
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of accurate transferable hydrofluorocarbon refrigerant force fields using a machine learning and optimization approach
Montana N.
Carlozo
 ,
Ning
Wang
,
Alexander W.
Dowling
* and
Edward J.
Maginn
*
,
Ning
Wang
,
Alexander W.
Dowling
* and
Edward J.
Maginn
*
Department of Chemical and Biomolecular Engineering, University of Notre Dame, Notre Dame, IN 46556, USA. E-mail: adowling@nd.edu; ed@nd.edu
First published on 28th January 2026
Abstract
Developing a transferable classical force field (FF) has historically been a lengthy, expert-informed process. In this work, we integrate optimization, machine learning, and data science techniques to accelerate the systematic design and parameterization of transferable FF models. As a demonstration, we create FF models which are transferable within one- and two-carbon (hydrofluorocarbon) refrigerants to accurately model diverse thermophysical properties including saturated liquid and vapor densities, vapor pressure, and enthalpy of vaporization. Estimability analysis and eigen-decomposition of the Fisher information matrix inform the number and identity of atom types in the final FF model. This model obtains an average mean absolute percent deviation (MAPD) between 2.92% (liquid density) and 31.5% (vapor density) on molecules not considered in the training set. This model (MAPD = 18.37%) also achieves a lower overall MAPD than an optimized version of the generalized AMBER FF (MAPD = 19.95%). Gaussian process surrogate models reduce the evaluation time associated with model selection and optimization from an order of months to minutes. This work suggests that the use of surrogate models combined with data science and optimization methods can greatly accelerate the development of accurate transferable force fields.
1 Introduction
Molecular simulation (MS) is a powerful tool in molecular design and discovery,1,2 but its predictive capabilities depend heavily on the accuracy of complex molecular models known as force fields (FFs), which define the Hamiltonian of the model.3 These FFs are often developed and parameterized for specific applications using a variety of different techniques.4 Designing and optimizing a FF is a time-consuming process that demands considerable expert intuition.1,4,5 This encourages the use of generalized FF models such as those shown in Table 1, in which the properties of many molecules can be predicted using a pre-defined set of chemical environments (atom types). Such models are especially useful in the case of molecular design and discovery before experimental data are available for each molecule of interest.1,6| FF name | # Atom types | Classes of molecules covered | # Citationsa |
|---|---|---|---|
| a Citation counts taken from referenced articles and citation metrics listed on journal websites accessed on 7/23/2025. b TraPPE-UA uses “pseudo-atom” types defined for groups of atoms. c OpenFF uses SMIRKS-based environments instead of traditional atom types, the number of which is dependent on the FF. | |||
| GAFF7 | 57 | Organic molecules, pharmaceuticals | 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 376 376 |
| OPLS-AA8 | 78 | Organic molecules, ions, peptides, nucleic acids | 12859 |
| UFF6 | 126 | All molecules | 8855 |
| CGenFF9 | 139 | Proteins, nucleic acids, lipids, carbohydrates | 5630 |
| COMPASS10 | 76 | Organic molecules, small inorganic molecules, polymers | 5203 |
| MMFF94 (ref. 11) | 99 | Small organic molecules | 2618 |
| TraPPE-UA12 | 150b | Hydrocarbons, sulfides, ketones, esters, etc. | 2536 |
| AMOEBA13 | Up to 134 | Water, ions, organics, proteins, ILs | 1170 |
| APPLE&P14 | 26 | Organics, energetics, ILs, fluoroalkanes, electrolytes | 605 |
| CVFF15 | 38 | Amino acids, hydrocarbons, organics | 214 |
| OpenFF16 | 35 | Small drug-like molecules | 95 |
1.1 Challenges creating generalized FFs
Table 1 lists eleven popular generalized FFs. While some generalized FFs such as the Universal Force Field (UFF) can model any compound, generalized FFs are often designed to model multiple or large classes of molecules. For example, the generalized AMBER force field (GAFF)7 and the optimized potentials for liquid simulations all-atom force field (OPLS-AA)8 have been optimized for studying drug-like molecules (e.g., organic compounds and proteins) and play a critical role in exploring the vast molecular design space that is otherwise inaccessible through experimental methods. Most of the FFs in Table 1 are traditional “class I” fixed charge models, although APPLE&P and AMOEBA account for polarizability, which can be important when modeling polar systems such as ionic liquids (ILs).14,17,18 Generalized FF development has evolved over decades6–15 as better parameterization methods have become available and new classes of molecules have been studied. For example, GAFF originally consisted of 57 chemical environments that were primarily informed by expert intuition on element type, hybridization, and aromaticity.7 GAFF2 improves on GAFF by re-optimizing select FF parameters to better reproduce liquid phase data, quantum mechanics (QM) calculations, and ab initio data for more compounds.19 Similarly, OPLS-AA initially consisted of 78 atom types, which has since been expanded to 124 in OPLS3 to increase its predictive performance by up to 30% and to utilize over an order of magnitude more reference data.20Often transferable FFs (not to be confused with generalized or customized FFs), designed to model a specific subclass of molecules rather than many classes of molecules, are initially postulated using generalized FFs as a base.21–23 Thus, similar development methods apply. For example, the transferable hydrofluoroolefin (HFO) FF of Raabe21 currently models 11 molecules using 11 atom types but was originally based on the AMBER24 FF. During development, atom types were added as more molecules were included in the FF on the basis of expert intuition and domain knowledge. As the number of atom types grew, the LJ parameters associated with them were periodically re-optimized to reproduce experimental property data. This process provided enough data to develop correlations to predict the values of LJ parameters from molar mass, facilitating the expansion of this HFO FF to fluorinated butenes and hydrochlorofluoroolefins (HCFOs).25 The resulting FF is very accurate, but required significant expert guidance and has been in development since 2010.21,26,27
Targeted efforts have streamlined the process of systematic FF development and optimization. An early example is the development of ForceBalance28 which uses gradient-based nonlinear regression (NLR) (via the Levenberg–Marquardt algorithm with a trust region) to optimize a set of user-defined FF parameters against either experimental or ab initio simulation data. In the original work, the TIP4P/2005 (ref. 29) and TIP3P30 water models were re-optimized from three different parameter sets (i.e., starting points) to avoid local optima during optimization.28 ForceBalance has been successfully used to reparameterize GAFF,31,32 OpenFF,16,31 the AMOEBA water model,33 and CHARMM.34 However, this method requires hundreds of pre-existing experimental and ab initio data points and can require up to 70 sequential molecular simulations32 (where each simulation can take several hours) to converge. This represents a limitation of applying NLR directly to expensive molecular simulation models. Other frameworks such as TAFFI35 have been developed to reduce the barrier to entry for FF design. For example, Seo et al. used TAFFI to design and optimize a generalized FF for 87 organic liquids from QM calculations and a set of expert-informed heuristics to speed up the atom type postulation process.35 This FF was comparable in accuracy to GAFF and OPLS on six properties not considered in training the model.35 However, OPLS generally outperformed TAFFI. Thus, the authors suggested that this model could be improved in two notable ways; either through more rigorous hyperparameter tuning or through more systematic selection of the molecules used to inform the atom typing scheme.35 Therefore, this example highlights the lack of transferability in generalized FFs to data outside of the training set.
1.2 machine-learned interatomic potentials (MLIPs)
Machine learning (ML) methods can decrease the time requirements of FF design and optimization. One popular method is the use of machine-learned interatomic potentials (MLIPs), which have been in use since the 1990s.36,37 The literature on this subject is vast and we recommend the review papers of others38–41 for a comprehensive analysis of the subject. Briefly, MLIPs attempt to bypass the complexities of atom typing (and traditional FF design) by using ML models, e.g., Gaussian approximation potentials (GAPs) or neural network potentials (NNPs), to learn the potential energy surface (PES) and forces of a system directly from reference data. MLIP development is generally less reliant on expert knowledge than traditional FF development.37 When good ab initio data are plentiful, MLIPs with high accuracy can be trained on small systems to access system sizes and timescales larger than what is possible with traditional QM calculations. This is because MLIPs can be used to calculate energies and forces many orders of magnitude faster than can be done with quantum chemical methods such as density functional theory (DFT).39,42 Particularly promising are GAPs that are more data-efficient than NNPs.37 Highly accurate GAPs have been generated for transition metals,43,44 amorphous carbon,45 graphene,46 silicon,47 liquid iron and sulfur (at the conditions of Earth's core),48 phase-change materials,49 and many other materials.39 In some cases, MLIPs can be more accurate than traditional FFs. For example, the general purpose silicon GAP reproduces the elastic properties, surface energies, and point-defect formation energies within 10% error (compared to DFT) while ReaxFF (a more conventional reactive FF) often models the same properties with more than 20% error.50 We also acknowledge the recent development of neuroevolution potentials (NEPs) which use evolutionary algorithms to build NN potentials and often achieve comparable accuracy to GAPs with greater computational efficiency.51 As a result of their success, NEPs have been applied to alloys,52–54 metal organic framework (MOF) crystals,55 perovskites,56 amorphous materials,52,57,58 GeTe,59 and many more.52,60In addition to solid state applications, MLIPs have recently been used to model a variety of liquid-phase systems, with water serving as the primary benchmark. Deep Potential (DP) models trained on DFT reference data have been used to map the phase diagram of water,61 to compute interfacial properties such as surface tension and cavitation rates,62 and to describe water and aqueous interfaces with near first principles accuracy.63 These studies highlight the promise of MLIPs for capturing subtle many-body effects in liquids while enabling system sizes and timescales inaccessible to ab initio molecular dynamics. Recent efforts have also pushed accuracy to the coupled-cluster level through transfer learning and advanced neural-network architectures.64 More recently, NEPs have also been used to accurately model the structural and thermodynamic properties of liquid water.65
Despite these successes, liquid-phase MLIPs still face important challenges. Their accuracy is strongly tied to the quality and diversity of the training data, and several models exhibit systematic deviations when predicting thermophysical properties. For example, the DP-SCAN water model reproduces coexistence densities only after a ∼40 K temperature shift and underestimates surface tension by roughly 20%.62 Similar problems arise in more chemically complex liquids. MLIPs developed for molten salts66 and reactive carbonate melts67 reproduce structural and transport properties near their training conditions, but may lose transferability when composition, oxidation state, or thermodynamic conditions shift.
Although MLIPs offer a compelling route for combining quantum accuracy with molecular simulation of liquids, their robustness across wide thermodynamic and chemical spaces remains uneven, motivating hybrid and physics-informed approaches that improve interpretability and extrapolation performance. In addition, MLIPs are expensive to train. For example, one of the latest foundation models required  quantum calculations to train.68 Even data-efficient GAPs require thousands of ab initio calculations to reach acceptable accuracy levels.69 Once trained, these models are still computationally expensive. For example, highly sophisticated MLIPs such as sGDML are up to three orders of magnitude slower to evaluate than traditional FFs.42 Furthermore, MLIP models are complicated and often have thousands and up to billions68 of parameters that must be trained. This significantly reduces the physical interpretability of these models.40 There is also a trade-off between accuracy and computational expense. If reference data generated with high levels of theory are used, the MLIP will be more accurate, but compute resources increase accordingly.38 We note that “on-the-fly” ML can reduce this requirement somewhat by intelligently sampling points based on model uncertainty information. We refer the reader to Schütt et al. for a detailed review on this topic.70 If instead low levels of theory are used to reduce the development time, the corresponding model accuracy can be limited.38 Nandi et al. demonstrated that MLIPs for ethanol corrected with higher levels of DFT functionals could reduce the root mean squared error (RMSE) of the predicted PES by up to 45%.71 Lastly, MLIPs are based on local QM data and therefore are generally not transferable to conditions outside of the reference data used to train them, motivating the use of physics-informed ML models.38,40,41 Pun et al. demonstrated that physics-informed neural networks (PINNs) for aluminum can reliably extrapolate energies outside of the training set, while traditional neural networks (NNs) instead generated unphysical predictions.72 However, even physics-informed ML models can have limited transferability. For example, the general-purpose silicon GAP referred to earlier was unable to accurately predict the planar defects of silicon, which were not part of the training set. Specifically, unstable stacking fault energies on the shuffle plane and the glide plane were predicted with less than 20% error (compared to DFT). In contrast, properties in the training set (i.e., bulk modulus, stiffness tensor components, unreconstructed surface energies, and various interstitials) were predicted with less than 10% error.50 Furthermore, the graphene MLIP of Rowe et al. is not transferable to other phases of carbon.46
quantum calculations to train.68 Even data-efficient GAPs require thousands of ab initio calculations to reach acceptable accuracy levels.69 Once trained, these models are still computationally expensive. For example, highly sophisticated MLIPs such as sGDML are up to three orders of magnitude slower to evaluate than traditional FFs.42 Furthermore, MLIP models are complicated and often have thousands and up to billions68 of parameters that must be trained. This significantly reduces the physical interpretability of these models.40 There is also a trade-off between accuracy and computational expense. If reference data generated with high levels of theory are used, the MLIP will be more accurate, but compute resources increase accordingly.38 We note that “on-the-fly” ML can reduce this requirement somewhat by intelligently sampling points based on model uncertainty information. We refer the reader to Schütt et al. for a detailed review on this topic.70 If instead low levels of theory are used to reduce the development time, the corresponding model accuracy can be limited.38 Nandi et al. demonstrated that MLIPs for ethanol corrected with higher levels of DFT functionals could reduce the root mean squared error (RMSE) of the predicted PES by up to 45%.71 Lastly, MLIPs are based on local QM data and therefore are generally not transferable to conditions outside of the reference data used to train them, motivating the use of physics-informed ML models.38,40,41 Pun et al. demonstrated that physics-informed neural networks (PINNs) for aluminum can reliably extrapolate energies outside of the training set, while traditional neural networks (NNs) instead generated unphysical predictions.72 However, even physics-informed ML models can have limited transferability. For example, the general-purpose silicon GAP referred to earlier was unable to accurately predict the planar defects of silicon, which were not part of the training set. Specifically, unstable stacking fault energies on the shuffle plane and the glide plane were predicted with less than 20% error (compared to DFT). In contrast, properties in the training set (i.e., bulk modulus, stiffness tensor components, unreconstructed surface energies, and various interstitials) were predicted with less than 10% error.50 Furthermore, the graphene MLIP of Rowe et al. is not transferable to other phases of carbon.46
1.3 Accelerating traditional FF optimization via ML
The drawbacks of MLIPs, combined with the physical interpretability and computational efficiency of classical FFs, along with the versatility of ML methods, motivate the creation of ML techniques to optimize classical FFs. In this work, the class I GAFF7 functional form with constrained bonds is used: | (1) |
 is the total potential energy, k parameters are force constants, θ0 is the nominal bond angle, γ is the nominal phase angle for dihedrals ϕ, n is the multiplicity, qi and qj are partial charges for atoms i and j, ε0 is the vacuum permittivity, εij and σij are the LJ parameters, and rij is the distance between atom i and atom j. van der Waals (vdW) interactions represent the attractive and repulsive forces between molecules and are particularly relevant for the calculation of liquid and vapor phase properties. However, it is difficult to calculate these interactions using standard QM calculations, since it requires large basis sets and electron correlation information.73 The LJ potential model described in eqn (1) instead treats vdW interactions using effective two-body potentials that capture the many-body effects of these interactions in a mean-field manner. One can approach the optimization of FF parameters in several ways. For example, ML techniques74–76 and genetic algorithms (GAs)77–80 have long been used to fit intramolecular parameters and partial charges81,82 of FFs to QM data. Similarly, while it is possible to accurately fit LJ parameters to QM data using ML and GAs,80 high levels of theory must be used and many-body effects must be accounted for to obtain accurate condensed phase predictions.10,83–85 To avoid this computational burden, it is often preferable to fit LJ parameters to existing experimental condensed phase data.10,86,87 Therefore, we focus on optimizing the LJ parameters using thermophysical property data.
is the total potential energy, k parameters are force constants, θ0 is the nominal bond angle, γ is the nominal phase angle for dihedrals ϕ, n is the multiplicity, qi and qj are partial charges for atoms i and j, ε0 is the vacuum permittivity, εij and σij are the LJ parameters, and rij is the distance between atom i and atom j. van der Waals (vdW) interactions represent the attractive and repulsive forces between molecules and are particularly relevant for the calculation of liquid and vapor phase properties. However, it is difficult to calculate these interactions using standard QM calculations, since it requires large basis sets and electron correlation information.73 The LJ potential model described in eqn (1) instead treats vdW interactions using effective two-body potentials that capture the many-body effects of these interactions in a mean-field manner. One can approach the optimization of FF parameters in several ways. For example, ML techniques74–76 and genetic algorithms (GAs)77–80 have long been used to fit intramolecular parameters and partial charges81,82 of FFs to QM data. Similarly, while it is possible to accurately fit LJ parameters to QM data using ML and GAs,80 high levels of theory must be used and many-body effects must be accounted for to obtain accurate condensed phase predictions.10,83–85 To avoid this computational burden, it is often preferable to fit LJ parameters to existing experimental condensed phase data.10,86,87 Therefore, we focus on optimizing the LJ parameters using thermophysical property data.
