
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDistilling system complexity to enable unbiased and predictive computational reaction investigations
Raphaël
Robidas
 *ab and
Claude Y.
Legault
*a
*ab and
Claude Y.
Legault
*a
aDepartment of Chemistry, Université de Sherbrooke, Centre in Green Chemistry and Catalysis Sherbrooke, Québec J1K 2R1, Canada. E-mail: raphael.robidas@usherbrooke.ca; claude.legault@usherbrooke.ca
bHexalence, Sherbrooke, Québec J1K 0A5, Canada. E-mail: raphael@hexalence.com
First published on 5th March 2026
Abstract
Computational modelling is a powerful tool to study chemical reactions. Currently, human guidance is nearly always required to avoid the intractable complexity of all a priori possible reaction steps, which consequently greatly limits automated predictive applications. In this work, we present novel theoretical strategies based on the concept of atomic reactivity, as well as a “neophile” kinetic model, demonstrating how they enable unbiased automated reaction modelling with molecules of size typically encountered in experimental methodologies. Our framework allows the identification of unlikely or redundant reaction steps based on first principles and previous analyses, while the neophile kinetic model separates crucial reaction intermediates from inconsequential ones. These advances significantly improved modelling efficiency, allowing us to automatically model 17 unimolecular gold(I)-catalyzed reactions of increasing complexity, starting only from the reactant and catalyst. In 10 reactions, the experimental product distribution is closely reproduced, with 5 additional cases yielding essentially correct reaction networks. Our results demonstrate that it is possible to predictively model catalytic reactions without human guidance through an innovative reformulation of the problem.
Discovering new chemical reactions by computational modelling is a long-standing chemist's dream.1 The ability to quickly and accurately predict the outcomes of chemical reactions without first empirically testing them in a laboratory would greatly benefit synthetic organic chemistry. Indeed, current limitations in synthetic methodologies can restrain access to organic compounds of interest, such as pharmaceuticals. However, developing new synthetic methodologies is a lengthy, iterative process that demands considerable resources. Computational modelling has the potential to significantly shorten this process by identifying plausible reactions through large-scale computational screenings.
Currently, the state of the art in computational chemistry modelling is quite far from this vision, but studies based on human interaction and intuition are widely successful in the study of known chemical reactions. Computational studies of reaction mechanisms are common in modern chemistry research, as they provide a microscopic view complementary to the experimentally observed outcomes. In addition to providing fundamental insight, the results can be leveraged to reduce side reactions, predict the compatibility of specific reactants, and consider new related transformations. The use of computational chemistry in this way, however, requires significant work by experts, making it too complex and time-consuming to achieve the ultimate objective of a fully automated in silico chemical exploration platform.
As computational chemistry grows more sophisticated and computing power increases, the limits of what can be modelled efficiently and rigorously keep getting pushed back. This means that failures in the theoretical models will keep getting less likely as a reason for discrepancies between theory and experiment. However, this increasing accuracy will not reduce the human time required; if anything, it will increase the complexity of computational studies as more possibilities become tractable and need to be evaluated, rather than being confined to small model systems.
To unlock the full potential of computational modelling, we must embrace a paradigm shift through the development of automated, and fully knowledge- and domain-agnostic (or “unbiased”) computational chemistry. Here, “bias” is defined as the modelling scope limitation based on chemical intuition, prior work and, sometimes, human error. This bias is currently necessary due to the overwhelming number of reaction processes that are a priori possible for any given chemical system. If we view elementary reaction steps as changes in atom bonding, we can enumerate all combinations of bond formation (F) and breakage (B), as shown in Table 1. This will be named the combinatorial approach.
|
|
|
|---|---|
| Bonds formed and broken | Number of possibilities |
| B 1 | 22 |
| F 1 | 231 |
| B 2 | 231 |
| F 1 B 1 | 5082 |
| F 2 | 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 565 565 |
| F 1 B 2 | 53361 |
| F 2 B 1 | 584430 |
| F 2 B 2 | 6136515 |
| Total | 6806437 |
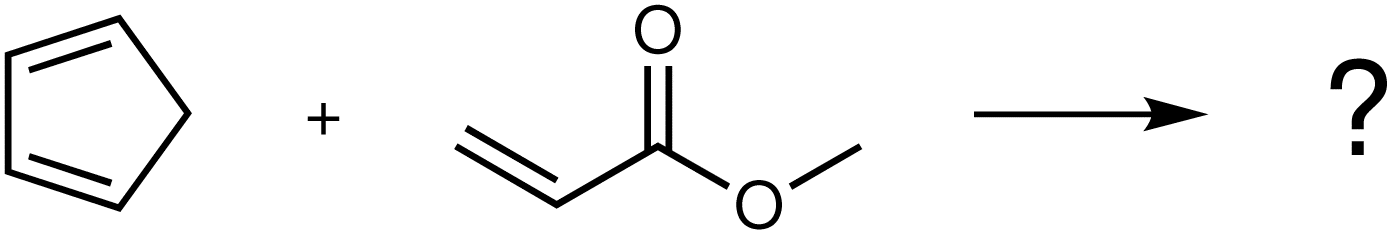
By assuming no prior knowledge about this chemical system, nearly 7 million elementary steps forming and breaking up to two bonds can be enumerated, with only one of them corresponding to the experimentally observed Diels–Alder (F2) reaction.2,3
Accordingly, while automatic and unbiased computational mechanism exploration has been an active field of research for at least 25 years,4 the applicability of such approaches has remained extremely limited. This automated exploration has also been formulated as a high-dimensional coordinate space exploration, although this does not limit the exponential growth of the system complexity with its size. Regardless of the approach, most developments until now involve almost exclusively small (≤25 atoms) molecules,5–15
To tackle larger systems, some combinatorial approaches use wisely chosen rules to consider only some atoms as reactive16 or require manual selection of reactive atoms,9,17–19 which sacrifices complete generality and automaticity. Reaction templates have also been used to reduce the combinatorial explosion, especially in the context of a limited diversity of possible mechanistic steps, like pyrolysis20,21 and polymerisation.22 Hammond's postulate has been used to eliminate some reaction possibilities once the product's thermochemistry is known.23 Additionally, manually curated mechanistic steps have been leveraged recently through an algorithmic approach in order to predict cationic rearrangement outcomes.24 Although encouraging, this approach cannot be easily generalised without substantial involvement from human experts. Different reactions would necessitate curating the relevant mechanistic steps from the ground up.
We believe that this shift towards fully automated, general, and unbiased computational chemistry is crucial for achieving remarkable progress in the field. In this study, we propose novel strategies to tame the exponential growth of the combinatorial approach. We then provide a proof-of-concept based on the automated investigation of catalytic organometallic reactions, which yields both insightful mechanistic details and reliable predictions of reaction outcomes. These results hold significant value as they can reveal outcomes that deviate from previously established patterns, which are often difficult to predict. This difficulty is intensified by the noticeable bias in the scientific literature towards successful reactions, with failed reactions seldom reported, and the common absence of identified and quantified side products. An unbiased reaction model offers valuable insights into the reactivity of the reactant in question, including unanticipated outcomes.
