
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
How far can you go? Extrapolating values of catalytic activity from known protein landscapes in natural and directed evolution
Douglas B. Kell
 *abc and
Ivayla Roberts
a
*abc and
Ivayla Roberts
a
aDepartment of Biochemistry, Cell and Systems Biology, Institute of Systems, Molecular and Integrative Biology, University of Liverpool, Crown St, Liverpool L69 7ZB, UK. E-mail: dbk@liv.ac.uk
bThe Novo Nordisk Foundation Centre for Biosustainability, Technical University of Denmark, Building 220, Søltofts Plads 200, 2800 Kongens Lyngby, Denmark
cDepartment of Physiological Sciences, Faculty of Science, Stellenbosch University, Stellenbosch Private Bag X1, Matieland, 7602, South Africa
First published on 13th May 2026
Abstract
The number of possible variants representing the landscape of a protein sequence of length N residues, made of the standard unmodified proteinogenic amino acids, is 20N; its exhaustive experimental analysis is consequently intractable. Our focus is on the real and perceived shapes of different fitness landscapes. Epistasis refers to a phenomenon by which the ‘best’ amino acid at a given residue depends on the nature of the amino acid at one or more other residues. Because of epistasis, real protein landscapes display peaks representing local maxima in which weak mutation/strong-selection regimes can cause evolution to become trapped, leading to landscapes that are rugged. Fortunately, although they are necessarily somewhat rugged, such protein landscapes possess regularities that admit their modelling from more limited experimental data, using the methods of statistics and machine learning. We provide a variety of arguments that for typical proteins of length 300–500 residues some 105 or 106 examples, and in favourable cases even fewer, are likely sufficient to allow a reasonable initial modelling (and accurate predictive exploration) of the entire 20N landscape for properties such as kcat. The distribution of fitness effects (DFE) around an existing wild type is usually reasonably fitted statistically by a gamma distribution. However, we also survey modern ideas, especially extreme value theory, that allow extrapolation from the known, with a focus on methods – especially the Generalised Pareto Distribution – that provide means for generating the statistical likelihood of obtaining activities or fitnesses far greater than those observed in existing populations as measured with what are small numbers. These likelihoods typically decrease exponentially, as do the decreases in errors as a function of the size of the network and of the training data as found by deep neural network models as ‘universal approximators’. This is entirely consistent with the large differences between the minuscule amount of available sequence-activity data, that are necessarily local in character, reflecting evolutionary contingency, and the overall distribution (20N, where N might usefully be decreased) that would be expected to contain examples that have much better properties than any observed thus far. This consequently requires careful choices of examples drawn from an extensive distribution (using active learning) for predictive modelling. For instance, a widespread view of a trade-off between catalytic activity and thermostability seems to follow directly from inadequate sampling. All of this has significant implications for the understanding, modelling, and optimisation of experiments in directed evolution and the biocatalysts they produce.
 Douglas B. Kell | Douglas B. Kell is Research Chair in Systems Biology at the University of Liverpool. Following an MA in Biochemistry and a DPhil in bioenergetics at the Oxford University Oxford he held various positions at the University of Aberystwyth, moving to UMIST (Manchester) in 2002. His interests are the use of novel experimental and computational methods for solving biological problems. He cofounded Aber Instruments (Queen's Award for Export Achievement, 1998) and Phenutest (now InnotiveDx). From 2008–2013 he was seconded as Chief Executive Officer of the UK BBSRC. He was awarded a CBE in 2014 ‘for services to science and research’. |
 Ivayla Roberts | Dr. Ivayla Roberts is a post-doctoral researcher at the University of Liverpool in Prof. Kell's system biology group. Originally coming from a computer science background (MSc, Montpellier France), Iva transitioned to molecular biology research via a Translational Molecular Medicine MRes (Manchester, UK) followed by a PhD in computational techniques applied to metabolomics in COVID-19 (Liverpool, UK). Iva's research interests are metabolomics, mass spectrometry, and the application of statistical and machine learning computational approaches to metabolomics. |
Introduction
The sequence of a protein determines its activities, and, following Sewall Wright,1 variations in sequences and their paired activities (here equated with fitnesses) are usually considered in terms of an evolutionary or fitness landscape (e.g., ref. 2–20). Although the true ‘dimensionality’ of this landscape is that of the protein's length, it is convenient (e.g., ref. 21–24) to visualise these landscapes, just as we do real geographical ones, with the position in sequence space determined by the X and Y coordinates and the fitness by height (Fig. 1). | ||
| Fig. 1 Cartoon illustrating a protein landscape and the evolution of a protein to greater fitnesses as a navigation or adaptive walk across this landscape. Multiple peaks, representing local maxima, are separated by troughs of lower fitness, creating a ‘rugged’ landscape. These features typically arise from epistasis1 caused by regimes of weak mutation and strong selection.22,25,26 Evidence that peaks are effectively somewhat rounded comes from the diminishing returns during ascent27,28 that are usually observed.29 The X and Y axes represent positions in sequence space while the Z (height) dimension represents the fitness of interest, e.g., a kcat value. | ||
As rehearsed in the legend to Fig. 1, the idea of ‘weak mutation/strong selection’22,25,26 describes a means of traversal of such fitness landscape in a manner that causes an ever increasing fitness in the desired property. As phrased by Reia and Campos,30 “In the simplistic view of evolutionary adaptation, namely strong-selection weak-mutation regime, the conditions NU ≪ 1 and Ns ≫ 1 hold, where N stands for population size, U is mutation rate and s the selective advantage conferred by the beneficial mutations. Those conditions ensure that selection proceeds much faster than mutations occur. According to this picture, the population is monomorphic most of the time, and the dynamics can be approximated by an adaptive walk, in which the population is depicted as a single entity that moves through the fitness landscape towards fitness peaks”.30 The consequence of the existence of such peaks is that populations become stuck in them.
Characterising these landscapes is consequently important if we are to understand them, but there is at once a combinatorial problem:31 for a protein of length N composed of the 20 common amino acids there are 20N possible variants. Just considering ‘local’ variation, the number of alternative variants M in a protein of length N is given by (N!19M)/(M!(N − M)!).4,22,32 For a protein of length 300 residues this equates to 5700 (=(N − 1) × M), 1·621 × 107 and 31.181 × 1010 for just 1, 2 and 3 mutations. Assessing even just the last of these exhaustively would already be seen as technically (and financially) out of reach.
Fig. 1 also illustrates the concept of ruggedness, which relates in general terms to the number and distribution of local maxima separated from each other and from the global maximum by areas or troughs of lower fitness. As with any combinatorial search problem, its ease of solution is effectively determined by some metric of ‘ruggedness’.33 A simple landscape, sometimes referred to as the ‘Mount Fuji’ landscape34 is defined as follows: “any points on the sequence space have at least one fitter neighbor sequence (an ascending path) which leads to the global optimum in the sequence space in question”.35 Thus, there is but a single global maximum, no or few local maxima, and the global maximum can be reached from anywhere just by selecting for increasing fitness; this would clearly be defined as a ‘smooth’ landscape.36 At the other end of the ruggedness spectrum would be something pathological (it may be referred to as a ‘bed of nails’ landscape) in which every peak (like a nail) is separated from every other peak by a deep and flat trough that itself gives no information about the potential location of the most adjacent peak(s). In this case an onward evolution to fitter variants would be effectively impossible. While no universal method is optimal for searching all landscapes37,38 (but cf. ref. 39), we nevertheless recognise that a high-level understanding of ruggedness can aid the selection of suitable algorithms. Unsurprisingly, it is harder to evolve to higher fitnesses by adaptive walks as landscapes become more rugged (e.g., ref. 33 and 40–48).
Note that we here focus mostly on enzymes and binding agents, though the points made apply equally to protein-based biomaterials such as those described in ref. 49–54. We also focus mostly on kcat, while recognising the importance of other elements of cell-based protein expression such as ensuring solubility (e.g., ref. 55) and the values of secretion (e.g., ref. 56–65). Equivalently, we do not really cover cell-free expression systems (e.g., ref. 66–73), though we recognise their potential benefits when they can be persuaded to work well.
For convenience, Fig. 2 shows the contents of this review as a Mind Map (see ref. 74 and 75).
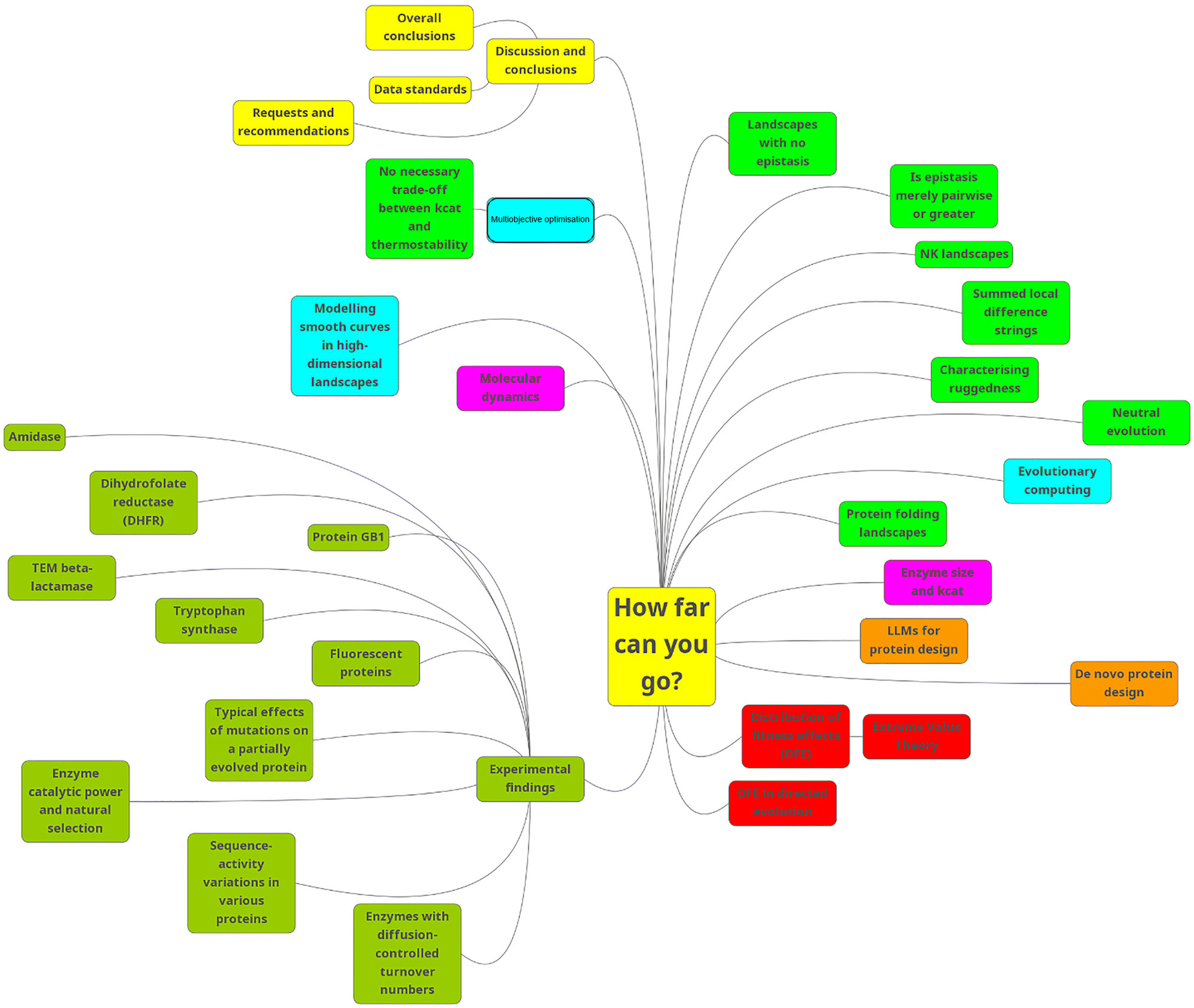 | ||
| Fig. 2 A mind map of the contents of this review. To read this, start at “1 am” and go clockwise. | ||
Landscapes with no epistasis
Epistasis describes the idea that the optimum amino acid at a given residue is not independent of the amino acid present in at least one other location.19,76,77 It is epistasis that essentially makes the prediction of function from sequence a generally unsolved problem.78–84 Reciprocal sign epistasis refers to a circumstance in which two sites distant in sequence space (but in this case close in structural space) need to bind to each other, e.g., a glutamate with a lysine. Thus a mutation at site 1 (from say glutamate to phenylalanine) makes a fitness worse, as does a mutation from a lysine to a phenylalanine at site 2, but mutation of both residues to phenylalanine restores or even enhances the fitness, for inter-residue binding reasons that are rather obvious when one knows the identity of the residues. It is the existence of reciprocal sign epistasis in particular that makes natural landscapes at least somewhat rugged.19,22,85–87The chemical space of small molecules that might harbour bioactivity (e.g., ref. 88–101) is also extremely large (often quoted as 1060 molecules or above102–104). Consequently, the ‘combinatorial’ search for a ‘good’ protein31 is in essence little different from those seeking a ‘good’ small molecule to act as a drug, and ruggedness, activity cliffs105–110 (small changes that destroy activity), ‘scaffold hopping’ (e.g., ref. 111–117) (equivalent to large jumps in the landscape), and broad areas of the search space with little or no activity95,100,118–121 are common to both. By analogy to the quantitative structure–activity relationship (QSAR) used in pharmacology, Fox and colleagues introduced the idea of ProSAR (protein sequence–activity relationship)122–127 to assess experimentally the effect of all possible single mutations on a fitness landscape. If there were no epistasis, evolution to the global maximum would then simply require taking the best amino acid at each residue, thereby reducing the search from 20N to 20N, effectively mimicking the purest Mt Fuji landscape. Unfortunately for scientists (but fortunately for evolution via reasonably stable and locally fit intermediates) biological landscapes do exhibit epistasis,17,128–131 for reasons of both structure and biophysics.19,132–134 The question is how much, and how does this affect our ability to understand and navigate these landscapes?
Is epistasis merely pairwise or greater?
The simplest level of epistasis beyond no epistasis is when it occurs between just two residues. So the first important question relates to the extent of epistasis between individual residues, e.g., is just pairwise or is it sometimes three-way or even greater. There is certainly some evidence for higher-order epistases at the organismal level,81,135–142 though this is certainly not always the case.143 However, for reasons that will become apparent later when we look at how many examples might be needed to model a single protein's landscape reasonably effectively, we will argue that a three-way epistasis can be modelled to a decent approximation as three two-way epistases, so for these purposes we will take it that data from pairwise epistases does allow a suitable approximation, a conclusion also highlighted by Thornton and colleagues.83 This leads immediately to the recognition that, for a protein of length N, rather than the 20N of ProSAR a suitable coverage including all pairwise epistases is given by 202N·(N − 1)/2, the exponent reflecting the pairwise nature of the epistasis. For a protein of 300 amino acids and for N = 20 this amounts to ∼18 M. If we accept that a reasonable landscape might be approximated by five classes (positive, negative, polar, apolar, proline) this drops to 1–2 M. Similarly, structures as estimated computationally will indicate residues that are unlikely to interact, so for modelling purposes N can also be decreased significantly, leading to numbers of 105 or even lower. Obviously these kinds of number, though large, are very far below 20N, and the whole principle of this review is that, while larger populations are inevitably more predictive,144 regularities are sufficiently exploitable that we can begin to model large landscapes with surprisingly few examples of mutations.145,146 What we are doing here is effectively anchoring points in the landscape for modelling with smooth curves, so these more restricted variants should indeed easily get us down to 105. A significant part of the present analysis is concerned with assessing how good or bad an approximation to the full landscape this is likely to offer.Distribution of fitness effects (DFE) and extreme value theory (EVT)
Highly related to the idea of epistatic landscapes, an important reflection of a protein's landscape is the distribution of fitness effects (DFE).28,147–149 It effectively describes the size distribution of peaks but without their location (so does not directly need sequence information). As reviewed by ref. 147, the proportion of mutations that are advantageous, effectively neutral and deleterious varies between species (perhaps unsurprisingly) (see also ref. 150), and the DFE also differs between coding and non-coding DNA. The commonest means of fitting such data to a statistical distribution uses the two-parameter gamma distribution family, of which one parameter is the mean while the other is a shape parameter that (as one would expect) determines the shape of the distribution.147 Fisher's geometric model151 naturally generates fitness epistasis, and the overall level of epistasis can be varied by adjusting the curvature of the rate at which fitness declines with distance from the optimum. A modified gamma distribution is in fact also expected on the basis of Fisher's geometric model,152,153 although conversely not all fitness landscapes are compatible with Fisher's geometric model.154In cases where expression levels are varied (e.g., ref. 155), beneficial and deleterious effects are observed in broadly equal measure. By contrast, when genes are deleted completely,156 or when mutations are made within an existing (already selected) protein, mutations are more commonly deleterious.157 This necessarily follows from the recognition that the ‘wild-type’ starting point has already been subjected to natural selection, and is likely to be in a local maximum as per Fig. 1. The DFE is consequently highly dependent on the extent of epistasis158 that thereby governs landscape ruggedness. Other examples that have been fitted to a modified gamma distribution include spontaneous mutations in Chlamydomonas reinhardtii159 and single mutations in RNA viruses.160 A large set of studies is available via MaveDB (https://www.mavedb.org).161,162
In contrast to deleterious, neutral or weakly beneficial mutations, highly advantageous mutations are not well fitted by the same curve.163 The DFE of advantageous mutations seems to be exponential in character, at least for strongly advantageous mutations,147 and this is entirely consistent with the expectations of extreme value theory, that we now describe.
Extreme value theory
Gillespie164 was probably the first to use extreme value theory (EVT) for predicting future mutational frequencies for fitter (faster) proteins based on the upper end of existing distributions. We think that EVT is highly appropriate for the problem of present interest (especially where weak mutation and strong selection are involved). EVT has been used to produce estimators for events that are in some sense more extreme or greater than those that have been previously observed, or observed increasingly infrequently as their magnitude increases.165–169 Balkema and de Haan170 and Pickands171 developed the Generalised Pareto distribution for such purposes, and in EVT the upper end of the existing distributions are indeed fit most frequently to the Generalised Pareto distribution.172–176 The GDP contains up to three parameters that can be set or optimised (e.g., ref. 177–183), viz. the location or threshold μ,179,184 the scale σ, and a shape parameter ξ.185 In the classic version165 (see also ref. 186), EVT recognises that exceedances above a high threshold are more or less well approximated to a generalised Pareto distribution, discusses how the threshold must be “high enough” (often around the 90th–95th percentile of the existing dataset) and develops likelihood-based methods of inference for σ and ξ once a threshold is chosen. It also discusses how the threshold must be “high enough” for the asymptotics to hold (non-asymptotic strategies are surveyed by Naess169). To minimise the inevitable bias-variance dilemma,179,187 most commonly (referred to as a conditional GPD) a threshold is first determined and then the other two parameters fitted.174,188,189 We have recently used just such a strategy for the prediction of the desirable concentration of a nutraceutical that seems to appear protective against the pregnancy disorder pre-eclampsia.190Among many applications, EVT using the GPD has shown promise in the prediction of drug safety in ever larger populations during the phases of pharmaceutical drug development.191–193 Most pertinently, it has been found useful in the prediction of fitnesses beyond an existing distribution,147,194–198 where these turn out to be exponentially decreasing in frequency as a function of fitness. As is stands, the availability of large experimental datasets is insufficient to understand where the variation of fitness with position in the landscape transitions from being fitted by a gamma distribution to being fitted by an exponential one, and given that this reflects landscape ruggedness it is likely to vary considerably with the protein of interest.