Befort et al. developed a semi-automatic workflow using Gaussian process (GP) ML models to optimize the LJ parameters for an ammonium perchlorate (AP) FF. In a time scale of weeks, 70 unique parameter sets were identified that reproduced the lattice structure of AP with a mean absolute percent error (MAPE) of less than 1%. In contrast, a manually tuned model identified one optimal parameter set with 1.42% MAPE. The accuracy of the GP-informed FFs was attributed to the ability of this workflow to quickly and exhaustively search parameter space with GP surrogate models. Manual tuning could only explore 15000 parameter sets and required a full molecular simulation for each candidate parameterization. In contrast, the workflow could evaluate 3 million parameter sets in minutes using the GP surrogates. The workflow only required 3000 molecular simulations, many of which were run in parallel, thereby significantly reducing the time required for parameter optimization.88 Chatterjee et al. developed a semi-automatic framework that uses deep neural networks (DNNs) to re-optimize the LJ parameters of ten atom types in the polarizable CHARMM Drude FF. The DNN was used to enable a brute-force search to down-select a single parameter set from 10 million possible parameter sets. The resulting FF accurately predicted liquid phase properties for 35 compounds.89 These two examples show that ML allows LJ parameter space to be searched much more thoroughly than what is possible with trial-and-error-based optimization.
GP models are a particularly popular choice for rapid FF parameter optimization. For example, Madin and Shirts used GPs and a differential evolution algorithm to re-optimize the LJ parameters of six atom types from OpenFF 2.0.0. Their approach required fewer than 40 molecular simulations to better reproduce the physical properties of 28 molecules.90 Rizzi et al. used Bayesian inference with GP surrogate models to regress the LJ parameters of the TIP4P water model. They obtained less than 1% error using only ten data points each for the enthalpy of vaporization, self-diffusivity, and density.91 Bayesian inference accurately identified parameters with limited data and provided insight into parameter uncertainty. More recently, Raabe et al. used a sequential Bayesian inference approach with GP models to quickly parameterize a FF for trans-1,2-dichloroethene (R-1130(E)), which showed improved performance over the current HFO/HCFO FF.92 The authors specifically recommend this method for tuning the parameters of transferable FFs in future work.92 GPs were also used to extend the transferable anisotropic Mie potential (TAMie) to alkanethiols93 and primary alkylamines.94 In both works, an objective function was minimized using a fitted perturbed chain statistical associating fluid theory (PC-SAFT) equation of state augmented by GPs to extrapolate to low temperatures.93,94 Property data were often reproduced with less than 1% mean absolute relative deviation (MARD).93 This work was later expanded to replace the entire objective function with GP models that could be cheaply optimized with Bayesian optimization (BO).94
BO uses an uncertainty aware ML surrogate, such as a GP, to perform derivative-free optimization while considering noisy “experiments”. For example, Müller et al. combined a pre-sampling step with GP-assisted covariance matrix adaption evolution in a BO framework to perform multiscale parameter optimization. This framework successfully optimized the LJ parameters of n-octane with less than 1% error in liquid density.95 In a more traditional approach, BO was used by McDonagh et al. to optimize ten non-bonded interaction strengths for a dissipative particle dynamics FF for alcohols and alkanes.96 Compared to the manually-tuned FF of Anderson et al.,97 the BO-optimized FF was more flexible, retained better accuracy on the testing set, and required only 1.5 weeks of development time compared to 16 weeks for the manually-tuned FF.96 Winget used BO to optimize the LJ parameters for rare earth elements and obtained less than 1% RMSE using  molecular simulations.98 We have shown99 that BO with GP surrogates that directly model property predictions (instead of objective functions98,100) can further increase the accuracy of FF optimization workflows using direct GP-based surrogates.101 These examples suggest that ML-based optimization methods are both faster and create more accurate and transferable models for a relatively small number of molecules than hand-tuning. We hypothesize that ML optimized FFs with atom types chosen on the basis of rigorous data analysis facilitated by ML models will be more transferable to new molecules and may assist in the development of generalized FFs with fewer atom types than current FFs.
molecular simulations.98 We have shown99 that BO with GP surrogates that directly model property predictions (instead of objective functions98,100) can further increase the accuracy of FF optimization workflows using direct GP-based surrogates.101 These examples suggest that ML-based optimization methods are both faster and create more accurate and transferable models for a relatively small number of molecules than hand-tuning. We hypothesize that ML optimized FFs with atom types chosen on the basis of rigorous data analysis facilitated by ML models will be more transferable to new molecules and may assist in the development of generalized FFs with fewer atom types than current FFs.
1.4 Customized FFs for refrigerants
When generalized (or transferable) FFs are unavailable or are not accurate enough, customized FFs, which model only one molecule, are often created instead. This is common in molecular simulation studies of hydrofluorocarbon (HFC) refrigerants.86,88,102–109 Although HFCs currently account for only about 1.5% of greenhouse gas emissions,110 demand for HFCs is expanding at a rate of 10–15% annually, which is projected to increase their share of greenhouse gas emissions to more than 19% by 2050.111 The EPA estimates a cumulative net benefit of $278.6 billion associated with the phase-out of HFCs by 2050,110 which has generated interest in systems that can separate existing near-azeotropic HFC-mixtures.17,112–115 However, data on these systems are often limited, which encourages the use of molecular simulation for the design of the separation process.17,107,116,117 From the studies of various all-atom FFs for HFCs listed in Table 2, we can draw multiple conclusions about the state of different FF parameterization methods and design techniques. Note that in Table 2, only atom types with different LJ parameters (even if partial charges are different) specifically used to model HFC refrigerants are counted.| Reference | HFC(s) covered | # Of atom types | LJ calibration method | # Of properties validated | Typical errora |
|---|---|---|---|---|---|
| a MSE: mean squared error, VLE: vapor–liquid equilibrium, RAD: relative absolute deviation. | |||||
| Befort et al.88 | HFC-32 | 3 | Semi-automatic GP algorithm | 4 | MAPE <2.5% |
| HFC-125 | 5 | ||||
| Befort et al.100 | HFC-32 | 3 | Bayesian optimization | 1 | MSE 1.20 × 10−4 g2 cm−1 |
| Wang et al.102 | R-50 | 2 | Semi-automatic GP algorithm | 4 | MAPE <2.7% |
| HFC-134a | 4 | ||||
| HFC-143a | 5 | ||||
| R-170 | 2 | ||||
| R-14 | 2 | ||||
| Potter et al.103 | HFC-32 | 4 | Manually-tuned from ref. 118 | 4 | 5% Error (VLE curves only) |
| HFC-23 | |||||
| R-14 | |||||
| Fermeglia et al.86 | HFC-32 | 3 (HFC-32, HFC-134a) | Manually-tuned | 3 | <2.35% RAD |
| All 7 two-carbon fluoroethanes | Not listed | ||||
| Peguin et al.104 | HFC-134a | 4 | Manually-tuned from OPLS-AA and AMBER | 6 | <6.2% Error |
| Yang et al.105 | HFC-152a | 3 | Manually-tuned from OPLS-AA | 7 | <5% Average RAD |
| Zhang et al.106 | HFC-161 | 3 | Manually-tuned from AMBER | 3 | <3.87% Average RAD |
| Alam and Jeong107 | HFC-32 | 4 | Taken from COMPASS | 5 | Good agreement |
| HFC-134a | |||||
| Higashi and Takada108 | HFC-32 | 3 | Manually-tuned | 5 | Very good agreement |
| Paulechka et al.109 | R-116 | 2 | Response surface mapping method | 4 | Very good agreement |
Unsurprisingly, customized FFs are usually more accurate than generalized FFs. Befort et al. demonstrated that while their FFs customized for difluoromethane (HFC-32) and pentafluoroethane (HFC-125) often exhibited MAPE values of less than 3% for all properties, GAFF often exhibited MAPE values of greater than 50%.88 Wang et al. reported similar results for other HFCs.102 Interestingly, many customized FFs shown in Table 2 either directly use the parameters of generalized FFs or other existing literature or otherwise use such works as a starting point for FF tuning and development. However, we highlight that all works in Table 2 use a pre-defined atom typing scheme and do not consider alternative atom typing schemes. Furthermore, we emphasize that FF parameterization using ML and optimization techniques is more accurate than traditional hand-tuning approaches. Wang et al.102 compared their FF for 1,1,1,2-tetrafluoroethane (HFC-134a) to that of Peguin et al.104 and demonstrated an improvement in MAPE for all thermophysical properties studied.102 The performance of FFs for methane (R-50), tetrafluoromethane (R-14) and ethane (R-170)102 are comparable to those developed by Potoff and Bernard-Brunel119 which use a three-parameter Mie potential instead of a traditional two-parameter LJ potential for each type of atom. This improved accuracy is directly related to the use of surrogate models to explore LJ parameter space more effectively.
In this work, we highlight the opportunity to parameterize transferable FFs with ML using HFC FF modeling as an example. Befort et al. used a semi-automatic GP-based workflow to identify 26 unique LJ parameter sets for HFC-32 and 45 unique parameter sets for HFC-125 on a time scale of weeks that accurately reproduced the thermophysical properties of interest with less than 2.5% mean absolute percent error (MAPE).88 This workflow was later refined and expanded to include five more refrigerants (R-50, R-170, R-14, HFC-134a, and 1,1,1-trifluoroethane (HFC-143a)) for which between 18 and 37 sets of unique and well-performing parameters were identified.102 These results suggest the possible existence of a transferable set of LJ parameters which can accurately reproduce the thermophysical properties of all one- and two-carbon HFC refrigerants. We highlight that there is no transferable FF in the literature that accurately models a significant number of both one- and two-carbon HFC refrigerants.
1.5 Paper contributions and organization
We hypothesize that ML and optimization techniques can be applied to design and optimize a transferable FF. The goal of this work is to demonstrate how ML methods and the effective leveraging of the information they provide can be used to improve the transferability of FFs for a small but critical class of molecules (HFCs). We present ML as a solution to inform FF structure (number and type of chemical environments) and automatically optimize a set of transferable FF parameters. In this work, we design and optimize a transferable FF for both one- and two-carbon refrigerants to:• Quantify the trade-off between the number and quality of atom types and FF accuracy.
• Demonstrate that applying standard data analysis techniques with ML optimization methods improves FF model accuracy.
• Demonstrate the benefits of ML optimized FFs for similar molecules over generalized FF models.
We highlight that a FF which accurately parametrizes one- and two-carbon refrigerants does not constitute a fully transferable model for all HFC refrigerants; this model is the first step in developing a fully transferable HFC refrigerant FF. We identify further validation of this model to larger molecules and more properties as a future priority in Section 4.
The remainder of the paper is organized as follows. Section 2 describes all aspects of the proposed workflow, including details on the relevant data, atom typing scheme generation, GP modeling, the objective function, data-science and estimability analysis tools, and molecular simulations. Section 3 compares the FFs developed via ML and optimization with each other and GAFF to demonstrate the usefulness of these techniques in systematic FF design. Finally, Section 4 synthesizes the conclusions and discusses future research opportunities.
2 Methods
2.1 Problem statement
Given a class of molecules (e.g., one- and two-carbon HFC refrigerants) and experimental physical property data (e.g., saturated liquid and vapor densities (ρl and ρv), vapor pressure (Pvap), and enthalpy of vaporization (ΔHvap)), determine an atom typing scheme and optimize its LJ parameters to accurately model the temperature dependence of the physical properties. In this work, our objective was to define a set of chemical environments (atom types) for a transferable FF that adequately describes the chemical environments present in one- and two-carbon HFCs. We then aim to mathematically optimize the LJ parameters of this transferable FF to accurately predict the thermophysical properties of each molecule. This work relies on using GP surrogate models and data analysis techniques to guide FF structure and facilitate the local optimization of the LJ parameters.Eqn (1) is used to compute the energies and forces of a molecular system, which can then be used in molecular simulations to estimate the thermophysical properties of interest. In this work, all bonds are constrained to their nominal lengths. For all FFs developed in this work, intramolecular parameters (kθ, kϕ, θ0, n, and γ) were taken directly from GAFF using GAFF atom types. We justify this on the basis that the VLE properties in which we are interested are relatively insensitive to intramolecular parameters.109 In addition, intramolecular parameters can be computed with high accuracy from gas phase quantum calculations, and so are not the focus of the present work. The partial charges (qi and qj) were calculated using DFT with RESP120 at the B3LYP/6-311++g(d,p) level of theory instead of the standard AM1-BCC121 level of theory commonly used for GAFF. We have found that the higher level of theory gives more reliable partial charges.122 These partial charges are used for both simulations using the LJ parameters (σi and εi) identified in this work and the simulations for the GAFF LJ parameters to which we compare our work. The LJ parameters for this work are optimized using the workflow defined in Section 2.8, based on the fact that they are critical to accurate prediction of VLE properties and are difficult to obtain from quantum calculations. The Lorentz–Berthelot combining rule was used to calculate unlike interactions. We highlight that the LJ parameters are fit using experimental data. In a sense, these are now “effective” LJ parameters which account for both vdW forces and other phenomena not directly considered by the model. For example, although the intramolecular parameters of highly-fluorinated molecules can impact their liquid phase properties,123,124 we hypothesize that optimization will implicitly account for these discrepancies for small one- and two-carbon HFC molecules. We recommend revisiting this assumption as larger molecules are investigated.