We have developed a conceptual framework based on the ansatz of “atomic reactivities” to determine plausible reactions and control the system-wise combinatorial explosion of possibilities without any human interaction. We have also developed the “neophile” kinetic model, which limits the network-wise possibility growth. In this work, we present how these innovative approaches enable the automated exploration of typical unimolecular gold(I)-catalyzed reactions with predictive accuracy. The studied reactions involve reactants with 22 to 42 (reactive) atoms as well as catalysts with 35 to 66 atoms, constituting system sizes significantly larger than previously possible. Historically, acquiring detailed mechanistic insights has required significant effort, and accurately predicting outcomes has often been elusive. Herein, we report a method that successfully achieves both.
1 Results and discussion
1.1 Atomic reactivity framework
Our approach uses bond formations and breakage as driving coordinates to find new elementary reactions. These coordinates are only used to bias the exploration of the potential energy surface through a relaxed scan; they do not restrict the reactions to be discovered, as opposed to methods that generate the product from the expected connectivity.
Thus, reactions that do not involve exactly the expected bond changes are also discovered. This enables the reduction of the number of explicitly considered reaction coordinates. While a common limit is the formation and breakage of up to two bonds each (denoted F0→2B0→2), we found that all relevant reactions were found with only four combinations: F1, B1, F1B1 and F2. As shown in Fig. 1, the number of possibilities is reduced by two orders of magnitude by this modification. In addition, possibilities of type F1 involving the catalyst and every other atom of the reactant were added in order to move the catalyst around and ensure that all complexation isomers are found. These possibilities can also lead to productive reactions due to implicit PES sampling.
 | ||
| Fig. 1 Example of reaction possibilities pruning through principles derived from atomic reactivity. (a) Pruning principle based on the lone reactant reactivity; (b) pruning principle based on atomic reactivity correlation; (c) reactant B1, whose reactions are presented in Subsection 1.3.2; (d) intermediate in the total synthesis of bryostatin 16 by Trost.27 Multiples in parentheses are calculated with respect to reactant B1. | ||
The foundation of our framework comes from what we know to be unreactive. In the case of gold(I)-catalyzed reactions, the reactant itself without a catalyst is stable and does not react by itself. This means that any imaginable reaction involving only the reactant has to be negligible. The role of the catalyst is to activate certain parts of the molecule to enable a reaction. The first step of the reaction then has to involve these activated parts of the molecule, since the rest has the same non reactivity as in the lone substrate.
In order to leverage those logical deductions, we propose the atomic reactivity ansatz, which is defined as such:
1. The molecular reactivity can be broken down into atomic reactivities, which are properties of the atoms.28
2. As a result, changing the reactivity of a given molecule inevitably implies alterations in the atomic properties of part of its atoms.
3. The combined atomic reactivities of the atoms involved in a reaction determine the barrier of the reaction.
In terms of atomic reactivities, the deductions above mean that any combination of atomic reactivities in the reactant is associated with a large barrier. In terms of notation, the atomic reactivity of atom i will be written as Ri. The formation of a bond between atom 1 and atom 2 will be denoted F1–2. If this bond formation occurs simultaneously with the breakage of a bond between atom 2 and atom 3, this overall reaction will be denoted F1–2B2–3.
Since our approach is combinatorial, these reactions can also be described by pairs of atomic reactivities, each pair corresponding to the reactivities of the atoms whose bond is being formed or broken. This formulation allows for the comparison between potential elementary reactions and the negligible reactions of the lone reactant. Thus, elementary reactions that are functionally equivalent to those of the lone reactant will be disregarded without any calculation.
In this work, we used the pair (q, α) as the formulation of R, where q is the atomic partial charge and α is the atomic polarisability. This formulation is derived in the Subsection 2.3.1 of the SI.
More formally, we define reactivity correlation as ∂Ri/∂Rj. If ∂Ri/∂Rj ≈ 0, then atoms i and j are said to be uncorrelated. This means that changes in the properties of atom i (because of a bonding change or other fluctuation in electronic density) will not influence how reactive atom j is. For the possibility Fi–kFj–l, if i and k are both uncorrelated to j and l, it is reasonable to assume that:
 | (1) |
 ). These results can be used to filter out negligible reactions without any additional calculation based on the equivalence of atomic reactivities. As the reaction network exploration progresses, these reference barriers accumulate and allow increasing pruning of reaction possibilities.
140 with standard pruning rules, yet decreases to only 3558 with the addition of atomic reactivity principles. Furthermore, this intermediate presents only 8.8 times more feasible possibilities despite containing 4.1 times more reactive atoms than reactant B1, indicating less than quadratic growth. Although larger reactants may have more relevant complexation isomers, the number of possibilities is still manageable.
). These results can be used to filter out negligible reactions without any additional calculation based on the equivalence of atomic reactivities. As the reaction network exploration progresses, these reference barriers accumulate and allow increasing pruning of reaction possibilities.
140 with standard pruning rules, yet decreases to only 3558 with the addition of atomic reactivity principles. Furthermore, this intermediate presents only 8.8 times more feasible possibilities despite containing 4.1 times more reactive atoms than reactant B1, indicating less than quadratic growth. Although larger reactants may have more relevant complexation isomers, the number of possibilities is still manageable.
Fig. 2a plots the reduction in the number of evaluated reaction possibilities for all the 17 considered reactions during the reaction network explorations. The number of possibilities that would have been considered without atomic reactivity pruning principles has been recalculated to quantify the reduction. Using the reactant's atomic reactivities reduces the possibilities by a factor of 1.5, on average. Considering atomic reactivity correlations and past data greatly increases the reduction factor, especially for large numbers of total possibilities. Explorations that would require more than 60000 possibility evaluations are reduced by a factor of 4 to 5 with both additional principles. This means that networks normally involving 140000 reasonable reaction possibilities (with baseline principles) will require only 28000 to 35000 possibilities to be evaluated. Fig. 2b shows that the percentage of pruned possibilities due to past data increases steadily with each possibility evaluation cycle, often reaching 60% beyond 14 cycles in the one case that reached that stage. The combinatorial explosion of possibilities is thus further contained by exploiting its limited novelty in terms of chemical reactivity.
 | ||
| Fig. 2 Global reduction in the number of evaluated reaction possibilities. (a) Reduction factor for entire network explorations depending on pruning principles and total possibilities; (b) pruning rate statistics across network exploration cycles, with the shaded area spans one standard deviation from the mean. | ||
1.2 Neophile kinetic simulations
Another key factor leading to the combinatorial explosion is the formation of the reaction network. Indeed, the elementary reactions starting from each new product also need to be considered; in nearly all systems, this number of elementary reactions will increase exponentially with the number of steps considered, since each product leads to multiple other products. Not all reactions will be significant in the final reaction network, but it is challenging to identify significant reactions in the incomplete reaction network. Reported approaches include considering all reactions with a barrier below a defined threshold29 and considering only the fastest reaction per intermediate.9 A more detailed review of the problem and suggested solutions has recently been published.30Alternatively, the changes in concentration of all intermediates can be modelled using a system of differential equations. The reaction rate for intermediate i is given by:
 | (2) |
With a full and exact reaction network, a kinetic simulation will determine which products accumulate and in which proportions. However, when the network is not quite complete, the kinetic simulation can be misleading. With a “short” simulation time, the most significant intermediates will be those who are kinetically favoured, while the true reaction pathway might involve a higher barrier that leads to a much more stable intermediate. In this case, the latter would be considered negligible, as it did not form sufficiently during the simulation time. With a “long” simulation time, the system tends towards the thermodynamic equilibrium, and the final distribution of products will depend only on their thermodynamic stabilities.
