
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visible-light driven H2 reductive elimination unlocks reactivity in polyhydrido niobium iridium clusters
Zachary
Dubrawski
 a,
Samy
Aïssiou
a,
Erwann
Jeanneau
b,
Chloé
Thieuleux
a and
Clément
Camp
*a
a,
Samy
Aïssiou
a,
Erwann
Jeanneau
b,
Chloé
Thieuleux
a and
Clément
Camp
*a
aLaboratory of Catalysis, Polymerization, Processes and Materials (CP2M UMR 5128), CNRS, Universite Claude Bernard Lyon 1, CPE-Lyon, Institut de Chimie de Lyon, 43 Bd du 11 Novembre 1918, F-69616 Villeurbanne, France. E-mail: clement.camp@univ-lyon1.fr
bCentre de Diffractométrie Henri Longchambon, Universite Claude Bernard Lyon 1, 5 Rue de la Doua, 69100 Villeurbanne, France
First published on 8th December 2025
Abstract
Heterobimetallic complexes have recently attracted considerable attention due to their interesting bonding patterns and reactivity. Here, we report the synthesis and characterization of a series of polyhydrido niobium–iridium heterobimetallic clusters using the Brønsted acidic Cp*IrH4 reagent. This series of complexes exhibits Ir–Nb intermetallic distances significantly shorter than the sum of atomic radii suggestive of multiple bonding character within the Nb–Ir interaction. When irradiated with visible light above 400 nm, a reductive elimination of dihydrogen occurs, which generates new species with very short Nb–Ir interactions. This photochemical transformation, absent in the Ta–Ir analogues, not only highlights a divergence in group-5 reactivity, but also unlocks substitution reactivity at the remaining dimethylamido ligand, which is inert in the parent complex.
Introduction
Early/late heterobimetallic complexes have seen a surge in popularity in recent years, in part due to their inherent asymmetry and unique bonding.1–12 This asymmetry lends itself nicely to unusual bond activations across the metals in a variety of mechanisms, opening avenues for new reactivity. Among these systems, iridium-containing heterobimetallic species have emerged as particularly effective,13–16 especially in C–H activation chemistry (Scheme 1).14–16 Related studies by our group on Ta–Ir and Mo–Ir species, as well as work by Suzuki on Zr–Ir systems, highlight how polarized early/late M–Ir bonds can engage in cooperative C–H activation.17–23 Building on these findings, our group recently developed efficient H/D exchange catalysts, with the Ta–Ir complex emerging as one of the most effective.23,24 | ||
| Scheme 1 Representative structures of multiple-bonded M–Ir species (M = Zr, Ta, Mo) reported in the literature that mediate C–H activation across the metal–metal bond. | ||
Building on the reactivity of the Ta–Ir system, we chose to turn our attention to its lighter congener, niobium. Although Nb is often regarded as chemically analogous to Ta, systematic comparisons of their organometallic complexes remain surprisingly scarce. Yet, differences do exist. A key distinction lies in redox behaviour: Nb is generally more reduction-prone, a trend that is exploited in Ta/Nb separation chemistry.25,26 In addition, the radial extension of 5d (Ta) versus 4d (Nb) orbitals imparts subtle but important differences in bonding, especially when looking at multiple M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) E (E = main group element) π-bonded systems.27,28 The latter enable notable stoichiometric and catalytic transformations through 1,2-addition and cycloaddition pathways, with each metal (Ta vs. Nb) outperforming the other in different cases.29–35
E (E = main group element) π-bonded systems.27,28 The latter enable notable stoichiometric and catalytic transformations through 1,2-addition and cycloaddition pathways, with each metal (Ta vs. Nb) outperforming the other in different cases.29–35
We therefore thought niobium to be an apt follow-up to this research as niobium based heterobimetallic complexes have long been underexplored in the literature (Scheme 2). Few Nb–M interactions have been reported,36–39 typically exhibiting intermetallic distances that are slightly longer than the sum of covalent radii (as represented by the Formal Shortness Ratio, FSR, defined as the ratio between the MM′ distance and the sum of MM′ covalent radii).40,41 Although some transition-metal pairs with FSR values slightly greater than one still exhibit clear evidence of metal–metal single bonding, these metal–metal distances may also result from the presence of bridging ligands. Multiple bonding between Nb and other transition metals remains, however, an exceptionally rare phenomenon. To our knowledge, the landmark phosphinamide supported Nb–Fe triple bond reported by Thomas in 2017 remains the only Nb heterobimetallic multiply bonded species that has been crystallographically characterized.39 Herein we report a family of Nb–Ir complexes which exhibit short inter-metal atom distances which we attribute to a multiple bonding character. Irradiation with visible light causes a reductive elimination of dihydrogen and a significant structural rearrangement. Further reactivity can be observed with the new photo-activated species where the parent complex was inert to substitution.
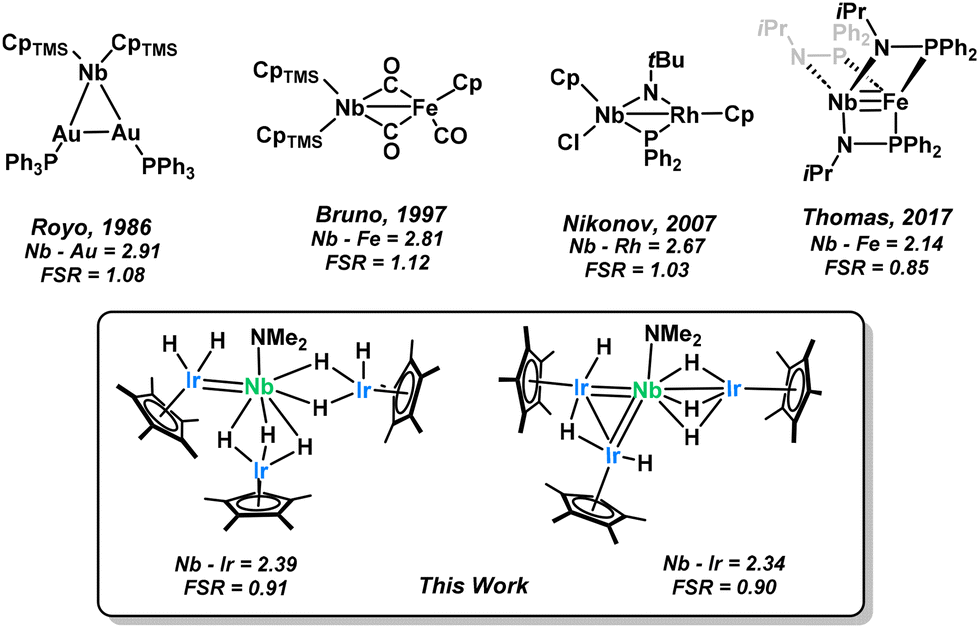 | ||
| Scheme 2 Niobium-based heterobimetallic complexes reported in the literature tend to have Nb–M distances longer than the sum of covalent radii, as represented by the FSR of the Nb–M interaction.36–39 | ||
Results and discussion
The synthesis of complex (Cp*IrH2)(Cp*IrH3)2Nb(NMe2), 1, follows the procedure developed in our research group exploiting the Brønsted acidity of the tetrahydridopentamethylcyclopentadienyliridium(V) complex (Cp*IrH4) with a basic amido complex, in this case, pentakis(dimethylamido)niobium(V).18,42 The reaction readily occurs at room temperature to yield the triiridoniobium(V) complex (Scheme 3), 1 in 86% isolated yield. 1H NMR spectroscopy in C6D6 solution reveals three sharp diamagnetic resonances at 3.31, 2.16 and −13.50 ppm corresponding to the dimethylamido, the Cp* ligands and 8 hydrides respectively (Fig. S1). We attribute the simplicity of the spectrum to some fluxionality of the Cp* and hydride positions, as observed in similar asymmetric Cp*IrHx complexes.43,44 Variable temperature NMR does not reveal any deconvolution of this hydride signal up to −35 °C (Fig. S2). | ||
| Scheme 3 The synthesis of the niobium iridium complexes. | ||
Diffuse reflectance infrared Fourier transform (DRIFT) spectroscopy reveals νC–H stretches at 3025–2724 and several bands in the 2256–1831 cm−1 range (Fig. S26), that we attribute to the presence of both terminal and bridging hydrides. In monometallic iridium complexes, terminal Ir–H bands typically appear in the 2100–1800 cm−1 region, while bridging hydrides generally fall below 1700 cm−1.45 Representative examples include the terminal Ir–H stretches in Cp*IrH4 (2150 cm−1),46 Cp*Ir(CO)H2 (2131 cm−1)47 and Cp*Ir(PMe3)(CH3)H (2090 cm−1).48,49 The homodimer [Cp*IrH3]2 exhibits both a terminal Ir–H stretching vibration at 2120 cm−1 and broad μ-H stretches at 1190 and 1120 cm−1 while complex {[Cp*Ir]2(μ-H)3}{PF6} is reported to have a μ-H band at 936 cm−1.46,50 However, in heterobimetallic systems, especially those featuring asymmetric three-center–two-electron (3c–2e) Ir–H(μ)–M bridges, the M–H stretching frequencies can deviate from this typical range. Both the asymmetry in the Ir–H–M bridge together with coupling between Ir–(μ-H) modes and other metal–ligand modes, can lead to frequency shifts.51 A relevant illustration is provided by the Ta–Ir and Hf–Ir complexes: the Ta–Ir species [Ta(CH2tBu)3IrH2Cp*] bearing terminal hydrides displays a ν(Ir–H) band at 2056 cm−1, while the Hf–Ir analogue [Hf(CH2tBu)3(μ-H)3IrCp*], which contains three bridging hydrides in a three-legged piano-stool geometry, shows a shifted ν(Ir–H) band at 1982 cm−1.22,52 Although this shift is significant, it is less pronounced than that typically observed for μ-H stretches in diiridium systems. For comparison, compounds [Cp*Ir(μ-H)3{Ln(η5-C5Me4SiMe3)2}] Ln = Y, Dy, Lu, in which the two metal centers are bridged by three hydride ligands, exhibit metal–hydride stretches at 1990, 1988, and 1994 cm−1, respectively.53 The complexity of the IR pattern for complex 1 is thus in agreement with the literature and the distribution of Ir–H environments in this compound.
Given the stoichiometry of the reaction, we assume that four dimethylamido ligands from Nb(NMe2)5 each abstract an iridium–hydride to give eight hydrides left on complex 1 and 4 equivalents of dimethylamine are released, analogous to the synthesis of tantalum iridium complexes well studied by our group from perhydrocarbyl Ta precursors.22,52 No further substitution of the final dimethylamido ligand could be obtained with excess Cp*IrH4 even under extended reaction times (7 days) nor elevated temperatures (75 °C), a result also seen with the Ta congeners.
In investigating the initial substitution chemistry of Nb(NMe2)5 with Cp*IrH4, the synthesis of bimetallic complexes with lower Nb![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ir stoichiometries was also attempted. Specially, the formation of species from 1:1 (NbIr) and 1:2 (NbIr2) stoichiometries could be observed by NMR spectroscopy (Fig. S6) but the resulting product distributions were very difficult to control. A significant concentration of 1, NbIr2 and NbIr were observed in the reactions aiming for lower stoichiometries even when the reactions were performed with slow addition of reagents (dropwise over 5 minutes), reduced temperature (−40 °C) or done in dilute (0.7 mM) pentane solutions. The purification of the materials was also very challenging as they coprecipitate from a saturated pentane solution and could never be cleanly isolated for characterization, despite repeated crystallization attempts. The reaction between complex 1 and Nb(NMe2)5 (2 equiv. or excess) was also attempted as an alternative route to access lower-nuclearity species, but no reactivity was observed (up to 100 °C) between the two reagents (Fig. S7). Therefore, these products were not explored further.
:Ir stoichiometries was also attempted. Specially, the formation of species from 1:1 (NbIr) and 1:2 (NbIr2) stoichiometries could be observed by NMR spectroscopy (Fig. S6) but the resulting product distributions were very difficult to control. A significant concentration of 1, NbIr2 and NbIr were observed in the reactions aiming for lower stoichiometries even when the reactions were performed with slow addition of reagents (dropwise over 5 minutes), reduced temperature (−40 °C) or done in dilute (0.7 mM) pentane solutions. The purification of the materials was also very challenging as they coprecipitate from a saturated pentane solution and could never be cleanly isolated for characterization, despite repeated crystallization attempts. The reaction between complex 1 and Nb(NMe2)5 (2 equiv. or excess) was also attempted as an alternative route to access lower-nuclearity species, but no reactivity was observed (up to 100 °C) between the two reagents (Fig. S7). Therefore, these products were not explored further.
Single crystals of 1 appropriate for X-ray diffraction could be grown from a saturated pentane solution (Fig. 1). The Nb1–Ir2 bond length (2.3973(6) Å, FSR = 0.91) is considerably shorter than the other Nb–Ir distances (2.6836(7) and 2.6901(4) Å, FSR = 1.03) and with a Formal Shortness Ratio below unity. We conclude that this is due to some niobium–iridium multiple bonding which we have represented here as a double bond for clarity. The long Nb–Ir interactions exhibit an FSR over one, suggestive of bridging hydrides between the metal centers rather than any formal Nb–Ir interaction. Hydrides adjacent to heavy transition metal centers are known to be very difficult to locate from X-ray diffraction data,54 however they can be qualitatively inferred from the Cp*–Ir–Nb angle, as used with great success in the literature.55 The Cp*centroid–Ir3–Nb angle of 176.2° suggests three hydrides bridges between the two metal centers. Conversely, we assign the Cp*centroid–Ir2–Nb (152°) and Cp*centroid–Ir1–Nb (141°) angles as indicative of the presence of one or two terminal hydrides on the Ir centers. These structures can be compared to the (Cp*IrH2)(Cp*IrH3)TaNp2(Np = neopentyl) structure previously reported by our group.24 The Ta–Ir distances (2.4291(9)–2.5891(9) Å, FSR = 0.93–0.99) also show this Cp*IrHx asymmetry with computational results confirming that a terminal hydride accounts for the non linearity of the Cp*centroid–Ir–Nb angle. From the attempted syntheses of lower stoichiometries of Nb and Ir, one X-ray quality crystal of NbIr2 could be obtained (Fig. S35). This structure also exhibits a low FSR on one of the Nb–Ir interactions but it is longer than that in 1 (Nb–Ir = 2.4284(6) Å, FSR = 0.93). As discussed above, this complex was not fully characterized and will not be discussed further in this article.
 | ||
| Fig. 1 Single crystal X-ray diffraction structures of complexes 1 (left) and 2 (right). Hydrogens are removed and the 2,6-diphenylphenol ligand is truncated for clarity; thermal ellipsoids are shown at 50% probability. Structural parameters (distances in Å and angles in °) for complex 1: Nb1–N1 = 1.982(3), Nb1–Ir1 = 2.6836(7) (FSR = 1.03), Nb1–Ir2 = 2.3973(6) (FSR = 0.91), Nb1–Ir3 = 2.6901(4) (FSR = 1.03), <Cp*Centroid–Ir1–Nb1 = 141.10, <Cp*Centroid–Ir2–Nb1 = 152.17, <Cp*Centroid–Ir3–Nb1 = 176.2, τ4 = 0.96. Structural parameters for complex 2: Nb1–O1 = 1.971(6), Nb1–Ir1 = 2.632(1) (FSR = 1.01), Nb1–Ir2 = 2.367(1) (FSR = 0.91), Nb1–Ir3 = 2.665(4) (FSR = 1.02), <Cp*Centroid–Ir1–Nb1 = 141.10, <Cp*Centroid–Ir2–Nb1 = 152.17, <Cp*Centroid–Ir3–Nb1 = 176.2, τ4 = 0.96. | ||
Much to our disappointment, complex 1 does not react with classical small molecules such as alkenes (vinyltrimethylsilane), alkynes (phenylacetylene and diphenylacetylene), heteroallenes (CO2, tBuNCO), carbon monoxide nor donor ligands like pyridine or N-heterocyclic carbenes (IMes). Attempts to abstract the dimethylamido ligand with strong Lewis acids (B(C6F5)3) led to decomposition of the material. Under a D2 atmosphere at 50 °C in the dark, complex 1 remains stable for several days. However, 1H and 2H NMR monitoring show no evidence of deuterium incorporation at either the hydride or Cp* positions under these conditions. Upon further heating, decomposition to unidentified products occurs, and deuterium exchange remains inefficient. These observations suggest that hydride ligand exchange with D2 does not take place under the tested conditions. The only substitution reactivity that could be observed with complex 1 was the protonolysis of the dimethylamido ligand with 2,6-diphenylphenol which cleanly liberates one equivalent of dimethylamine to generate the phenoxide substituted analogous complex, 2 (Scheme 3), in excellent yield (96%). Note that this protonolysis is pKa dependant as weaker acids (tris(tert-butoxy)silanol, pKa, MeCN, calculated = 34) does not show the same reactivity.56 Complex 2 is extremely light sensitive with even ambient light being enough to cleanly convert it into a new species (complex 4, see below), therefore 2 must be handled in the dark. Despite this challenge, 2 could be spectroscopically characterized before significant conversion. As expected, signals arising from the new diphenylphenoxide ligand can be observed in the 1H and 13C NMR spectra, with all resonances appropriately assigned (Fig. S8–S12). The reaction is quite selective with quantitative yields and no indication of hydride elimination to yield dihydrogen. A crystal suitable for X-ray crystallography could be grown from a saturated pentane solution cooled to −40 °C. The solid-state structure, shown in Fig. 1, is very similar to that for 1, with a short Nb1–Ir2 multiple bond (2.367(1) Å, FSR = 0.91 vs. 2.3973(6) Å, FSR = 0.91 for complex 1) and two [Cp*IrH3]− fragments with Nb–Ir bond lengths longer than the sum of atomic radii. While the light sensitivity is also observed with 2 in the solid-state, when the vial containing the material is tightly wrapped with aluminum foil, complex 2 is stable in the glovebox over several weeks.
Complex 1 is also sensitive to light, although less so, necessitating a more intense light source and longer reaction times. Exposing a C6D6 solution of 1 (approx. 17 mM) to full spectrum light (250 nm–1000 nm) from an unfiltered 300 W xenon light source (see SI for details) causes a darkening of the solution with total consumption of 1 in 18 h of exposure. This reaction can also be performed in pentane using thick wall glass Schlenk flasks without significant slowing of the reaction. The UV-Vis absorption profile of complex 1 is dominated by bands in the UV region with only one absorption band near the visible region at 389 nm (Fig. S32). This absorption band is very broad, extending into the visible region where the absorption begins at about 470 nm. Interestingly, when using a 400 nm longpass filter on the light source during the experiment, the reaction still readily occurs in the same period of time. This result confirms the reaction is visible-light mediated and likely due to the broad absorption centered at 389 nm.
The 1H NMR signals for the Cp* ligands of 3 in C6D6 appeared as two singlets at 2.32 ppm (integrating for 30H) and 1.98 ppm (integrating for 15H, Fig. S13), showing a distinct chemical environment of the Cp* ligands post-photolysis. The hydride signal shifted to −13.03 ppm, became very broad (1.5 ppm linewidth at half-height); and decreased in intensity from 8H in 1 to 6H in 3. Released dihydrogen can be observed from the reaction at 4.46 ppm through reaction monitoring via1H NMR in a tube completely full of C6D6 (see Fig. S15). Performing the 1H NMR spectroscopy experiment at −35 °C in d8-toluene allows some deconvolution of the hydride signal which splits into two broad singlets, each integrating for 3H, at −11.85 and −13.90 ppm (see Fig. S16). It is clear that, while the Nb–Ir interactions of 1 interconverted at room temperature, 3 has locked these positions at lower temperature, giving distinct environments for the two types of Nb–Ir interactions. UV-Vis spectroscopy on 3 shows a marked difference after photolysis (Fig. S33): the band at 389 nm has now broadened even further with absorbance starting at 600 nm.
Single crystals (grown from a saturated pentane solution cooled to −40 °C) suitable for X-ray diffraction reveal the new complex has undergone a significant structural change as a result of the reductive elimination of H2 (Fig. 2). Perhaps most obvious is the bending of Ir1 and Ir2 towards one another. Given the long distance between the two metal centers (2.7633(7) Å, FSR = 1.09), we propose that such structural rearrangement is driven by hydride ligands bridging the two iridium centers. Notably, the observed Ir–Ir distance closely matches those reported for other hydride-bridged polynuclear iridium clusters in the literature.57 A similar photo-induced structural change was also reported recently on polyhydride osmium–actinide multimetallic complexes.58 This change in geometry is reflected in the decrease in τ4 value from 0.96 in complex 1 to 0.85 in 3 indicative of a distorted tetrahedral geometry. The Nb–Ir2 distance has slightly shortened to 2.3479(7) Å (FSR = 0.90) with Nb–Ir1 showing the largest change from 2.6836(7) to 2.5620(7) Å (FSR in complex 1 = 1.03 to 0.98 in 3).
 | ||
| Fig. 2 Single crystal X-ray diffraction structures of complexes 3 (left) and 4 (right) with thermal ellipsoids at 50% and hydrogens and phenyl rings from the 1,6-diphenylphenoxy ligand removed for clarity. Relevant structural parameters (distances in Å and angles in °) for complex 3: Nb1–N1 = 1.996(6), Nb1–Ir1 = 2.5619(7) (FSR = 0.98), Nb1–Ir2 = 2.3477(7) (FSR = 0.90), Nb1–Ir3 = 2.6832(7) (FSR = 1.03), Ir1–Ir2 = 2.7633(4) (FSR = 1.09), <Cp*Centroid–Ir1–Nb1 = 136.24, <Cp*Centroid–Ir2–Nb1 = 154.77, <Cp*Centroid–Ir3–Nb1 = 178.02, τ4 = 0.85. Relevant structural parameters for complex 4: Nb1–O1 = 1.978(3), Nb1–Ir1 = 2.4395(4) (FSR = 0.93), Nb1–Ir2 = 2.4266(5) (FSR = 0.93), Nb1–Ir3 = 2.6598(6) (FSR = 1.02), Ir1–Ir2 = 2.8921(4) (FSR = 1.15), <Cp*Centroid–Ir1–Nb1 = 146.85, <Cp*Centroid–Ir2–Nb1 = 149.35, <Cp*Centroid–Ir3–Nb1 = 178.53, τ4 = 0.89. | ||
Addition of dihydrogen (1 bar) to complex 3 does not regenerate 1 even under prolonged reaction times, more elevated temperatures (75 °C), or light irradiation. The light sensitivity of these Nb–Ir species is especially intriguing, since this behaviour was not observed with the Ta–Ir congeners previously prepared in our group.15,22,24,52 Although examples of niobium-based photochemistry are rare,59,60 one particularly relevant precedent is a trihydridobis(cyclopentadienyl) niobium(V) complex which releases one equivalent of H2 upon irradiation from a xenon lamp.61,62 The authors of this work suggested a reductive elimination with oxidation state change from Nb(V) to Nb(III), a phenomenon documented in other high-valent Nb polyhydrides.63–65 Accordingly, we propose that the divergent reactivity between the Nb–Ir and the Ta–Ir systems arises from the easier reduction of Nb relative to Ta which predisposes the system to photochemical H2 elimination.25,26 Note that other heterobimetallic transition metal complexes based on a polyhydrido(pentamethylcyclopentadienylosmium(VI)) (Cp*OsHx) fragment have shown a photolytic dihydrogen elimination.58 Finally, the reducing character of the Cp*IrHx motif has also emerged in our recent work on Fe–Ir species, where Fe(III) precursors consistently yielded Fe(II)–Ir clusters, albeit not initiated by light.66
Addition of 1 equivalent of diphenylphenol to complex 3 generates complex 4 in quantitative yield (Scheme 3). This compound spectroscopically matches the product obtained when a C6D6 solution of 2 is intentionally exposed to ambient light. A crystal suitable for single crystal X-ray crystallography could be grown from a saturated pentane solution of 4 cooled to −40 °C. As anticipated, this crystal reveals a structure similar to that of 3 (Fig. 2) with two short Nb–Ir interactions (2.43955(4) Å, FSR = 0.93 and 2.4266(5) Å, FSR = 0.93) and one long (2.6598(6) Å, FSR = 1.02). The Ir1–Ir2 distance is perhaps the largest difference between these structures (Ir1–Ir2 = 2.8921(2) Å, FSR = 1.15 in 4versus 2.7633(4) Å, FSR 1.09 in 3) accompanied by the change in Cp*centroid–Ir1–Ir2–Cp*centroid dihedral angle to 4°, contrasted with the Cp*centroid–Ir1–Ir2–Cp*centroid dihedral angle in 3 of 87°. This marked change in Ir–Ir distance and dihedral angle may be explained by the steric demands of a diphenylphenoxide ligand.
After the loss of H2, complex 3 now exhibits reactivity at the dimethylamido ligand bound to niobium, unlike complex 1. Exposure of 3 with one equivalent of Cp*IrH4 very quickly generates the new tetrairidium complex, 5 (Scheme 4), in quantitative yield by NMR but only 75% after recrystallization as the complex is quite soluble in pentane. Complex 5 exhibits two sharp Cp* signals in 1H NMR, each integrating for 30H, along with a broad hydride signal at −14.17 ppm integrating for 9H (Fig. S22). Crystals appropriate for single-crystal X-ray diffraction could be obtained from a saturated pentane solution cooled to −40 °C. The solid-state structure (Fig. 3) shows the expected substitution of the [NMe2]− group by a [Cp*IrH3]− moiety, featuring an Ir–Nb distance of 2.6297(5) Å. This Cp*IrHx fragment shows a Cpcentroid–Ir4–Nb angle of 135(1)°, indicative of some terminal hydrides pointing away from the Nb center. This substitution is accompanied by a substantial geometric change, represented by the Ir1–Ir2 bond length, which expands from 2.7633(7) Å in complex 3 to 2.9383(7) Å (FSR 1.16) in complex 5. Similarly, the Cp*centroid–Ir1–Ir2–Cp*centroid dihedral angle falls from 87° in 3 to 1° in 5. Like in complex 4, this change again perhaps stems from the steric bulk of the fourth Cp*IrH3 group which pushes the iridium centers apart resulting in a cis orientation of the Cp*IrHx fragments. Exposing complex 5 to the same photolysis conditions described above do not cause another loss of dihydrogen although some decomposition is observed.
 | ||
| Scheme 4 Complex 3 exhibits protonolysis and insertion reactivity at the remaining dimethylamido ligand. This is exemplified by the synthesis of complexes 5 and 6. | ||
 | ||
| Fig. 3 Single crystal X-ray diffraction structures of complexes 5 (left) and 6 (right). Thermal ellipsoids set at 50% and hydrogens removed for clarity. Relevant structural parameters (distances in Å and angles in °) for complex 5: Nb1–Ir1 = 2.3709(3)(FSR = 0.91), Nb1–Ir2 = 2.4306(2) (FSR = 0.93), Nb1–Ir3 = 2.5799(3) (FSR = 0.99), Nb1–Ir4 = 2.6297(5) (FSR = 1.01), Ir1–Ir2 = 2.9383(3) (FSR = 1.16), <Cp*Centroid–Ir1–Nb1 = 151.08, <Cp*Centroid–Ir2–Nb1 = 151.25, <Cp*Centroid–Ir3–Nb1 = 175.69, <Cp*Centroid–Ir3–Nb1 = 134.99. τ4 = 0.86. Relevant structural parameters for complex 6: Nb1–O = 2.197(7)–2.198(7), Nb1–Ir1 = 2.347(1) (FSR = 0.90), Nb1–Ir2 = 2.5715(8) (FSR = 0.98), Nb1–Ir3 = 2.632(1) (FSR = 1.01), Ir1–Ir2 = 2.7527(6) (FSR = 1.09), <Cp*Centroid–Ir1–Nb1 = 153.16, <Cp*Centroid–Ir2–Nb1 = 139.49, <Cp*Centroid–Ir3–Nb1 = 147.25. | ||
Carbon dioxide (CO2) also inserts into the dimethylamido group of complex 3 to generate complex 6 (Scheme 4) in quantitative yields. However, the complex is very soluble in pentane, leading to a small recovered yield of 45%. The 1H NMR spectrum of this complex, recorded in C6D6 solution, exhibits four resonances, two for the inequivalent Cp* ligands (integrating for 15 H and 30 H respectively, as expected), a signal at 2.36 ppm for the dimethylcarbamato ligand integrating for 6H, and a hydride signal at −12.66 ppm integrating for 6H (Fig. S24). 13C NMR data confirms the insertion of CO2 into the dimethylamido ligand with the characteristic carbamato resonance found at 163 ppm (Fig. S25). DRIFT spectroscopy confirms the presence of the carbamate moiety, with a diagnostic CO stretching frequency at 1574 cm−1. Single crystals appropriate for X-ray diffraction could be grown from a saturated pentane solution of 6 cooled to −40 °C. The solid-state structure (Fig. 3) confirms the insertion of CO2 into the dimethylamido ligand. The Nb–O bond lengths of 2.197(7)–2.198(7) Å are within literature expectation and the Nb–Ir lengths are not altered significantly from those in complex 3 with only a moderate shortening of the Nb1–Ir3 bond length (from 2.6832(7) Å in complex 3 to 2.632(1) Å in 6).33,67 This is perhaps due to the change in Cp*centroid–Nb1–Ir3 angle which was almost linear in complex 3, moving to 147.25° in complex 6. We attribute this to the flexibility of this ligand architecture and a change in the hydride coordination mode, whereby the Ir hydrides can reorient to accommodate the coordination of the κ2–carbamato ligand. Addition of another equivalent or excess CO2 leads to rapid decomposition of the complex.
Conclusions
We have described herein a family of heterobimetallic niobium–iridium complexes which exhibit Nb–Ir intermetallic distances much shorter than the sum of atomic radii. Examples of Nb–M multiple bonding are extremely rare, and this research significantly expands the library of known examples. The Cp*IrHx fragment plays a central role in stabilizing these architectures by adapting its coordination mode. Besides shifting hydrides coordination from terminal to bridging, steric pressure can be alleviated through the lengthening of Ir–Ir interactions and Cp* bending. This inherent flexibility enables structural reorganization under photochemical conditions. The light sensitivity of compounds 1 and 2 is particularly striking, given that previously reported heterobimetallic complexes using the same Cp*IrHx motif show no photo-reorganization. These results thus highlight that the unique architecture of these niobium derivatives is needed for this reactivity to occur. Ongoing studies aim to explore how this photoreactivity can be exploited in catalysis, while future work will also target the use of these Nb–Ir complexes as versatile precursors for heterogeneous catalyst development.Conflicts of interest
There are no conflicts of interest to declare.Data availability
The additional data supporting this article have been included as part of the supplementary information (SI). Supplementary information: experimental procedures, NMR, DRIFT and UV-visible spectra. See DOI: https://doi.org/10.1039/d5dt02432c.CCDC 2487215–2487221 contain the supplementary crystallographic data for this paper.68a–g
Acknowledgements
Funded by the European Union (ERC StG DUO, 101041762). Views and opinions expressed are however those of the authors only and do not necessarily reflect those of the European Union or the European Research Council. Neither the European Union nor the granting authority can be held responsible for them.References
- J. Campos, Bimetallic Cooperation across the Periodic Table, Nat. Rev. Chem., 2020, 4(12), 696–702, DOI:10.1038/s41570-020-00226-5.
- A. M. Baranger and R. G. Bergman, Cooperative Reactivity in the Interactions of X-H Bonds with a Zirconium-Iridium Bridging Imido Complex, J. Am. Chem. Soc., 1994, 116(9), 3822–3835, DOI:10.1021/ja00088a019.
- M. J. Hostetler and R. G. Bergman, Synthesis and Reactivity of Cp2Ta(CH2)2Ir(CO)2: An Early-Late Heterobimetallic Complex That Catalytically Hydrogenates, Isomerizes and Hydrosilates Alkenes, J. Am. Chem. Soc., 1990, 112(23), 8621–8623, DOI:10.1021/ja00179a078.
- Y. Zhang, S. P. Roberts, R. G. Bergman and D. H. Ess, Mechanism and Catalytic Impact of Ir–Ta Heterobimetallic and Ir–P Transition Metal/Main Group Interactions on Alkene Hydrogenation, ACS Catal., 2015, 5(3), 1840–1849, DOI:10.1021/cs501884j.
- V. N. Setty, W. Zhou, B. M. Foxman and C. M. Thomas, Subtle Differences Between Zr and Hf in Early/Late Heterobimetallic Complexes with Cobalt, Inorg. Chem., 2011, 50(10), 4647–4655, DOI:10.1021/ic200445x.
- N. H. Hunter and C. M. Thomas, Polarized Metal–Metal Multiple Bonding and Reactivity of Phosphinoamide-Bridged Heterobimetallic Group IV/Cobalt Compounds, Dalton Trans., 2024, 53(38), 15764–15781, 10.1039/D4DT02064B.
- A. Lachguar, I. Del Rosal, L. Maron, E. Jeanneau, L. Veyre, C. Thieuleux and C. Camp, π-Bonding of Group 11 Metals to a Tantalum Alkylidyne Alkyl Complex Promotes Unusual Tautomerism to Bis-Alkylidene and CO2 to Ketenyl Transformation, J. Am. Chem. Soc., 2024, 146(27), 18306–18319, DOI:10.1021/jacs.4c02172.
- K. M. Gramigna, D. A. Dickie, B. M. Foxman and C. M. Thomas, Cooperative H2 Activation across a Metal–Metal Multiple Bond and Hydrogenation Reactions Catalyzed by a Zr/Co Heterobimetallic Complex, ACS Catal., 2019, 9(4), 3153–3164, DOI:10.1021/acscatal.8b04390.
- R. Srivastava, R. Moneuse, J. Petit, P.-A. Pavard, V. Dardun, M. Rivat, P. Schiltz, M. Solari, E. Jeanneau, L. Veyre, C. Thieuleux, E. A. Quadrelli and C. Camp, Early/Late Heterobimetallic Tantalum/Rhodium Species Assembled Through a Novel Bifunctional NHC-OH Ligand, Chem. – Eur. J., 2018, 24(17), 4361–4370, DOI:10.1002/chem.201705507.
- R. Srivastava, E. A. Quadrelli and C. Camp, Lability of Ta–NHC Adducts as a Synthetic Route towards Heterobimetallic Ta/Rh Complexes, Dalton Trans., 2020, 49(10), 3120–3128, 10.1039/D0DT00344A.
- K. Kawakita, Y. Kakiuchi, E. P. Beaumier, I. A. Tonks, H. Tsurugi and K. Mashima, Synthesis of Pyridylimido Complexes of Tantalum and Niobium by Reductive Cleavage of the NN Bond of 2,2′-Azopyridine: Precursors for Early–Late Heterobimetallic Complexes, Inorg. Chem., 2019, 58(22), 15155–15165, DOI:10.1021/acs.inorgchem.9b02043.
- M. L. Maiola and J. A. Buss, Accessing Ta/Cu Architectures via Metal-Metal Salt Metatheses: Heterobimetallic C–H Bond Activation Affords μ-Hydrides, Angew. Chem., Int. Ed., 2023, 62(48), e202311721, DOI:10.1002/anie.202311721.
- A. M. Baranger, T. A. Hanna and R. G. Bergman, Transfer of Oxygen and Sulfur from Organic Molecules to a Zr-Ir Bond. Evidence for an Unusually Rapid Atom Abstraction Reaction, J. Am. Chem. Soc., 1995, 117(40), 10041–10046, DOI:10.1021/ja00145a015.
- M. Oishi, M. Kino, M. Saso, M. Oshima and H. Suzuki, Early–Late Heterobimetallic Complexes with a Ta–Ir Multiple Bond: Bimetallic Oxidative Additions of C–H, N–H, and O–H Bonds, Organometallics, 2012, 31(13), 4658–4661, DOI:10.1021/om300429u.
- A. Lachguar, C. Z. Ye, S. N. Kelly, E. Jeanneau, I. D. Rosal, L. Maron, L. Veyre, C. Thieuleux, J. Arnold and C. Camp, CO2 Cleavage by Tantalum/M (M = Iridium, Osmium) Heterobimetallic Complexes, Chem. Commun., 2024, 60(61), 7878–7881, 10.1039/D4CC02207F.
- K. Yamamoto, K. Higashida, H. Nagae, H. Tsurugi and K. Mashima, Synthesis and Characterization of Heterobimetallic Tantalum–Rhodium and Tantalum–Iridium Complexes Connected by a Tantalacyclopentadiene Fragment, Helv. Chim. Acta, 2016, 99(11), 848–858, DOI:10.1002/hlca.201600180.
- A. Lachguar, A. V. Pichugov, T. Neumann, Z. Dubrawski and C. Camp, Cooperative Activation of Carbon–Hydrogen Bonds by Heterobimetallic Systems, Dalton Trans., 2024, 53(4), 1393–1409, 10.1039/D3DT03571A.
- Z. Dubrawski, I. D. Rosal, E. Jeanneau, L. Maron, C. Thieuleux and C. Camp, Synergistic C–H Bond Activation across Molybdenum–Iridium Multiply Bonded Complexes: A Cascade of Transformations, Chem. Sci., 2025, 16(32), 14564–14577, 10.1039/D5SC03465E.
- I. D. Rosal, S. Lassalle, C. Dinoi, C. Thieuleux, L. Maron and C. Camp, Mechanistic Investigations via DFT Support the Cooperative Heterobimetallic C–H and O–H Bond Activation across TaIr Multiple Bonds, Dalton Trans., 2021, 50(2), 504–510, 10.1039/D0DT03818K.
- M. Oishi and H. Suzuki, Zirconocene–Iridium Hydrido Complexes: Arene Carbon–Hydrogen Bond Activation and Formation of a Planar Square Zr2Ir2 Complex, Inorg. Chem., 2009, 48(6), 2349–2351, DOI:10.1021/ic8020113.
- M. Oishi, M. Oshima and H. Suzuki, A Study on Zr–Ir Multiple Bonding Active for C–H Bond Cleavage, Inorg. Chem., 2014, 53(13), 6634–6654, DOI:10.1021/ic500258g.
- S. Lassalle, R. Jabbour, P. Schiltz, P. Berruyer, T. K. Todorova, L. Veyre, D. Gajan, A. Lesage, C. Thieuleux and C. Camp, Metal–Metal Synergy in Well-Defined Surface Tantalum–Iridium Heterobimetallic Catalysts for H/D Exchange Reactions, J. Am. Chem. Soc., 2019, 141(49), 19321–19335, DOI:10.1021/jacs.9b08311.
- A. V. Pichugov, L. Escomel, S. Lassalle, J. Petit, R. Jabbour, D. Gajan, L. Veyre, E. Fonda, A. Lesage, C. Thieuleux and C. Camp, Highly Selective and Efficient Perdeuteration of N-Pentane via H/D Exchange Catalyzed by a Silica-Supported Hafnium-Iridium Bimetallic Complex, Angew. Chem., Int. Ed., 2024, 63(16), e202400992, DOI:10.1002/anie.202400992.
- S. Lassalle, R. Jabbour, I. Del Rosal, L. Maron, E. Fonda, L. Veyre, D. Gajan, A. Lesage, C. Thieuleux and C. Camp, Stepwise Construction of Silica-Supported Tantalum/Iridium Heteropolymetallic Catalysts Using Surface Organometallic Chemistry, J. Catal., 2020, 392, 287–301, DOI:10.1016/j.jcat.2020.10.016.
- M. H. Furigay, S. Chaudhuri, S. M. Deresh, A. B. Weberg, P. Pandey, P. J. Carroll, G. C. Schatz and E. J. Schelter, Selective Reduction of Niobium(V) Species to Promote Molecular Niobium/Tantalum Separation, Inorg. Chem., 2022, 61(1), 23–27, DOI:10.1021/acs.inorgchem.1c02976.
- L. G. Hubert-Pfalzgraf, Niobium & Tantalum: Inorganic & Coordination Chemistry, in Encyclopedia of Inorganic Chemistry, John Wiley & Sons, Ltd, 2006. DOI:10.1002/0470862106.ia153.
- A. B. Weberg, S. Chaudhuri, T. Cheisson, C. Uruburo, E. Lapsheva, P. Pandey, M. R. Gau, P. J. Carroll, G. C. Schatz and E. J. Schelter, Tantalum, Easy as Pi: Understanding Differences in Metal–Imido Bonding towards Improving Ta/Nb Separations, Chem. Sci., 2022, 13(23), 6796–6805, 10.1039/D2SC01926D.
- M. H. Furigay, S. Chaudhuri, C. Li, J. Zhou, P. Pandey, R. F. Higgins, H. Gupta, P. J. Carroll, M. R. Gau, J. M. Anna, G. C. Schatz and E. J. Schelter, Observing Similarities and Differences in the Properties of Isostructural Niobium(V)/Tantalum(V) Coordination Compounds with Strong Pi-Donor Ligands, Inorg. Chem., 2023, 62(47), 19238–19247, DOI:10.1021/acs.inorgchem.3c02514.
- F. R. Neururer, K. Huter, M. Seidl and S. Hohloch, Reactivity and Structure of a Bis-Phenolate Niobium NHC Complex, ACS Org. Inorg. Au, 2023, 3(1), 59–71, DOI:10.1021/acsorginorgau.2c00028.
- T. L. Gianetti, R. G. Bergman and J. Arnold, Carbon–Fluorine Bond Cleavage in Fluoroarenes via a Niobium(III) Imido Complex: From Stoichiometric to Catalytic Hydrodefluorination, Chem. Sci., 2014, 5(6), 2517–2524, 10.1039/C4SC00006D.
- T. R. Cundari, Methane Activation by Group VB Bis(Imido) Complexes, Organometallics, 1994, 13(8), 2987–2994, DOI:10.1021/om00020a014.
- A. H. Obenhuber, T. L. Gianetti, R. G. Bergman and J. Arnold, Regioselective [2 + 2] and [4 + 2] Cycloaddition Reactivity in an Asymmetric Niobium(Bisimido) Moiety towards Unsaturated Organic Molecules, Chem. Commun., 2014, 51(7), 1278–1281, 10.1039/C4CC07851A.
- J. I. Fostvedt, L. N. Grant, B. M. Kriegel, A. H. Obenhuber, T. D. Lohrey, R. G. Bergman and J. Arnold, 1,2-Addition and Cycloaddition Reactions of Niobium Bis(Imido) and Oxo Imido Complexes, Chem. Sci., 2020, 11(42), 11613–11632, 10.1039/D0SC03489D.
- N. C. Tomson, J. Arnold and R. G. Bergman, Halo, Alkyl, Aryl, and Bis(Imido) Complexes of Niobium Supported by the β-Diketiminato Ligand, Organometallics, 2010, 29(13), 2926–2942, DOI:10.1021/om1001827.
- J. I. Fostvedt, J. Mendoza, S. Lopez-Flores, D. Alcantar, R. G. Bergman and J. Arnold, Engendering Reactivity at Group 5-Heteroatom Multiple Bonds via π-Loading, Chem. Sci., 2022, 13(28), 8224–8242, 10.1039/D2SC02706B.
- M. Fajardo, M. P. Gómez-Sal, P. Royo, S. Martínez Carrera and S. García Blanco, Synthesis and Structural Characterisation of the Mixed-Metal Cluster Cation [Nb(H5-C5H4R)2{AuP(C6H5)3}2]+ with R = H or Si(CH3)3, J. Organomet. Chem., 1986, 312(2), c44–c46, DOI:10.1016/0022-328X(86)80310-2.
- B. Thiyagarajan, L. Michalczyk, V. G. Young and J. W. Bruno, Niobium–Mercury Heterometallic Compounds as Sources of Niobium(II): Radical Paths to Organoniobium Species, Organometallics, 1997, 16(26), 5884–5892, DOI:10.1021/om970779u.
- S. Leelasubcharoen, P. A. Zhizhko, L. G. Kuzmina, A. V. Churakov, J. A. K. Howard and G. I. Nikonov, Niobium/Rhodium Bimetallic Complexes: Synthesis, Structure, and Catalytic Hydrosilylation of Acetophenone and Benzaldehyde, Organometallics, 2009, 28(15), 4500–4506, DOI:10.1021/om900363r.
- G. Culcu, D. A. Iovan, J. P. Krogman, M. J. T. Wilding, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Heterobimetallic Complexes Comprised of Nb and Fe: Isolation of a Coordinatively Unsaturated NbIII/Fe0 Bimetallic Complex Featuring a Nb
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Fe Triple Bond, J. Am. Chem. Soc., 2017, 139(28), 9627–9636, DOI:10.1021/jacs.7b04151.
Fe Triple Bond, J. Am. Chem. Soc., 2017, 139(28), 9627–9636, DOI:10.1021/jacs.7b04151. - F. A. Cotton, Discovering and Understanding Multiple Metal-to-Metal Bonds, Acc. Chem. Res., 1978, 11(6), 225–232, DOI:10.1021/ar50126a001.
- Multiple Bonds Between Metal Atoms, ed. F. A. Cotton, C. A. Murillo and R. A. Walton, Springer US, Boston, MA, 2005. DOI:10.1007/b136230.
- Z. Dubrawski, B. Shearman, N. Soulé, E. Jeanneau, C. Thieuleux and C. Camp, Flash Communication: Well-Defined Paramagnetic Polyhydrido-Bridged Iron–Iridium Clusters, Organometallics, 2025, 44(19), 2151–2155, DOI:10.1021/acs.organomet.5c00301.
- L. Escomel, N. Soulé, E. Robin, I. Del Rosal, L. Maron, E. Jeanneau, C. Thieuleux and C. Camp, Rational Preparation of Well-Defined Multinuclear Iridium–Aluminum Polyhydride Clusters and Comparative Reactivity, Inorg. Chem., 2022, 61(15), 5715–5730, DOI:10.1021/acs.inorgchem.1c03120.
- C. Z. Ye, I. D. Rosal, M. A. Boreen, E. T. Ouellette, D. R. Russo, L. Maron, J. Arnold and C. Camp, A Versatile Strategy for the Formation of Hydride-Bridged Actinide–Iridium Multimetallics, Chem. Sci., 2023, 14(4), 861–868, 10.1039/D2SC04903A.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds. Part B: Applications in Coordination, Organometallic, and Bioinorganic Chemistry, Wiley, Hoboken, NJ, 2009 Search PubMed.
- T. M. Gilbert, F. J. Hollander and R. G. Bergman, (Pentamethylcyclopentadienyl)Iridium Polyhydride Complexes: Synthesis of Intermediates in the Mechanism of Formation of (Pentamethylcyclopentadienyl)Iridium Tetrahydride and the Preparation of Several Iridium(V) Compounds, J. Am. Chem. Soc., 1985, 107(12), 3508–3516, DOI:10.1021/ja00298a018.
- D. M. Heinekey, D. A. Fine, T. G. P. Harper and S. T. Michel, Dinuclear Dihydride Complexes of Iridium: A Study of Structure and Dynamics, Can. J. Chem., 1995, 73(7), 1116–1125, DOI:10.1139/v95-138.
- M. J. Wax, J. M. Stryker, J. M. Buchanan, C. A. Kovac and R. G. Bergman, Reversible Carbon-Hydrogen Bond Insertion/Reductive Elimination in (.Eta.5-Pentamethylcyclopentadienyl)(Trimethylphosphine)Iridium Complexes. Use in Determining Relative Metal-Carbon Bond Energies and Thermally Activating Methane, J. Am. Chem. Soc., 1984, 106(4), 1121–1122, DOI:10.1021/ja00316a054.
- T. M. Gilbert and R. G. Bergman, Synthesis of Trimethylphosphine-Substituted (Pentamethylcyclopentadienyl)Iridium Hydride Complexes; Protonation and Deprotonation of (Pentamethylcyclopentadienyl)(Trimethylphosphine)Iridium Dihydride, J. Am. Chem. Soc., 1985, 107(12), 3502–3507, DOI:10.1021/ja00298a017.
- C. White, A. J. Oliver and P. M. Maitlis, Pentamethylcyclopentadienyl-Rhodium and -Iridium Complexes. Part VII. Mono-, Di-, and Tri-µ-Hydrido-Complexes, J. Chem. Soc., Dalton Trans., 1973, 18, 1901–1907, 10.1039/DT9730001901.
- R. Fernández-Terán, J. Ruf and P. Hamm, Vibrational Couplings in Hydridocarbonyl Complexes: A 2D-IR Perspective, Inorg. Chem., 2020, 59(11), 7721–7726, DOI:10.1021/acs.inorgchem.0c00750.
- S. Lassalle, J. Petit, R. L. Falconer, V. Hérault, E. Jeanneau, C. Thieuleux and C. Camp, Reactivity of Tantalum/Iridium and Hafnium/Iridium Alkyl Hydrides with Alkyl Lithium Reagents: Nucleophilic Addition, Alpha-H Abstraction, or Hydride Deprotonation?, Organometallics, 2022, 41(13), 1675–1687, DOI:10.1021/acs.organomet.2c00158.
- Y. Takenaka and Z. Hou, Lanthanide Terminal Hydride Complexes Bearing Two Sterically Demanding C5Me4SiMe3 Ligands. Synthesis, Structure, and Reactivity, Organometallics, 2009, 28(17), 5196–5203, DOI:10.1021/om900453j.
- R. C. Stevens, M. R. Mclean, T. Wen, J. D. Carpenter, R. Bau and T. F. Koetzle, An X-Ray and Neutron Diffraction Structure Analysis of a Triply-Bridged Binuclear Iridium Complex, [[(C5(CH3)5Ir)2(μ-H)3]+ [ClO4]−˙ 2C6H6], Inorg. Chim. Acta, 1989, 161(2), 223–231, DOI:10.1016/S0020-1693(00)83097-2.
- T. Takao, S. Matsuzawa and M. Nagaoka, Synthesis of Bimetallic Dinuclear Hydrido Complexes of Re and Rh/Ir Supported by a Direct M–M′ Bond: The Role of the M–M′ Bond in the Site Exchange of Hydrides, Inorg. Chem., 2024, 63(46), 22214–22226, DOI:10.1021/acs.inorgchem.4c03837.
- F. Feige, L. A. Malaspina, E. Rychagova, S. Ketkov, S. Grabowsky, E. Hupf and J. Beckmann, Perfluorinated Trialkoxysilanol with Dramatically Increased Brønsted Acidity, Chem. – Eur. J., 2021, 27(64), 15898–15902, DOI:10.1002/chem.202103177.
- Y. Xu, M. A. Celik, A. L. Thompson, H. Cai, M. Yurtsever, B. Odell, J. C. Green, D. M. P. Mingos and J. M. Brown, Tetrameric Iridium Hydride-Rich Clusters Formed under Hydrogenation Conditions, Angew. Chem., Int. Ed., 2009, 48(3), 582–585, DOI:10.1002/anie.200804484.
- C. Z. Ye, I. D. Rosal, S. N. Kelly, I. J. Brackbill, L. Maron, C. Camp and J. Arnold, Photolysis-Driven Bond Activation by Thorium and Uranium Tetraosmate Polyhydride Complexes, Chem. Sci., 2024, 15(25), 9784–9792, 10.1039/D4SC02380C.
- A. C. Filippou, D. Hoffmann and G. Schnakenburg, Triple Bonds of Niobium with Silicon, Germaniun and Tin: The Tetrylidyne Complexes [(K3-Tmps)(CO)2NbE–R] (E = Si, Ge, Sn; Tmps = MeSi(CH2PMe2)3; R = Aryl), Chem. Sci., 2017, 8(9), 6290–6299, 10.1039/C7SC02708G.
- C. Camp, L. N. Grant, R. G. Bergman and J. Arnold, Photo-Activation of D0 Niobium Imido Azides: En Route to Nitrido Complexes, Chem. Commun., 2016, 52(32), 5538–5541, 10.1039/C6CC02081J.
- D. F. Foust, R. D. Rogers, M. D. Rausch and J. L. Atwood, Photoinduced Reactions of (.Eta.5-C5H5)2MH3 and (.Eta.5-C5H5)2M(CO)H (M = Nb, Ta) and the Molecular Structure of (.Eta.5-C5H5)2Ta(CO)H, J. Am. Chem. Soc., 1982, 104(21), 5646–5650, DOI:10.1021/ja00385a015.
- R. F. G. Baynham, J. Chetwynd-Talbot, P. Grebenik, R. N. Perutz and M. H. A. Powell, Photochemistry of M(H5-C5H5)2(H)CO and M(H5-C5H5)2H3 (M = Nb, Ta) in Low-Temperature Matrices, J. Organomet. Chem., 1985, 284(2), 229–242, DOI:10.1016/0022-328X(85)87214-4.
- T. L. Gianetti, G. Nocton, S. G. Minasian, N. C. Tomson, A. L. D. Kilcoyne, S. A. Kozimor, D. K. Shuh, T. Tyliszczak, R. G. Bergman and J. Arnold, Diniobium Inverted Sandwich Complexes with μ-H6:H6-Arene Ligands: Synthesis, Kinetics of Formation, and Electronic Structure, J. Am. Chem. Soc., 2013, 135(8), 3224–3236, DOI:10.1021/ja311966h.
- B. M. Kriegel, L. C. E. Naested, G. Nocton, K. V. Lakshmi, T. D. Lohrey, R. G. Bergman and J. Arnold, Redox-Initiated Reactivity of Dinuclear β-Diketiminatoniobium Imido Complexes, Inorg. Chem., 2017, 56(3), 1626–1637, DOI:10.1021/acs.inorgchem.6b02735.
- C. Camp, L. Maron, R. G. Bergman and J. Arnold, Activation of White Phosphorus by Low-Valent Group 5 Complexes: Formation and Reactivity of Cyclo-P4 Inverted Sandwich Compounds, J. Am. Chem. Soc., 2014, 136(50), 17652–17661, DOI:10.1021/ja5107282.
- Z. Dubrawski, B. Shearman, N. Soulé, E. Jeanneau, C. Thieuleux and C. Camp, Flash Communication: Well-Defined Paramagnetic Polyhydrido-Bridged Iron–Iridium Clusters, Organometallics, 2025, 44(19), 2151–2155, DOI:10.1021/acs.organomet.5c00301.
- P. B. Arimondo, F. Calderazzo, U. Englert, C. Maichle-Mössmer, G. Pampaloni and J. Strähle, Preparation and Characterization of Dialkylcarbamato Derivatives of Niobium and Tantalum, J. Chem. Soc., Dalton Trans., 1996, 3, 311–319, 10.1039/DT9960000311.
- (a) CCDC 2487215: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2ph4r3; (b) CCDC 2487216: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2ph4s4; (c) CCDC 2487217: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2ph4t5; (d) CCDC 2487218: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2ph4v6; (e) CCDC 2487219: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2ph4w7; (f) CCDC 2487220: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2ph4x8; (g) CCDC 2487221: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2ph4y9.
| This journal is © The Royal Society of Chemistry 2026 |