DFE in directed evolution
As mentioned above, deep mutational scanning has provided large datasets that (when provided in a usable format – a surprisingly rare event) can be analysed for assessing landscape ruggedness, but they also serve to provide the wherewithal for DFE studies.6,12,16,199,200 These provide important data for the understanding of fitness landscapes.NK landscapes
To seek to model (and learn to navigate) biopolymer landscapes, Kauffman201–206 introduced the idea of the NK landscape as a tunable fitness landscape in which the size (string or protein length) was determined by N and the ruggedness by K.207 More complex variants (e.g., ref. 208) exist, but the original is still the most widely used. As mentioned in the previous paragraph, natural evolution requires that landscapes be somewhat rugged to preserve local activities in the face of small amounts of mutation,47 but not pathologically so as evolution would then be impossible. A K of 0 implies something like a Mt Fuji landscape (no epistasis) with increasing values becoming more rugged. In terms of analysing (rather than creating) such landscapes, this is commonly done via correlation length or Fourier/amplitude spectra. An exhaustive search of all aptamers of length 10 (so 410 = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 048576) able to bind a target protein was consistent with a K below 1,209 while Aita and colleagues estimated protein folding landscapes to have a K in the range 1–3, and Reia and Campos30 observed something similar for the fitnesses of Hsp90 and Gb1 in yeast. Some protein landscapes are seen as not excessively rugged (e.g., ref. 143 and 210–213), though other protein landscapes are both expected26 and observed (e.g., ref. 17, 19, 85, 131 and 214–221) to be far more rugged, albeit some are easily navigated.84,212,222 This implies that predictability requires an appropriate knowledge of the type of landscape involved.
048576) able to bind a target protein was consistent with a K below 1,209 while Aita and colleagues estimated protein folding landscapes to have a K in the range 1–3, and Reia and Campos30 observed something similar for the fitnesses of Hsp90 and Gb1 in yeast. Some protein landscapes are seen as not excessively rugged (e.g., ref. 143 and 210–213), though other protein landscapes are both expected26 and observed (e.g., ref. 17, 19, 85, 131 and 214–221) to be far more rugged, albeit some are easily navigated.84,212,222 This implies that predictability requires an appropriate knowledge of the type of landscape involved.
Because actual measurements are comparatively hard and slow,223 other authors have used calculations to simulate the fitness of various sequences. du Plessis and colleagues87 used free energy calculations, while we224 have developed Summed Local Difference Strings that allow for a rugged landscape whose fitness is easily calculated from the sequence of amino acid letters alone.
Summed local difference strings
Imagine a string in which A = 1, M = 13 and Z = 26, etc. One can define, and easily calculate, the ‘fitness’ of a string of such letters by summing the alphabetic distances of adjacent letters.225 Thus AZAZAZ is 25 + 25 + 25 + 25 + 25, so the maximum fitness = 25 × (string length −1). AZAZAZ and ZAZAZA thus have the same fitnesses. If we confine ourselves to letters representing the 20 proteinogenic amino acids we can calculate the theoretical maximum fitness of a 100mer (AYAYAY… or YAYAYA.) as 24 × 99 = 2376. For illustrative purposes, we have generated a random string of length 100 (IPTDLWSPFITYSMVNELPWQYDHPKNVSAWHCNDHFWWIQPEFEHMMPTIVNGSKGAVCCNFCHIAIPTWYMTVICNTACRLVCMTQEGLTAVMKQMQN) which has a fitness of 771 and, of course, a search space of 20100. Then we randomly mutate between 1 and 100% of the letters and calculate their fitnesses in the same way, as well as the Hamming distance (number of mutated residues) between the starting string and all the others.Fig. 3 shows the results of such a simulation with 125 sequences. As expected, there is essentially no correlation between fitness and distance from the starting sequence (Fig. 3A) in what is potentially a rather rugged landscape. However, there is a fairly even spread of the number of mutations (Fig. 3B), as expected, while (Fig. 3C) the probability density function peaks near the starting fitness and, with just 125 examples, finding fitnesses over 950 is a significant rarity (Fig. 2A and C).
 | ||
| Fig. 3 Variation of fitness and Hamming distance from a starting sequence using the summed local distances algorithm when a random number of mutations are introduced into a starting sequence with a fitness of 771 in a landscape in which the maximum fitness is 2376. (A) Lack of fitness-distance correlation relative to the starting sequence, indicating the high overall landscape ruggedness. (B) Distribution of Hamming distances after random mutations. (C) Rarity of high fitnesses when mutating from starting sequences. PDF = probability density function and CDF = cumulative distribution function. | ||
In a similar vein, Fig. 4 illustrates the fitness of the best mutant, using the same starting sequence, as the population size is varied. Clearly the data are well fitted by an exponential relationship, requiring ever larger populations for each linear increment in fitness. Thus a population size of 1015 would have a best fitness, statistically, of just 1533, far below the maximum value. This exponential relationship appears multiple times below, and underscores the need for a good predictive fitness model.
 | ||
| Fig. 4 Effect of population size on the best fitness found in a population of random sequences in which the fitness is given by the summed local differences algorithm 132 as described above and in the text. | ||
Characterising ruggedness
Both low sampling226 and mistranslation227 tend to smooth the peaks (and raise troughs) in epistatic landscapes. Conversely, Song and Zhang23 (see also ref. 228) noted that fitness estimation error leads to overestimation of landscape ruggedness. They also rehearse the question of how best to characterise protein landscape ruggedness,23 suggesting that four methods are commonly used: (i) the number of fitness peaks in a landscape, (ii) the fraction of pairs of sites displaying reciprocal sign epistasis as defined above, (iii) the roughness to slope ratio (r/s), which quantifies the extent to which the landscape cannot be described by a linear model where mutations additively determine fitness, and (iv) the fraction of pathways that are blocked, in the sense that a pathway from genotype i to genotype j through single-mutation steps is considered blocked if the fitness is decreased in any of the steps (i.e. multiple mutations are required to increase fitness). Each of these methods is effectively a measure of epistasis, and in general, most sensible metrics of ruggedness lead to similar conclusions.229 Thus, the choice of method is a matter of taste, software availability, familiarity, and convenience. Another and intuitively obvious method involves fitness-distance correlations230 since these will clearly be high on a Mt Fuji landscape and negligible on one like a bed of nails. Note of course that in real landscapes such metrics will vary depending on the starting position in sequence space.A rather different method is that of Spence and colleagues,19 who propose a spectral graph theory approach to measure fitness landscape ruggedness in terms of speed of a heat diffusion analogue.
Early studies that sought to do exhaustive search were necessarily confined to small numbers of residues (e.g., ref. 231), but the availability of cheap(er) sequencing (sometimes referred to as deep mutational scanning (e.g., ref. 6, 12, 16, 18, 145, 200 and 232–248) has brought much more extensive analyses into play (e.g., ref. 10, 30 and 249–251 and see later).
Neutral evolution
One way to escape a local peak in the face of weak mutation-strong selection is by so-called neutral mutations (e.g., ref. 252–258), in which one finds routes that involve only small losses in fitness (relative to population size and selection pressure) en route to higher fitness peaks6, and neutral genetic drift has been considered useful for the purposes of directed evolution (e.g., ref. 259–268). This said, the great advantage of directed over natural evolution is that one can choose how to override the weak-mutation-strong-selection property of natural evolution in small, bounded populations, and we would argue that neutral evolution is not going to be of such major importance in modern directed evolution (where one has reasonably abundant sequence-activity data) if one has developed an understanding of the landscape that can effectively permit the equivalent of ‘scaffold hopping’.A note on evolutionary computing
Evolutionary computing describes a series of methods for addressing combinatorial search problems that are based on principles very similar to those of natural evolution.269 It is used explicitly to model, understand and navigate fitness landscapes of the type illustrated in Fig. 1. Here the individuals in the population are candidate solutions to a problem of interest, and they can be evolved by processes akin to mutation, recombination and selection. These methods can be highly efficient, and have been used to provide solutions for very high-dimensional problems that are seen as close to the global optimum. Evolutionary computing is not of recent origin and has proved of value in metabolic engineering270–273), and the following references may usefully be consulted.274–296 Evolutionary computing also admits variations of mutation and recombination (such as uniform crossover297) that are not likely to be exploited in natural evolution.A particularly important lesson from studies of evolutionary computing is that it is vital to maintain diversity in the evolving population. Simply selecting the best individual in each round effectively ensures that one will soon become trapped in a local peak from which it is effectively impossible to escape. This was regularly found in the early literature of directed evolution when cheap gene sequencing was not available, and can clearly be demonstrated when it is.298
Another issue in the variant of evolutionary computing known as genetic programming is referred to as ‘bloat’.299 ‘Bloat’ describes how the selection for improved activities is more easily done by making entities larger, in that making them smaller is usually associated locally with a lowered fitness and such variants are then selected out.
Evolutionary computing is also relevant to the question of active learning.18,84,300–303 For the present problem, this describes the idea that, armed with a model of a landscape, rather than picking a random sequence and asking what the paired fitness would be one can choose more rationally which sequences to interrogate the model with, thereby both exploiting, and increasing one's knowledge of, the better parts of the landscape.14 A classic article304 uses what amount to Bayesian methods that combine areas of the search space that seem promising with those that are seen as underexplored (or have greater uncertainty305), thus seeking simultaneously to improve both the objective function and the model of the landscape. Evolutionary computing methods are ideally suited for this purpose. (They can also be used to train neural networks, as optimising weights and architectures is effectively a combinatorial search problem too; this is known as neuroevolution.306–312)
The relationship of protein folding landscapes to the directed evolution problem
Protein folding provides another example in which there is a massive search space in that if each amino acid in a protein of N residues could adopt (for example) 10 conformations the number of conformations is 10N, which again cannot possibly be explored.313–315 It maps precisely onto the kind of landscape navigation problem that is our focus. The solution to this apparent paradox313 is of course that they are not explored, and that (modulo chaperone proteins) proteins use both the sequence itself316,317 and favoured kinetic pathways to fold up into stable structures as they emerge from the ribosome. These structures are not in fact the thermodynamically most stable (as was once assumed), and most proteins (see an analysis using AmyloGram318 for all human proteins at319) are capable of adopting so-called ‘amyloid’ forms with crossed-β motifs that are significantly more stable320. However, the usual state is separated from the amyloid form by high energy barriers, of maybe 36–38 kJ mol−1 in the case of prions.321Before the arrival of Alphafold,322–328 Rosettafold329,330 and similar, a significant advance was made in the de novo protein folding field through covariance analysis of series of sequences.331–338 Thus, if at a certain position there were multiple sequences that contained a negatively charge residue (glu or asp), while at another residue there was always a positive charge (lys or arg), but that for other orthologues the charges were reversed (so they always covaried), it could be assumed that these residues were in close proximity to each other in the 3D structure (also providing another clear example of reciprocal sign epistasis) (Fig. 5).
 | ||
| Fig. 5 The use of covariance analysis to provide a spatial constraint in de novo protein folding studies. See text for references and discussion. | ||
Using these interactions estimated from covariance analysis as anchors served to constrain the protein folding problem so significantly that with 100000 examples or so, it was possible to fold many proteins to within ∼4 Å of their correct values. In a sense this is a problem isomorphous to that of present interest, merely replacing an activity landscape by a folding landscape. Its importance for us is in the recognition (and see below) that ‘only’ some 105 examples or so were sufficient to model the (protein folding) landscape rather well.
Enzyme size and kcat
Eukaryotic proteins have an average size of 472 amino acid residues, whereas bacterial (320 residues) and archaeal proteins (283 residues) are typically smaller,339 but still large. For a given yield of recombinant protein a smaller enzyme with a given kcat obviously admits a greater activity, and is correspondingly easier to improve using directed evolution. Consequently, an often-asked question is “why are natural enzymes so big?” (e.g., ref. 340 and 341). A common answer (e.g., ref. 342) would be that this is necessary to form the 3D conformations that the active site requires for catalytic activity, but as well as being a self-defining prophecy this does not alone seem a satisfactory answer. Another notes a relationship between the molecular mass and the lowering of the activation energy for a reaction341 but again no systematic studies of homologues seem to have been performed. A third is related to the phenomenon of ‘bloat’ as described in the section on evolutionary computing; increasing a protein's size in a local landscape is far less likely to cause a significant decrease in fitness than removing residues that potentially mess up the structure considerably. Consequently, the question of (changes in) size is itself highly relevant to the understanding of fitness landscapes. Commonly, organisms sought to solve this fitness issue by gene duplication;343,344 one could evolve a separate gene while retaining a sufficient activity based on the retention of activity in the first gene.345 ‘Contingency’ or ‘historical contingency’346,347 decribes the fact that how evolution proceeds depends on earlier events, and not least since mutation is largely random evolution cannot be taken to be deterministic. Overall, then, we consider that certain sequences simply got fixed in inadequate maxima as a result of evolutionary contingency, and that large sizes are not de rigeur, for at least three classes of reason.First, some enzymes actually are very small. This said, the few enzymes under 10 kDa (ca. 90 amino acid residues) mostly remain poorly characterized.348 In addition, we know of course from any number of directed evolution studies that enzymes can be made much faster without getting bigger (∼1000-fold faster in one case – lovD – in which changes in the ground-state structure of the enzyme were undetectable349). The counterfactual is therefore that small(er) enzymes might be created/evolved to be just as fast as those produced by natural or directed evolution. While organocatalysis can be effected with a single amino acid (especially proline),350–353 we are here interested in polypeptides.354 The record small subunit is seemingly 62 amino acids (aa) for (the hexameric and highly active) 4-oxalocrotonate tautomerase355 and there is also a 6 kDa metalloprotease.356 As stated by ref. 357, “Two esterase enzymes have been isolated, one from Candida lipolytica and one from Bacillus stearothermophilus, which are characterised by an unusually small molecular weight. The Candida enzyme is 5.7 kDa, with 56 amino acid residues and the Bacillus enzyme is 1.57 kDa,358 with only 17 residues. It is also more thermostable. Both are metalloenzymes.” Similarly sized enzymes have been observed in fungi,359 which the authors refer to as microenzymes (a term possibly coined by ref. 360).
Other small enzymes include an ascorbate oxidase in barley with MW <10 kDa,361 a urease with 97 residues,362 the 74-residue ‘AlleyCat’ haem enzymes that can effect Kemp elimination363 (see also ref. 364–367), and a 98-mer acylphosphatase.368,369 Certainly ‘miniproteins’ of 40–50 residues do fold,370 and even a 29-mer can exhibit some reasonable catalytic activity.371–373
Secondly, the famously fast374,375 triose phosphate isomerase is normally a dimer, with a monomeric mutant having very low rates.376,377 However, directed evolution can be used to improve these,378 albeit not to wild-type levels.
Thirdly, there are a great many examples of convergent evolution in which entirely non-homologous proteins catalyse the same reaction,379–383 strongly implying the role of contingency in natural evolution (note that convergent evolution is not inevitable; many solute carriers, for instance, have evolved by divergent evolution384–388 by which a basic scaffold was found useful for a specific purpose, i.e. transmembrane transport, and modified to deal with different substrates).
All of the above speaks to the importance of contingency in natural evolution (e.g., ref. 28, 346 and 389–391), consistent with the ideas that we stress here of weak mutation and strong selection creating epistatic, rugged landscapes from whose peaks it is relatively hard (under these conditions) to escape.
Large language models (LLMs) for protein design
Large language models (LLMs) have come to dominate modern text-based AI and machine learning methods. They are trained on large sequences of words and learn to predict, statistically, the next letter or word (‘token’) in a sequence. The transformer architecture392 is the overwhelmingly dominant architecture used (it can be applied to many other problems, including predicting the structure of small molecules from their mass spectra393). Since protein design is such an important problem82, many LLMs for protein design have come to the fore in the public domain (and many others are proprietary). This purely computational protein design approach is a field that is very fast moving such that any survey will soon be superseded; however, we do recognise that it is highly germane to the general problem of protein landscape simulation, understanding, and even creation, so we provide a listing in Table 1. This is also the place to note the potential for biosecurity concerns that are now beginning to be addressed more openly.394| Model | Comments | Selected ref. |
|---|---|---|
| Anon (not named) | Early LLM including function | 395 |
| AntiFold | Antibody-specific inverse folding LLM, fine-tuned from ESM-IF1396 | 397 |
| BOM-POOLING | Optimising representations when input lengths vary | 398 |
| CrossDesign | Enzyme design under low-data regimes | 399 |
| ESM-IF1-combo | Design pipeline using Foldseek ESM-IF1 + AlphaFold2 for peptide binders to targets. | 400 |
| ESM3 | Combines Rosetta Sequence Design with protein language model predictions using evolutionary scale modeling (ESM) as a restraint | 401 and 402 |
| EVOLVEpro | Improved six proteins, including T7 RNA polymerase and a serine integrase | 403 |
| GENzyme | Reaction-driven enzyme design | 404 |
| IgHuAb | LLM-generated human antibody library (SynAbLib) | 405 |
| IgLM | Infilling language modelling for antibody sequence design; uses masked infilling | 406 and 407 |
| METL | Combines thermostability and protein engineering | 408 |
| MIF/MIF-ST | Structure-conditioned masked LM; masking improves inverse folding & design scoring | 409 |
| MSA transformer | Uses a masked LM to generate de novo sequences from a multiple sequence alignment transformer | 200 and 410 |
| p-IgGen | LLM for paired heavy–light chain antibody generation with desirable biophysical properties | 411 |
| PLMFit | Useful benchmarking study of ESM2, ProGen2, and ProteinBert | 412 |
| Pool PaRTI | Focus on variable sequence lengths | 413 |
| PRIME | Focus on thermostability | 414 |
| ProDualNet | Combines a protein language model with a structure model to co-design binders for dual targets | 415 |
| ProGen | Conditional LLM for de novo sequence generation with functional activity across families with sequence homologies as low as 31.4% | 416 |
| ProGen2 | Open-sourced 6.4Bn-parameter upscale of ProGen; improved zero-shot fitness and controllable generation | 407 and 417 |
| ProstT5 | ProtT5-derived bilingual Language model; large speed-up on AlphaFold to aid structure-aware design | 418 |
| Protein-as-Second-Language | Claims to avoid the notorious quadratic problem of standard transformers | 419 |
| ProteinBERT | Early BERT-style protein language model; has been widely fine-tuned for function and property prediction supporting design | 420 |
| ProteinGenerator | Not really an LLM as it uses denoising diffusion in sequence space | 330 |
| ProteinMPNN | A graph neural network rather than an LLM, for inverse folding | 421 |
| ProtFlash | Claims linear complexity by using a mixed chunk attention mechanism | 422 |
| ProtGPT2 | Autoregressive LLM for de novo sequence generation; samples unexplored regions of protein space | 423 |
| Reviews | 416 and 424–428 | |
| RFdiffusion | Another based on diffusion over structures (not an LLM), in combination with RosettaFold | 429 |
| RFDiffusion2 | Updated version of the above | 430 |
| RiffDiff | A hybrid machine learning and atomistic modelling strategy for scaffolding catalytic arrays in de novo proteins | 431 |
| SSRL | Structure-guided sequence representation learning | 432 |
| Unirep | Deep learning from sequence alone | 433 and 434 |
Many individual steps in synthetic biology have now been automated (including those in our own work, e.g., ref. 435–440). However, it is to be stressed that these AI-based methods441 are increasingly being accompanied by a fully closed-loop442–444 form of automation445–449 of the entire Design-Build-Test-Learn cycle of synthetic biology.
De novo fold and protein design
Much of the ethos of this review is based around the combinatorial fact that neither experimenters nor natural evolution have sampled anything other than a tiny fraction of possible sequence space, even for small proteins. In terms of protein folds, one may suppose that the more examples we can sample the more we shall find.450 Databases such as CATH451 have over 2000 folds (significantly increased by the addition of AlphaFold-predicted structures452) and hundreds of millions of domains.453 In addition, we note the significance of metamorphic454–457 or ‘fold-switching’458–460 proteins that can mediate major evolutionary transitions in scaffolds,461 while the existence of amyloidogenic proteins reflects the fact that the commonest structures as formed by the ribosome are not thermodynamically the most stable.462–465All of this said, the same issues we have highlighted, of weak mutation, strong selection, contingency,390 and epistasis, imply that natural evolution has sampled only a tiny fraction of possible folds,466 and there is now considerable interest, and some notable success, in designing completely novel folds, structures and functions de novo, leading to stable, soluble proteins with topologies absent from current structure databases (e.g., ref. 429 and 467–482). This very much highlights the importance of understanding protein sequence/activity landscapes.
Experimental findings
| Enzyme | Comments | Selected ref. |
|---|---|---|
| Acetylcholinesterase | kcat/Km > 108 M−1 s−1 | 485 and 486 |
| Carbonic anhydrase | kcat/Km ∼ 109 M−1 s−1 | 487 |
| Catalase | kcat/Km > 107 M−1 s−1 | 488 and 489 |
| Superoxide dismutase (Mn) | kcat/Km ∼ 8.108 M−1 s−1 | 490 |
| Triose phosphate isomerase | The original classic; kcat/Km > 108 M−1 s−1 | 374, 375, 483 and 484 |
While the reactions catalysed by these enzymes are relatively simple, it is not obvious for chemical reasons why virtually any enzyme could not be able to attain such rates, and we believe that in most cases they will be able to. Again, the simplest explanation (additional to lack of selection pressure) is that most proteins got stuck in areas of the landscape from which they could not simply escape.
Sequence-activity variations in various proteins
We next look at some experimental datasets that give an indication of the kinds of variation in (sequence and) activity that may be observed in real proteins. Obviously we are here starting with proteins that have themselves been selected (and likely been trapped in local fitness peaks) during natural evolution, so we are not modelling the very earliest stages in an evolution. We begin with an enzyme but look at the binding of an antibody thereto as an indication (in this case) of misfolding or effectiveness of secretion. Fowler and colleagues491 studied 44816 variant effects for 436 synonymous variants and 8528 of the 8759 possible missense variants of the serine protease (clotting) Factor IX (FIX). Their Table S4 contains 8964 examples, of which 5085 were encoded as wild type and 3879 as loss of function. We have plotted these data as a function of residue number in Fig. 6. This allows us to make the following observations, that are fairly generally true. First, almost no residue is completely immune from causing trouble (loss of function) when mutated, though some areas (such as the residues from ∼180 to ∼230) are much more resilient than others (see also ref. 134). For instance, surface residue are much more tolerant than are buried ones.199,492 Secondly, most mutations are deleterious (see also ref. 160, 493 and 494).
 | ||
| Fig. 6 Mutational spectrum of Factor IX. Data taken from ref. 491. | ||
Fig. 7 shows data495 on a large series of enzymes, where it is clear from the relative slopes of the line and best fit (continuous line) and the line of identity (dotted line) that mutations from better starting points tend to be deleterious, effectively reflecting peak in the system.
 | ||
| Fig. 7 Data from Yan et al.495 on a large series of enzymes and mutants thereof. Continuous line is line of best fit, while dotted line = line of identity. | ||
Enzyme catalytic power and natural selection
The usual metric for enzyme catalytic power is kcat/Km. In the more common Michaelis–Menten kinetics,496–499 diffusion of substrate to the enzyme active site is fast relative to the catalytic event500 (but see ref. 501). Diffusion-controlled enzymes, which it has been claimed can have catalytic powers as great as 1010 M−1 s−1, ref. 502 (108−9 M−1 s−1 is more commonly quoted) tend to exhibit Briggs–Haldane kinetics but are rare. Most values of catalytic power are far lower than this. In the case of directed evolution for industrial enzymes we normally have substrate concentrations far above most values of Km, and we are really more interested in optimising values of kcat or turnover number (that with stability determines space-time yield503–507), not least to avoid the burden of increasing protein synthesis.26,508–512 Protein levels even for a given amino acid sequence also depend strongly on codon usage, especially via RNA stability,513 as well as the thermodynamics of the reaction involved.514,515 Few of these enzyme turnover numbers naturally exceed 100 s−1, ref. 516 (the median for natural enzymes is ∼10 s−1, ref. 517), and in directed evolution most are far below this504,518,519. For instance the storied sitagliptin example520 improved an enzyme many thousandfold but the final kcat was still only ∼25 s−1. Tables of kcat, kcat/Km etc. are available at a variety of databases521 such as BRENDA522 and SABIO-RK.523,524 A distillation of the latter is given in Fig. 8. | ||
| Fig. 8 Distribution of 27757 ‘starting’ values of kcat as downloaded from SABIO-RK. | ||
Because extensive measurements covering many residues are often quite poorly available,18,223 it is increasingly possible to use the methods of machine learning to calculate kcat and other enzyme kinetic parameter values de novo.519,525–537 This will be very valuable for both creating and navigating activity landscapes.
Metabolic control analysis538–541 is a method of local sensitivity analysis542 in which the normalised effect of a small change in enzyme activity or concentration dE/E creates a normalised change in flux dJ/J. Their ratio (dJ/J)/(dE/E) is known as the flux-control coefficient and, importantly, the sum of the flux-control coefficients for a flux of interest, over all the enzymes in a system adds to 1. Consequently most flux-control coefficients have small values and increasing the rate of an individual enzyme simply means that it contributes increasingly less to flux control. Consequently, because (or when) they are embedded in metabolic networks, there is little or no selection pressure in natural evolution for individual enzymes to seek to achieve unusually high values of kcat. This is sufficient to explain why most mutations in diploid systems are recessive.345
Whether the chemistry of any enzymatic reaction (or at least most) could be converted to run at near diffusion-controlled rates is an open question, but our prejudice is that if we understood landscapes well enough there is no a priori reason why not. As stated above, the modest values for kcat seen in natural enzymes are easily seen to result from a combination of a lack of selection and the combination of weak mutation, strong selection and epistasis trapping enzymes in local optima that are far from the maximum. We give further arguments below based on the similarity of the exponential distributions of both extreme value theory and neural network scaling.
Typical effects of mutations on a partially evolved protein
Given that a starting protein of interest is some kind of wild type that has presumably evolved to a local maximum it is unsurprising that most initial mutations (commonly about one half434,495) result in lower fitnesses, meaning that one has to seek multiple mutations to find a better peak (‘reculer pour mieux sauter’543).544 The default view might be that if half of the single mutations are deleterious then only a quarter of double mutations and 1/(2n) of n mutations might be better. Fortunately this dismal view fails to take into account the existence of reciprocal sign epistasis and also that the improvements may be much better than this exponential relation would indicate.Fluorescent proteins
Fluorescence is a property that is easily measured, and the classical Green Fluorescent Protein (GFP) from Aequorea victoria, with just ∼250 residues, provides the archetype.545,546 Sarkisyan et al.547 studied the GFP landscape, finding that the effect of mutations on fluorescence was positively correlated with site conservation. They also commented that “The broad congruence of our data with the prevalence of epistasis from long-term evolution suggests that the shape of the local fitness landscape can be extrapolated to a larger scale”547. This challenge was effectively taken up in an important paper by Biswas et al.434 In this work, they studied both GFP and β-lactamase variants for which they had nearly 105 functional sequences. Neural networks require numerical rather than sequence inputs,548 so their own LLM UniRep was used to form a numerical representation, an embedding that effectively learned secondary structures from primary sequences. A supervised method was used to fine tune this representation, and the model landscape could then be navigated to produce variants with both high novelty in sequence space and high activity. This provided an important indication that it was indeed possible to generalise from landscapes trained with numbers far smaller than 10N. The same was true for TEM1 β-lactamase,434 and Wagner84 considers (with evidence) that this is likely to be generally true.An especially large dataset on GFP and related proteins was provided by Gonzalez Somermeyer and colleagues.218 It consisted of some 93925 variants with attendant fitnesses. Data were obtained from their supplementary information (SI). These are plotted in Fig. 9 as probability density (PDF) and cumulative (CDF) distributions, along with the fit to a gamma distribution. An interesting feature of this dataset, given its starting points in the GFP of four organisms, three with 18%, 59%, and 82% sequence divergence from the classical (Aequorea victoria) GFP, was the existence of four major fitness peaks, two sharp and two flatter, and these are evident in both the PDF and CDF of Fig. 9. Of especial interest in the context of this review is the fact that above a brightness of some 30000 or so the distribution of fitnesses evidently turns from something like a gamma distribution to an exponential one, an argument that is a central feature of this review.
 | ||
| Fig. 9 Probability density function, gamma distribution and cumulative distribution function of 93925 variants of green fluorescent protein studied by Gonzalez Somermeyer and colleagues.218. | ||
TEM1 β-lactamase
Gonzalez and Ostermeier549 also studied intragenic epistasis among several thousand pairs of mutations in adjacent residues (‘sequential mutations’) in TEM-1 β-lactamase. Negative epistasis (52%) occurred 7.6 times as frequently as did positive epistasis (6.8%). The distribution of activities is shown in Fig. 10, where the activity levels cover more than three orders of magnitude. | ||
| Fig. 10 Distribution of activities in 4507 mutants of TEM-β-lactamase. Data are taken from the SI of ref. 549. | ||
The same group550 then went on to study the effect of single indels, with the results for insertions shown in Fig. 11. Again the range covered more than three orders of magnitude, while insertions tended to be somewhat less deleterious than deletions, and the largest effects were seen in regions with significant secondary structure. Over half of insertions (51%) and deletions (59%) resulted in at least a 100-fold decrease in fitness relative to TEM-1. 9.8% of insertions and 11% of deletions retained 50% of wild-type fitness, although 40.9% of these were in the signal sequence (the first 23 aa), probably reflecting varying expression levels more than kcat changes. In a similar study, Macdonald and colleagues also found that deletions were most disruptive overall, that beta sheets are most sensitive to indels, and flexible loops are sensitive to deletions yet tolerate insertions.551 In general, disruptiveness depends on the local structural context;552 for instance, prolines are not tolerated inside α-helices.553
 | ||
| Fig. 11 Effect of insertions on the fitness levels of TEM-β-lactamase. Data are taken from the SI of ref. 550. | ||
Tryptophan synthase
Johnston et al.251 performed an exhaustive analysis of the effects of changing four amino acids (204 = 160000) in the active site of the beta subunit of a thermostable tryptophan synthase (TrpB). Jason Yang kindly provided the data via https://data.caltech.edu/records/h5rah-5z170. Just 10140 of the mutants were seen as active. Interestingly, the most-fit TrpB variants contained a substitution (K227) that is nearly absent in natural TrpB sequences, which could be seen as consistent with the view that once activity is sufficient for the needs of the organism in its environments there is little selection pressure to increase it.
This is illustrated in Fig. 12 in terms of the fitness of three of the amino acids at position 227. Clearly (as stated) K227 is the highest (other amino acids are excluded for clarity) but equally clearly there are other contexts in which K227 is very poor indeed, showing again how very epistatic is this particular landscape. Of course we do need to stress again that the starting sequence is a wild type that has been selected by natural evolution, albeit in a thermophile rather than the mesophilic recombinant host used (E. coli), and is to be seen as existing in some kind of a local peak, so it is not surprising that most mutations are less fit than the average.
 | ||
| Fig. 12 Illustration of the high fitness value of K227 (compared with A227 and G227) in some contexts in trpB. Data from ref. 251. | ||
In a similar vein, Fig. 13 shows the fitness and its probability density distribution for this large dataset251, indicating how most mutations are less fit, but that the cumulative distribution function is tolerably fitted by a gamma function (whose parameters are shape = 0.869 and scale (θ) = 0.034). Although the larger fitnesses are increasingly few in number, it is reasonable to state that above a fitness of about 0.5 there is a change in slope and these follow an exponential distribution as stressed throughout this review.
 | ||
| Fig. 13 Probability distribution function (PDF) and fit to a gamma distribution function for the cumulative fitnesses in the large dataset of Johnston et al.251 on trpB. | ||
Protein GB1
Wu et al. performed a similar exhaustive study at 4 sites (V39, D40, G41, V54; 160000 variants) in the 56-amino acid B1 domain of protein G, an immunoglobulin-binding protein expressed in certain streptococci. As expected, most mutants had a lower fitness compared to the wild type (VDGV, whose fitness was normalised to 1), 2.4% of mutants were beneficial. The best (FWAA) was nearly 9 times fitter.
Fig. 14 and 15 show some of the data (taken from Table S4 of ref. 10).
 | ||
| Fig. 14 Fitness values for various mutations in AA54 of GB1,10 coloured by the residue at AA39. The top 15 sequences are labelled. | ||
 | ||
| Fig. 15 Distribution of fitness values as taken from the data in Table S4 of Wu et al.10 | ||
These figures serve to illustrate several points. First, that the best 15 variants do not share a single residue in common with the VDGV wild type (see also ref. 554). Secondly, for D40 and V54 they are not even of the same classes (i.e. negatively charged and hydrophobic) as that of the original, and thirdly, above the wild-type fitness of 1 the frequency is evidently decreasing exponentially with increasing fitness.
Dihydrofolate reductase (DHFR)
D’Costa and colleagues performed a detailed study16 of the protein fitness landscape of DHFR, an enzyme whose ‘fitness’ can be both selected for and assessed in terms of resistance to trimethoprim. They found extensive epistasis but an overall global peak that was evolutionarily accessible from most starting sequences. In a similar vein, Papkou, Wagner and colleagues212 created a large biological fitness landscape (>260000 mutants) using CRISPR-Cas9 gene editing of the Escherichia coli dihydrofolate reductase, fitness again being assessed as growth rates in the presence of trimethoprim. In this case just 17774 of the strains (6.8%) were viable, encoding 1630 unique amino acid sequences.84 In this latter paper, 90% of the predictive power could be attained with just 7.8% (1400) of the genotypes.
Amidase
Wrenbeck et al.555 analysed over 7000 mutants of an amidase, representing over 96% of non-synonymous single mutations, for their activity against three different amides, with the distribution of beneficial mutations being quite different for each substrate while seen as ‘exponential’.The role of (molecular) dynamics in enzyme landscapes
Improvements in enzymatic rate constants normally occur far from the active site, and we have argued22 that this is because of the important role of dynamics in enzyme catalysis. Specifically, it is recognised that for Michaelis–Menten kinds of mechanism, any free energy of binding has already been ‘used up’, so the only source of conformational change that will drive the catalytic step comes from thermal fluctuations in both solvent and enzyme.556,557 The former necessarily transmits these to the enzyme active site from the protein's surface. Entirely consistent with this, Jiménez-Osés and colleagues349 studied two mutants of lovD whose ground state structures were indistinguishable but whose activities varied 1000-fold. Only differences in the dynamics could explain this, and Osuna (e.g., ref. 558–565) and others (e.g., ref. 526 and 566–586) have developed these ideas to exploit the methods of conformational selection and molecular dynamics (MD) to predict which residues might most usefully be mutated in order to navigate the fitnes landscapes more effectively. MD is also of value in the assessment of thermostability.587–595 Unsurprisingly, machine learning techniques are now being used in various ways to speed up these kinds of MD calculations.596–605Modelling methods that effectively draw smooth curves through high-dimensional landscapes
Thus far we have talked about modelling landscapes in rather abstract terms. Classical ‘supervised’ machine learning uses paired properties (here sequence and activity) to learn a mapping that uses the first to predict the second. However, it requires measurements of both sequence and activity that may not always be to hand.249 As mentioned above, normally this also requires sequence strings, which are discrete objects, to be converted to a numerical form or ‘embedding’ that allows such a continuous mapping.548 The easiest way is arguably to encode each residue with a small vector of numbers that represents its properties (e.g., hydrophobicity, polarity, α-helix-forming tendency, β-sheet-forming tendency, and tendency to be unstructured). This means that biophysically similar amino acids are also close in this vector space.The breakthrough of the large language models mentioned above was the recognition that much could be learned from sequence alone (known as unsupervised learning), as objects with meaningful structure – whether sentences or images – exhibited statistical regularities. This obviated the need for such large numbers of paired sequences and activity for supervised learning,606 and was consistent with the fact that human infants mainly learn languages in an unsupervised way. (For completeness there is also semi-supervised learning, which exploits a smaller number of supervised pairs to improve an otherwise unsupervised model.)
A straight line is of course given by the equation y = mx + c. Thus any of millions of values of x can be turned into equivalent values for y and if the nature of this curve (line) is known only the two parameters m and c need be stored. In a similar vein, the idea of this kind of modelling is to find a (complex) equation with a number of parameters far smaller than the number of possible examples (20N) that effectively plots the entire landscape for millions of sequence examples with which it might be interrogated. Importantly, it was long ago shown that classical artificial feedforward neural networks of the multilayer perceptron607–611 or radial basis function612–614 types can approximate any function, the so-called universal approximation theorem. Of course this theorem does not precisely state how many parameters are required for a certain degree of approximation,611 but the principle is important. More recently, similar arguments have been raised for transformer-based deep neural networks.615–619 Thus the first question becomes “can I model a landscape accurately using (deep) neural networks?”, and the answer is yes.
The second question is then, rather obviously, “how many parameters (i.e. neural network weights) will I require?”. The answer to this clearly depends on the landscape ruggedness, the efficiency and extent of sampling, the size of the training set, and the size and architecture of the transformer or other approximator. Consequently, there is no universal answer, but there is increasing knowledge of the scaling laws for deep networks: broadly errors decrease exponentially relative to both the number of examples and the number of parameters,620–623 and it is best to vary both in step but with substantial data examples per parameter.624,625 A useful summary is at https://lifearchitect.ai/chinchilla/.
