
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Benchmarking explainable AI methods for toxicophore detection and toxicity prediction
Dina Khasanova *abc and
Igor V. Tetko*ad
*abc and
Igor V. Tetko*ad
aHelmholtz Munich – German Research Center for Environmental Health (GmbH), Institute of Structural Biology, Molecular Targets and Therapeutics Center, 85764 Neuherberg, Germany. E-mail: igor.tetko@helmholtz-munich.de
bTUM School of Natural Sciences, Technical University of Munich, 85748 Garching, Germany. E-mail: dina.khasanova@tum.de
cMolecular Networks GmbH (MN-AM), 90411 Nuremberg, Germany
dBIGCHEM GmbH, Valerystr. 49, 85716 Unterschleißheim, Germany
First published on 14th May 2026
Abstract
Recent studies have reported inconsistent behavior across explainable AI (XAI) methods in molecular property prediction, raising concerns about their reliability. This work investigates whether such inconsistencies arise from the XAI methods themselves or from the accuracy of the underlying predictive model. A high-accuracy model was first trained on deterministic functional-group labels, where all evaluated XAI methods consistently highlighted the correct atoms corresponding to the true structural motifs. The analysis was extended to mutagenicity prediction, where the methods again identified known toxicophores and chemically meaningful scaffolds. Model performance was then systematically degraded by introducing controlled amounts of label noise. As predictive accuracy decreased, agreement between XAI methods weakened gradually, and the highlighted features became less chemically relevant. When accuracy reached around 0.65, this trend changed, with a much sharper loss of agreement, indicating an explainability cliff. These findings underline the importance of assessing model accuracy before drawing conclusions from XAI outputs.
Introduction
Deep learning models have gained a reputation for achieving high predictive accuracy in chemistry.1,2 However, these models remain black-box systems, meaning that experts cannot directly observe or understand which internal decisions the model makes to assign the class thus resulting in trade-off between predictivity and explainability.3 As a result, users are unable to obtain chemical insight from the model's learning process or to modify the initial molecular structure to enhance desired properties or reduce toxicity. Explainable artificial intelligence (XAI) therefore plays a crucial role in bridging this gap by making model predictions more interpretable and scientifically meaningful.4 XAI techniques can be broadly divided into model-agnostic and model-specific methods.5 Model-agnostic approaches estimate feature importance through perturbation or sampling strategies. Model-specific methods, in contrast, rely on internal signals such as gradients, activations, or convolutional feature maps, and their behavior depends strongly on the architecture. For instance, in convolutional neural networks with multiple parallel filters (as in Transformer-CNN6), methods like Grad-CAM (Gradient-weighted Class Activation Mapping7) must be adapted to capture contributions from all convolutional branches rather than a single layer. Recent studies have demonstrated that XAI methods can reveal chemically meaningful attributions in molecular property prediction and specifically toxicity modeling. SHAP (SHapley Additive exPlanations)8 has been applied to assess torsadogenic risk, successfully identifying electrophysiological biomarkers that improved both prediction and model design.9 Attention-based graph neural networks using multiple molecular graph representations have been used to detect toxicophores in mutagenic compounds, with explanations aligning with known structural alerts.10 Another approach aligned internal graph neural networks (GNN) activations with chemically interpretable features, resulting in a self-explaining framework for property prediction.11 These examples highlight the potential of XAI to extract human-interpretable insights from complex models and build trust in their outputs.Despite its potential, the application of XAI in chemistry faces notable challenges. Recent work by Hartog et al.12 evaluated multiple XAI methods in molecular property prediction and demonstrated that their explanations were often inconsistent. Different representations of the same molecule led to different sets of important features being highlighted, and the quantitative comparison showed low agreement among the explanation techniques. Similar inconsistencies were observed even in randomly initialized models, suggesting that some XAI outputs may be driven more by artifacts of molecular representation (such as SMILES tokenization) than by true learned chemical patterns. Additionally, Adebayo et al.13 demonstrated that several widely used attribution methods fail basic sanity checks, producing explanations that remain largely unchanged even when model parameters are randomized. This indicates that some explanations may be driven more by input statistics or architectural biases than by the learned decision function itself. Another study14 showed that saliency-based explanations can change substantially under simple input transformations, such as constant shifts or preprocessing operations, despite identical model predictions. These findings raise fundamental questions about the reliability and reproducibility of current XAI approaches in cheminformatics.
While such inconsistencies are often interpreted as a limitation of XAI itself, another possible explanation lies in the accuracy of the underlying predictive model. When the model's accuracy is low and its predictions are uncertain, the resulting explanations become unreliable. In this case, each method may attempt to rationalize unstable or incorrect predictions, leading to different sets of highlighted features. In this case, poor explainability may reflect the model's limited predictive power rather than a weakness of the XAI methods, making it misleading to assess explanations without considering model accuracy. To investigate this issue, we propose using a high-accuracy model trained on deterministic labels, such as functional groups. Because these labels are well-defined and directly linked to structural motifs, this controlled setup enables us to assess whether the inconsistencies observed in prior work arise from the XAI methods themselves or from the predictive model's performance.
Finally, the insights from this benchmark are extended to the more challenging task of toxicity prediction. For this purpose, the Ames dataset is used, a standard benchmark for mutagenicity prediction. It includes compounds with well-known structural alerts linked to mutagenicity, which makes it appropriate for evaluating toxicity prediction models and their explanations. Unlike functional groups, toxicity depends on diverse physicochemical features,2,15 including conjugated systems, bulky substituents, and reactive groups, which complicate both modeling and interpretation. Our goal is to produce explanations that are chemically interpretable and consistent, thereby enhancing confidence in predictive models.
Methods
In this section, the datasets used in this study are first introduced. Next, the applied XAI methods are presented, along with a discussion of their differences and domains of applicability. The model architecture, training procedure, and prediction of functional groups and toxicity are then described. Finally, we introduce the quantitative similarity metric used to assess agreement between token-level attributions produced by different XAI methods.Datasets
To pre-train the transformer encoder to convert non-canonical SMILES strings to their canonical SMILES, ChEMBL_V29 (ref. 16) dataset was used. ChEMBL provides a large collection of curated bioactive molecules, making it a suitable resource for learning robust SMILES representations. For predicting functional groups and the toxicity label, the Ames dataset was used.17,18 Ames test is a widely used biological assay that detects the mutagenic potential of chemical compounds by measuring their ability to induce mutations in Salmonella typhimurium strains. This dataset contains multiple well-characterized structural alerts associated with mutagenicity, making it a suitable benchmark for evaluating different XAI methods and their correctness. The dataset contains test results for 7255 drugs and class balance. Both these datasets were obtained from Therapeutics Data Commons.19 We pre-processed datasets to remove ambiguities and incorrect compounds. Molecules were standardised for correct bonding, aromaticity, and hybridisation. Salts were removed to isolate the primary compound and Simplified Molecular Input Line Entry System (SMILES)20 strings were converted to their canonical forms. Duplicate molecules and those with conflicting labels were removed. Molecules with canonical SMILES strings longer than 200 characters were also excluded. These steps were performed using the RDKit package version 2024.03.5. The reported dataset size is after cleaning. The ChEMBL_V29 dataset was randomly divided into 90% training and 10% validation data, while the Ames dataset was split into 70% training, 10% validation, and 20% test subsets.To support the interpretation of model predictions, a set of knowledge-driven structural alerts was included. These alerts correspond to well-established toxicophores associated with mutagenicity and genotoxic carcinogenicity, originally derived from systematic structure–activity analyses. Foundational contributions include the mutagenicity alerts curated and validated by Kazius et al.;21 the structural features associated with rodent carcinogenicity and bacterial mutagenicity reported by Ashby et al.;22 the optimized and mechanistically derived alerts compiled by Benigni et al.;23 and the SAR-based evaluation of carcinogenic hazards in food contact substances described by Bailey et al.24 The corresponding SMARTS patterns are available in OCHEM.25,26
XAI methods
Five XAI methods were employed in this study, including both model-agnostic and model-specific approaches. The model-agnostic methods included SHAP values (GradientExplainer)8 and Occlusion,27 which can be applied to any machine-learning model regardless of its architecture. These techniques estimate feature importance either through perturbation-based analyses (Occlusion) or by approximating Shapley values from cooperative game theory (SHAP). In contrast, the model-specific methods consisted of Integrated Gradients,28 DeepLIFT,29 and Grad-CAM.7 These gradient-based techniques rely on access to the internal structure of neural networks (activations, gradients or convolutional feature maps). Integrated Gradients and DeepLIFT attribute feature relevance by comparing predictions to a reference baseline, while Grad-CAM produces localization maps based on gradients propagated to the convolutional layers. All methods were implemented using the Captum library30 in Python, providing a unified and reproducible framework for comparing XAI techniques. Among the model-specific methods, gradient-based attribution techniques such as Integrated Gradients and DeepLIFT rely on two formal axioms. Sensitivity requires that features influencing the prediction compared to a chosen baseline receive non-zero attribution, and implementation invariance ensures that functionally identical models produce identical attributions. SHAP, in contrast, is based on the Shapley axioms from cooperative game theory: efficiency (the attributions sum to the model output relative to the baseline), symmetry (features contributing equally receive identical attributions), dummy or null-player (features that do not affect the prediction receive zero attribution), and additivity (attributions for combined models equal the sum of attributions for each model). Occlusion is a purely perturbation-based method and is not defined by attribution axioms, while Grad-CAM depends on convolutional feature maps in specific layers and therefore follows architectural constraints rather than general axiomatic requirements. In token-based language models, each feature corresponds to a discrete SMILES token whose embedding must remain consistent with the model vocabulary. Perturbation-based explanation methods that alter features directly in embedding space can therefore produce inputs that no longer correspond to valid chemical symbols or chemically meaningful molecular representations. This issue is particularly relevant for model-agnostic explanation methods such as Kernel SHAP8 and LIME,31 which approximate feature importance through evaluations on perturbed feature coalitions or locally sampled perturbations. Such perturbations can break the correspondence between embeddings and valid SMILES tokens, leading to unstable and chemically implausible attributions. For deep neural models, however, SHAP also provides gradient-based variants. GradientExplainer32 (often referred to as GradientSHAP) estimates attributions using expected gradients for differentiable models, avoiding arbitrary replacement of embedding dimensions. DeepExplainer is likewise designed for deep learning models, although it is based on Deep SHAP/DeepLIFT-style approximations rather than the model-agnostic Kernel SHAP formulation. In addition, Occlusion27 is more suitable for token-based molecular inputs because it replaces tokens with a valid mask or padding symbol, thereby preserving the discrete input structure expected by the model. In contrast, TreeExplainer and LinearExplainer are specialized for tree ensembles and linear models and are not directly applicable to token-based SMILES language models.Standard gradient-based attribution can suffer from the vanishing-gradient problem, where the gradient at the input becomes extremely small because of saturating nonlinearities or long chains of multiplications during backpropagation.31 As a result, important features may incorrectly appear to have little or no influence on the model's output. Integrated Gradients (IG) solve this issue by evaluating gradients along a path from a baseline input to the actual input, capturing changes in the model's sensitivity even when the final input gradient is near zero.28 For embedding-based models, several baselines can be used. A zero baseline sets all embedding values to zero and represents the absence of signal, while a mean baseline,33 computed as the average embedding across the training data, provides a smoother reference point. In this work, the zero baseline was chosen because it is computationally efficient and does not require additional embedding passes. Although IG overcomes the vanishing-gradient limitation of standard gradients, it remains computationally more demanding because it requires multiple gradient evaluations along the integration path.
DeepLIFT,29 on the other hand, compares each neuron's activation to a reference activation and assigns contribution scores based on these differences. This enables efficient computation of attributions in a single backward pass. Moreover, DeepLIFT separates positive and negative contributions, which helps avoid situations where gradient-based methods assign all importance to a single input feature. Integrated Gradients (IG), by comparison, computes feature attributions by integrating gradients along a path from a baseline input x′ to the actual input x, defined as:
 | (1) |
GradientSHAP32 is an extension of Integrated Gradients that incorporates SHAP's framework of attribution based on expected contributions. Unlike standard IG, which computes attributions along a single path from a baseline to the input, GradientSHAP samples multiple perturbed versions of the baseline by adding noise and averages the resulting integrated gradients. In our implementation, we use a zero baseline, corresponding to an input with no signal in the embedding space. By doing so, GradientSHAP approximates SHAP values,8 which quantify a feature's importance as its expected marginal contribution to the model output across different reference inputs. This combination allows GradientSHAP to capture both IG's path-based sensitivity and SHAP's distribution-aware feature contribution, providing robust attributions for our model.
While GradientSHAP extends Integrated Gradients by incorporating SHAP's framework of expected feature contributions, SHAP itself belongs to a broader family of perturbation-based explainability methods. These approaches estimate feature importance by measuring how the model's prediction changes when parts of the input are removed or replaced. Within this perturbation family, we adopt Occlusion,27 which operates at the token level rather than on individual embedding dimensions. In our setup, the method masks one token at a time by replacing its entire multidimensional embedding vector with a baseline value (zeros) and then measures the resulting change in the model output. This preserves the semantic structure of the learned representation and yields chemically meaningful perturbations. In contrast, classical SHAP attempts to remove individual embedding dimensions, even though these dimensions are not independent and altering a single component does not correspond to any meaningful chemical modification.
For comparison with gradient- and perturbation-based attributions, we additionally applied Gradient-weighted Class Activation Mapping (Grad-CAM), originally introduced by Selvaraju et al..7 Grad-CAM produces class-specific relevance maps by weighting the activations of a chosen convolutional layer with the gradients of the output score with respect to those activations, thereby highlighting the regions of the input that most strongly influence the prediction through higher-level convolutional features. Although Grad-CAM is applicable to a wide range of CNN architectures, it must be applied to a specific convolutional layer, typically the last one. Because our model contains multiple parallel convolutional layers with different kernel sizes, we compute Grad-CAM maps for each layer separately and merge them into a unified relevance representation (details provided in the Discussion section). Unlike GradientSHAP, IG, Occlusion, or DeepLIFT, which evaluate importance at the level of individual input features or perturbation units, Grad-CAM attributes importance at the level of learned feature maps, providing insight into how the network's convolutional filters integrate local sequence patterns into higher-level representations. This makes Grad-CAM a complementary XAI technique that captures model behavior not accessible through token-level or embedding-level attribution methods.
Model architecture
For property prediction, the model architecture proposed by Karpov et al.6 was adopted, combining a pre-trained transformer encoder with a convolutional neural network. This architecture has been successfully applied as a winning solution in several competitive benchmarks for molecular property prediction. For example, it was used by the winning models of the Tox24 challenge,1 where compounds were evaluated for activity against transthyretin (TTR), and by the top-performing model in the first EUOS/SLAS joint compound solubility challenge.34 All models were implemented in PyTorch35 (version 2.4.0). SMILES strings were processed at the character level, where each symbol was treated as an individual token with a learned embedding vector. The transformer encoder was pre-trained in a sequence-to-sequence architecture to convert non-canonical SMILES into their canonical form, a task that enables the model to learn chemically meaningful token-level representations. After pretraining, only the encoder block was retained and used as a fixed embedding model for downstream convolutional processing. For the transformer encoder, the same hyperparameters as in the original publication were used, except that the context length was increased from 110 to 202 tokens to enable the generation of embeddings for larger molecules. The hyperparameters of the convolutional neural networks were optimized using a Bayesian optimization approach, which is described in the next section. The model was first applied to functional group prediction, formulated as a multilabel classification task in which the output corresponds to a binary vector indicating the presence (1) or absence (0) of each functional group. The same architecture was subsequently used for toxicity prediction, formulated as a binary classification task to determine whether a molecule is toxic or non-toxic.Model training
The hyperparameters of the model were optimized using the Tree-structured Parzen Estimator (TPE) algorithm36 implemented in Optuna (version 4.1.0).37 The convolutional filter sizes from the original publication6 were kept, while dropout (0.0–1.0), learning rate (1e-5–1e-2), and weight decay (1e-6–1e-3) were optimized. The validation cross-entropy loss was evaluated for different hyperparameter combinations. Each training run was conducted for a maximum of 20 epochs. Training was terminated early if the validation loss did not improve for two consecutive epochs, or if, after 15 epochs, the validation loss fell below the median value for that epoch. After determining the optimal hyperparameters, the final models were trained until convergence on the validation set. Early stopping with a patience of 10 epochs was applied, and the model weights corresponding to the lowest validation loss were saved. All models were trained using a weighted binary cross-entropy loss function, where the weights accounted for class imbalance by being inversely proportional to class frequency. The original Adam (Adaptive Moment Estimation) optimizer38 was used and training was performed with a batch size of 64 samples.Cosine distance
With more than seven thousand molecules in the dataset, inspecting individual explanations becomes impractical. A quantitative metric is therefore needed to determine whether different XAI methods highlight similar molecular regions or identify entirely unrelated features. Cosine distance is applied for this purpose. Cosine similarity, and by extension cosine distance, is a standard measure for comparing high-dimensional vectors.39 The similarity between two vectors is defined as the cosine of the angle between them, and cosine distance is computed as: cosine distance = 1 − cos(θ). This metric captures whether two attribution vectors point in a similar direction (highlighting similar atoms), are orthogonal (unrelated), or point in opposite directions (highlighting contradictory atomic regions), as illustrated in Fig. 1. | ||
| Fig. 1 Geometric interpretation of cosine similarity and cosine distance for evaluating whether XAI methods highlight similar, unrelated, or different molecular regions. | ||
For attribution methods that produce embedding-level attributions per token (Integrated Gradients, GradientSHAP, DeepLIFT, and Occlusion), the original explanations are represented as token-by-embedding matrices, where each token is associated with a 64-dimensional attribution vector. To obtain a single attribution value per token for comparison, these embedding-level attributions were reduced by averaging across embedding dimensions:
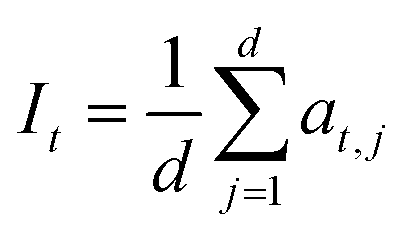 | (2) |
 | (3) |
Cosine distance was subsequently computed between pairs of normalized attribution vectors for each molecule, yielding one distance value per molecule for each pair of methods. Mean and standard deviation of these distances were then calculated across all molecules to quantify overall agreement or dissimilarity between the XAI methods.
Results
In the analysed dataset used in this study, functional groups were not pre-assigned and therefore had to be identified. This was accomplished using SMARTS pattern matching. Instead of relying on simple SMARTS definitions, which often capture only the core substructure, we used the Extended Functional Groups (EFG) SMARTS patterns.26 EFG defines both required and forbidden atomic environments (e.g., distinguishing ketones from carboxylic acids or amides that also contain a carbonyl group), resulting in more specific and chemically accurate functional group assignments.The number of occurrences of each functional group in the dataset is highly imbalanced: most molecules contain a group only once, while structures with multiple repetitions are rare (SI Table 1). From a toxicological perspective, the presence of a single toxicophore is generally sufficient to initiate mutagenicity, and additional copies do not proportionally increase the effect. Therefore, predicting exact counts provides little chemical value, and a regression objective would be dominated by the large number of low-count examples. For this reason, the task is formulated as a multi-label classification problem that focuses on the presence or absence of each functional group. Using this formulation, our model achieved a micro-averaged ROC-AUC of 1.0 on the test set, indicating perfect discrimination between the presence and absence of functional groups. We report the micro-average because it summarizes performance across all labels in a single metric, offering a clear overall view of how well the model detects functional groups in the multi-label setting.
The mean cosine distance was computed for each pair of the five XAI methods, and the results averaged over all functional groups are reported in Table 1. The per-group results are provided in the SI Table 2. Two metrics were evaluated: mean positive and mean all. The mean all metric considers all molecules, regardless of whether a given functional group is present, whereas mean positive includes only those cases where the corresponding bit in the output label vector is positive.
| Method pair | Mean distance (positive class) | Mean distance (all classes) |
|---|---|---|
| a SHAP – GradientSHAP; IG – Integrated Gradients; DeepLift – Deep Learning Important FeaTures; Occlusion – Occlusion-based attribution; Grad-CAM – Gradient-weighted Class Activation Mapping. | ||
| shap/ig | 0.0001 ± 0.00 | 0.0001 ± 0.00 |
| shap/deeplift | 0.0028 ± 0.00 | 0.0038 ± 0.00 |
| shap/occlusion | 0.0299 ± 0.01 | 0.0247 ± 0.01 |
| shap/gradcam | 0.3320 ± 0.07 | 0.2120 ± 0.02 |
| ig/deeplift | 0.0026 ± 0.00 | 0.0037 ± 0.00 |
| ig/occlusion | 0.0298 ± 0.01 | 0.0246 ± 0.01 |
| ig/gradcam | 0.3320 ± 0.07 | 0.2120 ± 0.02 |
| deeplift/occlusion | 0.0324 ± 0.01 | 0.0260 ± 0.01 |
| deeplift/gradcam | 0.3216 ± 0.07 | 0.2130 ± 0.02 |
| Occlusion/gradcam | 0.3578 ± 0.06 | 0.2173 ± 0.02 |
However, it is crucial to confirm that the agreed-upon attributions correspond to chemically meaningful features. Since our model is built to predict functional groups, the most influential fragments should indeed be the relevant functional groups rather than chemically unrelated parts of the molecule. To validate the chemical sense of these attributions, we visualized them on each molecule's structure using XSMILES:40 an interactive visualization technique that overlays attribution scores on a 2D molecular graph alongside the molecule's SMILES string. For visualization, token-level attribution values were computed by aggregating embedding-level attributions using the sum of absolute values across embedding dimensions:
 | (4) |
 | (5) |
 | ||
| Fig. 2 Comparison of atom-level attribution maps for the hydroxyl group using five explainability methods: Integrated Gradients (IG), SHAP, DeepLIFT, Occlusion, and Grad-CAM (ROC–AUC = 1). In this example, all methods highlight the hydroxyl (–OH) group as the key structural feature contributing to the prediction, while Grad-CAM produces a broader attribution pattern that extends to neighboring atoms beyond the hydroxyl group. The color scale encodes relative importance, with magenta indicating low-importance tokens and green indicating high-importance tokens; attribution values were normalized to [-1, 1], such that colors reflect relative magnitude rather than the direction of contribution. | ||
The attribution maps from IG, GradientSHAP, and DeepLIFT are almost identical after rounding the values to two decimal places. A similar correlation was observed by Ancona et al.41 when comparing gradient-based attribution methods. The occlusion method yields very similar importance scores at the token level but with slight deviations. It correctly highlights the key tokens corresponding to the hydroxyl group, although a few atoms appear with marginally lower emphasis. These minor differences arise because occlusion isolates each feature's impact by removal, which can cause subtle shifts in attribution when nonlinear feature interactions are present. In contrast, Grad-CAM produces a broader and more diffuse importance map. The tokens responsible for the functional group still exhibit positive attributions, but neighboring atoms also receive positive scores, some even higher than the target atom. This can be explained by Grad-CAM's tendency to spread importance across larger regions of the molecule, whereas the other methods concentrate it on more sharply defined atomic sites.
Once the methods are confirmed to highlight the correct token as the leading contributor to the prediction, all methods are applied to the same dataset for a different property: toxicity. Our model achieved an ROC–AUC of 0.85, indicating strong predictive performance given the challenging and heterogeneous nature of this property. This value is consistent with the best results reported in the literature,42–44 suggesting that the model operates near the dataset's performance ceiling, where further improvements are limited by data noise and experimental uncertainty (Table 2).
| Method pair | Method distance (positive class) | Mean distance (all classes) |
|---|---|---|
| shap/ig | 0.0001 ± 0.00 | 0.0001 ± 0.00 |
| shap/deeplift | 0.0008 ± 0.00 | 0.0012 ± 0.00 |
| shap/occlusion | 0.0175 ± 0.02 | 0.0199 ± 0.02 |
| shap/gradcam | 0.1761 ± 0.07 | 0.1784 ± 0.07 |
| ig/deeplift | 0.0007 ± 0.00 | 0.0011 ± 0.00 |
| ig/occlusion | 0.0174 ± 0.02 | 0.0197 ± 0.02 |
| ig/gradcam | 0.1761 ± 0.07 | 0.1785 ± 0.07 |
| deeplift/occlusion | 0.0171 ± 0.01 | 0.0197 ± 0.02 |
| deeplift/gradcam | 0.1761 ± 0.07 | 0.1788 ± 0.07 |
| Occlusion/gradcam | 0.1712 ± 0.07 | 0.1780 ± 0.07 |
The attribution patterns derived from IG, DeepLIFT, and GradientSHAP remain closely aligned, with mean cosine distances on the order of 1.0 × 10−4 between GradientSHAP and IG and around 7.0 × 10−4 between IG and DeepLIFT. These small values indicate a strong agreement among the gradient- and backpropagation-based approaches. Occlusion again shows slightly higher distances of approximately 1.7 × 10−2, reflecting small deviations caused by its perturbation-based mechanism. In contrast, Grad-CAM yields notably larger distances of roughly 1.8 × 10−1 from all other methods, confirming its broader and less localized attribution patterns. Overall, the consistency between IG, DeepLIFT, and GradientSHAP and the divergence of Grad-CAM follow the same pattern observed for the functional group prediction task, demonstrating that these relationships persist across different molecular endpoints.
Across all three case studies (Fig. 3, S4, and S5), the five explainability methods consistently recover chemically meaningful toxicophore regions, while differing in attribution sharpness and spatial spread. In Fig. 3 (epoxide toxicophore), all methods clearly identify the three-membered epoxide ring as the primary driver of toxicity. Integrated Gradients, GradientSHAP, and DeepLIFT produce highly localized attributions, concentrating almost exclusively on the oxygen atom and the two carbons forming the strained three-membered epoxide ring. Occlusion yields a similar pattern but with reduced contrast between important and non-important atoms. In contrast, Grad-CAM highlights a broader region, extending relevance beyond the epoxide ring into the neighboring aromatic system, suggesting a more diffuse representation of the same toxicophore. In Fig. S4 (aliphatic halide toxicophore), the methods consistently attribute importance to the terminal halogen (Br) and its adjacent carbon chain. IG, SHAP, and DeepLIFT sharply prioritize the bromine atom and the α-carbon, aligning with the expected electrophilic reactivity of alkyl halides. Occlusion again follows this trend but distributes importance more evenly along the carbon chain. Grad-CAM highlights the halogen while assigning negative contribution to the alcohol group at the opposite end of the molecule, indicating a broader contextual sensitivity. In Fig. S5 (nitroso toxicophore), all methods converge on the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O functional group as the primary driver of the prediction, with Integrated Gradients, GradientSHAP, and DeepLIFT specifically localizing importance to the oxygen and nitrogen atoms of the nitroso group. Occlusion reproduces this pattern with slightly reduced contrast. Grad-CAM again produces a more diffuse map, extending relevance to surrounding atoms while still capturing the nitroso group as the dominant signal. Overall, these examples demonstrate that while all methods reliably detect the same toxicophore across structurally diverse molecules, gradient-based methods (IG, SHAP, DeepLIFT) provide the most precise, atom-level localization, Occlusion offers smoother but consistent attribution, and Grad-CAM captures the correct region with reduced spatial specificity, often extending importance to neighboring atoms.
O functional group as the primary driver of the prediction, with Integrated Gradients, GradientSHAP, and DeepLIFT specifically localizing importance to the oxygen and nitrogen atoms of the nitroso group. Occlusion reproduces this pattern with slightly reduced contrast. Grad-CAM again produces a more diffuse map, extending relevance to surrounding atoms while still capturing the nitroso group as the dominant signal. Overall, these examples demonstrate that while all methods reliably detect the same toxicophore across structurally diverse molecules, gradient-based methods (IG, SHAP, DeepLIFT) provide the most precise, atom-level localization, Occlusion offers smoother but consistent attribution, and Grad-CAM captures the correct region with reduced spatial specificity, often extending importance to neighboring atoms.
 | ||
| Fig. 3 Comparison of atom-level attribution maps for the epoxide toxicophore using five explainability methods: integrated gradients (IG), SHAP, DeepLIFT, Occlusion, and Grad-CAM (ROC–AUC = 0.85). In this example, all methods correctly highlight the epoxide ring, while Grad-CAM highlights a broader region, extending relevance beyond the epoxide ring into the neighboring aromatic system, suggesting a more diffuse representation of the same toxicophore. The color scale encodes relative importance, with magenta indicating low-importance tokens and green indicating high-importance tokens; attribution values were normalized to [−1, 1], such that colors reflect relative magnitude rather than the direction of contribution. | ||
To investigate whether the agreement between XAI methods is influenced by the accuracy of the underlying model, the model's performance was intentionally reduced in a controlled way. A subset of training labels was randomly flipped to the opposite class, introducing symmetric label noise at seven levels (0, 5, 15, 25, 35, 40, and 50% of the training set). This type of synthetic label noise is widely used to assess the robustness of machine-learning models and the stability of their explanations.45 The chosen range applies a controlled reduction from clean labels to substantial corruption, resulting in a gradual decrease in ROC–AUC while ensuring that the prediction task remains learnable and meaningful.
Fig. 4 shows how the agreement between attribution methods changes when increasing amounts of noise (0–50%) are added to the toxicity labels. For each method pair, the mean cosine distance between their toxicity explanations is shown together with standard deviation bars. Smaller distances indicate higher similarity. Method pairs involving GradientSHAP, Integrated Gradients, and DeepLIFT remain highly consistent across all noise levels, while pairs that include Grad-CAM or Occlusion show substantially higher distances and increasingly divergent explanations as label noise rises. The dashed line indicates the model's ROC–AUC, which decreases with increasing noise, reflecting the expected decline in predictive performance. The apparent increase observed at low noise levels falls within the corresponding confidence intervals obtained via bootstrap resampling and does not represent a statistically significant improvement. In addition to the increase in the mean cosine distances, the standard deviations also grow with higher corruption levels, indicating that disagreement between attribution methods becomes not only stronger but also more variable across molecules. The overall trend is not strictly linear: several method pairs show local decreases or plateaus at intermediate noise levels. Such non-monotonic behavior can occur because moderate noise does not uniformly degrade the learned decision boundary. At certain noise levels, the model may still rely on partially stable patterns or may shift toward a simpler decision rule, temporarily reducing variability in the explanations. Only at higher noise levels does the combination of weaker predictive performance and less informative feature attributions lead to consistently larger cosine distances and increased standard deviation.
 | ||
| Fig. 4 Cosine distance comparisons for all XAI method pairs at different input noise levels. | ||
To examine how reduced model accuracy affects the resulting explanations, the same molecule used in the high-accuracy (ROC–AUC 0.85) setting was analyzed with XAI methods after degrading performance to a ROC–AUC of 0.66 (Fig. 5). The core attribution pattern remains detectable across methods, indicating that the model still captures some aspects of the underlying structure despite the added label noise. However, the attributions are noticeably less confident, with both positive and negative contribution values being substantially lower in magnitude. In addition, a larger number of tokens are highlighted as important, particularly in the case of Grad-CAM, suggesting that the model distributes relevance more diffusely when its predictions are less reliable. Taken together, these findings suggest the existence of a performance-dependent threshold below which XAI explanations lose reliability and increasingly capture noise rather than meaningful signal.
 | ||
| Fig. 5 Comparison of atom-level attribution maps for the epoxide toxicophore using five explainability methods: integrated gradients (IG), SHAP, DeepLIFT, Occlusion, and Grad-CAM (ROC–AUC = 0.66). In this example, all methods still identify the epoxide ring, but attributions are weaker and extend to an additional carbon not part of the toxicophore, with Grad-CAM showing reduced ability to distinguish the most relevant tokens. The color scale encodes relative importance, with magenta indicating low-importance tokens and green indicating high-importance tokens; attribution values were normalized to [−1, 1], such that colors reflect relative magnitude rather than the direction of contribution. | ||
Discussion
Impact of molecular representation on explainable AI
In this work, molecular structures are represented using SMILES strings, rather than alternative representations such as ECFPs or image-based encodings. This is important because XAI methods can behave differently depending on the type of molecular representation used. A major limitation of Extended Connectivity Fingerprints (ECFPs)46 lies in bit collisions, which occur when multiple distinct molecular substructures are mapped to the same bit during the fingerprint generation process. This becomes particularly problematic in the context of XAI, where attribution methods identify the contribution of individual bits to model predictions. When a specific bit is considered important, it is often unclear which exact molecular fragment it represents, as several may share the same bit due to collisions.47 There are ways to reduce bit collisions or retain RDKit's bitInfo to trace atom environments, but they are not straightforward.Another approach is to represent molecules as 2D images for model input, allowing the use of computer vision techniques for explainability. This enables pixel-space attribution maps that directly highlight relevant regions of a molecule's structure: from individual atoms and bonds to entire functional groups, thereby capturing both localized and higher-level chemical features. Such image-derived explanations remain consistent under rotations or reflections of the molecular depiction.48 However, image-based approaches often require substantially more computational resources and large, well-annotated datasets to achieve comparable performance to text- or graph-based methods. In contrast, SMILES representations are lightweight, widely available, and efficiently processed by transformer-based or sequence models, making them a practical choice for toxicity datasets and explainable AI studies.
Agreement between XAI methods
Pairwise cosine distances show that the gradient-based method: Integrated Gradients (IG) and the backpropagation-based method: DeepLIFT produce nearly identical token-level attributions (e.g., IG vs. DeepLIFT ≈ 0.0026). GradientSHAP, which combines gradient information with SHAP's perturbation weighting, also aligns closely with both (SHAP vs. IG ≈ 0.0001; SHAP vs. DeepLIFT ≈ 0.0028), confirming overall consistency among attribution methods that rely on model derivatives or internal backpropagation. Occlusion, a purely perturbation-based approach, shows slightly larger distances (≈0.03 vs. each), as expected since it measures the direct effect of masking tokens rather than propagating internal signals. In contrast, all comparisons involving Grad-CAM are an order of magnitude larger (≈0.33–0.36 on the positive subset and ≈ 0.21–0.22), indicating that Grad-CAM captures systematically different regions of importance than the other four techniques. In general, models with higher predictive performance tend to produce more consistent explanations across different XAI methods. When predictions are reliable, attribution techniques largely agree on which parts of the input are important, suggesting that the model relies on stable decision patterns. As performance decreases across the explored range, this agreement gradually weakens. At the lower end of this range, comparable to performance levels reported in the work of Hartog et al. (for different transformer architectures from 0.64 to 0.72),12 differences between explanation methods become more noticeable. This suggests that disagreement between XAI methods is primarily driven by reduced model reliability rather than by limitations of the explanation techniques themselves.The aggregation of Grad-CAM across all convolutional layers
The Transformer-CNN uses parallel convolutional layers with kernel sizes ranging from 1 to 20; each branch considers a specific number of tokens at a time (from single tokens up to 20-token fragments), and their activations are concatenated before the classifier. There is no single “last” convolutional layer whose feature maps alone summarize the model's decision-making process. If Grad-CAM is computed on only one layer (e.g., the last one with a filter size of 20), the explanation becomes biased toward that length and systematically misses evidence captured by the other layers with smaller filter sizes that can encode functional groups or other chemically relevant fragments shorter than 20 tokens. To reflect how the model truly forms its predictions, we compute Grad-CAM separately for each convolutional layer and project each layer's CAM back onto the token sequence by distributing the score of a filter window [j, j + k), where j is the start token index and k is the filter size (window length). We then accumulate the results across all convolutional layers and average overlapping contributions. The resulting token-level map is a multi-scale attribution that incorporates all evidence the classifier receives and is directly comparable to input-level methods (IG, DeepLIFT, GradientSHAP, and Occlusion) that already produce token-aligned scores.Grad-CAM difference from IG/DeepLIFT/SHAP/occlusion
Integrated Gradients (IG), DeepLIFT, GradientSHAP, and Occlusion operate on the model's full architecture, attributing the output back through all layers (convolutional, fully connected, dropout, and highway) to the input features. For example, IG ensures that the sum of attributions equals the entire output difference from a baseline,28 and DeepLIFT explicitly backpropagates contribution scores through all neurons in the network down to every input feature.29 Grad-CAM, on the other hand, computes importance at the level of the convolutional layers.7 This approach yields intuitive visual explanations quickly, but it comes at the cost of missing or diluting features whose importance only emerges after this mixing. Moreover, because Grad-CAM stops before the fully connected layers, it cannot pinpoint which exact input features within an activated region were ultimately most important.Additionally, Grad-CAM applies a ReLU to the weighted sum of feature maps, intentionally discarding negative values.5,7 Input-level attribution methods, on the other hand, preserve the sign of their attributions, allowing both positive and negative contributions to be expressed. For example, Integrated Gradients assigns negative importance scores to tokens that decrease the output score and positive scores to tokens that increase it. As a result, in the context of chemical interpretations, cosine similarity will penalize Grad-CAM's unipolar, partially suppressed map when compared to a bipolar input-level attribution map. Even if both methods highlight the same general region as important, Grad-CAM's attribution vector will differ from the attribution vectors produced by input-level methods.
Explainability cliff
For some model architectures, performance does not degrade linearly as the task becomes harder. Instead, it remains relatively high up to a certain point and then suddenly drops off. This phenomenon, known as an accuracy cliff,49 serves as an analogy for what can also occur in the explanatory quality of feature attributions, which we refer to as an “explainability cliff.” The explainability cliff describes a phenomenon in which the quality or faithfulness of a model's explanations is only modestly degraded at first, but once the model's accuracy falls below a critical threshold, the explanations rapidly become unreliable or even meaningless. Hartog et al.12 compared explanation outputs for models ranging from random-guess performance to moderate accuracy and found no significant differences in explanation quality across this range. In this work, we focus on the previously unexplored range between high (near-ideal) accuracy and moderate accuracy and observe a sharp change in explanation behavior. Specifically, around a ROC–AUC of approximately 0.65 for the Ames dataset, feature attributions become unstable, differ across explanation techniques, and are more diffusely distributed. A similar trend is observed for other endpoints (e.g., BBB permeability, Fig. S7), although the transition is less abrupt and occurs over a broader AUC range. This indicates that the degradation of explanation consistency is a general phenomenon, but its location and sharpness are task-dependent and influenced by dataset characteristics, such as the level of experimental noise, class balance, and whether the endpoint is driven by discrete toxicophores or more continuous physicochemical properties. Notably, when noise is structured rather than random, its effects can remain localized. For example, when labels associated with a specific toxicophore were systematically altered, attribution instability was primarily observed for that motif, while other chemical patterns remained comparatively stable. This suggests that, in realistic settings, loss of explanation consistency may occur in a region-specific manner rather than uniformly across the dataset. Below such performance regimes, when the model has not fully captured the underlying chemical patterns, its explanations become less consistent and less informative.Applications of explainable AI
Effective explainability should be both faithful (reflecting a model's true reasoning) and intelligible to humans. These principles apply across all XAI case studies. While our work emphasizes high predictive accuracy to support trustworthy toxicity explanations, it is important to note that explainable AI is also widely used for model validation, debugging, and generating user-oriented insights that contribute to more reliable AI systems.Beyond identifying which features drive predictions, XAI methods play a central role in model development and evaluation. For instance, explainability has been used as a debugging tool: Ross et al.50 proposed regularizing neural networks using explanation feedback, encouraging models to be “right for the right reasons” by penalizing reliance on irrelevant features without reducing accuracy. Similarly, feature-attribution methods have been applied to improve robustness by generating adversarial examples through perturbations of highly influential features (as identified by SHAP), followed by retraining on these challenging cases.51 Another important application is counterfactual explanations,52 which describe how specific changes to an input could alter a model's prediction. Counterfactual explanations resemble prediction-driven Matched Molecular Pairs, which shows a detailed map which molecular transformations were learned or not by the respective model.53 In chemistry, such explanations can provide actionable insights for molecular design. For example, if a model predicts a molecule to be toxic, a counterfactual explanation may suggest that removing a nitro group or reducing lipophilicity would change the prediction to non-toxic, offering hypotheses that can be explored in future drug design.
Conclusions
This study demonstrates that the consistency and chemical relevance of explainable AI (XAI) methods in molecular property prediction are dependent on the accuracy of the underlying predictive model. Using a deterministic functional-group annotation task, we first showed that a high-accuracy model yields stable and chemically correct explanations. All evaluated XAI methods: Integrated Gradients, GradientSHAP, DeepLIFT, Grad-CAM, and Occlusion consistently highlighted the exact atoms forming the functional groups. Extending the analysis to the more complex task of mutagenicity prediction, we again observed strong agreement between methods. Each technique successfully identified known toxicophores and key scaffolds commonly associated with mutagenic activity, while also highlighting chemically neighboring atoms whose roles require further investigation. XAI should therefore be interpreted in the context of both model performance and established chemical knowledge, rather than in isolation.Author contributions
Dina Khasanova: conceptualization, methodology, software, validation, formal analysis, investigation, data curation, writing – original draft, writing – review & editing, visualization. Igor Tetko: conceptualization, writing – review & editing, supervision, project administration, funding acquisition.Conflicts of interest
The authors declare that there are no conflicts of interest.Data availability
The code for the models and XAI methods, together with the data used in this study, is available on GitHub: https://github.com/DinaKhasanova/xai_benchmark. Supplementary information is available. See DOI: https://doi.org/10.1039/d5dd00576k.Acknowledgements
This editorial was partially funded by the European Union's Horizon Europe programme under the Marie Skłodowska-Curie Actions Doctoral Networks grant agreement No. 101120466 “Explainable AI for Molecules” (AiChemist). The authors thank Drs Katya Ahmad and Peter Hartog for their comments and suggestions.References
- S. A. Eytcheson and I. V. Tetko, Which Modern AI Methods Provide Accurate Predictions of Toxicological End Points? Analysis of Tox24 Challenge Results, Chem. Res. Toxicol., 2025, 38(9), 1443–1451 Search PubMed.
- S. Seal, M. Mahale, M. García-Ortegón, C. K. Joshi, L. Hosseini-Gerami, A. Beatson, M. Greenig, M. Shekhar, A. Patra, C. Weis, A. Mehrjou, A. Badré, B. Paisley, R. Lowe, S. Singh, F. Shah, B. Johannesson, D. Williams, D. Rouquie, D.-A. Clevert, P. Schwab, N. Richmond, C. A. Nicolaou, R. J. Gonzalez, R. Naven, C. Schramm, L. R. Vidler, K. Mansouri, W. P. Walters, D. D. Wilk, O. Spjuth, A. E. Carpenter and A. Bender, Machine Learning for Toxicity Prediction Using Chemical Structures: Pillars for Success in the Real World, Chem. Res. Toxicol., 2025, 38(5), 759–807, DOI:10.1021/acs.chemrestox.5c00033.
- L. Wu, R. Huang, I. V. Tetko, Z. Xia, J. Xu and W. Tong, Trade-off Predictivity and Explainability for Machine-Learning Powered Predictive Toxicology: An in-Depth Investigation with Tox21 Data Sets, Chem. Res. Toxicol., 2021, 34(2), 541–549 Search PubMed.
- J. Jiménez-Luna, F. Grisoni and G. Schneider, Drug Discovery with Explainable Artificial Intelligence, Nat. Mach. Intell., 2020, 2(10), 573–584, DOI:10.1038/s42256-020-00236-4.
- C. Molnar, Interpretable Machine Learning, Lulu.com, 2020 Search PubMed.
- P. Karpov, G. Godin and I. V. Tetko, Transformer-CNN: Swiss Knife for QSAR Modeling and Interpretation, J. Cheminf., 2020, 12(1), 17, DOI:10.1186/s13321-020-00423-w.
- R. R. Selvaraju; M. Cogswell; A. Das; R. Vedantam; D. Parikh and D. Batra, Grad-Cam: Visual Explanations from Deep Networks via Gradient-Based Localization, 2017; pp pp 618–626 Search PubMed.
- S. M. Lundberg and S.-I. Lee, A Unified Approach to Interpreting Model Predictions, Adv. Neural Inf. Process. Syst., 2017, 30, 4765–4774 Search PubMed.
- M. A. Pramudito, Y. N. Fuadah, A. I. Qauli, A. Marcellinus and K. M. Lim, Explainable Artificial Intelligence (XAI) to Find Optimal in-Silico Biomarkers for Cardiac Drug Toxicity Evaluation, Sci. Rep., 2024, 14(1), 24045 CrossRef CAS PubMed.
- A. Kengkanna and M. Ohue, Enhancing Property and Activity Prediction and Interpretation Using Multiple Molecular Graph Representations with MMGX, Commun. Chem., 2024, 7(1), 74 CrossRef PubMed.
- M. Proietti, A. Ragno, B. L. Rosa, R. Ragno and R. Capobianco, Explainable AI in Drug Discovery: Self-Interpretable Graph Neural Network for Molecular Property Prediction Using Concept Whitening, Mach. Learn., 2024, 113(4), 2013–2044 CrossRef.
- P. B. R. Hartog, F. Krüger, S. Genheden and I. V. Tetko, Using Test-Time Augmentation to Investigate Explainable AI: Inconsistencies between Method, Model and Human Intuition, J. Cheminf., 2024, 16(1), 39, DOI:10.1186/s13321-024-00824-1.
- J. Adebayo, J. Gilmer, M. Muelly, I. Goodfellow, M. Hardt and B. Kim, Sanity Checks for Saliency Maps, Adv. Neural Inf. Process. Syst., 2018, 31, 9505–9515 Search PubMed.
- P.-J. Kindermans, S. Hooker, J. Adebayo, M. Alber, K. T. Schütt, S. Dähne, D. Erhan and B. Kim, The (Un) Reliability of Saliency Methods, in Explainable AI: Interpreting, explaining and visualizing deep learning, Springer, 2019, pp 267–280 Search PubMed.
- C. N. Cavasotto and V. Scardino, Machine Learning Toxicity Prediction: Latest Advances by Toxicity End Point, ACS Omega, 2022, 7(51), 47536–47546, DOI:10.1021/acsomega.2c05693.
- B. Zdrazil, E. Felix, F. Hunter, E. J. Manners, J. Blackshaw, S. Corbett, M. de Veij, H. Ioannidis, D. M. Lopez, J. F. Mosquera, M. P. Magarinos, N. Bosc, R. Arcila, T. Kizilören, A. Gaulton, A. P. Bento, M. F. Adasme, P. Monecke, G. A. Landrum and A. R. Leach, The ChEMBL Database in 2023: A Drug Discovery Platform Spanning Multiple Bioactivity Data Types and Time Periods, Nucleic Acids Res., 2024, 52(D1), D1180–D1192, DOI:10.1093/nar/gkad1004.
- K. Hansen, S. Mika, T. Schroeter, A. Sutter, A. ter Laak, T. Steger-Hartmann, N. Heinrich and K.-R. Müller, Benchmark Data Set for in Silico Prediction of Ames Mutagenicity, J. Chem. Inf. Model., 2009, 49(9), 2077–2081, DOI:10.1021/ci900161g.
- C. Xu, F. Cheng, L. Chen, Z. Du, W. Li, G. Liu, P. W. Lee and Y. Tang, In Silico Prediction of Chemical Ames Mutagenicity, J. Chem. Inf. Model., 2012, 52(11), 2840–2847, DOI:10.1021/ci300400a.
- K. Huang, T. Fu, W. Gao, Y. Zhao, Y. Roohani, J. Leskovec, C. W. Coley, C. Xiao, J. Sun, M. Zitnik, Therapeutics Data Commons: Machine Learning Datasets and Tasks for Drug Discovery and Development, arXiv, 2021, preprint, arXiv:2102.09548, DOI:10.48550/arXiv.2102.09548.
- D. Weininger, SMILES, a Chemical Language and Information System. 1. Introduction to Methodology and Encoding Rules, J. Chem. Inf. Comput. Sci., 1988, 28(1), 31–36, DOI:10.1021/ci00057a005.
- J. Kazius, R. McGuire and R. Bursi, Derivation and Validation of Toxicophores for Mutagenicity Prediction, J. Med. Chem., 2005, 48(1), 312–320, DOI:10.1021/jm040835a.
- J. Ashby and R. W. Tennant, Chemical Structure, Salmonella Mutagenicity and Extent of Carcinogenicity as Indicators of Genotoxic Carcinogenesis among 222 Chemicals Tested in Rodents by the US NCI/NTP, Mutat. Res. Toxicol., 1988, 204(1), 17–115 CrossRef CAS PubMed.
- R. Benigni and C. Bossa, Structure Alerts for Carcinogenicity, and the Salmonella Assay System: A Novel Insight through the Chemical Relational Databases Technology, Mutat. Res. Mutat. Res., 2008, 659(3), 248–261 CrossRef CAS PubMed.
- A. B. Bailey, R. Chanderbhan, N. Collazo-Braier, M. A. Cheeseman and M. L. Twaroski, The Use of Structure–Activity Relationship Analysis in the Food Contact Notification Program, Regul. Toxicol. Pharmacol., 2005, 42(2), 225–235 CrossRef CAS PubMed.
- I. Sushko, S. Novotarskyi, R. Körner, A. K. Pandey, M. Rupp, W. Teetz, S. Brandmaier, A. Abdelaziz, V. V. Prokopenko, V. Y. Tanchuk, R. Todeschini, A. Varnek, G. Marcou, P. Ertl, V. Potemkin, M. Grishina, J. Gasteiger, C. Schwab, I. I. Baskin, V. A. Palyulin, E. V. Radchenko, W. J. Welsh, V. Kholodovych, D. Chekmarev, A. Cherkasov, J. Aires-de-Sousa, Q.-Y. Zhang, A. Bender, F. Nigsch, L. Patiny, A. Williams, V. Tkachenko and I. V. Tetko, Online Chemical Modeling Environment (OCHEM): Web Platform for Data Storage, Model Development and Publishing of Chemical Information, J. Comput.-Aided Mol. Des., 2011, 25(6), 533–554, DOI:10.1007/s10822-011-9440-2.
- I. Sushko, E. Salmina, V. A. Potemkin, G. Poda and I. V. Tetko, ToxAlerts: A Web Server of Structural Alerts for Toxic Chemicals and Compounds with Potential Adverse Reactions, J. Chem. Inf. Model., 2012, 52(8), 2310–2316, DOI:10.1021/ci300245q.
- M. D. Zeiler and R. Fergus, Visualizing and Understanding Convolutional Networks, Springer, 2014, pp. 818–833 Search PubMed.
- M. Sundararajan, A. Taly and Q. Yan, Axiomatic Attribution for Deep Networks, PMLR, 2017, pp. 3319–3328 Search PubMed.
- A. Shrikumar, P. Greenside and A. Kundaje, Learning Important Features through Propagating Activation Differences; PMlR, 2017, pp 3145–3153 Search PubMed.
- N. Kokhlikyan, V. Miglani, M. Martin, E. Wang, B. Alsallakh, J. Reynolds, A. Melnikov, N. Kliushkina, C. Araya and S. Yan, arXiv, 2020, preprint, arXiv:2009.07896, DOI:10.48550/arXiv.2009.07896.
- S. R. Dubey, S. K. Singh and B. B. Chaudhuri, Activation Functions in Deep Learning: A Comprehensive Survey and Benchmark, Neurocomputing, 2022, 503, 92–108 CrossRef.
- E. Mosca, F. Szigeti, S. Tragianni, D. Gallagher and G. Groh, SHAP-Based Explanation Methods: A Review for NLP Interpretability, 2022, pp. 4593–4603 Search PubMed.
- P. Sturmfels, S. Lundberg and S.-I. Lee, Visualizing the Impact of Feature Attribution Baselines, Distill, 2020, 5(1), e22 Search PubMed.
- A. Hunklinger, P. Hartog, M. Šícho, G. Godin and I. V. Tetko, The openOCHEM Consensus Model Is the Best-Performing Open-Source Predictive Model in the First EUOS/SLAS Joint Compound Solubility Challenge, SLAS Discovery, 2024, 29(100123) CAS.
- A. Paszke, S. Gross, F. Massa, A. Lerer, J. Bradbury, G. Chanan, T. Killeen, Z. Lin, N. Gimelshein, L. Antiga, A. Desmaison, A. Kopf, E. Yang, Z. DeVito, M. Raison, A. Tejani, S. Chilamkurthy, B. Steiner, L. Fang, J. Bai and S. Chintala, PyTorch: An Imperative Style, High-Performance Deep Learning Library, 2019 Search PubMed.
- J. Bergstra, R. Bardenet, Y. Bengio and B. Kégl, Algorithms for Hyper-Parameter Optimization, Adv. Neural Inf. Process. Syst., 2011, 24, 2546–2554 Search PubMed.
- T. Akiba, S. Sano, T. Yanase, T. Ohta and M. Koyama, Optuna: A next-Generation Hyperparameter Optimization Framework, 2019, pp. 2623–2631 Search PubMed.
- D. P. Kingma: Adam: A Method for Stochastic Optimization, arXiv, 2014, preprint, arXiv:1412.6980, DOI:10.48550/arXiv.1412.6980.
- C. D. Manning, Introduction to Information Retrieval, Syngress Publishing, 2008 Search PubMed.
- H. Heberle, L. Zhao, S. Schmidt, T. Wolf and J. Heinrich, XSMILES: Interactive Visualization for Molecules, SMILES and XAI Attribution Scores, J. Cheminf., 2023, 15(1), 2 Search PubMed.
- M. Ancona, E. Ceolini, C. Öztireli and M. Gross, Towards Better Understanding of Gradient-Based Attribution Methods for Deep Neural Networks, arXiv, 2017, preprint, arXiv:1711.06104, DOI:10.48550/arXiv.1711.06104.
- T. T. Van Tran, H. Tayara and K. T. Chong, AMPred-CNN: Ames Mutagenicity Prediction Model Based on Convolutional Neural Networks, Comput. Biol. Med., 2024, 176, 108560 CrossRef PubMed.
- J. Hu, X. Yang, C. Yao, M. Zhang, S. Shen, L. Na and Q. Zhao, AMPred-MFG: Investigating the Mutagenicity of Compounds Using Motif-Based Graph Combined with Molecular Fingerprints and Graph Attention Mechanism, Interdiscip. Sci.:Comput. Life Sci., 2025, 1–17 Search PubMed.
- L. A. Thompson, J. G. Evans and S. T. Matthews, AmesFormer: State-of-the-Art Mutagenicity Prediction with Graph Transformers, Chem. Res. Toxicol., 2025, 38(7), 1167–1182 Search PubMed.
- B. Frénay and M. Verleysen, Classification in the Presence of Label Noise: A Survey, IEEE Trans. Neural Netw. Learn. Syst., 2013, 25(5), 845–869 Search PubMed.
- D. Rogers and M. Hahn, Extended-Connectivity Fingerprints, J. Chem. Inf. Model., 2010, 50(5), 742–754, DOI:10.1021/ci100050t.
- S. Riniker and G. A. Landrum, Similarity Maps - a Visualization Strategy for Molecular Fingerprints and Machine-Learning Methods, J. Cheminf., 2013, 5(1), 43, DOI:10.1186/1758-2946-5-43.
- M. Bertolini, L. Zhao, F. Montanari and D.-A. Clevert, Enhancing Interpretability in Molecular Property Prediction with Contextual Explanations of Molecular Graphical Depictions, in AI in Drug Discovery, ed. Clevert, D.-A., Wand, M., Malinovská, K., Schmidhuber, J. and Tetko, I. V., Springer Nature Switzerland, Cham, 2025, pp. 1–12 Search PubMed.
- D. Tsipras, S. Santurkar, L. Engstrom, A. Turner and A. Madry, Robustness May Be at Odds with Accuracy arXiv, 2018, preprint, arXiv:1805.12152, DOI:10.48550/arXiv.1805.12152.
- A. S. Ross, M. C. Hughes and F. Doshi-Velez, Right for the Right Reasons: Training Differentiable Models by Constraining Their Explanations, arXiv, 2017, preprint, arXiv:1703.03717, DOI:10.48550/arXiv.1703.03717.
- R. Tomsett, A. Widdicombe, T. Xing, S. Chakraborty, S. Julier, P. Gurram, R. Rao and M. Srivastava, Why the Failure? How Adversarial Examples Can Provide Insights for Interpretable Machine Learning, IEEE, 2018, 838–845 Search PubMed.
- J. Jiang, F. Leofante, A. Rago and F. Toni, Robust Counterfactual Explanations in Machine Learning: A Survey, arXiv, 2024, preprint, arXiv:2402.01928, DOI:10.48550/arXiv.2402.01928.
- Y. Sushko, S. Novotarskyi, R. Körner, J. Vogt, A. Abdelaziz and I. V. Tetko, Prediction-Driven Matched Molecular Pairs to Interpret QSARs and Aid the Molecular Optimization Process, J. Cheminf., 2014, 6(1), 48 Search PubMed.
| This journal is © The Royal Society of Chemistry 2026 |