DOI:
10.1039/D5DT02044A
(Paper)
Dalton Trans., 2026,
55, 865-884
Mono- and bimetallic homo- and heterodinuclear Pd and Pt complexes bridged by diphosphines: synthesis, characterisation and cytotoxicity
Received
25th August 2025
, Accepted 5th December 2025
First published on 10th December 2025
Abstract
Herein we report a series of monometallic complexes: [(κ2-dppe)MCl(κ1-dppx)](OTf) (where dppe = 1,2-bis(diphenylphosphino)ethane; dppx = 1,4-bis(diphenylphosphino)butane (dppb) (M = Pd (Pd-1) or Pt (Pt-1)), 1,5-bis(diphenylphosphino)pentane (dpppent) (M = Pd (Pd-2) or Pt (Pt-2)), or 1,6-bis(diphenylphosphino)hexane (dpph) (M = Pd (Pd-3) or Pt (Pt-3))). We also report the homobimetallic complexes [((κ2-dppe)PdCl)2(μ-dppx)](2OTf) (dppx = dppb (PdPd-1), dpppent (PdPd-2) or dpph (PdPd-3)). In addition, the Ru(II)-based heterobimetallic complexes [(κ2-dppe)ClM(μ-dppx)RuCl2(η6-p-cym)](OTf) (M = Pd and dppx = dppb (PdRu-1), M = Pd and dppx = dpppent (PdRu-2), M = Pd and dppx = dpph (PdRu-3), M = Pt and dppx = dppb (PtRu-1), M = Pt and dppx = dpppent (PtRu-2), M = Pt and dppx = dpph (PtRu-3), and Os-based heterobimetallic complexes [(κ2-dppe)ClPd(μ-dppx)OsCl2(η6-p-cym)](OTf) (dppx = dppb (PdOs-1), dpppent (PdOs-2) and dpph (PdOs-3)) are also reported. All complexes were fully characterised by spectroscopic means and their stability in DMSO was determined via time-dependent NMR spectroscopy. Aquation studies were performed in a PBS buffer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMSO solution (90:10 v/v) at 37 °C to determine the various entities which could form in biological media and are discussed. The solid-state structures of complexes PdPd-0, PdPd-1 and PdRu-1 were further characterised by single crystal X-ray diffraction analyses and are reported. The cytotoxicity of all complexes was evaluated in vitro against a wide panel of cancer cell lines. While the monometallic and homobimetallic complexes showed sub-optimal activity, the heterobimetallic complexes all displayed good to excellent cytotoxicity. PdRu-1 displayed an IC50 value of 3.43 ± 1.43 µM and a selectivity index of 6.16 on the Ca Ski cell line. The results from this study highlight the increased efficacy of heterobimetallic systems with regards to their monometallic or homobimetallic counterparts for Pd(II) and Pt(II) type complexes.
:DMSO solution (90:10 v/v) at 37 °C to determine the various entities which could form in biological media and are discussed. The solid-state structures of complexes PdPd-0, PdPd-1 and PdRu-1 were further characterised by single crystal X-ray diffraction analyses and are reported. The cytotoxicity of all complexes was evaluated in vitro against a wide panel of cancer cell lines. While the monometallic and homobimetallic complexes showed sub-optimal activity, the heterobimetallic complexes all displayed good to excellent cytotoxicity. PdRu-1 displayed an IC50 value of 3.43 ± 1.43 µM and a selectivity index of 6.16 on the Ca Ski cell line. The results from this study highlight the increased efficacy of heterobimetallic systems with regards to their monometallic or homobimetallic counterparts for Pd(II) and Pt(II) type complexes.
Introduction
Cancer is one of the predominant health challenges in the world, with more than 2 million new cases and more than 600000 deaths projected in 2025 in the United States alone.1 The number of cases worldwide is predicted to reach 35 million by 2050, given the overall increase in population and life expectancy.2 Pt-based drugs, which are linked to a plethora of side effects, are still the forefront chemotherapeutic drugs (Scheme 1a–d).3–8 However some Ru(II)-based complexes have been studied and are linked to promising results (Scheme 1e–i).9–16 Another class of compounds which have been somewhat underexplored in comparison to the multitude of mononuclear complexes reported in the literature are bimetallic complexes. These compounds make use of two metals, where it can be further differentiated into the homobimetallic (same metals) or heterobimetallic (two different metals) class. The nature of the bridging ligand can be varied, and diphosphines have shown to be suitable for this purpose due to the simple finetuning of both steric and electronic properties.17 Upon addition of a diphosphine to a halide-bridged dimer precursor, dimer cleavage and binding to the metal centre by the donor phosphine occurs readily to form a monomeric structure bearing a pendant P atom. This intermediate complex can then be used in a subsequent dimer-cleavage reaction, generating a homo- or heterobimetallic complex. We have recently reviewed all homo- and heteromultimetallic complexes with a group 8 metal bridged by diphosphines,18 as well as Pd and Pt complexes of analogous motifs,17 all complexes bridged by dppm (1,1-bis(diphenylphosphino)methane)19 and osmium-based complexes bearing a η6-arene and a phosphine co-ligand20 involved in biological studies.
 |
| | Scheme 1 Chemical structures of complexes which reached human clinical trials. a: Cisplatin, b: carboplatin, c: oxaliplatin, d: nedaplatin, e. RAPTA-C, f. RAED-C, g: NAMI-A, h: KP1019 (Z = indazoleH+), KP1339 (Z = Na+), and i. TLD1433. | |
Bennet et al. have reported some examples of homobimetallic Pd(II) and Pt(II) complexes bridged by diphosphines and showed suboptimal activity of the Pd(II) examples compared to their Pt(II) analogues.21 Another study has uncovered enhanced activity of a cyclometallated Pd(II)–(µ-dppe)–Pd(II) complex compared to cisplatin in a wide array of cell lines.22 A more recent study reported encouraging cytotoxic activity of dinuclear palladacycles in MCF-7 and MDA-MB-231 cell lines, even though cisplatin was not used as control.23 It is worth mentioning that some monometallic Pd(II) and Pt(II) complexes also displayed good to excellent cytotoxic activities against several cancer cell lines,24–29 and several reviews highlight the relevance of monometallic complexes.30–33 Homobimetallic complexes of the type [(η6-arene)RuCl2]2(µ-diphosphine) have been shown to be underwhelmingly active,34,35 with the exception of two complexes reported by Klaimanee et al., making use of the methyl-substituted diphosphines 1,2-bis(diphenylphosphino)propane and 2,4-bis(diphenylphosphino)pentane, which displayed considerably higher cytotoxic activities than cisplatin.36
Several accounts report increased cytotoxic activity in line with the heterobimetallic nature of the complexes studied. In fact, some examples of Pt(II)–(µ-dppm)–Au(I) complexes were reported to be more effective than their monometallic Pt(II) precursors.37 In addition, previous research conducted by us on monometallic Fe(II) and their analogous heterobimetallic Fe(II)–Ru(II) complexes showed that the bimetallic examples displayed higher level of DNA interactions and their cytotoxic activities were found similar to that of cisplatin on the cell lines tested.38 We also reported increased activity of [(η5-Cp)Fe(CO)I(µ-dppm)RuCl2(η6-arene)] (where arene = p-cymene or C6H6) compared to homobimetallic [(η6-C6H6)RuCl2]2(µ-dppm),35 and some comprehensive reviews highlight multiple heterobimetallic complexes with various ligand scaffolds and their anticancer activities.39,40
As part of our ongoing research programme into the development and anticancer testing of bimetallic complexes, with a view to establish a large library of compounds for further biological studies, we report herein eighteen novel complexes subdivided in three categories: (a) monometallic Pd or Pt-based complexes bearing a κ1-diphosphine with the general formula [(κ2-dppe)MCl(κ1-dppx)](OTf), where dppx = 1,4-bis(diphenylphosphino)butane (dppb), 1,5-bis(diphenylphosphino)pentane (dpppent), or 1,6-bis(diphenylphosphino)hexane (dpph) and M = Pd or Pt; (b) homobimetallic Pd-based complexes bridged by a diphosphine with the general formula [((κ2-dppe)PdCl)2(μ-dppx)](2OTf); and (c) heterobimetallic complexes of the type [(κ2-dppe)ClM(μ-dppx)RuCl2(p-cym)] (where M = Pd or Pt), and [(κ2-dppe)ClPd(μ-dppx)OsCl2(p-cym)](OTf) (Fig. 1). The rationale behind this collection of complexes is to evaluate multiple aspects in the drug design process: Firstly, analogous complexes of which the only structural difference stems from one same group transition metal (i.e. Pd(II) vs. Pt(II), Ru(II) vs. Os(II)) are tested to determine which is most active. Secondly, as seen in multiple reports, monometallic complexes tend to be less active than their bimetallic counterparts, as well as homobimetallic complexes displaying less activity than their heterobimetallic complexes.18 Hence, the monomer precursors and homobimetallic complexes were also evaluated to further test these observations. Thirdly, a previous study reported by us has seen a correlation between spacer length (i.e. number of CH2 groups within the bridging ligand backbone) and cytotoxic activity.38 Hence, the length of the diphosphine spacer chain was varied systematically for all classes of compounds reported herein to evaluate whether this trend can be observed in the three different types of complexes. All complexes were characterised spectroscopically, three of which were further characterised by single crystal X-ray diffraction analysis. Rates of aquation and tentative speciation were derived for selected examples in a PBS buffer:DMSO solution (90:10 v/v) and are presented. The cell viability was evaluated on eight human cancer cell lines, including cervical (HeLa and Ca Ski), pancreatic (PANC-1 and CFPAC-1), rhabdomyosarcoma (RD and RH30), and breast (MCF-7 and MDA-MB-231). The most effective compounds were also tested against non-malignant MRC-5 cell line. The IC50 values of the two best-performing complexes were determined on cervical and pancreatic cancer cell lines, and their selectivity indices were derived by cross-testing on FG0 non-malignant cells.
 |
| | Fig. 1 Schematic representation of the complexes discussed herein, and their respective labels. | |
Results and discussion
Synthesis and spectroscopic properties of precursor dimer complexes PdPd-0 and PtPt-0
Dimer complexes similar in structure as PdPd-0 have been previously reported in the literature,41–45 with the first example dating back to 1979.41 However, these complexes contained either ClO4 or BF4 counteranions. Herein we report the synthesis of an analogous dimer complex containing the triflate (SO3CF3) counteranion. The dimer precursor was synthesised using adapted methods,46 by mixing [(dppe)PdCl2] with AgOTf in dichloromethane at room temperature (Scheme 2). The desired product was isolated in good yields (75%) as an air stable solid. Interestingly, the dimer exhibited different reactivities towards deuterated solvents upon NMR analysis (Scheme 3). In fact, in CDCl3, a signal corresponding to [(dppe)PdCl2] starting material progressively increases in intensity over time in the 31P{1H} NMR spectrum, revealing chlorination at Pd(II) in CDCl3. However, in DMSO-d6, dimer cleavage to form the DMSO adduct is observed through the appearance of two sets of doublets in the 31P{1H} NMR spectrum (δ = 70.9 and 69.0 ppm), corresponding to the chemically inequivalent P atoms of dppe upon DMSO binding to the Pt(II) centre. In acetone-d6, a singlet signal is observed at δ = 76.9 ppm in the 31P{1H} NMR spectrum, supporting the chemical equivalence of each P atom within the structure, as well as a doublet signal in the aliphatic region in the 13C{1H} NMR spectrum, corresponding to the C atoms within the dppe backbone (Fig. S1 and S20). In addition, a singlet signal can be observed at δ = −77.6 ppm in the 19F NMR spectrum, confirming the presence of the triflate anions. Crystals suitable for X-ray diffraction analyses were grown through slow evaporation of a concentrated dichloromethane solution at 4 °C and the structure is reported in the SI (Fig. S193). The synthesis of PtPt-0 was optimised compared to previous reports.47 The precursor complex [(dppe)PtCl2] is obtained via reaction of [(COD)PtCl2] and dppe in dichloromethane.48 The intermediate monomer complex is subsequently reacted with AgOTf in dichloromethane at room temperature for 1.5 h in the absence of light, yielding the desired dimer complex PtPt-0 in 70% yield (Scheme 2).
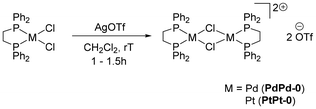 |
| | Scheme 2 Synthesis of the dimer precursors PdPd-0 and PtPt-0. | |
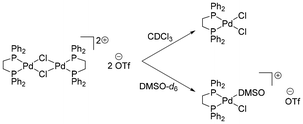 |
| | Scheme 3 Decomposition products observed in solution upon NMR analysis of PdPd-0 in different solvents. | |
Synthesis and spectroscopic properties of monometallic precursor complexes Pd-x and Pt-x (x = 1, 2 or 3)
The monometallic complexes Pd-x and Pt-x were synthesised through slow addition of a solution containing the respective dimer precursors PdPd-0 or PtPt-0 to another solution containing two equivalents of the respective diphosphine ligands (1,4-bis(diphenylphosphino)butane (dppb, Pd-1 or Pt-1), 1,5-bis(diphenylphosphino)pentane (dpppent, Pd-2 or Pt-2) and 1,6-bis(diphenylphosphino)hexane (dpph, Pd-3 or Pt-3)) in dichloromethane (Scheme 4). The slow addition of the dimer complex prevents the formation of dinuclear species where each P atoms on the ligand would bind to a different Pd(II)/Pt(II) centre. Reactions attempted with shorter chain length diphosphines (dppm, dppe or dppp) resulted in the formation of the desired products as well as a cyclic side-product where the diphosphine bound in a κ2 fashion to the M(II) centre. The mononuclear complexes were obtained in good yields (65–81%) as pale-yellow powders, stable in air for extended periods of time.
 |
| | Scheme 4 Synthesis of the monometallic complexes Pd-x and Pt-x (x = 1, 2 or 3). The letters circled in grey represent the labeling of the P atoms used in-text. | |
FT-IR spectroscopy revealed very similar spectra along all monometallic complexes, with C–H stretching vibrations observed in the range of 3100–2800 cm−1 being the only assignable signals (Fig. S103–S108). The UV-Vis spectra of Pd-x were very similar to one another, with the main features being an intense absorption at 226 nm, a shoulder at 270 nm and a minor absorption at 331 nm (Fig. S125–S127). In Pt-3, the shoulder shifted to 274 nm, and no absorption was observed at 331 nm (Fig. S129). 1H NMR spectra revealed a doublet signal in the aliphatic region in all six cases, corresponding to the κ2-dppe ethylene bridgehead protons. Additional sets of multiplets in the aromatic region, as well as extra signals in the aliphatic region confirmed the binding of the diphosphine ligand to the Pd(II) centre. The aromatic signals integrate to 40 H, corresponding to the four Ph rings attached to P, whereas the aliphatic protons of the tether integrate to the expected values in all three cases (Fig. S2–S7). Further proof of the binding of diphosphine in a κ1-fashion to Pd(II) was revealed by the 31P{1H} NMR spectra: an upfield singlet signal is observed corresponding to the pendant P atom. The three P atoms bound to Pd(II) or Pt(II) were all displayed as doublet of doublets. PA and PC exhibit large trans-coupling to each other, while PB exhibits smaller cis-couplings to PA and PC (see Scheme 4 for atoms labelling) (Fig. S39–S44). These complexes exist as conformers in solution, as can be seen especially in the PC signal, which displays two sets of doublet of doublets. This is not uncommon for such complexes, as conformational changes were observed in other phosphine-containing complexes.49–53 DMSO-stability studies performed via1H and 31P NMR spectroscopy in DMSO-d6 revealed that the complexes maintain their structural integrity in solution over time (Fig. S67–S78). ESI-TOF-MS confirmed the presence of the expected complexes, as the molecular peak of the cation was observed in all cases, with the expected isotope pattern (Fig. S155–S160). Complexes Pd-3 and Pt-x (x = 1, 2 or 3) also displayed an [M + O]+ fragment, likely due to oxidation at the pendant P, which is typical of such complexes under ESI-MS ionisation conditions.38
Synthesis and spectroscopic properties of homobimetallic complexes PdPd-x (x = 1, 2 or 3)
The syntheses of the homodinuclear complexes PdPd-x (x = 1, 2 or 3) were carried out by reacting the dimer precursor PdPd-0 with the respective diphosphine in a 1:1 equivalent in dichloromethane to selectively afford through dimer-cleavage the desired bimetallic complexes in good yields (Scheme 5). Binding of the diphosphine in a μ-fashion was confirmed through the absence of the upfield singlet in the 31P{1H} NMR spectrum, which is observed in the case of complexes Pd-x and Pt-x bearing the pendant P atom. On the other hand, three resonance signals are observed, corresponding to Pd-bound PA, PB and PC (Fig. S45–S47). The 1H NMR spectra are distinct from Pd-x, as the homobimetallic systems are symmetrical. The aromatic region integrates to 60 protons, in line with the presence of a second κ2-dppe moiety. The aliphatic region displays two signals assignable to the κ2-dppe moiety, each integrating to four protons. In addition, resonance signals assigned to the dppx bridging ligand are also observed, with the respective integration values matching depending on the ligand length (Fig. S8–S10). A singlet peak at δ = −77.7 ppm in the 19F NMR spectrum corresponds to the presence of the triflate anion. Similar to the mononuclear complexes, the homobimetallic complexes displayed good stability in DMSO-d6, as no noticeable change was observed in the 1H and 31P NMR spectra over a 180 minutes period (Fig. S79–S84). ESI-MS confirmed the formation of the desired products through the [M-OTf]+ peak with expected isotope pattern in all cases (Fig. S161–S163). The UV-Vis spectra exhibit very similar features as the monometallic complexes Pd-x, with an intense absorption around 225 nm, followed by a clear shoulder at 265 nm and a lower absorption around 335 nm (Fig. S131, S133 and S135).
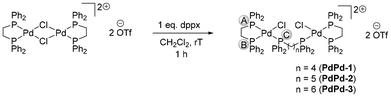 |
| | Scheme 5 Synthesis of the homobimetallic complexes PdPd-x (x = 1, 2 or 3). The letters circled in grey represent the labeling of the P atoms used in-text. | |
Synthesis and characterisation of the heterobimetallic complexes PdRu-x, PtRu-x and PdOs-x (x = 1, 2 or 3)
The heterodinuclear complexes were synthesised in similar fashion, through the addition of dimer precursors [(p-cym)MCl2]2 (M = Ru(II), Os(II)) to a solution of the respective monometallic complexes Pd-x or Pt-x in dichloromethane and stirring at room temperature for 1.5 to 2 hours (Schemes 6 and 7).
 |
| | Scheme 6 Synthesis of the heterobimetallic complexes PdRu-x and PtRu-x (x = 1, 2 or 3). The letters circled in grey represent the labeling of the P atoms used in-text. | |
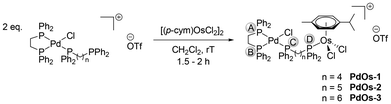 |
| | Scheme 7 Synthesis of the heterobimetallic complexes PdOs-x (x = 1, 2 or 3). The letters circled in grey represent the labeling of the P atoms used in-text. | |
The complexes were obtained as orange to brick-red powders in good yields (61 to 85%) and are air stable over extended periods of time. The 1H NMR spectra of all complexes showed similar signals compared to the monometallic counterparts, with the addition of extra resonance signals corresponding to the 4H of the p-cymene moiety, a doublet corresponding to the CH3 groups of the isopropyl unit, as well as a singlet peak corresponding to the CH3 on p-cymene ring. The expected septet from methine CH could not be observed due to overlapping signals from the alkyl chain of the bridging ligand, nevertheless its presence was determined through 1H–1H 2D COSY NMR experiments (Fig. S11–S19). 13C{1H} NMR spectroscopy further corroborated the incorporation of Ru(II)/Os(II) into the structure, with seven extra signals observed, in line with the seven distinct C atoms present within the p-cymene moiety (Fig. S29–S37). In the case of PdRu-x and PtRu-x, binding of Ru(II) was further confirmed by the presence of a lowfield shifted resonance signal in the 31P{1H} NMR spectrum, in comparison to the precursors bearing a pendant P atom. This observation is not the case for PdOs-x, as the signal remains upfield, in line with the greater shielding effect of Os(II) compared to Ru(II) (Fig. S48–S56). The presence of conformers was observed in all heterobimetallic complexes. In order to further corroborate this, variable temperature NMR spectroscopic investigations was performed on RuPt-3, where coalescence of the PC signals into a clear doublet of doublet was observed from 333 K on (Fig. 3), revealing the existence of conformers in solution. In line with the previously discussed Pd-x, Pt-x and PdPd-x series, the heterobimetallic complexes seem to maintain their structural integrity in DMSO-d6 over time, as no obvious change in the 1H and 31P NMR spectra were observed (Fig. S85–S100). The formation of the desired products was further supported by ESI-MS, where the M+ peak of all complexes was observed (Fig. 2 and Fig. S164–S178). Regarding UV-Vis spectroscopy, a clear difference can be observed when comparing the heterobimetallic complexes to the monometallic precursors and the homobimetallic complexes PdPd-x. In fact, discreet low intensity absorption maxima can be seen at higher concentrations, corresponding to symmetry forbidden electronic d–d transitions occurring at the Ru(II) or Os(II) centres (Fig. S137–S153).
 |
| | Fig. 2 Stacked mass spectra of PdOs-1 (bottom), PdOs-2 (middle) and PdOs-3 (top) zoomed in on the [M+] peak. | |
 |
| | Fig. 3 Variable temperature 31P{1H} NMR studies of PtRu-3, zoomed in on Pc. The FID data was further processed with an exponential apodization function with a line broadening of 1 Hz in order to improve the signal to noise ratio. | |
Single crystal X-ray diffraction investigations
Crystals of PdPd-0, PdPd-1 and PdRu-1, suitable for X-ray diffraction analysis, were grown via slow evaporation of dichloromethane at 4 °C (PdPd-0), or slow diffusion of Et2O into a concentrated solution of the respective complexes in dichloromethane at room temperature (PdPd-1 and PdRu-1) and are the results are presented in Fig. 4–6.
 |
| | Fig. 4 ORTEP representation of complex PdPd-0 in the solid state determined by single crystal X-ray diffraction analysis. Thermal ellipsoids represented at the 30% probability level. H atoms and solvent molecule omitted for clarity. Selected bond lengths [Å]: Pd(1)–Cl(1) 2.4014(5), Pd(1)–Cl(1′) 2.4040(5), Pd(1)–P(1) 2.2257(6), Pd(1)–P(2) 2.2471(5). Selected bond angles [°]: P(1)–Pd(1)–Cl(1) 178.38(2), P(1)–Pd(1)–Cl(1′) 91.661(19), P(2)–Pd(1)–Cl(1′) 174.967(19), P(2)–Pd(1)–Cl(1) 96.039(19), P(2)–Pd(1)–P(1) 84.80(2). | |
 |
| | Fig. 5 ORTEP representation of the PdPd-1 cation in the solid state determined by single crystal X-ray diffraction analysis. Thermal ellipsoids represented at the 30% probability level. H atoms and counteranions omitted for clarity. The phenyl rings of the bridging dppb entity are represented as wireframes for clarity. Selected bond lengths [Å]: Pd(1)–Cl(1) 2.3458(5), Pd(1)–P(1) 2.2980(6), Pd(1)–P(2) 2.2651(6), Pd(1)–P(3) 2.3734(6). Selected bond angles [°]: Cl(1)–Pd(1)–P(3) 89.78(2), P(1)–Pd(1)–Cl(1) 88.64(2), P(1)–Pd(1)–P(3) 175.60(2), P(2)–Pd(1)–Cl(1) 172.88(2), P(2)–Pd(1)–P(1) 85.14(2), P(2)–Pd(1)–P(3) 96.18(2). | |
 |
| | Fig. 6 ORTEP representation of the PdRu-1 cation in the solid state determined by single crystal X-ray diffraction analysis. Thermal ellipsoids represented at the 30% probability level. H atoms and counteranion omitted for clarity. The phenyl rings of the bridging dppb entity are represented as wireframes for clarity. Selected bond lengths [Å]: Pd(1)–P(1) 2.2472(17), Pd(1)–Cl(1) 2.3401(18), Pd(1)–P(2) 2.3065(18), Pd(1)–P(3) 2.3493(18), Ru(1)–Cl(3) 2.419(3), Ru(1)–Cl(2) 2.412(3), Ru(1)–P(4) 2.344(2), Ru(1)–C(58) 2.208(8). Selected bond angles[°]: P(1)–Pd(1)–Cl(1) 169.33(7), P(1)–Pd(1)–P(2) 83.77(6), P(1)–Pd(1)–P(3) 98.87(6), Cl(1)–Pd(1)–P(3) 90.46(7), P(2)–Pd(1)–Cl(1) 86.79(7), P(2)–Pd(1)–P(3) 177.03(7), Cl(2)–Ru(1)–Cl(3) 88.03(11), P(4)–Ru(1)–Cl(3) 88.21(8), P(4)–Ru(1)–Cl(2) 84.21(8). | |
The dimer complexes [(dppe)PdCl]2(2ClO4) and [(dppe)PdCl]2(2BF4) have previously been characterised via single crystal X-ray diffraction analysis.44,45 Herein we report the solid state crystal structure of the analogous complex [(dppe)PdCl]2(2OTf). PdPd-0 crystallised in the triclinic crystal system with the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (Fig. 4). The cationic species has an inversion centre in between the two Pd atoms and presents strikingly similar features than the previously reported analogous structures, which is to be expected.
(Fig. 4). The cationic species has an inversion centre in between the two Pd atoms and presents strikingly similar features than the previously reported analogous structures, which is to be expected.
PdPd-1 crystallises in the monoclinic crystal system with the space group P21/c (Fig. 5). The molecule bears an inversion centre between the two central C atoms of the bridging dppb ligand. The complex displays a distorted square planar geometry at Pd(II), with bond angles of 88.63° (Cl1–Pd1–P1), 85.14° (P1–Pd1–P2), 96.18° (P2–Pd1–P3) and 89.78° (P3–Pd1–Cl1). This is similar to the complex [(κ2-dppe)Pd(Ph)]2(µ-dppe), reported by Zhuravel and co-workers, where the bond angle P2–Pd1–P3 was found to be 100.95°, whereas the remaining angles were all below 90°.54 This large difference is likely due to steric strain between the phenyl rings on each P atoms. The main difference between the herein reported structure and that of Zhuravel et al. is the shorter Pd(1)–P(2) bond length in PdPd-1, due to the lesser electron donating abilities of Cl compared to Ph. Another noteworthy detail are the more obtuse bond angles around P(3) in the bridging dppb moiety compared to the free ligand. Indeed, while the bond angle between C(Ph)–P–C(Ph) remains similar between free dppb and PdPd-1, both C(Ph)–P–C(chain) angles average 105.51° in PdPd-1, where they average 101.54° in free dppb.55 The complete crystal data for PdPd-1 can be found in Tables S8–S15.
Complex PdRu-1 crystallises in the monoclinic crystal system with the space group P21/n (Fig. 6). The geometry and metrical parameters around Pd(II) is analogous to PdPd-1 within narrow limits. The geometry around the Ru(II) centre is distorted octahedral, where the p-cymene unit occupies three binding sites, while the remaining ligands occupy the remaining three. This “piano-stool” configuration for Ru(II) is common and multiple examples of [(p-cym)RuCl2(PPh2R)] complexes have been characterised via X-ray diffraction analyses.56–59 The complete crystal data for PdRu-1 can be found in Tables S16–S22.
Cell viability studies
All complexes were tested at 10 µM in vitro against cervical cancer (HeLa and Ca Ski), pancreatic cancer (PANC-1 and CFPAC-1), rhabdomyosarcoma (RD and RH30), and breast cancer (MCF-7 and MDA-MB-231) cell lines, and one non-malignant cell line (MRC-5). The initial concentration of 10 µM was selected based on the National Cancer Institute (NCI, USA) screening protocol, which uses this threshold to identify compounds with promising anticancer activity.60–62 Compounds that inhibit cell viability by more than 50% at this concentration are considered sufficiently active to warrant further investigation. The results are tabulated in Table 1 and the related plots can be found in the SI (Fig. S179–S183). The most notable results were observed on the two pancreatic cancer cell lines, where all heterobimetallic complexes reduced cell viability by more than 50%, as well as outperformed cisplatin. The two homobimetallic complexes PdPd-1 and PdPd-2 displayed similar results in PANC-1 cells. All monometallic complexes, except for Pd-3, reduced cell viability by more than 50% in PANC-1 cells. Also worth mentioning is that all heterobimetallic complexes outperformed cisplatin in RH30 cells, with only PdRu-2 and PdOs-2 reducing cell viability by less than 50%, albeit by a narrow margin (51.86% and 52.65%, respectively). Regarding the Ca Ski cell line, all heterobimetallic complexes reduced cell viability by more than 50%, with PdRu-1, PtRu-3 and PdOs-3 outperforming cisplatin. On HeLa cells, three complexes (PdRu-1, PtRu-1 and PdOs-3) reduced cell viability by more than 50%, whereas all heterobimetallic complexes except PdOs-2 outperformed cisplatin, as well as Pd-1 and PdPd-2. Most of the complexes tested outperformed cisplatin in RD cells. However, none reduced cell viability by more than 50%. In MCF-7 cells, only PdRu-1 reduced cell viability by more than 50%, whereas all heterobimetallic complexes, except PtRu-1, as well as Pd-1, Pt-3 and PdPd-1, outperformed cisplatin. Finally, in MDA-MB-231 cells, the heterobimetallic complexes generally perform better, with PdRu-1, PtRu-3 and PdOs-2 reducing cell viability by more than 50% and outperforming cisplatin. None of the complexes tested at 10 µM inhibited non-malignant MRC-5 cell viability by more than 50%, and none were more cytotoxic than cisplatin, which indicates the potential for selectivity in this molecular design.
Table 1 Summary of percentage (%) cell viability of various cancer cell lines after 48 h 10 µM treatment with complexes
| The complexes that reduced % cell viability by more than 50% are highlighted in red, and the complexes outperforming cisplatin are highlighted in bold. |

|
PdRu-1 and PdOs-3 were determined to be the best-performing complexes overall through averaging of the percentage cell viability values obtained over the eight cancer cell lines. These two complexes were therefore further investigated in cervical and pancreatic cell lines because they were most cytotoxic in those, while displaying minimal cytotoxicity against the non-malignant cell line. The IC50 values were determined by multidose experiments in the cervical cancer and pancreatic cancer cell lines as well as in the non-malignant FG0 cell line. The related selectivity indices were derived by dividing the IC50 values in the non-malignant cells by the IC50 values in the cancer cells (Table 2 and Fig. S184–S186). Both complexes outperformed cisplatin in the four cell lines tested.63,64 In the cervical cancer cells, PdRu-1 showed high selectivity (SI ≥ 5) for Ca Ski cells and moderate selectivity (2 ≤ SI ≤ 5) for HeLa cells, whereas PdOs-3 showed moderate selectivity for Ca Ski cells only. In pancreatic cancer cell lines, both PdRu-1 and PdOs-3 displayed moderate selectivity for PANC-1 cells but low selectivity (SI ≤ 2) towards CFPAC-1 cells.
Table 2 IC50 values (µM) determined for the most active complexes, PdRu-1 and PdOs-3, in cervical, pancreatic and non-malignant cell lines
| Complex |
|
Cervical cancer |
Pancreatic cancer |
Non-malignant |
| HeLa |
Ca-Ski |
PANC-1 |
CFPAC-1 |
FG0 |
| Selectivity indices (SI) derived by the following equation: IC50 FG0 cells/IC50 cancer cells, where SI ≤ 2: low selectivity, 2 ≤ SI ≤ 5: moderate selectivity, and SI ≥ 5: high selectivity. |
|
PdRu-1
|
IC50 |
6.87 ± 0.97 |
3.43 ± 1.32 |
5.69 ± 1.87 |
17.56 ± 1.14 |
21.12 ± 1.75 |
|
SI
|
3.07 |
6.16 |
3.71 |
1.20 |
— |
|
PdOs-3
|
IC50 |
23.84 ± 2.59 |
12.05 ± 1.99 |
12.53 ± 1.61 |
28.07 ± 1.08 |
30.41 ± 2.09 |
|
SI
|
1.28 |
2.52 |
2.43 |
1.08 |
— |
| Cisplatin |
IC50 |
66.03 (ref. 63) |
17.46 (ref. 63) |
87.86 ± 2.29 (ref. 64) |
41.31 (ref. 63) |
— |
Surprisingly, a correlation could not be drawn between diphosphine spacer length and biological activity, which contrasts with our previous report on some heterobimetallic Fe–Ru(II) complexes.38 However, a clear trend can be observed in terms of activity of the different types of complexes reported herein: the heterobimetallic complexes perform better than their homo- and monometallic counterparts across all cancer cell lines tested, apart from few exceptions. Apart from CFPAC-1, where the Os-based heterobimetallic complexes seemed somewhat less active than the Ru-containing complexes, having Ru(II) or Os(II) does not seem to greatly affect activity. In Ca Ski and RH30, the homobimetallic complexes generally outperform the monometallic complexes. In the other cell lines, activities are roughly comparable. Details on the cell-lines: PANC-1 (ATCC; CRL-1469), CFPAC-1 (ATCC; CRL-1918), MRC-5 (ATCC; CCL-171), HeLa (ATCC; CCL-2), Ca Ski (ATCC; CRL-1550); RD (ATCC; CCL-136TM), MCF-7 (ATCC; HTB-22TM), MDA-MB-231 (ATCC; HTB-26TM).
Aquation studies
Aquation studies were conducted using UV-Vis spectroscopy on one example complex per homologous series at C = 5 × 10−6 M in a PBS buffer:DMSO solution (90:10 v/v) in order to identify the potential species forming in biological media. All the kinetic plots can be found in the SI (Fig. S187–S192). In the case of monometallic complex Pd-2, biphasic spectral changes are observed, which fit a double-exponential decay function, indicating two processes occurring one after the other. The first process (k1 = 2.38 × 10−2 min−1) is tentatively attributed to hydrolysis at the Pd(II) centre, followed by HCl elimination (k2 = 2.58 × 10−6 min−1) to form a hydroxo complex. Phosphane dissociation, to account for the second process can be entirely ruled out, as NMR experiments performed over this time period did not show any phosphane dissociation. Similar results were observed in the case of Pt-2, even though the rates are comparatively, faster than Pd-2. The initial ligand exchange rate is in the same order of magnitude (k1 = 7.15 × 10−2 min−1), which comes as a surprise, owing to Pd's typically faster ligand exchange kinetics.65 HCl elimination is seemingly much faster in Pt-2 (k2 = 1.15 × 10−2 min−1) compared to Pd-2. Regarding PdPd-3, a single exponential decay (k1 = 2.13 × 10−3 min−1) is observed on the time scale of the experiment (Fig. 7). This likely indicates either one of two things: a. aquation step occurs at one of the Pd(II) centers, generating a monoaqua complex bearing a +3 charge and a Cl counteranion, or b. aquation occurs, followed by fast HCl elimination (faster than the experimental time scale) in order to restore the more stable +2 charged complex. The rate of aquation is somewhat similar to k1 in Pd-2, which supports aquation at Pd(II) as the first/sole step in PdPd-3. Concerning the heterobimetallic complexes, the speciation is more complicated. Regarding PdRu-2, a two steps process with first a decrease in absorbance (k1 = 4.46 × 10−2 min−1), followed by an increase (k2 = 9.17 × 10−2 min−1) is observed. However, PtRu-2 follows exclusively a double exponential decay indicating also a two-step mechanism (k1 = 2.97 × 10−1 min−1 and k2 = 2.71 × 10−2 min−1). Finally, PdOs-2 undergoes a three steps process, with first a single exponential increase in absorbance (k1 = 2.88 × 10−1 min−1), followed by a double exponential decay (k2 = 2.52 × 10−2 min−1 and k3 = 3.57 × 10−2 min−1). We tentatively assign hydrolysis at Ru(II), followed by a second hydrolysis at Ru(II) and HCl elimination in the case of PtRu-2 to afford an Ru(II) aqua hydroxo complex as with previous findings reported by us (Scheme 8, bottom).38 In the case of PdRu-2, the decrease in absorbance indicates likely hydrolysis at Ru(II), while the increase in absorbance is indicative of hydrolysis occurring at Pd(II), as is also observed in PdOs-2. It is hence the case in PdOs-2, that hydrolysis occurs first at Pd(II), which would explain the increase in absorbance observed. Hydrolysis at Os(II) happens afterwards due to Os increased inertness. The third process can either be HCl elimination to form a Pd-hydroxo complex, or else hydrolysis followed by HCl elimination at Os(II), in a similar fashion as PtRu-2.
 |
| | Fig. 7 UV-Vis spectral changes over time of PdPd-3 (C = 5 × 10−6 M) in a 90:10 PBS buffer (pH = 7.45):DMSO solution at 37 °C (top left), plot of the change in absorbance over time at 326 nm (top right), and hypothesised mechanism (bottom). | |
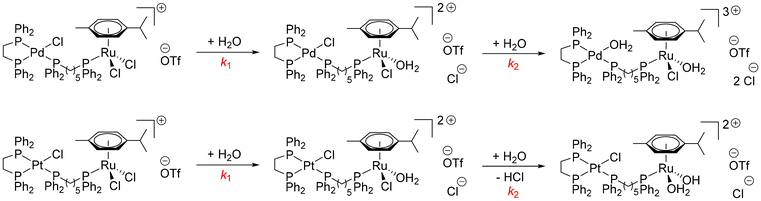 |
| | Scheme 8 Putative product formation upon hydrolysis of PdRu-2 (top) and PtRu-2 (bottom) in biological media. | |
A tentative link to activity in vitro can be derived from the above speciation experiments. Indeed, the mononuclear complexes were determined to be rather inactive in vitro, which is consistent with the formation of a more biologically inert hydroxo complex. The homodinuclear complexes showed slightly higher biological activity than their mononuclear counterparts, which can be attributed to one Pd(II) still bearing a labile Cl co-ligand which can later be substituted for binding to a suitable biological target, as is the case with the mechanism of cisplatin.66 Regarding the heterobimetallic complexes, the PtRu series showed slightly less activity than the Pd-based examples, which might be due to a more inert Pt(II) center, whereas a labile OH2 bound to Ru(II) is present. Regarding the PdOs series, in both possible species a labile OH2 is present at Os(II), whereas in the case of Pd, either a Pd-hydroxo or Pd-aqua complex is possible, Pd-aqua likely being more active. Finally, the PdRu series was determined to be the most active of all, which is consistent with the formation of a doubly aquated complex, with labile OH2 groups on both Pd(II) and Ru(II) (Scheme 8, top).
Conclusions
Mononuclear Pd(II) and Pt(II)-based complexes, bearing pendant diphosphines, their corresponding homodinuclear Pd–Pd, and heterodinuclear Pd(II)–Ru(II), Pt(II)–Ru(II) and Pd(II)–Os(II) complexes bearing bridging diphosphines have been reported and fully characterised by spectroscopic means. Their in vitro cytotoxic activity was determined on eight cancerous cell lines and a non-malignant cell line, as well as IC50 values and selectivity indices of the most active complexes in the series. No obvious relationship between the diphosphine tether or spacer length and activity could be derived, although a strong correlation between the heterobimetallic nature of the complexes and higher biological activity was observed, possibly due to the different species formed upon aquation in biological media, as demonstrated by our tentative aquation studies. The monometallic and homobimetallic complexes displayed suboptimal activities in vitro, which suggests the need for hetero metals in future bimetallic anticancer drug designs for complexes containing Pd(II) and Pt(II). The complexes reported herein highlight a novel class of compounds which encourages the pursuit of heterobimetallic scaffolds as an enhanced molecular design for the next generation of anticancer drugs.
Experimental section
General considerations
The chemicals were handled using standard Schlenk techniques, unless otherwise stated. Solvents used were dried and degassed using standard techniques prior to use. [(κ2-dppe)PdCl2] (TCI), AgOTf (Sigma-Aldrich), 1,4-bis(diphenylphosphino)butane (dppb) (Sigma-Aldrich), 1,5-bis(diphenylphosphino)pentane (dpppent) (Sigma-Aldrich), 1,6-bis(diphenylphosphino)hexane (dpph) (Sigma-Aldrich), [(p-cym)RuCl2]2 (Strem), [(COD)PtCl2] (Strem), were used as received. EtOH (100%), acetonitrile, dichloromethane (stab. amylene), THF, Et2O were purchased from Biosolve. DMSO-d6, CDCl3 and acetone-d6 were purchased from VWR chemicals. [(p-cym)OsCl2]2 and [(κ2-dppe)PdCl2] were synthesised according to literature proceedings.48,67 NMR spectra were recorded on a JEOL JNM-ECZ400s FT NMR spectrometer. NMR data was processed using MestReNova v14.3.0, and reported in parts per million (ppm) referenced to TMS (1H and 13C{1H}), 85% phosphoric acid (31P{1H}) or CFCl3 (19F{1H}). The peak multiplicities abbreviations are as follows: s = singlet, d = doublet, t = triplet, q = quartet, sept = septet, m = multiplet, ps = pseudo, br = broad. FT-IR spectra were recorded on a Shimadzu IRSpirit FTIR spectrometer, using the following parameters: 64 scans, resolution 4, Happ-Genzel apodization, with a scan range of 4000–400 cm−1. The signals reported as follows: s = strong, m = medium, w = weak, v = very, br = broad. UV-Vis absorption spectra were recorded using 1 cm pathlength quartz cuvettes on a Shimadzu UV-3600 iPlus-1 spectrometer within a range of 190–800 nm. OriginPro 2018 SR1 b9.5.1.195 was used for data processing of IR and UV-Vis data. Mass spectrometry was conducted using a Waters Syanpt G2S ESI-TOF-MS (3 kV) and the spectra processed using MassLynx™ v4.2 and the experimentally determined spectra accuracy and isotope patterns were checked using the built-in isotope calculator. Melting points were obtained on a Stuart SMP10 Digital Melting Point apparatus, performed in duplicate and the average reported in °C. Single crystal X-ray diffraction analysis was performed on a Rigaku XtaLAB Synergy, Dualflex, HyPix-Arc 100 diffractometer using Cu radiation (λ = 1.54184 Å). CrysAlisPro was used to reduce data to Fo2 and corrected for absorption effects. Structures were solved using SHELXT and refined with SHELXL as implemented in OLEX2.
Synthesis of [(κ2-dppe)PdCl]2(2OTf) (PdPd-0).
2.05 g (3.56 mmol, 1.00 eq.) of [(κ2-dppe)PdCl2] were dissolved in dichloromethane (35 mL). 0.914 g (3.56 mmol, 1.00 eq.) of AgOTf was added to the solution while stirring at room temperature. The reaction vessel was covered in aluminium foil and the mixture was left to stir at room temperature for 1 hour. The resulting solution was filtered through a Schlenk-frit, and the filtrate evaporated and dried in vacuo. Properties: reverts to [(κ2-dppe)PdCl2] in chloroform, forms [(κ2-dppe)PdCl(DMSO)](OTf) in dimethyl sulfoxide, insoluble in tetrahydrofuran and soluble in acetone. 1.99 g (81%) of bright yellow solid, air stable. 1H NMR (400.130 MHz, acetone-d6, 298 K): δ 7.91–7.81 (16H, m, PPh), 7.77–7.70 (8H, m, PPh), 7.65–7.57 (16H, m, PPh), 3.14 (8H, ps d, CH2, κ2-dppe). 13C{1H} NMR (100.613 MHz, acetone-d6, 298 K): δ 134.6–134.3 (m, PPh), 130.8–130.4 (m, PPh), 28.2 (ps d, CH2, κ2-dppe). 31P{1H} NMR (161.976 MHz, acetone-d6, 298 K): δ 76.9 (br s, Δν1/2 = 158.7 Hz, PPh2). 31P{1H} NMR (161.976 MHz, DMSO-d6, 298 K): δ 70.9 (br ps d, Δν1/2 = 60.0 Hz, PA), 69.0 (br ps d, Δν1/2 = 60.8 Hz, 2JPB–PA = 11.5 Hz, PB). 19F NMR (376.498 MHz, DMSO-d6, 298 K): δ −77.6 (OTf). FT-IR (cm−1): 3058 (vw), 2963 (vw), 1436 (m), 1256 (br s), 1223 (m), 1148 (m), 1099 (m), 1027 (s), 997 (w), 815 (m), 707 (m), 687 (s), 635 (vs), 527 (vs), 480 (m). UV/Vis λmax (dichloromethane): 227 nm (major, ε = 120952 M−1 cm−1), 276 nm (ε = 60533 M−1 cm−1), 346 (minor, ε = 27924 M−1 cm−1). ESI-TOF-MS (3 kV, dichloromethane): m/z calcd for [M-OTf]+, 1229.0; expt., 1228.9. m/z calcd for [M-2OTf + Cl]+, 1115.0; expt., 1114.9. m/z calcd for [(κ2-dppe)Pd(OTf)]+, 653.0; expt., 653.0. m/z calcd for [(κ2-dppe)PdCl]+, 541.0; expt., 541.0 (100%).
Synthesis of [(κ2-dppe)PtCl]2(2OTf) (PtPt-0).
450 mg of [Pt(κ2-dppe)Cl2] (0.677 mmol, 1.00 eq.) and 174 mg of AgOTf (0.677 mmol, 1.00 eq.) were added as solids in a Schlenk tube, and dissolved in 50 mL of dichloromethane. The solution was stirred in the dark for 1.5 h at room temperature, and subsequently filtered, washed with Et2O and dried in vacuo. 381 mg (70%) of white powder, air stable. Melting point: progressive browning from 160 °C to 180 °C. 1H NMR (400.130 MHz, DMSO-d6, 293 K): δ 7.94–7.55 (40 H, m, PPh), 2.73 (8 H, m, 4 × CH2, κ2-dppe). 1H NMR (400.130 MHz, acetone-d6, 293 K): δ 8.48–7.14 (40 H, unresolved m, PPh), 3.06 (8 H, br s, κ2-dppe). 13C{1H}NMR (100.613 MHz, DMSO-d6, 293 K): δ 133.7 (unresolved m, PPh), 132.7 (dd, xJC–P = 17.95 Hz, xJC–P = 2.71 Hz, PPh), 129.8–129.6 (unresolved m, PPh), 129.3 (dd, xJC–P = 26.64 Hz, xJC–P = 11.60 Hz, PPh), 126.1 (d, xJC–P = 66.71 Hz, PPh), 125.9 (d, xJC–P = 64.45 Hz, PPh), 120.7 (d, xJC–P = 322.79 Hz, PPh), 27.4 (ps s, κ2-dppe). 31P{1H} NMR (161.976 MHz, DMSO-d6, 293 K): δ 45.9 (br s, 1JP–Pt = 3636 Hz, Δν1/2 = 66.3 Hz). 31P{1H} NMR (161.976 MHz, acetone-d6, 293 K): δ 49.0 (s, 1JP–Pt = 3780.8 Hz). 19F NMR (376.498 MHz, DMSO-d6, 293 K): δ −77.7 (s, OTf). FT-IR (cm−1): 3057 (w), 2963 (w), 2922 (w), 1438 (m), 1258 (s), 1224 (m), 1143 (m), 1104 (s), 1027 (s), 798 (s), 720 (s), 708 (s), 688 (s), 635 (s), 532 (s), 517 (s), 480 (s). UV/Vis λmax (dichloromethane): 226 nm (major, ε = 55924 M−1 cm−1), 302 nm (minor, ε = 3676 M−1 cm−1). ESI-TOF-MS (3 kV, dichloromethane): m/z calcd for [(κ2dppe)PtCl]2Cl+, 1293.1; expt., 1293.1 (<1%); m/z calcd for [(κ2dppe)PtCl]+, 629.1; expt., 629.1 (100%).
General procedure for the synthesis of [(κ2-dppe)PdCl(κ1-dppx)](OTf) (Pd-x)
200 mg (0.145 mmol, 1.00 eq.) of PdPd-0 were dissolved in dichloromethane (5 mL). Separately, the respective diphosphine (0.29 mmol, 2.00 eq.) was dissolved in dichloromethane (5 mL) in a Schlenk flask. The solution of PdPd-0 was added dropwise to the diphosphine solution and the resulting solution was stirred at room temperature for one hour. The solvent was removed in vacuo, after which the resulting solid was washed with diethyl ether (10 mL). The product was subsequently dried in vacuo overnight. (In the case of Pd-3, 142 mg (0.103 mmol, 1.00 eq.) of PdPd-0 were used in the synthesis).
Characterisation of [(κ2-dppe)PdCl(κ1-dppb)](OTf) (Pd-1)
235 mg (72%) of pale yellow solid, air stable. Melting point: 125–130 °C (melt), 150 °C + (dec). 1H NMR (400.130 MHz, DMSO-d6, 298 K): δ 7.90–7.10 (40H, m, PPh), 2.85 (2H, br d, 2JH–P = 35 Hz, PCH2ACH2BP, κ2-dppe), 2.46 (obscured by solvent, 2H, br d, PCH2ACH2BP, κ2-dppe), 2.18 (2H, br s, Δν1/2 = 27.3 Hz, 1 × CH2, dppb), 2.10–1.80 (2H, br m, 1 × CH2, dppb), 1.47 (2H, br s, Δν1/2 = 27.1 Hz, 1 × CH2, dppb), 1.27 (2H, br s, Δν1/2 = 27.7 Hz, 1 × CH2, dppb). 13C{1H} NMR (100.613 MHz, DMSO-d6, 298 K, aliphatic signals not visible, assigned from 1H–13C 2D HSQC spectrum): δ 134.3–128.9 (unresolved m, PPh), 33.2 (PCAH2CBH2P, κ2-dppe), 26.8 (1 × CH2, dppb), 26.6 (1 × CH2, dppb), 26.5 (1 × CH2, dppb), 25.1 (1 × CH2, dppb), 24.1 (PCAH2CBH2P, κ2-dppe). 31P{1H} NMR (161.976 MHz, DMSO-d6, 298 K): δ 65.5–66.4 (br d, PB, conformer A + B), 64.8 (dd, 2JPA–PC = 411 Hz, 2JPA–PB = 7.3 Hz, PA, conformer A + B), 18.1 (dd, 2JPC–PA = 411.2 Hz, 2JPC–PB = 17.5 Hz, PC, conformer B), 17.8 (dd, 2JPC–PA = 410.2 Hz, 2JPC–PB = 17.9 Hz, PC, conformer A), −15.9 (br s, Δν1/2 = 205 Hz, PD, conformer B), −17.2 (br s, Δν1/2 = 32.5 Hz, PD, conformer A). 19F NMR (376.498 MHz, DMSO-d6, 298 K): δ −77.6 (OTf). FT-IR (cm−1): 3055 (vw), 2933 (vw), 1436 (m), 1262 (vs), 1223 (m), 1147 (m), 1101 (m), 1029 (s), 999 (m), 741 (m), 690 (vs), 635 (vs), 530 (s), 516 (s), 480 (s). UV/Vis λmax (dichloromethane): 227 (major), 267 (shoulder), 331 (minor). ESI-TOF-MS (3 kV, dichloromethane): m/z calcd for [(κ2-dppe)PdCl(κ1-dppb)]+, 965.2; expt., 965.4 (100%). m/z calcd for [(κ2-dppe)PdCl]+, 541.0; expt., 541.1 (7%).
Characterisation of [(κ2-dppe)PdCl(κ1-dpppent)](OTf) (Pd-2)
216 mg (76%) of pale-yellow solid, air stable. 1H NMR (400.130 MHz, DMSO-d6, 298 K): δ 7.85–7.14 (40 H, unresolved m, PPh), 2.84 (2 H, br d, 2JH–P = 34.9 Hz, PCH2ACH2BP, κ2-dppe), 2.47 (obscured by solvent) (2 H, obscured br d, PCH2ACH2BP, κ2-dppe), 2.34–2.16 (2 H, br s, Δν1/2 = 27.8 Hz, 1 × CH2, dpppent), 2.03–1.88 (2 H, br m, Δν1/2 = 28.6 Hz 1 × CH2, dpppent), 1.55–1.26 (4 H, m, 2 × CH2, dpppent), 1.26–1.13 (2 H, br s, Δν1/2 = 24.4 Hz, 1 × CH2, dpppent). 13C{1H} NMR (signals assigned from HSQC spectrum) (100.613 MHz, DMSO-d6, 298 K): δ 128.9–134.3 (unresolved m, PPh), 33.3 (s, PCH2ACH2BP, κ2-dppe), 31.9 (1 × CH2, dppp), 31.8 (1 × CH2, dppp), 26.5 (1 × CH2, dppp), 25.3 (1 × CH2, dppp), 25.2 (1 × CH2, dppp), 24.3 (1 × Ph2PCAH2CBH2PPh2). 31P{1H} NMR (161.976 MHz, DMSO-d6, 298 K): δ 66.5–66.1 (br m, Δν1/2 = 36.5 Hz, PB, conformer A + B), 64.7 (dd, 2JPA–PC = 411.9 Hz, 2JPA–PB = 6.8 Hz, PA, conformer B), 64.6 (dd, 2JPA–PC = 411.1 Hz, 2JPA–PB = 5.1 Hz, PA, conformer A), 18.1 (dd, 2JPC–PA = 411.8 Hz, 2JPC–PB = 19.7 Hz, PC, conformer B), 17.8 (dd, 2JPC–PA = 408.6 Hz, 2JPC–PB = 19.5 Hz, PC, conformer A), −16.1 (br s, (in baseline), PD, conformer B), −16.9 (br s, Δν1/2 = 16.6 Hz, PD, conformer A). 19F NMR (376.498 MHz, DMSO-d6, 298 K): δ −77.6 (OTf). FT-IR (cm−1): 3054 (vw), 2926 (vw), 2855 (vw), 1434 (m), 1262 (s), 1221 (w), 1147 (m), 1099 (m), 1030 (s), 999 (w), 741 (m), 690 (vs), 635 (vs), 530 (s), 514 (s), 481 (s). UV/Vis λmax (dichloromethane): 227 (major), 261 (shoulder), 332 (minor). ESI-TOF-MS (3 kV, dichloromethane): m/z calcd for [(κ1-dppe)PdCl(κ1dppp)]+, 979.2; expt., 979.3 (100%). m/z calcd for [(κ2-dppe)PdCl]+, 541.0; expt., 541.1 (24%). Temperature studies: 1H NMR (400.130 MHz, DMSO-d6, 343 K): δ 7.91–7.09 (40 H, unresolved m, PPh), 2.82 (2 H, br d, 2JH–P = 33.3 Hz, PCH2ACH2BP, κ2-dppe), 2.46 (obscured by solvent) (2 H, obscured br d, PCH2ACH2BP, κ2-dppe), 2.34–1.85 (4 H, ps m, 2 × CH2, dpppent, pseudo coalescent signals), 1.53–1.15 (6 H, ps br s, Δν1/2 = 55.1 Hz, 3 × CH2, dpppent, coalescent signals). 1H NMR (400.130 MHz, DMSO-d6, 353 K): δ 7.91–7.12 (40 H, unresolved m, PPh), 2.82 (2 H, br d, 2JH–P = 32.5 Hz, PCH2ACH2BP, κ2-dppe), 2.45 (obscured by solvent) (2 H, obscured br d, PCH2ACH2BP, κ2-dppe), 2.30–1.90 (4 H, ps m, 2 × CH2, dpppent, pseudo coalescent signals), 1.53–1.15 (6 H, br s, Δν1/2 = 55.8 Hz, 3 × CH2, dpppent, coalescent signals). 31P{1H} NMR (161.976 MHz, DMSO-d6, 343 K): δ 66.0 (br s, Δν1/2 = 30.8 Hz, PB, coalescent signals), 64.7 (ps dd, 2JPA–PC = 412.0 Hz, PA, coalescent signals), 18.2 (ps d, 2JPC–PA = 416.0 Hz, PC, coalescent signals), −14.8 (m, PD, conformer B), −15.5 (br s, Δν1/2 = 72.4 Hz, PD, conformer A). 31P{1H} NMR (161.976 MHz, DMSO-d6, 353 K): δ 66.0 (br s, Δν1/2 = 30.1 Hz, PB, coalescent signals), 64.8 (ps dd, 2JPA–PC = 406.8 Hz, PA, coalescent signals), 18.2 (ps d, 2JPC–PA = 411.9 Hz, PC, coalescent signals), −14.2 (m, in baseline, PD, conformer B), −15.3 (br s, Δν1/2 = 64.4 Hz, PD, conformer A).
Characterisation of [(κ2-dppe)PdCl(κ1-dpph)](OTf) (Pd-3)
159 mg (77%) of pale-yellow solid, air stable. 1H NMR (400.130 MHz, DMSO-d6, 298 K): δ 7.91–7.10 (40 H, unresolved m, PPh), 2.84 (2 H, br d, 2JH–P = 34.7 Hz, PCH2ACH2BP, κ2-dppe), 2.44 (obscured by solvent) (2 H, obscured br d, PCH2ACH2BP, κ2-dppe), 2.29 (2 H, br s, Δν1/2 = 27.4 Hz, 1 × CH2, dpph), 2.01 (2 H, br s, Δν1/2 = 21.8 Hz, 1 × CH2, dpph), 1.40–1.00 (8 H, unresolved m, 4 × CH2, dpph). 13C{1H} NMR (signals assigned from HSQC spectrum) (100.613 MHz, DMSO-d6, 298 K): δ 134.3–128.9 (unresolved m, PPh), 33.2 (PCH2ACH2BP, κ2-dppe), 30.6 (1 × CH2, dpph), 30.3 (1 × CH2, dpph), 30.1 (1 × CH2, dpph), 26.7 (1 × CH2, dpph), 25.6 (1 × CH2, dpph), 25.3 (1 × CH2, dpph), 24.3 (PCH2ACH2BP, κ2-dppe). 31P{1H} NMR (161.976 MHz, DMSO-d6, 298 K): δ 66.5–66.1 (br d, Δν1/2 = 38.3 Hz, PB, conformer A + B), 64.7 (dd, 2JPA–PC = 415 Hz, 2JPA–PB = 5 Hz, PA, conformer B), 64.6 (dd, 2JPA–PC = 411.2 Hz, 2JPA–PB = 5.4 Hz, PA, conformer A), 18.0 (dd, 2JPC–PA = 410.7 Hz, 2JPC–PB = 19.5 Hz, PC, conformer B), 17.6 (dd, in baseline, PC, conformer A), −16.5 (br s, in baseline PD, conformer B), −16.9 (br s, Δν1/2 = 16.6 Hz, PD, conformer A). 19F NMR (376.498 MHz, DMSO-d6, 298 K): δ −77.6 (OTf). FT-IR (cm−1): 3054 (vw), 2924 (vw), 2854 (vw), 1434 (s), 1260 (s), 1223 (w), 1145 (m), 1099 (m), 1029 (s), 999 (m), 816 (m), 741 (m), 688 (vs), 635 (vs), 530 (s), 516 (s), 481 (s). UV/Vis λmax (dichloromethane): 226 nm (major, ε = 47609 M−1 cm−1), 259 nm (shoulder, ε = 32171 M−1 cm−1), 332 nm (minor, ε = 21752 M−1 cm−1). ESI-TOF-MS (3 kV, dichloromethane): m/z calcd for [(κ1-dppe)PdCl(κ1dpph)]+, 993.2; expt., 993.3 (100%). m/z calcd for [(κ2-dppe)PdCl]+, 541.0; expt., 541.0 (43%).
General procedure for the synthesis of [(κ2-dppe)PtCl(κ1-dppx)](OTf) (Pt-x)
A solution of 200 mg (0.125 mmol, 1.00 eq.) of PtPt-0 in 6 mL dichloromethane was added to a solution containing two equivalents of the respective diphosphine in 6 mL of dichloromethane. It was then left to stir for 1 h at room temperature. The solvents were removed in vacuo, the solids were then washed with diethyl ether and dried in vacuo. The products are obtained as off white solids.
Characterisation of [(κ2-dppe)PtCl(κ1-dppb)](OTf) (Pt-1)
220 mg (72%) of off-white solid, air stable. Melting point: 117 °C. 1H NMR (400.130 MHz, DMSO-d6, 293 K): δ 7.90–7.06 (40 H, unresolved m, PPh), 2.73 (2 H, unresolved m, PCH2ACH2BP, κ2-dppe), 2.46 (signal obscured by solvent) (2 H, obscured m, PCH2ACH2BP, κ2-dppe), 2.30 (3 H, unresolved m, CH2, dppb), 1.92 (1 H, t, xJH–X = 7.81 Hz, CH2, dppb), 1.62–1.19 (4 H, unresolved m, 2 × CH2, dppb). 13C{1H} (100.613 MHz, DMSO-d6, 293 K): δ 138.3 (d, xJC–P = 13.6 Hz, PPh), 132.9 (unresolved m, PPh), 131.2 (d, xJC–P = 12.9 Hz, PPh), 128.8 (unresolved m, PPh), 124.7 (d, xJC–P = 17.3 Hz, PPh), 124.1 (d, xJC–P = 17.3 Hz, PPh), 120.7 (d, xJC–P = 322.3 Hz, PPh), 32.7 (br s, PCAH2CBH2P, κ2-dppe), 26.8 (s, dppb), 26.2 (d, xJC–P = 12.3 Hz, dppb), 24.2 (br s, dppb + PCAH2CBH2P, κ2-dppe). 31P{1H} (161.976 MHz, DMSO-d6, 293 K): δ 53.82 (dd, 1JPA–Pt = 2369.0 Hz, 2JPA–PC = 380.8 Hz, 2JPA–PB = 5.53 Hz, PA, conformer A), 53.79 (dd, 1JPA–Pt = 2369.0 Hz, 2JPA–PC = 380.2 Hz, 2JPA–PB = 5.57 Hz, PA, conformer B), 44.84 (dd, 1JPB–Pt = 3512.2 Hz, 2JPB–PC = 16.14 Hz, 2JPB–PA = 5.61 Hz, PB, conformer X), 44.77 (dd, 1JPB–Pt = 3512.2 Hz, 2JPB–PC = 16.14 Hz, 2JPB–PA = 5.61 Hz, PB, conformer X), 17.3 (dd, 1JPC–Pt = 2374.6 Hz, 2JPC–PA = 380.40 Hz, 2JPC–PB = 16.24 Hz, PC, conformer A), 17.16 (dd, 1JPC–Pt = 2374.6 Hz, 2JPC–PA = 380.02 Hz, 2JPC–PB = 16.1 Hz, PC, conformer B), −16.72 (br s, Δν1/2 = 51.28 Hz, PD, conformer A), −17.48 (s, PD, conformer B). 19F (376.498 MHz, DMSO-d6, 293 K): δ −77.6 (OTf). FTIR (cm−1): 3054 (w), 1482 (w), 1435 (m), 1260 (s), 1146 (m), 1102 (s), 1030 (s), 744 (s), 690 (s), 636 (s), 532 (s), 515 (s), 486 (s), 442 (m). ESI-TOF-MS (3 kV, dichloromethane): m/z calcd [(κ2-dppe)PtCl(κ1-dppb)O]+, 1071.2; expt., 1071.1 (36.2%); calcd [(κ2-dppe)PtCl(κ1-dppb)]+, 1055.2; expt., 1055.1 (100%); calcd [PtCl(κ1-dppb)]OTf-H+, 842.0; expt., 842.1 (3.67%).
Characterisation of [(κ2-dppe)PtCl(κ1-dpppent)](OTf) (Pt-2)
248 mg (81%) of white powder, air stable. 1H NMR (400.130 MHz, DMSO-d6, 293 K): δ 7.87–7.13 (40 H, unresolved m, PPh), 2.73 (2 H, unresolved m, PCH2ACH2BP, κ2-dppe), 2.37 (4 H, unresolved m, 1 × CH2, dpppent + PCH2ACH2BP, κ2-dppe), 1.98 (2 H, ps q, 1 × CH2, dpppent), 1.30 (6 H, unresolved m, 3 × CH2, dpppent). 13C{1H} NMR (100.613 MHz, DMSO-d6, 293 K): δ 138.8–128.0 (unresolved m, PPh), 126.9 (d, xJC–P = 57.3 Hz, PPh), 124.4 (d, xJC–P = 62.4 Hz, PPh), 122.3 (s, PPh), 119.1 (s, PPh), 32.8 (br s, PCAH2CBH2P, κ2-dppe), 31.5 (br s, dpppent), 26.4 (unresolved m, dpppent), 24.6 (unresolved m, dpppent + PCAH2CBH2P, κ2-dppe). 31P{1H} NMR (161.976 MHz, DMSO-d6, 293 K): δ 53.93 (dd, 1JPA–Pt = 2366.1 Hz, 2JPA–PC = 379.0 Hz, 2JPA–PB = 5.9 Hz, PA, conformer A), 53.88 (dd, 1JPA–Pt = 2366.1 Hz, 2JPA–PC = 380.1 Hz, 2JPA–PB = 5.6 Hz, PA, conformer B), 45.0 (dd,1JPB–Pt = 3514.0 Hz, 2JPB–PC = 15.4 Hz, 2JPB–PA = 5.7 Hz, PB, conformer A), 44.9 (dd, 1JPB–Pt = 3514.0 Hz, 2JPB–PC = 16.1 Hz, 2JPB–PA = 5.7 Hz, PB, conformer B), 17.2 (dd, 1JPC–Pt = 2368.5 Hz, 2JPC–PA = 379.8 Hz, 2JPC–PB = 16.1 Hz, PC, conformer B), 16.9 (dd, 1JPC–Pt = 2374.9 Hz, 2JPC–PA = 379.0 Hz, 2JPC–PB = 16.0 Hz, PC, conformer A), −16.87 (s, PD, conformer B), −16.94 (obscured s, PD, conformer A). 19F NMR (376.498 MHz, DMSO-d6, 293 K): δ −77.6 (OTf). FTIR (cm−1): 3056 (w), 2931 (w), 2852 (w), 1435 (m), 1261 (s), 1223 (m), 1147 (m), 1102 (m), 1030 (s), 999 (m), 742 (m), 690 (s), 636 (s), 533 (s), 488 (s). ESI-TOF-MS (3 kV, dichloromethane): m/z calcd [(κ2-dppe)PtCl(κ1-dppp)O]+, 1085.2; expt., 1085.2 (10.0%); calcd [(κ2-dppe)PtCl(κ1-dppp)]+, 1069.2; expt., 1069.2 (100%); calcd [PtCl(κ1-dppp)]OTf-H+, 842.0; expt., 842.1 (3.67%).
Characterisation of [(κ2-dppe)PtCl(κ1-dpph)](OTf) (Pt-3)
1H NMR (400.130 MHz, DMSO-d6, 293 K): δ 7.88–7.14 (40 H, unresolved m, PPh), 2.73 (2 H, unresolved m, 1 × CH2, PCH2ACH2BP, κ2-dppe), 2.46 (signal obscured by solvent) (2 H, obscured m, 1 × CH2, PCH2ACH2BP, κ2-dppe), 2.34 (3 H, unresolved m, CH2, dpph), 2.01 (2 H, unresolved m, 1 × CH2, dpph), 1.26 (7 H, unresolved m, CH2, dpph). 13C{1H} NMR (100.613 MHz, acetone-d6, 293 K): δ 135.2–128.5 (unresolved m, PPh), 31.2 (unresolved m, CH2, dpph), 26.1 (br s, PCH2CH2P, κ2-dppe). 31P{1H} NMR (161.976 MHz, acetone-d6, 293 K): δ 53.6 (dd, 1JPA–Pt = 2353.4 Hz, 2JPA–PC = 383.5, 2JPA–PB = 6.53 Hz, PA, conformer A), 53.5 (dd, 1JPA–Pt = 2353.4 Hz, 2JPA–PC = 382.5 Hz, 2JPA–PB = 6.15 Hz, PA, conformer B), 45.0 (dd, 1JPB–Pt = 3506 Hz, 2JPB–PC = 16.11 Hz, 2JPB–PA = 6.25, PB), 17.2 (dd, 2JPC–Pt = 2349.8 Hz, 2JPC–PA = 382.0 Hz, 2JPC–PB = 16.55, PC, conformer A + B), −15.8 (s, PD, conformer A), −16.4 (s, PD, conformer B). 19F NMR (376.498 MHz, acetone-d6, 293 K): δ −78.6 (OTf). FTIR (cm−1): 3055 (w), 2924 (w), 2857 (w), 1742 (w), 1435 (m), 1260 (s), 1223 (m), 1147 (s), 1101 (s), 1030 (s), 999 (m), 822 (m), 743 (s), 690 (s), 636 (s), 532 (s), 516 (s), 485 (s). UV/Vis λmax (dichloromethane): 226 nm (major, ε = 154941 M−1 cm−1), 274 nm (shoulder, ε = 61601 M−1 cm−1). ESI-TOF-MS (3 kV, dichloromethane): m/z calcd [(κ2-dppe)PtCl(κ1-dpph)O]+, 1099.3; expt., 1099.2 (36.2%); calcd [(κ2-dppe)PtCl(κ1-dpph)]+, 1083.3; expt., 1083.2 (100%).
General procedure for the synthesis of [((κ2-dppe)PdCl)2(μ-dppx)](2OTf) (PdPd-x)
200 mg (0.140 mmol, 1.00 eq.) of PdPd-0 and 0.140 mmol (1.00 eq.) of diphosphine were added as solids in a Schlenk flask, which was subsequently purged through vacuum/N2 cycles. 30 mL of dried, degassed DCM were then added and the solution was stirred at room temperature for 1 h. The solvents were removed in vacuo, after which the solids were washed with 3 × 10 mL dry Et2O, and subsequently dried in vacuo.
Characterisation of [((κ2-dppe)PdCl)2(μ-dppb)](2OTf) (PdPd-1)
169.3 mg (67%) of light yellow solid, air stable. Melting point: 211 °C + dec. 1H NMR (400.130 MHz, DMSO-d6, 298 K): δ 7.83–7.14 (60 H, unresolved m, PPh), 2.84 (4 H, ps dd, 2JH–P = 34.9 Hz, 3JH–P = 6.2 Hz, PCH2ACH2BP, κ2-dppe), 2.47 (obscured by solvent, 4 H, ps d, PCH2ACH2BP, κ2-dppe), 2.17 (4 H, br s, Δν1/2 = 24.2 Hz, 2 × CH2, dppb), 1.28 (4 H, br s, Δν1/2 = 20.4 Hz, 2 × CH2, dppb). 13C NMR (100.613 MHz, DMSO-d6, 298 K): δ 133.5 (d, xJC–P = 10.7 Hz, PPh), 133.3 (d, xJC–P = 11.7 Hz, PPh), 132.9 (d, xJC–P = 10.6 Hz, PPh), 132.3 (s, PPh), 131.1 (s, PPh), 129.4 (d, xJC–P = 11.1 Hz, PPh), 129.2 (d, xJC–P = 11.0 Hz, PPh), 128.7 (d, xJC–P = 10.2 Hz, PPh), 128.0 (d, xJC–P = 43.5 Hz, PPh), 127.8 (d, xJC–P = 49.6 Hz, PPh), 125.2 (d, xJC–P = 52.6 Hz, PPh), 120.7 (d, xJC–P = 322.3 Hz, PPh), 32.7 (ps dd, 1JC–P = 37.1 Hz, 2JC–P = 18.1 Hz, PCAH2CBH2P, κ2-dppe), 26.2 (d, 2JC–P = 14.3 Hz, μ-dppb-PCAH2CBH2), 24.6 (d, 1JC–P = 26.4 Hz, μ-dppb-PCAH2CBH2), 24.3–23.8 (m, PCAH2CBH2P, κ2-dppe). 31P NMR (161.976 MHz, DMSO-d6, 298 K): δ 66.0 (br s, Δν1/2 = 24.2 Hz, PB), 64.81 (dd, 2JPA–PC = 431.2 Hz, 2JPA–PB = 6.8 Hz, PA), 17.9 (dd, 2JPC–PA = 411.3, 2JPC–PB = 17.8, PC). 19F NMR (376.498 MHz, DMSO-d6, 298 K): δ −77.7 (OTf). FT-IR (cm−1): 3057 (w), 1484 (w), 1436 (m), 1262 (s), 1223 (m), 1147 (m), 1101 (m), 1030 (m), 999 (m), 879 (w), 813 (w), 742 (m), 690 (s), 635 (s). UV-Vis λmax (dichloromethane): 226 nm (major, ε = 103695 M−1 cm−1), 267 nm (shoulder, ε = 42876 M−1 cm−1), 332 nm (minor, ε = 38438 M−1 cm−1). ESI-TOF-MS: m/z calcd for [M-OTf]+, 1656.1; expt., 1656.3.
Characterisation of [((κ2-dppe)PdCl)2(μ-dpppent)](2OTf) (PdPd-2)
170.2 mg (67%) of light yellow solid, air stable. 1H NMR (400.130 MHz, DMSO-d6, 298 K): δ 7.84–7.15 (60 H, unresolved m, PPh), 2.83 (4 H, ps dd, 2JH–P = 34.9 Hz, 3JH–P = 6.4 Hz, PCH2ACH2BP, κ2-dppe), 2.48 (obscured by solvent, 4 H, ps d, PCH2ACH2BP, κ2-dppe), 2.27 (4 H, br s, Δν1/2 = 25.1 Hz, 2 × CH2, dpppent-CH2A), 1.21 and 1.16 (6 H, br s, overlapped signals, 2 × CH2, dpppent-CH2B and dpppent-CH2C). 13C NMR (100.613 MHz, DMSO-d6, 298 K): δ 133.5 (d, xJC–P = 10.5 Hz, PPh), 133.3 (d, xJC–P = 11.8 Hz, PPh), 132.9 (d, xJC–P = 9.1 Hz, PPh), 132.3 (s, PPh), 131.1 (s, PPh), 129.4 (d, xJC–P = 11.1 Hz, PPh), 129.1 (d, xJC–P = 11.1 Hz, PPh), 128.6 (d, xJC–P = 10.0 Hz, PPh), 128.0 (s, PPh), 128.0 (d, xJC–P = 95.1 Hz, PPh), 125.2 (d, xJC–P = 52.7 Hz, PPh), 120.7 (d, xJC–P = 322.3 Hz, PPh), 32.7 (ps dd, 1JC–P = 34.5 Hz, 2JC–P = 14.5 Hz, PCAH2CBH2P, κ2-dppe), 31.7–31.3 (m, dpppent-PCH2CH2CH2), 25.0 (d, 2JC–P = 25.1 Hz, dpppent-PCH2CH2CH2), 24.2 (s, dpppent-PCH2CH2CH2), 24.4–23.9 (m, PCAH2CBH2P, κ2-dppe). 31P NMR (161.976 MHz, DMSO-d6, 298 K): δ 66.1 (dd, 2JPX–PX = 38.9 Hz, 2JPX–PX = 7.4 Hz, PB), 64.9 (dd, 2JPA–PC = 467.5 Hz, 2JPA–PB 7.3 Hz, PA), 17.8 (dd, 2JPC–PA = 410.1 Hz, 2JPC–PB = 19.0, PC). 19F NMR (376.498 MHz, DMSO-d6, 298 K): δ −77.6 (OTf). FTIR (cm−1): 3056 (w), 1586 (w), 1574 (w), 1436 (m), 1410 (w), 1312 (w), 1262 (s), 1223 (m), 1147 (m), 1101 (m), 1030 (s), 999 (m), 879 (w), 844 (w), 813 (w), 741 (m), 716 (m), 704 (m), 688 (s), 657 (m), 635 (s), 616 (m). UV-Vis λmax (dichloromethane): 225 nm (major, ε = 101105 M−1 cm−1), 266 nm (shoulder, ε = 42676 M−1 cm−1), 331 nm (minor, ε = 34038 M−1 cm−1).
Characterisation of [((κ2-dppe)PdCl)2(μ-dpph)](2OTf) (PdPd-3)
182 mg (71%) of light yellow solid, air stable. 1H NMR (400.130 MHz, DMSO-d6, 298 K): δ 7.85–7.17 (60 H, unresolved m, PPh), 2.85 (4 H, ps dd, 2JH–P = 34.9 Hz, 3JH–P = 6.4 Hz, 2 × CH2, PCH2ACH2BP, κ2-dppe), 2.50 (obscured by solvent, 4 H, ps d, 2 × CH2, PCH2ACH2BP, κ2-dppe), 2.30 (4 H, br s, Δν1/2 = 25.6 Hz, 2 × dpph-CH2), 1.23 (4 H, br s, Δν1/2 = 26.2 Hz, 2 × dpph-CH2), 1.06 (obscured by Et2O signal, 4 H, br s, Δν1/2 = 14.8 Hz, 2 × dpph-CH2). 13C NMR (100.613 MHz, DMSO-d6, 298 K): δ 133.5 (d, xJC–P = 10.6 Hz, PPh), 133.3 (d, xJC–P = 11.7 Hz, PPh), 132.8 (d, xJC–P = 10.3 Hz, PPh), 132.6 (d, xJC–P = 58.7 Hz, PPh), 131.0 (s, PPh), 129.5 (d, xJC–P = 11.1 Hz, PPh), 129.2 (d, xJC–P = 11.0 Hz, PPh), 128.6 (d, xJC–P = 10.3 Hz, PPh), 127.8 (d, xJC–P = 50.5 Hz, PPh), 125.2 (d, xJC–P = 51.3 Hz, PPh), 120.7 (d, xJC–P = 321.7 Hz, PPh), 33.1–32.3 (m, PCAH2CBH2P, κ2-dppe), 29.8 (d, 2JC–P = 14.2 Hz, dpph-PCH2CH2CH2), 25.2–24.6 (m, overlapping signals, dpph-PCH2CH2CH2 and dpph-PCH2CH2CH2), 24.4–23.8 (m, PCAH2CBH2P, κ2-dppe). 31P NMR (161.976 MHz, DMSO-d6, 298 K): δ 66.1 (dd, 2JPX–PX = 32.7 Hz, 2JPX–PX = 7.3 Hz, PB), 64.9 (dd, 2JPA–PC = 462.1 Hz, 2JPA–PB = 7.3 Hz, PA), 18.0 (dd, 2JPC–PA = 411.0 Hz, 2JPC–PB = 19.1, PC). 19F NMR (376.498 MHz, DMSO-d6, 298 K): δ −77.6 (OTf). FTIR (cm−1): 3055 (w), 2927 (w), 1586 (w), 1574 (w), 1483 (w), 1436 (m), 1408 (w), 1311 (w), 1259 (s), 1223 (m), 1188 (w), 1145 (m), 1099 (m), 1029 (s), 999 (m), 881 (w), 819 (w), 743 (m), 714 (m), 704 (m), 688 (s), 657 (m), 635 (s), 616 (m). UV-Vis λmax (dichloromethane): 226 nm (major, ε = 104209 M−1 cm−1), 266 nm (shoulder, ε = 44857 M−1 cm−1), 331 nm (minor, ε = 35485 M−1 cm−1). TOF-MS: m/z calcd for [M-OTf]+, 1684.2; expt., 1684.3.
General procedure for the synthesis of [(κ2-dppe)ClPd(μ-dppx)RuCl2(η6-p-cym)](OTf) (PdRu-x)
26.8 mg (0.044 mmol, 1.00 eq.) of [(η6-p-cym)RuCl2]2 dimer was dissolved in dichloromethane (10 mL) in a Schlenk flask. Similarly, Pd-x (0.087 mmol, 2.00 eq.) was dissolved in dichloromethane (10 mL) and added rapidly to the Ru dimer solution while stirring. The resulting solution was left to stir at room temperature for two hours. The solvent was removed in vacuo, after which the resulting solid was washed with diethyl ether (10 mL), and the washings discarded. The product was subsequently dried in vacuo overnight.
Characterisation of [(κ2-dppe)ClPd(μ-dppx)RuCl2(η6-p-cym)](OTf) (PdRu-1)
117 mg (69%) of brick-red solid, air stable. Melting point: 165–170 °C, 180 °C + (dec). 1H NMR (400.130 MHz, DMSO-d6, 298 K): δ 7.92–7.10 (40 H, unresolved m, PPh), 5.42 (2 H, ps d, 2 × CH, –C3,5–H, p-cymene, conformer B), 5.36 (d, 3JH–H = 5.9 Hz, 2 × CH, –C3,5–H, p-cymene, conformer A), 5.21 (2 H, ps d, 2 × CH, –C2,6–H, p-cymene conformer B), 5.17 (d, 3JH–H = 5.6 Hz, 2 × CH, –C2,6–H, p-cymene, conformer A), 2.81 (2 H, ps d, 2JH–P = 36.7 Hz, PCH2ACH2BP, κ2-dppe), 2.45 (obscured by solvent) (2 H, obscured br d, PCH2ACH2BP, κ2-dppe), 2.35–1.85 (5 H, unresolved m, 2 × CH2 + 1 × –CH(CH3)2, dppb + p-cymene), 1.76 (3 H, s, 1 × –C(CH3), p-cymene, conformer B), 1.70 (s, 1 × –C(CH3), p-cymene, conformer A), 1.32–1.11 (2 H, ps d, 1 × CH2, dppb), 0.96–0.84 (2 H, br s, Δν1/2 = 23.6 Hz, 1 × CH2, dppb), 0.74 (6 H, ps d, 3JH–H = 7.4 Hz, 1 × –CH(CH3)2, p-cymene, conformer B), 0.72 (ps d, 3JH–H = 7.2 Hz, 1 × –CH(CH3)2, p-cymene, conformer A). 13C{1H} NMR (100.613 MHz, DMSO-d6, 298 K): δ 133.8–127.4 (unresolved m, PPh), 106.3 (br ps d, Δν1/2 = 10.1 Hz, –C1, p-cymene, conformer A + B), 93.5 (br ps d, Δν1/2 = 10.5 Hz, –C4, p-cymene, conformer A + B), 90.2 (br s, Δν1/2 = 9.8 Hz, 2 × CH, –C3,5–H, p-cymene, conformer A + B), 85.5 (br s, Δν1/2 = 10.9 Hz, 2 × CH, –C2,6–H, p-cymene, conformer A + B), 32.6 (PCH2ACH2BP, κ2-dppe), 29.5 (br m, Δν1/2 = 9.7 Hz, 1 × –CH(CH3)2, p-cymene, conformer A + B), 26.6 (1 × CH2, dppb), 26.2 (1 × CH2, dppb), 24.4 (1 × CH2, dppb), 24.1 (1 × CH2, dppb), 23.2 (PCH2ACH2BP, κ2-dppe), 21.0 (br s, Δν1/2 = 9.9 Hz, 1 × –CH(CH3)2, p-cymene, conformers A + B), 17.0 (br s, Δν1/2 = 10.0 Hz, 1 × –C(CH3), conformer A + B). 31P{1H} NMR (161.976 MHz, DMSO-d6, 298 K): δ 66.2–65.9 (ps dd, PB, conformer A + B), 64.7 (dd, 2JPA–PC = 390.9 Hz, 2JPA–PB = 7.2 Hz, PA, conformer B), 64.5 (ps dd, 2JPA–PC = 329.9 Hz, 2JPA–PB = 7.3 Hz, PA, conformer A), 23.7 (br s, Δν1/2 = 3.9 Hz, PD, conformer B), 23.3 (br s, Δν1/2 = 2.7 Hz, PD, conformer A), 17.8 (ps dd, 2JPC–PA = 412.2 Hz, 2JPC–PB = 20.3 Hz, PC, conformer B), 17.7 (dd, 2JPC–PA = 415.4 Hz, 2JPC–PB = 20.9 Hz, PC, conformer A). 19F NMR (376.498 MHz, DMSO-d6, 298 K): δ = −77.6 (OTf). FTIR (cm−1): 3055 (vw), 2959 (vw), 2868 (vw), 1434 (m), 1260 (s), 1147 (m), 1099 (m), 1030 (s), 997 (w), 743 (m), 690 (vs), 637 (vs), 529 (vs), 516 (s), 483 (m). UV/Vis λmax (dichloromethane): 226 nm (major), 266 nm (shoulder), 332 nm (minor), 488 (weak, forbidden d–d). ESI-TOF-MS (3 kV, dichloromethane): m/z calcd for [(κ2-dppe)PdCl(μ-dppb)RuCl2(η6-p-cymene)]+, 1273.1; expt., 1273.3 (55%). m/z calcd for [(κ2-dppe)PdCl(κ1-dppb)O]+, 981.2; expt., 981.3 (31%). m/z calcd for [(κ2-dppe)PdCl(κ1-dppb)]+, 965.2 expt., 965.3 (100%). m/z calcd for [(η6-p-cymene)RuCl(κ1-dppb)O]+, 713.2; expt. 713.2 (4%). m/z calcd for [(η6-p-cymene)RuCl(κ1-dppb)]+, 697.2; expt. 697.2 (12%). m/z calcd for [(κ2-dppe)PdCl]+, 541.0; expt., 541.0 (28%).
Characterisation of [(κ2-dppe)ClPd(μ-dppx)RuCl2(η6-p-cym)](OTf) (PdRu-2)
235.5 mg (85%) of brick-red solid, air stable. 1H NMR (400.130 MHz, DMSO-d6, 298 K): δ 7.90–7.10 (40 H, unresolved m, PPh), 5.42 (2 H, d, 3JH–H = 5.8 Hz, 2 × CH, –C3,5–H, p-cymene, conformer B), 5.37 (d, 3JH–H = 6.1 Hz, 2 × CH, –C3,5–H, p-cymene, conformer A), 5.23 (2 H, d, 3JH–H = 6.2 Hz, 2 × CH, –C2,6–H, conformer B), 5.19 (d, 3JH–H = 6.2 Hz, 2 × CH, –C2,6–H, conformer A), 2.82 (2 H, m, PCH2ACH2BP, κ2-dppe), 2.47 (obscured by solvent) (2 H, obscured br m, PCH2ACH2BP, κ2-dppe), 2.41–2.00 (5 H, unresolved m, 2 × CH2 + 1 × –CH(CH3)2, dppp + p-cymene), 1.77 (3 H, s, 1 × –C(CH3), p-cymene, conformer B), 1.71 (s, 1 × –C(CH3), p-cymene, conformer A), 1.28–0.98 (4 H, unresolved m, 2 × CH2, dppp), 0.94–0.81 (2 H, br m, Δν1/2 = 22.1 Hz, 1 × CH2, dppp), 0.74 (6 H, ps d, 3JH–H = 7.4 Hz, 1 × –CH(CH3)2, p-cymene, conformer B), 0.72 (ps d, 3JH–H = 7.2 Hz, 1 × –CH(CH3)2, p-cymene, conformer A). 13C{1H} NMR (100.613 MHz, DMSO-d6, 298 K): δ 133.7–127.3 (unresolved m, PPh), 106.3 (s, 1 × –C1, p-cymene, conformer B), 106.2 (s, 1 × –C1, p-cymene, conformer A), 93.3 (s, 1 × –C4, p-cymene, conformer B), 93.2 (s, 1 × –C4, p-cymene, conformer A), 90.34 (ps d, 2JC–P = 4.3 Hz, 2 × CH, –C3,5–H, p-cymene, conformer B), 90.28 (ps d, 2JC–P = 4.3 Hz, 2 × CH, –C3,5–H, p-cymene, conformer A), 85.4 (d, 2JC–P = 6.0 Hz, 2 × CH, –C3,5–H, p-cymene, conformer B), 85.2 (d, 2JC–P = 6.0 Hz, 2 × CH, –C3,5–H, p-cymene, conformer B), 32.6 (PCH2ACH2BP, κ2-dppe), 31.4 (1 × CH2, dppp), 29.49 (s, 1 × –CH(CH3)2, p-cymene, conformer B), 29.48 (s, 1 × –CH(CH3)2, p-cymene, conformer A), 24.8 (s, 1 × CH2, dppp), 23.4 (PCH2ACH2BP, κ2-dppe), 23.1 (1 × CH2, dppp), 22.3 (1 × CH2, dppp), 22.1 (1 × CH2, dppp), 20.9 (br s, Δν1/2 = 1.4 Hz, 1 × –CH(CH3)2, p-cymene, conformers A + B), 17.0 (s, 1 × –C(CH3), p-cymene, conformer B), 16.9 (s, 1 × –C(CH3), p-cymene, conformer A). 31P{1H} NMR (161.976 MHz, DMSO-d6, 298 K): δ 66.3 (ps dd (partially hidden by conformer A), 2JPB–PC = 19.1 Hz, 2JPB–PA = 7.5 Hz, PB, conformer B), 66.2 (dd, 2JPB–PC = 19.1 Hz, 2JPB–PA = 7.5 Hz, PB, conformer A), 64.7 (dd, 2JPA–PC = 410.7 Hz, 2JPA–PB = 7.2 Hz, PA, conformer B), 64.6 (dd, 2JPA–PC = 410.8 Hz, 2JPA–PB = 7.3 Hz, PA, conformer B), 23.7 (s, PD, conformer B), 23.3 (s, PD, conformer A), 17.8 (dd, 2JPC–PA = 410.8 Hz, 2JPC–PB = 19.1 Hz, PC, conformer B), 17.7 (dd, 2JPC–PA = 411.3 Hz, 2JPC–PB = 19.7 Hz, PC, conformer A). 19F NMR (376.498 MHz, DMSO-d6, 298 K): δ −77.6 (1 × CF3SO3, OTf−). FTIR (cm−1): 3054 (vw), 2924 (vw), 1436 (m), 1262 (s), 1148 (m), 1099 (m), 1030 (s), 997 (w), 743 (m), 691 (vs), 637 (vs), 529 (vs), 516 (s), 483 (m). UV/Vis λmax (dichloromethane): 226 nm (major, ε = 105543 M−1 cm−1), 268 nm (shoulder, ε = 38971 M−1 cm−1), 331 nm (minor, ε = 25124 M−1 cm−1), 487 (weak, forbidden d–d). ESI-TOF-MS (3 kV, dichloromethane): m/z calcd for [(κ2-dppe)PdCl(μ-dppp)RuCl2(η6-p-cymene)]+, 1287.1; expt., 1287.1 (77%). m/z calcd for [(κ2-dppe)PdCl(κ1-dppp)O]+, 995.2; expt., 995.2 (100%). m/z calcd for [(κ2-dppe)PdCl(κ1-dppp)]+, 979.2; expt., 979.2 (8%). m/z calcd for [(η6-p-cymene)RuCl(κ1-dppp)]+, 711.2; expt. 711.2 (76%). m/z calcd for [(κ2-dppe)PdCl]+, 541.0; expt., 541.1 (39%).