One approach would be to aim to be in the intermediate regime where both kinetic and thermodynamic effects are balanced. However, this can be problematic if the next intermediate in the reaction pathway is relatively high in energy and thus will not accumulate meaningfully. Additionally, determining the appropriate simulation time is challenging and depends on the overall reaction barrier. Any systematic errors in the reaction barrier will also affect the optimal simulation time. Consequently, we opted to abandon this approach and developed an alternative kinetic model instead, coined neophile kinetic simulation.
The key challenge to address is the incompleteness of the network. The edges of the nascent network induce errors, since further reactions are missing. The matter that reaches the edge intermediates is kept within the limited network, although they might undergo a very rapid and favourable reaction in the fully explored network. Neophile kinetic simulations thus use one simple rule: intermediates whose reactions have not been fully evaluated are formed irreversibly. Thus, these intermediates will accumulate during the simulation proportionally to their ease of formation, and their final concentrations can be used to identify significant intermediates. This approach is extremely robust, and we were unable to find any scenario where it induced errors. By design, it is suitable for reaction networks of any energy span.
A related kinetic strategy has been independently developed by the Savoie group this year.31 In their case, a graphical distance to the nearest unexplored intermediate is defined and used to further modify the reaction kinetics in combination with an exploration hyperparameter.
Quantifying the computational savings from neophile kinetic simulations is challenging, as conducting complete network explorations without them would be intractable. Nevertheless, the substantial number of energetically favourable intermediates identified, yet not prioritised in the neophile simulations, suggests that the potential reaction pathways are diminished by at least an order of magnitude compared to a simplistic threshold-based approach.
1.3 Reaction results
To assess the viability and scope of automated reaction studies using these new concepts, we selected 17 known gold(I)-catalyzed reactions that had a single reactant as starting material, with the goal of reproducing product distributions and automatically elucidating their formation mechanism. We categorised them into three classes based on their anticipated mechanistic complexity. We chose these reactions to assess the accuracy of the predictions across a wide range of transformations. Reactions producing more than one identified and quantified product were particularly significant to verify how accurately the product distribution can be reproduced. All reactions resulted in good to quantitative yields of identified products and were reported in reputable journals.The A-class category is composed of 1,6-enyne and 1,5-enyne isomerisations, well-known gold(I)-catalyzed reactions which form multiple new carbon–carbon bonds. The expected reaction mechanisms are likely to involve relatively few steps (2–4) and are not anticipated to require intermolecular proton transfers. The reactants bear only one or two functional groups other than the enyne motif.
The B-class category displays more complexity and chemical transformation diversity. The reactants generally contain multiple reactive functional groups. One or multiple proton transfers, and dehydration, are expected to be important in the mechanisms.
Finally, the C-class category may involve considerable skeletal changes and larger reactants. Sizable alkyl groups were modelled as-is without simplification and many reactive functional groups may be present. Proton transfers and unfavoured tautomers are also expected to be important in multiple mechanisms. Multiple reactants bear relative or absolute stereochemistry, although in this first proof-of-concept we did not address this explicitly.
In every case, starting from only the reactant and the experimental ligand, the entire relevant reaction network was explored until no further reaction was predicted to occur significantly. Products with predicted yields below 1% are not shown. All quantum chemistry calculations with more than one electron are imperfect, and shortcomings in the theory levels used may affect the results. As such, a perfect match between experimental and calculated results is exceedingly rare. In some cases, errors linked to the theory level could explain incorrect predictions, and such cases will be discussed.
This unbiased exploration provides an unprecedented level of insight into the mechanistic study of complex systems (Fig. 3). Indeed, our approach now allows for the investigation of both productive and unproductive pathways, enabling access to the true energetic span of complex catalytic processes. This will be crucial for addressing, for example, the structure–activity relationship in ligand design.
 | ||
| Fig. 3 Transformation of reactant A1-1 (ref. 32) and selected portion of its reaction network (relative Gibbs free energies in kJ mol−1). Some complexation isomers and negligible reaction pathways have been removed to reduce clutter (view the full networks in the SI). The blue node corresponds to the lone reactant, light blue nodes indicate complexation isomerisation dummy transition states, light purple nodes indicate tautomerization pseudo-transition states, red nodes indicate intermediates that were formed in negligible amounts and not explored further, and light green nodes indicate the most significant intermediates at the end of the kinetic simulation. The thickness of the edges represents the final net flux that has passed through those edges. | ||
In this graph, the lone reactant A1-1 is fixed to have an energy of 0 kJ mol−1. Multiple complexation sites are available for the catalyst (alkene, alkyne, esters), leading to multiple complexation pathways to intermediates A1-i1 to A1-i3. The most stable complexation isomer A1-i2 is also defined to have an energy of 0 kJ mol−1 since the complexation energy of the catalyst is not relevant in the case of pseudo-unimolecular reactions. All other relative energies are calculated with respect to this intermediate. The complexation barriers are considered to be negligible. All nodes between the identified intermediates correspond to transition states, their structure not being illustrated in the figure but available in the SI within the full networks.
Starting from the complexation isomers, cyclisations on the esters are found to be possible (A1-i4 and A1-i5), but endergonic and not ultimately significant. The expected 6-endo-dig and 5-exo-dig cyclopropanations are also found, leading to A1-i6 and A1-i7 respectively, with the latter possessing a much lower formation barrier. This favoured product rearranges mainly to the bicycle A1-i8, which has other complexation isomers (A1-i9, A1-i16). Those intermediates can notably rearrange to carbocation A1-i12, which can further rearrange to the non-classical carbocation A1-i13. This intermediate directly leads to the final product bound to the catalyst (A1-i17). No further reaction is predicted to be significant and this product accumulates. In addition to this product, trace amounts of allene A1-i18 are formed through the elimination and reprotonation of intermediate A1-i12.
