Comparison of oxidation products generated from the reaction of α-pinene with hydroxyl radicals, chlorine atoms, and bromine atoms measured using ammonium adduct chemical ionization mass spectrometry
Received
4th August 2025
, Accepted 6th January 2026
First published on 8th January 2026
Abstract
Halogen atoms play important but undercharacterized roles in atmospheric oxidation chemistry. Here, we report laboratory measurements of gas- and condensed-phase products formed from the oxidation of α-pinene by hydroxyl radicals (OH), chlorine atoms (Cl), and bromine atoms (Br) in an oxidation flow reactor (OFR). Products were detected using a Vocus proton-transfer time-of-flight reaction mass spectrometer (PTR-ToF-MS) operated with low-pressure ammonium adduct (NH4+) ionization and a Vaporization Inlet for Aerosols (VIA). We applied Positive Matrix Factorization (PMF) to classify precursor and product ions into early and later-generation oxidation products. While some common products were observed across all oxidants, significant compositional differences were also apparent. Vocus![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :VIA signal ratios were used to estimate volatility trends, revealing that more highly oxygenated compounds and many halogenated products contributed to SOA formation. Cl and Br oxidation led to the formation of oxygenated volatile organic compounds (OVOCs) and secondary organic aerosol (SOA), which retained halogen atoms, with Br-derived products exhibiting the lowest carbon oxidation state and the highest halogen retention. Halogenated oxidation products were less volatile than their non-halogenated counterparts. Photochemical modeling suggests that the fates of organic peroxy radicals (RO2) were primarily influenced by RO2 + HO2 reactions for α-pinene/OH, RO2 + Cl, RO2 + HO2, and potentially RO2 isomerization/autooxidation reactions for α-pinene/Cl, and RO2 + Br reactions for α-pinene/Br.
:VIA signal ratios were used to estimate volatility trends, revealing that more highly oxygenated compounds and many halogenated products contributed to SOA formation. Cl and Br oxidation led to the formation of oxygenated volatile organic compounds (OVOCs) and secondary organic aerosol (SOA), which retained halogen atoms, with Br-derived products exhibiting the lowest carbon oxidation state and the highest halogen retention. Halogenated oxidation products were less volatile than their non-halogenated counterparts. Photochemical modeling suggests that the fates of organic peroxy radicals (RO2) were primarily influenced by RO2 + HO2 reactions for α-pinene/OH, RO2 + Cl, RO2 + HO2, and potentially RO2 isomerization/autooxidation reactions for α-pinene/Cl, and RO2 + Br reactions for α-pinene/Br.
Environmental significance
Chlorine (Cl) and bromine (Br) atoms are increasingly recognized as important atmospheric oxidants, especially in coastal, marine, and polar environments. This study provides a comprehensive molecular-level comparison of products generated from the oxidation of α-pinene by OH, Cl, and Br. Our analysis reveals distinct chemical pathways and product distributions for each oxidant. By identifying unique halogenated products and quantifying their volatility, this work highlights the need to account for halogen-specific chemistry in models. Our results contribute to an improved understanding of how halogen initiated oxidation of volatile organic compounds influences SOA formation and oxidative aging and support the development of more accurate atmospheric chemical mechanisms.
|
1 Introduction
Atmospheric oxidation processes play a central role in the transformation of organic and inorganic compounds, with gas-phase oxidants such as ozone (O3), hydroxyl radicals (OH), nitrate radicals (NO3), chlorine atoms (Cl), and bromine atoms (Br) initiating these reactions. The relative importance of these oxidants varies with regional meteorology, emissions, and photochemical conditions. Among them, OH is the most globally significant due to its rapid daytime production and its broad reactivity with atmospheric constituents. OH initiates key processes, including the oxidation of sulfur dioxide (SO2) to sulfuric acid and the transformation of volatile organic compounds (VOCs) into low-volatility products that contribute to secondary organic aerosol (SOA) formation. While NO3 dominates nighttime oxidation in certain regions, halogen atoms offer unique daytime oxidation pathways, especially in marine, polar, and urban atmospheres.1,2 Cl is especially relevant in polluted coastal,3–5 urban regions,6,7 and other inland sources8,9 and can react with many VOCs with rate coefficients that are 10 to 100 times greater than OH (Fig. 1a). This reactivity is particularly enhanced for alkanes and siloxanes, where oxidation proceeds exclusively via hydrogen abstraction. Only a small subset of VOCs, such as benzene and naphthalene, exhibit lower reactivity toward Cl than OH. However, Cl does not significantly react with SO2, limiting its role in sulfate aerosol formation. Br is especially active in the polar boundary layer,10 where heterogeneous reactions on snow and ice surfaces release reactive bromine species that drive episodic O3 depletion and oxidant cycling.11,12 While Br is highly reactive with specific VOC classes such as alkenes and aldehydes, it is generally less reactive with alkanes, aromatics, and alcohols (Fig. 1b). Although Br has a high electron affinity, its H-abstraction reactions are typically endothermic, making them less favorable due to added reaction enthalpy and activation energy. Aldehydes are an exception: the formyl C–H bond is much weaker than other C–H bonds, making H-abstraction by Br nearly thermoneutral.
 |
| | Fig. 1 Rate coefficients for reactions of (a) Cl and (b) Br versus OH with various classes of VOCs.13–120 Solid and dashed lines indicate fixed ratios of halogen-to-OH reactivity: 100:1, 10:1, and 1:1 in (a), and 1:1, 1:10, and 1:100 in (b). | |
The role of Cl and Br in the oxidation of biogenic VOCs (BVOCs) has received far less attention than O3, OH, and NO3, particularly for α-pinene – a globally relevant BVOC often used as a model compound due to its well-characterized OH oxidation chemistry. α-Pinene reacts with Cl at near gas-kinetic rates53,121 and with Br at a rate roughly half that of OH.37 While early studies primarily focused on reaction kinetics, more recent work has begun to examine the chemical composition of SOA formed from Cl-and Br-initiated α-pinene oxidation. Cl-driven oxidation has been shown to generate highly oxygenated molecules – including chlorinated products – with SOA yields that are comparable to or greater than those from OH.122–125 Far fewer studies have investigated Br-initiated α-pinene oxidation, but existing results indicate very low SOA yields.125,126 These findings highlight the potential for halogen atoms to significantly alter SOA composition and yield in environments influenced by reactive halogen chemistry, while also underscoring the limited availability of comparative data across oxidants under consistent experimental conditions.
In a companion study, we characterized the chemical composition and yield of laboratory SOA generated from the OH and Cl oxidation of n-dodecane and toluene, and the OH, Cl, and Br oxidation of isoprene and α-pinene.125 While OH and Cl produced comparable SOA yields with oxygen-to-carbon (O/C) ratios indicative of multigenerational aging, Br-mediated SOA formation was more limited, yielding lower O/C values. These findings challenge model assumptions that Cl and Br produce SOA with similar efficiency.127 To build on that work, this study characterizes the detailed molecular composition of gas- and condensed-phase products obtained from the reaction of α-pinene with OH, Cl, and Br in an oxidation flow reactor (OFR). Using an ammonium-adduct Vocus proton-transfer reaction time-of-flight mass spectrometer (PTR-ToF-MS) equipped with a vaporization inlet for aerosols (VIA), we detected a wide range of gas- and condensed-phase oxidation products. We applied PMF to classify these products and compared their composition and volatility across the three oxidants. These measurements provide new insight into the distinct pathways and product distributions associated with halogen- versus OH-mediated oxidation, with implications for the treatment of halogen chemistry in atmospheric models.
2 Experimental
2.1 Oxidation flow reactor setup
Experiments were conducted inside a Potential Aerosol Mass (PAM) OFR (Aerodyne Research), a 13 L horizontal aluminum cylindrical chamber (46 cm long × 22 cm ID) operated in continuous flow mode with a total flow of 6.0–6.8 L min−1, yielding a calculated mean residence time (τOFR) of 114–130 s. An electroconductive Teflon coating was applied to the OFR to improve chemical compatibility with halogen precursors while maintaining high gas and particle transmission.125,128 Two low-pressure mercury (Hg) lamps housed in type 214 quartz sleeves were used to photolyze oxidant precursors, with UV output regulated by a fluorescent dimming ballast (IZT-2S28-D, Advance Transformer Co.). The UV irradiance was controlled by adjusting the ballast control voltage (1.5–10 VDC) and measured using a TOCON-GaP6 photodetector (sglux GmbH). The corresponding actinic flux ranged from approximately 1 × 1014 to 3 × 1015 photons cm−2 s−1.129,130
2.2 Oxidant, OVOC and SOA generation
OH was generated by photolyzing O2 and H2O at λ = 185 nm and O3 at λ = 254 nm using two low-pressure Hg lamps. The relative humidity (RH) was controlled to 31–43% using a Nafion humidifier (Perma Pure); corresponding H2O mixing ratios were 1.0–1.5% at OFR temperatures of 26–29 °C. The integrated OH exposure (OHexp) in the OFR, defined as the product of the mean OH concentration and τOFR, was calculated using an empirical estimation equation130 and ranged from 6.7 × 1010 to 1.0 × 1012 molecules cm−3 s (hereafter “cm−3 s”) – equivalent to 0.5–8 days of atmospheric oxidation at OH = 1.5 × 106 cm−3.131 The estimated uncertainty in calculated OHexp values was ±50%. Cl was produced by photolyzing 4.2 ppm oxalyl chloride (C2Cl2O2) at λ = 254 or 313 nm,125,132,133 and Br was generated by photolyzing 1.8 ppm oxalyl bromide (C2Br2O2) at λ = 254 nm.125,134,135 These C2Cl2O2 and C2Br2O2 mixing ratios were chosen to ensure >99.5% consumption of α-pinene by reaction with Cl or Br at maximum actinic flux in the OFR. The RH in Cl and Br experiments was maintained at 1.1–4.1%; corresponding H2O mixing ratios were 0.04–0.12% at OFR temperatures between 24–28 °C. Integrated Cl and Br exposures (Clexp, Brexp) were characterized via offline calibration using O3 decay measurements, with estimated uncertainties of ±70%.125 Estimated Clexp values ranged from 4.6 × 108 to 1.3 × 1011 cm−3 s, and Brexp values ranged from 4.5 × 1011 to 3.7 × 1012 cm−3 s, corresponding to 2 h – 25 days ([Cl] = 6 × 104 cm−3) and 0.7–6 days ([Br] = 7 × 106 cm−3) of equivalent atmospheric exposure.136 Gas-phase OH, Cl, or Br oxidation of α-pinene led to the formation of OVOCs and SOA via homogeneous nucleation. α-Pinene (10% (v/v) in carbon tetrachloride) was injected into the OFR carrier gas flow at 0.94–2.8 µL h−1 using a syringe pump, yielding ∼30 ppbv for OH and Cl experiments and ∼90 ppbv for Br experiments to ensure nucleation.125
2.2.1 Caution.
This study involved preparing polyfluorotetraethylene (PTFE) permeation tubes filled with liquid C2Cl2O2 and C2Br2O2,125 which are toxic, corrosive, and release harmful gases upon decomposition. C2Cl2O2 and C2Br2O2 permeation tubes were prepared in a well-ventilated fume hood with appropriate personal protective equipment, including gloves, lab coats, and eye protection. O3 is a strong oxidant and respiratory irritant. Exhaust flows containing O3, C2Cl2O2 or C2Br2O2 were vented to a laboratory fume exhaust system to prevent exposure and accumulation.
2.3 Instrumentation
Aerosol number concentrations and size distributions were measured using a scanning mobility particle sizer (SMPS; TSI), and aerosol mass spectra were acquired using an Aerodyne long high-resolution time-of-flight aerosol mass spectrometer (L-ToF-AMS). Gas- and condensed-phase organic compounds were measured with a Vocus 2R PTR-ToF-MS137 (hereafter referred to as “Vocus”) following low-pressure ammonium adduct (NH4+) ionization.138,139 The system alternated between gas-phase and particle-phase sampling using a PTFE solenoid valve manifold (Fig. 2). During gas sampling, the Vocus sampled the OFR through an unheated 0.25 in. o.d. fluorinated ethylene propylene (FEP) line with a PTFE membrane filter to remove particles. For particle sampling, the Vocus sampled the OFR through a charcoal denuder that removed gas-phase organics before evaporation of aerosols in a heated (220 °C) Sulfinert-coated stainless-steel VIA140–142 at a flow of 1.5 slpm.
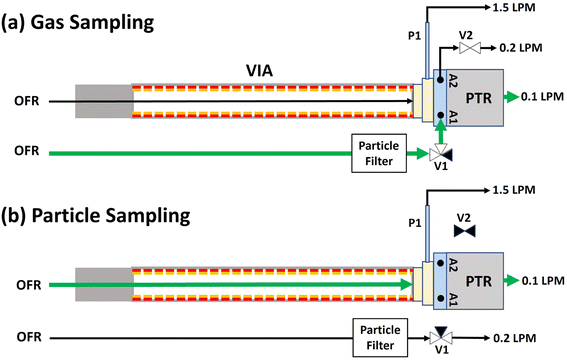 |
| | Fig. 2 Schematic of (a) gas and (b) particle sampling Vocus configurations used in these experiments. During gas sampling (a), the OFR was sampled through an unheated 0.25 in. o.d. FEP inlet line that was connected to Vocus atmospheric pressure ports A1 and A2. Aerosols were removed using a PTFE membrane filter upstream of three-way valve V1, and sample flow pulled through the VIA was removed through inlet pump port P1. During particle sampling (b), the OFR was sampled through an activated charcoal denuder and the VIA (T = 220 °C), while the unheated FEP inlet line was isolated from the Vocus by switching V1 and closing on/off valve V2. Constant flows were continuously pulled through P1 and either A1 and A2 or pumped to exhaust, while 0.1 L min−1 flow was continuously subsampled into the Vocus PTR reactor. | |
2.4 Data analysis
L-ToF-AMS mass spectra were analyzed using SQUIRREL version 1.63I and PIKA version 1.23I143 software, and elemental analysis of high-resolution L-ToF-AMS spectra was performed using the “Improved-Ambient” method.143,144 High-resolution Vocus mass spectra were analyzed using Tofware version 3.2.5.145 To aid in the interpretation of Vocus mass spectra, PMF146 was applied to NH4+ adduct signals with even m/z values between m/z = 150 and 350, exported from Tofware. This range and selection minimized interference from low-mass background and fragment ions in the mass spectra. Ion-specific errors were calculated as 1.28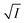 ,147 where I is the high-resolution peak height. PMF was used primarily to group ions with similar temporal behavior trends and to qualitatively apportion ions contributing to multiple factors. Separate PMF analyses were performed for the α-pinene/OH, α-pinene/Cl, and α-pinene/Br systems using the PMF Evaluation Tool (PET) version 3.08C,148 each yielding a four-factor solution representing precursors and early- and late-generation products. Three-factor PMF tended to combine precursor and product signals, often with higher residuals. Solutions with five or more factors led to the splitting of existing factors without identifying new product groups of products or significantly reducing residuals. To avoid artificially forcing ion signals to zero in specific factors, no rotations were applied.
,147 where I is the high-resolution peak height. PMF was used primarily to group ions with similar temporal behavior trends and to qualitatively apportion ions contributing to multiple factors. Separate PMF analyses were performed for the α-pinene/OH, α-pinene/Cl, and α-pinene/Br systems using the PMF Evaluation Tool (PET) version 3.08C,148 each yielding a four-factor solution representing precursors and early- and late-generation products. Three-factor PMF tended to combine precursor and product signals, often with higher residuals. Solutions with five or more factors led to the splitting of existing factors without identifying new product groups of products or significantly reducing residuals. To avoid artificially forcing ion signals to zero in specific factors, no rotations were applied.
2.5 Effective saturation concentration (C°) calculations
C° values for C10H14,16Ox compounds were calculated using the empirical formula: 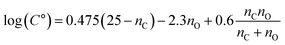 ,149 where nC and nO are the carbon and oxygen numbers of each compound. Vocus:VIA-Vocus ratios of measured C10H14,16Ox compounds were plotted against their calculated C° values and fit to sigmoid or polynomial regression equations. These equations were then used to infer C° for C10H13,15ClOx and C10H13,15BrOx from their measured Vocus:VIA-Vocus ratios. This analysis was restricted to C10H14,16Ox, C10H13,15ClOx and C10H13,15BrOx signals to minimize the effect of possible thermal decomposition reactions in the VIA on the calculated C° values.
,149 where nC and nO are the carbon and oxygen numbers of each compound. Vocus:VIA-Vocus ratios of measured C10H14,16Ox compounds were plotted against their calculated C° values and fit to sigmoid or polynomial regression equations. These equations were then used to infer C° for C10H13,15ClOx and C10H13,15BrOx from their measured Vocus:VIA-Vocus ratios. This analysis was restricted to C10H14,16Ox, C10H13,15ClOx and C10H13,15BrOx signals to minimize the effect of possible thermal decomposition reactions in the VIA on the calculated C° values.
2.6 Peroxy radical fate modeling
To explore the fate of RO2 generated from OH, Cl, and Br oxidation of α-pinene, we used the KinSim chemical kinetic solver150 with a simplified mechanism adapted from prior OFR studies.125,130,151,152 In the model, RO2 were generated from the reactions
While RO2 composition varies across the OH, Cl, and Br systems, we assumed that rate coefficients for RO2 remained the same due to the lack of kinetic data. Following the approach introduced by Peng et al.153 and used in previous studies by our group141,154 we applied kinetic data and reaction pathways associated with reactions between Cl/Br and the methylperoxy radical (CH3O2) as a surrogate RO2 species. These RO2 were assumed to undergo autooxidation via isomerization, react with other RO2, and/or react with hydroperoxyl radicals (HO2):
| RO2 + RO2 → ROH + R(O) + O2 |
| RO2 + RO2,isom → 2RO + O2 |
| RO2 + RO2,isom → ROH + R(O) + O2 |
Here, RO, ROH, R(O), and ROOH denote generic alkoxy radical, alcohol, carbonyl, and organic peroxide species, respectively. In the α-pinene/OH system, we assumed RO
2 reacted with OH to generate ROH and O
2,
155 and that RO isomerized and decomposed to generate pinonaldehyde (PINAL) and HO
2:
156
In the α-pinene/Cl system, we assumed RO2 reacted with Cl to generate RO and ClO,44 and that RO2 reacted with ClO to generate RO and ClO2 or organic chlorides (ROCl) and O2.59 We also assumed that all RO generated from RO2 + RO2 and RO2 + Cl reactions were chlorinated, and that they subsequently isomerized and decomposed to yield either chloropinonaldehyde (CHLOROPINAL) and HO2, or pinonaldehyde and Cl,122 with branching ratios of 0.33 and 0.67 respectively:126
| RO2,isom + ClO → RO + ClO2 |
| RO2,isom + ClO → ROCl + O2 |
| RO + O2 → CHLOROPINAL + HO2 |
In the α-pinene/Br system, we assumed RO2 reacted with Br to generate RO and BrO,157 and that RO2 reacted with BrO to generate RO2H and HOBr.158,159 We also assumed all RO generated from RO2 + RO2 and RO2 + Br were brominated, and that they isomerized and decomposed to produce either bromopinonaldehyde (BROMOPINAL) and HO2, or pinonaldehyde and Br, with branching ratios of 0.66 and 0.34 respectively:126
| RO2,isom + BrO → RO2H + HOBr |
Finally, RO2 were regenerated following reaction of ROH, R(O), ROOH, RO2H, ROCl, PINAL, CHLOROPINAL, and BROMOPINAL with OH, Cl, and Br:
The kinetic parameters used in these calculations16,19,37,44,50,53,59,79,107,156,158,160–165 are listed in Table S2, with the following additional assumptions:
1. A bimolecular rate coefficient of 1.6 × 10−10 cm3 s−1 for RO2 + Br → RO + BrO.
2. Rate coefficients for ROH + Cl, R(O) + Cl, and ROOH + Cl reactions were 10 times faster than the corresponding ROH + OH, R(O) + OH, and ROOH + OH rate coefficients.
3. Rate coefficients for ROH + Br, R(O) + Br, and ROOH + Br reactions were 100 times slower than the α-pinene + Br rate coefficient.
4. Secondary OH production from halogen-initiated α-pinene oxidation was negligible.
Simulations were run with and without isomerization pathways, assuming first-order isomerization rate coefficients of 0, 0.1, 1, and 4 s−1.163,165 Results from these simulations were used to calculate the fractional loss of generic RO2 (denoted FRO2) as a function of OHexp, Clexp, and Brexp.
3 Results & discussion
3.1 Overview of results obtained from alternating Vocus and VIA-Vocus measurements
Fig. S1 shows example time series of Vocus signals collected during an α-pinene/Cl experiment, alternating between Vocus and VIA-Vocus sampling modes every 15 min. Fig. S1a displays Clexp, which was varied in discrete steps from 0 to 1.2 × 1011 cm−3 s throughout the experiment, and Fig. S1b–e show representative ammonium adduct ion signals exhibiting distinct trends with Clexp. In Fig. S1b, the signal for unreacted α-pinene (NH4+·C10H16) was highest at low Clexp and decreased with increasing Clexp, as expected. At low Clexp, NH4+·C10H16 signal was enhanced during Vocus sampling periods but was negligible during VIA-Vocus sampling periods due to its removal by the charcoal denuder upstream of the VIA. Fig. S1c shows that NH4+·C10H15ClO2 also peaked during Vocus sampling and reached maximum intensity at intermediate Clexp, suggesting it is an early-generation gas-phase oxidation product. Its decline at higher Clexp likely reflects further oxidation to more functionalized compounds. In Fig. S1d, both NH4+·C3H6O and its chlorinated analog NH4+·C3H5ClO plateau at intermediate-to-high Clexp, consistent with their formation as fragmentation products from α-pinene and/or its early-generation oxidation products. Finally, Fig. S1e shows NH4+·C10H14O7 and NH4+·C10H13ClO7, which peaked at higher Clexp and were enriched during VIA-Vocus sampling, indicating their low volatility and likely contribution to SOA. Similar trends were observed in α-pinene/OH and α-pinene/Br experiments. Additional compounds with analogous temporal behavior were identified with PMF and are discussed in the next section.
To examine how Vocus:VIA-Vocus signal ratios relate to compound volatility, Fig. 3 presents three panels showing the ratio of Vocus to VIA-Vocus signals for C10 species detected in α-pinene/OH, α-pinene/Cl, and α-pinene/Br experiments, plotted as a function of oxygen number. Symbols are colored by C°, ranging from approximately 107 µg m−3 (purple) to 10−8 µg m−3 (red). In Fig. 3a, α-pinene/OH products show a clear trend: as oxygen number (nO) increases from 0 to 8, the Vocus:VIA-Vocus ratio decreases from approximately 137 to 0.4, C° decreases. Compounds with nO ≈ 0–2 exhibit high C° values (104–107 µg m−3), consistent with dominant gas-phase behavior, and compounds with nO = 3–5 and C° ≈ 0.1–10 µg m−3 show comparable signals in both configurations, consistent with semivolatile behavior. Highly oxygenated species (nO ≥ 6) exhibit C° < 10−3 µg m−3 and low Vocus:VIA-Vocus ratios, indicating low volatility and likely contribution to SOA.
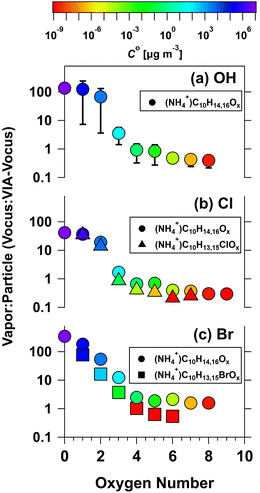 |
| | Fig. 3 Ratio of Vocus to VIA-Vocus signal intensities (Vocus:VIA) for C10 ammonium adducts as a function of oxygen number, measured during α-pinene oxidation experiments with (a) OH, (b) Cl, and (c) Br as the primary oxidants. Circle symbols represent non-halogenated species (NH4+·C10H14,16Ox), triangles represent chlorinated species (NH4+·C10H13,15ClOx), and squares represent brominated species (NH4+·C10H13,15BrOx). Colors indicate estimated volatility (C°) from 10−9 to 107 µg m−3. C° values were calculated using methods described in Section 2.5. | |
Fig. 3b compares Vocus:VIA-Vocus ratios for non-halogenated (circles) and chlorinated (triangles) species from α-pinene/Cl experiments. Both groups show decreasing Vocus:VIA-Vocus ratios and C° with higher nO. Chlorinated compounds generally have similar or slightly lower C° than their non-halogenated counterparts. Cl-containing species with nO = 1–2 behave predominantly as gases, nO = 3 are semivolatile, and nO ≥ 4 mostly partition to the condensed phase. Fig. 3c shows analogous results for brominated products (squares), alongside non-halogenated compounds (circles) from α-pinene/Br experiments. Similar trends are observed: Vocus:VIA-Vocus ratios and C° values decrease with nO. Br-containing compounds with nO = 1–2 are mainly in the gas phase, nO = 3 are semivolatile, and nO ≥ 4 are low-volatility and likely mostly in the condensed-phase. Overall, Fig. 3 demonstrates a strong correlation between nO and volatility. Higher nO corresponds to lower Vocus:VIA-Vocus ratios and C° values, indicating enhanced partitioning to the condensed phase. The presence of Cl or Br further modulates this trend, generally lowering volatility and increasing the likelihood of SOA formation relative to non-halogen species with equal nO. To extend the volatility estimation framework of Donahue et al.,149 which relates log(C°) to molecular composition for CHO compounds, we adapted the formulation to include chlorinated (CHOBr) and brominated (CHOBr) species. The modified expression is:
| | 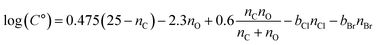 | (1) |
where
nCl and
nBr are the numbers of Cl and Br atoms, and
bCl and
bBr represent their respective contributions to log(
C°). For compounds with
nO ≥3,
Fig. 3b and c suggest that Cl and Br decrease volatility comparably to the addition of ∼2 and ∼4 oxygen atoms, respectively; that is,
bCl ≈ 2
bO and
bBr ≈ 4
bO. Assuming
bO ≈ 2.3,
149 we arrive at the following revised formulation:
| | 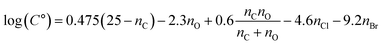 | (2) |
While beyond the scope of this work, a more comprehensive characterization of bCl and bBr using the approach of Li et al.166 should be explored in future studies.
3.2 Molecular characterization of α-pinene/OH, α-pinene/Cl, and α-pinene/Br oxidation products
3.2.1 α-Pinene/OH.
Fig. 4 presents results from PMF analysis of α-pinene/OH oxidation products detected with the Vocus. Four factors were identified, each characterized by its evolution with OHexp (Fig. 4a, c, e and g) and corresponding Kendrick Mass Defect (KMD) plots (Fig. 4b, d, f and h), where symbol size is proportional to signal intensity. Factor 1 (“VOC/OVOC1”) consists primarily of gas-phase species detected in Vocus mode (Fig. 4a). Its signal peaks at the lowest OHexp and decreases rapidly, approaching zero by approximately 4 × 1011 cm−3 s. The dominant signals include α-pinene (NH4+·C10H16), NH4+·C10H14O and NH4+·C10H16O (Fig. 4b). These signals likely represent unreacted precursor and either α-pinene impurities that the Vocus is highly sensitive to, or early-generation oxidation products that PMF could not fully separate from α-pinene due to their fast formation and consumption. Factor 2 (“OVOC2”) also comprises gas-phase compounds, peaking at OHexp ≈ 1 × 1011 cm−3 s before decreasing (Fig. 4c.). Major signals include NH4+·C10H16O2 – possibly contributed from pinonaldhyde, 2-hydroxy-3-pinanone, and/or α-pinene hydroperoxide156,167 – and NH4+·C9H14O4, likely representing pinic acid167 (Fig. 4d). Other species include NH4+·C10H14O2, NH4+·C10H16O, and various C8–C10 compounds. Factor 3 (“OVOC3”) peaks at OHexp ≈ 4 × 1011 cm−3 s and then gradually declines (Fig. 4e). Like the previous factors, it primarily consists of gas-phase species. Although NH4+·C3H6O was excluded from the PMF analysis because it was below m/z 150 (Sect. 2.4), its temporal trend matches this factor. Major constituents include NH4+·C5H8O4 and NH4+·C5H8O5, with additional contributions from NH4+·C4H6O5–6, NH4+·C6,7H8O4–7, NH4+·C7H10O4–6, and NH4+·C8H12O2–4 (Fig. 4f). These species generally have lower nC and higher nO than those in Factors 1 and 2. Factor 4 (“SOA”) includes 70 signals dominated by low-volatility, condensed-phase compounds and peaks at OHexp = 2.3 × 1011 cm−3 s (Fig. 4g), suggesting formation via continued oxidation of Factor 2 compounds. The KMD plot shows a complex mixture of highly oxygenated C4 to C10 compounds (Fig. 4h).
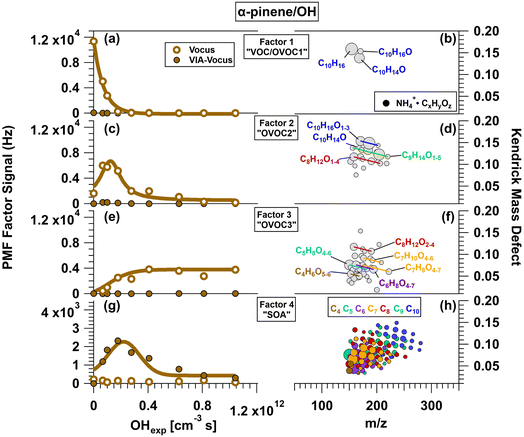 |
| | Fig. 4 Results from PMF analysis of Vocus and VIA-Vocus measurements of the α-pinene + OH system. (a, c, e and g) Signals of four distinct factors as a function of OH exposure (OHexp): initial α-pinene precursor (Factor 1), gas-phase oxidation products (Factors 2 and 3), and condensed-phase oxidation products (Factor 4). (b, d, f and h) Corresponding Kendrick Mass Defect (KMD) plots showing the chemical composition of each factor. Symbol size is proportional to signal intensity. For clarity, only species with relative abundances ≥ 0.040 (Factor 1), 0.0080 (Factor 2), 0.0044 (Factor 3), and 0.002 (Factor 4) are shown. | |
The Factor 4 elemental composition is further examined in Fig. 5, which overlays the H/C and O/C ratios of individual factor compounds on a Van Krevelen diagram, allowing qualitative assessment of their contributions to ensemble O/C and H/C ratios derived from L-ToF-AMS measurements of α-pinene OH-SOA. Several prominent homologous series of C5–C10 compounds are evident. To contextualize observed oxidation products in terms of known α-pinene/OH chemistry, we compared measured molecular formulas to those predicted by the Master Chemical Mechanism (MCM), a near-explicit gas-phase chemical model that describes the degradation of VOCs like α-pinene through detailed reaction pathways.156 Here, diamond symbols denote molecular formulas represented in the MCM, including the largest signal, NH4+·C5H8O4, which may originate from 3-hydroperoxy-4-oxo-pentanal (“C511OOH” in the MCM) and/or 3-hydroxy-4-oxopentanoic acid (“H3C2C4CO2H”). Similarly, NH4+·C6H8O4 may arise from 6-hydroxyhexane-2,3,5-trione (“C614CO”) and/or 4-hydroxy-2,5-dioxi-hexanal (“H3C25C5CHO”). Structures and formulas for MCM-predicted and detected compounds are shown in Fig. S2. Triangle symbols represent known α-pinene products that are not included in the MCM, such as terpenylic and 2-hydroxyterpenylic acids (NH4+·C8H12O4–5), diaterpenylic acid acetate (NH4+·C10H16O6),168 and 3-methyl-1,2,3-butanetricarboxylic acid (NH4+·C8H12O6),169,170 along with others.171–174 Circle symbols denote 39 molecular formulas not previously reported in α-pinene/OH oxidation studies. Some may result from thermal degradation in the VIA, especially those with low H/C ratios (0.5 ≤ H/C < 1). Others may represent later-generation oxidation products formed via functionalization or fragmentation of known precursors. For example, NH4+·C6H6,8O5–6 may result from alcohol addition to C6H6,8O4-type compounds like “CO235C5CHO”, “C614CO” and/or “H3C25C5CHO”, while NH4+·C10H12O5–7 may result from addition of two carbonyl groups to known C10H16O3–5 products,171 along with other possible pathways. Additional signals such as NH4+·C4H4,6O5 likely stem from OH-initiated fragmentation of earlier-generation α-pinene/OH oxidation products.
 |
| | Fig. 5 Van Krevelen diagram showing H/C and O/C ratios of SOA generated from the OH oxidation of α-pinene. L-ToF-AMS data (gray squares) show bulk SOA elemental composition, while VIA-Vocus data (diamonds/triangles/circles) identify individual molecular formulas of Factor 4 components (Fig. 4). Colored lines show homologous series of oxidation products. Additional figure notes: 1molecular formulas of oxidation products included in the Master Chemical Mechanism (MCM);1562molecular formulas of previously-reported oxidation products that are not included in the MCM,168–1743molecular formulas not previously reported in α-pinene/OH studies. | |
3.2.2 α-Pinene/Cl.
Fig. 6 presents the PMF analysis of α-pinene/Cl oxidation products measured with the Vocus. As with the α-pinene/OH system, four distinct factors were identified, each characterized by unique mass spectral signatures and trends with Clexp. Factor 1 closely resembles Factor 1 from the α-pinene/OH system. It consists mainly of unreacted α-pinene plus NH4+·C10H14O and NH4+·C10H16O, and its signal decreases steadily with increasing Clexp. Factor 2 contains gas-phase species and peaks at Clexp ≈ 5.3 × 1010 cm−3 (Fig. 6c). The largest contributor is NH4+·C10H14O, followed by chlorinated compounds including NH4+·C10H15ClO, NH4+·C10H17ClO, and NH4+·C10H15ClO2, along with additional chlorinated and non-chlorinated adducts. Factor 3 also contains gas-phase compounds, but its signal increases continuously with Clexp. It includes NH4+·C5H6Cl2O2, NH4+·C6H9ClO2, and NH4+·C7H9ClO3, along with 33 other C3–C9 chlorinated and non-chlorinated species. NH4+·C3H6O and its NH4+·C3H5ClO chlorinated analog followed a similar trend, but plateaued at lower Clexp than Factor 3; additionally, the yield of NH4+·C3H6O was lower than in the α-pinene/OH system. Factor 4 is composed primarily of low-volatility, condensed-phase products and peaks at Clexp ≈ 8.2 × 1010 cm−3 s (Fig. 6g), suggesting it results from continued oxidation of compounds in Factor 2. Like the α-pinene OH-SOA factor, this Cl-SOA factor is chemically complex, containing 45 chlorinated and 87 non-chlorinated C4 to C10 ammonium adducts.
 |
| | Fig. 6 Results from PMF analysis of Vocus and VIA-Vocus measurements of the α-pinene + Cl system. (a, c, e and g) Signals of four distinct factors as a function of Cl exposure (Clexp): initial α-pinene precursor (Factor 1), gas-phase oxidation products (Factors 2 and 3), and condensed-phase oxidation products (Factor 4). (b, d, f and h) Corresponding KMD plots showing the chemical composition of each factor. Symbol size is proportional to signal intensity. For clarity, only species with relative abundances ≥ 0.043 (Factor 1), 0.010 (Factor 2), 0.0059 (Factor 3), and 0.0015 (Factor 4) are shown. | |
Factor 4 components are examined in detail in Fig. S3a and b, which plot their H/C and O/C ratios on Van Krevelen diagrams, alongside ensemble AMS-derived O/C and H/C ratios for α-pinene Cl-SOA. Given the greater complexity of α-pinene Cl-SOA compared to α-pinene OH-SOA, formulas for non-chlorinated (NH4+·CxHyOz) and chlorinated (NH4+·CxHyClwOz) compounds are shown separately. Triangle symbols represent species previously reported by Masoud and Hildebrandt Ruiz,124 including 37 of the 87 non-chlorinated compounds and 10 of the 45 chlorinated compounds. The only other study characterizing α-pinene/Cl oxidation products used a method selective for highly oxygenated organic molecules (HOM)123 which NH4+ CIMS is less sensitive to. Circle symbols represent compounds not previously reported in α-pinene/Cl oxidation studies. As with the α-pinene/OH system, species with low H/C ratios (0.5 ≤ H/C < 1) may result from thermal decomposition in the VIA. Non-chlorinated species detected in Cl-SOA include many also observed in α-pinene OH-SOA, though with different relative abundances. The most intense were NH4+·C7H8O4 and NH4+·C6H8O4. Species present in OH-SOA but largely absent in Cl-SOA include: C4H6O5, C7H12O5, C8H10O6–7, C8H12O6, C8H14O2, C10H16O5–6, and C10H18O5. Conversely, non-chlorinated species enriched in Cl-SOA include C8H10,12O2, C9H10O1–3, C9H12,14O2–3, C10H12O2–4, and C10H14O1–3. These are likely thermal or fragmentation artifacts, given their volatilty and absence in OH-SOA. Some may also result from HCl eliminations not present in the α-pinene/OH system. Chlorinated adducts in the Cl-SOA include NH4+·C6H7ClO3 (most abundant), NH4+·C5H7ClO2–3, NH4+·C7H9ClO3–4, and NH4+·C10H13ClO5. In α-pinene OH-SOA, NH4+·C6H8O4 was hypothesized to originate from “C614CO” and/or “H3C25C5CHO” based on MCM predictions. The presence of the corresponding chlorinated analog NH4+·C6H7ClO3 suggests Cl-initiated oxidation of similar precursors. Analogous relationships likely exist between NH4+·C5H8O4 and NH4+·C5H7ClO3, and between NH4+·C7H10O4–5 and NH4+·C7H9ClO3–4.
3.2.3 α-Pinene/Br.
Fig. 7 presents the PMF analysis of α-pinene/Br oxidation products measured with the Vocus. Factor 1 closely resembles the first factor in the OH and Cl systems – it is dominated by unreacted α-pinene, NH4+·C10H14O, and NH4+·C10H16O, and its signal decreases steadily with Brexp. Factor 2 contains gas-phase species, peaking at Brexp ≈ 2 × 1012 cm−3 s before decreasing (Fig. 7c). The largest signal is NH4+·C10H14O, followed by NH4+·C9H10O, NH4+·C10H13BrO, NH4+·C10H14,16O2, and NH4+·C10H15BrO1,2. Factor 3 includes predominantly C3–C6 brominated gas-phase species whose signals increase with Brexp, suggesting progressive fragmentation or oxidation of larger molecules. The dominant signal is NH4+·C3H5BrO, along with lesser signals from NH4+·C9H10O and C4–C6 species such as NH4+·C4H5,7BrO2, NH4+·C5H5,7BrO2, NH4+·C5H7BrO3, and NH4+·C6H7,9BrO2. NH4+·C3H6O plateaued at lower Brexp than Factor 3 and at lower yield than in the α-pinene/OH system, similar to its behavior in the α-pinene/Cl system.
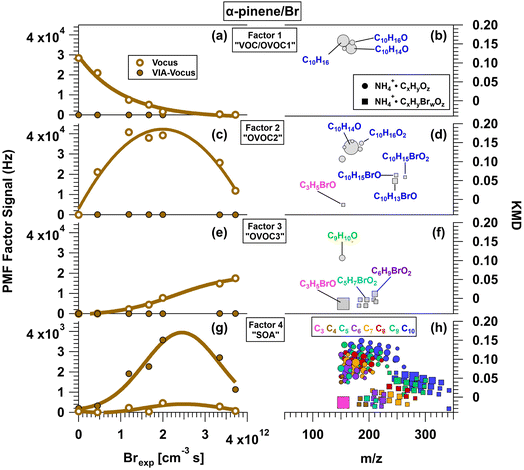 |
| | Fig. 7 Results from PMF analysis of Vocus and VIA-Vocus measurements of the α-pinene + Br system. (a, c, e and g) Signals of four distinct factors as a function of Br exposure (Brexp): initial α-pinene precursor (Factor 1), gas-phase oxidation products (Factors 2 and 3), and condensed-phase oxidation products (Factor 4). (b, d, f and h) Corresponding KMD plots showing the chemical composition of each factor. Symbol size is proportional to signal intensity. For clarity, only species with relative abundances ≥0.031 (Factor 1), 0.014 (Factor 2), 0.012 (Factor 3), and 0.0026 (Factor 4) are shown. | |
Factor 4 represents condensed-phase products and peaks at Brexp ≈ 2.4 × 1012 cm−3 s (Fig. 7g), suggesting formation via further oxidation of Factor 2 species. This factor includes 57 brominated and 77 non-brominated C3 to C10 ammonium adducts. Their H/C and O/C ratios are shown in Van Krevelen space in Fig. S4a and b alongside α-pinene Br-SOA elemental ratios measured by L-ToF-AMS. As with Cl-SOA, brominated and non-brominated formulas are plotted separately. To our knowledge, this is the first study to report the molecular composition of α-pinene/Br oxidation products. As with the OH and Cl systems, species with 0.5 ≤ H/C < 1 are likely artifacts of thermal decomposition in the VIA. Many of the nonbrominated compounds shown in Fig. S4a overlap with those found in OH- and Cl-SOA, though in different proportions. The most abundant are NH4+·C10H14O4, NH4+·C6H6,8O4, and NH4+·C7H8O3,4. Unique to the Br system are NH4+·C6H12O3 and NH4+·C6H10O4, which were not significant in OH-/Cl-SOA. Fig. S4b shows that the most intense brominated signal is NH4+·C3H5BrO – likely bromoacetone – which is too volatile to be a true condensed-phase species. Its Vocus:VIA-Vocus ratio was nearly 100, qualitatively consistent with other volatile C10 species (Fig. 3) and suggesting possible charcoal denuder breakthrough from the gas phase. After NH4+·C3H5BrO, the next largest brominated adducts are NH4+·C10H13,15BrO3,4. While the Br-SOA composition differs markedly from OH- and Cl-SOA, some chemical parallels remain. For instance, α-pinene/OH pathways in the MCM leading to“PINALOH” (C10H16O3) and “C106OH” (C10H16O4) can be adapted to α-pinene/Br chemistry. Br-initiated oxidation of bromopinonaldehyde (C10H15BrO2) may produce a C10H14BrO4 peroxy radical, followed by RO2 + RO2 forming a C10H14BrO3 alkoxy radical and subsequent C10H13BrO3 (carbonyl) and C10H15BrO3 (alcohol) products. Further isomerization and O2 addition yield a C10H15BrO5 peroxy radical, which could react with RO2 again to produce C10H13,15BrO4 carbonyl and alcohol products.
3.3 Comparison of α-pinene/OH, α-pinene/Cl, and α-pinene/Br OVOC and SOA factors
Comparison of Fig. 4d, 6d and 7d reveals notable differences in the composition of the “OVOC2” factors derived from α-pinene oxidation by OH, Cl and Br. In the Cl and Br systems, we hypothesize that NH4+·C10H15ClO, NH4+·C10H17ClO and NH4+·C10H15BrO are first-generation products formed via halogen addition to the α-pinene endocyclic double bond, followed by RO2 + RO2 reactions. This is analogous to OH-initiated oxidation in the MCM, which generates C10H16O2 (“APINBCO”) and C10H18O2 (“APINBOH”) through similar pathways.156 All three OVOC2 factors contain NH4+·C10H14O, though its relative abundance is lower in the OH system. While pinonaldehyde (C10H16O2) is known to undergo dehydration to form C10H14O in H3O+ PTR-MS instruments,175–177 separate measurements of a pinonaldehyde standard using NH+4 Vocus show that this dehydration product is much less prominent. Specifically, NH+4-adduct signals for C10H14O were more than 250 times weaker than those for C10H16O2 (NH4+·C10H14O: NH4+·C10H16O2 = 0.0038; Fig. S5). In contrast, H3O+ Vocus measurements of the same standard showed a much higher relative abundance of C10H14O (H+·C10H14O: H+·C10H16O2 = 0.47), consistent with known dehydration artifacts during proton-transfer ionization. This suggests that NH4+·C10H14O represents something else.
Given the greater complexity of the OVOC3 and SOA factors, Fig. 8 summarizes their composition, grouped by carbon and oxygen number, with OVOC2 factors included for reference. Solid bars denote non-halogenated species, vertically striped bars represent chlorinated species, and diagonally striped bars denote brominated species. Several trends are evident:
 |
| | Fig. 8 Carbon number distributions for OVOC2, OVOC3, and SOA factors resulting from oxidation of α-pinene by (a–c) OH, (d–f) Cl, and (g–i) Br. Fill patterns distinguish non-halogenated (CxHyOz, solid), chlorinated (CxHyClOz, vertical stripes), and brominated (CxHyBrOz, diagonal stripes) compounds, while the colors indicate the number of oxygen atoms. Pie charts in the halogen experiments quantify the total contribution of halogenated products to each fraction. | |
1. Carbon oxidation state increases in the order Br < Cl < OH across all systems.
2. Within each system, OVOC3 and SOA factors are more oxidized than the OVOC2 factor.
3. Carbon number distributions are broader and more complex for OH and Cl systems than for Br, suggesting more extensive fragmentation and multigenerational oxidative aging.
4. The fraction of halogenated species, shown in the pie chart insets of Fig. 8d–i, increased in the order SOA < OVOC2 < OVOC3 for Cl and OVOC2 < SOA < OVOC3 for Br. These trends suggest that Cl and Br atoms are more likely retained in gas-phase products than in lower-volatility species that contribute to SOA.
5. Br-SOA contained a higher fraction of halogenated signals than Cl-SOA, which is qualitatively consistent with previous findings from α-pinene ozonolysis SOA exposed to Cl and Br.126These trends are complicated by potential HCl and HBr elimination via chemical processes in the OFR and/or thermal degradation in the VIA, making quantitative interpretation uncertain. Nonetheless, the enrichment of halogenated signals in the OVOC3 factors combined with lower levels of halogenated signals in the SOA factors supports the idea that Cl and Br are preferentially retained in more volatile fragmentation products.
Differences in OVOC and SOA composition among the α-pinene/OH, α-pinene/Cl, and α-pinene/Br systems are linked to differences in the fate of RO2 formed from OH, Cl and Br oxidation. Fig. 9 shows FRO2 in the absence of isomerization/autooxidation. In the OH system, RO2 loss is dominated by reactions with HO2 and OH: FRO2+HO2 decreased from 0.96 to 0.73 and FRO2+OH increased from 0.02 to 0.27 with rising OHexp (Fig. 9a). In contrast, RO2 produced from Cl and Br oxidation show more complex RO2 fates. In the α-pinene/Cl system (Fig. 9b), FRO2+RO2 decreases from 0.50 to 0.11, FRO2+HO2 decreases from 0.36 to 0.22, FRO2+Cl increases from 0.08 to 0.37, and FRO2+ClO rises from 0.06 to 0.30. In the α-pinene/Br system (Fig. 9c), FRO2+RO2 drops from 0.23 to 0.002, FRO2+HO2 decreases from 0.23 to 0.05, FRO2+Br increases from 0.30 to 0.90, and FRO2+BrO decreases from 0.24 to 0.05. To assess the role of RO2 isomerization/autooxidation, we ran separate simulations assuming isomerization rate coefficients (kisom) of 0.1, 1, and 4 s−1 (Fig. S6–S8). These values span the range of experimentally observed rates,163 with kisom = 4 s−1 corresponding to the isomerization rate coefficient of the “APINCO2” peroxy radical156,165 derived from H-abstraction on α-pinene's terminal methyl group. In the α-pinene/OH system, RO2 loss remains dominated by RO2 + HO2 reactions at kisom = 0.1 s−1. Isomerization becomes competitive with RO2 + HO2 at kisom = 1 s−1 and becomes dominant at kisom = 4 s−1. In the α-pinene/Cl system, isomerization is competitive with RO2 + Cl at kisom = 0.1 s−1 and becomes dominant at kisom ≥ 1 s−1. Finally, in the α-pinene/Br system, RO2 + Br remains dominant at kisom = 0.1 s−1 and under most kisom = 1 s−1 conditions. However, isomerization plays an important role at kisom = 1 s−1 and becomes dominant at kisom = 4 s−1. In summary, while exact contributions are uncertain, this analysis suggests that RO2 + HO2 reactions dominate in the α-pinene/OH system, multiple pathways (including RO2 + Cl and isomerization) are important in the α-pinene/Cl system, and RO2 + Br reactions dominate in the α-pinene/Br system, with increasing isomerization at higher at higher kisom values. These mechanistic differences contribute to the observed variability in product distributions and SOA formation across the three oxidant systems.
 |
| | Fig. 9 Modeled fates of the organic peroxy radical (RO2) as a function of oxidant exposure during the (a) OH, (b) Cl, and (c) Br oxidation of α-pinene in the absence of isomerization/autooxidation reactions. Reactions and kinetic rate coefficients used in these calculations are provided in Table S2. | |
4 Conclusions
In this study, we presented a comprehensive laboratory investigation into the gas and condensed-phase products generated from the oxidation of α-pinene by OH, Cl, and Br. By employing a Vocus equipped with a VIA and NH4+ reagent ion chemistry, we characterized a wide array of oxidation products, including multifunctional organic compounds and previously challenging-to-measure low-volatility species, using a single reagent ion. PMF analysis revealed consistent formation of four distinct factors corresponding to early-generation VOCs/OVOCs, later-generation oxidized products, and low-volatility species contributing to SOA.
Our results demonstrate distinct chemical pathways and product distributions for each oxidant. While the initial oxidation of α-pinene by all three oxidants yielded C10H14,16O and C10H16O2 products, among others, subsequent reactions led to significantly different compound classes. OH-initiated oxidation produced a complex mixture of OVOC and SOA components that were consistent with established α-pinene/OH chemistry. In contrast, α-pinene reactions with Cl and Br atoms led to the formation of a substantial number of halogenated organic compounds. Key early-generation gas-phase products identified included C10H15ClO, and C10H15ClO2 from the Cl reaction, and C10H15BrO and C10H15BrO2 from the Br reaction. The product mass spectra obtained for each oxidation system provide unique fingerprints that can aid in identifying the chemical history and sources of ambient organic aerosol. Cl- and Br-initiated oxidation produced condensed-phase products that were generally less oxidized than those from OH oxidation, with Br-SOA being the least oxidized overall. However, Br-SOA retained a higher fraction of halogenated products than Cl-SOA. Photochemical modeling demonstrated oxidant-specific differences in RO2 fate, with RO2 + HO2 reactions dominating under OH oxidation, a more complex interplay of reactions under Cl oxidation, and RO2 + Br reactions dominating under Br oxidation. These differences help explain the observed variation in SOA composition and oxidative aging between systems.
Altogether, these results underscore that oxidation by halogen atoms leads to distinct chemical pathways and product distributions compared to OH oxidation. This highlights the need for updated chemical mechanisms and SOA yield treatments that explicitly account for Cl- and Br-mediated oxidation chemistry, especially in regions where halogen atoms are abundant. The presence of these species may alter the physicochemical properties of atmospheric aerosols, including their hygroscopicity, optical properties, and potential toxicity, particularly in marine and coastal environments, with important implications for air quality, climate, and human health. This work highlights the power and utility of NH4+ CIMS as a tool for elucidating complex atmospheric oxidation mechanisms, from initial gas-phase reactions to the formation and chemical evolution of aerosols. Future studies should focus on extending this methodology to other precursors and exploring the evolution of these distinct SOA types under a broader range of atmospheric conditions to better constrain their impact in regional and global models.
Author contributions
AL and JK conceived, planned, and carried out the experiments. AL performed data analysis. AL, MC, AA, MA and ND contributed to the interpretation of the results. AL took the lead in writing the manuscript. All authors provided feedback on the manuscript.
Conflicts of interest
There are no conflicts to declare.
Data availability
Data for this article, including source files used to generate Fig. 1 and 3–9, are available at Open Science Framework at [https://osf.io/6fc3q/overview].
Supplementary information (SI): additional experimental time series, molecular formula assignments and proposed structures, Van Krevelen analyses of α-pinene Cl-SOA and Br-SOA composition, data obtained from Vocus measurements of pinonaldehyde, and detailed chemical mechanisms and kinetic parameters used in the OFR modeling. See DOI: https://doi.org/10.1039/d5ea00091b.
Acknowledgements
This work was supported by the Atmospheric Chemistry Program of the United States National Science Foundation: grant AGS-1934352 to Aerodyne Research. AL thanks Lea Hildebrandt Ruiz (University of Texas at Austin) and Harald Stark (Aerodyne Research) for helpful discussions.
References
- X. Wang, D. J. Jacob, S. D. Eastham, M. P. Sulprizio, L. Zhu, Q. Chen, B. Alexander, T. Sherwen, M. J. Evans, B. H. Lee, J. D. Haskins, F. D. Lopez-Hilfiker, J. A. Thornton, G. L. Huey and H. Liao, Atmos. Chem. Phys., 2019, 19, 3981–4003 CrossRef CAS.
- X. Peng, W. Wang, M. Xia, H. Chen, A. R. Ravishankara, Q. Li, A. Saiz-Lopez, P. Liu, F. Zhang, C. Zhang, L. Xue, X. Wang, C. George, J. Wang, Y. Mu, J. Chen and T. Wang, Natl. Sci. Rev., 2020, 8, nwaa304 CrossRef PubMed.
- A. K. Baker, C. Sauvage, U. R. Thorenz, P. van Velthoven, D. E. Oram, A. Zahn, C. A. M. Brenninkmeijer and J. Williams, Sci. Rep., 2016, 6, 36821 CrossRef CAS PubMed.
- X. Yi, G. Sarwar, J. Bian, L. Huang, Q. Li, S. Jiang, H. Liu, Y. Wang, H. Chen, T. Wang, J. Chen, A. Saiz-Lopez, D. C. Wong and L. Li, J. Geophys. Res. Atmos., 2023, 128, e2023JD038898 Search PubMed.
- G. Chen, Z. Chen, Y. Zhang, X. Fan, L. Xu, Z. Lin, X. Ji and J. Chen, npj Clim. Atmos. Sci., 2025, 8, 248 Search PubMed.
- P. L. Tanaka, D. D. Riemer, S. Chang, G. Yarwood, E. C. McDonald-Buller, E. C. Apel, J. J. Orlando, P. J. Silva, J. L. Jimenez, M. R. Canagaratna, J. D. Neece, C. Mullins and D. T. Allen, Atmos. Environ., 2003, 37, 1393–1400 CrossRef CAS.
- C. B. Faxon and D. T. Allen, Environ. Chem., 2013, 10, 221–233 Search PubMed.
- C. G. Masoud, M. Modi, N. Bhattacharyya, L. G. Jahn, K. N. McPherson, P. Abue, K. Patel, D. T. Allen and L. Hildebrandt Ruiz, Environ. Sci. Technol., 2023, 57, 15454–15464 Search PubMed.
- D. Chang, Q. Li, Z. Wang, J. Dai, X. Fu, J. Guo, L. Zhu, D. Pu, C. A. Cuevas, R. P. Fernandez, W. Wang, M. Ge, J. C. H. Fung, A. K. H. Lau, C. Granier, G. Brasseur, A. Pozzer, A. Saiz-Lopez, Y. Song and T. Wang, Natl. Sci. Rev., 2024, 11, nwae285 CrossRef CAS PubMed.
- W. R. Simpson, R. von Glasow, K. Riedel, P. Anderson, P. Ariya, J. Bottenheim, J. Burrows, L. J. Carpenter, U. Frieß, M. E. Goodsite, D. Heard, M. Hutterli, H.-W. Jacobi, L. Kaleschke, B. Neff, J. Plane, U. Platt, A. Richter, H. Roscoe, R. Sander, P. Shepson, J. Sodeau, A. Steffen, T. Wagner and E. Wolff, Atmos. Chem. Phys., 2007, 7, 4375–4418 CrossRef CAS.
- C. R. Thompson, P. B. Shepson, J. Liao, L. G. Huey, C. Cantrell, F. Flocke and J. Orlando, Atmos. Chem. Phys., 2017, 17, 3401–3421 CrossRef CAS.
- S. Wang, S. M. McNamara, C. W. Moore, D. Obrist, A. Steffen, P. B. Shepson, R. M. Staebler, A. R. W. Raso and K. A. Pratt, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 14479–14484 CrossRef CAS PubMed.
- J. H. Knox and R. L. Nelson, Trans. Faraday Soc., 1959, 55, 937–946 RSC.
- K. C. Ferguson and E. Whittle, Trans. Faraday Soc., 1971, 67, 2618–2628 RSC.
- K. H. Homann, Ber. Bunsenges. Phys. Chem., 1973, 77, 223–224 CrossRef.
- D. L. Baulch, J. Duxbury, S. J. Grant and D. C. Montague, J. Phys. Chem. Ref. Data, 1981, 10, Suppl. No. 1 CrossRef.
- T. Ohta, Int. J. Chem. Kinet., 1984, 16, 879–886 CrossRef CAS.
- D. H. Semmes, A. R. Ravishankara, C. A. Gump-Perkins and P. H. Wine, Int. J. Chem. Kinet., 1985, 17, 303–313 CrossRef CAS.
- R. Atkinson, Chem. Rev., 1986, 86, 69–201 Search PubMed.
- S. W. Benson, O. Kondo and R. M. Marshall, Int. J. Chem. Kinet., 1987, 19, 829–839 Search PubMed.
- R. Atkinson and S. M. Aschmann, J. Phys. Chem., 1988, 92, 4008 Search PubMed.
- F. Nolting, W. Behnke and C. Zetzsch, J. Atmos. Chem., 1988, 6, 47–59 Search PubMed.
- J. J. Russell, J. A. Seetula and D. Gutman, J. Am. Chem. Soc., 1988, 110, 3092–3099 Search PubMed.
- T. J. Wallington, L. M. Skewes, W. O. Siegl, C.-H. Wu and S. M. Japar, Int. J. Chem. Kinet., 1988, 20, 867–875 CrossRef CAS.
- R. Atkinson and S. M. Aschmann, Int. J. Chem. Kinet., 1989, 21, 355–365 Search PubMed.
- I. Barnes, V. Bastian, K. H. Becker, R. Overath and Z. Tong, Int. J. Chem. Kinet., 1989, 21, 499–517 Search PubMed.
- T. J. Wallington, L. M. Skewes, W. O. Siegl and S. M. Japar, Int. J. Chem. Kinet., 1989, 21, 1069–1076 Search PubMed.
- L. Nelson, O. Rattigan, R. Neavyn, H. Sidebottom, J. Treacy and O. J. Nielsen, Int. J. Chem. Kinet., 1990, 22, 1111–1126 Search PubMed.
- O. J. Nielsen, H. W. Sidebottom, L. Nelson, O. Rattigan, J. J. Treacy and D. J. O'Farrell, Int. J. Chem. Kinet., 1990, 22, 603–612 CrossRef CAS.
- R. Atkinson, D. L. Baulch, R. A. Cox, J. Hampson, R. F., J. A. Kerr and J. Troe, J. Phys. Chem. Ref. Data, 1992, 21, 1125–1568 Search PubMed.
- D. L. Baulch, C. J. Cobos, R. A. Cox, C. Esser, P. Frank, T. Just, J. A. Kerr, M. J. Pilling, J. Troe, R. W. Walker and J. Warnatz, J. Phys. Chem. Ref. Data, 1992, 21, 411 Search PubMed.
- A. Bierbach, I. Barnes and K. Becker, Atmos. Environ. Gen. Top, 1992, 26, 813–817 Search PubMed.
- D. M. Rowley, R. Lesclaux, P. D. Lightfoot, B. Noziere, T. J. Wallington and M. D. Hurley, J. Phys. Chem., 1992, 96, 4889–4894 Search PubMed.
- B. Noziere, R. Lesclaux, M. D. Hurley, M. A. Dearth and T. J. Wallington, J. Phys. Chem., 1994, 98, 2864–2873 Search PubMed.
- S. M. Aschmann and R. Atkinson, Int. J. Chem. Kinet., 1995, 27, 613–622 CrossRef CAS.
- A. Bierbach, I. Barnes and K. Becker, Atmos. Environ., 1995, 29, 2651–2660 Search PubMed.
- A. Bierbach, I. Barnes and K. H. Becker, Int. J. Chem. Kinet., 1996, 28, 565–577 Search PubMed.
- P. A. Hooshiyar and H. Niki, Int. J. Chem. Kinet., 1995, 27, 1197–1206 Search PubMed.
- E. W. Kaiser and T. J. Wallington, J. Phys. Chem., 1996, 100, 4111–4119 Search PubMed.
- D. J. Kinnison, W. Mengon and J. A. Kerr, J. Chem. Soc., Faraday Trans., 1996, 92, 369–372 RSC.
- S. Teton, A. Mellouki, G. Le Bras and H. Sidebottom, Int. J. Chem. Kinet., 1996, 28, 291–297 Search PubMed.
- A. A. Turnipseed, S. B. Barone and A. R. Ravishankara, J. Phys. Chem., 1996, 100, 14703–14713 Search PubMed.
- R. Atkinson, D. L. Baulch, R. A. Cox, R. F. Hampson, J. A. Kerr, M. J. Rossi and J. Troe, J. Phys. Chem. Ref. Data, 1997, 26, 521–1011 CrossRef CAS.
-
W. B. DeMore, S. P. Sander, D. M. Golden, R. F. Hampson, M. J. Kurylo, C. J. Howard, A. R. Ravishankara, C. E. Kolb, M. J. Molina, Chemical Kinetics and Photochemical Data for Use in Stratospheric Modeling, JPL Publication 97-4, 1997 Search PubMed.
- M. L. Ragains and B. J. Finlayson-Pitts, J. Phys. Chem. A, 1997, 101, 1509–1517 Search PubMed.
- J. Shi and M. J. Bernhard, Int. J. Chem. Kinet., 1997, 29, 349–358 Search PubMed.
- G. S. Tyndall, J. J. Orlando, T. J. Wallington, M. Dill and E. W. Kaiser, Int. J. Chem. Kinet., 1997, 29, 43–55 CrossRef CAS.
- J. T. Jodkowski, M.-T. Rayez, J.-C. Rayez, T. Bérces and S. Dóbé, J. Phys. Chem. A, 1998, 102, 9230–9243 Search PubMed.
- S. Le Calvé, D. Hitier, G. Le Bras and A. Mellouki, J. Phys. Chem. A, 1998, 102, 4579–4584 Search PubMed.
- B. Nozière and I. Barnes, J. Geophys. Res. Atmos., 1998, 103, 25587–25597 CrossRef.
- I. Szilágyi, K. Imrik, S. Dóbé and T. Bérces, Ber. Bunsenges. Phys. Chem., 1998, 102, 79–84 CrossRef.
- A. Bierbach, I. Barnes and K. Becker, Atmos. Environ., 1999, 33, 2981–2992 CrossRef CAS.
- B. J. Finlayson-Pitts, C. J. Keoshian, B. Buehler and A. A. Ezell, Int. J. Chem. Kinet., 1999, 31, 491–499 CrossRef CAS.
- T. Maurer, I. Barnes and K. H. Becker, Int. J. Chem. Kinet., 1999, 31, 883–893 CrossRef CAS.
- C. Sauer, I. Barnes and K. Becker, Atmos. Environ., 1999, 33, 2969–2979 Search PubMed.
- R. Atkinson, E. C. Tuazon and S. M. Aschmann, Environ. Sci. Technol., 2000, 34, 623–631 CrossRef CAS.
- B. Ramacher, J. J. Orlando and G. S. Tyndall, Int. J. Chem. Kinet., 2000, 32, 460–465 CrossRef CAS.
- R. Thévenet, A. Mellouki and G. Le Bras, Int. J. Chem. Kinet., 2000, 32, 676–685 Search PubMed.
-
R. Atkinson, D. Baulch, R. Cox, J. Crowley, J. Hampson, R. F., J. Kerr, M. Rossi and J. Troe, IUPAC Subcommittee on Gas Kinetic Data Evaluation for Atmospheric Chemistry - Web Version, 2001 Search PubMed.
- W. R. Bradley, S. E. Wyatt, J. R. Wells, M. V. Henley and G. M. Graziano, Int. J. Chem. Kinet., 2001, 33, 108–117 CrossRef CAS.
- J. Albaladejo, B. Ballesteros, E. Jiménez, P. Martín and E. Martínez, Atmos. Environ., 2002, 36, 3231–3239 CrossRef CAS.
- M. J. Ezell, W. Wang, A. A. Ezell, G. Soskin and B. J. Finlayson-Pitts, Phys. Chem. Chem. Phys., 2002, 4, 5813–5820 RSC.
- K. J. Gill and R. A. Hites, J. Phys. Chem. A, 2002, 106, 2538–2544 CrossRef CAS.
- W. Wang, M. J. Ezell, A. A. Ezell, G. Soskin and B. J. Finlayson-Pitts, Phys. Chem. Chem. Phys., 2002, 4, 1824–1831 RSC.
- R. Atkinson, Atmos. Chem. Phys. Discuss., 2003, 3, 2233–2307 CrossRef CAS.
- M. D. Hurley, W. F. Schneider, T. J. Wallington, D. J. Mann, J. D. DeSain and C. A. Taatjes, J. Phys. Chem. A, 2003, 107, 2003–2010 CrossRef CAS.
- J. Moriarty, H. Sidebottom, J. Wenger, A. Mellouki and G. Le Bras, J. Phys. Chem. A, 2003, 107, 1499–1505 CrossRef CAS.
- J. J. Orlando, G. S. Tyndall, E. C. Apel, D. D. Riemer and S. E. Paulson, Int. J. Chem. Kinet., 2003, 35, 334–353 CrossRef CAS.
- R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, R. G. Hynes, M. E. Jenkin, M. J. Rossi and J. Troe, Atmos. Chem. Phys., 2004, 4, 1461–1738 CrossRef CAS.
- C. A. Cuevas, A. Notario, E. Martínez and J. Albaladejo, Phys. Chem. Chem. Phys., 2004, 6, 2230–2236 RSC.
- T. Imamura, Y. Iida, K. Obi, I. Nagatani, K. Nakagawa, I. Patroescu-Klotz and S. Hatakeyama, Int. J. Chem. Kinet., 2004, 36, 379–385 Search PubMed.
- B. Caba nas, F. Villanueva, P. Martín, M. Baeza, S. Salgado and E. Jiménez, Atmos. Environ., 2005, 39, 1935–1944 CrossRef CAS.
- E. Jiménez, B. Ballesteros, E. Martínez and J. Albaladejo, Environ. Sci. Technol., 2005, 39, 814–820 CrossRef PubMed.
- D. Rodríguez, A. Rodríguez, A. Notario, A. Aranda, Y. Díaz-de Mera and E. Martínez, Atmos. Chem. Phys., 2005, 5, 3433–3440 CrossRef.
- C. A. Cuevas, A. Notario, E. Martínez and J. Albaladejo, Atmos. Environ., 2006, 40, 3845–3854 CrossRef CAS.
- R. S. Anderson, L. Huang, R. Iannone and J. Rudolph, J. Phys. Chem. A, 2007, 111, 495–504 CrossRef CAS PubMed.
- M. Baasandorj and P. S. Stevens, J. Phys. Chem. A, 2007, 111, 640–649 CrossRef CAS PubMed.
- B. Ballesteros, A. Garzón, E. Jiménez, A. Notario and J. Albaladejo, Phys. Chem. Chem. Phys., 2007, 9, 1210–1218 RSC.
- M. E. Davis, R. K. Talukdar, G. Notte, G. B. Ellison and J. B. Burkholder, Environ. Sci. Technol., 2007, 41, 3959–3965 CrossRef CAS PubMed.
- E. Jiménez, B. Lanza, E. Martínez and J. Albaladejo, Atmos. Chem. Phys., 2007, 7, 1565–1574 CrossRef.
- B. Caba nas, A. Tapia, F. Villanueva, S. Salgado, E. Monedero and P. Martín, Int. J. Chem. Kinet., 2008, 40, 670–678 CrossRef CAS.
- P. M. Cometto, P. R. Dalmasso, R. A. Taccone, S. I. Lane, F. Oussar, V. Daële, A. Mellouki and G. L. Bras, J. Phys. Chem. A, 2008, 112, 4444–4450 CrossRef CAS PubMed.
- D. Rodriguez, A. Rodriguez, A. Soto, A. Aranda, Y. Diaz-de Mera and A. Notario, J. Atmos. Chem., 2008, 59, 187–197 Search PubMed.
- B. R. Giri and J. M. Roscoe, J. Phys. Chem. A, 2009, 113, 8001–8010 Search PubMed.
- G. Oksdath-Mansilla, A. B. Pe né nory, M. Albu, I. Barnes, P. Wiesen and M. A. Teruel, Chem. Phys. Lett., 2009, 477, 22–27 CrossRef CAS.
- R. Sivaramakrishnan and J. V. Michael, J. Phys. Chem. A, 2009, 113, 5047–5060 Search PubMed.
- B. R. Giri and J. M. Roscoe, J. Phys. Chem. A, 2010, 114, 8369–8375 CrossRef CAS PubMed.
- M. E. Jenkin, M. D. Hurley and T. J. Wallington, J. Phys. Chem. A, 2010, 114, 408–416 CrossRef CAS PubMed.
- E. W. Kaiser, T. J. Wallington and M. D. Hurley, J. Phys. Chem. A, 2010, 114, 343–354 CrossRef CAS PubMed.
- S. M. Aschmann, J. Arey and R. Atkinson, J. Phys. Chem. A, 2011, 115, 14452–14461 CrossRef CAS PubMed.
- M. E. Davis and J. B. Burkholder, Atmos. Chem. Phys., 2011, 11, 3347–3358 CrossRef CAS.
- Y. Gai, M. Ge and W. Wang, Atmos. Environ., 2011, 45, 53–59 CrossRef CAS.
- A. A. Ceacero-Vega, B. Ballesteros, I. Bejan, I. Barnes, E. Jiménez and J. Albaladejo, J. Phys. Chem. A, 2012, 116, 4097–4107 CrossRef CAS PubMed.
- L. Renbaum-Wolff and G. D. Smith, J. Phys. Chem. A, 2012, 116, 6664–6674 CrossRef CAS PubMed.
- D. Rodríguez, A. Rodríguez, A. Garzón, J. M. Granadino-Roldán, A. Soto, A. Aranda and A. N., Mol. Phys., 2012, 110, 2941–2950 CrossRef.
- E. Szabo, G. L. Zuegner, M. Farkas, I. Szilagyi and S. Dobe, Oxid. Commun., 2012, 35, 538–544 CAS.
- S. M. Aschmann and R. Atkinson, Int. J. Chem. Kinet., 2013, 45, 52–58 CrossRef CAS.
- R. G. Gibilisco, A. N. Santiago and M. A. Teruel, Atmos. Environ., 2013, 77, 358–364 CrossRef CAS.
- J. A. Liljegren and P. S. Stevens, Int. J. Chem. Kinet., 2013, 45, 787–794 CrossRef CAS.
- M. R. Dash and B. Rajakumar, Atmos. Environ., 2014, 99, 183–195 CrossRef CAS.
- R. G. Gibilisco, I. Bejan, I. Barnes, P. Wiesen and M. A. Teruel, Atmos. Environ., 2014, 94, 564–572 CrossRef CAS.
- M. Riva, R. M. Healy, P.-M. Flaud, E. Perraudin, J. C. Wenger and E. Villenave, J. Phys. Chem. A, 2014, 118, 3535–3540 Search PubMed.
- A. Lauraguais, I. Bejan, I. Barnes, P. Wiesen, C. Coeur-Tourneur and A. Cassez, J. Phys. Chem. A, 2014, 118, 1777–1784 CrossRef CAS PubMed.
- R. G. Gibilisco, M. B. Blanco, I. Bejan, I. Barnes, P. Wiesen and M. A. Teruel, Environ. Sci. Technol., 2015, 49, 7717–7725 CrossRef CAS PubMed.
- A. Lauraguais, I. Bejan, I. Barnes, P. Wiesen and C. Coeur, J. Phys. Chem. A, 2015, 119, 6179–6187 CrossRef CAS PubMed.
- A. Safron, M. Strandell, A. Kierkegaard and M. Macleod, Int. J. Chem. Kinet., 2015, 47, 420–428 CrossRef CAS PubMed.
- B. Srinivasulu and G. Rajakumar, J. Chem. Sci., 2016, 128, 977–989 CrossRef.
- S. Ausmeel, C. Andersen, O. J. Nielsen, F. F. Østerstrøm, M. S. Johnson and E. J. K. Nilsson, J. Phys. Chem. A, 2017, 121, 4123–4131 CrossRef CAS PubMed.
- J. A. Barrera, P. R. Dalmasso, R. A. Taccone and S. I. Lane, Environ. Sci. Pollut. Res., 2017, 24, 26049–26059 CrossRef CAS PubMed.
- J. A. Barrera, M. de los, A. Garavagno, P. R. Dalmasso and R. A. Taccone, Atmos. Environ., 2019, 202, 28–40 CrossRef CAS.
- B. Shi, W. Wang, L. Zhou, J. Li, J. Wang, Y. Chen, W. Zhang and M. Ge, Atmos. Environ., 2019, 207, 75–81 CrossRef CAS.
- M. W. Alton and E. C. Browne, Environ. Sci. Technol., 2020, 54, 5992–5999 Search PubMed.
- I. Colmenar, S. Salgado, P. Martín, I. Aranda, A. Tapia and B. Caba nas, Atmos. Environ., 2020, 224, 117367 CrossRef CAS.
- K. H. Møller, R. V. Otkjær, J. Chen and H. G. Kjaergaard, J. Phys. Chem. A, 2020, 124, 2885–2896 Search PubMed.
- J. T. Shaw, A. R. Rickard, M. J. Newland and T. J. Dillon, Atmos. Chem. Phys., 2020, 20, 9725–9736 CrossRef CAS.
- S. Szymański and D. S. Sarzyński, Int. J. Chem. Kinet., 2020, 52, 957–963 CrossRef.
- Z. Tan, L. Hantschke, M. Kaminski, I.-H. Acir, B. Bohn, C. Cho, H.-P. Dorn, X. Li, A. Novelli, S. Nehr, F. Rohrer, R. Tillmann, R. Wegener, A. Hofzumahaus, A. Kiendler-Scharr, A. Wahner and H. Fuchs, Atmos. Chem. Phys., 2021, 21, 16067–16091 CrossRef CAS.
- A. Allani and M. N. Romanias, Int. J. Chem. Kinet., 2022, 54, 424–434 Search PubMed.
- M. Asensio, M. Anti nolo, S. Blázquez, J. Albaladejo and E. Jiménez, Atmos. Chem. Phys., 2022, 22, 2689–2701 CrossRef CAS.
- D. Van Hoomissen, A. Chattopadhyay and J. B. Burkholder, Int. J. Chem. Kinet., 2025, 57, 213–231 CrossRef CAS.
- Q. K. Timerghazin and P. A. Ariya, Phys. Chem. Chem. Phys., 2001, 3, 3981–3986 RSC.
- X. Cai and R. J. Griffin, J. Geophys. Res., 2006, 111, D14206 CrossRef.
- Y. Wang, M. Riva, H. Xie, L. Heikkinen, S. Schallhart, Q. Zha, C. Yan, X.-C. He, O. Peräkylä and M. Ehn, Atmos. Chem. Phys., 2020, 20, 5145–5155 CrossRef CAS.
- C. G. Masoud and L. Hildebrandt Ruiz, ACS Earth Space Chem., 2021, 5, 2307–2319 CrossRef CAS.
- A. T. Lambe, A. M. Avery, N. Bhattacharyya, D. S. Wang, M. Modi, C. G. Masoud, L. H. Ruiz and W. H. Brune, Environ. Sci.: Atmos., 2022, 2, 687–701 CAS.
- J. Ofner, N. Balzer, J. Buxmann, H. Grothe, P. Schmitt-Kopplin, U. Platt and C. Zetzsch, Atmos. Chem. Phys., 2012, 12, 5787–5806 CrossRef CAS.
- Q. Li, X. Fu, X. Peng, W. Wang, A. Badia, R. P. Fernandez, C. A. Cuevas, Y. Mu, J. Chen, J. L. Jimenez, T. Wang and A. Saiz-Lopez, Environ. Sci. Technol., 2021, 55, 13625–13637 CrossRef CAS PubMed.
- B. L. Deming, D. Pagonis, X. Liu, D. A. Day, R. Talukdar, J. E. Krechmer, J. A. de Gouw, J. L. Jimenez and P. J. Ziemann, Atmos. Meas. Tech., 2019, 12, 3453–3461 CrossRef CAS.
- A. T. Lambe, J. E. Krechmer, Z. Peng, J. R. Casar, A. J. Carrasquillo, J. D. Raff, J. L. Jimenez and D. R. Worsnop, Atmos. Meas. Tech., 2019, 12, 299–311 CrossRef CAS.
- J. P. Rowe, A. T. Lambe and W. H. Brune, Atmos. Chem. Phys., 2020, 20, 13417–13424 Search PubMed.
- J. Mao, X. Ren, W. Brune, J. Olson, J. Crawford, A. Fried, L. Huey, R. Cohen, B. Heikes and H. Singh, Atmos. Chem. Phys., 2009, 9, 163–173 CrossRef CAS.
- A. V. Baklanov and L. N. Krasnoperov, J. Phys. Chem. A, 2001, 105, 97–103 Search PubMed.
- B. Ghosh, D. K. Papanastasiou and J. B. Burkholder, J. Chem. Phys., 2012, 137, 164315 CrossRef PubMed.
- C.-C. Wu, H.-C. Lin, Y.-B. Chang, P.-Y. Tsai, Y.-Y. Yeh, H. Fan, K.-C. Lin and J. S. Francisco, J. Chem. Phys., 2011, 135, 234308 CrossRef PubMed.
- D. Paul, H. K. Kim, M. M. Rahman and T. K. Kim, J. Appl. Spectrosc., 2021, 88, 737–743 CrossRef CAS.
- C. R. Stephens, P. B. Shepson, A. Steffen, J. W. Bottenheim, J. Liao, L. G. Huey, E. Apel, A. Weinheimer, S. R. Hall, C. Cantrell, B. C. Sive, D. J. Knapp, D. D. Montzka and R. S. Hornbrook, J. Geophys. Res., 2012, 117, D14 CrossRef.
- J. Krechmer, F. Lopez-Hilfiker, A. Koss, M. Hutterli, C. Stoermer, B. Deming, J. Kimmel, C. Warneke, R. Holzinger, J. Jayne, D. Worsnop, K. Fuhrer, M. Gonin and J. de Gouw, Anal. Chem., 2018, 90, 12011–12018 CrossRef CAS PubMed.
- P. Khare, J. E. Krechmer, J. E. Machesky, T. Hass-Mitchell, C. Cao, J. Wang, F. Majluf, F. Lopez-Hilfiker, S. Malek, W. Wang, K. Seltzer, H. O. T. Pye, R. Commane, B. C. McDonald, R. Toledo-Crow, J. E. Mak and D. R. Gentner, Atmos. Chem. Phys., 2022, 22, 14377–14399 CrossRef CAS PubMed.
- L. Xu, M. M. Coggon, C. E. Stockwell, J. B. Gilman, M. A. Robinson, M. Breitenlechner, A. Lamplugh, J. D. Crounse, P. O. Wennberg, J. A. Neuman, G. A. Novak, P. R. Veres, S. S. Brown and C. Warneke, Atmos. Meas. Tech., 2022, 15, 7353–7373 CrossRef CAS.
- E. Häkkinen, J. Zhao, F. Graeffe, N. Fauré, J. Krechmer, D. Worsnop, H. Timonen, M. Ehn and J. Kangasluoma, Atmos. Meas. Tech., 2022, 2022, 1–29 Search PubMed.
- A. M. Avery, M. W. Alton, M. R. Canagaratna, J. E. Krechmer, D. T. Sueper, N. Bhattacharyya, L. Hildebrandt Ruiz, W. H. Brune and A. T. Lambe, ACS Earth Space Chem., 2023, 7, 218–229 CrossRef CAS.
- J. Zhao, V. Mickwitz, Y. Luo, E. Häkkinen, F. Graeffe, J. Zhang, H. Timonen, M. Canagaratna, J. E. Krechmer, Q. Zhang, M. Kulmala, J. Kangasluoma, D. Worsnop and M. Ehn, Atmos. Meas. Tech., 2024, 17, 1527–1543 CrossRef CAS.
-
D. Sueper, 2022, http://cires.colorado.edu/jimenez-group/ToFAMSResources/ToFSoftware.
- M. R. Canagaratna, J. L. Jimenez, J. H. Kroll, Q. Chen, S. H. Kessler, P. Massoli, L. Hildebrandt Ruiz, E. Fortner, L. R. Williams, K. R. Wilson, J. D. Surratt, N. M. Donahue, J. T. Jayne and D. R. Worsnop, Atmos. Chem. Phys., 2015, 15, 253–272 CrossRef.
- H. Stark, R. L. Yatavelli, S. L. Thompson, J. R. Kimmel, M. J. Cubison, P. S. Chhabra, M. R. Canagaratna, J. T. Jayne, D. R. Worsnop and J. L. Jimenez, Int. J. Mass Spectrom., 2015, 389, 26–38 Search PubMed.
- P. Paatero and U. Tapper, Environmetrics, 1994, 5, 111–126 CrossRef.
- C. Yan, W. Nie, M. Äijälä, M. P. Rissanen, M. R. Canagaratna, P. Massoli, H. Junninen, T. Jokinen, N. Sarnela, S. A. K. Häme, S. Schobesberger, F. Canonaco, L. Yao, A. S. H. Prévôt, T. Petäjä, M. Kulmala, M. Sipilä, D. R. Worsnop and M. Ehn, Atmos. Chem. Phys., 2016, 16, 12715–12731 Search PubMed.
- I. Ulbrich, M. Canagaratna, Q. Zhang, D. Worsnop and J. Jimenez, Atmos. Chem. Phys., 2009, 9, 2891–2918 CrossRef CAS.
- N. M. Donahue, S. A. Epstein, S. N. Pandis and A. L. Robinson, Atmos. Chem. Phys., 2011, 11, 3303–3318 CrossRef CAS.
- Z. Peng and J. L. Jimenez, J. Chem. Educ., 2019, 96, 806–811 CrossRef CAS.
- R. Li, B. B. Palm, A. M. Ortega, J. Hlywiak, W. Hu, Z. Peng, D. A. Day, C. Knote, W. H. Brune, J. A. De Gouw and J. L. Jimenez, J. Phys. Chem. A, 2015, 119, 150406123535006 Search PubMed.
- Z. Peng and J. L. Jimenez, Chem. Soc. Rev., 2020, 49, 2570–2616 RSC.
- Z. Peng, J. Lee-Taylor, J. J. Orlando, G. S. Tyndall and J. L. Jimenez, Atmos. Chem. Phys., 2019, 19, 813–834 CrossRef CAS.
- A. T. Lambe, C. K. Glenn, A. M. Avery, T. Xu, J. C. Ditto, M. R. Canagaratna, D. R. Gentner, K. S. Docherty, M. Jaoui, J. Zaks, A. K. Bertram, N. L. Ng and P. Liu, ACS Earth Space Chem., 2025, 9(3), 545–559 CrossRef CAS PubMed.
- W. Tsang and R. F. Hampson, J. Phys. Chem. Ref. Data, 1986, 15, 1087–1279 CrossRef CAS.
- M. Jenkin, S. Saunders and M. Pilling, Atmos. Environ., 1997, 31, 81–104 CrossRef CAS.
- J. S. Francisco and J. N. Crowley, J. Phys. Chem. A, 2006, 110, 3778–3784 CrossRef CAS PubMed.
- S. Enami, T. Yamanaka, T. Nakayama, S. Hashimoto, M. Kawasaki, D. E. Shallcross, Y. Nakano and T. Ishiwata, J. Phys. Chem. A, 2007, 111, 3342–3348 CrossRef CAS PubMed.
- D. E. Shallcross, K. E. Leather, A. Bacak, P. Xiao, E. P. F. Lee, M. Ng, D. K. W. Mok, J. M. Dyke, R. Hossaini, M. P. Chipperfield, M. A. H. Khan and C. J. Percival, J. Phys. Chem. A, 2015, 119, 4618–4632 CrossRef CAS PubMed.
- R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, R. G. Hynes, M. E. Jenkin, M. J. Rossi and J. Troe, Atmos. Chem. Phys., 2007, 7, 981–1191 Search PubMed.
- P. J. Ziemann and R. Atkinson, Chem. Soc. Rev., 2012, 41, 6582–6605 RSC.
- J. J. Orlando and G. S. Tyndall, Chem. Soc. Rev., 2012, 41, 6294–6317 RSC.
- F. Bianchi, T. Kurtén, M. Riva, C. Mohr, M. P. Rissanen, P. Roldin, T. Berndt, J. D. Crounse, P. O. Wennberg, T. F. Mentel, J. Wildt, H. Junninen, T. Jokinen, M. Kulmala, D. R. Worsnop, J. A. Thornton, N. Donahue, H. G. Kjaergaard and M. Ehn, Chem. Rev., 2019, 119, 3472–3509 Search PubMed.
- C. Fittschen, Chem. Phys. Lett., 2019, 725, 102–108 Search PubMed.
- L. Xu, K. H. Møller, J. D. Crounse, R. V. Otkjær, H. G. Kjaergaard and P. O. Wennberg, J. Phys. Chem. A, 2019, 123, 1661–1674 CrossRef CAS PubMed.
- Y. Li, U. Pöschl and M. Shiraiwa, Atmos. Chem. Phys., 2016, 16, 3327–3344 CrossRef CAS.
- N. C. Eddingsaas, C. L. Loza, L. D. Yee, J. H. Seinfeld and P. O. Wennberg, Atmos. Chem. Phys., 2012, 12, 6489–6504 CrossRef CAS.
- M. Claeys, Y. Iinuma, R. Szmigielski, J. D. Surratt, F. Blockhuys, C. Van Alsenoy, O. Böge, B. Sierau, Y. Gómez-González, R. Vermeylen, P. Van der Veken, M. Shahgholi, A. W. H. Chan, H. Herrmann, J. H. Seinfeld and W. Maenhaut, Environ. Sci. Technol., 2009, 43, 6976–6982 CrossRef CAS PubMed.
- R. Szmigielski, J. D. Surratt, Y. Gómez-González, P. Van der Veken, I. Kourtchev, R. Vermeylen, F. Blockhuys, M. Jaoui, T. E. Kleindienst, M. Lewandowski, J. H. Offenberg, E. O. Edney, J. H. Seinfeld, W. Maenhaut and M. Claeys, Geophys. Res. Lett., 2007, 34, L24811 CrossRef.
- L. Müller, M.-C. Reinnig, K. H. Naumann, H. Saathoff, T. F. Mentel, N. M. Donahue and T. Hoffmann, Atmos. Chem. Phys., 2012, 12, 1483–1496 CrossRef.
- N. C. Eddingsaas, C. L. Loza, L. D. Yee, M. Chan, K. A. Schilling, P. S. Chhabra, J. H. Seinfeld and P. O. Wennberg, Atmos. Chem. Phys., 2012, 12, 7413–7427 CrossRef CAS.
- T. Jokinen, T. Berndt, R. Makkonen, V.-M. Kerminen, H. Junninen, P. Paasonen, F. Stratmann, H. Herrmann, A. B. Guenther, D. R. Worsnop, M. Kulmala, M. Ehn and M. Sipilä, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7123–7128 CrossRef CAS PubMed.
- L. Poulain, A. Tilgner, M. Brüggemann, P. Mettke, L. He, J. Anders, O. Böge, A. Mutzel and H. Herrmann, J. Geophys. Res. Atmos., 2022, 127, e2021JD036414 CrossRef CAS.
- H. Luo, Y. Guo, H. Shen, D. D. Huang, Y. Zhang and D. Zhao, Environ. Sci.: Atmos., 2024, 4, 519–530 CAS.
- A. Wisthaler, N. Jensen, R. Winterhalter, W. Lindinger and J. Hjorth, Atmos. Environ., 2001, 35, 6181–6191 Search PubMed.
- A. Lee, A. H. Goldstein, J. H. Kroll, N. L. Ng, V. Varutbangkul, R. C. Flagan and J. H. Seinfeld, J. Geophys. Res. Atmos., 2006, 111, D07302 Search PubMed.
- B. Rosati, R. Teiwes, K. Kristensen, R. Bossi, H. Skov, M. Glasius, H. B. Pedersen and M. Bilde, Atmos. Environ., 2019, 199, 15–31 CrossRef CAS.
Footnote |
| † ‘Present address:’ Osmo Labs, PBC, Somerville, Massachusetts, United States. |
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Jordan E.
Krechmer†
a,
Anita M.
Avery
*a,
Jordan E.
Krechmer†
a,
Anita M.
Avery


 ,147 where I is the high-resolution peak height. PMF was used primarily to group ions with similar temporal behavior trends and to qualitatively apportion ions contributing to multiple factors. Separate PMF analyses were performed for the α-pinene/OH, α-pinene/Cl, and α-pinene/Br systems using the PMF Evaluation Tool (PET) version 3.08C,148 each yielding a four-factor solution representing precursors and early- and late-generation products. Three-factor PMF tended to combine precursor and product signals, often with higher residuals. Solutions with five or more factors led to the splitting of existing factors without identifying new product groups of products or significantly reducing residuals. To avoid artificially forcing ion signals to zero in specific factors, no rotations were applied.
,147 where I is the high-resolution peak height. PMF was used primarily to group ions with similar temporal behavior trends and to qualitatively apportion ions contributing to multiple factors. Separate PMF analyses were performed for the α-pinene/OH, α-pinene/Cl, and α-pinene/Br systems using the PMF Evaluation Tool (PET) version 3.08C,148 each yielding a four-factor solution representing precursors and early- and late-generation products. Three-factor PMF tended to combine precursor and product signals, often with higher residuals. Solutions with five or more factors led to the splitting of existing factors without identifying new product groups of products or significantly reducing residuals. To avoid artificially forcing ion signals to zero in specific factors, no rotations were applied.
 ,149 where nC and nO are the carbon and oxygen numbers of each compound. Vocus:VIA-Vocus ratios of measured C10H14,16Ox compounds were plotted against their calculated C° values and fit to sigmoid or polynomial regression equations. These equations were then used to infer C° for C10H13,15ClOx and C10H13,15BrOx from their measured Vocus:VIA-Vocus ratios. This analysis was restricted to C10H14,16Ox, C10H13,15ClOx and C10H13,15BrOx signals to minimize the effect of possible thermal decomposition reactions in the VIA on the calculated C° values.
,149 where nC and nO are the carbon and oxygen numbers of each compound. Vocus:VIA-Vocus ratios of measured C10H14,16Ox compounds were plotted against their calculated C° values and fit to sigmoid or polynomial regression equations. These equations were then used to infer C° for C10H13,15ClOx and C10H13,15BrOx from their measured Vocus:VIA-Vocus ratios. This analysis was restricted to C10H14,16Ox, C10H13,15ClOx and C10H13,15BrOx signals to minimize the effect of possible thermal decomposition reactions in the VIA on the calculated C° values.