2.2 Atom typing schemes
The LJ model relies on the definition of separate chemical environments, for which many definitions are reasonable. Although all HFCs are comprised of only three distinct atoms (hydrogen, fluorine, and carbon), the chemical environments in which these atoms interact within a molecule can vary significantly. Conventional FFs (as opposed to MLIPs) treat different chemical environments using distinct atom types. The accuracy of the FF model is thus governed by the number and definition of these atom types. In this work, we compare five atom typing schemes, described in the following paragraphs and summarized in Table 3. For this work, italics are used when referring to atom types and regular text is used when referring to a specific atom. For example, the hydrogen atom H is represented by atom type H in atom typing scheme four (AT-4).| Scheme | Number | Unique types |
|---|---|---|
| AT-4 | 4 | C 1 , C2, H, F |
| GAFF (baseline) | 6 | C, Hc, H1, H2, H3, F |
| AT-6a | 6 | C, Hc, H1, H2, H3, F |
| AT-6b (recommended) | 6 | C m , C1, C2, Hc, H, F |
| AT-8 | 8 | C m , C1, C2, H, F1, F2, F3, F4 |
 | ||
| Fig. 1 AT-6b uses six data-informed atom types (CM, C1, C2, F, H, and Hc). | ||
We highlight that the GP models designed in our previous work88,102 require the LJ parameters of the maximum number of chemically reasonable environments used to design a molecule as inputs. This number is different for each molecule. For example, methane (R-50) and ethane (R-170) both require only two atom types while molecules such as R-134a can require up to five. Thus, parameter optimization is facilitated by mapping the distinct atom types to their transferable FF counterparts during optimization via atom type transformation matrices Am. In this way, the LJ parameters from the transferable models (defined as θ = [σ, ε]) can be mapped to the distinct atom types that are used by the GP models  for each molecule m. SI Fig. S5 shows the distinct atom types for each molecule and SI Table S1 shows the transformation matrix for each molecule in the training set.
for each molecule m. SI Fig. S5 shows the distinct atom types for each molecule and SI Table S1 shows the transformation matrix for each molecule in the training set.
2.3 Experimental and simulation data
Experimental data for the 13 refrigerantss studied in this work are crucial for optimization and validation of the transferable FF models for one- and two-carbon (HFC) refrigerants. Reference data for all molecules except HFC-143127 were taken from the REFPROP package.128 REFPROP is based on an equation of state fit to experimental data and has a very low uncertainty. Therefore, we use 2% of the value of the REFPROP values as a conservative estimate of experimental uncertainty. We are unaware of any existing thermophysical property data for HFC-134 or the toxic molecule HFC-152 (ref. 129) and therefore they are excluded from this work. For the remainder of this paper, we will refer to a set of molecules  and set of properties
and set of properties  at the set of temperatures under study
at the set of temperatures under study  as data set
as data set  and define all reference thermophysical property data
and define all reference thermophysical property data  .
.
All experimental data in set  are further partitioned by molecule into either the training subset or the testing subset according to Fig. 1. For brevity, we refer to these as sets instead of subsets henceforth. The training set thus consists of the experimental data for all properties in the set
are further partitioned by molecule into either the training subset or the testing subset according to Fig. 1. For brevity, we refer to these as sets instead of subsets henceforth. The training set thus consists of the experimental data for all properties in the set  for the eight refrigerant molecules (R-50, HFC-32, R-14, R-170, HFC-41, HFC-134a, HFC-143a, and HFC-125) identified as the training set in Fig. 1. These data are used in the optimization of the LJ parameters. Since GP models are used to complete optimization, GP models only exist for the molecules defined in the training set. The testing set is then defined as the experimental data for all properties in
for the eight refrigerant molecules (R-50, HFC-32, R-14, R-170, HFC-41, HFC-134a, HFC-143a, and HFC-125) identified as the training set in Fig. 1. These data are used in the optimization of the LJ parameters. Since GP models are used to complete optimization, GP models only exist for the molecules defined in the training set. The testing set is then defined as the experimental data for all properties in  for the five molecules identified as the testing set (HFC-23, R-116, HFC-161, HFC-152a, and HFC-143). These data are only used to validate the simulation results of the LJ parameter sets identified in this work and do not have GP models associated with them.
for the five molecules identified as the testing set (HFC-23, R-116, HFC-161, HFC-152a, and HFC-143). These data are only used to validate the simulation results of the LJ parameter sets identified in this work and do not have GP models associated with them.
We use NLR to compute locally optimal sets of LJ parameters (σ and ε) for each atom type for the transferable FFs in this work. This optimization relies on the availability of GP surrogate models, which map the LJ parameters and temperature to the thermophysical properties of interest for each molecule in the training set. Our previous work details a method for generating such GP models.88,102 For this work, we use this method to build GP models for HFC-41 and reuse the GP models for the other HFCs trained in our previous work.88,102 Note that there is one GP model for each molecule and property combination considered in the training set, for a total of 32 GP models.
Finally, we compare experimental and predicted values using the mean absolute percent deviation (MAPD) metric. MAPD for each molecule and property is defined as:
 | (2) |
 for a given molecule and property.
for a given molecule and property.
2.4 Primer on GPs
A GP is a probabilistic surrogate model that includes uncertainty information about its predictions; it is particularly well suited for low-dimensional and low-data applications, such as the present problem.130–132 To explain GP models, consider the case of training a GP with the predictions of molecular simulations. Consider a molecular simulation data set with inputs of temperature and LJ parameters denoted and outputs of predictions for a thermophysical property
and outputs of predictions for a thermophysical property  for an arbitrary molecule
for an arbitrary molecule  and property
and property  over all temperature state points
over all temperature state points  . For simplicity, consider the full data set that we wish to predict using a GP as where
. For simplicity, consider the full data set that we wish to predict using a GP as where  and
and  . Note that hereafter the subscripts p, m, and T are omitted for brevity.
. Note that hereafter the subscripts p, m, and T are omitted for brevity.
Furthermore, let y* correspond to predictions at new input values Z*. The GP surrogate then assumes that y and y* follow a multivariate normal (MVN) distribution which can be used to estimate the uncertainty in the model.131,133 This distribution is defined by the mean function  , and positive semi-definite covariance matrix K(z, z|h) constructed using the kernel function
, and positive semi-definite covariance matrix K(z, z|h) constructed using the kernel function  . Each element ki,j in this matrix can be evaluated using a kernel function with respect to GP hyperparameters, h. Therefore, the distribution can be written:
. Each element ki,j in this matrix can be evaluated using a kernel function with respect to GP hyperparameters, h. Therefore, the distribution can be written:
 | (3) |
For any GP model, the mean function m(·) reflects prior beliefs about how input characteristics affect a function. For this work, we use a linear mean function. The covariance matrix K describes the extent to which model parameters are related. GP models are created using an anisotropic kernel function of the form:
| k(zi, zj) = τ2k*(·) + cδnm | (4) |
For all the examples in this work, either an anisotropic Matèrn 5/2 kernel or a radial basis function (RBF) kernel is used.131
The Matèrn 5/2 kernel is given by the following expression:
 | (5) |
This kernel is defined by its smoothness,  , and length scales
, and length scales  . It is a particularly good choice for functions that are twice differentiable. This kernel was chosen as the default for modeling Pvap and ρv based on higher empirical accuracy compared to the RBF and Matèrn 3/2 kernel. The length scale hyperparameters
. It is a particularly good choice for functions that are twice differentiable. This kernel was chosen as the default for modeling Pvap and ρv based on higher empirical accuracy compared to the RBF and Matèrn 3/2 kernel. The length scale hyperparameters  indicate the importance of each input feature by scaling the Euclidean distance:
indicate the importance of each input feature by scaling the Euclidean distance:
 | (6) |
The RBF kernel is defined as:
 | (7) |
 . The RBF kernel is a good choice for infinitely differentiable functions and was chosen as the default for modeling ΔHvap and ρl, based on its higher empirical accuracy compared to the Matèrn 3/2 and Matèrn 5/2 kernels. In eqn (5) and (7) the hyperparameter τ2 scales the entire base kernel, and we model white noise by applying hyperparameter c on the diagonal using the Kronecker delta δnm.
. The RBF kernel is a good choice for infinitely differentiable functions and was chosen as the default for modeling ΔHvap and ρl, based on its higher empirical accuracy compared to the Matèrn 3/2 and Matèrn 5/2 kernels. In eqn (5) and (7) the hyperparameter τ2 scales the entire base kernel, and we model white noise by applying hyperparameter c on the diagonal using the Kronecker delta δnm. | (8) |
Hyperparameters  are inferred through maximum likelihood estimation with the training data, in which the logarithmic likelihood function of the GP is maximized using derivative-based nonlinear optimization. Note that this problem is nonconvex and thus there are likely to be multiple local maxima.131,134 Therefore, we implemented a multistart procedure in which hyperparameter optimization was repeated three times using different initial guesses for h to find the most suitable set of hyperparameters. After conditioning on the training data y and Z, the GP follows a (multivariate) normal distribution with mean:
are inferred through maximum likelihood estimation with the training data, in which the logarithmic likelihood function of the GP is maximized using derivative-based nonlinear optimization. Note that this problem is nonconvex and thus there are likely to be multiple local maxima.131,134 Therefore, we implemented a multistart procedure in which hyperparameter optimization was repeated three times using different initial guesses for h to find the most suitable set of hyperparameters. After conditioning on the training data y and Z, the GP follows a (multivariate) normal distribution with mean:
| µGP(Z*) = K(Z*, Z)(K(Z, Z))−1(y − m) | (9) |
| ΣGP(Z*, Z*) = K(Z*, Z*) − K(Z*, Z)(K(Z, Z))−1K(Z, Z*) | (10) |
 . We also eliminate the arguments for convenience.
. We also eliminate the arguments for convenience.
We reiterate that we train one GP model for each molecule and property combination considered in the training set. These GPs are used to optimize a set of LJ parameters for each transferable refrigerant FF according to the atom typing schemes defined in Section 2.2.
2.5 Primer on NLR
Consider a mathematical model f(·,·),| y = f(X, θ) + ε, | (11) |
Next, we assume the random measurement error (and thus experimental data) can be accurately described by a probability distribution, such as the MVN distribution with mean 0 and covariance Σexp:
 | (12) |
Using the assumed probability distribution, we can construct the likelihood function:
 | (13) |
 is the probability of observing data y for a given instance of the model parameters θ. Taking the log yields the log-likelihood function
is the probability of observing data y for a given instance of the model parameters θ. Taking the log yields the log-likelihood function  :
: | (14) |
In Frequentist statistics, unknown/uncertain model parameters θ are inferred by maximizing  (or equivalently
(or equivalently  ), which gives the maximum likelihood estimate (MLE)
), which gives the maximum likelihood estimate (MLE) ![[small straight theta, Greek, circumflex]](https://www.rsc.org/images/entities/b_i_char_e12d.gif) . In the special case where Σexp is known a priori, the first term of eqn (14) is a constant. The second term is the weighted SSE (WSSE). Thus, under these conditions, minimizing WSSE is equivalent to maximizing
. In the special case where Σexp is known a priori, the first term of eqn (14) is a constant. The second term is the weighted SSE (WSSE). Thus, under these conditions, minimizing WSSE is equivalent to maximizing  :
:
 | (15) |
 | (16) |
Often, this expectation is approximated by evaluating the Hessian of  at the MLE . Furthermore, when eqn (15) holds, the FIM is approximated using the Hessian of the WSSE:
at the MLE . Furthermore, when eqn (15) holds, the FIM is approximated using the Hessian of the WSSE:
 | (17) |
For the MVN distribution in eqn (12), each element of the FIM is defined as follows:136
 | (18) |
 | (19) |
It is convenient to collect these partial derivatives into the local sensitivity matrix,
 | (20) |
 | (21) |
In this work, we scale all parameters θ between 0 and 1 when computing the FIM to improve interpretability.
. The Cramér-Rao bound states that the inverse of the FIM is a lower bound of the variance of an unbiased estimate:137 | (22) |
is the covariance of the estimate . It is common to approximate V ≈ FIM−1.
Structural identifiability is a theoretical property of the mathematical model f(·,·), independent of a particular dataset.138 A parameter vector θ is structurally identifiable if, under noise-free conditions, there exists a set of experiment designs X such that there is a unique mapping between parameters and model outputs:
| f(X, θ1) = f(X, θ2) ⇒ θ1 = θ2 | (23) |
If this condition fails, two or more parameter sets can reproduce identical outputs, and no amount of data or optimization, i.e., changing X, can distinguish them. Structural identifiability thus depends only on the model form and experiment observability. Symbolic or analytic methods (e.g., Taylor series methods,142 similarity transformation,143 differential algebra,144 and identifiability tableaus145) are typically used to assess structure identifiability.146–148 However, these models are often difficult to implement and limited to small systems.148,149
Practical identifiability, also called estimability, concerns the numerical precision and stability of parameter estimates when experimental data are finite and noisy.148,150 Even if parameters are structurally identifiable, correlations between them or insufficient data sensitivity can make the estimation ill-conditioned.
In a local sense, practical identifiability is often quantified by the FIM.148 The relationship between the FIM and parameter precision V−1, see eqn (22), can be further explained with a geometric interpretation of the likelihood function, see eqn (13) and (14). The log-likelihood function is said to be sharp near the MLE if small perturbations in θ cause large differences between the measured data and model predictions, i.e., the log-likelihood function (and WSSE under certain conditions) increases significantly. Mathematically, this means the log-likelihood function exhibits strong curvature, the eigenvalues of the Hessian matrix of the log-likelihood are large, and per eqn (13) the FIM is also large.140 In other words, if the model predictions are strongly sensitive to the model parameters θ, the experimental measurements at conditions X contain significant information and the parameter estimates are precise, i.e., they have small uncertainty.
| FIM = QΛQ−1 | (24) |
Expert intuition is often required to inspect the eigen-decomposition and determine actionable modeling outcomes (e.g., fix parameters, adjust model equations). To help streamline this process, we introduce the following heuristic:
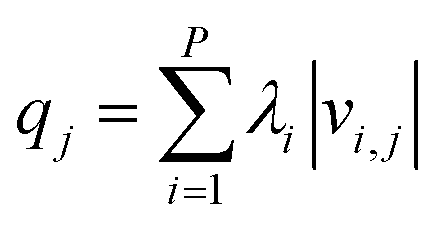 | (25) |
Here, qj approximates the contributions of parameter j to the information contained in the FIM. λi are the eigenvalues and vi,j are the corresponding eigenvectors. The values of qj can be found on GitHub151 for each atom typing scheme. See SI Section S2 for additional discussion of this heuristic.
where αn,j are user-selected scaling factors. We highlight that the unscaled version of the matrix B (where αn,j = 1 ∀ n ∈ N, j ∈ P) is the matrix F by definition. In this work, instead of using arbitrary scaling factors α, we scale all parameters and property predictions between 0 and 1 to ensure that all parameters and properties are equally weighted.