This unbiased exploration of the mechanistic manifolds of this transformation provides an unprecedented level of insight on other reasonable reactions, albeit negligible in this case. The starting adduct A1-i2 can directly form bicycle A1-i8 with a barrier of 53 kJ mol−1, altihough the 5-exo cyclisation followed by a rearrangement (A1-i2 → A1-i7 → A1-i8) has a much lower rate-determining step of 28 kJ mol−1. Similarly, bicycles A1-i8 and A1-i9 can directly form the final product A1-i17, although those reactions are not favoured. The formation of intermediate A1-i15 was also investigated, although it ended up being unproductive and inconsequential. Intermediate A1-i14, which has been identified but not significantly formed, is analogous to the main product A2-3 of the second reaction (see Fig. 4). The reaction network thus shows that the selectivity between those two possible products is entirely determined by the reaction barriers of intermediate A1-i13.
 | ||
| Fig. 4 Other modelled 1,6-enyne isomerisation reactions.33–35 | ||
This subgraph thus shows a glimpse of the profound mechanistic understanding that is inherently accessible from the generated data results.
Fig. 4 shows other 1,6-enyne isomerisation reactions that were explored in the same way. Gratifyingly, the predicted product distributions closely follow the experimental ones in every case. These results indicate that the influence of functional group changes has been accurately modelled.
The reactions of 1,5-enynes A5-1 (ref. 36) and A6-1 (ref. 37) were explored with similar success (see Fig. 5). The reaction from A5-1 to A5-2 was also found to be the only significant reaction, in agreement with the high experimental yield of 99%.
 | ||
| Fig. 5 Results of modelled 1,5-enyne isomerisation reactions.36,37 | ||
The reaction of A6-1 is slightly less straightforward. Experimentally, the aldehyde direct product A6-2 was not isolable without significant degradation, so the authors carried out a reduction step to isolate the alcohol A6-3. Of course, the results do not take into account the reduction step, so the expected product is the aldehyde A6-2. This product is indeed formed, but quickly undergoes proton transfers to form the thermodynamically favored products A6-4 as a mixture of cis/trans isomers. The proton transfers are estimated to have a low barrier of 32 kJ mol−1, making them much faster than the formation of the aldehyde itself (barrier of 68 kJ mol−1). The intermediate immediately preceding the aldehyde can also undergo a proton transfer to ultimately yield A6-4. As explained in Subsection 3.4, proton transfers are not modeled completely explicitly, in part due to the uncertain and variable species involved. In particular, the approximate proton transfer barriers are calculated with the reactant as proton transfer agent. In dilute solutions, this intermolecular process will be significantly slower, which could explain the apparent discrepancy. Indeed, the effect of substrate concentration during the reaction was not taken into account at this time.
If tautomerizations are considered to be too slow and thus effectively impossible, the automated exploration does predict the sole formation and accumulation of aldehyde A6-2. It is thus unclear whether the calculated Gibbs free energies are affected by an error or if the overall kinetic modelling requires additional sophistication and experimental parameters.
 | ||
| Fig. 6 First set of B-class reactions.38–40 | ||
The conversion of sulfoxide B2-1 was predicted to yield mostly the expected product B2-2 (ref. 39) with a small amount of ylide B2-3. Although not experimentally identified and quantified, it is possible that ylide B2-3 is indeed formed and could explain part of the lower experimental yield, as B2-3 could even react and degrade with B2-2.
The transformation from B3-1 to B3-2 was automatically identified,40 in addition to the formation of biphenyl B3-3. Although the predicted yield is somewhat too low, the lost mass balance might be explained in parts by the formation of the biphenyl B3-3, which is very apolar and could go unnoticed during purification. Additional insight is obtained from the reaction network (Fig. 7).
 | ||
| Fig. 7 Selected portion of the reaction network of B3-1 under gold(I) catalysis (relative Gibbs free energies in kJ mol−1). Some complexation isomers and negligible reaction pathways have been removed to reduce clutter (view the full network in the SI). | ||
The major product B4-2 of the transformation of B4-1 (ref. 41) has been correctly predicted to be formed, albeit as minor product (Fig. 8). The experimentally observed secondary product B4-3 was not predicted computationally at all. Instead, product B4-4 was predicted to be the main product, while it was not obtained experimentally in this specific case. However, this transformation has also been reported for very similar substrates,42 suggesting that a minor deviation in reaction barriers could explain this partially correct result.
 | ||
| Fig. 8 Last B-class reaction.41 | ||
 | ||
| Fig. 9 First set of C-class reactions.43–46 | ||
Errors associated with the theory level cannot explain such a large difference in result, which might indicate that the reaction involves an element missing from the modelled system. It has been previously shown47 that the autoionisation of water is poorly reproduced when using only continuum solvation models, while small water clusters yield results much closer to experiment. Based on this hypothesis, we calculated the energy associated with the formation of hydronium and hydroxide ions microsolvated by four water molecules.
Using those values to calibrate the tautomerization barriers, the exploration of the reaction was carried out again. With these parameters, other proton transfers become possible. In particular, alkene isomerisations become more accessible, leading to a mixture of terminal and internal alkenes on the side chain. In addition, intermediates with the same core as C1-3 can lose a proton to form a gold alkoxide, which can then be reprotonated at the exocyclic enol position. This process forms an activated alkoxide-carboxonium species which readily undergoes the semi-pinacol rearrangement and leads to C1-6 as major product. This product is correct except for the alkene position, which suggests that alkene isomerisation barriers are underestimated. The formation of the identified minor side products (C1-4, C1-7, C1-8 and C1-9) is a possible reason for the lost mass balance, although it is not possible to confirm this hypothesis from the available data.
The reaction of C2-1 (ref. 44) yields a zwitterion bearing a quaternary centre, which could plausibly be thought to react further to a more thermodynamic product. However, the automated exploration correctly predicts the sole and complete formation of the main product, in agreement with the experimental yield of 94%. It is worth specifying that the hexyl chain of the reactant has not been simplified in the modelled reactant, as is common practice. The modelling approach keeps computational costs sufficiently low that such a chain can be modelled as-is, thus preventing discrepancies caused by (unknowingly) altering the reactivity with simplifications. Moreover, this result suggests that our conformational sampling process does not become an issue with larger, more flexible reactants.
Reactant C3-1 can cyclize nearly quantitatively under gold(I) catalysis into product C3-2, presumably through an enol intermediate.45 The automated exploration did not find any reasonable reaction, predicting that the reactants remain unreacted. We once again considered the influence of traces of water on the reaction. With this modification, tautomerizations become accessible, leading to the expected enol intermediates. Moreover, the automated exploration lead to the discovery of 5 possible cyclisation transition states, which vary in terms of internal hydrogen bonding and of alkene position. All transition states were associated with significant barriers (112 kJ mol−1 to 147 kJ mol−1), and thus did not lead to any significant reaction.
It is possible that a second molecule of reactant could be involved in the reaction. The catalyst's counterion, ClO−4, may also play a role in the transformation. In the original work, the authors screened several counter-ions and found that BF−4 lead to no conversion, SbF−6 lead to a yield of 19%, while PF−6, TfO− and ClO−4 lead to nearly quantitative yields (96–99%), with ClO−4 reducing the required reaction time greatly. Although not entirely conclusive, these results indicate that the counterion affects the reaction, which was not in the scope of our current implementation. A dimeric catalyst could also be involved in the reaction, which is not unheard of in gold(I) catalysis.51 In any case, these factors may complexify the exploration process beyond what is handled by our current implementation. Nonetheless, the robustness observed in A- and B-class methodologies suggests that our platform could be used to uncover uncommon mechanistic behaviours in developing methodologies, in which case additional experimental studies would be required to address the true nature of the active catalytic species or trace species involved in the overall process.
Under gold(I) catalysis, reactant C4-1 selectively cyclizes on the exocyclic ketone, resulting in the formation of product C4-3.46 Our automated exploration of the reaction network indicates that intermediate C4-2 is formed faster than C4-3 and initially accumulates (Fig. 11). However, because of the greater thermodynamic stability of C4-3, interconversion occurs at a slower rate, and may account for the increased temperature requirement (see Fig. 10). With this additional mechanistic insight regarding the lifetime of intermediate C4-2, one could envision exploiting its formation and reactivity to develop novel methodologies to access new compounds bearing multiple quaternary centres. While outside the scope of the current implementation of the platform, bimolecular automated reaction explorations could be used to identify valuable transformations from C4-2. Furthermore, the lost mass balance may stem from side reactions between C4-2 and either C4-1 or C4-3, which were not examined computationally. The mechanism automatically found also differs from the authors' proposed mechanism,46 which omits C4-2, preventing further exploration of this intermediate. This exemplifies the insights provided by such unbiased automated studies.
 | ||
| Fig. 10 Result of the kinetic simulation of the C4 reaction network. | ||
 | ||
| Fig. 11 Second set of C-class reactions.48–50 | ||
The reaction of C5-1 experimentally yields product C5-2,48 while only product C5-3 was found significant in silico. Considering the length of the pathways leading to this product (at least 7 elementary steps), it is difficult to pinpoint the error source. Despite the accuracy of current computational methods, they remain subject to small intrinsic errors, which can shift the course of reactions. While such errors can be identified in mechanistic studies with known products, predictive applications require highly accurate results at each step. The outcome of reaction C5 exemplifies the challenge of outcome prediction with errors compounding over 7+ reaction steps.
Reasonable reaction pathways of C6-1 into C6-2 and its isomer C6-3 were found,49 but the formation of C6-4 was found to be more favourable. In this reaction, the selectivity between the two kinds of products comes from a competition between a proton transfer and the cyclopropyl ring opening. As proton transfers are modelled semi-explicitly (see Methods), their barriers are only approximate. The error linked to this approximation influences a key step and can change the reaction outcome. In these conditions, however, it is not obvious what the true proton-transfer agent would be, and so fully explicit modelling is quite challenging. Even with this approximation, the actual product is expected to be significant, representing approximatively 10% of the predicted products.
Finally, reactant C7-1 is highly enantioenriched and can cyclize under gold(I) catalysis to form enantioenriched products C7-2 and C7-3.50 The experimental ratio between the two products relies on specific conditions. The authors of this study found that the reaction benefitted from adding water, likely because it facilitated the hydrolysis of a reaction intermediate. The modelled conditions did not involve explicit water molecules, and so this hydrolysis was naturally not accounted for. However, product C7-3 is still found as the sole product of the reaction. As mentioned before, we elected not to strictly enforce stereochemistry considerations in our current implementation. Nonetheless, the predicted product bears the correct absolute stereochemistry at all three chiral centres. This suggests that the rigorous consideration of stereochemistry will require only modest changes to our approach.
The shortest reaction pathway from C7-1 to C7-2 involves 6 elementary steps, confirming that the accuracy of computed energies is generally excellent and can also be applied to complex transformations with many elementary steps.
2 Conclusion
In this work, we have demonstrated that it is possible to address the combinatorial explosion of possibilities in unbiased mechanistic explorations through the use of innovative pruning strategies. The use of an atomic reactivity framework and neophile kinetic simulations reduces the number of reaction possibilities sufficiently to render completely tractable automated investigations of organometallic reactions involving organic reactants of typical size. These improvements become increasingly valuable as the molecular size grows, as they bring the reaction possibilities growth under control. They have been proven very effective in systems with up to 42 reactive atoms, much more than was previously tractable. We even already used our approach to automatically discover a complex reaction mechanism, which served as a starting point for a DFT study we independently published.52 Moreover, the improvements are very general and a priori applicable to any molecular system. Limitations in the accuracy of the predictions have been linked to approximations in treating tautomerizations and unspecified experimental details, such as traces of water, which are challenging to account for. A more sophisticated approach to handling tautomerization barriers would reduce those limitations and further narrow down the possible sources of error.The atomic reactivity framework introduced here has been applied to organometallic catalysis, but we expect to develop it further to create pruning principles for both catalysed and uncatalysed polymolecular reactions. Developing more accurate atomic reactivity formulations will enable greater reaction clustering and reuse of existing data. A sufficiently precise formulation would enable the reuse of reaction data across reaction networks, thus greatly minimizing the occurrence of unexplored unproductive reactions to evaluate. We also envision exploiting machine learning to leverage this data to provide a very fast way to estimate reaction barriers, thus further accelerating reaction network explorations. The resulting capabilities would have a remarkable potential in many fields of science which involve studying how organic molecules react and transform.
3 Computational methodology
3.1 Reaction modeling
Gold(I)-catalyzed reactions involve an organic reactant, a gold(I) catalyst (e.g., Ph3PAuNTf2) or a gold(I) precatalyst (e.g., Ph3PAuCl) with an activator (e.g., AgOTf), and usually a somewhat polar, aprotic and non-coordinating solvent, often dichloromethane or dichloroethane. Due to the noncoordinating nature of the catalyst counterion, we proposed in this study that it is safe to neglect it. Although it has been proposed to be involved as proton transfer agent in some reactions,53 we aimed to address this effect using an alternative approach to explicit modeling (see Section 2.2 of the SI), to limit model size and keep it unimolecular.Gold catalysts involve a ligand, which has a great influence on the reaction outcome54 and can be used to induce stereoselectivity.55 However, the size of the ligand (typically 35 to 90 atoms) signifies a substantial increase in computational costs when using DFT and post-Hartree–Fock methods, which is difficult to manage, particularly in this context. Indeed, many computational studies involving such ligands rely on smaller model ligands, such as PH3 or PMe3, to simplify calculations.56
To address this issue, we aimed to create a composite scheme for calculating Gibbs free energies with reasonable computational costs and sufficient accuracy. In particular, recent semi-empirical tight-binding methods such as GFN2-xTB57,58 have been reported as much faster alternatives to DFT methods for many applications.59 Although those methods are typically not as accurate and reliable as a carefully selected DFT functional paired with a large basis set, they prove extremely useful when high accuracy is unnecessary or when DFT methods are simply impractical. Solvation is taken into account implicitly for every GFN2-xTB calculation using the ALPB solvation model with parameters for dichloromethane.60
Through a preliminary benchmark (see Section 2.4 of the SI), we found that GFN2-xTB geometries were adequate for achieving good results, provided that the electronic energies are calculated using DFT. This made large complexes computationally manageable while ensuring the accuracy of the results. Single-point energy calculations, even with large ligands, were found to be perfectly reasonable. Furthermore, we devised pruning strategies to minimize the number of such calculations, as outlined in subsequent sections. After evaluating several computational methods, we decided on B2K-PLYP-D3/def2-mTZVP/SMD(DCM) with a reduced integration grid to perform single-point calculations energies. This method is expected to provide sufficiently accurate results compared to the more accurate and greatly more computationally expensive coupled-cluster methods.61–63
We have not explicitely evaluated the impact of different free energy corrections on the results. By default, the xtb package uses Grimme's entropy correction for low-lying frequency modes,64 and Gibbs free energy corrections were only calculated at the GFN2-xTB theory level.
