Effects of sulfur on dehydrogenation of methylcyclohexane over platinum catalysts
Received
12th February 2026
, Accepted 13th April 2026
First published on 24th April 2026
Abstract
Liquid organic hydrogen carriers (LOHCs) enable storage and transport of H2 using existing liquid fuel infrastructure, through reversible catalytic hydrogenation and dehydrogenation of organic molecules. A viable, commercially deployed LOHC system is the methylcyclohexane (C7H14)/toluene (C7H8) system. However, Pt-based catalysts, which are used for both hydrogenation of C7H8 and dehydrogenation of C7H14, also promote unwanted C–C bond scission, leading to demethylation and LOHC degradation. One route to prevent C–C bond scission over Pt catalysts is to mildly poison the catalyst with sulfur. Using density functional theory (DFT) calculations and mean-field microkinetic modeling, we show that the presence of sulfur, besides suppressing demethylation, also improves the C7H14 dehydrogenation activity. The enhanced performance is a result of sulfur destabilizing neighboring co-adsorbates, which promotes C7H8 desorption. The computational results for dehydrogenation over Pt surfaces reproduce experimental trends for alumina-supported Pt nanoparticles, suggesting that controlled, moderate sulfur poisoning is a universal means to improve the overall performance of Pt catalysts for LOHC dehydrogenation.
1 Introduction
As hydrogen is gaining traction as an energy vector, more practical storage and transport solutions are required for economic viability.1 Liquid organic hydrogen carriers (LOHCs) offer a sustainable and infrastructure-compatible approach to hydrogen storage, enabling reversible incorporation of hydrogen in organic liquids. Several LOHC systems have been explored, including perhydrobenzyltoluene (H12-BT)/benzyltoluene (H0-BT), perhydrodibenzyltoluene (H18-DBT)/dibenzyltoluene (H0-DBT), and methylcyclohexane (C7H14)/toluene (C7H8).2,3 The C7H14/C7H8 system, shown in Fig. 1, has a reasonable hydrogen storage capacity (6.1 wt%), is comparatively inexpensive,2 and has already been commercialized.4,5 However, dehydrogenation of C7H14 requires high reaction temperatures (approximately 320 °C), opening up the possibility of unwanted side-reactions.3 A key challenge is therefore to increase the selectivity of dehydrogenation catalysts.
 |
| | Fig. 1 Investigated LOHC system. | |
Hydrogenation and dehydrogenation reactions are commonly performed over Pt-based catalysts. However, Pt also promotes C–C scission, including demethylation.3 Interestingly, moderate sulfur poisoning of Pt catalysts has been shown to improve the dehydrogenation selectivity of C7H14 (ref. 6) and H18-DBT7 over Pt/Al2O3, as well as several other reforming reactions.8–11 A suggested reason for the enhanced selectivity is that sulfur primarily adsorbs at under-coordinated atoms, effectively blocking these sites.7,12,13 Under-coordinated sites are experimentally known to facilitate C–C scission,14 meaning that restricted access to these sites could increase the selectivity toward the desired dehydrogenation reaction.
In addition to improving selectivity, sulfur poisoning of Pt/Al2O3 has also been reported to enhance the dehydrogenation activity for C7H14 (ref. 6) and H18-DBT.7 The enhanced activity cannot be explained by poisoning of under-coordinated sites alone, as a maximum in H18-DBT dehydrogenation activity was recorded at the same ratio of accessible metal atoms after sulfur poisoning on three different catalyst samples.7 This implies that the impact of sulfur on dehydrogenation is not strongly dependent on the catalyst surface geometry. Because sulfur oxidizes Pt, it has been suggested that sulfur modifies the interaction between Pt and other adsorbates.15,16 However, the underlying mechanisms and the consequences for dehydrogenation of C7H14 are unknown.
Here, we use density functional theory (DFT) calculations to investigate how adsorbed sulfur interacts with co-adsorbates, and how it influences dehydrogenation of C7H14 and demethylation of C7H8. Calculations are performed for flat and stepped surfaces, including Pt(100), Pt(111), Pt(223), and Pt(553). We show that the distribution of adsorbed sulfur depends on the Pt surface and that sulfur exhibits a destabilizing effect on neighboring co-adsorbates. The effect of sulfur on the dehydrogenation reaction is two-fold: by substantially decreasing the adsorption energy of C7H8, sulfur i) improves reaction kinetics and ii) prevents demethylation of C7H8.
2 Computational methods
2.1 First principles calculations
DFT calculations were performed using the plane-wave Vienna ab initio simulation package (VASP) 6.3,17–19 within the generalized gradient approximation (GGA) using the Perdew–Burke–Ernzerhof (PBE)20 exchange–correlation functional. Van der Waals interactions were accounted for by the zero-damping Grimme D3 dispersion correction.21 Spin-polarization was applied to relevant gas phase atoms and molecules, including H, S, C7H13, C7H11, and C7H9. Valence–core interactions were described by the projector-augmented wave (PAW)22 scheme, treating the following valence electrons explicitly: 1s1 (H), 2s22p2 (C), 2s22p4 (O), 3s23p4 (S) and 6s15d9 (Pt).
Pt(100), Pt(111), Pt(223), and Pt(553) surface slabs were constructed using the calculated lattice constant of 3.92 Å, which coincides with the experimental value. The Pt bulk calculation was performed by applying a (12 × 12 × 12) k-point grid to the Pt primitive unit cell. p(4 × 4) surface cells were used for Pt(100) and Pt(111), whereas p(4 × 1) surface cells were used for Pt(223) and Pt(553). All slabs were constructed using five atomic layers, where the bottom two layers were constrained to the bulk positions. Periodic images in the z-direction were separated by at least 15 Å. Following a convergence study of the adsorption energy of CO in an atop configuration on Pt(111), the plane-wave cutoff was set to 450 eV and the k-point density to a (4 × 4 × 1) gamma-centered grid for all surfaces. With these settings, the absolute adsorption energy of CO is converged within 0.04 eV. A denser k-point grid of (8 × 8 × 1) was used when evaluating the electronic density of states. Gas phase molecules were modeled in a (30 × 30 × 30) Å box, using only the gamma point.
Structural relaxations were performed using a conjugate gradient force optimizer until all forces were converged within 0.01 eV Å−1. The convergence criterion of the electronic self-consistent loop was set to 10−6 eV. First order Methfessel–Paxton smearing of 0.1 eV was applied to all surfaces. Transition states were obtained through the nudged elastic band (NEB) and climbing image tools developed by the Henkelman group23 using the FIRE24 optimizer with default settings. The bottom two layers of each slab were removed during the NEB calculations, leaving three layers, of which the third was constrained. Trajectories were interpolated using seven images between fully relaxed initial and final configurations. After partial conversion without using the climbing image scheme, the image immediately following the apparent transition state was selected as the new final state. The new final state was relaxed and a new trajectory involving three to seven images was created. Thereafter, a NEB optimization with climbing images was performed until all forces throughout the trajectory were converged within 0.02 eV Å−1. Vibrational modes were calculated for the transition state structure to ensure that a saddle point has been reached, identified through the appearance of a single imaginary frequency (or possibly two, if one of them corresponded to a frustrated rotation of methyl). Vibrational frequencies of adsorbates were calculated using the harmonic approximation and central finite differences with 0.015 Å steps.
The adsorption energy of a single adsorbate X on the surface cell was calculated as:
| | |
Eads,X = EX/slab − Eslab − EX(g)
| (1) |
where
EX/slab,
Eslab and
EX(g) are the electronic energies of the modified slab, the pristine slab and the gas phase adsorbate molecule, respectively. The differential adsorption energy was calculated according to:
| | |
Ediff,X = EX,nY/slab − EnY/slab − EX(g)
| (2) |
where
n is the number of species Y,
EX,nY/slab is the electronic energy of the slab modified with both X and
nY, and
EnY/slab is the electronic energy of the slab modified with
nY.
The dehydrogenation reaction from C7H14 to C7H8 was modeled on Pt(111) using a sulfur coverage of 3/16 (∼0.19) monolayers (ML). H was removed from one carbon site of C7H14 at a time, and the lowest energy configuration was chosen as the C7H13 species. The same procedure was repeated to obtain C7H12/Pt(111), C7H11/Pt(111), C7H10/Pt(111), C7H9/Pt(111) and C7H8/Pt(111). Co-adsorption of the carbon-containing molecules with H was investigated by placing H on each available site close to the dehydrogenated atom. The energy landscape for the pristine Pt(111) surface was obtained by structural relaxation after removing sulfur from the corresponding sulfur-modified structures.
2.2 Microkinetic modeling
The reaction kinetics were investigated using a simplified version of the microkinetic mean-field model developed in ref. 25. In the simplified model, the dehydrogenation of C7H14 and the demethylation of C7H8 were modeled as lumped reactions. The considered reaction network includes the following reaction steps:| |
 | (R1) |
| |
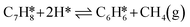 | (R2) |
| |
 | (R3) |
| |
 | (R4) |
| |
 | (R5) |
where * denotes an adsorption site, which can be composed of one or more metal sites. Reaction and adsorption energies for reactions (R1)–(R6) were obtained from the DFT calculations performed in the present work. Activation energies and partition functions were obtained from ref. 25. The partition function of the simplified dehydrogenation reaction (R1) was approximated as the partition function for the dehydrogenation of C7H14 to C7H13 + H. The activation energy of the forward reaction of (R1) was taken as the highest activation energy among the dehydrogenation steps. The backward activation energy was taken as the sum of the forward activation energy (1.20 eV) and the reaction energy (0.68 eV). For the lumped demethylation reaction (R2), the partition function and the activation energy were modeled by the first step of C7H8 demethylation, as reported in ref. 25. All kinetic parameters of the model are reported in Table S9.
In the microkinetic model, Pt(111) modified with a 0.19 ML sulfur coverage was used as a reference system. Accordingly, a change in the sulfur coverage was represented by a change in the reaction energies. Sulfur was assumed to not compete for sites with the reaction intermediates. The maximum total coverage of carbon-based molecules was fixed to 0.11 ML. The simulations were performed in the temperature range of 573–923 K, using p[C7H14] = 1.00 bar, considering 5% conversion to C7H8 + H2 and 1% conversion to C6H6 + CH4. Further details of the model are given in the SI.
3 Results and discussion
3.1 Sulfur adsorption
Adsorption of sulfur was investigated on Pt(100), Pt(111), Pt(223) and Pt(553), considering the adsorption sites indicated in Fig. 2. Sulfur adsorbs preferentially at hollow sites, fcc hollow sites, step edge hcp hollow sites and step edge fcc hollow sites on Pt(100), Pt(111), Pt(223) and Pt(553), respectively (results for Pt(111) and Pt(553) are shown in Fig. 3). The preferred adsorption sites on Pt(100) and Pt(111) agree with experimental findings,26,27 and it has previously been suggested that sulfur preferably adsorbs at under-coordinated sites on nanoparticles.7 The adsorption energies with respect to atomic sulfur for the preferred adsorption sites are −6.16 eV on Pt(100), −5.73 eV on Pt(111), −5.67 eV on Pt(223) and −5.56 eV on Pt(553).
 |
| | Fig. 2 Investigated adsorption sites on Pt(100), Pt(111), Pt(223) and Pt(553). The letters in the headings indicate the axis or plane along which the image is oriented. The atomic models were made using VESTA.28 | |
 |
| | Fig. 3 Adsorption configurations of sulfur on Pt(111) (left) and Pt(553) (right). The adsorption energies are indicated. | |
The adsorption energy is higher in the fcc site on Pt(111) than at the step edges of both Pt(223) and Pt(553). This is not unexpected, considering that the stepped surfaces are subjected to higher internal compressive strain, and that an increase in strain is associated with a decrease in adsorption energy.29,30 Defining strain as the average shift in interatomic distance, relative to the equilibrium interatomic distance d0 in bulk Pt ((d − d0)/d0), among the Pt atoms coordinated to sulfur at the considered adsorption site, we find that the strain is −0.1%, −1.7% and −2.3% for adsorption in fcc sites on Pt(111), step edge fcc sites on Pt(553) and terrace fcc sites on Pt(553), respectively (see Table 1). There is no obvious trend between the preferred adsorption sites and strain on Pt(553) itself, as variations in, for example, bond length and coordination number will also influence the adsorption energies. However, it is clear that the strain is higher for Pt(553) than for Pt(111). The adsorption energy scales linearly with strain on Pt(111), as shown in the SI.
Table 1 Adsorption energies and strain at the considered adsorption sites for sulfur on Pt(111) and Pt(553)
| Surface |
Adsorption site |
Eads (eV) |

(%) |
| Pt(111) |
fcc |
−5.73 |
−0.1 |
| hcp |
−5.53 |
−0.1 |
| Top |
−3.57 |
+0.3 |
| Pt(553) |
Step edge fcc |
−5.56 |
−1.7 |
| Terrace fcc |
−5.45 |
−2.3 |
| Terrace hcp |
−5.32 |
−2.3 |
| Step edge hcp |
−5.30 |
−1.7 |
| Step bottom fcc |
−5.22 |
−1.5 |
| Step three-fold hollow |
−5.18 |
−1.6 |
| Step edge bridge |
−5.04 |
0.0 |
| Step four-fold hollow |
−4.75 |
−1.8 |
The distribution of sulfur on Pt nanoparticles will depend on how the adsorption energy is affected by strain. On nanoparticles with large terraces, where the local strain approaches that of Pt(111), sulfur would adsorb on the terrace rather than at under-coordinated sites. In contrast, on nanoparticles with small terraces, where the strain is higher, sulfur would primarily adsorb at under-coordinated sites. Consequently, the nature of sulfur poisoning of Pt catalysts – previously often attributed to blocking of under-coordinated sites12 – depends on the catalyst surface geometry.
The presence of sulfur on the surface will affect the adsorption properties of other adsorbates. In ref. 7, CO was used as a probe molecule for sulfur adsorption on Pt nanoparticles. Here we investigate adsorption of sulfur and CO on Pt(100), Pt(111), Pt(223), and Pt(553). Considering first only CO adsorption, we find that CO preferentially adsorbs at bridge sites on Pt(100) (−2.49 eV) and at fcc hollow sites on Pt(111) (−2.16 eV). These results are in agreement with previous computational studies at low CO coverages.31–35 We note that the adsorption energies are overestimated compared to experimental results, according to which the adsorption energy of CO is approximately −1.9 eV on Pt(111),36 and that the experimentally favored top site on Pt(111) is not reproduced. These discrepancies are well-known limitations of using the PBE functional,37–39 although the inclusion of van der Waals corrections mitigates the site-ordering error between the top and fcc sites.40
CO preferentially adsorbs at step bridge sites (−2.44 eV) and at step top sites (−2.26 eV), on Pt(223) and Pt(553), respectively. As noted above, sulfur also favors adsorption at step edges on Pt(223) and Pt(553), implying that CO and sulfur compete for the under-coordinated sites on these surfaces. This result agrees with the diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) observations of Auer et al.7 Upon sulfur exposure, spectral features associated with CO at defect or corner/apex sites disappeared and all CO-related peaks shifted to higher wavenumbers.
The effect of sulfur on CO adsorption can be examined in greater detail by evaluating the CO adsorption energy in different sulfur co-adsorption configurations. On all studied surfaces, the adsorption energy and preferred adsorption site of CO are unaffected by the presence of sulfur as long as CO and sulfur are separated by at least one Pt atom. However, when sulfur and CO are positioned as nearest neighbors, such that they share at least one Pt atom, the adsorption energy of CO decreases on all four surfaces. On Pt(111), sharing the same Pt atom does not correspond to a minimum on the potential energy surface and CO relaxes to a bridge site during the geometry optimization. Similarly, CO moves from its preferred step edge top site to a step edge bridge site on Pt(553). These results are shown in Fig. 4.
 |
| | Fig. 4 Adsorption energies and configurations of CO with and without sulfur on Pt(111) (left column) and Pt(553) (right column). The first row from the top shows CO adsorption on pristine Pt. The second (third) row from the top shows non-nearest-neighbor (nearest neighbor) configurations of CO and sulfur. The markers in the third row indicate the adsorption sites of CO before structural relaxation. The fourth row shows the sulfur-induced charge density difference relative to the pristine Pt surface (isosurface 0.001 Å−3). Blue and purple indicate electron accumulation and depletion, respectively. | |
The decrease in CO adsorption energy in the immediate vicinity of sulfur is rationalized by the traditional d-band model.41 For both Pt(111) and Pt(553), the d-band centers εd of the sulfur-coordinated Pt atoms are shifted to lower energy (by 0.64 and 0.45 eV, respectively, see the SI), which is associated with a weakening of the chemisorption energy of nearby species. Notably, only the d-band centers of the Pt atoms directly bound to sulfur are strongly affected, clearly showing that the effect of sulfur on co-adsorbates is local.
The local character of the electronic influence of sulfur on the Pt surface is further illustrated by the sulfur-induced charge density difference relative to the pristine Pt surface, as shown in the bottom panel of Fig. 4. From this analysis, it is evident that sulfur partially oxidizes the Pt surface and that the lateral extension of the effect is limited to the Pt atoms directly bound to sulfur. Although the lateral effect of sulfur is local, the induced charge-density difference normal to the surface extends to the fourth atomic layer of the slab. This is understood considering that it is the dyz, dxz and dz2 orbitals that are involved in bonding to the sulfur orbitals. It is noteworthy that the appearance and spatial extent of the induced charge-density difference are nearly identical on the Pt(111) facet and the step edge of Pt(553).
The spatial extent of the electronic influence determines which sulfur coverage is attainable at under-coordinated sites. The adsorption energy of sulfur at the step edge of Pt(553) is the same for a 25% and a 50% sulfur coverage of these sites, −5.56 eV. At a 75% sulfur coverage, some sulfur atoms must share Pt atoms, which lowers the adsorption energy to −4.94 eV. As the adsorption energy on the terrace of Pt(553) is −5.45 eV, sulfur will start to occupy terrace sites at higher coverages than 50%. Thus, a 50% coverage of the step edge is sufficient to destabilize all co-adsorbates at the step.
3.2 Effect of sulfur on dehydrogenation of C7H14 and demethylation of C7H8
3.2.1 Adsorption of C7H14 and C7H8. The effect of sulfur on the adsorption properties of C7H14 and C7H8 was evaluated for Pt(111) and Pt(553). On Pt(111), C7H14 is adsorbed in a slightly buckled configuration in which the carbon ring is positioned above an fcc site and the methyl group is aligned above an hcp site. The adsorption energy for this configuration is −1.49 eV. On Pt(553), C7H14 is adsorbed over an hcp hollow site at the bottom of the step, with the CH3 group partially angled toward the step. The adsorption energy is in this case −1.57 eV. These configurations are shown in Fig. 5. C7H14 is physisorbed on both surfaces, i.e. the bonding is governed by electrostatic and van der Waals interactions. Because C7H14 is physisorbed, sulfur does not significantly impact its adsorption energy. On Pt(111), the presence of 1/16 ML sulfur (corresponding to a single sulfur atom in the supercell) leaves the adsorption energy of C7H14 unchanged, and on Pt(553), a 50% coverage of sulfur along the step edge decreases the adsorption energy from −1.57 eV to −1.51 eV. A 50% coverage of sulfur at the step edge is therefore not enough to change the adsorption site of C7H14 to the terrace of Pt(553), where the adsorption energy is −1.42 eV.
 |
| | Fig. 5 Adsorption energies of C7H8 (upper panel) and C7H14 (lower panel) on pristine and sulfur-modified Pt(111) and Pt(553). Two configurations are shown for Pt(553); the overall most thermodynamically favorable, and the most favorable configuration on the terrace. | |
The situation is different for C7H8. On Pt(111), C7H8 adsorbs in a bridge configuration similar to the most favorable orientation of benzene on Pt(111).42 The molecule is chemisorbed with an adsorption energy of −2.78 eV. The adsorption energy is higher on Pt(553), −3.07 eV. C7H8 is in this case preferentially adsorbed at the step with the methyl group aligned along the step edge. In comparison, the adsorption energy at the most stable configuration on the terrace of Pt(553) is −2.40 eV. Thus, C7H8 exhibits a strong preference for adsorption at under-coordinated sites, which are known to promote C–C scission.14
In contrast to the case of C7H14, sulfur has a notable effect on the adsorption energy of C7H8 on both Pt(111) and Pt(553), even if sulfur and C7H8 do not share Pt atoms directly on Pt(111). In the studied configuration on Pt(111), C7H8 binds to three Pt atoms that are nearest neighbors to sulfur-coordinated Pt sites, resulting in an electronic interaction between chemisorbed C7H8 and sulfur, mediated through the surface. In the presence of 1/16 ML (∼6%) of sulfur on Pt(111), the adsorption energy of C7H8 is decreased from −2.78 eV to −2.56 eV. On Pt(553), a 50% sulfur coverage along the step edge decreases the adsorption energy of C7H8 from −3.07 eV to −2.24 eV. Consequently, on Pt(553), C7H8 is less stable at the step in the presence of sulfur than on the terrace. That is, a moderate coverage of sulfur on the step edge could, in principle, change the preferred adsorption site of C7H8 to the terrace, where C–C bond scission is less likely. However, as will be discussed below, when taking the entire dehydrogenation reaction into account, the most significant effect of sulfur is that it destabilizes C7H8 adsorption and in this way promotes C7H8 desorption over demethylation.