Characterisation of [(κ2-dppe)ClPd(μ-dppx)RuCl2(η6-p-cym)](OTf) (PdRu-3)
83 mg (65%) of brick-red solid, air stable. 1H NMR (400.130 MHz, DMSO-d6, 298 K): δ 7.85–7.15 (40 H, unresolved m, PPh), 5.42 (2 H, d, 3JH–H = 6.2 Hz, 2 × CH, –C3,5–H, conformer B), 5.38 (d, 3JH–H = 6.5 Hz, 2 × CH, –C3,5–H, conformer A), 5.21 (2 H, d, 3JH–H = 6.0 Hz, 2 × CH, C2,6–H, p-cymene, conformer B), 5.19 (d, 3JH–H = 6.1 Hz, 2 × CH, –C2,6–H, conformer A), 2.84 (2 H, m, PCH2ACH2BP, κ2-dppe), 2.44 (obscured by solvent) (2 H, obscured br d, PCH2ACH2BP, κ2-dppe), 2.39–1.88 (5 H, unresolved m, 2 × CH2 + 1 × –CH(CH3)2, dpph + p-cymene), 1.76 (3 H, s, 1 × –C(CH3), p-cymene, conformer B), 1.73 (s, 1 × –C(CH3), p-cymene, conformer A), 1.29–1.19 (2 H, br s, Δν1/2 = 19.9 Hz, 1 × CH2, dpph), 1.18–1.02 (2 H, br m, Δν1/2 = 14.2 Hz, 1 × CH2, dpph), 0.89 (4 H, ps d, 2 × CH2, dpph), 0.72 (6 H, d, 3JH–H = 7.0 Hz, 1 × –CH(CH3)2, p-cymene, conformer B), 0.70 (d, 3JH–H = 6.9 Hz, 1 × –CH(CH3)2, p-cymene, conformer A). 13C{1H} NMR (100.613 MHz, DMSO-d6, 298 K): δ 133.7–127.3 (unresolved m, PPh), 106.2 (br m, 1 × –C1, p-cymene, conformer A + B), 93.1 (br m, 1 × –C4, p-cymene, conformer A + B), 90.3 (br s, Δν1/2 = 11.8 Hz, 2 × CH, –C3,5–H, p-cymene, conformer A + B), 85.3 (br m, Δν1/2 = 22.7 Hz, 2 × CH, –C2,6–H, p-cymene, conformer A + B), 32.6 (unresolved m, PCH2ACH2BP, κ2-dppe), 29.8 (s, CH2, dpph), 29.7 (s, CH2, dpph), 29.5 (br s, Δν1/2 = 10.1 Hz, 1 × –CH(CH3)2, p-cymene, conformer A + B), 25.3–21.5 (unresolved m, 4 × CH2, dpph), 23.4 (PCH2ACH2BP, κ2-dppe), 20.9 (br s, Δν1/2 = 9.8 Hz, 1 × –CH(CH3)2, p-cymene, conformer A + B), 16.9 (br m, 1 × –C(CH3), p-cymene, conformer A + B). 31P{1H} NMR (161.976 MHz, DMSO-d6, 298 K): δ 66.4–66.1 (br ps d, Δν1/2 = 34.7 Hz, PB, conformer A + B), 64.7 (ps dd, 2JPA–PC = 413.5 Hz, 2JPA–PB = 6.8 Hz, PA, conformer B), 64.6 (ps dd, 2JPA–PC = 413.7 Hz, PA, conformer A), 23.9 (br s, Δν1/2 = 6.5 Hz, PD, conformer B), 23.6 (br s, Δν1/2 = 6.5 Hz, PD, conformer A), 18.0 (dd, 2JPC–PA = 410.3 Hz, 2JPC–PB = 19.1 Hz, PC, conformer B), 17.8 (dd, 2JPC–PA = 411.6 Hz, 2JPC–PB = 19.1 Hz, PC, conformer A). 19F NMR (376.498 MHz, DMSO-d6, 298 K): δ = −77.6 (OTf). FTIR (cm−1): 3054 (vw), 2920 (vw), 1434 (m), 1262 (s), 1147 (m), 1099 (m), 1030 (s), 997 (w), 743 (m), 690 (vs), 637 (vs), 530 (vs), 516 (s), 483 (s). UV/Vis λmax (dichloromethane): 226 nm (major, ε = 102476 M−1 cm−1), 267 nm (shoulder, ε = 39295 M−1 cm−1), 332 nm (minor, ε = 24133 M−1 cm−1), 487 (weak, forbidden d–d). ESI-TOF-MS (3 kV, dichloromethane): m/z calcd for [(κ2-dppe)PdCl(μ-dpph) RuCl2(η6-p-cymene)]+, 1301.2; expt., 1301.1 (70%). m/z calcd for [(κ2-dppe)PdCl(κ1-dpph)O]+, 1009.2; expt. 1009.1 (12%). m/z calcd for [(κ2-dppe)PdCl(κ1-dpph)]+, 993.2; expt., 993.2 (100%). m/z calcd for [(κ2-dppe)PdCl]+, 541.0; expt., 541.0 (7%). Temperature studies: 31P{1H} NMR (161.976 MHz, DMSO-d6, 313 K): δ 66.1 (ps d, 2JPB–PC = 20.3 Hz, PB, conformer A + B), 64.7 (ps dd, 2JPA–PC = 413.5 Hz, 2JPA–PB = 6.8 Hz, PA, pseudo coalescent signals), 23.7 (br s, Δν1/2 = 7.7 Hz, PD, conformer B), 23.3 (br s, Δν1/2 = 6.9 Hz, PD, conformer A), 17.9 (ps dd, 2JPC–PA = 412.6 Hz, 2JPC–PB = 19.5 Hz, PC, pseudo coalescent signals). 31P{1H} NMR (161.976 MHz, DMSO-d6, 333 K): δ 66.7 (m, PB, coalescent signals), 64.5 (ps dd, 2JPA–PC = 406.3 Hz, PA, pseudo coalescent signals), 23.4 (br s, Δν1/2 = 11.4 Hz, PD, conformer B), 23.1 (br s, Δν1/2 = 7.3 Hz, PD, conformer A), 18.2 (ps dd, 2JPC–PA = 411.6 Hz, 2JPC–PB = 19.5 Hz, PC, pseudo coalescent signals). 31P{1H} NMR (161.976 MHz, DMSO-d6, 353 K): δ 65.9 (m, PB/PA, coalescent signals, PA dd in baseline), 23.1 (br s, Δν1/2 = 8.0 Hz, PD, conformer B), 22.8 (br s, Δν1/2 = 7.1 Hz, PD, conformer A). 31P{1H} NMR (161.976 MHz, DMSO-d6, 373 K): δ 65.8 (m, PB/PA, coalescent signals, PA dd in baseline), 22.9 (br s, Δν1/2 = 7.8 Hz, PD, conformer B), 22.7 (br s, Δν1/2 = 7.4 Hz, PD, conformer A). 31P{1H} NMR (161.976 MHz, DMSO-d6, 393 K): δ 23.4 (s, PD, coalescent signal).
General procedure for the synthesis of [(κ2-dppe)ClPt(μ-dppx)RuCl2(η6-p-cym)](OTf) (PtRu-x)
A solution of 0.123 mmol (2.00 eq.) of Pt-x in 15 mL dichloromethane was added to a solution containing 1 equivalent of [(p-cym)RuCl2]2 in 10 mL of dichloromethane. It was stirred at room temperature for 2 h. The solvents were removed in vacuo before washing with Et2O. The resulting powder was subsequently dried in vacuo.
Characterisation of [(κ2-dppe)ClPt(μ-dppb)RuCl2(η6-p-cym)](OTf) (PtRu-1)
139 mg (73%) of red powder, air stable. Melting point: decomposition at 182 °C. 1H NMR (400.130 MHz, DMSO-d6, 293 K): δ 7.99–7.01 (40 H, unresolved m, PPh, conformer A + B), 5.43 (2 H, d, 3JH–H = 5.9 Hz, 2 × CH, –C3,5–H, p-cymene, conformer B), 5.36 (d, 3JH–H = 6.0 Hz, 2 × CH, p-cymene, –C3,5–H, conformer A), 5.22 (2 H, d, 3JH–H = 6.0 Hz, 2 × CH, –C2,6–H, p-cymene, conformer B), 5.17 (d, 3JH–H = 6.0 Hz, 2 × CH, –C2,6–H, p-cymene, conformer A), 2.71 (2 H, unresolved m, CH2, PCHA2CHB2P, κ2-dppe, conformer A + B), 2.44 (2 H, unresolved m, CH2, PCHA2CHB2P, κ2-dppe, conformer A + B), 2.35–2.22 (5 H, unresolved m, 2 × CH2, dppb + 1 × –CH(CH3)2, p-cymene, conformer A + B), 2.08 (1 H, unresolved m, CH2, dppb, conformer A + B), 1.78 (3 H, s, –C(CH3), p-cymene, conformer B), 1.70 (s, –C(CH3), p-cymene, conformer A), 1.29–1.15 (3 H, unresolved m, CH2, dppb, conformer A + B), 0.74 (6 H, d, 3JH–H = 6.9 Hz, –CH(CH3)2, p-cymene, conformer B), 0.69 (d, 3JH–H = 6.9 Hz, –CH(CH3)2, p-cymene, conformer A). 13C{1H} NMR (100.613 MHz, DMSO-d6, 293 K): δ 134.0128.2 (unresolved m, PPh), 126.9 (d, xJC–P = 57.7 Hz, PPh), 124.4 (d, xJC–P = 62.3 Hz, PPh), 122.3 (s, PPh), 119.1 (s, PPh), 106.3 (s, C1, p-cymene, conformer A + B), 93.5 (s, C4, p-cymene, conformer A + B), 90.2 (ps d, 2JH–P = 3.9 Hz, –C3,5–H, p-cymene, conformer A + B), 85.5 (d, 2JH–P = 5.9 Hz, –C2,6–H, p-cymene, conformer A + B), 32.8 (br s, PCAH2CBH2P, κ2-dppe), 29.5 (s, –CH(CH3)2), 26.6 (s, dppb), 24.3 (unresolved m, dppb + PCAH2CBH2P, κ2-dppe), 21.5 (s, dppb), 20.9 (s, –CH(CH3)2), 17.0 (s, –C(CH3), p-cymene,). 31P{1H} NMR (161.976 MHz, DMSO-d6, 293 K): δ 53.8 (obscured dd, 2JPA–Pt = 2377.5 Hz, 2JPA–PB = 380 Hz, PA, conformer A), 53.7 (dd, 2JPA–Pt = 2377.5 Hz, 2JPA–PB = 381 Hz, 2JPA–PB = 5.6 Hz, PA, conformer B), 44.8 (dd, 2JPB–Pt = 3517.3 Hz, 2JPB–PC = 16.2 Hz, 2JPB–PA = 5.4 Hz, PB, conformer A), 44.5 (dd, 2JPB–Pt = 3509.0 Hz, 2JPB–PC = 16.3 Hz, 2JPB–PA = 5.6 Hz, PB, conformer A), 23.8 (s, PD, conformer A), 23.3 (s, PD, conformer B), 17.2 (dd, 2JPC–Pt = 2375.5 Hz, 2JPC–PA = 380.4 Hz, 2JPC–PB = 16.0 Hz, PC, conformer A), 17.1 (dd, 2JPC–Pt = 2375.5 Hz, 2JPC–PA = 380.8 Hz, 2JPC–PB = 16.3 Hz, PC, conformer B). 19F (376.498 MHz, DMSO-d6, 293 K): δ −77.6 (OTf). FTIR (cm−1): 3055 (w), 2962 (w), 1436 (m), 1260 (s), 1147 (m), 1101 (s), 1030 (s), 999 (m), 800 (m), 744 (s), 691 (s), 637 (s), 532 (s), 517 (s), 486 (s). ESI-TOF-MS (3 kV, acetonitrile): m/z calcd [(κ2-dppe)PtCl(µ-dppb)RuCl2(η6-p-cymene)]+, 1361.2; expt., 1361.3 (100%); [(κ2-dppe)PtCl (κ1-dppb)O]+, 1055.2; expt., 1055.3 (54.1%); [PtCl(κ1-dppb)]OTf-H+, 842.0; expt., 842.2 (86.1%).
Characterisation of [(κ2-dppe)ClPt(μ-dpppent)RuCl2(η6-p-cym)](OTf) (PtRu-2)
116 mg (61%) of red solid, air stable. 1H NMR (400.130 MHz, DMSO-d6, 293 K): δ 7.95–7.03 (40 H, unresolved m, PPh, conformer A + B), 5.43 (2 H, d, 3JH–H = 5.40 Hz, 2 × CH, –C3,5–H, p-cymene, conformer B), 5.37 (d, 3JH–H = 5.30 Hz, 2 × CH, –C3,5–H, p-cymene, conformer A), 5.23 (2 H, d, 3JH–H = 6.10 Hz, 2 × CH, –C2,6–H, p-cymene, conformer B), 5.19 (d, 3JH–H = 6.10 Hz, 2 × CH, –C2,6–H, p-cymene, conformer A), 2.69 (2 H, unresolved m, 1 × CH2, PCH2ACH2BP, κ2-dppe, conformer A + B), 2.33 (6 H, unresolved m, –CH(CH3)2 + PCH2ACH2BP, κ2-dppe + dpppent, conformer A + B), 2.15 (1 H, m, dpppent, conformer A + B), 1.77 (3 H, s, –C(CH3), p-cymene, conformer B), 1.71 (2 H, s, –C(CH3), p-cymene, conformer A), 1.23 (3 H, m, dpppent, conformer A + B), 1.06 (1.86 H, m, dpppent, conformer A + B), 0.74 (6 H, d, 3JH–H = 6.95 Hz, –CH(CH3)2, p-cymene, conformer B), 0.72 (4.5 Hz, d, 3JH–H = 7.05 Hz, –CH(CH3)2, p-cymene, conformer A). 13C{1H} NMR (100.613 MHz, DMSO-d6, 293 K): δ 133.7–127.9 (unresolved m, PPh, conformer A + B), 119.1 (s, PPh), 106.3 (s, –C1–H, p-cymene, conformer A + B), 93.4 (s, –C4–H, p-cymene, conformer B), 93.2 (s, –C4–H, p-cymene, conformer A), 90.3 (unresolved m, –C3,5–H, p-cymene, conformer A + B), 85.4 (d, 3JC–P = 4.93 Hz, –C2,6–H, p-cymene, conformer B), 85.2 (d, 3JC–P = 5.39 Hz, –C2,6–H, p-cymene, conformer A), 33.1 (unresolved m, PCAH2CBH2P, κ2-dppe), 31.5 (unresolved m, dpppent), 29.5 (s, –CH(CH3)2), 24.7 (unresolved m, dpppent + PCAH2CBH2P, κ2-dppe), 21.0 (s, –CH(CH3)2), 17.0 (s, –C(CH3), conformer B), 16.9 (s, –C(CH3), conformer A). 31P{1H} NMR (161.976 MHz, DMSO-d6, 293 K): δ 53.93 (overlapping dd, 2JPA–Pt = 2368.6 Hz, 2JPA–PC = 379.9 Hz, 2JPA–PB = 5.59 Hz, PA, conformer A), 53.89 (overlapping dd, 1JPA–Pt = 2368.6 Hz, 2JPA–PC = 379.9 Hz, 2JPA–PB = 5.65 Hz, PA, conformer B), 45.04 (overlapping dd, 1JPB–Pt = 3514.4 Hz, 2JPB–PC = 15.5 Hz, 2JPB–PA = 5.6 Hz, PB, conformer A), 44.94 (overlapping dd, 1JPB–Pt = 3514.4 Hz, 2JPB–PC = 15.6 Hz, 2JPB–PA = 5.7 Hz, PB, conformer B), 23.8 (s, PD, conformer B), 23.3 (s, PD, conformer A), 17.00 (dd, 1JPC–Pt = 2372.6 Hz, 2JPC–PA = 380.1 Hz, 2JPC–PB = 15.9 Hz, PC, conformer A), 16.87 (dd, 2JPC–PB = 16.2 Hz, 2JPC–PA = 380.2 Hz, 1JPC–Pt = 2372.6 Hz, PC, conformer B). 19F NMR (376.498 MHz, DMSO-d6, 293 K): δ −77.6 (OTf). FTIR (cm−1): 3055 (w), 2928 (w), 2864 (w), 1436 (m), 1261 (s), 1223 (m), 1147 (m), 1100 (m), 1030 (s), 998 (m), 744 (m), 691 (s), 636 (s), 532 (s), 517 (s), 488 (s). ESI-TOF-MS (3 kV, acetonitrile): m/z calcd [(κ2-dppe)PtCl(µ-dppp)RuCl2(η6-p-cymene)]+, 1375.2; expt., 1375.2 (67.3%); calcd [(κ2-dppe)PtCl(κ1-dppp)]+, 1069.2; expt., 1069.3 (76.6%).
Characterisation of [(κ2-dppe)ClPt(μ-dpph)RuCl2(η6-p-cym)](OTf) (PtRu-3)
94 mg (74%) of red solid, air stable. 1H NMR (400.130 MHz, DMSO-d6, 293 K): δ 7.88–7.15 (40 H, unresolved m, PPh, conformer A + B), 5.42 (2 H, d, 3JH–H = 5.24 Hz, 2 × CH, –C3,5–H, p-cymene, conformer B), 5.38 (d, 3JH–H = 5.29 Hz, 2 × CH, –C3,5–H, p-cymene, conformer A), 5.22 (2 H, d, 3JH–H = 6.04 Hz, 2 × CH, –C2,6–H, p-cymene, conformer B), 5.19 (d, 3JH–H = 6.14 Hz, 2 × CH, –C2,6–H, p-cymene, conformer A), 2.73 (2 H, unresolved m, 1 × CH2, PPh2CH2ACH2BP, κ2-dppe, conformer A + B), 2.46 (signal obscured by solvent) (2 H, obscured m, 1 × CH2, PPh2CH2ACH2BP, κ2-dppe, conformer A + B), 2.40–2.22 (5 H, unresolved m, 2 × CH2 + –CH(CH3)2, dpph + p-cymene, conformer A + B), 2.15 (1 H, unresolved m, 12 × CH2, dpph, conformer A + B), 1.77 (3 H, s, –C(CH3), p-cymene, conformer B), 1.73 (s, –C(CH3), p-cymene, conformer A), 1.35–1.13 (3 H, unresolved m, 32 × CH2, dpph, conformer A + B), 0.89 (4 H, ps d, xJH–X = 25 Hz, 2 × CH2, dpph, conformer A + B), 0.72 (6 H, d, 3JH–H = 6.9 Hz, –CH(CH3)2, p-cymene, conformer B), 0.71 (d, 3JH–H = 6.9 Hz, –CH(CH3)2, p-cymene, conformer A). 13C{1H} NMR (100.613 MHz, acetone-d6, 293 K): δ 135.0–128.7 (unresolved m, PPh), 107.7 (s, –C1, p-cymene, conformer A + B), 94.8 (s, –C4, p-cymene, conformer A + B), 91.3 (s, –C3,5, p-cymene, conformer A + B), 86.7 (s, –C2,6, p-cymene, conformer A + B), 34.4 (br s, PCH2CH2P, κ2-dppe) 30.8 (s, dpph), 25.9 (s, –CH(CH3)2, p-cymene), 21.6 (s, dpph), 17.5 (s, –C(CH3), p-cymene). 31P{1H} NMR (161.976 MHz, DMSO-d6, 293 K): δ 54.0 (dd, 1JPA–Pt = 2366.8 Hz, 2JPA–PC = 380.0 Hz, 2JPA–PB = 5.61 Hz, PA, conformer A), 53.9 (dd, 1JPA–Pt = 2366.8 Hz, 2JPA–PC = 380.2 Hz, 2JPA–PB = 5.61 Hz, PA conformer B), 45.0 (dd, 1JPB–Pt = 3490.7 Hz, 2JPB–PC = 16.2 Hz, 2JPB–PA = 5.60 Hz, PB conformer A), 44.8 (dd, 1JPB–Pt = 3526.4 Hz, 2JPB–PC = 16.3 Hz, 2JPB–PA = 5.60 Hz, PB, conformer B), 24.0 (s, PD, conformer B), 23.6 (s, PD, conformer A), 17.1 (dd, 1JPC–Pt = 2366.7 Hz, 2JPC–PA = 379.0 Hz, 2JPC–PB = 16.1 Hz, PC, conformer A), 17.0 (dd, 1JPC–Pt = 2366.7 Hz, 2JPC–PA = 380.4 Hz, 2JPC–PB = 16.3 Hz, PC, conformer B). 19F (376.498 MHz, acetone-d6, 293 K): δ −78.6 (OTf). FT-IR (cm−1): 3056 (w), 2928 (w), 2868 (w), 1436 (m), 1261 (s), 1223 (m), 1147 (m), 1101 (s), 1030 (s), 999 (m), 745 (m), 690 (s), 636 (s), 532 (s), 517 (s), 487 (s). UV/Vis λmax (dichloromethane): 226 nm (major, ε = 199139 M−1 cm−1), 264 nm (shoulder, ε = 67970 M−1 cm−1), 363 nm (minor), 489 (weak, forbidden d–d). ESI-TOF-MS (3 kV, acetonitrile): m/z calcd [(κ2-dppe)PtCl(μ-dpph)RuCl2(η6-p-cymene)]+, 1389.2; expt., 1389.3 (20.3%); [(κ2-dppe)PtCl(κ1-dpph)O]+, 1099.3; expt., 1099.3 (0.53%); [(κ2-dppe)PtCl(κ1-dpph)]+, 1083.3; expt., 1083.3 (12.9%). Temperature studies: 1H NMR (400.130 MHz, DMSO-d6, 313 K): δ 7.90–7.15 (40 H, unresolved m, PPh, conformer A + B), 5.40 (2 H, d, 3JH–H = 5.9 Hz, 2 × CH, –C3,5–H, p-cymene, conformer B), 5.36 (d, 3JH–H = 6.0 Hz, 2 × CH, –C3,5–H, p-cymene, conformer A), 5.23 (2 H, d, 3JH–H = 5.43 Hz, 2 × CH, –C2,6–H, p-cymene, conformer B), 5.20 (d, 3JH–H = 5.95 Hz, 2 × CH, –C2,6–H, p-cymene, conformer A), 2.73 (2 H, unresolved m, 1 × CH2, PCH2ACH2BP, κ2-dppe, conformer A + B), 2.45 (signal obscured by solvent) (2 H, obscured m, 1 × CH2, PCH2ACH2BP, κ2-dppe, conformer A + B), 2.34 (4 H, unresolved m, CH2, dpph + –CH(CH3)2, p-cymene, conformer A + B), 2.15 (2 H, unresolved m, 1 × CH2, dpph, conformer A + B), 1.76 (3 H, s, –C(CH3), p-cymene, conformer B), 1.72 (s, –C(CH3), p-cymene, conformer A), 1.37–1.13 (3 H, unresolved m, CH2, dpph, conformer A + B), 0.90 (4 H, ps d, xJH–X = 17.2 Hz, 2 × CH2, dpph, conformer A + B), 0.77 (6 H, d, 3JH–H = 6.00 Hz, –CH(CH3)2, p-cymene, conformer A + B). 31P{1H} NMR (161.976 MHz, DMSO-d6, 313 K): δ 53.93 (dd, 2JPA–PC = 380.5 Hz, 2JPA–PB = 5.46 Hz, PA, conformer A), 53.84 (dd, 2JPA–PC = 381.8 Hz, 2JPA–PB = 5.6 Hz, PA, conformer B), 44.9 (dd, 1JPB–Pt = 3541.1 Hz, 2JPB–PC = 16.1 Hz, 2JPB–PA = 5.52 Hz, PB conformer A), 44.7 (dd, 1JPB–Pt = 3541.1 Hz, 2JPB–PC = 16.2 Hz, 2JPB–PA = 5.54 Hz, PB conformer B), 23.7 (s, PC, conformer B), 23.3 (s, PC, conformer A), 17.095 (pdd, 1JPC–Pt = 2366.2 Hz, 2JPC–PA = 381.6 Hz, 2JPC–PB = 16.3 Hz, PC, conformer A), 17.0695 (dd, 1JPC–Pt = 2366.2 Hz, 2JPC–PA = 381.8 Hz, 2JPC–PB = 16.3 Hz, PC, conformer B). 1H NMR (400.130 MHz, DMSO-d6, 333 K): δ 7.90–7.17 (40 H, unresolved m, PPh, conformer A + B), 5.37 (2 H, s, 2 × CH, –C3,5–H, p-cymene, conformer B), 5.34 (d, 3JH–H = 5.7 Hz, 2 × CH, –C3,5–H, p-cymene, conformer A), 5.23 (2 H, br s, Δν1/2 = 20.79 Hz, –C2,6–H, p-cymene, conformer A + B), 2.72 (2 H, unresolved m, PCH2ACH2BP, κ2-dppe, conformer A + B), 2.35 (7 H, br s, Δν1/2 = 36.02 Hz, 3 × CH2, dpph + –CH(CH3)2, p-cymene + PCH2ACH2BP, κ2-dppe, conformer A + B), 2.15 (1 H, unresolved m, CH2, dpph, conformer A + B), 1.76 (3 H, s, –C(CH3), p-cymene, conformer B), 1.72 (s, –C(CH3), p-cymene, conformer A), 1.38–1.13 (3 H, unresolved m, CH2, dpph, conformer A + B), 0.92 (4 H, unresolved m, 2 × CH2, dpph, conformer A + B), 0.81 (6 H, s, –CH(CH3)2, p-cymene, conformer A + B). 31P{1H} NMR (161.976 MHz, DMSO-d6, 333 K): δ 53.9 (ps dd, 2JPA–PC = 381.1 Hz, PA, conformer A), 53.8 (ps dd, 2JPA–PC = 382.5 Hz, PA, conformer B), 44.8 (ps dd, 2JPB–PC = 16.1 Hz, PB, conformer A), 44.6 (ps dd, 2JPB–PC = 16.2 Hz, 2JPB–PA = 5.85 Hz, PB, conformer B), 23.4 (s, PC, conformer B), 23.1 (s, PC, conformer A), 17.4 (dd, 2JPC–PA = 382.0 Hz, 2JPC–PB = 16.3 Hz, PC, conformer A + B). 1H NMR (400.130 MHz, DMSO-d6, 353 K): δ 7.90–7.18 (40 H, unresolved m, PPh, conformer A + B), 5.34 (2 H, br s, Δν1/2 = 21.20 Hz, –C3,5–H, p-cymene, conformer A + B), 5.23 (2 H, br s, Δν1/2 = 17.63 Hz, –C2,6–H, p-cymene, conformer A + B), 2.72 (2 H, ps d, xJH–X = 31.9 Hz, PCH2ACH2BP, κ2-dppe, conformer A + B), 2.45–2.25 (7 H, unresolved m, 3 × CH2, dpph + –CH(CH3)2, p-cymene + PCH2ACH2BP, κ2-dppe, conformer A + B), 2.15 (1 H, unresolved m, CH2, dpph, conformer A + B), 1.75 (3 H, s, –C(CH3), p-cymene, conformer B), 1.72 (s, –C(CH3), p-cymene, conformer A), 1.22 (3 H, unresolved m, CH2, dpph, conformer A + B), 0.92 (4 H, 2 × CH2, dpph, conformer A + B), 0.84 (6 H, s, –CH(CH3)2, p-cymene, conformer A + B). 31P{1H} NMR (161.976 MHz, DMSO-d6, 353 K): δ 53.8 (ps dd, 2JPA–PC = 380.1 Hz, PA, conformer A + B), 44.8 (resolved m, PB, conformer A), 44.6 (dd, 2JPB–PC = 16.0 Hz, 2JPB–PA = 5.79 Hz, PB, conformer B), 23.2 (s, PC, conformer B), 23.1 (s, PC, conformer A), 17.2 (ps dd, 2JPC–PA = 381.3 Hz, 2JPC–PB = 16.5 Hz, PC, conformer A + B).
General procedure for the synthesis of [(κ2-dppe)ClPd(μ-dppx)OsCl2(η6-p-cym)](OTf) (PdOs-x)
Two equivalents of the respective diphosphine (0.253 mmol, 2.00 eq.) were dissolved in dichloromethane (5 mL) in a Schlenk flask. Similarly, 174.4 mg (0.1265 mmol, 1.00 eq.) of PdPd-0 was dissolved in dichloromethane (5 mL) and added dropwise to the diphosphine solution. The resulting solution was left to stir at room temperature for one hour. 100 mg (0.1265 mmol, 1.00 eq.) of [(η6-p-cymene)OsCl2]2 dimer was dissolved in dichloromethane (5 mL), under the same conditions as above, and transferred to a Schlenk flask. The Pd-x solution was then added rapidly to the Os dimer solution while stirring. The resulting solution was left to stir at room temperature for two hours. The solvent was removed in vacuo, after which the resulting solid was washed with Et2O (10 mL), and the washings discarded. The product was subsequently dried in vacuo overnight.
Characterisation of [(κ2-dppe)ClPd(μ-dppx)OsCl2(η6-p-cym)](OTf) (PdOs-1)
283 mg (74%) of grey-brown-yellow solid. Melting point: 160–165 °C (melt), 170 °C + (dec). 1H NMR (400.130 MHz, DMSO-d6, 298 K): δ 7.84–7.11 (40 H, unresolved m, PPh), 5.56 (2 H, d, 3JH–H = 5.8 Hz, 2 × CH, –C3,5–H, p-cymene, conformer B), 5.50 (d, 3JH–H = 5.9 Hz, 2 × CH, –C3,5–H, p-cymene, conformer A), 5.41 (2 H, d, 3JH–H = 5.6 Hz, 2 × CH, –C2,6–H, p-cymene, conformer B), 5.38 (d, 3JH–H = 5.5 Hz, 2 × CH, –C2,6–H, p-cymene, conformer A), 2.82 (2 H, ps d, PCH2ACH2BP, κ2-dppe), 2.45–2.26 (obscured by solvent) (4 H, obscured m, PCH2ACH2BP, κ2-dppe + 1 × CH2, dppb), 2.15 (1 H, ps sept, 3JH–H = 6.7 Hz, 1 × –CH(CH3)2, p-cymene), 1.90 (2 H, unresolved m, 1 × CH2, dppb), 1.88 (3 H, br s, Δν1/2 = 3.6 Hz, –C(CH3), p-cymene, conformer B), 1.82 (br s, Δν1/2 = 3.3 Hz, –C(CH3), p-cymene, conformer A), 1.33–1.13 (2 H, br m, 1 × CH2, dppb), 0.98–0.84 (2 H, br m, 1 × CH2, dppb), 0.79 (6 H, br d, Δν1/2 = 2.8 Hz, 3JH–H = 6.8 Hz, –CH(CH3)2, p-cymene, conformer A), 0.75 (br d, Δν1/2 = 2.7 Hz, 3JH–H = 6.8 Hz, –CH(CH3)2, p-cymene, conformer B). 13C{1H} NMR (100.613 MHz, DMSO-d6, 298 K): δ 133.9–127.1 (unresolved m, PPh), 97.2 (s, –C1, p-cymene, conformer A + B), 86.0 (s, –C4, p-cymene, conformer B), 85.7 (s, –C4, p-cymene, conformer A), 81.9 (br s, Δν1/2 = 12.4 Hz, –C3,5–H, p-cymene, conformer A + B), 78.0 (br s, Δν1/2 = 15.6 Hz, –C2,6–H, p-cymene, conformer B), 77.8 (br s, Δν1/2 = 8.7 Hz, –C2,6–H, p-cymene, conformer A), 32.6 (ps d, 1JC–P = 45.0 Hz, PCH2ACH2BP, κ2-dppe), 29.3 (br s, Δν1/2 = 13.2 Hz, –CH(CH3)2, p-cymene, conformer A + B), 26.2 (1 × CH2, dppb), 24.2 (1 × CH2, dppb), 23.9 (1 × CH2, dppb), 22.2 (PCH2ACH2BP, κ2-dppe), 21.8 (1 × CH2, dppb), 21.3 (br s, Δν1/2 = 10.0 Hz, –CH(CH3)2, p-cymene, conformer A + B), 16.8 (br m, –C(CH3), p-cymene, conformer A + B). 31P{1H} NMR (161.976 MHz, DMSO-d6, 298 K): δ 66.2–65.9 (unresolved m, PB, conformer A + B), 64.7 (ps dd, 2JPA–PC = 385.5 Hz, 2JPA–PB = 7.8 Hz, PA, conformer B), 64.5 (ps dd, 2JPA–PC = 397.0 Hz, 2JPA–PB = 7.4 Hz, PA, conformer A), 17.9 (dd, 2JPC–PA = 412.4 Hz, 2JPC–PB = 20.4 Hz, PC, conformer B), 17.8 (dd, 2JPC–PA = 411.8 Hz, 2JPC–PB = 20.7 Hz, PC, conformer A), −17.8 (br s, Δν1/2 = 7.0 Hz, PD, conformer B), −18.4 (br s, Δν1/2 = 6.9 Hz PD, conformer A). 19F NMR (376.498 MHz, DMSO-d6, 298 K): δ −77.6 (OTf). FTIR (cm−1): 3054 (vw), 2960 (vw), 1436 (m), 1260 (s), 1148 (m), 1099 (m), 1030 (s), 999 (w), 743 (m), 690 (vs), 637 (vs), 530 (vs), 516 (vs), 483 (s). UV/Vis λmax (dichloromethane): 226 nm (major, ε = 133371 M−1 cm−1), 267 nm (shoulder, ε = 47219 M−1 cm−1), 332 nm (minor ε = 35676 M−1 cm−1), 421 (weak, forbidden d–d). ESI-TOF-MS (3 kV, dichloromethane): m/z calcd for [(κ2-dppe)PdCl(μ-dppb)OsCl2(η6-p-cymene)]+, 1361.2; expt., 1361.7 (26%). m/z calcd for [(κ2-dppe)PdCl(κ1-dppb)]+, 965.2; expt. 965.5 (46%). m/z calcd for [(η6-p-cymene)OsCl(κ1-dppb)]+, 787.2; expt. 787.5 (100%). m/z calcd for [(κ2-dppe)PdCl]+, 541.0; expt., 541.2 (3%).
Characterisation of [(κ2-dppe)ClPd(μ-dppx)OsCl2(η6-p-cym)](OTf) (PdOs-2)
311 mg (81%) of brown-yellow (turmeric) solid, air stable. 1H NMR (400.130 MHz, DMSO-d6, 298 K): δ 7.84–7.12 (40 H, unresolved m, PPh), 5.56 (2 H, d, 3JH–H = 5.7 Hz, –C3,5–H, p-cymene, conformer B), 5.51 (d, 3JH–H = 5.8 Hz, –C3,5–H, p-cymene, conformer A), 5.43 (2 H, d, 3JH–H = 5.8 Hz, –C2,6–H, p-cymene, conformer B), 5.39 (d, 3JH–H = 6.0 Hz, –C2,6–H, p-cymene, conformer A), 2.81 (2 H, br m, PCH2ACH2BP, κ2-dppe), 2.48 (obscured by solvent) (2 H, obscured br m, PCH2ACH2BP, κ2-dppe), 2.41–2.03 (5 H, unresolved m (ps sept.), 2 × CH2, dppp + 1 × –CH(CH3)2 + p-cymene), 1.87 (3 H, s, 1 × –C(CH3), p-cymene, conformer B), 1.83 (s, 1 × –C(CH3), p-cymene, conformer A), 1.25–0.83 (6 H, unresolved m, 3 × CH2, dppp), 0.80 (6 H, ps d, 3JH–H = 7.0 Hz, CH(CH3)2, p-cymene, conformer B), 0.78 (ps d, 3JH–H = 7.0 Hz, –CH(CH3)2, p-cymene, conformer A). 13C{1H} NMR (100.613 MHz, DMSO-d6, 298 K): δ 133.7–127.4 (unresolved m, PPh), 97.3 (s, –C1, p-cymene, conformer A + B), 85.9 (s, –C4, p-cymene, conformer B), 85.7 (s, –C4, p-cymene, conformer A), 81.9 (br m, Δν1/2 = 7.4 Hz, –C3,5–H, p-cymene, conformer A + B), 77.9 (ps d, –C2,6–H, p-cymene, conformer B), 77.7 (ps d, 2JC–P = 5.2 Hz, –C2,6–H, p-cymene, conformer A), 32.6 (PCH2ACH2BP, κ2-dppe), 31.3 (1 × CH2, dppp), 29.2 (br s, Δν1/2 = 2.84 Hz, –CH(CH3)2, p-cymene, conformer A + B), 24.7 (1 × CH2, dppp), 24.6 (1 × CH2, dppp), 22.2 (PCH2ACH2BP, κ2-dppe), 21.9 (1 × CH2, dppp), 21.4 (br s, Δν1/2 = 1.5 Hz, –CH(CH3)2, p-cymene, conformer A + B), 16.8 (s, –C(CH3), p-cymene, conformer B), 16.7 (s, –C(CH3), p-cymene, conformer A). 31P{1H} NMR (161.976 MHz, DMSO-d6, 298 K): δ 66.3 (ps dd (hidden by conformer A), PB, conformer B), 66.2 (dd, 2JPB–PC = 19.3 Hz, 2JPB–PA = 7.6 Hz, PB, conformer A), 64.7 (ps dd, 2JPA–PC = 410.7 Hz, PA, conformer B), 64.6 (dd, 2JPA–PC = 411.3 Hz, 2JPA–PB = 7.7 Hz, PA, conformer A), 17.8 (dd, 2JPC–PA = 411.0 Hz, 2JPC–PB = 18.8 Hz, PC, conformer B), 17.7 (dd, 2JPC–PA = 411.0 Hz, 2JPC–PB = 18.8 Hz, PC, conformer A), −17.9 (s, PD, conformer B), −18.4 (s, PD, conformer A). 19F NMR (376.498 MHz, DMSO-d6, 298 K): δ −77.6 (OTf). FT-IR (cm−1): 3055 (vw), 2927 (vw), 1434 (m), 1262 (s), 1147 (m), 1099 (m), 1030 (s), 997 (w), 743 (m), 690 (vs), 637 (vs), 530 (vs), 516 (s), 481 (m). UV/Vis λmax (dichloromethane): 226 (major), 270 (shoulder), 331 (minor), 430 (weak, forbidden d–d). ESI-TOF-MS (3 kV, dichloromethane): m/z calcd for [(κ2-dppe)PdCl(μ-dppp)OsCl2(η6-p-cymene)]+, 1375.2; expt., 1375.1 (78%). m/z calcd for [(κ2-dppe)PdCl(κ1-dppp)]+, 979.2; expt. 979.2 (100%). m/z calcd for [(η6-p-cymene)OsCl(κ1-dppp)]+, 801.2; expt. 801.2 (7%). m/z calcd for [(κ2-dppe)PdCl]+, 541.0; expt., 541.0 (60%).
Characterisation of [(κ2-dppe)ClPd(μ-dppx)OsCl2(η6-p-cym)](OTf) (PdOs-3)
318 mg (82%) of golden-orange solid, air stable. 1H NMR (400.130 MHz, DMSO-d6, 298 K): δ 7.84–7.16 (40 H, unresolved m, PPh), 5.56 (2 H, d, 3JH–H = 5.7 Hz, –C3,5–H, p-cymene, conformer B), 5.52 (d, 3JH–H = 5.8 Hz, –C3,5–H, conformer A), 5.42 (2 H, d, 3JH–H = 5.1 Hz, –C2,6–H, p-cymene, conformer B), 5.39 (d, 3JH–H = 5.8 Hz, –C2,6–H, p-cymene, conformer A), 2.83 (2 H, br m, PCH2ACH2BP, κ2-dppe), 2.47 (obscured by solvent) (2 H, obscured br m, PCH2ACH2BP, κ2-dppe), 2.35–1.89 (5 H, unresolved m (ps sept.), 2 × CH2, dpph + –CH(CH3)2, p-cymene), 1.87 (3 H, s, –C(CH3), p-cymene, conformer B), 1.84 (s, –C(CH3), p-cymene, conformer A), 1.36–0.84 (8 H, unresolved m, 4 × CH2, dpph), 0.78 (6 H, br d, Δν1/2 = 11.6 Hz, –CH(CH3)2, p-cymene, conformer A + B). 13C{1H} NMR (100.613 MHz, DMSO-d6, 298 K): δ 133.7–127.4 (unresolved m, PPh), 97.2 (s, –C1, p-cymene, conformer A + B), 85.5 (s, –C4, p-cymene, conformer A + B), 82.0 (br m, Δν1/2 = 8.3 Hz, C3,5, p-cymene, conformer A + B), 77.9 (m, –C2,6, p-cymene, conformer A + B), 32.6 (PCH2ACH2BP, κ2-dppe), 29.8 (s, 1 × CH2, dpph), 29.7 (s, 1 × CH2, dpph), 29.2 (br s, Δν1/2 = 3.3 Hz, –CH(CH3)2, p-cymene, conformer A + B), 26.5 (1 × CH2, dpph), 25.2 (1 × CH2, dpph), 24.7 (1 × CH2, dpph), 24.4 (1 × CH2, dpph), 22.1 (PCH2ACH2BP, κ2-dppe), 21.3 (br s, Δν1/2 = 2.9 Hz, –CH(CH3)2, p-cymene, conformer A + B), 16.81 (s, –C(CH3), p-cymene, conformer B), 16.76 (s, –C(CH3), p-cymene, conformer A). 31P{1H} NMR (161.976 MHz, DMSO-d6, 298 K): δ 66.4–66.1 (br m, Δν1/2 = 37.3 Hz, PB, conformer A + B), 64.8 (ps dd, 2JPA–PC = 412.3 Hz, 2JPA–PB = 7.4 Hz, PA, conformer B), 64.6 (ps dd, 2JPA–PC = 415.0 Hz, PA, conformer A), 18.0 (dd, 2JPC–PA = 410.7 Hz, 2JPC–PB = 19.3 Hz, PC, conformer B), 17.9 (dd, 2JPC–PA = 411.3 Hz, 2JPC–PB = 19.4 Hz, PC, conformer A), −17.7 (br s, Δν1/2 = 6.8 Hz, PD, conformer B), −18.1 (br s, Δν1/2 = 6.2 Hz, PD, conformer A). 19F NMR (376.498 MHz, DMSO-d6, 298 K): δ −77.6 (OTf). FT-IR (cm−1): 3054 (vw), 2930 (vw), 1434 (m), 1263 (s), 1148 (w), 1099 (m), 1030 (s), 998 (w), 743 (m), 691 (vs), 637 (vs), 530 (vs), 516 (s), 486 (m). UV/Vis λmax (dichloromethane): 267 nm (shoulder, ε = 51010 M−1 cm−1), 331 nm (minor ε = 37695 M−1 cm−1), 421 (weak, forbidden d–d). ESI-TOF-MS (3 kV, dichloromethane): m/z calcd for [(κ2-dppe)PdCl(μ-dpph)OsCl2(η6-p-cymene)]+, 1389.2; expt., 1389.1 (33%). m/z calcd for [(κ2-dppe)PdCl(μ-dpph)O]+, 1009.2; expt., 1009.1 (8%). m/z calcd for [(κ2-dppe)PdCl(κ1-dpph)]+, 993.1; expt. 993.1 (100%). m/z calcd for [(η6-p-cymene)OsCl(κ1-dpph)]+, 815.2; expt. 815.2 (35%). m/z calcd for [(κ2-dppe)PdCl]+, 541.0; expt., 541.0 (12%).
Cell viability assays
The effect of all complexes on the viability of various human cancer cell lines was evaluated using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltrazolium bromide (MTT) assay (11465007001; Sigma Aldrich, Missouri, USA) following the manufacturer's instructions. Briefly, cells were seeded in 96-well plates at low densities of 3000–10000 and incubated for 24 to 48 hours. After incubation, cells were treated with vehicle, and all ligands and complexes at 10 μM, and the clinically approved cisplatin drug (CDDP, 10 μM) for 48 hours. Following treatment, 10 μl of MTT solution was added to each well, and the plates were incubated at 37 °C for 4 hours. The resulting purple formazan crystals were solubilised by adding 100 μl of solubilising reagent to each well. Absorbance was measured at 600 nm using a Glomax® microplate spectrophotometer. The cytotoxic effect of each complex was quantified based on the percentage inhibition of cell viability compared to the vehicle-treated control. At least three independent experiments were performed in quadruplicate to determine the IC50 concentration of the respective complexes. The IC50 values were calculated from sigmoidal plots with GraphPad Prism version 7.0 (GraphPad Software, California, USA).
Author contributions
B. R. – conceptualisation, data curation, formal analysis, investigation, methodology, software, validation, visualisation, writing – original draft, writing – review and editing. T. B., M. P., L. W. M., K. S., M. V. M., T. T. M., C. M. – data curation, investigation, methodology, resources, validation, visualisation, writing – review and editing. S. P., T. J. C. – funding acquisition, project administration, supervision. B. B. – conceptualisation, data curation, formal analysis, funding acquisition, methodology, project administration, resources, supervision, validation, writing – review and editing.
Conflicts of interest
There are no conflicts to declare.
Data availability
Supplementary information (SI): Experimental procedures and full characterisation of the complexes, including all spectra and X-ray data; detailed biological studies information. See DOI: https://doi.org/10.1039/d5dt02044a.
CCDC 2470481–2470483 contain the supplementary crystallographic data for this paper.68a–c
Acknowledgements
Prof. Prince gratefully acknowledges and thank the University of Cape Town under the UCT Vision 2030 Grand Challenges Programme, the National Research Foundation of South Africa under a Competitive Programme for Rated Researchers, the International Centre for Genetic Engineering and Biotechnology (ICGEB) under a Collaborative Research Programme, the South African Medical Research Council (SAMRC) under a Self-Initiated Research Grant for financial support, as well as the SAMRC Gynaecological Cancer Research Centre (GCRC). The views and opinions expressed are those of the author(s) and do not necessarily represent the official views of the SAMRC. We also wish to express thanks to Prof. M. Honing and his co-workers for access to and assistance with the HRMS at Maastricht University, Faculty of Science and Engineering.
References
- R. L. Siegel, T. B. Kratzer, A. N. Giaquinto, H. Sung and A. Jemal, CA Cancer J. Clin., 2025, 75, 10–45 Search PubMed.
- F. Bray, M. Laversanne, H. Sung, J. Ferlay, R. L. Siegel, I. Soerjomataram and A. Jemal, CA Cancer J. Clin., 2024, 74, 229–263 Search PubMed.
- M. Peyrone, Justus Liebigs Ann. Chem., 1844, 51, 1–29 CrossRef.
- B. Rosenberg, L. Van Camp and T. Krigas, Nature, 1965, 205, 698–699 CrossRef CAS PubMed.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- G. Y. Ho, N. Woodward and J. I. G. Coward, Crit. Rev. Oncol. Hematol., 2016, 102, 37–46 CrossRef PubMed.
- T. Alcindor and N. Beauger, Curr. Oncol., 2011, 18, 18–25 CrossRef CAS PubMed.
- M. Shimada, H. Itamochi and J. Kigawa, Cancer Manage. Res., 2013, 5, 67–76 CrossRef CAS PubMed.
-
G. Mestroni, E. Alessio and G. Sava, SIGEA S.R.L., World Intellectual Property Organization (WIPO), WO9800431, 1998.
-
B. K. Keppler, Deutsches Patentamt, DE19612291, 1997.
-
B. K. Keppler, M. Henn, U. M. Juhl, M. R. Berger, R. Niebl and F. E. Wagner, New ruthenium complexes for the treatment of cancer, in Progress in clinical biochemistry and medicine, 1989, pp. 41–69 Search PubMed.
-
S. McFarland, Theralase INC., World Intellectual Property Organization (WIPO), WO2013158550, 2013.
- C. S. Allardyce, P. J. Dyson, D. J. Ellis and S. L. Heath, Chem. Commun., 2001, 1396–1397 RSC.
- B. S. Murray, M. V. Babak, C. G. Hartinger and P. J. Dyson, Coord. Chem. Rev., 2016, 306, 86–114 CrossRef CAS.
- R. E. Morris, R. E. Aird, P. del Socorro Murdoch, H. Chen, J. Cummings, N. D. Hughes, S. Parsons, A. Parkin, G. Boyd, D. I. Jodrell and P. J. Sadler, J. Med. Chem., 2001, 44, 3616–3621 CrossRef CAS PubMed.
- R. E. Aird, J. Cummings, A. A. Ritchie, M. Muir, R. E. Morris, H. Chen, P. J. Sadler and D. I. Jodrell, Br. J. Cancer, 2002, 86, 1652–1657 CrossRef CAS PubMed.
- T. Kuijpers and B. Blom, Eur. J. Med. Chem., 2021, 223, 113651 CrossRef CAS PubMed.
- B. Roufosse, C. Serbu, C. Marschner, S. Prince and B. Blom, Eur. J. Med. Chem., 2024, 274, 116528 CrossRef CAS PubMed.
- M. Odachowski, C. Marschner and B. Blom, Eur. J. Med. Chem., 2020, 204, 112613 CrossRef CAS PubMed.
- T. Nabiyeva, C. Marschner and B. Blom, Eur. J. Med. Chem., 2020, 201, 112483 CrossRef CAS PubMed.
- J. Bennett, A. D. Rae, G. Salem, N. C. Ward, P. Waring, K. Wells and A. C. Willis, J. Chem. Soc., Dalton Trans., 2002, 234–243 RSC.
- F. A. Serrano, A. L. Matsuo, P. T. Monteforte, A. Bechara, S. S. Smaili, D. P. Santana, T. Rodrigues, F. V. Pereira, L. S. Silva, J. Machado, E. L. Santos, J. B. Pesquero, R. M. Martins, L. R. Travassos, A. C. F. Caires and E. G. Rodrigues, BMC Cancer, 2011, 11, 296 CrossRef CAS PubMed.
- A. van Niekerk, A. Blanckenberg, S. Kimani, S. Chakraborty, S. Prince, P. Chellan and S. Mapolie, J. Inorg. Biochem., 2023, 243, 112191 CrossRef CAS PubMed.
- W. Villarreal, L. Colina-Vegas, C. Rodrigues de Oliveira, J. C. Tenorio, J. Ellena, F. C. Gozzo, M. R. Cominetti, A. G. Ferreira, M. A. B. Ferreira, M. Navarro and A. A. Batista, Inorg. Chem., 2015, 54, 11709–11720 CrossRef CAS PubMed.
- V. T. Yilmaz, C. Icsel, M. Aygun, M. Erkisa and E. Ulukaya, Eur. J. Med. Chem., 2018, 158, 534–547 CrossRef CAS PubMed.
- C. Cullinane, G. B. Deacon, P. R. Drago, A. P. Erven, P. C. Junk, J. Luu, G. Meyer, S. Schmitz, I. Ott, J. Schur, L. K. Webster and A. Klein, Dalton Trans., 2018, 47, 1918–1932 RSC.
- C. Icsel, V. T. Yilmaz, M. Aygun, B. Cevatemre, P. Alper and E. Ulukaya, Dalton Trans., 2018, 47, 11397–11410 RSC.
- J. L. Dutra, J. Honorato, A. Graminha, C. A. F. Moraes, K. T. de Oliveira, M. R. Cominetti, E. E. Castellano and A. A. Batista, Dalton Trans., 2024, 53, 18902–18916 RSC.
- G. Tonon, M. Mauceri, E. Cavarzerani, R. Piccolo, C. Santo, N. Demitri, L. Orian, P. A. Nogara, J. B. T. Rocha, V. Canzonieri, F. Rizzolio, F. Visentin and T. Scattolin, Dalton Trans., 2024, 53, 8463–8477 RSC.
- R. Czarnomysy, D. Radomska, O. K. Szewczyk, P. Roszczenko and K. Bielawski, Int. J. Mol. Sci., 2021, 22, 8271 CrossRef CAS PubMed.
- T. Scattolin, V. A. Voloshkin, F. Visentin and S. P. Nolan, Cell Rep. Phys. Sci., 2021, 2, 100446 CrossRef CAS.
- C. Zhang, C. Xu, X. Gao and Q. Yao, Theranostics, 2022, 12, 2115–2132 CrossRef CAS PubMed.
- D. Sahoo, P. Deb, T. Basu, S. Bardhan, S. Patra and P. K. Sukul, Bioorg. Med. Chem., 2024, 112, 117894 CrossRef CAS PubMed.
- S. Das, S. Sinha, R. Britto, K. Somasundaram and A. G. Samuelson, J. Inorg. Biochem., 2010, 104, 93–104 CrossRef CAS PubMed.
- B. Herry, L. K. Batchelor, B. Roufosse, D. Romano, J. Baumgartner, M. Borzova, T. Reifenstahl, T. Collins, A. Benamrane, J. Weggelaar, M. C. Correia, P. J. Dyson and B. Blom, J. Organomet. Chem., 2019, 901, 120934 CrossRef CAS.
- E. Klaimanee, T. Nhukeaw, S. Saithong, A. Ratanaphan, S. Phongpaichit, Y. Tantirungrotechai and N. Leesakul, Polyhedron, 2021, 204, 115244 CrossRef CAS.
- H. R. Shahsavari, N. Giménez, E. Lalinde, M. T. Moreno, M. Fereidoonnezhad, R. B. Aghakhanpour, M. Khatami, F. Kalantari, Z. Jamshidi and M. Mohammadpour, Eur. J. Inorg. Chem., 2019, 2019, 1360–1373 CrossRef CAS.
- M. Odachowski, R. Neven, G. Perversi, D. Romano, C. A. Slabber, M. Hadiji, M. Honing, Y. Zhao, O. Q. Munro and B. Blom, J. Inorg. Biochem., 2023, 242, 112156 CrossRef CAS PubMed.
- B. Tűz, I. Correia and P. N. Martinho, J. Inorg. Biochem., 2025, 264, 112813 CrossRef PubMed.
- E. Giorgi, F. Binacchi, C. Marotta, D. Cirri, C. Gabbiani and A. Pratesi, Molecules, 2023, 28, 273 CrossRef CAS PubMed.
- J. A. Davies, F. R. Hartley and S. G. Murray, J. Chem. Soc., Dalton Trans., 1979, 1705–1708, 10.1039/DT9790001705.
- J. A. Davies, F. R. Hartley and S. G. Murray, Inorg. Chem., 1980, 19, 2299–2303 CrossRef CAS.
- F. R. Hartley, S. G. Murray and A. Wilkinson, Inorg. Chem., 1989, 28, 549–554 CrossRef CAS.
- J. S. Kumar, A. K. Singh, J. Yang and J. E. Drake, J. Coord. Chem., 1998, 44, 335–342 CrossRef CAS.
- A. S. Abu-Surrah, T. Debaerdemaeker, W. Huhn, B. Rieger, M. Klinga, T. Repo and M. Leskelä, Acta Crystallogr. Sect. C, 2000, 56, e42–e43 CrossRef.
- Y. Zhou, X. Xu, H. Sun, G. Tao, X.-Y. Chang, X. Xing, B. Chen and C. Xu, Nat. Commun., 2021, 12, 1953 CrossRef CAS PubMed.
- F. E. Hahn, D. Klusmann and T. Pape, Eur. J. Inorg. Chem., 2008, 2008, 4420–4424 CrossRef.
- G. Berton, T. Lorenzetto, G. Borsato, P. Sgarbossa, C. Santo, F. Visentin, F. Fabris and A. Scarso, Tetrahedron Lett., 2019, 60, 151202 CrossRef CAS.
- P. A. W. Dean, J. J. Vittal and R. S. Srivastava, Can. J. Chem., 1987, 65, 2628–2633 CrossRef CAS.
- T. Grell, P. Wonneberger, D. M. Yufanyi, P. Lönnecke and E. Hey-Hawkins, Chem. – Eur. J., 2025, 31, e202500746 CrossRef CAS PubMed.
- J. W. Faller and N. Sarantopoulos, J. Organomet. Chem., 2023, 1000, 122846 CrossRef CAS.
- Y. Otero, D. Peña, A. Arce, M. Hissler, R. Réau, Y. De Sanctis, E. Ocando-Mavárez, R. Machado and T. González, J. Organomet. Chem., 2015, 799–800, 45–53 CrossRef CAS.
- C. Gourlaouen, F. Elaieb, E. Brenner, D. Matt, J. Harrowfield and L. Ricard, Dalton Trans., 2023, 52, 9202–9207 RSC.
- M. A. Zhuravel, D. S. Glueck, C. D. Incarvito and A. L. Rheingold, Organometallics, 1999, 18, 4673–4676 CrossRef CAS.
- J. Bruckmann and C. Krüger, J. Organomet. Chem., 1997, 536–537, 465–472 CrossRef.
- A. B. Chaplin and P. J. Dyson, Eur. J. Inorg. Chem., 2007, 2007, 4973–4979 CrossRef.
- O. Volkov, N. P. Rath and L. Barton, Organometallics, 2002, 21, 5505–5514 CrossRef CAS.
- J. Fernández-Gallardo, B. T. Elie, M. Sanaú and M. Contel, Chem. Commun., 2016, 52, 3155–3158 RSC.
- V. Cadierno, P. Crochet, J. Díez, J. García-Álvarez, S. E. García-Garrido, J. Gimeno, S. García-Granda and M. A. Rodríguez, Inorg. Chem., 2003, 42, 3293–3307 CrossRef CAS PubMed.
- M. R. Boyd and K. D. Paull, Drug Dev. Res., 1995, 34, 91–109 CrossRef CAS.
-
A. M. Burger and H. H. Fiebig, Preclinical screening for new anticancer agents, in Handbook of Anticancer Pharmacokinetics and Pharmacodynamics, Cancer drug discovery and develogment, 2014, pp. 23–38 Search PubMed.
- A. Welsh, K. Serala, S. Prince and G. S. Smith, J. Med. Chem., 2024, 67, 6673–6686 CrossRef CAS PubMed.
- W. Yang, J. Soares, P. Greninger, E. J. Edelman, H. Lightfoot, S. Forbes, N. Bindal, D. Beare, J. A. Smith, I. R. Thompson, S. Ramaswamy, P. A. Futreal, D. A. Haber, M. R. Stratton, C. Benes, U. McDermott and M. J. Garnett, Nucleic Acids Res., 2012, 41, D955–D961 CrossRef PubMed.
- E. Stefàno, L. G. Cossa, F. De Castro, E. De Luca, V. Vergaro, G. My, G. Rovito, D. Migoni, A. Muscella, S. Marsigliante, M. Benedetti and F. P. Fanizzi, Bioinorg. Chem. Appl., 2023, 2023, 1–14 CrossRef PubMed.
- Ž. D. Bugarčić, J. Bogojeski and R. van Eldik, Coord. Chem. Rev., 2015, 292, 91–106 CrossRef.
- S. Dasari and P. B. Tchounwou, Eur. J. Pharmacol., 2014, 740, 364–378 CrossRef CAS PubMed.
- R. Castarlenas, M. A. Esteruelas and E. Oñate, Organometallics, 2005, 24, 4343–4346 CrossRef CAS.
-
(a)
CCDC 2470481: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2nxqy8;
(b)
CCDC 2470482: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2nxqz9;
(c)
CCDC 2470483: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2nxr0c.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Tomas
Brice
a,
Marco
Papaldo
a,
Lucy W.
Macharia
b,
Karabo
Serala
b,
Mihlali V.
Mlaza
b,
Thato T.
Medupe
b,
Sharon
Prince
a,
Tomas
Brice
a,
Marco
Papaldo
a,
Lucy W.
Macharia
b,
Karabo
Serala
b,
Mihlali V.
Mlaza
b,
Thato T.
Medupe
b,
Sharon
Prince