3.2 Automated reaction evaluation
The reaction evaluation process starts with the enumeration of all a priori reasonable reaction possibilities. We consider only σ bond formation and breakage, which also corresponds to atoms coming together or moving apart. This means that a Diels–Alder reaction would be a F2 reaction, and not F3B3. Relaxed coordinate scans are carried out to bring together or push apart the reactive atoms. This can be seen as an exploration of the potential energy surface (PES) biased by the expected reaction coordinates. The direct consequence of this is that the net transformation that occurs is not limited to the expected one. For example, by considering an F1 reaction approaching Cl− from the carbon of CH3I, we will discover an SN2 reaction, which is formally an F1B1 reaction. Similarly, any formation or breakage of π bonds will occur naturally as the electron density moves during the reaction. This means that the considered reaction coordinates do not need to be complete and can have reduced dimensionality. Indeed, with this approach, we found that it was sufficient to consider B1, F1, F1B1 and F2 reactions only.In order to reduce the error rate related to spurious GFN2-xTB geometries, DFT gradients are calculated for each new product and compared to a threshold value determined from preliminary spurious and valid GFN2-xTB structures. If above this threshold, the structure is perturbed once following the steepest energy descent according to the DFT gradient, then reoptimized using the same theory level and loose convergence parameters. The optimized structure is reoptimized by GFN2-xTB with the expectation of converging to the same local minimum. This final structure is not modified further, even if it converges back to its initial state. For this process, the M06-2X/def2-mSVP/CPCM(DCM) theory level combined with geometrical counter-poise65 (GCP(DFT/SVP)) was found to be adequate. Since this theory level is only used to break out of a potentially spurious local minimum, having a high accuracy is not necessary.
Relaxed scans provide crude reaction barriers to eliminate certain reaction possibilities at this stage. If a product emerges, its geometry will be optimized, and its energy will be computed. It can then be used with the reactant to perform a transition state search using the double-ended growing string method (GSM) developed by Chakraborty66 and Zimmerman.67,68 We used its implementation in the Pysisyphus Python package69 combined with the GFN2-xTB level of theory. The approximate transition state obtained with the GSM is fully optimized using the RS-P-RFO algorithm as implemented in Pysisyphus. The vibrational modes of transition state are expected to contain one negative frequency below −75 cm−1 and that this frequency mode involves the movement of the expected reactive atoms. Spurious transition states are thus identified and filtered.
The immediate reactant and product are verified by carrying out an IRC calculation on the transition state, also with GFN2-xTB as level of theory. It is possible that the true reactant or product are not equivalent to the initial species that lead to this transition state. This is not an issue as long as the correct connectivities are considered in the reaction network.
3.3 Data-driven pruning
The reaction Fi–j for intermediate A will be associated with a reaction barrier once evaluated. To reuse this data, we just need to consider the atomic reactivities as the relevant dimensions instead of the atom indices and nature of the intermediate. In other words, instead of storing we will store
we will store  The major difference is that the former is necessarily unique (there is just one intermediate A and one possibility Fi–j), while the latter is not (Ri and Rj stem from the reactivity of the system, not its label). However, in both cases, the same key will be linked with the same value (i.e., reaction barrier).
The major difference is that the former is necessarily unique (there is just one intermediate A and one possibility Fi–j), while the latter is not (Ri and Rj stem from the reactivity of the system, not its label). However, in both cases, the same key will be linked with the same value (i.e., reaction barrier).
In practice, the implementation adds the following modifications (see extended discussion in Section 2.3 of the SI):
• To maximize the error cancellation, two reactions are considered equivalent if they involve equivalent atomic reactivities and the same atom indices (or atoms equivalent by symmetry). This partially accounts for geometrical constraints, which are difficult to account for in R. For example, hydrogens at different positions of a substituted cyclohexane might have equivalent atomic reactivities, but will differ in their feasible reactions due to their different positions on the ring (for example due to ring strain). However, if the steric effects around an atom or its ring strain change significantly, it implies that a reaction has taken place nearby, and so its electronic properties are expected to change. As such, changes in steric and ring strain effects are expected to also involve changes in R. This restriction related to atom indices then reduces the number of reaction possibilities that can be pruned, but is expected to avoid a very large percentage of invalid prunings due to steric and ring strain effects.
• The atomic reactivities are considered equivalent if they are within a certain range of each other (see threshold determination in Subsection 2.3.2 of the SI).
• When using the barrier of an equivalent reaction to determine whether a given reaction should be evaluated, a certain margin of error is permitted. Depending on the estimated reliability, a barrier may exceed the exploration threshold by a specific margin without ruling out the considered reaction. Therefore, if the previously evaluated equivalent reaction was linked to an exceptionally large barrier, the reaction considered will be discarded. If that barrier was only slightly above the exploration threshold, it might not be sufficient to eliminate the considered reaction. The tolerance thresholds used in this work are presented and explained in the Subsection 2.1.1 of the SI.
3.4 Complexation isomers, conformers and tautomers
In non-covalent adducts, the catalyst can move from one Lewis basic complexation site to another, which is explicitly considered in our approach. The complexation isomers are chosen to sample the whole range of possible activations by the catalyst, as measured through the R descriptor (see Section 2.2 of the SI for the complete methodology). Conformers of covalent adducts are also sampled to find geometries that might exhibit a different reactivity.In addition to reaction possibilities, proton transfers are considered possible and are explicitly considered. However, in nearly all reaction conditions, there is no strong acid or base, and so the reactant is considered to be the species allowing proton transfers. The proton transfer barriers are approximated through the Gibbs free energy of the transient protonated/deprotonated species. The relative Gibbs free energy of this species is calculated by coupling the (de)protonation process with the inverse process involving the starting material. This models the net reaction of a proton transfer between the intermediate and a molecule of starting material. Water clusters have also been considered as acid and base, as indicated in the text. The technical approach is described in detail in the Section 2.2 of the SI.
3.5 Network exploration cycles
The network exploration is carried out in a series of cycles, during which a set of relevant intermediates are considered as reactants of possible new reactions. At the start of the exploration, the complexation isomers of the starting materials are generated (see Section 2.2 of the SI) and considered as the reactive intermediates of the first cycle. All their reaction possibilities are generated and evaluated during this cycle. Tautomerizations are also considered for these intermediates. Once all those reactions have been evaluated, the updated reaction network undergoes a neophile kinetic simulation which will determine the new set of intermediates to consider in the following cycle. In addition to reactions and tautomerizations, complexation isomerisation is evaluated for all intermediates who aren't complexation isomers themselves.The exploration stops when there is no new reactive intermediate to consider according to the neophile kinetic simulation. The product distribution is calculated using a normal kinetic simulation.
Author contributions
R. R. conceived the project, lead the methodology development, obtained literature data, developed the software implementation, developed the visualization approach and wrote the original draft of the manuscript. C. Y. L. obtained funding and computational resources, contributed to the methodology development and provided supervision. Both authors co-revised and co-edited the manuscript.Conflicts of interest
R. R. is developing a commercial service based on the described methodology. All queries regarding this service should be sent to E-mail: raphael@hexalence.com.Data availability
The data supporting this article have been included as part of the supplementary information (SI). The specific software implementation used in this work is not available due to its importance in the commercial endeavour of one of the authors. Pseudocode can also be found in the supplementary information, and an archived version on Zenodo (https://doi.org/10.5281/zenodo.18635813). Supplementary information: all final reaction networks, including molecular structures, Gibbs free energy and original labels. They can be explored interactively using the provided static web pages, which were also used to produce the network illustrations of this paper. Additionally, the same raw data is provided in JSON format. The results of the preliminary theory level benchmark are also provided. See DOI: https://doi.org/10.1039/d5dd00366k.Acknowledgements
C. Y. L. and R. R. thank the Natural Science and Engineering Research Council (NSERC) of Canada, the Canada Foundation for Innovation (CFI), the FRQNT Centre in Green Chemistry and Catalysis (CGCC), and the Université de Sherbrooke for their support. Computational resources were provided by Calcul Québec and the Digital Research Alliance of Canada. R. R. is grateful to FRQNT for a Graduate Scholarship (B1X/B2X), Hydro-Québec and Université de Sherbrooke for a doctoral scholarship, and NSERC for a Canada Graduate Scholarship (CGS M) as well as a Vanier Canada Graduate Scholarship. R. R. thanks Johannes Steinmetzer for providing help and prompt bug fixes for Pysisyphus.Notes and references
- S. Grimme and P. R. Schreiner, Angew. Chem., Int. Ed., 2018, 57, 4170–4176 CrossRef CAS PubMed.
- C. A. Dvorak and V. H. Rawal, Chem. Commun., 1997, 2381–2382 RSC.
- M. Alaboalirat, L. Qi, K. J. Arrington, S. Qian, J. K. Keum, H. Mei, K. C. Littrell, B. G. Sumpter, J.-M. Y. Carrillo, R. Verduzco and J. B. Matson, Macromolecules, 2019, 52, 465–476 CrossRef CAS.
- K. K. Irikura and R. D. Johnson, J. Phys. Chem. A, 2000, 104, 2191–2194 CrossRef CAS.
- P. M. Zimmerman, Mol. Simul., 2015, 41, 43–54 CrossRef CAS.
- M. Jafari and P. M. Zimmerman, Phys. Chem. Chem. Phys., 2018, 20, 7721–7729 RSC.
- A. L. Dewyer, A. J. Argüelles and P. M. Zimmerman, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2018, 8, e1354 CrossRef.
- S. Maeda, Y. Harabuchi and K. Saita, Molecular Technology, John Wiley & Sons, Ltd, 2018, ch. 8, pp. 173–199 Search PubMed.
- Q. Zhao and B. M. Savoie, Nat. Comput. Sci., 2021, 1, 479–490 CrossRef PubMed.
- A. Hashemi, S. Bougueroua, M.-P. Gaigeot and E. A. Pidko, J. Chem. Theory Comput., 2022, 18, 7470–7482 CrossRef CAS PubMed.
- A. Stan, B. von der Esch and C. Ochsenfeld, J. Chem. Theory Comput., 2022, 18, 6700–6712 CrossRef CAS PubMed.
- J. P. Unsleber, S. A. Grimmel and M. Reiher, J. Chem. Theory Comput., 2022, 18, 5393–5409 CrossRef CAS PubMed.
- S. Maeda, Y. Harabuchi, H. Hayashi and T. Mita, Annu. Rev. Phys. Chem., 2023, 74, 287–311 CrossRef CAS PubMed.
- P. Ramos-Sánchez, J. N. Harvey and J. A. Gámez, J. Comput. Chem., 2023, 44, 27–42 CrossRef PubMed.
- H. Hayashi, S. Maeda and T. Mita, Chem. Sci., 2023, 14, 11601–11616 RSC.
- M. Bensberg and M. Reiher, J. Phys. Chem. A, 2024, 128, 4532–4547 CrossRef CAS PubMed.
- I. M. Pendleton, M. H. Perez-Temprano, M. S. Sanford and P. M. Zimmerman, J. Am. Chem. Soc., 2016, 138, 6049–6060 CrossRef CAS PubMed.
- A. L. Dewyer and P. M. Zimmerman, ACS Catal., 2017, 7, 5466–5477 CrossRef CAS.
- M. Bensberg and M. Reiher, Isr. J. Chem., 2023, 63, e202200123 CrossRef CAS.
- C. W. Gao, J. W. Allen, W. H. Green and R. H. West, Comput. Phys. Commun., 2016, 203, 212–225 CrossRef CAS.
- M. Liu, A. Grinberg Dana, M. S. Johnson, M. J. Goldman, A. Jocher, A. M. Payne, C. A. Grambow, K. Han, N. W. Yee, E. J. Mazeau, K. Blondal, R. H. West, C. F. Goldsmith and W. H. Green, J. Chem. Inf. Model., 2021, 61, 2686–2696 CrossRef CAS PubMed.
- J. P. Unsleber, J. Chem. Inf. Model., 2023, 63, 3392–3403 CrossRef CAS PubMed.
- D. Rappoport, C. J. Galvin, D. Y. Zubarev and A. Aspuru-Guzik, J. Chem. Theory Comput., 2014, 10, 897–907 CrossRef CAS PubMed.
- T. Klucznik, L.-D. Syntrivanis, S. Baś, B. Mikulak-Klucznik, M. Moskal, S. Szymkuć, J. Mlynarski, L. Gadina, W. Beker, M. D. Burke, K. Tiefenbacher and B. A. Grzybowski, Nature, 2024, 625, 508–515 CAS.