3.2.2 Demethylation of C7H8. Preventing C–C bond scission is relevant at all stages of dehydrogenation. However, the strong preference of C7H8 for adsorption at under-coordinated sites warrants a discussion on, in particular, demethylation of C7H8. Focusing on demethylation is further motivated by the fact that desorption of C7H8 has proven to be kinetically important for dehydrogenation of C7H14 over Pt(111).25 The demethylation reaction proceeds according to:| |
 | (R7) |
| |
 | (R8) |
| |
 | (R9) |
Over Pt(111), the first reaction step (R7) is endothermic by 1.21 eV and associated with an energy barrier of 2.26 eV, see Fig. 6. However, the hydrogenation step (R8) makes the surface-bound demethylation reaction (R7) and (R8) exothermic by −0.73 eV.
 |
| | Fig. 6 The first step of demethylation of C7H8 over Pt(111) and Pt(553). The upper and lower panels show the most stable configurations of C7H8 and C6H5 + CH3, respectively. The middle panels show identified transition states and barrier heights in eV. For Pt(553), the three identified transition states are denoted by A, B and C. | |
Cleaving C7H8 to C6H5 + CH3 (R7) is considerably less endothermic over Pt(553) than over Pt(111). Over Pt(553), the dissociation is endothermic by 0.69 eV. The difference is mainly attributed to the stabilization of C6H5, which on Pt(553) preferentially adsorbs in an fcc site at the step with the radical center bound to the bottom of the step. CH3 adsorbs C-down at a top site at the step edge, but its preference for this under-coordinated site over adsorption on the terrace is marginal. In addition to being less endothermic, the first step of demethylation has a slightly lower barrier over Pt(553) than over Pt(111). Three transition states were identified, along the following trajectories: A) rotation of C7H8 toward the bottom of the step and CH3 occupying a top site at the lower terrace after C–C scission, B) rotation of C7H8 toward the upper terrace and C–C scission over a bridge site along the step edge, and C) rotation of C7H8 toward the upper terrace and CH3 occupying a top site at the upper terrace. The trajectories are shown in the right panel of Fig. 6. The associated activation energies are 2.50 eV, 2.54 eV and 2.20 eV, respectively. Note that additional transition states over Pt(553) with lower barriers may exist that were not captured by the NEB approach. These observations support experimental evidence that C–C scission is favored at under-coordinated sites.14
3.2.3 Effect of sulfur on dehydrogenation of C7H14. The preferential adsorption and demethylation of C7H8 on Pt(553) relative to Pt(111) are consistent with experimental observations showing enhanced dehydrogenation selectivity when under-coordinated sites are poisoned by sulfur.7 In addition to improving selectivity, sulfur poisoning has also been reported to enhance the dehydrogenation activity of H18-DBT7 and C7H14.6 Our calculations indicate that C7H14 does not adsorb directly at step sites and that C7H8 preferentially binds to facet sites rather than sulfur-poisoned steps. Moreover, as the degree of chemisorption (the number of Pt–C covalent bonds) increases during dehydrogenation from C7H14 to C7H8, the initial adsorption site of C7H14 can be expected to determine the primary reaction site. Consequently, dehydrogenation should preferentially occur on facet sites, and the positive effect of sulfur is likely electronic in nature, arising from sulfur-induced modification of the Pt surface.Here we considered the full dehydrogenation reaction from C7H14 to C7H8 over Pt(111), with stepwise scission of C–H bonds and desorption of H2 to the atmosphere:
| |
 | (R10) |
| |
 | (R11) |
| |
 | (R12) |
| |
 | (R13) |
| |
 | (R14) |
| |
 | (R15) |
| |
 | (R16) |
| |
 | (R17) |
| |
 | (R18) |
| |
 | (R19) |
| |
 | (R20) |
| |
 | (R21) |
| |
 | (R22) |
| |
 | (R23) |
where, in the case of demethylation, step
(R23) is replaced by
reactions (R7)–(R9).
Fig. 7 shows the potential energy diagram for dehydrogenation over Pt(111) modified with 3/16 ML sulfur (∼19% of available surface fcc sites) and over pristine Pt(111). The adsorption of C7H14 is close to unaffected by the presence of sulfur. In subsequent reaction steps, sulfur increasingly destabilizes the carbon-containing molecules from physisorbed (C7H14) to chemisorbed (C7H8). Sulfur also increases the reaction energy of the first demethylation step (R7) from 1.28 eV to 1.32 eV, but leaves the activation energy of this step mostly unaffected (2.25 eV vs. 2.26 eV) (we note that the transition state found for the sulfur-modified surface did not fully converge under the chosen criteria, but oscillated by ∼0.1 eV over the last 100 ionic steps). Desorption of C7H8 (R23) is significantly less endothermic in the presence of sulfur (2.18 eV vs. 2.78 eV). Thus, sulfur inverts the preference of steps (R7) and (R23): the first demethylation step (R7) is favored over desorption (R23) on pristine Pt(111), whereas the opposite applies on the sulfur-modified surface.
 |
| | Fig. 7 Dehydrogenation of C7H14 to C7H8 and demethylation of C7H8 to C6H6 + CH4 on Pt(111) in the presence of 0% and 19% S. The insets show the obtained configuration of C7H14 (left) and C7H8 (right) on Pt(111) modified with 19% S. Labels a, b and c indicate C7H8(g) + 3H2(g),  + +  and C6H6(g) + CH4(g) + 2H2(g), respectively. and C6H6(g) + CH4(g) + 2H2(g), respectively. | |
The energy diagram in Fig. 7 displays the purely electronic influence of sulfur on dehydrogenation, as diffusion and rotation beyond what occurred during structure relaxation were not considered for any of the intermediates. In particular for the pristine case, more favorable configurations may exist, as the starting configuration for the optimization was obtained from the sulfur-modified structure. Consequently, the difference in stability between the pristine and sulfur-modified surface is underestimated.
The fact that sulfur affects the first and last steps of dehydrogenation differently is beneficial for the reaction kinetics. In ref. 25, it was demonstrated that decreasing the adsorption energy of C7H14 lowers the turnover frequency (TOF), whereas decreasing the adsorption energy of C7H8 increases the TOF. Because sulfur does not alter the adsorption energy of C7H14 but destabilizes C7H8, introducing a small amount of sulfur to Pt(111) should not affect the reaction kinetics of C7H14 adsorption, but likely increase the C7H8 desorption rate. Therefore, moderate sulfur poisoning should enhance catalyst selectivity by favoring product desorption over a side reaction, and also improve activity by facilitating a kinetically important step.
It should be noted that modifying the catalyst surface with sulfur introduces a risk of forming SH and subsequently H2S. However, the formation of SH from S + H is calculated to be endothermic by 1.08 eV (calculated using a p(3 × 3) surface cell with 5 atomic layers). Interestingly, the endothermicity of the reaction contrasts with the behavior of oxygen and hydrogen, as water is known to form on Pt(111).43 The difference is attributed to the larger size of sulfur, which causes sulfur to bind more strongly to the surface than does oxygen – their respective adsorption energies are −5.60 eV and −4.28 eV. This result agrees with previous computational studies showing that H2S formation is endothermic over Pt(111),44 as well as with experimental dehydrogenation of H18-DBT, where no H2S was detected in the exhaust after catalyst pretreatment in a reducing hydrogen atmosphere.7
4 Sulfur coverage and dehydrogenation kinetics
The energy landscape in Fig. 7 was evaluated at a sulfur coverage of 3/16 ML (∼19%). This coverage was chosen to have the highest effect on C7H8 adsorption, while still allowing adsorption of C7H14. The dependence of C7H8 and C7H14 adsorption on sulfur coverage is shown in Fig. 8. The adsorption energy of C7H8 decreases from −2.78 eV to −2.26 eV when the sulfur coverage is increased from 0 to 3/16 ML. The clear coverage dependence originates from the fact that C7H8 is chemisorbed on the surface. Conversely, the adsorption energy of C7H14, which is physisorbed, remains unaffected up to a sulfur coverage of 3/16 ML. At higher sulfur coverages, the molecules cannot access enough metal sites and the adsorption energies decrease significantly.
 |
| | Fig. 8 Adsorption energy of C7H14 and C7H8 as a function of sulfur coverage on Pt(111). | |
There is also a limit to the amount of sulfur that can be adsorbed on the surface. At about 5/16 ML (∼31%), some sulfur atoms must share metal sites, resulting in a significantly lower average adsorption energy. According to the above discussion on sulfur-induced charge density difference, each sulfur atom can be assumed to prevent co-adsorption on three metal sites. At 4/16 ML (25%), 12/16 ML or 75% of all metal sites on the surface would be effectively blocked by sulfur. This result aligns with the experimental findings by Auer et al.,7 suggesting that there is an upper limit to sulfur loading on Pt/Al2O3 corresponding to 26–35% available metal sites.
The 3/16 ML coverage of sulfur used to construct the potential energy diagram in Fig. 7 corresponds to 44% available metal sites. This value agrees well with ref. 7, where the maximum H18-DBT dehydrogenation activity was achieved at 44–48% available metal sites for all their investigated catalyst samples. In fact, through microkinetic mean-field modeling, we accurately reproduce the experimentally observed activity as a function of accessible metal sites, Fig. 9. Specifically, we find that the TOF increases between 100% and 44% available metal sites, corresponding to sulfur coverages of 0 to 19%. When the sulfur coverage reaches 25%, the TOF decreases dramatically, which coincides with the calculated sulfur-induced drop in adsorption energy of both C7H8 and C7H14 at this coverage.
 |
| | Fig. 9 Maximum TOF as a function of sulfur coverage and accessible metal atoms. The simulations were performed using T = 573–923 K, p[C7H14] = 1.00 bar, 5% conversion to C7H8 + H2, 1% conversion to C6H6 + CH4, and a saturation coverage of 11% for the carbon-containing species. The maximum TOF is reached at temperatures of 817 K, 787 K, 773 K, 769 K and 787 K, from lowest to highest sulfur coverage. Each sulfur atom is assumed to block three metal sites. C7Hn indicates C7H8 or C7H14. | |
As shown in Fig. 9, the dependence of the TOF on the sulfur coverage reflects the surface coverages of C7H8 and C7H14 (reported with respect to the number of Pt surface sites). The coverage of C7H8 decreases with increasing sulfur coverage, whereas the coverage of C7H14 follows the trend in the TOF. In particular, the coverage of C7H14 increases between 0 and 19% sulfur and drops at higher sulfur coverages. The kinetic results highlight that sulfur coverages of up to 19% promotes dehydrogenation activity by facilitating desorption of C7H8, which frees sites for C7H14 adsorption. As we reach the same conclusions as ref. 7 regarding both maximum and optimum sulfur coverage, using Pt(111) as a model surface, we conclude that the most important contribution of sulfur to dehydrogenation performance is not to block under-coordinated sites but rather to electronically modify the Pt surface.
Conclusions
We have investigated the effects of under-coordinated sites and sulfur on the dehydrogenation of C7H14 to C7H8 over Pt surfaces. The strongest sulfur adsorption energy is obtained on extended Pt(111). On stepped Pt(223) and Pt(553) surfaces, sulfur preferentially adsorbs at under-coordinated sites; however, the adsorption is weaker than on Pt(111). Compressive strain makes the adsorption energies on the (111) micro-facets of Pt(223) and Pt(553) weaker than on Pt(111). Irrespective of the surface geometry, sulfur locally modifies the charge density and d-band center of neighboring Pt atoms, thereby reducing the adsorption energies of co-adsorbates in its immediate vicinity.
Sulfur influences the adsorption and reaction properties of C7H14 and C7H8 in two main ways. First, controlled sulfur poisoning suppresses undesired demethylation of C7H8 by blocking access to step sites, which are both the preferred adsorption sites of chemisorbed C7H8 and the primary reaction sites for demethylation. Second, sulfur coverages from 0 to 19% reduces the adsorption energy of C7H8, approximately linearly. In contrast, sulfur coverages of up to 19% leave C7H14 — preferentially physisorbed on (111) facets — largely unaffected. At sulfur coverages above 19%, the adsorption energies of both C7H8 and C7H14 decrease substantially.
The sulfur-induced changes in adsorption and reaction properties have important consequences for the dehydrogenation of C7H14 to C7H8. The primary effects are facilitated C7H8 desorption and suppression of demethylation. Our DFT-based mean-field microkinetic model shows that decreasing the C7H8 adsorption energy leads to an increased turnover frequency, with a maximum at a sulfur coverage of 19%, corresponding to 44% available metal sites. Our computational results for Pt(111) are consistent with ref. 7, where H18-DBT dehydrogenation over Pt nanoparticles was found to reach maximum activity at 44–48% available metal sites. Our study suggests that an important effect of adsorbed sulfur is its electronic modification of the catalyst surface. In conclusion, controlled and moderate sulfur poisoning of Pt catalysts prevents LOHC degradation and improves overall catalyst performance.
Author contributions
Felicia Zaar: investigation, formal analysis, visualization, writing – original draft. Alvaro Posada-Borbón: methodology, investigation, writing – review & editing. Henrik Grönbeck: conceptualization, methodology, funding acquisition, supervision, writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Data availability
Data for this article, including vasprun.xml files for structure relaxations, vibrational modes, charge densities, densities of states and NEB trajectories, are available at Zenodo at https://www.doi.org/10.5281/zenodo.18601893.
Supplementary information (SI): convergence tests, d-band center analysis, additional adsorption energy results, detailed description of the micro-kinetic model and additional kinetic results. See DOI: https://doi.org/10.1039/d6cy00183a.
Acknowledgements
The Competence Centre for Catalysis (KCK) is hosted by Chalmers University of Technology and is financially supported by the Swedish Energy Agency (52689-1) and the member companies Johnson Matthey, Perstorp, Powercell, Preem, Traton, Umicore, and Volvo Group. Additional support is acknowledged from the Swedish Research Council (2024-05250) and the Wallenberg Initiative Material Science for Sustainability (WISE). The calculations were performed at NSC via a NAISS grant (NAISS 2024/3-26).
Notes and references
- M. Niermann, S. Drünert, M. Kaltschmitt and K. Bonhoff, Energy Environ. Sci., 2019, 12, 290–307 RSC.
- J. Cashel, D. Yan, R. Han, H. Jeong, C. W. Yoon, J. A. Ambay, Y. Liu, A. T. Ung, L. Yang and Z. Huang, Angew. Chem., Int. Ed., 2025, 64, e202423661 CrossRef CAS PubMed.
- P. Preuster, C. Papp and P. Wasserscheid, Acc. Chem. Res., 2017, 50, 74–85 CrossRef CAS PubMed.
- Y. Okada, T. Mikuriya and M. Yasui, Kemikaru Enjiniyaringu, 2015, 60, 187 CAS.
- Y. Okada and M. Yasui, Hyomen Kagaku, 2015, 36, 577–582 CrossRef CAS.
- Y. Okada, K. Imagawa and M. Yasui, 8th Tokyo Conference on Advanced Catalytic Science and Technology (TOCAT8), 2018 Search PubMed.
- F. Auer, D. Blaumeiser, T. Bauer, A. Bösmann, N. Szesni, J. Libuda and P. Wasserscheid, Catal. Sci. Technol., 2019, 9, 3537–3547 RSC.
- C. Apesteguía and J. Barbier, J. Catal., 1982, 78, 352–359 CrossRef.
- P. van Trimpont, G. Marin and G. Froment, Appl. Catal., 1985, 17, 161–173 CrossRef CAS.
- W. J. Doolittle, N. D. Skoularikis and R. W. Coughlin, J. Catal., 1987, 107, 490–502 CrossRef CAS.
- J. Parera and J. Beltramini, J. Catal., 1988, 112, 357–365 CrossRef CAS.
- F. Alhumaidan, D. Cresswell and A. Garforth, Energy Fuels, 2011, 25, 4217–4234 CrossRef CAS.
- R. Streber, C. Papp, M. P. A. Lorenz, A. Bayer, S. Wickert, M. Schöppke, R. Denecke and H.-P. Steinrück, J. Phys.: Condens. Matter, 2009, 21, 134018 CrossRef CAS PubMed.
- G. Somorjai and D. Blakely, Nature, 1975, 258, 580–583 CrossRef CAS.
- T. Tri, J. Massardier, P. Gallezot and B. Imelik, Metal-Support and Metal-Additive Effects in Catalysis, Elsevier, 1982, vol. 11, pp. 141–148 Search PubMed.
- C. Apesteguia, C. Brema, T. Garetto, A. Borgna and J. Parera, J. Catal., 1984, 89, 52–59 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- E. Bitzek, P. Koskinen, F. Gähler, M. Moseler and P. Gumbsch, Phys. Rev. Lett., 2006, 97, 170201 CrossRef PubMed.
- A. Posada-Borbón, T. Möslinger and H. Grönbeck, ChemRxiv, 2026, preprint, DOI:10.26434/chemrxiv.10001989/v1.
- T. E. Fischer and S. R. Kelemen, Surf. Sci., 1977, 69, 1–22 CrossRef CAS.
- K. Hayek, H. Glassl, A. Gutmann, H. Leonhard, M. Prutton, S. Tear and M. Welton-Cook, Surf. Sci., 1985, 152–153, 419–425 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- M. Mavrikakis, B. Hammer and J. K. Nørskov, Phys. Rev. Lett., 1998, 81, 2819–2822 CrossRef.
- W. Vanmoerkerke, R. Svensson and H. Grönbeck, Surf. Sci., 2025, 754, 122678 CrossRef CAS.
- S. Yamagishi, T. Fujimoto, Y. Inada and H. Orita, J. Phys. Chem. B, 2005, 109, 8899–8908 CrossRef CAS PubMed.
- K. Doll, Surf. Sci., 2004, 573, 464–473 CrossRef CAS.
- M. Gajdoš, A. Eichler and J. Hafner, J. Phys.: Condens. Matter, 2004, 16, 1141 CrossRef.
- A. Stroppa, K. Termentzidis, J. Paier, G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 195440 CrossRef.
- Z. Yang, R. Wu and J. A. Rodriguez, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 155409 CrossRef.
- Y. Y. Yeo, L. Vattuone and D. A. King, J. Chem. Phys., 1997, 106, 392–401 CrossRef CAS.
- P. J. Feibelman, B. Hammer, J. K. Nørskov, F. Wagner, M. Scheffler, R. Stumpf, R. Watwe and J. Dumesic, J. Phys. Chem. B, 2001, 105, 4018–4025 CrossRef CAS.
- R. A. Olsen, P. H. T. Philipsen and E. J. Baerends, J. Chem. Phys., 2003, 119, 4522–4528 CrossRef CAS.
- K. G. Lakshmikanth, I. Kundappaden and R. Chatanathodi, Surf. Sci., 2019, 681, 143–148 CrossRef.
- P. Janthon, F. Viñes, J. Sirijaraensre, J. Limtrakul and F. Illas, J. Phys. Chem. C, 2017, 121, 3970–3977 CrossRef CAS.
- B. Hammer and J. Nørskov, Impact of Surface Science on Catalysis, Academic Press, 2000, vol. 45, pp. 71–129 Search PubMed.
- M. Saeys, M.-F. Reyniers, G. B. Marin and M. Neurock, J. Phys. Chem. B, 2002, 106, 7489–7498 CrossRef CAS.
- K. M. Ogle, J. R. Creighton, H. S. Luftman and J. M. White, J. Chem. Phys., 1983, 78, 5839–5840 CrossRef CAS.
- A. Michaelides and P. Hu, J. Chem. Phys., 2001, 115, 8570–8574 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *,
Alvaro Posada-Borbón
*,
Alvaro Posada-Borbón





























 +
+  and C6H6(g) + CH4(g) + 2H2(g), respectively.
and C6H6(g) + CH4(g) + 2H2(g), respectively.