In step 1 of Table 4, the most important column of B (which corresponds to the most important parameter) is identified, and in steps 2 and 3, it is used to create a linear reconstruction of the original matrix. Step 4 calculates the difference between this prediction and the original matrix, and step 5 uses this to estimate the next most important parameter while also accounting for parameter coupling. Thus parameters that appear at the bottom of this list do not dramatically affect the data either because the parameter is unimportant or because it correlates with other more important parameters.
| Algorithm 1 orthogonalization-based parameter estimability ranking algorthim139 | |
|---|---|
| Given: scaled sensitivity matrix B | |
| 1: | Compute the magnitude of each column in B. The most estimable parameter is the one corresponding to the column with the highest magnitude. Set k = 1 for the first iteration |
| 2: | Construct matrix Xk using k selected columns from B. Each column corresponds to a ranked parameter |
| 3: | Calculate Bk, the prediction of matrix B, Bk = (X⊤kXk)−1X⊤kB |
| 4: | Calculate the residual matrix Rk = B − Bk |
| 5: | Compute the magnitude of each column in Rk. The next most estimable parameter is the one corresponding to the column with the highest magnitude |
| 6: | Increase counter k by one and repeat steps 2 to 5 until all parameters have been ranked or step 3 fails due to matrix singularity |
2.6 Calibration and atom type selection via data science techniques
We hypothesize that considering estimability when designing FFs helps improve transferability and reliability. However, most prior efforts create generalized FFs by starting with a base set of atom types, and adjusting the atom types based on expert intuition and empirical evidence.7,21,156 While systematic optimization of FF models is common, parameter estimability insights are generally not considered.28,32,90,92,101 Therefore in this work we use eigen-decomposition of the FIM and orthogonalization based estimability analysis techniques to inform a set of atom types for a transferable HFC FF. These techniques inform both the number of necessary atom types and define which chemical environments each should cover to maximize transferability and accuracy after optimization of the LJ parameters with NLR. objective function.
We minimize the expected value of SSE
objective function.
We minimize the expected value of SSE  objective function99 to calibrate the FF parameters for a given atom typing scheme:
objective function99 to calibrate the FF parameters for a given atom typing scheme: | (26) |
 | (27) |
and


A full derivation of eqn (27) (ref. 157) is reproduced in SI Section S3 for completeness. In eqn (26) and (27),  is the set of molecules in the training set. yexpm,p,T and σexp,m,p,T2 are the experimental property values and measurement error (variance), respectively. The global (transferable) FF parameters, θ = [σ, ε] are optimized between bounds of
is the set of molecules in the training set. yexpm,p,T and σexp,m,p,T2 are the experimental property values and measurement error (variance), respectively. The global (transferable) FF parameters, θ = [σ, ε] are optimized between bounds of  and
and ![[small theta, Greek, macron]](https://www.rsc.org/images/entities/b_i_char_e0c9.gif) . This objective function relies on the transformation matrix Am that maps the transferable FF parameters defined by a given atom typing scheme to the different FF parameters used as GP input for a given molecule (see Section 2.2). We note that this transformation is possible only because the GP models use the maximum number of chemically reasonable LJ parameters (and therefore atom types) as inputs. For example, the GPs for HFC-134a accept ten LJ parameters as inputs (five atom types) which allows fluorine atom types to remain distinct from each other when AT-8 is applied via matrix AHFC-134a. Σ−1exp is the inverse covariance matrix of the experimental data. In this work, Σ−1exp is a diagonal matrix with the values of
. This objective function relies on the transformation matrix Am that maps the transferable FF parameters defined by a given atom typing scheme to the different FF parameters used as GP input for a given molecule (see Section 2.2). We note that this transformation is possible only because the GP models use the maximum number of chemically reasonable LJ parameters (and therefore atom types) as inputs. For example, the GPs for HFC-134a accept ten LJ parameters as inputs (five atom types) which allows fluorine atom types to remain distinct from each other when AT-8 is applied via matrix AHFC-134a. Σ−1exp is the inverse covariance matrix of the experimental data. In this work, Σ−1exp is a diagonal matrix with the values of  .
.
 :
: | (28) |
 | (29) |
 (see eqn (27)) evaluated at :
(see eqn (27)) evaluated at :
We now explore when eqn (29) is a reasonable approximation. Let us define vectors, µGP = µGP;m,p,T and ΣGP = σGP;m,p,T2 ∀ n: = {m, p, T} ∈ N where  .
.
With these definitions, the Hessian of eqn (27) included here for convenience and derived in SI Section S4 is as follows:
 | (30) |
 and we assume that measurements are independent across different molecules and temperatures.
and we assume that measurements are independent across different molecules and temperatures.
Finally, let us consider the case where the GP model is a good emulator of our original model and µGP(·,·) ≈ f(·,·). Moreover, assume the GP variance is smooth and  , and the second term in eqn (30) is zero. Under these conditions, eqn (30) is a reasonable approximation for eqn (19).
, and the second term in eqn (30) is zero. Under these conditions, eqn (30) is a reasonable approximation for eqn (19).
We compute H with a central finite difference method implemented in  . Similarly, we approximate the refrigerant property predictions with GP models, i.e., f(·,·) ≈ µGP(·,·), and compute the matrix B for Table 4 using a finite difference method (
. Similarly, we approximate the refrigerant property predictions with GP models, i.e., f(·,·) ≈ µGP(·,·), and compute the matrix B for Table 4 using a finite difference method ( ).
).
2.7 Molecular simulation specifics and computing environment
The Cassandra (version 1.3.1) package158 was used to perform Gibbs ensemble Monte Carlo (GEMC) simulations to calculate the VLE properties in set at five temperature state points for each molecule using the unique optimal parameter sets defined by the transferable FF optimization schemes described in Section 2.2. The initial number of liquid and vapor molecules was 640 and 160, respectively, and the liquid and vapor boxes were randomly initialized using Packmol (version 20.16.1)159 through the Foyer software (version 0.12.1).160 The liquid box was pre-equilibrated for 2500 canonical ensemble (NVT) sweeps and 5000 isothermal-isobaric ensemble (NPT) sweeps before starting an equilibration GEMC simulation with at least 10000 sweeps. The cutoffs of Coulombic and LJ interactions were 1.2 nm for the liquid box and 0.4 times the length of the vapor box for the vapor box. As in our previous work, the following settings were used:102 Ewald summation was applied for long-range electrostatics with a relative accuracy of 10−5; all bonds were fixed at the nominal bond length; standard LJ tail corrections were applied to pressure and energy; all other MC settings can be found on the GitHub151 repository associated with this paper.
at five temperature state points for each molecule using the unique optimal parameter sets defined by the transferable FF optimization schemes described in Section 2.2. The initial number of liquid and vapor molecules was 640 and 160, respectively, and the liquid and vapor boxes were randomly initialized using Packmol (version 20.16.1)159 through the Foyer software (version 0.12.1).160 The liquid box was pre-equilibrated for 2500 canonical ensemble (NVT) sweeps and 5000 isothermal-isobaric ensemble (NPT) sweeps before starting an equilibration GEMC simulation with at least 10000 sweeps. The cutoffs of Coulombic and LJ interactions were 1.2 nm for the liquid box and 0.4 times the length of the vapor box for the vapor box. As in our previous work, the following settings were used:102 Ewald summation was applied for long-range electrostatics with a relative accuracy of 10−5; all bonds were fixed at the nominal bond length; standard LJ tail corrections were applied to pressure and energy; all other MC settings can be found on the GitHub151 repository associated with this paper.
We note that all the FFs developed here use fixed bond lengths, making them compatible with the Cassandra package158 used to compute vapor–liquid phase equilibrium properties. If flexible bonds are desired, we recommend the use of standard GAFF harmonic bond parameters in conjunction with the LJ parameters proposed here, although care should be taken since the optimization did not account for bond flexibility. To test how reliable this approximation is, we computed liquid densities as a function of temperature with the GROMACS package161 for all of the molecules in this study using the LJ parameters from AT-6b and either rigid bonds or flexible bonds modeled with the GAFF harmonic bond potential. The results of this study are available in SI Section S5. We observed that differences in liquid densities between the flexible and rigid models are often less than 1%, but can be up to 3% for some molecules (see SI Table S2), suggesting that the use of GAFF bond parameters is reasonable. SI Fig. S6–S18 show the results for this study. For R-14, however, we observe that the density of the flexible bond model is significantly higher than both experiment and the rigid bond model (see SI Fig. S6). We therefore do not recommend the use of GAFF flexible bonds for R-14. In future work, it would be useful to include bond flexibility in the parameter optimization workflow.
We used the  package (version 1.0.21)162 to create an on-the-fly algorithm that detects when the simulations had equilibrated. We considered an equilibrated simulation to be any simulation in which at least 25% of the total number of sweeps were considered stationary through 99% confidence in rejection of the Augmented Dickey–Fuller test null hypothesis.163 This test approximates the time-series molecular simulation data as a high-order autoregressive model with a constant mean and no trend and uses statistics to reject the null hypothesis that a unit root is present in the data (i.e., the data are non-stationary). When this test was not passed, sweeps were added in increments of 2500 (10000 sweeps for systems that took more than 100000 sweeps to equilibrate) until equilibrium was reached. This method performs best when the minimum number of equilibrium sweeps is large enough to pass through metastable states. The
package (version 1.0.21)162 to create an on-the-fly algorithm that detects when the simulations had equilibrated. We considered an equilibrated simulation to be any simulation in which at least 25% of the total number of sweeps were considered stationary through 99% confidence in rejection of the Augmented Dickey–Fuller test null hypothesis.163 This test approximates the time-series molecular simulation data as a high-order autoregressive model with a constant mean and no trend and uses statistics to reject the null hypothesis that a unit root is present in the data (i.e., the data are non-stationary). When this test was not passed, sweeps were added in increments of 2500 (10000 sweeps for systems that took more than 100000 sweeps to equilibrate) until equilibrium was reached. This method performs best when the minimum number of equilibrium sweeps is large enough to pass through metastable states. The  module is designed to split molecular simulation data into equilibration and production (stationary) data. Therefore, our in-house method risks failure when 7500 sweeps (75% of the total initial equilibrium sweeps) are insufficient to reach equilibrium or when this method is applied to production series data (in which all data are already equilibrated). To account for this detail, this check was bypassed when the simulations were restarted from already equilibrated data. In addition, all simulations were manually checked retroactively to ensure that liquid and vapor densities had both equilibrated. When 10000 equilibrium GEMC sweeps were deemed too short, the initial number of sweeps was increased to 50000. For future applications, we suggest a minimum of 50000 sweeps to avoid apparent convergence problems.
module is designed to split molecular simulation data into equilibration and production (stationary) data. Therefore, our in-house method risks failure when 7500 sweeps (75% of the total initial equilibrium sweeps) are insufficient to reach equilibrium or when this method is applied to production series data (in which all data are already equilibrated). To account for this detail, this check was bypassed when the simulations were restarted from already equilibrated data. In addition, all simulations were manually checked retroactively to ensure that liquid and vapor densities had both equilibrated. When 10000 equilibrium GEMC sweeps were deemed too short, the initial number of sweeps was increased to 50000. For future applications, we suggest a minimum of 50000 sweeps to avoid apparent convergence problems.
After the system was equilibrated, a 100000 sweep production phase was completed. Three molecular simulations per FF, molecule, and temperature were performed to obtain property averages and standard deviations, which were used as estimates of uncertainty. Usually, all three runs were started from different initial configurations. However, if at any point the system from one simulation completely vaporized or liquidated or was otherwise unstable, a previous configuration was used to collect production data for that run using a different random number seed. Additionally, if production run densities showed that the vapor and liquid GEMC boxes were exchanging identities at any point for a given configuration, which happens as the critical point is approached, the production run data for that configuration were not used during analysis. If two of the three simulations for a given temperature were unstable, the system at that temperature was considered unstable and was not included in this analysis. These systems can be identified through their omission in the property prediction plots available at GitHub.151
All GP models are built in GPflow version 2.9.2 (ref. 164) and all kernels are implemented with a trainable Gaussian likelihood variance. Maximum likelihood estimation is used to infer hyperparameters using the L-BFGS-B optimizer165via the Scipy version 1.15.2  function.166 All runs were completed using the University of Notre Dame Center for Research Computing (CRC) on either dual 12-core Intel(R) Xeon(R) CPU E5-2680 v3 processors at 2.50 GHz with 256 GB of RAM or dual 32-core AMD EPYC 7543 processors at 2.80 GHz with 256 GB of RAM.
function.166 All runs were completed using the University of Notre Dame Center for Research Computing (CRC) on either dual 12-core Intel(R) Xeon(R) CPU E5-2680 v3 processors at 2.50 GHz with 256 GB of RAM or dual 32-core AMD EPYC 7543 processors at 2.80 GHz with 256 GB of RAM.
2.8 Workflow
Fig. 2 shows the workflow used in this paper. We reuse the GP models obtained in our previous work102 for each property of interest for each molecule in the training set.102 Additionally, GP models for R-41 were generated using the workflow of Wang et al. specifically for this work.102 These pre-existing models are represented by Fig. 2A. We begin the process of transferable FF development and optimization by postulating an atom typing scheme for the transferable FF (Fig. 2B). For the purpose of this demonstration, AT-4, AT-8, and AT-6a were simultaneously evaluated as examples of common approaches to transferable FF design (start small, start highly customized, and start with a pre-defined model). We caution that each atom type present in the testing set should appear at least twice in the training set and that atom types should be independent of each other. | ||
| Fig. 2 The workflow for systematically creating and calibrating a transferable force field. | ||
Given an atom typing scheme, we generate a Latin Hypercube Sample (LHS) of 105 LJ parameter sets and evaluate the  objective function at each of these sets using GPs as a proxy for molecular simulations. We randomly select up to 25 nondominated parameter sets as starting guesses for minimization of the
objective function at each of these sets using GPs as a proxy for molecular simulations. We randomly select up to 25 nondominated parameter sets as starting guesses for minimization of the  objective using the L-BFGS-B optimizer (Fig. 2C). We then select all unique local minima, where uniqueness is defined by an L2-norm of at most 0.05 between any two local minima when the parameter values are scaled between 0 and 1 based on the parameter bounds (Fig. 2D). Thus, when the same local solution is found multiple times, only the first instance is considered unique.
objective using the L-BFGS-B optimizer (Fig. 2C). We then select all unique local minima, where uniqueness is defined by an L2-norm of at most 0.05 between any two local minima when the parameter values are scaled between 0 and 1 based on the parameter bounds (Fig. 2D). Thus, when the same local solution is found multiple times, only the first instance is considered unique.
We then advance all parameter sets with an average MAPD (predicted using GPs) over all properties of interest smaller than that of GAFF for at least half of the training set molecules to the validation step (Fig. 2E). For this study, we only select the parameter set with the lowest objective value for validation. We validate the fit for this parameter set by comparing molecular simulation predictions with experimental data using MAPD (Fig. 2F). For future work attempting to converge on a single transferable FF model, if no locally optimal parameter set is found where average MAPD values for at least three molecules in the testing set are smaller than those of GAFF, the atom typing scheme should be re-evaluated.
3 Results and discussion
This section is organized by five key findings summarized in the subsection titles.3.1 Six (data science informed) atom types empirically minimize MAPD
Table 5 shows that AT-6b and AT-6a (six atom types) best balance overall error compared to AT-4 (four atom types) and AT-8 (eight atom types). For completeness, the data in Table 5 are visualized by molecule and property in SI Fig. S19. Similarly, property predictions for each individual molecule can be found on GitHub.151 AT-6b (data-informed) is more accurate than AT-6a (optimized GAFF) and demonstrates the benefit of data-informed atom typing scheme development. Overall, AT-4 is approximately 25% less accurate than AT-6b since it uses fewer parameters, but performs within 5% accuracy of AT-6a on the testing set, suggesting that using fewer optimized parameters can be as effective as starting with a preexisting model. As a result of having more parameters, AT-8 performs very well on the training set, but often performs poorly on molecules in the testing set. This demonstrates the consequences of a model with too many parameters. Unsurprisingly, GAFF is outperformed by every atom typing scheme developed in this work.| Atom typing scheme | Overall | Train | Test |
|---|---|---|---|
| AT-6b | 18.37 | 18.68 | 17.88 |
| AT-6a | 19.95 | 19.04 | 21.40 |
| AT-4 | 23.94 | 25.83 | 20.91 |
| AT-8 | 27.32 | 9.33 | 56.12 |
| GAFF | 42.74 | 50.37 | 30.52 |
Fig. 3 suggests that six (data-informed) atom types empirically balance training and testing set errors. As expected, the accuracy on the training set increases with the number of parameters, since all atom typing schemes are optimized to fit the same data. Notably, SI Fig. S19 shows that on the training set, AT-8 can approach the accuracy of the customized FFs for R-14 and R-50 generated by Wang et al. for all properties.102 This observation highlights the well-known benefits of improving FF fit by adding atom types as the scope of the FF expands.
 | ||
| Fig. 3 Average MAPD for each thermophysical property of interest (ρl, ρv, Pvap, and ΔHvap) for AT-4, AT-6a, AT-6b, and AT-8. Panel (A) shows the results of the training set and Panel (B) shows the results of the testing set. | ||
For the testing set, the advantages of optimization and data-informed FFs are evident. Fig. 3B shows that AT-6b (average MAPD = 18.68%) predicts almost all properties more accurately for both the testing and training set. As expected, AT-4 shows an increased error on average across both training (average MAPD = 25.83%) and testing set molecules (average MAPD = 20.91%) compared to AT-6b. However, AT-6a and AT-4 perform similarly on the testing set, indicating that using more parameters does not necessarily increase accuracy. In fact, these results demonstrate that it is often just as reliable to use a simple model as to use an optimized preexisting model, which drastically reduces the expert intuition required for transferable FF design. Further supporting this claim, while AT-8 performs exceptionally well on the training set (average MAPD = 9.33%), it drastically underperforms on the testing set (average MAPD = 56.12%). In fact, SI Fig. S19 demonstrates that property predictions for AT-8 are often an order of magnitude less accurate than other optimized models. This highlights that data-informed atom types are more transferable to molecules outside of the training set and highlights the potentially severe consequences of adding atom types as needed to account for a growing FF model.
We recommend AT-6b for a transferable one- and two-carbon atom typing scheme because of its superior performance compared against other atom typing schemes considered in this work (see Table 3). Table 6 shows the LJ parameters associated with this scheme. All FF parameters for each atom type and molecule are available on GitHub.151
| Atom type | σ (Å) |
|
|---|---|---|
| C m | 3.61 | 40.55 |
| C 1 | 3.32 | 55.50 |
| C 2 | 3.48 | 45.21 |
| H c | 2.60 | 11.57 |
| H | 2.22 | 11.74 |
| F | 2.94 | 26.57 |

3.2 Data science techniques improve the transferability of LJ parameters for one- and two-carbon HFCs
Table 7 shows the results of the estimability analysis and eigen decomposition of the FIM for all postulated FFs. SI Table S4 shows the full numerical eigen-decomposition of the FIM for the best performing atom typing scheme (AT-6b) and SI Tables S5–S8 show the estimability analysis rankings for each AT scheme compared to the rankings from eigen-decomposition of the FIM. Finally, the numerical values of eigen-decomposition of the FIM, the estimability analysis rankings, and the optimized LJ parameters for all atom types generated in this work are available on GitHub.151 Using Table 7 and the insights below, we explain how the results of AT-4, AT-6a, and AT-8 were used to propose AT-6b.

|
3.3 Property prediction accuracy is maximized by optimizing data-informed models
Fig. 4 compares the computed coexistence densities of R-14 (Fig. 4A) and HFC-143 (Fig. 4B) with experiments. This, along with the results in Table 5 demonstrates the benefit of optimizing LJ parameters. Recall, GAFF and AT-6a use the same atom types. However, the LJ parameters of AT-6a were tuned to HFC data using this workflow, while GAFF is an off-the-shelf generalized FF. As expected, AT-6a reduces the overall MAPD over all properties by approximately 53% compared to GAFF. Fig. 4B and 5 suggest that the F atom type LJ parameters for GAFF are less accurate for one- and two-carbon HFCs than those of other FFs in this work. For example, the predictions for R-14 are worse for GAFF compared to AT-6a and AT-4, despite the C atom type parameters of each being nearly identical. However, we also highlight that AT-4, which uses four instead of six atom types and required minimal expert intuition to inform the structure, reduced the overall average MAPD values by 44% compared to six non-optimized atom types (GAFF) and had MAPD values overall within 5% of those of AT-6a. | ||
| Fig. 4 Vapor–liquid coexistence curves with 95% confidence intervals for R-14 (Panel (A)) and HFC-143 (Panel (B)) as representatives from the training and testing set, respectively. | ||
 | ||
| Fig. 5 The parameters for R-14 for AT-6a, AT-4, and GAFF and the corresponding MAPD for Pvap and ΔHvap for the parameter sets. | ||
These results highlight that the proposed framework is a simple and systematic method of creating transferable FFs. GAFF took decades7 to develop and specific manually-tuned FFs for HFOs have been in development for years.26 However, the process of re-optimizing the GAFF parameters to create AT-6a based on existing HFC data was performed on a timescale of days (once the workflow was built) and only required an expert to specify reasonable parameter bounds. The creation of AT-4, which was similarly accurate to AT-6a, required even less effort in terms of atom type postulation and optimization time. Therefore, even when expert-informed models such as GAFF exist, less complicated models can be quickly created without sacrificing accuracy by optimizing either a very simple model or the existing parameters of an expert-informed model, as long as some experimental data are available.
The AT-6b atom typing scheme was developed using data science techniques and delivers more accurate predictions on average (average MAPD = 18.37%) than AT-6a (average MAPD = 19.95%). We visualize this performance increase with Fig. 6 using Pvap and ΔHvap predictions for training (HFC-125 – Fig. 6(A and B)) and testing (HFC-23 – Fig. 6(C and D)) set molecules. There is a marked improvement from AT-6b over AT-6a in both of these properties and molecules, highlighting the benefit of applying data analysis to inform the selection of atom types.
 | ||
| Fig. 6 Property predictions with 95% confidence intervals for Pvap and ΔHvap for HFC-125 and HFC-23 as representatives from the training and testing set, respectively. Panels (A) and (C) show the (natural logarithm) of the predicted vapor pressure (ln(Pvap)) as a function of the inverse of temperature (1/T). Panels (B) and (D) show the predicted enthalpy of vaporization (ΔHvap) as a function of temperature T. | ||
Comparison of MAPD values across properties can be misleading. For example, we note that the MAPD values for Pvap and ρv are higher in both the training and testing sets than for other property values. This is an artifact of the magnitude of these properties; large MAPDs tend to be associated with small values  . Therefore, it is more important to compare the MAPD values for a given property across different models when evaluating model accuracy. SI figures on GitHub151 show the property predictions for all molecules and FFs and suggest that the predictions for Pvap and ρl are generally reasonable, despite their high MAPD values.
. Therefore, it is more important to compare the MAPD values for a given property across different models when evaluating model accuracy. SI figures on GitHub151 show the property predictions for all molecules and FFs and suggest that the predictions for Pvap and ρl are generally reasonable, despite their high MAPD values.
3.4 Trade-offs in prediction quality across molecules suggests further opportunities for atom type optimization
Fig. 7 shows the LJ parameter values and corresponding MAPD values for ρl and ΔHvap for AT-4, AT-6a, and GAFF models of methane (R-50). Despite the improved prediction accuracy of optimized FFs compared to GAFF for most HFCs, Fig. 7 indicates that GAFF predicts ρl and ΔHvap significantly more accurately than either AT-6a or AT-4 for R-50. This exception highlights the importance of adequate atom type representation and special attention to parameter estimability for well-rounded optimization. Fig. 7 thus provides direct insights into the physical meaning of these parameters and the consequences of poor estimability. For example, consider the values of the εH parameter employed by AT-6a to model R-50, which represents the attractive strength of the atom. This parameter is the second least estimable parameter overall for this molecule, despite being the fifth most estimable parameter and the fifth most important parameter to the objective overall (out of 12). This parameter increases 37% from its GAFF value upon optimization, but all other parameters between GAFF and AT-6a are within 5% of each other. This suggests that the accuracy loss of AT-6a compared to GAFF for R-50 is completely attributable to the relative inestimability of εH. Furthermore, since R-50 consists mainly of H atoms, prediction errors resulting from less-estimable H atom type LJ parameters (and less realistic atomistic behavior) are magnified for AT-6a, leading to the increase in error for R-50 observed in Fig. 7 (despite this molecule appearing in the training set). These problems are compounded in AT-4, in which the H atom type is both the most important to optimization and the least estimable. In this example, εH is optimized to a value 46% larger than its GAFF value, while the σH parameter is 14% smaller. This represents a smaller H atom with a significantly higher attractive strength. As a result of this optimized, but less realistic LJ model, the errors for R-50 for AT-4 in Fig. 7 are much more pronounced than they are for AT-6a. In support of this finding, Fig. 5 shows that the predictions for R-14 for both AT-4 and AT-6a are considerably more accurate than the R-50 predictions (especially compared to GAFF) since they are independent of the less-estimable H atom type parameters. | ||
| Fig. 7 The parameters for R-50 for AT-6a, AT-4, and GAFF and the corresponding MAPD for Pvap and ΔHvap for the parameter sets. | ||
During optimization, ρv and Pvap errors are minimized at the cost of ρl and ΔHvap errors. We attribute this behavior to the relative accuracy of the underlying GP models. Since the GP models predict Pvap and ρv (average MAPD 4.67%) less accurately than ΔHvap and ρl (average MAPD 0.28%) on the GP testing set (see SI Table S9), the objective function assigns a larger penalty for increasing the predicted error of these properties according to eqn (27). As a consequence, these parameters are more influential during optimization. Interestingly, SI Fig. S19 demonstrates a few notable exceptions where GAFF, despite being highly general, exhibits favorable MAPDs for both Pvap and ρv compared to other atom typing schemes. These examples include HFC-23 and HFC-143 for both properties for AT-4 and AT-6a and ρv for HFC-23 with AT-6b. This suggests that errors in the GP models are prone to propagation when using an  objective function, especially on molecules outside of the testing set. As future work, transformations of GP outputs as 1/ln(Pvap) and 1/ρv, which both exhibit a linear relationship with respect to temperature T, may improve GP accuracy.
objective function, especially on molecules outside of the testing set. As future work, transformations of GP outputs as 1/ln(Pvap) and 1/ρv, which both exhibit a linear relationship with respect to temperature T, may improve GP accuracy.
SI Fig. S19 highlights an intriguing trade-off in the predictive capabilities of AT-4 as a result of poor H atom type estimability; AT-4 achieves better accuracy than AT-6a for HFC-23 and HFC-143, yet performs worse on R-50. This finding motivates future efforts to improve parameter estimability through further restructuring of the FF. SI Fig. S19 offers a possible solution for this case study as an example. AT-8, which has poor out-of-sample predictions (testing set), best predicts the properties of HFC-32 and HFC-41 in the training set. Further inspection reveals the pattern that except for AT-8, all models studied in this work, including GAFF, have lower MAPD values for two-carbon refrigerants. As an alternative to using available data to inform the design of a new set of LJ parameters for all molecules, as is done in the development of AT-6b, one could imagine using this information to design multiple transferable models to increase the estimability of the atom types in each. This line of thought is particularly interesting in the context of GAFF, which despite its flexibility, still experiences accuracy trade-offs even after optimization via AT-6a. For future work, we therefore recommend the testing of two transferable models for HFCs which are fit to the data of one- and two-carbon refrigerants, respectively, but use only three atom types (C, H, and F) each.
3.5 Gaussian process models significantly speed up FF calibration
The previous sections report the improved reliability and accuracy of the optimized transferable HFC FF models. We now quantify the computational benefits of the proposed method.Table 8 shows that, while evaluation speed decreases with the number of parameters, one loss evaluation takes on the order of seconds or less. For clarity, we note that the number of loss evaluations here includes the number used to compute the Jacobian for optimization with a two-point finite difference estimation.
| Atom typing scheme | Estimable atom types | Loss evaluations | Optimization time (min) | Time per loss evaluation (s) |
|---|---|---|---|---|
| AT-4 | 4 | 844 | 8.8 | 0.63 |
| AT-6a | 5 | 1211 | 13.6 | 0.67 |
| AT-6b | 6 | 2075 | 22.8 | 0.66 |
| AT-8 | 8 | 4048 | 69.6 | 1.03 |
The largest model under consideration (AT-8) required approximately 70 minutes to optimize 16 parameters. The smallest model (AT-4) requires less than ten minutes to optimize its eight parameters, demonstrating the ability of this method to rapidly calibrate FF models even as the number of parameters increases. During optimization, the loss function is evaluated many times, since traditional gradient-based optimization techniques use finite differences to approximate derivatives.167–169 However, we note that the time per loss evaluation (calculated as the total optimization time divided by the number of loss evaluations) is still on the order of seconds. For example, AT-8 takes approximately 70 minutes to optimize using approximately 4050 loss evaluations for an average of one second per evaluation of the objective function. Furthermore, in this workflow, one loss evaluation corresponds to 160 GP evaluations (five temperatures, eight training molecules, and four properties), which otherwise would need to be predicted from 40 (five temperatures and eight training molecules) molecular simulations.
To maximize FF accuracy, traditional NLR techniques could be used to directly fit LJ parameters in a FF to reproduce the experimental data of interest using molecular simulations. However, if molecular simulations had been used in place of GP emulators during optimization of these FFs, the optimization workflow would have taken significantly longer. For example, consider AT-4 which required the fewest number of loss evaluations to optimize. If all loss evaluations were run in series then approximately 34000 molecular simulations (40 molecular simulations times 844 loss evaluations) would be necessary. Assuming that each simulation takes approximately eight hours, this optimization would require approximately 31 years of wall-clock time to complete. However, molecular simulations are often parallelized. If we consider the conditions of this work, where 100 simulations are parallelized at once, this optimization time is reduced to 3.70 months. However, loss evaluations can also be parallelized. Therefore, let us assume optimistically that this optimization could be completed in 100 optimization iterations and that all loss evaluations and molecular simulations could be parallelized. In this optimistic case, FF optimization would still require approximately one month. In contrast, this workflow requires less than a second to evaluate the loss function and on the order of minutes to complete LJ parameter optimization. Thus, these results highlight the benefit of using GP surrogate models to facilitate the optimization of LJ FF parameters with NLR.
4 Conclusions and recommendations
Systematically building a transferable FF is difficult because it requires simultaneously determining the best atom typing scheme and the LJ parameter values. This paper demonstrates the efficacy of ML and data science techniques to systematically design and optimize transferable FFs. We stress that this workflow uses GP surrogate models to develop transferable FFs for one- and two-carbon (HFC) refrigerants in weeks compared to months or years for alternative approaches (e.g., gradient-based optimization or stochastic heuristic search). For one- and two-carbon HFC refrigerants, we identified the best performing atom type (AT-6b: average MAPD = 18.37%) using estimability analysis and eigen-decomposition of the FIM for three alternative FF atom typing schemes (AT-4, AT-8, and AT-6a). We highlight that this best model performs favorably compared to both “off-the-shelf” GAFF (average MAPD = 42.74%) and the GAFF atom typing scheme with re-optimized parameters (AT-6a: average MAPD = 19.95%). In particular, considering the FIM helps select atom types with good predictive performance on molecules omitted from the training data, as demonstrated by the out-of-sample predictions for AT-6b (average MAPD = 17.88%) and AT-6a (average MAPD = 21.40%). Moreover, applying data science techniques allows smaller models (AT-4: average MAPD = 20.91%) to achieve better out-of-sample predictions than optimized expert-informed models with more atom types (AT-6a: average MAPD = 21.40%). On the other hand, choosing an arbitrarily large number of atom types can have significant accuracy losses (AT-8: average MAPD = 56.12%). This encourages the practice of using available data to design small transferable FF models with purposely selected parameters. This practice not only promotes more defensible models, but also reduces the necessary expert intuition and time requirements of FF design.Finally, we address the limitations of the study and directions for future work, organized into three themes.
4.1 Future applications
Other promising applications of this workflow include materials such as ILs and deep eutectic solvents (DESs), which are chemically diverse17,170 and commonly studied for separations, as well as electrolyte solvents used in battery applications. Because experimental screening across these large chemical spaces is challenging, molecular simulation is frequently employed to guide material selection and predict properties, although such predictions require accurate force fields.117,171–177 However, even well known molecules in these systems, such as glycerol (a popular DES component) and diethylcarbonate (a common electrolyte), lack accurate FF models.178,179 This encourages the prompt adoption of the presented methods to develop better FFs for these important molecules. We highlight that the same techniques applied in this work for transferable FFs are also useful for the creation of customized FF for other systems, although we anticipate several challenges for molecules with more complexity than the relatively simple molecules studied in the present work. Regardless, as the number of molecular classes studied using this workflow increases and more GP models become available, the extension of this workflow to creating a generalized FF becomes more promising. However, the VLE data which are required for the workflow presented in this paper is often limited for other systems. Therefore, we recommend the extension of this method to surface tension and viscosity data, which are more common. Although surface tension and viscosity simulations often yield high variances and are computationally demanding, uncertainties can likely be accounted for through GP modeling best practices. We similarly encourage extending this method to use both existing QM and physical property data, which could allow for other FF parameters such as partial charges to be optimized if desired. Outside of new molecules and properties, this study also suggested that poor parameter estimability resulted in FF models that predicted the thermophysical properties of two-carbon HFCs better than one-carbon HFCs. These findings justify the existence of two-carbon HFC FFs in the literature86 and suggest that designing separate transferable FFs for one- vs. two-carbon HFCs is promising. Finally, we also acknowledge the importance of validating the FF developed in this work on other molecules and properties. For example, future work should consider validation for larger refrigerant molecules (such as propane derivatives) and the accuracy of structural or dynamic properties.4.2 GP enhancements
The GP models could be improved in several ways. For simplicity, this study neglects correlations between the emulated physical properties by using scalar-output GPs. However, thermophysical property values are often correlated and vector-output (multi-output) GPs would allow the correlations between properties to be captured. Similarly, this study only examines linear GP mean functions, although the relationship between the LJ parameters and the properties of interest is unknown. Exploring nonlinear GP mean functions and alternative feature or output transformations (e.g., 1/ln(Pvap) vs. Pvap) could result in more accurate GP models. Additionally, we recommend iteratively retraining the GP models during optimization as in BO180 to add targeted molecular simulation data to the model and improve local surrogate accuracy.4.3 Algorithm enhancements
We first acknowledge the availability of other methods for approximating the FIM181–183 and other objective functions, such as the acquisition functions used in BO180 as potential improvements to the current objective function. We further recommend replacing L-BFGS-B, which was chosen for convenience, with deterministic global optimization with embedded GPs.184 Additionally, combining GPBO with multi-objective optimization185 or regularization to FFs in literature28 would allow for the explicit weighting of each property in the context of optimization while preventing overfitting. Lastly, future work should consider automating atom typing and transferable FF construction from the numerical estimability results. This would further decrease the expert effort required to propose, implement, and test new models.
objective function. We further recommend replacing L-BFGS-B, which was chosen for convenience, with deterministic global optimization with embedded GPs.184 Additionally, combining GPBO with multi-objective optimization185 or regularization to FFs in literature28 would allow for the explicit weighting of each property in the context of optimization while preventing overfitting. Lastly, future work should consider automating atom typing and transferable FF construction from the numerical estimability results. This would further decrease the expert effort required to propose, implement, and test new models.
Author contributions
Montana N. Carlozo: conceptualization (equal); data curation (lead); formal analysis (lead); investigation (lead); methodology (equal); project administration (lead); software (lead); validation (lead); visualization (lead); writing – original draft preparation (lead); writing – review & editing (lead). Ning Wang: conceptualization (equal); methodology (equal); software (supporting). Alexander W. Dowling: conceptualization (equal); funding acquisition (equal); methodology (equal); resources (equal); supervision (equal); writing – review & editing (equal). Edward J. Maginn: conceptualization (equal); funding acquisition (equal); methodology (equal); resources (equal); supervision (equal); writing – review & editing (equal).Conflicts of interest
There are no conflicts to declare.Nomenclature
Bold capital letters are matrices, bold lowercase letters are vectors, and unbolded lowercase letters are scalars. The exception is variable T which is a scalar temperature.Abbreviations
| AT | Atom type |
| AP | Ammonium perchlorate |
| BO | Bayesian optimization |
| DFT | Density functional theory |
| DNN | Deep neural network |
| DP | Deep potential |

| Expected value of SSE |
| FF | Force field |
| FIM | Fisher information matrix |
| GAP | Gaussian approximation potential |
| GAFF | Generalized AMBER force field |
| GEMC | Gibbs ensemble Monte Carlo |
| GP | Gaussian process |
| GPBO | Gaussian process Bayesian optimization |
| HFC | Hydrofluorocarbon |
| HFO | Hydrofluoroolefin |
| HCFO | Hydrochlorofluoroolefin |
| L-BFGS-B | Limited-memory Broyden–Fletcher–Goldfarb–Shanno with bounds |
| LHS | Latin hypercube sampling |
| LJ | Lennard-Jones |
| MAPE | Mean absolute percent error |
| MAPD | Mean absolute percent deviation |
| ML | Machine learning |
| MLE | Maximum likelihood estimate |
| MLIP | machine-learned interatomic potential |
| MSE | Mean squared error |
| MVN | Multivariate normal |
| NEP | Neuroevolution potential |
| NLR | Nonlinear regression |
| NN | Neural network |
| NNP | Neural network potential |
| PES | Potential energy surface |
| PINN | Physics informed neural network |
| QM | Quantum mechanics |
| RAD | Relative absolute deviation |
| RMSE | Root mean squared deviation |
| SI | Supplementary information |
| SSE | Sum of squared errors |
| VLE | Vapor–liquid equilibrium |
| vdW | van der Waals |
Experimental data
| ΔHvap | Enthalpy of vaporization |

| A set of molecules |
| N | Number of experimental data points |
| ρ l | Liquid density |
| ρ v | Vapor density |
| P vap | Vapor pressure |

| The set of properties ρl, ρv, Pvap, ΔHvap |
| Σ exp(·,·) | Experimental data covariance matrix |
| σ exp 2(·,·) | Experimental variance of a single experimental data point |
| T | Temperature |

| The set of temperatures |
| ω | Number of temperature data points for a given molecule and property |
| y exp | Experimental data |
Gaussian processes and kernels
| c | Kernel function variance parameter |
| d(·) | Euclidean distance |
| δ nm | Kronecker delta |

| Gaussian process model |
| h | Gaussian process hyperparameters |
| k(·,·) | Gaussian process kernel function |
| K(·,·) | Gaussian process kernel matrix |

| Kernel function length scales |
| µ GP(·) | Gaussian process prediction mean (vector) |

| Normal distribution |
| Σ GP(·,·) | Gaussian process prediction covariance matrix |
| τ 2 | Kernel function scale parameter |
| y | GP outputs |
| Z | GP inputs |
Force field parameters
| ε | LJ parameter for well-depth |
| ε 0 | Vacuum permittivity |
| γ | Nominal phase angle for dihedrals |
| k θ | Angle force constant |
| k ϕ | Torsion force constant |
| n | Multiplicity |
| ϕ | Phase angle for dihedrals |
| q | Partial charge |
| r i,j | Distance between two particles i and j |
| σ | LJ parameter for van der Waals radius |
| θ | Bond angle |
| θ 0 | Nominal bond angle |

| Potential energy |
Optimization and data analysis symbols
| A | Transformation matrix mapping transferable LJ parameters to the LJ parameters of distinct molecules |
| B | Sensitivity matrix for Algorithm 1 |
| F | Parameter sensitivity Matrix for FIM |
| f(·) | Predictions of an arbitrary model |
| f(·) | An arbitrary model |
| g(θ) |
 objective function objective function |
| H | Hessian of g(θ) |

| A likelihood function (and log-likelihood function  ) ) |
| λ e | Eigenvalue |
| θ | Transferable LJ parameters for optimization of the objective, θ = [σ, ε] |

| Lower bounds for optimization for θ |
|
| Upper bounds for optimization for θ |
| q | Importance metric for eigen-decomposition |
| v j | Eigenvector |
Data availability
All data underlying this study and all versions of the code with which it was produced are available at genFF_public on GitHub at https://github.com/MaginnGroup/genFF_public151 – https://doi.org/10.5281/zenodo.17713215.Supplementary information (SI): a PDF file containing the atom typing scheme figures, preliminary results for AT-3, relevant derivations and justifications, results for the rigid bond and flexible bond implementations for AT-6b, eigen-decomposition and estimability analysis results, the MAPD results broken down by molecule and model, and the MAPD of the testing set of the GP predictions for each molecule and property. See DOI: https://doi.org/10.1039/d5dd00537j.
Acknowledgements
This research is based on work supported by the National Science Foundation under award number EEC-2330175 for the Engineering Research Center EARTH and under award number EFMA-2029354 for the EFRI DChem project Next-generation Low Global Warming Refrigerants. Computing resources were provided by the Center for Research Computing (CRC) at the University of Notre Dame. MC acknowledges support from the Graduate Assistance in Areas of National Need fellowship from the Department of Education, grant number P200A210048. During the preparation of this work the authors used Chat GPT to properly format Table 7. After using this tool/service, the authors reviewed and edited the content as needed and take full responsibility for the content of the published article.References
- E. J. Maginn, AIChE J., 2009, 55, 1304–1310 CrossRef CAS.
- B. M. Wood, M. Dzamba, X. Fu, M. Gao, M. Shuaibi, L. Barroso-Luque, K. Abdelmaqsoud, V. Gharakhanyan, J. R. Kitchin, D. S. Levine, K. Michel, A. Sriram, T. Cohen, A. Das, A. Rizvi, S. J. Sahoo, Z. W. Ulissi and C. L. Zitnick, The Open Molecules 2025 (OMol25) Dataset, Evaluations, and Models, arXiv, 2025, preprint, arXiv:2505.08762, DOI:10.48550/arXiv.2505.08762.
- D. Frenkel and B. Smit, Understanding Molecular Simulations: From Algorithms to Applications, Academic Press, 2002 Search PubMed.
- L. Wang, P. K. Behara, M. W. Thompson, T. Gokey, Y. Wang, J. R. Wagner, D. J. Cole, M. K. Gilson, M. R. Shirts and D. L. Mobley, J. Phys. Chem. B, 2024, 128, 7043–7067 CrossRef CAS PubMed.
- X. He, B. Walker, V. H. Man, P. Ren and J. Wang, Curr. Opin. Struct. Biol., 2022, 72, 187–193 CrossRef CAS PubMed.
- A. K. Rappé, C. J. Casewit, K. S. Colwell, W. A. Goddard III and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes, I. Vorobyov and A. D. Mackerell, J. Comput. Chem., 2010, 31, 671–690 CrossRef CAS PubMed.
- H. Sun, J. Phys. Chem. B, 1998, 102, 7338–7364 CrossRef CAS.
- T. A. Halgren, J. Comput. Chem., 1996, 17, 490–519 Search PubMed.
- M. G. Martin and J. I. Siepmann, J. Phys. Chem. B, 1998, 102, 2569–2577 Search PubMed.
- J. W. Ponder, C. Wu, P. Ren, V. S. Pande, J. D. Chodera, M. J. Schnieders, I. Haque, D. L. Mobley, D. S. Lambrecht, R. A. Distasio, M. Head-Gordon, G. N. Clark, M. E. Johnson and T. Head-Gordon, J. Phys. Chem. B, 2010, 114, 2549–2564 CrossRef CAS PubMed.
- O. Borodin, J. Phys. Chem. B, 2009, 113, 11463–11478 CrossRef CAS PubMed.
- P. Dauber-Osguthorpe, V. A. Roberts, D. J. Osguthorpe, J. Wolff, M. Genest and A. T. Hagler, Proteins: Struct., Funct., Bioinf., 1988, 4, 31–47 CrossRef CAS.
- S. Boothroyd, P. K. Behara, O. C. Madin, D. F. Hahn, H. Jang, V. Gapsys, J. R. Wagner, J. T. Horton, D. L. Dotson, M. W. Thompson, J. Maat, T. Gokey, L.-P. Wang, D. J. Cole, M. K. Gilson, J. D. Chodera, C. I. Bayly, M. R. Shirts and D. L. Mobley, J. Chem. Theory Comput., 2023, 19, 3251–3275 CrossRef CAS PubMed.
- K. R. Baca, K. Al-Barghouti, N. Wang, M. G. Bennett, L. M. Valenciano, T. L. May, I. V. Xu, M. Cordry, D. M. Haggard, A. G. Haas, A. Heimann, A. N. Harders, H. G. Uhl, D. T. Melfi, A. D. Yancey, R. Kore, E. J. Maginn, A. M. Scurto and M. B. Shiflett, Chem. Rev., 2024, 124, 5167–5226 CrossRef CAS.
- S. Mallakpour and M. Dinari, in Ionic Liquids as Green Solvents: Progress and Prospects, ed. A. Mohammad and D. Inamuddin, Springer Netherlands, Dordrecht, 2012, pp. 1–32 Search PubMed.
- X. He, V. H. Man, W. Yang, T. S. Lee and J. Wang, J. Chem. Phys., 2020, 153, 114502 CrossRef CAS PubMed.
- E. Harder, W. Damm, J. Maple, C. Wu, M. Reboul, J. Y. Xiang, L. Wang, D. Lupyan, M. K. Dahlgren, J. L. Knight, J. W. Kaus, D. S. Cerutti, G. Krilov, W. L. Jorgensen, R. Abel and R. A. Friesner, J. Chem. Theory Comput., 2016, 12, 281–296 CrossRef CAS PubMed.
- G. Raabe, J. Phys. Chem. B, 2012, 116, 5744–5751 CrossRef CAS PubMed.
- K. Goloviznina, J. N. Canongia Lopes, M. Costa Gomes and A. A. Pádua, J. Chem. Theory Comput., 2019, 15, 5858–5871 CrossRef CAS PubMed.
- N. Ferrando, V. Lachet, J. M. Teuler and A. Boutin, J. Phys. Chem. B, 2009, 113, 5985–5995 CrossRef CAS PubMed.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 5179–5197 CrossRef CAS.
- G. Raabe, J. Chem. Eng. Data, 2020, 65, 1234–1242 CrossRef CAS.
- G. Raabe and E. J. Maginn, J. Phys. Chem. B, 2010, 114, 10133–10142 CrossRef CAS PubMed.
- G. Raabe, J. Chem. Eng. Data, 2015, 60, 2412–2419 CrossRef CAS.
- L. P. Wang, T. J. Martinez and V. S. Pande, J. Phys. Chem. Lett., 2014, 5, 1885–1891 CrossRef CAS PubMed.
- J. L. Abascal and C. Vega, J. Chem. Phys., 2005, 123, 234505 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- S. Boothroyd, L.-P. Wang, D. L. Mobley, J. D. Chodera and M. R. Shirts, J. Chem. Theory Comput., 2022, 18, 3566–3576 CrossRef CAS PubMed.
- S. M. Kantonen, H. S. Muddana, M. Schauperl, N. M. Henriksen, L.-P. Wang and M. K. Gilson, J. Chem. Theory Comput., 2020, 16, 1115–1127 CrossRef CAS PubMed.
- L. P. Wang, T. Head-Gordon, J. W. Ponder, P. Ren, J. D. Chodera, P. K. Eastman, T. J. Martinez and V. S. Pande, J. Phys. Chem. B, 2013, 117, 9956–9972 CrossRef CAS PubMed.
- K. Claridge and A. Troisi, J. Phys. Chem. B, 2019, 123, 428–438 CrossRef CAS PubMed.
- B. Seo, Z.-Y. Lin, Q. Zhao, M. A. Webb and B. M. Savoie, J. Chem. Inf. Model., 2021, 61, 5013–5027 CrossRef CAS PubMed.
- T. B. Blank, S. D. Brown, A. W. Calhoun and D. J. Doren, J. Chem. Phys., 1995, 103, 4129–4137 CrossRef CAS.
- J. Behler, J. Chem. Phys., 2016, 145, 170901 CrossRef.
- O. T. Unke, S. Chmiela, H. E. Sauceda, M. Gastegger, I. Poltavsky, K. T. Schütt, A. Tkatchenko and K. R. Müller, Chem. Rev., 2021, 121, 10142–10186 CrossRef CAS PubMed.
- V. L. Deringer, M. A. Caro and G. Csányi, Adv. Mater., 2019, 31, 1902765 CrossRef CAS.
- Y. Mishin, Acta Mater., 2021, 214, 116980 CrossRef CAS.
- T. Mueller, A. Hernandez and C. Wang, J. Chem. Phys., 2020, 152, 050902 CrossRef CAS PubMed.
- S. Chmiela, H. E. Sauceda, I. Poltavsky, K. R. Müller and A. Tkatchenko, Comput. Phys. Commun., 2019, 240, 38–45 CrossRef CAS.
- J. Byggmästar, K. Nordlund and F. Djurabekova, Phys. Rev. Mater., 2020, 4, 093802 CrossRef.
- D. Dragoni, T. D. Daff, G. Csányi and N. Marzari, Phys. Rev. Mater., 2018, 2, 013808 CrossRef.
- V. L. Deringer and G. Csányi, Phys. Rev. B: Condens. Matter Mater. Phys., 2017, 95, 094203 CrossRef.
- P. Rowe, G. Csányi, D. Alfè and A. Michaelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2018, 97, 054303 CrossRef.
- N. Bernstein, B. Bhattarai, G. Csányi, D. A. Drabold, S. R. Elliott and V. L. Deringer, Angew. Chem., Int. Ed. Engl., 2019, 131, 7131–7135 CrossRef.
- Z. Zhang, G. Csányi and D. Alfè, Geochim. Cosmochim. Acta, 2020, 291, 5–18 CrossRef CAS.
- F. C. Mocanu, K. Konstantinou, T. H. Lee, N. Bernstein, V. L. Deringer, G. Csányi and S. R. Elliott, J. Phys. Chem. B, 2018, 122, 8998–9006 CrossRef CAS PubMed.
- A. P. Bartók, J. Kermode, N. Bernstein and G. Csányi, Phys. Rev. X, 2018, 8, 041048 Search PubMed.
- Z. Fan, Z. Zeng, C. Zhang, Y. Wang, K. Song, H. Dong, Y. Chen and T. Ala-Nissila, Phys. Rev. B, 2021, 104, 104309 CrossRef CAS.
- P. Ying, C. Qian, R. Zhao, Y. Wang, K. Xu, F. Ding, S. Chen and Z. Fan, Chem. Phys. Res., 2025, 6, 011310 Search PubMed.
- Z. Fan, J. Phys.: Condens. Matter, 2022, 34, 125902 CrossRef CAS PubMed.
- R. Zhao, S. Wang, Z. Kong, Y. Xu, K. Fu, P. Peng and C. Wu, Mater. Des., 2023, 231, 112012 CrossRef CAS.
- P. Ying and Z. Fan, J. Phys.: Condens. Matter, 2023, 36, 125901 CrossRef PubMed.
- R. Cheng, Z. Zeng, C. Wang, N. Ouyang and Y. Chen, Phys. Rev. B, 2024, 109, 054305 CrossRef CAS.
- Y. Li, Y. Guo, S. Xiong and H. Yi, Int. J. Heat Mass Transfer, 2024, 222, 125167 CrossRef CAS.
- T. Liang, P. Ying, K. Xu, Z. Ye, C. Ling, Z. Fan and J. Xu, Phys. Rev. B, 2023, 108, 184203 CrossRef CAS.
- J. Zhang, H.-C. Zhang, W. Li and G. Zhang, Chin. Phys. B, 2024, 33, 047402 CrossRef CAS.
- H. Dong, Y. Shi, P. Ying, K. Xu, T. Liang, Y. Wang, Z. Zeng, X. Wu, W. Zhou, S. Xiong, S. Chen and Z. Fan, J. Appl. Phys., 2024, 135, 161101 CrossRef CAS.
- L. Zhang, H. Wang, R. Car and W. E, Proc. Natl. Acad. Sci. U. S. A, 2021, 118, e2025270118 Search PubMed.
- I. Sánchez-Burgos, Q. Zeng, R. Car, W. E and P. G. Debenedetti, J. Chem. Phys., 2023, 158, 104504 CrossRef PubMed.
- H. Omranpour, O. Akin-Ojo and T. D. Kühne, J. Chem. Phys., 2024, 160, 080901 CrossRef.
- M. Chen and H.-Y. Ko, et al. , Nat. Commun., 2023, 14, 2655 CrossRef PubMed.
- K. Xu, T. Liang, N. Xu, P. Ying, S. Chen, N. Wei, J. Xu and Z. Fan, npj Comput. Mater., 2025, 11, 279 CrossRef.
- C. Glover, T. Li and R. Shannon, et al. , npj Comput. Mater., 2023, 9, 25 CrossRef.
- Z. Lu, Y. Zhang and X. Liu, et al. , J. Phys. Chem. C, 2023, 127, 21654–21667 Search PubMed.
- B. M. Wood, M. Dzamba, X. Fu, M. Gao, M. Shuaibi, L. Barroso-Luque, K. Abdelmaqsoud, V. Gharakhanyan, J. R. Kitchin, D. S. Levine, K. Michel, A. Sriram, T. Cohen, A. Das, A. Rizvi, S. J. Sahoo, Z. W. Ulissi and C. L. Zitnick, UMA: A Family of Universal Models for Atoms, arXiv, 2025, preprint, arXiv:2506.23971, DOI:10.48550/arXiv.2506.23971.
- V. L. Deringer, A. P. Bartók, N. Bernstein, D. M. Wilkins, M. Ceriotti and G. Csányi, Chem. Rev., 2021, 121, 10073–10141 CrossRef CAS PubMed.
- K. T. Schütt, S. Chmiela, O. A. v. Lilienfeld, A. Tkatchenko, K. Tsuda and K.-R. Müller, Machine Learning Meets Quantum Physics, Springer International Publishing, Cham, 1st edn, 2020 Search PubMed.
- A. Nandi, P. Pandey, P. L. Houston, C. Qu, Q. Yu, R. Conte, A. Tkatchenko and J. M. Bowman, J. Chem. Theory Comput., 2024, 20, 8807–8819 CrossRef CAS PubMed.
- G. P. Pun, R. Batra, R. Ramprasad and Y. Mishin, Nat. Commun., 2019, 10, 2339 CrossRef PubMed.
- A. R. Leach, Molecular Modeling Principles and Applications, Addison Wesley Longman, Singapore, 1996, pp. 1–587 Search PubMed.
- J. Hunger and G. Huttner, J. Comput. Chem., 1999, 20, 455–471 CrossRef CAS.
- R. Galvelis, S. Doerr, J. M. Damas, M. J. Harvey and G. De Fabritiis, J. Chem. Inf. Model., 2019, 59, 3485–3493 CrossRef CAS PubMed.
- J. M. Sestito, M. L. Thatcher, L. Shu, T. A. L. Harris and Y. Wang, J. Phys. Chem. A, 2020, 124, 58 CrossRef.
- J. Wang and P. A. Kollman, J. Comput. Chem., 2001, 22, 1219–1228 CrossRef CAS.
- S. Mostaghim, M. Hoffmann, P. Konig, T. Frauenheim and J. Teich, Proceedings of the 2004 Congress on Evolutionary Computation (IEEE Cat. No. 04TH8753), 2004, vol. 1, pp. 212–219 Search PubMed.
- R. M. Betz and R. C. Walker, J. Comput. Chem., 2015, 36, 79–87 CrossRef CAS PubMed.
- Y. Li, H. Li, F. C. Pickard, B. Narayanan, F. G. Sen, M. K. Chan, S. K. Sankaranarayanan, B. R. Brooks and B. Roux, J. Chem. Theory Comput., 2017, 13, 4492–4503 CrossRef CAS PubMed.
- M. V. Ivanov, M. R. Talipov and Q. K. Timerghazin, J. Phys. Chem. A, 2015, 119, 1422–1434 CrossRef CAS PubMed.
- R. Islam, M. F. Kabir, S. R. Dhruba and K. Afroz, Comput. Mater. Sci., 2021, 200, 110759 CrossRef.
- L. Pereyaslavets, I. Kurnikov, G. Kamath, O. Butin, A. Illarionov, I. Leontyev, M. Olevanov, M. Levitt, R. D. Kornberg and B. Fain, Proc. Natl. Acad. Sci. U. S. A, 2018, 115, 8878–8882 CrossRef PubMed.
- K. Kříž, P. J. van Maaren and D. van der Spoel, J. Chem. Theory Comput., 2024, 20, 2362–2376 CrossRef PubMed.
- S. Izvekov, M. Parrinello, C. J. Bumham and G. A. Voth, J. Chem. Phys., 2004, 120, 10896–10913 CrossRef CAS PubMed.
- M. Fermeglia, M. Ferrone and S. Pricl, Fluid Phase Equilib., 2003, 210, 105–116 CrossRef CAS.
- E. K. Watkins and W. L. Jorgensen, J. Phys. Chem. A, 2001, 105, 4118–4125 CrossRef CAS.
- B. J. Befort, R. S. Defever, G. M. Tow, A. W. Dowling and E. J. Maginn, J. Chem. Inf. Model., 2021, 19, 28 Search PubMed.
- P. Chatterjee, M. Y. Sengul, A. Kumar and A. D. MacKerell, J. Chem. Theory Comput., 2022, 18, 2388–2407 CrossRef CAS PubMed.
- O. C. Madin and M. R. Shirts, Digital Discovery, 2023, 2, 828–847 RSC.
- F. Rizzi, H. N. Najm, B. J. Debusschere, K. Sargsyan, M. Salloum, H. Adalsteinsson and O. M. Knio, Multiscale Model. Simul., 2012, 10, 1460–1492 CrossRef.
- G. Raabe, V. P. Chheda and U. Römer, Ind. Eng. Chem. Res., 2025, 121, 511–522 Search PubMed.
- M. Fleck, S. Darouich, N. Hansen and J. Gross, J. Phys. Chem. B, 2024, 128, 9544–9552 CrossRef CAS PubMed.
- M. Fleck, R. Katsuta, T. Esper, N. Hansen and J. Gross, Ind. Eng. Chem. Res., 2025, 64, 6170–6179 CrossRef CAS.
- M. Müller, A. Hagg, R. Strickstrock, M. Hülsmann, A. Asteroth, K. N. Kirschner and D. Reith, J. Chem. Inf. Model., 2023, 63, 1872–1881 CrossRef PubMed.
- J. L. McDonagh, A. Shkurti, D. J. Bray, R. L. Anderson and E. O. Pyzer-Knapp, J. Chem. Inf. Model., 2019, 59, 4278–4288 CrossRef CAS PubMed.
- R. L. Anderson, D. J. Bray, A. S. Ferrante, M. G. Noro, I. P. Stott and P. B. Warren, J. Chem. Phys., 2017, 147, 094503 CrossRef PubMed.
- A. C. Winget, PhD thesis, University of Dayton, Dayton, OH, 2024.
- M. N. Carlozo, K. Wang and A. W. Dowling, Ind. Eng. Chem. Res., 2025, 64, 18277–18297 CrossRef CAS.
- B. J. Befort, R. S. Defever, E. J. Maginn and A. W. Dowling, Comput.-Aided Chem. Eng., 2022, 49, 1249–1254 CAS.