- P. M. Zimmerman, J. Comput. Chem., 2013, 34, 1385–1392 CrossRef CAS PubMed.
- Q. Zhao and B. M. Savoie, Angew. Chem., Int. Ed., 2022, 134, e202210693 CrossRef.
- B. M. Trost and G. Dong, Nature, 2008, 456, 485–488 CrossRef CAS PubMed.
- S. A. Grimmel and M. Reiher, Chimia, 2021, 75, 311 CrossRef CAS PubMed.
- M. Bergeler, G. N. Simm, J. Proppe and M. Reiher, J. Chem. Theory Comput., 2015, 11, 5712–5722 CrossRef CAS PubMed.
- M. S. Johnson, H.-W. Pang, M. Liu and W. H. Green, Int. J. Chem. Kinet., 2025, 57, 93–107 CrossRef CAS.
- M. Woulfe and B. M. Savoie, J. Chem. Theory Comput., 2025, 21, 1276–1291 CrossRef CAS PubMed.
- N. Mézailles, L. Ricard and F. Gagosz, Org. Lett., 2005, 7, 4133–4136 CrossRef PubMed.
- C. Nieto-Oberhuber, M. P. Muñoz, E. Buñuel, C. Nevado, D. J. Cárdenas and A. M. Echavarren, Angew. Chem., Int. Ed., 2004, 43, 2402–2406 CrossRef CAS PubMed.
- Y. T. Lee, Y. K. Kang and Y. K. Chung, J. Org. Chem., 2009, 74, 7922–7934 CrossRef CAS PubMed.
- A. Simonneau, Y. Harrak, L. Jeanne-Julien, G. Lemière, V. Mouriès-Mansuy, J.-P. Goddard, M. Malacria and L. Fensterbank, ChemCatChem, 2013, 5, 1096–1099 CrossRef CAS.
- M. R. Luzung, J. P. Markham and F. D. Toste, J. Am. Chem. Soc., 2004, 126, 10858–10859 CrossRef CAS PubMed.
- B. D. Sherry and F. D. Toste, J. Am. Chem. Soc., 2004, 126, 15978–15979 CrossRef CAS PubMed.
- X. Li, J. Zhao, X. Xie and Y. Liu, Org. Biomol. Chem., 2017, 15, 8119–8133 RSC.
- N. D. Shapiro and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 4160–4161 CrossRef CAS PubMed.
- V. Mamane, T. Gress, H. Krause and A. Fürstner, J. Am. Chem. Soc., 2004, 126, 8654–8655 CrossRef CAS PubMed.
- D. Garayalde, E. Gómez-Bengoa, X. Huang, A. Goeke and C. Nevado, J. Am. Chem. Soc., 2010, 132, 4720–4730 CrossRef CAS PubMed.
- S. Wang and L. Zhang, J. Am. Chem. Soc., 2006, 128, 8414–8415 CrossRef CAS PubMed.
- Y.-Q. Gu, P.-P. Zhang, J.-K. Fu, S. Liu, Y. Lan, J.-X. Gong and Z. Yang, Adv. Synth. Catal., 2016, 358, 1392–1397 CrossRef CAS.
- C. Wang, Q. Cui, Z. Zhang, Z.-J. Yao, S. Wang and Z.-X. Yu, Chem.–Eur. J., 2019, 25, 9821–9826 CrossRef CAS PubMed.
- Y.-P. Xiao, X.-Y. Liu and C.-M. Che, Angew. Chem., Int. Ed., 2011, 50, 4937–4941 CrossRef CAS PubMed.
- C. Zhang, H. Jiang and S. Zhu, Chem. Commun., 2017, 53, 2677–2680 RSC.
- S. Dhillon and A. L. L. East, Int. J. Quantum Chem., 2018, 118, e25703 CrossRef.
- M.-C. Cordonnier, A. Blanc and P. Pale, Org. Lett., 2008, 10, 1569–1572 CrossRef CAS PubMed.
- J. M. Fernández-García, H. A. Garro, L. Fernández-García, P. García-García, M. A. Fernández-Rodríguez, I. Merino and E. Aguilar, Adv. Synth. Catal., 2017, 359, 3035–3051 Search PubMed.
- M. Brandstätter, N. Huwyler and E. M. Carreira, Chem. Sci., 2019, 10, 8219–8223 RSC.
- A. S. K. Hashmi, Acc. Chem. Res., 2014, 47, 864–876 CrossRef CAS PubMed.
- R. Robidas and C. Y. Legault, Chem.–Eur. J., 2023, 29, e202301510 CrossRef CAS PubMed.
- J. Shi, S. Bi, Y.-Y. Jiang, Y. Liu, B. Ling and X.-A. Yuan, J. Organomet. Chem., 2018, 864, 136–142 CrossRef CAS.
- Z. Lu, G. B. Hammond and B. Xu, Acc. Chem. Res., 2019, 52, 1275–1288 CrossRef CAS PubMed.
- W. Zi and F. D. Toste, Chem. Soc. Rev., 2016, 45, 4567–4589 RSC.
- O. N. Faza and C. S. López, Homogeneous Gold Catalysis, Springer International Publishing, Cham, 2015, pp. 213–283 Search PubMed.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- H. Neugebauer, B. Bädorf, S. Ehlert, A. Hansen and S. Grimme, J. Comput. Chem., 2023, 44, 2120–2129 CrossRef CAS PubMed.
- C. Bannwarth, E. Caldeweyher, S. Ehlert, A. Hansen, P. Pracht, J. Seibert, S. Spicher and S. Grimme, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2021, 11, e1493 Search PubMed.
- S. Ehlert, M. Stahn, S. Spicher and S. Grimme, J. Chem. Theory Comput., 2021, 17, 4250–4261 CrossRef CAS PubMed.
- A. Karton and L. Goerigk, J. Comput. Chem., 2015, 36, 622–632 CrossRef CAS PubMed.
- L.-J. Yu, F. Sarrami, R. J. O'Reilly and A. Karton, Chem. Phys., 2015, 458, 1–8 CrossRef CAS.
- A. Karton, J. Phys. Chem. A, 2019, 123, 6720–6732 CrossRef CAS PubMed.
- S. Grimme, Chem.–Eur. J., 2012, 18, 9955–9964 CrossRef CAS PubMed.
- H. Kruse and S. Grimme, J. Chem. Phys., 2012, 136, 154101 CrossRef PubMed.
- B. Peters, A. Heyden, A. T. Bell and A. Chakraborty, J. Chem. Phys., 2004, 120, 7877–7886 CrossRef CAS PubMed.
- P. M. Zimmerman, J. Chem. Phys., 2013, 138, 184102 CrossRef PubMed.
- P. M. Zimmerman, J. Chem. Theory Comput., 2013, 9, 3043–3050 CrossRef CAS PubMed.
- J. Steinmetzer, S. Kupfer and S. Gräfe, Int. J. Quantum Chem., 2021, 121, e26390 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |