DOI:
10.1039/D5CY01319D
(Review Article)
Catal. Sci. Technol., 2026,
16, 3014-3057
Recent trends in palladium-catalysed isocyanide chemistry: from heterocyclic frameworks to mechanistic understanding
Received
6th November 2025
, Accepted 27th March 2026
First published on 27th March 2026
Abstract
Heterocycles represent a fundamental class of structural motifs that are integral to a wide array of pharmaceuticals, natural products, and functional organic materials, owing to their unique chemical and biological properties. The development of efficient methods for constructing these scaffolds remains a central focus in modern synthetic chemistry. Among the various strategies, palladium-catalyzed reactions have emerged as highly versatile tools, enabling selective C–H functionalization and facilitating the formation of complex molecular architectures through both inter- and intramolecular pathways. Additionally, isocyanides have attracted considerable attention as reagents due to their ambident reactivity, which allows them to act simultaneously as electrophiles and nucleophiles, making them exceptionally useful in multicomponent and cascade reactions. In recent years, Pd-catalyzed isocyanide insertion cascade reactions have witnessed significant advancements, demonstrating remarkable efficiency and broad substrate scope in the synthesis of diverse heterocyclic frameworks. This review aims to provide an overview of the recent progress in Pd-catalysed isocyanide insertion strategies, with a focus on the construction of five-membered, six-membered, and fused heterocyclic systems, highlighting their mechanistic aspects, synthetic utility, and potential applications in medicinal and materials chemistry.
Introduction
Over the past several decades, transition metal complexes have emerged as an important class of catalysts, owing to their remarkable properties such as the ability to access multiple oxidation states. Also, the presence of vacant d-orbitals that facilitate electron donation and acceptance, variable coordination numbers, and high tolerance toward a wide range of ligands make them a good choice for catalysis.1 Moreover, the homogeneous and heterogeneous nature of these catalysts enables efficient and rapid synthesis of novel organic molecules, which find diverse applications across fields such as medicine, environmental science, energy storage, and polymer synthesis.2 In particular, transition metals are widely employed to catalyse a broad spectrum of reactions, including coupling reactions, hydrogenation, alkylation, cyclopropanation, hydrogenolysis, annulation, C–X and C–H bond activation, insertion reactions, cyclization, metathesis, and carbene insertion, among others.3 Despite their long-standing role in synthetic organic chemistry, the development of novel transition metal-catalysed methodologies remains an active and highly regarded area of contemporary research.4 Among various transition metal catalysts, palladium-based catalysts have stood out as some of the most versatile and widely used in organic synthetic chemistry in recent years.5 In addition to traditional methodologies, palladium catalysts have been employed to facilitate a wide range of reactions, including coupling reactions, multicomponent reactions, etc.6 Furthermore, palladium-catalysed cross-coupling reactions between organic electrophiles and organometallic reagents have become an invaluable tool for the synthesis of C–C, C–N, and C–O bonds, enabling the development of novel and complex molecules.7 Also, Pd-based catalysts are primarily employed in tandem processes, where multiple bond-forming events occur in a single reaction sequence.8 The cascade or tandem reactions have become increasingly important in modern synthetic chemistry, offering a wide range of applications across various fields.9 These reactions enable multiple transformations to occur in a single reaction vessel without the need to isolate intermediates, and they often allow the formation of two or more chemical bonds in one step under unified conditions.10 Despite their benefits, cascade reactions also present certain challenges. These include the need for substrate compatibility under common reaction conditions, the stability of reactive intermediates, functional group tolerance, and the potential formation of undesired side products.11 Therefore, careful planning is essential when designing a cascade strategy to minimize these limitations. Nevertheless, due to their numerous advantages, cascade reactions have gained widespread popularity over traditional stepwise methods.12 As a result, considerable research efforts continue to focus on developing and optimizing such synthetic methodologies.
On the other hand, isocyanides, also known as isonitriles or carbylamines, are a unique class of organic compounds renowned for their remarkable versatility in chemical synthesis.13 Their dual reactivity—as both nucleophiles and electrophiles—makes them valuable intermediates in a wide range of transformations.14 Beyond their well-established roles in multicomponent reactions such as the Ugi and Passerini reactions, isocyanides also serve as key building blocks in various transition metal-catalyzed coupling processes.15 One of the most notable features of isocyanides is their ability to insert between two coupling partners, enabling the development of innovative cascade reactions to synthesize heterocyclic compounds.16 Among the different transition metals explored, such as Ni, Cu, Fe, and Co, palladium has emerged as particularly effective in catalysing isocyanide insertion reactions as it provides an optimal balance of strong yet tunable coordination and redox cycling with isocyanides; while alternative metals often suffer from weak activation, poor selectivity, or the need for harsh conditions, palladium enables smooth isocyanide insertion with broad functional-group tolerance under mild conditions, making it uniquely suited for this transformation.17 Over the years, palladium has emerged as a particularly powerful catalyst for isocyanide-based transformations and this success arises from the strong and selective coordination of isocyanides to palladium, their high efficiency in undergoing migratory insertion into Pd–C and Pd–X bonds, and the flexible redox behaviour of palladium, which enables smooth progression through the fundamental steps of oxidative addition, insertion, and reductive elimination.18 Alongside this, palladium-catalysed isocyanide insertion reactions have shown remarkable advancement in recent years and continue to expand rapidly. In this context, Orru et al. presented a comprehensive review summarizing the progress in Pd-catalysed isocyanide insertion reactions reported up to 2020; however, no updated review focusing specifically on this area has appeared thereafter. In this review, we highlight the significant developments reported from 2020 to the present, with particular emphasis on Pd-catalysed cascade reactions utilizing isocyanides as one of the primary starting materials.19 The review primarily covers transformations involving two or more reactants, highlighting their potential to facilitate the efficient synthesis of structurally diverse 5,6-membered, benzo-fused, and polycyclic heterocycles and quinazoline derivatives, which serve as part of many pharmaceuticals and biologically active moieties. Furthermore, these synthetic strategies are robust and can withstand various conditions to further functionalise or transform the moiety. Alongside, the scope of various isocyanide substituents' tolerance and the detailed mechanistic studies for the Pd-catalysed reactions are well discussed in this review for the better understanding of the readers.
Synthesis of five-membered heterocycles
Five-membered heterocycles, in particular, consist of at least one heteroatom such as O, N or S and have emerged prominently in pharmaceutical chemistry because of reinforced stability, physiological availability and good solubility.20 These heterocycles hold exceptional physicochemical characteristics and biological activity, which makes them a competent motif in several therapeutically potent molecules.21 In this context, we have discussed recent developments in palladium-catalysed isocyanide insertion reactions for the efficient synthesis of five-membered heterocycles. An interesting multicomponent approach toward the synthesis of biologically relevant 5-aminoimidazole scaffolds was reported by Pan et al. (Scheme 1).22 This Pd-catalyzed reaction involves amidoximes, isocyanides, and amines as substrates for the tandem formation of C–C and C–N bonds to provide imidazole derivatives in moderate to good yields. After screening various palladium-based catalysts like PdCl2, Pd(acac)2, PdBr2, Pd(dba)2, Pd(PPh3)4, Pd(OAc)2, PdCl2(PPh3)2, etc., they found Pd(OAc)2 along with the PPh3 ligand in DMSO as the best catalyst to synthesize the desired product within 6 h at elevated temperature. Further, various electron-donating and withdrawing functional groups were examined to explore the feasibility of the reaction and obtain corresponding products in good to excellent yields, which clearly demonstrates the applicability of the reported protocol. A gram-scale reaction was also performed, which appropriately justified the synthetic utility of the protocol.
 |
| | Scheme 1 (a) General reaction scheme for the Pd-catalysed synthesis of 5-aminoimidazole scaffolds. (b) Substrate scope showing selected examples with corresponding yields (c) gram-scale synthesis. | |
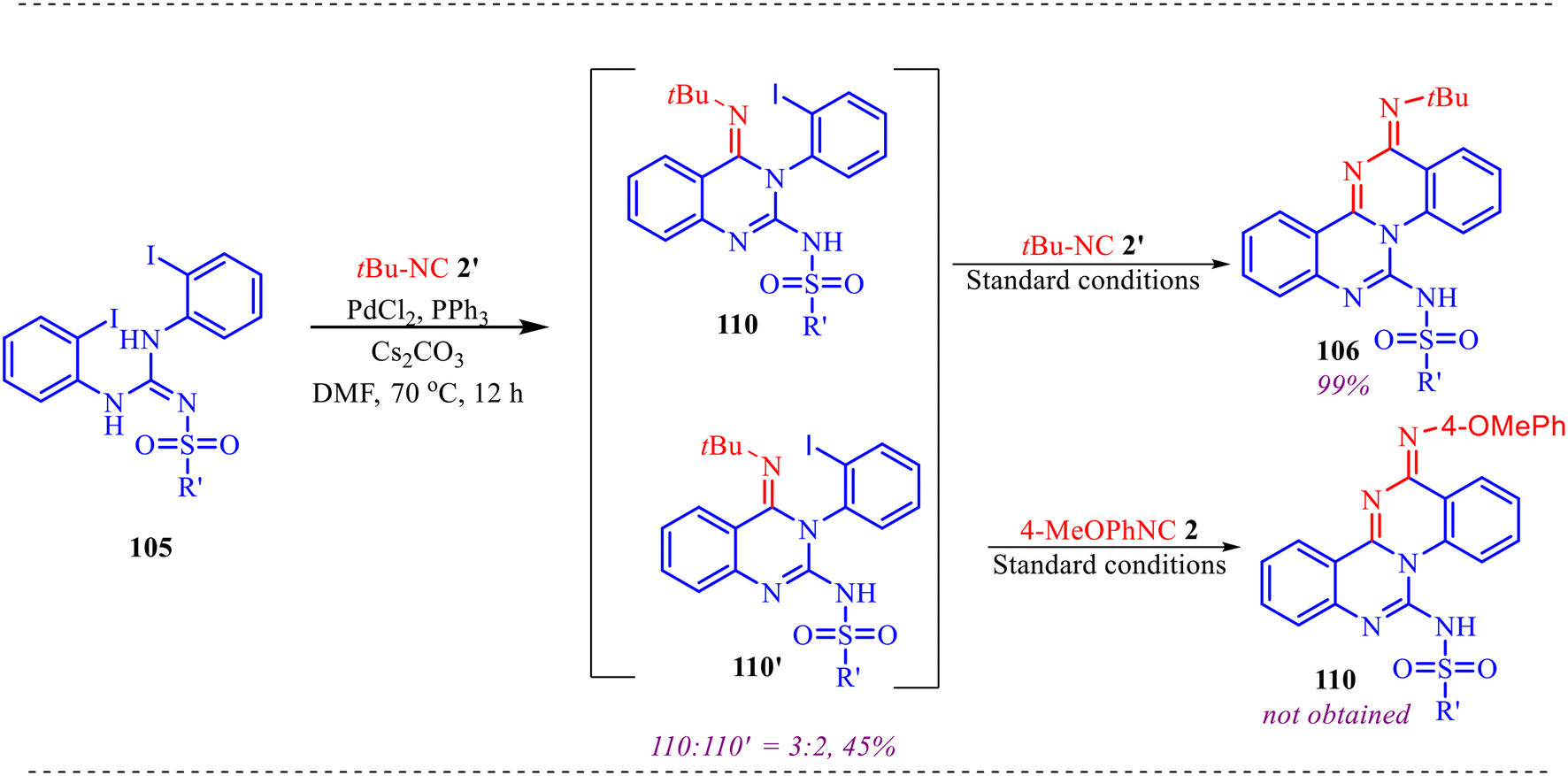
To investigate the reaction mechanism, several control experiments were performed as shown in Scheme 2. Under the optimized conditions, 1 was converted to amidine 5 in 93% yield (Scheme 2a). However, subjecting substituted amidine 5, isonitrile 2, and aniline 3 to the standard reaction conditions did not afford the desired product 4 (Scheme 4b), indicating that 5 is unlikely to be an intermediate. When 1 was reacted with 2 under standard conditions, intermediate 6 ([M + Na]+ = 307.1884) was detected in 94% GC yield (Scheme 4c). Subsequently, addition of aniline 3 followed by heating at 100 °C for 1 h furnished the target product 4 in 91% yield, confirming that 6 serves as a key intermediate in the transformation.
 |
| | Scheme 2 (a) Formation of amidine from substrate 1 under optimized conditions. (b) Control experiment to prove amidine as intermediate (c) detection of key intermediate 6 in the reaction for forming product 4. | |
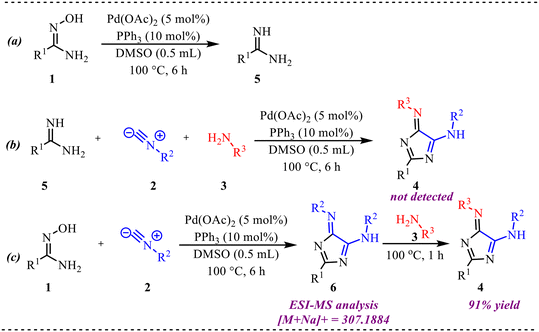
Furthermore, a plausible mechanism for this transformation is presented in Scheme 3. The catalytic cycle is proposed to initiate with the reduction of Pd(II)L2 to the catalytically active Pd(0)L2 species in the presence of PPh3. The generated Pd(0) complex undergoes oxidative addition with substrate 1 to form Pd(II) intermediate (I), thereby activating the substrate for subsequent transformations. Sequential double insertion of isocyanide 2 into the Pd–substrate bond then produces intermediate (II), forming the C–N bond. Subsequently, intermediate (II) undergoes intramolecular cyclopalladation to generate the palladacyclic complex (III), which enables regioselective heterocycle formation. Reductive elimination from intermediate (III) furnishes intermediate 6 while regenerating the Pd(0) catalyst, thus completing the catalytic cycle. Intermediate 6 further undergoes isomerization to afford compound (IV), which ultimately undergoes an amine exchange reaction with aniline 3 to deliver the desired product 4. Overall, this transformation underscores the cooperative role of palladium catalysis and sequential isocyanide insertion in facilitating efficient heterocyclic framework construction.
 |
| | Scheme 3 Possible reaction mechanism. | |
Ren et al. reported an imine-directed triple isocyanide insertion approach to synthesize poly-substituted pyrrole derivatives involving the palladium-catalysed aza-Nazarov cyclization reaction of alkenyl bromide, isocyanides, and amines (Scheme 4).23 A broad substrate scope was achieved by employing diverse substituted substrates, and it was observed that the reaction showed high efficiency with various electron-donating and withdrawing substituents at alkenyl bromide and provided good yields of corresponding products. However, the reaction didn't proceed when an alkyl group was taken in place of the aromatics at the alkenyl bromide. Similarly, a trace amount of product formation was observed when aliphatic amines were employed in the reaction. Also, various aromatic and aliphatic rings containing isocyanides gave the desired outcome for the proposed reaction and provided good to moderate yields. Along with this, further transformations were performed to show the application of the palladium-catalysed isocyanide reaction.
 |
| | Scheme 4 (a) General reaction scheme for the synthesis of poly-substituted pyrrole derivatives via triple isocyanide insertion. (b) Substrate scope showing selected examples with corresponding yields. (c) Further transformation of pyrrole derivatives. | |
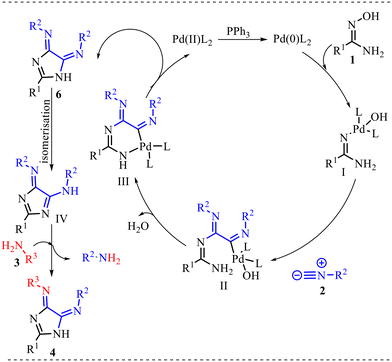
Additionally, to gain insight into the reaction mechanism, several control experiments were performed, as shown in Scheme 5. The imide intermediate 12 was synthesised by treating aldehyde 7 and amine 8 in methanol. Then 12 was reacted with isocyanide 2 under standard reaction conditions which furnished product 9 in good yield, proving 12 as an intermediate in the reaction process (Scheme 5a). The formation of the olefin product was confirmed by HRMS analysis of the reaction mixture, suggesting that the transformation likely proceeds through a sequential β-carbon elimination followed by β-hydride elimination from a tert-butyl palladium intermediate (Scheme 5b).
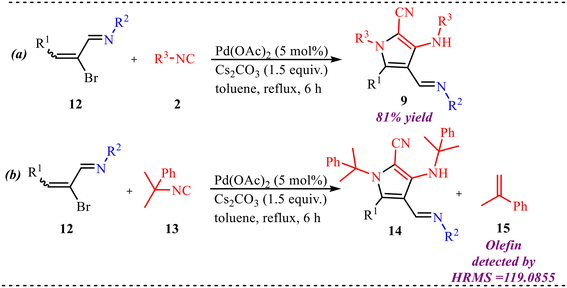 |
| | Scheme 5 (a) Determination of imide intermediate as key species in the reaction. (b) Determination of a sequential β-carbon elimination–β-hydride elimination pathway. | |
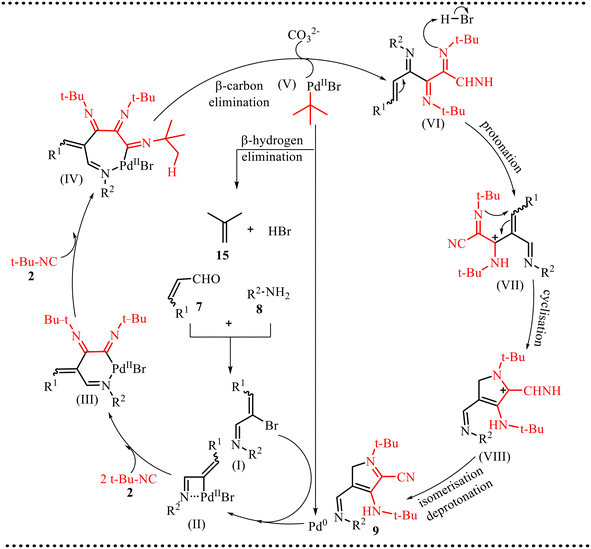
Based on experimental and literature precedents, a plausible catalytic pathway is illustrated in Scheme 6. The transformation is initiated by condensation of aldehyde 7 with amine 8 to generate imine intermediate (I), which serves as the key electrophilic component of the reaction. Coordination of (I) to the palladium catalyst followed by oxidative addition forms a strained four-membered palladacycle (II). This step is crucial, as it activates the imine functionality and forms a reactive Pd–C/N bond that enables subsequent migratory insertion reactions. Further, intermediate (II) undergoes sequential insertion of two isocyanide molecules to afford intermediate (III). These insertion events are central to the transformation, as they promote rapid molecular complexity, establish multiple C–N bonds, and simultaneously relieve ring strain within the palladacyclic framework. A third isocyanide insertion further expands the coordination sphere of palladium to generate the seven-membered complex (IV), which serves as a pivotal intermediate enabling the elimination steps. Under basic conditions, intermediate (IV) undergoes β-carbon elimination followed by β-hydride elimination, resulting in extrusion of isobutene 15 and formation of intermediate (VII). This elimination sequence is mechanistically significant as it facilitates structural reorganization, rearomatization, and regeneration of the catalytically active Pd(0) species, thereby sustaining the catalytic cycle. Subsequently, intermediate (VII) undergoes protonation and intramolecular cyclization to furnish intermediate (VIII), establishing the heterocyclic core. Finally, intermediate (VIII) undergoes isomerization and deprotonation to afford the thermodynamically stable product 9. Overall, the transformation proceeds through a cascade sequence involving oxidative addition, multiple isocyanide insertions, elimination, and cyclization, collectively enabling efficient assembly of structurally complex nitrogen-containing heterocycles.
