
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermodynamics of metal–organic frameworks: from fundamentals to advanced applications
Grace C.
Thaggard
,
Danielle N.
Smith
,
Buddhima K. P.
Maldeni Kankanamalage
,
Serena M.
Jentz
and
Natalia B.
Shustova
 *
*
Department of Chemistry and Biochemistry, University of South Carolina, Columbia, South Carolina 29208, USA. E-mail: shustova@sc.edu
First published on 14th April 2026
Abstract
The fundamental thermodynamics of metal–organic frameworks (MOFs) is a key aspect that defines the suitability of MOFs for a wide range of applications, including as sorbents for gas storage and water harvesting, solid-state electrolytes, radionuclide storage materials, and heterogeneous catalysts. However, reliable experimental evaluation of thermodynamic parameters related to, for example, framework stability, guest adsorption or binding, and phase transitions can be challenging due to MOF insolubility, limited diffusion of analytes through framework pores, and potential structural defects. The presented review aims to discuss practical methods and accessible experimental techniques, such as calorimetry, thermal measurements, absorbance and emission spectroscopies, and electrochemical experiments, for determining various thermodynamic aspects that are useful for predicting and tailoring MOF performance. By highlighting the advantages and potential outcomes of each class of considered techniques, this review serves as a guide for selecting and combining experimental methods to evaluate the detailed thermodynamics and kinetics of MOF-based processes, ranging from framework formation, post-synthetic modification, diffusion and binding of guest molecules, phase changes, and energy storage. In addition, this review highlights how theoretical modeling could potentially complement experimental work to provide a more comprehensive overview of the MOF thermodynamic landscape. Overall, each of the discussed case studies emphasizes how fundamental thermodynamics can shed light on the design of functional materials to address emergent global challenges in the technological, energy, and biomedical sectors and beyond.
 Grace C. Thaggard | Grace C. Thaggard is currently pursuing her PhD degree in inorganic chemistry at the University of South Carolina after completing her BSc degree in chemistry from the University of Alabama at Birmingham in 2021. Her research interests include the design of novel stimuli-responsive materials exhibiting directional energy transfer. She was selected as an NSF Graduate Research Fellow. |
 Danielle N. Smith | Danielle N. Smith is currently pursing her PhD degree in inorganic chemistry at the University of South Carolina after completing her BSc degree in chemistry from Radford University in 2024. Her research interests include the synthesis of stimuli-responsive frameworks for nuclear waste administration and actinide-based materials. |
 Buddhima K. P. Maldeni Kankanamalage | Buddhima K. P. Maldeni Kankanamalage is currently pursuing her PhD in inorganic chemistry at the University of South Carolina after completing her BSc degree in chemistry from the University of Colombo in 2019. Her research interests include metal–organic framework- and cage-based catalysis, as well as design of photoresponsive materials. |
 Serena M. Jentz | Serena M. Jentz is currently pursuing her PhD degree in inorganic chemistry at the University of South Carolina after completing her BSc degree in chemistry at the University of Nebraska-Lincoln in 2025. Her research interests include the synthesis of novel photochromic compounds and materials exhibiting stimuli-modulated properties. |
 Natalia B. Shustova | Natalia B. Shustova is a Fred M. Weissman Palmetto Professor of Chemistry and Biochemistry at the University of South Carolina and an Associate Editor of ACS Materials Letters. She received her MSc degree in materials science, two PhD degrees in physical and inorganic chemistry, and postdoctoral training at Massachusetts Institute of Technology. Her research program focuses on materials for sustainable energy conversion, the chemistry of f-block elements, and stimuli-responsive materials. Her research accolades include the Humboldt, Fischer, McCausland, and Sloan Fellowships as well as Dreyfus Teaching-Scholar, Cottrell Scholar, Breakthrough, Russel Research, Friedrich Wilhelm Bessel Research, and NSF Career Awards. |
Introduction
Due to their synthetic tunability, high surface areas, thermochemical stability, and well-defined structures, metal–organic frameworks (MOFs) have already been applied as advanced functional materials that act as multifaceted and “smart” adsorbents for gas storage and water harvesting, membranes for selective gas separation, efficient solid-state electrolytes, heterogeneous catalysts exhibiting sieving effects, and radionuclide storage materials, among many others.1–31 However, a given framework's suitability for the mentioned applications is in many cases defined by fundamental MOF thermodynamics. For example, understanding the thermodynamics of water harvesting in MOFs, covalent-organic frameworks (COFs), or any other porous systems, is the foundation for addressing one of the important global challenges of clean water scarcity in arid regions through the development of efficient atmospheric water harvesters.1,9,12,13,16,18,20,32–35 Similarly, the design of biocompatible MOFs capable of on-demand drug delivery and/or medical imaging is based on an understanding of the thermodynamic parameters associated with framework-guest interactions (e.g., binding energy between MOF linkers/nodes and solvent molecules, pharmaceuticals, or imaging agents), as well as predictions of framework stability in complex media.36–42However, the importance of understanding MOF thermodynamics extends far beyond the described applications in the areas of water harvesting and drug delivery. Indeed, many of the common metrics used to evaluate MOF performance can be tied to fundamental thermodynamic parameters (e.g., changes in enthalpy, entropy, and Gibbs free energy).
For instance, framework stability could be related to enthalpy of formation, guest storage and/or separation can be described by heat of adsorption, and even catalytic activity can often be tied to the reduction potentials of metal nodes.26,32,43–57 Overall, it is important to note that all of these emergent applications are based on a detailed understanding of fundamental parameters, including a broad range of kinetics and thermodynamics aspects.
One of the potential challenges in applying fundamental thermodynamic values (e.g., enthalpy of formation, binding energy, or enthalpy of phase transitions) to predict MOF properties is that many of the listed parameters have been obtained through theoretical modeling rather than experimental measurements.54,58,59 The use of computational analysis to probe MOF formation/degradation and performance is certainly an irreplaceable catalyst for progress in the MOF field, and some advanced computational methods have already been reviewed in depth recently.45,48,49,52 For example, DFT studies have already provided valuable predictions of thermodynamically stable MOF structures and topologies, as well as shed light on MOF electronic structures which is a key component for the design of multifunctional catalysts, semiconductors, and light-harvesting devices.45,48,49,52 However, experimental evaluation of MOF thermodynamic properties is equally important, yet often technically challenging.34,40,43,60–63 For instance, the insolubility of MOFs creates limitations with the use of conventional solution-based experimental techniques, such as solution calorimetry, UV-vis spectroscopy, or solution-based nuclear magnetic resonance spectroscopy.59,60,64–66 Moreover, careful experimental design and control of all variables is essential for accurately evaluating MOF thermodynamic data, especially considering the wide range of processes that can occur simultaneously within a given framework and which must be deconvoluted for accurate data interpretation.26,27,54,59,62,67–71 For example, the outcomes of an experiment designed to probe host–guest interactions in a MOF could be influenced by many competing factors, such as the presence of defects, variable particle size/morphology, slow diffusion of guest molecules throughout MOF channels, and multiple possible guest binding sites (i.e., at the organic linker, defects, and/or metal nodes).15,17,26,36,40–43,56,65,72–81 Thus, rigorous experimental design and selection of appropriate thermodynamic models for data analysis are critical and can often be validated through theoretical modeling.22,24,44,48,49,52,71,75,82–85 Moreover, the morphology of MOF samples (e.g., single crystals or particles with variable sizes) is highly dependent on the used synthetic protocols, which could significantly impact critical parameters, such as the rate of guest diffusion and availability of binding sites, further complicating the reproducibility of thermodynamic measurements.
Herein, we showcase the experimental toolbox available for uncovering critical thermodynamic parameters used to describe and predict the behavior of MOFs, specifically those designed to address global challenges, including the production of safe drinking water, nuclear waste remediation, and energy storage (Scheme 1).5,6,9,10,26,39,41,46,59,70,72,77,82,86–92 This review also focuses on the utility of relatively accessible laboratory techniques, including solution calorimetry, thermal analysis, spectroscopy, and electrochemical measurements, to deepen fundamental understanding of MOF thermodynamics, with an emphasis on using the obtained thermodynamic data to guide functional material design. In addition, we consider how different experimental methods (e.g., calorimetry versus spectroscopy) offer unique advantages that can be carefully selected and/or combined to comprehensively evaluate MOF performance or structure–property relationships. For example, depending on the specific properties of the targeted MOF, this review illustrates how either calorimetric or spectroscopic methods (as well as many others9,10,24,26,34,46,47,50,59,61,64,65,69,93–96) could be applied to evaluate hydrolytic stability, selective binding of guest molecules or gases, and possible phase transitions.7,16,18,22,26,39–41,54,56,58,63,70,77,79,87,95,97,98 As a result, the present work targets a practical discussion of how the design and interpretation of thermodynamic experiments can play a role in nearly all avenues of research in the MOF field, ranging from synthesis of novel frameworks and development of material preparation protocols to fabrication of MOF-based devices, ultimately opening the door for reticular chemistry to address upcoming urgent needs in the energy, technological, and biomedical sectors and beyond.
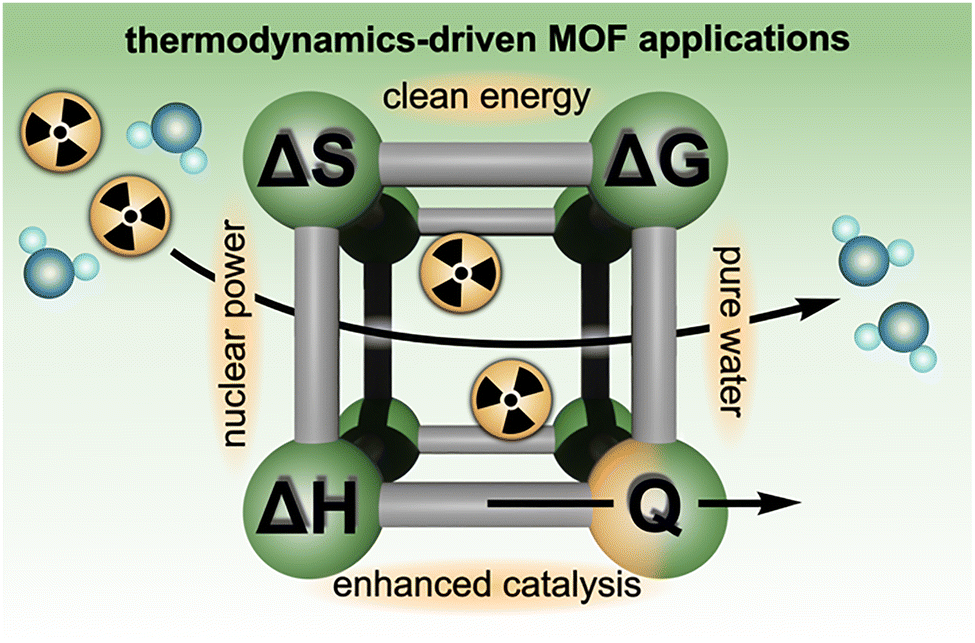 | ||
| Scheme 1 Schematic representation of essential thermodynamic parameters, such as entropy (ΔS), enthalpy (ΔH), Gibbs free energy (ΔG), and heat (Q), as well as examples of global challenges, the potential solutions for which are tied to thermodynamic aspects, as discussed below in this review. | ||
Calorimetry
The following discussion of experimental evaluation of MOF thermodynamics is organized based on several common classes of techniques used in many laboratory settings, including calorimetry, thermal measurements, and spectroscopy, as well as some less frequently utilized approaches. Each class of experimental techniques is highlighted using several case studies, which were selected based on the variety of fundamental thermodynamic parameters, processes (i.e., MOF formation, degradation, guest adsorption, phase transitions, etc.), and applications which they target. As a starting point, calorimetric measurements will be discussed.Calorimetric measurements are powerful, but often experimentally challenging, for understanding a wide range of MOF fundamentals and upcoming applications, including, for instance, CO2 capture from industrial flue gas, post-synthetic modification (PSM) strategies targeting novel structures, and design of biocompatible frameworks.46,50,89,92,93,99–102 Some of the technical challenges associated with calorimetric methods may be attributed to the insolubility of MOF-based materials, which introduces additional sample preparation steps (e.g., framework degradation in acidic media) prior to analysis by conventional solution calorimetry routes.93 In response, recent reports have developed attractive methods to extract valuable information either from measurements performed directly on solids (e.g., isothermal titration calorimetry (ITC) experiments or drop combustion calorimetry) or through strategic degradation of frameworks leading to their dissolution (e.g., solution calorimetry).15,36,39,40,43,50,59,86 In this review, the discussion of several practical experimental approaches for understanding the thermodynamic aspects of MOFs (or any other porous materials) will begin with three different calorimetric techniques: solution calorimetry, high-temperature drop combustion calorimetry, and ITC, with an emphasis on the wide variety of MOF-based applications and properties that could be studied using these methods.39,40,43,59,86
One of the fundamental thermodynamic parameters that can be used to predict and tailor MOF stability toward specific applications is the interaction enthalpy associated with the binding of solvent molecules to MOF linkers and metal nodes.43 In particular, understanding solvent-metal node interactions can shed light on the framework's susceptibility to degradation in organic or aqueous environments, as well as solvent-dependent MOF properties (discussed in detail below).26,43,44,50 For example, one series of studies by Navrotsky and co-workers utilized solution calorimetry experiments (Fig. 1) to reveal how not only the choice of organic solvent but also the degree of framework solvation (i.e., the number of solvent molecules per pore) governs the strength of solvent⋯framework interactions.26,43,47,50 The authors hypothesized that the degree of solvent saturation (e.g., MOFs stored under an organic solvent versus being evacuated under vacuum) could significantly impact the magnitude of reported thermodynamic values for solvent⋯framework interactions. To address their concern, Navrotsky and co-workers measured the enthalpy of formation (ΔHf) for MOF-5 (Zn4O(C8H4O4)3) with varying amounts of incorporated N,N-dimethylformamide (DMF) or N,N-diethylformamide (DEF) molecules (i.e., either coordinated to the metal nodes or non-coordinatively held within the pore volume). Specifically, the MOF-5 powders were first washed with either DMF or DEF. Then, the powders were washed with dichloromethane prior to evacuation. The values of ΔHf derived from solution calorimetry experiments performed in aqueous NaOH can be used to estimate the enthalpy of the interactions (ΔHint) between solvent molecules and the [Zn4O]6+ node of MOF-5.43 As a result, the authors determined ΔHint = −83 and −89 kJ mol−1 (per mole of DMF and DEF, respectively).43 However, the exact values of ΔHint were highly dependent on the amount of solvent contained within the pores. For example, samples that were saturated with DMF or DEF exhibited less favorable changes in enthalpy due to the fact that a majority of solvent molecules are not directly bound to the metal node but rather are non-coordinatively held in the pore volume.43,63 In other words, the experimentally-determined energy values for solvent⋯framework interactions at high concentrations of solvent can be influenced by competing solvent⋯solvent interactions in the pore space. Notably, the described results demonstrating the influence of the degree of solvent saturation on the outcome of calorimetric experiments emphasizes one of the challenges that must be considered in order to obtain reproducible thermodynamic values from calorimetric experiments. Careful attention to synthetic protocols, particle size and morphology, MOF activation conditions, sample preparation, and humidity is critical when comparing data across multiple measurements, and all relevant experimental procedures must be reported accurately. For instance, detailed studies by Long and co-workers identified that eight different preparation methods reported for MOF-5 resulted in samples possessing a wide variety of surface areas (570–3800 m2 g−1), highlighting the importance of reporting synthetic conditions in detail alongside careful description of experimental results.18
 | ||
| Fig. 1 Schematic representation of a typical solution calorimetry setup for experimental determination of the enthalpy of dissolution of MOF samples in acidic environments. Created in BioRender. Thaggard, G. (2025) https://BioRender.com/3ou2gpz. | ||
Navrotsky and co-workers were also able to use their calorimetric experiments to demonstrate that the solvent molecules within the MOF not only fill the pores but also strongly coordinate to the metal nodes.43 One example of an application of such findings is the rational choice of solvent to promote PSM of frameworks through either transmetallation or solvent-assisted linker exchange.26 Specifically, the authors explain that the rate of transmetallation in MOFs is strongly related to the strength of the metal node⋯solvent interactions (i.e., relatively labile solvent molecules corresponding to a smaller value of ΔHint can result in dynamic metal node coordination environments that facilitate transmetallation),37,43 and, therefore, accurate experimental measurements of solvent⋯framework interactions (i.e., experiments that consider solvent⋯metal node and solvent⋯solvent interactions as separate variables) are crucial steps toward designing synthetic pathways for the formation of novel structures via PSM. Moreover, these results highlight the importance of using consistent synthetic protocols for comparison of thermodynamic experiments since variables, such as solvent content, could significantly alter the outcomes of the measurements.
Building on the idea that solvent⋯metal node interactions can govern the pathways for PSM of MOF-based materials, the Navrotsky group has also utilized an alternative calorimetric technique, high-temperature drop combustion calorimetry, to explain and predict the water stability of several MOFs (Fig. 2).47,50 Unlike conventional solution calorimetry, drop combustion calorimetry can be performed directly on dry powders (i.e., MOF powders which were evacuated under vacuum to remove adsorbed water molecules) by combusting samples at high temperature (∼800 °C) in an oxygen atmosphere.50 As a result, drop combustion calorimetry may be a more straightforward approach than conventional solution calorimetry in the case of MOFs that are difficult to dissolve in acidic or basic conditions at room temperature, thereby broadening the pool of structures that can be analyzed via calorimetric experiments. However, it is also important to consider that experiments performed on dry powders (i.e., drop combustion calorimetry) may result in different values in comparison with thermodynamic parameters measured using solvated samples. Thus, it is critical that comparison of experimental thermodynamic values with reported ones is only done for samples that were synthesized and prepared through similar protocols.
 | ||
| Fig. 2 (top) Schematic representation demonstrating the role of thermodynamics in determining MOF stability in water. Specifically, increasing values of ΔHf are correlated with increased framework susceptibility toward hydrolysis as demonstrated for the MOF-74 family. (bottom) A part of the X-ray crystal structure showing a fragment of the MOF metal node (large dark green sphere) and the structure of the organic linker (gray rod). The small gray, red, and light green spheres represent carbon, oxygen, and magnesium atoms, respectively. Hydrogen atoms have been omitted for clarity. Reproduced from ref. 50 with permission from the American Chemical Society under the terms of the ACS Author Choice/Editors’ Choice usage agreement. Copyright 2020, American Chemical Society. | ||
The authors applied drop combustion calorimetry toward understanding the hydrolytic stability of Mg-MOF-74 ((M2(C8H2O6), M = divalent metal), as a promising candidate for CO2 capture from water-containing flue gas produced by coal-fired power plants.50 Specifically, the authors tested whether the water stability of MOF-74 derivatives was controlled primarily by thermodynamic or kinetic factors.50
Analysis of the thermodynamics of MOF-74 water stability was carried out based on the combustion of activated MOFs (i.e., MOFs containing no solvent molecules in the pore volume or coordinated to the metal nodes) yielding the corresponding metal oxide (e.g., MgO in the case of Mg-MOF-74), CO2, and H2O.50 Construction of an appropriate thermochemical cycle allowed for conversion of the experimentally determined heat of combustion to the enthalpy of reaction 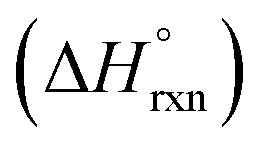 for the formation of MOF-74 samples from the corresponding metal oxide and organic linker.50 In particular, the authors determined that the
for the formation of MOF-74 samples from the corresponding metal oxide and organic linker.50 In particular, the authors determined that the 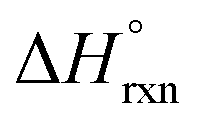 value of the Mg-MOF-74 formation reaction (304 kJ mol−1) is more positive (i.e., the reaction is more endothermic) than in the case of Ni-MOF-74 (264 kJ mol−1). As a result, Mg-MOF-74 is more susceptible to degradation in the presence of water than Ni-MOF-74.50 The acquired data could also be expressed as ΔHf (enthalpy of MOF formation from metal oxides and organic linkers), resulting in a general trend that more positive values of ΔHf of MOF-74 formation are related to decreased water stability (Fig. 2). Thus, the authors concluded that the stability of the MOF-74 series toward hydrolysis is a strongly thermodynamically driven process. The fact that fundamental thermodynamics (e.g., changes in ΔHf as a function of metal node) plays a significant role in determining the hydrolytic stability of the MOF-74 family suggests that the suitability of MOFs for capture of CO2 from industrial flue gas containing water vapor can be synthetically tailored through choice of metal node and/or organic linker.50,103 In a broader sense, selection of a MOF for any studies targeting, for instance, water harvesting or biomedical applications could be guided by reported values of ΔHf to predict MOF hydrolytic stability. Specifically, selecting a framework with a relatively low reported value of ΔHf is likely to result in better hydrolytic stability.
value of the Mg-MOF-74 formation reaction (304 kJ mol−1) is more positive (i.e., the reaction is more endothermic) than in the case of Ni-MOF-74 (264 kJ mol−1). As a result, Mg-MOF-74 is more susceptible to degradation in the presence of water than Ni-MOF-74.50 The acquired data could also be expressed as ΔHf (enthalpy of MOF formation from metal oxides and organic linkers), resulting in a general trend that more positive values of ΔHf of MOF-74 formation are related to decreased water stability (Fig. 2). Thus, the authors concluded that the stability of the MOF-74 series toward hydrolysis is a strongly thermodynamically driven process. The fact that fundamental thermodynamics (e.g., changes in ΔHf as a function of metal node) plays a significant role in determining the hydrolytic stability of the MOF-74 family suggests that the suitability of MOFs for capture of CO2 from industrial flue gas containing water vapor can be synthetically tailored through choice of metal node and/or organic linker.50,103 In a broader sense, selection of a MOF for any studies targeting, for instance, water harvesting or biomedical applications could be guided by reported values of ΔHf to predict MOF hydrolytic stability. Specifically, selecting a framework with a relatively low reported value of ΔHf is likely to result in better hydrolytic stability.
The discussed studies by the Navrotsky group showcase how the metal node can play an important role in determining MOF stability, yet the selection of an appropriate organic linker is an equally important factor that dictates MOF performance which was investigated by, for instance, Friščić and co-workers.44 For their studies, the authors prepared eight isostructural MOFs from the ZIF family (ZIF = zeolitic imidazolate framework) based on a series of imidazolate linkers (Fig. 3).44 Each of the eight ZIF samples was prepared through mechanochemical synthesis and evacuated prior to experimental estimation of ΔHf values. To approximate the changes in enthalpy of MOF formation (used as a metric to predict MOF stability), the authors experimentally measured the dissolution enthalpies (ΔHs) of the eight MOF samples using solution calorimetry performed in hydrochloric acid.44
 | ||
| Fig. 3 (top) Schematic representation of MOF formation from metal nodes (green spheres) and organic linkers (gray rods). The enthalpy of MOF formation is described as ΔHf. (middle) Structures of six different organic linkers used for preparation of a series of ZIFs. The gradient from green to yellow indicates an increase in ΔHf correlated with the decrease in framework stability. (bottom) Structures of the metal node (large dark green sphere) and organic linker (gray rod). The gray tetrahedron and blue spheres represent zinc and nitrogen atoms, respectively. Reproduced with permission from ref. 44. Copyright 2020, American Chemical Society. | ||
Combining the values of ΔHs with an appropriate thermochemical cycle yielded values of ΔHf for all eight MOFs, which varied over 30 kJ mol−1 as a function of the used organic linker (Fig. 3). The authors hypothesized that the stability of the prepared ZIFs (related to the ΔHf values) could be predicted based on the strength of the bond between the Zn-based metal node and the imidazolate linker (e.g., Zn–N bond). To confirm this hypothesis, they plotted the determined values of ΔHf against reported Hammett parameters, revealing a linear correlation and providing power to predict the stability of ZIFs. As a result, these studies44 are one of the first examples demonstrating how the use of Hammett parameters allows for predicting the properties of framework solids, thereby bringing the principles of molecular chemistry to the supramolecular arena. In addition, the authors showcase how the choice of organic linker can significantly impact the outcomes of thermodynamic measurements even for a series of frameworks with identical metal nodes, coordination environments, and ligand binding modes.
Outside of predicting MOF structure–property relationships through thermodynamic analysis described above, one of the unique phenomena that has been explored through solution calorimetry is the concept of “structural memory”, as demonstrated on the example of actinide-containing frameworks.99,104 As shown in Fig. 4, the thorium-based framework, Th6O4(OH)4(NO3)2(Me2BPDC)5 (Me2BPDC2− = 2,2′-dimethylbiphenyl-4,4′-dicarboxylate), was observed to undergo reversible crystalline ↔ amorphous structural transitions upon its exposure to organic solvents.99 Interestingly, such dynamic structural behavior, allowing for cycling between ordered and disordered phases, is hard to replicate using transition metal-based analogs (e.g., Zr-MOFs) containing the same organic linkers.99 Solution calorimetry measurements were employed to probe the possible mechanism for the observed solvent-dependent structural transitions and the effect of the metal node on the framework's behavior.99,105
 | ||
| Fig. 4 (top) Schematic representation of the “structural memory” effect observed for the Th-based framework, Th6O4(OH)4(NO3)2(Me2BPDC)5, driven by the enthalpy-favorable solvent-framework interactions. (bottom) Structures of the actinide-based metal node (large dark green sphere) and organic linker (gray rod). Small light green, red, and gray spheres represent thorium, oxygen, and carbon atoms, respectively. Reproduced with permission from ref. 99. Copyright 2019, American Chemical Society. | ||
In particular, the stabilizing effect of organic solvent (e.g., DMF) for Th-based MOFs was evaluated using experimentally determined enthalpies of dissolution (ΔHs) for either evacuated MOF samples (i.e., containing no solvent molecules in the framework pores) or solvated samples that had been stored under DMF.99 Specifically, ΔHs for Th-MOF samples was analyzed by placing the MOF powders in a mixture of HCl and DMF in a solution microcalorimeter (resulting in framework decomposition). The Th-based framework was estimated to be stabilized by DMF by approximately −22 kJ mol−1 of Th, while the reported values for transition metal-based analogs were only in the range of −3 to −5 kJ mol−1 of transition metal which could explain the difference in the observed framework behavior.63,99,105,106 Overall, these data demonstrate that the presence of the DMF molecules within the framework pores may be one of the key driving forces for the “structural memory” of Th-MOFs, in contrast to MOFs based on transition metals.99 For instance, highly favorable solvent-framework interactions (i.e., a negative value of ΔHint) for DMF molecules contained in the Th6O4(OH)4(NO3)2(Me2BPDC)5 structure could facilitate the transition from a relatively amorphous material to a highly crystalline framework.99
In addition to solution and drop combustion calorimetry, ITC has been more recently recognized in the MOF field as a powerful tool for understanding the thermodynamic aspects, such as the energy changes related to host–guest interactions in MOFs (Fig. 5).15,22,39,40,42,70,86 Primarily pioneered by the Farha group, ITC experiments involving MOFs were inspired by the more conventional use of ITC to analyze biological processes, such as protein folding and/or binding.59,86,87,107,108 One of the primary advantages of applying ITC toward understanding MOF thermodynamics is that values of ΔH, ΔS, ΔG, and binding stoichiometry (n) can all be extracted simultaneously by fitting an ITC thermogram with an appropriate thermodynamic model (Fig. 5). For example, ITC has already proven to be a valuable technique for understanding the thermodynamic driving forces for non-covalent surface modification of MOFs,108 polyoxometalate encapsulation,87,109 detoxification of agrochemicals,59 and many classes of host–guest interactions.59,110 Despite all listed advantages, analysis of data acquired through ITC experiments requires careful selection of a thermodynamic model for data fitting, which is a nontrivial step in utilizing ITC as a tool to predict MOF performance.
 | ||
| Fig. 5 (top) Schematic representation of a typical isothermal titration calorimetry (ITC) setup for experimental determination of host–guest interactions in MOFs. Created in BioRender. Thaggard, G. (2025) https://BioRender.com/oq3frbw. (bottom) Example of a thermogram that can be obtained through ITC experiments and the thermodynamic values that can be extracted by data fitting. Reproduced from ref. 22 with permission from the Royal Society of Chemistry under the terms of the Creative Commons Attribution-NonCommerical 3.0 Unported Licsense. Copyright 2025, Royal Society of Chemistry. | ||
For instance, Farha and co-workers, as well as many others, have applied ITC to obtain values of ΔH, ΔS, ΔG, and binding affinity (Ka) for a variety of processes occurring either within MOF pores or at a particle surface.15,36,40,59,78,86,87,108
Specifically, ITC experiments have been utilized to demonstrate how several structural aspects of MOFs (e.g., hydrophobicity, pore size, and topology) influence protein encapsulation within MOF pores (Fig. 6). Notably, immobilization in MOFs is a promising strategy for stabilizing fragile proteins in vitro (i.e., by preventing protein degradation outside of a biological environment), potentially allowing for enzymes to be applied to catalyze chemical reactions in the pharmaceutical and food industries, for example.86,87,111,112 However, in order to use this approach in the mentioned industries, fundamental knowledge of the binding interactions between guest proteins and host frameworks, as well as the ways in which confinement within MOF pores could impact protein folding, must first be established.
 | ||
| Fig. 6 (top) Schematic representation of entropy-driven diffusion of insulin (blue helix) through MOF channels followed by enthalpy-driven immobilization within the MOF pores. (bottom) Structures of the MOF metal node (large dark green sphere) and organic linker (gray rod). Small light green, red, and gray spheres represent zirconium, oxygen, and carbon atoms, respectively. Reproduced with permission from ref. 86. Copyright 2022, John Wiley and Sons. | ||
As a representative protein, the Farha group selected insulin for immobilization within a series of mesoporous Zr-MOFs (Fig. 6).86 The authors hypothesized that the relatively hydrophobic nature of insulin would result in thermodynamically favored interactions with highly conjugated MOF linkers (e.g., pyrene-based linkers).86 In addition, the authors anticipated that decreasing the MOF pore size would facilitate favorable, close interactions between encapsulated insulin and the framework walls.86 Specifically, a close match between the diameter of the MOF channels and insulin would optimize the number and strength of intermolecular interactions (e.g., π–π stacking) which are dependent on the distance between molecules.86
To test these hypotheses, the authors titrated suspensions of several Zr-MOFs, including NU-1000 (Zr6O8(OH)4(H2O)4(TBAPy)2, TBAPy4− = 4,4′,4″,4‴-(pyrene-1,3,6,8-tetrayl)-tetrabenzoate; NU = Northwestern University) with an insulin solution and observed strong negative (exothermic) peaks in the ITC thermogram (Fig. 5 and 6). Importantly, the suspensions of all studied MOFs were prepared in identical buffer solutions of pH = 3.9 ± 0.1 to ensure consistency between all measurements. Based on these data, the authors concluded that the encapsulation of insulin within NU-1000 was thermodynamically favored (i.e., exothermic, ΔG < 0). Further fitting of the acquired data with an appropriate model revealed that the encapsulation process (i.e., diffusion of insulin through the channels of NU-1000, Fig. 6) is entropically driven (TΔS > 0), while the immobilization of insulin via protein-framework interactions, such as π–π stacking between the pyrene-based linker and aromatic amino acids in insulin, is driven by a favorable enthalpy change (ΔH < 0). Thus, the authors confirmed the hypothesis that thermodynamically favorable protein-linker interactions result in the stabilization of insulin, and possibly other proteins, in MOFs. Moreover, insights provided by ITC experiments allowed the authors to elucidate key design criteria for MOFs targeted toward protein immobilization: organic linkers possessing extended π-conjugation (contributing toward a more negative ΔH) and appropriately sized channels that allow for diffusion of the protein (contributing to a more positive TΔS) while also maximizing protein-framework interactions.86
Outside of understanding protein encapsulation in porous materials, studies by Tan, Ghosh, and co-workers have demonstrated the broader applicability of ITC experiments toward the design of an anionic MOF for selective capture of uranyl cations from seawater (Fig. 7).15 While materials for uranium extraction from seawater are highly desirable due to the possibility of using uranium in sustainable fuel cycles, low natural abundance in seawater, as well as high salinity (i.e., presence of competing ions), make selective uranium extraction challenging.15
 | ||
| Fig. 7 (top) Schematic representation of entropically-driven capture of uranyl cations (yellow spheres) by an anionic framework. A more positive value for TΔS for uranyl binding in comparison with competing cations results in high selectivity for uranyl even in solutions simulating the rich cation content of seawater. (bottom) X-ray crystal structure of the metal node (large dark green sphere) and structure of the organic linker (gray rod). Small light green, red, gray, and blue spheres represent nickel, oxygen, carbon, and nitrogen atoms, respectively. The pyrazine moiety of the organic linker for binding cationic species is highlighted in blue. Reproduced with permission from ref. 15. Copyright 2023, John Wiley and Sons. | ||
Motivated by the challenge of selective uranyl capture, the authors designed a novel anionic framework constructed from pyrazine-functionalized organic linkers and dimethylammonium cations that occupied the framework pores (Fig. 7). The latter are utilized for charge balance. The designed anionic framework was prepared on the gram scale via solvothermal synthetic methods and analyzed by both single-crystal and powder X-ray diffraction prior to evaluation of the MOF's ability to bind uranyl cations. Upon exposure to a solution mimicking seawater, the dimethylammonium cations could be displaced by other cations in aqueous solution, including uranyl.15 However, the high selectivity for uranyl over competitive ions was achieved via strong coordination of UO22+ to the Lewis-basic nitrogen sites of the pyrazine group of the organic linker. To shed light on the possible mechanism of such selectivity, ITC experiments were carried out.
They revealed that UO22+ adsorption is thermodynamically favorable in general (ΔG < 0) and entropically more favorable than adsorption of other competing cations (e.g., Na+ or Ca2+). In addition to determining values of TΔS and ΔG, fitting of the obtained ITC thermograms for titration of the anionic framework with solutions of UO22+, Na+, or Ca2+ showed nearly 100% occupancy (i.e., ratio between the number of pyrazine sites and adsorbed cations) for UO22+, while only partial occupancy could be obtained for Na+ and Ca2+ solutions. These experiments demonstrate that ITC is a powerful tool, providing fundamental thermodynamic values to explain experimental observations and allowing for logical structure–property relationships to be derived for the rational design of functional materials.
While the studies presented above demonstrate how ITC experiments can be applied to understand the details of radionuclide sequestration by MOFs, an equally interesting direction to pursue has been demonstrated by the Biswas group, who employed a combination of ITC experiments and photoluminescence spectroscopy in their studies of recyclable MOF-based sensors for heavy metals that pollute rivers, lakes, and tap water (Fig. 8).78 In particular, Biswas and co-workers targeted the design of a MOF-based sensor for Pd2+ ions, which was motivated by the wide use of palladium in automotive and chemical industries (e.g., to catalyze Sonogashira, Suzuki, and Heck coupling reactions). It is important to note that even small amounts of Pd2+ contamination pose significant human health risks, including paralysis, memory loss, stomatitis, and gum diseases.78,113 Therefore, control of Pd2+ levels and its detection, including the detailed understanding of the mechanism by which MOFs can bind and sense Pd2+ ions, is an essential industry-driven process.
 | ||
| Fig. 8 (top) Schematic representation of fluorescence quenching of a MOF-based sensor upon binding of Pd2+ ions in solution. Binding of Pd2+ is enthalpically driven, associated with negative values of ΔH and ΔS, as determined by ITC experiments. (bottom) Structures of the metal node (large dark green sphere) and organic linker (gray rod). The propynyloxy binding moiety is highlighted in blue. Small light green, red, and gray spheres represent aluminum, oxygen, and carbon atoms, respectively. Hydrogen atoms are omitted for clarity. Reproduced with permission from ref. 78. Copyright 2023, American Chemical Society. | ||
To prepare MOF-based materials exhibiting high sensitivity toward Pd2+ cations, the authors synthesized a MOF possessing propynyloxy functionalities on the organic linker, which could bind Pd2+ cations through previously reported alkyne-π-Pd2+ interactions.78 The targeted MOF-based sensor shown in Fig. 8 was prepared in the form of a powder through solvothermal methods carried out in a mixture of water and DMF. Moreover, the resulting parent MOF exhibited strong fluorescence centered around 350 nm, which was effectively quenched in the presence of Pd2+ (Fig. 8), allowing the designed framework to simultaneously extract and sense Pd2+.78 Mechanistic details of Pd2+ binding and subsequent fluorescence quenching were uncovered by ITC experiments and photoluminescence spectroscopy.78 For this, the authors incrementally injected a solution of Pd2+ salt into a solution of the MOF linker possessing the propynyloxy binding moiety.78 As a result, they were able to determine that the initial injections were exothermic (−ΔH) and associated with a decrease in entropy.78 The negative values of ΔH and ΔS are consistent with favorable binding interactions and “ordering” (i.e., localization of Pd2+ ions at specific binding sites) of the Pd2+ bound to the propynyloxy functionalities. However, continued titration of the linker solution with Pd2+ resulted in more positive values of ΔH and ΔS, suggesting saturation of the binding sites. As a result, the authors concluded that binding of Pd2+ by the prepared MOF occurred through interactions with the propynyloxy moiety of the organic linker and was a primarily enthalpy-driven process.78
As summarized in Table 1, calorimetric measurements have been used in the mentioned studies to experimentally determine values of ΔG, ΔH, ΔS, n, and Ka for MOFs used in water harvesting, radionuclide sequestration, protein encapsulation, metal ion sensing, and CO2 capture. However, the reliability and errors associated with calorimetric measurements are heavily dependent on sample purity (e.g., possible presence of unreacted metal salts or organic linkers remaining in MOF pores after synthesis) and activation conditions. Therefore, careful consideration of all experimental protocols is necessary before comparing data obtained for multiple frameworks or from different calorimetric techniques.
| Examples of techniques | Possible thermodynamic parameters | Sample requirements | Sample preparation | Ref.a |
|---|---|---|---|---|
| a References related to MOF synthesis.ΔH = change in enthalpy, ΔG = change in Gibbs free energy, ΔS = change in entropy, n = binding stoichiometry, Ka = association constant, W = work, Q = heat, ΔU = change in internal energy, Cp = heat capacity, Keq = equilibrium constant, k = rate constant, qe = adsorption capability, Kd = distribution coefficient, Ttr = phase transition temperature, Tdecomp = decomposition temperature, EOCP = open circuit potential, E° = standard reduction potential, ICP-AES = inductively-coupled plasma atomic emission spectroscopy, ICP-OES = inductive-coupled plasma optical emission spectroscopy, ICP-MS = inductively-coupled plasma mass spectrometry. | ||||
| Solution calorimetry | ΔH, ΔG | MOFs decomposed in acidic or basic media | Optional framework activation under vacuum and/or elevated temperature | 43 , 105, 143–146a and 159a |
| Isothermal titration calorimetry | ΔG, ΔH, ΔS, n, Ka | Suspensions in aqueous or organic media | Framework activation and suspension in a buffer of known pH | 15, 22, 36, 40, 59, 71 , 86 and 159a |
| Drop combustion calorimetry | ΔH, ΔG | Dry powders | Framework activation under vacuum and/or elevated temperature | 147 , 148 and 159a |
| Differential scanning calorimetry | ΔH, ΔG, Ttr, W, Q, ΔU, Cp | Powders or single crystals | Optional framework activation under vacuum and/or elevated temperature | 93, 114 , 125, 147–149 and 159a |
| Thermogravimetric analysis | ΔH, Tdecomp | Powders or single crystals | Optional framework activation under vacuum and/or elevated temperature | 89, 93, 127, 147, 149 and 159–161 |
| UV-vis absorbance spectroscopy | K eq, ΔG, ΔH, ΔS | Suspensions or thin films | Deposition of thin films on quartz substrates or MOF suspension in an organic solvent | 22, 46, 66, 95, 115, 150–154, 159 and 160a |
| ICP-AES/ICP-OES/ICP-MS | K eq, Ka, ΔG, k Kd, qe | MOF samples decomposed in acidic media | Acid treatment and dilution to known concentrations of MOF components | 15, 132, 155, 156, 159 and 160a |
| Gas sorption analysis | Q, ΔH, ΔG, ΔS | Dry (activated) powders or single crystals | Framework activation under vacuum, elevated temperature, or supercritical CO2 drying | 10, 18, 29, 55, 56, 81, 126 and 157–161 |
| Electrochemical measurements | E OCP, E° | Powders, single crystals, or thin films | Deposition of MOF powders, thin films, or crystals on electrodes | 40, 95 and 159a |
Thermal measurements
Thermal analysis techniques, including those based on differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA), offer complementary strategies to the aforementioned ones which were utilized for fundamental understanding of thermodynamically-driven processes. In particular, thermal measurements can be used to evaluate framework stability as well as the energy associated with structural transitions.24,26,47,88,92,93,109,114 For example, TGA is commonly used to determine the thermal decomposition temperatures of materials, but it can also be extended to probe, for instance, changes in enthalpy associated with water or gas sorption processes through variable-temperature and variable-humidity experiments.47,82,88,115 At the same time, DSC measurements have provided valuable insights into the “breathing” behavior of flexible MOFs (associated with significant changes in enthalpy and entropy) as well as the tailorable gas sorption properties.74,75,116 In many cases, data from TGA and DSC measurements are combined to offer a more comprehensive view of the structural changes that occur for some frameworks upon heating, exposure to external pressure, or in the presence of different gases. For instance, simultaneous monitoring of TGA and DSC data allows for correlation of distinct structural changes (e.g., removal of solvent coordinated to metal nodes or held within the pore volume, metal node oxidation, ligand decomposition, etc.) detected by TGA with the temperature and related enthalpy changes derived from DSC experiments.74,75,116Among the applications reported for DSC is the evaluation of mechanical energy storage in flexible MOFs, as demonstrated by Llewellyn and co-workers (Fig. 9).116 For instance, derivatives of MIL-53(Cr), which were typically prepared as polycrystalline powders via hydrothermal synthesis at 220 °C, exhibit significant flexibility and structural sensitivity toward an external pressure.116Fig. 9 shows how MIL-53(Cr)-Br(Cr(OH)(Br-BDC), Br-BDC2− = 2-bromo-1,4-benzenedicarboxylate) undergoes reversible transitions between “large pore” and “narrow pore” states under an applied pressure, allowing the framework to store and release mechanical energy on demand.116 Despite many attractive applications for structures exhibiting reversible mechanical energy storage, such as materials for vehicle dampers116–118 and shock absorbers,116–119 much of the data reported for mechanical energy storage in MOFs has been obtained via mercury porosimetry rather than direct measurements of heat exchange during MOF structural transitions.51,57,74,100,116,120 To address this gap, the Llewellyn group utilized a combination of high-pressure calorimetry, DSC, and extended X-ray absorption spectroscopy (XAS) to understand the energy of both pressure- and temperature-induced phase transitions in the MIL series of MOFs.116 In particular, the authors employed high-pressure calorimetry to determine the internal energy changes and work (Fig. 9) associated with MOF compression/decompression cycles, while DSC allowed for calculation of the phase transition enthalpy and mechanical work (W).
 | ||
| Fig. 9 (top) Schematic representation of a pressure-driven mechanical energy storage cycle in a flexible MOF, MIL-53(Cr)-Br. Applying pressure (yellow arrows) induces a transition from the large pore state to the narrow pore state, resulting in storage of mechanical energy (+W). (bottom) X-ray crystal structure of the metal node (large dark green sphere) and structure of the organic linker (gray rod). Small light green, red, and gray spheres represent chromium, oxygen, and carbon atoms, respectively. Reproduced with permission from ref. 116. Copyright 2016, Royal Society of Chemistry. | ||
As a result, the “large-to-narrow” pore transition in MIL-53(Cr) was associated with a positive value for work (W = 9.5 J g−1, mechanical energy storage, Fig. 9), while the reverse transition (decompression of MIL-53(Cr)-Br resulting in the large pore geometry) involves a release of mechanical energy (W = −5.4 J g−1). Moreover, the authors determined that the changes in internal energy (i.e., changes in the sum of heat (Q) and work (W) upon a phase transition) of the MOF system increased upon compression and decreased upon decompression. While the total internal energy of the MOF increased upon compression, it is worth noting that the pressure-induced large-to-narrow pore transition was surprisingly exothermic (Q = −9.1 J g−1). The detected release of heat (−ΔQ) upon compression of MIL-53(Cr)-Br is critical for applying MOFs as dampers since it suggests that, in contrast to more conventional lyophobic systems, flexible MOFs may be resistant to excessive heating upon their use in multiple compression-decompression cycles. Furthermore, coupling DSC with XAS measurements allowed for the phase transitions (and related changes in W) detected by DSC to be directly correlated with changes in the electronic structure and local geometry around the bromine atom of the organic linker.116 As a result, the authors could confirm that the phase change detected by DSC corresponded to the NP-to-LP transition based on changes in the local geometry around the bromine atom detected by XAS. This specific example demonstrates how the combination of high-pressure calorimetry and DSC with XAS could be used to characterize both the structural and energetic changes occurring during the MOF's breathing transitions.116
Notably, an alternative method that could be useful for characterizing a MOF's potential to behave as a mechanical energy storage or energy dissipation material is to determine the ratio of work done to the system (W) to the quantity of heat produced (Q). As suggested by Grosu and coworkers, who measured the heats of water intrusion/extrusion into ZIF-8 through scanning transitiometry, materials exhibiting a ratio of Q/W ≫ 1 may be suitable for thermal energy storage.121–123 In contrast, a measured value of Q/W ≪ 1 is more favorable for applying flexible MOFs as mechanical systems that exchange very little heat with their environments. When designing frameworks for thermal or mechanical energy storage applications, it may also be valuable to employ in operando experiments alongside calorimetric methods to evaluate the structural and energetic changes that occur when exposing the MOF to real working conditions (i.e., repeated exposure to relevant temperatures and pressures for a specific application).121–123 In this direction, calorimetric measurements of the heat of water intrusion/extrusion in flexible frameworks could be supported by in operando high-pressure neutron scattering, as demonstrated by the Grosu group. For instance, in operando high-pressure powder neutron diffraction could be used to correlate the mechanism of water intrusion in MOF pores to the energy changes detected by calorimetry.
In addition to a fundamental understanding of the changes in energy associated with phase transitions of flexible MOFs, a combination of DSC and TGA has also been applied to evaluate the water adsorption enthalpy (ΔHads) for MOF-801 (Zr6O4(OH)4(fumarate)6) as well as zeolites, highlighting the utility of the mentioned techniques across several porous material classes.88 Although accurate determination of ΔHads is crucial for applying MOFs in the areas of thermal energy storage, climate control, and water purification systems,9,32,88,116 conventional gas sorption experiments for ΔHads evaluation124 are often challenged by minimal separation between isotherms at low relative pressures, potentially leading to poor data fitting.88 To address the mentioned challenges, Yaghi and co-workers presented a method relying on relatively accessible DSC and TGA setups which can be applied as an alternative strategy for determining ΔHads.88 To demonstrate the power of the presented method, the adsorption enthalpy of two classes of materials, hydrophilic MOFs and zeolites, was analyzed.88 In particular, samples of MOF-801 and zeolite MgY were saturated with water vapor prior to analysis by DSC and TGA.88 Subsequent DSC and TGA measurements provided the enthalpy and mass loss associated with water desorption from the framework, respectively. Combining the experimental enthalpies of desorption, heat transfer during “cool down” (i.e., thermal equilibration of the sample and instrument after water desorption), and mass loss with an appropriate thermodynamic cycle resulted in calculated values for ΔHads. Importantly, the developed methodology could also be expanded to measure the changes in ΔHads of porous materials at various water uptakes and at different temperature regimes.88
In addition to gaining a thermodynamic understanding of vapor adsorption processes and structural transitions as described in the previous studies, thermal measurements can also be applied to evaluate a unique concept known as “gate opening” exhibited by some flexible MOFs. As shown in Fig. 10, “gate opening” describes the phenomenon of rapid adsorption of gas molecules by flexible MOFs as a result of a phase transition.92 Such behavior is particularly important for applications of MOFs in selective gas sorption, storage, and separation from mixtures, where different gas molecules induce “gate opening” at distinct pressures.92 In order to predictably design flexible frameworks that selectively adsorb target gases, methods for accurately measuring values of ΔH and ΔG, as well as the entropic contributions to ΔG, for “gate opening” transitions must be developed. For example, ZIF-7 (Zn(BIm)2, BIm = benzimidazolate) undergoes reversible changes between rhombohedral and triclinic structures upon adsorption of CO2 (Fig. 10), which is made possible by an overall negative value of ΔG for the combined structural change and gas sorption processes as described in more detail below.
 | ||
| Fig. 10 (top) Schematic representation of “gate opening” by ZIF-7 upon adsorption of CO2, associated with a decrease in enthalpy. (bottom) Structures of the metal node (large dark green sphere) and organic linker (gray rod) of ZIF-7. The gray tetrahedron and blue spheres represent zinc and nitrogen atoms, respectively. Reproduced from ref. 92 with permission from the Royal Society of Chemistry under the terms of the Creative Commons Attribution-NonCommercial 3.0 Unported License. Copyright 2024, Royal Society of Chemistry. | ||
In a similar vein, Ohtani and co-workers utilized DSC measurements under either CO2 or N2 atmosphere to provide a detailed mechanistic analysis of CO2 adsorption in ZIF-7.92 While no endothermic or exothermic events (i.e., no phase transition) could be detected by DSC upon heating a sample of ZIF-7 under a constant flow of N2, a distinct endothermic event was detected upon heating ZIF-7 under CO2.92 Likewise, cooling the MOF under CO2 resulted in an exothermic transition associated with desorption of CO2 from the framework, suggesting that the “gate opening” transition was fully reversible in the presence of CO2 but did not occur at all under N2 atmosphere (Fig. 10).92 Since the energy of the observed “gate opening” transition is related to both a phase change for the framework and gas adsorption, the energy changes detected by DSC experiments could not be directly assigned to a single process.92 Therefore, the authors deconvoluted the energy contributions of gas sorption and structural changes through a series of DSC and theoretical modelling studies. Specifically, a positive value of ΔH was associated with the structural transition or “gate opening” process, and a negative value of ΔH was associated with CO2 adsorption (Fig. 10). Moreover, the overall Gibbs free energy of the reaction (ΔGrxn) was estimated through a series of DSC experiments conducted under a mixed atmosphere of CO2 and N2 where the ratio between the two gases was systematically varied but the overall pressure was held constant.92 As a result, the authors determined ΔGrxn = −15.1 kJ mol−1, which was consistent with previously reported values based on DFT studies.92 Overall, this work demonstrated an accessible strategy to experimentally determine thermodynamic parameters associated with the “gate opening” or “breathing” behavior of frameworks which could be applied to compare important material performance metrics, such as adsorption capacity and heat of adsorption, across a variety of flexible materials.
Spectroscopy
While the previously discussed techniques for probing the thermodynamics of MOFs mainly rely on direct measurement of heat exchange during a chemical process (e.g., calorimetry or thermal measurements), spectroscopic techniques can offer complementary pathways toward measuring MOF thermodynamic parameters. For example, UV-vis absorbance, Fourier transform infrared (FTIR), photoluminescence, and inductively-coupled plasma atomic emission spectroscopies (ICP-AES) have all been applied in the MOF field to evaluate thermodynamic aspects, such as the driving force for water adsorption, mechanism of selective metal ion sensing, energetics of cation exchange, and PSM of MOF-based nanoparticles.15,22,34,71,74,75,78,79,84,95,98,115,125–131 In many cases, these spectroscopic techniques are applied to understand guest binding events in MOFs which are associated with, for instance, distinct shifts in the material's optical (e.g., absorbance or emission) profile. Very often, the energy and/or binding constants for MOF-guest interactions can be derived through comparison of the optical profiles of the “free” (unbound) and MOF-bound guest molecules. Moreover, spectroscopic techniques offer the advantage of high sensitivity (i.e., low detection limits), and they can often be performed in a time-resolved manner to gain information about kinetics in addition to thermodynamic aspects.36,41–43,56,73–76,79,81 As a result, the different spectroscopic techniques mentioned below can provide both thermodynamic and kinetic insights into processes occurring in MOFs, even at very low analyte concentrations in comparison with other experimental techniques (e.g., calorimetry or thermal measurements requiring larger sample sizes).26,27,36,43,110One example of how spectroscopic experiments can be used to probe MOF thermodynamic parameters was demonstrated by Dincă and co-workers, who applied ICP-AES measurements to determine the thermodynamic driving forces for solid-state metathesis (transmetallation processes) occurring in MOFs.132 Motivated by the synthetic power of cation exchange for material design (which allows for predictable insertion of metal cations into crystallographically-defined coordination environments), the authors developed a general method to determine ΔH and ΔS of cation exchange processes on the example of MOF-5 and MFU-4l (Zn5Cl4(BTDD)3; BTDD = bis(1H-1,2,3-triazolo-[4,5-b],[4′,5′-i]dibenzo-[1,4]-dioxin)).132 As a starting point, polycrystalline powders of MOF-5 and MFU-4l were prepared via solvothermal synthesis (Fig. 11).
 | ||
| Fig. 11 (top) Schematic representation of the Zn-to-Ni exchange occurring in metal nodes of MOF-5, which is associated with both an increase in enthalpy and entropy. (bottom) Structures of the zinc- (large yellow sphere) and nickel-based (large dark green sphere) metal nodes and organic linker of MOF-5 (gray rod). Small yellow, light green, red, and gray spheres represent zinc, nickel, oxygen, and carbon atoms, respectively. Reproduced with permission from ref. 132. Copyright 2015, Royal Society of Chemistry. | ||
To promote the exchange of zinc cations to nickel ones in the MOF metal nodes, the authors suspended MOF-5 in a Ni(NO3)·6H2O solution in DMF for over two months.132 By analyzing the concentrations of Zn2+ and Ni2+ in solution at equilibrium by ICP-AES, the equilibrium constant (Keq) for the cation metathesis reaction was estimated.132 Combining Keq with the temperature of the MOF suspension (maintained precisely throughout the course of the experiment) allowed for calculation of ΔG for the Zn-to-Ni exchange process.132 Furthermore, plotting the values of ln(Keq) as a function of 1/T was also used to estimate the values of ΔH and ΔS for the Zn-to-Ni cation exchange process in MOF-5.132 As a result, the authors concluded that Zn-to-Ni exchange in MOF-5 is favorable in terms of ΔS and unfavorable in terms of ΔH. The favorable value of ΔS is counterbalanced by the increase in ΔH of the system, leading to a relatively low absolute free energy value (i.e., the Zn-to-Ni cation exchange in MOF-5 is essentially thermoneutral).132 Thus, cation exchange in MOF-5 is likely primarily driven by a large excess of Ni2+ cations in the starting MOF suspension according to Le Chatelier's principle. The authors extended their thermodynamic analysis of cation exchange in MOFs by testing the effects of different solvents (e.g., DMF versus acetonitrile) and cations (e.g., Zn2+, Ni2+, and Co2+) during solid-state metathesis using MOF-5 and MFU-4l as model systems. As a result, it was determined that some of the factors that significantly impact cation exchange thermodynamics are the geometry of the metal node, the presence of labile ligands (e.g., DMF or other solvent molecules), and the degree of covalency of the linker-metal node interactions.132 Notably, the covalency of the metal node-linker interactions is a parameter that could also be tailored through synthetic modification of the organic linker (e.g., with electron-donating or withdrawing groups) in addition to the identity of the metal node to control the coordination strength. However, even though many variables can impact the specific values of Keq, ΔG, ΔH, and ΔS, the collected data suggest that, in general, cation exchange is an endergonic process that is primarily promoted by a large excess of the inserting cation but can be tailored by the choice of framework and metal salt.132
The thermodynamics of solid-state metathesis on actinide-containing frameworks (including transuranic ones) was performed by Shustova and co-workers who demonstrated that only certain actinide-frameworks could serve as a platform for the preparation of isostructural actinide-based analogs.46 This approach is especially critical for the synthesis of transuranic MOFs since the quantity and high radioactivity of the used actinides imply significant restrictions on the number of synthetic attempts and amounts of available radioactive precursors.22,46 For instance, the authors demonstrated that uranium-based MOFs could be used as a convenient precursor for incorporation of transuranic elements, such as Pu, within the MOF matrix.46 As an example, crystals of the uranium-based MOF, U-Me2BPDC-8 (U6O4(OH)8(Me2BPDC)4, Me2BPDC2− = 2,2′-dimethylbiphenyl-4,4′-dicarboxylate), were used as a precursor for transmetallation reactions.46 Upon its exposure to a Th(NO3)4 solution in air, complete transmetallation to a Th-based analog was achieved after 56 hours.46 However, U-Me2BPDC-8 remained intact (i.e., no transmetallation occurred) when the same procedure was repeated under an inert atmosphere, even after several months. Based on the experimental observations, the authors hypothesized that oxidation of U4+ to UO22+ in the presence of oxygen could be the thermodynamic driving force for efficient U-to-Th exchange. To probe this hypothesis, a combination of UV-vis spectroscopy and thermochemical calculations was employed to comprehensively study both the thermodynamics and kinetics of the transmetallation processes.46 Specifically, the authors employed UV-vis spectroscopy to monitor the formation of UO22+ in the supernatant above U-Me2BPDC-8 during the transmetallation procedure (i.e., release of UO22+ from the framework upon U-to-Th exchange in the presence of oxygen). Kinetics data could then be extracted by plotting the absorbance of the supernatant at 424 nm (corresponding to the π–π* transitions of UO22+) as a function of time. Fitting the acquired spectroscopic data with a first-order rate model resulted in the first transmetallation rate constant in actinide-based MOFs of k = 5.64 × 10−6 s−1.46
In addition to spectroscopic data correlating UO22+ formation with U-to-Th exchange in actinide-based MOFs, Shustova and co-workers also utilized solution calorimetry experiments to evaluate the standard enthalpy of the transmetallation reaction 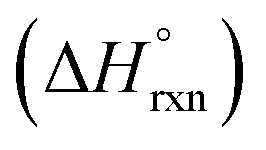 .46 For this, the authors measured the enthalpy of dissolution for monometallic U-Me2BPDC-8 and heterometallic U/Th-Me2BPDC-8 with varying degrees of Th incorporation. Applying an appropriate thermochemical cycle to the acquired calorimetric data revealed that the U-to-Th exchange process is exothermic in the presence of oxygen with
.46 For this, the authors measured the enthalpy of dissolution for monometallic U-Me2BPDC-8 and heterometallic U/Th-Me2BPDC-8 with varying degrees of Th incorporation. Applying an appropriate thermochemical cycle to the acquired calorimetric data revealed that the U-to-Th exchange process is exothermic in the presence of oxygen with 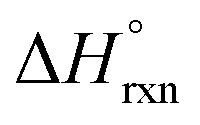 = −587.57 kJ mol−1 (i.e., assuming a change in uranium oxidation state from +4 to +6, associated with formation of UO22+). In contrast, the same calculations without considering the changes in the uranium oxidation state (i.e., no UO22+ formation) resulted in a positive value of
= −587.57 kJ mol−1 (i.e., assuming a change in uranium oxidation state from +4 to +6, associated with formation of UO22+). In contrast, the same calculations without considering the changes in the uranium oxidation state (i.e., no UO22+ formation) resulted in a positive value of 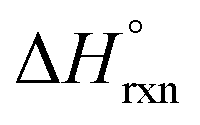 of approximately 177 kJ mol−1. Thus, both spectroscopic and calorimetric experiments supported that formation of a stable uranyl species was the primary driving force for U-to-Th exchange in MOFs. The acquired thermodynamic data (i.e., favorable formation of UO22+ in the presence of air) can be used to guide the synthesis of actinide-based frameworks by using U-containing MOFs as precursors for isostructural transuranic analogs. For example, U-based MOFs can be used for preparation of Pu-containing frameworks based on the fact that UO22+ formation in air is strongly exothermic (
of approximately 177 kJ mol−1. Thus, both spectroscopic and calorimetric experiments supported that formation of a stable uranyl species was the primary driving force for U-to-Th exchange in MOFs. The acquired thermodynamic data (i.e., favorable formation of UO22+ in the presence of air) can be used to guide the synthesis of actinide-based frameworks by using U-containing MOFs as precursors for isostructural transuranic analogs. For example, U-based MOFs can be used for preparation of Pu-containing frameworks based on the fact that UO22+ formation in air is strongly exothermic (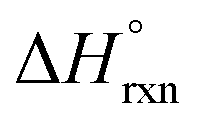 = −587.57 kJ mol−1).
= −587.57 kJ mol−1).
Outside of ICP-AES and UV-vis absorbance spectroscopy, FTIR spectroscopy has also been applied to understand thermodynamic and mechanistic aspects of water adsorption in frameworks such as NU-1500-Cr (Cr3(μ3-O)(H2O)2(Cl)(PET); H6PET = 4,4′,4″,4‴,4‴′,4‴″-(9,10-dihydro-9,10-[1,2]benzenoanthracene-2,3,6,7,14,15-hexayl)hexabenzoic acid), which is a high-performance MOF capable of atmospheric water harvesting.34 For example, Paesani and co-workers applied a combination of FTIR spectroscopy and molecular dynamics simulations to probe the thermodynamic aspects of water adsorption in NU-1500-Cr as a function of relative humidity.34 In their studies, molecular dynamics simulations were employed to estimate ΔHads for water adsorption and the entropy of the adsorbed water molecules (ΔSwater), while FTIR spectroscopy revealed how the thermodynamic parameters correlated with the water adsorption mechanism.34 In particular, ΔHads was estimated to be −14.5 kcal mol−1 at relative humidities (RH) between 15–31%, which corresponds to favorable framework-water interactions and hydrogen bond formation.34 At the same time, saturation of the framework above 36% RH resulted in a decrease in ΔSwater from ∼20 to ∼17 cal mol−1 K−1, which was attributed to confinement effects and restricted motion of the adsorbed water molecules upon formation of a hydrogen-bonded network.34
The values determined from molecular dynamics simulations could then be correlated with FTIR spectroscopic analysis to shed light on the detailed mechanism of water adsorption in NU-1500-Cr.34 In particular, the combination bend + libration stretching modes of water molecules are a strong indicator of the local environment and hydrogen-bonded network of adsorbed water molecules, as it shifts to lower frequencies for a highly ordered network.34 In contrast, the bend + libration band shifts to higher frequencies when the hydrogen-bonded network is disrupted. Upon increasing RH from 30 to 40%, the network of water molecules becomes less disrupted and more similar to that of liquid water, supporting the idea that the first step of water adsorption is coordination of individual water molecules to MOF metal nodes, followed by formation of H2O⋯H2O interactions. Thus, FTIR spectroscopy combined with molecular dynamics simulations could be used to determine that the “pore filling” step (i.e., formation of a hydrogen-bonded network of water molecules within the pore volume) was entropically unfavorable, but the decrease in entropy was compensated by formation of energetically favorable hydrogen bonds.34 Importantly, the authors also note that coordination of water molecules to open metal sites in the framework is not the only mechanism for efficient water adsorption in MOFs. For instance, MOF-801 adsorbs water through binding with μ3-OH groups followed by formation of a hydrogen-bonded network of water molecules, and water harvesting COFs are reported to adsorb water primarily through hydrogen bonding interactions with the organic linkers.88
Other techniques
Although a majority of studies have chosen to employ calorimetry, gas sorption analysis, thermal measurements, and spectroscopic techniques to experimentally probe the thermodynamics of MOF-based processes, the utilization of alternative techniques can greatly expand fundamental knowledge toward desirable applications.10,11,15,22,26,29,34,36,37,39,40,43,45,50,59,70,71,74,75,78,79,82,84,86–89,92,93,95,98,100,102,103,108,114,115,120,127–130,133,134 For example, electrochemical measurements and advanced X-ray diffraction studies are less frequently applied toward thermodynamic analysis but can offer comprehensive mechanistic insights into the structure-energy landscape for a variety of frameworks.8,40,51,58,59,71,75,77,82,94,98,114,115For example, electrochemical measurements have recently been applied toward evaluation of proton, electron, and hydrogen atom transfer processes which are at the heart of many MOF-based catalytic transformations.17,40 Specifically, proton-coupled electron transfer (PCET) events occurring at the surface of redox-active MOF particles or at metal nodes can be investigated using electrochemical open-circuit potential (EOCP) measurements as well as potentiometric acid–base titrations.94,135,136 The Noh group used the mentioned electrochemical techniques to establish correlations between the thermodynamics of PCET, structural disorder, and particle size of catalytically active Ti-MIL-125 (Ti8O8(OH)4(BDC)6, MIL = Materials of Institute Lavoisier).94 In their studies, the authors prepared three distinct batches of Ti-MIL-125 with different particle sizes and measured the PCET thermodynamics as a function of MOF particle size.94 As a first step, EOCP measurements were employed to establish a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 proton:electron stoichiometry for redox events occurring within Ti-MIL-125 regardless of particle size and structural disorder. However, despite the fact that the proton:electron stoichiometry did not vary as a result of particle size, distinct differences in the thermodynamics of PCET as a function of particle size were revealed by analyzing the Ti3+O–H/Ti4+O redox couple (representing proton and electron transfer occurring at the Ti-based metal nodes) via cyclic voltammetry. In particular, the authors compared the values of E°(Ti3+O–H/Ti4+O) for each of the three particle sizes and detected a cathodic shift associated with increasing size. In other words, increasing the particle size of Ti-MIL-125 resulted in more thermodynamically favored reduction events (i.e., the H atoms of larger particles were stronger reductants than those of smaller particles). The chemical basis for the described change in thermodynamics of proton and electron transfer could be elucidated by theoretical calculations, which indicated that significant local geometric distortion of the Ti cations in the metal nodes of smaller particles was a key factor that dictated the thermodynamics of PCET. The hypothesized geometric distortions were also supported by UV-vis spectroscopy and potentiometric acid–base titrations.94 To summarize, the Noh group combined electrochemical measurements, UV-vis spectroscopy, and simulations to conclude that reducing MOF particle size leads to stronger structural disorder, significantly shifting the thermodynamics of PCET, as well as proton and electron transfer events at the heart of MOF-based heterogeneous catalysis. The implications of the Noh group's results are profound, suggesting that careful control of particle size (i.e., through choice of MOF synthetic conditions) is essential for not only reproducible thermodynamic measurements, but also for predictable catalytic performance for redox-active frameworks.
:1 proton:electron stoichiometry for redox events occurring within Ti-MIL-125 regardless of particle size and structural disorder. However, despite the fact that the proton:electron stoichiometry did not vary as a result of particle size, distinct differences in the thermodynamics of PCET as a function of particle size were revealed by analyzing the Ti3+O–H/Ti4+O redox couple (representing proton and electron transfer occurring at the Ti-based metal nodes) via cyclic voltammetry. In particular, the authors compared the values of E°(Ti3+O–H/Ti4+O) for each of the three particle sizes and detected a cathodic shift associated with increasing size. In other words, increasing the particle size of Ti-MIL-125 resulted in more thermodynamically favored reduction events (i.e., the H atoms of larger particles were stronger reductants than those of smaller particles). The chemical basis for the described change in thermodynamics of proton and electron transfer could be elucidated by theoretical calculations, which indicated that significant local geometric distortion of the Ti cations in the metal nodes of smaller particles was a key factor that dictated the thermodynamics of PCET. The hypothesized geometric distortions were also supported by UV-vis spectroscopy and potentiometric acid–base titrations.94 To summarize, the Noh group combined electrochemical measurements, UV-vis spectroscopy, and simulations to conclude that reducing MOF particle size leads to stronger structural disorder, significantly shifting the thermodynamics of PCET, as well as proton and electron transfer events at the heart of MOF-based heterogeneous catalysis. The implications of the Noh group's results are profound, suggesting that careful control of particle size (i.e., through choice of MOF synthetic conditions) is essential for not only reproducible thermodynamic measurements, but also for predictable catalytic performance for redox-active frameworks.
Alternative parameters that can be evaluated through electrochemical methods include charge transfer kinetics/resistance (i.e., through electrochemical impedance spectroscopy), electrochemically active surface areas, and electrocatalytic stability (i.e., through cyclic voltammetry). For instance, Qian, Kirillov, and co-workers have demonstrated how the mentioned electrochemical techniques can be used to guide the design of multimetallic MOFs for small molecule oxidation. The authors specifically use the example of a Mn- and Co-based MOF electrocatalyst that couples urea oxidation with the hydrogen evolution reaction as an energetically favorable hydrogen production pathway.100–102
While the Noh and Qian groups above have utilized diverse electrochemical methods to probe MOF thermodynamics, an equally important direction is to couple X-ray diffraction techniques with theoretical modeling (e.g., DFT studies) and molecular dynamics simulations, especially for understanding larger structural changes in the framework backbone. For example, Kieslich, Schmid, and co-workers investigated how the high-pressure behavior of flexible MOFs is related to the system's configurational entropy, which could be tailored through side-chain modification of the organic linkers (Fig. 12).51 As an example, the authors analyzed the phase transitions of two flexible MOFs, Cu2(BDC)(dabco) and Cu2(DB-BDC)(dabco), using high-pressure PXRD (HP-PXRD). Importantly, the two selected MOFs differed only by functionalization of the BDC2− linker with a 2,5-dibutoxy (DB) moiety. The authors hypothesized that such modification would increase the vibrational and configurational entropy of the framework due to side chain flexibility. Interestingly, parent Cu2(BDC)(dabco), possessing no flexible side chains on the organic linker, exhibited an irreversible phase transition from a large- to narrow-pore state upon increasing pressure, which was visualized by HP-PXRD experiments.51 In contrast, side-chain-modified Cu2(DB-BDC)(dabco) exhibited a fully reversible phase transition from the large- to narrow-pore state upon increasing pressure followed by a return to the large-pore state when the pressure was released. Therefore, the authors hypothesized that modification of the organic linkers through incorporation of flexible side chains altered the thermodynamic free energy landscape that governed reversible phase transitions. Such behavior is also consistent with the recently reported concept of “molecular grease” in which incorporation of long aliphatic chains within extended structures can facilitate reversible phase transitions by promoting molecular conformational flexibility.24
 | ||
| Fig. 12 (top) Schematic representation of the large- to narrow-pore transition of Cu2(DB-BDC)(dabco) upon an increase in pressure. The side chains (blue) exhibit higher configurational entropy in the large-pore phase than the narrow-pore phase. (bottom) Structures of the metal node (large dark green sphere), organic linker (gray rod), and side chain (blue). Small light green, red, gray, and blue spheres represent copper, oxygen, carbon, and nitrogen atoms, respectively. Hydrogen atoms are omitted for clarity. Reproduced with permission from ref. 51 under the terms of the Creative Commons CC BY license. Copyright 2020, the Authors, published by John Wiley and Sons. | ||
To further probe the hypothesis that side chain flexibility could facilitate reversible phase transitions, Kieslich, Schmid, and co-workers combined HP-PXRD experiments with force field-based molecular dynamics simulations.51 As a result, the irreversible phase transition of Cu2(BDC)(dabco) was attributed to bistability of the large- and narrow-pore phases (i.e., the two phases represented two local thermodynamic minima based on calculated values of Helmholtz free energy and internal energy). Due to the absence of flexible side chains, the free energy of Cu2(BDC)(dabco) was not significantly influenced by configurational entropy.51 In contrast, the large-pore phase of Cu2(DB-BDC)(dabco) was calculated to have a large entropic contribution from the configurational freedom of the side chains.51 As a result, formation of the dense narrow-pore phase of Cu2(DB-BDC)(dabco) coincided with a significant loss of configurational entropy.51 The thermodynamically unfavorable decrease in entropy prevented the narrow-pore phase of Cu2(DB-BDC)(dabco) from being in a deep thermodynamic minimum, ultimately allowing for a reversible transition back to the large-pore phase (Fig. 12). To summarize, the reversible phase transitions of Cu2(DB-BDC)(dabco) were made possible by small chemical modifications of the organic linker (e.g., incorporation of butoxy groups), which could be used to tailor the overall configurational entropy of the system.51
As an alternative to understanding the Helmholtz free energy and entropy changes in a system through X-ray diffraction and computational approaches, the Yaghi and Gagliardi research groups have applied a combination of single-crystal X-ray diffraction, DFT calculations, and gas sorption experiments to uncover the mechanism and energy of water adsorption in water harvesting materials.137 Specifically, the authors have applied synchrotron single-crystal X-ray diffraction to establish relationships between the hydrophilic nature of an organic linker, water sorption mechanism, and the energy of the interactions between adsorbed water molecules and the framework. For this, the authors grew large single crystals of MOF-303 (Al(OH)(PZDC), PZDC2− = 1-H-pyrazole-3,3-dicarboxylate), which was selected as an example of a state-of-the-art water harvesting material, and collected X-ray diffraction data upon gradual desorption of water from the framework. Surprisingly, the crystallographic data showed that the initial water adsorption started not at the metal node, which is commonly assumed to be the strongest binding site for water molecules in most frameworks, but rather at a hydrophilic pocket formed by neighboring PZDC2− linkers.137 These results emphasize the importance of organic linker design, which ultimately can shift both the thermodynamic landscape and performance of framework materials. Strong hydrogen-bonding interactions between the organic linker and water molecules allowed for the first three adsorbed water molecules to bind in the mentioned hydrophilic pocket, while subsequent water molecules were bound through water⋯water interactions rather than to the framework itself. Thus, the water adsorption process could be broken into a “seeding stage” where water molecules interacted with the organic linkers and a “pore filling” stage involving formation of a hydrogen-bonded network of water molecules within the pore volume. Moreover, the authors detected an increase in the unit cell parameters of MOF-303 upon water adsorption, which prompted an investigation into the relationship between the binding energy of water molecules and framework deformation energy (i.e., MOF “strain” upon water adsorption).137
DFT calculations revealed that the “seeding” stage (i.e., adsorption of the first four water molecules) is associated with a deformation energy of ∼58.5 kJ mol−1 per crystallographic asymmetric unit. However, the unfavorable thermodynamic penalty associated with MOF deformation was compensated by formation of hydrogen bonding interactions between neighboring water molecules and the organic linkers, resulting in an overall favorable binding energy of −74.1 kJ mol−1 per water molecule.138 Moreover, water sorption isotherms collected for MOF-303 and a series of eight isoreticular analogs containing varying ratios of a less hydrophilic organic linker, FDC2− (2,4-furandicarboxylate) revealed how the choice of an organic linker influences the adsorption enthalpy of the material.
By systematically varying the ratio of organic linkers in the framework, the authors were able to tailor ΔHads from −53 to −50 kJ mol−1 (determined from gas sorption isotherm fitting). The differences in ΔHads due to increasing the content of less hydrophilic FDC2− could be associated with changes in the number of hydrogen bonds formed during water adsorption.135 Overall, the mentioned correlations provide a thermodynamic basis for precise molecular-level design of water harvesting frameworks with highly tunable water sorption properties (e.g., onset position, uptake capacity, ΔHads, and desorption temperature).137
Conclusions
As demonstrated by the studies highlighted in this review, the experimental toolbox for evaluating the thermodynamic aspects of MOFs is rich, providing many opportunities to understand and predict the properties of several classes of porous materials. For example, framework stability, structural transitions, possibility for PSM, gas/vapor adsorption, guest encapsulation, redox activity, and many other aspects of MOF chemistry are directly tied to a detailed fundamental understanding of associated thermodynamics.4,18,36,37,40,45,55,67,71,72,79,86,87,89,91,94,96,120,123,124,127,134,135,137,138 As a result, emergent applications of MOFs, including in the areas of atmospheric water harvesting, controlled drug delivery, heterogeneous catalysis, gas separation, and radionuclide sequestration/storage, all rely on an understanding of MOF thermodynamics.1,12,13,20–22,34,36,37,46,73,80,87,88,94,139 Therefore, experiments designed to probe thermodynamics are integral parts of all MOF research, ranging from ligand and framework design to long-term applications, stability testing, and even material recycling/repurposing.10,11,22,25,26,60,61,70,77,78,80,81,90,96,139–141As a first step in this review, we demonstrated how conventional experimental techniques, such as solution calorimetry, can be applied to evaluate the enthalpy of formation for a variety of materials by measuring the heat evolved upon framework decomposition. However, alternative calorimetric methods, such as drop combustion calorimetry or ITC, can allow for direct measurements performed on MOF solids (as opposed to solution calorimetry in which MOF samples are typically decomposed in acidic or basic media). For example, drop combustion calorimetry can be applied to predict the hydrolytic stability of materials targeting CO2 capture from industrial flue gas.29,50,124,125 While drop combustion calorimetry is advantageous because it allows for analysis of dry powders, it also requires high temperatures (>800 °C), and experiments need to be carried out under an oxidizing atmosphere. In contrast, ITC experiments can be performed on MOF suspensions under milder conditions (e.g., room temperature aqueous or organic suspensions), which may be more suitable for evaluating MOF performance in biomedical applications, such as protein encapsulation.86,102,112,142 Moreover, the ability to deconvolute the contributions of ΔH and ΔS to processes occurring within MOFs makes ITC a powerful technique that can be used to guide, and at the same time, “fine tune” material design.
Outside of conventional solution-based calorimetry, drop combustion calorimetry, and ITC, more conventional thermal techniques, such as DSC or TGA, allow for direct measurement of heat exchange occurring during a chemical reaction or phase transition. As a result, DSC and TGA experiments can often be more straightforward to perform than the mentioned calorimetric methods. Relative accessibility and the possibility to perform measurements under a variety of experimental constraints (e.g., variable humidity, temperature regimes, gases, and pressures) make DSC and TGA valuable techniques to gain both preliminary thermodynamic data, such as thermal stability and decomposition temperatures, and phase transition enthalpies. For example, DSC is an extremely powerful method for evaluating the energy and entropic contributions to “gate opening” or “breathing” transitions of flexible frameworks, especially when combined with PXRD or gas sorption studies.51,75,92
While calorimetric and thermal experiments can provide thermodynamic insights through direct measurement of heat exchange occurring during a chemical process, spectroscopic techniques can provide access to thermodynamic parameters like equilibrium constants and dissociation/binding constants by monitoring changes in a material's optical profile as a function of analyte concentration (Table 1).22,40,53,59 As a separate advantage, the equilibrium and/or binding constants extracted from spectroscopic data can be mathematically converted into values of ΔG, allowing for direct comparison with other experimental methods or with literature reports, assuming identical sample preparation methods, including choice of solvent, activation conditions, and phase purity.39,59,86,124 Moreover, spectroscopic methods, especially photoluminescence spectroscopy, generally offer high sensitivity and low detection limits, potentially allowing for evaluation of binding events in MOFs even at low analyte concentrations. At the same time, the high sensitivity of spectroscopic methods, which could be used to probe metal coordination environments for both adsorbed metal ions and metal nodes, also requires thorough analysis of subtle spectral features and rigorous control experiments.
This review also surveys a few less frequently used methods for probing MOF thermodynamics, such as advanced X-ray diffraction and electrochemical techniques.8,40,58,59,91,94,114 For example, EOCP measurements can shed light on the detailed mechanisms for electron and/or proton transfer occurring at MOF metal nodes, which are not readily accessible through other experimental techniques.8,40,94 In particular, some electrochemical techniques offer the potential advantage of allowing for MOFs to remain intact during analysis under carefully selected experimental conditions, thereby preserving the coordination environment of the metal node, in contrast to solution calorimetry which usually requires MOF samples to be degraded in acidic media. At the same time, HP-PXRD and synchrotron single-crystal X-ray diffraction can allow for structural changes in response to an external stimulus (e.g., applied pressure or presence of water vapor) to be monitored in situ or in operando, providing an experimental basis for further molecular dynamics simulations or theoretical modeling.51
To summarize, there is no single experimental technique that is sufficient to map the detailed thermodynamic landscape of MOF formation, applications, and degradation. Instead, the entire library of experimental techniques supported by computational analysis must be utilized in order to rationally design new materials and address upcoming challenges and current knowledge gaps. For instance, many of the future applications of MOFs described in this review rely on their integration within larger device architectures, and the optimization of the synthesis and fabrication of such devices will rely on understanding thermodynamics, for instance, behind the processes occurring at the interfaces between MOF particles and device components. Access to the necessary thermodynamic information (e.g., energy associated with charge transfer across device interfaces and/or MOF stability as a function of material shaping) will likely require extensive in operando calorimetric, thermal, or electrochemical measurements. For example, the field of MOF-based photo- and electrocatalysis is continually evolving, and further advancements will require detailed studies of the reaction mechanisms involving both a catalytically active MOF and a conductive support (in the case of hybrid or composite materials). In a similar vein, the electrochemical stability of hybrid materials and interfaces at variable current densities (e.g., >100 mA cm−2) and long reaction times is a critical parameter to carefully study moving forward.101 Thermodynamic measurements could also be applied to predict and design energetically favorable small-molecule oxidation reactions with suitable potentials to facilitate, for instance, hydrogen evolution as a clean energy source.100,101
Moreover, upcoming practical applications of MOFs in the areas of radioactive/hazardous waste management, drug delivery, and solid-state electrolytes for efficient batteries require more in-depth thermodynamic information, allowing for predicting and tailoring the long-term stability of frameworks under harsh and multicomponent environments. Alternatively, defect engineering in MOFs has already been proven in the literature to be a valuable pathway for radionuclide capture, tunable thermal conductivity, and enhanced catalytic activity, yet almost no thermodynamic data exists to describe defect formation and post-synthetic modification. For instance, current literature lacks fundamental knowledge related to entropy changes associated with post-synthetic coordination to open metal sites (i.e., “missing linker” defects), which will likely require development of new methods for both mapping and measuring random defects in an otherwise crystalline material.
Finally, continual development of theoretical models (e.g., DFT or other methods) that are more directly correlated with experimental reality will be vital for accessing next-generation MOF applications. In particular, new theoretical models should account for the dynamic nature of frameworks related to structural evolution upon exposure to material operational conditions. For instance, presence of labile/exchangeable linkers, structural changes upon repeated exposure of a material to industrially relevant conditions, partial pore blockage by guest molecules, and incomplete activation all significantly impact the outcome of thermodynamic measurements. Advanced computational studies that begin to account for “imperfect” MOF structures could provide important insights and clarify more precise structure–property relationships. As a result, the combination of experimental and computational thermodynamic aspects will continue to play a vital role in the development of the entire MOF “life cycle,” from design and structure–property analysis to commercialization and recycling.
Author contributions
G. C. T. writing – original draft: lead; writing – review & editing: lead visualization: supporting. D. N. S. visualization: lead; writing – review & editing: supporting. B. K. P. M. K. visualization: supporting; writing – review & editing: supporting. S. M. J. visualization: supporting; writing – review & editing: supporting. N. B. S. conceptualization: lead; funding acquisition: lead; supervision: lead; writing – review & editing: lead.Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software, or code have been included, and no new data were generated or analyzed as part of this review.Acknowledgements
The authors are grateful for support from the NSF awards DMR-2535079 and DMR-2103722. N. B. S. would also like to acknowledge support from the Dreyfus Teaching-Scholar Supplementary Award, supported by the Camille and Henry Dreyfus Foundation, as well as the ACS Bridge Award. B. K. P. M. K. is grateful for a SPARC Graduate Research Award from the Office of the Vice President for Research at the University of South Carolina.References
- W. Song, Z. Zheng, A. H. Alawadhi and O. M. Yaghi, Nat. Water, 2023, 1, 626–634 CrossRef CAS.
- H.-C. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 RSC.
- B.-N. T. Nguyen, T. T. Y. Tan, K.-I. Otake, S. Kitagawa and J. Y. C. Lim, Angew. Chem., Int. Ed., 2025, 64, e202504017 CrossRef CAS PubMed.
- T. Islamoglu, S. Goswami, Z. Li, A. J. Howarth, O. K. Farha and J. T. Hupp, Acc. Chem. Res., 2017, 50, 805–813 CrossRef CAS PubMed.
- K. Ma, M. C. Wasson, X. Wang, X. Zhang, K. B. Idrees, Z. Chen, S.-J. Lee, R. Cao, Y. Chen, L. Yang, F. A. Son, T. Islamoglu, G. W. Peterson, J. J. Mahle and O. K. Farha, Chem. Catal., 2021, 1, 721–733 CAS.
- J. Y. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. B. T. Nguyena and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- Y. Wu, X. Wang, K. O. Kirlikovali, X. Gong, A. Atilgan, K. Ma, N. M. Schweitzer, N. C. Gianneschi, Z. Li, X. Zhang and O. K. Farha, Angew. Chem., Int. Ed., 2022, 61, e202117528 CrossRef CAS PubMed.
- Z. Wang, Z. Yang, K.-I. Otake, J. Guo, X. Yang, Z. Xue, M.-S. Yao and S. Kitagawa, ChemElectroChem, 2025, 12, e202400525 CrossRef CAS.
- H. Furukawa, F. Gándara, Y.-B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369–4381 CrossRef CAS PubMed.
- C. A. Trickett, A. Helal, B. A. Al-Maythalony, Z. H. Yamani, K. E. Cordova and O. M. Yaghi, Nat. Rev. Mater., 2017, 2, 17045 CrossRef CAS.
- M. Ding, R. W. Flaig, H.-L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC.
- W. Xu and O. M. Yaghi, ACS Cent. Sci., 2020, 6, 1348–1354 CrossRef CAS PubMed.
- A. H. Alawadhi, S. Chheda, G. D. Stroscio, Z. Rong, D. Kurandina, H. L. Nguyen, N. Rampal, Z. Zheng, L. Gagliardi and O. M. Yaghi, J. Am. Chem. Soc., 2024, 146, 2160–2166 CrossRef CAS PubMed.
- C. R. Kim, T. Uemura and S. Kitagawa, Chem. Soc. Rev., 2016, 45, 3828–3845 RSC.
- Y. D. More, S. Mollick, S. Saurabh, S. Fajal, M. Tricarico, S. Dutta, M. M. Shirolkar, W. Mandal, J.-C. Tan and S. K. Ghosh, Small, 2024, 20, 2302014 CrossRef CAS PubMed.
- K. Ravin, P. Sarver, B. Dinakar, L. Palatinus, P. Müller, J. Oppenheim and M. Dincă, J. Am. Chem. Soc., 2025, 147, 11407–11411 CrossRef CAS PubMed.
- A. Y. Su and M. Dincă, J. Am. Chem. Soc., 2025, 147, 46720–46728 CrossRef CAS PubMed.
- S. S. Kaye, A. Dailly, O. M. Yaghi and J. R. Long, J. Am. Chem. Soc., 2007, 129, 14176–14177 CrossRef CAS PubMed.
- E. M. Miner, S. S. Park and M. Dincă, J. Am. Chem. Soc., 2019, 141, 4422–4427 CrossRef CAS PubMed.
- H. L. Nguyen, A. Darù, S. Chheda, A. H. Alawadhi, S. E. Neumann, L. Wang, X. Bai, M. O. Alawad, C. Borgs, J. T. Chayes, J. Sauer, L. Gagliardi and O. M. Yaghi, ACS Cent. Sci., 2025, 11, 665–671 CrossRef CAS PubMed.
- K. C. Park, C. R. Martin, G. A. Leith, G. C. Thaggard, G. R. Wilson, B. J. Yarbrough, B. K. P. Maldeni Kankanamalage, P. Kittikhunnatham, A. Mathur, I. Jatoi, M. A. Manzi, J. Lim, I. Lehman-Andino, A. Hernandez-Jimenez, J. W. Amoroso, D. P. DiPrete, Y. Liu, J. Schaeperkoetter, S. T. Misture, S. R. Phillpot, S. Hu, Y. Li, A. Leydier, V. Proust, A. Grandjean, M. D. Smith and N. B. Shustova, J. Am. Chem. Soc., 2022, 144, 16139–16149 CrossRef CAS PubMed.
- K. C. Park, J. Lim, G. C. Thaggard, B. K. P. Maldeni Kankanamalage, I. Lehman-Andino, Y. Liu, J. M. Burrell, C. R. Martin, A. T. Ta, A. B. Greytak, J. W. Amoroso, D. D. DiPrete, M. D. Smith, S. R. Phillpot and N. B. Shustova, Chem. Sci., 2025, 16, 14115–14126 RSC.
- J. Haimerl, G. C. Thaggard, B. K. P. Maldeni Kankanamalage, R. Bühler, J. Lim, K. C. Park, J. Warnan, R. A. Fischer and N. B. Shustova, J. Am. Chem. Soc., 2025, 147, 19918–19930 CrossRef CAS PubMed.
- G. C. Thaggard, B. K. P. Maldeni Kankanamalage, J. Lim, X. Shi, V. S. Arnold, M. Badiger, R. Mulkerrin, R. D. Broskey, E. Irizarry, G. Atim, A. Azmy, M. D. Smith, S. Garashchuk, I. Spanopoulos and N. B. Shustova, J. Am. Chem. Soc., 2025, 147, 42591–42606 CAS.
- G. C. Thaggard, B. K. P. Maldeni Kankanamalage, K. C. Park, J. Lim, M. A. Quetel, M. Naik and N. B. Shustova, Adv. Mater., 2024, 2410067 Search PubMed.
- D. Wu and A. Navrotsky, J. Solid State Chem., 2015, 223, 53–58 CrossRef CAS.
- H. Jeong, G. Park, J. Jeon and S. S. Park, Acc. Chem. Res., 2024, 57, 2336–2346 CrossRef CAS PubMed.
- S. Chakraborty, H. P. Q. Nguyen, J. Usuba, J. Y. Choi, Z. Sun, C. Raju, G. Sigelmann, Q. Qiu, S. Cho, S. M. Tenney, K. E. Shulenberger, K. Schmidt-Rohr, J. Park and G. G. D. Han, Chem, 2024, 10, 3309–3322 CAS.
- W. Liang, P. M. Bhatt, A. Shkurenko, K. Adil, G. Mouchaham, H. Aggarwal, A. Mallick, A. Jamal, Y. Belmabkhout and M. Eddaoudi, Chem, 2019, 5, 950–963 CAS.
- J. R. L. Hong-Cai Zhou and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef PubMed.
- M. Kim, Y. Lee and H. R. Moon, Acc. Chem. Res., 2024, 57, 2347–2357 CrossRef CAS PubMed.
- X. Li, S. Narayanan, V. K. Michaelis, T.-C. Ong, E. G. Keeler, H. Kim, I. S. Mckay, R. G. Griffin and E. N. Wang, Microporous Mesoporous Mater., 2015, 201, 151–159 CrossRef CAS PubMed.
- Y. Lin, X. Jiang, S. T. Kim, S. B. Alahakoon, X. Hou, Z. Zhang, C. M. Thompson, R. A. Smaldone and C. Ke, J. Am. Chem. Soc., 2017, 139, 7172–7175 CrossRef CAS PubMed.
- C.-H. Ho, M. L. Valentine, Z. Chen, H. Xie, O. Farha, W. Xiong and F. Paesani, Commun. Chem., 2023, 6, 70 CrossRef CAS PubMed.
- G. C. Thaggard and N. B. Shustova, ACS Cent. Sci., 2025, 11, 653–655 CrossRef CAS PubMed.
- A. Aykaç, M. Noiray, M. Malanga, V. Agostoni, J. M. Casas-Solvas, É. Fenyvesi, R. Gref and A. Vargas-Berenguel, Biochim. Biophys. Acta – Gen. Subj., 2017, 1861, 1606–1616 CrossRef PubMed.
- M. Kalaj and S. M. Cohen, ACS Cent. Sci., 2020, 6, 1046–1057 CrossRef CAS PubMed.
- A. C. McKinlay, R. E. Morris, P. Horcajada, G. Férey, R. Gref, P. Couvreur and C. Serre, Angew. Chem., Int. Ed., 2010, 49, 6260–6266 CrossRef CAS PubMed.
- F. Sha, T.-Y. Tai, M. A. Gaidimas, F. A. Son and O. K. Farha, Langmuir, 2022, 38, 6771–6779 CrossRef CAS PubMed.
- Z. Wang, R. Zhang, W. Zhang, L. Liao, J. Han, G. Xie and S. Chen, Anal. Chem., 2025, 97, 15895–15903 CrossRef CAS PubMed.
- G.-R. Si, T. He, X.-J. Kong, L.-H. Xie and J.-R. Li, Acc. Mater. Res., 2025, 6, 1006–1019 CrossRef CAS.
- H. Cai, J.-W. Wu, X.-J. Cai, Z. Lu, Y.-L. Lai, J.-X. Sun, Z.-L. Yuan, Y.-Y. Huang, J.-W. Cai, W. Lu, Y.-H. Lu, H.-Y. Zhang and D. Li, Inorg. Chem. Front., 2023, 10, 5144–5151 RSC.
- Z. Akimbekov, D. Wu, C. K. Brozek, M. Dincă and A. Navrotsky, Phys. Chem. Chem. Phys., 2016, 18, 1158–1162 RSC.
- N. Novendra, J. M. Marrett, A. D. Katsenis, H. M. Titi, M. Arhangelskis, T. Friščić and A. Navrotsky, J. Am. Chem. Soc., 2020, 142, 21720–21729 CrossRef CAS PubMed.
- Y. Tu, S. Yousaf, S. Tariq, F. M. Tawfiq, A. Aslam and K. A. Tola, Sci. Rep., 2025, 15, 37311 CrossRef CAS PubMed.
- K. C. Park, P. Kittikhunnatham, J. Lim, G. C. Thaggard, Y. Liu, C. R. Martin, G. A. Leith, D. J. Toler, A. T. Ta, N. Birkner, I. Lehman-Andino, A. Hernandez-Jimenez, G. Morrison, J. W. Amoroso, H.-C. zur Loye, D. P. DiPrete, M. D. Smith, K. S. Brinkman, S. R. Phillpot and N. B. Shustova, Angew. Chem., Int. Ed., 2023, 62, e202216349 CrossRef CAS PubMed.
- G. J. Leonel, M. Verma, G. A. Agbanga, L. Bonatti, H. Boukhalfa, A. Navrotsky and H. Xu, Phys. Chem. Miner., 2025, 52, 11 CrossRef CAS.
- J. Chen, R. Abazari, K. A. Adegoke, N. W. Maxakato, O. S. Bello, M. Tahir, S. Tasleem, S. Sanati, A. M. Kirillov and Y. Zhou, Coord. Chem. Rev., 2022, 469, 214664 CrossRef CAS.
- P. Z. Moghadam, Y. G. Chung and R. Q. Snurr, Nat. Energy, 2024, 9, 121–133 CrossRef CAS.
- A. A. Voskanyan, V. G. Goncharov, N. Novendra, X. Guo and A. Navrotsky, ACS Omega, 2020, 5, 13158–13163 CrossRef CAS PubMed.
- P. Vervoorts, J. Keupp, A. Schneemann, C. L. Hobday, D. Daisenberger, R. A. Fischer, R. Schmid and G. Kieslich, Angew. Chem., Int. Ed., 2021, 60, 787–793 CrossRef CAS PubMed.
- G. A. McCarver, T. Rajeshkumar and K. D. Vogiatzis, Coord. Chem. Rev., 2021, 436, 213777 CrossRef CAS.
- J. A. Mason, K. Sumida, Z. R. Herm, R. Krishna and J. R. Long, Energy Environ. Sci., 2011, 4, 3030–3040 RSC.
- J. L. Mancuso, A. M. Mroz, K. N. Le and C. H. Hendon, Chem. Rev., 2020, 120, 8641–8715 CrossRef CAS PubMed.
- I. A. Ibarra, A. Mace, S. Yang, J. Sun, S. Lee, J.-S. Chang, A. Laaksonen, M. Schröder and X. Zou, Inorg. Chem., 2016, 55, 7219–7228 CrossRef CAS PubMed.
- Q.-G. Zhai, X. Bu, C. Mao, X. Zhao and P. Feng, J. Am. Chem. Soc., 2016, 138, 2524–2527 CrossRef CAS PubMed.
- Q.-G. Zhai, X. Bu, X. Zhao, D.-S. Li and P. Feng, Acc. Chem. Res., 2017, 50, 407–417 CrossRef CAS PubMed.
- R. Abazari, S. Sanati, W. K. Fan, M. Tahir, S. Nayak, K. Parida, M. El-Shahat, R. M. Abdelhameed, D. S. Nesterov, A. M. Kirillov and J. Qian, Coord. Chem. Rev., 2025, 523, 216256 CrossRef CAS.
- A. K. Cheetham, G. Kieslich and H. H. M. Yeung, Acc. Chem. Res., 2018, 51, 659–667 CrossRef CAS PubMed.
- R. J. Drout, S. Kato, H. Chen, F. A. Son, K.-I. Otake, T. Islamoglu, R. Q. Snurr and O. K. Farha, J. Am. Chem. Soc., 2020, 142, 12357–12366 CrossRef CAS PubMed.
- R. R. Bhawnani, O. M. Barreto, P. K. R. Podupu, Y. Colón, G. Giri and M. R. Singh, ACS Mater. Lett., 2025, 7, 906–927 CrossRef CAS.
- Z. Han, Y. Yang, J. Rushlow, J. Huo, Z. Liu, Y.-C. Hsu, R. Yin, M. Wang, R. Liang, K.-Y. Wang and H.-C. Zhou, Chem. Soc. Rev., 2025, 54, 367–395 RSC.
- D. Tassé, V. Quezada-Novoa, C. Copeman, A. J. Howarth and A. Rochefort, Chem. Phys. Chem., 2025, 26, e202401050 CrossRef PubMed.
- C. K. Brozek, V. K. Michaells, T.-C. Ong, L. Bellarosa, N. Lopez, R. G. Griffin and M. Dinca, ACS Cent. Sci., 2015, 1, 252–260 CrossRef CAS PubMed.
- T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS PubMed.
- M. Gutiérrez, Y. Zhang and J.-C. Tan, Chem. Rev., 2022, 122, 10438–10483 CrossRef PubMed.
- T. C. Maponya, K. Makgopa, T. R. Somo and K. D. Modibane, Polymers, 2022, 14, 3613 CrossRef CAS PubMed.
- H. T. B. Pham, J. Y. Choi, S. Huang, X. Wang, A. Claman, M. Stodolka, S. Yazdi, S. Sharma, W. Zhang and J. Park, J. Am. Chem. Soc., 2022, 144, 10615–10621 CrossRef CAS PubMed.
- G. Park, M. C. Demuth, C. H. Hendon and S. S. Park, J. Am. Chem. Soc., 2024, 146, 11493–11499 CAS.
- J. L. Obeso, D. R. Amaro, C. V. Flores, A. Gutiérrez-Alejandre, R. A. Peralta, C. Leyva and I. A. Ibarra, Coord. Chem. Rev., 2023, 485, 215135 CrossRef CAS.
- K. M. Fahy, S. Lee, I. Akpinar, F. Sha, M. Ahmadi Khoshooei, S. Su, T. Islamoglu, N. C. Gianneschi and O. K. Farha, J. Am. Chem. Soc., 2024, 146, 5661–5668 CrossRef CAS PubMed.
- R. T. Mogharbel, O. Alaysuy, I. S. S. Alatawi, N. M. Alshammari, S. Sallam, A. M. Almutairi, A. M. Al-Bonayan and N. M. El-Metwaly, ACS Omega, 2025, 10, 15096–15115 CrossRef CAS PubMed.
- S. Krause, V. Bon, I. Senkovska, U. Stoeck, D. Wallacher, D. M. Többens, S. Zander, R. S. Pillai, G. Maurin, F.-X. Coudert and S. Kaskel, Nature, 2016, 532, 348–352 CrossRef CAS PubMed.
- E. A. Dolgopolova, O. A. Ejegbavwo, C. R. Martin, M. D. Smith, W. Setyawan, S. G. Karakalos, C. H. Henager, H.-C. zur Loye and N. B. Shustova, J. Am. Chem. Soc., 2017, 139, 16852–16861 CrossRef CAS PubMed.
- S. Leubner, R. Stäglich, J. Franke, J. Jacobsen, J. Gosch, R. Siegel, H. Reinsch, G. Maurin, J. Senker, P. G. Yot and N. Stock, Chem. – Eur. J., 2020, 26, 3877–3883 CrossRef CAS PubMed.
- H. Wang, M. Warren, J. Jagiello, S. Jensen, S. K. Ghose, K. Tan, L. Yu, T. J. Emge, T. Thonhauser and J. Li, J. Am. Chem. Soc., 2020, 142, 20088–20097 CrossRef CAS PubMed.
- P. M. Stanley, J. Haimerl, C. Thomas, A. Urstoeger, M. Schuster, N. B. Shustova, A. Casini, B. Rieger, J. Warnan and R. A. Fischer, Angew. Chem., Int. Ed., 2021, 60, 17854–17860 CrossRef CAS PubMed.
- P. Chakraborty, A. Rana, S. Mukherjee and S. Biswas, Inorg. Chem., 2023, 62, 802–809 CrossRef CAS PubMed.
- J. Samanta, Y. Zhang, M. Zhang, A. D. Chen and C. Ke, Acc. Mater. Res., 2022, 3, 1186–1200 CrossRef CAS.
- Y. Chen, W. Lu, M. Schröder and S. Yang, Acc. Chem. Res., 2023, 56, 2569–2581 CrossRef CAS PubMed.
- H. Lin, Y. Yang, B. G. Diamond, T.-H. Yan, V. I. Bakhmutov, K. W. Festus, P. Cai, Z. Xiao, M. Leng, I. Afolabi, G. S. Day, L. Fang, C. H. Hendon and H.-C. Zhou, J. Am. Chem. Soc., 2024, 146, 1491–1500 CrossRef CAS PubMed.
- X. Luo, R. Abazari, M. Tahir, W. K. Fan, A. Kumar, T. Kalhorizadeh, A. M. Kirillov, A. R. Amani-Ghadim, J. Chen and Y. Zhou, Coord. Chem. Rev., 2022, 461, 214505 CrossRef CAS.
- R. Abazari, S. Sanati, M. A. Bajaber, M. S. Javed, P. C. Junk, A. K. Nanjundan, J. Qian and D. P. Dubal, Angew. Chem., Int. Ed., 2024, 20, 2306353 CAS.
- E. A. Dolgopolova and N. B. Shustova, MRS Bull., 2016, 41, 890–896 CrossRef CAS.
- J. Lim, K. C. Park, G. C. Thaggard, Y. Liu, B. K. P. Maldeni Kankanamalage, D. J. Toler, A. T. Ta, P. Kittikhunnatham, M. D. Smith, S. R. Phillpot and N. B. Shustova, J. Am. Chem. Soc., 2024, 146, 12155–12166 CrossRef CAS PubMed.
- T. Y. Fai, F. Sha, X. Wang, X. Wang, K. Ma, K. O. Kirlikovali, S. Su, T. Islamoglu, S. Kato and O. K. Farha, Angew. Chem., Int. Ed., 2022, 61, e202209110 CrossRef PubMed.
- P. Saini, N. Chakinala, P. K. Surolia and A. Gupta Chakinala, Sep. Purif. Technol., 2025, 354, 128730 CrossRef CAS.
- K. M. Fahy, F. Sha, S. Reischauer, S. Lee, T.-Y. Tai and O. K. Farha, ACS Appl. Mater. Interfaces, 2024, 16, 30296–30305 CrossRef CAS PubMed.
- H. Kim, H. J. Cho, S. Narayanan, S. Yang, H. Furukawa, S. Schiffres, X. Li, Y.-B. Zhang, J. Jiang, O. M. Yaghi and E. N. Wang, Sci. Rep., 2016, 6, 19097 CrossRef CAS PubMed.
- X. Song, T. K. Kim, H. Kim, D. Kim, S. Jeong, H. R. Moon and M. S. Lah, Chem. Mater., 2012, 24, 3065–3073 CrossRef CAS.
- M. Kim, M. Pander and H. R. Moon, ACS Appl. Electron. Mater., 2024, 6, 3024–3038 CrossRef CAS.
- S. Kannaka, A. Ohmiya, C. Ozaki and M. Ohtani, Chem. Commun., 2024, 60, 4170–4173 RSC.
- H. Yang, F. Peng, A. N. Hong, Y. Wang, X. Bu and P. Feng, J. Am. Chem. Soc., 2021, 143, 14470–14474 CrossRef CAS PubMed.
- G. Zelenková, T. Zelenka and M. Almáši, J. Therm. Anal. Calorim., 2024, 149, 12675–12683 CrossRef.
- N. G. Altınçekiç, C. W. Lander, J. Yu, A. Roslend, Y. Shao and H. Noh, J. Am. Chem. Soc., 2025, 147, 34777–34790 CrossRef PubMed.
- J. J. Oppenheim, C.-H. Ho, D. Alezi, J. L. Andrews, T. Chen, B. Dinakar, F. Paesani and M. Dincă, Chem. Mater., 2024, 36, 3395–3404 CrossRef CAS.
- Z. Wang and S. M. Cohen, Chem. Soc. Rev., 2009, 38, 1315–1329 RSC.
- X. Li, J. Liu, K. Zhou, S. Ullah, H. Wang, J. Zou, T. Thonhauser and J. Li, J. Am. Chem. Soc., 2022, 144, 21702–21709 CrossRef CAS PubMed.
- O. A. Ejegbavwo, C. R. Martin, O. A. Olorunfemi, G. A. Leith, R. T. Ly, A. M. Rice, E. A. Dolgopolova, M. D. Smith, S. G. Karakalos, N. Birkner, B. A. Powell, S. Sandy, R. J. Koch, S. T. Misture, H.-C. zur Loye, S. R. Phillpot, K. S. Brinkman and N. B. Shustova, J. Am. Chem. Soc., 2019, 141, 11628–11640 CrossRef CAS PubMed.
- S. Sanati, R. Abazari, J. Shariati, Z. Guo, P. C. Junk, A. M. Kirillov and J. Qian, Inorg. Chem., 2025, 64, 20507–20516 CrossRef CAS PubMed.
- S. Sanati, R. S. Varma, M. Liu and R. Abazari, Energy Environ. Sci., 2025, 18, 7733–7755 RSC.
- R. Abazari, J. Goscianska, M. Naderi, M. Liu and S. Sanati, J. Mater. Chem. A, 2026, 14, 9347–9359 RSC.
- C. Xiao, J. Tian, Q. Chen and M. Hong, Chem. Sci., 2024, 15, 1570–1610 RSC.
- Y. Cui and J. Lin, Cell Rep. Phys. Sci., 2025, 6, 102846 CrossRef.
- M. K. Bhunia, J. T. Hughes, J. C. Fettinger and A. Navrotsky, Langmiur, 2013, 29, 8140–8145 CrossRef CAS PubMed.
- M. G. Goesten, P. C. M. M. Magusin, E. A. Pidko, B. Mezari, E. J. M. Hensen, F. Kapteijn and J. Gascon, Inorg. Chem., 2014, 53, 882–887 CrossRef CAS PubMed.
- F. Sha, H. Xie, F. A. Son, K. S. Kim, W. Gong, S. Su, K. Ma, X. Wang, X. Wang and O. K. Farha, J. Am. Chem. Soc., 2023, 145, 16383–16390 CrossRef CAS PubMed.
- T. R. Sheridan, M. A. Gaidimas, B. V. Kramar, S. Goswami, L. X. Chen, O. K. Farha and J. T. Hupp, Langmuir, 2022, 38, 11199–11209 CrossRef CAS PubMed.
- K. Yan, Z. Yu, Y. Wang, M. Guo, J. Xiong, R. Zhang, X. Li and X. Lu, ChemSusChem, 2025, 18, e202401456 CrossRef CAS PubMed.
- L. A. M. Mahmoud, R. A. dos Reis, X. Chen, V. P. Ting and S. Nayak, ACS Omega, 2022, 7, 45910–45934 CrossRef CAS PubMed.
- X. Wang, P. C. Lan and S. Ma, ACS Cent. Sci., 2020, 6, 1497–1506 CrossRef CAS PubMed.
- Jack D. Wright, T. Zhang, X. Wang and I. A. Riddell, Chem. Commun., 2025, 61, 7945 RSC.
- A. Sadrolvaezin, A. Pezhman, I. Zare, S. Z. Nasab, S. Chamani, A. Naghizadeh and E. Mostafavi, MedComm, 2023, 4, e386 CrossRef CAS PubMed.
- D. Banerjee, H. Wang, Q. Gong, A. M. Plonka, J. Jagiello, H. Wu, W. R. Woerner, T. J. Emge, D. H. Olson, J. B. Parise and J. Li, Chem. Sci., 2016, 7, 759–765 RSC.
- N. Kumar, P. Verma and A. Thakur, Results Chem., 2025, 14, 102144 CrossRef CAS.
- J. Rodriguez, I. Beurroies, M. V. Coulet, P. Fabry, T. Devic, C. Serre, R. Denoyel and P. L. Llewellyn, Dalton Trans., 2016, 45, 4274–4282 RSC.
- C. V. Suciu, T. Iwatsubo and S. Deki, J. Colloid Interface Sci., 2003, 259, 62–80 CrossRef CAS PubMed.
- B. Lefevre, A. Saugey, J. L. Barrat, L. Bocquet, E. Charlaix, P. F. Gobin and G. Vigier, Colloids Surf., A, 2004, 241, 265–272 CrossRef CAS.
- S. K. Jha, J. Sound Vib., 1976, 47, 543–558 CrossRef.
- Y.-F. Liu, H. He, L. Gao, R.-R. Liang, J. Li, J. Huang, Y. Yin, Y. Meng, Y. Zhong, R. Luo, L.-L. Zhang, H.-C. Zhou and S. Yuan, Chem. Sci., 2025, 16, 14995–15003 RSC.
- L. J. W. Johnson, A. R. Lowe, A. Le Donne, E. Arkan, S. Merchiori, L. Bartolomé, E. Amayuelas, D. Mirani, G. A. López, G. Grancini, M. Chorążewski, S. Meloni and Y. Grosu, J. Phys. Chem. Lett., 2025, 16, 2089–2096 CrossRef CAS PubMed.
- M. Chorążewski, P. Zajdel, T. Feng, D. Luo, A. R. Lowe, C. M. Brown, J. B. Leão, M. Li, M. Bleuel, G. Jensen, D. Li, A. Faik and Y. Grosu, ACS Nano, 2021, 15, 9048–9056 CrossRef PubMed.
- A. R. Lowe, P. Ślęczkowski, E. Arkan, A. Le Donne, L. Bartolomé, E. Amayuelas, P. Zajdel, M. Chorążewski, S. Meloni and Y. Grosu, ACS Appl. Mater. Interfaces, 2024, 16, 5286–5293 CrossRef CAS PubMed.
- J. Liu, A. I. Benin, A. M. B. Furtado, P. Jakubczak, R. R. Willis and M. D. LeVan, Langmuir, 2011, 27, 11451–11456 CrossRef CAS PubMed.
- T. M. McDonald, W. R. Lee, J. A. Mason, B. M. Wiers, C. S. Hong and J. R. Long, J. Am. Chem. Soc., 2012, 134, 7056–7065 CrossRef CAS PubMed.
- Q.-G. Zhai, N. Bai, S. N. Li, X. Bu and P. Feng, Inorg. Chem., 2015, 54, 9862–9868 CrossRef CAS PubMed.
- H. Fei, J. F. Cahill, K. A. Prather and S. M. Cohen, Inorg. Chem., 2013, 52, 4011–4016 CrossRef CAS PubMed.
- A. M. Hastings, D. Ray, W. Jeong, L. Gagliardi, O. K. Farha and A. E. Hixon, J. Am. Chem. Soc., 2020, 142, 9363–9371 CrossRef CAS PubMed.
- P. Kittikhunnatham, G. A. Leith, A. Mathur, J. K. Naglic, C. R. Martin, K. C. Park, K. McCullough, H. D. A. C. Jayaweera, R. E. Corkill, J. Lauterbach, S. G. Karakalos, M. D. Smith, S. Garashchuk, D. A. Chen and N. B. Shustova, Angew. Chem., Int. Ed., 2022, 61, e202113909 CrossRef CAS PubMed.
- H. Woo, A. M. Devlin and A. J. Matzger, J. Am. Chem. Soc., 2023, 145, 18634–18641 CrossRef CAS PubMed.
- W. J. Thompson, B. K. P. Maldeni Kankanamalage, G. C. Thaggard, K. C. Park, C. R. Martin, J. Niu, J. A. Byers and N. B. Shustova, Angew. Chem., Int. Ed., 2025, 64, e202416695 CrossRef CAS PubMed.
- C. K. Brozek and M. Dincă, Chem. Commun., 2015, 51, 11780–11782 RSC.
- Q. Wang, J. Bai, Z. Lu, Y. Pan and X. You, Chem. Commun., 2016, 52, 443–452 RSC.
- P. Mondal, Z. Neuschuler, D. Mandal, R. E. Hernandez and S. M. Cohen, Angew. Chem., Int. Ed., 2024, 63, e202317062 CrossRef CAS PubMed.
- C. T. Saouma, S. Richard, S. Smolders, M. F. Delley, R. Ameloot, F. Vermoortele, D. E. De Vos and J. M. Mayer, J. Am. Chem. Soc., 2018, 140, 16184–16189 CrossRef CAS PubMed.
- P. J. Jabalera-Ortiz, A. M. Rodriguez-Jimenez, R. Vismara, P. Delgado, N. M. Padial, J. A. R. Navarro and P. Garrido-Barros, ChemSusChem, 2025, 18, e202500219 CrossRef CAS PubMed.
- N. Hanikel, X. Pei, S. Chheda, H. Lyu, W. Jeong, J. Sauer, L. Gagliardi and O. M. Yaghi, Science, 2021, 374, 454–459 CrossRef CAS PubMed.
- J. Y. Choi, B. Check, X. Fang, S. Blum, H. T. B. Pham, K. Tayman and J. Park, J. Am. Chem. Soc., 2024, 146, 11319–11327 CAS.
- V. Quezada-Novoa, H. M. Titi, A. A. Sarjeant and A. J. Howarth, Chem. Mater., 2021, 33, 4163–4169 CrossRef CAS.
- A. E. B. S. Stone, A. Fortunato, X. Wang, E. Saggioro, R. Q. Snurr, J. T. Hupp, F. Arcudi and L. Dordevic, Adv. Mater., 2025, 37, 2408658 CrossRef CAS PubMed.
- M. A. Pearson, S. Bhagchandani, M. Dincă and J. A. Johnson, Mol. Syst. Des. Eng., 2023, 8, 591–597 RSC.
- A. Zulys, F. Yulia, N. Muhadzib and Nasruddin, Ind. Eng. Chem. Res., 2021, 60, 37–51 CrossRef CAS.
- R. L. Nuttall, K. L. Churney and M. V. Kilday, J. Res. Natl. Bur. Stand., 1978, 83, 335–345 CrossRef CAS PubMed.
- J. T. Hughes and A. Navrotsky, J. Am. Chem. Soc., 2011, 133, 9184–9187 CrossRef CAS PubMed.
- A. K. Cheetham, G. Kieslich and H. H. M. Yeung, Acc. Chem. Res., 2018, 51, 659–667 CrossRef CAS PubMed.
- J. T. Hughes, T. D. Bennett, A. K. Cheetham and A. Navrotsky, J. Am. Chem. Soc., 2013, 135, 598–601 CrossRef CAS PubMed.
- J. Li, V. G. Goncharov, A. C. Strzelecki, H. Xu, X. Guo and Q. Zhang, Inorg. Chem., 2022, 61, 15152–15165 CrossRef CAS PubMed.
- E. Y. Mertsoy, X. Zhang, C. B. Cockreham, V. G. Goncharov, X. Guo, J. Wang, N. Wei, H. Sun and D. Wu, J. Phys. Chem. C, 2021, 125, 14039–14047 CrossRef CAS.
- C. Chen, H. Li, J. Yi, Z. Qin, C. Wang, Z. Sun, Y. Xu, S. Xu, K. Xu and F. Zhao, FirePhysChem, 2022, 2, 295–302 CrossRef CAS.
- L. Tong, Q. Chen, A. A. Wong, R. Gómez-Bombarelli, A. Aspuru-Guzik, R. G. Gordon and M. J. Aziz, Phys. Chem. Chem. Phys., 2017, 19, 31684–31691 RSC.
- J. Yue, Q. Zheng, S. Ding, Y. Yin, X. Zhang, L. Wang, Y. Gu, J. Li, Y. Zhang, Y. Shi, Y. Dong, Q. Zhu and H. Duo, Sci. Rep., 2024, 14, 17607 CrossRef CAS PubMed.
- R. Liu, H. Wu, H. Y. Chung and Y. H. Ng, Adv. Funct. Mater., 2025, 35, 2421318 CrossRef CAS.
- J. Xing, Y. Liu, G. Mathew, Q. He, J. Aghassi-Hagmann, S. Schweidler and B. Breitung, Adv. Sci., 2025, 12, 2411175 CrossRef CAS PubMed.
- C. Nanthamathee, C. Chantarangkul, C. Jakkrawhad, A. Payaka and P. Dechatiwongse, Heliyon, 2022, 8, e08961 CrossRef CAS PubMed.
- S. Almuqhawi, R. AL-Dadah, S. Mahmoud, C. Waldron and M. Walker, Next, Materials, 2026, 10, 101445 CAS.
- I. Ijaz, A. Bukhari, E. Gilani, A. Nazira and H. Zainb, RSC Adv., 2023, 13, 5643–5655 RSC.
- R. Goeminne, S. Krause, S. Kaskel, T. Verstraelen and J. D. Evans, J. Am. Chem. Soc., 2021, 143, 4143–4147 CrossRef CAS PubMed.
- H. Li, L. Li, R.-B. Lin, W. Zhou, Z. Zhang, S. Xiang and B. Chen, EnergyChem, 2019, 1, 100006 CrossRef PubMed.
- C. Pathak, S. Mistry and S. Seth, Coord. Chem. Rev., 2025, 543, 216928 CrossRef CAS.
- B. Achenbach, A. Yurdusen, N. Stock, G. Maurin and C. Serre, Adv. Mater., 2025, 37, 2411359 CrossRef CAS PubMed.
- C. McKinstry, E. J. Cussen, A. J. Fletcher, S. V. Patwardhan and J. Sefcik, Cryst. Growth Des., 2013, 13, 5481–5486 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |