
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptical probes for bioimaging of tumor-infiltrating immune cells and their applications in cancer immunotherapy
Xinglong
Chen†
a,
Cong
Hu†
b,
Heejeong
Kim
 c,
Lemeng
Zhang
*a,
Zhipengjun
Zhang
b,
Hongwen
Liu
*b and
Juyoung
Yoon
*cd
c,
Lemeng
Zhang
*a,
Zhipengjun
Zhang
b,
Hongwen
Liu
*b and
Juyoung
Yoon
*cd
aThe Affiliated Cancer Hospital of Xiangya School of Medicine, Central South University/Hunan Cancer Hospital, Changsha 410013, P. R. China. E-mail: zhanglemeng@hnca.org.cn
bKey Laboratory of Chemical Biology and Traditional Chinese Medicine Research (Ministry of Education), College of Chemistry and Chemical Engineering, Hunan Normal University, Changsha 410081, P. R. China. E-mail: liuhongwen@hnu.edu.cn
cDepartment of Chemistry and Nanoscience, Ewha Womans University, Seoul 03760, Republic of Korea. E-mail: jyoon@ewha.ac.kr
dGraduate Program in Innovative Biomaterials Convergence, Ewha Womans University, Seoul 03760, South Korea
First published on 5th May 2026
Abstract
The role of tumor-infiltrating immune cells (TIICs) in the tumor microenvironment (TME) is crucial in tumor development. The precise localization of TIICs within tumor tissues and the elucidation of their biological processes are critical for tumor diagnosis, treatment, and prognosis. Currently, optical probes with high specificity, sensitivity, and spatiotemporal resolution have emerged as powerful tools for the precise localization of TIICs and the investigation of their biological functions. In addition, the modifiability of optical biomaterials with these characteristics enables them to facilitate diverse and precise localization of TIICs and the monitoring of biological processes according to different design strategies. This review summarizes recent advancements in optical probes derived from optical biomaterials, including design strategies for TIIC-targeted optical probes, their applications in precise TIIC localization, and biological process monitoring. Furthermore, we emphasize the importance of their integration with immunotherapy, analyze the underlying factors contributing to the limited development of optical probes for regulatory T cells (Tregs), and systematically discuss the opportunities and challenges associated with their rapid advancement in tumor diagnosis and treatment.
 From left to right: Xinglong Chen and Lemeng Zhang | Dr Lemeng Zhang (right) is the master's supervisor of Xinglong Chen (left). Dr Lemeng Zhang received his MD and PhD degrees in 2012 from Xiangya Hospital of Central South University. He served as a postdoctoral fellow in Dr Allan Tsung's Lab. Currently, he works at Hunan Cancer Hospital as a tenured professor. His current research interests include the resistance mechanism of lung cancer immunotherapy and precise in vivo imaging of fluorescent probes. In 2025, Mr. Chen joined Dr Hongwen Liu's research group as a doctoral student, and under the joint guidance of Dr Liu and Dr Zhang, he is engaged in the development of activatable molecular probes and innovative lung cancer diagnosis and treatment technologies, and is committed to promoting the application research of advanced in vivo imaging technologies. |
 Cong Hu | Cong Hu earned his Master's degree in Chemistry from Hunan Normal University, where he conducted research under the guidance of Prof. Ronghua Yang and Prof. Hongwen Liu. In 2025, he joined the research group of Prof. Xiaojun Peng as a PhD candidate under the supervision of Prof. Mingle Li. His current research focuses on the design and development of Nile blue-based Type I photosensitizers for advanced photodynamic therapy applications. |
 Heejeong Kim | Heejeong Kim graduated from Ewha Womans University in 2020 with a bachelor's degree and with a doctoral degree in 2025 under the guidance of Professor Juyoung Yoon. She is currently pursuing postdoctoral research in the same group. Her main research focuses on the design and construction of supramolecular nanomedicines and the development and application of AIE therapeutic drugs, dedicated to fundamental research and cutting-edge exploration in these fields. |
 From left to right: Hongwen Liu and Zhipengjun Zhang | Dr Hongwen Liu (left) is the doctoral supervisor of Zhipengjun Zhang (right). Dr Liu received his PhD degree in Analytic Chemistry from the Department of Chemistry, Hunan University, in 2017, under the supervision of Prof. Dr Xiao-Bing Zhang. He then served as a postdoctoral fellow in Prof. Dr Weihong Tan's Lab. Currently, he works at Hunan Normal University as a professor in the College of Chemistry. His current research interests include the construction of novel small molecules for biosensing, bioimaging, and cancer therapy. Under Dr Liu's guidance, Mr. Zhang is engaged in the innovative development of high-performance fluorescent dyes and various fluorescent probes, and is committed to promoting the application research of advanced in vivo imaging technology. |
 Juyoung Yoon | Juyoung Yoon is a distinguished professor in the Department of Chemistry and Nanoscience at Ewha Womans University. In 1994, he obtained a doctoral degree from Ohio State University. Subsequently, he completed his postdoctoral research at the University of California, Los Angeles, and the Scripps Research Institute. His main research focuses are on fluorescence imaging, phototherapy, and integrated research on diagnosis and treatment. From 2014 to 2024, he has continuously been included in the list of highly cited researchers in the field of chemistry. |
1. Introduction
TIICs are a fundamental component of the TME.1,2 These include a variety of cell types—such as neutrophils, macrophages, T cells, natural killer (NK) cells, dendritic cells (DCs), and B cells (BCs)—that interact with each other and with other elements of the TME, collectively shaping the evolution of the tumor immune landscape in both established and yet-to-be-defined ways.3–12 In recent years, the antitumor immune response within the TME has garnered increasing research attention. Accumulating evidence indicates that various cancer therapies, including chemotherapy and immune checkpoint inhibitors (ICIs), can directly or indirectly stimulate endogenous antitumor immunity. Specifically, chemotherapy induces immunogenic tumor cell death, prompting the release of tumor-associated antigens (TAAs) and damage-associated molecular patterns (DAMPs), thereby activating antigen-presenting cells and subsequent antitumor immune cascades. In contrast, immune checkpoint inhibitors (ICIs) directly enhance effector T cell-mediated antitumor responses. Nevertheless, the efficacy of these interventions is often limited by immunosuppressive factors in the TME, such as Tregs. Despite this pivotal role, the functional diversity and dynamic nature of TIICs present a major challenge to fully deciphering their complex interactions and net impact on therapy.Currently, a range of bioimaging techniques have been utilized in the study of TIICs, although these approaches are constrained by inherent limitations. Conventional clinical modalities—such as magnetic resonance imaging (MRI), positron emission tomography (PET), single-photon emission computed tomography (SPECT), computed tomography (CT), and X-ray—are often constrained by resolution (MRI: 1–2 mm; PET: 6–10 mm; SPECT: 7–15 mm; CT/X-ray: 0.1–0.5 mm), sensitivity, or radiation exposure, hindering practical utility.13–17 Emerging optical bioimaging offers a promising alternative.18–24 Optical probes are highly programmable, allowing adaptation to TIIC-specific metabolic features (e.g., enzyme or receptor expression) while providing superior targeting, sensitivity, and biosafety. Moreover, they support diverse imaging modalities, which can be categorized into fluorescence (FL), photoacoustic (PA), afterglow luminescence, bioluminescence (BL), chemiluminescence (CL), and upconversion luminescence (UCL), collectively enabling precise in vivo visualization of TIICs (Table 1). This, in turn, elucidates their behavior, interactions, and impact on immunotherapeutic responses, significantly advancing research in tumor diagnosis, treatment, and prognosis.25–31 Furthermore, multimodal bioimaging, particularly the integration of optical and clinical bioimaging modalities (e.g., FL/CT imaging, PA/MRI, and FL/MRI), has successfully overcome the inherent constraints of single-modality imaging.32 This approach is poised to revolutionize medical imaging and propel advancements in TIIC-targeted tumor diagnosis and treatment.
| Imaging mode | Principle | Sensitivity | Penetration depth | Resolution | Background signal | Applicable scenarios |
|---|---|---|---|---|---|---|
| FL | External light excitation | High | Shallow | High | Medium-high | Cells/epithelial tissues |
| PA | Photoacoustic effect | Medium | Medium | Medium | Medium | Deep tissue/vessel imaging |
| CL/BL | FL mediated by chemical/biological reactions | High | Shallow | Medium-high | Low | Molecular event detection |
| Afterglow | Long afterglow effect after external light excitation | High | Shallow | Medium-high | Low | Low-background imaging without excitation |
| X-ray imaging and CT | X-ray penetration | Low | Deep | Medium | High | Bone injury/tumor burden assessment |
| MRI | Relaxation signal imaging | Medium-low | Deep | Medium-low | Medium-low | Soft tissue imaging |
| PET and SPECT | Positron/single-photon emission imaging | High | Deep | Low | High | Functional/metabolic imaging |
In light of these capabilities, optical probes offer significant advantages over conventional clinical bioimaging across multiple dimensions.33–39 By combining high resolution, specificity, sensitivity, and real-time imaging capabilities, these tailored optical probes enable the in vivo monitoring of complex biological processes within TIICs throughout tumorigenesis (Fig. 1).60 This capacity is pivotal for advancing cancer immunotherapy, as it permits real-time assessment of therapeutic efficacy and dynamic tracking of immune cell subsets in response to treatment.61–66 These attributes effectively overcome the limitations of clinical bioimaging through high-precision, real-time biological monitoring and enhanced biosafety. Furthermore, optical probes are a powerful tool for enhancing the efficacy of cancer diagnosis and treatment based on TIICs. They present the information of TIICs in the TME via real-time readable optical signals by utilizing the specific biomarkers (such as enzymes, reactive species, special structures, receptors, etc.) metabolized by TIICs. This method not only depicts the tumor morphology but also provides real-time information of TIICs in the TME, which paves the way for optimizing tumor diagnosis and treatment strategies in clinical practice, improving treatment outcomes, and ultimately enhancing patient prognosis.67–74 Notably, the programmability of optical probes gives them true design flexibility, allowing uses that go well beyond conventional tumor imaging. Many groups have now built multifunctional probes that merge real-time diagnosis with therapy—something traditional methods cannot achieve.75–77 Owing to high specificity and tumor-targeting efficiency, these probes let us monitor treatment as it happens while reducing the ineffectiveness and adverse effects typical of single-modality therapies.78–81
 | ||
| Fig. 1 Optical probes and their applications in biomedicine. | ||
This review summarizes the latest advancements in TIIC-targeted optical probes and discusses their diverse applications, including but not limited to tumor immunotherapy, immune status assessment, tumor imaging, and diagnosis. First, we explain the critical role of TIICs in tumor development, thereby enhancing the understanding of TIICs.
Subsequently, we outline the targeting and design strategies of these probes to offer insights into the development of TIIC-targeted optical probes in the future. Furthermore, we classify and introduce existing probes based on target type, focusing on their composition and interaction mechanisms with target biomolecules. More importantly, we emphasize the significance and feasibility of integrating TIIC-targeted optical probes with immunotherapy, offering guidance for the future development of multifunctional TIIC-targeted optical probes. The combination of optical probes with immunotherapy directly links diagnosis to treatment. This integration gives us tight control over when and where therapy is delivered, improving selectivity and safety. We can also track treatment responses in real time and adjust regimens as needed. Beyond that, this strategy can boost the effects of immunotherapy and close the loop from diagnosis and treatment to outcome assessment—an appealing tool for precision oncology. By merging optical probes with immune-based therapies, this integrated diagnostic and therapeutic framework aims to support more effective, personalized cancer care. We also summarize the shortcomings of current research and look forward to the development of this field. This review aims to articulate an interdisciplinary framework integrating expertise from diverse fields, including tumor biology, immunology, biomedical engineering, analytical chemistry, and biomedicine. The meticulous design of optical probes is paramount, ensuring the accuracy and reliability of bioimaging data, ultimately guiding the evolution of precision medicine in oncology.
2. Immune function, infiltration process and classification of TIICs
2.1. Immune function of TIICs
TIICs constitute a central component of the TME, orchestrating anti-tumor immunity and immune escape while exhibiting pronounced functional plasticity and bidirectional regulatory functions. Canonical effector populations include cytotoxic T lymphocytes (CTLs) and NK cells, which induce tumor cell apoptosis through perforin-granzyme release and Fas-FasL signalling pathways; DCs, which initiate adaptive immune responses via antigen presentation; and M1-type tumor-associated macrophages (TAMs), which secrete pro-inflammatory cytokines such as IL-12 and TNF-α to promote Th1-type immunity. Together, these populations mediate immune surveillance and tumor clearance. Conversely, under the influence of tumor-derived factors, certain TIIC subsets undergo polarization toward immunosuppressive or pro-tumor phenotypes. Tregs suppress effector T-cell activity through IL-10 and TGF-β secretion; M2-type TAMs facilitate angiogenesis and tissue remodeling; and myeloid-derived suppressor cells (MDSCs) inhibit T-cell function via the production of reactive oxygen and nitrogen species (ROS and RNS) and the depletion of essential metabolic substrates. In addition, neutrophils display functional heterogeneity within the TME: N1-type tumor-associated neutrophils (TANs) exert anti-tumor activity by enhancing cytotoxicity and immune activation, whereas N2-type TANs secrete factors such as vascular endothelial growth factor (VEGF) and matrix metalloproteinases (MMPs), thereby promoting angiogenesis, extracellular matrix degradation, and metastasis. Collectively, the cellular composition, polarization states, metabolic milieu, and spatial organization of TIICs determine the overall immune tone of the TME and critically influence tumor progression as well as responsiveness to immunotherapy.2.2. Infiltration process of TIICs
All immune cells in the body originate from hematopoietic stem cells, which first differentiate within the bone marrow into two primary progenitor lineages: lymphoid and myeloid progenitors. As these progenitor cells develop and mature, they further differentiate into innate immune cells, including macrophages, neutrophils, DCs, and NK cells, and adaptive immune cells, including T cells and BCs (Fig. 2A).82–86 When cytokine receptors on immune cells—including chemokine receptors (e.g., CCR, CXCR, XCR, and CX3CR) and non-chemokine receptors (e.g., TNFR, IFNGR, and CSF1R)—bind to cytokines secreted by tumor and stromal cells, this binding directs their migration toward the tumor site. Upon reaching the tumor site, adhesion molecules, such as intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1), expressed on the tumor vascular endothelium facilitate immune cell entry via the sequential processes of scrolling, adhesion, and transendothelial migration. Once within the tumor tissue, immune cells utilize enzymes (e.g., MMPs and neutrophil elastase [NE]) and adhesion molecules (e.g., integrins) to degrade the extracellular matrix and migrate toward the tumor core.87–89 Through these coordinated steps, immune cells successfully infiltrate the tumor tissue from the circulation (Fig. 2B).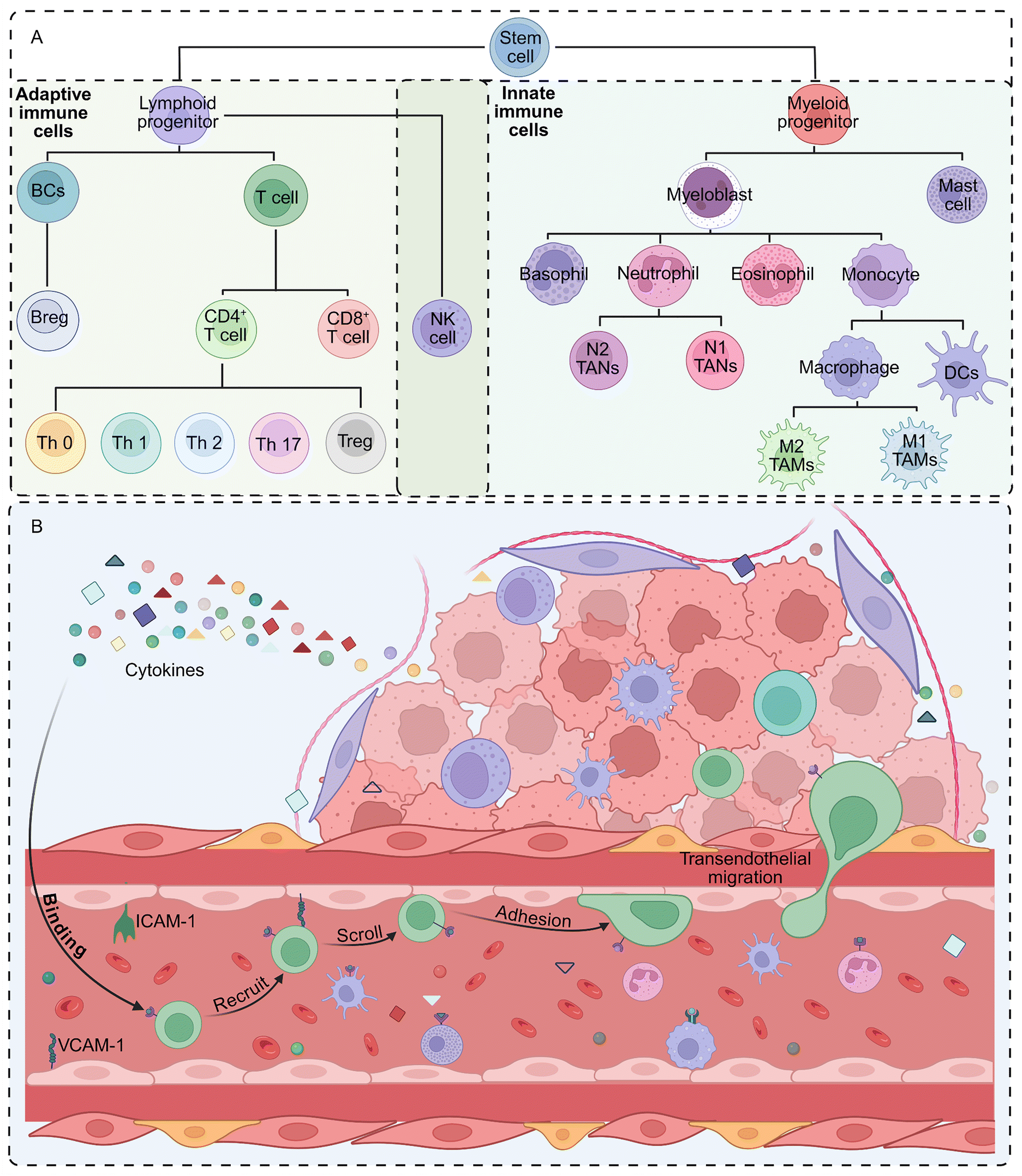 | ||
| Fig. 2 (A) Generation and differentiation of immune cells. (B) Mechanisms of immune cell infiltration into tumor tissue. Created in BioRender. ss, A. (2026) https://BioRender.com/epgcy08. | ||
2.3. Classification of TIICs
Although immune cells are essential for maintaining homeostasis, they can paradoxically promote tumor progression after becoming TIICs.90–92 The impact of TIICs on tumor fate is context-dependent, exhibiting both anti-tumor and pro-tumor functions. This duality is determined by their specific subtype and activation status within the TME, as exemplified by the classic N1/N2-type TANs and the M1/M2-type TAMs. In the early stages of tumor development, TANs and TAMs primarily cluster at the tumor periphery, suppressing tumor progression by releasing apoptosis-inducing factors and ROS.93,94 However, as the tumor advances, they gradually polarize into N2 and M2 type and infiltrate deeper into the tumor. At this stage, TANs and TAMs primarily promote angiogenesis and tumor progression by releasing VEGF and MMPs.95–97 Notably, because MDSCs can differentiate into myeloid cells, some studies suggest that TAMs and TANs in the TME may originate from MDSCs.98–100 Compared to MDSCs, DCs and NK cells both possess natural anti-tumor functions. DCs recognize and capture tumor antigens and subsequently present them to T cells, thereby activating T cell-mediated cytotoxicity. NK cells, which are typically present in the blood, induce tumor cell apoptosis through the release of perforins and granzymes (Grs) and are highly effective in eliminating circulating tumor cells, thereby inhibiting metastasis.101,102 Interestingly, although NK cells are traditionally classified as innate immune cells, accumulating evidence indicates that they also exhibit adaptive immune-like features within the TME. Specifically, NK cells can acquire long-term antitumor memory and enhance cytotoxic efficacy. This finding suggests that NK cells represent a promising target for tumor diagnosis and immunotherapy.103–105 However, in the TME, the anti-tumor activity of DCs and NK cells is frequently suppressed by immunosuppressive factors (such as TGF-β, IL-10, and VEGF), immunosuppressive cells (such as Tregs and MDSCs), metabolic disorders (including lactic acid accumulation, hypoxia, and nutrient competition), and tumor cell evasion of immune mechanisms (such as receptor-ligand regulation and exosome interference).106–109Adaptive immune cells exert potent inhibitory effects on tumor progression and display extensive intercellular interactions. T cell subsets comprise CTLs, CD4+ helper T cells, and Tregs.110 CTLs recognize tumor cells via their T cell receptors by binding to tumor-specific antigens presented on the cell surface and induce apoptosis through the secretion of perforins and Grs. CD4+ helper T cells secrete cytokines, such as IL-2, IFN-γ, and TNF-α, that promote the activation of CTLs and NK cells, thereby enhancing anti-tumor immune responses. BCs contribute to tumor inhibition by presenting antigens that activate and enhance CTL-mediated cytotoxicity.111–113 Although adaptive immune cells have a strong inhibitory effect on tumor development, there seem to be “traitors” among them. For example, Tregs and regulatory B cells (Bregs) induce immunosuppressive phenotypes in TAMs, TANs, and CTLs by secreting cytokines such as IL-10 and TGF-β, thereby supporting tumor immune escape.114–117
In brief, TIICs and their surrounding milieu collectively constitute a highly complex tumor immune microenvironment (TIME). Accordingly, the development of optical probes targeting TIICs and incorporating immunotherapeutic functionalities holds substantial scientific and translational value. This strategy transcends conventional tumor cell-centric imaging paradigms by enabling real-time, dynamic, and functional visualization of the TIME. It allows non-invasive characterization of TIIC activation/polarization states and the immunosuppressive network in vivo, thereby enhancing the prediction of immunotherapeutic responses and facilitating patient stratification. Furthermore, by engineering theranostic optical probes that integrate imaging and immune modulation, precise activation and localized immune reprogramming within specific tumor microenvironments can be achieved, amplifying systemic antitumor immunity while mitigating systemic toxicity. Collectively, this emerging direction provides critical tools for elucidating the dynamic evolution of the TIME and establishes a robust foundation for the advancement of precision immunotherapy and non-invasive, real-time monitoring technologies, underscoring its substantial interdisciplinary innovation and clinical potential.
3. Response mechanisms of TIIC-targeted optical probes
Optical probes targeting TIICs typically comprise three core functional units: (1) an imaging unit responsible for generating optical signals (e.g., FL dyes, FL proteins, metal nanoparticles, quantum dots [QDs], upconversion nanoparticles [UCNPs], metal–organic frameworks [MOFs] and others); (2) a recognition unit that confers target specificity (e.g., antibodies, peptide sequences, ligands, responsive chemical groups, and others); and (3) a signal transduction unit that converts target recognition into measurable signal changes, such as intramolecular charge transfer (ICT), twisted intramolecular charge transfer (TICT), photoinduced electron transfer (PeT), chemiluminescence resonance energy transfer (CRET), Förster resonance energy transfer (FRET), disassembly, aggregation and others (Fig. 3).118–124 The selection of imaging units dictates the corresponding imaging modality, including FL, PA, CL, BL, afterglow, surface plasmon resonance (SPR), metal-to-ligand charge transfer (MLCT) and others. The rational integration of these three core functional modules constitutes the fundamental design principle for constructing optical probes targeting TIICs. Distinct modes of integration define the probe's response mechanism, namely, the manner in which biological interactions are translated into quantifiable optical outputs. This integration ultimately governs the critical performance parameters of TIIC-targeted optical probes, including specificity, signal-to-background ratio (SBR), and spatiotemporal imaging dynamics. And with continued technological advances, multiple imaging strategies can be rationally integrated to construct multimodal optical probes, thereby overcoming the intrinsic limitations of single-modality imaging. In particular, multimodal probes combined with clinical bioimaging technologies enable complementary signal acquisition, providing multidimensional information encompassing both tumor function and anatomical structure. Beyond multimodal designs, multi-lock optical probes capable of responding to multiple biomarkers have been increasingly developed. By incorporating multiple recognition elements into a single probe architecture, specificity and diagnostic accuracy can be substantially enhanced.125–127 Based on target characteristics and signal activation mechanisms, current TIIC-targeted optical probes can be broadly categorized into two types: (1) probes activated by specific biomarkers secreted or expressed by TIICs; and (2) probes that directly label TIICs through binding to membrane proteins. Furthermore, the incorporation of therapeutic functionalities represents a critical extension of probe design, transforming purely diagnostic systems into theranostic platforms. This section systematically reviews recent advances within these mechanistic frameworks.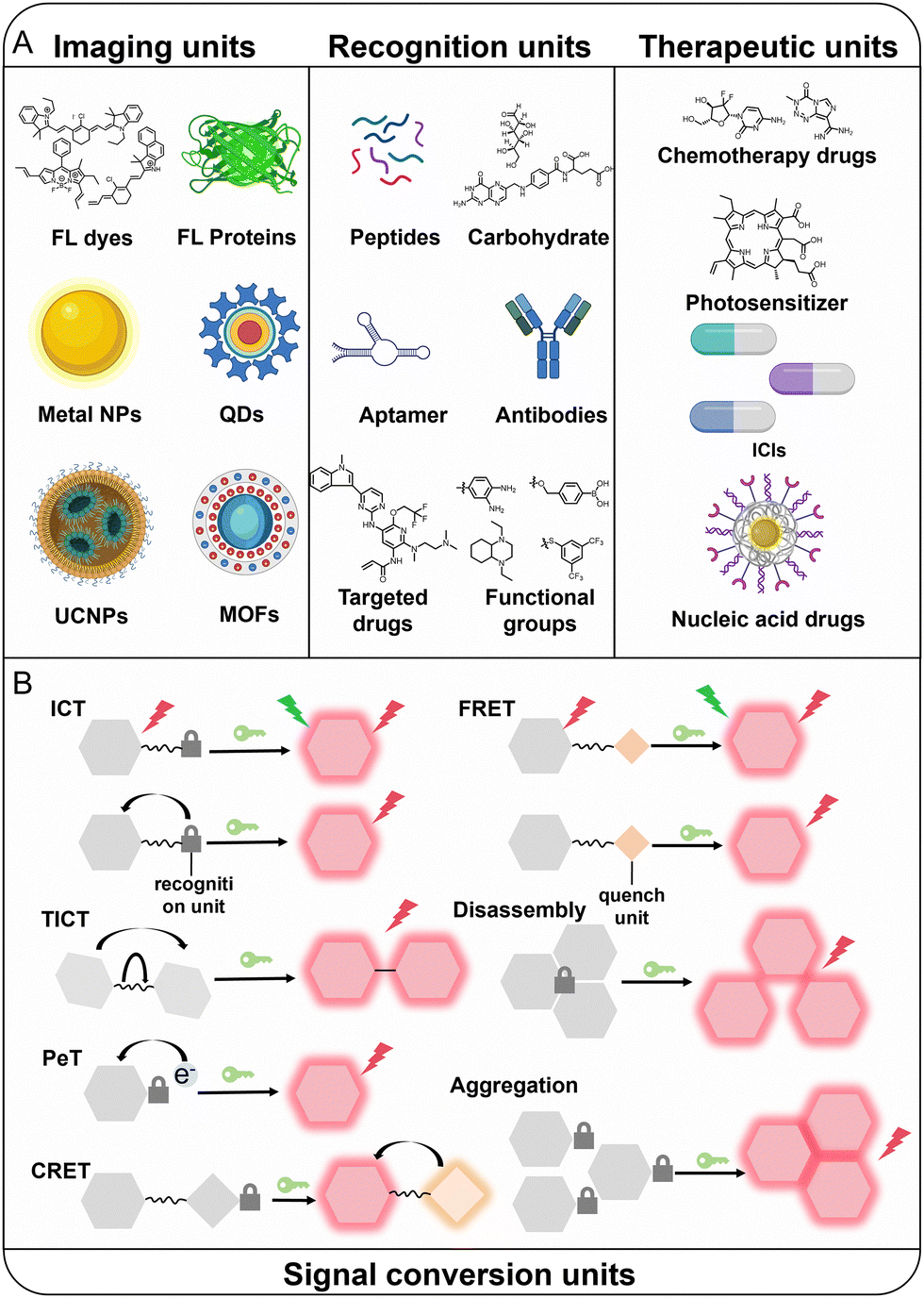 | ||
| Fig. 3 (A) Components of TIIC-targeted optical probes. (B) Signal conversion mechanism of TIIC-targeted optical probes. | ||
3.1. Probes activated by TIIC-associated biomarkers
Metabolites produced by TIICs not only reflect their biological functions but also provide critical molecular cues for the rational design of enzyme/reactive species-activated optical probes (Fig. 4A).128,129 These metabolites include diverse enzymes capable of promoting or inhibiting tumor growth or metastasis—such as NE highly expressed in TANs, myeloperoxidase (MPO) expressed in TANs and TAMs, MMPs and cathepsins (CTS) expressed by various cells within the TME, and Grs released by CTLs and NK cells—as well as reactive species including ROS, RNS, and reactive sulfur species (RSS).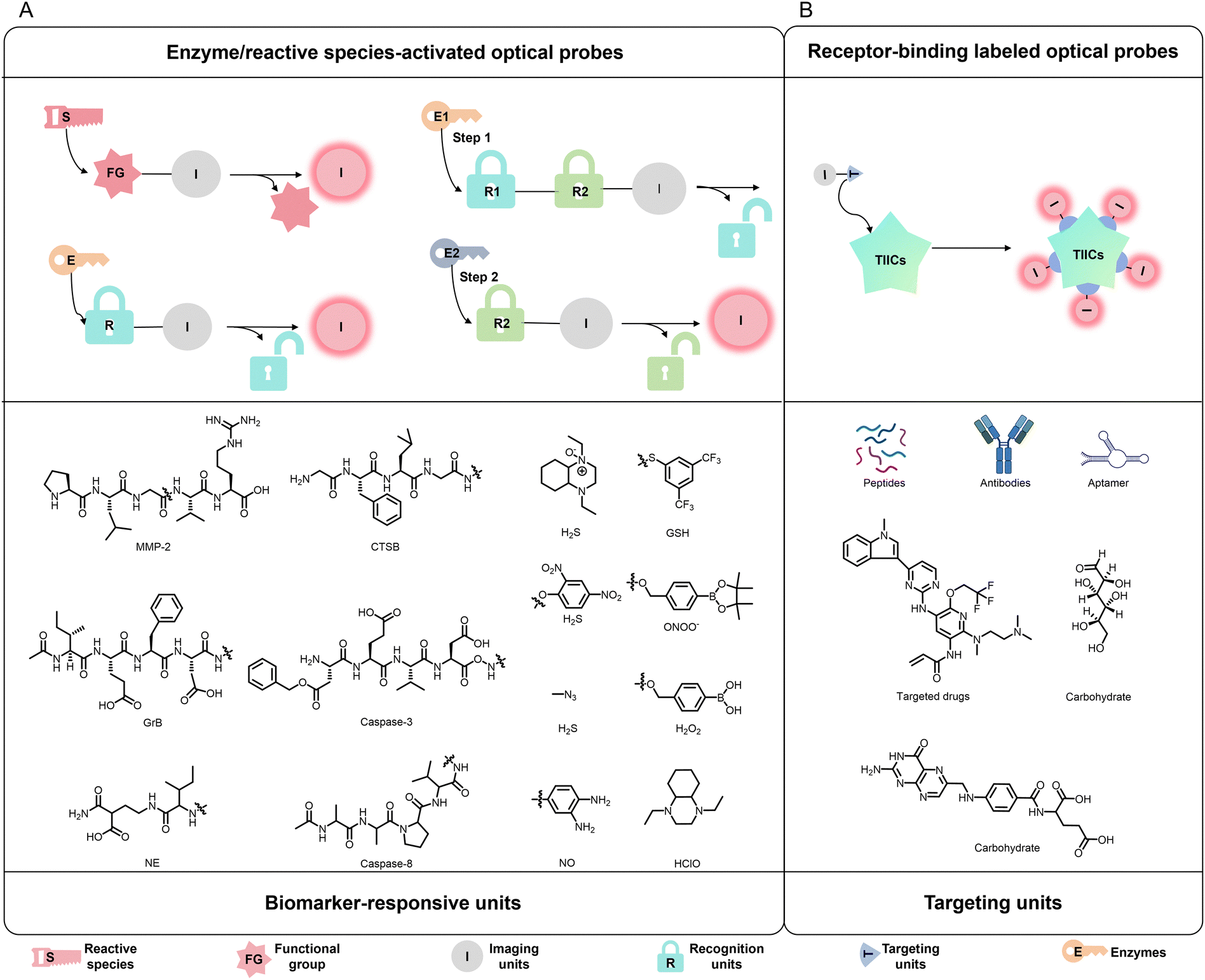 | ||
| Fig. 4 TIIC-targeted optical probes of different types. (A) Signal conversion process and common biomarker response units of enzyme/reactive species-activated optical probes. (B) Signal conversion process and common targeting units of receptor-binding labelled optical probes. | ||
These metabolites are recognized by tailored recognition units (e.g., peptide sequences, amide bonds, or reactive functional groups), triggering a signal conversion unit that activates the imaging unit to produce quantifiable optical signals for molecular readout. This type of molecular recognition-based active-targeting strategy is widely used in the design and construction of small-molecule probes for targeting TIICs. Through the rational selection of signal transduction units and the covalent integration of imaging moieties, recognition elements, linkers, and auxiliary components via organic synthesis, target-specific probes can be constructed. Small-molecule probes typically exhibit low molecular weight, rapid response kinetics, high sensitivity, and structural tunability. The primary advantage of this active-targeting strategy lies in its high specificity and rich information output. Specifically, when a particular receptor is overexpressed in lesions, a protease is aberrantly activated, or a specific cell subpopulation is significantly enriched, small-molecule probes can generate a distinct signal differential between diseased and normal tissues, thereby enhancing imaging contrast and improving the molecular diagnostic value of the results.130–133
Macromolecular optical probes are better equipped to handle the complexity of the TME. They take advantage of the enhanced permeability and retention (EPR) effect—leaky tumor vessels and poor drainage let macromolecules build up passively in tumor tissue.134,135 But for probes aimed at TIICs, passive accumulation alone rarely works.133,134 The EPR effect helps enrich probes in tumor but offers no molecular recognition, so it can't pick out specific immune cell types. That leaves key problems unsolved: deep tumor penetration, precise uptake by target cells, and stable intracellular retention.136,137 That's why we still need the active targeting mechanisms discussed earlier—such as sensing TIIC-specific enzymes or reactive species—to get accurate, efficient imaging. Passive and active targeting actually work hand in hand. Passive accumulation boosts probe levels in the TME, while active targeting adds cell-type specificity, together extending probe retention where it matters.138,139 A thoughtful blend of both strategies is therefore essential for building high-performance TIIC-targeted optical probes.140–148
3.2. Probes labelled by TIIC-specific membrane proteins
In addition to metabolites reflective of TIICs’ biological activity that serve as the foundation for enzyme/reactive species-activated optical probe design, a range of TIIC-expressed proteins, such as receptors, glycoproteins and others involved in cell recruitment and functional regulation, also provide rational targets for the development of receptor-binding labelled optical probes (Fig. 4B). These optical probes typically employ conventional imaging units, such as FL dyes, FL proteins, commercially available fluorophores and others, while their recognition modules consist of ligands capable of binding specific membrane receptors expressed on TIICs, including polysaccharides, antibodies, nucleic acid aptamers and others. Representative examples include mannose targeting the M2-type TAM surface receptor CD206 and IgG antibodies directed against the TAN surface receptor Ly6G. Accordingly, CD206 and Ly6G have emerged as widely utilized targets for receptor-based probe design. Beyond these classical markers, chemokine receptors (e.g., CXCR5 and CXCR3) that regulate TIIC recruitment provide an additional strategy for probe development. These receptors are broadly expressed across distinct TIIC subsets and exhibit subpopulation-specific patterns. Therefore, rational selection of membrane receptors according to the intended application enables the development of highly selective probes capable of elucidating the biological roles of specific TIIC subpopulations within the TME. Notably, although these receptors are membrane-localized, many undergo receptor-mediated endocytosis, facilitating intracellular probe uptake and signal amplification. Moreover, their expression is dynamically regulated and functionally linked to immune activation or immunosuppression within the TME, rendering receptor-targeted probes particularly advantageous for evaluating immune status and TIIC interactions. Another noteworthy development is the rapid advancement of biotechnology in recent years, which has provided strong support for TIIC-targeted optical probes. In particular, gene cloning techniques and aptamer screening platforms—most notably systematic evolution of ligands by exponential enrichment (SELEX)—enable the identification of high-affinity nucleic acid aptamers, including ssDNA aptamers, RNA aptamers, Spiegelmers and others. These technologies significantly enhance optical probe's recognition specificity and targeting precision toward defined TIIC subpopulations, promoting the refinement and diversification of receptor-binding labelled optical probes.3.3. Immunotherapy integration of TIIC-targeted optical probes
While the targeting and response mechanisms described in Sections 3.1 and 3.2 primarily focus on imaging TIICs, the incorporation of therapeutic units introduces an additional interventional layer of functionality to TIIC-targeted optical probes. The synchronization of diagnosis and therapy—often conceptualized as “see and treat”—represents a significant paradigm shift in cancer management. Accordingly, beyond their core sensing mechanisms, the integration of therapeutic functionalities has transformed TIIC-targeted optical probes from purely diagnostic tools into multifunctional theranostic platforms, constituting a rapidly expanding frontier in probe design. For example, multifunctional optical probes enriched at tumor sites can mediate the activation of the body's anti-tumor immune response directly or indirectly (stimulated by TAAs and DAMPs) through localized and precise delivery of chemotherapy, ICIs, immune signalling pathway modulators, etc., significantly improving the shortcomings of traditional systemic drug administration, such as large side effects, low efficiency, and untimely prediction/monitoring of efficacy, thereby achieving spatiotemporal control of TME immune status.149–153 As of now, strategies for integrating therapeutic units include (1) incorporating therapeutic agents into nanoparticles alongside optical probes; (2) formulating them as prodrugs and co-administering them with optical probes to enhance therapeutic efficacy; and (3) utilizing optical probes as carriers to load and transport therapeutic agents directly to the TME.154–156 Notably, the rational selection and integration of therapeutic modules must be carefully tailored according to the clinical context, including treatment regimen, targeted immune cell subtype, and potential synergistic or antagonistic interactions with imaging functions. For instance, optical probes targeting tumor cells or programmed death-ligand 1 (PD-L1) expressed on T cells can be combined with immune checkpoint blockade agents to achieve simultaneous visualization of target engagement and localized immunotherapeutic delivery. Such integrated diagnostic–therapeutic systems not only enable image-guided intervention but also permit real-time assessment of therapeutic response, thereby advancing the development of personalized cancer immunotherapy.4. Applications of optical probes in TIIC detection and imaging
Optical probes constructed using the design principles outlined above are now widely employed for the detection and imaging of TIICs. These probes typically target TIIC-associated metabolites (e.g., enzymes and reactive species), specialized structures such as neutrophil extracellular traps (NETs), and other relevant biomolecules (e.g., surface receptors), and they commonly exhibit turn-on or always-on FL signals (Table 2). Each class of probe is discussed in detail in the following sections.| Number | Name | Imaging mode | Target molecule | Cell type | Application level | Application model | Ref. |
|---|---|---|---|---|---|---|---|
| 01 | NEP | FL | NE | — | Cell and animal | Acute lung injury model (mouse) | 162 |
| 02 | NanoNEP | FL | NE | — | Cell and animal | Diabetes model (mouse) | 163 |
| 03 | LET-8 | FL/PA | NE | — | Cell and animal | A549 cancer model (mouse; subcutaneous) | 164 |
| 04 | NEP3 | FL/PA | NE | — | Cell and animal | Acute inflammatory injury model (mouse) | 165 |
| 05 | Cou-HN | NE/ROS (HClO) | — | Cell and animal | Osteoarthritis model (mouse and zebrafish) | 166 | |
| 06 | SPCy | FL/PA | NE | TANs | Cell (contains immune cells extracted from mice) and animal | 4T1 cancer model (mouse; subcutaneous) | 167 |
| 07 | TAMPs (TAMPM1/TAMPCTL/TAMPNE) | FL | Cas-1/GrB/NE | TAMs/CTLs/TANs | Cell (contains immune cells extracted from mice) and animal | 4T1 and CT26 cancer model (mouse; subcutaneous) | 168 |
| 08 | FAR | FL/afterglow | Cas-3/RNS (NO) | TAMs (M1) | Cell and animal | MC38, CT26, and 4T1 cancer model (mouse; subcutaneous) | 169 |
| 09 | Lu-bCD NPs | FL | MPO/ROS | Neutrophils | Cell (contains immune cells extracted from mice) and animal | Peritonitis model (mouse) | 171 |
| 10 | BRET-FRET NBs | BL/UL | MPO | Neutrophils/monocytes/macrophages | Cell (contains immune cells extracted from rabbit) and animal | Peritonitis and 4T1 cancer model (mouse; subcutaneous) | 172 |
| 11 | LAD-PGP NPs | CL | MPO | TANs | Cell and animal | 4T1 cancer model (mouse; lung metastasis) | 173 |
| 12 | heMAMP | MR | MPO | Neutrophils/macrophages (M1) | Animal | Subcutaneous inflammation and atherosclerosis model (mouse) | 174 |
| 13 | MABS | FL/CT | MPO | Neutrophils/monocytes/macrophages | Animal | Inflammation model (mouse; feet and brain) | 175 |
| 14 | 5HFeC NPs | FL/MP/CTA | MPO | Macrophages | Animal | Atherosclerosis model (mouse) | 176 |
| 15 | MP/NPs-SLIPS | FL | MMP-2 | — | — | — | 180 |
| 16 | Probe 0 | Multicolor FL | MMP-2/MMP-7/MMP-9 | — | Cell | — | 181 |
| 17 | PMPSD | MR | MMP-9 | — | Cell and animal | 4T1 cancer model (mouse; subcutaneous) | 182 |
| 18 | MMP2cNPs | PET/MR | MMP-2 | Macrophages | Animal | Atherosclerosis model (mouse; neck) | 183 |
| 19 | [18F]11 | FL/PET | Broad spectrum (mainly MMP-12 and MMP-13) | — | Animal | Dermatitis model (mouse; ear) | 184 |
| 20 | PEG-PepMMP2-MNP-Gd | PA/MR | MMP-2 | — | Animal | 4T1 cancer model (mouse; subcutaneous) | 185 |
| 21 | [89Zr]Zr-DFO-anti-MT1-MMP-BOD665 | FL | MMP-14 | — | — | HT1080 cancer model (mouse, in situ) | 186 |
| 22 | MP-CB-2 | FL | CTSB | — | Cell and clinical samples (non-small cell lung cancer) | — | 190 |
| 23 | Pep SQ@USPIO | FL/MR | CTSB | — | Cell and animal | MDA-MB-231 and MCF-7 cancer model (mouse; subcutaneous) | 191 |
| 24 | HCy-Cit-Val /HCy-Gly-Leu-Phe-Gly | FL/PA | CTSB | — | Cell and animal | HeLa cancer model (mouse; subcutaneous) | 192 |
| 25 | OFS-1 | FL | CTSK | — | Cell and animal | RPMI-8226 cancer model (mouse; left femur, pelvis and lower lumbar vertebrae) | 197 |
| 26 | MP-cL3 | FL | CTSL | — | Cell | — | 198 |
| 27 | Non-lipidated probe 2/lipidated probe 3 | FL | CTSS | Macrophages | Cell and animal | 4T1 cancer model (mouse) | 199 |
| 28 | MORs | CL/FL | CTSS/iP | DCs | Cell (contains immune cells extracted from mice) and animal | 4T1 cancer model (mouse; subcutaneous) | 200 |
| 29 | SK15.5 | FL | GrA | NK92 cells | Cell | — | 203 |
| 30 | GzmA-CL | CL | GrA | T cell | Clinical samples (inflammatory bowel disease) | — | 204 |
| 31 | Probe 1 | FL | GrB | NK cells | Cell and animal | MDA-MB-231 cancer model (mouse; subcutaneous) | 205 |
| 32 | CyGbPF/CyGbPP | FL | GrB | CTLs/CD4+ T cells | Cell and animal | 4T1 cancer model (mouse; subcutaneous) | 206 |
| 33 | SPNP | FL/PA | GrB | T cells | Cell and animal | 4T1 cancer model (mouse; subcutaneous) | 207 |
| 34 | DRET | FL/MR | GrB/Cas-3 | CTLs | Cell and animal | CT26 cancer model (mouse; subcutaneous) | 208 |
| 35 | Cy5.5-CBT-NPs | FL | GrB | CTLs | Cell and animal | B16-OVA cancer model (mouse; subcutaneous) | 209 |
| 36 | TAD-BHQ | Afterglow | GrB | CTLs/NK cells | Cell and animal | CT-26, U87 and Pan02 cancer model (mouse; subcutaneous or in situ) | 210 |
| 37 | ncRuPA | Sonoafterglow | APN (ncRuPAAPN) | — | Cell and animal | 4T1 cancer model (mouse; subcutaneous) | 211 |
| 38 | PSiR | FL | hROS | — | Cell and animal | HeLa cancer model (mouse; subcutaneous) | 213 |
| 39 | Probe 4 | FL | MMP-2/ROS (H2O2) | — | Cell | — | 214 |
| 40 | BTPE-NO2@F127 | FL/PA | ROS (H2O2) | — | Cell and animal | Interstitial cystitis, liver injury and ischemia-reperfusion injury model (mouse) | 215 |
| 41 | MPN@CeOx | MR/CT | ROS (H2O2) | — | Cell and animal | Acute colitis model (mouse) | 216 |
| 42 | PN910 | FL | RNS (ONOO−)/ROS (H2O2) | — | Animal | Colitis and cystitis model (mouse) | 218 |
| 43 | NTG | FL | RNS (ONOO−)/RSS (GSH) | — | Cell and animal | Inflammation model (zebrafish) | 219 |
| 44 | NRP@M-PHCQ | FL | RNS (NO) | TAMs | Cell and animal | 4T1 cancer model (mouse; subcutaneous, lung and lymph node metastasis) | 220 |
| 45 | NRMP | MR | RNS (NO) | TAMs | Animal | 4T1 cancer model (mouse; subcutaneous) | 221 |
| 46 | FHMI | FL | RSS (SO2) | — | Cell and animal | Exogenous SO2 elevation models (zebrafish) | 226 |
| 47 | MI-H2S | FL | RSS (H2S) | — | Cell and animal | Exogenous H2S elevation (zebrafish and mouse) and inflammation (mouse, subcutaneous and peritoneal) models | 227 |
| 48 | Lyso-RC | FL | RSS (H2S/Cys/Hcy, GSH) | — | Cell | — | 228 |
| 49 | HNE-FQ | FL | NE | NETs (from neutrophils) | Cell | — | 231 |
| 50 | HNE-3F1Q | FL | NE | NETs (from neutrophils) | Cell | — | 232 |
| 51 | TNR1/TNR2 | FL | NE/CTSG | NETs (NETosis process, from neutrophils) | Cell (contains immune cells extracted from mice) and animal | 4T1 cancer model (mouse; subcutaneous) | 233 |
| 52 | H-NE/H-CG | FL | NE/CTSG | NETs (from neutrophils) | Clinical samples (pulmonary cystic fibrosis) and animal | Pulmonary cystic fibrosis model (mouse) | 234 |
| 53 | CDr15 (commercially available DNA dyes) | FL | DNA | NETs (from neutrophils) | Cell and clinical samples (lung, esophagus, stomach, pancreas, breast, urinary bladder, kidney, thyroid, ovary, prostate. colorectal and liver cancer) | — | 235 |
| 54 | TPE-Man | FL | CD206 | TAMs | Cell (immune cells extracted from mice) and tumor tissue section | — | 236 |
| 55 | DN-ICG | FL | SIGNR1 | TAMs | Cell (contains immune cells extracted from mice) and animal | SW1990 cancer models (mouse; subcutaneous and in situ) | 237 |
| 56 | Ly6G NPs | FL/MP/CT | Ly6G | TANs | Animal | Abdominal aortic aneurysms (mouse; in situ) | 238 |
| 57 | DCPD7-Ly6G | FL | Ly6G | Neutrophils | Cell and animal | Stroke model (mouse, in situ) | 239 |
| 58 | BODIPY-aa-tRNA | FL | ST2 receptors | Mast cells/innate lymphocytes/Tregs | Cell | — | 240 |
| 59 | Probe 5 | FL | CXCR5/CXCR3 | BCs | Cell | — | 241 |
| 60 | Probe 6 | FL | Fc receptors | Monocytes/granulocytes | Animal | FaDu and A-431 cancer models (mouse; subcutaneous) | 242 |
| 61 | AIE-Apt-99 | FL | S100A8&A9 heterodimer | TANs | Cell (TANs derived from clinical samples of ascites from ovarian cancer patients) | — | 243 |
| 62 | Gd@BSA-BODIPY | FL/MR | Self-targeting | Neutrophils | Animal | LLC cancer model (mouse; subcutaneous) | 245 |
| 63 | GCZM | FL | Self-targeting | Neutrophils | Cell and animal | 4T1 cancer model (mouse; subcutaneous and lung metastasis) | 246 |
| 64 | PAN | FL | Self-targeting | Neutrophils | Cell and animal | B16F10 and oral cancer models (mouse; subcutaneous and in situ) | 247 |
| 66 | TFML-NE | PA | Self-targeting | TANs | Animal | U87 cancer models (mouse; in situ) | 248 |
| 67 | DCNP@786s | FL | Self-targeting | NK cells | Animal | HCC cancer models (mouse; in situ) | 249 |
| 68 | aNeu | FL/MR | Self-targeting | Neutrophils | Cell and animal | E0771 cancer models (mouse; subcutaneous) | 250 |
| 69 | Az-ROS@MB/CSSC@DBCO | FL | Self-targeting | Cancer cell | Cell and animal | 4T1 cancer model (mouse; subcutaneous) | 251 |
4.1. Probes based on enzymes
Enzymes are essential biomolecules that regulate physiological processes. They participate in biochemical reactions and regulate cell growth and metabolic processes. Under physiological conditions, enzyme activity and expression remain relatively stable; however, during disease or tissue damage, the expression and activity of certain enzymes undergo abnormal changes, creating opportunities for their utilization in optical probe development. In TIICs, key enzymes frequently involved in TIIC-targeted optical probe development include NE in TANs, MPO and MMPs in both TANs and TAMs, and Grs in T cells and NK cells.157–159Optical probes such as FL probes, which are functionalized derivatives of fundamental fluorescent dyes, are widely used in the detection and imaging of biomacromolecules, particularly enzymes with essential biological functions such as NE. In 2019, Liu et al.162 developed an optical probe (NEP) for detecting NE (Fig. 5A). NEP exhibits exceptional specificity and sensitivity and enables real-time monitoring of NE transport, exogenous NE uptake, and endogenous NE upregulation at the cellular level with high spatial resolution. Building on this work, Yang et al.163 conjugated another NE probe to bovine serum albumin (BSA) to enhance the biocompatibility, solubility, and diffusivity of the optical probe, thereby constructing a nanostructured optical probe termed NanoNEP (Fig. 5B). This probe was successfully applied to wound and serum detection in a diabetic mouse model. Notably, in contrast to the single-modality FL imaging employed in NEP, NIR fluorescence (NIRF)/PA dual-modal imaging significantly enhances imaging depth and signal-to-noise ratio. Zhang et al.164 synthesized an optical probe (LET-8) structurally similar to NEP for in vivo imaging of lung cancer (Fig. 5C). They found that, upon interaction of the newly synthesized LET-8 with NE, both the absorption and emission wavelengths of FL were red-shifted, leading to a 20-fold enhancement in NIRF intensity at 725 nm and a 10.5-fold increase in PA signal intensity at 700 nm, thus enabling in vivo NIRF/PA dual-modal imaging of lung cancer. The output of multiple optical signals facilitates cross-validation of imaging results, offering more accurate diagnostic information. A similar probe is the NIRF/PA dual-mode imaging optical probe (NEP3) reported by Huang et al.165 (Fig. 5D). To further enhance biocompatibility, a triethylene glycol group was incorporated, and, owing to the NE recognition site, NEP3 exhibited exceptional specificity and sensitivity in both cell and animal studies. Notably, tumors typically exhibit a pronounced inflammatory response, and therefore, in addition to elevated NE expression, multiple inflammation-associated biomarkers are commonly present within tumor sites. To further increase specificity in complex inflammatory milieus, Deng et al.166 proposed a “double-lock” strategy with the probe Cou-HN, which requires sequential activation by NE and HClO. (Fig. 5E). This strategy effectively minimizes false-positive signals associated with single-responder probes and offers a robust design paradigm for the development of highly specific NE-responsive optical probes.
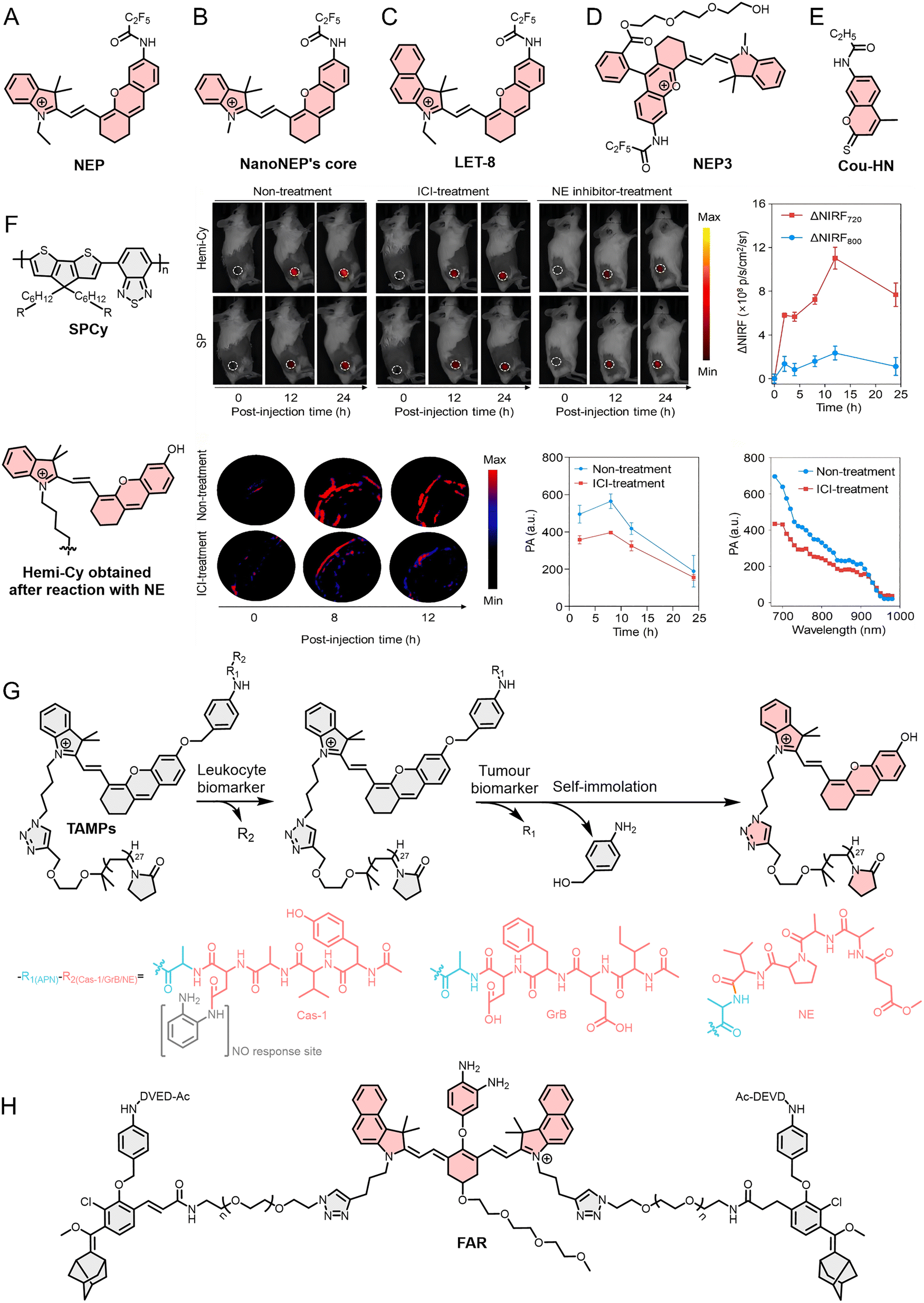 | ||
| Fig. 5 (A)–(E), (G) and (H) Molecular structure of NEP, NanoNEP's core, LET-8 NEP3, Cou-HN, TAMPs, and FAR. (F) The molecular structure of SPCy, and hemi-Cy formed after its reaction with NE, and the application of SPCy in monitoring immunotherapy in a tumor animal model via NIRF and PA imaging. Reproduced from ref. 167 with permission from John Wiley and Sons, Copyright 2022. | ||
Beyond small-molecule scaffolds, semiconducting polymers (SPs) have emerged as versatile platforms for constructing stable and multifunctional NE probes. Their excellent processability and structural flexibility further facilitate the integration of dyes, targeting ligands, and responsive elements, making SPs particularly well suited for the development of multifunctional TIIC-targeted optical probes. For example, Zhang et al.167 used SPs to develop an NIRF/PA dual-mode optical probe (SPCy) for detecting TANs (Fig. 5F). Distinct from previous probes, the SP core of SPCy exhibits exceptional stability and does not react with analytes or other species. Therefore, its NIRF and PA signals remain stable regardless of SPCy's reaction with NE, allowing it to serve as an internal reference for calibrating the NIRF and PA signals, thereby enhancing the accuracy of conventional optical probe imaging. This property enables SPCy to perform ratiometric NIRF/PA dual-mode imaging of TANs. In tumor-bearing animal model experiments, SPCy exhibited significantly reduced NIRF and PA signals in both ICI and NE inhibitor treatment groups, indicating that SPCy can specifically recognize and respond to NE in vivo, and ICI treatment significantly reduced TAN infiltration within tumor tissue. The experimental results showed that SPCy enabled real-time tracking of immunotherapy efficacy by monitoring dynamic changes in TAN populations within the TME.
In addition to optical probes with multiple imaging modes, optical probes capable of simultaneously monitoring multiple TIICs offer distinct advantages in evaluating tumor immune status. For example, He et al.168 reported in 2021 the development of an optical probe (TAMPs) specifically activated by tumor-infiltrating leukocytes (TILs) (Fig. 5G). They modified the probe's R2 group (with caspase-1 [cas-1] peptide substrate YVAD, GrB peptide substrate IEFD, and NE peptide substrate AAPV) to enable specific detection of different immune cells (TAMs, CTLs, and TANs). They designed three TAMPs—TAMPM1, TAMPCTL, and TAMPNE—to image M1 TAMs, CTLs, and TANs, respectively. Importantly, they also introduced the recognition site (R1, CyA) for a broad-spectrum tumor biomarker, aminopeptidase N (APN). The double verification mechanism prevents nonspecific activation of TAMPs in normal or inflammatory tissues, ensuring the specific detection of TIICs by TAMPs.
In addition, cas-3, another central executioner caspase in the apoptotic cascade, is a key factor for assessing tumor immune status. For example, Li et al.169 developed FAR, a dual-channel probe that integrates an NO-responsive NIRF module and a cas-3-responsive afterglow module (Fig. 5H). By interpreting the combined signal patterns (NIRF on/off and afterglow on/off), FAR can non-invasively classify tumor into three immune phenotypes: immune-desert (both signals off), immune-excluded (only NIRF on), and inflamed (both signals on). This approach translates complex tumor-immune interactions into straightforward optical readouts, providing a valuable tool for predicting immunotherapy responsiveness and guiding treatment decisions. In summary, NE-targeted optical probes have evolved from simple FL sensors to sophisticated multimodal, ratiometric, and logic-gated systems. This section has focused on NE as a neutrophil-specific enzyme; the relationship between NE and NETs will be discussed subsequently. These advances not only improve the accuracy and depth of TAN imaging but also enable real-time assessment of immune cell dynamics and therapy efficacy, offering powerful tools for both basic research and translational oncology.
Initial efforts in MPO probe development focused on improving sensitivity and biocompatibility. This focus advanced significantly in 2017 when Guo et al.171 reported Lu-bCD, a cyclodextrin-luminol-based probe capable of real-time imaging of inflammation in animal models (Fig. 6A). This probe responds specifically to MPO activity while maintaining excellent biocompatibility. Its optical signal intensity correlates positively with neutrophil abundance, allowing precise tracking of neutrophil dynamics in both superficial and deep tissues across multiple models of inflammatory disease, including peritonitis, paw edema, colitis, and acute lung injury. Moreover, Lu-bCD self-assembles into nanoparticles (Lu-bCD NPs) with an average diameter of 228 ± 19 nm, thereby addressing the limitations commonly associated with small-molecule probes—such as rapid absorption, fast clearance, and signal decay—and supporting prolonged, high-contrast imaging. Lu-bCD reacts with MPO and ROS in vivo, leading to enhanced FL output reflecting the abundance of neutrophils.
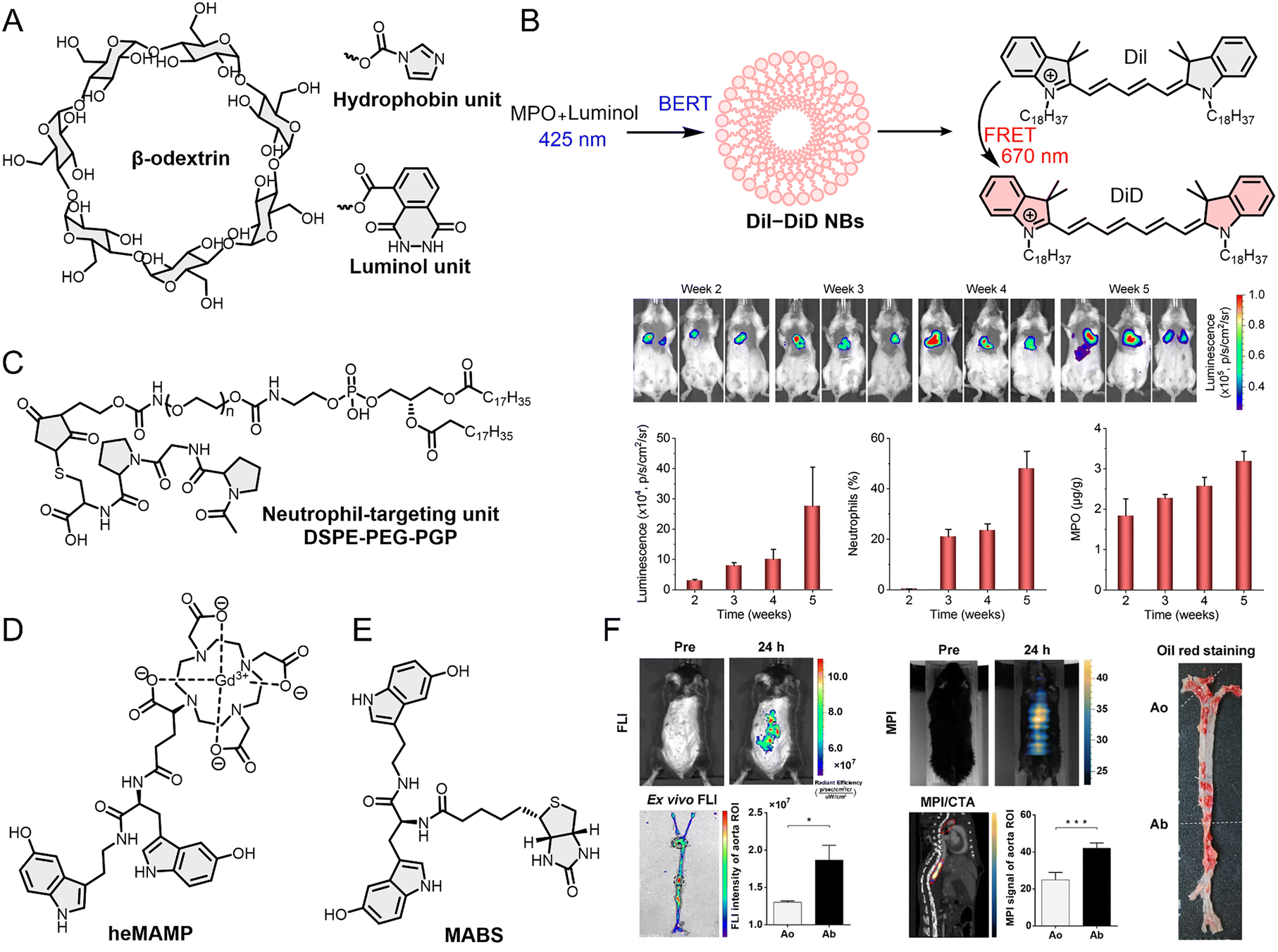 | ||
| Fig. 6 (A) Molecular structures of organic materials used to construct three optical probes, Lu-bCD, BRET-FRET NPs and LAD-PGP NPs. (B) BRET and FRET processes of BRET-FRET NBs. (C) Molecular structure of neutrophil targeting units and application of LAD-PGP NPs in the 4T1 breast cancer lung metastasis animal model. (D) Molecular structure of heMAMP. (E) Molecular structure of MABS. (F) 5HFeC NPs performed FL/MP/CTA imaging of vulnerable plaques in the abdominal aorta of an atherosclerotic mouse model via MPO. Reproduced from ref. 173 and 176 with permission from Springer Nature, Copyright 2022 and Ivyspring International, Copyright 2021, respectively. | ||
Optical probes combined with clinical bioimaging hold greater potential for clinical translation. For instance, Liu et al.172 developed a BL/ultrasound (UL) dual-mode imaging optical probe (BRET-FRET NBs) using BL resonance energy transfer (BRET) and FRET mechanisms to detect MPO activity (Fig. 6B). Luminol reacts with the peroxide generated by MPO and induces NIRF signals at 670 nm via the FRET process. This strategy leverages the FRET process to convert luminol's reaction with MPO-generated peroxide into a 670 nm NIRF signal, thereby circumventing the tissue-penetration issues associated with luminol's shorter-wavelength emission. Subsequent experiments demonstrated that the NIRF signal intensity correlates strongly with MPO activity. Simultaneously, the BRET-FRET NBs exhibit outstanding UL contrast properties, clearly revealing tissue microvascular structures. Therefore, combined analysis of BL and UL imaging results enables more accurate assessment of inflammatory responses. Compared to Lu-bCD, the introduction of UL enables BRET-FRET NBs to image deeper tissues; combined with the high resolution of BL imaging, BRET-FRET NBs are capable of targeting TANs as well as accurately identifying monocytes and TAMs in animal tissue. Chemiluminescence resonance energy transfer (CRET) has also been utilized in the development of TIIC-targeted optical probes. For example, Zheng et al.173 reported an optical probe (LAD-PGP NPs) that quantifies the number of TANs and MPO activity (Fig. 6C). LAD-PGP NPs clearly showed FL signals at the lung metastasis site in the 4T1 breast cancer lung metastasis animal model, with signal intensity correlating with TAN count and MPO activity, suggesting a strong correlation between TAN infiltration and tumor cell numbers during lung metastasis. In parallel to its use in emerging optical technologies, MPO is also being harnessed to refine established clinical imaging methods. The enzyme's selective expression in neutrophils and macrophages makes it an ideal target for probes designed to bridge the gap between molecular imaging and clinical translation, offering a route to enhance the diagnostic power of traditional modalities. For example, heMAMP, reported by Wang et al.,174 leveraged MPO to enhance traditional MR imaging (Fig. 6D). When MPO reacts with 5-hydroxyindole in heMAMP and forms free radicals, it undergoes self-polymerization or polymerization to enhance the MR signal. The free radicals can also covalently bind to nearby proteins, further enhancing the signal and enabling high-contrast long-term imaging. They further developed a FL/CT dual-modal imaging probe based on MPO to achieve the integration of emerging optical bioimaging and traditional bioimaging (Fig. 6E).175 This probe consists of a biotin sensor (MABS) that can be activated by MPO and a secondary component. The secondary probe is composed of FL materials and gold nanoparticles, providing both FL and CT imaging capabilities. The modular design enhances the probe's versatility in applications. MABS can be combined with different secondary probes to enable the FL or CT imaging of MPO. Other optical probes capable of integrating with traditional biological imaging include 5-HT-Fe3O4-Cy7 (5HFeC NPs) reported by Tong et al.176 5HFeC NPs achieve the integration of magnetic particle imaging (MPI), FL imaging, and computed tomographic angiography (CTA) for quad-modality imaging (Fig. 6F). These probes integrated with traditional biological imaging hold significant potential for clinical translation.
The above examples demonstrate that MPO-targeting optical probes can directly report the activity of TIICs, particularly through the biochemical process of MPO-catalyzed HClO generation, and thus serve as effective tools for noninvasively assessing tumor immune status. And emerging evidence suggests that MPO-targeting multimodal optical probes are increasingly integrated with clinical bioimaging modalities, thereby conferring enhanced potential for clinical translation.
Pushing the limits of sensitivity for early detection, Wu et al.180 developed MP/NPs-SLIPS, an innovative probe system designed for the ultra-sensitive quantification of MMP-2 (Fig. 7A). MP/NPs-SLIPS achieves precise modulation of the aggregation state of aggregation-induced emission (AIE) moieties via the synergistic interplay between electrostatic adsorption and nanoconfinement, thereby markedly enhancing FL signal intensity. This “two-step aggregation enhancement” strategy lowers the detection limit from 46.8 ng mL−1 (conventional solution-based assay) to 3.7 ng mL−1, representing an approximately 12.6-fold enhancement in sensitivity and thereby enabling the detection of low-abundance MMP-2found in early tumors at a concentration of approximately 5 ng mL−1, enabling sensitive-enough imaging of MMP-2 in early-stage tumors. This offers a solution to the challenge of detecting TIIC-related biomarkers with extremely low abundance in early tumors using conventional detection methods, potentially improving early cancer diagnosis rates.
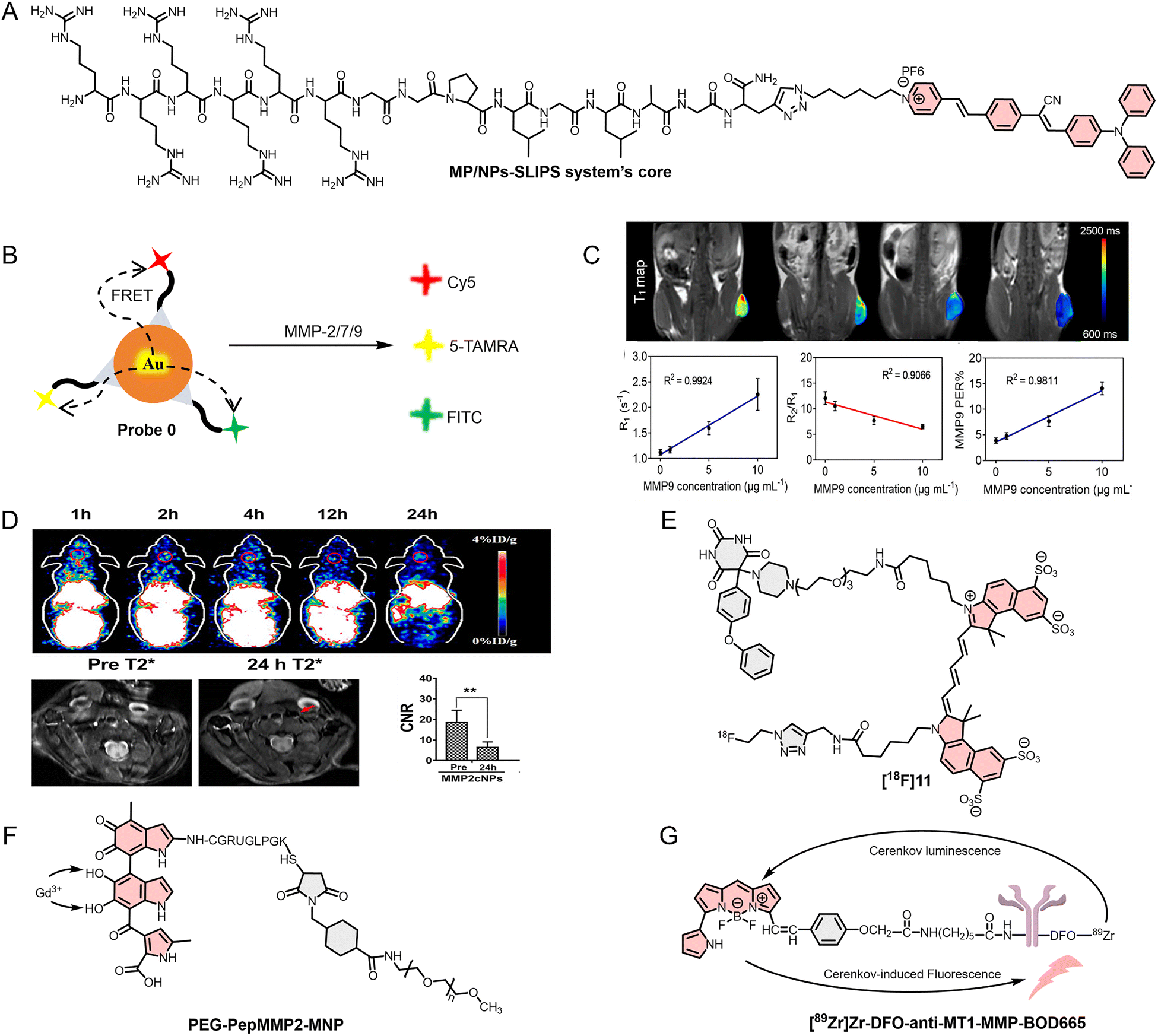 | ||
| Fig. 7 (A) and (E)–(G) Molecular structures of the MP/NPs-SLIPS system's core, [18F]11, PEG-PepMMP2-MNP and [89Zr]Zr-DFO-anti-MT1-MMP-BOD665. (B) Construction principle of the tricolor optical probe (Probe 0) based on Au–Se bonds. (C) Application of PMPSD in a tumor mouse model in vivo (quantification of MMP-9 in tumors). (D) Application of MMP2cNPs in an animal model of carotid atherosclerosis. Reproduced from ref. 182 and 183 with permission from Elsevier, Copyright 2022 and DOVE Medical Press, Copyright 2022, respectively. | ||
Accurately distinguishing different types of MMPs helps to clarify the relationship between MMPs and TIICs, and a multicolor imaging strategy that links various imaging and signal conversion tags to a metal material scaffold provides an ideal approach to achieve this. Guo et al.181 substituted the gold–thiol (Au–S) bond with a gold–selenium (Au–Se) bond to create a tricolor FL probe (Probe 0) that can simultaneously respond to MMP-2, MMP-7, and MMP-9 (Fig. 7B). This probe not only achieves the simultaneous detection of MMP-2, MMP-7, and MMP-9, but also addresses the issue of Au–S bond instability caused by GSH in the body, which can lead to false positive signals in Au–S-based FL probes. The surface of gold nanoparticles is conjugated with MMP-2 peptide substrates (labelled with FITC), MMP-7 (labelled with 5-TAMRA), and MMP-9 (labelled with Cy5). Upon encountering the target enzymes, it specifically cleaves the respective peptide substrates, releasing the FL groups from the gold nanoparticle surface and restoring the previously quenched FL via the FRET mechanism, resulting in clear tricolor imaging, which facilitates cancer imaging and classification.
Using MMPs to enhance the accuracy of clinical bioimaging and broaden its range of applications is an effective and feasible approach. For example, Chen et al.182 developed an MMP-9 responsive MRI nanoplatform (PMP@USPIO/DOX, PMPSD) for quantitative tumor bioimaging and for synergistic chemo-photothermal therapy (Fig. 7C). Furthermore, Tu et al.183 also used 1,4,7-triazacyclononane-1,4,7-triacetic acid (NOTA) derivatives for nuclear tracer 64Cu labeling, and incorporated MMP-2 peptide substrate-modified polyethylene glycol 2000 into IONP to create a dual-mode imaging optical probe (64Cu-NOTA-IONP@MMP2c-PEG2K, MMP2cNPs) for PET/MR imaging (Fig. 7D). Compared with PMPSD, the MMP2cNPs simultaneously utilized the advantages of PET and MR imaging, allowing MMP2cNPs to better address the challenges of the in vivo environment. Clinical bioimaging modalities such as MRI and PET provide valuable anatomical and metabolic information about tumors; however, they are limited by spatial resolution and, in certain contexts, insufficient sensitivity to fine structural details. Consequently, integration with optical imaging modalities such as FL and PA imaging can effectively compensate for these limitations, enabling the acquisition of multidimensional and complementary tumor information. For example, in 2020, Schwegmann et al.184 developed a novel FL/PET dual-mode imaging probe ([18F]11) for MMP detection by radiolabeling Cy5-barbituric acid conjugates (Fig. 7E). Similarly, in 2022, Meng et al.185 reported a PA/MR dual-mode optical probe (PEG-PepMMP2-MNP-Gd) (Fig. 7F). These ingeniously designed multimodal imaging probes serve as valuable references for the development of new TIIC-targeted optical probes in the future.
Most existing optical probes require external light sources for excitation, which leads to two major limitations: scattering and attenuation of the excitation light within biological tissues, as well as the introduction of additional background signals, ultimately resulting in suboptimal imaging contrast. To address these challenges, Gristwood et al.186 introduced a self-activating imaging strategy in 2026. In this approach, 89Zr and BODIPY665 were conjugated through an anti-MT1-MMP antibody—an MMP-14 specific antibody—to construct a self-activating optical probe ([89Zr]Zr-DFO-anti-MT1-MMP-BOD665), for in situ imaging of osteosarcoma mouse models (Fig. 7G). Because MMP-14 is highly expressed in osteosarcoma, it specifically binds to the anti-MT1-MMP moiety, leading to preferential probe accumulation at the tumor site. The radioactive decay of 89Zr within the probe generates Cherenkov blue FL, which subsequently excites BODIPY665, thereby inducing NIRF emission at approximately 665 nm. Furthermore, owing to the incorporation of both 89Zr and BODIPY665, this probe also exhibits PET imaging capability, enabling in situ FL/PET dual-modal imaging in osteosarcoma mouse models.
CTSB is a key focus in the development of TIIC-targeted optical probes. For example, Poreba et al.190 used hybrid combinatorial substrate library (HyCoSuL) technology (a technique combining combinatorial chemistry and biochemistry, which enables the efficient screening of a large number of substrate molecules and the determination of a protease's amino acid preferences) to rapidly screen a large array of non-natural amino acid combinations to determine the optimal reaction substrate for CTSB, and based on this, utilized the Cy5-labeled MP-CB-2 probe for subcellular localization of CTSB (Fig. 8A). Furthermore, the FL/MR dual-mode imaging probe (Pep SQ@USPIO) reported by Wang et al.191 in 2020 enabled imaging triple-negative breast cancer in vivo (Fig. 8B). The superparamagnetic iron oxide nanoparticles (USPIO) in the probe provide MRI, while the rectangular hydrazone photosensitizer (SQ) enables FL imaging after cleavage by CTSB. Chen et al.192 also reported two FL/PA dual-mode imaging probes (HCy-Cit-Val and HCy-Gly-Leu-Phe-Gly), for real-time monitoring of CTSB activity in vivo (Fig. 8C). The two peptide sequences in the probe undergo specific enzymatic cleavage by CTSB, resulting in signal conversion, thereby enabling the visualization of CTSB. Although both optical probes can detect and image endogenous CTSB in vivo, HCy-Cit-Val exhibits greater catalytic efficiency and imaging contrast, attributable to its reduced steric hindrance, as demonstrated in cellular studies and tumor-bearing animal model imaging experiments.
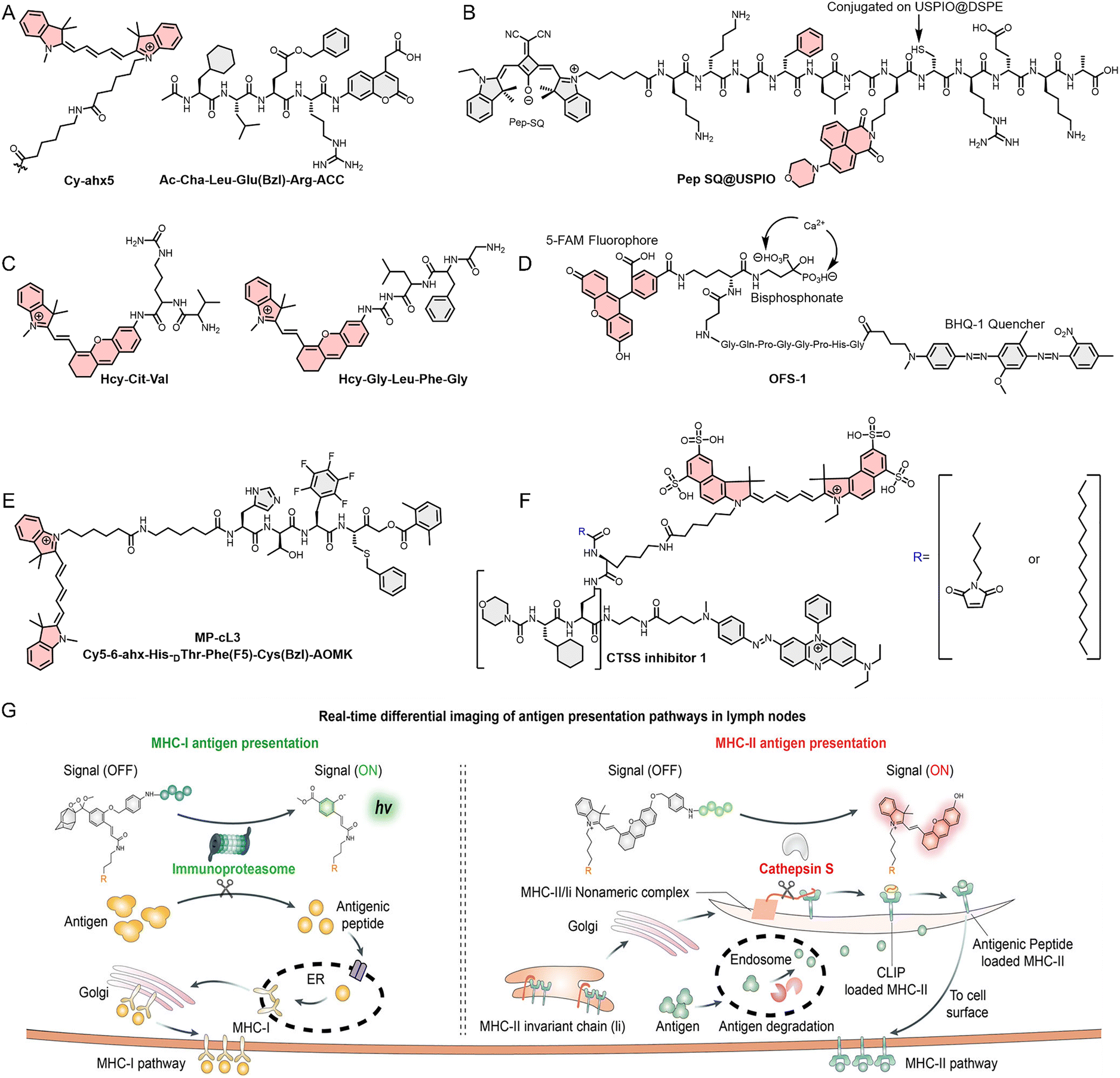 | ||
| Fig. 8 (A) Molecular structures of the optimal substrates and Cy5 derivatives for CTSB screened using HyCoSuL technology. (B)–(F) Molecular structure of Pep-SQ, HCy-Cit-Val, HCy-Gly-Leu-Phe-Gly OSF-1, MP-cL3, and non-lipidated probe 2 or lipidated probe 3. (G) The mechanism by which MORs distinguish and predict the intensity of different immune response types (MHC-I or MHC-II). Reproduced from ref. 200 with permission from John Wiley and Sons, Copyright 2025. | ||
In addition to CTSB, several other CTS that are highly expressed in the TME can likewise serve as targets for the construction of optical probes. Their elevated expression in the TME is closely associated with tumor growth and metastasis; particularly CTSG, as a key enzyme in NETs,193–196 has been utilized for the development of optical probes for NET imaging, and will be discussed in detail in the NET imaging section later. For CTSK, the optical probe (OFS-1) reported by Richard et al.197 was employed for in vivo imaging of multiple myeloma mouse models (Fig. 8D). The high affinity of the bisphosphonate group in OFS-1 with bone minerals, such as hydroxyapatite, directs the probe to specifically adsorb on the bone surface, where CTSK cleaves the peptide substrate (GHPGGPQG), leading to the separation of the FRET donor fluorescein (FAM) from the FRET acceptor (BHQ-1), thus activating the FL signal at 520 nm. Furthermore, Poreba et al.198 used an optical probe (MP-cL3) for imaging CTSL. Similar to their work in 2019, the tetrapeptide substrate (MP-cL3, Ac-His-dThr-Phe (F5)-Arg-ACC) screened by HyCoSuL technology is recognized and cleaved by CTSL (Fig. 8E).
Long-term imaging is particularly valuable for analyzing the in vivo biological dynamics of TIICs, especially for cysteine cathepsins such as CTSS that are enriched on the surface of TAMs. Furthermore, leveraging the membrane-enriched localization of CTSS, the development of membrane-anchored optical probes can substantially prolong the imaging window and enhance imaging specificity. On the basis of this property, Hu et al.199 reported that two probes (non-lipidated probe 2 and lipidated probe 3) were developed for visualizing it in TAMs (Fig. 8F). Although both probes can be recognized and specifically cleaved by CTSS, the lipidated probe 3 exhibits stronger binding to the TAM membrane as a result of lipidation, thereby achieving localized long-term retention within tumor tissue. Cellular experiments demonstrated that, following cleavage by CTSS on the surface of TAMs, the activated FL moiety enters the cell and emits a strong FL signal. In contrast, the non-lipidated probe 2 (control probe) enters TAMs directly and is subsequently cleaved by CTSS. In animal experiments, lipidated probe 3 exhibited prolonged imaging duration and high contrast. Notably, even 5 days post-injection—when the non-lipidated probe 2 group displayed almost no detectable FL—the tumor-to-skin signal ratio remained approximately 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, while the tumor-to-muscle signal ratio reached as high as 18:1. This study provides a novel strategy for the development of TIIC-targeted optical probes. For certain secreted enzymes, initial immobilization of optical probes on the cell surface may represent an effective approach to extending imaging duration. Furthermore, antigen presentation is a crucial process in the anti-tumor immune response. Monitoring the antigen presentation capacity of antigen-presenting cells (APCs), such as DCs, is an effective means of assessing the immune system's anti-tumor activity and predicting the efficacy of tumor immunotherapy. To achieve this goal, He et al.200 developed optical probes (MORs, MORC-I and MORF-II) based on CTSS and the immunoproteasome (iP), which are closely associated with the major histocompatibility complex class I (MHC-I) and MHC-II pathways (Fig. 8G). The incorporation of DSPE-PEG into MORs facilitates their passive accumulation within lymph nodes, enabling differential cleavage according to MHC-I or MHC-II activity. By detecting activated CL and FL signals, the intensity and type of immune responses can be distinguished. These studies demonstrate that CTS-specific probes not only enable the imaging of distinct TIIC subtypes but also serve as robust tools for dynamically monitoring enzymatic activity within the TME.
:1, while the tumor-to-muscle signal ratio reached as high as 18:1. This study provides a novel strategy for the development of TIIC-targeted optical probes. For certain secreted enzymes, initial immobilization of optical probes on the cell surface may represent an effective approach to extending imaging duration. Furthermore, antigen presentation is a crucial process in the anti-tumor immune response. Monitoring the antigen presentation capacity of antigen-presenting cells (APCs), such as DCs, is an effective means of assessing the immune system's anti-tumor activity and predicting the efficacy of tumor immunotherapy. To achieve this goal, He et al.200 developed optical probes (MORs, MORC-I and MORF-II) based on CTSS and the immunoproteasome (iP), which are closely associated with the major histocompatibility complex class I (MHC-I) and MHC-II pathways (Fig. 8G). The incorporation of DSPE-PEG into MORs facilitates their passive accumulation within lymph nodes, enabling differential cleavage according to MHC-I or MHC-II activity. By detecting activated CL and FL signals, the intensity and type of immune responses can be distinguished. These studies demonstrate that CTS-specific probes not only enable the imaging of distinct TIIC subtypes but also serve as robust tools for dynamically monitoring enzymatic activity within the TME.
Kolt et al.203 reported an optical probe (SK15.5) for monitoring GrA activity in NK92 cells. The active site of GrA remains concealed until the leader peptide is cleaved through processing and shearing, and the diphenyl phosphonate in SK15.5 covalently binds only to the active site of GrA, allowing SK15.5 to act exclusively on GrA in its active form (homodimer) (Fig. 9A). In cell experiments, active GrA in living cells and lysates was successfully labelled. Besides NK cells, GrA-specific activatable optical probes also serve as powerful tools for assessing T cell activity. For example, in 2025, Scott et al.204 reported the development of a CL probe (GzmA-CL), for the detection and analysis of T cell activity in intestinal diseases (Fig. 9B). In this study, the authors applied GzmA-CL to analyze the supernatants from approximately 150 clinical stool samples. The results demonstrated that GrA activity in the stool of patients with intestinal diseases was significantly higher than that observed in healthy individuals. Notably, GzmA-CL exhibited exceptionally high sensitivity toward GrA, enabling detection at picomolar concentration levels. Furthermore, correlation analyses revealed that GrA activity was positively associated with endoscopic scores and T-cell-related clinical indices. Collectively, these findings indicate that GzmA-CL provides a robust and direct means of assessing intestinal T cell activity in patients with intestinal diseases. In addition to GrA, they reported in a separate study the development of an optical probe (Probe 1) that leverages GrB activity to enable in vivo imaging of NK cell-mediated antitumor activity (Fig. 9C).205 The GrB substrate peptide (Ac-IEPD) in the probe facilitates efficient GrB targeting. When the probe interacts with GrB released by NK cells, it activates FL with an emission peak at approximately 520 nm within minutes. Cell and animal model experiments validated the probe's targeting ability, and by detecting GrB activity, direct optical bioimaging of NK cell-mediated anti-tumor activity was successfully achieved.
 | ||
| Fig. 9 (A)–(H) Molecular structure of SK15.5, Probe 1, GzmA-CL, CyGbPF or CyGbPP, SPP, CGB (GrB peptide substrate)/DEVD (cas-3 peptide substrate), Cy5.5-CBT, and TAD (afterglow luminescent core)/BHQ-NHS (BHQ3, quencher). (I) The sonoafterglow mechanism of ncRuPA. (J) and (K) Imaging applications of SPNP and TAD-BHQ in tumor animal models. Reproduced from ref. 207 and 210 with permission from John Wiley and Sons, Copyright 2021 and Springer Nature, Copyright 2024, respectively. | ||
GrB can also be used to design optical bioimaging probes targeting T cells. For example, He et al.206 reported two optical probes (CyGbPF and CyGbPP) in 2020 for in vivo imaging and urine testing to evaluate cancer immunotherapy efficacy. The researchers linked two GrB-specific peptide substrates to the FL group CyOH, forming the complete CyGbPF and CyGbPP (Fig. 9D). In the initial state, the FL remains off due to the weak electron-donating ability of the oxygen atoms in CyOH. Upon reaction with GrB, the electron-donating ability of the oxygen atoms in CyOH is enhanced, leading to a 24-fold and 22-fold increase in FL intensity at 717 nm. The in vivo imaging results correlate strongly with the infiltration levels of CTLs and CD4+ T cells in tumor tissues, demonstrating that the probe can assess immunotherapy efficacy based on optical signal variations. Additionally, the probe can also be utilized for optical urine testing to assess immune activation levels. To improve quantification and depth, dual-modal GrB probes have been engineered. Zhang et al.207 developed a polymer-based nano-optical probe (SPNP) for FL/PA dual-modal imaging of T cells (Fig. 9E and J). SPNP is a nanoparticle formed through the self-assembly of SPP monomers, where SPP consists of a polymer core (SP) and a GrB peptide substrate labeled with the IR800 FL dye. SPNP accumulates in the TME in vivo via the EPR effect. The action of GrB leads to a reduction in both the NIRF and PA signals of SPNP, while the signals from the SP core remain unchanged. The change in their ratio provides insight into GrB expression levels and further evaluates the activation state of T cells. Cell and animal model experiments have shown that SPNP can achieve real-time monitoring of immune cell activation, with signal intensity correlating closely with immune cell activation. As noted above, GrB-induced cell death is caspase-dependent. Accordingly, Ma et al.208 simultaneously employed GrB and cas-3 to develop an AND-gate logic dual resonance energy transfer nano-optical probe (DRET) for dynamic monitoring of programmed CTL activation and tumor cell apoptosis, evaluating the anti-tumor immune effect (Fig. 9F). The probe incorporates a GrB-responsive FRET group (composed of Cy5 and BHQ-3, connected to GrB's peptide substrate IEFD) and a Cas-3 (caspase-3)-responsive MR tuning group (composed of superparamagnetic iron oxide nanoparticles and Gd3+, connected to Cas-3's peptide substrate DEVD). Due to its innovative design, the probe enables early prediction and classification, predicting efficacy and performing immune stratification before tumor morphology changes. It also distinguishes between “responders” and “non-responders”, as well as “acquired resistance” and “maintained responders”, providing clinicians with timely and accurate information to adjust treatment plans.
In addition to the dual-mode and AND-gate logic optical probes, Xu et al.209 introduced, in 2022, a “double quenching” mechanism into the optical probe (Cy5.5-CBT-NPs) for imaging CTL activity (Fig. 9G). In contrast to the high specificity achieved by multi-responder activation strategies, the double quenching mechanism—combining intramolecular and intermolecular quenching—significantly reduces background signals and enhances imaging performance. Specifically, 2-cyanobenzothiazole (CBT) in the Cy5.5-CBT monomer undergoes intramolecular quenching with Cy5.5, reducing its FL intensity. When two Cy5.5-CBT molecules form a cyclic dimer (Cy5.5-CBT-Dimer), a second intermolecular quenching takes place. Upon GrB activation, the FL emission of Cy5.5-CBT-NPs at 702 nm is significantly enhanced, and the FL intensity is positively correlated with the concentration of GrB. Cell and tumor model experiments demonstrated that Cy5.5-CBT-NPs can specifically image the CTL-mediated tumor cell killing process and thereby reflect the activity of CTLs.
Ultrabright afterglow offers significant advantages in deep tissue imaging, ultrafast imaging, and minimizing photobleaching. For instance, Wang et al.210 developed a novel organic afterglow nanoparticle (TAD-BHQ) for ultrabright and ultrafast in vivo imaging (Fig. 9H and K). TAD-BHQ operates via a bioorthogonal activation strategy, where specific interaction with GrB triggers the separation of the afterglow luminogen (TAD) from the quencher (BHQ-3), restoring afterglow emission. Due to the correlation between the intensity of TAD-BHQ afterglow light and GrB concentration, TAD-BHQ can monitor GrB levels in tumors in real time and assess immunotherapy efficacy after chemotherapy, radiotherapy, or immune checkpoint blockade. Although traditional photoactivated afterglow optical probes minimize background interference by eliminating the need for continuous external light excitation during signal acquisition, tissue-induced scattering and absorption of pre-irradiation excitation light still limit afterglow activation in deep tissues, thereby constraining imaging contrast, sensitivity, and accuracy. To overcome these inherent limitations, Wang et al.211 developed a sono-afterglow probe (ncRuPA) in 2026 as an alternative to conventional photoactivated afterglow imaging (Fig. 9I). In the ncRuPA, ultrasound activates the sonosensitizer [Ru(bpy)3], leading to the generation of 1O2. The generated 1O2 oxidizes the afterglow substrate, thereby forming a stable dioxane intermediate. Owing to the presence of a non-conjugated amide linkage, rapid electron transfer is inhibited, thereby inducing cleavage of the intermediate through an electron exchange-induced chemiluminescence mechanism. The resulting luminescence is efficiently transferred to Ru(bpy)3via intramolecular CRET, resulting in the emission of a delayed NIR afterglow signal. This “sonic-chemiluminescence-energy transfer” cascade mechanism enables optical imaging without the need for an external light source, allows deep tissue activation, and achieves a high signal-to-background ratio. Furthermore, the authors further developed an APN-activated sonoafterglow probe (ncRuPAAPN), which enables highly sensitive tumor imaging and mediates efficient sonodynamic therapy, thereby providing a promising platform for tumor diagnosis and treatment.
4.2. Probes based on reactive nitrogen and oxygen species (RNOS)
Beyond probes directed at immune cell markers, a distinct class of optical probes target reactive small molecules—specifically reactive oxygen, nitrogen, and sulfur species (collectively RNOS)—within the TME. Although TIICs are a key source, RNOS are also produced by stromal cells and cancer cells themselves, positioning them as broader, cell-agnostic indicators of TME activity.212 In contrast to the above MPO-directed probes, which primarily track neutrophil or macrophage populations, RNOS-responsive sensors report on global oxidative stress and redox signalling. These species exert dual effects on tumor progression, promoting proliferation and therapeutic resistance at certain levels while contributing to immune-mediated tumor suppression in other contexts. Imaging RNOS dynamics thus enables investigation of their context-dependent roles in tumor biology. Toward this end, a range of optical probes have been developed to track RNOS fluctuations, offering insight into how these species shape TIIC function, tumor growth, and therapy response.For ROS, Zhang et al.213 synthesized an optical probe (PSiR) based on the PeT mechanism, capable of imaging highly reactive oxygen species (hROS) in real time (Fig. 10A). hROS reacts rapidly with the 2,4-dimethyl-3-ethylpyrrole moiety, and the oxidation product formed breaks the electron transfer balance enabling PSiR to be activated within seconds, achieving efficient real-time imaging of hROS. Furthermore, Wen et al.214 reported a multicolor optical probe (Probe 4) based on the FRET mechanism, simultaneously responding to changes in H2O2 and MMP-2 levels (Fig. 10B). When only H2O2 is present, the probe emits NIRF with a wavelength of 672 nm, but when MMP-2 is present, the probe emits green FL with a wavelength of 555 nm due to peptide cleavage and the activation of H2O2. Multimodal imaging optical probes targeting ROS have also been reported. For example, BTPE-NO2@F127 reported by Chen et al.215 can be used for FL/PA dual-mode imaging of H2O2 (Fig. 10C). On the other hand, MPN@CeOx reported by Deng et al.216 can monitor real-time changes in ROS levels in vivo through MR/CT imaging (Fig. 10D). Trivalent iron ions and CeOx materials confer MR and CT imaging capabilities to MPN@CeOx, respectively. More importantly, MPN@CeOx enables regulation of macrophage polarization through ROS scavenging, thereby effectively alleviating inflammatory symptoms.
 | ||
| Fig. 10 (A)–(C), (E)–(G), (I) and (J) Molecular structures of PSiR, Probe 4, BTPE-NO2 monomer, PN910, NTG, NRP, FHMI, and MI-H2S. (D) Application of MPN@CeOx in an animal model of intestinal inflammation (MR and CT imaging). (H) The mechanism of NRMP imaging in response to RNS. (K) Signal conversion processes of Lyso-RC. Reproduced from ref. 216 and 221 with permission from Elsevier, Copyright 2023, Springer Nature, Copyright 2024, respectively. | ||
In the TME, the interaction between RNS and TIICs significantly influences tumor initiation and progression.217 RNS-based TIIC-targeting optical probes serve as valuable tools for elucidating the intricate mechanisms underlying these effects. For example, Zhang et al.218 developed a multi-biomarker detection optical probe (PN910) based on the ICT mechanism, capable of responding to both ONOO− and H2O2 in an alkaline environment (pH > 7.4) (Fig. 10E). Furthermore, Luo et al.219 reported a multi-detector optical probe (NTG) capable of simultaneously responding to endogenous ONOO− and GSH (Fig. 10F). The probe, NTG, generates distinct red and green FL signals upon reaction with GSH and ONOO−, respectively, via nucleophilic aromatic substitution and oxidative cleavage of its 2,4-dinitrobenzenesulfonyl and the extended conjugated double bond of 1,8-naphthalimide moieties. This differential response enables NTG to discriminate between normal, inflammatory, and tumor cells based on their characteristic ONOO− and GSH levels. Using RNS to monitor the repolarization of TIICs is highly innovative, especially for TIICs with different phenotypes such as TAMs. For instance, Yuan et al.220 reported an optical probe (NRP@M-PHCQ) to track and monitor the migration and polarization of M2-type TAMs (Fig. 10G). The M-PHCQ in NRP@M-PHCQ targets CD206 and enters TAMs through endocytosis, thereby polarizing M2- to M1-type TAMs. The NO released in this process activates the FL of NRP. Similarly, Lu et al.221 reported an improved MR imaging probe (NRMP) (Fig. 10H). Like NRP@M-PHCQ, NRMP also detects NO with high sensitivity and selectivity in vivo, enabling studies on the role of NO in tumor progression, immune response, and TAMs’ responses to cancer immunotherapy. NRP@M-PHCQ not only enables intratumoral imaging of NO, but also induces the repolarization of M2-type TAMs toward the M1 phenotype and remodels the immune status of the TME, highlighting the close integration of optical imaging and immune regulation. This represents a promising paradigm for the future development of TIIC-targeted optical probes.
The specific role of RSS in tumor development remains elusive, but it has been implicated in various diseases. RSS can regulate a variety of physiological processes, including vasodilation, antioxidant defense, cell proliferation, apoptosis, differentiation, development, and immune response. These biological processes are closely linked to tumor progression.222 For example, a series of GSH-responsive optical probes developed by our team—including NIRF/PA dual-modal and visible/NIR-II dual-channel probes—enabled real-time in vivo monitoring of GSH fluctuations during tumor ferroptosis, as well as prolonged in vivo imaging.223–225 Furthermore, Chen et al.226 reported an optical probe (FHMI) based on the ICT mechanism for mitochondrial SO2 detection (Fig. 10I). In addition, Gong et al.227 introduced an optical probe (MI-H2S) for mitochondrial H2S detection (Fig. 10J). Pyridium targets MI-H2S to mitochondria, and the MI-OH released after the thiolysis reaction between H2S and dinitrophenyl ether produces a strong FL signal for H2S detection. In order to achieve imaging detection of multiple biothiols using the same optical probe, Zhang et al.228 reported a novel optical probe (Lyso-RC), which targets lysosomes and simultaneously detects three biothiols (Cys, Hcy, GSH) and H2S (Fig. 10K). Different RSS react with Lyso-RC to generate distinct FL signal patterns, with response patterns for Cys/Hcy, GSH, and H2S being blue-red, green-red, and red, respectively.
4.3. Probes for NETs
The focus of neutrophil imaging has recently expanded from labelling intracellular enzymes to targeting NETs—extracellular DNA scaffolds released by TANs during NETosis. These structures actively drive tumor metastasis by promoting local invasion, vascular anchoring, immune evasion, and organ colonization.229 To elucidate these NET-specific roles in the TME, researchers have developed a suite of optical probes capable of visualizing NETs directly, thereby enabling studies of their contribution to tumor progression distinct from that of the parent neutrophils. For this case, Hao et al.230 summarized NET research from 1996 to 2024 in a review, suggesting that NETs may become a promising alternative for tumor imaging and treatment. The DNA fragments and various proteins (such as NE and CTS) contained in NETs serve as critical elements for the development of TIIC-targeted optical probes.Rios et al. have made significant advancements in the development of optical probes targeting TAN-related enzymes, developing a series of activatable optical probes for NE, protease 3, and CTSG. However, these optical probes fail to achieve effective NET imaging owing to high background FL, limited signal amplification capacity, and challenges in penetrating NET structures and binding effectively to their associated enzymes. Thence, they developed an optical probe (HNE-FQ) using an optimized design strategy to enable precise imaging of NETs (Fig. 11A and E).231 The peptide substrate (Glu–Glu–Ile–Nle–Arg–Arg) attached to the tripeptide branch scaffold is cleaved by NE, separating the FL group (5-FAM) from the quencher (methyl red), thereby restoring FL. Due to the trivalent scaffold and FRET effect, HNE-FQ achieves a 420-fold enhancement in FL intensity. Furthermore, they used the sulfonated Cy5 FL group to further improve the deep tissue penetration ability of the new optical probe (HNE-3F1Q) enabling NET imaging in deep tissues (Fig. 11B and F).232
 | ||
| Fig. 11 (A)–(D) and (G) Molecular structure of HNE-FQ, HNE-3F1Q, TNR1 or TNR2, H-NE or H-CG, and CDr15. (E) and (F) HNE-FQ and HNE-3F1Q's application in NET imaging. Reproduced from ref. 231 and 232 with permission from Royal Society of Chemistry, Copyright 2019 and Royal Society of Chemistry, Copyright 2023, respectively. | ||
Effective monitoring of the NETosis process is essential for elucidating the interaction mechanisms between TANs and tumors. For example, Cheng et al.233 reported the development of two optical probes (TNR1 and TNR2) for imaging NETosis. TNR1 and TNR2 activate FL signals only when both NE and CTSG are simultaneously present (Fig. 11C). Unlike single-lock designs that cannot discriminate between NETosis and neutrophil activation, this tandem-locking approach leverages CTSG as a specific marker of NETosis to achieve selective detection. The results of animal experiments in tumor models demonstrated a significant enhancement in the NIRF signal at the tumor site of mice treated with NETosis inducers, consistent with histological findings. At the same time, the activated FL signals correlate with the tumor growth following immunotherapy and can also be used to evaluate the effect of cancer immunotherapy.
In addition to these enzymes in NETs, the DNA fragments of NETs also serve as valuable factors for developing optical probes targeting NETs. For example, Guerra et al.234 simultaneously employed DNA, NE, and CTSG to develop two optical probes (H-NE and H-CG) for imaging NETs (Fig. 11D). Hoechst 33258 in both H-NE and H-CG binds tightly to the minor groove of DNA. NE and CTSG bound to DNA cleave the peptide sequences in H-NE and H-CG, disrupting the original FRET system and restoring FL. The experimental results of H-NE and H-CG revealed that NE and CTSG are only enzymatically active when bound to DNA, which aids in clarifying the functional relationship between exogenous DNA content, protease activity, and disease progression. In addition to these precisely developed optical probes, certain DNA-specific FL dyes can also detect NETs. Kim et al.235 confirmed that CDr15 can be used for imaging NET structures in human tumor tissue samples, exhibiting lower background FL and higher specificity than DAPI and SYTOX DNA dyes (Fig. 11G). Experimental results of tumor tissue imaging demonstrated that CDr15 could accurately assess NETosis extent.
4.4. Probes based on receptors
The specific binding between ligands and their cognate receptors provides a robust foundation for designing optical probes with high targeting specificity toward TIICs. For example, CD206, a canonical biomarker of M2-type TAMs, has emerged as a widely investigated target for targeted imaging and therapeutic applications. Moreover, the diverse receptors abundantly expressed on TIIC subtypes—such as chemokine receptors—represent important targets for probe design, and probes developed on this basis have shown considerable application potential.Based on this, Gao et al.236 employed the CD206 ligand (mannose) and the AIE mechanism to design an optical probe (TPE-Man) for in vivo tumor imaging and photodynamic therapy (PDT) (Fig. 12A). TPE-Man enters TAMs through CD206-mediated endocytosis and emits FL with a wavelength of 600 nm due to the AIE mechanism. After irradiation with white light, TPE-Man produces ROS that significantly reduce TAM survival through the PDT, enabling precision treatment. In addition, Luo et al.237 reported a metabolizable optical probe (DN-ICG) for imaging TAMs in pancreatic cancer (Fig. 12B). The carboxymethyl dextran in DN-ICG interacts with ICAM-3-grabbing nonintegrin related 1 (SIGNR1), enabling DN-ICG to specifically target TAMs. The experimental results demonstrated that the emission intensity of DN-ICG is approximately 279% higher than that of free indocyanine green (ICG), and the NIR-II emission wavelength of 1000–1700 nm ensures deep tissue penetration, enabling monitoring of TAM changes during pancreatic cancer treatment. The probe that remains outside tumor tissues is decomposed into non-toxic small molecules via enzymatic hydrolysis and cleared through liver metabolism, thereby reducing the background FL of in vivo imaging. Unlike optical probes targeting CD206, probes directed against Ly6G are primarily designed for in vivo dynamic tracking of neutrophils. For example, Wang et al.238 employed Ly6G, predominantly expressed on neutrophil surfaces, to design an FL/MP/CT multimodal imaging optical probe (PEG-Fe3O4-Ly6G-Cy7 nanoparticles, Ly6G NPs), for monitoring neutrophil infiltration in abdominal aortic aneurysm (AAA) (Fig. 12C). The superparamagnetic nanoparticle material (Fe3O4@PEG-COOH) and Cy7 FL dye in Ly6G NPs provide MP/CT and FL imaging capabilities, respectively. In the mouse model, FL/MP/CT multimodal imaging results offered detailed insights into AAA, intuitively reflecting the correlation between neutrophil infiltration and AAA severity. Alongside this, in 2026, Chen et al.239 conjugated Ly6G with an NIR-II probe (DCPD7) to construct an ultra-bright NIR-II optical probe (DCPD7-Ly6G) for high-resolution imaging of individual neutrophils in deep tissues (Fig. 12D). In subsequent stroke mouse model experiments, the authors developed a multicolor imaging system incorporating DCPD7-Ly6G, DCPD3-KTP5—an NIR-II probe specifically targeting mesenchymal stem cells—and ICG to track neutrophil dynamics in real time. This approach revealed fine spatiotemporal features of neutrophil migration and stem cell–immune cell interactions following stroke, thereby providing a powerful tool for elucidating complex biological processes and informing the development of novel therapeutic strategies.
 | ||
| Fig. 12 (A), (B), (D), (E) and (G) Molecular structure of TPE-Man, DN-ICG, DCPDs, Trp-BODIPY, and IRDye800CW NHS ester. (C) Application of Ly6G NPs in an animal model of abdominal aortic aneurysm via TAN imaging. (F) Probe 5 interacts via the CXCL10-CXCR3 and CXCL13-CXCR5 pathways. (H) Molecular structure of DCBT and application of AIE-Apt-99 in visualizing TANs in ascites smears from ovarian cancer patients. Reproduced from ref. 238 and 243 with permission from Springer Nature, Copyright 2024 and Elsevier, Copyright 2024. | ||
Beyond targeting these classic membrane surface receptors, receptors for cytokines—such as interleukins and chemokines—are highly expressed on TIICs and represent promising targets for probe development. For example, Reese et al.240 initially conjugated BODIPY to tRNA (transfer RNA) to create BODIPY-aa-tRNA and subsequently incorporated BODIPY-aa-tRNA into the optimal modification site of IL-33 for FL labeling (Fig. 12E). The newly developed IL-33 FL analog retains the biological activity of wild-type IL-33, binds effectively to the ST2 receptor on TIICs’ surfaces, and can be used to monitor IL-33/ST2 signal transduction in real time. Moreover, Bertolini et al.241 labeled CXCL13 and CXCL10 cytokines with BODIPY and endo-9-hydroxymethyl-bicyclo[6.1.0]non-4-yne (BCN), prepared two chemically modified cytokine FL analogs (Probe 5), and employed a combination of CXCL13 and CXCL10 with CXCR5 and CXCR3 for real-time imaging of drug-resistant leukemia BCs (Fig. 12F). The work of Reese and Bertolini has provided new tools for studying immune cell signalling and enables a deeper understanding of the interactions between TIICs.
In addition to the above traditional strategies, advanced biological technologies (such as gene cloning and aptamer screening) have significantly advanced the development of TIIC-targeted optical probes. For example, Bernhard et al.242 employed genetic engineering to clone the Fc domain from human immunoglobulin (IgG) to design an optical probe (Probe 6) for monitoring the infiltration of immune cells within the TME. They cloned the VHH-Fc domain, which specifically targets Fc receptors on the surfaces of monocytes and granulocytes (such as FcγRI [CD64], FcγRII [CD32], FcγRIII [CD16], etc.). VHH-Fc was subsequently labeled with the IRDye800CW FL dye at a 1:3 (protein to dye) ratio (Fig. 12G). Animal experiments on tumor models showed that the probe specifically images tumor tissues and can evaluate TIIC infiltration. In addition to genetic engineering technology, Wang et al.243 first employed the aptamer selection strategy of FL-activated cell sorting to identify Apt-99, a DNA aptamer that binds with high affinity and selectivity to the S100A8&A9 heterodimer on the surface of TANs in ovarian cancer ascites smears. They subsequently developed an optical probe, AIE-Apt-99, by labelling Apt-99 with fluorescein DCBT (Fig. 12H). In ovarian cancer ascites smears, AIE-Apt-99 specifically binds to TANs, where it aggregates and activates FL via the AIE mechanism.
4.5. Probes based on artificial TIICs
The preceding sections have summarized recent advances in optical probes based on enzymes, reactive species, and specialized structures, together with their design strategies and applications. Beyond these approaches, additional strategies have been explored for TIIC-targeted probe development. For example, in vitro reprogramming of immune cells to generate optical imaging-capable “artificial TIICs” (ATCs) enables real-time tracking of immune cell migration in the bloodstream, offering insights into the biological roles of TIICs in tumor progression.Most TIICs demonstrate an inherent ability to migrate to tumor tissues. Therefore, this property can be exploited to create ATCs—cells endowed with optical imaging capabilities through strategic design—for effective in vivo tracking of immune activity. When constructing ATCs, researchers typically select homologous TIICs as the basis for ATCs to ensure optimal biocompatibility and evade immune clearance.244 These homologous TIICs are then co-incubated with optical probes, or their cell membranes are extracted and employed to encapsulate the optical probes. ATCs constructed using these methods typically retain either complete or partial biological characteristics of the homologous TIICs, thereby enabling precise delivery and targeted accumulation of optical probes at tumor sites.
The most direct approach involves chemically conjugating imaging probes onto the surface of natural immune cells. Qiu et al.245 synthesized an optical probe (Gd@BSA-BODIPY) for FL/MR dual-mode imaging (Fig. 13A). They immobilized gadolinium ions (Gd3+) and BODIPY onto the BSA skeleton via coordination and covalent bonds, forming Gd@BSA-BODIPY nanoparticles, which were subsequently activated with 5-thio-2-nitrobenzoate (TNB) and further conjugated to the neutrophil membrane surface via covalent bonds. Experiments on animal models of lung cancer demonstrated that FL and MR signals in tumor tissues of mice receiving ATCs combined with Gd@BSA-BODIPY were significantly amplified. Likewise, a more integrated method entails internalizing nanoprobes into immune cells, transforming them into living, trackable microcarriers. Zhang et al.246 developed an artificial “super neutrophil” (GCZM). GCZM targets and eliminates malignant tumor cells and pathogens. Cy5.5 integrated into GCZM enables the imaging of its tumor-targeting capability. Additionally, they synthesized an optical probe (BClO) for real-time monitoring of GCZM toxicity by detecting ROS (Fig. 13B). Moreover, Xu et al.247 developed a novel class of photoactive neutrophils (PANs) by bioengineering living neutrophils to encapsulate a multifunctional nanocomplex (RA/Ce6), enabling self-amplified, multistage tumor targeting that triggers mitochondria-specific photochemotherapy in mouse models of melanoma and oral cancer (Fig. 13C).
 | ||
| Fig. 13 (A) Construction process of Gd@BSA-BODIPY. (B) GCZM's application (in vitro (left) cytotoxicity to 4T1 cells and its damage to DNA and mitochondria, and in vivo (right) therapeutic applications in animal models of tumors). (C) Application of PAN in a B16F10 melanoma cell subcutaneous tumor animal model. (D)–(F) Molecular structure of photoacoustic contrast agents TFML, IR786s, and IR780. (G) Application of aNeu in a tumor animal model. (H) Imaging mechanism of the biological orthogonal imaging system constructed by Az-ROS@MB and CSSC@DBCO. Reproduced from ref. 246, 247, 250, and 251 with permission from John Wiley and Sons, Copyright 2019, Elsevier, Copyright 2021, Elsevier, Copyright 2024, and John Wiley and Sons, Copyright 2025, respectively. | ||
To circumvent challenges associated with live cell manipulation—such as variable viability, batch-to-batch inconsistency, and complex manufacturing—cell membrane coating technology has emerged as a robust alternative. This approach extracts membranes from source cells (e.g., neutrophils) and uses them to cloak synthetic nanoprobes, bestowing the probes with the source cell's surface antigens and biological tropism while enabling scalable production. In 2021, Liu et al.248 developed a lipid-modified optical probe (TFML) and incorporated it into the neutrophil membrane via membrane insertion to form ATCs (TFML-NE) (Fig. 13D). They used TFML-NE to detect the PA signal of TANs infiltrating tumor tissues. A similar strategy was adopted by Liao et al.,249 who loaded NK cells in vitro with nanoparticles (DCNP@786s) containing FL dyes and rare earth material cores to construct ATCs with imaging capabilities (Fig. 13E). They employed these ATCs for quantitative tracking and real-time visualization of NK cell survival in a liver cancer mouse model. DCNP@786s is formed by IR786s (ROS-sensitive NIRF dye) encapsulating rare earth nanoparticles (DCNP, synthesized from lanthanide rare elements, with a diameter of about 50 nm). When intracellular ROS degrade IR786s, the FL emission of DCNP@786s under 808 nm excitation light is significantly enhanced, whereas the FL emission under 980 nm laser excitation remains largely unchanged. This differential response enables ratiometric imaging for quantitative assessment of intracellular ROS levels, which serves as an indicator of NK cell activity.
The aforementioned approaches for direct in vitro editing of immune cells present several practical limitations, including the risk of cellular contamination, compromised cell viability, low optical probe loading efficiency, restricted editing flexibility, and unintended polarization of TIICs during manipulation. Accordingly, the approach of directly extracting cell membranes and encapsulating optical probes in vitro provides an alternative strategy to overcome the above limitations. In contrast to direct immune cell editing strategies, TIIC-derived cell membranes can be isolated in vitro using techniques such as low-temperature ultrasonic disruption and ultracentrifugation. Standardized processing protocols help preserve the intrinsic biological characteristics of the source TIICs, thereby facilitating subsequent in vivo applications. Furthermore, those ATCs can be modularly designed and constructed to suit diverse practical applications, thereby providing enhanced application flexibility. Thus, Huang et al.250 proposed a more straightforward strategy for constructing ATC optical probes (Fig. 13F and G). They developed ATCs (aNeu), comprising porcine pancreatic elastase (PPE), IR780 FL dye, and hollow manganese dioxide nanoparticles (hMnO2 NPs). The cell membrane derived from neutrophils confers aNeu with strong tumor tropism. In addition, aNeu possesses FL/MR dual-mode imaging capabilities. PPE, Mn2+, and IR780 respectively endowed aNeu with tumor cell killing, MRI, and FL imaging ability. Besides utilizing immune cell membranes for tumor targeting, leveraging the tumor-homing and self-recognition capabilities of tumor cell membranes represents another feasible strategy for developing ATC-type optical probes. Similar to immune cell membrane encapsulation approaches, this strategy mitigates the limited tumor accumulation associated with the low EPR efficiency of non-cell membrane-coated nano-optical probes. For example, Bian et al.251 encapsulated the ROS-responsive azide precursor Ac4ManNAz within 4T1-derived tumor cell membranes to construct a pre-targeting nanocarrier (Az-ROS@MB), thereby enhancing tumor-selective accumulation of Ac4ManNAz. This approach enabled efficient azide labelling of tumor cells via metabolic glycoengineering and provided high-density reaction sites for subsequent bioorthogonal reactions. Furthermore, the GSH-responsive imaging probe (CSSC@DBCO) efficiently recognizes Ac4ManNAz-labeled tumor cells through bioorthogonal click reactions and emits detectable optical signals under elevated intracellular GSH conditions, achieving high-fidelity and high-contrast imaging in vivo (Fig. 13H). Together, Az-ROS@MB and CSSC@DBCO constitute a spatiotemporally controllable, multistage-validated, and signal-amplified precision tumor imaging platform. Through the synergy of homologous biomembrane encapsulation and bioorthogonal chemistry, this strategy effectively addresses key bottlenecks in the practical application of optical probes and provides a versatile platform with promising clinical translational potential for precision tumor diagnosis and image-guided surgery.
Although ATCs have demonstrated promising performance in preclinical animal models, this strategy requires careful evaluation. This approach is associated with several risks, including increased immunogenic potential, which may trigger host immune responses and compromise imaging reliability; significant batch-to-batch variability arising from differences in donor cell sources and in vitro expansion conditions, thereby affecting cell phenotype, functional activity, and imaging signal consistency; and reduced cell viability during in vitro manipulations (e.g., transduction, membrane coating, and prolonged culture), leading to impaired proliferation, migration, and immune function, and consequently diminishing imaging accuracy.252–254 These limitations hinder the clinical application and broader translational potential of ATCs. Systematic investigations are urgently needed in areas such as immunogenicity assessment, batch consistency control, cell viability preservation, manufacturing process optimization, and regulatory strategy development to enhance the clinical translational potential of TIIC-targeted optical probes.
5. Application of TIIC-based optical probes for cancer immunotherapy
The multifunctionalization of TIIC-targeted optical probes represents a key future direction. In particular, optical probes with integrated therapeutic functions possess broader application prospects and greater potential for clinical translation, and are expected to become indispensable tools in the precise and personalized diagnosis and treatment of tumors in the future. The integration of therapeutic functions into TIIC-targeted optical probes represents a key direction for advancing cancer immunotherapy. Unlike conventional immunotherapeutic approaches, which lack real-time feedback on treatment response, optical probes with integrated therapeutic capabilities enable simultaneous diagnosis and intervention. Through quantifiable optical signals, these probes provide real-time feedback on therapeutic efficacy—such as tumor suppression, TIIC infiltration dynamics, and antitumor immune activation—thereby serving as reliable tools for optimizing immunotherapy regimens, enhancing treatment precision, enabling patient stratification, and dynamically adjusting treatment strategies. When designing such probes, therapeutic units (e.g., chemotherapeutic drugs, immune checkpoint inhibitors, and photosensitizers) can be incorporated into TIIC-targeted optical platforms to achieve seamless integration of diagnostic imaging and immunotherapy.255,2565.1. Chemotherapy-antitumor immune response synergy
The mechanism of chemotherapy is to inhibit the proliferation of tumor by targeting and killing rapidly dividing tumor cells across a broad range. Its key characteristics include rapid onset and broad applicability (Fig. 14A). It is widely used in the treatment of various cancers at all stages, though its limitations are also obvious. For instance, chemotherapy drugs typically lack specificity and fail to effectively target tumor tissues. As a result, chemotherapy drugs harm normal cells and tissues, leading to serious side effects, including bone marrow suppression and hair loss. A major advantage of integrating chemotherapeutic functions into TIIC-targeted optical probes lies in exploiting their tumor-targeting capability to reduce the nonspecific accumulation of chemotherapeutic agents in normal tissues, thereby mitigating the adverse effects associated with systemic chemotherapy. An additional advantage is that the chemotherapeutic drugs concentrated within tumor tissue can efficiently eliminate tumor cells, leading to the release of TAAs and thereby activating TIIC-mediated anti-tumor immune responses. This process facilitates localized activation of anti-tumor immunity within the TME, ultimately achieving synergistic therapeutic effects between chemotherapy and immune-mediated tumor suppression. | ||
| Fig. 14 (A) Examples of common chemotherapy drugs integrated with optical probes. (B) Application of S&D@Ag2Se in the MDA-MB-231 cell orthotopic tumor animal model (tumor volume growth in different treatment groups). (C), (D) and (F) Molecular structures of Olsa-RVRR, IR820-α-CD, PACDx and PARx. (E) In vivo imaging application of PEG/αCD25-Cy7/TMZ in animal tumor models (tumor growth of different treatment regimens). Reproduced from ref. 257 and 154 with permission from John Wiley and Sons, Copyright 2018 and American Chemical Society, Copyright 2023. | ||
Combining chemotherapy with adoptive cell therapy holds real promise for cancer treatment. Yet standard protocols often fall short because we cannot track drug pharmacokinetics or immune cell dynamics in real time. That forces clinicians to rely on empirical dosing, which rarely gives optimal synergy. Nanoparticle platforms offer a way to integrate chemotherapy with imaging. Take Hao and colleagues’ work: they built a programmable system (S&D@Ag2Se) using NIR-II multiplex fluorescence.257 One probe (emitting at 1350 nm) carries doxorubicin (DOX) plus the chemokine SDF-1α on Ag2Se dots, letting them monitor drug release. Another probe (1050 nm emission) labels NK-92 cells with Ag2S dots, so they can track immune cells. In tumor models, NIR-II imaging showed the best time window for dosing. Compared with simple co-injection, this imaging guided schedule ensures DOX and NK cells both hit the tumor at peak levels—overcoming a major drawback of traditional regimens (Fig. 14B). Unlike simply mixing chemotherapy with NK cells, the NIR-II imaging-guided programmed schedule lets DOX and NK cells reach the tumor together at peak levels—bypassing key drawbacks of conventional dosing. This work shows how optical imaging can help fine-tune therapies in real time.
Multidrug resistance often stems from small chemotherapeutic drugs being pumped out of cells by P-glycoprotein. Even with nanocarriers, less than 1% of the injected dose reaches solid tumors; most ends up in the liver and spleen, causing toxicity. Yuan et al.258 developed an “enzyme-triggered in situ self-assembly” strategy to get around this.255 They made a small precursor probe, Olsa-RVRR, by linking olsalazine (a DNA methylation inhibitor) to a cell-penetrating peptide. Once inside cells, high glutathione cleaves a disulfide bond, then furin protease cuts the peptide, exposing thiol groups that drive self-assembly into 20–50 nm nanoparticles. Those particles avoid efflux pumps and stay inside cells. Olsa-RVRR also works for CEST-MRI (chemical exchange saturation transfer magnetic resonance imaging) via olsalazine's hydroxyl protons. In mice, the probe gave a 4.3% CEST signal at 2 h that lasted 24 h, while free olsalazine faded in 0.5 h. The probe mainly accumulated in tumors and kidneys—little in the liver, suggesting renal clearance. Importantly, the 2 h CEST signal correlated strongly with tumor shrinkage at day 33 (R2 = 0.97), so you can predict treatment response just two hours after injection (Fig. 14C). Thus, therapeutic outcomes can be predicted as early as 2 h after systemic administration, allowing timely strategy adjustments. Modulating the immunosuppressive TME is central to improving immunotherapy, and chemotherapy can directly support this process. Dong et al.259 demonstrated the synergistic effect of chemotherapy and immunotherapy by reducing the immunosuppressive environment of the TME in their 2020 study. The CpG NPs and DOX contained in the delivery system they developed have FL properties, providing two fluorescence signals that track the distribution and dynamic changes of both drugs in tumor, offering real-time insights into TME changes (Fig. 14D). Systemic chemotherapy with temozolomide (TMZ), used as the first-line treatment for glioblastoma (GBM), can damage the bone marrow and impair both the number and functional activity of immune cells; therefore, local chemotherapy is essential to mitigate the side effects associated with TMZ. The diagnostic and therapeutic optical probe (PEG/αCD25-Cy7/TMZ) developed by Zeng et al.154 successfully accomplished this objective (Fig. 14E). αCD25-Cy7 has Treg cell targeting ability (targeting efficiency up to 92.3%), enabling real-time monitoring of Tregs through Cy7's FL/PA dual-mode imaging ability. Notably, their study indicated that after TMZ treatment, Treg infiltration within tumor tissues was markedly increased, while treatment with the indoleamine 2,3-dioxygenase inhibitor suppressed Treg infiltration while enhancing CTL infiltration, thereby inhibiting tumor growth. This offers new and valuable evidence for the treatment of GBM. Real-time monitoring of Treg infiltration during chemotherapy can help evaluate the immunosuppressive status of GBM, and based on this information, allow timely adjustment of the treatment plan to effectively improve patient survival. Chemically modifying chemotherapy drugs into prodrugs with targeting capabilities is also an effective method. In 2021, Lucero et al.155 integrated PA imaging with chemotherapy. While developing the GSH-responsive PA imaging probe (PACDx), they modified gemcitabine into a GSH-responsive prodrug (PARx) (Fig. 14F). Notably, in contrast to conventional strategies, this study strategically elevated the activation threshold of the GSH-responsive optical probe, thereby minimizing unintended activation by basal GSH levels in normal tissues. This approach reduced false-positive signals and significantly enhanced the probe's capacity for accurate tumor discrimination, yielding excellent imaging contrast in a murine lung cancer model. PACDx and PARx further demonstrated robust performance in both in situ imaging and therapeutic intervention within the same model. Importantly, this work offers a valuable design paradigm for the future development of TIIC-targeted optical probes, particularly when targeting biomarkers expressed in both normal and immune cells. By rationally tuning the probe activation threshold according to differential expression levels between TIICs and normal cells, TIIC selectivity can be substantially improved. Moreover, the incorporation of responsive prodrug design markedly reduced collateral damage to normal cells compared with non-selective therapeutics. Collectively, this strategy represents a promising framework for enhancing the specificity and safety of next-generation multifunctional optical probes.
5.2. PDT/PTT-antitumor immune response synergy
PDT and PTT are two treatment methods based on light energy, which have gained significant attention for their precision and minimally invasive characteristics.260–263 Both PDT and PTT offer more precise treatments compared to traditional methods via localized light irradiation. They result in less damage to normal tissues and allow for quicker postoperative recovery compared to surgical resection, chemotherapy, and radiotherapy. Furthermore, PDT/PTT directly mediates tumor cell death through the generation of ROS and localized hyperthermia. The resulting release of TAAs and DAMPs can activate the patient's adaptive immune response. This process is characterized by calreticulin (CRT) exposure and the release of ATP and high-mobility group box 1 (HMGB1). These DAMPs facilitate DC phagocytosis via the CRT-CD91 axis, trigger NLRP3 inflammasome activation and IL-1β secretion through ATP-P2X7 signalling, and promote DC maturation through HMGB1-TLR4-mediated NF-κB activation. Concurrently, cytosolic DNA activates the cGAS-STING pathway, inducing type I interferon production and enhancing antigen cross-presentation capacity. Following antigen uptake and processing of TAAs, mature DCs activate CD4+ T cells through major histocompatibility complex class II (MHC II) presentation and initiate CTLs’ responses via MHC I cross-presentation. With the support of co-stimulatory molecules and pro-inflammatory cytokines, T cells undergo clonal expansion and effector differentiation. Ultimately, this cascade establishes a systemic adaptive immune response against the tumor, suppressing the growth of primary lesions and distant metastases while laying the foundation for durable immune memory, thereby enhancing the anti-tumor activity of the immune system and establishing a coordinated PDT/PTT–anti-tumor immune response network.Integrating photodynamic (PDT) and photothermal (PTT) therapies into a single optical probe has become a clear direction for theranostic platforms. Sun et al. developed a liposomal nanoporphyrin probe (Pp18-lipos, Fig. 15A).264 By adjusting the Pp18 lipid ratio, the platform combines FL imaging, PA imaging, MRI, PDT/PTT, and antitumor immune activation. FL picks up drug distribution at µg mL−1 levels and tracks probe buildup in tumors and major organs from 0.5 to 48 hours, confirming targeting. PA gives deep-tissue views of tumor anatomy and vascularization while also following probe accumulation—handy for real-time efficacy checks. MRI delivers millimeter-scale anatomy with excellent soft-tissue contrast, clearly separating tumor from normal tissue, which helps with both pre-op planning and post-op monitoring. This all-in-one design cuts down on the complexity and instability of multi-component nanomedicines, making clinical translation simpler, safer, and more efficient. Imaging feedback lets users adjust laser timing and dose on the fly. In mouse subcutaneous tumor models, the combined PDT/PTT slowed tumor growth and triggered a stronger antitumor immune response: more DCs and CTLs infiltrated the tumor, cytokine levels (IFN-γ, TNF-α, IL-12p70) went up, and Treg infiltration dropped. That shifted the tumor from an immunosuppressive “cold” state to an immunostimulatory “hot” one, making it more responsive to immunotherapy.
 | ||
| Fig. 15 (A) Construction of the molecular structure of the monomer (Pp18-lipid) of Pp18-lipos, and the mechanism of Pp18-lipos in tumor immunotherapy. (B) Molecular structure of DDTB and its application (quantitative analysis of ex vivo tumor images and related data from different treatment groups in a subcutaneous animal model of B16F10 and 4T1). (C) Schematic diagram of the photoimmunotherapy mechanism of PBNO-Ce6. (D) The mechanism of FAL-ICG-HAuNS + FAL-Hb-lipo in tumor treatment and application in the subcutaneous tumor model. Reproduced from ref. 264–266 and 269 with permission from John Wiley and Sons, Copyright 2020, John Wiley and Sons, Copyright 2021, Editorial Office of Opto-Electron. Adv.; Opto-Electron. Adv., Copyright 2024, and Springer Nature, Copyright 2019. | ||
In another study, Jiang et al.265 combined PDT/PTT with fluorescence guided surgery (FGS) and anti-PD-L1 therapy in animal tumor models, pushing survival rates up to 90% (Fig. 15B). The NIR imaging capability of DDTB-DP NPs gave reliable data for preoperative diagnosis and for picking the best phototherapy time window. NIRF signals told them when the nanoparticles had accumulated optimally, improving treatment efficiency and lowering the risk of overtreatment. Under NIRF guidance, residual microtumors with strong FL signals stood out clearly, allowing a second precise resection. Ex vivo NIRF imaging also let them assess nanoparticle accumulation and clearance in major organs and tumors, helping refine treatment strategies. Notably, the FGS group showed effective tumor accumulation of DDTB-DP NPs, and after resection, the combined PDT/PTT wiped out residual microtumors completely. In the non-FGS group, peripheral blood levels of TNF-α and IFN-γ, along with CD4+ T cell and CTL infiltration in tumors, all rose markedly—showing that DDTB-DP NPs effectively elicited adaptive antitumor immunity and greatly improved the response to PD-L1 blockade. Furthermore, Xu et al.266 showed that optical probes combining PDT and PTT strongly boost CTL infiltration in triple-negative breast cancer, a tumor type known for severe immunosuppression and hypoxia, while cutting Treg infiltration—essentially reversing the immunosuppressive tumor immune microenvironment (Fig. 15C). Compared with controls, intratumoral CTLs went up 2.7-fold and Tregs dropped 62% after PBNO-Ce6 treatment. The CTL/Treg ratio in the spleen also rose significantly, hinting that local PDT/PTT might spark a systemic antitumor immune response and possibly curb metastasis. It is worth mentioning that the hypoxic nature of the TME is a major factor limiting the efficacy of PDT.267,268 Overcoming low PDT efficiency under hypoxia would be a big step forward. Li et al.269 tackled this with a nano-optical probe (Fal-ICG-HAuNS + Fal-Hb-lipo) (Fig. 15D). The probe uses hemoglobin liposomes (Hb-lipo) as oxygen carriers, releasing oxygen locally to boost PDT. Unlike conventional tumor-targeting approaches, this one sticks FAL peptides onto multiple components to hit the endoplasmic reticulum (ER) precisely. It releases oxygen right inside the ER, where ER-localized PDT triggers ER stress and immunogenic cell death (ICD), which in turn activates antitumor immunity. In B16F10 tumor-bearing mice, depleting CTLs with specific antibodies sharply reduced the probe's antitumor effect. This tells us that the improved efficacy depends largely on adaptive immune activation, not just the direct killing from PDT/PTT—highlighting the value of “dual ER targeting” for driving antitumor immunity. Also, FAL-ICG-HAuNS and FAL-Hb-lipo emit at different wavelengths, so you can monitor drug distribution in real time and nail down the best PDT/PTT window. This work offers fresh insights for designing multifunctional TIIC-targeted optical probes, making clear that precise targeting is key to getting a strong local antitumor immune response.
5.3. Activation of immune-related signalling pathways
Signalling pathways involve a series of molecules, such as proteins, enzymes, receptors, and second messengers, that transmit signals through orderly interactions. These signals are typically initiated by extracellular stimuli, including hormones, cytokines, and drugs, ultimately influencing biological functions, including cell proliferation, differentiation, apoptosis, and immune responses, through the regulation of gene expression and protein activity. In cancer immunotherapy, the A2AR, cGAS-STING, and JAK-STAT signalling pathways have emerged as key targets and have been extensively studied for their roles in modulating immune cell activity, tumor immune evasion, and the TME.A2AR is a receptor on immune cells. When engaged, its signalling dampens immune activation and function, reducing anti-tumor responses in the TME and helping tumors dodge immune attack. In 2024, Liu et al.270 designed a nanotheranostic platform (SSPNiNO) that delivers A2AR inhibitors straight into glioblastoma (GBM) while providing in situ treatment (Fig. 16A). SSPNiNO first targets neutrophils in the bloodstream. These cells naturally cross the blood–brain barrier, so they carry the probe into GBM tissue. NIRF irradiation then generates localized heat, killing tumor cells and triggering NO release from a thermosensitive donor inside SSPNiNO. That NO helps repolarize tumor-associated macrophages. At the same time, the released A2AR inhibitors act on multiple immune cell subsets in the TME, reshaping the immune landscape and reversing GBM's immunosuppressive state. In tumor-bearing mice, SSPNiNO plus NIRF irradiation gave 100% survival over the whole experiment, a big jump from controls (23 days) or free A2AR inhibitors (24 days). Owing to the SP2 component, SSPNiNO also enables sensitive NIRF imaging in an orthotopic GBM model, with FL signals closely matching the neutrophil marker Ly6G. Longitudinal imaging showed that nanoparticle accumulation in the tumor peaked at 24 h post-injection, giving a clear window for image-guided phototherapy. In the same year, Gao et al.271 developed a NIR-II PTT agent (DB4CaP NPs) for PTT and activation of the cGAS-STING immune pathway (Fig. 16B). Experimental results show that DB4CaP NPs effectively promote DC maturation and infiltration of tumor-infiltrating CTLs, enhance anti-tumor immune responses, and eliminate both local and metastatic tumors. Similar to the work of Liu et al., DB4CaP NPs can be activated by an 808 nm laser to emit NIR-II fluorescence at 1200 nm, thereby enabling determination of the time at which the nanoparticles reach peak accumulation at the tumor site, as well as their relatively prolonged circulation half-life. This provides reliable in vivo imaging information to support the precise implementation of therapeutic strategies.
 | ||
| Fig. 16 (A) Molecular structures of three organic materials used to construct SSPNiNO, and the mechanism of SSPNiNO in tumor treatment. (B) The mechanism of DB4CaP NPs in GBM treatment, and the molecular structure of the BODIPY tetramer (B4). (C) Signal conversion process of BN-O under hypoxia, and (D) the mechanism of BC@Z-M in tumor treatment. (E) The mechanism of OA-siSTAT1@FA-PNBs in myocardial infarction, and the molecular structure of the anti-inflammatory small molecule compound 9 and 10-nitro-octadec-9(E)-enoic acid [nitro-oleic acid (OA-NO2)]. Reproduced from ref. 271–274 with permission from John Wiley and Sons, Copyright 2024, John Wiley and Sons, Copyright 2024, Springer Nature, Copyright 2024, and The American Association for the Advancement of Science, Copyright 2025. | ||
The hypoxic TME significantly affects cancer treatment, particularly oxygen-dependent therapies like PDT. However, the hypoxic TME offers a foundation for developing new nano-diagnostic and therapeutic platforms. For example, the nano-diagnostic and therapeutic platform (BC@Z-M) developed by Song et al.272 in 2024 enables precise multimodal imaging and treatment under hypoxic conditions (Fig. 16C and D). Importantly, BC@Z-M contains Zn2+, which will be released into the TME after stimulation under acidic conditions (TME pH ≈ 6.5). This, in turn, activates the cGAS-STING pathway, promotes the production and release of type I interferons, enhances the innate anti-tumor immune response, and ultimately promotes DC maturation and CTL activation. In addition, they adopted a biomembrane delivery system, encapsulating the hypoxia-responsive optical probe (BN-O) within M1-type TAM membranes to enhance its cumulative efficiency in the TME. The hypoxic TME induces the transformation of BN-O from an “AA” structure to a “DA” structure, thereby activating NIRF and PA signals, as well as phototherapeutic effects. This enables noninvasive, high-contrast tumor imaging via NIRF and PA modalities, thereby providing crucial image guidance and precise navigation for subsequent photoimmunotherapy.
CD86 is a key biomarker for screening various cancers. It is widely expressed on the cell membrane surface of various TIICs (such as DCs, TAMs) and plays a critical role in T cell activation. Therefore, in 2024, Ma et al.273 developed a nanotherapeutic delivery system (OA-si@FA-PNBs) that can specifically target CD86-positive macrophages and has ultrasound-responsive release capability, and regulates the immune microenvironment via siSTAT1, a nucleic acid drug used to activate the STAT1 immune signalling pathway. Experimental results from animal models of myocardial infarction showed that the extent of fibrosis in the cardiac tissue of mice treated with OA-si@FA-PNBs was significantly reduced. Perfluorohexane in OA-si@FA-PNBs undergoes a liquid–gas phase transition under low-intensity focused ultrasound, forming microbubbles that enhance UL imaging and promote the release of small molecules (nitrated oil acid [OA-NO2] and siSTAT1) (Fig. 16E). Both work synergistically to promote the phenotypic transition of CD86-positive macrophages (M1 type) toward Cx3cr1-positive, thereby facilitating myocardial infarction recovery and alleviating post-infarction cardiac dysfunction.
The three therapeutic modules discussed above constitute the principal strategies currently integrated into multifunctional TIIC-targeted optical probes, reflecting the prevailing developmental trajectory of such platforms. Furthermore, tumor cells exploit endogenous immune checkpoint pathways—physiological negative regulatory mechanisms that maintain immune homeostasis—by overexpressing immune checkpoint ligands (e.g., PD-L1), thereby inducing T-cell exhaustion or functional suppression and facilitating immune evasion. ICIs restore anti-tumor immunity by blocking inhibitory signalling pathways, thereby reactivating effector T cells and promoting durable immune memory formation. ICIs have consequently become one of the most widely implemented immunotherapeutic modalities in clinical oncology. However, when administered as monotherapy, their therapeutic efficacy remains suboptimal, largely due to limited tissue penetration and insufficient accumulation within tumor sites. To address these limitations, several studies have explored the integration of PD-L1-targeting strategies into multifunctional nanoplatforms. For example, Zhang et al.,274 Deng et al.,275 and Jing et al.276 achieved enhanced immunotherapeutic outcomes in tumor models such as GL261, CT26, and MDA-MB-231 by incorporating PD-L1 modulation into multifunctional systems. These investigations provide valuable design paradigms for the development of TIIC-targeted optical probes endowed with immunotherapeutic functionality. Given the established clinical relevance of ICIs, their integration with TIIC-targeted optical probes holds substantial promise for improving translational potential and advancing precision immunotherapy.
The above work points to a clear trend: packing multiple therapeutic functions into TIIC-targeted optical probes is not just an add-on—it is becoming essential. These probes are moving beyond simply telling us where immune cells are. Instead, they are turning into closed-loop systems that do “identification–localization–intervention–feedback” right inside the tumor immune microenvironment. Why does that matter? Because TIICs do more than predict outcomes. Their functional state, spatial arrangement, heterogeneity, and cross-talk with tumor cells directly shape how well ICIs, cancer vaccines, and other immunotherapies work. These probes are already used for real-time in vivo imaging, drug screening, predicting efficacy, and patient stratification. Adding therapeutic modules—chemotherapy, PDT/PTT, ICIs, activating anti-tumor pathways, reprogramming TIICs—isn’t just stacking features. It turns immune cells from passive observation targets into active handles for intervention. That lets these probes directly help design precision immunotherapy protocols.
A major plus is real-time imaging, which speeds up decision making. Traditional biopsies or single time-point ex vivo analyses give only static snapshots. TIIC-targeted probes, in contrast, track changes in the TIME dynamically. They can spot specific TIIC subsets in vivo, with sensitivity and specificity approaching that of flow cytometry on biopsy samples. They also monitor immunotherapy responses in real time and predict how tumors will grow under different treatments. Shifting from morphological endpoints to early functional endpoints is a game-changer for optimizing immunotherapy regimens inside multifunctional platforms. Then there's spatial precision. Unlike systemic, non-selective immune activation, these probes are typically activated only when two or more independent conditions are met. For example, the probe can be activated by simultaneously binding to tumor and TIIC markers, or by recognizing a tumor marker (or a TIIC marker) and simultaneously responding to key physicochemical conditions of the TME such as hypoxia and low pH. As a result, therapy lands where it is needed: at lesions, in immune-rich areas, or inside key immunosuppressive niches. That cuts down non-specific background signals and off-target effects. Equally important, functional monitoring helps with patient stratification and efficacy prediction. Optical readouts from integrated platforms can also guide dynamic regimen adjustments. A persistent problem in immunotherapy (both in the clinic and in preclinical work) is that some patients don’t mount a strong enough effector immune response early on. If you stick to the initial plan without adjusting, you may miss the best window for combination therapy or switching treatments. Multifunctional TIIC-targeted probes—by combining imaging, drug delivery, and functional feedback—can tell you early whether immune activation is sufficient. That information can drive decisions like adding checkpoint blockade, boosting innate immunity, tweaking light dose, or reordering treatment steps.
So, the real value of integrating therapeutic functions into TIIC-targeted optical probes is this: they move cancer immunotherapy away from the old linear “treat first, evaluate later” model, toward a closed loop of “identify, locate, treat and monitor at the same time, then dynamically optimize.” That doesn’t just improve precision and local immune remodeling. It also gives direct functional evidence for patient stratification, predicting efficacy, and personalizing adaptive treatments (Table 3).
| Name | Administration method | Dosage | Treatment type | Tumor types | Tumor location | Anti-tumor effect | Ref. |
|---|---|---|---|---|---|---|---|
| S&D@Ag2Se | Intravenous injection | 1 or 2 mg kg−1 (DOX) | Chemotherapy | MB-231 breast cancer (human-derived) | In situ (mammary fat pad) | Significant inhibition | 257 |
| Olsa-RVRR | Intravenous injection | 0.1 mmol kg−1 (139 mg kg−1) | Chemotherapy | HCT116 and LoVo colorectal cancer (both human-derived) | Left (HCT116) and right (LoVo) ventral side | Tumor inhibition rate: 62% (HCT116); 39% (LoVo) | 258 |
| GEL-CpG NP-DOX | Intratumoral injection | 5 mg kg−1 (DOX); 50 µg (CpG) | Chemotherapy | B16F10 melanoma (mouse-derived) | Subcutaneous | Significant inhibition | 259 |
| PEG/αCD25-Cy7/TMZ | Intravenous injection | 1.5 mM, 200 µL | Chemotherapy | GL261 glioblastoma (mouse-derived) | In situ and subcutaneous | Significant inhibition | 154 |
| PACDx/PARx | Intratumoral and retroorbital injection | PACDx (400 µM)/PARx (100 µM or 400 µM) | Chemotherapy | A549 lung cancer (human-derived) | In situ; liver metastasis; subcutaneous | Complete suppression | 155 |
| Pp18-lipids | Intravenous injection | 6 mg kg−1 | PDT/PTT | 4T1 breast cancer cells (mouse-derived) | Subcutaneous | Tumor inhibition rate: 90.1% | 264 |
| DDTB-DP NPs | Intravenous injection | 1 mg mL−1, 200 µL | PDT/PTT and ICIs | B16F10 melanoma and 4T1 breast cancer cells (both mouse-derived) | Subcutaneous | Survival rate: up to 90% | 265 |
| PBNO-Ce6 | Intravenous injection | 15 mg mL−1, 100 µL | PDT/PTT | 4T1 breast cancer cells (mouse-derived) | Subcutaneous | Median survival > 60 days (compared to 25 days in the control group). | 266 |
| FAL-ICG-HAuNS + FAL-Hb-lipo | Intravenous injection | ICG: 0.5 mg kg−1, HAuNS: 1 mg kg−1, Hb: 20 mg kg−1 | PDT/PTT | CT26 colon cancer and B16F10 melanoma (both mouse-derived) | Subcutaneous | Significant inhibition | 269 |
| SSPNiNO | Intravenous injection | SSPNiNO: 300 µg mL−1, 200 µL; A2AR: 1 mg kg−1, 200 µL | Immune pathway stimulation and PDT/PTT | C6 glioma (mouse-derived) | In situ | Almost completely suppressed | 270 |
| DB4CaP NPs | Intravenous injection | B4: 1 mg kg−1; dsDNA: 0.625 mg kg−1; anti-PD-1: 20 µg | Immune pathway stimulation, PDT/PTT, and ICIs | CT26 colon cancer (mouse-derived) | Subcutaneous tumor/metastatic tumor/recurrent tumor | Complete remission rate: 83.3% | 271 |
| BC@Z-M | Intraperitoneal and subcutaneous injection | 1 mg mL−1, 200 µL (intraperitoneal) and 50 µL (subcutaneous) | Immune pathway stimulation and PDT/PTT | 4T1 breast cancer cells (mouse-derived) | Subcutaneous tumor/metastatic tumor | Tumor inhibition rate: 81% (subcutaneous tumor) and 85% (metastatic tumor) | 272 |
| OA-si@FA-PNBs | Intravenous injection | 200 µg mL−1 | Immune pathway stimulation | Non-tumor model (myocardial infarction model) | — | Significantly inhibits cardiac fibrosis- | 273 |
| d-K@γ-PGA@5-ALA@MUCNP@aPDL1 | Intravenous injection | — | ICIs and PDT | GL261 glioblastoma (mouse-derived) | In situ | Significant inhibition | 274 |
| αPDL1@MnO2 | Intravenous injection | 10 mg kg−1 | Immune pathway stimulation, radiotherapy, and ICIs | CT26 colon cancer (mouse-derived) | Subcutaneous | Significant inhibition | 275 |
| Pt@RHC-LXL-1/TPP/CLP002 | Intravenous injection | 100 µg mL−1, 100 µL | ICIs and immune pathway stimulation | MB-231 breast cancer (human-derived) | Subcutaneous | Tumor inhibition rate: 90% | 276 |
6. Conclusion and perspectives
Optical probes that target tumor-infiltrating immune cells (TIICs) have emerged as versatile tools in cancer immunotherapy. By tracking TIICs in vivo, they enable specific, sensitive, and non-invasive visualization of immune cell trafficking, infiltration, and functional state within the TME. Their current applications range from basic TIIC biology and immunotherapy response monitoring to theranostic integration and image-guided surgery. In this review, we have categorized existing TIIC-targeted optical probes by their response mechanisms, summarized how they are being integrated with immunotherapy, and outlined strategies for embedding therapeutic functions into probe design. Our aim is to provide a practical framework for designing next-generation probes that can handle the complexity of the tumor immune microenvironment.What optical probes should bring to immunotherapy—but rarely do. A common problem: most studies only report probe performance (brightness, stability, specificity) without actually showing an improved immunotherapeutic outcome. We see plenty of imaging data but few clear links to better treatment decisions. In principle, optical probes could actively guide therapy. Real-time imaging could track TIIC infiltration and activation as treatment unfolds, not just at isolated time points. Spatial targeting could deliver immune modulators precisely where needed, reducing off-target effects. Functional readouts could tell us early whether a patient mounts a useful effector response—information that would support rational adjustments (adding a checkpoint blocker, changing light dose, altering sequence). Probe-based signals could even inform patient stratification and dynamic regimen changes, moving beyond the old “treat first, evaluate later” model. Yet almost none of this is done systematically. Most work stops at probe validation in animal models, never integrating the readout into a closed-loop therapeutic strategy. The field needs to shift from designing brighter probes to designing smarter ones—where imaging feedback directly drives intervention. That is the real value of probe-integrated immunotherapy, and it remains largely unrealized.
Despite great advances, several critical hurdles persist. At the most fundamental level: can current probes reliably distinguish TIICs from tumor cells? Many biomarkers used for TIIC imaging—ROS, NE, certain MMPs—are also expressed or produced by tumor cells themselves. This overlap raises genuine concerns about signal specificity and misidentification. This challenge has spurred strategies such as multi-lock probes that incorporate multiple responders to prevent erroneous activation. Unfortunately, multi-lock probes, while highly specific, often suffer from complex synthesis, slow activation kinetics, and reduced signal amplitude due to multiple energy transfer steps. Meanwhile, ATC-based strategies (homologous cell membrane coating or in vitro reprogramming of TIICs) primarily address probe enrichment in tumors and prolonged circulation, getting more probes to the tumor site. But these approaches have their own limitations: potential immunogenicity, batch-to-batch variability, and constrained cargo capacity. More importantly, even with enhanced delivery, probes entering the TME still face the core problem of distinguishing immune cells from their malignant neighbors.
Addressing this will require a shift from recognition-only designs to recognition-and-retention mechanisms that achieve spatially constrained activation.277 For instance, in situ self-assembling probes leverage TIIC-associated enzymes to trigger local aggregation, enhancing retention specifically within immune cells.278–280 Alternatively, TME-anchored responsive probes undergo conformational change and covalent binding only upon encountering immune-rich tumor regions.281 Target-activated covalent binding strategies couple signal generation with permanent retention at the target site, allowing unbound probes to be washed out and significantly improving imaging fidelity. Looking ahead, resolving immune–tumor interactions will demand probes that can spatially resolve multiple targets at once. NIR multi-color platforms engineered for compartment-specific sensing could integrate orthogonal sensors on a single nanocarrier, each tuned to a distinct biomarker combination with well-separated spectral signatures.239,282 Going further, activatable probes with molecular logic gates (AND, OR, NOT) could discriminate TIICs from tumor cells based on combinatorial biomarker expression. A probe that requires both NE and CTSG (enriched in neutrophil extracellular traps) for activation, for instance, would light up NETs specifically while remaining dark in the presence of viable neutrophils or tumor cells alone. Ultimately, achieving single-cell resolution across the NIR spectrum would transform our understanding of TIIC–tumor crosstalk. Spatially mapping TIIC subsets and their functional states relative to surrounding cancer cells could reveal how local immune architecture shapes therapeutic response and identify spatially defined biomarkers for precision immuno-oncology.
Beyond cellular specificity, clinical translation also faces technical hurdles related to imaging performance. The need to image deep-seated tumors (lung, liver, ovarian) imposes stringent requirements on probe design in two areas: deep tissue penetration and long-term monitoring. NIR-II fluorescence reduces photon scattering and autofluorescence, but it still suffers from photobleaching and repeated excitation during long-term monitoring. Emerging NIR-II afterglow imaging probes address this by providing a prolonged imaging window after a single excitation, minimizing optical background and allowing repeated sessions without continuous illumination.283 Still, these advanced optical methods largely provide only molecular information. To understand the complex spatial architecture of TIICs within tumors, we also need complementary anatomical and structural context. This has motivated the development of multimodal optical probes that combine fluorescence with PA, CT, MRI, UL, MPI, or PET. These integrations trade off sensitivity, penetration depth, and spatial resolution. By combining modalities, we can correlate TIIC distribution with detailed anatomical features, giving multidimensional insights into tumor immune architecture. Nevertheless, current multimodal probes bring their own problems: complex synthesis, batch-to-batch variability, and potential interference between imaging modalities.225,284 Additionally, larger size and altered surface chemistry can also affect pharmacokinetics, biodistribution, and tumor penetration. Future efforts should focus on simpler, robust designs with orthogonal signal outputs that minimize mutual interference and maintain favorable in vivo behavior.
In parallel with these technical challenges, the current landscape of TIIC-targeted optical probes is characterized by a significant imbalance in research focus. The majority of probes have been developed for abundant myeloid populations such as macrophages and neutrophils, with ROS-responsive probes being particularly prevalent due to the relative ease of designing chemical sensors for small molecules. In contrast, many lymphoid subsets—particularly Tregs, memory T cells, and innate lymphoid cells—remain underexplored. This disparity stems primarily from the limited availability of robust, specific surface markers for these populations, as well as the challenge of designing probes that can distinguish functionally distinct subsets (e.g., activated versus exhausted T cells) within the same lineage. Tregs exemplify this gap. Increasing evidence indicates that Tregs exhibit pronounced functional heterogeneity, metabolic plasticity, and context-dependent roles in tumors, closely correlating with prognosis and immunotherapy outcomes. Current clinical detection of Tregs relies largely on conventional methods such as tissue biopsy, while imaging-based approaches remain limited by high cost and technical complexity. The absence of Treg-specific optical probes prevents real-time visualization of these cells within the TME, substantially impeding the development and assessment of Treg-based immunotherapies. Therefore, addressing this imbalance by developing probes for underrepresented TIIC subsets represents an important and promising direction for future research.
To overcome both the biological and technical challenges outlined above, future progress will increasingly depend on cross-disciplinary collaboration. Beyond traditional recognition element screening techniques, innovative platforms such as the diversity-oriented fluorescence library (DOFL) approach offer powerful strategies for discovering novel probes.285,286 DOFL enables systematic generation and screening of large libraries of fluorescent small molecules, allowing identification of probes that can distinguish subtle phenotypic differences between immune cell subsets without predefined biomarkers. This phenotype-based discovery paradigm is particularly valuable for targeting currently “undruggable” or poorly characterized TIIC populations, opening new avenues for immune cell visualization. Moreover, the advanced analytical and pattern-recognition capabilities of artificial intelligence (AI) will be crucial for future probe development and application. AI can accurately match existing data, correlate imaging readouts with disease characteristics, systematically analyze datasets, and predict potential risks with higher accuracy.
Ultimately, successful translation of TIIC-targeted optical probes from bench to bedside depends on addressing challenges in both probe design and instrumentation. Many well-designed probes still require extensive clinical trials before they can be used in patients. Researchers developing new probes should prioritize FDA-approved biocompatible materials, such as Pluronic F127, PLGA, and ICG. These materials have excellent biocompatibility, biodegradability, and low immunogenicity, and are already widely used in clinical practice and research. Using them can speed up regulatory review. On the instrumentation side, the trend toward multimodal and multifunctional probes demands parallel development of highly compatible, portable, and user-friendly imaging devices. Image quality depends as much on the instrument as on the probe itself. Close collaboration between probe chemists and biomedical engineers will be essential for successful clinical adoption. In short, the road to high-performance tumor diagnosis and treatment is long and hard, but with continued innovation and interdisciplinary work, optical probes will undoubtedly become indispensable allies in this fight.
Author contributions
Prof. Lemeng Zhang, Mr. Xinglong Chen, Mr. Cong Hu, and Prof. Hongwen Liu: conceptualization, formal analysis, investigation, writing – original draft. Heejeong Kim and Zhipengjun Zhang: investigation. Prof. Juyoung Yoon, Hongwen Liu and Lemeng Zhang: supervision, project administration, funding acquisition, review & editing.Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Acknowledgements
This work was supported by the National Research Foundation of Korea (Grants 2022R1A2C3005420 and RS-2024-00407093), Natural Science Foundation of Hunan Province (Grants 2026JJ30157 and 2025JJ40014), National Natural Science Foundation of China (Grant 22474036), Scientific Research Project of Hunan Provincial Health Commission (Grant No. 20256533), Hunan Cancer Hospital Climb Plan (Grant No. 2023NFSC-A006), High-Level Talent Support Program of Hunan Cancer Hospital (20250801-1009) and High-Level Talent Support Program of Hunan Province (20250905-1002).References
- J. Fang, Y. Lu, J. Zheng, X. Jiang, H. Shen, X. Shang, Y. Lu and P. Fu, Cell Death Dis., 2023, 14, 586 CrossRef PubMed.
- R. Wang, C. Lan, K. Benlagha, N. O. S. Camara, H. Miller, M. Kubo, S. Heegaard, P. Lee, L. Yang, H. Forsman, X. Li, Z. Zhai and C. Liu, MedComm, 2024, 5, e714 CrossRef CAS PubMed.
- W. Li, F. Li, X. Zhang, H.-K. Lin and C. Xu, Signal Transduct. Target. Ther., 2021, 6, 422 CrossRef PubMed.
- Y. Cao, B. Qiao, Q. Chen, Z. Xie, X. Dou, L. Xu, H. Ran, L. Zhang and Z. Wang, Acta Biomater., 2023, 160, 239–251 CrossRef CAS PubMed.
- Y. Yang, J. Guo and L. Huang, Trends Pharmacol. Sci., 2020, 41, 701–714 CrossRef CAS PubMed.
- S. Camargo, O. Moskowitz, A. Giladi, M. Levinson, R. Balaban, S. Gola, A. Raizman, K. Lipczyc, A. Richter, N. Keren-Khadmy, O. Barboy, Y. Dugach, Y. Carmi, A. Sonnenblick and M. Cohen, Nat. Cancer, 2025, 6, 540–558 CrossRef CAS PubMed.
- E. Eruslanov, Y. Nefedova and D. I. Gabrilovich, Nat. Immunol., 2025, 26, 17–28 CrossRef CAS PubMed.
- J. K. Mandula, R. A. Sierra-Mondragon, R. V. Jimenez, D. Chang, E. Mohamed, S. Chang, J. A. Vazquez-Martinez, Y. Cao, C. M. Anadon, S. B. Lee, S. Das, L. Rocha-Munguba, V. M. Pham, R. Li, A. A. Tarhini, M. Furqan, W. Dalton, M. Churchman, C. M. Moran-Segura, J. Nguyen, B. Perez, D. J. Kojetin, A. Obermayer, X. Yu, A. Chen, T. I. Shaw, J. R. Conejo-Garcia and P. C. Rodriguez, Immunity, 2024, 57, 1124–1140 CrossRef CAS PubMed.
- X. Pan and L. Zheng, Cell. Mol. Immunol., 2020, 17, 940–953 CrossRef CAS PubMed.
- S. Qin, B. Xie, Q. Wang, R. Yang, J. Sun, C. Hu, S. Liu, Y. Tao and D. Xiao, MedComm, 2024, 5, e551 CrossRef CAS PubMed.
- Y. Liu, Y. Zhao, H. Song, Y. Li, Z. Liu, Z. Ye, J. Zhao, Y. Wu, J. Tang and M. Yao, Cancer Lett., 2024, 597, 217076 CrossRef CAS PubMed.
- A. Hu, L. Sun, H. Lin, Y. Liao, H. Yang and Y. Mao, Signal Transduct. Target. Ther., 2024, 9, 68 CrossRef PubMed.
- P. J. Gawne, F. Man, P. J. Blower and R. T. M. de Rosales, Chem. Rev., 2022, 122, 10266–10318 CrossRef CAS PubMed.
- M. B. Cooley, D. Wegierak and A. A. Exner, WIREs Nanomed. Nanobiotechnol., 2024, 16, e1957 CrossRef CAS PubMed.
- A. Rizwan, B. Sridharan, J. H. Park, D. Kim, J.-C. Vial, K. Kyhm and H. G. Lim, J. Nanobiotechnol., 2025, 23, 170 CrossRef PubMed.
- W. L. Bi, A. Hosny, M. B. Schabath, M. L. Giger, N. J. Birkbak, A. Mehrtash, T. Allison, O. Arnaout, C. Abbosh, I. F. Dunn, R. H. Mak, R. M. Tamimi, C. M. Tempany, C. Swanton, U. Hoffmann, L. H. Schwartz, R. J. Gillies, R. Y. Huang and H. J. W. L. Aerts, CA: A Cancer J. Clin., 2019, 69, 127–157 Search PubMed.
- J. Pellico, P. J. Gawne and R. T. M. de Rosales, Chem. Soc. Rev., 2021, 50, 3355–3423 RSC.
- B. Li, M. Zhou, C. Zhao, L. Xiao, T. Qi, H. Xu, L. Guo, G. Ning, X. Lu, K. Zhu, H. Zhao and C.-P. Li, J. Anal. Test., 2025, 9, 21–31 CrossRef CAS.
- F. Yu, Y. Wang, C. Yu, W. Zhang and X. Bai, J. Anal. Test., 2024, 8, 237–244 CrossRef CAS.
- B. Fan, S. Wang, S. Huang, S. Ma, H. Zuo, S. Li and Z. Chen, J. Anal. Test., 2026, 10, 83–101 CrossRef CAS.
- Y. Yu, Z. Li, S. Liao, B. Yin, Q. Zhang, J. Fu, C. Zhang, Y. Zhou and G. Song, Research, 2025, 8, 0834 CrossRef CAS.
- L. Gong, H. Wang, L. Zhang, J. Li, H. Zhang, Y. Sun and L.-A. Fan, J. Anal. Test., 2025, 9, 232–239 CrossRef CAS.
- X. He, Y. Dong, P. He, C. Liu and W. Ren, J. Anal. Test., 2024, 8, 270–277 CrossRef CAS.
- S.-S. Zhang, H. Lee, Y. Zhou and J. Yoon, Coord. Chem. Rev., 2026, 549, 217255 CrossRef CAS.
- M. Kazim, A. Ganguly, S. M. Malespini, L. Thang, N. L. Patel, C. Kim, K. Thakur, J. D. Kalen, C. Calcagno, S. Difilippantonio and E. Yoo, J. Am. Chem. Soc., 2025, 147, 37926–37939 CrossRef CAS PubMed.
- Y. Li, J. Lin, J. Zou, Y. Zeng and X. Liu, Nat. Commun., 2025, 16, 10880 CrossRef CAS PubMed.
- Y. Feng, Y. Hu, J. Liu, X. Zhang, Y. Zhang, Z. Wang and K. Pu, Angew. Chem., Int. Ed., 2025, 64, e202507765 CrossRef CAS PubMed.
- L. Ding, X. Zhang, P. Wang, J. Ke, Y. Zhou, M. Wu, Z. Wei, Y. Cao, H. Li, G. Chen, G. Zheng, Y. Zeng, M. Hong and X. Liu, Nat. Commun., 2025, 16, 8054 CrossRef CAS PubMed.
- D. Gong, Z. Xiao, Y. Hu, S. Wu, Z. Yi, T. Sheng, Z. Zha, Y. Ma and Z. Miao, Chem. Eng., 2024, 499, 156099 CrossRef CAS.
- D. Hu, M. Zha, H. Zheng, D. Gao and Z. Sheng, Research, 2025, 8, 0583 CrossRef CAS PubMed.
- Y. Zhou, X. Kuang, X. Yang, J. Li, X. Wei, W. J. Jang, S.-S. Zhang, M. Yan and J. Yoon, Chem. Sci., 2024, 15, 19669–19697 RSC.
- Y. Chen, Q. Chen, Y. Ma, X. Su, C. Zhang, K. Li, M. Liang, M. Sun, L. Zhang, X. Kuang, T. Zhang, J. Zhang, R. Yan, S. Ju and W. Fan, J. Am. Chem. Soc., 2025, 147, 24884–24899 CrossRef CAS PubMed.
- Y. Hang, A. Wang and N. Wu, Chem. Soc. Rev., 2024, 53, 2932–2971 RSC.
- J. Li, J. Wang, L. Xu, H. Chi, X. Liang, J. Yoon and W. Lin, Angew. Chem., Int. Ed., 2024, 63, e202312632 CrossRef CAS PubMed.
- J. Mei and H. Tian, Aggregate, 2021, 2, e32 CrossRef CAS.
- H. Fang, Y. Chen, Z. Jiang, W. He and Z. Guo, Acc. Chem. Res., 2023, 56, 258–269 CrossRef CAS PubMed.
- Y. Ji, K. Bao, L. Mei, Y. Su, Y. Wu, C. Li, Y. Wu, Z. Ge, S. Choi, Z. Wang and H. S. Choi, Biomater. Res., 2025, 29, 0275 CrossRef CAS PubMed.
- H. Cao, X. Meng, Y. Ma, Z. Li, X. Xu, J. Fu, Q. Zhang, H. Wei, P. Liang, D. Lu, J. Li, B. Yin, S. Huan, X.-B. Zhang and G. Song, Nat. Commun., 2025, 16, 9635 CrossRef CAS.
- J. Zhu, L. Zhao, W. An and Q. Miao, Chem. Soc. Rev., 2025, 54, 1429–1452 RSC.
- G. Hong, A. L. Antaris and H. Dai, Nat. Biomed. Eng., 2017, 1, 0010 CrossRef CAS.
- E. L. Schmidt, Z. Ou, E. Ximendes, H. Cui, C. H. C. Keck, D. Jaque and G. Hong, Nat. Rev. Methods Primers, 2024, 4, 23 CrossRef CAS.
- F. Wang, Y. Zhong, O. Bruns, Y. Liang and H. Dai, Nat. Photonics, 2024, 18, 535–547 CrossRef CAS.
- S. Choi, J. Kim, H. Jeon, C. Kim and E.-Y. Park, npj Acoust., 2025, 1, 1 CrossRef.
- L. Lin and L. V. Wang, Nat. Rev. Clin. Oncol., 2022, 19, 365–384 CrossRef.
- M. Yang, J. Huang, J. Fan, J. Du, K. Pu and X. Peng, Chem. Soc. Rev., 2020, 49, 6800–6815 RSC.
- Y. Yang, S. Wang, L. Lu, Q. Zhang, P. Yu, Y. Fan and F. Zhang, Angew. Chem., Int. Ed., 2020, 59, 18380–18385 CrossRef CAS PubMed.
- J. Huang, Y. Jiang, J. Li, J. Huang and K. Pu, Angew. Chem., Int. Ed., 2021, 60, 3999–4003 CrossRef CAS PubMed.
- S. Liu, Y. Su, M. Z. Lin and J. A. Ronald, ACS Chem. Biol., 2021, 16, 2707–2718 CrossRef CAS PubMed.
- T. Kuchimaru, S. Iwano, M. Kiyama, S. Mitsumata, T. Kadonosono, H. Niwa, S. Maki and S. Kizaka-Kondoh, Nat. Commun., 2016, 7, 11856 CrossRef CAS PubMed.
- L. Lu, B. Li, S. Ding, Y. Fan, S. Wang, C. Sun, M. Zhao, C.-X. Zhao and F. Zhang, Nat. Commun., 2020, 11, 4192 CrossRef PubMed.
- C. Xu, Y. Zhang, G. Liang and K. Pu, Nat. Mater., 2025, 24, 1694–1708 CrossRef CAS PubMed.
- C. Xu, J. Huang, Y. Jiang, S. He, C. Zhang and K. Pu, Nat. Biomed. Eng., 2023, 7, 298–312 CrossRef CAS PubMed.
- P. Pei, Y. Chen, C. Sun, Y. Fan, Y. Yang, X. Liu, L. Lu, M. Zhao, H. Zhang, D. Zhao, X. Liu and F. Zhang, Nat. Nanotechnol., 2021, 16, 1011–1018 CrossRef CAS PubMed.
- J. Zhou, P. J. Schuck and D. Jin, Nat. Rev. Phys., 2026, 8, 195–207 CrossRef.
- C. Chen, F. Wang, S. Wen, Q. P. Su, M. C. L. Wu, Y. Liu, B. Wang, D. Li, X. Shan, M. Kianinia, I. Aharonovich, M. Toth, S. P. Jackson, P. Xi and D. Jin, Nat. Commun., 2018, 9, 3290 CrossRef PubMed.
- P. J. Withers, C. Bouman, S. Carmignato, V. Cnudde, D. Grimaldi, C. K. Hagen, E. Maire, M. Manley, A. Du Plessis and S. R. Stock, Nat. Rev. Methods Primers, 2021, 1, 18 CrossRef CAS.
- S. Azhar and L. R. Chong, Postgrad. Med. J., 2022, 99, 894–903 CrossRef PubMed.
- J. J. Vaquero and P. Kinahan, Annu. Rev. Biomed. Eng., 2015, 17, 385–414 CrossRef CAS PubMed.
- M. Ljungberg and P. H. Pretorius, Br. J. Radiol., 2018, 91, 20160402 CrossRef PubMed.
- J. Zhu, W. Chen, L. Yang, Y. Zhang, B. Cheng, W. Gu, Q. Li and Q. Miao, Angew. Chem., Int. Ed., 2024, 63, e202318545 CrossRef CAS PubMed.
- J. Huang, M. Xu, P. Cheng, J. Yu, J. Wu and K. Pu, Angew. Chem., Int. Ed., 2024, 63, e202319780 CrossRef CAS PubMed.
- Y. Hu, J. Yu, M. Xu and K. Pu, J. Am. Chem. Soc., 2024, 146, 12656–12663 CrossRef CAS PubMed.
- S. Zhang, M. Ma, C. Zhao, J. Li, L. Xu, Z. Zhang, Q. Diao, P. Ma and D. Song, Biosens. Bioelectron., 2024, 261, 116514 CrossRef CAS PubMed.
- X. Wang, B. Song, M. Wu, L. Qin and W. Liang, Mater. Today Bio, 2025, 32, 101694 CrossRef CAS PubMed.
- T. J. M. Manoharan, K. Ravi, A. P. Suresh, A. P. Acharya and M. Nikkhah, Adv. Healthcare Mater., 2024, 13, 2303658 CrossRef CAS PubMed.
- M. Kazim and E. Yoo, Angew. Chem., Int. Ed., 2024, 63, e202310694 CrossRef CAS PubMed.
- J. Cui, F. Zhang, D. Yan, T. Han, L. Wang, D. Wang and B. Z. Tang, Adv. Mater., 2023, 35, 2302639 CrossRef CAS PubMed.
- L. Li, A. Liu, H. Liang, X. Li, M. D. Nešić, H. Guo, Z. Wang and Q. Lin, Chem. Eng., 2025, 518, 164692 CrossRef CAS.
- L. Ding, Z. Lyu, T.-A. Perles-Barbacaru, A. Y.-T. Huang, B. Lian, Y. Jiang, T. Roussel, C. Galanakou, S. Giorgio, C.-L. Kao, X. Liu, J. Iovanna, M. Bernard, A. Viola and L. Peng, Adv. Mater., 2024, 36, 2308262 CrossRef CAS PubMed.
- J. Li, N. Niu, D. Wang, J. Zhu, X. Li, Q. Kong, B. Zhong Tang and D. Wang, Angew. Chem., Int. Ed., 2025, 64, e202413219 CrossRef CAS PubMed.
- Q. Yue, Q. Zeng, Q. Guo, X. Zhao, Y. Yuan, Y. Yang, W. Jiang and X. Zhou, Adv. Healthcare Mater., 2024, 13, 2402915 CrossRef CAS PubMed.
- W. Zhang, M. Kang, X. Li, H. Yang, Z. Zhang, Z. Li, Y. Zhang, M. Fan, C. Liao, C. Liu, G. Xu, D. Wang, Z. Xu and B. Z. Tang, Adv. Mater., 2024, 36, 2406474 CrossRef CAS PubMed.
- G. Shu, T. Jiang, X. Zhang and H. Zhao, Chem. Eng., 2024, 494, 153086 CrossRef CAS.
- M. Zhang, Y. Wu, X. Lu, Y. Qin, C. Zhang, W. Gu, R. Gao, Y. Qi, M. Wang and Q. Huang, Chem. Eng., 2024, 494, 152953 CrossRef CAS.
- S. Qin, Q. Wang, Z. Zhang, J. Gu, G. He, F. Zeng, R. Chen, B. He, Y. Wang, M. Wang and Y. Song, J. Am. Chem. Soc., 2025, 147, 38180–38194 CrossRef CAS PubMed.
- S. Zuo, H. Liu, G. Jiang, Y. Chen, T. Ren, Z. Zhou, X.-B. Zhang and L. Yuan, J. Am. Chem. Soc., 2025, 147, 42825–42838 CAS.
- H. Xu, H. Hong, C. Kim, Y. Lee, Y. Li, Y. S. Zhang, P. Makvandi, G. Song, H. Zhang, H. Kang and J. Yoon, Chem. Rev., 2025, 125, 11461–11523 CrossRef CAS PubMed.
- X. Lin, R. Zhu, Z. Hong, X. Zhang, S. Chen, J. Song and H. Yang, Adv. Funct. Mater., 2021, 31, 2101278 CrossRef CAS.
- Z. Li, Y. Yu, W. Zeng, F. Ding, D. Zhang, W. Cheng, M. Wang, H. Chen, G. Pan, L. Mei, X. Zeng and N. Gao, Small, 2022, 18, 2201803 CrossRef CAS PubMed.
- B. Qiao, Y. Luo, H.-B. Cheng, J. Ren, J. Cao, C. Yang, B. Liang, A. Yang, X. Yuan, J. Li, L. Deng, P. Li, H.-T. Ran, L. Hao, Z. Zhou, M. Li, Y. Zhang, P. S. Timashev, X.-J. Liang and Z. Wang, ACS Nano, 2020, 14, 12652–12667 CrossRef CAS PubMed.
- K. Du, P. Lei, L. Dong, M. Zhang, X. Gao, S. Yao, J. Feng and H. Zhang, Appl. Mater. Today, 2020, 18, 100497 CrossRef.
- R. D. Leone and J. D. Powell, Nat. Rev. Cancer, 2020, 20, 516–531 CrossRef CAS PubMed.
- C. Suo, E. Dann, I. Goh, L. Jardine, V. Kleshchevnikov, J.-E. Park, R. A. Botting, E. Stephenson, J. Engelbert, Z. K. Tuong, K. Polanski, N. Yayon, C. Xu, O. Suchanek, R. Elmentaite, C. Domínguez Conde, P. He, S. Pritchard, M. Miah, C. Moldovan, A. S. Steemers, P. Mazin, M. Prete, D. Horsfall, J. C. Marioni, M. R. Clatworthy, M. Haniffa and S. A. Teichmann, Science, 2022, 376, eabo0510 CrossRef CAS PubMed.
- S. Hillion, M. I. Arleevskaya, P. Blanco, A. Bordron, W. H. Brooks, J. Y. Cesbron, S. Kaveri, E. Vivier and Y. Renaudineau, Clin. Rev. Allergy Immunol., 2020, 58, 151–154 CrossRef PubMed.
- B. Ruf, B. Heinrich and T. F. Greten, Cell. Mol. Immunol., 2021, 18, 112–127 CrossRef CAS PubMed.
- M. Chen, X. Shi, Y. Wang, J. Zhang, J. Guo, X. Guan, Y. Sun, W. Wu, C. Li, Y. Tian, Y. Ou, T. Li, K. Jiang, M. D. Taylor, X. Liao, L. Zhang and T. Sun, eBioMedicine, 2025, 122, 106043 CrossRef CAS PubMed.
- H. Que, Q. Fu, T. Lan, X. Tian and X. Wei, BBA-Rev. Cancer, 2022, 1877, 188762 CAS.
- W.-J. Qian, J.-S. Yan, X.-Y. Gang, L. Xu, S. Shi, X. Li, F.-J. Na, L.-T. Cai, H.-M. Li and M.-F. Zhao, BBA-Rev. Cancer, 2024, 1879, 189187 CAS.
- H. Won Jun, H. Kyung Lee, I. Ho Na, S. Jeong Lee, K. Kim, G. Park, H. Sook Kim, D. Ju Son, Y. Kim, J. Tae Hong and S.-B. Han, Int. Immunopharmacol., 2022, 113, 109332 CrossRef CAS PubMed.
- L. Sun, Y. Su, A. Jiao, X. Wang and B. Zhang, Signal Transduct. Target. Ther., 2023, 8, 235 CrossRef CAS PubMed.
- C. Domínguez Conde, C. Xu, L. B. Jarvis, D. B. Rainbow, S. B. Wells, T. Gomes, S. K. Howlett, O. Suchanek, K. Polanski, H. W. King, L. Mamanova, N. Huang, P. A. Szabo, L. Richardson, L. Bolt, E. S. Fasouli, K. T. Mahbubani, M. Prete, L. Tuck, N. Richoz, Z. K. Tuong, L. Campos, H. S. Mousa, E. J. Needham, S. Pritchard, T. Li, R. Elmentaite, J. Park, E. Rahmani, D. Chen, D. K. Menon, O. A. Bayraktar, L. K. James, K. B. Meyer, N. Yosef, M. R. Clatworthy, P. A. Sims, D. L. Farber, K. Saeb-Parsy, J. L. Jones and S. A. Teichmann, Science, 2022, 376, eabl5197 CrossRef PubMed.
- M. G. Netea, J. Domínguez-Andrés, L. B. Barreiro, T. Chavakis, M. Divangahi, E. Fuchs, L. A. B. Joosten, J. W. M. van der Meer, M. M. Mhlanga, W. J. M. Mulder, N. P. Riksen, A. Schlitzer, J. L. Schultze, C. Stabell Benn, J. C. Sun, R. J. Xavier and E. Latz, Nat. Rev. Immunol., 2020, 20, 375–388 CrossRef CAS PubMed.
- C. Silvestre-Roig, L. Kalafati and T. Chavakis, Signal Transduct. Target. Ther., 2024, 9, 77 CrossRef CAS PubMed.
- X. Chu, Y. Tian and C. Lv, Mol. Cancer, 2024, 23, 150 CrossRef PubMed.
- L. Cassetta and J. W. Pollard, Curr. Biol., 2020, 30, R246–R248 CrossRef CAS PubMed.
- J. Xu, L. Ding, J. Mei, Y. Hu, X. Kong, S. Dai, T. Bu, Q. Xiao and K. Ding, Signal Transduct. Target. Ther., 2025, 10, 268 CrossRef CAS PubMed.
- J. Kzhyshkowska, J. Shen and I. Larionova, Cell. Mol. Immunol., 2024, 21, 1376–1409 CrossRef CAS PubMed.
- L. Akkari, I. Amit, V. Bronte, Z. G. Fridlender, D. I. Gabrilovich, F. Ginhoux, C. C. Hedrick and S. Ostrand-Rosenberg, Nat. Rev. Immunol., 2024, 24, 850–857 CrossRef CAS PubMed.
- L. You, Q. Wang, T. Zhang, H. Xiao, M. Lv, H. Lv, L. Deng, X. Zhang and Y. Zhang, Oncogene, 2025, 44, 2413–2426 CrossRef CAS PubMed.
- H. J. Lee, Y. R. Choi, J. H. Ko, J. S. Ryu and J. Y. Oh, Mol. Ther., 2024, 32, 1970–1983 CrossRef CAS PubMed.
- S. K. Wculek, F. J. Cueto, A. M. Mujal, I. Melero, M. F. Krummel and D. Sancho, Nat. Rev. Immunol., 2020, 20, 7–24 CrossRef CAS PubMed.
- M. Shanley, M. Daher, J. Dou, S. Li, R. Basar, H. Rafei, M. Dede, J. Gumin, J. Pantaleon Garcia, A. K. Nunez Cortes, S. He, C. M. Jones, S. Acharya, N. W. Fowlkes, D. Xiong, S. Singh, H. Shaim, S. C. Hicks, B. Liu, A. Jain, M. F. Zaman, Q. Miao, Y. Li, N. Uprety, E. Liu, L. Muniz-Feliciano, G. M. Deyter, V. Mohanty, P. Zhang, S. E. Evans, E. J. Shpall, F. F. Lang, K. Chen and K. Rezvani, Cancer Cell, 2024, 42, 1450–1466 CrossRef CAS PubMed.
- N. Kirchhammer, M. P. Trefny, M. Natoli, D. Brücher, S. N. Smith, F. Werner, V. Koch, D. Schreiner, E. Bartoszek, M. Buchi, M. Schmid, D. Breu, K. P. Hartmann, P. Zaytseva, D. S. Thommen, H. Läubli, J. P. Böttcher, M. A. Stanczak, A. S. Kashyap, A. Plückthun and A. Zippelius, Sci. Transl. Med., 2022, 14, eabm9043 CrossRef CAS PubMed.
- S. Chen, H. Zhu and Y. Jounaidi, Signal Transduct. Target. Ther., 2024, 9, 302 CrossRef CAS PubMed.
- N. A. Maskalenko, D. Zhigarev and K. S. Campbell, Nat. Rev. Drug Discovery, 2022, 21, 559–577 CrossRef CAS PubMed.
- I. Dean, C. Y. C. Lee, Z. K. Tuong, Z. Li, C. A. Tibbitt, C. Willis, F. Gaspal, B. C. Kennedy, V. Matei-Rascu, R. Fiancette, C. Nordenvall, U. Lindforss, S. M. Baker, C. Stockmann, V. Sexl, S. A. Hammond, S. J. Dovedi, J. Mjösberg, M. R. Hepworth, G. Carlesso, M. R. Clatworthy and D. R. Withers, Nat. Commun., 2024, 15, 683 CrossRef CAS PubMed.
- F. Portale, R. Carriero, M. Iovino, P. Kunderfranco, M. Pandini, G. Marelli, N. Morina, M. Lazzeri, P. Casale, P. Colombo, G. De Simone, C. Camisaschi, E. Lugli, G. Basso, J. Cibella, S. Marchini, M. Bordi, G. Meregalli, A. Garbin, M. Dambra, E. Magrini, W. Rackwitz, F. Cecconi, A. Corbelli, F. Fiordaliso, J. Eitler, T. Tonn and D. Di Mitri, Nat. Commun., 2024, 15, 10343 CrossRef CAS PubMed.
- R. Saddawi-Konefka, R. A. Msari, S. Tang, C. Philips, S. Sadat, L. M. Clubb, S. Luna, S. Fassardi, R. Jones, I. F. Pietryga, F. Faraji, S. Schokrpur, B. S. Yung, M. M. Allevato, K. E. Decker, C. A. Nasamran, D. Chilin-Fuentes, S. B. Rosenthal, S. M. Jensen, B. A. Fox, R. B. Bell, J. S. Gutkind, A. Sharabi and J. A. Califano, Nat. Commun., 2025, 16, 6578 CrossRef CAS PubMed.
- M. P. Plebanek, Y. Xue, Y.-V. Nguyen, N. C. DeVito, X. Wang, A. Holtzhausen, G. M. Beasley, B. Theivanthiran and B. A. Hanks, Sci. Immunol., 2024, 9, eadi4191 CrossRef CAS PubMed.
- H. Chi, M. Pepper and P. G. Thomas, Cell, 2024, 187, 2052–2078 CrossRef CAS PubMed.
- M. E. Hoekstra, M. Slagter, J. Urbanus, M. Toebes, N. Slingerland, I. de Rink, R. J. C. Kluin, M. Nieuwland, R. Kerkhoven, L. F. A. Wessels and T. N. Schumacher, Cancer Cell, 2024, 42, 157–167 CrossRef CAS PubMed.
- C. C. Zebley, D. Zehn, S. Gottschalk and H. Chi, Nat. Immunol., 2024, 25, 1344–1354 CrossRef CAS PubMed.
- Y. Ji, C. Xiao, T. Fan, Z. Deng, D. Wang, W. Cai, J. Li, T. Liao, C. Li and J. He, Mol. Cancer, 2025, 24, 66 CrossRef PubMed.
- T. L. Murphy and K. M. Murphy, Cell. Mol. Immunol., 2022, 19, 3–13 CrossRef CAS PubMed.
- J. Ma, Y. Wu, L. Ma, X. Yang, T. Zhang, G. Song, T. Li, K. Gao, X. Shen, J. Lin, Y. Chen, X. Liu, Y. Fu, X. Gu, Z. Chen, S. Jiang, D. Rao, J. Pan, S. Zhang, J. Zhou, C. Huang, S. Shi, J. Fan, G. Guo, X. Zhang and Q. Gao, Science, 2024, 384, eadj4857 CrossRef CAS PubMed.
- K. Ichiyama, J. Long, Y. Kobayashi, Y. Horita, T. Kinoshita, Y. Nakamura, C. Kominami, K. Georgopoulos and S. Sakaguchi, Immunity, 2024, 57, 2043–2060 CrossRef CAS PubMed.
- C. Chen, J. Ma, C. Pi, W. Huang, T. Zhang, C. Fu, W. Liu and Y.-G. Yang, Cell Discovery, 2023, 9, 54 CrossRef CAS PubMed.
- Z. Chen, Y. Yang, Y. Tian, J. Yang and H. Xiong, Anal. Chem., 2024, 96, 9236–9243 CrossRef CAS PubMed.
- L. Yang, M. Zhao, W. Chen, J. Zhu, W. Xu, Q. Li, K. Pu and Q. Miao, Angew. Chem., Int. Ed., 2024, 63, e202313117 CrossRef CAS PubMed.
- G. Li, L. Zhang, H. Zheng and W. Lin, Coord. Chem. Rev., 2024, 517, 215975 CrossRef CAS.
- D. Ren, W. Dong, S. Shen, W. Zhao, T. Qian, X. Xia, Y. Li, F. Xu, X. Tang, X. Li, Y. Chen, J. Zhang, X. Cheng, J. Yang and P. Yu, J. Anal. Test., 2026, 10, 342–351 CrossRef CAS.
- W. Zhu, Q. Li, S. Gong and G. Feng, Anal. Chim. Acta, 2023, 1278, 341748 CrossRef CAS PubMed.
- J. Yin, X. Kong and W. Lin, Anal. Chem., 2021, 93, 2072–2081 CrossRef CAS PubMed.
- X. Wang, Q. Ding, R. R. Groleau, L. Wu, Y. Mao, F. Che, O. Kotova, E. M. Scanlan, S. E. Lewis, P. Li, B. Tang, T. D. James and T. Gunnlaugsson, Chem. Rev., 2024, 124, 7106–7164 CrossRef CAS PubMed.
- Y. Ma, K. Wang, F. Yang, H. Wei, C.-Y. Li, H. Mao, J. Ying, J. Yu, S. Ying, J. Shen and H. Cui, Anal. Chem., 2025, 97, 22359–22367 CrossRef CAS PubMed.
- Y. Hu, J. Liu, M. Xu and K. Pu, J. Am. Chem. Soc., 2025, 147, 7148–7157 CrossRef CAS PubMed.
- Y. Ma, H. Cao, B. Chen, X. Xu, Q. Zhang, H. Chen, X.-B. Zhang and G. Song, Angew. Chem., Int. Ed., 2024, 63, e202411840 CrossRef CAS PubMed.
- J. Zhang, X. Chai, X.-P. He, H.-J. Kim, J. Yoon and H. Tian, Chem. Soc. Rev., 2019, 48, 683–722 RSC.
- S. Pan, A. Ding, Y. Li, Y. Sun, Y. Zhan, Z. Ye, N. Song, B. Peng, L. Li, W. Huang and H. Shao, Chem. Soc. Rev., 2023, 52, 5706–5743 RSC.
- D. Rosenblum, N. Joshi, W. Tao, J. M. Karp and D. Peer, Nat. Commun., 2018, 9, 1410 CrossRef PubMed.
- H. Cabral, J. Li, K. Miyata and K. Kataoka, Nat. Rev. Bioeng., 2024, 2, 214–232 CrossRef CAS.
- H. M. Schouw, L. A. Huisman, Y. F. Janssen, R. H. J. A. Slart, R. J. H. Borra, A. T. M. Willemsen, A. H. Brouwers, J. M. van Dijl, R. A. Dierckx, G. M. van Dam, W. Szymanski, H. H. Boersma and S. Kruijff, Eur. J. Nucl. Med. Mol. Imaging, 2021, 48, 4272–4292 CrossRef CAS PubMed.
- J. Liu, P. Cheng, C. Xu and K. Pu, Nat. Biomed. Eng., 2025, 9, 618–637 CrossRef PubMed.
- V. R. Shinde, N. Revi, S. Murugappan, S. P. Singh and A. K. Rengan, Photodiagn. Photodyn. Ther., 2022, 39, 102915 CrossRef CAS PubMed.
- Y. Zi, K. Yang, J. He, Z. Wu, J. Liu and W. Zhang, Adv. Drug Delivery Rev., 2022, 188, 114449 CrossRef CAS PubMed.
- L. N. M. Nguyen, W. Ngo, Z. P. Lin, S. Sindhwani, P. MacMillan, S. M. Mladjenovic and W. C. W. Chan, Nat. Rev. Bioeng., 2024, 2, 201–213 CrossRef CAS.
- A. Dasgupta, A. M. Sofias, F. Kiessling and T. Lammers, Nat. Rev. Bioeng., 2024, 2, 714–716 CrossRef CAS PubMed.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. W. Chan, Nat. Rev. Mater., 2016, 1, 16014 CrossRef CAS.
- R. Sun, J. Xiang, Q. Zhou, Y. Piao, J. Tang, S. Shao, Z. Zhou, Y. H. Bae and Y. Shen, Adv. Drug Delivery Rev., 2022, 191, 114614 CrossRef CAS PubMed.
- W. Ngo, S. Ahmed, C. Blackadar, B. Bussin, Q. Ji, S. M. Mladjenovic, Z. Sepahi and W. C. W. Chan, Adv. Drug Delivery Rev., 2022, 185, 114238 CrossRef CAS PubMed.
- R. Brusini, M. Varna and P. Couvreur, Adv. Drug Delivery Rev., 2020, 157, 161–178 CrossRef CAS PubMed.
- A. Schudel, D. M. Francis and S. N. Thomas, Nat. Rev. Mater., 2019, 4, 415–428 CrossRef PubMed.
- L. Chen, F. Zang, H. Wu, J. Li, J. Xie, M. Ma, N. Gu and Y. Zhang, Nanoscale, 2018, 10, 1788–1797 RSC.
- H. Kang, S. Rho, W. R. Stiles, S. Hu, Y. Baek, D. W. Hwang, S. Kashiwagi, M. S. Kim and H. S. Choi, Adv. Healthcare Mater., 2020, 9, 1901223 CrossRef CAS PubMed.
- Y. Zhou, G. Shen, X. Zhou and J. Li, Signal Transduct. Target. Ther., 2025, 10, 178 CrossRef CAS PubMed.
- Y. Zheng, Y. Han, Q. Sun and Z. Li, Exploration, 2022, 2, 20210166 CrossRef CAS PubMed.
- W. Yi, P. Xiao, X. Liu, Z. Zhao, X. Sun, J. Wang, L. Zhou, G. Wang, H. Cao, D. Wang and Y. Li, Signal Transduct. Target. Ther., 2022, 7, 386 CrossRef CAS PubMed.
- W. Chen, Z. Sun and L. Lu, Angew. Chem., Int. Ed., 2021, 60, 5626–5643 CrossRef CAS PubMed.
- H. Wang, W. Zhang, C. Huang, X. Yang, Q. Yu, H. Wang, W. Li, L. Zhang and D. Zhu, Chin. Chem. Lett., 2025, 36, 111092 CrossRef CAS.
- Z. Wang, X. Huang, L. Duan, S. Tian, C. Shi, Y. Mou, H. Dong, Y. Gao and L. Weng, Mol. Ther., 2025, 33, 5162–5176 CrossRef CAS PubMed.
- V. Singh, S. Ubaid, M. Kashif, T. Singh, G. Singh, R. Pahwa and A. Singh, J. Exp. Clin. Cancer Res., 2025, 44, 109 CrossRef CAS PubMed.
- X. Wang, J. Qian, Z. Yang, Y. Song, W. Pan, Y. Ye, X. Qin, X. Yan, X. Huang, X. Wang, M. Gao and Y. Zhang, Adv. Mater., 2024, 36, 2310964 CrossRef CAS PubMed.
- C. Xiang, Y. Liu, Q. Ding, T. Jiang, C. Li, J. Xiang, X. Yang, Y. Wang, T. Yang, W. Tong, K. Qian, Q. Zhao, Z. Lu, Z. Cheng and P. Gong, Biomaterials, 2026, 324, 123490 CrossRef CAS PubMed.
- F. Zeng, Z. Fan, S. Li, L. Li, T. Sun, Y. Qiu, L. Nie and G. Huang, ACS Nano, 2023, 17, 19753–19766 CrossRef CAS PubMed.
- M. Y. Lucero and J. Chan, Nat. Chem., 2021, 13, 1248–1256 CrossRef CAS PubMed.
- F. Tang, J.-Y. Liu, C.-Y. Wu, Y.-X. Liang, Z.-L. Lu, A.-X. Ding and M.-D. Xu, ACS Appl. Mater. Interfaces, 2021, 13, 23384–23395 CrossRef CAS PubMed.
- Y. Xiao, M. Cong, J. Li, D. He, Q. Wu, P. Tian, Y. Wang, S. Yang, C. Liang, Y. Liang, J. Wen, Y. Liu, W. Luo, X. Lv, Y. He, D.-D. Cheng, T. Zhou, W. Zhao, P. Zhang, X. Zhang, Y. Xiao, Y. Qian, H. Wang, Q. Gao, Q.-C. Yang, Q. Yang and G. Hu, Cancer Cell, 2021, 39, 423–437 CrossRef CAS PubMed.
- P. Valadez-Cosmes, S. Raftopoulou, Z. N. Mihalic, G. Marsche and J. Kargl, Pharmacol. Ther., 2022, 236, 108052 CrossRef CAS PubMed.
- S. Quintero-Fabián, R. Arreola, E. Becerril-Villanueva, J. C. Torres-Romero, V. Arana-Argáez, J. Lara-Riegos, M. A. Ramírez-Camacho and M. E. Alvarez-Sánchez, Front. Oncol., 2019, 9, 1370 CrossRef PubMed.
- Z. Qu, Y. Han, Q. Zhu, W. Ding, Y. Wang, Y. Zhang, W. Wei, Y. Lei, M. Li, Y. Jiao, K. Gu and Y. Zhang, J. Inflammation Res., 2023, 16, 3419–3436 CrossRef CAS PubMed.
- M. Okamoto, R. Mizuno, K. Kawada, Y. Itatani, Y. Kiyasu, K. Hanada, W. Hirata, Y. Nishikawa, H. Masui, N. Sugimoto, T. Tamura, S. Inamoto, Y. Sakai and K. Obama, Int. J. Mol. Sci., 2023, 24, 1118 CrossRef CAS PubMed.
- S.-Y. Liu, H. Xiong, R.-R. Li, W.-C. Yang and G.-F. Yang, Anal. Chem., 2019, 91, 3877–3884 CrossRef CAS PubMed.
- Y. Yang, J. Zhang, Z. Li, F. Li, K. Wang, B. Zhang, G. Lyu and M. Yang, Anal. Chem., 2025, 97, 19349–19359 CrossRef CAS PubMed.
- X. Zhang, C. Jiang, T. He, F. Zhao, J. Qu, P. Huang and J. Lin, Anal. Chem., 2022, 94, 3227–3234 CrossRef CAS PubMed.
- L. Huang, W. Su, L. Zhu, J. Li, W. Quan, J. Yoon and W. Lin, Angew. Chem., Int. Ed., 2023, 62, e202217508 CrossRef CAS PubMed.
- M. Deng, S. Ai, Y. Liu, H. Zhang, Z. Zhai, D. Cheng, L. He and S. Li, Spectrochim. Acta, Part A, 2026, 344, 126623 CrossRef CAS PubMed.
- Y. Zhang, S. He, C. Xu, Y. Jiang, Q. Miao and K. Pu, Angew. Chem., Int. Ed., 2022, 61, e202203184 CrossRef CAS PubMed.
- S. He, P. Cheng and K. Pu, Nat. Biomed. Eng., 2023, 7, 281–297 CrossRef CAS PubMed.
- H. Li, R. Qu, L. Chen, M. Wu, W. Zhou, S. Liu, Y. Liu, X. Jiang and X. Zhen, J. Am. Chem. Soc., 2026, 148, 1116–1128 CrossRef CAS PubMed.
- H. Luyang, F. Zeng, Y. Lei, Q. He, Y. Zhou and J. Xu, Mol. Cancer, 2025, 24, 22 CrossRef PubMed.
- J. Guo, H. Tao, Y. Dou, L. Li, X. Xu, Q. Zhang, J. Cheng, S. Han, J. Huang, X. Li, X. Li and J. Zhang, Materials Today, 2017, 20, 493–500 CrossRef CAS.
- R. Liu, J. Tang, Y. Xu and Z. Dai, ACS Nano, 2019, 13, 5124–5132 CrossRef CAS PubMed.
- H. Zheng, C. Yuan, J. Cai, W. Pu, P. Wu, C. Li, G. Li, Y. Zhang, J. Zhang, J. Guo and D. Huang, J. Nanobiotechnol., 2022, 20, 134 CrossRef CAS PubMed.
- C. Wang, D. Cheng, N. Jalali Motlagh, E. G. Kuellenberg, G. R. Wojtkiewicz, S. P. Schmidt, R. Stocker and J. W. Chen, J. Med. Chem., 2021, 64, 5874–5885 CrossRef CAS PubMed.
- C. Wang, B. Pulli, N. Jalali Motlagh, A. Li, G. R. Wojtkiewicz, S. P. Schmidt, Y. Wu, M. W. G. Zeller and J. W. Chen, Theranostics, 2019, 9, 7525–7536 CrossRef CAS PubMed.
- W. Tong, H. Hui, W. Shang, Y. Zhang, F. Tian, Q. Ma, X. Yang, J. Tian and Y. Chen, Theranostics, 2021, 11, 506–521 CrossRef CAS PubMed.
- G. A. Conlon and G. I. Murray, J. Pathol., 2019, 247, 629–640 CrossRef PubMed.
- B. Grillet, R. V. S. Pereira, J. Van Damme, A. Abu El-Asrar, P. Proost and G. Opdenakker, Nature Rev. Rheumatol., 2023, 19, 363–377 CrossRef PubMed.
- S. Shoucair, J. Chen, J. R. Martinson, J. R. Habib, B. Kinny-Köster, N. Pu, A. F. van Oosten, A. A. Javed, E. J. Shin, S. Z. Ali, K. J. Lafaro, C. L. Wolfgang, J. He and J. Yu, JAMA Surgery, 2022, 157, e221362 CrossRef PubMed.
- F. Wu, Y. Huang, X. Yang, J.-J. Hu, X. Lou, F. Xia, Y. Song and L. Jiang, Anal. Chem., 2021, 93, 16257–16263 CrossRef CAS PubMed.
- W. Guo, X. Gao, R. Zhan, Z. Zhao, K. Xu and B. Tang, Talanta, 2021, 222, 121525 CrossRef CAS PubMed.
- A. Chen, H. Lu, R. Cao, Y. Zhu, Y. Li, R. Ge, S. Zhang, Y. Li, L. Xiao, L. Su, J. Zhao, H. Hu and Z. Wang, Nano Today, 2022, 45, 101524 CrossRef CAS.
- Y. Tu, X. Ma, H. Chen, Y. Fan, L. Jiang, R. Zhang and Z. Cheng, Int. J. Nanomed., 2022, 17, 6773–6789 CrossRef CAS PubMed.
- K. Schwegmann, M. Hohn, S. Hermann, M. Schäfers, B. Riemann, G. Haufe, S. Wagner and H.-J. Breyholz, Bioconjugate Chem., 2020, 31, 1117–1132 CrossRef CAS PubMed.
- T. Meng, B. Fan, Q. Li, X. Peng, J. Xu and R. Zhang, J. Mater. Chem. B, 2020, 8, 9888–9898 RSC.
- K. Gristwood, S. Luli, H. J. Blair, K. S. Rankin and J. C. Knight, Bioconjugate Chem., 2026, 37, 93–99 CrossRef CAS PubMed.
- T. Jakoš, A. Pišlar, A. Jewett and J. Kos, Front. Immunol., 2019, 10, 2037 CrossRef PubMed.
- A. Caliò, M. Brunelli, S. Gobbo, P. Argani, E. Munari, G. Netto and G. Martignoni, Cancers, 2021, 13, 2441 CrossRef PubMed.
- D. Bararia, J. A. Hildebrand, S. Stolz, S. Haebe, S. Alig, C. P. Trevisani, F. Osorio-Barrios, M. D. Bartoschek, M. Mentz, A. Pastore, E. Gaitzsch, M. Heide, V. Jurinovic, K. Rautter, J. Gunawardana, M. B. Sabdia, M. Szczepanowski, J. Richter, W. Klapper, A. Louissaint, Jr., C. Ludwig, S. Bultmann, H. Leonhardt, S. Eustermann, K.-P. Hopfner, W. Hiddemann, M. von Bergwelt-Baildon, C. Steidl, R. Kridel, J. W. D. Tobin, M. K. Gandhi, D. M. Weinstock, M. Schmidt-Supprian, M. B. Sárosi, M. Rudelius, V. Passerini, J. Mautner and O. Weigert, Cell Rep., 2020, 31, 107522 CrossRef CAS PubMed.
- M. Poreba, K. Groborz, M. Vizovisek, M. Maruggi, D. Turk, B. Turk, G. Powis, M. Drag and G. S. Salvesen, Chem. Sci., 2019, 10, 8461–8477 RSC.
- Y. Wang, L. Jiang, Y. Zhang, Y. Lu, J. Li, H. Wang, D. Yao and D. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 33564–33574 CrossRef CAS PubMed.
- X. Chen, X. Ren, Y. Zhu, Z. Fan, L. Zhang, Z. Liu, L. Dong and Z. Hai, Anal. Chem., 2021, 93, 9304–9308 CrossRef CAS PubMed.
- R. Li, R. Zhou, H. Wang, W. Li, M. Pan, X. Yao, W. Zhan, S. Yang, L. Xu, Y. Ding and L. Zhao, Cell Death Differ., 2019, 26, 2447–2463 CrossRef CAS PubMed.
- Z. Wang, Z. Xiang, T. Zhu, J. Chen, M. Z. Zhong, J. Huang, K. S. Wang, L. Li, L. Q. Sun and W. B. Zhou, Oncol Lett, 2020, 19, 167–176 CAS.
- S. Taheri Baghmisheh, C.-H. Chen, Y.-M. Yeh, P.-C. Lin, P.-C. Chen, R.-H. Chan, J.-W. Kang, C.-T. Lee, H.-J. Tsai, Y.-C. Fang, C. H. A. Cheung, K.-Y. Chang, J.-Y. Chang and S.-H. Chen, Cancer Immunol., Immunother., 2025, 74, 287 CrossRef CAS PubMed.
- Y. Li, S. Wu, Y. Zhao, T. Dinh, D. Jiang, J. E. Selfridge, G. Myers, Y. Wang, X. Zhao, S. Tomchuck, G. Dubyak, R. T. Lee, B. Estfan, M. Shapiro, S. Kamath, A. Mohamed, S. C.-C. Huang, A. Y. Huang, R. Conlon, S. Krishnamurthi, J. Eads, J. E. Willis, A. A. Khorana, D. Bajor and Z. Wang, J. Clin. Invest., 2024, 134, e175031 CrossRef CAS PubMed.
- E. T. Richard, K. Morinaga, Y. Zheng, O. Sundberg, A. Hokugo, K. Hui, Y. Zhou, H. Sasaki, B. A. Kashemirov, I. Nishimura and C. E. McKenna, Bioconjugate Chem., 2021, 32, 916–927 CrossRef CAS PubMed.
- M. Poreba, W. Rut, M. Vizovisek, K. Groborz, P. Kasperkiewicz, D. Finlay, K. Vuori, D. Turk, B. Turk, G. S. Salvesen and M. Drag, Chem. Sci., 2018, 9, 2113–2129 RSC.
- H.-Y. Hu, D. Vats, M. Vizovisek, L. Kramer, C. Germanier, K. U. Wendt, M. Rudin, B. Turk, O. Plettenburg and C. Schultz, Angew. Chem., Int. Ed., 2014, 53, 7669–7673 CrossRef CAS PubMed.
- S. He, J. Yu, P. Cheng, J. Liu, C. Zhang, C. Xu, K. Pu and Y. Zhang, Adv. Mater., 2025, 37, 2420393 CrossRef CAS PubMed.
- Z. Zhou, H. He, K. Wang, X. Shi, Y. Wang, Y. Su, Y. Wang, D. Li, W. Liu, Y. Zhang, L. Shen, W. Han, L. Shen, J. Ding and F. Shao, Science, 2020, 368, eaaz7548 CrossRef CAS PubMed.
- S. Hiroyasu, M. R. Zeglinski, H. Zhao, M. A. Pawluk, C. T. Turner, A. Kasprick, C. Tateishi, W. Nishie, A. Burleigh, P. A. Lennox, N. Van Laeken, N. J. Carr, F. Petersen, R. I. Crawford, H. Shimizu, D. Tsuruta, R. J. Ludwig and D. J. Granville, Nat. Commun., 2021, 12, 302 CrossRef CAS PubMed.
- S. Kołt, T. Janiszewski, D. Kaiserman, S. Modrzycka, S. J. Snipas, G. Salvesen, M. Dr
![[a with combining cedilla]](https://www.rsc.org/images/entities/char_0061_0327.gif) g, P. I. Bird and P. Kasperkiewicz, J. Med. Chem., 2020, 63, 3359–3369 CrossRef PubMed.
g, P. I. Bird and P. Kasperkiewicz, J. Med. Chem., 2020, 63, 3359–3369 CrossRef PubMed. - J. I. Scott, Z. Cheng, E. J. Thompson, U. Karmakar, V. Cowell, M. David, D. Gordon, L. Mendive-Tapia, A. Le Saint-Grant, P. Volkmer, C. S. Chuah, P. Lau, A. G. Rossi, W. B. Nagengast, D. Shabat, G.-T. Ho and M. Vendrell, Nat. Biomed. Eng., 2026 DOI:10.1038/s41551-025-01588-1.
- J. I. Scott, S. Gutkin, O. Green, E. J. Thompson, T. Kitamura, D. Shabat and M. Vendrell, Angew. Chem., Int. Ed., 2021, 60, 5699–5703 CrossRef CAS PubMed.
- S. He, J. Li, Y. Lyu, J. Huang and K. Pu, J. Am. Chem. Soc., 2020, 142, 7075–7082 CrossRef CAS PubMed.
- Y. Zhang, S. He, W. Chen, Y. Liu, X. Zhang, Q. Miao and K. Pu, Angew. Chem., Int. Ed., 2021, 60, 5921–5927 CrossRef CAS PubMed.
- X. Ma, M. Mao, Z. Liu, C. Liang, J. He, Y. Qu, L. Xu, R. Cheng, W. Zhuang, Y. Lei, W. Nie, L. Yuan, D.-W. Pang and H.-Y. Xie, J. Am. Chem. Soc., 2024, 146, 31873–31884 CrossRef CAS PubMed.
- L. Xu, N. Liu, W. Zhan, Y. Deng, Z. Chen, X. Liu, G. Gao, Q. Chen, Z. Liu and G. Liang, ACS Nano, 2022, 16, 19328–19334 CrossRef CAS PubMed.
- Y. Wang, J. Guo, M. Chen, S. Liao, L. Xu, Q. Chen, G. Song and X.-B. Zhang, Nat. Biomed. Eng., 2025, 9, 656–670 CrossRef CAS PubMed.
- T. Wang, J. Huang, C. Xu, L. Zhang, Y. Lin and K. Pu, Angew. Chem., Int. Ed., 2026, 65, e21145 CrossRef CAS PubMed.
- G. Feng, H. Zhang, H. Liu, X. Zhang, H. Jiang, S. Liao, X. Luo, H. Yao, B. Xiang, S. Liu, J. Zhang, J. Zhang and J. Fang, Research, 2024, 7, 0464 CrossRef CAS PubMed.
- H. Zhang, J. Liu, C. Liu, P. Yu, M. Sun, X. Yan, J.-P. Guo and W. Guo, Biomaterials, 2017, 133, 60–69 CrossRef CAS PubMed.
- Y. Wen, F. Huo, J. Wang and C. Yin, Anal. Chem., 2019, 91, 15057–15063 CrossRef CAS PubMed.
- J. Chen, L. Chen, Y. Wu, Y. Fang, F. Zeng, S. Wu and Y. Zhao, Nat. Commun., 2021, 12, 6870 CrossRef CAS PubMed.
- Z. Deng, W. Ma, C. Ding, C. Wei, B. Gao, Y. Zhu, Y. Zhang, F. Wu, M. Zhang, R. Li and M. Zhang, Nano Today, 2023, 53, 102044 CrossRef CAS.
- Z. Che, Z. Zhou, S.-Q. Li, L. Gao, J. Xiao and N.-K. Wong, Trends Mol. Med., 2023, 29, 951–967 CrossRef CAS PubMed.
- X. Zhang, Y. Chen, H. He, S. Wang, Z. Lei and F. Zhang, Angew. Chem., Int. Ed., 2021, 60, 26337–26341 CrossRef CAS PubMed.
- X. Luo, C. Zhang, F. Yuan, S. Cheng, Y. Zhu, M. Xiang, X. Hu and Y. Xian, Anal. Chem., 2022, 94, 15790–15800 CrossRef CAS PubMed.
- P. Yuan, X. Xu, D. Hu, Y. Chen, J. Fan, S. Yao, Y. Piao, Z. Zhou, S. Shao, N. K. H. Slater, Y. Shen and J. Tang, J. Am. Chem. Soc., 2023, 145, 7941–7951 CrossRef CAS PubMed.
- C. Lu, S. Liao, B. Chen, L. Xu, N. Wu, D. Lu, H. Kang, X.-B. Zhang and G. Song, Nat. Mater., 2025, 24, 133–142 CrossRef CAS PubMed.
- M. Iciek, A. Bilska-Wilkosz, M. Kozdrowicki and M. Górny, Antioxid. Redox Signaling, 2023, 39, 1000–1023 CrossRef CAS PubMed.
- H. Ge, H. Huang, S. He, K. Chen, M. Dai, Z. Zhang, X. Chen, L. Zhang, G. Mao and H. Liu, Anal. Chem., 2025, 97, 17111–17120 CrossRef CAS PubMed.
- L. Li, Z. Zhang, L. Zhou, H. Ge, Y. Zhao, Y. Gong, G.-J. Mao and H. Liu, Anal. Chem., 2024, 96, 7248–7256 CrossRef CAS PubMed.
- Y. Zhao, L. Li, Q. Ye, Y. Gong, R. Yang and H. Liu, Anal. Chem., 2023, 95, 14043–14051 CrossRef CAS PubMed.
- G. Chen, W. Zhou, C. Zhao, Y. Liu, T. Chen, Y. Li and B. Tang, Anal. Chem., 2018, 90, 12442–12448 CrossRef CAS PubMed.
- S. Gong, Z. Zheng, X. Guan, S. Feng and G. Feng, Anal. Chem., 2021, 93, 5700–5708 CrossRef CAS PubMed.
- H. Zhang, L. Xu, W. Chen, J. Huang, C. Huang, J. Sheng and X. Song, ACS Sens., 2018, 3, 2513–2517 CrossRef CAS PubMed.
- M. H. Shahzad, R. F. Rayes, J. Cools-Lartigue and J. D. Spicer, Nat. Rev. Cancer, 2026, 26, 104–117 CrossRef CAS PubMed.
- Y. Hao, D. Liu, K. Wang, Q. Liu, H. Chen, S. Ji and D. Ding, Small Sci., 2024, 4, 2400212 CrossRef CAS PubMed.
- M. R. Rios, G. Garoffolo, G. Rinaldi, A. Megia-Fernandez, S. Ferrari, C. T. Robb, A. G. Rossi, M. Pesce and M. Bradley, Chem. Commun., 2021, 57, 97–100 RSC.
- M. Rodriguez-Rios, G. Rinaldi, A. Megia-Fernandez, A. Lilienkampf, C. T. Robb, A. G. Rossi and M. Bradley, Chem. Commun., 2023, 59, 11660–11663 RSC.
- P. Cheng, S. He, C. Zhang, J. Liu and K. Pu, Angew. Chem., Int. Ed., 2023, 62, e202301625 CrossRef CAS PubMed.
- M. Guerra, V. S. Halls, J. Schatterny, M. Hagner, M. A. Mall and C. Schultz, J. Am. Chem. Soc., 2020, 142, 20299–20305 CrossRef CAS PubMed.
- S.-J. Kim, J. Kim, B. Kim, W.-W. Lee, X. Liu, Y.-T. Chang and J.-W. Park, Biochem. Biophys. Res. Commun., 2020, 527, 646–653 CrossRef CAS PubMed.
- X. Gao, D. Mao, X. Zuo, F. Hu, J. Cao, P. Zhang, J. Z. Sun, J. Liu, B. Liu and B. Z. Tang, Anal. Chem., 2019, 91, 6836–6843 CrossRef CAS PubMed.
- X. Luo, D. Hu, D. Gao, Y. Wang, X. Chen, X. Liu, H. Zheng, M. Sun and Z. Sheng, ACS Nano, 2021, 15, 10010–10024 CrossRef CAS PubMed.
- H. Wang, R. Zhang, X. Jia, S. Gao, T. Gao, K. Fan, Y. Li, S. Wang, M. Qiao, S. Yan, H. Hui and H. Dong, Cell Death Discovery, 2024, 10, 395 CrossRef CAS PubMed.
- Y. Chen, Y. Yang, B. Yun, Z. He, H. Zhang, Q. Jiang, W. Li, A. Liang, L. Wang, B.-Z. Qian and F. Zhang, Adv. Mater., 2026, 38, e16197 CrossRef CAS PubMed.
- A. E. Reese, F. de Moliner, L. Mendive-Tapia, S. Benson, E. Kuru, T. Bridge, J. Richards, J. Rittichier, T. Kitamura, A. Sachdeva, H. J. McSorley and M. Vendrell, ACS Cent. Sci., 2024, 10, 143–154 CrossRef CAS PubMed.
- M. Bertolini, L. Mendive-Tapia, U. Karmakar and M. Vendrell, J. Am. Chem. Soc., 2024, 146, 30565–30572 CrossRef CAS PubMed.
- W. Bernhard, K. Barreto, A. El-Sayed, J. DeCoteau and C. R. Geyer, Cancers, 2022, 14, 300 CrossRef CAS PubMed.
- J. Wang, X. Fang, J. Yang, Q. Tang, Y. Yang, Z. Li, F. Zhang, Q. Wu, Z. Luo and Z. Zheng, Sens. Actuators, B Chem., 2024, 405, 135385 CrossRef CAS.
- K. P. Kubelick, J. Kim, M. Kim, X. Huang, C. Wang, S. Song, Y. Xia and S. Y. Emelianov, ACS Nano, 2025, 19, 6079–6094 CrossRef CAS PubMed.
- Q. Qiu, Y. Wen, H. Dong, A. Shen, X. Zheng, Y. Li and F. Feng, Biomater. Sci., 2019, 7, 5211–5220 RSC.
- C. Zhang, L. Zhang, W. Wu, F. Gao, R.-Q. Li, W. Song, Z.-N. Zhuang, C.-J. Liu and X.-Z. Zhang, Adv. Mater., 2019, 31, 1901179 CrossRef PubMed.
- Y. Xu, X. Zhang, G. Hu, X. Wu, Y. Nie, H. Wu, D. Kong and X. Ning, Biomaterials, 2021, 279, 121224 CrossRef CAS PubMed.
- X. Liu, Y. Duan, D. Hu, M. Wu, C. Chen, P. B. Ghode, G. Magarajah, N. Thakor, X. Liu, C. Liu, Z. Sheng, H. Zheng and B. Liu, ACS Mater. Lett., 2021, 3, 1284–1290 CrossRef CAS.
- N. Liao, L. Su, Y. Zheng, B. Zhao, M. Wu, D. Zhang, H. Yang, X. Liu and J. Song, Angew. Chem., Int. Ed., 2021, 60, 20888–20896 CrossRef CAS PubMed.
- X. Huang, Z. Yi, T. Zhang, M. He, C. Nie, T. Chen, J. Jiang and X. Chu, Chem. Eng., 2024, 495, 153436 CrossRef CAS.
- Y. Bian, P. Liang, Q. Yuan, Y. Li, X. An, J. Liu, X. Gao and D. Su, Adv. Funct. Mater., 2026, 36, e15395 CrossRef CAS.
- A. Volpe, P. S. Adusumilli, H. Schöder and V. Ponomarev, J. ImmunoTher. Cancer, 2022, 10, e004902 CrossRef PubMed.
- A. Aalipour, H.-Y. Chuang, S. Murty, A. L. D’Souza, S.-M. Park, G. S. Gulati, C. B. Patel, C. Beinat, F. Simonetta, I. Martinić, G. Gowrishankar, E. R. Robinson, E. Aalipour, Z. Zhian and S. S. Gambhir, Nat. Biotechnol., 2019, 37, 531–539 CrossRef CAS PubMed.
- L. Dan and L. Kang-Zheng, J. Transl. Med., 2025, 23, 501 CrossRef PubMed.
- S. Janssens, S. Rennen and P. Agostinis, Immunol. Rev., 2024, 321, 350–370 CrossRef CAS PubMed.
- C. Zhao, C. Wang, W. Shan, Z. Wang, X. Chen and H. Deng, Acc. Chem. Res., 2024, 57, 905–918 CrossRef CAS PubMed.
- X. Hao, C. Li, Y. Zhang, H. Wang, G. Chen, M. Wang and Q. Wang, Adv. Mater., 2018, 30, 1804437 CrossRef PubMed.
- Y. Yuan, J. Zhang, X. Qi, S. Li, G. Liu, S. Siddhanta, I. Barman, X. Song, M. T. McMahon and J. W. M. Bulte, Nat. Mater., 2019, 18, 1376–1383 CrossRef CAS PubMed.
- X. Dong, A. Yang, Y. Bai, D. Kong and F. Lv, Biomaterials, 2020, 230, 119659 CrossRef CAS PubMed.
- Q. Wang, T. Yang, S. Li, C. Xu, C. Wang, Y. Xiong, X. Wang, J. Wan, X. Yang and Z. Li, Research, 2023, 6, 0223 CrossRef CAS PubMed.
- R. Xiao, F. Zheng, K. Kang, L. Xiao, A. Bi, Y. Chen, Q. Zhou, X. Feng, Z. Chen, H. Yin, W. Wang, Z. Chen, X. Cheng and W. Zeng, Biomater. Res., 2023, 27, 112 CrossRef CAS PubMed.
- F. Chen, X.-F. Tian, T. Yang, Y.-J. Dai, D.-Y. Chen, H.-B. Chen, T. Shimura, X.-F. Li, C.-L. Sha, Q. Ji, J. Cao, M.-Y. Fang, J.-B. Shang, J.-M. Fang, Y. Lu, W.-H. Zheng, P. Guo and W.-H. Tan, Adv. Sci., 2025, 12, e04851 CrossRef CAS PubMed.
- Y.-Y. Zhao, L. Lu, H. Jeong, H. Kim, X. Li, H. Zhang and J. Yoon, Chem. Soc. Rev., 2025, 54, 7749–7768 RSC.
- Y. Sun, Y. Zhang, Y. Gao, P. Wang, G. He, N. T. Blum, J. Lin, Q. Liu, X. Wang and P. Huang, Adv. Mater., 2020, 32, 2004481 CrossRef CAS PubMed.
- R. Jiang, J. Dai, X. Dong, Q. Wang, Z. Meng, J. Guo, Y. Yu, S. Wang, F. Xia, Z. Zhao, X. Lou and B. Z. Tang, Adv. Mater., 2021, 33, 2101158 CrossRef CAS PubMed.
- Z. Xu, Y. Kang, J. Zhang, J. Tang, H. Sun, Y. Li, D. He, X. Sha, Y. Tang, Z. Fu, F. Wu and S. Wang, Opto-Electron. Adv., 2024, 7, 240013 CrossRef CAS.
- Y.-Y. Zhao, Y. Xu, J. Wen, H. Jeong, H. Kim, X. Li and J. Yoon, J. Am. Chem. Soc., 2025, 147, 48072–48086 CrossRef CAS PubMed.
- J. Liu, Y. Liu, S. Zhi, Y. Yang, H. Kim, D. Wu, G. Wang, T. D. James, J. Yoon and H. Zhang, Angew. Chem., Int. Ed., 2025, 64, e202425631 CrossRef CAS PubMed.
- W. Li, J. Yang, L. Luo, M. Jiang, B. Qin, H. Yin, C. Zhu, X. Yuan, J. Zhang, Z. Luo, Y. Du, Q. Li, Y. Lou, Y. Qiu and J. You, Nat. Commun., 2019, 10, 3349 CrossRef PubMed.
- J. Liu, D. Cheng, A. Zhu, M. Ding, N. Yu and J. Li, Adv. Sci., 2024, 11, 2406750 CrossRef CAS PubMed.
- J. Gao, Q. Wu, Y. Yan, M. Chen, Q. Li, Y. Xu, C. Wang, E. Hao, Z. Liu and L. Feng, Adv. Funct. Mater., 2024, 34, 2401830 CrossRef CAS.
- J. Song, H. Wang, X. Meng, W. Li and J. Qi, Nat. Commun., 2024, 15, 10395 CrossRef CAS PubMed.
- Z. Ma, M. Li, R. Guo, Y. Tian, Y. Zheng, B. Huang, Y. You, Q. Xu, M. Cui, L. Shen, F. Lan, H. Yang, R. Liu, T. Yang, F. Wan, Q. He, X. Huo, Y. Bi, Y. Zhang and Y. Ling, Sci. Adv., 2025, 11, eadp7126 CrossRef CAS PubMed.
- M. Zhang, X. Jiang, Q. Zhang, T. Zheng, M. Mohammadniaei, W. Wang, J. Shen and Y. Sun, Adv. Funct. Mater., 2021, 31, 2102274 CrossRef CAS.
- Z. Deng, M. Xi, C. Zhang, X. Wu, Q. Li, C. Wang, H. Fang, G. Sun, Y. Zhang and G. Yang, ACS Nano, 2023, 17, 4495–4506 CrossRef CAS PubMed.
- D. Jing, J. Zhang, Z. Li, W. Yan and Y. Guo, Talanta, 2025, 284, 127258 CrossRef CAS PubMed.
- L. Zhou, C. Hu, H. Huang, H. Ge, Z. Zhang, W. Cheng, H. Liu and R. Yang, Angew. Chem., Int. Ed., 2025, 64, e202509372 CrossRef CAS PubMed.
- J. Yan, H. Liu, Y. Wu, B. Niu, X. Deng, L. Zhang, Q. Dang, Y. Wang, X. Lu, B. Zhang and W. Sun, Biomaterials, 2023, 301, 122281 CrossRef CAS PubMed.
- K. Li, S. Xu, M. Xiong, S.-Y. Huan, L. Yuan and X.-B. Zhang, Chem. Soc. Rev., 2021, 50, 11766–11784 RSC.
- H.-W. Liu, K. Li, X.-X. Hu, L. Zhu, Q. Rong, Y. Liu, X.-B. Zhang, J. Hasserodt, F.-L. Qu and W. Tan, Angew. Chem., Int. Ed., 2017, 56, 11788–11792 CrossRef CAS PubMed.
- Y. Miao, Y. Tian and D. Ye, Chem. Soc. Rev., 2025, 54, 11624–11658 RSC.
- P. Kasperkiewicz, Y. Altman, M. D’Angelo, G. S. Salvesen and M. Drag, J. Am. Chem. Soc., 2017, 139, 10115–10125 CrossRef CAS PubMed.
- Z. Li, H. Liu and X.-B. Zhang, Chem. Soc. Rev., 2024, 53, 11207–11227 RSC.
- C. Hu, H. Liu, Z. Zhang, L. Li, G.-J. Mao, W. Cheng and L. Zhou, Small, 2025, 21, 2408527 CrossRef CAS PubMed.
- H. Cho, H.-Y. Kwon, S. H. Lee, H.-G. Lee, N.-Y. Kang and Y.-T. Chang, J. Am. Chem. Soc., 2023, 145, 2951–2957 CrossRef CAS PubMed.
- M. Gao, S. H. Lee, S. H. Park, L. M. Ciaramicoli, H.-Y. Kwon, H. Cho, J. Jeong and Y.-T. Chang, Angew. Chem., Int. Ed., 2021, 60, 23743–23749 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2026 |