
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHybrid quantum algorithm for simulating real-time thermal correlation functions
Elliot C. Eklund *ab and
Nandini Ananth*b
*ab and
Nandini Ananth*b
aSchool of Chemistry, University of Sydney, NSW 2006, Australia. E-mail: elliot.eklund@sydney.edu.au
bDepartment of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, USA. E-mail: ananth@cornell.edu
First published on 29th May 2026
Abstract
We introduce a hybrid Path Integral Monte Carlo (hPIMC) algorithm to calculate real-time quantum thermal correlation functions for condensed-phase systems. The hPIMC algorithm builds on the successes of classical PIMC as a computational tool for high-dimensional system studies by exactly simulating dissipation using the Feynman-Vernon influence functional on a classical computer. This method does not rely on variational quantum algorithms and offers an alternative path for near-term quantum algorithm design. We show that by using a quantum computer to compute short-time matrix elements of the quantum propagator, hPIMC achieves an asymptotic quantum speedup over its classical counterpart, given an efficient oracle for the system Hamiltonian. To enable practical realization of hPIMC, we employ the recently developed Probabilistic Imaginary-Time Evolution (PITE) algorithm to simulate imaginary-time evolution accurately, and we introduce a novel low-depth circuit that simulates real-time evolution under the kinetic energy operator using an approximate Discrete Variable Representation (DVR). We investigate the accuracy of combining PITE with an approximate DVR by computing the position–position thermal correlation function of a proton transfer reaction on a classical computer. We estimate the circuit depth and CNOT gate count to compute short-time matrix elements for a 1D system with our method to be on the order of 105 with a space overhead of 30 qubits.
1 Introduction
Real-time thermal correlation functions (TCF) are a valuable tool for chemists as they provide insight into the dynamics of physical systems and are used to interpret spectroscopic data. However, computing real-time TCFs is difficult because one must propagate the system of interest under its time evolution operator (TEO), which is generally expensive to compute exactly using classical computers. As a result, the computation of exact, real-time TCFs is often limited to low-dimensional systems.1–4One approach to compute real-time TCFs is to use a path integral formulation in which the TEOs are split into products of many short-time TEOs that can be evaluated accurately. In the case of open quantum systems, the path integral framework includes the effects of the environment degrees of freedom exactly through the Feynman-Vernon influence functional.5–7 The numerical success of the path integral relies on accurate evaluation of the TEO through the short-time Trotter approximation that becomes exact in the limit of a very small time interval.8,9 Additionally, the path integral formulation allows one to use a host of Path Integral Monte Carlo (PIMC) methods to compute the necessary integrals of the TCF.10–13 Since their introduction, significant advances have been made in the numerical implementations of classical PIMC approaches for real-time quantum TCFs.14,15 However, this typically corresponds to an increase in the number of environment degrees of freedom with the system TEO being computationally feasible for only a relatively low dimensional model with, at most, a couple of continuous degrees of freedom coupled to multiple electronic states.
Within the field of quantum computing the situation is different. Here, several well-known quantum algorithms exist that evolve a given quantum system under an approximate TEO using a polynomial amount of resources, providing a quantum speedup over classical algorithms.16–23 In principle, one could construct an algorithm to compute real-time TCFs in which the TEO matrix elements are evaluated using these quantum algorithms. However, algorithms designed for currently available quantum computers, referred to as noisy intermediate-scale quantum (NISQ) computers, have to account for strict hardware limitations, such as small qubit counts and short coherence times.24–28 Hybrid quantum algorithms offer an alternate strategy in which inexpensive subroutines are performed classically while expensive, quantum-in-nature calculations are offloaded to a quantum computer. This allows quantum circuits to be kept shallow and mitigates error accumulation due to hardware noise.27,29–37
We propose the first hybrid PIMC (hPIMC) algorithm and show that it achieves an asymptotic speedup for computing real-time TCFs of open quantum systems assuming an efficient oracle for the system Hamiltonian. The central ideas of the hPIMC approach are to use a quantum computer to push the boundaries of the system size limitations working within the path integral representation where only short-time TEO matrix elements are required. Computing these matrix elements using relatively shallow quantum circuits can mitigate errors that result from deep circuits. The resulting hybrid Monte Carlo algorithm performs operations such as sampling random paths and evaluating acceptance/rejection criteria classically, while using a low-depth quantum circuit to perform the expensive task of computing TEO matrix elements.
The fundamental algorithm design of hPIMC is similar to a recently proposed hybrid algorithm by Layden et al. for quantum-enhanced Monte Carlo simulations.38 Specifically, they introduce a hybrid algorithm based on Markov chain Monte Carlo and Metropolis–Hastings acceptance/rejection criteria for sampling the Boltzmann distribution of an Ising spin model and demonstrate a significant speedup over classical methods. Both our method and Layden's represent a new class of hybrid algorithm design that is characterized by two important features: they are constructed starting from classical Monte Carlo-based methods, and they do not call subroutines that use variational quantum algorithms (VQA).27,30–37,39–41 As a result, these hybrid methods do not face the same limitations as VQA-based methods, such as the emergence of barren plateaus and strict limitations on circuit depth.42–45
In this paper, we introduce the hPIMC algorithm to compute real-time quantum TCFs in an arbitrary representation and show that it achieves an asymptotic speedup compared with classical PIMC. We then adapt the hPIMC algorithm to condensed-phase systems modeled by the Caldeira–Leggett (CL) Hamiltonian in position space and introduce an influence functional to capture dissipative effects.46 We use the Probabilistic Imaginary Time Evolution (PITE) algorithm47,48 for imaginary-time evolution of the TEO matrix elements. We further propose a new approach to approximate real-time evolution of the system under the kinetic energy operator using a discrete variable representation (DVR).49–51 We test the accuracy of the TEOs against an exact classical simulations of the position TCF of a proton transfer reaction and estimate the circuit depth of hPIMC.