- X. Wang and Y.-L. S. Tse, J. Chem. Theory Comput., 2022, 18, 7155–7165 CrossRef CAS PubMed.
- N. Wang, M. N. Carlozo, E. Marin-Rimoldi, B. J. Befort, A. W. Dowling and E. J. Maginn, J. Chem. Theory Comput., 2023, 19, 4546–4558 CrossRef CAS PubMed.
- S. C. Potter, D. J. Tildesley, A. Burgess and S. C. Rogers, Mol. Phys., 1997, 92, 825–834 CrossRef CAS.
- R. P. S. Peguin, G. Kamath, J. J. Potoff and S. R. P. da Rocha, J. Phys. Chem. B, 2009, 113, 178–187 CrossRef CAS PubMed.
- Z. Yang, M. Gong, X. Dong, X. Li and J. Wu, Fluid Phase Equilib., 2015, 394, 93–100 CrossRef CAS.
- N. Zhang, P. Hu, L. Chen and L. Zhi, J. Mol. Liq., 2020, 306, 112896 CrossRef CAS.
- M. S. Alam and J. H. Jeong, Int. J. Refrig., 2019, 104, 311–320 CrossRef CAS.
- S. Higashi and A. Takada, Mol. Phys., 1997, 92, 641–650 CrossRef CAS.
- E. Paulechka, K. Kroenlein, A. Kazakov and M. Frenkel, J. Phys. Chem. B, 2012, 116, 14389–14397 CrossRef CAS PubMed.
- U.S. Environmental Protection Agency, Draft Regulatory Impact Analysis for Phasing Down Production and Consumption of Hydrofluorocarbons (HFCs), 2021 Search PubMed.
- B. K. Sovacool, S. Griffiths, J. Kim and M. Bazilian, Renewable Sustainable Energy, 2021, 141, 110759 CrossRef CAS.
- K. R. Baca, G. M. Olsen, L. M. Valenciano, M. G. Bennett, D. M. Haggard, B. J. Befort, A. Garciadiego, A. W. Dowling, E. J. Maginn and M. B. Shiflett, ACS Sustain. Chem. Eng., 2022, 10, 816–830 CrossRef CAS.
- E. A. Finberg, T. L. May and M. B. Shiflett, Ind. Eng. Chem. Res., 2022, 61, 9795–9812 Search PubMed.
- J. M. Sousa, J. F. Granjo, A. J. Queimada, A. G. Ferreira, N. M. Oliveira and I. M. Fonseca, J. Chem. Thermodyn., 2014, 73, 36–43 Search PubMed.
- S. Asensio-Delgado, M. Viar, F. Pardo, G. Zarca and A. Urtiaga, Fluid Phase Equilib., 2021, 549, 113210 CrossRef CAS.
- N. Wang, Y. Zhang, K. S. Al-Barghouti, R. Kore, A. M. Scurto and E. J. Maginn, J. Phys. Chem. B, 2022, 126, 8309–8321 CrossRef CAS PubMed.
- K. S. Al-Barghouti, R. Kore, B. Agbodekhe, D. Trevisan Melfi, E. Marin-Rimoldi, M. B. Shiflett, E. J. Maginn and A. M. Scurto, J. Phys. Chem. B, 2025, 129, 7311–7326 CrossRef CAS PubMed.
- R. D. Mountain and G. Morrison, Mol. Phys., 1988, 64, 91–95 CrossRef CAS.
- J. J. Potoff and D. A. Bernard-Brunel, J. Phys. Chem. B, 2009, 113, 14725–14731 CrossRef CAS PubMed.
- C. I. Bayly, P. Cieplak, W. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS.
- A. Jakalian, B. L. Bush, D. B. Jack and C. I. Bayly, J. Comput. Chem., 2000, 21, 132–146 CrossRef CAS.
- Y. Zhang, L. Xue, F. Khabaz, R. Doerfler, E. L. Quitevis, R. Khare and E. J. Maginn, J. Phys. Chem. B, 2015, 119, 14934–14944 CrossRef CAS PubMed.
- R. Pollice and P. Chen, J. Am. Chem. Soc., 2019, 141, 3489–3506 CrossRef CAS PubMed.
- A. Manayil Parambil, E. Priyadarshini, S. Paul, A. Bakandritsos, V. K. Sharma and R. Zbořil, J. Mater. Chem. A, 2025, 13, 8246–8281 RSC.
- G. Raabe, Sci. Technol. Built. Environ., 2016, 22, 1077–1089 CrossRef.
- K. Wang, M. Zeng, J. Wang, W. Shang, Y. Zhang, T. Luo and A. W. Dowling, Digit. Chem. Eng., 2023, 6, 100076 CrossRef.
- C. D. Holcomb and L. J. V. Poolen, Fluid Phase Equilib., 1994, 100, 223–239 CrossRef CAS.
- E. W. Lemmon, I. H. Bell, M. L. Huber and M. O. McLinden, NIST Standard Reference Database 23: Reference Fluid Thermodynamic and Transport Properties-REFPROP, Version 10.0, National Institute of Standards and Technology, 2018, https://www.nist.gov/srd/refprop Search PubMed.
- D. A. Keller, C. D. Row and P. H. Lieder, Toxicol. Sci., 1996, 30, 213–219 CrossRef CAS.
- C. E. Rasmussen and C. K. I. Williams, Gaussian Processes for Machine Learning, MIT Press, 2006, pp. 1–248 Search PubMed.
- R. B. Gramacy, Surrogates: Gaussian Process Modeling, Design and Optimization for the Applied Sciences, Chapman Hall/CRC, Boca Raton, Florida, 2020, pp. 117–376 Search PubMed.
- W. Shang, M. Zeng, A. Tanvir, K. Wang, M. Saeidi-Javash, A. Dowling, T. Luo and Y. Zhang, Adv. Mater., 2023, 35, 2212230 CrossRef CAS PubMed.
- P. I. Frazier, INFORMS TutORials in Operations Research, 2018, pp. 255–278 Search PubMed.
- K. Wang and A. W. Dowling, Curr. Opin. Chem. Eng., 2022, 36, 100728 CrossRef.
- Y. Bard, Nonlinear Parameter Estimation, Academic Press, 1974, pp. 1–340 Search PubMed.
- L. Malagò and G. Pistone, FOGA 2015 – Proceedings of the 2015 ACM Conference on Foundations of Genetic Algorithms XIII, 2015, pp. 150–162 Search PubMed.
- C. R. Rao, in Information and the Accuracy Attainable in the Estimation of Statistical Parameters, ed. S. Kotz and N. L. Johnson, Springer New York, New York, NY, 1992, pp. 235–247 Search PubMed.
- R. Bellman and K. Åström, Math. Biosci., 1970, 7, 329–339 CrossRef.
- K. Z. Yao, B. M. Shaw, B. Kou, K. B. McAuley and D. W. Bacon, Polym. React. Eng., 2003, 11, 563–588 CrossRef CAS.
- J. Wang and A. W. Dowling, AIChE J., 2022, 68, 1–24 Search PubMed.
- O. T. Chis, A. F. Villaverde, J. R. Banga and E. Balsa-Canto, Math. Biosci., 2016, 282, 147–161 CrossRef PubMed.
- H. Pohjanpalo, Math. Biosci., 1978, 41, 21–33 CrossRef.
- S. Vajda, K. R. Godfrey and H. Rabitz, Math. Biosci., 1989, 93, 217–248 CrossRef CAS PubMed.
- L. Ljung and T. Glad, Automatica, 1994, 30, 265–276 CrossRef.
- E. Balsa-Canto, A. A. Alonso and J. R. Banga, BMC Syst. Biol., 2010, 4, 1–18 CrossRef PubMed.
- O. T. Chis, J. R. Banga and E. Balsa-Canto, PLoS One, 2011, 6, 1–16 CrossRef PubMed.
- H. Miao, X. Xia, A. S. Perelson and H. Wu, SIAM Rev., 2011, 53, 3–39 CrossRef PubMed.
- K. A. McLean and K. B. McAuley, Can. J. Chem. Eng., 2012, 90, 351–366 CrossRef CAS.
- A. F. Villaverde, N. Tsiantis and J. R. Banga, J. R. Soc., Interface, 2019, 16, 20190043 CrossRef PubMed.
- A. Holmberg, Math. Biosci., 1982, 62, 23–43 CrossRef.
- M. Carlozo, genFF_public, 2025, DOI:10.5281/zenodo.17713215.
- J. A. Jacquez and P. Greif, Math. Biosci., 1985, 77, 201–227 CrossRef.
- M. Rodriguez-Fernandez, J. A. Egea and J. R. Banga, BMC Bioinf., 2006, 7, 483 CrossRef PubMed.
- R. Brun, P. Reichert and H. R. Künsch, Water Resour. Res., 2001, 37, 1015–1030 CrossRef.
- R. Brun, M. Kühni, H. Siegrist, W. Gujer and P. Reichert, Water Res., 2002, 36, 4113–4127 CrossRef CAS PubMed.
- S. J. Keasler, S. M. Charan, C. D. Wick, I. G. Economou and J. I. Siepmann, J. Phys. Chem. B, 2012, 116, 11234–11246 CrossRef CAS PubMed.
- A. C. Rencher and G. B. Schaalje, Linear Models in Statistics, John Wiley & Sons, Inc., 2nd edn, 2008, pp. 105–115 Search PubMed.
- J. K. Shah, E. Marin-Rimoldi, R. G. Mullen, B. P. Keene, S. Khan, A. S. Paluch, N. Rai, L. L. Romanielo, T. W. Rosch, B. Yoo and E. J. Maginn, J. Comput. Chem., 2017, 38, 1727–1739 CrossRef CAS PubMed.
- L. Martinez, R. Andrade, E. G. Birgin and J. M. Martínez, J. Comput. Chem., 2009, 30, 2157–2164 CrossRef CAS PubMed.
- C. Klein, A. Z. Summers, M. W. Thompson, J. B. Gilmer, C. McCabe, P. T. Cummings, J. Sallai and C. R. Iacovella, Comput. Mater. Sci., 2019, 167, 215–227 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1, 19–25 CrossRef.
- F. L. Oliveira, B. Luan, P. M. Esteves, M. Steiner and R. Neumann Barros Ferreira, J. Chem. Theory Comput., 2024, 20, 8559–8568 CrossRef CAS PubMed.
- D. A. Dickey and W. A. Fuller, J. Am. Stat. Assoc., 1979, 74, 427 Search PubMed.
- A. G. d. G. Matthews, M. van der Wilk, T. Nickson, K. Fujii, A. Boukouvalas, P. León-Villagrá, Z. Ghahramani and J. Hensman, J. Mach. Learn. Res., 2017, 18, 1–6 Search PubMed.
- C. Zhu and R. H. Byrd, ACM Trans. Math. Softw., 1997, 23, 550–560 CrossRef.
- P. Virtanen, R. Gommers, T. E. Oliphant, M. Haberland, T. Reddy, D. Cournapeau, E. Burovski, P. Peterson, W. Weckesser, J. Bright, S. J. van der Walt, M. Brett, J. Wilson, K. J. Millman, N. Mayorov, A. R. J. Nelson, E. Jones, R. Kern, E. Larson, C. J. Carey, I. Polat, Y. Feng, E. W. Moore, J. VanderPlas, D. Laxalde, J. Perktold, R. Cimrman, I. Henriksen, E. A. Quintero, C. R. Harris, A. M. Archibald, A. H. Ribeiro, F. Pedregosa, P. van Mulbregt and SciPy 1.0 Contributors, Nat. Methods, 2020, 17, 261–272 CrossRef CAS PubMed.
- C. W. Hopkins and A. E. Roitberg, J. Chem. Inf. Model., 2014, 54, 1978–1986 CrossRef CAS PubMed.
- M. Devereux, N. Gresh, J.-P. Piquemal and M. Meuwly, J. Comput. Chem., 2014, 35, 1577–1591 CrossRef CAS PubMed.
- S. Izvekov, M. Parrinello, C. J. Bumham and G. A. Voth, J. Chem. Phys., 2004, 120, 10896–10913 CrossRef CAS PubMed.
- B. B. Hansen, S. Spittle, B. Chen, D. Poe, Y. Zhang, J. M. Klein, A. Horton, L. Adhikari, T. Zelovich, B. W. Doherty, B. Gurkan, E. J. Maginn, A. Ragauskas, M. Dadmun, T. A. Zawodzinski, G. A. Baker, M. E. Tuckerman, R. F. Savinell and J. R. Sangoro, Chem. Rev., 2021, 121, 1232–1285 CrossRef CAS PubMed.
- H. Qin, Z. Wang, J. Ruan, F. Wei, Z. Yuan, W. Jiao, G. Qi and Y. Liu, Sep. Purif. Technol., 2025, 356, 129796 CrossRef CAS.
- M. Mu, G. Yu, B. Liu, B. Chen, Y. Hu and C. Dai, J. Mol. Liq., 2024, 409, 125596 CrossRef CAS.
- F. Castillo-Borja, U. I. Bravo-Sánchez, R. Vázquez-Román and C. O. Díaz-Ovalle, J. Mol. Liq., 2020, 297, 111904 CrossRef CAS.
- A. Boruń and A. Wypych-Stasiewicz, J. Mol. Liq., 2021, 344, 117695 CrossRef.
- P. Zittlau, S. Mross, D. Gond and M. Kohns, J. Chem. Phys., 2024, 161, 124118 CrossRef CAS PubMed.
- K. Tasaki, J. Phys. Chem. B, 2005, 109, 2920–2933 CrossRef CAS PubMed.
- Y. Zhang, E. J. Maginn, S. Tepavcevic, E. Carino, N. T. Hahn, N. Becknell, J. Mars, K. S. Han, K. T. Mueller and M. Toney, J. Phys. Chem. Lett., 2023, 14, 11393–11399 CrossRef CAS PubMed.
- D. A. Jahn, F. O. Akinkunmi and N. Giovambattista, J. Phys. Chem. B, 2014, 118, 11284–11294 CrossRef CAS PubMed.
- V. García-Melgarejo, J. Alejandre and E. Núñez-Rojas, J. Phys. Chem. B, 2020, 124, 4741–4750 CrossRef PubMed.
- R. Garnett, Bayesian Optimization, Cambridge University Press, 2023, pp. 123–158 Search PubMed.
- J. C. Spall, J. Comput. Graph Stat., 2005, 14, 889–909 CrossRef.
- T. A. Louis, J. R. Stat. Soc. Ser. B, 2018, 44, 226–233 CrossRef.
- B. Efron and D. V. Hinkley, Biometrika, 1978, 65, 457–483 CrossRef.
- A. M. Schweidtmann, D. Bongartz, D. Grothe, T. Kerkenhoff, X. Lin, J. Najman and A. Mitsos, Mathematical Programming Computation, 2021, vol. 13, pp. 553–581 Search PubMed.
- Z. Cheng, K. Wang, A. M. Tanvir, W. Shang, T. Luo, Y. Zhang, A. W. Dowling and D. B. Go, ACS Appl. Mater. Interfaces, 2024, 16, 46897–46908 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |