DOI:
10.1039/D5DT02243F
(Paper)
Dalton Trans., 2026,
55, 653-662
Chemically bonded Bi@C in porous carbon bundles for ultrafast and stable sodium storage
Received
19th September 2025
, Accepted 28th November 2025
First published on 3rd December 2025
Abstract
Metallic bismuth is one of the promising sodium ion anodes due to its exceptional conductivity, high theoretical capacity and suitable redox potential. However, severe volume changes (∼240%) during alloying reactions hinder its further development. In this study, carbon coated Bi nanoparticles embedded in micro-sized porous carbon bundles (Bi@C) are successfully fabricated. The dual-carbon confinement superstructure with inner covalent anchoring of C–O–Bi bonds significantly relieves the volume expansion of the Bi alloy anode and improves the electron transfer rate for the Bi@C anode. This unique structure promotes a pseudocapacitive sodium storage mechanism. As a result, Bi@C exhibits an excellent rate capability of 342.2 mAh g−1 at 15 A g−1 and long cyclability with 320.2 mAh g−1 after 1000 cycles at 2 A g−1 and 82.5% capacity retention. This strategy could be applied to other alloy-type anode materials for optimizing the rate performance and cycling stability.
1. Introduction
Sodium-ion batteries (SIBs) are widely regarded as promising commercial alternatives owing to their cost-effectiveness, superior low-temperature performance, and enhanced safety.1–3 Recent breakthroughs in energy density and cycle life, particularly through maturation of layered oxide cathodes and hard carbon anodes, have propelled SIBs into initial commercialization phases.4–7 Nevertheless, hard carbon anodes face critical limitations: low specific capacity, inadequate rate capability, and compromised tap density (∼1.07 g cm−3).8–10 These constraints severely restrict volumetric energy density, impeding further performance advancement in demanding applications.8,11–14
Driven by the pursuit of higher energy density in sodium-ion batteries (SIBs), alloy-type anodes, particularly bismuth (Bi) with its high theoretical capacity (386 mAh g−1), suitable redox potential (0.4–0.6 V vs. Na+/Na), environmental compatibility, and exceptional conductivity, have emerged as promising candidates, enabling enhanced rate capability through reduced polarization.5,15–20 However, severe volume changes (∼240%) during alloying reactions trigger structural degradation: (1) particle pulverization and electrostatic agglomeration form electrochemically inactive “dead Bi”, causing rapid capacity decay;5,21 (2) the unstable SEI undergoes cyclic fracture/regeneration, depleting active Na+ and amplifying interfacial impedance, collectively impeding practical implementation.6,22,23
Recent research has pioneered innovative Bi-composite architectures to solve the above issues: Yu and Chen embedded Bi nanoparticles within carbon hollow spheres to form a yolk@shell structure to buffer volume changes, achieving exceptional electrode stability.17,24,25 Xiong and Wei synthesized carbon-encapsulated Bi through calcining a Bi-MOF structure, significantly enhancing its cycling life.18,19,26 Beyond carbon-based composites, bismuth has been integrated into diverse heterostructures including Bi@SnSb, Bi/BiOCl, and Bi/TiO2;5,24,27–29 these architectures leverage built-in interfacial electric fields to accelerate ion transport, thus improving the rate performance. Nevertheless, persistent challenges also remain: (1) low tap density compromises volumetric capacity;16,17,24 (2) weak interfacial adhesion from physical mixing or van der Waals interactions causes protective layer slippage during cycling.16,17,25
To overcome these limitations, we propose a dual-mechanism stabilization strategy integrating porous carbon encapsulation with C–O–Bi covalent anchoring to improve the structural stability of Bi composites. In this article, we demonstrate how tuning the trimesic acid/Bi source ratio to optimize the C–O–Bi bond density results in a multi-level confinement architecture that more effectively mitigates volume expansion than single-mechanism systems, thereby accounting for the superior performance. Results show that the C–O–Bi bond density plays an important role in the strength of interfacial chemical bonds and the sodium ion storage mechanism. The Bi@C-2 sample with a “dual confinement” strategy demonstrates an excellent sodium ion storage performance with a capacity of 320.2 mAh g−1 after 1000 cycles at 2 A g−1 and 82.5% capacity retention. Notably, the anode could deliver an excellent rate capability of 342.2 mAh g−1 at 15 A g−1. The outstanding sodium storage ability could be attributed to the unique architecture with physical confinement and chemical bonding, which not only alleviate volume expansion but also promote charge transfer. Finally, the Bi@C anode realizes a pseudocapacitive sodium storage mechanism to achieve excellent sodium ion storage performance.
2. Experimental
2.1 Synthesis of Bi@C samples
Bi(NO3)3·5H2O (1.455 g, 3 mmol) and 1,3,5-benzenetricarboxylic acid (1.890 g, 9 mmol) at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 molar ratio were dissolved in 50 mL of N,N-dimethylformamide (DMF)/methanol (1:1 v/v) with 30 min stirring. The mixture was transferred to a Teflon-lined autoclave and reacted at 120 °C for 24 h. The resultant precipitate was washed with DMF and ethanol three times, respectively, and then dried at 80 °C for 12 h. Subsequent carbonization was performed in a tube furnace under an Ar flow: heating to 600 °C at 5 °C min−1 and maintaining for 4 h, followed by natural cooling to room temperature. The black product was labeled Bi@C-2. For comparative carbon content analysis, a control sample (Bi@C-1) was synthesized identically except with reduced 1,3,5-benzenetricarboxylic acid (1.26 g, 6 mmol; Bi/ligand molar ratio = 1:2).
:3 molar ratio were dissolved in 50 mL of N,N-dimethylformamide (DMF)/methanol (1:1 v/v) with 30 min stirring. The mixture was transferred to a Teflon-lined autoclave and reacted at 120 °C for 24 h. The resultant precipitate was washed with DMF and ethanol three times, respectively, and then dried at 80 °C for 12 h. Subsequent carbonization was performed in a tube furnace under an Ar flow: heating to 600 °C at 5 °C min−1 and maintaining for 4 h, followed by natural cooling to room temperature. The black product was labeled Bi@C-2. For comparative carbon content analysis, a control sample (Bi@C-1) was synthesized identically except with reduced 1,3,5-benzenetricarboxylic acid (1.26 g, 6 mmol; Bi/ligand molar ratio = 1:2).
2.2 Materials characterization
X-ray diffraction (XRD) patterns were collected using a Rigaku D/max-A diffractometer with Cu Kα radiation (λ = 1.5406 Å), scanning the 2θ range from 10° to 80° at a step size of 0.02°. The material morphology was examined by field-emission scanning electron microscopy (FESEM, Hitachi S-4800) at 5 kV accelerating voltage and transmission electron microscopy (TEM, Tecnai G2F20S-TWIN, FEI) operated at 200 kV. The surface chemical composition and states were analyzed via X-ray photoelectron spectroscopy (XPS) on a Kratos Axis Ultra system, with binding energies referenced to the adventitious carbon C 1s peak at 284.8 eV. Thermogravimetric analysis (TGA) was performed in air using a Mettler Toledo TGA/SDTA851 thermal analyzer. The surface area and porosity were evaluated via nitrogen physisorption at 77 K (Micromeritics ASAP 2460).
2.3 Electrochemical measurements
The working electrode was prepared by blending active materials, Super P, Styrene Butadiene Rubber (SBR), and carboxymethyl cellulose (CMC) (7:1:1:1 weight ratio). The mixture was ground, mixed with deionized water to form a slurry, coated onto pre-cleaned copper foil with a thickness of 15 μm, and vacuum-dried at 80 °C for 24 h. Active material mass loading values for each tested electrode typically ranged between 1.2 and 1.5 mg cm−2. Circular discs (12 mm diameter) were punched from the dried copper foil. Electrochemical testing employed 1 M NaPF6 in diglyme electrolyte and a Neware battery tester (0.01–1.80 V voltage window). Sodium-ion diffusion kinetics were analyzed using the galvanostatic intermittent titration technique (GITT). Cyclic voltammetry (CV, 0.2–1.0 mV s−1) and electrochemical impedance spectroscopy (EIS, 100 kHz–0.01 Hz) measurements were performed on a CHI660E workstation to probe the electrochemical behavior and charge transfer mechanisms.
3. Results and discussion
The XRD characteristic peaks align perfectly with the standard card (JCPDS Card No. 85-1329), confirming high phase purity for Bi@C-1 and Bi@C-2 (Fig. 1a). The lower diffraction peak intensity of Bi@C-2 relative to Bi@C-1 indicates reduced crystallinity or smaller particle size. TGA profiles (Fig. 1b) reveal distinct oxidation behaviors. The mass gain between 200 and 325 °C corresponds to Bi → Bi2O3 oxidation, where greater mass increments indicate higher proportions of exposed bismuth particles. Subsequent mass loss (>325 °C) stems from carbon oxidation to CO2, yielding carbon contents of 79.0% (Bi@C-1) and 73.8% (Bi@C-2) according to the mass loss.19 Raman spectra (Fig. 1c) reveal distinct ID/IG ratios. Compared with Bi@C-1, an elevated ID/IG ratio with increased carbon content signifies a higher proportion of amorphous carbon in Bi@C-2. XPS was employed to investigate the surface chemistry of Bi@C, which shows the presence of Bi, C and O elements on the surface of the composite material (Fig. S1a and d). The two samples show similar high-resolution C 1s spectra which are divided into 284.6, 285.3 and 288.6 eV, representing C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–O and CO (Fig. S1b and e).20,30 High-resolution O 1s XPS spectra (Fig. 1d and e) were deconvoluted into Bi–O (530.1 eV) and C–O–Bi (531.7 eV) bonds.19 The Bi–O bonds originate from Bi2O3 (pyrolysis products of the precursor Bi(NO3)3), while C–O–Bi bonds indicate interfacial bonding between Bi and carbon layers. The decreased Bi–O and increased C–O–Bi intensities in Bi@C-2 suggest conversion to Bi–O–C linkages, likely due to enhanced carbon coverage strengthening the bismuth–carbon interactions. The Bi 4f spectrum shows two peaks at 163.8 and 158.2 eV, which are assigned to Bi 4f5/2 and Bi 4f7/2, respectively (Fig. S1c and f). To further confirm the existence of the C–O–Bi bonds, we conducted FT-IR measurements on the samples. As shown in Fig. 1f, the distinct peak at 1235 cm−1 can be assigned to the Bi–O–C stretching vibration.19 The N2 adsorption/desorption isotherms for Bi@C-1 and Bi@C-2 are ascribed to type IV,31 combined with the obvious hysteresis loop at P/P0 from 0.4 to 1.0, indicating the existence of a large percentage of mesopores (Fig. S2a and b). The specific surface areas for Bi@C-1 and Bi@C-2 are 86.3 and 88.5 m2 g−1. Their pore size distributions also confirm abundant 15–20 nm mesopores (Fig. S2 inset). The higher surface area of Bi@C-2 is related to the higher content of carbon and uniformly carbon coated structures. The well-defined mesoporous structure with a high surface area of Bi@C-2 could facilitate ion accessibility and shorten the solid-state diffusion distance. This structural characteristic may synergistically contribute to the dominant pseudocapacitive behavior and the outstanding rate performance.
C, C–O and CO (Fig. S1b and e).20,30 High-resolution O 1s XPS spectra (Fig. 1d and e) were deconvoluted into Bi–O (530.1 eV) and C–O–Bi (531.7 eV) bonds.19 The Bi–O bonds originate from Bi2O3 (pyrolysis products of the precursor Bi(NO3)3), while C–O–Bi bonds indicate interfacial bonding between Bi and carbon layers. The decreased Bi–O and increased C–O–Bi intensities in Bi@C-2 suggest conversion to Bi–O–C linkages, likely due to enhanced carbon coverage strengthening the bismuth–carbon interactions. The Bi 4f spectrum shows two peaks at 163.8 and 158.2 eV, which are assigned to Bi 4f5/2 and Bi 4f7/2, respectively (Fig. S1c and f). To further confirm the existence of the C–O–Bi bonds, we conducted FT-IR measurements on the samples. As shown in Fig. 1f, the distinct peak at 1235 cm−1 can be assigned to the Bi–O–C stretching vibration.19 The N2 adsorption/desorption isotherms for Bi@C-1 and Bi@C-2 are ascribed to type IV,31 combined with the obvious hysteresis loop at P/P0 from 0.4 to 1.0, indicating the existence of a large percentage of mesopores (Fig. S2a and b). The specific surface areas for Bi@C-1 and Bi@C-2 are 86.3 and 88.5 m2 g−1. Their pore size distributions also confirm abundant 15–20 nm mesopores (Fig. S2 inset). The higher surface area of Bi@C-2 is related to the higher content of carbon and uniformly carbon coated structures. The well-defined mesoporous structure with a high surface area of Bi@C-2 could facilitate ion accessibility and shorten the solid-state diffusion distance. This structural characteristic may synergistically contribute to the dominant pseudocapacitive behavior and the outstanding rate performance.
 |
| | Fig. 1 (a) XRD patterns; (b) TG curves; (c) Raman spectra; (d and e) XPS spectra for O 1s and (f) FTIR spectra for the obtained samples. | |
Fig. 2 presents the SEM images of both samples. Bi@C-2 forms uniform fibrous architectures (Fig. 2a), featuring homogeneously embedded ∼100 nm Bi particles within carbon fibers (Fig. 2b) with minimal surface exposure (Fig. 2c). Conversely, Bi@C-1 exhibits significant surface-exposed Bi particles (up to ∼250 nm diameter) and prevalent fiber cracking (Fig. 2d–f), indicative of structural instability. This morphological disparity may originate from the lower carbon content of 26.2% for Bi@C-1, causing incomplete Bi encapsulation. The enhanced surface exposure accelerates oxidation, correlating with the pronounced 200–325 °C mass gain in TGA for Bi@C-1 (Fig. 1b). TEM analysis (Fig. 2g–i) further verifies superior Bi dispersion uniformity in Bi@C-2. Bismuth nanoparticles are encapsulated by 7 nm carbon layers and embedded within porous carbon bundles. This dual-functional carbon coating simultaneously mitigates volume variation during sodiation/desodiation and enhances charge transfer kinetics. Fig. 2j shows the TEM image of Bi@C-1, revealing a broad particle size distribution of Bi particles ranging from 30 to 200 nm. Furthermore, the carbon layer encapsulating the Bi particles is thinner than that in Bi@C-2, measuring approximately 5 nm.