 |
| | Scheme 6 Proposed mechanism for poly-substituted pyrrole derivatives via an imine-directed triple isocyanide insertion. | |
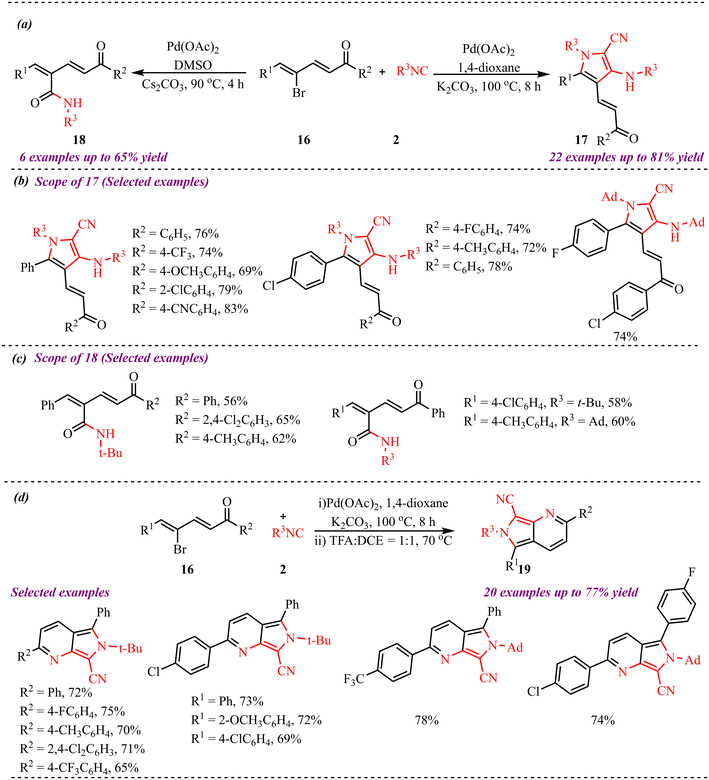
Liu et al. reported an efficient palladium-catalyzed route for the synthesis of pyrrole derivatives involving isocyanide insertion, enabled by ring strain and supported by non-covalent interactions (Scheme 7).24 The reaction conditions were successfully optimized, and the substrate scope of both α,β-unsaturated ketones and isocyanides was thoroughly investigated. At the R1-position of the unsaturated ketones, a range of electron-donating and electron-withdrawing substituents were well tolerated, affording the desired products in moderate to high yields. Similarly, variation at the R2-position with electron-donating and halogen substituents also led to the formation of the corresponding products in good yields. The scope of isocyanides was further explored. In this context, the use of adamantyl (Ad) isocyanide resulted in the successful synthesis of the corresponding pyrrole derivatives. In contrast, cyclohexyl and n-butyl isocyanides did not participate in the reaction, and no desired products were obtained. Additionally, the use of substituted phenyl isocyanide led to a complex reaction mixture with no clean product formation. During optimisation, compound 18 was identified, formed via monomolecular isocyanide insertion involving water as the nucleophile. The substrate scope for this transformation was also examined. Subsequently, the compound was reacted with trifluoroacetic acid (TFA) and 1,2-dichloroethane (DCE) as a solvent in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, leading to the synthesis of pyridine derivatives. Finally, the influence of electron-rich and electron-deficient substituents at the R1 position was systematically studied in this transformation, resulting in the formation of the corresponding products in moderate to good yields.
:1 ratio, leading to the synthesis of pyridine derivatives. Finally, the influence of electron-rich and electron-deficient substituents at the R1 position was systematically studied in this transformation, resulting in the formation of the corresponding products in moderate to good yields.
 |
| | Scheme 7 (a) General reaction scheme for palladium-catalysed synthesis of pyrrole derivatives via isocyanide insertion reaction. (b) Substrate scope showing selected examples for synthesis of pyrrole derivatives (c) substrate scope showing selected examples of mono-substituted isocyanide insertion product. (d) Palladium-catalysed synthesis of pyridine derivatives. | |
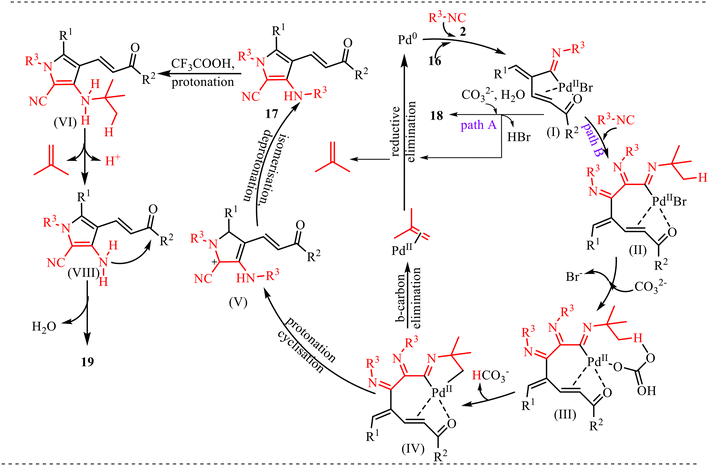
Based on experimental observations and precedents from the literature, the plausible reaction mechanism for the formation of products 17, 18 and 19 is depicted in Scheme 8. The catalytic cycle is initiated by the generation of an active Pd(0) species, which undergoes oxidative addition with substrate 16, followed by coordination of isocyanide 2. Subsequent 1,1-migratory insertion of the isocyanide into the Pd–C bond affords intermediate (I), which serves as a common reaction intermediate for two possible reaction pathways. In pathway A, intermediate (I) undergoes direct nucleophilic attack by water, leading to hydrolysis of the palladium–carbon bond and formation of product 18, along with regeneration of the palladium catalyst. Alternatively, intermediate (I) proceeds via pathway B, which involves a cascade transformation. In this pathway, insertion of a second molecule of isocyanide generates a seven-membered palladacyclic intermediate (II). This intermediate subsequently undergoes anion exchange, followed by base-assisted deprotonation, to produce the key intermediate (IV). Further, the intermediate (IV) undergoes β-carbon elimination, releasing isobutylene and forming (VIII) and a palladium species that facilitates an aza-Nazarov cyclisation, yielding 17, accompanied by reductive elimination and regeneration of the Pd(0) catalyst. Subsequently, 17 undergoes protonation, which generates intermediate (VI). Finally, nucleophilic addition affords the desired product 19. Overall, the transformation highlights the dual role of isocyanide as both a coupling and cyclisation partner, while the palladium catalyst orchestrates multiple elementary steps including oxidative addition, migratory insertion, β-carbon elimination, and reductive elimination. The extrusion of isobutylene serves as a key thermodynamic driving force that facilitates the cascade cyclisation process leading to product formation.
 |
| | Scheme 8 Plausible mechanism for palladium-catalysed synthesis of pyrrole derivatives via an isocyanide insertion reaction. | |
Synthesis of benzo-fused five-membered heterocycles
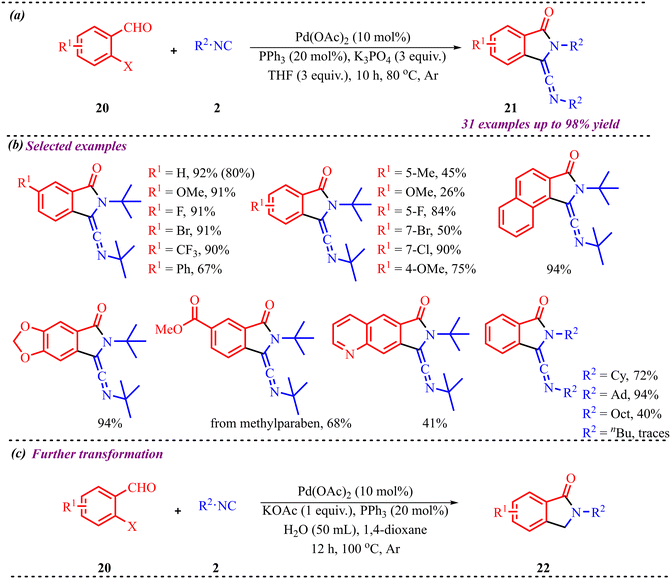
Benzo-fused five-membered heterocycles, in general, consist of a five-membered ring containing heteroatoms such as N, O, or S fused with another ring system. These motifs have applications ranging from pharmaceutical chemistry to agrochemistry, extending up to material chemistry.25 Concerning this, we have discussed a few studies for the synthesis of five-membered fused heterocycles using palladium as a catalyst. In addition to the above, Ji and co-workers developed a straightforward and efficient tandem strategy for synthesizing biologically relevant isoindolinone derivatives via palladium-catalyzed isocyanide insertion into o-bromobenzaldehydes (Scheme 9).26 This method accommodates a variety of substituents on both the o-bromobenzaldehyde and the isocyanide components. Notably, electron-donating and electron-withdrawing groups at the meta-position of 2-bromobenzaldehyde yielded products in good to excellent yields, whereas para-substituted electron-donating groups resulted in reduced efficiency compared to electron-deficient or halogen substituents. This approach was also successfully employed for late-stage functionalization of bioactive compounds such as methyl paraben and estrone, and it enabled gram-scale synthesis of a model compound with an 80% yield.
 |
| | Scheme 9 (a) General scheme for the synthesis of isoindolinone derivatives via Pd-catalysed isocyanide insertion. (b) Substrate scope showing selected examples with corresponding yield. (c) Further transformations. | |
Additionally, to better understand the reaction mechanism, several control experiments were performed, as illustrated in Scheme 10. The palladium complex 23 was synthesized and subjected to the standard reaction conditions, leading to the formation of product 24 in 72% yield (Scheme 10a). To further verify the formation of product 22, isotopic labelling experiments were conducted using varying amounts of D2O. An increase in D2O concentration resulted in a corresponding increase in deuterium incorporation. These results suggest that the hydrogen atom in the CH2 group of 22 likely originates from water present in the reaction medium (Scheme 10b).
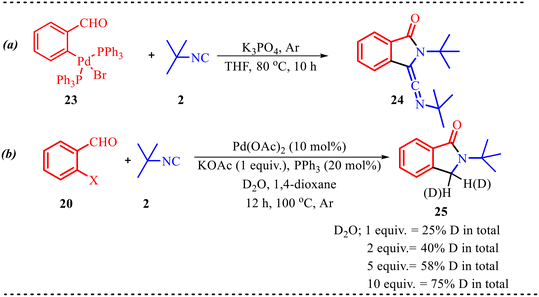 |
| | Scheme 10 (a) Determination of palladium complex formed in the reaction. (b) Determination of role of water in the reaction medium. | |
Based on the findings from the control experiments, the plausible reaction mechanism is shown in Scheme 11. The proposed catalytic cycle provides a coherent mechanistic rationale for the observed transformation, highlighting the critical roles of palladium catalysis, base assistance, and sequential isocyanide insertion. The reaction is initiated by oxidative addition of the aryl C–Br bond to Pd(0), generating the organopalladium intermediate (I), which activates the substrate toward further transformations. The intermediate (I) subsequently undergoes coordination and migratory insertion of isocyanide 2 to afford intermediate (II), establishing the key C–C bond framework. The formation of the cyclopalladated species (III) from (II) through a concerted metalation–deprotonation (CMD) process supports the involvement of base-assisted C–H activation. Protonation of the Pd–C bond in intermediate (III) then furnishes intermediate (IV), releasing palladium from the cyclometalated framework and generating a reactive organic intermediate. Intermediate (IV) undergoes irreversible intramolecular imine insertion via intermediate (V) to produce species (VI), which drives the reaction forward and prevents reversibility of earlier steps. In the presence of K3PO4, intermediate (VI) reacts with a second equivalent of isocyanide 2 to form intermediate (VII), where the base likely facilitates deprotonation and stabilizes the intermediate. Finally, intermediate (VII) undergoes β-hydride elimination to furnish ketenimine 21 with concurrent regeneration of Pd(0), completing the catalytic cycle.
 |
| | Scheme 11 Plausible mechanism for the synthesis of isoindolinone derivatives. | |
Sharma et al. reported an efficient strategy for the synthesis of 2,3-difunctionalized indole derivatives via palladium-catalyzed isocyanide insertion and triple bond-activation methodology (Scheme 12).27 During reaction condition optimization, palladium acetate as a catalyst, along with different ligands, bases, and additives, was screened. However, they couldn't achieve more than 77% yield for the model reaction. Interestingly, the reaction proceeded smoothly with freshly prepared 2-(2-phenylethynyl)phenyl isocyanides using a continuous flow process without direct exposure and odour. To check the synthetic feasibility of this protocol, a range of substituents was tested on both substrates i.e. 2-(2-phenylethynyl)phenyl isocyanide and N-(2-iodophenyl)-N-methyl acrylamide. This protocol was found feasible for electron-rich species over some electron-withdrawing substrates and provided moderate to good yields (Scheme 12b). Further, the optimized conditions were also applied for enantioselective synthesis of the desired product using a chiral ligand (Scheme 12c). However, this protocol provided an enantioselective product only in 36–77% yield, along with low enantiomeric access. A plausible catalytic cycle evidenced the formation of the palladium complex of N-(2-iodophenyl)-N-methyl acrylamide, which subsequently undergoes intramolecular carbopalladation, followed by isocyanide insertion and intramolecular addition to the triple bond, to afford the desired product.
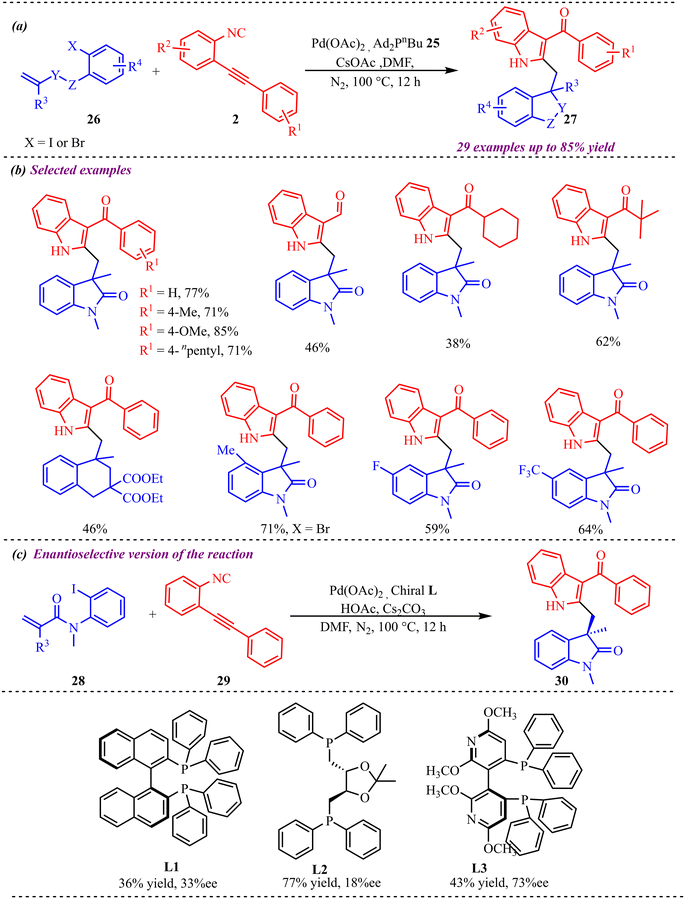 |
| | Scheme 12 (a) General scheme for the synthesis of 2,3-difunctionalized indole derivatives via palladium-catalysed isocyanide insertion. (b) Substrate scope of selected examples with corresponding yield. (c) Enantioselective version of the reaction using various chiral ligands. | |
To gain deeper insight into the reaction mechanism, an isotopic labelling study was conducted to determine the oxygen source in 27 (Scheme 13). The 18O-incorporating product 31 was obtained when H218O was used as an additive under standard reaction conditions, affording a mixture of 27 and 31 in 20% isolated yield.
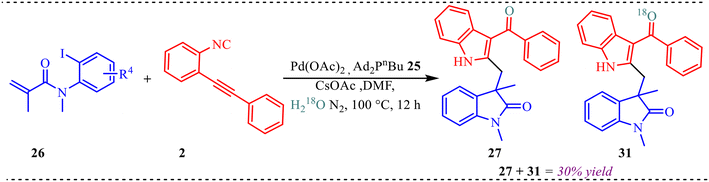 |
| | Scheme 13 Control experiment. | |
Additionally, a plausible mechanism for the synthesis of 2,3-difunctionalized indole derivatives is illustrated in Scheme 14. The catalytic cycle begins with oxidative addition of Pd(0) into the C–I bond of N-(2-iodophenyl)-N-methyl acrylamide 26, generating the aryl palladium intermediate (I), which activates the substrate for subsequent intramolecular transformation. Intermediate (I) then undergoes intramolecular carbopalladation across the olefin to afford intermediate (II), establishing the indole core. The resulting σ-alkyl palladium species (II) undergoes migratory insertion with 2-(2-phenylethynyl)phenyl isocyanide 2 to produce the imidoyl palladium intermediate (III), representing a key carbon–carbon bond-forming step that contributes to molecular complexity. Thereafter, intermediate (III) undergoes intramolecular addition to the alkyne, followed by nucleophilic attack of the oxygen-based nucleophile to furnish intermediate (IV). Subsequent reductive elimination releases the intermediate (V) while regenerating the Pd(0) catalyst, thereby continuing the catalytic cycle. Finally, intermediate (V) undergoes isomerization to deliver the thermodynamically stable 2,3-difunctionalized indole product 27. Overall, the transformation highlights the sequential interplay of oxidative addition, carbopalladation, isocyanide insertion, and intramolecular cyclization steps, which collectively govern the efficiency and regioselectivity of the reaction.
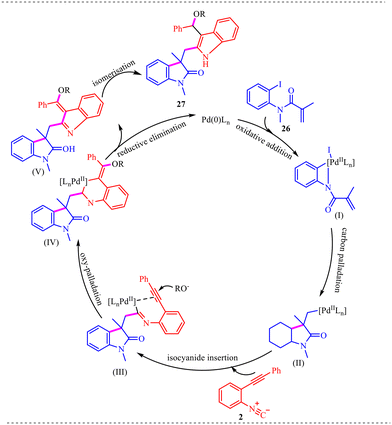 |
| | Scheme 14 Proposed mechanism of the synthesis of 2,3-difunctionalized indole derivatives. | |
Guchhait et al. reported an intramolecular cascade cyclization/isocyanide insertion strategy to synthesize bicyclic indolylisoindolinone motifs involving an indolyl migration, redox-neutral process in the presence of palladium acetate and the P(o-tolyl)3 ligand (Scheme 15a).28 Interestingly, for the first time, an alkyl isocyanide C–H acts as a hydride source for promoting rearrangement in the reaction. The substrate scope investigation showed good functional group tolerance and was independent of the electronic nature of the substituents (Scheme 15b). Also, various isocyanides were compatible in this reaction. However, the reaction didn't work with aromatic isocyanides. Further modification via de-tert butylation of the synthesized heterocycle also provides the products in moderate yields, i.e., up to 47% (Scheme 15c). Also, the reaction yield was not so much affected when the reaction was performed at the gram-scale level.
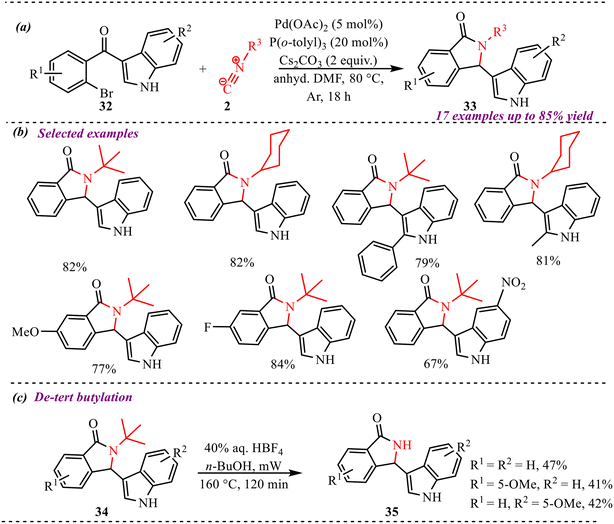 |
| | Scheme 15 (a) General scheme for the synthesis of bicyclic indolylisoindolinones. (b) Substrate scope of selected examples (c) de-tert butylation of indolylisoindolinones. | |
Furthermore, a plausible reaction mechanism was proposed, as shown in Scheme 16. The catalytic cycle is initiated by the generation of the active Pd(0) species, which undergoes oxidative addition with the aryl bromide substrate to form aryl-Pd(II) intermediate (I). Subsequent coordination and migratory insertion of the isocyanide into the Pd-aryl bond affords the imidoyl-Pd(II) species (II). This intermediate is characterised by a polarised C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N functionality, furnishing the imidoyl carbon highly electrophilic. Next, intramolecular nucleophilic attack of the 3-indolyl moiety onto the imidoyl carbon takes place, leading to cyclization and formation of palladacyclic intermediate (III). Reductive elimination from (III) delivers the spirocyclic framework (IV) and regenerates the Pd(0) catalyst. The formation of (IV) establishes the key spiro-quaternary center and represents a crucial branching point in the mechanism. Subsequently, a second molecule of isocyanide participates in an isocyanide-triggered rearrangement. Coordination of the isocyanide to the spirocyclic intermediate activates the adjacent carbon–nitrogen bond, promoting indolyl migration through intermediate (V). This step likely proceeds via a cationic or nitrilium-type species stabilized by resonance with the indole ring. The rearrangement furnishes intermediate (VI), in which the indolyl group has migrated and an alkylnitrilium ion is generated. In the final stage, the alkylnitrilium species undergoes a bromide-mediated redox process. Bromide attack forms a transient ion pair that proceeds through a favourable six-membered cyclic transition state. This arrangement facilitates intramolecular hydride transfer from the adjacent alkyl group to the electron-deficient nitrilium carbon. Concurrently, elimination of an alkene and liberation of BrCN occur, leading to rearomatization and formation of the final product 33.
N functionality, furnishing the imidoyl carbon highly electrophilic. Next, intramolecular nucleophilic attack of the 3-indolyl moiety onto the imidoyl carbon takes place, leading to cyclization and formation of palladacyclic intermediate (III). Reductive elimination from (III) delivers the spirocyclic framework (IV) and regenerates the Pd(0) catalyst. The formation of (IV) establishes the key spiro-quaternary center and represents a crucial branching point in the mechanism. Subsequently, a second molecule of isocyanide participates in an isocyanide-triggered rearrangement. Coordination of the isocyanide to the spirocyclic intermediate activates the adjacent carbon–nitrogen bond, promoting indolyl migration through intermediate (V). This step likely proceeds via a cationic or nitrilium-type species stabilized by resonance with the indole ring. The rearrangement furnishes intermediate (VI), in which the indolyl group has migrated and an alkylnitrilium ion is generated. In the final stage, the alkylnitrilium species undergoes a bromide-mediated redox process. Bromide attack forms a transient ion pair that proceeds through a favourable six-membered cyclic transition state. This arrangement facilitates intramolecular hydride transfer from the adjacent alkyl group to the electron-deficient nitrilium carbon. Concurrently, elimination of an alkene and liberation of BrCN occur, leading to rearomatization and formation of the final product 33.
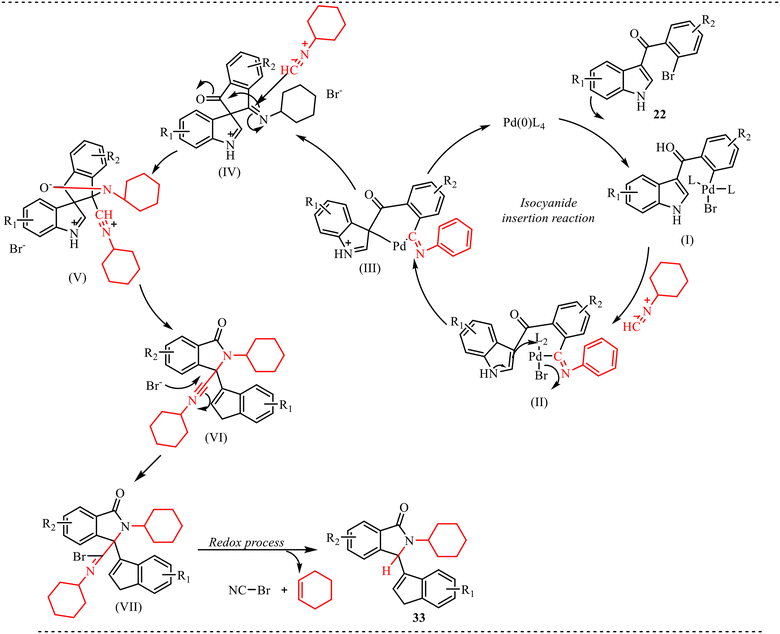 |
| | Scheme 16 Plausible mechanism for synthesis of bicyclic indolylisoindolinones. | |
A novel proximal C–H activation strategy using isocyanide insertion was reported by Huang and co-workers (Scheme 17).29 The reaction provided three different types of products under slightly different reaction conditions. Additionally, the role of the palladium catalyst, along with various ligands and additives, determines the reaction route; consequently, the reaction yields different products. Interestingly, when palladium acetate was employed as a catalyst with CsF as a base and PivOH as an additive, a mono isocyanide insertion took place and provided a 3-unsubstituted isoindolinone derivative in moderate to good yield. Next, when [Pd2(dba)3]CHCl3 with PPh3 as a ligand, cesium carbonate as a base, and PivOH as a ligand were used, then a double isocyanide insertion product, i.e., 3-cyano-substituted isoindolinone derivative, was obtained in excellent yield. Finally, when only the (4-CF3C6H6)3P ligand was used, disubstituted isoindolinone derivatives were formed in good yields.
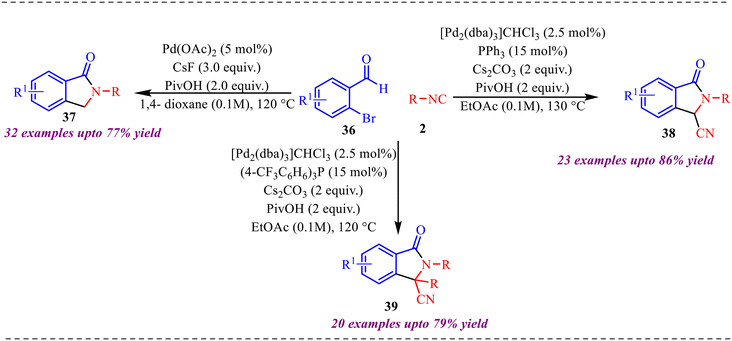 |
| | Scheme 17 Synthesis of isoindolinone derivatives via C–H activation. | |
Further transformation of the 3-cyano substituted isoindolinone derivative by hydrogenation reaction in the presence of Pd/C and ring expansion in the presence of NaOH was performed, which provided 55% and 78% of the desired product. Also, 3-unsubstituted isoindolinone was further modified to access biologically relevant indobufen and indoprofen molecules (Scheme 18).
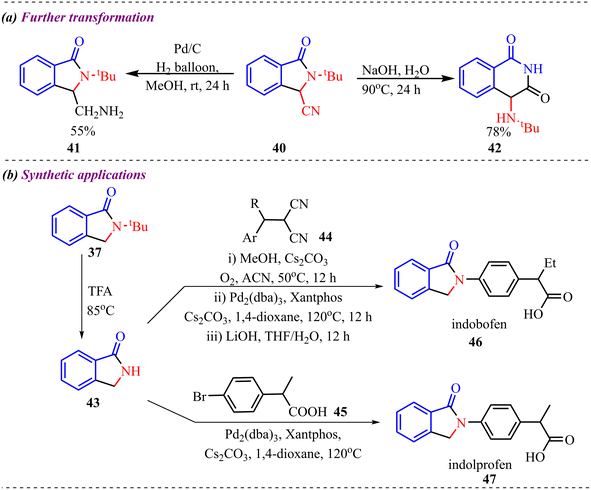 |
| | Scheme 18 (a) Further transformations (b) synthetic applications. | |
Furthermore, to have insight into the reaction mechanism, several control experiments were carried out, as shown in Scheme 19. To determine the reactive intermediate in the reaction mechanism, the reaction of palladium complex 48 with isocyanide furnished products 2 synthesised compounds, 37 and 38, in 53% and 70% isolated yields, respectively (Scheme 19a). Along with this, deuterium labelling experiments were conducted in the presence of 10 equiv. of D2O, synthesising deuterated products 37 and 38, incorporating deuterium at the benzylic position (Scheme 19b).
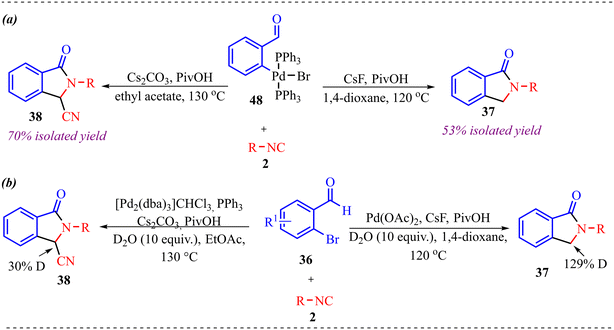 |
| | Scheme 19 (a) Determination of reactive intermediate in the reaction mechanism. (b) Isotopic labelling experiment. | |
Additionally, a plausible catalytic pathway is outlined in Scheme 20. The cycle is initiated by oxidative addition of Pd(0) to aryl bromide 36, generating aryl–Pd(II) intermediate (I). Subsequent migratory insertion of isocyanide 2 into the Pd–aryl bond affords the imidoyl–Pd(II) species (II), in which the polarized CN unit renders the carbon electrophilic and predisposed to further transformation. A base-assisted concerted metalation–deprotonation (CMD) step then forms palladacycle (III), a key chelated intermediate that stabilises the Pd(II) centre and promotes intramolecular reorganisation. Further, protonation of (III) produces the acyl–Pd intermediate (IV), which undergoes intramolecular CN insertion to furnish the isoindolinone-containing Pd(II) species (V). This intermediate is a crucial branching point because it can evolve along multiple pathways. The insertion of a second equivalent of isocyanide into (V) gives intermediate (VI), which upon base-mediated deprotonation forms a reactive ketene–imine species (VII). Owing to its cumulated CCN system, (VII) readily proceeds to products 38 (path b, R = tBu) or 39 (path c, R = Bn), with the substituent influencing the reaction course. Alternatively, direct protodepalladation of (V) leads to product 37 (path d), bypassing the second isocyanide insertion. In all cases, reductive (or decarboxylative reductive) elimination regenerates Pd(0), completing the catalytic cycle. Thus, the mechanism highlights sequential isocyanide insertions, CMD-driven cyclopalladation, and divergence from a common isoindolinone–Pd intermediate that dictates product selectivity.
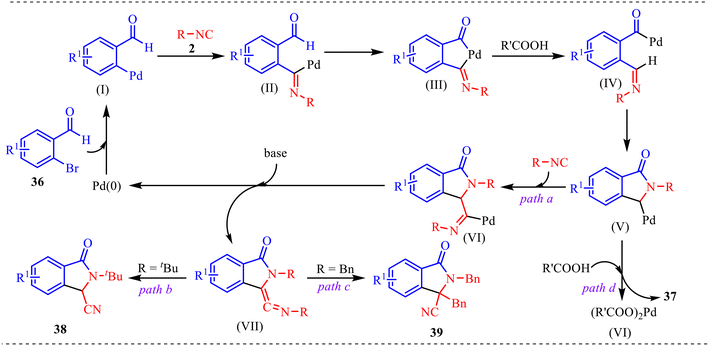 |
| | Scheme 20 Plausible pathway synthesis of isoindolinone derivatives. | |
Liu et al. reported a palladium-catalysed one-pot synthesis of 3-acyl-substituted isoindolinone derivatives by reacting o-bromobenzaldehyde, isocyanides, and carboxylic acids in the presence of a base (Scheme 21).30 This method utilizes readily accessible starting materials and provides the desired isoindolinone with high yields up to 90%. Further, the substrate scope was investigated for the synthesis of 3-diacyl-substituted isoindolinone derivatives, and it was found that o-bromobenzaldehydes bearing electron-donating groups at various positions afforded moderate to good yields. However, electron-donating substituents at the para-position of the benzaldehyde ring led to a notable decrease in product yield. Similarly, halide substituents at the 3-position of o-bromobenzaldehyde were found to negatively affect the formation of the desired products. The study also explored substitutions at the benzaldehyde ring, including the replacement of the benzene core with other aromatic rings, showing the formation of the desired product with high efficiency, whereas substrates containing thiophene and pyridine rings did not form the desired products. The generality of isocyanides was thoroughly examined, with bulky isocyanide substituents showing better compatibility and higher reactivity compared to smaller or aromatic ones. Additionally, various carboxylic acids were evaluated, and the desired products were obtained in moderate yields across a diverse range of acid substrates.
 |
| | Scheme 21 (a) General scheme for the synthesis of diacyl-substituted isoindolinone. (b) Substrate scope of selected examples. (c) Further transformation of diacyl-substituted isoindolinone. | |
To better understand the reaction mechanisms, several control experiments were conducted, as shown in Scheme 22. Firstly, the ketenimine compound (I) was synthesised using 49 and 2, which was then subjected to standard reaction conditions in the presence of acid and base, yielding product 51 in 98% yield (Scheme 22a). Additionally, an 18O-labelling experiment using H2O18 indicated that the oxygen in product 51 does not come from water (Scheme 22b).
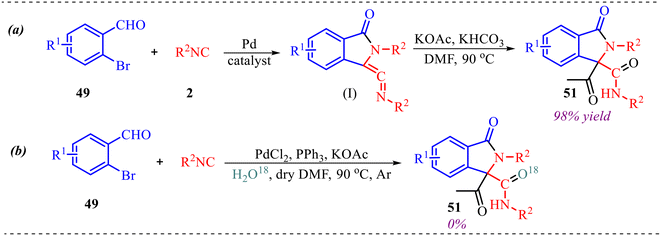 |
| | Scheme 22 (a) Determination of ketenimine as intermediate in the reaction pathway. (b) Isotopic labelling experiment. | |
Based on the control experiments conducted, a plausible reaction mechanism is proposed in Scheme 23. The reaction is initiated by oxidative addition of Pd(0) into the C–Br bond of o-bromobenzaldehyde 49, generating aryl–Pd(II) intermediate (I). This oxidative addition step produces a σ-aryl palladium species in which the metal center is activated toward migratory insertion. Subsequent coordination and insertion of tert-butyl isocyanide into the Pd–aryl bond affords an imidoyl-Pd(II) intermediate. In this species, the CN bond is highly polarized, rendering the imidoyl carbon strongly electrophilic. The acetate ion then acts as a nucleophile and attacks the electrophilic imidoyl carbon, leading to intermediate (II). This step forms a new C–O bond and generates a stabilized acyl-type species, while palladium remains coordinated to the aryl framework. The intermediate (II) contains both the aldehyde functionality and the newly introduced imidoyl fragment in close proximity. Subsequently, intramolecular nucleophilic addition occurs, wherein the nitrogen centre attacks the aldehyde carbonyl group, promoting cyclisation and formation of the isoindolinone core. This step proceeds through a rearranged intermediate featuring a hemiaminal-type structure, which undergoes proton transfer and rearomatization to furnish compound 51. The formation of 51 establishes the lactam ring and represents the key ring-closing event of the sequence. Finally, under acidic conditions, intermediate 51 undergoes protonation and stabilization to deliver the substituted isoindolinone derivative 52 as the acidic medium facilitates proton transfers and ensures complete conversion to the thermodynamically stable isoindolinone framework.
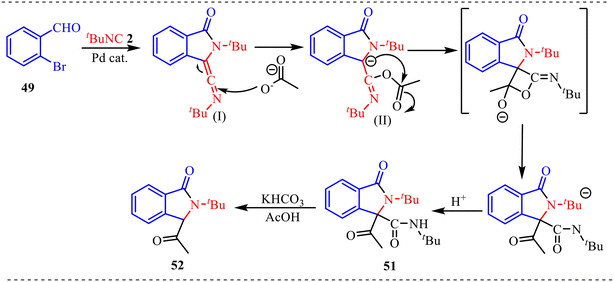 |
| | Scheme 23 Proposed mechanism for the synthesis of diacyl-substituted isoindolinone. | |
Zheng et al. reported a palladium-catalyzed synthesis of isoindoline derivatives through the reaction of aziridines with isocyanides, where the aziridine acts as a nucleophile (Scheme 24).31 The transformation proceeded with good regioselectivity and Z-type stereoselectivity, affording the desired products in moderate to good yields. The substrate scope of the reaction was thoroughly investigated using a variety of substituted aziridines and isocyanides, affording the corresponding products in isolated yields ranging from 62% to 81%. Further, aziridines bearing electron-neutral, electron-donating, and electron-withdrawing groups were all well tolerated under the palladium-catalyzed conditions, delivering the desired products in moderate to good yields. Additionally, the isocyanide scope was examined: isocyanides with aliphatic substituents underwent the insertion reaction smoothly under the optimized conditions. In contrast, isocyanides bearing cyclic or sterically bulky groups proved incompatible, failing to deliver the desired products. The structure of the isoindoline derivative was unambiguously confirmed by single-crystal X-ray diffraction, which revealed the presence of an intramolecular hydrogen bond that contributes to the formation of the final product with Z-type stereoselectivity.
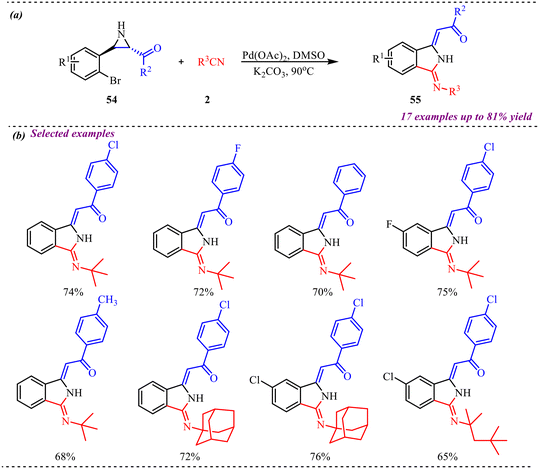 |
| | Scheme 24 (a) General scheme for the Pd-catalysed isocyanide insertion reaction for the synthesis of isoindoline. (b) Substrate scope of the selected examples. | |
To rationalize the experimental findings, a detailed mechanism is proposed in Scheme 25 for the formation of isoindoline derivatives. The catalytic cycle is initiated by oxidative addition of substrate 54 to the Pd(0) species, affording aryl–Pd(II) intermediate (I), in which palladium inserts into the C–Br bond to generate a σ-aryl palladium complex; this step increases the oxidation state of palladium to +2 and activates the ortho-positioned framework for intramolecular transformation. Subsequent coordination of isocyanide 2 and 1,1-migratory insertion into the Pd–aryl bond produces the imidoyl–Pd intermediate (II), a key species bearing a Pd–C(imidoyl) linkage that introduces the carbon atom required for annulation and enhances electrophilicity at the imidoyl carbon. In the presence of base, intramolecular nucleophilic attack occurs, leading to cyclization and formation of a six-membered palladacycle (III); this chelated intermediate is conformationally rigid and thermodynamically stabilized through metal coordination, which precisely orients the reacting centers for bond construction. Further, the reductive elimination from (III) then forges the crucial C–N bond, delivering intermediate (IV) while regenerating the Pd(0) catalyst to complete the catalytic cycle, which undergoes structural reorganization stabilized by intramolecular hydrogen bonding, which activates the strained aziridine ring toward regioselective C–N bond cleavage, forming intermediate (V); the relief of ring strain serves as a significant driving force at this stage. A subsequent intramolecular 1,5-hydrogen atom transfer generates intermediate (VI), enabling redistribution of electron density and formation of a conjugated imine-type system. Finally, tautomerization of (VI) affords the isoindoline product 55 predominantly as the thermodynamically favoured Z-isomer, while a minor amount of the corresponding E-isomer (VII) is also formed, with the stereochemical outcome governed by conjugative stabilisation and minimisation of steric repulsion.
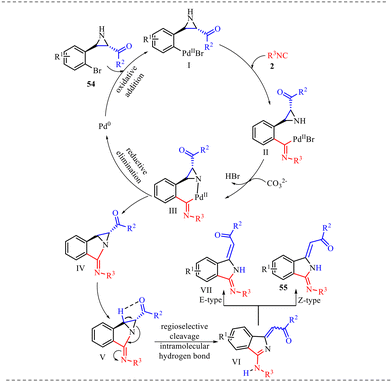 |
| | Scheme 25 Proposed mechanism for the synthesis of isoindoline derivatives. | |
Synthesis of six-membered heterocycles
Six-membered heterocycles, most significantly, refer to molecules that consist of a ring with six atoms having at least one heteroatom. These rings have natural abundance and act as a pillar of many drug designs.32 These rings are versatile in nature because of their physicochemical and electronic properties.33 There are several methods reported for synthesising six-membered heterocycles, including cyclisation reaction, oxidative coupling and cycloaddition reactions.34 In this section, we focus on the palladium-catalysed strategies developed for the construction of six-membered heterocycles.
Synthesis of Pd-catalysed atroposelective quinazoline heterocycles
In this context, Balalaie's group reported an interesting ring-expanding approach to afford polysubstituted pyrimidines heterocycles via palladium-catalyzed cascade consisting C–C and C–N bond formation through ring opening of 2H-azirines and simultaneous insertion of isocyanide species (Scheme 26).35 This methodology was applicable to successfully synthesize 20 derivatives in moderate to excellent yields by using substituted azirines having different electron-withdrawing and electron-donating groups. However, they observed that the electronic effect on azirines doesn't provide any significant effect on the reaction outcome. Moreover, various aliphatic isocyanides were found to be superior to this cyclization reaction, while aromatic isocyanides provide lower yields.
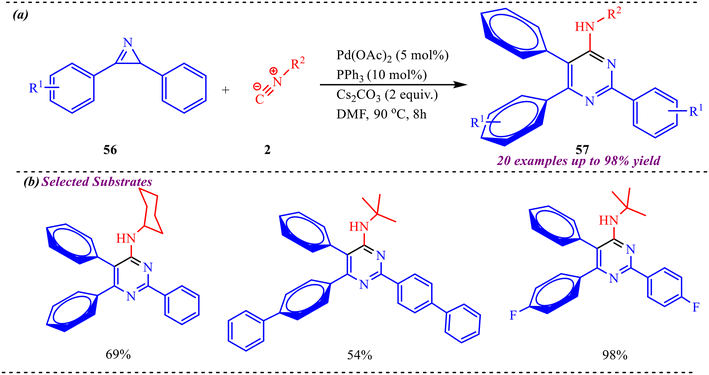 |
| | Scheme 26 (a) General scheme for the synthesis of polysubstituted pyrimidines heterocycles via a palladium-catalysed reaction. (b) Selected substrate scope. | |
Further, to acquire better mechanistic insight, a competitive experiment was conducted in which two 2H-azirines (56a and 56b) and cyclohexyl isocyanide 2a were introduced simultaneously into a single reaction vessel under identical conditions (Scheme 27). This transformation afforded three products, 57a, 57b, and 57c, in 35%, 25%, and 38% yields, respectively.
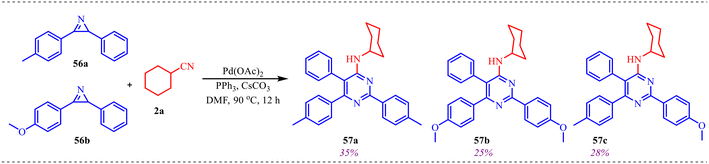 |
| | Scheme 27 Control reaction. | |
Based on the control experiment the reaction mechanism was proposed as shown in Scheme 28. The proposed catalytic cycle begins with the in situ generation of an active Pd(0) species, which undergoes oxidative addition into the strained C–N bond of azirine 56 to form a four-membered palladacyclic intermediate (I). This step is driven by the high ring strain and polarisation of the azirine, and it oxidises Pd(0) to Pd(II). Intermediate (I) is crucial as it activates the azirine within a square-planar Pd(II) framework, making the Pd–C bond susceptible to migratory insertion. Subsequently, isocyanide coordinates to palladium and undergoes migratory insertion into the Pd–C bond to afford a five-membered palladacycle (II). This step expands the ring, reduces strain, and incorporates the isocyanide carbon into the growing skeleton. Intermediate (II) then interacts with a second azirine molecule to generate intermediate (III), where palladium acts as a template to assemble all reacting components in proximity for C–C bond formation. Finally, reductive elimination from (III) regenerates Pd(0) and releases a zwitterionic intermediate (IV), featuring iminium and carbanionic character. Upon aqueous workup, proton transfer and stabilization lead to the formation of the final product 57. Overall, the mechanism features strain-driven oxidative addition, efficient isocyanide insertion, and palladium-mediated bond construction via well-defined metallacyclic intermediates.
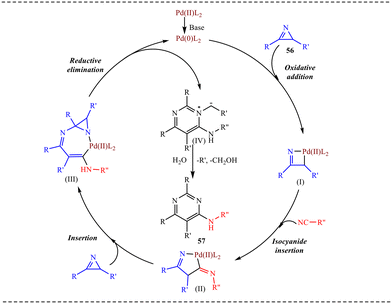 |
| | Scheme 28 Proposed mechanism for the synthesis of polysubstituted pyrimidines. | |
Concerning this, in this section, we will discuss the studies related to palladium-catalysed synthesis of fused six-membered heterocycles. In addition to this, an interesting methodology towards the development of axially chiral 2-aryl and 2,3-diaryl quinazolinones via Pd-catalysed atroposelective coupling-cyclization of 2-isocyanobenzamides and 2-iodo-3-methylbenzoate substrates was reported by Zhu et al. (Scheme 29a).36 This reaction provided a novel coupling strategy when sterically hindered coupling partners were taken into consideration for providing atroposelective biaryl derivatives. To get the maximum enantiomeric excess, various SPINOL-derive phosphoramidite chiral ligands were tested. Out of five ligands, methyl and ethyl substituted ligands showed the best enantioselectivity of 94% and provided a maximum yield of 93% (Scheme 29b). It was observed that sterically hindered ligands were not found to be good for the reaction and diminished the atroposelectivity as well as the yield of the product. To examine the feasibility of the reaction, various 2-isocyanobenzamides having electron-rich, electron-deficient, and bulky substituents were tested, and a negligible effect of substituents was observed on the yield and enantioselectivity of the reaction. However, a significant effect on the yield of the product was observed while different substituents at 2-iodo-3-methyl benzoate were installed. This methodology was successfully applied for the synthesis of diastereoselective quinazolinone derivatives, and two desired derivatives were formed in 70% and 21% yield, having 93% and 89% ee (Scheme 29c).
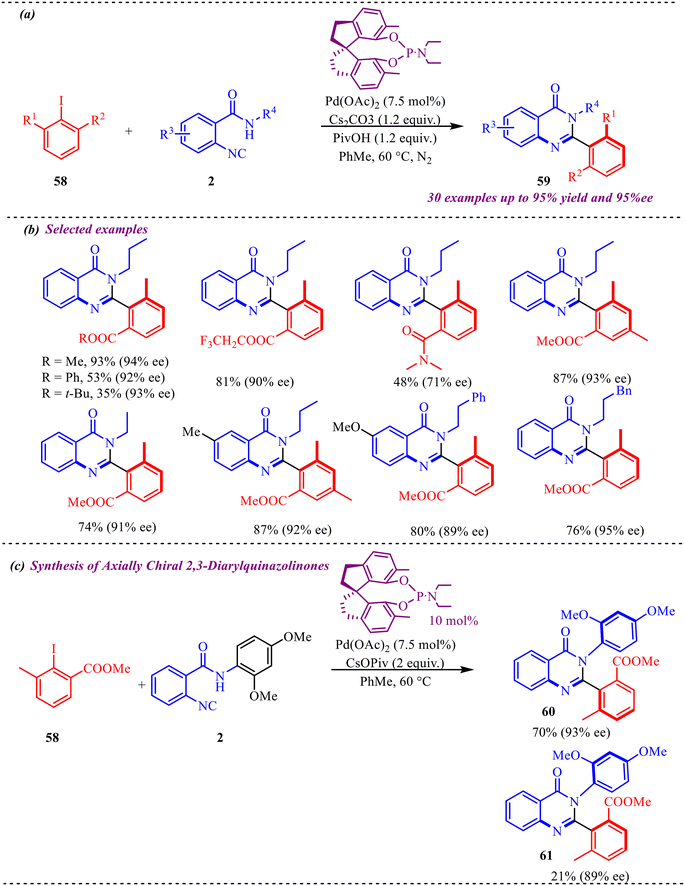 |
| | Scheme 29 (a) General scheme for the synthesis of axially chiral 2-aryl and 2,3-diaryl quinazolinones. (b) Substrate scope of selected examples. (c) Synthesis of axially-chiral 2,3-diarylquinazolinones. | |
Additionally, the plausible mechanism under the standard conditions is depicted in Scheme 30. The reaction is initiated by the in situ generation of an active Pd(0) species, which undergoes oxidative addition into the C–I bond of 2-iodo-N-propylbenzamide 58 to form aryl–Pd(II) intermediate (I). In this step, palladium is oxidized from Pd(0) to Pd(II), giving a square-planar complex bearing the aryl group, iodide, and ligand framework. This oxidative addition is a crucial activation step, as it converts the relatively inert aryl iodide into a highly reactive organopalladium species. Next, the isocyanide substrate 2 coordinates to the Pd(II) center and undergoes migratory insertion into the Pd–aryl bond. This step forms an imidoyl–Pd(II) intermediate, in which the isocyanide carbon is inserted between palladium and the aryl moiety. The efficiency of this transformation, as evidenced by the high yield (up to 95%) of product 59, indicates that isocyanide insertion into the Pd(II) species is a highly favourable and kinetically competent step. Following insertion, intramolecular cyclisation takes place. The amide nitrogen (or its activated form under basic conditions) nucleophilically attacks the electrophilic imidoyl carbon, forming a new C–N bond and generating a five-membered palladacyclic intermediate. This cyclisation step forms the iminoisoindolinone core. Finally, reductive elimination from the Pd(II) center regenerates the Pd(0) catalyst and releases the five-membered iminoisoindolinone derivative 59. The regeneration of Pd(0) completes the catalytic cycle.
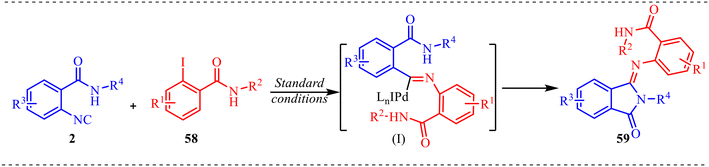 |
| | Scheme 30 Plausible mechanistic pathway. | |
Synthesis of Pd-catalysed tetrazolyl quinazoline derivatives
Recently, Yao et al. reported a sequential Ugi-azide coupling along with palladium-catalyzed azide-isocyanide cross-coupling followed by cyclization without purifying intermediate species for the synthesis of biologically valuable 4-tetrazolyl-3,4-dihydroquinazoline derivatives (Scheme 31a).37 This reaction strategy involves two different isocyanide units for the Ugi reaction and Pd-coupling reaction. This multicomponent reaction showed good functional group tolerance and provided a yield of up to 90%. However, the reaction provided lower yields when strong electron-donating substituents were employed at benzaldehyde. Similarly, the reaction was feasible with both aliphatic and aromatic amines, but lower yields were observed with electron-donating amines (Scheme 31b). To evaluate the biological potential of the synthesised moiety, the compound's anticancer activity was tested for the least cytotoxicity. The Edu method detected its activity via the proliferation of breast cancer cells treated with synthesized molecules. It also shows comparable results for glioma cell apoptosis. A gram-scale synthesis was also performed, yielding the desired product in 80% yield, demonstrating the synthetic utility of the reported strategy.
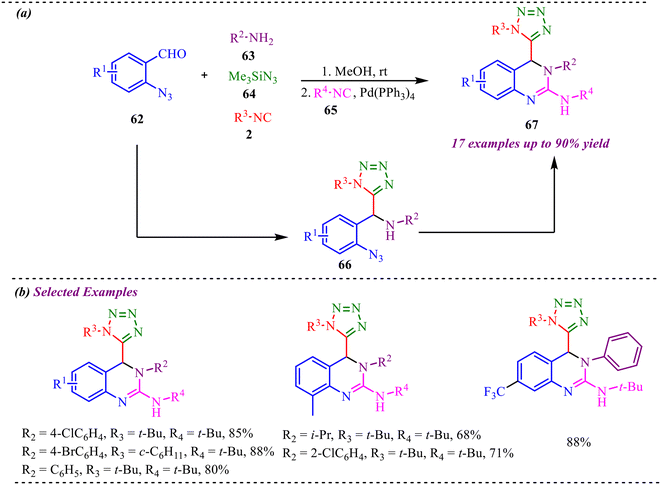 |
| | Scheme 31 (a) General scheme for the synthesis of biologically valuable 4-tetrazolyl-3,4-dihydroquinazoline derivatives. (b) Substrate scope of selected examples. | |
To gain deeper insight into the reaction mechanism, a control experiment was performed as illustrated in Scheme 32. In this study, the reaction was carried out in the presence of 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO), a well-known radical scavenger commonly employed to probe the involvement of radical intermediates. Notably, the formation of product 67 proceeded smoothly and in comparable yield even in the presence of TEMPO. The absence of any inhibition, suppression, or TEMPO-trapped adducts clearly indicates that free radical species are not generated during the course of the reaction.
 |
| | Scheme 32 Control experiment. | |
In addition, a plausible mechanism for the one-pot formation of 4-tetrazolyl-3,4-dihydroquinazolines 67 is outlined in Scheme 33. The one-pot synthesis of 4-tetrazolyl-3,4-dihydroquinazolines 67 begins with the coordination of the Ugi-azide adduct 66 and isocyanide 2 to the active Pd(0)/Pd(II) catalyst system to generate intermediate (I). In 1this complex, palladium binds to the isocyanide carbon and the azide functionality of 66, organising both components within its coordination sphere. This chelation not only activates the azide moiety but also properly orients the isocyanide for subsequent bond reorganisation. Next, intermediate (I) undergoes N2 release from the azide fragment to generate a highly reactive palladium-associated nitrene intermediate (II). The loss of thermodynamically stable N2 serves as a strong driving force for this step. Further, the nitrene intermediate (II) then participates in a concerted intramolecular nitrene transfer, leading to C–N bond formation and generating intermediate (III). In this step, palladium plays a crucial templating role, positioning the nitrene and the reacting carbon center in close proximity, thereby enabling selective insertion and ring construction. Intermediate (III) represents a key organopalladium species in which the new C–N linkage has been established within a partially cyclized framework. Subsequently, reductive elimination from intermediate (III) yields a carbodiimide (IV) and regenerates the active palladium catalyst, completing the catalytic cycle. The carbodiimide functionality in (IV) is highly electrophilic and predisposed toward nucleophilic attack. Finally, intermediate (IV) undergoes intramolecular cyclization, where a nucleophilic nitrogen atom attacks the carbodiimide carbon, forming the dihydroquinazoline core and delivering the target 4-tetrazolyl-3,4-dihydroquinazoline 67.
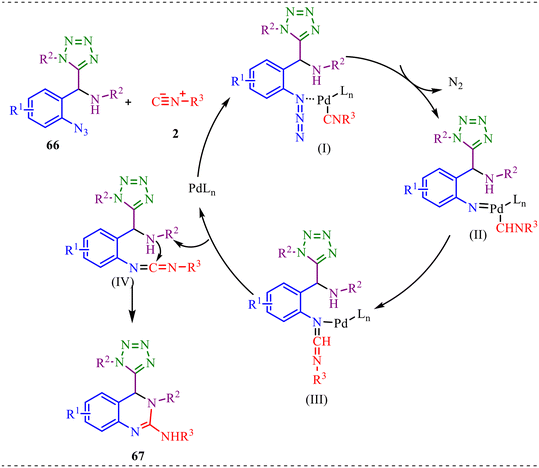 |
| | Scheme 33 Proposed mechanism for the formation of 4-tetrazolyl-3,4-dihydroquinazoline derivatives. | |
Synthesis of benzo-fused six-membered heterocycles
Fused six-membered heterocycles, consisting of a six-membered ring containing one heteroatom such as O, N or S, are fused with another ring system. These fused systems hold the core of many alkaloids, antibiotics, essential amino acids and various pharmaceuticals.38 These motifs possess an outstanding ability to act as biologically reactive scaffolds contributing to drug discovery and development.39 Zhang and co-workers reported another palladium-catalysed isocyanide insertion approach to synthesize N-substituted benz[c,d]indol-2-imine and N-substituted amino-1-napthylamide derivatives in the absence and presence of water molecules, respectively (Scheme 34a).40 After optimising various palladium-based catalysts, such as Pd(OAc)2, Pd(TFA)2, Pd(PPh3)4, Pd2(dba)3, etc., Pd(TFA)2 was found to be superior to other catalysts. Also, DCE was found to be the best solvent among the solvents screened for this reaction. The reaction provided moderate to good yields with both substrates and was found to be slightly dependent on the electronic nature of the substituents (Scheme 34b). This methodology was successfully applied to the synthesis of antiproliferative agents, i.e., N-substituted (benzo[c,d]-indol-2(1H)-ylidene)-4-nitroaniline derivatives, yielding 56% and 73% in 56% and 73% yield, respectively (Scheme 34c). Further transformation towards the synthesis of the BET bromodomain inhibitor was successfully achieved, which showed the synthetic applicability of the protocol.
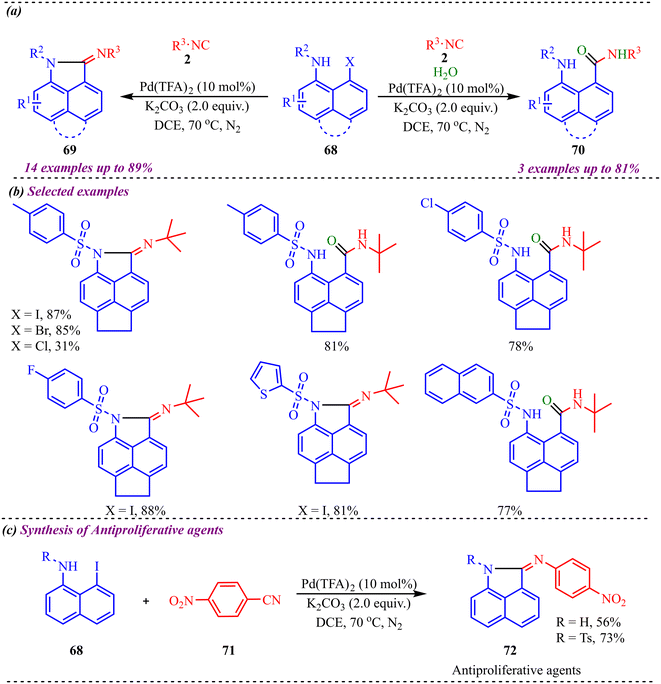 |
| | Scheme 34 (a) General scheme for the synthesis of N-substituted benz[c,d]indol-2-imines and N-substituted amino-1-napthylamides. (b) Substrate scope of selected examples. (c) Synthesis of antiproliferative agents. | |
To gain deeper mechanistic insight, a plausible catalytic pathway for the formation of products 69 and 70 is illustrated in Scheme 35. The cycle begins with oxidative addition of N-substituted 8-halonaphthylamines 68 to the active Pd(0) species, generating aryl–Pd(II) intermediate (I). In this step, palladium inserts into the C–X bond (X = halogen), forming a square-planar Pd(II) complex bearing the naphthyl fragment and halide ligand. This oxidative addition is a key activation step, converting the relatively inert aryl halide into a reactive organopalladium species. Subsequently, isocyanide 2 coordinates to the Pd(II) center and undergoes migratory insertion into the Pd–aryl bond, affording imidoyl–Pd(II) intermediate (II). This step incorporates the isocyanide carbon into the molecular framework and generates an electrophilic imidoyl moiety bound to palladium. The formation of (II) is crucial, as it establishes the structural backbone required for subsequent annulation. Next, intramolecular coordination of the pendant amino nitrogen to the Pd(II) center takes place, producing intermediate (III). This chelation step increases the proximity between reactive centres and facilitates cyclisation. Under basic conditions, hydrogen halide (HX) elimination occurs, promoting C–N bond formation within the coordinated complex. Finally, reductive elimination releases the annulated product 69 and regenerates the Pd(0) catalyst, thus completing the primary catalytic cycle. Alternatively, in the presence of base and water, halide substitution by hydroxide can occur at intermediate (II), generating hydroxo–Pd(II) species (IV). Reductive elimination from (IV) forms an imino alcohol intermediate, which is typically unstable and undergoes tautomerization/isomerisation to furnish product 70. This divergent pathway highlights the influence of reaction conditions—particularly the presence of water and base—on product selectivity.
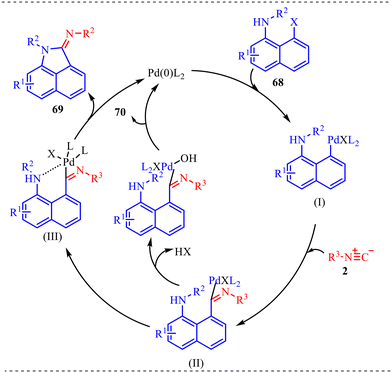 |
| | Scheme 35 Proposed mechanism N-substituted benz[c,d]indol-2-imines and N-substituted amino-1-napthylamides. | |
Bolm et al. developed a green and novel approach to synthesize 3-amino substituted benzothiadiazine oxides in good to excellent yields via a Pd-catalyzed tandem reaction of 2-azido sulfoximines with isonitriles (Scheme 36a).41 Interestingly, the reaction was completed in a very short time in the presence of 0.25 mol% Pd catalyst at room temperature, making this protocol more efficient and economical. However, the reaction was found to be slightly selective towards 2-azidosulfoximine substrates (Scheme 36b). The reaction involves isocyanide insertion into the N–H and azide bonds, with multiple intermediate steps. The applicability of the derived product was explored by further transformations, including removal of the tert-butyl group to form amine-substituted derivatives, which can be readily transformed via simple organic reactions (Scheme 36c).
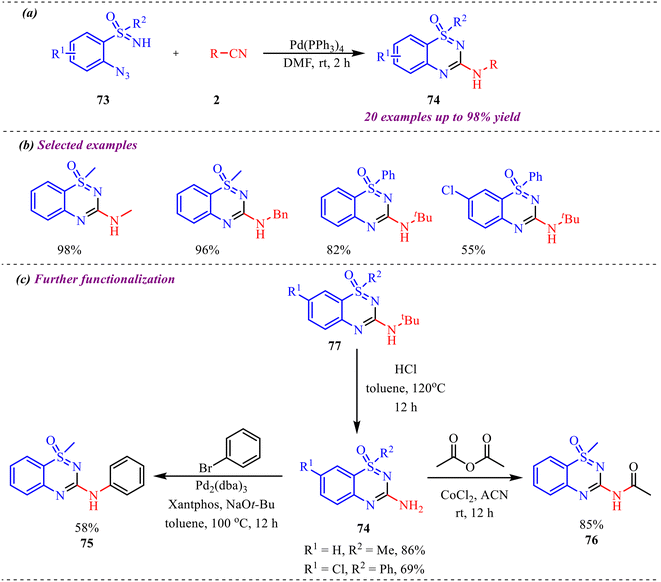 |
| | Scheme 36 (a) General scheme for the synthesis of 3-amino-substituted benzothiadiazine oxides (b) substrate scope of selected examples. (c) Further transformation of 3-amino-substituted benzothiadiazine oxides. | |
Further, to gain a deep understanding of the reaction pathway, a control experiment was carried out to identify the key intermediate (Scheme 37). Concerning this, Boc-protected azido sulfoximine 78 was treated with isonitrile 2, leading to an interrupted cascade process that selectively afforded the Boc-protected carbodiimide 79 in 88% yield. This outcome indicates that carbidamide is the intermediate in the reaction pathway. Subsequently, the cyclization of intermediate 79 to the corresponding heterocycle 74 was examined. Given that this transformation might proceed without a metal catalyst, carbodiimide 79 was treated with HCl in dioxane. Gratifyingly, the reaction smoothly furnished heterocycle 74 in an excellent 99% yield, thereby confirming that the cyclisation step occurs efficiently under metal-free acidic conditions.
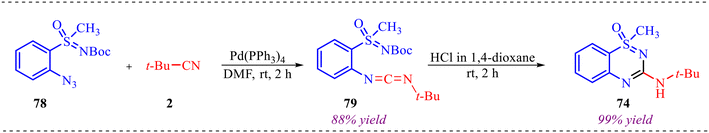 |
| | Scheme 37 Control experiment. | |
A plausible catalytic cycle for the Pd-catalysed synthesis of 3-amino-substituted benzothiadiazine oxides is depicted in Scheme 38. The transformation proceeds through a sequence of coordination, nitrene generation, migratory transfer, and cyclization steps. The cycle is initiated by the coordination of 2-azidosulfoximine 73 to the active palladium complex (I), forming the coordination complex (II). In this intermediate, the azide functionality interacts with the Pd center, which activates the substrate toward subsequent nitrogen evolution. This coordination step is essential, as it brings the reactive azide moiety into the metal's coordination sphere, enabling controlled reactivity. Subsequent ligand exchange with isonitrile 2 generates complex (III), in which both the azide-derived fragment and the isonitrile are bound to palladium. This arrangement positions the reacting partners in close proximity. Complex (III) then undergoes loss of dinitrogen (N2) from the azide group to furnish a palladium–imido intermediate (IV). The removal of molecular nitrogen provides a strong thermodynamic driving force for the formation of this key species. Intermediate (IV) can be described as a metal-stabilised nitrene (imido–Pd) complex, in which palladium modulates the high reactivity of the electron-deficient nitrogen centre. The palladium–imido intermediate (IV) subsequently undergoes a concerted nitrene transfer to the coordinated isonitrile through a cyclic transition state (V). This step results in the insertion of the nitrene into the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond of the isonitrile, generating the carbodiimide complex (VI). This transformation is crucial because it constructs the central carbodiimide functionality that ultimately drives heterocycle formation. Palladium plays a dual role here—stabilising the nitrene and orchestrating selective bond formation via a well-organised transition state. Following the formation of complex (VI), release of the palladium catalyst (I) regenerates the active species and liberates the free carbodiimide intermediate (VII), thereby closing the catalytic cycle. The carbodiimide moiety in (VII) is highly electrophilic and predisposed toward nucleophilic attack. Finally, intermediate (VII) undergoes intramolecular nucleophilic addition, typically by a neighbouring nitrogen atom, leading to cyclisation and formation of the 3-amino-substituted benzothiadiazine oxide 74.
N bond of the isonitrile, generating the carbodiimide complex (VI). This transformation is crucial because it constructs the central carbodiimide functionality that ultimately drives heterocycle formation. Palladium plays a dual role here—stabilising the nitrene and orchestrating selective bond formation via a well-organised transition state. Following the formation of complex (VI), release of the palladium catalyst (I) regenerates the active species and liberates the free carbodiimide intermediate (VII), thereby closing the catalytic cycle. The carbodiimide moiety in (VII) is highly electrophilic and predisposed toward nucleophilic attack. Finally, intermediate (VII) undergoes intramolecular nucleophilic addition, typically by a neighbouring nitrogen atom, leading to cyclisation and formation of the 3-amino-substituted benzothiadiazine oxide 74.
 |
| | Scheme 38 Proposed mechanism for synthesis of 3-amino-substituted benzothiadiazine oxides. | |
Dai et al. reported a Pd-catalysed synthesis of quinoxaline-2,3-diamines via a cyclisation reaction between diamines and isocyanides, which involves the formation of C–N and C–C bonds via double insertion of isocyanide (Scheme 39).42 The substrate scope for the synthesis of quinoxaline derivatives was explored, and the desired products were obtained with moderate to excellent yields. First, the reaction scope was investigated using various phenylenediamines and tert-butyl isocyanide. The reaction was strongly influenced by the electronic properties of o-phenylene diamine; electron-rich groups enhanced the yield, whereas electron-poor groups decreased it. In addition, o-phenylene diamine, which contains both electron-donating and withdrawing groups, afforded the corresponding products in moderate yields. Further, the scope of isocyanide was investigated with o-phenylene diamine. It was observed that both aliphatic and aromatic isocyanides were well tolerated in the reaction, yielding the desired products in moderate to high yields. To show the practicality of the methodology, the reaction was carried out on a gram scale under the optimal reaction conditions, synthesizing the desired product 81a with 89% isolated yield.
 |
| | Scheme 39 (a) General scheme for palladium-catalysed synthesis of quinoxaline-2,3-diamines via double isocyanide insertion. (b) Substrate scope of selected examples. (c) Reaction scale-up. (d) Further transformation of quinoxaline-2,3-diamines. | |
Moreover, a control experiment was conducted to have a clear insight into the reaction mechanism (Scheme 40). The reaction of o-phenylenediamines 80 with two different isocyanides was carried out under the optimized conditions. However, only trace amounts of the non-symmetric quinoxaline-2,3-diamine products were detected. Additionally, during reaction optimization and substrate scope studies, no intermediates corresponding to single isocyanide insertion were observed. These findings indicate that the reaction most likely proceeds through a simultaneous double isocyanide insertion rather than a stepwise single insertion pathway.
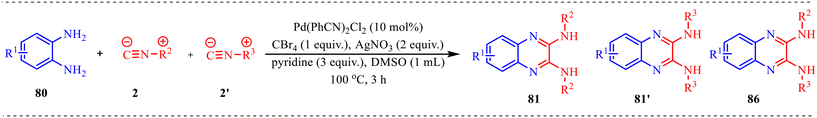 |
| | Scheme 40 Control experiment. | |
A plausible mechanism for this transformation is outlined in Scheme 41. The process initiates with the coordination of Pd(II) to the o-phenylenediamine substrate 80. In the presence of a base, which facilitates deprotonation of the amine groups, the palladium center anchors to the nitrogen atoms to form the first key intermediate, a five-membered palladacycle (I). This intermediate is essential because it activates the N–H bond and creates a stable environment for subsequent carbon–nitrogen bond-forming steps. The mechanism then proceeds through a double isocyanide insertion phase, in which the two molecules of isocyanide 2 undergo successive 1,1-insertions into the metal–nitrogen bonds of the palladacycle. This leads to the formation of intermediate (II), a more complex seven-membered palladacycle. Following the assembly of the skeleton, the metal center facilitates reductive elimination. In this step, palladium is expelled from the organic framework, forming the dihydro-intermediate (III) and reducing the metal from Pd(II) to Pd(0). This regeneration allows the catalyst to re-enter the cycle and process another molecule of the diamine. The final stage of the transformation involves isomerization of intermediate (III). In this step, a prototropic shift occurs, wherein hydrogen atoms migrate from the ring nitrogens to the exocyclic imine nitrogens. This hydrogen transfer serves as the key driving force of the reaction, enabling the system to attain a more thermodynamically stable and fully aromatic structure. Consequently, the desired 2,3-diaminoquinoxaline 81 is formed as the final product.
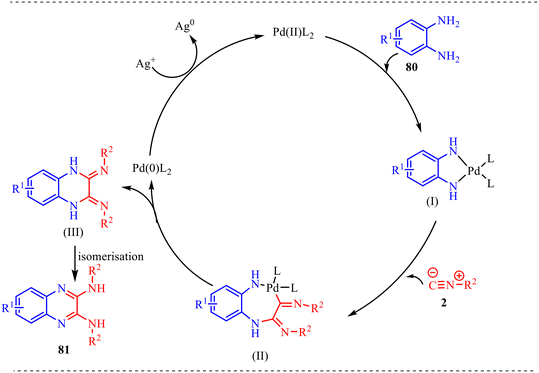 |
| | Scheme 41 Proposed reaction mechanism for synthesis of quinoxaline-2,3-diamines. | |
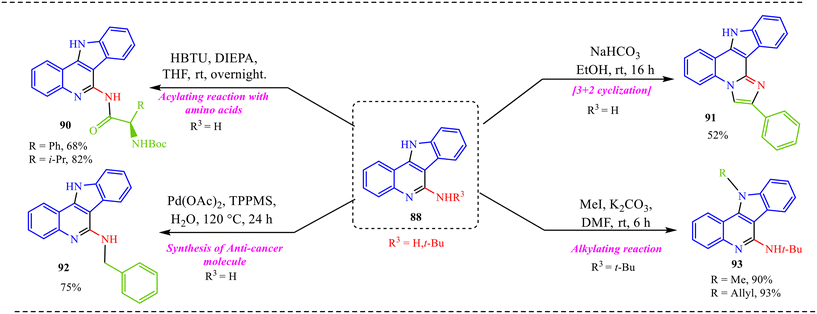
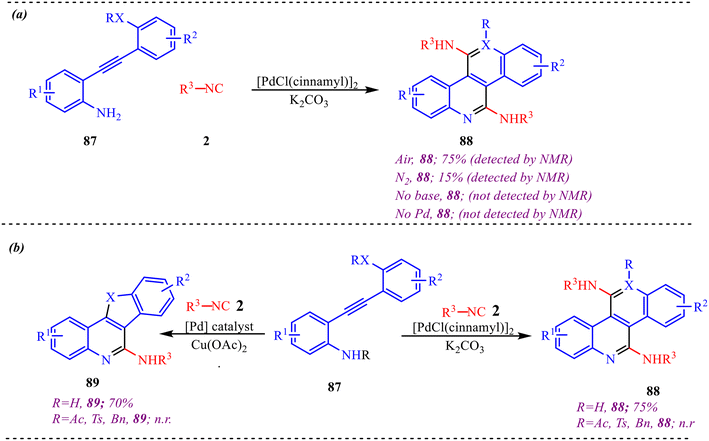
Synthesis of polycyclic heterocycles
Polycyclic heterocycles are vital structural scaffolds composed of two or more fused rings, at least one of which contains a heteroatom such as nitrogen (N), oxygen (O), or sulfur (S). These motifs are widely distributed in nature and, owing to their structural complexity, exhibit diverse agrochemical, pharmacological, and biological activities.43 In this context, Wu's group reported a novel and diverse synthesis of polysubstituted fused tetracyclic heterocyclic scaffolds (Scheme 42a).44 This methodology relied on the nature of the Pd-catalyst to provide two distinct product types via mono- or double-isocyanide insertion. A wide range of substrates having electron-rich and electron-deficient substitutions was tested for synthesizing both types of heterocycles (Scheme 42b). Also, different isocyanide units were tested, and it was observed that steric hindrance played a significant role, resulting in lower yields. To demonstrate the practical utility of the reaction, a gram-scale reaction was performed, which provided 58% and 55% for the corresponding polyheterocycles 88 and 89.
 |
| | Scheme 42 (a) General scheme for the synthesis of poly substituted fused tetracyclic heterocyclic scaffolds. (b) Substrate scope of the selected examples. | |
Next, to further demonstrate the synthetic utility and practical applicability of the developed protocol, a series of post-functionalization reactions were carried out. These transformations highlight the versatility of the obtained scaffold and confirm its suitability for downstream structural diversification. As illustrated in Scheme 43, a variety of structurally modified derivatives were successfully synthesised, delivering the corresponding products in moderate to good yields.
 |
| | Scheme 43 Further transformations. | |
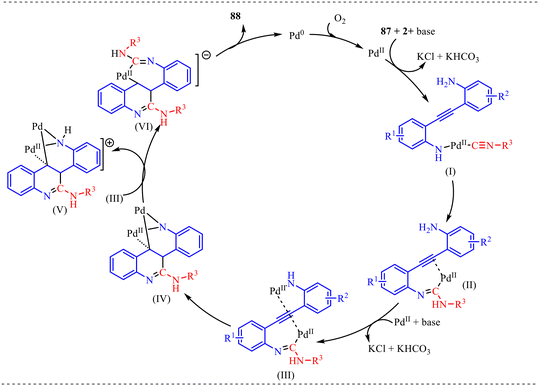
To gain deeper insight into the mechanistic pathway of the reaction, a series of control experiments were performed, as depicted in Scheme 44. When the reaction was conducted under an inert nitrogen (N2) atmosphere in the absence of oxygen, product 88 was obtained in only 15% yield, clearly highlighting the crucial role of molecular oxygen in the transformation. Furthermore, both the base and the catalyst were found to be essential for the reaction, as no formation of product 88 was observed in their absence (Scheme 44a). In addition, the influence of various amino-protecting groups was examined under standard reaction conditions (Scheme 44b). It was observed that no formation of product 88 occurred when the amino group was protected. Similarly, the synthesis of product 89 also failed when N-protected substrates were employed. These results strongly suggest that the free primary amine is indispensable for the cyclization process and plays a key role in the reaction pathway.
 |
| | Scheme 44 (a) Determination of role of oxygen, base and palladium in the reaction. (b) Effect of N-protection on amines. | |
Based on the above control experiments, the mechanistic pathway leading to 88 is illustrated in Scheme 45. The cycle is initiated by a base-mediated direct aminopalladation, which anchors the palladium catalyst to the amino group 87 with 2 of the substrate while coordinating an isocyanide molecule to form intermediate (I). This is followed by a migratory insertion of the isocyanide into the Pd–N bond and a subsequent 1,3-H shift, yielding the imidoyl–Pd intermediate (II). The reactivity continues to escalate as a second aminopalladation leads to intermediate (III), which then undergoes a pivotal anti-carbopalladation to produce the dinuclear Pd(II) species (IV)—a key step that transitionally bridges two metal centres to stabilise the growing heterocyclic core. As the mechanism progresses, a second round of isocyanide migratory insertion and another 1,3-H shift occur, furnishing a heterocyclic dinuclear species that evolves into a trinuclear Pd(II) species (V). This high-order cluster eventually collapses into a seven-membered mononuclear Pd(II) intermediate (VI). The final transformation is achieved through reductive elimination from intermediate (VI), which expels the target quinoline product 88 and leaves the metal in a Pd(0) state. To complete the catalytic cycle, this Pd(0) species is re-oxidized by molecular oxygen (O2) back to the active Pd(II) oxidation state.
 |
| | Scheme 45 Proposed mechanism for the synthesis of 88. | |
Further, a novel Pd-catalyzed coupling reaction between azide and isocyanides was reported by Xiong's group (Scheme 46a).45 The reaction involves the Groebke–Blackburn–Bienayme multicomponent reaction followed by azide-isocyanide coupling by using another substrate of isocyanide to afford imidazo [1,2-a] pyridine-fused 1,3-benzodiazepine heterocycles in moderate to excellent yields. A number of substrates were screened, featuring different electron-donating and withdrawing substituents on amino pyridines and benzaldehydes, along with different isocyanides, to demonstrate the generality of the protocol (Scheme 46b). In addition, the synthesised derivatives were screened for their anticancer activities, and the results showed good activity of compound 82 in inducing apoptosis of glioma cells, making this methodology more attractive for drug development and synthetic chemistry. Interestingly, the gram-scale synthesis of compound 83 yielded 78%, demonstrating the applicability of this methodology (Scheme 46c).
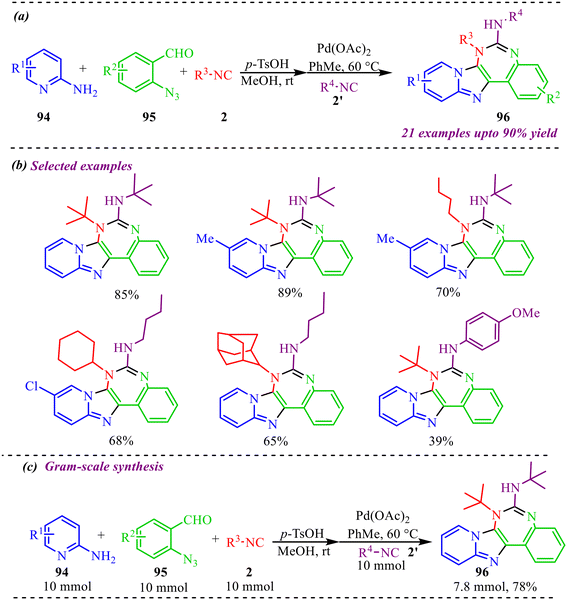 |
| | Scheme 46 (a) General scheme for synthesis of Imidazo [1,2-a] pyridine-fused 1,3-benzodiazepine heterocycles. (b) Substrate scope of the reaction (c) gram scale-up of reaction. | |
Furthermore, to confirm the reaction mechanism, a control experiment was conducted, as shown in Scheme 47. To determine whether the reaction follows the radical pathway or not, we have added the radical scavenger TEMPO to the standard reaction conditions on 97 and 2′, with no significant effect on the yield of 96, showing no radical formed in the reaction.
 |
| | Scheme 47 Control experiment. | |
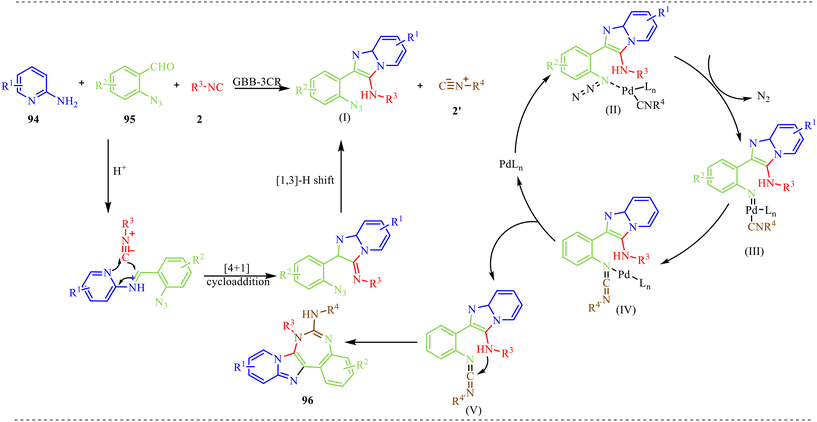
In addition to the control experiment, based on its results, the reaction mechanism presented in Scheme 48 illustrates multicomponent coupling with a palladium-catalysed nitrogen-release cycle to construct the complex heterocycle 96. The sequence initiates with an acid-catalyzed condensation between the amino-substituted pyridine 94 and the azide-containing aldehyde 95 to form a protonated Schiff base. This electrophilic species then undergoes a [4 + 1] cycloaddition with the first isocyanide 2, which, upon a subsequent [1,3]-hydrogen shift, yields the Groebke–Blackburn–Bienaymé (GBB) adduct (I) as a stable intermediate. This adduct then enters the catalytic phase by coordinating with a palladium complex and a second, distinct isocyanide 2′ to generate intermediate (II). A pivotal step occurs when this complex facilitates the extrusion of molecular nitrogen (N2), transforming the azide functionality into a highly reactive palladium-nitrene intermediate (III). This nitrene species undergoes an intramolecular rearrangement to produce intermediate (IV), which is then positioned for a reductive elimination. This elimination step is crucial, as it simultaneously expels the carbodiimide intermediate (V) and regenerates the active palladium catalyst, allowing it to re-enter the cycle. The synthesis reaches its conclusion through a final, spontaneous intramolecular cyclization of the carbodiimide (V), which effectively stitches the final rings together to deliver the target polycyclic product 96.
 |
| | Scheme 48 Plausible mechanism for the synthesis of imidazo[1,2-a]pyridine-fused 1,3-benzodiazepine heterocycles. | |
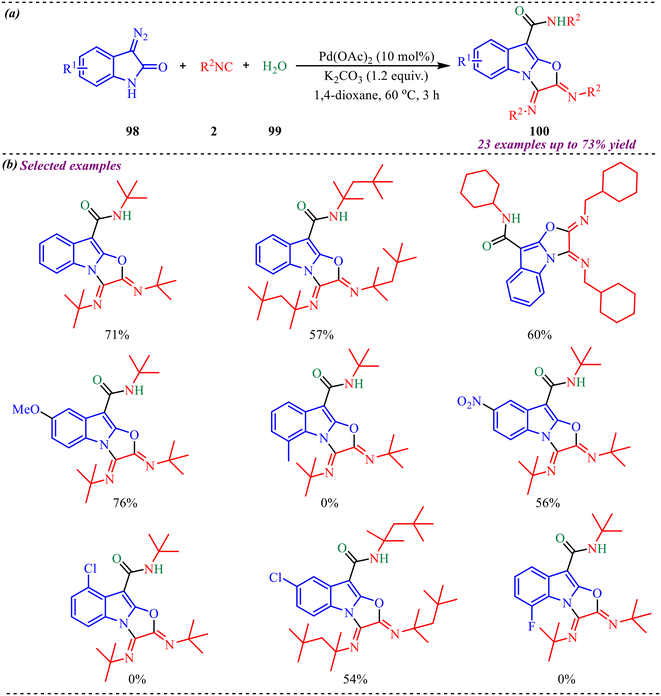
Also, we have reported a multicomponent cascade reaction for the synthesis of N-fused polycyclic indoles via a palladium-catalyzed process involving 3-diazooxindoles and isocyanides (Scheme 49).46 The substrate scope of the Pd-catalyzed reaction was thoroughly investigated. Moreover, various substitutions on the isocyanide component were examined, including bulky groups, which led to reduced yields due to increased steric hindrance. The influence of electron-donating, electron-withdrawing, and halogen substituents at different positions of the 3-diazo-oxindole core was also systematically studied. Next, electron-donating groups at the C5-position afforded the desired products in good yields. However, when these substituents were placed at the C6 and C7 positions, a noticeable decrease in yield was observed, likely due to steric hindrance near the reactive centre. In contrast, electron-withdrawing groups at the C5 position led to lower yields, while their presence at the C7 position completely suppressed product formation. The halogen substituents at the C4, C5, and C6-positions were generally well tolerated, yielding the corresponding products in moderate yields. However, substitution at the C7-position with a halogen failed to yield the desired compound.
 |
| | Scheme 49 Pd-catalyzed multicomponent cascade reaction for synthesis of N-fused polycyclic indoles. | |
Alongside, several control experiments were performed to verify the role of amide formation in the synthesis. The reaction was performed on 98 under standard reaction conditions, and the product, substituted oxoindoline-3-carboxamide 101, was obtained in 78% yield (Scheme 50a). Consequently, the reaction of 101 with isocyanide resulted in the formation of product 100 in good yield (Scheme 50b). To further validate the involvement of the in situ generated amide intermediate, the reaction was examined using oxindole, indolin-2-one 102 (Scheme 50c). However, no product formation was observed under the standard reaction conditions. This outcome reinforces the crucial role of the in situ-generated amide species in driving the transformation. Subsequently, the reaction was performed with D2O instead of H2O to probe deuterium incorporation during the transformation. The formation of the product 104 was confirmed by HRMS analysis. The HRMS spectrum of the crude reaction mixture displayed a prominent peak at m/z 398.2648, corresponding to the deuterated amide-based product, thereby supporting the involvement of water in the reaction pathway (Scheme 50d).
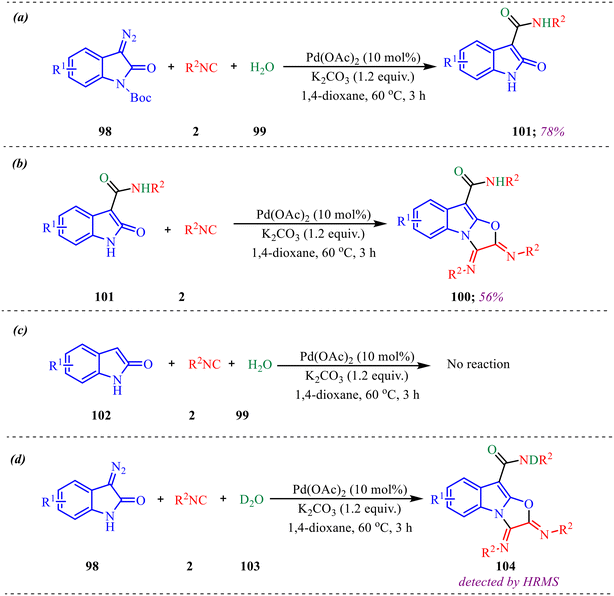 |
| | Scheme 50 (a) Reaction with N-boc protected 3-diazooxindoles. (b) To verify the amide intermediate in the reaction mechanism. (c) Reaction with indolin-2-one. (d) Role of water in the reaction medium. | |
In addition to this, the formation of tricyclic oxazolo[3,2-a]indole scaffolds 100 proceeds through a highly detailed sequence of organometallic transformations involving multiple isocyanide insertions and palladium-mediated cyclizations as depicted in Scheme 51. The mechanism initiates with the coordination of an isocyanide 2 to Pd(OAc)2 to form complex (I), which then binds 3-diazo oxindole 98 to yield the square-planar complex (II). The subsequent extrusion of molecular nitrogen from this species generates a reactive Pd–carbene species (III), which undergoes migratory isocyanide insertion to afford the Pd–ketenimine complex (IV). Upon the release of the palladium catalyst, the resulting ketenimine intermediate (V) undergoes hydrolysis and tautomerization to yield the stable amide (VII). This amide then re-coordinates with Pd(OAc)2 to form the six-membered complex (VIII), which is converted to the activated complex (IX) via deprotonation at the C-3 position. The coordination and insertion of a second isocyanide molecule into the amide carbonyl bond expands the metal center into the seven-membered complex (XI), which subsequently rearranges into the palladacycle (XII). A third isocyanide insertion into the Pd–C bond then progresses through intermediate (XIII) to form (XIV), where an acetate-assisted N–H deprotonation and loss of acetic acid lead to the formation of species (XVI). Finally, the mechanism concludes with intramolecular C–N bond formation to generate (XVII), followed by reductive elimination via (XVIII) to deliver the final tricyclic product 100 and regenerate the active Pd(OAc)2 catalyst.
 |
| | Scheme 51 Proposed mechanism for the multicomponent cascade reaction. | |
Ge et al. reported Pd-catalysed synthesis of 5- or 6-membered heterocyclic fused quinazolines efficiently and selectively using di-ortho-iodophenyl sulfonylguanidines with isocyanides (Scheme 52).47 The reaction conditions were thoroughly optimized across various parameters to achieve the efficient synthesis of both 5- and 6-membered heterocyclic fused quinazolines. Under optimized conditions, the reaction scope was thoroughly examined for the synthesis of fused quinazoline frameworks. In this context, iodophenyl guanidines with electron-donating (–CH3) and electron-withdrawing (–CF3) groups at the C-4 position afforded 6-membered fused products in good yields. Fluorine and chlorine were well tolerated, while bromine gave no product, and C-5 substitution also proved effective. The sulfonamide moiety was then explored; halogens and electron-donating groups provided good yields, while electron-withdrawing groups showed satisfactory performance. Moreover, heteroaryl-, naphthyl-, and alkyl-substituted sulfonamides were compatible, and the reaction provided the desired products in excellent yields. A notable shift in selectivity was observed with aryl isocyanides, which exclusively led to 5-membered fused quinazolines. Next, guanidines bearing halide or methyl groups at the C-4 position gave moderate to good yields, except for bromine, which showed only trace conversion. Similar substituent trends on sulfonamides favoured the formation of the 5-membered product. Beyond aryl isocyanides, the structure–reactivity relationship was further evaluated using various isocyanides. Adamantyl isocyanide exclusively yielded the 6-membered product, cyclohexyl led to the 5-membered product, while n-butyl showed no conversion. Finally, the reaction demonstrated good scalability and synthetic applicability, affording products 106 and 109 in 86% and 90% isolated yields, respectively.
 |
| | Scheme 52 (a) General scheme for palladium-catalysed synthesis of heterocyclic fused quinazoline. (b) Substrate scope of the reaction. (c) Further transformation of quinazoline derivative. | |
To gain mechanistic insight, a series of control experiments were carried out (Scheme 53). When substrate 105 was treated with two equivalents of tert-butyl isocyanide 2′ at a reduced temperature, a mixture of intermediates 110 and 110′ was obtained in a 3:2 ratio, with a combined yield of 45%. Subsequently, the isolated mixture of 110 and 110′ was subjected to two equivalents of tert-butyl isocyanide under the optimized reaction conditions, which led to the formation of product 106 in nearly quantitative yield. In contrast, no reaction was observed when an aryl isocyanide was employed under similar conditions. These findings strongly suggest that 110 and 110′ serve as key intermediates in the reaction pathway and indicate that aryl isocyanides exhibit significantly lower reactivity in this transformation.
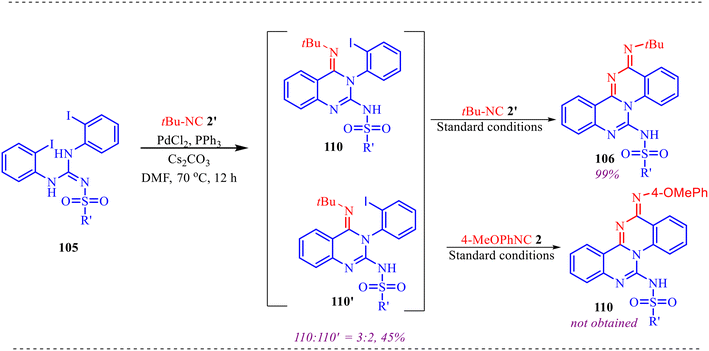 |
| | Scheme 53 Control experiments. | |
Based on the control experiments, the palladium-catalysed sequential isocyanide insertion and cyclisation is described in Scheme 54. The process initiates with the oxidative addition of an in situ-generated Pd(0) species to the aryl–iodine bond of the di-o-iodophenylguanidine substrate 105. In the presence of an isocyanide, this step yields the aryl–palladium intermediate (I), which subsequently undergoes a migratory isocyanide insertion into the carbon–palladium bond to form the Pd(II) complex (II). Base-assisted elimination of hydrogen iodide then facilitates the formation of the cyclopalladated species (III), a critical step that prepares the framework for reductive elimination to generate the pivotal intermediate (IV). At this juncture, the reaction pathway diverges based on the isocyanide used: when aryl isocyanides are employed, the intermediate (IV) readily undergoes a Pd(0)-mediated intramolecular cyclization to furnish the five-membered heterocycle-fused quinazoline 107. In contrast, the use of tert-butyl isocyanide—likely due to its distinct steric profile—promotes a second isocyanide insertion. This leads to the formation of a seven-membered palladium intermediate (V), which eventually undergoes a final reductive elimination to deliver the six-membered heterocycle-fused quinazoline 106 while simultaneously regenerating the active palladium catalyst to restart the cycle.
 |
| | Scheme 54 Plausible mechanism for Pd-catalysed synthesis of heterocyclic fused quinazoline. | |
Liu et al. reported a Pd-catalysed C–H functionalisation/[4 + 1] cyclisation reaction to synthesise quinoline derivatives using substituted imine and aryl isocyanides (Scheme 55).48 After obtaining the best reaction conditions, the functional group tolerance of this transformation was thoroughly explored, which consisted of the employment of various electron-donating, electron-withdrawing, and halogen substituents on imines as starting materials, resulting in the desired products up to 66% yields. However, no product formation was observed when heterocyclic groups like quinoline or pyridine were attached to the imine nitrogen. Further, aryl isocyanides were also investigated, revealing that sterically bulky groups were compatible and led to efficient product formation. In contrast, aliphatic isocyanides such as tert-butyl isocyanide failed to yield quinoline derivatives.
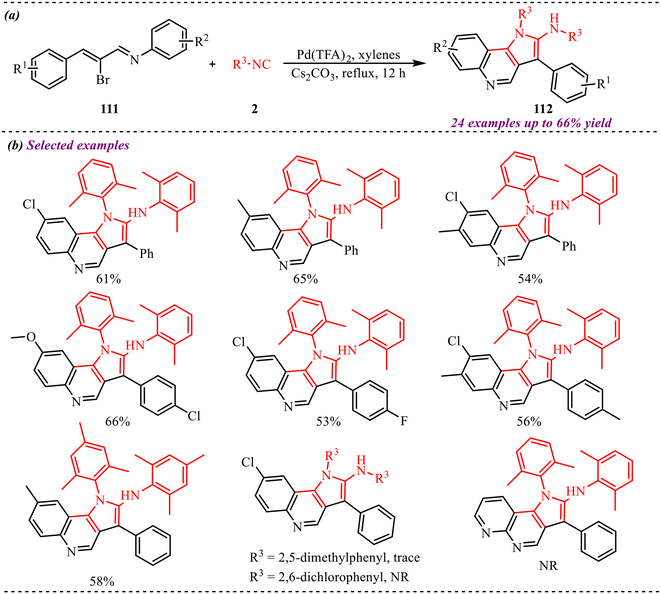 |
| | Scheme 55 (a) General scheme for palladium-catalysed synthesis of quinoline derivatives via isocyanide insertion. (b) Substate scope of the reaction. | |
Moreover, to obtain deeper mechanistic insight, a series of carefully designed control experiments were performed as shown in Scheme 56. For this, the radical scavenger TEMPO was added under standard reaction conditions. No significant change in reaction yield was observed (Scheme 56a). This result strongly suggests that the transformation does not proceed through a radical pathway. Moreover, in the reaction system comprising substrates 111 and 2, the characteristic HRMS peak corresponding to intermediate 114 (m/z = 371.1318) was successfully detected (Scheme 56b). This finding indicates that the reaction likely initiates with the insertion of a single isocyanide molecule, accompanied by C(sp2)–H functionalization, thereby providing key evidence for the proposed mechanistic pathway.
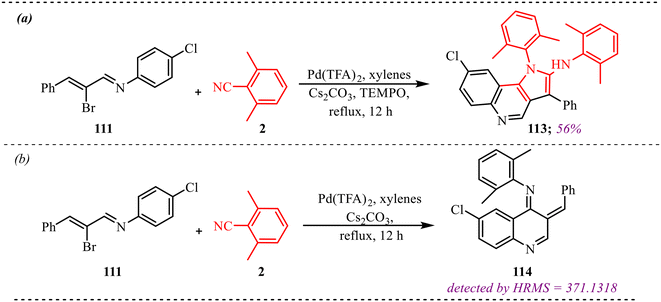 |
| | Scheme 56 (a) Radical scavenging experiment. (b) Determination of key intermediates in the reaction pathway. | |
Based on the results from control experiments, a plausible catalytic cycle is depicted in Scheme 57. The mechanism is initiated by the oxidative addition of Pd(0) into the appropriate bond of the substrate 111, generating an organopalladium complex (I). This activated palladium species subsequently undergoes migratory insertion of the isocyanide into the Pd–C bond, furnishing intermediate (II). Intramolecular coordination and cyclization of (II) then lead to the formation of cyclic palladium intermediates (III) and (IV). These intermediates undergo reductive elimination to release Pd(0), thereby regenerating the active catalyst and delivering intermediate (V). Upon heating, intermediate (V) undergoes configurational inversion, leading to the formation of the thermodynamically more favourable intermediate (VI). This rearranged species then participates in an intramolecular [4 + 1] cyclization process, generating the cyclic intermediate (VII). Subsequently, intermediate (VII) undergoes a 1,3-hydrogen shift, facilitating rearomatization and structural stabilization, ultimately delivering the final product 112. Collectively, these observations suggest that the transformation proceeds via initial single isocyanide insertion followed by C–H functionalization, ultimately leading to product formation through a palladium-mediated organometallic pathway.
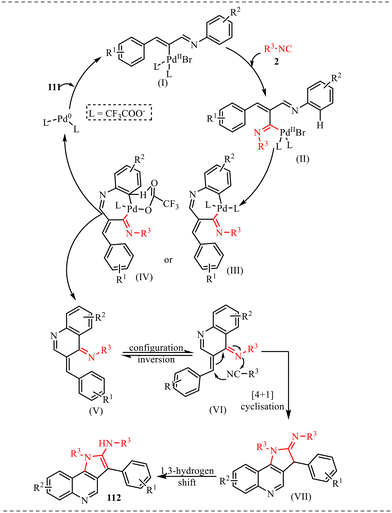 |
| | Scheme 57 Plausible mechanism for palladium-catalysed quinoline derivative synthesis. | |
Functionalisation of 5,6-membered heterocycles
The functionalization of five- and six-membered heterocycles constitutes a crucial area of modern organic synthetic chemistry, owing to the widespread occurrence of these frameworks in natural products, pharmaceuticals, including anti-inflammatory and anticancer agents and advanced materials, particularly in the development of organic light-emitting diodes (OLEDs) and organic semiconductors.49 Consequently, considerable efforts have been devoted to the development of efficient and selective methodologies for their modification. Among these, transition-metal-catalysed C–H bond activation has emerged as a powerful strategy, enabling site-selective functionalization, especially in structurally complex and fused heterocyclic systems.50 Concerning this, we have also reported a palladium-catalysed synthesis of indole-N-carboxamide derivatives via isocyanide insertion into the N–H bond of 3-phenylhydrazone-functionalized oxindoles Scheme 58.51 After optimizing the reaction conditions, the substrate scope was explored. In this context, replacing tert-butyl isocyanide with tetramethylbutyl and cyclohexyl isocyanides yielded comparable results, indicating minimal steric effects of isocyanide substitution. Further, substituent effects on the oxindole moiety were also studied. In this context, electron-donating groups at the C-5 position of the oxindole moiety reduced the reaction yield, while electron-withdrawing groups slightly improved it. Moreover, halides at C-5 were well tolerated, but those at C-6 led to modest reaction yields. Next, electron-donating groups on the phenyl ring of the hydrazone provided high yields, especially when combined with electron-donating or halogen groups on the oxindole. Notably, substitutions at C-4 and C-5 of the oxindole ring completely suppressed product formation, highlighting the crucial roles of electronic effects and substitution patterns in this transformation.
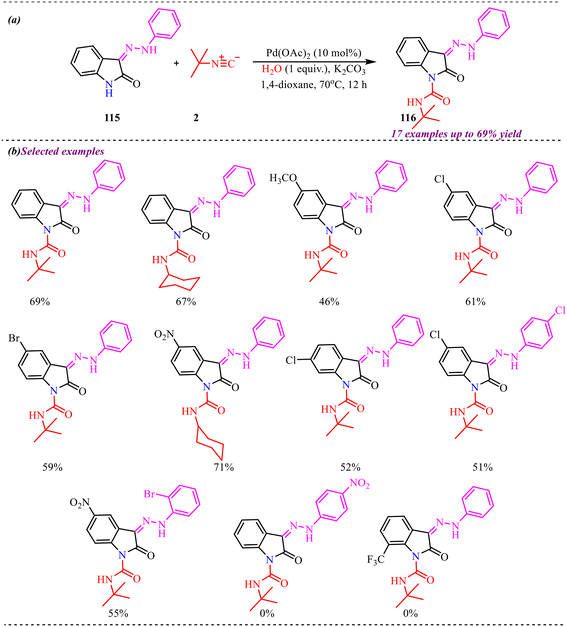 |
| | Scheme 58 (a) General scheme for palladium-catalysed synthesis of indole-N-carboxamide derivatives. (b) Substrate scope of the reaction. | |
Moreover, the palladium-catalysed transformation of indolin-2-one derivative 115 into the densely functionalized product 116 proceeds through a well-orchestrated catalytic cycle involving directed C–H activation and sequential isocyanide insertions, as illustrated in Scheme 59. The process is initiated by coordination of tert-butyl isocyanide 2 to Pd(OAc)2, generating the catalytically active Pd(II)–isocyanide complex (I). Assisted by the azo-directing group, electrophilic palladation occurs selectively at the ortho C–H bond of the indolin-2-one framework, accompanied by the loss of acetic acid, to furnish the cyclometalated palladacycle intermediate (II). This well-defined five-membered palladacycle serves as a pivotal species in the catalytic sequence. Subsequent coordination and migratory insertion of a second isocyanide molecule into the Pd–C bond of (II) leads to the formation of the ring-expanded, seven-membered palladium intermediate (III), representing a key C–C bond-forming event. Further acetate-assisted deprotonation and elimination of another molecule of acetic acid promote structural reorganization through intermediates (IV), (V), and (VI), in which the palladium center remains coordinated to the nitrogen-rich scaffold, enabling sequential C–C and C–N bond construction while stabilizing the evolving framework. The catalytic cycle proceeds to the intermediate (VIII), where coordination of two acetate ligands facilitates reductive elimination and the release of the palladium catalyst, generating the open-chain intermediate (IX). This species equilibrates with its tautomeric form (X), which undergoes spontaneous intramolecular cyclization to re-establish the five-membered ring core and furnish the final product 116. Throughout the transformation, the azo-directing group ensures high regioselectivity in C–H activation, while the sterically demanding tert-butyl substituents help stabilize key palladium intermediates, thereby promoting efficient progression through the catalytic cycle.
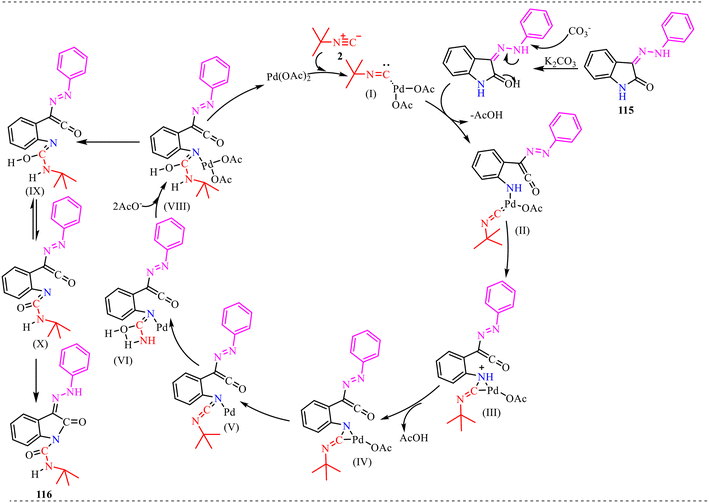 |
| | Scheme 59 Proposed reaction mechanism for synthesis of indole-N-carboxamide derivatives. | |
Wang and co-workers recently extended isocyanide insertion methodology towards the synthesis of biologically active naphthalen-2-amines in moderate to high yields via a Pd-catalysed tandem cyclization reaction of 2-(2-vinylarene) acetonitrile with tert-butyl isocyanide (Scheme 60a).52 Interestingly, this methodology yielded good results with various electron-rich and electron-poor groups attached to 2-(2-vinylarene) acetonitrile, along with differently substituted isocyanides. Besides, the in vitro antitumor activity of synthesized derivatives was tested against various tumour cell lines, i.e., U87, 4T1, HeLa, PC-3, HCT116, HepG-2, and A549. Among them, the compounds showed significant antitumor activity compared with 5-fluorouracil (5-FU) and amonafide, a positive control (Scheme 60b).
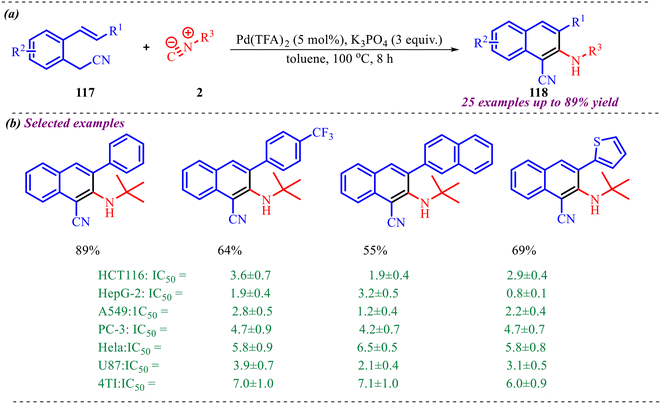 |
| | Scheme 60 (a) General scheme for synthesis of naphthalen-2-amine derivatives via isocyanide insertion. (b) Substrate scope of the reaction. | |
Notably, a series of control experiments were performed to gain mechanistic insight into the transformation depicted in Scheme 61. When (E)-2-(2-styrylphenyl)acetonitrile 117 was subjected to the optimized reaction conditions, no cyclized products 119 or 119′ were detected (Scheme 61a). In contrast, product 119 was observed in the GC–MS analysis upon addition of isocyanide (Scheme 61b), highlighting the essential role of isocyanide in promoting catalysis. Moreover, the presence of radical scavengers such as TEMPO and BHT did not significantly affect the reaction yield (Scheme 61c), suggesting that a radical pathway is unlikely to be involved in the reaction mechanism.
 |
| | Scheme 61 (a) Reaction in the absence of isocyanide. (b) Role of isocyanide as catalyst in the reaction. (c) Radical scavenging experiments. | |
A plausible catalytic cycle for this transformation, as depicted in Scheme 62, begins with the coordination of Pd(II) to isocyanide 2, generating a Pd(II)–isocyanide complex that serves as the active catalytic species. In the presence of K3PO4, substrate deprotonation facilitates the formation of intermediate (I), likely through base-assisted activation of the C–H bond and subsequent palladation. This activated species then coordinates to the Pd(II)–isocyanide complex to furnish intermediate (II), in which the metal center is simultaneously ligated to the aryl moiety and the isocyanide ligand. Subsequent migratory insertion of the coordinated isocyanide into the Pd–C bond leads to intermediate (III), forming a new imidoyl–Pd species that constitutes a key carbon–carbon bond-forming step in the catalytic cycle. The tethered alkene then undergoes intramolecular migratory insertion into the Pd–C bond, affording the alkyl–Pd intermediate (IV), which is a crucial branching point of the mechanism. Finally, β-hydride elimination from intermediate (IV) delivers the desired product 118 while regenerating the Pd(II) catalyst, thereby completing the catalytic cycle. The sequence of isocyanide insertion, alkene migratory insertion, and β-hydride elimination collectively accounts for the formation of the functionalized product and highlights the pivotal roles of intermediates (III) and (IV) in driving the transformation forward.
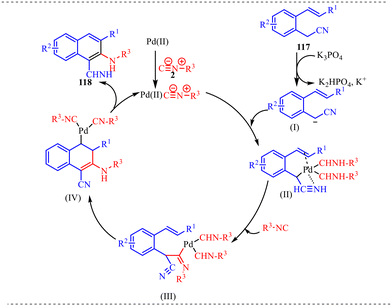 |
| | Scheme 62 Plausible mechanism synthesis of naphthalen-2-amine derivatives. | |
Conclusion
Palladium-catalyzed cascade reactions employing isocyanides as key starting materials have emerged as a powerful and versatile synthetic platform for the construction of diverse heterocyclic frameworks. The ambident reactivity of isocyanides, coupled with the tunable catalytic behaviour of palladium complexes, enables the efficient formation of multiple C–C and C–N bonds within a single synthetic operation. This streamlined approach minimizes purification steps and waste generation, making it inherently aligned with the principles of green and sustainable chemistry. Recent advances have demonstrated that these methodologies can efficiently access a wide array of heterocyclic architectures, including five-, six-, and fused-ring systems. Furthermore, detailed mechanistic studies have elucidated the roles of key intermediates, facilitating the rational design of catalysts and ligands. The broad substrate scope and excellent functional-group tolerance underscore the potential of these strategies for the synthesis of structurally complex, biologically active, and pharmaceutically relevant scaffolds. Overall, the continued evolution of Pd-mediated isocyanide cascade reactions highlights their pivotal role in modern heterocyclic synthesis and their growing impact on synthetic, medicinal, and materials chemistry. Future efforts should prioritise the design of more efficient catalytic platforms, reduction of palladium usage, and incorporation of electrochemical or photochemical strategies to broaden the applicability and sustainability of these transformations.
Conflicts of interest
The authors declare no conflict of interest.
Data availability
No primary research results, software, or code have been included, and no new data were generated or analysed as part of this review.
Acknowledgements
The financial support from the Department of Biotechnology, Ministry of Science & Technology, Government of India (BT/PR55062/BSA/33/294/2024), Centre of Excellence for Emerging Materials, Thapar Institute of Engineering and Technology, Patiala (TIET/CEEMS/Regular/2022/042) and the DST/INSPIRE Fellowship/2020/IF200048 is greatly acknowledged.
References
-
(a)
B. M. Gardner, C. C. C. Johansson Seechurn and T. J. Colacot, Organometallic Chemistry in Industry: A Practical Approach, 2020, pp. 1–22 Search PubMed;
(b) Y. Xia, D. Qiu and J. Wang, Chem. Rev., 2017, 117, 13810–13889 CrossRef CAS PubMed;
(c) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417–1492 CrossRef CAS PubMed;
(d) J. Cheng, L. Wang, P. Wang and L. Deng, Chem. Rev., 2018, 118, 9930–9987 CrossRef CAS PubMed;
(e) D. Wang and D. Astruc, Chem. Soc. Rev., 2017, 46, 816–854 RSC;
(f) A. Rajeev, M. Balamurugan and M. Sankaralingam, ACS Catal., 2022, 12, 9953–9982 CrossRef CAS;
(g) K. D. Vogiatzis, M. V. Polynski, J. K. Kirkland, J. Townsend, A. Hashemi, C. Liu and E. A. Pidko, Chem. Rev., 2018, 119, 2453–2523 CrossRef PubMed.
-
(a)
M. L. Crawley and B. M. Trost, Applications of Transition Metal Catalysis in Drug Discovery and Development: An Industrial Perspective, John Wiley & Sons, 2012 CrossRef;
(b) A. Piontek, E. Bisz and M. Szostak, Am. Ethnol., 2018, 130, 11284–11297 Search PubMed;
(c) S. Wacławek, V. V. Padil and M. Černík, Ecol. Chem. Eng. S, 2018, 25, 9 Search PubMed;
(d)
R. Sarkar, A. Pal and B. Saha, Homogeneous Catalysis: Concepts and Basics, Elsevier, 2024, pp. 299–331 Search PubMed;
(e) X. Cheng, A. Lei, T. S. Mei, H. C. Xu, K. Xu and C. Zeng, CCS Chem., 2022, 4, 1120–1152 CrossRef CAS;
(f) P. Lakhani, D. Bhanderi and C. K. Modi, Discov. Catal., 2024, 1, 2 CrossRef;
(g) C. H. Mak, X. Han, M. Du, J. J. Kai, K. F. Tsang, G. Jia and H. Y. Hsu, J. Mater. Chem. A, 2021, 9, 4454–4504 RSC;
(h) M. H. Sun, S. Z. Huang, L. H. Chen, Y. Li, X. Y. Yang, Z. Y. Yuan and B. L. Su, Chem. Soc. Rev., 2016, 45, 3479–3563 RSC.
-
(a) S. K. Sinha, P. Ghosh, S. Jain, S. Maiti, S. A. Al-Thabati, A. A. Alshehri and D. Maiti, Chem. Soc. Rev., 2023, 52, 7461–7503 RSC;
(b) Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Transition-metal-catalysed C–H alkylation using alkenes, Chem. Rev., 2017, 117(13), 9333–9403 CrossRef CAS PubMed;
(c) D. Mandal, S. Roychowdhury, J. P. Biswas, S. Maiti and D. Maiti, Chem. Soc. Rev., 2022, 51, 7358–7426 RSC;
(d) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS PubMed; S. W. Roh, K. Choi and C. Lee, Chem. Rev., 2019, 119, 4293–4356 Search PubMed;
(e) D. W. Gao, Q. Gu, C. Zheng and S. L. You, Acc. Chem. Res., 2017, 50, 351–365 CrossRef CAS PubMed;
(f) A. H. Elwahy, M. R. Shaaban and I. A. Abdelhamid, Appl. Organomet. Chem., 2022, 36, e6772 CrossRef CAS;
(g) J. S. S. Neto and G. Zeni, Org. Chem. Front., 2020, 7, 155–210 RSC;
(h) S. Bera, L. M. Kabadwal and D. Banerjee, Chem. Soc. Rev., 2024, 53, 4607–4647 RSC;
(i) I. Khan, S. Zaib and A. Ibrar, Org. Chem. Front., 2020, 7, 3734–3791 RSC;
(j) A. Banerjee, S. Kundu, A. Bhattacharyya, S. Sahu and M. S. Maji, Org. Chem. Front., 2021, 8, 2710–2771 RSC.
-
(a) A. F. P. Biajoli, C. S. Schwalm, J. Limberger, T. S. Claudino and A. L. Monteiro, J. Braz. Chem. Soc., 2014, 25, 2186–2214 CAS;
(b) A. Lauria, R. Delisi, F. Mingoia, A. Terenzi, A. Martorana, G. Barone and A. M. Almerico, Eur. J. Org. Chem., 2014, 2014, 3289–3306 CrossRef CAS.
-
(a) S. M. Shakil Hussain, M. S. Kamal and M. K. Hossain, J. Nanomater., 2019, 2019, 1562130 Search PubMed;
(b) A. Chen and C. Ostrom, Chem. Rev., 2015, 115, 11999–12044 CrossRef CAS PubMed;
(c) Ó. López and J. M. Padrón, Iridium-and palladium-based catalysts in the pharmaceutical industry, Catalysts, 2022, 12(2), 164 CrossRef;
(d) R. Chinchilla and C. Nájera, Chem. Rev., 2014, 114, 1783–1826 CrossRef CAS PubMed;
(e) B. M. Trost and J. T. Masters, Chem. Soc. Rev., 2016, 45, 2212–2238 RSC;
(f) I. Saldan, Y. Semenyuk, I. Marchuk and O. Reshetnyak, J. Mater. Sci., 2015, 50, 2337–2354 CrossRef CAS;
(g) J. Fan, H. Du, Y. Zhao, Q. Wang, Y. Liu, D. Li and J. Feng, ACS Catal., 2020, 10, 13560–13583 CrossRef CAS;
(h) A. Cabré, X. Verdaguer and A. Riera, Chem. Rev., 2021, 122, 269–339 CrossRef PubMed.
-
(a) S. Saranya, K. R. Rohit, S. Radhika and G. Anilkumar, Org. Biomol. Chem., 2019, 17, 8048–8061 RSC;
(b) A. Biffis, P. Centomo, A. Del Zotto and M. Zecca, Chem. Rev., 2018, 118, 2249–2295 CrossRef CAS PubMed;
(c) P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed;
(d) C. Shen and X. F. Wu, Chem. – Eur. J., 2017, 23, 13 Search PubMed;
(e) A. S. Díaz-Marta, C. R. Tubío, C. Carbajales, C. Fernández, L. Escalante, E. Sotelo, F. Guitián, V. L. Barrio, A. Gil and A. Coelho, ACS Catal., 2018, 8, 392–404 CrossRef;
(f) N. Kaur, Catal. Rev.: Sci. Eng., 2015, 57, 1–78 CrossRef CAS;
(g) K. Sakthivel, R. J. Gana, T. Shoji, N. Takenaga, T. Dohi and F. V. Singh, Front. Chem., 2023, 11, 1217744 CrossRef CAS PubMed;
(h) S. Perrone, L. Troisi and A. Salomone, Eur. J. Org. Chem., 2019, 2019, 4626–4643 CrossRef CAS;
(i) R. Tandon, N. Tandon and S. M. Patil, RSC Adv., 2021, 11, 29333–29353 RSC;
(j) S. Majhi and S. K. Jash, Synth. Commun., 2023, 53, 2061–2087 CrossRef CAS;
(k) M. N. Chen, L. P. Mo, Z. S. Cui and Z. H. Zhang, Curr. Opin. Green Sustainable Chem., 2019, 15, 27–37 CrossRef; P. X. T. Rinu, S. Saranya and G. Anilkumar, Eur. J. Org. Chem., 2025, 28, e202401429 Search PubMed.
-
(a) J. Rayadurgam, S. Sana, M. Sasikumar and Q. Gu, Org. Chem. Front., 2021, 8, 384–414 RSC;
(b) X. Ma, B. Murray and M. R. Biscoe, Nat. Rev. Chem., 2020, 4, 584–599 CrossRef CAS PubMed;
(c) S. Sain, S. Jain, M. Srivastava, R. Vishwakarma and J. Dwivedi, Curr. Org. Synth., 2019, 16, 1105–1142 CrossRef CAS PubMed;
(d) S. S. Ng, W. H. Pang, O. Y. Yuen and C. M. So, Org. Chem. Front., 2023, 10, 4408–4436 RSC;
(e) J. Perez Sestelo and L. A. Sarandeses, Molecules, 2020, 25, 4500 CrossRef PubMed.
-
(a) S. J. Firsan, V. Sivakumar and T. J. Colacot, Chem. Rev., 2022, 122, 16983–17027 CrossRef CAS PubMed;
(b) S. J. Firsan, V. Sivakumar and T. J. Colacot, Chem. Rev., 2022, 122, 16983–17027 CrossRef CAS PubMed;
(c) T. Patra and D. Maiti, Chem. – Eur. J., 2017, 23, 7382–7401 CrossRef CAS PubMed.
-
(a) K. C. Nicolaou, T. Montagnon and S. A. Snyder, Chem. Commun., 2003, 551–564 RSC;
(b) L.-Q. Lu, J.-R. Chen and W.-J. Xiao, Acc. Chem. Res., 2012, 45, 1278–1293 CrossRef CAS PubMed;
(c) D. E. Fogg and E. N. dos Santos, Coord. Chem. Rev., 2004, 248, 2365–2379 CrossRef CAS.
-
(a) M. J. Climent, A. Corma, S. Iborra and M. J. Sabater, ACS Catal., 2014, 4, 870–891 CrossRef CAS;
(b) J. C. Wasilke, S. J. Obrey, R. T. Baker and G. C. Bazan, Chem. Rev., 2005, 105, 1001–1020 CrossRef CAS PubMed;
(c) A. Behr, A. J. Vorholt, K. A. Ostrowski and T. Seidensticker, Green Chem., 2014, 16, 982–1006 RSC;
(d) J. Kuchhadiya and K. Kapadiya, Mini-Rev. Org. Chem., 2025, 22, 85–98 CrossRef CAS;
(e) S. Brauch, S. S. van Berkel and B. Westermann, Chem. Soc. Rev., 2013, 42, 4948–4962 RSC.
-
(a) S. Schmidt, K. Castiglione and R. Kourist, Chem. – Eur. J., 2018, 24, 1755–1768 CrossRef CAS PubMed;
(b) Y. Wang, H. Lu and P. F. Xu, Acc. Chem. Res., 2015, 48, 1832–1844 CrossRef CAS;
(c) S. F. Mayer, W. Kroutil and K. Faber, Chem. Soc. Rev., 2001, 30, 332–339 RSC;
(d) J. Muschiol, C. Peters, N. Oberleitner, M. D. Mihovilovic, U. T. Bornscheuer and F. Rudroff, Chem. Commun., 2015, 51, 5798–5811 RSC;
(e) A. Dhakshinamoorthy and H. Garcia, ChemSusChem, 2014, 7, 2392–2410 CrossRef CAS PubMed;
(f) L.-Q. Lu, J.-R. Chen and W.-J. Xiao, Acc. Chem. Res., 2012, 45, 1278–1293 CrossRef CAS PubMed;
(g) S. Gonzalez-Granda, J. Albarran-Velo, I. Lavandera and V. Gotor-Fernández, Chem. Rev., 2023, 123, 5297–5346 CrossRef CAS PubMed.
-
(a) Y. Wang, H. Lu and P. F. Xu, Acc. Chem. Res., 2015, 48, 1832–1844 CrossRef CAS PubMed;
(b) M. K. Yadav and S. Chowdhury, Green Chem., 2023, 25, 10144–10181 RSC;
(c) S. Gonzalez-Granda, J. Albarran-Velo, I. Lavandera and V. Gotor-Fernández, Chem. Rev., 2023, 123, 5297–5346 CrossRef CAS PubMed;
(d) B. A. Farhan, L. Zhihe, S. Ali, T. A. Shah, L. Zhiyu, A. Zhang, S. Javed and M. Asad, Environ. Sci. Pollut. Res., 2023, 30, 64904–64931 CrossRef CAS PubMed.
-
(a) M. L. Bode, D. Gravestock and A. L. Rousseau, Org. Prep. Proced. Int., 2016, 48, 89–221 CrossRef CAS;
(b) M. Giustiniano, A. Basso, V. Mercalli, A. Massarotti, E. Novellino, G. C. Tron and J. Zhu, Chem. Soc. Rev., 2017, 46, 1295–1357 RSC;
(c) A. Shaabani, R. Mohammadian, R. Afshari, S. E. Hooshmand, M. T. Nazeri and S. Javanbakht, Mol. Diversity, 2021, 25, 1145–1210 CrossRef CAS PubMed;
(d) M. Heravi and N. Nazari, Curr. Org. Chem., 2017, 21, 1440–1529 CAS;
(e) Y. Zhu, J. Y. Liao and L. Qian, Front. Chem., 2021, 9, 670751 CrossRef CAS PubMed.
-
(a) G. Qiu, Q. Ding and J. Wu, Chem. Soc. Rev., 2013, 42, 5257–5269 RSC;
(b) J. W. Collet, T. R. Roose, E. Ruijter, B. U. Maes and R. V. Orru, Angew. Chem., Int. Ed., 2020, 59, 540–558 CrossRef CAS PubMed;
(c) Z. Tashrifi, M. Mohammadi Khanaposhtani, F. Gholami, B. Larijani and M. Mahdavi, Adv. Synth. Catal., 2023, 365, 926–947 CrossRef CAS.
-
(a) Q. Wang, D. X. Wang, M. X. Wang and J. Zhu, Acc. Chem. Res., 2018, 51, 1290–1300 CrossRef CAS PubMed;
(b) F. De Moliner, L. Banfi, R. Riva and A. Basso, Comb. Chem. High Throughput Screening, 2011, 14, 782–810 CrossRef CAS PubMed;
(c) Z. Q. Liu, Curr. Org. Chem., 2014, 18, 719–739 CrossRef CAS;
(d) A. Dömling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168–3210 CrossRef;
(e) Y. Shan, X. Zhang, G. Liu, J. Li, Y. Liu, J. Wang and D. Chen, Chem. Commun., 2024, 60, 1546–1562 RSC.
-
(a) M. Heravi and N. Nazari, Curr. Org. Chem., 2017, 21, 1440–1529 CAS;
(b) A. Váradi, T. C. Palmer, R. Notis Dardashti and S. Majumdar, Molecules, 2015, 21, 19 CrossRef PubMed;
(c) T. Nasiriani, S. Javanbakht, M. T. Nazeri, H. Farhid, V. Khodkari and A. Shaabani, Top. Curr. Chem., 2022, 380, 50 CrossRef CAS PubMed;
(d) D. R. Mishra, B. S. Panda, S. Nayak, J. Panda and S. Mohapatra, ChemistrySelect, 2022, 7, e202200531 CrossRef CAS;
(e) T. R. Roose, D. S. Verdoorn, P. Mampuys, E. Ruijter, B. U. W. Maes and R. V. A. Orru, Chem. Soc. Rev., 2022, 51, 5842–5877 RSC;
(f) M. Gao, S. Lu and B. Xu, Chem. Soc. Rev., 2024, 53, 10147–10170 RSC.
-
(a) G. Qiu, Q. Ding and J. Wu, Chem. Soc. Rev., 2013, 42, 5257–5269 RSC;
(b) V. P. Boyarskiy, N. A. Bokach, K. V. Luzyanin and V. Y. Kukushkin, Chem. Rev., 2015, 115, 2698–2779 CrossRef CAS PubMed;
(c) J. W. Collet, T. R. Roose, B. Weijers, B. U. Maes, E. Ruijter and R. V. Orru, Molecules, 2020, 25, 4906 CrossRef CAS PubMed;
(d) V. P. Boyarskiy, N. A. Bokach, K. V. Luzyanin and V. Y. Kukushkin, Chem. Rev., 2015, 115, 2698–2779 CrossRef CAS PubMed;
(e) R. Zhang, N. Liu, Z. Lei and B. Chen, Chem. Rev., 2016, 116, 3658–3721 CrossRef CAS PubMed;
(f) B. Song and B. Xu, Chem. Soc. Rev., 2017, 46, 1103–1123 RSC;
(g) M. Z. Lu, J. Goh, M. Maraswami, Z. Jia, J. S. Tian and T. P. Loh, Chem. Rev., 2022, 122, 17479–17646 CrossRef CAS PubMed.
-
(a) F. Nahra and C. S. Cazin, Chem. Soc. Rev., 2021, 50, 3094–3142 RSC;
(b) T. R. Roose, D. S. Verdoorn, P. Mampuys, E. Ruijter, B. U. W. Maes and R. V. A. Orru, Chem. Soc. Rev., 2022, 51, 5842–5877 RSC;
(c) M. Markovič, P. Koóš, T. Čarný and T. Gracza, Tetrahedron Lett., 2020, 61, 152370 CrossRef;
(d) S. Hosseininezhad, S. P. A. Abad and A. Ramazani, RSC Adv., 2025, 15, 1163–1204 RSC;
(e) I. Khan, J. Singh, I. Khan, S. Dutt, S. Khan and V. Tyagi, Org. Chem., 2019, 279–291 CAS;
(f) G. Qiu, Q. Ding and J. Wu, Chem. Soc. Rev., 2013, 42, 5257–5269 RSC;
(g) W. Kong, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2016, 55, 9866–9870 CrossRef.
- J. W. Collet, T. R. Roose, B. Weijers, B. U. Maes, E. Ruijter and R. V. Orru, Molecules, 2020, 25, 4906 CrossRef CAS PubMed.
-
(a) A. Rusu, I. M. Moga, L. Uncu and G. Hancu, Pharmaceutics, 2023, 15, 2554 CrossRef CAS PubMed;
(b) A. Omar, Al-Azhar J. Pharm. Sci., 2020, 62, 39–54 CrossRef;
(c) A. Kumar, B. Nehra, D. Singh, D. Kumar and P. A. Chawla, Curr. Top. Med. Chem., 2023, 23, 1277–1306 CrossRef CAS PubMed;
(d) Y. Hu, C. Y. Li, X. M. Wang, Y. H. Yang and H. L. Zhu, Chem. Rev., 2014, 114, 5572–5610 CrossRef CAS PubMed.
-
(a) A. A. Abbas, T. A. Farghaly and K. M. Dawood, RSC Adv., 2024, 14, 19752–19779 RSC;
(b) S. L. Albino, J. M. da Silva, M. S. de C. Nobre, Y. M. de M. e Silva, M. B. Santos, R. S. de Araújo, M. do C. A. de Lima, M. Schmitt and R. O. de Moura, Curr. Pharm. Des., 2020, 26, 4112–4150 CrossRef CAS PubMed;
(c) A. Adamczyk-Wozniak, K. M. Borys and A. Sporzynski, Chem. Rev., 2015, 115, 5224–5247 CrossRef CAS PubMed.
- X. Wang, J. P. Fu, J. H. Mo, Y. H. Tian, C. Y. Liu, H. T. Tang, Z. J. Sun and Y. M. Pan, Adv. Synth. Catal., 2021, 363, 2762–2766 CrossRef CAS.
- J. Li, Z. W. Zhao, S. Zheng, P. He, J. Y. Qiu, Q. Q. Zhou and Z. L. Ren, Org. Chem. Front., 2023, 10, 3252–3258 RSC.
- S. S. Liu, J. N. Zheng, Z. W. Zhao, Y. Zhang, P. He, Y. J. Wu and Z. L. Ren, Org. Chem. Front., 2025, 12, 2643–2650 RSC.
-
(a) M. Kumar, S. Verma, M. Sharma, Poonam and B. Rathi, Eur. J. Org. Chem., 2023, 26(46), e202300877 CrossRef CAS;
(b) C. J. Zhou, H. Gao, S. L. Huang, S. S. Zhang, J. Q. Wu, B. Li, X. Jiang and H. Wang, ACS Catal., 2018, 9(1), 556–564 CrossRef;
(c) F. Gu, B. Lin, Z. H. Peng, S. Liu, Y. Wu, M. Luo, N. Ding, Q. Zhan, P. Cao, Z. Zhou and T. Cao, Adv. Sci., 2024, 11(40), 2407931 CrossRef CAS PubMed.
- Y. M. Zhu, Y. Fang, H. Li, X. P. Xu and S. J. Ji, Org. Lett., 2021, 23, 7342–7347 CrossRef CAS PubMed.
- S. Chen, M. Oliva, L. Van Meervelt, E. V. Van der Eycken and U. K. Sharma, Adv. Synth. Catal., 2021, 363, 3220–3226 CrossRef CAS.
- S. Sisodiya, A. Acharya, M. Nagpure, N. Roy, S. K. Giri, H. R. Yadav, A. R. Choudhury and S. K. Guchhait, Chem. Commun., 2022, 58, 11827–11830 RSC.
- F. Zhang, R. Zhao, L. Zhu, Y. Yu, S. Liao, Z. X. Wang and X. Huang, Cell Rep., 2022, 3, 100776 CAS.
- J. Liu, Y. M. Zhu, X. P. Xu and S. J. Ji, Chin. J. Chem., 2024, 42, 259–263 CrossRef CAS.
- S. Zheng, H. J. Fan, S. S. Liu, Y. Xu, Z. W. Zhao, H. H. Kong and Z. L. Ren, Org. Chem. Front., 2024, 11, 1775–1781 RSC.
-
(a) N. Kumar and N. Goel, Anti-Cancer Agents Med. Chem., 2022, 22(19), 3196–3207 CrossRef CAS PubMed;
(b) A. Frühauf, M. Behringer and F. J. Meyer-Almes, Molecules, 2023, 28(15), 5686 CrossRef PubMed.
- N. A. Meanwell, Adv. Heterocycl. Chem., 2017, 123, 245–361 CrossRef CAS.
- A. Chaudhary and R. Srivastava, Asian J. Org. Chem., 2025, 14(5), e202400785 CrossRef CAS.
- L. Saeifard, K. Amiri, F. Rominger, T. J. J. Müller and S. Balalaie, J. Org. Chem., 2023, 88, 12519–12525 CrossRef CAS PubMed.
- F. Teng, T. Yu, Y. Peng, W. Hu, H. Hu, Y. He, S. Luo and Q. Zhu, J. Am. Chem. Soc., 2021, 143, 2722–2728 CrossRef CAS PubMed.
- J. Xiong, H. T. He, H. Y. Yang, Z. G. Zeng, C. R. Zhong, H. Shi, M. L. Ouyang, Y. Y. Tao, Y. L. Pang, Y. H. Zhang, B. Hu, Z. X. Fu, X. L. Miao, H. L. Zhu and G. Yao, J. Org. Chem., 2022, 87, 9488–9496 CrossRef CAS PubMed.
- N. Jangir, Poonam, S. Dhadda and D. K. Jangid, ChemistrySelect, 2022, 7(6), e202103139 CrossRef CAS.
- E. Teli-Kokalari, V. Stefanou, D. Matiadis, G. Athanasellis, O. Igglessi-Markopoulou, S. Hamilakis and J. Markopoulos, Fresenius Environ. Bull., 2012, 21(11), 3215–3223 CAS.
- Y. Zhang, T. Liu, L. Liu, H. Guo, H. Zeng, W. Bi, G. Qiu, W. Gao, X. Ran, L. Yang, G. Du and L. Zhang, J. Org. Chem., 2022, 87, 8515–8524 CrossRef CAS PubMed.
- R. Hommelsheim, S. Bausch, R. van Nahl, J. S. Ward, K. Rissanen and C. Bolm, Green Chem., 2023, 25, 3021–3026 RSC.
- B. Dai, Y. Pan, X. Wang, J. Fu, H. Tang, Y. Yi and Y. Pan, Adv. Synth. Catal., 2025, 367, e202400851 CrossRef CAS.
-
(a) M. H. A. Al-Jumaili, A. A. Hamad, H. E. Hashem, A. D. Hussein, M. J. Muhaidi, M. A. Ahmed, A. H. A. Albanaa, F. Siddique and E. A. Bakr, J. Mol. Struct., 2023, 1271, 133970 CrossRef CAS;
(b)
C. Lamberth and J. Dinges, Bioactive Heterocyclic Compound Classes: Agrochemicals, 2012, pp. 1–20 Search PubMed.
- M. Li, R. Zhang, Q. Gao, H. Jiang, M. Lei and W. Wu, Angew. Chem., Int. Ed., 2022, 61, e202208203 CrossRef CAS PubMed.
- C. R. Zhong, Y. H. Zhang, G. Yao, H. L. Zhu, Y. D. Hu, Z. G. Zeng, C. Z. Liao, H. T. He, Y. T. Luo and J. Xiong, J. Org. Chem., 2023, 88, 13125–13134 CrossRef CAS PubMed.
- P. Soam, D. Mandal and V. Tyagi, New J. Chem., 2024, 48, 2639–2648 RSC.
- S. Ge, Y. M. Zhu, X. P. Xu, Y. Zi and S. J. Ji, Chem. Commun., 2024, 60, 14613–14616 RSC.
- S. S. Liu, Y. J. Wu, J. N. Zheng, Z. L. Ren, S. J. Jiang, J. Wu and L. Wang, Org. Chem. Front., 2025, 12, 4757–4763 RSC.
-
(a) E. A. Gyrgenova, Y. Y. Titova and A. V. Ivanov, Molecules, 2025, 30, 3264 CrossRef CAS PubMed;
(b) N. Mazin Zeki and Y. Fakri Mustafa, Chem. Biodiversity, 2024, 21, e202301855 CrossRef CAS PubMed;
(c) A. N. Singh Chauhan, G. Mali, G. Dua, P. Samant, A. Kumar and R. D. Erande, ACS Omega, 2023, 8, 27894–27919 CrossRef CAS PubMed.
-
(a) U. Dutta, S. Maiti, T. Bhattacharya and D. Maiti, Science, 2021, 372, eabd5992 CrossRef CAS PubMed;
(b) A. Fanourakis and R. J. Phipps, Chem. Sci., 2023, 14, 12447–12476 RSC;
(c) L. Guillemard, L. Ackermann and M. J. Johansson, Nat. Commun., 2024, 15, 3349 CrossRef CAS PubMed;
(d) S. Gupta, M. Sravani Galla, K. Prasad Suhas and N. Shankaraiah, Adv. Synth. Catal., 2025, e202500093 CrossRef CAS.
- P. Soam, D. Mandal and V. Tyagi, Eur. J. Org. Chem., 2024, 27, e202400439 CrossRef CAS.
- H. Lin, Y. Pan, J. Fu, Y. Yi, H. Tang, Y. Pan, W. Yu and X. Wang, J. Org. Chem., 2023, 88, 12409–12420 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*