2 Theory
We start this section by briefly reviewing the classical PIMC formalism for computing TCFs, then introduce our algorithm for hPIMC. Next, we specialize hPIMC to open quantum systems described by the Caldeira–Leggett Hamiltonian. Finally, we provide the subroutines used during hPIMC to compute TEO matrix elements on a quantum computer.The symmetrized real-time TCF between two operators, A and B, at time t for a system at equilibrium is52
 | (1) |
Within the classical PIMC framework, U(tc) and U†(tc) are both discretized into a product of N short-time TEOs, U(Δtc) and U†(Δtc), respectively, called Trotter steps. Here, we use the terms Trotter step and short-time TEO interchangeably. The discretized TCF is then
 | (2) |
 | (3) |
Eqn (3) can be expressed in the conventional classical PIMC form of an expectation value over a distribution W13
 | (4) |
 | (5) |
 | (6) |
At a high level, CAB(t) is computed using classical PIMC by the following steps. To begin, the short-time TEO matrix elements are computed by diagonalizing the matrix U(Δtc). The computational cost of this step typically limits classical PIMC to low-dimensional systems. Next, a Monte Carlo importance sampling subroutine is implemented as follows. First, a random path J is sampled from the distribution W. Second, Θ(ϕ) is evaluated using the short-time TEO matrix elements. Next, W(J) is computed and compared to the most recently accepted path using the standard Metropolis–Hastings acceptance/rejection criterion. If the path is accepted, then the sign, sgn(Θ(ϕ)) and the matrix elements of A and B are computed and accumulated in an estimator. Otherwise, the path is rejected and these quantities are not accumulated Monte Carlo sampling is repeated until a user-specified convergence criterion is met that concludes the subroutine.
We propose to implement hPIMC following the same steps described in the preceding paragraph to implement classical PIMC, with one key modification. Instead of obtaining the short-time TEO matrix elements by constructing and diagonalizing the matrix representation of U(Δtc), we use a quantum computer to directly compute individual matrix elements as they are sampled. Specifically, during each iteration of the Monte Carlo importance sampling subroutine, after J is sampled, each of the corresponding matrix elements are computed individually on the quantum computer, then Θ(ϕ) is computed by multiplying the matrix elements on a classical computer. Quantities that depend on Θ(ϕ) are also computed on the classical computer.
We now analyze the performance of classical PIMC compared with hPIMC. We derive asymptotic expressions for the computational resources needed to compute the TCF to within a specified error. Throughout, we assume that the operators A and B can be computed on a classical computer with negligible cost.
In general, we cannot compute CAB(t) exactly using either classical PIMC or hPIMC. Instead, we compute an approximation ![[C with combining tilde]](https://www.rsc.org/images/entities/i_char_0043_0303.gif) AB(t) to CAB(t) such that the error of the approximation ε = |CAB(t) − AB(t)| is bounded. For classical PIMC, ε consists of two terms,
AB(t) to CAB(t) such that the error of the approximation ε = |CAB(t) − AB(t)| is bounded. For classical PIMC, ε consists of two terms,
| ε = εMC + εC. | (7) |
The first term arises from approximating CAB(t) using Monte Carlo and depends on the Monte Carlo sampling rate of convergence. In general, for M iterations of the Monte Carlo importance sampling subroutine, this error is given by ref. 13
 | (8) |
Thus, to achieve an accuracy of εMC, we require M = O(1/εMC2) iterations. Although eqn (8) holds in the asymptotic limit, the true rate of convergence is more complicated and not easy to express since for small systems with no significant dissipation, the well-established sign problem in quantum dynamics must be considered.54 Additionally, this error will include a contribution from computing the partition function in eqn (4).
The second term results from the fact that we generally cannot compute U(tc) exactly and must instead approximate it using Ũ(tc). We show in (SI) Section I that
| εC = O(‖A‖1‖B‖1‖e−βH/2‖1εU/Z), | (9) |
| ‖U(tc) − Ũ(tc)‖1 ≤ εU | (10) |
| N = O((‖A‖1‖B‖1‖e−βH/2‖1Ω2k+1|tc|2k+1/ZεC)1/2k), | (11) |
| Ω = max{‖H1‖,‖H2‖}, | (12) |
The space and runtime complexities of approximately computing CAB(t) for a d-dimensional system and a basis set of size P using classical PIMC are
| PIMC space = O(Pd × Pd), | (13) |
| PIMC runtime = O(P3d + MN). | (14) |
The space complexity comes from constructing the matrix representation of Ũ(Δtc), and the leading order term in the runtime complexity comes from the one-time cost of diagonalizing this matrix. The second term in the runtime complexity comes from performing M Monte Carlo iterations. For each iteration, two random strings each consisting of N matrix elements, {〈ϕjk+1|Ũ(Δtc)|ϕjk〉} and {〈ϕjk+1|Ũ†(Δtc)|ϕjk〉}, referred to as the forward and backward paths, respectively, are sampled and the TCF estimators are computed. We assume that the cost to compute and store 〈ϕjk+1|A|ϕjk〉 and 〈ϕjk+1|B|ϕjk〉 is at most on the same order of Ũ(Δtc).
For hPIMC, ε consists of three terms
| ε = εC + εMC + εQ. | (15) |
The first two terms are the same as eqn (8) and (9). The third term comes from approximately computing the matrix elements 〈ϕi|Ũ(Δtc)|ϕj〉 on a quantum computer. We propose to compute the short-time TEO matrix elements using a Hadamard test as this approach often yields shallow quantum circuits.56 Given access to an oracle that prepares the states |ϕj〉 on the quantum computer and an oracle that implements Ũ(Δtc), the Hadamard test estimates matrix elements by repeatedly performing measurements on an ancilla qubit.56 Because the Hadamard test is shot-noise limited, the error of a given matrix element is  , where S is the number of circuit repetitions.57 Each path that contributes to the sum in eqn (3) consists of a product of 2N matrix elements. Propagating the error from each matrix element results in a total error for each path εP given by ref. 58
, where S is the number of circuit repetitions.57 Each path that contributes to the sum in eqn (3) consists of a product of 2N matrix elements. Propagating the error from each matrix element results in a total error for each path εP given by ref. 58
 | (16) |
 | (17) |
 | (18) |
Because there are M iterations, each contributing an error εP, we have that εQ = O(MεP).
All three errors that contribute to ε are controllable. To decrease εC, we increase the number of Trotter steps N, to decrease εMC, we increase M and run more Monte Carlo iterations, and to decrease εQ, we increase S and run more repetitions of the Hadamard test circuit. Hence, the total error made by hPIMC can be systematically reduced by using more classical and quantum computational resources.
The space and runtime complexities of hPIMC are
| hPIMC space = O(dn) + Anc.(Ũ) | (19) |
| hPIMC runtime = O(MNSQ(Ũ)). | (20) |
The first term of the space complexity is the result of using n = log(P) qubits to represent a given |ϕj〉 on the quantum computer. Because we assume a d-dimensional system, we use d quantum registers each consisting of n qubits. The second term represents the number of ancilla qubits needed to implement the oracles used in the Hadamard test circuit. The runtime complexity comes from performing M Monte Carlo iterations, in which 2N matrix elements are computed per iteration. Each matrix element uses S repetitions of the Hadamard test circuit, and Q(Ũ) represents the runtime cost to implement the oracles used in the Hadamard test circuit. We make the same assumption about the cost to compute matrix elements of A and B as for classical PIMC.
The efficiency of hPIMC depends on the efficiency of the oracles used by the Hadamard test. For many chemical systems, efficient implementations of Ũ(Δtc) are known.16–23,30,36,47,48,59–62 In Sections 2.2 and 2.3, we provide an efficient algorithm to implement Ũ(Δtc) for a 1D proton transfer reaction modeled by a symmetric double-well potential. Assuming that Q(Ũ) and Anc.(Ũ) scale polynomially, we see that hPIMC scales exponentially better than classical PIMC in terms of space and runtime complexity.
The performance of hPIMC can be improved by storing TEO matrix elements in a classical look-up table and querying the table each Monte Carlo iteration. If the table contains a queried matrix element, the value is pulled from the table instead of recomputing the matrix element on the quantum computer. Thus, the distribution between classical and quantum resources becomes increasingly more classical as the calculation progresses and the table fills up. This method is only successful when a small subset of matrix elements are needed to converge the Monte Carlo simulation. If all P2d matrix elements are required, then an exponential amount of memory will be required to store the table. In the next section, we argue that this situation is avoided for condensed-phase systems at finite temperature when working in a position space representation.
2.1 Open quantum systems
Much of the success of classical PIMC has been in the context of dissipative systems at finite temperature where the rapid damping of quantum system oscillations by coupling to a large number of environment degrees of freedom significantly reduces the dynamical sign problem. This motivates us to adapt hPIMC for condensed-phase dynamics described by the CL Hamiltonian using a position space representation.The CL Hamiltonian describes a primary system consisting of η modes in d physical dimensions bilinearly coupled to an isotropic bath approximated by f harmonic modes,46
 | (21) |
 is the system sub-Hamiltonian shifted along the adiabatic path,
is the system sub-Hamiltonian shifted along the adiabatic path,
 | (22) |
 | (23) |
 | (24) |
The bath sub-Hamiltonian is modified to include the system-bath coupling term and is given by ref. 2
 | (25) |
It has been shown that for this choice of sub-Hamiltonians the error made by approximating the TEO as
 | (26) |
Expressing the CL Hamiltonian this way, in a position space representation, enables all terms involving contributions from the bath to be collected and integrated out analytically, to obtain the Feynman–Vernon influence functional.1–3,53,63 Assuming A and B are functions only of position, the TCF of eqn (21) becomes
 | (27) |
 | (28) |
 | (29) |
Integrating out the bath degrees of freedom via the influence functional results in large computational savings. Before integration, the integral in eqn (27) is 2Nd(η + f) dimensional. Afterwards, the dimensionality is reduced to 2Ndη. This reduction in dimensionality comes at the cost of evaluating the influence functional. For most spectral densities, I(x) can be evaluated analytically or numerically on a classical computer at a cost of O(dN2), which tends to be negligible compared to the cost of computing matrix elements.65,66 Because f ≫ η is typically required to accurately model the bath using eqn (21), this reduction in dimensionality generally outweighs the cost of computing I(x). The influence functional also mediates dissipation from the system to the bath, damping oscillatory phases in the integrand and thereby alleviating the dynamical sign problem, which improves Monte Carlo convergence.
We can compute eqn (27) using either classical PIMC or hPIMC by modifying eqn (5):
 | (30) |
| Θ(x) = Pf(xf)Pb(xb). | (31) |
Algorithmically, the procedure to compute eqn (30) using classical PIMC is the same as for eqn (5), as described in the paragraph following eqn (5), except that now the product A(xN)B(x0)I(x) includes the influence functional that is computed and accumulated in the estimator whenever a path is accepted. To compute eqn (30) with hPIMC, the same procedure as for classical PIMC is followed, but the TEO matrix elements of Θ(x) are evaluated using a quantum computer.
For bound systems at finite temperature, we only need to sample matrix elements that connect nearby points rather than sample all possible displacements in position space, leading to enhanced sampling efficiency. This is because, at finite temperatures, the TEO matrix elements contain an imaginary time contribution that causes their magnitude, as well as the magnitudes of Pf(xf) and Pb(xb), to decay exponentially with increasing energy. Additionally, for many bound systems, localized states tend to be lower in energy, causing Pf(xf) and Pb(xb) to be relatively large, while delocalized states tend to be higher in energy, causing Pf(xf) and Pb(xb) to be relatively small.63 As a result, only paths containing TEO matrix elements that connect points close in position space contribute significantly to eqn (27).
We note that after the arXiv version of this paper was published,67 a path integral quantum algorithm that leverages the influence functional formalism has been reported for spin-boson systems that, unlike the present work, cannot be used in the context of Hamiltonians where the system includes continuous degrees of freedom.68
2.2 Quantum circuit
We can approximate the continuous integrals in eqn (27) as discrete sums over a finite-width grid of evenly spaced grid points. In one dimension, let L be the total length of the grid and D the number of grid points. Any position on the grid is given by , where Δx = L/D, j ∈ G, and G is the set of integers G = [0, D − 1]. We represent the grid on a quantum computer by mapping j to the computational basis:
, where Δx = L/D, j ∈ G, and G is the set of integers G = [0, D − 1]. We represent the grid on a quantum computer by mapping j to the computational basis:| |j〉 = |bjn−1〉 ⊗ |bjn−2〉 ⊗ …|bj0〉, | (32) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D⌉ (the ceiling log used here returns the least integer greater than or equal to base 2 logD). Here, |j〉 represents a single quantum register. For ηd dimensions, each position coordinate is mapped to its own quantum register and the overall state is the direct product of all ηd registers. The constant offset, −L/2, and grid spacing, Δx, are absorbed into the implementation of the potential energy.69 Because we are mapping positions on a grid directly to the computational basis, each register can be efficiently initialized with a single layer of Pauli-X gates.
D⌉ (the ceiling log used here returns the least integer greater than or equal to base 2 logD). Here, |j〉 represents a single quantum register. For ηd dimensions, each position coordinate is mapped to its own quantum register and the overall state is the direct product of all ηd registers. The constant offset, −L/2, and grid spacing, Δx, are absorbed into the implementation of the potential energy.69 Because we are mapping positions on a grid directly to the computational basis, each register can be efficiently initialized with a single layer of Pauli-X gates.
We compute the short-time TEO matrix elements 〈xk+1|U(Δtc)|xk〉, where now  , on a quantum computer using a Hadamard test.56 A circuit diagram that implements the Hadamard test is given in Fig. 1. Within the Hadamard test, we split U(Δtc) into real- and imaginary-time components,
, on a quantum computer using a Hadamard test.56 A circuit diagram that implements the Hadamard test is given in Fig. 1. Within the Hadamard test, we split U(Δtc) into real- and imaginary-time components,
 | (33) |
 | ||
Fig. 1 Hadamard test to compute TEO matrix elements. Here, H is a Hadamard gate, Uxj is a unitary that prepares the state |xj〉, and U(Δtc) performs time evolution under  for an amount Δtc. To measure the real or imaginary component of the matrix element, A is either set to identity or S†, respectively, where S is the phase gate. for an amount Δtc. To measure the real or imaginary component of the matrix element, A is either set to identity or S†, respectively, where S is the phase gate. | ||
For an initial state |ψ〉 and a single ancilla qubit, the PITE algorithm takes as input a circuit that executes real-time evolution,  , and approximates evolution in imaginary-time by the unitary UPITE such that47,48
, and approximates evolution in imaginary-time by the unitary UPITE such that47,48
 | (34) |
 , s1 = m0/γ, and m0 is a real parameter. The PITE quantum circuit diagram is given in Fig. 2. When the ancillary qubit is measured in the state |0〉, |ψ〉 is evolved in imaginary-time by a first-order approximation to
, s1 = m0/γ, and m0 is a real parameter. The PITE quantum circuit diagram is given in Fig. 2. When the ancillary qubit is measured in the state |0〉, |ψ〉 is evolved in imaginary-time by a first-order approximation to  .
.
 | ||
Fig. 2 PITE quantum circuit diagram for evolving an input state |ψ〉 in imaginary time by Δβ/2. The gate W implements  , Rz(2θ0) is a rotation about the z-axis of the Bloch sphere by an angle 2θ0, where , Rz(2θ0) is a rotation about the z-axis of the Bloch sphere by an angle 2θ0, where  . .  and m0 is an adjustable real parameter with conditions 0 < m0 < 1 and and m0 is an adjustable real parameter with conditions 0 < m0 < 1 and  . U(τ) performs evolution under . U(τ) performs evolution under  for time for time  , where s1 = m0/γ. , where s1 = m0/γ. | ||
We note that the probability of successfully measuring |0〉 increases for smaller values of τ. PITE was originally designed for finding the ground state of an input Hamiltonian where successive applications of the circuit are needed to reach larger τ, each of which lowers the overall probability of success. However, in hPIMC, τ is inherently small to ensure that eqn (26) is a good approximation. Thus, we will only require a single application of PITE per TEO matrix element, and the additional overhead from calling PITE until a successful outcome is measured is minimal.
For evolution in real-time, we approximate the TEO using a second-order Trotter splitting,8
 | (35) |
2.3 DVR approximation
We propose to evaluate time evolution under the kinetic energy operator with a discrete variable representation (DVR). The kinetic energy operator, T, can be expressed using a sinc basis set as49
 | (36) |
| TDVRα = I⊗d1 ⊗ …I⊗dα−1 ⊗ DVR⊗dα ⊗ I⊗dα+1 ⊗ … ⊗ I⊗dη. | (37) |
 | (38) |
Ordinarily, one diagonalizes the DVR Hamiltonian, HDVR = TDVR + V, and performs dynamics in the basis of DVR eigenvectors. Instead, we propose to approximate time evolution under the kinetic energy operator in the position basis by directly applying e−iTDVRΔt to a state |ψ〉. This approximation is justified because, in the limit that Δx goes to zero, the sinc basis becomes the position basis.50 This allows us to avoid having to diagonalize HDVR. The resulting unitary is given by
 | (39) |
We present results in the next section that validate the use of this approximation.
In general, we cannot efficiently implement e−iDVRαΔt on a quantum computer because DVRα is dense. At the same time, we can see from eqn (38) that the matrix elements decrease in magnitude moving away from the main diagonal, and should eventually become small enough to safely neglect. In SI Section II, we prove that this is indeed the case and find that for a given error tolerance δ, DVRα can be made sparse by neglecting all diagonals, ν, where  , and
, and  is given by
is given by
 | (40) |
We can then simulate time evolution under DVRα using a first-order Trotter splitting about the diagonals of DVRα:
 | (41) |
 | (42) |
In SI Section V, we construct a quantum circuit that performs time evolution under eqn (42) using the sparse matrix simulation approach detailed in Aharonov and Ta-Shma.70 The main idea of this approach is to use the fact that 1-sparse Hermitian matrices can always be represented in block-diagonal form where each block acts on a two-dimensional subspace of the overall Hilbert space. Time evolution can be performed by constructing an oracle that identifies each subspace, then applying the appropriate two-dimensional time evolution operator for a given subspace. We show how to construct the necessary oracles and two-dimensional time evolution operators for eqn (42) in SI Section V, and demonstrate that simulating a single diag(DVRα,ν)σ for a 1D system can be performed with O(n) ancilla qubits, O(nlog(n)) CNOT gates, and a circuit depth of O(n).
3 Results
Our proposed method to simulate the system TEO involves several approximations: imaginary-time evolution is approximated by PITE, real-time evolution is approximated using a second-order Trotter splitting, the kinetic energy operator is approximated using a DVR, the DVR is approximated using a truncated number of diagonals, and the Hadamard test is approximated using a finite number of circuit repetitions. Although the error from some of these approximations can be analyzed individually, rigorously studying the cumulative error is not straightforward.Instead, we investigate the resulting error numerically by computing the position–position TCF of a 1D model proton transfer reaction using the approach outlined above to compute TEO matrix elements and compare the approximate results with exact results obtained by diagonalizing the DVR matrix. We model the donor and acceptor sites of the reaction using a standard symmetric double-well potential:
 | (43) |
Real-time evolution is approximated with a second-order Trotter splitting, as in eqn (35), and imaginary-time evolution is approximated with a first-order Trotter splitting to replicate PITE.
Our focus is to study how the error introduced by approximating TEO matrix elements using the algorithm proposed in the previous section propagates through the calculation of the overall TCF. To isolate the error from approximating the TEO matrix elements, we use quadrature on a classical computer to compute all TCF integrals in eqn (1). Computing the TCF this way eliminates the error that would be introduced from Monte Carlo sampling or using a finite number of circuit repetitions. The results presented here are equivalent to what one would obtain from a fully converged Monte Carlo and a fully converged Hadamard test using noiseless hardware.
Initially, we used eqn (42) to approximate evolution under the kinetic energy operator for both real- and imaginary-time. However, we found that N > 100 was needed to achieve accurate results, which is relatively large. Instead, we found that using eqn (42) for imaginary-time only, and using a standard Quantum Fourier Transform (QFT) approach to diagonalize the kinetic energy operator in real-time achieved the same accuracy with a smaller N. Because a smaller N results in a shorter hPIMC runtime, we used this mixed approach for evolution under the kinetic energy operator to obtain the results presented in Fig. 3. Interestingly, we also found that using QFT for both real- and imaginary-time evolution required an even larger N than using eqn (42) for both, and that the results do not systematically improve with increasing N as one would expect.
 | ||
Fig. 3 TCF of symmetric double-well potential. We evaluate real-time TEO matrix elements using a second-order Trotter splitting about T and V, and QFT for evolution under the kinetic energy operator. For imaginary-time evolution, we use a first-order Trotter splitting about T and V, and eqn (42) for evolution under the kinetic energy operator. For each plot, m = 1836 a.u., 1/β = 350 K, ωb = 500 cm−1, V0 = 1500 cm−1, and L = 30 a.u. In (a) we vary the number of Trotter steps N, (b) we approximate the DVR by neglecting all diagonals  , and in (c) we vary the number of grid points, D = 2n. In (d), we plot the TCF using optimized parameters so that the resource requirements are minimized while maintaining reasonable error. Each plot is normalized by the magnitude of its maximum value. , and in (c) we vary the number of grid points, D = 2n. In (d), we plot the TCF using optimized parameters so that the resource requirements are minimized while maintaining reasonable error. Each plot is normalized by the magnitude of its maximum value. | ||
In Fig. 3, panel (a), we see that for sufficiently large N the approximate results match the exact results. As N decreases, the error from Trotter splitting increases, and we see that the amplitude of the TCF begins to deteriorate, although the frequency of the oscillations remains accurate. In panel (b), we approximate T by neglecting all diagonals ν, where  from DVRα. We see that the approximate results lie directly on top of the exact results up to
from DVRα. We see that the approximate results lie directly on top of the exact results up to  . In other words, 87.5% of the diagonals in DVRα can be neglected without impacting the results of the calculation. This is important because each additionally neglected diagonal results in a shallower circuit.
. In other words, 87.5% of the diagonals in DVRα can be neglected without impacting the results of the calculation. This is important because each additionally neglected diagonal results in a shallower circuit.
In panel (c), we compare approximate and exact results for different numbers of grid points, D = 2n. For n = 8 and 7, the difference is indistinguishable. At n = 6, the approximate results slightly deviate, and for n = 5 the grid spacing becomes too large and all features of the TCF are lost. In panel (d), we pick a set of parameters that is optimized to use minimal computational resources: N = 40, n = 6, and  . We see that our method of computing the TEO matrix elements is robust to the cumulative error resulting from varying multiple parameters simultaneously. These results are encouraging and demonstrate that complex-time evolution can be well approximated by directly exponentiating DVRα to simulate imaginary-time evolution and simulating real-time evolution using a QFT approach. Further, we see that the Trotter error inherent in eqn (42) and introduced through PITE is manageable for N = 40, and only 6 qubits and
. We see that our method of computing the TEO matrix elements is robust to the cumulative error resulting from varying multiple parameters simultaneously. These results are encouraging and demonstrate that complex-time evolution can be well approximated by directly exponentiating DVRα to simulate imaginary-time evolution and simulating real-time evolution using a QFT approach. Further, we see that the Trotter error inherent in eqn (42) and introduced through PITE is manageable for N = 40, and only 6 qubits and  diagonals are needed to achieve results that recover the amplitude and frequency of the position TCF of a proton transfer model.
diagonals are needed to achieve results that recover the amplitude and frequency of the position TCF of a proton transfer model.
In SI Section VI, we estimate the resources needed to simulate time evolution under eqn (43) on a quantum computer. Our approach uses the same method described by Beneti and Strini69 for simulating quadratic potentials. We find that simulating eqn (43) for a 1D system can be performed with a depth and CNOT gate count of O(n4), and a constant number of ancilla qubits. Because time evolution under the kinetic energy operator using the DVR approximation and conventional QFT approach can be performed in  and O(n2), respectively, we approximate the cost of the hPIMC circuit as the cost of simulating the double-well potential. Using the constant factor scalings that we derive in SI Section VI, we estimate that a 1D proton transfer reaction using n = 6 qubits can be simulated with a depth and CNOT count of 2.96 × 105, and 24 ancilla qubits. As a comparison, accounting for the DVR circuit only increases the CNOT count by 4.2 × 104, and the depth by 3.0 × 104 for
and O(n2), respectively, we approximate the cost of the hPIMC circuit as the cost of simulating the double-well potential. Using the constant factor scalings that we derive in SI Section VI, we estimate that a 1D proton transfer reaction using n = 6 qubits can be simulated with a depth and CNOT count of 2.96 × 105, and 24 ancilla qubits. As a comparison, accounting for the DVR circuit only increases the CNOT count by 4.2 × 104, and the depth by 3.0 × 104 for  .
.
4 Discussion
Our results show that for a 1D proton transfer reaction, computing the TEO matrix elements to be used in hPIMC requires circuits with depth and CNOT gate count of 2.96 × 105. For context, this is comparable to other recently proposed NISQ algorithms which estimate O(107) CNOT gates to simulate a spin-boson system using Trotterization.39 For currently available quantum computers with two-qubit error rates on the order of 10−3, a circuit of depth O(105) is likely infeasible without significant error accumulation.71 For reference, an error rate of 10−3 means that we expect on average one error to occur for every 103 two-qubit operations.71 Thus, we can expect on average O(102) errors to occur for a circuit consisting of O(105) CNOT gates. However, there are many strategies to further reduce the costs of the present algorithm that we will explore moving forward.The dominant cost to compute the TEO for a proton transfer reaction comes from our method of computing a double-well potential, which scales as O(n4). An alternative approach is to first compute x2 into a new register, then evolve under the quadratic term via phase kickback.19,72 Next, the x4 term can be computed into an additional register using quantum arithmetic to square x2, and then evolving under the quartic term via phase kickback again. The ensuing algorithm would be O(n2). The circuit we propose in SI Section VI does not explicitly include coupling to the bath degrees of freedom. However, adding this effect does not increase the cost of the circuit. For linear, isotropic couplings, we see from eqn (22) that incorporating coupling to the bath adds a term that is quadratic in the system position coordinates. This term can be grouped with the other quadratic terms and simulated in one go without additional resources.
Our circuit for time evolution under the DVR kinetic energy approximation is more encouraging with 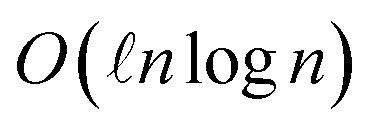 and
and  scaling in terms of CNOT gates and depth, respectively. We have demonstrated in Fig. 3 and SI Section II that
scaling in terms of CNOT gates and depth, respectively. We have demonstrated in Fig. 3 and SI Section II that  can be made quite small while retaining accurate results. We point out that the numerical study in Section 3 was for a proton. However, from eqn (40), we see that for a given δ,
can be made quite small while retaining accurate results. We point out that the numerical study in Section 3 was for a proton. However, from eqn (40), we see that for a given δ,  can be made smaller for particles with larger mass. Thus, we expect our approach to perform better for heavier particles. Despite the relatively low asymptotic scaling, the associated constant factors found in SI Section V are quite high. This is because many of the primitive circuits used to construct the circuit are not optimal. Improvements can be made by replacing them with the currently best known methods. Additionally, optimizing the circuit over all
can be made smaller for particles with larger mass. Thus, we expect our approach to perform better for heavier particles. Despite the relatively low asymptotic scaling, the associated constant factors found in SI Section V are quite high. This is because many of the primitive circuits used to construct the circuit are not optimal. Improvements can be made by replacing them with the currently best known methods. Additionally, optimizing the circuit over all  of the diag(DVRα, ν) operators simultaneously rather than on a case-by-case basis could make it easier to detect and remove redundant operations.
of the diag(DVRα, ν) operators simultaneously rather than on a case-by-case basis could make it easier to detect and remove redundant operations.
In Section 3, we report that the best results were obtained by simulating evolution under the kinetic energy operator using a QFT approach for real time and DVR for imaginary time. We suggest using this pairing when implementing hPIMC as it performed better than using QFT or DVR for both real and imaginary time evolution, or DVR for real time and QFT for imaginary time. Further investigation is needed to understand why this particular combination gives the best results.
hPIMC experiences a steep penalty from the error εS of computing matrix elements using a Hadamard test. This is because εS is compounded both from multiplying TEO matrix elements together, then adding these products together to compute AB(t). Ideally, we would like εS to be as small as possible, but this comes at the expense of more circuit repetitions. We note that several error mitigation schemes have been designed to help offset the effect of noise and decoherence that contributes to the error of performing NISQ calculations.29,73–75 Applying these schemes to hPIMC will help to keep εS as small as possible. Although hPIMC was originally conceived for NISQ computing, it is worth considering its application for near-term fault tolerant quantum computing. In addition, coherent techniques such as amplitude estimation, which scale more favorably with the error of the computation, can be explored.76 This would significantly reduce the overall error from performing hPIMC.
We do not estimate the number of shots needed to converge the Hadamard test to a given accuracy in this work. Although hPIMC performs asymptotically better than classical PIMC, this estimate is needed to conduct a more rigorous comparison at the level of constant factors. Taking this into consideration, it is likely that the turnover point where hPIMC begins to outperform classical PIMC occurs for higher-dimensional systems. We leave this analysis to future work.
An important distinction between hPIMC and VQA-based hybrid approaches is that hPIMC is non-variational and therefore avoids trainability issues such as barren plateaus.42 Additionally, the dominant sources of systematic error in hPIMC, Trotterization and the approximation of the DVR operator, are systematically controllable. Decreasing Δtc reduces the Trotter error, and including more diagonals in eqn (41) reduces the error associated with the DVR approximation. For VQAs, there is typically no controllable parameter that guarantees convergence to the exact result because the error depends on the expressiveness of the chosen ansatz.
The CL Hamiltonian described in this paper involves a single electronic state potential energy surface. However, hPIMC can easily be extended to nonadiabatic systems that employ a diabatic representation for multiple potential energy surfaces.30 We expect hPIMC to provide a useful tool for studies of condensed-phase adiabatic systems as well as nonadiabatic systems in which the electronic state transitions are strongly coupled to multiple vibrational degrees of freedom. Extending such systems to multiple dimensions challenges current classical methodology.
5 Conclusion
In this initial paper, we propose and develop the theory of hPIMC for computing real-time TCFs. hPIMC is based on Monte Carlo simulation and does not use variational methods to perform simulations. We also propose a new method to perform time evolution under the kinetic energy operator using a DVR approximation. We provide an efficient algorithm for our DVR approach with scaling in CNOT count and
scaling in CNOT count and  in circuit depth. We perform classical numerical studies on a 1D proton transfer reaction and show that accurate results can be obtained using a DVR approximation. We estimate that the space overhead needed to simulate the DVR circuit is low, using only 6 state qubits and 24 ancilla qubits.
in circuit depth. We perform classical numerical studies on a 1D proton transfer reaction and show that accurate results can be obtained using a DVR approximation. We estimate that the space overhead needed to simulate the DVR circuit is low, using only 6 state qubits and 24 ancilla qubits.
Moving forward, additional algorithm optimizations and tests are needed to enable hPIMC for use on currently available NISQ machines. We identify several opportunities to optimize our circuit and reduce the circuit depth and CNOT gate count from O(105). In particular, error mitigation schemes, optimal circuit primitives, and an O(n2) algorithm for simulating the double-well potential have all yet to be explored. We are optimistic that these improvements will allow for the study of higher-dimensional condensed-phase systems via hPIMC that are currently inaccessible with classical algorithms.
Author contributions
EE and NA conceptualized the main algorithm and wrote and edited the manuscript. EE wrote the software and prepared figures for visualization. NA supervised the project and secured funding.Conflicts of interest
There are no conflicts to declare.Data availability
The code used to simulate a proton transfer reaction, create Fig. 3, and analyze the DVR error in supplementary information (SI) Fig. 1 can be found at https://github.com/AnanthGroup/hPIMC_figures with DOI https://doi.org/10.5281/zenodo.16900433. Supplementary information is available. See DOI: https://doi.org/10.1039/d5dd00381d.Acknowledgements
The authors acknowledge funding for this work through NSF EAGER: QCA-QSA grant number CHE-2038005. The authors thank Peter McMahon for insightful discussion and guidance.References
- M. Topaler and N. Makri, J. Chem. Phys., 1992, 97, 9001–9015 CrossRef CAS.
- M. Topaler and N. Makri, Chem. Phys. Lett., 1993, 210, 285–293 CrossRef CAS.
- M. Topaler and N. Makri, Chem. Phys. Lett., 1993, 210, 448–457 CrossRef CAS.
- F. Matzkies and U. Manthe, J. Chem. Phys., 1997, 106, 2646–2653 CrossRef CAS.
- R. P. Feynman, Rev. Mod. Phys., 1948, 20, 367–387 CrossRef.
- R. Feynman and A. H. Q. Mechanics, Lect. Notes Phys., 1979, 106, 64 Search PubMed.
- R. P. Feynman, Int. J. Theor. Phys., 1982, 21, 467–488 CrossRef.
- H. F. Trotter, Proc. Am. Math. Soc., 1959, 10, 545–551 CrossRef.
- H. De Raedt and B. De Raedt, Phys. Rev. A, 1983, 28, 3575–3580 CrossRef.
- B. J. Berne and D. Thirumalai, Annu. Rev. Phys. Chem., 1986, 37, 401–424 CrossRef CAS.
- V. Zamalin and G. Norman, USSR Comput. Math. & Math. Phys., 1973, 13, 169–183 CrossRef.
- B. Bernu and D. M. Ceperley, Quantum simulations of complex many-body systems: From theory to algorithms, 2002, vol. 10, pp. 51–61 Search PubMed.
- M. E. Tuckerman, Statistical Mechanics: Theory and Molecular Simulation, Oxford University Press, 2023 Search PubMed.
- N. Makri, Annu. Rev. Phys. Chem., 1999, 50, 167–191 CrossRef CAS PubMed.
- S. Kundu and N. Makri, Annu. Rev. Phys. Chem., 2022, 73, 349–375 CrossRef CAS.
- N. C. Rubin, D. W. Berry, A. Kononov, F. D. Malone, T. Khattar, A. White, J. Lee, H. Neven, R. Babbush and A. D. Baczewski, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2317772121 CrossRef CAS PubMed.
- R. Babbush, W. J. Huggins, D. W. Berry, S. F. Ung, A. Zhao, D. R. Reichman, H. Neven, A. D. Baczewski and J. Lee, Nat. Commun., 2023, 14, 4058 CrossRef CAS PubMed.
- Y. Su, D. W. Berry, N. Wiebe, N. Rubin and R. Babbush, PRX Quantum, 2021, 2, 040332 CrossRef.
- I. Kassal, S. P. Jordan, P. J. Love, M. Mohseni and A. Aspuru-Guzik, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18681–18686 Search PubMed.
- M. E. S. Morales, P. C. S. Costa, G. Pantaleoni, D. K. Burgarth, Y. R. Sanders and D. W. Berry, Quant. Inf. Comput., 2025, 25, 1–35 Search PubMed.
- G. H. Low and I. L. Chuang, Quantum, 2019, 3, 163 CrossRef.
- E. Campbell, Phys. Rev. Lett., 2019, 123, 070503 CrossRef CAS PubMed.
- D. Motlagh, R. A. Lang, P. Jain, J. A. Campos-Gonzalez-Angulo, T. Zeng, A. Aspuru-Guzik and J. M. Arrazola, Quantum Algorithm for Vibronic Dynamics: Case Study on Singlet Fission Solar Cell Design, arXiv, 2025, preprint, arXiv:2411.13669, DOI:10.48550/arXiv.2411.13669.
- J. Preskill, Quantum, 2018, 2, 79 CrossRef.
- K. Bharti, A. Cervera-Lierta, T. H. Kyaw, T. Haug, S. Alperin-Lea, A. Anand, M. Degroote, H. Heimonen, J. S. Kottmann, T. Menke, W.-K. Mok, S. Sim, L.-C. Kwek and A. Aspuru-Guzik, Rev. Mod. Phys., 2022, 94, 015004 CrossRef CAS.
- F. D. Malone, R. M. Parrish, A. R. Welden, T. Fox, M. Degroote, E. Kyoseva, N. Moll, R. Santagati and M. Streif, Chem. Sci., 2022, 13, 3094–3108 RSC.
- X. Xing, A. Gomez Cadavid, A. F. Izmaylov and T. V. Tscherbul, J. Phys. Chem. Lett., 2023, 14, 6224–6233 CrossRef CAS PubMed.
- Y. Wang, E. Mulvihill, Z. Hu, N. Lyu, S. Shivpuje, Y. Liu, M. B. Soley, E. Geva, V. S. Batista and S. Kais, J. Chem. Theory Comput., 2023, 19, 4851–4862 CrossRef CAS PubMed.
- S. Endo, Z. Cai, S. C. Benjamin and X. Yuan, J. Phys. Soc. Jpn., 2021, 90, 032001 CrossRef.
- P. J. Ollitrault, G. Mazzola and I. Tavernelli, Phys. Rev. Lett., 2020, 125, 260511 CrossRef CAS PubMed.
- P. J. Ollitrault, A. Miessen and I. Tavernelli, Acc. Chem. Res., 2021, 54, 4229–4238 CrossRef CAS PubMed.
- P. J. Ollitrault, S. Jandura, A. Miessen, I. Burghardt, R. Martinazzo, F. Tacchino and I. Tavernelli, Quantum, 2023, 7, 1139 CrossRef.
- J. R. McClean, J. Romero, R. Babbush and A. Aspuru-Guzik, New J. Phys., 2016, 18, 023023 CrossRef.
- D. Wang, O. Higgott and S. Brierley, Phys. Rev. Lett., 2019, 122, 140504 Search PubMed.
- A. Peruzzo, J. McClean, P. Shadbolt, M.-H. Yung, X.-Q. Zhou, P. J. Love, A. Aspuru-Guzik and J. L. O'Brien, Nat. Commun., 2014, 5, 4213 CrossRef CAS PubMed.
- C.-K. Lee, C.-Y. Hsieh, S. Zhang and L. Shi, J. Chem. Theory Comput., 2022, 18, 2105–2113 CrossRef CAS PubMed.
- Y.-X. Yao, N. Gomes, F. Zhang, C.-Z. Wang, K.-M. Ho, T. Iadecola and P. P. Orth, PRX Quantum, 2021, 2, 030307 CrossRef.
- D. Layden, G. Mazzola, R. V. Mishmash, M. Motta, P. Wocjan, J.-S. Kim and S. Sheldon, Nature, 2023, 619, 282–287 CrossRef CAS PubMed.
- A. Miessen, P. J. Ollitrault and I. Tavernelli, Phys. Rev. Res., 2021, 3, 043212 CrossRef CAS.
- M.-H. Yung, J. Casanova, A. Mezzacapo, J. McClean, L. Lamata, A. Aspuru-Guzik and E. Solano, Sci. Rep., 2014, 4, 3589 CrossRef PubMed.
- F. Benfenati, G. Mazzola, C. Capecci, P. K. Barkoutsos, P. J. Ollitrault, I. Tavernelli and L. Guidoni, J. Chem. Theory Comput., 2021, 17, 3946–3954 CrossRef CAS.
- S. Wang, E. Fontana, M. Cerezo, K. Sharma, A. Sone, L. Cincio and P. J. Coles, Nat. Commun., 2021, 12, 6961 CrossRef CAS PubMed.
- S. H. Sack, R. A. Medina, A. A. Michailidis, R. Kueng and M. Serbyn, PRX Quantum, 2022, 3, 020365 CrossRef.
- M. Cerezo, A. Arrasmith, R. Babbush, S. C. Benjamin, S. Endo, K. Fujii, J. R. McClean, K. Mitarai, X. Yuan, L. Cincio and P. J. Coles, Nat. Rev. Phys., 2021, 3, 625–644 CrossRef.
- A. Tikku and I. H. Kim, Circuit depth versus energy in topologically ordered systems, arXiv, 2022, preprint, arXiv:2210.06796, DOI:10.48550/arXiv.2210.06796.
- A. O. Caldeira and A. J. Leggett, Phys. Rev. Lett., 1981, 46, 211–214 CrossRef.
- T. Kosugi, Y. Nishiya, H. Nishi and Y.-i. Matsushita, Phys. Rev. Res., 2022, 4, 033121 CrossRef CAS.
- H. Nishi, K. Hamada, Y. Nishiya, T. Kosugi and Y.-i. Matsushita, Phys. Rev. Res., 2023, 5, 043048 Search PubMed.
- J. C. Light and T. Carrington Jr, Adv. Chem. Phys., 2000, 114, 263–310 CrossRef.
- R. G. Littlejohn, M. Cargo, T. Carrington, K. A. Mitchell and B. Poirier, J. Chem. Phys., 2002, 116, 8691–8703 Search PubMed.
- D. T. Colbert and W. H. Miller, J. Chem. Phys., 1992, 96, 1982–1991 CrossRef CAS.
- W. H. Miller, S. D. Schwartz and J. W. Tromp, J. Chem. Phys., 1983, 79, 4889–4898 CrossRef CAS.
- M. Topaler and N. Makri, J. Chem. Phys., 1994, 101, 7500–7519 Search PubMed.
- S. Caratzoulas and P. Pechukas, J. Chem. Phys., 1996, 104, 6265–6277 Search PubMed.
- D. W. Berry, G. Ahokas, R. Cleve and B. C. Sanders, Commun. Math. Phys., 2007, 270, 359–371 CrossRef.
- D. Aharonov, V. Jones and Z. Landau, Algorithmica, 2009, 55, 395–421 Search PubMed.
- S. Polla, G.-L. R. Anselmetti and T. E. O'Brien, Phys. Rev. A, 2023, 108, 012403 CrossRef CAS.
- J. R. Taylor, An Introduction to Error Analysis: The Study of Uncertainties in Physical Measurements, University Science Books, Sausalito, CA, 2nd edn, 1997 Search PubMed.
- Y. Cao, J. Romero, J. P. Olson, M. Degroote, P. D. Johnson, M. Kieferová, I. D. Kivlichan, T. Menke, B. Peropadre, N. P. D. Sawaya, S. Sim, L. Veis and A. Aspuru-Guzik, Chem. Rev., 2019, 119, 10856–10915 CrossRef CAS PubMed.
- R. Babbush, N. Wiebe, J. McClean, J. McClain, H. Neven and G. K.-L. Chan, Phys. Rev. X, 2018, 8, 011044 CAS.
- R. Babbush, D. W. Berry, J. R. McClean and H. Neven, npj Quantum Inf., 2019, 5, 92 CrossRef.
- A. M. Childs, J. Leng, T. Li, J.-P. Liu and C. Zhang, Quantum, 2022, 6, 860 Search PubMed.
- N. Makri, J. Phys. Chem. A, 1998, 102, 4414–4427 CrossRef CAS.
- N. Makri, J. Math. Phys., 1995, 36, 2430–2457 CrossRef.
- K. Allinger and M. A. Ratner, Phys. Rev. A, 1989, 39, 864–880 Search PubMed.
- T. C. Allen, P. L. Walters and N. Makri, J. Chem. Theory Comput., 2016, 12, 4169–4177 CrossRef CAS PubMed.
- E. C. Eklund and N. Ananth, Hybrid Quantum Algorithm for Simulating Real-Time Thermal Correlation Functions, arXiv, 2025, preprint, arXiv:2405.19599, DOI:10.48550/arXiv.2405.19599.
- A. Seneviratne, P. L. Walters and F. Wang, J. Chem. Phys., 2025, 162, 194101 CrossRef CAS PubMed.
- G. Beneti and G. Strini, Am. J. Phys., 2008, 76, 657–662 CrossRef.
- D. Aharonov and A. Ta-Shma, Proceedings of the Thirty-Fifth Annual ACM Symposium on Theory of Computing, 2003, pp. 20–29 Search PubMed.
- D. C. McKay, I. Hincks, E. J. Pritchett, M. Carroll, L. C. G. Govia and S. T. Merkel, Benchmarking Quantum Processor Performance at Scale, 2023 Search PubMed.
- R. Cleve, A. Ekert, C. Macchiavello and M. Mosca, Proc. R. Soc. London, Ser. A, 1998, 454, 339–354 CrossRef.
- K. Temme, S. Bravyi and J. M. Gambetta, Phys. Rev. Lett., 2017, 119, 180509 CrossRef.
- Z. Cai, R. Babbush, S. C. Benjamin, S. Endo, W. J. Huggins, Y. Li, J. R. McClean and T. E. O'Brien, Rev. Mod. Phys., 2023, 95, 045005 CrossRef.
- D. Bultrini, M. H. Gordon, P. Czarnik, A. Arrasmith, M. Cerezo, P. J. Coles and L. Cincio, Quantum, 2023, 7, 1034 CrossRef.
- G. Brassard, P. Høyer, M. Mosca and A. Tapp, Quantum Amplitude Amplification and Estimation, 2002, DOI:10.1090/conm/305/05215.
| This journal is © The Royal Society of Chemistry 2026 |