 |
| | Fig. 2 SEM images for (a–c) Bi@C-2 and (d–f) Bi@C-1; TEM images of (g–i) Bi@C-2 and (j–l) Bi@C-1. | |
In order to investigate the electrochemical properties of the two samples, cyclic voltammetry (CV) curves were recorded. Fig. 3a shows the CV curve of Bi@C-2, which reveals anodic peaks at 0.96 V (electrolyte decomposition) and 0.58/0.34 V (stepwise alloying: Bi → NaBi → Na3Bi), with the corresponding cathodic dealloying peaks at 0.66/0.79 V.5,6 The weak peaks at 0.05/0.08 V can be attributed to intercalation/deintercalation of hard carbon.32 The peak intensity is enhanced in the 2nd and 3rd cycles which confirms the activation of the anode materials, and the anodic peaks at 0.34 and 0.58 V shift to 0.39 and 0.61 V in the 2nd and 3rd cycles. Bi@C-2 exhibits a morphological change after the first cycle.33 Bi@C-1 exhibits similar CV curves but amplified side reactions at 0.14 V due to bismuth exposure. The corresponding charge/discharge curves (Fig. 3b) corroborate these mechanisms through three discharge plateaus (1.11, 0.65 and 0.42 V) and two charge plateaus (0.59 and 0.76 V), yielding first discharge/charge capacities of 497.6/457.8 mAh g−1 with an initial coulombic efficiency of 92% for Bi@C-2. Rate performance is further tested for both samples. As shown in Fig. 3c, Bi@C-2 demonstrates superior capacities of 381.2, 362.5, 359.8, 356.7, 350.1 and 347.6 mAh g−1 at 0.2, 0.5, 1.0, 2.0, 5.0 and 10.0 A g−1. Even at 15 A g−1, 342.2 mAh g−1 can be maintained for Bi@C-2, with a capacity retention of 89.6% (compared with the 381.7 mAh g−1 at 0.2 A g−1). When the current density went back to 0.2 A g−1, the capacity can recover to 361.4 mAh g−1. In contrast, Bi@C-1 delivers lower capacities of 276.1, 249.2, 243.4, 237.7, 230.6, 218.3 and 211.2 mAh g−1 from 0.2 to 15 A g−1. When the current went back to 0.2 A g−1, the capacity could recover to only 245.6 mAh g−1. The high rate performance (342.2 mAh g−1 at 15 A g−1) of Bi@C-2 stems from the synergistic effect of the dual-confinement strategy and optimized electrode kinetics. As shown in the EIS data of Fig. 3e, the Bi@C-2 material exhibits extremely low charge transfer resistance and fast sodium ion diffusion from the more vertical line. These factors ensure highly efficient electron/ion transport even at high current densities, thereby contributing to exceptional high-rate capacity. To ensure the statistical reliability of the findings, all data in Fig. 3c and d were obtained from three independent synthesis experiments and electrochemical tests for both Bi@C-1 and Bi@C-2 with active material mass loading between 1.2 and 1.5 mg cm−2. Long-term cycling at 2 A g−1 further highlights the performance difference. The Bi@C-2 electrode maintains a capacity of 320.2 mAh g−1 after 1000 cycles (82.5% retention compared to the charge capacity of the 2nd cycle). In contrast, Bi@C-1 degrades rapidly to 190.5 mAh g−1 after only 500 cycles (74.8% retention compared to the charge capacity of the 2nd cycle), with an accelerated capacity fade in the initial stages. This contrast underscores the critical role of carbon encapsulation in stabilizing volume changes, suppressing side reactions and enhancing charge transfer kinetics. The electrochemical performance of Bi@C-2 is also much better than that of Bi-based anodes previously reported (Table S1 and Fig. S4 in the SI). To elucidate the superior cycling stability of Bi@C-2 over Bi@C-1, electrochemical impedance spectroscopy (EIS) was performed. The Nyquist plots in Fig. 3e consist of a depressed semicircle in the high-frequency region, corresponding to the charge-transfer resistance (Rct), and an inclined line in the low-frequency region, representing sodium-ion diffusion. The fresh Bi@C-2 electrode exhibits an Rct of 56 Ω. After 10 cycles, however, the Rct drops sharply to 3 Ω due to electrode activation in the DME-based electrolyte. It then increases only slightly to 4 Ω after 200 cycles, indicating a highly stable and fast charge-transfer kinetics. Concurrently, the low-frequency line becomes more vertical, suggesting enhanced capacitive behavior.34 In contrast, the fresh Bi@C-1 electrode shows a significantly larger Rct of 102 Ω. Although it decreases to 10 Ω after 10 cycles, it subsequently rises to 15 Ω after 200 cycles, reflecting an inferior electron transfer rate and stability compared to Bi@C-2.
 |
| | Fig. 3 Electrochemical performance for Bi@C-1 and Bi@C-2: (a) CV curves of Bi@C-2; (b) charge–discharge curves; (c) rate performance with error bars; (d) long-term cycling test at 2 A g−1 with error bars and (e) Nyquist plots of the fresh and cycled Bi@C-2 electrodes. | |
To investigate the electrochemical reaction kinetics of Bi@C-1 and Bi@C-2, cyclic voltammetry (CV) curves at varying scan rates (Fig. 4a and d) were analyzed via the following formulas:35,36
where
i is the peak current and
ν is the scan rate. According to the above formulas
(1) and (2), the
b value derived from the slope of ln(
i)
versus ln(
ν) plots determines the sodium ion storage mechanism:
b ≈ 0.5 signifies diffusion-controlled processes, while
b ≈ 1.0 indicates pseudocapacitive-dominated kinetics. Fitting results for four redox peaks (
Fig. 4b and e) reveal distinct behaviors: Bi@C-1 exhibits
b-values of 0.759, 0.693, 0.653, and 0.803, consistent with a dual mechanism of diffusion and pseudocapacitive control. In contrast, Bi@C-2 shows
b-values approaching 1.0 (0.816, 0.899, 0.936, and 0.967), and the high
b-values of Bi@C-2 confirm a typical pseudocapacitive predominance. In order to further quantify the pseudocapacitive contribution, corresponding calculations were carried out according to
eqn (3) and (4) put forward by Bruce Dunn:
37,38| | | i (V)/v1/2 = k1v1/2 + k2 | (4) |
where
k1v represents the pseudocapacitance and
k2v1/2 represents the diffusion contribution.
k1 and
k2 can be obtained from the slope of
i (V)/
v1/2versus v1/2 according to
eqn (4).
3 Accordingly, the pseudocapacitance contributions for Bi@C-1 and Bi@C-2 were calculated to be 73.8% and 87.9% at 0.8 mV s
−1, respectively (
Fig. 4c and f), revealing higher pseudocapacitive storage of Bi@C-2. This phenomenon also agrees well with the conclusion obtained from the EIS evolution. The more-vertical line (closer to the 90° phase angle) in Nyquist plots also indicates a capacitive behavior for Bi@C-2. The pseudocapacitive mechanism endows Bi@C-2 with excellent rate performance and high cycling stability. Furthermore, the sodium ion diffusion rate (
D) of both samples is qualified using the galvanostatic intermittent titration technique (GITT), following the below equation:
25| | 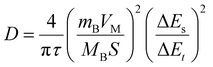 | (5) |
where
τ denotes the relaxation time and
mB,
VM,
MB and
S represent the mass, molar volume, molar mass and effective electrode/electrolyte interfacial area of Bi@C, respectively. Δ
Es and Δ
Et correspond to the steady potential shift between pulses and the pulse-induced potential difference.
25 According to
eqn (5), the obtained Na
+ diffusion coefficient (
DNa+) of Bi@C-2 (5.68 × 10
−15–5.78 × 10
−12 cm
2 s
−1) is much higher than that of Bi@C-1 (9.55 × 10
−16–6.30 × 10
−12 cm
2 s
−1), especially in the alloying reaction stage from the NaBi to the Na
3Bi phase, confirming that fast ion transport promotes the pseudocapacitive mechanism of Bi@C-2.
 |
| | Fig. 4 (a and d) CV curves at different scan rates; (b and e) ln(sweep rate) versus ln(peak current); (c and f) CV curve of Bi@C at 0.8 mV s−1 marked in green with the pseudocapacitance-contributed part marked in orange and (g and h) diffusion coefficients of the Na-ion for (g) Bi@C-1 and (h) Bi@C-2. | |
In order to further investigate the root cause of the different cycling performances of the two samples, post-cycling electrodes after 500 cycles were disassembled for morphological analysis. Fig. 5 reveals distinct structural evolution: Bi@C-2 maintains integrated bundle-like architectures (1–2 μm in length and 200–500 nm in diameter) with minimal pulverization (Fig. 5a and b). Compared with the Bi@C-2 electrode before cycling, the length and diameter of the bundles both decreased. This morphological reconstruction may be from the repeated expansion and contraction of Bi@C-2 during charge/discharge. However, there are hardly any pulverized Bi nanoparticles that detached from the porous carbon bundle, indicating that pulverized Bi nanoparticles are trapped in the porous carbon due to effective carbon confinement and strong chemical bonding between Bi and the carbon layer. In contrast, cycled Bi@C-1 exhibits fragmented rods with 300–500 nm length and 100 nm diameter, alongside extensive pulverized particles (Fig. 5c and d). The ex situ TEM images of Bi@C-1 and Bi@C-2 in Fig. 5e and f confirm this phenomenon. The Bi particles in Bi@C-2 maintain their spherical morphology with a diameter of 30–50 nm, whereas those in Bi@C-1 are largely pulverized into much finer particles. Most of these pulverized Bi nanoparticles originate from surface-exposed Bi, where insufficient carbon confinement and deficient C–O–Bi interfacial bonding fail to accommodate volume expansion during alloying reactions. Consequently, particle detachment from carbon matrices occurs, accelerating capacity degradation. Therefore, it can be concluded that the ultralong cycle life of Bi@C-2 results from stable physical confinement and effective chemical bonding. Bi@C-2 with this dual-mechanism stabilization strategy exhibits outstanding rate capability and long-term cycling stability, which can be attributed to three aspects: (1) a rapid electron transfer rate from the abundant C–O–Bi bonds, supported by the EIS evidence; (2) a fast ion Na+ diffusion coefficient from the stable porous carbon confined Bi structure, supported by the GITT results; and (3) high-proportion and sustainable pseudocapacitive contribution, supported by the b-value and CV fitting evidence (Fig. 5g).
 |
| | Fig. 5 SEM and TEM images of post-cycling electrodes after 500 cycles: (a, b and e) Bi@C-2; (c, d and f) Bi@C-1. (g) Schematic illustration of the morphological evolution of the Bi@C electrode after cycling. | |
4. Conclusion
In summary, chemically bonded Bi nanoparticles embedded in porous carbon bundles (Bi@C) were successfully fabricated. Bi@C with this dual-mechanism stabilization strategy exhibits an excellent rate capability of 342.2 mAh g−1 at 15 A g−1 and an extended cycle life of 1000 cycles with 320.2 mAh g−1 at 2 A g−1 and 82.5% capacity retention. The carbon confinement effectively accommodates volume expansion during Bi alloying and stabilizes the architecture. Meanwhile, EIS evolution has proved that abundant C–O–Bi covalent bonds at Bi/carbon interfaces facilitate rapid electron transport, accelerating alloying kinetics and improving rate capability. The porous carbon bundles decrease the sodium ion diffusion paths and promote fast sodium ion diffusion, evidenced by the GITT data. Consequently, the Bi@C composite exhibits pseudocapacitive-dominated reaction mechanisms, and pseudocapacitance contributes 87.9% of the total capacity. This work establishes an effective dual-mechanism strategy of physical confinement and chemical bonding which can be applied to other alloy anode materials for electrochemical performance enhancement.
Author contributions
Hui Qi: funding acquisition, resources, and writing – original draft; Bin Gao: software and investigation; Siqin Zheng: investigation; Jianghui Song: software; Hongwei Yuan: initial data analysis; Weipeng Luo: conceptualization and supervision; Ze Zhang: methodology; Haoyu Liu: data curation; Xiaojing Yuan: writing review & editing, resources, and project administration; Wenfeng Wu: formal analysis.
Conflicts of interest
The authors declare no conflict of interest.
Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. The supplementary information includes XPS survey, Nitrogen adsorption-desorption isotherms of Bi@C-1 and Bi@C-2, CV curves for Bi@C-1, Comparison of Bi-based anodes reported in literatures for SIBs, Nyquist plots of the fresh and cycled Bi@C-1 electrodes, and Rate performance of two samples with error bars. See DOI: https://doi.org/10.1039/d5dt02243f.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 52002305), the Natural Science Basic Research Program in Shanxi Province of China (Grant No. 202403021221184 and 202403021222281), and the College Students’ Innovation Program of Taiyuan Normal University (No. CXCY2443).
References
- F. Zhang, B. He, Y. Xin, T. Zhu, Y. Zhang, S. Wang, W. Li, Y. Yang and H. Tian, Emerging chemistry for wide-temperature sodium-ion batteries, Chem. Rev., 2024, 124(8), 4778–4821 CrossRef CAS PubMed.
- S. S. A. Kumar, M. Nujud Badawi, J. Liew, T. Prasankumar, K. Ramesh, S. Ramesh, S. Ramesh and S. K. Tiong, High-Performance Sodium-Ion Batteries with Graphene: An overview of recent developments and design, ChemSusChem, 2025, 18(2), e202400958 CrossRef CAS PubMed.
- H. Qi, C. Zhao, J. Huang, C. He, L. Tang and W. Deng, Metastable FeCN2@nitrogen-doped carbon with high pseudocapacitance as an anode material for sodium ion batteries, Nanoscale, 2022, 14(3), 780–789 RSC.
- Y. Dai, X. Li, X. Sang, Y. He, S. Gu, J. Li, G. Zhou, X. Wang, B. Sun and Y. He, Cobalt-doped iron-based selenide for sodium-ion battery anode: Insight into the doping and sodium storage mechanism, Chem. Eng. J., 2025, 511, 161949 CrossRef CAS.
- L. Fan, B. Liu, Q. Ge, Q. Ai, N. Miao and X. Ding, Hierarchical Bi@SnSb nanofibers as high-rate and long-cycle sodium ions battery anodes, Electrochim. Acta, 2025, 535, 146574 CrossRef CAS.
- G. Li, Y. Tang, Y. Wang, S. Cui, H. Chen, Y. Hu, H. Pang and L. Han, Single Atomic Cu-C3 sites catalyzed interfacial chemistry in Bi@C for ultra-stable and ultrafast sodium-ion batteries, Angew. Chem., Int. Ed., 2024, e202417602 Search PubMed.
- Q. Mao, R. Gao, Q. Li, D. Ning, D. Zhou, G. Schuck, G. Schumacher, Y. Hao and X. Liu, O3-type NaNi0.5Mn0.5O2 hollow microbars with exposed {0 1 0} facets as high performance cathode materials for sodium-ion batteries, Chem. Eng. J., 2020, 382, 122978 CrossRef CAS.
- K. Wang, F. Sun, H. Wang, D. Wu, Y. Chao, J. Gao and G. Zhao, Altering thermal transformation pathway to create closed pores in coal–derived hard carbon and boosting of Na+ plateau storage for high–performance sodium–ion battery and sodium–ion capacitor, Adv. Funct. Mater., 2022, 32(34), 2203725 CrossRef CAS.
- H. Chen, N. Sun, Q. Zhu, R. A. Soomro and B. Xu, Microcrystalline hybridization enhanced coal-based carbon anode for advanced sodium-ion batteries, Adv. Sci., 2022, 9(20), e2200023 CrossRef PubMed.
- Z. Lu, J. Wang, W. Feng, X. Yin, X. Feng, S. Zhao, C. Li, R. Wang, Q. A. Huang and Y. Zhao, Zinc single–atom–regulated hard carbons for high–rate and low–temperature sodium–ion batteries, Adv. Mater., 2023, 35(26), 2211461 CrossRef CAS.
- Z. Zheng, S. Hu, W. Yin, J. Peng, R. Wang, J. Jin, B. He, Y. Gong, H. Wang and H. J. Fan, CO2−etching creates abundant closed pores in hard carbon for high–plateau–capacity sodium storage, Adv. Energy Mater., 2023, 14(3), 2303064 CrossRef.
- Z. Tang, R. Liu, D. Jiang, S. Cai, H. Li, D. Sun, Y. Tang and H. Wang, Regulating the pore structure of biomass-derived hard carbon for an advanced sodium-ion battery, ACS Appl. Mater. Interfaces, 2024, 16(36), 47504–47512 CrossRef CAS.
- Y. Li, D. Xia, L. Tao, Z. Xu, D. Yu, Q. Jin, F. Lin and H. Huang, Hydrothermally assisted conversion of switchgrass into hard carbon as anode materials for sodium-ion batteries, ACS Appl. Mater. Interfaces, 2024, 16(22), 28461–28472 CrossRef CAS PubMed.
- Z. Yan, Q.-W. Yang, Q. Wang and J. Ma, Nitrogen doped porous carbon as excellent dual anodes for Li- and Na-ion batteries, Chin. Chem. Lett., 2020, 31(2), 583–588 CrossRef CAS.
- J. Pan, Z. Sun, X. Wu, T. Liu, Y. Xing, J. Chen, Z. Xue, D. Tang, X. Dong, H. Zhang, H. Liu, Q. Wei, D. L. Peng, K. Amine and Q. Zhang, Mechanically robust bismuth-embedded carbon microspheres for ultrafast charging and ultrastable sodium-ion batteries, J. Am. Chem. Soc., 2025, 147, 3047–3061 CrossRef CAS PubMed.
- W. Yang, Q. Zhou, H. Dong, R. Hu, A. Kang, L. Liu, X. Liang, H. Zhang, H. Lu, C. Wei, A. C. Y. Yuen and W. Yang, Core-shell structured Bi@graphdiyne nanospheres enable high rate capacity and outstanding stability for sodium-ion batteries, Chem. Eng. J., 2025, 512, 162747 CrossRef CAS.
- W. Yang, Z.-w. Yang, C.-h. Wang, H.-n. Bian, Y.-d. Pan, G. Li, K.-y. Wang and J.-X. Wang, Core−shell Bi@ mesoporous carbon nanospheres as high rate and long life anodes for sodium ion batteries, Trans. Nonferrous Met. Soc. China, 2025, 35(6), 1996–2007 CrossRef CAS.
- X. Zhang, M. Zheng, C. Wu, S. Li, B. Li, J. Guo, M. Wei and L. Chen, Tannic acid-derived carbon-coated Bi nanodiscs for high-performance sodium-ion batteries, Chem. Commun., 2025, 61(29), 5483–5486 RSC.
- Y. Liang, N. Song, Z. Zhang, W. Chen, J. Feng, B. Xi and S. Xiong, Integrating Bi@C nanospheres in porous hard carbon frameworks for ultrafast sodium storage, Adv. Mater., 2022, 34(28), 2202673 CrossRef CAS.
- Y. Cheng, H. Xie, L. Zhou, B. Shi, L. Guo and J. Huang,
In situ liquid-phase transformation of SnS2/CNTs composite from SnO2/CNTs for high performance lithium-ion battery anode, Appl. Surf. Sci., 2021, 566, 150645 CrossRef CAS.
- L. Shi, R. Jiao, D. Lan, L. Wang, Y. Gao, M.-F. Lin, Z. Lang, W. He, G. Jia, J. Cui and S. Li, Boosting the initial Coulombic efficiency of Bi2S3-based anode for practical sodium-ion batteries, Chem. Eng. J., 2025, 519, 165074 CrossRef CAS.
- J. Xiang, Z. Liu and T. Song, Bi@C Nanoplates derived from (BiO)2CO3 as an enhanced electrode material for lithium/sodium-ion batteries, ChemistrySelect, 2018, 3(31), 8973–8979 CrossRef CAS.
- Z. Zhao, G. Sun, Y. Zhang, R. Hua, X. Wang, N. Wu, J. Li, G. Liu, D. Guo, A. Cao, X. Liu and H. Hou, Introduction of SnS2 to regulate the ferrous disulfide phase evolution for the construction of triphasic heterostructures enabling kinetically accelerated and durable sodium storage, Adv. Funct. Mater., 2024, 34, 2314679 CrossRef CAS.
- Z. Li, R. Yuan, Y. Hou, X. Wei, Z. Zhu, Y. Wang, S. Xu, L. Chen, Y. Wang, R. Wu, Q. Kong and J. S. Chen, Spatial confinement of Bi/TiO2 nanoparticles with an abundant heterointerface in hollow carbon spheres for durable sodium storage, Chem. Eng. J., 2025, 519, 165239 CrossRef CAS.
- H. Yang, R. Xu, Y. Yao, S. Ye, X. Zhou and Y. Yu, Multicore–shell Bi@N–doped carbon nanospheres for high power density and long cycle life sodium– and potassium–ion anodes, Adv. Funct. Mater., 2019, 29(13), 1809195 CrossRef.
- J. Xu, Y. Xie, J. Zheng, C. Liu, Y. Lai and Z. Zhang, A sodiophilic carbon cloth decorated with Bi-MOF derived porous Bi@C nanosheets for stable Na metal anode, J. Electroanal. Chem., 2021, 903, 115853 CrossRef CAS.
- S. L. Wei, Y. L. Yang, J. G. Chen, X. L. Shi, Y. Sun, P. Li, X. F. Tian, H. J. Chen, Z. Luo and Z. G. Chen, A fast–charging and ultra–stable sodium–ion battery anode enabled by N–Doped Bi/BiOCl in a Carbon Framework, Adv. Energy Mater., 2024, 14, 2401825 CrossRef CAS.
- J. Zhao, J. Xu, Q. Li, W. Yao, C. Yu, N. Zhang, X. Chen and X. Ding, BiSbx nanoalloys encapsulated by carbon fibers as high rate sodium ions storage anodes, J. Electroanal. Chem., 2023, 939, 117452 CrossRef CAS.
- G. Lakshmi Sagar, K. Brijesh, P. Mukesh, A. P. Hegde, A. Kumar, A. Kumar, K. S. Bhat and H. S. Nagaraja, Dual storage mechanism of Bi2O3/Co3O4/MWCNT composite as an anode for lithium-ion battery and lithium-ion capacitor, J. Electroanal. Chem., 2024, 975, 118777 CrossRef CAS.
- H. Qi, J. Huang, L. Tang, M. Ma, W. Deng and C. Zhang, Confined pulverization promoting durable pseudocapacitance for FeOOH@PEDOT anode in Li-ion battery, J. Electroanal. Chem., 2021, 882, 115005 CrossRef CAS.
- Y. Yun, B. Xi, F. Tian, W. Chen, W. Sun, H. Pan, J. Feng, Y. Qian and S. Xiong, Zero–strain structure for efficient potassium storage: nitrogen–enriched carbon dual–confinement CoP composite, Adv. Energy Mater., 2021, 12(3), 2103341 CrossRef.
- H. Qi, C. He, C. Song, J. Gao, Q. Gao, W. Luo, Z. Zhang, H. Liu, X. Yuan, W. Wu, B. Zhao, L. Kong, Y. Cheng and L. Guo, Tailoring the exposure of active facets of FeNCN towards enhanced pseudocapacitive behavior for sodium storage, Chin. Chem. Lett., 2025, 36, 111591 CrossRef CAS.
- H. Qi, Y. Hou, W. Wang, L. Tang, C. Zhang, W. Deng, Y. Cheng and J. Zhang, Controlled phase and crystallinity of FeNCN/NC dominating sodium storage performance, Dalton Trans., 2022, 51(21), 8223–8233 RSC.
- Z. Hu, Z. Zhu, F. Cheng, K. Zhang, J. Wang, C. Chen and J. Chen, Pyrite FeS2 for high-rate and long-life rechargeable sodium batteries, Energy Environ. Sci., 2015, 8(4), 1309–1316 RSC.
- T. Brezesinski, J. Wang, S. H. Tolbert and B. Dunn, Ordered mesoporous α-MoO3 with iso-oriented nanocrystalline walls for thin-film pseudocapacitors, Nat. Mater., 2010, 9(2), 146–151 CrossRef CAS PubMed.
- Y. Cheng, J. Huang, F. Yu, Y. Zhou, G. Li, W. Cheng, P. Duan, H. Qi and H. Xie, Chemically bonded MXene/SnSe2 composite with special structural transformation as a high-performance anode for lithium and potassium ions battery, Chem. Eng. J., 2024, 481, 148737 CrossRef CAS.
- J. Wang, J. Polleux, J. Lim and B. Dunn, Pseudocapacitive contributions to electrochemical energy storage in TiO2 (Anatase) nanoparticles, J. Phys. Chem. C, 2007, 111(40), 14925–14931 CrossRef CAS.
- P. Simon, Y. Gogotsi and B. Dunn, Where do batteries end and supercapacitors begin?, Science, 2014, 343(6176), 1210–1211 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Bin
Gao
a,
Siqin
Zheng
a,
Jianghui
Song
a,
Hongwei
Yuan
a,
Weipeng
Luo
b,
Ze
Zhang
b,
Haoyu
Liu
b,
Xiaojing
Yuan
*b and
Wenfeng
Wu
c
*a,
Bin
Gao
a,
Siqin
Zheng
a,
Jianghui
Song
a,
Hongwei
Yuan
a,
Weipeng
Luo
b,
Ze
Zhang
b,
Haoyu
Liu
b,
Xiaojing
Yuan
*b and
Wenfeng
Wu
c





