the crucial role of kinetic measurements and quantum-chemical predictions in assessing the radiative forcing and climate significance of halogenated esters.34 The vibrational properties of halogenated esters within the atmospheric infrared window (700–1400 cm−1) greatly influence their GWPs. Since this area has considerable IR absorption, compounds with C–F, C–Cl, C–Br, S–F, and N–F bonds exhibit enhanced GWPs, with fluorine substitution having the significant impact. While chlorinated equivalents show noticeably less absorption, increasing the amount of fluorine atoms increases the intensity of IR absorption (Table S4) and, thus, the radiative efficiency (RE). CHCl2C(O)OCH3, for instance, has a comparatively low intensity (approximately 618 km per mole), which is in line with its lower RE and GWP. We also calculated the instantaneous and lifetime-corrected REs and GWPs of the 17 halogenated esters. However, these aspects have not yet been reported in the literature. The calculated GWP values for unsaturated halogenated esters over 20-, 100-, and 500-year time horizons were negligible because of their short atmospheric lifetimes. Tables S3 and S5 show that both RE and GWP exhibits a positive dependence on atmospheric lifetime. This behavior is strongly influenced by the intrinsic characteristics of the XOVOCs. In particular, substitution of hydrogen with fluorine alters the molecular dipole moment, vibrational modes, and chemical reactivity. These changes generally decrease the OH reaction rate coefficient, thereby extending the atmospheric lifetime while enhancing RE and GWP. In addition to the ODPs of four chlorine-containing esters and the APs of forty-one fluorinated esters, the GTPs of forty-one halogenated esters were assessed (Table S6) across time horizons of 20, 50, and 100 years. For the first time, 19 halogenated esters were reported, and both instantaneous and lifetime-corrected GTPs were measured. The GTP values scale with atmospheric lifetime (τ) in accordance with GWP trends, whereas unsaturated fluorinated acrylates and methacrylates exhibit insignificant GTPs because of their short lifetimes. Because the acidification potentials of fluorinated esters are identical to or higher than that of SO2, the atmospheric breakdown of these esters may lead to acidification of rainfall. These results emphasize the broad climatic and environmental consequences of halogenated esters, which extend beyond greenhouse forcing to induce phenomena such as acidification and ozone depletion.
 |
| | Fig. 1 HC(O)OCHFCF2CF3 molecular descriptor. (a) Optimized geometry. (b) Absorption cross-section. (c) Molecular electrostatic potential. (d) Frontier molecular orbital. | |
3.2 Halogenated alcohols
3.2.1 Rate coefficient of halogenated alcohols.
Halogenated alcohols are mainly eliminated from the atmosphere via oxidation by OH radicals and Cl atoms, particularly in coastal and marine boundary layer areas. According to rate coefficients and lifetimes reported in the literature (Tables S9 and S10), H abstraction at sites close to the hydroxyl group is often less favorable than at other locations. This is because the carbon-centered radicals formed are more polarizable, which made them more stable than oxygen-centered radicals. Reaction rates for CF3(CH2)nOH (n = 0–3) show the combined effects of both chain extension and electron-withdrawing substitution, increasing with carbon chain length while decreasing with more fluorine substitution.129–132 Electrostatic potential maps (Fig. S7) indicate that the most electron-deficient locations are hydrogen atoms, which makes them more susceptible to abstraction than F or Cl atoms. Although the reactivity of –OCH2– marginally decreases with increasing distance from the OH group, the higher reactivity of the β- and γ-CH2 groups compared to unsubstituted alkanes further emphasizes the activating impact of the hydroxyl functionality. These mechanistic trends, which relate electronic structure to the observed reactivity patterns of halogenated alcohols, are supported by comparisons of calculated frontier molecular orbital (FMO) energy gaps with experimental rate coefficient data. The FMO diagram is presented in Fig. S8, and the FMO energy gaps are listed in Table S8.
3.2.2 Atmospheric fate of halogenated alcohols.
The formation of fluorinated aldehydes and perfluorinated carboxylic acids through the tropospheric oxidation of halogenated alcohols by OH radicals and Cl atoms has been reported in previous studies.20,31,129–133 Based on earlier research, the OH-initiated oxidation reaction mechanisms of CF3C(CH3)OHCF3, (CF3)2CHOH, and (CF3)3COH were examined here.130,134–136 The radical product (CF3)2COHC(˙)H2 is the initial byproduct of the oxidation of CF3CXOHCF3 (X = CH3) with atmospheric oxidants. H abstraction from the –CH3 group was preferred over abstraction from the –OH position. These alkyl radicals like (CF3)2COHC(˙)H2 react with ambient oxygen in the atmosphere to produce peroxy radicals, such as (CF3)2COHCH2OO(˙) moiety. In NO-rich environments, they react with peroxy radicals to produce alkoxy radicals (CF3)2COHCH2O(˙), which then break down the C–O bond to produce formaldehyde and (CF3)2C(˙)OH. The presence of an O2 molecule causes (CF3)2C(˙)OH to undergo HO2 elimination to produce CF3C(O)CF3via the following reaction pathways. When CF3CXOHCF3 (X = H) reacts with oxidizing radicals, the alkyl radical product (CF3)2C(˙)OH is formed primarily because H abstraction from the –CH group is easier than that from the –OH position. Alkyl radicals react with ambient O2 to eliminate HO2 and generate CF3C(O)CF3. A minor reaction pathway can produce fluorinated peroxy radicals (CF3)COO(˙)OH; if these are persistent, they can form fluorinated carboxylic acids. Detailed degradation pathways for CF3CXOHCF3 (X = H and CH3) are shown in Scheme 5. We utilized information gathered from previous research to determine the atmospheric degradation pathways of CF3(CX2)nCH2OH (X = H and F; n = 0, 1, and 2) and CF3(CF2)nCH2CH2OH (n = 4, 6, and 8) by oxidizing radicals such as Cl atoms and OH radicals.131,137 In these molecules, two positions are susceptible to H abstraction: one from –CH2O and another from –OH. Reaction channels involving the –CH2O position are more frequently observed than those involving the –OH position. The first stage is the production of alkyl radicals, which then break down in four distinct pathways: reaction with oxygen, C–C bond cleavage, C–H bond cleavage, and C–O bond cleavage. Data from earlier research indicate that the degradation processes of CHF2CH2OH, CH2FCH2OH, and CHF2CF2CH2OH follows a similar sequence to that of CF3(CX2)nCH2OH (X = H and F; n = 0, 1, and 2), proceeding via these four distinct pathways (Scheme 6).18,31
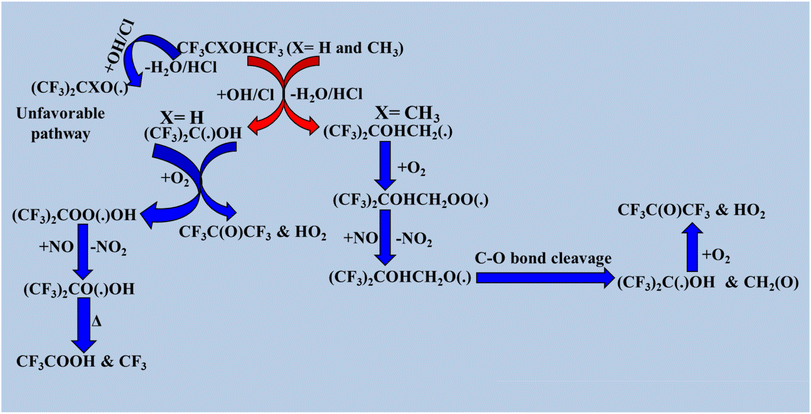 |
| | Scheme 5 Degradation process of CF3CXOHCF3, where X = H and CH3. | |
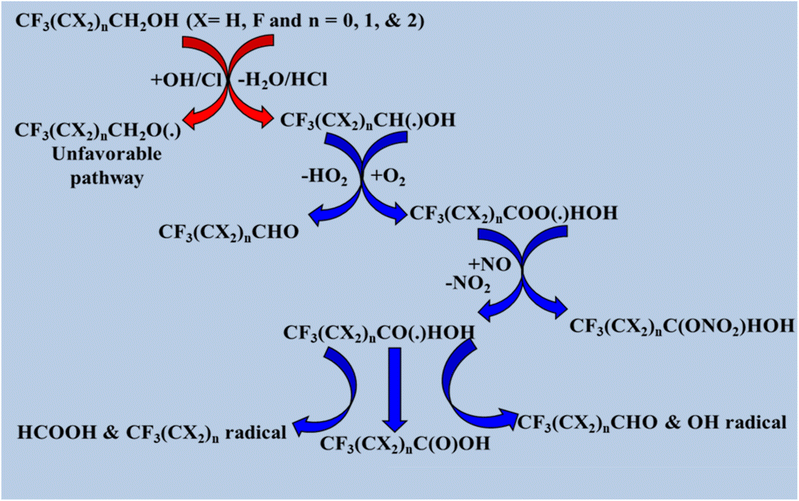 |
| | Scheme 6 Degradation process of CF3(CX2)nCH2OH. | |
Mabury et al.133 previously discussed the degradation of C4F9SO2N(CH3)CH2CH2OH by OH radicals. As shown in Scheme 7, when alkyl radicals react with ambient oxygen, they remove HO2, producing fluorinated aldehydes, and a second pathway results in the addition of OH radicals. In this pathway, the major byproducts are perfluorinated acids and alcohols. Perfluorinated acids are environmentally persistent and resistant to oxidation, hydrolysis, and reduction in biotic and abiotic environments. Fluorinated aldehydes are the primary oxidation products of fluorinated alcohols. Secondary oxidation products such as CHFO or CF2O, are absorbed into droplets/aerosols and hydrolyzed within days to generate CO, CO2, and HF.137
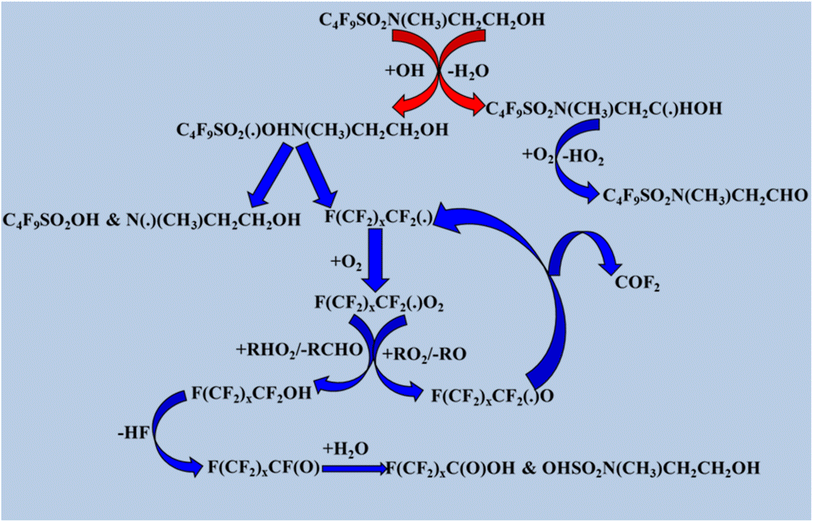 |
| | Scheme 7 Degradation process of C4F9SO2N(CH3)CH2CH2OH. | |
3.2.3 Atmospheric implications of halogenated alcohols.
Rajakumar et al.,136 Garzon et al.,129 Pan et al.,131 and Chandra et al.138 have reported atmospheric lifetimes for several types of halogenated alcohols reacting with both OH radicals and Cl atoms, with Cl-driven life times assessed under two conditions: global mean chlorine concentrations and elevated concentrations typical of the marine boundary layer. As shown in Table S10, the total lifetime of each compound was determined by combining the OH- and Cl-mediated pathways. Based on the global average [Cl], the shortest reported total lifetime value is 10 days for CF3CH2CH2OH, and the highest lifetime is 46 years for (CF3)3COH. Considering the chlorine concentration in marine boundary layers, the shortest estimated atmospheric lifetime is 0.0028 years for CF3CH2CH2CH2OH, as reported by Pan et al.131 The longest value is 2.9 years for CF3C(CH3)OHCF3, reported by Rajakumar et al.136 These findings emphasize the importance of local halogen chemistry in determining atmospheric persistence. We estimated the POCP values for 19 halogenated alcohols, which are listed in Table S9. Table S9 shows that halogenated alcohols have considerably lower POCP values compared to ethene (100).
IR absorption spectra and the subsequent calculation of absorption cross-sections, REs, and GWPs are presented in Tables S11–S13 and Fig. S6, S9. Fluorine substitution, as predicted, increased RE values because of stronger absorption (Fig. S8) within the atmospheric window (700–1400 cm−1). Using the absorption cross-section calculations, we summarized the instantaneous and lifetime-corrected REs and GWPs in Table S12. Rajakumar et al.,136 Pan et al.,132 and Chandra et al.18 calculated two different GWP values at two different [Cl] atom concentrations, one at the global average concentration (620 molecules per cm3)139 and the other in marine boundary layers (1200 molecules per cm3)139 for CF3C(CH3)OHCF3, CF3CH2CH2CH2OH, and CHF2CF2CH2OH. The GWP values of most halogenated alcohols are significantly lower, but CF2CF2CF2CH2OH and (CF3)3COH molecules exhibit comparatively high GWP values, which are comparable to CFC-11, with a GWP value of 6330 over a 20-year time horizon, highlighting their possible climatic significance owing to their long atmospheric lifetimes. The GTP values of 19 halogenated alcohols over 20-, 50-, and 100-year time horizons are listed in Table S11 in both instantaneous and lifetime-corrected forms, among which we report GTP values of seven halogenated alcohols for the first time. Furthermore, the APs of 19 halogenated alcohols and the ozone depletion potential (ODP) of CHCl2CH2OH were assessed. Rainwater acidification may be caused by halogenated alcohol breakdown products, as their AP values continuously exceed those of SO2. These results demonstrate that, although the majority of halogenated alcohols have no direct greenhouse or ozone-depleting effects, highly fluorinated congeners are hazardous to the climate and environment.
3.3 Halogenated aldehydes
3.3.1 Rate coefficient of halogenated aldehydes.
The rate coefficients and lifetime data for halogenated aldehyde reacting with the OH radicals and Cl atoms are presented in Tables S16 and S17, respectively. The rate coefficient for F(CF2)nCHO (n = 1, 2, 3, 4, and 6) is determined by abstracting the hydrogen atom from the –CH(O) position, whereas for CF3(CH2)nCHO (n = 1 and 2), H abstraction can occur from both the –CH2 and –CH(O) positions. Previous studies indicate that the –CH(O) position is more favorable than the –CH2 position in the case of CF3(CH2)nCHO (n = 1 and 2).7 The reactivity of oxidants with halogenated aldehydes, such as CClF2CHO, CCl2FCHO, CHClFCHO, CCl3CHO, CHCl2CHO, and CH2ClCHO, was examined by Sidebottom et al.140 Their findings indicated that the rate coefficient for the reaction was reduced when halogen atoms were substituted for methyl hydrogens in acetaldehyde. Electrostatic potential (ESP) maps (Fig. S11) reveal that the most electron-deficient locations are the aldehydic hydrogens, which prefer radical attacks over halogen abstraction. To validate the ESP analysis, we computed the bond dissociation energies (BDEs) of C6F13CH2CHO at the B3LYP/6-311+G(d,p) level of theory. We found that the BDEs for aldehydic C–H bonds were approximately 93 kcal mol−1, whereas those for C–F bonds were approximately 107 kcal mol−1. Overall, these findings explain the observed decreases in reactivity with successive halogen substitution and highlight the crucial role of aldehydic H abstraction in determining the atmospheric degradation of halogenated aldehydes.
The fluoroaldehydes F(CF2)nCHO (n = 1, 2, 3, 4, and 6) react with OH radicals at rates that are over 30 times lower than acetaldehyde, according to published kinetic data, indicating substantial substituent effects.141 Rayez et al.142 identified two factors responsible for this reduced reactivity: (i) a slight strengthening of the aldehydic C–H bond and (ii) destabilization of the transition state caused by the potent electron-withdrawing inductive effect of the fluoroalkyl group. Similarly, in the case of CF3(CH2)nCHO (n = 1 and 2), the rate coefficient with OH radicals decreased by five times compared to CH3(CH2)nCHO (n = 1 and 2).143 The reduced reactivity of β- and γ-substituted fluorinated aldehydes is likely caused by the long-range destabilizing inductive action of the –CF3 group in the transition state, as the aldehydic C–H bond strengths in these two molecules are relatively comparable.144 These trends are further supported by our FMO calculations, which show that fluorination in F(CF2)nCHO (n = 2, 3, 4, and 6) progressively increases the HOMO–LUMO energy gap, which is associated with decreased chemical reactivity. Together, our results highlight that stronger C–H bonds are not the primary cause of the reduced atmospheric degradation of fluorinated aldehydes; electronic factors that destabilize the transition state and limit radical accessibility also play a role. SI data, including Table S15 and Fig. S12, provide additional details.
3.3.2 Atmospheric fate of halogenated aldehydes.
Under atmospheric conditions, the fate of the byproducts formed during the photolysis and OH radical-initiated oxidation of halogenated aldehydes is of significant interest. Reactions with OH radicals produce CX3CO radicals (X = H, F, and Cl), whereas photolysis is predicted to form CX3 radicals. In the troposphere, fully halogenated formaldehydes are photolytically stable, and their reactions with OH radicals are negligible.140 The degradation mechanisms of halogenated aldehydes, such as F(CF2)nCHO (n = 1, 2, 3, 4, and 6) and CF3(CH2)nCHO (n = 1 and 2), have been demonstrated in previous studies. For F(CF2)nCHO (n = 1, 2, 3, 4, and 6), alkyl radicals such as F(CF2)nC(˙)O (n = 1, 2, 3, 4, and 6) are produced via H abstraction. Scheme 8 illustrates the degradation mechanism of F(CF2)nCHO (n = 1, 2, 3, 4, and 6) via a single H abstraction route involving an OH radical and a Cl atom. The following schematic is Scheme 8. Degradation process of F(CF2)nCHO (n = 1, 2, 3, 4, and 6).
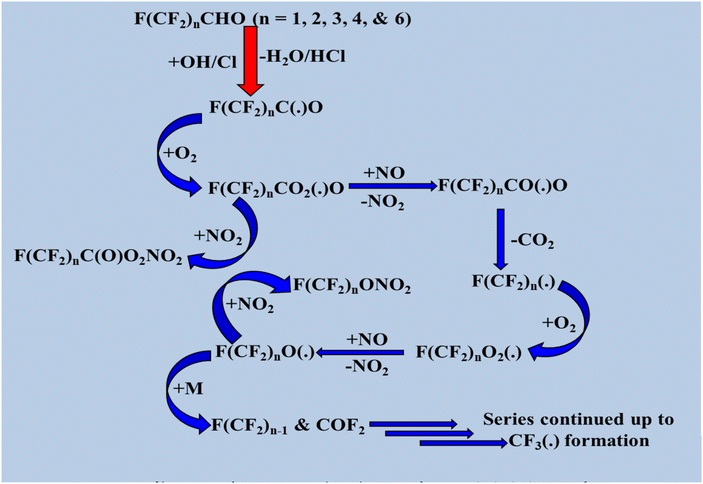 |
| | Scheme 8 Degradation process of F(CF2)nCHO (n = 1, 2, 3, 4, and 6). | |
Furthermore, previous studies have found that the degradation of CF3(CH2)nCHO (n = 1 and 2) compounds occurs via aqueous-phase atmospheric reactions and UV photolysis in the actinic region (>290 nm).7,145 Similar to reactions with oxidizing radicals like Cl atoms and OH radicals, CF3(CH2)nCHO (n = 1 and 2) can be removed from the environment through UV photolysis.7,145 In addition, Sidebottom et al.140 investigated the atmospheric degradation of various halogenated aldehydes (CClF2CHO, CCl2FCHO, CHClFCHO, CCl3CHO, CHCl2CHO, and CH2ClCHO), focusing on the reaction mechanism with OH radicals and Cl atoms. Initially, hydrogen abstraction from the halogenated aldehydic site by oxidizing radicals formed the corresponding alkyl radicals. Two different mechanisms were observed: the first was decomposition, which was preferred if X = Cl, and the second was addition of O2 to the alkyl radical, as depicted in Scheme 9.
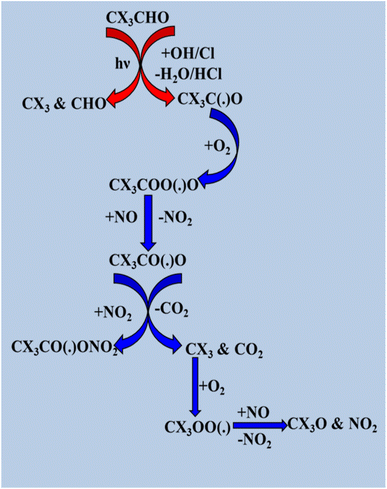 |
| | Scheme 9 Degradation process of CX3CHO. | |
3.3.3 Atmospheric implications of halogenated aldehydes.
The atmospheric lifetimes of halogenated aldehydes containing C–H bonds are primarily governed by oxidation reactions involving OH radicals and Cl atoms. The lifetimes of the 15 halogenated aldehydes are listed in Table S18. The estimated total atmospheric lifetime of different halogenated aldehydes varies from a minimum of 0.00026 years for CF2CH2CH2CHO, as reported by Albaladejo et al.,7 to a maximum of 0.36 years for CF3CHO, as reported by Nielsen et al.141 In Table S16, we present the POCP values for halogenated aldehydes calculated in this study. Almost all the studied halogenated aldehydes exhibit negligible POCP values. Although CF3CH2CH2CHO shows a moderate POCP value of 31.7, its atmospheric emissions and ambient concentration were extremely low. Consequently, its overall contribution to tropospheric ozone formation is expected to be negligible, despite the moderate POCP value. To evaluate their radiative characteristics, GWP, and GTP, IR absorption cross-sections were calculated using vibrational frequencies and intensities, and molecular geometries were optimized (Fig. S10 and Table S18). We found that C6F13CH2CHO had the most fluorine atoms, and its absorption intensity in the atmospheric window (700–1400 cm−1) was higher than that of the other compounds, at approximately 2175 km mol−1, whereas that of CH2ClCHO was relatively low, at approximately 39 km mol−1. We calculated the instantaneous and lifetime-corrected REs and GWPs for halogenated aldehydes over 20-, 100-, and 500-year time horizons, and the results are listed in Table S18. For most of the halogenated aldehydes, both the instantaneous and lifetime-corrected GWP values remains low. This is primarily because the presence of the aldehydic (H–C(O)–) hydrogen leads to a relatively fast reactions with atmospheric oxidants, resulting in short atmospheric lifetimes and consequently lower GWP values. The GTP values (instantaneous and lifetime-corrected) of 15 halogenated aldehydes over 20-, 50-, and 100-year time horizons were calculated for the first time and are reported in Table S19. Because of their short atmospheric lifetimes, most halogenated aldehydes exhibit negligible GTP values at 20-, 50-, and 100-year time horizons. However, six compounds (CClF2CHO, CCl2FCHO, CHClFCHO, CCl3CHO, CHCl2CHO, and CH2ClCHO) show detectable ozone-depletion potentials (ODPs) (Table S19). Rainwater acidification may be caused by the breakdown products of halogenated aldehydes, as their acidification potentials (APs) are higher than those of SO2. These results show that while the majority of halogenated aldehydes have an insignificant effect on climate and ozone because of their short lifetimes, their contribution to atmospheric acidification is also negligible due to their very low ambient concentrations.
3.4 Halogenated carboxylic acids
3.4.1 Rate coefficient of halogenated acids.
The rate coefficients of halogenated acids were explored via hydrogen abstraction from the –C (O)OH position of halogenated acids using OH radicals. Experimental studies by Hurley et al.146 and Chen et al.147 reported rate coefficients for F(CF2)nCOOH (n = 1, 2, 3, and 4) with OH radicals, which are summarized in SI Table S23. According to earlier research,147 the transition states involve a hydrogen atom centered between the attacking electrophilic OH radical and the CF3C(O)O(˙) radical, and the process proceeds via hydrogen atom abstraction. When the CH3 group is replaced with fluorine, the electron density on the H atom decreases, destabilizing the transition state compared to CH3C(O)OH. ESP calculations (Fig. S15) show that hydrogen atoms occupy electron-deficient regions (red isosurfaces), making them more susceptible to H abstraction by OH radicals. In contrast, fluorine substituents are surrounded by electron-rich areas, making abstraction even more difficult. The frontier molecular orbital (FMO) energy gaps were analyzed, and the results are reported in Table S22 and illustrated in Fig. S13, respectively.
3.4.2 Atmospheric fate of halogenated acids.
Hurley et al.146 and Chen et al.147 previously examined how OH radicals break down halogenated acids in the troposphere. We used their overall reaction scheme from the preliminary findings, which shows that radical products are formed via hydrogen abstraction from halogenated acids, as illustrated in Scheme 10. Owing to their thermodynamic stability, CO2 molecules are released from halogenated acids following the formation of alkyl radicals, which form F(CF2)n radicals. Finally, CF3ONO2, FNO, and COF2 molecules are formed, as shown in Scheme 10.
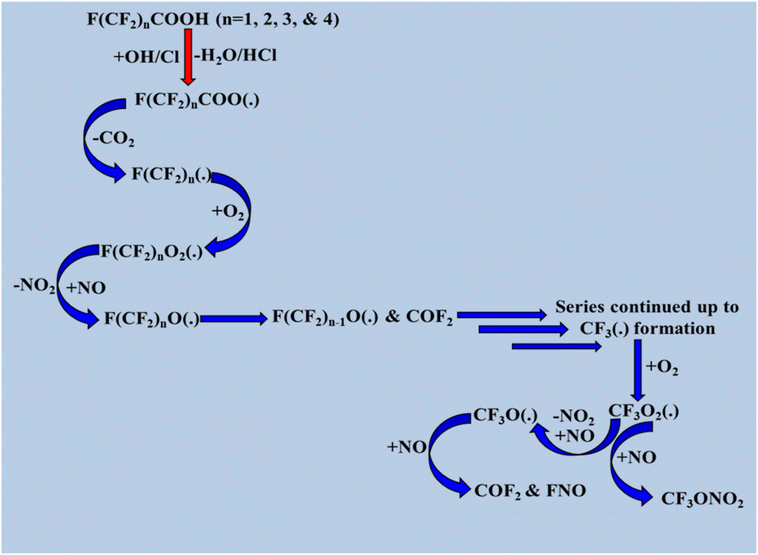 |
| | Scheme 10 Degradation process of F(CF2)nCOOH (n = 1, 2, 3, and 4). | |
3.4.3 Atmospheric implications of halogenated acids.
The atmospheric lifetimes of halogenated carboxylic acids containing C–H bonds are mainly governed by OH- and Cl-driven oxidation reactions. The lifetimes of five halogenated carboxylic acids are listed in Table S24. These data were obtained from Hurley et al.,146 who studied the lifetime of F(CF2)nCOOH (n = 1, 2, 3, and 4) and found that the lifetime ranges between 130 and 230 days. Chen et al.147 also calculated the lifetime of i-C4F9COOH and reported it as 0.84 years. The reaction of F(CF2)nCOOH with OH radicals contributes only minimally to atmospheric removal. Wet and dry deposition are considered the main atmospheric removal mechanism for F(CF2)nCOOH, occurring on a timescale of approximately ten days.146 Table S23 presents the POCP values for halogenated carboxylic acids, which are negligible; therefore, they have the least probability of creating tropospheric ozone. Fig. S17 shows the IR absorption cross-section spectra of these halogenated carboxylic acids. According to Table S25, the predicted IR intensity within the atmospheric window increases with the number of fluorine atoms in the halogenated acids, leading to a corresponding increase in RE. For the first time, we report the instantaneous and lifetime-corrected REs and GWP values for five halogenated carboxylic acids over 20-, 100-, and 500-year time horizons (Table S26). The GTP values (lifetime-corrected and instantaneous) of the five halogenated carboxylic acids were calculated over 20-, 50-, and 100-year time horizons for the first time and are tabulated in Table S27. In both cases, the instantaneous and lifetime-corrected GWP and GTP values were significant and may have impacted the atmosphere. The estimated AP values of halogenated carboxylic acids were higher than those of the reference compound SO2.
3.5 Halogenated ketones and diketones
3.5.1 Rate coefficients of halogenated ketones and diketones.
The rate coefficients of halogenated ketones and diketones were determined through H abstraction via oxidants. Earlier research has found that the rate constants of halogenated aliphatic ketones are lower than those of non-halogenated aliphatic ketones.148,149 Wayne et al.150 computed the rate coefficients (SI Table S30) and discovered that the rate constant value for the reaction with oxidizing radicals decreased when –CH3 was entirely substituted by –CX3 (X = F and Cl). It is possible that destabilizing polar effects in the transition state or modifications in the total enthalpy are responsible for the observed reduction in the reactivity upon acetone halogenation. The bond dissociation energy of the remaining C–H bonds in a methyl group decreases when an F/Cl atom is added in place of a H atom.151 We computed the BDEs of CHCl2COCH3 at the B3LYP/6-311+G(d,p) level of theory, and our results are consistent with experimental findings. The BDEs for the dissociation of substituted methyl C–H bonds were approximately 88.2 kcal mol−1, and the BDEs for unsubstituted methyl C–H bonds were 99.8 kcal mol−1.
Additionally, we computed the ESP diagrams for the halogenated ketones and diketones examined in this study (SI Fig. S19). ESP maps were created for the halogenated ketones and diketones studied (Fig. S19). These maps clearly demonstrate that H abstraction is preferable at electron-deficient regions rather than at halogen sites, which are surrounded by electron-rich zones. Consequently, hydrogen-atom abstraction is projected to occur preferentially from halogenated methyl groups rather than from unsubstituted methyl groups, consistent with the trends indicated by the reaction enthalpies. According to previous studies, CF3CF2CF2C(O)CH3 and CF3CF2CF2CF2C(O)CH3 have almost identical rate coefficients, which is expected because the size of the CnF2n+1 group typically has insignificant impact on the reactivity of halogenated ketones towards oxidizing radicals.152 Regarding the rate coefficient of halogenated diketones, previous studies have demonstrated that the enol form exhibits significantly greater reactivity owing to the addition of oxidizing radicals to the CC double bond, which is a dominant pathway compared to the H atom abstraction from the diketone forms. We also calculated the FMO energy gaps, which are provided in SI Table S29. Here, the enol form exhibits electron density concentrated around the CC bonds, as shown in Fig. 2.
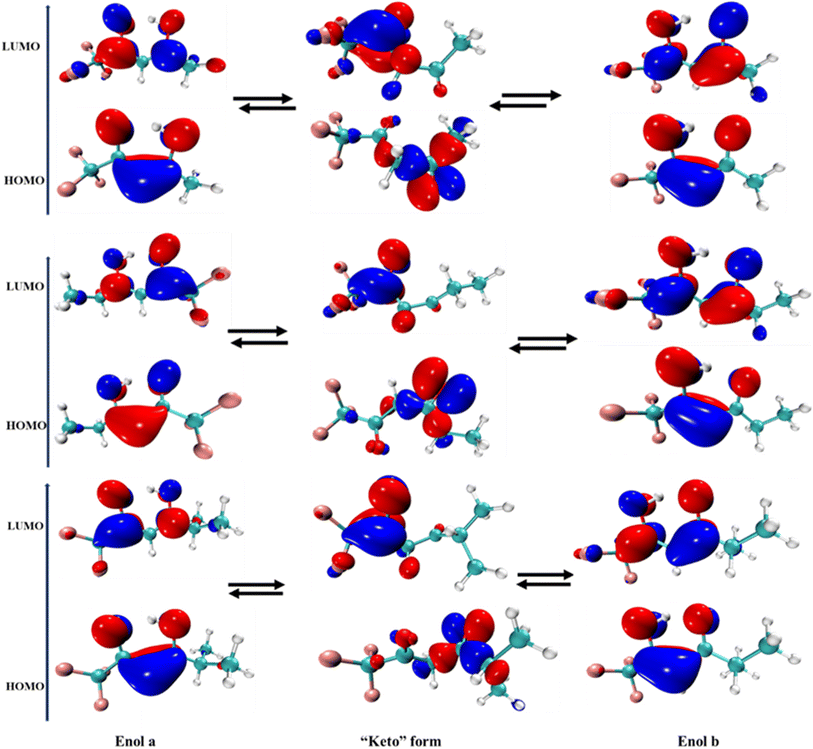 |
| | Fig. 2 Analysis of frontier molecular orbitals (FMO) in the halogenated diketone (keto–enol tautomerization) compounds calculated at the B3LYP/6-311+G(d,p) level of theory. | |
3.5.2 Atmospheric fate of ketones and diketones.
Following previous studies,150 there are two ways in which OH radicals and Cl atoms attack partly halogenated substituted ketones: first, they extract hydrogen from the halogenated substituted methyl group, and second, they abstract hydrogen from the –CH3 group. Both substituted and unsubstituted methyl groups in a monohalogenated ketone produced the same result, as shown in the Scheme 11. However, in the case of disubstituted halogenated ketones, the predicted outcome is HCOX when –H abstraction occurs through the methyl position, as shown in the schematic diagram of degradation pathways below, and COX2 when a reaction occurs with a substituted –CH3 group (Scheme 12).
 |
| | Scheme 11 Degradation process of CH2XC(O)CH3 (X = F and Cl). | |
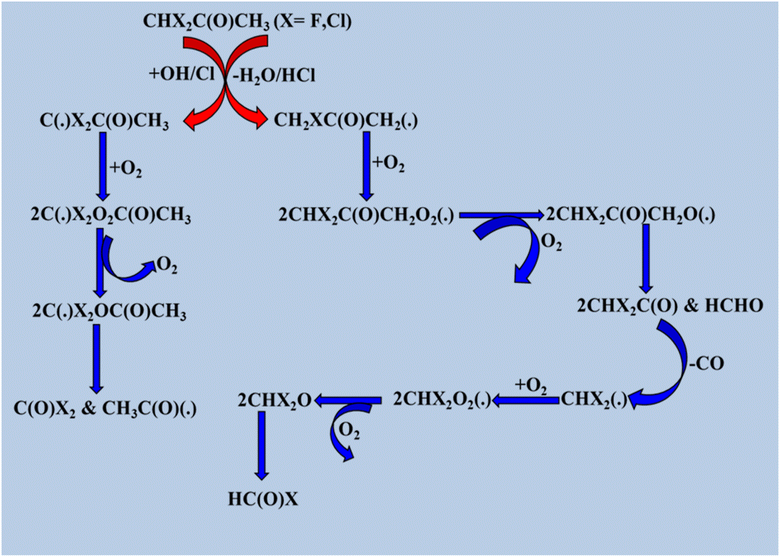 |
| | Scheme 12 Degradation process of CHX2C(O)CH3 (X = F and Cl). | |
The degradation of CX3COCH3 (X = F and Cl) was also examined. Here, only one pathway is possible; that is, the hydrogen at the –CH3 position is abstracted by the oxidizing radical to create the alkyl radical product CX3COCH2(˙). According to the schematic diagram, if X = Cl, CCl3COCH3 yields COCl2 as its main byproduct, with a yield of nearly 100%. Moreover, trifluoromethyl-glyoxal was identified as a significant product based on spectral analysis, which revealed significant absorption bands at 1100–1300 cm−1 and 1813 cm−1. The degradation processes of HC(O)F and CH3C(O)F involve OH radicals and Cl atoms, and both reactions with oxidizing radicals have low atmospheric relevance.153,154 The ability of halogenated diketones to change the position of double bonds inside their molecules by replacing an enol group with a single carbonyl moiety makes them noteworthy. The OH moiety and the carbonyl oxygen produce an intramolecular H bond that stabilizes the enolic state. This type of isomerism is known as keto–enol tautomerism.29 We conducted an NCI analysis to validate the experimental results. Green spikes displayed in Fig. 3 are associated with the hydrogen bonding between the OH moiety and the carbonyl oxygen. As a result, the OH radical can react with these compounds can occur either by attaching the OH radical to the double bond and H abstraction in the enolic form or H abstraction in the keto form of the compound. Acetic acid, methyl glyoxal, and pentane-2,3,4-trione were identified as main products of OH radical-induced oxidation of enolic compounds in the gas phase (Scheme 13).29
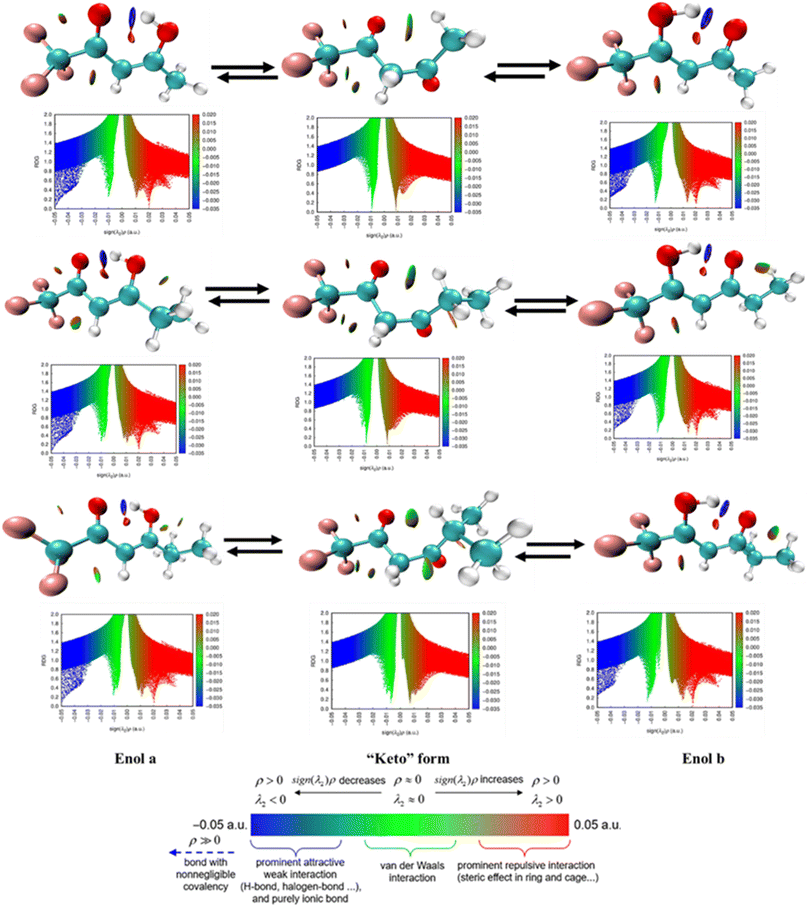 |
| | Fig. 3 Analysis of non-covalent interactions (NCI) of halogenated diketone (keto–enol tautomerization) compounds calculated using the B3LYP method and the 6-311+G(d,p) basis set. | |
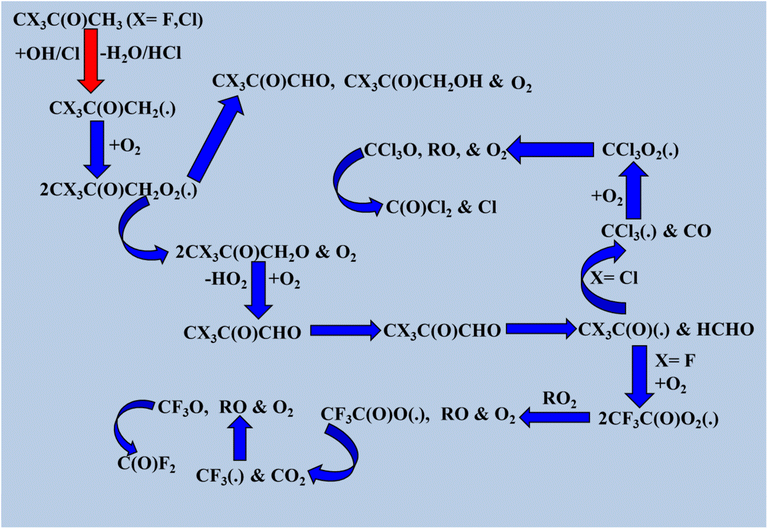 |
| | Scheme 13 Degradation process of CHX2C(O)CH3 (X = F and Cl). | |
3.5.3 Atmospheric implications of halogenated ketones and diketones.
The oxidation reaction of halogenated ketones and diketones with C–H bonds by OH radicals and Cl atoms in the atmosphere is their primary removal process. The lifetimes of the 13 halogenated ketones and diketones are listed in Table S31. Wayne et al.150 calculated rate coefficients for CF3C(O)CH3, CH2FC(O)CH3, CCl3C(O)CH3, CHCl2C(O)CH3, and CH2ClC(O)CH3. The atmospheric lifetimes of these compounds were calculated using eqn (S1A) and (S1B). The total estimated lifetimes of different halogenated ketones and diketones range from a minimum of 8 h for CF3C(O)CH2C(O)CH(CH3)2, as reported by Blanco et al.,29 to a maximum of 9.31 years for CF3C(O)CH3, as reported by Wayne et al.150 Our calculated POCP values for halogenated ketones and POCP values for diketones reported by Blanco et al.29 are listed in Table S30. POCP values for ketones are negligible, but in the case of diketones (CF3C(O)CH2C(O)CH3, CF3C(O)CH2C(O)CH2CH3, and CF3C(O)CH2C(O)CH(CH3)2), POCP values are significantly moderate based on the reference compound ethene (100).
The optimization and frequency calculations reported in Table S32 indicate that successive fluorination systematically increases the RE. This pattern can be attributed to the fact that a greater number of fluorine atoms increases the absorption intensities in the atmospheric window (700–1400 cm−1). The optimized molecular geometries of the halogenated ketones and diketones (Fig. S18) reflect this tendency, and the predicted IR spectra demonstrate that higher fluorine substitution leads to increased absorption characteristics in this important spectral region. The estimated absorption cross-section profiles (Fig. S21) confirmed this improvement. We measured the instantaneous and lifetime-corrected REs and GWPs for 13 halogenated ketones and diketones over 20-, 100-, and 500-year time horizons (Table S33). Table S28 shows the instantaneous and lifetime-corrected global temperature potentials (GTPs) of the same chemicals over 20-, 50-, and 100-year time horizons. Overall, our data show a strong relationship between increased fluorine substitution, enhanced IR absorption in the atmospheric window, and higher climate-relevant indices. We also calculated the ODP values of three chlorinated ketones (CCl3C(O)CH3, CHCl2C(O)CH3, and CH2ClC(O)CH3), and these values are summarized in Table S34. Table S34 shows that the AP values calculated and reported by Blanco et al.29 were similar to those of SO2. This indicates that halogenated ketones and diketones, as well as the products of their atmospheric degradation, may be involved in acid rain and are known to degrade water and soil quality, impacting the biota and human health.155–157
3.6 Summary of atmospheric metrics across XOVOC classes
The findings in Table 1 show prominent structure–function correlations among the different XOVOC classes. Halogenated alcohols have the highest atmospheric persistence (2 days to 46 years), often exceeding the lifetimes of typical hydrofluorocarbons, and thus contribute disproportionately to long-term climate forcing (GWP of up to 3788). Halogenated aldehydes exhibit relatively short atmospheric lifetimes (4 days to approximately 0.36 years). Their reported POCP values (2.5–31.7), reference to ethene = 100, clearly indicate a low photochemical ozone creation potential. Moreover, their atmospheric concentrations are generally very low. Therefore, these compounds are not expected to contribute meaningfully to local or regional ozone formation, even under favorable photochemical conditions. Halogenated esters display moderate lifetimes and GWPs, indicating a balance between structural stability and reactivity, whereas halogenated ketones and diketones have the greatest variability in lifetimes (8 h to 10 y), highlighting their dual potential as both short-lived ozone precursors and longer-lived climate forcers. Halogenated acids, despite having modest lifetimes (130 days to 0.84 years), exhibit comparatively higher acidification potential; however, their low atmospheric concentrations imply that they are not expected to contribute significantly to large-scale atmospheric acidification. Overall, halogenated alcohols appear to be the most relevant for short-term effects on local air quality, whereas some carbonyl-containing species may also exert longer-term climatic impacts depending on their atmospheric persistence and radiative properties.
Table 1 Typical ranges of atmospheric lifetimes (τ), GWP, POCP, and AP values for the representative classes of XOVOCs. These OVOC classes are ordered based on lifetime ranges and their atmospheric matrices
| OVOC class |
Lifetime (τ) |
GWP (100 year) |
POCP |
AP |
| Halogenated alcohols |
2 days to 46 years |
Less than 1 to 3788 |
0.4–17 |
0.50–1.22 |
| Halogenated ketone and diketones |
8 hours to 10 years |
Less than 1 to 1291 |
0.7–33.4 |
0.35–1.10 |
| Halogenated esters |
5 hours to 3.2 years |
Less than 1 to 849 |
0.4–32 |
0.35–1.09 |
| Halogenated acids |
130 days to 0.84 years |
23–73 |
0.4–1.4 |
0.84–1.09 |
| Halogenated aldehydes |
4 days to 0.36 years |
Less than 1 to 33 |
2.5–31.7 |
0.57–1.16 |
4. Dielectric strength (DS)
One of the main issues with climate change is finding an environmentally friendly insulating gas to replace sulfur hexafluoride (SF6), which has been an ongoing study in the past few years. The concern now is, why should alternatives replace SF6? This is because the electric grid sector uses SF6 extensively because of its exceptional insulating capabilities and chemical durability.158 However, its GWP value is high, approximately 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 times that of CO2. Therefore, some suitable replacements that have confirmed exceptional qualities, such as high insulation strengths and low GWPs, include c-C4F8, CF3I, C5F10O, C6F12O (NOVEC 1230), and C4F7N.158 A new sustainable insulating gas called NOVEC 1230 has been developed recently.159 In this review, our chosen high-thought-out computational approaches include the B3LYP/6-311+G(d,p) level of theory, which has been shown to outperforms methods such as PBE1PBE, M06-2X, and M11 for predicting density and structure-related properties of a broad range of organic compounds for DS prediction.106 Furthermore, the halogenated OVOCs of interest require the inclusion of polarization and diffuse functions in the minimal basis sets for the following reasons. (i) Polarization function: these compounds have multiple chemical bonds and exhibit soft electronic nature. (ii) Diffuse function: for neutral and cationic systems, DFT calculations predict one of the important variables, namely the ionization energies. (iii) Computational accuracy: to balance the computational accuracy and efficiency, the diffuse functions of substantial elements must be used preferentially. Considering all factors, the 6-311+G(d,p) basis set was the most appropriate for this investigation.106 As shown in the figures, there is a wide range of halogenated esters, alcohols, aldehydes, acids, ketones, and diketones. Polarizability increases linearly with molecular weight and backbone length (number of carbon atoms), as shown in Fig. 4 and 5, whereas the ionization energy is insensitive to these structural characteristics. Furthermore, dielectric strength shows a linear relationship with molecular weight and backbone length owing to the unique characteristics of the two fundamental variables (polarizability and ionization energy) that define DS. SI Tables S7, S14, S21, S28 and S35 list the theoretically determined DSs, polarizabilities, and ionization energies for XOVOCs.
500 times that of CO2. Therefore, some suitable replacements that have confirmed exceptional qualities, such as high insulation strengths and low GWPs, include c-C4F8, CF3I, C5F10O, C6F12O (NOVEC 1230), and C4F7N.158 A new sustainable insulating gas called NOVEC 1230 has been developed recently.159 In this review, our chosen high-thought-out computational approaches include the B3LYP/6-311+G(d,p) level of theory, which has been shown to outperforms methods such as PBE1PBE, M06-2X, and M11 for predicting density and structure-related properties of a broad range of organic compounds for DS prediction.106 Furthermore, the halogenated OVOCs of interest require the inclusion of polarization and diffuse functions in the minimal basis sets for the following reasons. (i) Polarization function: these compounds have multiple chemical bonds and exhibit soft electronic nature. (ii) Diffuse function: for neutral and cationic systems, DFT calculations predict one of the important variables, namely the ionization energies. (iii) Computational accuracy: to balance the computational accuracy and efficiency, the diffuse functions of substantial elements must be used preferentially. Considering all factors, the 6-311+G(d,p) basis set was the most appropriate for this investigation.106 As shown in the figures, there is a wide range of halogenated esters, alcohols, aldehydes, acids, ketones, and diketones. Polarizability increases linearly with molecular weight and backbone length (number of carbon atoms), as shown in Fig. 4 and 5, whereas the ionization energy is insensitive to these structural characteristics. Furthermore, dielectric strength shows a linear relationship with molecular weight and backbone length owing to the unique characteristics of the two fundamental variables (polarizability and ionization energy) that define DS. SI Tables S7, S14, S21, S28 and S35 list the theoretically determined DSs, polarizabilities, and ionization energies for XOVOCs.
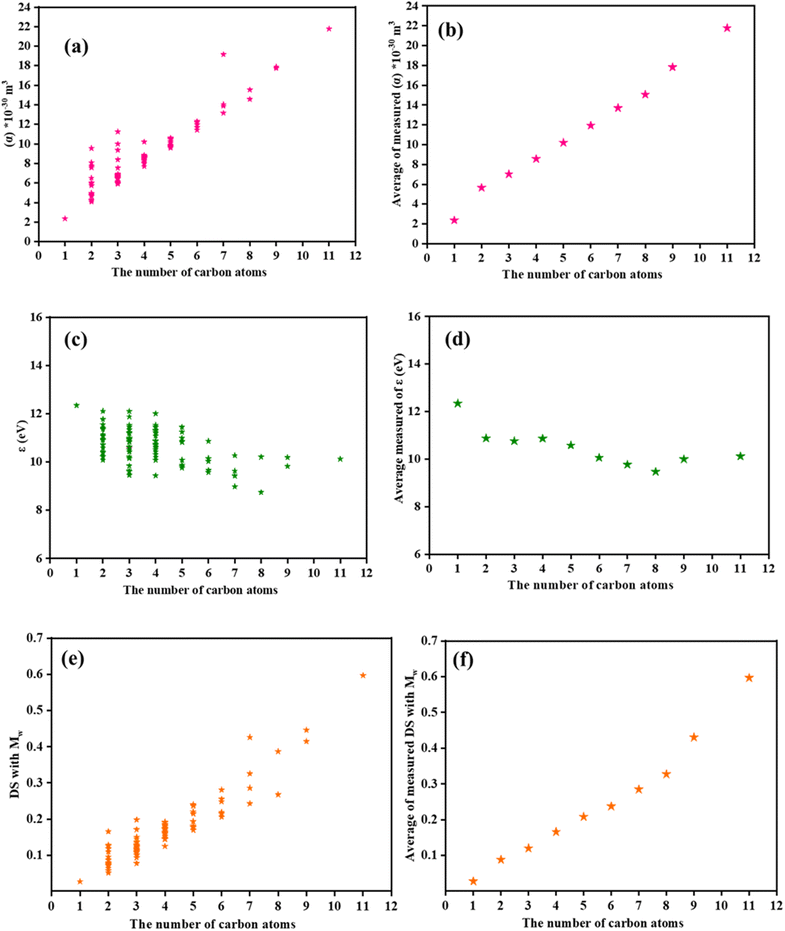 |
| | Fig. 4 Correlations between the physical characteristics of the compounds. (a and b) Polarizabilities, (c and d) ionization energies, and (e and f) and dielectric strengths of the halogenated OVOCs correlated with the number of carbon atoms. | |
 |
| | Fig. 5 Correlations between the physical characteristics of the compounds. (a and b) Polarizabilities, (c and d) ionization energies, and (e and f) dielectric strengths of the halogenated OVOCs as a function of their molecular weight. | |
4.1 Survey of SF6 alternatives
The demand for insulating gases in industrial applications are extremely high and includes aspects such as dielectric performance, chemical and thermal stability, non-flammability, cost-effectiveness, durability, and ecologically friendly design.160 An ideal insulating gas would be a substance that minimally impacts the atmospheric environment, endures electric fields corresponding to the highest voltage levels, and satisfies safety, risk, and toxicity requirements for equipment operating under all conceivable circumstances.161 In this context, our assessment of XOVOCs offers key metrics, including atmospheric lifetimes, RE, GWP, and POCP, that are specifically relevant for preliminary environmental examinations such as minimal impact on the atmosphere. Efforts to replace SF6 in electrical equipment have yielded compounds that can be utilized as pure gases. However, no single gas has been identified as the most suitable option for all requirements. Extensive screening studies, such as those assessing halogenated alcohols, ketones, and associated compounds, have investigated a wide array of candidates for SF6 substitution. None of them fulfils the complete set of dielectric performance, chemical and thermal stability, safety, and environmental compatibility needed for dependable use in high-voltage equipment, even if some have qualities that make them appropriate for use as pure gases.
5. Conclusion
We reviewed the reactions of halogenated oxygenated volatile organic compounds (XOVOCs) with Cl atoms and OH radical oxidants. However, based on our prior findings, we concluded that hydroxyl radicals often control daytime chemistry in the troposphere and, to a certain extent, in natural water. The high oxidative capability of OH radicals makes them the “detergent of the atmosphere.” Compared to other oxidative species, hydroxyl radicals are highly reactive, causing most tropospheric trace chemicals and contaminants to undergo oxidation and chemical conversion. This review presents an extensive database on the kinetics and mechanistic degradation pathways of gas-phase oxidation of XOVOCs. Based on these data, we concluded that some significant general predictions regarding the reaction rate coefficients and degradation pathway mechanisms for OVOCs could be established. From this reviewed data, we have found their atmospheric implications; we provide an extensive evaluation of the lifetimes of 94 oxygenated hydrofluorocarbons, including fluorinated and chlorinated (esters, alcohols, aldehydes, and ketones), fluorinated carboxylic acids, and fluorinated diketones, which absorb IR radiation and disturb the radiation budget of the Earth. For halogenated esters, the lifetime value decreased with the addition of hydrogen atoms to the molecules. Halogenated esters had the highest lifetime values among the 94 oxygenated hydrofluorocarbons considered in this study. The instantaneous and lifetime-corrected REs of 105 compounds are summarized, among which we calculated the REs of 46 compounds for the first time, which have not been studied yet. We also summarized the GWP values in both cases, instantaneous and lifetime-corrected values of 94 oxygenated hydrofluorocarbons over 20-, 100-, and 500-year time horizons, among which the GWP values of 46 compounds were reported for the first time. The GWP values of some oxygenated hydrofluorocarbons are significantly high over a 20-year time horizon, which is a concern. This review article also contains a thorough compilation of instantaneous and lifetime-corrected GTP values of 94 oxygenated hydrofluorocarbon compounds over 20-, 50-, and 100-year time horizons, among which the GTPs of 59 compounds were calculated.
The GTP values of some halogenated ketones are considerably high. The ODPs of 14 chlorine-containing oxygenated hydrofluorocarbons were also calculated by us and reported for the first time. The calculated ODP values for chlorine-containing oxygenated hydrofluorocarbons were considerably low. In this review, we calculated the POCP and AP values for almost all the OVOCs considered. The POCP values were negligible for the considered halogenated compounds, and some had moderate POCP values such as CF3CH2CH2CHO, diketones, and unsaturated halogenated esters. Despite their moderate POCP values, the atmospheric impact of these XOVOCs is negligible. Their low emission rates from industrial sources prevent any significant contribution to tropospheric ozone or smog formation; thus, they may not fall under stringent air quality regulations. Overall, the structural class of XOVOCs determines their atmospheric effects; alcohols and ketones with longer lives exert greater climatic forcing (GWP), whereas esters and aldehydes are more significant for short-term air quality (POCP and AP) than alcohols and ketones. Although the high acidification potential (AP) of XOVOCs is a noted characteristic, implying a theoretical risk of contributing to water acidification after degradation, their environmental impact is negligible. This is because of their very low atmospheric concentration. This review covers XOVOC chemicals, their rate coefficients, mechanistic pathways, and their atmospheric implications. Although we determined their dielectric strength (DS), which is crucial for identifying substitutes for SF6, a molecule with high GWP values that negatively impacts the atmosphere, no current XOVOC can substitute this SF6 molecule based on our calculations.
Author contributions
Kanika Guleria: conceptualization, methodology, writing-original draft preparation, formal analysis, reviewing, and editing. Suresh Tiwari: conceptualization, methodology, writing-original draft preparation, formal analysis, reviewing, and editing. Ranga Subramanian: conceptualization, methodology, formal analysis, supervision, reviewing, and editing.
Conflicts of interest
There are no conflicts to declare.
Data availability
All required data are provided in the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5ea00138b.
Acknowledgements
S. T. and K. G. thank the Indian Institute of Technology Patna for providing research facilities and financial support to carry out this work.
References
- Ø. Hodnebrog, M. Etminan, J. Fuglestvedt, G. Marston, G. Myhre, C. Nielsen, K. P. Shine and T. Wallington, Rev. Geophys., 2013, 51, 300–378 CrossRef.
- J. E. Hansen and M. Sato, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14778–14783 CrossRef CAS.
- V. Papadimitriou, J. Sci. Educ. Technol., 2004, 13, 299–307 CrossRef CAS.
- S. Solomon and J. S. Daniel, Clim. Change, 1996, 32, 7–17 CrossRef CAS.
- G. Ghani, Econ. Bull., 2007, 17, 1–5 Search PubMed.
-
M. Grubb, C. Vrolijk and D. Brack, Routledge Revivals: Kyoto Protocol (1999): A Guide and Assessment, Routledge, 2018 Search PubMed.
- M. Antiñolo, E. Jiménez, A. Notario, E. Martínez and J. Albaladejo, Atmos. Chem. Phys., 2010, 10, 1911–1922 CrossRef.
- N. K. Gour, K. Borthakur, S. Paul and R. C. Deka, Chemosphere, 2020, 238, 124556 CrossRef CAS.
- G. J. Velders, S. Solomon and J. S. Daniel, Atmos. Chem. Phys., 2014, 14, 4563–4572 CrossRef.
-
D. Gielen and T. Kram, OECD Workshop Report, 1998, pp. 17–18 Search PubMed.
- Y. Ninomiya, M. Kawasaki, A. Guschin, L. Molina, M. Molina and T. Wallington, Environ. Sci. Technol., 2000, 34, 2973–2978 CrossRef CAS.
- L. Chen, S. Kutsuna, K. Tokuhashi and A. Sekiya, J. Phys. Chem. A, 2006, 110, 12845–12851 CrossRef CAS PubMed.
- M. Sulbaek Andersen, O. Nielsen, T. Wallington, M. Hurley and W. DeMore, J. Phys. Chem. A, 2005, 109, 3926–3934 CrossRef CAS.
- D. Zehavi and J. N. Seiber, Anal. Chem., 1996, 68, 3450–3459 CrossRef CAS PubMed.
- H. Frank, A. Klein and D. Renschen, Nature, 1996, 382, 34 CrossRef CAS.
- A. Jordan and H. Frank, Environ. Sci. Technol., 1999, 33, 522–527 CrossRef CAS.
- C. E. Wujcik, D. Zehavi and J. N. Seiber, Chemosphere, 1998, 36, 1233–1245 CrossRef CAS.
- B. Baidya, M. Lily and A. K. Chandra, ChemistrySelect, 2019, 4, 7134–7143 CrossRef CAS.
- A. Garzón, M. Moral, A. Notario, A. A. Ceacero-Vega, M. Fernández-Gómez and J. Albaladejo, ChemPhysChem, 2010, 11, 442–451 CrossRef PubMed.
- T. Kelly, V. Bossoutrot, I. Magneron, K. Wirtz, J. Treacy, A. Mellouki, H. Sidebottom and G. Le Bras, J. Phys. Chem. A, 2005, 109, 347–355 CrossRef CAS PubMed.
- M. Hurley, T. Wallington, M. Sulbaek Andersen, D. Ellis, J. Martin and S. Mabury, J. Phys. Chem. A, 2004, 108, 1973–1979 CrossRef CAS.
- M. D. Hurley, J. A. Misner, J. C. Ball, T. J. Wallington, D. A. Ellis, J. W. Martin, S. A. Mabury and M. Sulbaek Andersen, J. Phys. Chem. A, 2005, 109, 9816–9826 CrossRef CAS PubMed.
- S. R. Sellevåg, C. J. Nielsen, O. A. Søvde, G. Myhre, J. K. Sundet, F. Stordal and I. S. Isaksen, Atmos. Environ., 2004, 38, 6725–6735 CrossRef.
-
T. J. Wallington, M. P. Sulbaek Andersen and O. J. Nielsen, in Advances in atmospheric chemistry, World Scientific, 2017, pp. 305–402 Search PubMed.
- M. De Rosa, D. Arnold, D. Hartline, L. Truong, R. Verner, T. Wang and C. Westin, J. Org. Chem., 2015, 80, 12288–12299 CrossRef CAS.
- R. Ilmi, A. Haque, I. J. Al-Busaidi, N. K. Al Rasbi and M. S. Khan, Dyes Pigm., 2019, 162, 59–66 CrossRef CAS.
- V. G. Isakova, S. K. Tat'yana and F. A. Lakhvich, Russ. Chem. Rev., 2010, 79, 849 CrossRef CAS.
- V. Babain, V. Romanovskii, V. Starchenko, A. Shadrin, G. Kudinov, S. Podoinitsyn and Y. Revenko, J. Nucl. Sci. Technol., 2002, 39, 267–269 CrossRef.
- P. L. Lugo, V. Straccia, C. B. Rivela, I. Patroescu-Klotz, N. Illmann, M. A. Teruel, P. Wiesen and M. B. Blanco, Chemosphere, 2022, 286, 131562 CrossRef CAS PubMed.
- D. A. Ellis, S. A. Mabury, J. W. Martin and D. C. Muir, Nature, 2001, 412, 321–324 Search PubMed.
- M. Lily, S. Hynniewta, B. Muthiah, W. Wang, A. K. Chandra and F. Liu, Atmos. Environ., 2021, 249, 118247 CrossRef.
- D. Good, J. Francisco, A. Jain and D. J. Wuebbles, J. Geophys. Res.:Atmos., 1998, 103, 28181–28186 CrossRef.
- Ø. Hodnebrog, B. Aamaas, J. S. Fuglestvedt, G. Marston, G. Myhre, C. J. Nielsen, M. Sandstad, K. P. Shine and T. J. Wallington, Rev. Geophys., 2020, 58, e2019RG000691 CrossRef PubMed.
- I. Bravo, Y. Díaz-de-Mera, A. Aranda, E. Moreno, D. R. Nutt and G. Marston, Phys. Chem. Chem. Phys., 2011, 13, 17185–17193 RSC.
- P. Zhu, X.-m. Duan and J.-y. Liu, J. Fluorine Chem., 2015, 176, 61–70 Search PubMed.
- G. Srinivasulu, S. Vijayakumar and B. Rajakumar, ChemistrySelect, 2018, 3, 4480–4489 CrossRef.
- G. Solignac, A. Mellouki, G. Le Bras, M. Yujing and H. Sidebottom, Phys. Chem. Chem. Phys., 2007, 9, 4200–4210 RSC.
- L. P. Viegas, Atmosphere, 2022, 13, 1256 CrossRef.
- J. B. Burkholder, R. Cox and A. Ravishankara, Chem. Rev., 2015, 115, 3704–3759 CrossRef.
- K. Tokuhashi, K. Takizawa and S. Kondo, Environ. Sci. Pollut. Res., 2018, 25, 15204–15215 CrossRef PubMed.
-
V. Schmidt, G.-Y. Zhu, K. Becker and E. Fink, Proceedings of the third European Symposium held in Varese, Italy, 10-12, April, 1984, pp. 177–187 Search PubMed.
- T. Schaefer, J. Schindelka, D. Hoffmann and H. Herrmann, J. Phys. Chem. A, 2012, 116, 6317–6326 CrossRef PubMed.
- H. Herrmann, Chem. Rev., 2003, 103, 4691–4716 CrossRef PubMed.
- B. Ervens, S. Gligorovski and H. Herrmann, Phys. Chem. Chem. Phys., 2003, 5, 1811–1824 RSC.
- D. Hoffmann, B. Weigert, P. Barzaghi and H. Herrmann, Phys. Chem. Chem. Phys., 2009, 11, 9351–9363 RSC.
- H. Herrmann, D. Hoffmann, T. Schaefer, P. Bräuer and A. Tilgner, ChemPhysChem, 2010, 11, 3796–3822 CrossRef PubMed.
- K. Tokuhashi, A. Takahashi, M. Kaise, S. Kondo, A. Sekiya and E. Fujimoto, Chem. Phys. Lett., 2000, 325, 189–195 CrossRef.
-
K. Tokuhashi, L. Chen, K. Takizawa, A. Takahashi, T. Uchimaru, M. Sugie, S. Kondo and A. Sekiya, Fluorine Chemistry Research Advances, 2007, pp. 143–241 Search PubMed.
- R. Simonaitis and J. Heicklen, Int. J. Chem. Kinet., 1973, 5, 231–241 CrossRef.
- G. J. Doyle, A. C. Lloyd, K. Darnall, A. M. Winer and J. N. Pitts Jr, Environ. Sci. Technol., 1975, 9, 237–241 CrossRef.
- J. G. Calvert, A. Lazrus, G. L. Kok, B. G. Heikes, J. G. Walega, J. Lind and C. A. Cantrell, Nature, 1985, 317, 27–35 CrossRef.
- H. Niki, P. Maker, C. Savage and L. Breitenbach, J. Phys. Chem., 1978, 82, 132–134 CrossRef.
- K. R. Darnall, R. Atkinson and J. N. Pitts Jr, J. Phys. Chem., 1978, 82, 1581–1584 CrossRef.
- C. J. Young, M. D. Hurley, T. J. Wallington and S. A. Mabury, J. Geophys. Res.:Atmos., 2008, 113, D24301–D24309 Search PubMed.
- P. J. Godin, A. Cabaj, S. Conway, A. C. Hong, K. Le Bris, S. A. Mabury and K. Strong, J. Mol. Spectrosc., 2016, 323, 53–58 CrossRef.
- G. Myhre, C. Nielsen, D. Powell and F. Stordal, Atmos. Environ., 1999, 33, 4447–4458 CrossRef.
- P. J. Godin, A. Cabaj, L.-H. Xu, K. Le Bris and K. Strong, J. Quant. Spectrosc. Radiat. Transfer, 2017, 186, 150–157 CrossRef.
- F.-Y. Liu, Z.-W. Long, X.-F. Tan and B. Long, Comput. Theor. Chem., 2014, 1038, 33–39 CrossRef.
- L. Yang, J. Y. Liu, L. Wang, H. Q. He, Y. Wang and Z. S. Li, J. Comput. Chem., 2008, 29, 550–561 CrossRef.
- L. Yang, J. Y. Liu, S. Q. Wan and Z. S. Li, J. Comput. Chem., 2009, 30, 565–580 CrossRef.
- F.-Y. Bai, G. Sun, X. Wang, Y.-Q. Sun, R.-S. Wang and X.-M. Pan, J. Phys. Chem. A, 2015, 119, 1256–1266 CrossRef.
- T.-X. Chi, X.-X. Li, S. Ni, F.-Y. Bai, X.-M. Pan and Z. Zhao, Phys. Chem. Chem. Phys., 2024, 26, 24821–24832 RSC.
- S. Tiwari and R. Subramanian, Phys. Chem. Chem. Phys., 2025 10.1039/D5CT02637G.
-
M. Frisch, G. Trucks, H. B. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, G. Petersson and H. Nakatsuji, 2016.
-
R. Dennington, T. Keith and J. Millam, Gaussview, 2009 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101–194119 CrossRef PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef.
- K. Guleria and R. Subramanian, ACS Earth Space Chem., 2023, 7, 947–959 CrossRef.
- K. Guleria and R. Subramanian, Theor. Chem. Acc., 2022, 141, 77 Search PubMed.
- K. Guleria and R. Subramanian, Comput. Theor. Chem., 2022, 1208, 113547 Search PubMed.
- S. Begum and R. Subramanian, RSC Adv., 2015, 5, 39110–39121 RSC.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- J. Pal and R. Subramanian, Phys. Chem. Chem. Phys., 2019, 21, 6525–6534 RSC.
- C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154–2161 CrossRef.
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef.
- J. A. Pople, M. Head-Gordon and K. Raghavachari, J. Chem. Phys., 1987, 87, 5968–5975 CrossRef.
- B. C. Garrett and D. G. Truhlar, J. Chem. Phys., 1979, 70, 1593–1598 CrossRef.
- B. C. Garrett and D. G. Truhlar, J. Am. Chem. Soc., 1979, 101, 4534–4548 CrossRef.
- B. C. Garrett, D. G. Truhlar, R. S. Grev and A. W. Magnuson, J. Phys. Chem., 1980, 84, 1730–1748 CrossRef.
- D. G. Truhlar and A. Kuppermann, J. Am. Chem. Soc., 1971, 93, 1840–1851 CrossRef.
- A. Fernandez-Ramos, B. A. Ellingson, B. C. Garrett and D. G. Truhlar, Rev. Comput. Chem., 2007, 23, 125 Search PubMed.
- D.-h. Lu, T. N. Truong, V. S. Melissas, G. C. Lynch, Y.-P. Liu, B. C. Garrett, R. Steckler, A. D. Isaacson, S. N. Rai and G. C. Hancock, Comput. Phys. Commun., 1992, 71, 235–262 CrossRef.
- Y. P. Liu, G. C. Lynch, T. N. Truong, D. H. Lu, D. G. Truhlar and B. C. Garrett, J. Am. Chem. Soc., 1993, 115, 2408–2415 CrossRef.
- A. Virmani, M. P. Walavalkar, A. Sharma, S. Sengupta, A. Saha and A. Kumar, Atmos. Environ., 2020, 237, 117709 CrossRef.
- K. Guleria and R. Subramanian, ACS Earth Space Chem., 2022, 6, 1596–1611 CrossRef.
- I. Bravo, G. Marston, D. R. Nutt and K. P. Shine, J. Quant. Spectrosc. Radiat. Transfer, 2011, 112, 1967–1977 CrossRef.
- I. Bravo, A. Aranda, M. D. Hurley, G. Marston, D. R. Nutt, K. P. Shine, K. Smith and T. J. Wallington, J. Geophys. Res.:Atmos., 2010, 115, D24317–D24328 CrossRef.
- P. Blowers and K. Hollingshead, J. Phys. Chem. A, 2009, 113, 5942–5950 CrossRef PubMed.
- V. C. Papadimitriou and J. B. Burkholder, J. Phys. Chem. A, 2016, 120, 6618–6628 CrossRef PubMed.
- P. J. Godin, K. Le Bris and K. Strong, J. Quant. Spectrosc. Radiat. Transfer, 2017, 203, 522–529 CrossRef.
-
K. Strong, P. J. Godin, K. Le Bris, H. Johnson, R. Piunno, A. Cabaj, C. MacDougall and L.-H. Xu, AGU Fall Meeting Abstracts, 2018, pp. A33L–3349 Search PubMed.
- P. Blowers, K. F. Tetrault and Y. Trujillo-Morehead, Theor. Chem. Acc., 2008, 119, 369–381 Search PubMed.
- Z. Li, Z. Tao, V. Naik, D. A. Good, J. C. Hansen, G. R. Jeong, J. S. Francisco, A. K. Jain and D. J. Wuebbles, J. Geophys. Res.:Atmos., 2000, 105, 4019–4029 CrossRef.
- K. Le Bris, J. DeZeeuw, P. J. Godin and K. Strong, J. Quant. Spectrosc. Radiat. Transfer, 2017, 203, 538–541 CrossRef.
- P. J. Godin, H. Johnson, R. Piunno, K. Le Bris and K. Strong, J. Quant. Spectrosc. Radiat. Transfer, 2019, 225, 337–350 CrossRef.
- S. Tiwari and R. Subramanian, Theor. Chem. Acc., 2024, 143, 1–15 Search PubMed.
-
K. Guleria, S. Tiwari, D. Barman, S. Daschakraborty and R. Subramanian, Electron Density: Concepts, Computation and DFT Applications, 2024, pp. 527–549 Search PubMed.
- A. D. Becke, Phys. Rev. A:At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef.
- H. William, J. Mol. Graphics, 1996, 14, 33–38 CrossRef PubMed.
- J. Tirado-Rives and W. L. Jorgensen, J. Chem. Theory Comput., 2008, 4, 297–306 CrossRef PubMed.
- K. E. Riley, B. T. Op't Holt and K. M. Merz, J. Chem. Theory Comput., 2007, 3, 407–433 CrossRef PubMed.
- C. Y. Go and K. C. Kim, J. Phys. Chem. A, 2024, 128, 2245–2252 CrossRef.
- J. Jang, K. H. Jung and K. C. Kim, Sci. Rep., 2022, 12, 7027 CrossRef PubMed.
- T. Wallington, W. Schneider, J. Sehested, M. Bilde, J. Platz, O. Nielsen, L. Christensen, M. Molina, L. Molina and P. Wooldridge, J. Phys. Chem. A, 1997, 101, 8264–8274 CrossRef.
- L. Chen, S. Kutsuna, K. Tokuhashi and A. Sekiya, Chem. Phys. Lett., 2004, 400, 563–568 CrossRef.
- L. Chen, S. Kutsuna, K. Tokuhashi and A. Sekiya, Int. J. Chem. Kinet., 2004, 36, 337–344 CrossRef.
- S. Urata, T. Uchimaru, A. K. Chandra, A. Takada and A. Sekiya, Int. J. Chem. Kinet., 2002, 34, 524–530 CrossRef CAS.
- A. K. Chandra, T. Uchimaru, M. Sugie and A. Sekiya, Chem. Lett., 2002, 31, 132–133 CrossRef.
- N. Oyaro, S. R. Sellevåg and C. J. Nielsen, Environ. Sci. Technol., 2004, 38, 5567–5576 CrossRef CAS PubMed.
- L. Christensen, J. Sehested, O. Nielsen, M. Bilde, T. Wallington, A. Guschin, L. Molina and M. Molina, J. Phys. Chem. A, 1998, 102, 4839–4845 CrossRef CAS.
- C. M. Tovar and M. A. Teruel, Atmos. Environ., 2014, 94, 489–495 CrossRef.
- M. B. Blanco and M. A. Teruel, Atmos. Environ., 2007, 41, 7330–7338 CrossRef.
- M. B. Blanco, I. Bejan, I. Barnes, P. Wiesen and M. A. Teruel, Chem. Phys. Lett., 2008, 453, 18–23 CrossRef.
- N. K. Gour, R. C. Deka, H. J. Singh and B. K. Mishra, J. Fluorine Chem., 2014, 160, 64–71 CrossRef.
- T. J. Wallington, P. Dagaut, R. Liu and M. J. Kurylo, Int. J. Chem. Kinet., 1988, 20, 177–186 CrossRef.
- A. El Boudali, S. Le Calvé, G. Le Bras and A. Mellouki, J. Phys. Chem., 1996, 100, 12364–12368 CrossRef.
- S. Le Calve, G. Le Bras and A. Mellouki, J. Phys. Chem. A, 1997, 101, 5489–5493 CrossRef.
- K. G. Kambanis, Y. G. Lazarou and P. Papagiannakopoulos, J. Phys. Chem. A, 1998, 102, 8620–8625 CrossRef.
- I. W. Smith and A. Ravishankara, J. Phys. Chem. A, 2002, 106, 4798–4807 CrossRef.
- M. B. Blanco, C. Rivela and M. A. Teruel, Chem. Phys. Lett., 2013, 578, 33–37 CrossRef.
- T. Stein, L. Christensen, J. Platz, J. Sehested, O. Nielsen and T. Wallington, J. Phys. Chem. A, 1999, 103, 5705–5713 CrossRef.
- M. Gnanaprakasam, L. Sandhiya and K. Senthilkumar, J. Phys. Chem. A, 2018, 122, 9316–9325 CrossRef PubMed.
- V. G. Straccia C, A. L. Cardona, M. a. B. Blanco, O. N. Ventura and M. Teruel, ACS Earth Space Chem., 2024, 8, 2599–2610 CrossRef.
- A. Rodríguez, I. Bravo, D. Rodríguez, M. Tajuelo, Y. Diaz-de-Mera and A. Aranda, RSC Adv., 2016, 6, 21833–21843 RSC.
- C. M. Butt, C. J. Young, S. A. Mabury, M. D. Hurley and T. J. Wallington, J. Phys. Chem. A, 2009, 113, 3155–3161 CrossRef PubMed.
- A. Garzon, M. Antinolo, M. Moral, A. Notario, E. Jimenez, M. Fernandez-Gomez and J. Albaladejo, Mol. Phys., 2013, 111, 753–763 CrossRef.
- G. Srinivasulu and B. Rajakumar, J. Phys. Chem. A, 2013, 117, 4534–4544 CrossRef PubMed.
- F.-Y. Bai, Y.-J. Liu, X. Wang, Y.-Q. Sun and X.-M. Pan, RSC Adv., 2016, 6, 63954–63964 RSC.
- F.-Y. Bai, X. Wang, Y.-Q. Sun, R.-S. Wang and X.-M. Pan, RSC Adv., 2016, 6, 36096–36108 RSC.
- J. C. Deon,
et al.
, Environ. Sci. Technol., 2006, 40(6), 1862–1868 CrossRef PubMed.
- V. L. Orkin, V. G. Khamaganov and M. J. Kurylo, J. Phys. Chem. A, 2012, 116, 6188–6198 CrossRef PubMed.
- M. Lily and A. K. Chandra, J. Fluorine Chem., 2015, 175, 185–192 CrossRef.
- A. Kumar, S. Gonu, S. Vijayakumar, C. Ramya and B. Rajakumar, J. Phys. Chem. A, 2021, 125, 523–535 CrossRef PubMed.
- F.-Y. Bai, M.-S. Deng, M.-Y. Chen, L. Kong, S. Ni, Z. Zhao and X.-M. Pan, Phys. Chem. Chem. Phys., 2021, 23, 13115–13127 RSC.
- B. Baidya, M. Lily and A. K. Chandra, ChemistrySelect, 2018, 3, 6136–6144 CrossRef.
- X. Wang, D. J. Jacob, S. D. Eastham, M. P. Sulprizio, L. Zhu, Q. Chen, B. Alexander, T. Sherwen, M. J. Evans and B. H. Lee, Atmos. Chem. Phys., 2019, 19, 3981–4003 CrossRef.
- D. Scollard, J. Treacy, H. Sidebottom, C. Balestra-Garcia, G. Laverdet, G. LeBras, H. MacLeod and S. Teton, J. Phys. Chem., 1993, 97, 4683–4688 CrossRef.
- S. R. Sellevåg, T. Kelly, H. Sidebottom and C. J. Nielsen, Phys. Chem. Chem. Phys., 2004, 6, 1243–1252 RSC.
- M. T. Rayez, J. C. Rayez, T. Berces and G. Lendvay, J. Phys. Chem., 1993, 97, 5570–5576 CrossRef.
- R. Atkinson, J. Phys. Chem. Ref. Data, Monogr., 1994, 2, 1–216 Search PubMed.
- D. F. McMillen and D. M. Golden, Annu. Rev. Phys. Chem., 1982, 33, 493–532 CrossRef.
- M. Antiñolo, E. Jimenez and J. Albaladejo, J. Photochem. Photobiol., A, 2012, 231, 33–40 CrossRef.
- M. Hurley, M. Sulbaek Andersen, T. Wallington, D. Ellis, J. Martin and S. Mabury, J. Phys. Chem. A, 2004, 108, 615–620 CrossRef CAS.
- L. Chen, T. Uchimaru, S. Kutsuna, K. Tokuhashi and A. Sekiya, Chem. Phys. Lett., 2011, 514, 207–213 CrossRef CAS.
- G. Vasvári, I. Szilágyi, Á. Bencsura, S. Dóbe, T. Bérces, E. Henon, S. Canneaux and F. Bohr, Phys. Chem. Chem. Phys., 2001, 3, 551–555 RSC.
- S. Vandenberk, L. Vereecken and J. Peeters, Phys. Chem. Chem. Phys., 2002, 4, 461–466 RSC.
- S. Carr, D. E. Shallcross, C. E. Canosa-Mas, J. C. Wenger, H. W. Sidebottom, J. J. Treacy and R. P. Wayne, Phys. Chem. Chem. Phys., 2003, 5, 3874–3883 RSC.
-
D. R. Lide, CRC Handbook of Chemistry and Physics, CRC press, 2004 Search PubMed.
-
J. Calvert, Mechanisms of Atmospheric Oxidation of the Oxygenates, 2011 Search PubMed.
- X. Song, G. b. L. Zügner, M. Farkas, Á. Illés, D. Sarzynski, T. Rozgonyi, B. Wang and S. Dóbé, J. Phys. Chem. A, 2015, 119, 7753–7765 CrossRef PubMed.
- T. J. Wallington and M. D. Hurley, Environ. Sci. Technol., 1993, 27, 1448–1452 CrossRef.
- A. Bouwman, D. Van Vuuren, R. Derwent and M. Posch, Water, Air, Soil Pollut., 2002, 141, 349–382 CrossRef.
- D. Granados Sánchez, G. López Ríos and M. Hernández García, Revista Chapingo Serie Ciencias Forestales y del Ambiente, 2010, 16, 187–206 CrossRef.
- H. Huang, Int. J. Environ. Stud., 1992, 41, 267–275 CrossRef.
- Y. Yang, K. Gao, L. Ding, J. Bi, S. Yuan and X. Yan, High Volt., 2021, 6, 733–749 CrossRef.
- J. Pagliaro and G. Linteris, Fire Saf. J., 2017, 87, 10–17 CrossRef.
- M. Rabie and C. M. Franck, Environ. Sci. Technol., 2018, 52, 369–380 CrossRef PubMed.
- X. Li, H. Zhao and A. B. Murphy, J. Phys. D: Appl. Phys., 2018, 51, 153001 CrossRef.
Footnote |
| † Presently at Department of Chemistry, University Institute of Sciences, Chandigarh University, Mohali-140413, Punjab, India. E-mail: E-mail: kanika.e19098@cumail.in |
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*


