The reason for this exponentiality actually comes from the bias-variance trade-off in high-dimensional space.626 Gratifyingly and importantly, this result fits exactly with the exponential fall off predicted by extreme value theory as rehearsed above, where we seek to fit a population far larger than the small and often biased/local one represented by the samples within the existing population. Consequently interest is shifting towards means of ‘beating’ the standard scaling laws (e.g., ref. 624), that evidently suffer from diminishing returns (not to say massive environmental energy costs).627
Multiobjective optimisation
Thus far we have focused more or less explicitly on single fitness objectives, especially kcat, but almost all optimisations actually have more than one objective, e.g., specificity (or lack of it), pH dependence, solvent tolerance, and/or, the one we shall focus on, thermostability. Thermostability is desirable not only for reasons of stability but because running reactors at a higher temperature when they contain enzymes with higher values of kcat helps avoid cooling costs. Thermostability is also of value when processes are intensive628,629 or continuous506,630–632 or both.633 Obviously reaction rates tend to increase with temperature (for reasons based on basic chemical kinetics, fluctuations, and internal protein mobility given above and by634) but protein stability commonly does not.635 Consequently there is seen to be some kind of a trade-off595,636–642 (also for solubility643), although it is not at all obvious that this is inevitable for any kind of fundamental physico-chemical reason.635 In fact it is not.Fig. 16 illustrates the key general idea of multiobjective optimisation, in which individuals may be especially good at one of the objectives but not the other. Those filled symbols in Fig. 15 represent individuals that are seen as the best in at least one objective for a given value of the other objective, and they occupy what is referred to as the non-dominated or Pareto front. During an adaptive walk it is then a matter of preference as to how (in directed evolution) the experimenter chooses to trade off the two objectives (e.g., ref. 443 and 644). One effective and well-established class of algorithm (e.g., ref. 644–658) seeks to improve individuals by recombining elements of those on the Pareto front in the hope that the Pareto front will effectively move ‘north-east’ in Fig. 15. Memetic algorithms659–670 do something similar.
 | ||
| Fig. 16 General principle of multiple objectives and the existence of a Pareto front. Individual sequences are represented by symbols, with the filled symbols being on the Pareto or non-dominated front. Redrawn from the CC-By 4.0 Open Access paper.22 | ||
No necessary trade-off between kcat and thermostability
Ancient proteins in an evolutionary sequence tend to be more stable, something considered (assumed) to be reflecting a variety of conditions prevalent at the time,671 though they tend to evolve towards more mesophilic or psychrophilic temperature optima and greater catalytic power when the temperature is in the relevant range.672 This said, there does not seem to be any a priori reason for such a trade-off when sequences are considered more globally and in principle these properties can be decoupled414,635,641,673–680 so as to achieve high catalytic activity together with thermostability. Arguably some of the basis for the earlier view was effectively an artefact caused by the fact that many mutations are destabilising so that it appears that activity is in conflict with stability. In addition, comparing thermophilic and mesophilic enzymes at a single temperature erroneously conflates temperature adaptation with activity. Consequently, much as in the spirit of the rest of this review, we consider that the earlier ‘trade-off’ view most likely simply reflects evolutionary contingency, local search, weak mutation/strong selection, and epistasis trapping proteins in local maxima.Following the multiobjective principle, much as in other areas (e.g., ref. 681) we consider that speeding up directed evolution is best done with a suite of orthogonal methods (including sequence and structural, dynamics, assay-based, and so on). This is because the scientific philosophy principle of coherence682–685 states that the more that different and orthogonal methods lead to the same conclusion, the more likely is that conclusion to be correct.
Discussion and conclusions
Requests and recommendations
We do not think that this is otherwise the place to go into the necessary level of detail, but it does seem that an RO-Crate strategy https://www.researchobject.org/ro-crate/697 might be one way forward, while the Analytical Information Markup Language (AniML) (https://www.animl.org/)698 would seem to offer the necessary framework for including the necessary data and metadata in a standardised, machine-readable format.
We also note that the Organic Reaction database699 bears many similarities to what is desired, while RetroBioCat700 is another but far less open reaction database. The Open Reaction Database (ORD) (https://open-reaction-database.org)699 is an emerging standard for structuring and sharing organic reaction data in a machine-readable format. The ORD already has some capabilities to include enzymes and proteins as reaction participants, with UniProt ID,701 Protein Data Bank,702 Hierarchical Editing Language for Macromolecules (HELM) (https://www.pistoiaalliance.org/project/helm-project/), and amino acid sequences being currently supported component identifiers. The ORD is an evolving standard, and the synbio community is encouraged to identify additional data features that may be required for the appropriate description of biocatalytic reactions. Finally, the emergence of ‘vibe coding’ or agentic software703–705 that produces code to perform analyses when presented with requests in natural language form offers the oppportunity to democrastise these kinds of analyses.
Overall conclusions
Existing enzymes selected via natural or directed evolution commonly follow a path of weak mutation/strong selection, and the epistatic landscapes that they thereby inhabit necessarily lead to entrapment in local maxima and to ruggedness. The distribution of local fitnesses commonly follows a gamma distribution, but that of the whole landscape is more nearly exponential, indicating that the first is likely to be a poor model for the second. Without models that can cover the landscapes more widely it is hard to extrapolate beyond the known, and active learning is needed to choose wisely the examples with which to populate models that can generalise well to properties far better than those on which they were initially trained. Having such models will potentially usher in an era of enzymes that are designed de novo, and are small, thermostable, and highly active. All of these are highly desirable goals.Author contributions
Conceptualization, DBK; formal analysis, IR, DBK; resources, DBK; writing – original draft preparation, DBK; writing – review and editing, IR, DBK; visualization, IR, DBK; funding acquisition, DBK.Conflicts of interest
There are no conflicts of interest to declare.Data availability
Data used here are in the public domain and their sources full referenced.Acknowledgements
DBK thanks the BBSRC (grant BB/Y009258/1) and the Novo Nordisk Foundation (grant NNF20CC0035580) for funding. The content and findings reported and illustrated are the sole deduction, view and responsibility of the researchers and do not reflect the official position and sentiments of the funders. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We thank Dr Ben Deadman for useful inputs regarding open databases and data standards and Jason Yang for assistance with the provision of data for trpB.References
- The roles of mutation, inbreeding, crossbreeding and selection in evolution, in Proc Sixth Int Conf Genetics, ed. S. Wright, Genetics Society of America, Austin TX, Ithaca, NY, 1932 Search PubMed.
- J. M. Smith, Natural selection and the concept of a protein space, Nature, 1970, 225(5232), 563–564 CrossRef CAS PubMed.
- C. A. Voigt, S. Kauffman and Z. G. Wang, Rational evolutionary design: the theory of in vitro protein evolution, Adv. Prot. Chem., 2001, 55, 79–160 CrossRef PubMed.
- F. J. Poelwijk, D. J. Kiviet, D. M. Weinreich and S. J. Tans, Empirical fitness landscapes reveal accessible evolutionary paths, Nature, 2007, 445(7126), 383–386 CrossRef CAS PubMed.
- B. Calcott, Assessing the fitness landscape revolution, Biol. Philos., 2008, 23, 639–657 CrossRef.
- P. A. Romero and F. H. Arnold, Exploring protein fitness landscapes by directed evolution, Nat. Rev. Mol. Cell Biol., 2009, 10(12), 866–876 CrossRef CAS PubMed.
- M. Carneiro and D. L. Hartl, Adaptive landscapes and protein evolution, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(suppl 1), 1747–1751 CrossRef CAS PubMed.
- P. A. Romero, A. Krause and F. H. Arnold, Navigating the protein fitness landscape with Gaussian processes, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(3), E193–E201 CrossRef CAS.
- J. A. G. M. de Visser and J. Krug, Empirical fitness landscapes and the predictability of evolution, Nat. Rev. Genet., 2014, 15(7), 480–490 CrossRef CAS PubMed.
- N. C. Wu, L. Dai, C. A. Olson, J. O. Lloyd-Smith and R. Sun, Adaptation in protein fitness landscapes is facilitated by indirect paths, eLife, 2016, 5, e16965 CrossRef.
- I. Fragata, A. Blanckaert, M. A. Dias Louro, D. A. Liberles and C. Bank, Evolution in the light of fitness landscape theory, Trends Ecol. Evol., 2019, 34(1), 69–82 CrossRef PubMed.
- E. C. Hartman and D. Tullman-Ercek, Learning from protein fitness landscapes: a review of mutability, epistasis, and evolution, Curr. Opin. Struct. Biol., 2019, 14, 25–31 CrossRef.
- C. B. Ogbunugafor, A Reflection on 50 Years of John Maynard Smith's “Protein Space”, Genetics, 2020, 214(4), 749–754 CrossRef CAS PubMed.
- C. R. Freschlin, S. A. Fahlberg and P. A. Romero, Machine learning to navigate fitness landscapes for protein engineering, Curr. Opin. Biotechnol, 2022, 75, 102713 CrossRef CAS.
- R. J. McLure, S. E. Radford and D. J. Brockwell, High-throughput directed evolution: a golden era for protein science, Trends Chem., 2022, 4, 278–291 Search PubMed.
- S. D'Costa, E. C. Hinds, C. R. Freschlin, H. Song and P. A. Romero, Inferring protein fitness landscapes from laboratory evolution experiments, PLoS Comput. Biol., 2023, 19(3), e1010956 CrossRef PubMed.
- A. T. Meger, M. A. Spence, M. Sandhu, D. Matthews, J. Chen and C. J. Jackson, et al., Rugged fitness landscapes minimize promiscuity in the evolution of transcriptional repressors, Cell Syst., 2024, 15(4), 374–387 CrossRef CAS PubMed.
- F. Z. Li, J. Yang, K. E. Johnston, E. Gursoy, Y. Yue and F. H. Arnold, Evaluation of machine learning-assisted directed evolution across diverse combinatorial landscapes, Cell Syst., 2025, 16(9), 101387 CrossRef CAS PubMed.
- M. A. Spence, M. Sandhu, D. S. Matthews, J. Nichols and C. J. Jackson, Fitness landscape ruggedness arises from biophysical complexity, bioRxiv, 2025, 2025.04.12.648556.
- E. Firnberg, J. W. Labonte, J. J. Gray and M. Ostermeier, A comprehensive, high-resolution map of a gene's fitness landscape, Mol. Biol. Evol., 2014, 31(6), 1581–1592 CrossRef CAS PubMed.
- D. M. McCandlish, Visualizing fitness landscapes, Evolution, 2011, 65(6), 1544–1558 CrossRef.
- A. Currin, N. Swainston, P. J. Day and D. B. Kell, Synthetic biology for the directed evolution of protein biocatalysts: navigating sequence space intelligently, Chem. Soc. Rev., 2015, 44(5), 1172–1239 RSC.
- S. Song and J. Zhang, Unbiased inference of the fitness landscape ruggedness from imprecise fitness estimates, Evolution, 2021, 75(11), 2658–2671 CrossRef PubMed.
- J. L. Payne and A. Wagner, The causes of evolvability and their evolution, Nat. Rev. Genet., 2019, 20(1), 24–38 CrossRef CAS PubMed.
- J. H. Gillespie, Some properties of finite populations experiencing strong selection and weak mutation, Am. Nat., 1983, 121, 691–708 CrossRef.
- D. Heckmann, D. C. Zielinski and B. O. Palsson, Modeling genome-wide enzyme evolution predicts strong epistasis underlying catalytic turnover rates, Nat. Commun., 2018, 9(1), 5270 CrossRef CAS PubMed.
- H. H. Chou, H. C. Chiu, N. F. Delaney, D. Segrè and C. J. Marx, Diminishing returns epistasis among beneficial mutations decelerates adaptation, Science, 2011, 332(6034), 1190–1192 CrossRef CAS PubMed.
- D. Aggeli, Y. Li and G. Sherlock, Changes in the distribution of fitness effects and adaptive mutational spectra following a single first step towards adaptation, Nat. Commun., 2021, 12(1), 5193 CrossRef CAS PubMed.
- J. Diaz-Colunga, A. Skwara, K. Gowda, R. Diaz-Uriarte, M. Tikhonov and D. Bajic, et al., Global epistasis on fitness landscapes, Philos. Trans. R. Soc. London, B: Biol. Sci., 2023, 378(1877), 20220053 CrossRef.
- S. M. Reia and P. R. A. Campos, Analysis of statistical correlations between properties of adaptive walks in fitness landscapes, R. Soc. Open Sci., 2020, 7(1), 192118 CrossRef PubMed.
- D. B. Kell, Scientific discovery as a combinatorial optimisation problem: how best to navigate the landscape of possible experiments?, BioEssays, 2012, 34(3), 236–244 CrossRef PubMed.
- J. C. Moore, H. M. Jin, O. Kuchner and F. H. Arnold, Strategies for the in vitro evolution of protein function: Enzyme evolution by random recombination of improved sequences, J. Mol. Biol., 1997, 272(3), 336–347 CrossRef CAS PubMed.
- M. Sandhu, A. C. Mater, D. S. Matthews, M. A. Spence, A. A. Lenskiy and C. Jackson, Investigating the determinants of performance in machine learning for protein fitness prediction, Protein Sci., 2025, 34(8), e70235 CrossRef CAS PubMed.
- T. Aita and Y. Husimi, Adaptive Walks by the Fittest among Finite Random Mutants on a Mt. Fuji-type Fitness Landscape, J. Theor. Biol., 1998, 193(3), 383–405 CrossRef PubMed.
- T. Aita and Y. Husimi, Fitness spectrum among random mutants on Mt. Fuji-type fitness landscape, J. Theor. Biol., 1996, 182(4), 469–485 CrossRef CAS PubMed.
- J. Neidhart, I. G. Szendro and J. Krug, Adaptation in tunably rugged fitness landscapes: the rough Mount Fuji model, Genetics, 2014, 198(2), 699–721 CrossRef.
- N. J. Radcliffe and P. D. Surry, Fundamental limitations on search algorithms: evolutionary computing in perspective, Comput. Sci. Today, 1995, 1995, 275–291 Search PubMed.
- D. H. Wolpert and W. G. Macready, No Free Lunch theorems for optimization, IEEE Trans. Evol. Comput., 1997, 1, 67–82 CrossRef.
- J. McDermott, When and Why Metaheuristics Researchers can Ignore “No Free Lunch” Theorems, SN Comput. Sci., 2020, 1, 60 CrossRef.
- T. Smith, P. Husbands, P. Layzell and M. O'Shea, Fitness landscapes and evolvability, Evol. Comput., 2002, 10(1), 1–34 CrossRef PubMed.
- M. Kogenaru, M. G. de Vos and S. J. Tans, Revealing evolutionary pathways by fitness landscape reconstruction, Crit. Rev. Biochem. Mol. Biol., 2009, 44(4), 169–174 CrossRef PubMed.
- D. B. Saakian and J. F. Fontanari, Evolutionary dynamics on rugged fitness landscapes: exact dynamics and information theoretical aspects, Phys. Rev. E: Stat. Nonlin. Soft Matter Phys., 2009, 80(4 Pt 1), 041903 CrossRef PubMed.
- J. Van Cleve and D. B. Weissman, Measuring ruggedness in fitness landscapes, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(24), 7345–7346 CrossRef CAS PubMed.
- J. A. G. M. de Visser, S. F. Elena, I. Fragata and S. Matuszewski, The utility of fitness landscapes and big data for predicting evolution, Heredity, 2018, 121(5), 401–405 CrossRef PubMed.
- U. Obolski, Y. Ram and L. Hadany, Key issues review: evolution on rugged adaptive landscapes, Rep. Prog. Phys., 2018, 81(1), 012602 CrossRef PubMed.
- C. Bank, Epistasis and Adaptation on Fitness Landscapes, Annu. Rev. Ecol. Evol. Syst., 2022, 53, 457–479 CrossRef.
- L. Trujillo, P. Banse and G. Beslon, Getting higher on rugged landscapes: Inversion mutations open access to fitter adaptive peaks in NK fitness landscapes, PLoS Comput. Biol., 2022, 18(10), e1010647 CrossRef CAS PubMed.
- Y. Li and J. Zhang, On the Probability of Reaching High Peaks in Fitness Landscapes by Adaptive Walks, Mol. Biol. Evol., 2025, 42(4) CrossRef PubMed.
- A. Pena-Francesch, H. Jung, M. C. Demirel and M. Sitti, Biosynthetic self-healing materials for soft machines, Nat. Mater., 2020, 19(11), 1230–1235 CrossRef CAS PubMed.
- J. A. Tomko, A. Pena-Francesch, H. Jung, M. Tyagi, B. D. Allen and M. C. Demirel, et al., Tunable thermal transport and reversible thermal conductivity switching in topologically networked bio-inspired materials, Nat. Nanotechnol., 2018, 13(10), 959–964 CrossRef CAS PubMed.
- N. A. Carter and T. Z. Grove, Functional protein materials: beyond elastomeric and structural proteins, Polym. Chem., 2019, 10, 2952 RSC.
- J. A. Doolan, L. S. Alesbrook, K. Baker, I. R. Brown, G. T. Williams and K. L. F. Hilton, et al., Next-generation protein-based materials capture and preserve projectiles from supersonic impacts, Nat. Nanotechnol., 2023, 18, 1060–1066 CrossRef CAS PubMed.
- W. Lu, N. A. Lee and M. J. Buehler, Modeling and design of heterogeneous hierarchical bioinspired spider web structures using deep learning and additive manufacturing, Proc. Natl. Acad. Sci. U. S. A., 2023, 120(31), e2305273120 CrossRef CAS PubMed.
- K. Singhal, H. E. Adamson, T. M. Baer, H. M. Salis and M. C. Demirel, Microcapillary Array-Based High Throughput Screening for Protein Biomanufacturability, ACS Synth. Biol., 2025, 14, 2328–2340 CrossRef CAS PubMed.
- S. Mital, G. Christie and D. Dikicioglu, Recombinant expression of insoluble enzymes in Escherichia coli: a systematic review of experimental design and its manufacturing implications, Microb. Cell Fact., 2021, 20(1), 208 CrossRef CAS PubMed.
- F. Segato, A. R. L. Damásio, T. A. Gonçalves, R. C. de Lucasa, F. M. Squina and S. R. Decker, et al., High-yield secretion of multiple client proteins in Aspergillus, Enz. Micr. Technol., 2012, 51, 100–106 CrossRef CAS PubMed.
- T. R. Costa, C. Felisberto-Rodrigues, A. Meir, M. S. Prevost, A. Redzej and M. Trokter, et al., Secretion systems in Gram-negative bacteria: structural and mechanistic insights, Nat. Rev. Microbiol., 2015, 13(6), 343–359 CrossRef CAS PubMed.
- E. R. Green and J. Mecsas, Bacterial Secretion Systems: An Overview, Microbiol. Spectr., 2016, 4(1) CrossRef CAS PubMed.
- L. A. Burdette, S. A. Leach, H. T. Wong and D. Tullman-Ercek, Developing Gram-negative bacteria for the secretion of heterologous proteins, Microb. Cell Fact., 2018, 17(1), 196 CrossRef CAS PubMed.
- R. Freudl, Signal peptides for recombinant protein secretion in bacterial expression systems, Microb. Cell Fact., 2018, 17(1), 52 CrossRef PubMed.
- S. Chai, Z. Zhu, E. Tian, M. Xiao, Y. Wang and G. Zou, et al., Building a Versatile Protein Production Platform Using Engineered Trichoderma reesei, ACS Synth. Biol., 2021, 11, 486–496 CrossRef PubMed.
- J. Neef, J. M. van Dijl and G. Buist, Recombinant protein secretion by Bacillus subtilis and Lactococcus lactis: pathways, applications, and innovation potential, Essays Biochem., 2021, 65, 187–195 CrossRef CAS PubMed.
- H. Yang, J. Qu, W. Zou, W. Shen and X. Chen, An overview and future prospects of recombinant protein production in Bacillus subtilis, Appl. Microbiol. Biotechnol., 2021, 105, 6607–6626 CrossRef CAS PubMed.
- R. Jadhav, R. L. Mach and A. R. Mach-Aigner, Protein secretion and associated stress in industrially employed filamentous fungi, Appl. Microbiol. Biotechnol., 2024, 108, 92 CrossRef CAS PubMed.
- S. R. Lokireddy, S. R. Kunchala and R. Vadde, Advancements in Escherichia coli secretion systems for enhanced recombinant protein production, World J. Microbiol. Biotechnol., 2025, 41, 90 CrossRef PubMed.
- L. E. Contreras-Llano and C. Tan, High-throughput screening of biomolecules using cell-free gene expression systems Open Access, Synth. Biol., 2018, 1, ysy012 CrossRef PubMed.
- N. Laohakunakorn, Cell-Free Systems: A Proving Ground for Rational Biodesign, Front. Bioeng. Biotechnol., 2020, 8, 788 CrossRef PubMed.
- A. Maharjan and Jung-Ho Park1, Cell-free protein synthesis system: A new frontier for sustainable biotechnology-based products, Biotechnol. Appl. Biochem., 2023, 70, 2136–2149 CrossRef CAS PubMed.
- K. Yue, J. Chen, Y. Li and L. Kai, Advancing synthetic biology through cell-free protein synthesis, Comput. Struct. Biotechnol. J., 2023, 21, 2899–2908 CrossRef CAS PubMed.
- A. C. Hunt, B. J. Rasor, K. Seki, H. M. Ekas, K. F. Warfel and A. S. Karim, et al., Cell-Free Gene Expression: Methods and Applications, Chem. Rev., 2024, 125, 91–149 CrossRef PubMed.
- E. L. Thornton, S. M. Paterson, M. J. Stam, C. W. Wood, N. Laohakunakorn and L. Regan, Applications of cell free protein synthesis in protein design, Prot. Sci., 2024, 33, e5148 CrossRef CAS PubMed.
- A. Clark-ElSayed, I. M. Harrison and M. L. Olsen, John T. Lazar, Jewett MC, Ellington AD. LDBT instead of DBTL: combining machine learning and rapid cell-free testing, Nat. Commun., 2025, 16, 9782 CrossRef CAS PubMed.
- B. J. Rasor and T. J. Erb, Cell-Free Systems to Mimic and Expand Metabolism, ACS Synth. Biol., 2025, 14, 316–322 CrossRef CAS PubMed.
- T. Buzan, How to mind map, Thorsons, London, 2002 Search PubMed.
- D. Hull, S. R. Pettifer and D. B. Kell, Defrosting the digital library: bibliographic tools for the next generation web, PLoS Comput. Biol., 2008, 4(10), e1000204 CrossRef PubMed.
- M. S. Breen, C. Kemena, P. K. Vlasov, C. Notredame and F. A. Kondrashov, Epistasis as the primary factor in molecular evolution, Nature, 2012, 490(7421), 535–538 CrossRef CAS PubMed.
- Epistasis: methods and protocols, ed. K.-C. Wong, Humana Press, Berlin, 2021 Search PubMed.
- C. M. Miton and N. Tokuriki, How mutational epistasis impairs predictability in protein evolution and design, Protein Sci., 2016, 25(7), 1260–1272 CrossRef CAS PubMed.
- D. M. Lyons, Z. Zou, H. Xu and J. Zhang, Idiosyncratic epistasis creates universals in mutational effects and evolutionary trajectories, Nat. Ecol. Evol., 2020, 4(12), 1685–1693 CrossRef PubMed.
- Y. Park, B. P. H. Metzger and J. W. Thornton, Epistatic drift causes gradual decay of predictability in protein evolution, Science, 2022, 376(6595), 823–830 CrossRef CAS PubMed.
- K. Buda, C. M. Miton and N. Tokuriki, Pervasive epistasis exposes intramolecular networks in adaptive enzyme evolution, Nat. Commun., 2023, 14(1), 8508 CrossRef CAS PubMed.
- R. Lipsh-Sokolik and S. J. Fleishman, Addressing epistasis in the design of protein function, Proc. Natl. Acad. Sci. U. S. A., 2024, 121(34), e2314999121 CrossRef CAS PubMed.
- Y. Park, B. P. H. Metzger and J. W. Thornton, The simplicity of protein sequence-function relationships, Nat. Commun., 2024, 15(1), 7953 CrossRef CAS PubMed.
- A. Wagner, Genotype sampling for deep-learning assisted experimental mapping of a combinatorially complete fitness landscape, Bioinformatics, 2024, 40(5), btae317 CrossRef CAS PubMed.
- D. A. Kondrashov and F. A. Kondrashov, Topological features of rugged fitness landscapes in sequence space, Trends Genet., 2015, 31(1), 24–33 CrossRef CAS PubMed.
- M. S. Packer and D. R. Liu, Methods for the directed evolution of proteins, Nat. Rev. Genet., 2015, 16(7), 379–394 CrossRef CAS PubMed.
- L. du Plessis, G. E. Leventhal and S. Bonhoeffer, How Good Are Statistical Models at Approximating Complex Fitness Landscapes?, Mol. Biol. Evol., 2016, 33(9), 2454–2468 CrossRef CAS PubMed.
- R. S. Bon and H. Waldmann, Bioactivity-guided navigation of chemical space, Acc. Chem. Res., 2010, 43(8), 1103–1114 CrossRef CAS PubMed.
- A. L. Hopkins and G. R. Bickerton, Know your chemical space, Nat. Chem. Biol., 2010, 6(7), 482–483 CrossRef CAS PubMed.
- K. L. M. Drew, H. Baiman, P. Khwaounjoo, B. Yu and J. Reynisson, Size estimation of chemical space: how big is it?, J. Pharm. Pharmacol., 2012, 64(4), 490–495 CrossRef CAS PubMed.
- J. L. Reymond, The Chemical Space Project, Acc. Chem. Res., 2015, 48(3), 722–730 CrossRef CAS PubMed.
- M. Koch, T. Duigou, P. Carbonell and J. L. Faulon, Molecular structures enumeration and virtual screening in the chemical space with RetroPath2.0, J. Cheminform., 2017, 9(1), 64 CrossRef PubMed.
- A. Lin, D. Horvath, V. Afonina, G. Marcou, J. L. Reymond and A. Varnek, Mapping of the Available Chemical Space versus the Chemical Universe of Lead-Like Compounds, ChemMedChem, 2018, 13(6), 540–554 CrossRef CAS PubMed.
- P. S. Gromski, A. B. Henson, J. M. Granda and L. Cronin, How to explore chemical space using algorithms and automation, Nat. Rev. Chem., 2019, 3(2), 119–128 CrossRef.
- T. Hoffmann and M. Gastreich, The next level in chemical space navigation: going far beyond enumerable compound libraries, Drug Discovery Today, 2019, 24(5), 1148–1156 CrossRef CAS PubMed.
- D. B. Kell, S. Samanta and N. Swainston, Deep learning and generative methods in cheminformatics and chemical biology: navigating small molecule space intelligently, Biochem. J., 2020, 477, 4559–4580 CrossRef CAS PubMed.
- H. Öztürk, A. Özgür, P. Schwaller, T. Laino and E. Ozkirimli, Exploring chemical space using natural language processing methodologies for drug discovery, Drug Discovery Today, 2020, 25(4), 689–705 CrossRef PubMed.
- C. W. Coley, Defining and Exploring Chemical Spaces, Trends Chem., 2021, 3(2), 133–145 CrossRef CAS.
- A. Lavecchia, Navigating the frontier of drug-like chemical space with cutting-edge generative AI models, Drug Discovery Today, 2024, 29(9), 104133 CrossRef CAS PubMed.
- A. A. Kattuparambil, D. K. Chaurasia, S. Shekhar, A. Srinivasan, S. Mondal and R. Aduri, et al., Exploring chemical space for “druglike” small molecules in the age of AI, Front. Mol. Biosci., 2025, 12, 1553667 CrossRef CAS PubMed.
- J. L. Reymond, Chemical space as a unifying theme for chemistry, J. Cheminform., 2025, 17(1), 6 CrossRef CAS PubMed.
- R. S. Bohacek, C. McMartin and W. C. Guida, The art and practice of structure-based drug design: A molecular modeling perspective, Med. Res. Rev., 1996, 16(1), 3–50 CrossRef CAS PubMed.
- A. M. Virshup, J. Contreras-García, P. Wipf, W. Yang and D. N. Beratan, Stochastic voyages into uncharted chemical space produce a representative library of all possible drug-like compounds, J. Am. Chem. Soc., 2013, 135(19), 7296–7303 CrossRef CAS PubMed.
- M. Orsi and J. L. Reymond, Navigating a 1E + 60 Chemical Space of Peptide/Peptoid Oligomers, Mol. Inform., 2025, 44(1), e202400186 CrossRef PubMed.
- M. Cruz-Monteagudo, J. L. Medina-Franco, Y. Pérez-Castillo and O. Nicolotti, Cordeiro MNDS, Borges F. Activity cliffs in drug discovery: Dr Jekyll or Mr Hyde?, Drug Discovery Today, 2014, 19(8), 1069–1080 CrossRef CAS PubMed.
- D. Stumpfe, H. Hu and J. Bajorath, Advances in exploring activity cliffs, J. Comput. Aided Mol. Des., 2020, 34(9), 929–942 CrossRef CAS PubMed.
- H. Hu and J. Bajorath, Activity cliffs produced by single-atom modification of active compounds: Systematic identification and rationalization based on X-ray structures, Eur. J. Med. Chem., 2020, 207, 112846 CrossRef CAS PubMed.
- D. Stumpfe, H. Hu and J. Bajorath, Computational method for the identification of third generation activity cliffs, Methods X, 2020, 7, 100793 CAS.
- T. Janela and J. Bajorath, Anatomy of Potency Predictions Focusing on Structural Analogues with Increasing Potency Differences Including Activity Cliffs, J. Chem. Inf. Model., 2023, 63(22), 7032–7044 CrossRef CAS PubMed.
- S. Tamura, T. Miyao and J. Bajorath, Large-scale prediction of activity cliffs using machine and deep learning methods of increasing complexity, J. Cheminform., 2023, 15(1), 4 CrossRef PubMed.
- A. Jahn, G. Hinselmann, N. Fechner and A. Zell, Optimal assignment methods for ligand-based virtual screening, J. Cheminform., 2009, 1, 14 CrossRef PubMed.
- H. Sun, G. Tawa and A. Wallqvist, Classification of scaffold-hopping approaches, Drug Discovery Today, 2012, 17(7–8), 310–324 CrossRef CAS PubMed.
- B. Zdrazil and R. Guha, The Rise and Fall of a Scaffold: A Trend Analysis of Scaffolds in the Medicinal Chemistry Literature, J. Med. Chem., 2018, 61, 4688–4703 CrossRef CAS PubMed.
- T. B. Callis, T. R. Garrett, A. P. Montgomery, J. J. Danon and M. Kassiou, Recent Scaffold Hopping Applications in Central Nervous System Drug Discovery, J. Med. Chem., 2022, 65(20), 13483–13504 CrossRef CAS PubMed.
- A. Acharya, M. Yadav, M. Nagpure, S. Kumaresan and S. K. Guchhait, Molecular medicinal insights into scaffold hopping-based drug discovery success, Drug Discovery Today, 2024, 29(1), 103845 CrossRef CAS PubMed.
- Shivani, T. A. Abdul Rahaman and S. Chaudhary, Targeting cancer using scaffold-hopping approaches: illuminating SAR to improve drug design, Drug Discovery Today, 2024, 29(9), 104115 CrossRef CAS PubMed.
- M. R. Viana, A. C. B. Galeriani and W. P. Almeida, Approaches to Scaffold Hopping for Identifying New Bioactive Compounds and the Contribution of Artificial Intelligence, Curr. Med. Chem., 2025 Search PubMed.
- R. Guha, Exploring Structure–Activity Data Using the Landscape Paradigm, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2(6), 829–841 Search PubMed.
- M. A. Skinnider, R. G. Stacey, D. S. Wishart and L. J. Foster, Deep generative models enable navigation in sparsely populated chemical space, Nat. Mach. Intell., 2021, 3, 759–770 CrossRef.
- J. L. Medina-Franco, N. Sánchez-Cruz, E. López-López and B. I. Díaz-Eufracio, Progress on open chemoinformatic tools for expanding and exploring the chemical space, J. Comput. Aided Mol. Des., 2022, 36(5), 341–354 CrossRef CAS PubMed.
- J. I. Espinoza-Castañeda and J. L. Medina-Franco, MAYA (Multiple ActivitY Analyzer): An Open Access Tool to Explore Structure-Multiple Activity Relationships in the Chemical Universe, Mol. Inform., 2025, 44(2), e202400306 CrossRef PubMed.
- M. Berland, B. Offmann, I. Andre, M. Remaud-Simeon and P. Charton, A web-based tool for rational screening of mutants libraries using ProSAR, Protein Eng., Des. Sel., 2014, 27(10), 375–381 CrossRef CAS PubMed.
- H. Chen, U. Borjesson, O. Engkvist, T. Kogej, M. A. Svensson and N. Blomberg, et al., ProSAR: a new methodology for combinatorial library design, J. Chem. Inf. Model., 2009, 49(3), 603–614 CrossRef CAS PubMed.
- R. Fox, A. Roy, S. Govindarajan, J. Minshull, C. Gustafsson and J. T. Jones, et al., Optimizing the search algorithm for protein engineering by directed evolution, Protein Eng., 2003, 16(8), 589–597 CrossRef CAS PubMed.
- R. J. Fox, S. C. Davis, E. C. Mundorff, L. M. Newman, V. Gavrilovic and S. K. Ma, et al., Improving catalytic function by ProSAR-driven enzyme evolution, Nat. Biotechnol., 2007, 25(3), 338–344 CrossRef CAS PubMed.
- C. Savile, Evolving new catalysts for more efficient and cost-effective pharmaceuticals, Chim. Oggi, 2013, 31(5), 49–52 Search PubMed.
- J. Zaugg, Y. Gumulya, E. M. Gillam and M. Boden, Computational tools for directed evolution: a comparison of prospective and retrospective strategies, Methods Mol. Biol., 2014, 1179, 315–333 CrossRef PubMed.
- J. R. Nahum, P. Godfrey-Smith, B. N. Harding, J. H. Marcus, J. Carlson-Stevermer and B. Kerr, A tortoise-hare pattern seen in adapting structured and unstructured populations suggests a rugged fitness landscape in bacteria, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(24), 7530–7535 CrossRef CAS PubMed.
- T. N. Starr and J. W. Thornton, Epistasis in protein evolution, Protein Sci., 2016, 25(7), 1204–1218 CrossRef CAS PubMed.
- D. W. Anderson, F. Baier, G. Yang and N. Tokuriki, The adaptive landscape of a metallo-enzyme is shaped by environment-dependent epistasis, Nat. Commun., 2021, 12(1), 3867 CrossRef CAS PubMed.
- M. Sandhu, J. Z. Chen, D. S. Matthews, M. A. Spence, S. B. Pulsford and B. Gall, et al., Computational and Experimental Exploration of Protein Fitness Landscapes: Navigating Smooth and Rugged Terrains, Biochemistry, 2025, 64(8), 1673–1684 CrossRef CAS PubMed.
- C. A. Olson, N. C. Wu and R. Sun, A comprehensive biophysical description of pairwise epistasis throughout an entire protein domain, Curr. Biol., 2014, 24(22), 2643–2651 CrossRef CAS PubMed.
- A. S. Canale, P. A. Cote-Hammarlof, J. M. Flynn and D. N. A. Bolon, Evolutionary mechanisms studied through protein fitness landscapes, Curr. Opin. Struct. Biol., 2018, 48, 141–148 CrossRef CAS PubMed.
- A. S. Dunham and P. Beltrao, Exploring amino acid functions in a deep mutational landscape, Mol. Syst. Biol., 2021, 17(7), e10305 CrossRef CAS PubMed.
- X. Pang, Z. Wang, J. S. Yap, J. Wang, J. Zhu and W. Bo, et al., A statistical procedure to map high-order epistasis for complex traits, Brief. Bioinform., 2013, 14(3), 302–314 CrossRef CAS PubMed.
- D. M. Weinreich, Y. Lan, C. S. Wylie and R. B. Heckendorn, Should evolutionary geneticists worry about higher-order epistasis?, Curr. Opin. Genet. Dev., 2013, 23(6), 700–707 CrossRef CAS PubMed.
- Z. R. Sailer and M. J. Harms, High-order epistasis shapes evolutionary trajectories, PLoS Comput. Biol., 2017, 13(5), e1005541 CrossRef PubMed.
- Z. R. Sailer and M. J. Harms, Detecting High-Order Epistasis in Nonlinear Genotype-Phenotype Maps, Genetics, 2017, 205(3), 1079–1088 CrossRef CAS PubMed.
- G. Yang, D. W. Anderson, F. Baier, E. Dohmen, N. Hong and P. D. Carr, et al., Higher-order epistasis shapes the fitness landscape of a xenobiotic-degrading enzyme, Nat. Chem. Biol., 2019, 15(11), 1120–1128 CrossRef CAS PubMed.
- C. M. Miton, J. Z. Chen, K. Ost, D. W. Anderson and N. Tokuriki, Statistical analysis of mutational epistasis to reveal intramolecular interaction networks in proteins, Methods Enzymol., 2020, 643, 243–280 CAS.
- J. Chen and K. C. Wong, Analyzing High-Order Epistasis from Genotype-Phenotype Maps Using 'Epistasis' Package, Methods Mol. Biol., 2021, 2212, 265–275 CrossRef CAS PubMed.
- J. Zhou, M. S. Wong, W. C. Chen, A. R. Krainer, J. B. Kinney and D. M. McCandlish, Higher-order epistasis and phenotypic prediction, Proc. Natl. Acad. Sci. U. S. A., 2022, 119(39), e2204233119 CrossRef CAS PubMed.
- J. Otwinowski and I. Nemenman, Genotype to phenotype mapping and the fitness landscape of the E. coli lac promoter, PLoS One, 2013, 8(5), e61570 CrossRef CAS PubMed.
- I. G. Szendro, J. Franke, J. A. G. M. de Visser and J. Krug, Predictability of evolution depends nonmonotonically on population size, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(2), 571–576 CrossRef CAS PubMed.
- H. Kemble, P. Nghe and O. Tenaillon, Recent insights into the genotype-phenotype relationship from massively parallel genetic assays, Evol. Appl., 2019, 12(9), 1721–1742 CrossRef PubMed.
- W. P. Russ, M. Figliuzzi, C. Stocker, P. Barrat-Charlaix, M. Socolich and P. Kast, et al., An evolution-based model for designing chorismate mutase enzymes, Science, 2020, 369(6502), 440–445 CrossRef CAS PubMed.
- A. Eyre-Walker and P. D. Keightley, The distribution of fitness effects of new mutations, Nat. Rev. Genet., 2007, 8(8), 610–618 CrossRef CAS PubMed.
- J. Chen, T. Bataillon, S. Glémin and M. Lascoux, What does the distribution of fitness effects of new mutations reflect? Insights from plants, New Phytol., 2022, 233(4), 1613–1619 CrossRef PubMed.
- M. Sane, S. Parveen and D. Agashe, Mutation bias alters the distribution of fitness effects of mutations, PLoS Biol., 2025, 23(7), e3003282 CrossRef CAS PubMed.
- J. James, C. Kastally, K. B. Budde, S. C. González-Martínez, P. Milesi and T. Pyhäjärvi, et al., Between but Not Within-Species Variation in the Distribution of Fitness Effects, Mol. Biol. Evol., 2023, 40(11) CrossRef PubMed.
- R. Fisher, The genetical theory of natural selection, Clarendon Press, Oxford, 1930 Search PubMed.
- O. Cotto and T. Day, A null model for the distribution of fitness effects of mutations, Proc. Natl. Acad. Sci. U. S. A., 2023, 120(23), e2218200120 CrossRef CAS PubMed.
- G. Martin and T. Lenormand, A general multivariate extension of Fisher's geometrical model and the distribution of mutation fitness effects across species, Evolution, 2006, 60(5), 893–907 CrossRef PubMed.
- F. Blanquart and T. Bataillon, Epistasis and the Structure of Fitness Landscapes: Are Experimental Fitness Landscapes Compatible with Fisher's Geometric Model?, Genetics, 2016, 203(2), 847–862 CrossRef CAS PubMed.
- D. Delneri, D. C. Hoyle, K. Gkargkas, E. J. Cross, B. Rash and L. Zeef, et al., Identification and characterization of high-flux-control genes of yeast through competition analyses in continuous cultures, Nat. Genet., 2008, 40(1), 113–117 CrossRef CAS PubMed.
- J. W. Thatcher, J. M. Shaw and W. J. Dickinson, Marginal fitness contributions of nonessential genes in yeast, Proc. Natl. Acad. Sci. U. S. A., 1998, 95(1), 253–257 CrossRef CAS PubMed.
- R. T. Hietpas, J. D. Jensen and D. N. Bolon, Experimental illumination of a fitness landscape, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(19), 7896–7901 CrossRef CAS PubMed.
- D. P. Rice, B. H. Good and M. M. Desai, The evolutionarily stable distribution of fitness effects, Genetics, 2015, 200(1), 321–329 CrossRef PubMed.
- K. B. Böndel, T. Samuels, R. J. Craig, R. W. Ness, N. Colegrave and P. D. Keightley, The distribution of fitness effects of spontaneous mutations in Chlamydomonas reinhardtii inferred using frequency changes under experimental evolution, PLoS Genet., 2022, 18(6), e1009840 CrossRef PubMed.
- R. Sanjuán, A. Moya and S. F. Elena, The distribution of fitness effects caused by single-nucleotide substitutions in an RNA virus, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(22), 8396–8401 CrossRef PubMed.
- A. F. Rubin, J. Stone, A. H. Bianchi, B. J. Capodanno, E. Y. Da and M. Dias, et al., MaveDB 2024: a curated community database with over seven million variant effects from multiplexed functional assays, Genome Biol., 2025, 26(1), 13 CrossRef PubMed.
- D. Esposito, J. Weile, J. Shendure, L. M. Starita, A. T. Papenfuss and F. P. Roth, et al., MaveDB: an open-source platform to distribute and interpret data from multiplexed assays of variant effect, Genome Biol., 2019, 20(1), 223 CrossRef PubMed.
- T. R. Booker, Inferring Parameters of the Distribution of Fitness Effects of New Mutations When Beneficial Mutations Are Strongly Advantageous and Rare, G3, 2020, 10(7), 2317–2326 CrossRef PubMed.
- J. H. Gillespie, Molecular Evolution over the Mutational Landscape, Evolution, 1984, 38(5), 1116–1129 CrossRef CAS PubMed.
- A. C. Davison and R. L. Smith, Models for Exceedances over High Thresholds, J. R. Stat. Soc., 1990, 52, 393–442 CrossRef.
- S. G. Coles, An Introduction to Statistical Modeling of Extreme Values, Springer, London, 2001 Search PubMed.
- J. Hüsler, Extreme value analysis in biometrics, Biomed. J., 2009, 51(2), 252–272 Search PubMed.
- K. Hayashi, N. Takamatsu and S. Takaramoto, Extreme-value analysis in nano-biological systems: applications and implications, Biophys. Rev., 2024, 16(5), 571–579 CrossRef PubMed.
- A. Naess, Applied Extreme Value Statistics, Springer, Berlin, 2024 Search PubMed.
- A. A. Balkema and L. de Haan, Residual Life Time at Great Age, Ann. Probab., 1974, 2, 792–804 Search PubMed.
- J. Pickands, Statistical Inference Using Extreme Order Statistics, Ann. Statist., 1975, 3, 119–131 Search PubMed.
- H. T. Davis and M. L. Feldstein, The generalized Pareto law as a model for progressively censored survival data, Biometrika, 1979, 66, 299–306 CrossRef.
- A. Alvarez-Iglesias, J. Newell, C. Scarrott and J. Hinde, Summarising censored survival data using the mean residual life function, Stat. Med., 2015, 34(11), 1965–1976 CrossRef PubMed.
- J. del Castillo and I. Serra, Likelihood inference for generalized Pareto distribution, Comput. Stat. Data Anal., 2015, 83, 116–128 CrossRef.
- N. Hanayama and M. Sibuya, Estimating the Upper Limit of Lifetime Probability Distribution, Based on Data of Japanese Centenarians, J. Gerontol. A: Biol. Sci. Med. Sci., 2016, 71(8), 1014–1021 CrossRef PubMed.
- Y. He, L. Peng, D. Zhang and Z. Zhao, Refining Kaplan-Meier Estimation with the Generalized Pareto Model for Survival Analysis, 2024.
- J. Beirlant, E. Joossens and J. Segers, Second-order refined peaks-over-threshold modelling for heavy-tailed distributions, arXiv, 2009, arXiv:2009:0901.1518 DOI:10.48550/arXiv.0901.1518.
- I. Papastathopoulos and J. A. Tawn, Extended Generalised Pareto Models for Tail Estimation, arXiv, 2011, arXiv:2011:1111.6899 DOI:10.48550/arXiv.1111.6899.
- C. Scarrott and A. MacDonald, A review of extreme value threshold estimation and uncertainty quantification, Stat. J., 2012, 10, 33–60 Search PubMed.
- S. Solari, M. Egüen, M. J. Polo and M. A. Losada, Peaks Over Threshold (POT): A methodology for automaticthreshold estimation using goodness of fit p-value, Water Res., 2017, 53, 2833–2849 CrossRef.
- B. Liang, Z. Shao, H. Li, M. Shao and D. Lee, An automated threshold selection method based on the characteristic of extrapolated significant wave heights, Coastal Eng., 2019, 144, 22–32 CrossRef.
- J. Martín, M. I. Parra, M. M. Pizarro and E. L. Sanjuán, Baseline Methods for the Parameter Estimation of the Generalized Pareto Distribution, Entropy, 2022, 24(2), 178 CrossRef PubMed.
- R. Mínguez, Automatic Threshold Selection for Generalized Pareto and Pareto–Poisson Distributions in Rainfall Analysis: A Case Study Using the NOAA NCDC Daily Rainfall Database, Atmosphere, 2025, 16, 61 CrossRef.
- A. Langousis, A. Mamalakis, M. Puliga and R. Deidda, Threshold detection for the generalized Pareto distribution:Review of representative methods and application to theNOAA NCDC daily rainfall database, Water Res., 2016, 52, 2659–2681 CrossRef.
- W. A. Pels, A. O. Adebanji, S. Twumasi-Ankrah and R. Minkah, Shrinkage Methods for Estimating the Shape Parameter of the Generalized Pareto Distribution, J. Appl. Math., 2023, 2023, 9750638 CrossRef.
- R. C. de Fondeville and A. C. Davison, High-dimensional peaks-over-threshold inference, Biometrika, 2018, 105, 575–592 CrossRef.
- T. Hastie, R. Tibshirani and J. Friedman, The elements of statistical learning: data mining, inference and prediction, Springer-Verlag, Berlin, 2nd edn, 2009 Search PubMed.
- S. D. Grimshaw, Computing Maximum Likelihood Estimates for the Generalized Pareto Distribution, Technometrics, 1993, 35, 185–191 CrossRef.
- M. H. Pham, C. Tsokos and B.-J. Choi, Maximum likelihood estimation for the generalized Pareto distribution and goodness-of-fit test with censored data, J. Mod. Appl. Stat. Meth., 2018, 17, eP2608 CrossRef.
- I. Roberts, L. C. Kenny, A. Merriel, J. B. Moore, E. Pretorius and C. Waitt, et al., Determining the desired concentration of a nutraceutical based on the variation of its concentration with the incidence of a disease: application to ergothioneine and pre-eclampsia, Pregnancy Hypertens, 2026, 44, 101450 CrossRef PubMed.
- H. Southworth and J. E. Heffernan, Multivariate extreme value modelling of laboratory safety data from clinical studies, Pharm. Stat., 2012, 11(5), 367–372 CrossRef PubMed.
- H. Southworth and J. E. Heffernan, Extreme value modelling of laboratory safety data from clinical studies, Pharm. Stat., 2012, 11(5), 361–366 CrossRef PubMed.
- H. Southworth, Predicting potential liver toxicity from phase 2 data: a case study with ximelagatran, Stat. Med., 2014, 33(17), 2914–2923 CrossRef PubMed.
- H. A. Orr, The distribution of fitness effects among beneficial mutations in Fisher's geometric model of adaptation, J. Theor. Biol., 2006, 238(2), 279–285 CrossRef PubMed.
- D. R. Rokyta, C. J. Beisel and P. Joyce, Properties of adaptive walks on uncorrelated landscapes under strong selection and weak mutation, J. Theor. Biol., 2006, 243(1), 114–120 CrossRef PubMed.
- J. Neidhart and J. Krug, Adaptive Walks and Extreme Value Theory, Phys. Rev. Lett., 2011, 107, 178102 CrossRef PubMed.
- T. Bataillon and S. F. Bailey, Effects of new mutations on fitness: insights from models and data, Ann. N. Y. Acad. Sci., 2014, 1320(1), 76–92 CrossRef PubMed.
- S. Boyer, D. Biswas, A. Kumar Soshee, N. Scaramozzino, C. Nizak and O. Rivoire, Hierarchy and extremes in selections from pools of randomized proteins, Proc. Natl. Acad. Sci. U. S. A., 2016, 113(13), 3482–3487 CrossRef CAS PubMed.
- N. Tokuriki, F. Stricher, J. Schymkowitz, L. Serrano and D. S. Tawfik, The stability effects of protein mutations appear to be universally distributed, J. Mol. Biol., 2007, 369(5), 1318–1332 CrossRef CAS PubMed.
- L. Chen, Z. Zhang, Z. Li, R. Li, R. Huo and L. Chen, et al., Learning protein fitness landscapes with deep mutational scanning data from multiple sources, Cell Syst, 2023, 14(8), 706–721 CrossRef CAS PubMed.
- S. Kauffman and S. Levin, Towards a general theory of adaptive walks on rugged landscapes, J. Theor. Biol., 1987, 128(1), 11–45 CrossRef CAS PubMed.
- S. A. Kauffman and E. D. Weinberger, The NK model of rugged fitness landscapes and its application to maturation of the immune response, J. Theor. Biol., 1989, 141(2), 211–245 CrossRef CAS PubMed.
- S. A. Kauffman and S. Johnsen, Coevolution to the edge of chaos: coupled fitness landscapes, poised states, and coevolutionary avalanches, J. Theor. Biol., 1991, 149(4), 467–505 CrossRef CAS PubMed.
- S. A. Kauffman, The origins of order, Oxford University Press, Oxford, 1993 Search PubMed.
- S. A. Kauffman and W. G. Macready, Search strategies for applied molecular evolution, J. Theor. Biol., 1995, 173(4), 427–440 CrossRef CAS PubMed.
- W. Hordijk, S. A. Kauffman and P. F. Stadler, Average Fitness Differences on NK Landscapes, Theory Biosci., 2020, 139(1), 1–7 CrossRef CAS PubMed.
- L. Kallel, B. Naudts and C. R. Reeves, Properties of fitness functions and search landscapes, in Theoretical aspects of evolutionary computing, ed. L. Kallel, B. Naudts and A. J. Rogers, Springer, Berlin, 2001, pp. 175–206 Search PubMed.
- Ruggedness and neutrality: the NKp family of fitness landscapes, in Proc6th Int'l Conf on Artificial Life, ed. L. Barnett, MIT Press, 1998 Search PubMed.
- W. Rowe, M. Platt, D. Wedge, P. J. Day, D. B. Kell and J. Knowles, Analysis of a complete DNA-protein affinity landscape, J. R. Soc., Interface, 2010, 7(44), 397–408 CrossRef CAS PubMed.
- J. Franke, A. Klözer, J. A. G. M. de Visser and J. Krug, Evolutionary accessibility of mutational pathways, PLoS Comput. Biol., 2011, 7(8), e1002134 CrossRef CAS PubMed.
- C. Fraïsse and J. J. Welch, The distribution of epistasis on simple fitness landscapes, Biol. Lett., 2019, 15(4), 20180881 CrossRef PubMed.
- A. Papkou, L. Garcia-Pastor, J. A. Escudero and A. Wagner, A rugged yet easily navigable fitness landscape, Science, 2023, 382(6673), eadh3860 CrossRef CAS PubMed.
- T. Stadelmann, D. Heid, M. Jendrusch, J. Mathony, S. Aschenbrenner and S. Rosset, et al., A deep mutational scanning platform to characterize the fitness landscape of anti-CRISPR proteins, Nucleic Acids Res., 2024, 52(22), e103 CrossRef CAS PubMed.
- Y. Hayashi, T. Aita, H. Toyota, Y. Husimi, I. Urabe and T. Yomo, Experimental rugged fitness landscape in protein sequence space, PLoS One, 2006, 1, e96 CrossRef PubMed.
- D. W. Anderson, A. N. McKeown and J. W. Thornton, Intermolecular epistasis shaped the function and evolution of an ancient transcription factor and its DNA binding sites, eLife, 2015, 4, e07864 CrossRef PubMed.
- M. R. Meini, P. E. Tomatis, D. M. Weinreich and A. J. Vila, Quantitative Description of a Protein Fitness Landscape Based on Molecular Features, Mol. Biol. Evol., 2015, 32(7), 1774–1787 CrossRef CAS PubMed.
- J. Otwinowski, D. M. McCandlish and J. B. Plotkin, Inferring the shape of global epistasis, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(32), E7550–E7558 CrossRef CAS PubMed.
- L. Gonzalez Somermeyer, A. Fleiss, A. S. Mishin, N. G. Bozhanova, A. A. Igolkina and J. Meiler, et al., Heterogeneity of the GFP fitness landscape and data-driven protein design, eLife, 2022, 11 CrossRef PubMed.
- V. O. Pokusaeva, D. R. Usmanova, E. V. Putintseva, L. Espinar, K. S. Sarkisyan and A. S. Mishin, et al., An experimental assay of the interactions of amino acids from orthologous sequences shaping a complex fitness landscape, PLoS Genet., 2019, 15(4), e1008079 CrossRef CAS PubMed.
- S. Towers, J. James, H. Steel and I. Kempf, Learning-Based Estimation of Fitness Landscape Ruggedness for Directed Evolution, bioRxiv, 2024, 2024.02.28.582468.
- S. Schulz, T. J. C. Tan, N. C. Wu and S. Wang, Epistatic hotspots organize antibody fitness landscape and boost evolvability, Proc. Natl. Acad. Sci. U. S. A., 2025, 122(2), e2413884122 CrossRef CAS PubMed.
- C. A. Westmann, L. Goldbach and A. Wagner, The highly rugged yet navigable regulatory landscape of the bacterial transcription factor TetR, Nat. Commun., 2024, 15(1), 10745 CrossRef CAS PubMed.
- D. H. Brookes, A. Aghazadeh and J. Listgarten, On the sparsity of fitness functions and implications for learning, Proc. Natl. Acad. Sci. U. S. A., 2022, 119(1) CrossRef CAS PubMed.
- H. S. Pannu and D. B. Kell, Hyperparameter optimisation in differential evolution using Summed Local Difference Strings, a rugged but easily calculated landscape for combinatorial search problems, bioRxiv, 2023, 2023.07.11.548503v1.
- H. S. Pannu and D. B. Kell, Hyperparameter optimisation in differential evolution using Summed Local Difference Strings, a rugged but easily calculated landscape for combinatorial search problems, Comput. Sci. Inf. Syst., 2025, 22, 1555–1575 CrossRef.
- J. Otwinowski and J. B. Plotkin, Inferring fitness landscapes by regression produces biased estimates of epistasis, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(22), E2301–E2309 CrossRef CAS PubMed.
- M. Schmutzer and A. Wagner, Not Quite Lost in Translation: Mistranslation Alters Adaptive Landscape Topography and the Dynamics of Evolution, Mol. Biol. Evol., 2023, 40(6), msad136 CrossRef CAS PubMed.
- V. Sundar, B. Tu, L. Guan and K. Esvelt, FLIGHTED: Inferring Fitness Landscapes from Noisy High-Throughput Experimental Data, bioRxiv, 2024.
- I. G. Szendro, M. F. Schenk, J. Franke, J. Krug and J. A. G. M. de Visser, Quantitative analyses of empirical fitness landscapes, J. Stat. Mech., 2013, P01005 Search PubMed.
- Fitness distance correlation as a measure of problem difficulty for genetic algorithms, in Proceedings of the Sixth International Conference on Genetic Algorithms, ed. T. Jones and S. Forrest, Morgan Kaufmann, 1995 Search PubMed.
- M. T. Reetz and J. D. Carballeira, Iterative saturation mutagenesis (ISM) for rapid directed evolution of functional enzymes, Nat. Protoc., 2007, 2(4), 891–903 CrossRef CAS PubMed.
- C. L. Araya and D. M. Fowler, Deep mutational scanning: assessing protein function on a massive scale, Trends Biotechnol., 2011, 29(9), 435–442 CrossRef CAS PubMed.
- J. Fernandez-de-Cossio-Diaz, G. Uguzzoni and A. Pagnani, Unsupervised Inference of Protein Fitness Landscape from Deep Mutational Scan, Mol. Biol. Evol., 2021, 38(1), 318–328 CrossRef CAS PubMed.
- D. M. Fowler and S. Fields, Deep mutational scanning: a new style of protein science, Nat. Methods, 2014, 11(8), 801–807 CrossRef CAS PubMed.
- D. T. Harris, N. Wang, T. P. Riley, S. D. Anderson, N. K. Singh and E. Procko, et al., Deep Mutational Scans as a Guide to Engineering High Affinity T Cell Receptor Interactions with Peptide-bound Major Histocompatibility Complex, J. Biol. Chem., 2016, 291(47), 24566–24578 CrossRef CAS PubMed.
- M. Leander, Z. Liu, Q. Cui and S. Raman, Deep mutational scanning and machine learning reveal structural and molecular rules governing allosteric hotspots in homologous proteins, eLife, 2022, 11, e79932 CrossRef CAS PubMed.
- R. W. Newberry, J. T. Leong, E. D. Chow, M. Kampmann and W. F. DeGrado, Deep mutational scanning reveals the structural basis for alpha-synuclein activity, Nat. Chem. Biol., 2020, 16(6), 653–659 CrossRef CAS PubMed.
- A. Nikoomanzar, D. Vallejo and J. C. Chaput, Elucidating the Determinants of Polymerase Specificity by Microfluidic-Based Deep Mutational Scanning, ACS Synth. Biol., 2019, 8(6), 1421–1429 CrossRef CAS PubMed.
- P. A. Romero, T. M. Tran and A. R. Abate, Dissecting enzyme function with microfluidic-based deep mutational scanning, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(23), 7159–7164 CrossRef CAS PubMed.
- H. Shin and B. K. Cho, Rational Protein Engineering Guided by Deep Mutational Scanning, Int. J. Mol. Sci., 2015, 16(9), 23094–23110 CrossRef CAS PubMed.
- C. K. Sruthi and M. Prakash, Deep2Full: Evaluating strategies for selecting the minimal mutational experiments for optimal computational predictions of deep mutational scan outcomes, PLoS One, 2020, 15(1), e0227621 CrossRef CAS PubMed.
- L. M. Starita and S. Fields, Deep Mutational Scanning: A Highly Parallel Method to Measure the Effects of Mutation on Protein Function, Cold Spring Harb. Protoc., 2015, 2015(8), 711–714 Search PubMed.
- J. B. Kinney and D. M. McCandlish, Massively Parallel Assays and Quantitative Sequence-Function Relationships, Annu. Rev. Genomics Hum. Genet., 2019, 20, 99–127 CrossRef CAS PubMed.
- A. S. Dunham, P. Beltrao and M. AlQuraishi, High-throughput deep learning variant effect prediction with Sequence UNET, Genome Biol., 2023, 24(1), 110 CrossRef CAS PubMed.
- H. Wei and X. Li, Deep mutational scanning: A versatile tool in systematically mapping genotypes to phenotypes, Front. Genet., 2023, 14, 1087267 CrossRef CAS PubMed.
- M. Sun, A. Stoltzfus and D. M. McCandlish, A fitness distribution law for amino-acid replacements, bioRxiv, 2024.
- M. J. Call, M. E. Call and X. Wu, Insights from deep mutational scanning in the context of an emerging pathogen, Biochem. Soc. Trans., 2025 Search PubMed.
- J. M. Lee, J. Huddleston, M. B. Doud, K. A. Hooper, N. C. Wu and T. Bedford, et al., Deep mutational scanning of hemagglutinin helps predict evolutionary fates of human H3N2 influenza variants, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(35), E8276–E8285 CrossRef CAS PubMed.
- S. O'Hagan, J. Knowles and D. B. Kell, Exploiting genomic knowledge in optimising molecular breeding programmes: algorithms from evolutionary computing, PLoS One, 2012, 7(11), e48862 CrossRef PubMed.
- C. Blanco, E. Janzen, A. Pressman, R. Saha and I. A. Chen, Molecular Fitness Landscapes from High-Coverage Sequence Profiling, Annu. Rev. Biophys., 2019, 48, 1–18 CrossRef CAS PubMed.
- K. E. Johnston, P. J. Almhjell, E. J. Watkins-Dulaney, G. Liu, N. J. Porter and J. Yang, et al., A combinatorially complete epistatic fitness landscape in an enzyme active site, Proc. Natl. Acad. Sci. U. S. A., 2024, 121(32), e2400439121 CrossRef CAS PubMed.
- M. Kimura, The neutral theory of molecular evolution, Cambridge University Press, Cambridge, 1983 Search PubMed.
- G. Amitai, R. D. Gupta and D. S. Tawfik, Latent evolutionary potentials under the neutral mutational drift of an enzyme, HFSP J., 2007, 1(1), 67–78 CrossRef CAS PubMed.
- D. L. Hartl and C. H. Taubes, Compensatory nearly neutral mutations: selection without adaptation, J. Theor. Biol., 1996, 182(3), 303–309 CrossRef CAS PubMed.
- J. Noirel and T. Simonson, Neutral evolution of proteins: The superfunnel in sequence space and its relation to mutational robustness, J. Chem. Phys., 2008, 129(18), 185104 CrossRef PubMed.
- E. van Nimwegen, J. P. Crutchfield and M. Huynen, Neutral evolution of mutational robustness, Proc. Natl. Acad. Sci. U. S. A., 1999, 96(17), 9716–9720 CrossRef CAS PubMed.
- C. M. Reidys and P. F. Stadler, Neutrality in fitness landscapes, Appl. Math. Comput., 2001, 117(2–3), 321–350 CrossRef.
- J. D. Jensen, B. A. Payseur, W. Stephan, C. F. Aquadro, M. Lynch and D. Charlesworth, et al., The importance of the Neutral Theory in 1968 and 50 years on: A response to Kern and Hahn 2018, Evolution, 2019, 73(1), 111–114 CrossRef PubMed.
- J. D. Bloom, J. J. Silberg, C. O. Wilke, D. A. Drummond, C. Adami and F. H. Arnold, Thermodynamic prediction of protein neutrality, Proc. Natl. Acad. Sci. U. S. A., 2005, 102(3), 606–611 CrossRef CAS PubMed.
- J. D. Bloom, A. Raval and C. O. Wilke, Thermodynamics of neutral protein evolution, Genetics, 2007, 175(1), 255–266 CrossRef CAS PubMed.
- J. D. Bloom, P. A. Romero, Z. Lu and F. H. Arnold, Neutral genetic drift can alter promiscuous protein functions, potentially aiding functional evolution, Biol. Direct, 2007, 2, 17 CrossRef PubMed.
- R. D. Gupta and D. S. Tawfik, Directed enzyme evolution via small and effective neutral drift libraries, Nat. Methods, 2008, 5(11), 939–942 CrossRef CAS PubMed.
- F. H. Arnold, How proteins adapt: lessons from directed evolution, Cold Spring Harb. Symp. Quant. Biol., 2009, 74, 41–46 CrossRef CAS PubMed.
- J. D. Bloom and F. H. Arnold, In the light of directed evolution: pathways of adaptive protein evolution, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(Suppl 1), 9995–10000 CrossRef CAS PubMed.
- C. A. Tracewell and F. H. Arnold, Directed enzyme evolution: climbing fitness peaks one amino acid at a time, Curr. Opin. Chem. Biol., 2009, 13(1), 3–9 CrossRef CAS PubMed.
- W. S. Smith, J. R. Hale and C. Neylon, Applying neutral drift to the directed molecular evolution of a beta-glucuronidase into a beta-galactosidase: Two different evolutionary pathways lead to the same variant, BMC Res. Notes, 2011, 4, 138 CrossRef CAS PubMed.
- M. Goldsmith and D. S. Tawfik, Directed enzyme evolution: beyond the low-hanging fruit, Curr. Opin. Struct. Biol., 2012, 22(4), 406–412 CrossRef CAS PubMed.
- E. T. Liechty, A. Hren, L. Kramer, G. Donovan, A. J. Friedman and M. R. Shirts, et al., Analysis of neutral mutational drift in an allosteric enzyme, Protein Sci., 2023, 32(8), e4719 CrossRef CAS PubMed.
- J. H. Holland, Adaption in natural and artificial systems: an introductory analysis with applications to biology, control, and artificial intelligence, MIT Press, 1992 Search PubMed.
- K. R. Patil, I. Rocha, J. Förster and J. Nielsen, Evolutionary programming as a platform for in silico metabolic engineering, BMC Bioinf., 2005, 6(1), 308 CrossRef PubMed.
- M. Rocha, P. Maia, R. Mendes, J. P. Pinto, E. C. Ferreira and J. Nielsen, et al., Natural computation meta-heuristics for the in silico optimization of microbial strains, BMC Bioinformat., 2008, 9, 499 CrossRef PubMed.
- I. Rocha, P. Maia, P. Evangelista, P. Vilaca, S. Soares and J. P. Pinto, et al., OptFlux: an open-source software platform for in silico metabolic engineering, BMC Syst. Biol., 2010, 4, 45 CrossRef PubMed.
- P. Vilaça, P. Maia, H. Giesteira, I. Rocha and M. Rocha, Analyzing and Designing Cell Factories with OptFlux, Methods Mol. Biol., 2018, 1716, 37–76 CrossRef PubMed.
- D. Ashlock, Evolutionary computation for modeling and optimization, Springer, New York, 2006 Search PubMed.
- Handbook of evolutionary computation, ed. T. Bäck, D. B. Fogel and Z. Michalewicz, IOPPublishing/Oxford University Press, Oxford, 1997 Search PubMed.
- W. Banzhaf, P. Nordin, R. E. Keller and F. D. Francone, Genetic programming: an introduction, Morgan Kaufmann, San Francisco, 1998 Search PubMed.
- New ideas in optimization, ed. D. Corne, M. Dorigo and F. Glover, McGraw Hill, London, 1999 Search PubMed.
- A. Darwish, A. E. Hassanien and S. Das, A survey of swarm and evolutionary computing approaches for deep learning, Artif. Intell. Rev., 2020, 53(3), 1767–1812 CrossRef.
- K. De Jong, Evolutionary Computation – A Unified Approach, MIT Press, Cambridge, MA, 2006 Search PubMed.
- A. E. Eiben and J. E. Smith, Introduction to evolutionary computing, Springer, Berlin, 2017 Search PubMed.
- D. B. Fogel, Evolutionary computation: toward a new philosophy of machine intelligence, IEEE Press, Piscataway, 2nd edn, 2000 Search PubMed.
- J. A. Foster, Evolutionary computation, Nat. Rev. Genet., 2001, 2(6), 428–436 CrossRef CAS PubMed.
- D. E. Goldberg, Genetic algorithms in search, optimization and machine learning, Addison-Wesley, 1989 Search PubMed.
- Multiobjective Problem Solving from Nature, ed. J. Knowles, D. Corne and K. Deb, Springer, Berlin, 2008 Search PubMed.
- J. R. Koza, Genetic programming: on the programming of computers by means of natural selection, MIT Press, Cambridge, MA, 1992 Search PubMed.
- J. R. Koza, Genetic programming II: automatic discovery of reusable programs, MIT Press, Cambridge, MA, 1994 Search PubMed.
- J. R. Koza, F. H. Bennett, M. A. Keane and D. Andre, Genetic Programming III: Darwinian Invention and Problem Solving, Morgan Kaufmann, San Francisco, 1999 Search PubMed.
- W. B. Langdon and R. Poli, Foundations of genetic programming, Springer-Verlag, Berlin, 2002 Search PubMed.
- Z. Michalewicz, Nonstandard methods in evolutionary computation, Stat. Comput., 1994, 4(2), 141–155 Search PubMed.
- M. Mitchell and C. E. Taylor, Evolutionary computation: An overview, Annu. Rev. Ecol. Systemat, 1999, 30, 593–616 CrossRef.
- K. V. Price, R. Storn and J. A. Lampinen, Differential evolution: a practiical approach to global optimization, Springer, Berlin, 2005 Search PubMed.
- K. V. Price, Differential evolution. Handbook of optimization, Springer, Berlin, 2013, pp. 187–214 Search PubMed.
- J. Rönkkönen, S. Kukkonen and K. V. Price, Real-parameter optimization with differential evolution, Proc. IEEE Congr. Evol. Comput., 2005, 506–513 Search PubMed.
- L. A. Scardua, Applied Evolutionary Algorithms for Engineers Using Python, CRC Press, Boca Raton, FL, 2021 Search PubMed.
- M. Schoenauer and Z. Michalewicz, Evolutionary computation, Control Cybernetics, 1997, 26(3), 307–338 Search PubMed.
- D. B. Kell, Genotype:phenotype mapping: genes as computer programs, Trends Genet., 2002, 18(11), 555–559 CrossRef CAS PubMed.
- Uniform crossover in genetic algorithms, in Proc 3rd Int Conf on Genetic Algorithms, ed. G. Syswerda, Morgan Kaufmann, 1989 Search PubMed.
- B. A. Alpay and M. M. Desai, Effects of selection stringency on the outcomes of directed evolution, PLoS One, 2024, 19(10), e0311438 CrossRef CAS PubMed.
- W. B. Langdon and R. Poli, Fitness causes bloat: mutation, in Proceedings of the First European Workshop on Genetic Programming, ed. W. Banzhaf, R. Poli, M. Schoenauer and T. C. Fogarty, Springer-Verlag, Berlin, 1998, pp. 37–48 Search PubMed.
- D. B. Kell and R. D. King, On the optimization of classes for the assignment of unidentified reading frames in functional genomics programmes: the need for machine learning, Trends Biotechnol., 2000, 18(3), 93–98 CrossRef CAS PubMed.
- B. Settles, From Theories to Queries: Active Learning in Practice, JMLR, 2011, 16, 1–18 Search PubMed.
- R. Godin, S. Hejazi, B. Lange, B. Aldamak and N. F. Reuel, Rapid Cell-Free Combinatorial Mutagenesis Workflow Using Small Oligos Suitable for High-Iteration, Active Learning-Guided Protein Engineering, bioRxiv, 2025.
- J. Yang, R. G. Lal, J. C. Bowden, R. Astudillo, M. A. Hameedi and S. Kaur, et al., Active learning-assisted directed evolution, Nat. Commun., 2025, 16(1), 714 CrossRef CAS PubMed.
- J. Sacks, W. Welch, T. Mitchell and H. Wynn, Design and analysis of computer experiments (with discussion), Stat. Sci., 1989, 4, 409–435 Search PubMed.
- K. P. Greenman, A. P. Amini and K. K. Yang, Benchmarking uncertainty quantification for protein engineering, PLoS Comput. Biol., 2025, 21(1), e1012639 CrossRef CAS PubMed.
- X. Yao, Evolving artificial neural networks, Proc. IEEE, 1999, 87(9), 1423–1447 CrossRef.
- D. Floreano, P. Dürr and C. Mattiussi, Neuroevolution: from architectures to learning, Evol. Intell., 2008, 1, 47–62 CrossRef.
- E. Galván and P. Mooney, Neuroevolution in Deep Neural Networks: Current Trends and Future Challenges, arXiv, 2020, 2006.05415v1 DOI:10.48550/arXiv.2006.05415v1.
- Deep Neural Evolution: Deep Learning with Evolutionary Computation, ed. H. Iba and N. Noman, Springer, Berlin, 2020 Search PubMed.
- R. Miikkulainen, J. Liang, E. Meyerson, A. Rawal, D. Fink, O. Francon, et al., Evolving Deep Neural Networks, arXiv, 2007, 2017:1703.00548 DOI:10.48550/arXiv.1703.00548.
- K. O. Stanley, J. Clune, J. Lehman and R. Miikkulainen, Designing neural networks through neuroevolution, Nat. Mach. Intell., 2019, 1, 24–35 CrossRef.
- S. Whitelam, V. Selin, S. W. Park and I. Tamblyn, Correspondence between neuroevolution and gradient descent, Nat. Commun., 2021, 12(1), 6317 CrossRef CAS PubMed.
- C. Levinthal, Are there pathways for protein folding?, J Chim Phys, 1968, 65, 44–45 CrossRef.
- B. Honig, Protein folding: from the Levinthal paradox to structure prediction, J. Mol. Biol., 1999, 293(2), 283–293 CrossRef CAS PubMed.
- S. W. Englander and L. Mayne, The nature of protein folding pathways, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(45), 15873–15880 CrossRef CAS PubMed.
- C. B. Anfinsen, E. Haber, M. Sela and F. H. White, The kinetics of formation of native ribonuclease during oxidation of the reduced polypeptide chain, Proc. Natl. Acad. Sci., 1961, 47, 1309–1314 CrossRef CAS PubMed.
- C. B. Anfinsen, Principles that govern the folding of protein chains, Science, 1973, 181, 223–230 CrossRef CAS PubMed.
- M. Burdukiewicz, P. Sobczyk, S. Rödiger, A. Duda-Madej, P. Mackiewicz and M. Kotulska, Amyloidogenic motifs revealed by n-gram analysis, Sci. Rep., 2017, 7(1), 12961 CrossRef PubMed.
- D. B. Kell, K. M. Doyle, E. Salcedo-Sora, A. Sekhar, M. Walker and E. Pretorius, AmyloGram reveals amyloidogenic potential in stroke thrombus proteomes, Biochem. J., 2025, 482 Search PubMed.
- D. B. Kell and E. Pretorius, Proteins behaving badly. Substoichiometric molecular control and amplification of the initiation and nature of amyloid fibril formation: lessons from and for blood clotting, Progr. Biophys. Mol. Biol., 2017, 123, 16–41 CrossRef CAS PubMed.
- F. E. Cohen and S. B. Prusiner, Pathologic conformations of prion proteins, Annu. Rev. Biochem., 1998, 67, 793–819 CrossRef CAS PubMed.
- A. W. Senior, R. Evans, J. Jumper, J. Kirkpatrick, L. Sifre and T. Green, et al., Protein structure prediction using multiple deep neural networks in the 13th Critical Assessment of Protein Structure Prediction (CASP13), Proteins, 2019, 87(12), 1141–1148 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov and O. Ronneberger, et al., Highly accurate protein structure prediction with AlphaFold, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- J. Jumper and D. Hassabis, Protein structure predictions to atomic accuracy with AlphaFold, Nat. Methods, 2022, 19(1), 11–12 CrossRef CAS PubMed.
- M. Varadi, S. Anyango, M. Deshpande, S. Nair, C. Natassia and G. Yordanova, et al., AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models, Nucleic Acids Res., 2022, 50(D1), D439–D444 CrossRef CAS PubMed.
- J. Abramson, J. Adler, J. Dunger, R. Evans, T. Green and A. Pritzel, et al., Accurate structure prediction of biomolecular interactions with AlphaFold 3, Nature, 2024, 630(8016), 493–500 CrossRef CAS PubMed.
- J. Fleming, P. Magana, S. Nair, M. Tsenkov, D. Bertoni and I. Pidruchna, et al., AlphaFold Protein Structure Database and 3D-Beacons: New Data and Capabilities, J. Mol. Biol., 2025, 168967 CrossRef CAS PubMed.
- J. Lyu, N. Kapolka, R. Gumpper, A. Alon, L. Wang and M. K. Jain, et al., AlphaFold2 structures guide prospective ligand discovery, Science, 2024, 384(6702), eadn6354 CrossRef CAS PubMed.
- R. Krishna, J. Wang, W. Ahern, P. Sturmfels, P. Venkatesh and I. Kalvet, et al., Generalized biomolecular modeling and design with RoseTTAFold All-Atom, Science, 2024, 384(6693), eadl2528 CrossRef CAS PubMed.
- S. L. Lisanza, J. M. Gershon, S. W. K. Tipps, J. N. Sims, L. Arnoldt and S. J. Hendel, et al., Multistate and functional protein design using RoseTTAFold sequence space diffusion, Nat. Biotechnol., 2025, 43(8), 1288–1298 CrossRef CAS PubMed.
- W. R. Taylor, D. T. Jones and M. I. Sadowski, Protein topology from predicted residue contacts, Protein Sci., 2011, 21(2), 299–305 CrossRef PubMed.
- T. A. Hopf, L. J. Colwell, R. Sheridan, B. Rost, C. Sander and D. S. Marks, Three-dimensional structures of membrane proteins from genomic sequencing, Cell, 2012, 149(7), 1607–1621 CrossRef CAS PubMed.
- D. S. Marks, L. J. Colwell, R. Sheridan, T. A. Hopf, A. Pagnani and R. Zecchina, et al., Protein 3D structure computed from evolutionary sequence variation, PLoS One, 2011, 6(12), e28766 CrossRef CAS PubMed.
- D. S. Marks, T. A. Hopf and C. Sander, Protein structure prediction from sequence variation, Nat. Biotechnol., 2012, 30(11), 1072–1080 CrossRef CAS PubMed.
- T. Nugent and D. T. Jones, Accurate de novo structure prediction of large transmembrane protein domains using fragment-assembly and correlated mutation analysis, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(24), E1540–E1547 CrossRef CAS PubMed.
- D. Cozzetto, D. W. Buchan, K. Bryson and D. T. Jones, Protein function prediction by massive integration of evolutionary analyses and multiple data sources, BMC Bioinf., 2013, 14(Suppl 3), S1 CrossRef CAS PubMed.
- T. Kosciolek and D. T. Jones, De novo structure prediction of globular proteins aided by sequence variation-derived contacts, PLoS One, 2014, 9(3), e92197 CrossRef PubMed.
- D. T. Jones, T. Singh, T. Kosciolek and S. Tetchner, MetaPSICOV: combining coevolution methods for accurate prediction of contacts and long range hydrogen bonding in proteins, Bioinformatics, 2015, 31(7), 999–1006 CrossRef CAS PubMed.
- A. Tiessen, P. Pérez-Rodríguez and L. J. Delaye-Arredondo, Mathematical modeling and comparison of protein size distribution in different plant, animal, fungal and microbial species reveals a negative correlation between protein size and protein number, thus providing insight into the evolution of proteomes, BMC Res Notes, 2012, 5, 85 CrossRef CAS PubMed.
- D. B. Kell, Enzymes As Energy Funnels, Trends Biochem. Sci., 1982, 7(10), 349 CrossRef.
- P. H. Pawlowski and P. Zielenkiewicz, Theoretical model explaining the relationship between the molecular mass and the activation energy of the enzyme revealed by a large-scale analysis of bioinformatics data, Acta Biochim. Pol., 2013, 60(2), 239–247 CrossRef CAS PubMed.
- P. K. Robinson, Enzymes: principles and biotechnological applications, Essays Biochem., 2015, 59, 1–41 CrossRef PubMed.
- W. Qian and J. Zhang, Genomic evidence for adaptation by gene duplication, Genome Res., 2014, 24(8), 1356–1362 CrossRef CAS PubMed.
- S. D. Copley, Evolution of new enzymes by gene duplication and divergence, FEBS J., 2020, 287(7), 1262–1283 CrossRef CAS PubMed.
- H. Kacser and J. A. Burns, The molecular basis of dominance, Genetics, 1981, 97(3–4), 639–666 CrossRef CAS PubMed.
- Z. D. Blount, C. Z. Borland and R. E. Lenski, Historical contingency and the evolution of a key innovation in an experimental population of Escherichia coli, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 7899–7906 CrossRef CAS PubMed.
- Z. D. Blount, R. E. Lenski and J. B. Losos, Contingency and determinism in evolution: Replaying life's tape, Science, 2018, 362(6415), eaam5979 CrossRef PubMed.
- D. Monti and S. Riva, Natural and artificial microenzymes: Is it possible to have small and efficient biocatalysts?, Biocatal. Biotransform., 2001, 19(4), 251–266 CrossRef CAS.
- G. Jiménez-Osés, S. Osuna, X. Gao, M. R. Sawaya, L. Gilson and S. J. Collier, et al., The role of distant mutations and allosteric regulation on LovD active site dynamics, Nat. Chem. Biol., 2014, 10, 431–436 CrossRef PubMed.
- E. R. Jarvo and S. J. Miller, Amino acids and peptides as asymmetric organocatalysts, Tetrahedron, 2002, 58, 2481–2495 CrossRef CAS.
- V. da Gama Oliveira, M. F. do Carmo Cardoso and L. da Silva Magalhães Forezi, Organocatalysis: A Brief Overview on Its Evolution and Applications, Catalysis, 2018, 8, 605 Search PubMed.
- K. Kamanna, Amino Acids and Peptides Organocatalysts: A Brief Overview on Its Evolution and Applications in Organic Asymmetric Synthesis, Curr. Organocatal., 2021, 8, 126–146 CAS.
- P. Pecchini, M. Fochi, F. Bartoccini, G. Piersanti and L. Bernardi, Enantioselective organocatalytic strategies to access noncanonical alpha-amino acids, Chem. Sci., 2024, 15, 5832 RSC.
- E. Zandvoort, E. M. Geertsema, B. J. Baas, W. J. Quax and G. J. Poelarends, Bridging between organocatalysis and biocatalysis: asymmetric addition of acetaldehyde to beta-nitrostyrenes catalyzed by a promiscuous proline-based tautomerase, Angew. Chem., Int. Ed., 2012, 51(5), 1240 CrossRef CAS PubMed.
- L. H. Chen, G. L. Kenyon, F. Curtin, S. Harayama, M. E. Bembenek and G. Hajipour, et al., 4-Oxalocrotonate tautomerase, an enzyme composed of 62 amino acid residues per monomer, J. Biol. Chem., 1992, 267(25), 17716–17721 CrossRef CAS PubMed.
- W. H. Yu, P. T. Huang, K. L. Lou, S. S. Yu and C. Lin, A smallest 6 kda metalloprotease, mini-matrilysin, in living world: a revolutionary conserved zinc-dependent proteolytic domain- helix-loop-helix catalytic zinc binding domain (ZBD), J. Biomed. Sci., 2012, 19, 54 CrossRef CAS PubMed.
- M. Mattey, D. Simoes, A. Brown and X. Fan, Enzymes with a low molecular weight, Acta Chim. Slov., 1998, 45(1), 45–57 CAS.
- Mc. Neill D. Simoes DdCM and B. Kristiansen, Mattey M. Purification and partial characterisation of a 1.57 kDa thermostable esterase from Bacillus stearothermophilus, FEMS Microbiol. Lett., 1997, 147(1), 151–156 CrossRef.
- X. L. Fan and M. Mattey, Small enzymes with esterase activities from two thermophilic fungi, Emericella nidulans and Talaromyces emersonii, Biotechnol. Lett., 1999, 21(12), 1071–1076 CrossRef CAS.
- R. U. Schenk and J. Bjorksten, Search for Microenzymes - Enzyme of Bacillus cereus, Finska Kemistsamfundets Meddelanden, 1973, 82(2), 26–46 CAS.
- M. Kanauchi, K. J. Simon and C. W. Bamforth, Ascorbic Acid Oxidase in Barley and Malt and Its Possible Role During Mashing, J. Am. Soc. Brew. Chem, 2014, 72(1), 30–35 CrossRef CAS.
- P. Chellapandi and J. Balachandramohan, Implication of molecular conservation on computational designing of haloarchaean urease with novel functional diversity, Turk. J. Biochem., 2012, 37(2), 110–119 CrossRef CAS.
- O. V. Moroz, Y. S. Moroz, Y. Wu, A. B. Olsen, H. Cheng and K. L. Mack, et al., A single mutation in a regulatory protein produces evolvable allosterically regulated catalyst of nonnatural reaction, Angew. Chem., Int. Ed. Engl., 2013, 52(24), 6246 CrossRef CAS PubMed.
- V. Vaissier, S. C. Sharma, K. Schaettle, T. R. Zhang and T. Head-Gordon, Computational Optimization of Electric Fields for Improving Catalysis of a Designed Kemp Eliminase, ACS Catal., 2018, 8(1), 219–227 CrossRef CAS.
- R. Blomberg, H. Kries, D. M. Pinkas, P. R. Mittl, M. G. Grutter and H. K. Privett, et al., Precision is essential for efficient catalysis in an evolved Kemp eliminase, Nature, 2013, 503(7476), 418–421 CrossRef CAS PubMed.
- I. V. Korendovych, D. W. Kulp, Y. Wu, H. Cheng, H. Roder and W. F. DeGrado, Design of a switchable eliminase, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(17), 6823–6827 CrossRef CAS PubMed.
- S. Bhattacharya, E. G. Margheritis, K. Takahashi, A. Kulesha, A. D'Souza and I. Kim, et al., NMR-guided directed evolution, Nature, 2022, 610(7931), 389–393 CrossRef CAS PubMed.
- M. Stefani, N. Taddei and G. Ramponi, Insights into acylphosphatase structure and catalytic mechanism, Cell. Mol. Life Sci., 1997, 53(2), 141–151 CrossRef CAS PubMed.
- A. V. Gribenko, M. M. Patel, J. Liu, S. A. McCallum, C. Wang and G. I. Makhatadze, Rational stabilization of enzymes by computational redesign of surface charge-charge interactions, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(8), 2601–2606 CrossRef CAS PubMed.
- G. J. Rocklin, T. M. Chidyausiku, I. Goreshnik, A. Ford, S. Houliston and A. Lemak, et al., Global analysis of protein folding using massively parallel design, synthesis, and testing, Science, 2017, 357(6347), 168–175 CrossRef CAS PubMed.
- W. R. Lindemann, E. D. Evans, A. J. Mijalis, O. M. Saouaf, B. L. Pentelute and J. H. Ortony, Quantifying residue-specific conformational dynamics of a highly reactive 29-mer peptide, Sci. Rep., 2020, 10(1), 2597 CrossRef CAS PubMed.
- E. D. Evans and B. L. Pentelute, Discovery of a 29-Amino-Acid Reactive Abiotic Peptide for Selective Cysteine Arylation, ACS Chem. Biol., 2018, 13(3), 527–532 CrossRef CAS PubMed.
- E. D. Evans, Z. P. Gates, Z. J. Sun, A. J. Mijalis and B. L. Pentelute, Conformational Stabilization and Rapid Labeling of a 29-Residue Peptide by a Small Molecule Reaction Partner, Biochemistry, 2019, 58(10), 1343–1353 CrossRef CAS PubMed.
- S. C. Blacklow, R. T. Raines, W. A. Lim, P. D. Zamore and J. R. Knowles, Triosephosphate isomerase catalysis is diffusion controlled. Appendix: Analysis of triose phosphate equilibria in aqueous solution by 31P NMR, Biochemistry, 1988, 27(4), 1158–1167 CrossRef CAS PubMed.
- J. A. Gerlt, Evolution of Enzyme Function and the Development of Catalytic Efficiency: Triosephosphate Isomerase, Jeremy R. Knowles, and W. John Albery, Biochemistry, 2021, 60(46), 3529–3538 CrossRef CAS PubMed.
- M. Alahuhta, M. Salin, M. G. Casteleijn, C. Kemmer, I. El-Sayed and K. Augustyns, et al., Structure-based protein engineering efforts with a monomeric TIM variant: the importance of a single point mutation for generating an active site with suitable binding properties, Protein Eng., Des. Sel., 2008, 21(4), 257–266 CrossRef CAS PubMed.
- M. Alahuhta, M. G. Casteleijn, P. Neubauer and R. K. Wierenga, Structural studies show that the A178L mutation in the C-terminal hinge of the catalytic loop-6 of triosephosphate isomerase (TIM) induces a closed-like conformation in dimeric and monomeric TIM, Acta Crystallogr., D: Biol. Crystallogr., 2008, 64(Pt 2), 178–188 CrossRef CAS PubMed.
- G. Saab-Rincón, V. R. Juárez, J. Osuna, F. Sánchez and X. Soberón, Different strategies to recover the activity of monomeric triosephosphate isomerase by directed evolution, Prot. Eng. Des. Sel., 2001, 14, 149–155 CrossRef PubMed.
- M. Y. Galperin, D. R. Walker and E. V. Koonin, Analogous Enzymes: Independent Inventions in Enzyme Evolution, Genome Res., 1998, 8, 778–790 CrossRef PubMed.
- M. V. Omelchenko, M. Y. Galperin, Y. I. Wolf and E. V. Koonin, Homologous isofunctional enzymes: A systematic analysis of alternative solutions in enzyme evolution, Biol. Direct, 2010, 5, 31 CrossRef PubMed.
- K. E. Medvedev, L. N. Kinch, R. D. Schaeffer and N. V. Grishin, Functional analysis of Rossmann-like domains reveals convergent evolution of topology and reaction pathways, PLoS Comput. Biol., 2019, 15, e1007569 CrossRef PubMed.
- A. J. M. Ribeiro, I. G. Riziotis, N. Borkakoti and J. M. Thornton, Enzyme function and evolution through the lens of bioinformatics, Biochem. J., 2023, 480, 1845–1863 CrossRef CAS PubMed.
- I. G. Riziotis, J. C. Kafas, G. Ong, N. Borkakoti, A. J. M. Ribeiro and J. M. Thornton, Paradigms of convergent evolution in enzymes, FEBS J., 2024, 292, 537–555 CrossRef PubMed.
- P. J. Höglund, K. J. V. Nordström, H. B. Schiöth and R. Fredriksson, The solute carrier families have a remarkably long evolutionary history with the majority of the human families present before divergence of Bilaterian species, Mol. Biol. Evol., 2011, 28(4), 1531–1541 CrossRef PubMed.
- B. Darbani, D. B. Kell and I. Borodina, Energetic evolution of cellular transportomes, BMC Genomics, 2018, 19, 418 CrossRef PubMed.
- L. Schaller and V. M. Lauschke, The genetic landscape of the human solute carrier (SLC) transporter superfamily, Hum. Genet., 2019, 138(11–12), 1359–1377 CrossRef CAS PubMed.
- M. D. Pizzagalli, A. Bensimon and G. Superti-Furga, A guide to plasma membrane solute carrier proteins, FEBS J., 2021, 288(9), 2784–2835 CrossRef CAS PubMed.
- E. Ferrada and G. Superti-Furga, A structure and evolutionary-based classification of solute carriers, iScience, 2022, 25(10), 105096 CrossRef CAS PubMed.
- D. M. Weinreich, N. F. Delaney, M. A. Depristo and D. L. Hartl, Darwinian evolution can follow only very few mutational paths to fitter proteins, Science, 2006, 312(5770), 111–114 CrossRef CAS PubMed.
- T. N. Starr, L. K. Picton and J. W. Thornton, Alternative evolutionary histories in the sequence space of an ancient protein, Nature, 2017, 549(7672), 409–413 CrossRef CAS PubMed.
- T. N. Starr, J. M. Flynn, P. Mishra, D. N. A. Bolon and J. W. Thornton, Pervasive contingency and entrenchment in a billion years of Hsp90 evolution, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4453–4458 CrossRef CAS PubMed.
- A. Vaswani, N. Shazeer, N. Parmar, J. Uszkoreit, L. Jones, A. N. Gomez, et al., Attention is all you need, arXiv, 2017, 1706.03762 DOI:10.48550/arXiv.1706.03762.
- A. D. Shrivastava, N. Swainston, S. Samanta, I. Roberts, M. Wright Muelas and D. B. Kell, MassGenie: a transformer-based deep learning method for identifying small molecules from their mass spectra, Biomolecules, 2021, 11, 1793 CrossRef CAS PubMed.
- B. J. Wittmann, T. Alexanian, C. Bartling, J. Beal, A. Clore and J. Diggans, et al., Strengthening nucleic acid biosecurity screening against generative protein design tools, Science, 2025, 390(6768), 82–87 CrossRef CAS PubMed.
- A. Rives, J. Meier, T. Sercu, S. Goyal, Z. Lin and J. Liu, et al., Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, 15 CrossRef PubMed.
- C. Hsu, R. Verkuil, J. Liu, Z. Lin, B. Hie and T. Sercu, et al., Learning inverse folding from millions of predicted structures, Proc Machine Learning Res, 2022, 162, 8946–8970 Search PubMed.
- M. H. Høie, A. M. Hummer, T. H. Olsen, B. Aguilar-Sanjuan, M. Nielsen and C. M. Deane, AntiFold: improved structure-based antibody design using inverse folding, Bioinform Adv., 2025, 5(1), vbae202 CrossRef PubMed.
- M. Hoang and M. Singh, Locality-aware pooling enhances protein language model performance across varied applications, Bioinformatics, 2025, 41, i217–i226 CrossRef CAS PubMed.
- J. Zheng, G. Wang, H. Zhang and S. Z. Li, Pan-protein Design Learning Enables Task-adaptive Generalization for Low-resource Enzyme Design, arXiv, 2024, 2024:2411.17795 DOI:10.48550/arXiv.2411.17795.
- P. Bryant and A. Elofsson, Peptide binder design with inverse folding and protein structure prediction, Commun Chem, 2023, 6(1), 229 CrossRef CAS PubMed.
- M. Ertelt, J. Meiler and C. T. Schoeder, Combining Rosetta Sequence Design with Protein Language Model Predictions Using Evolutionary Scale Modeling (ESM) as Restraint, ACS Synth. Biol., 2024, 13, 1085–1092 CrossRef CAS PubMed.
- T. Hayes, R. Rao, H. Akin, N. J. Sofroniew, D. Oktay and Z. Lin, et al., Simulating 500 million years of evolution with a language model, Science, 2025, 387(6736), 850–858 CrossRef CAS PubMed.
- K. Jiang, Z. Yan, M. Di Bernardo, S. R. Sgrizzi, L. Villiger and A. Kayabolen, et al., Rapid in silico directed evolution by a protein language model with EVOLVEpro, Science, 2025, 387(6732), eadr6006 CrossRef CAS PubMed.
- C. Hua, J. Lu, Y. Liu, O. Zhang, J. Tang, R. Ying, et al., Reaction-conditioned De Novo Enzyme Design with GENzyme, arXiv, 2024, 2024:2411.16694 DOI:10.48550/arXiv.2411.16694.
- T. M. Marinov, P. T. Wasdin, G. Jordaan, A. K. Janke, A. A. Abu-Shmais and I. S. Georgiev, An expandable synthetic library of human paired antibody sequences, PLoS Comput. Biol., 2025, 21(4), e1012932 CrossRef CAS PubMed.
- R. W. Shuai, J. A. Ruffolo and J. J. Gray, IgLM: Infilling language modeling for antibody sequence design, Cell Syst., 2023, 14(11), 979–989 CrossRef CAS PubMed.
- J. Leem and J. D. Galson, Becoming fluent in proteins, Cell Syst., 2023, 14(11), 923–924 CrossRef CAS PubMed.
- S. Gelman, B. Johnson, C. R. Freschlin, A. Sharma, S. D’Costa and J. Peters, et al., Biophysics-based protein language models for protein engineering, Nat. Methods, 2025, 22, 1868–1879 CrossRef CAS PubMed.
- K. K. Yang, N. Zanichelli and H. Yeh, Masked inverse folding with sequence transfer for protein representation learning, Protein Eng., Des. Sel., 2023, 36, 1–10 CrossRef CAS PubMed.
- D. Sgarbossa, U. Lupo and A. F. Bitbol, Generative power of a protein language model trained on multiple sequence alignments, eLife, 2023, 12, e79854 CrossRef CAS PubMed.
- O. M. Turnbull, D. Oglic, R. Croasdale-Wood and C. M. Deane, p-IgGen: a paired antibody generative language model, Bioinformatics, 2024, 40(11), btae659 CrossRef CAS PubMed.
- T. Bikias, E. Stamkopoulos and S. T. Reddy, PLMFit: benchmarking transfer learning with protein language models for protein engineering, Brief Bioinform., 2025, 26(4) CrossRef CAS PubMed.
- A. Tartici, G. Nayar and R. B. Altman, Pool PaRTI: a PageRank-based pooling method for identifying critical residues and enhancing protein sequence representations, Bioinformatics, 2025, 41, btaf330 CrossRef CAS PubMed.
- F. Jiang, M. Li, J. Dong, Y. Yu, X. Sun and B. Wu, et al., A general temperature-guided language model to design proteins of enhanced stability and activity, Sci. Adv., 2024, 10, eadr2641 CrossRef CAS PubMed.
- L. Cheng, T. Wei, X. Cui, H. F. Chen and Z. Yu, ProDualNet: dual-target protein sequence design method based on protein language model and structure model, Brief. Bioinform., 2025, 26(4), bbaf391 CrossRef CAS PubMed.
- A. Madani, B. Krause, E. R. Greene, S. Subramanian, B. P. Mohr and J. M. Holton, et al., Large language models generate functional protein sequences across diverse families, Nat. Biotechnol., 2023, 41, 1099–1106 CrossRef CAS PubMed.
- E. Nijkamp, J. A. Ruffolo, E. N. Weinstein, N. Naik and A. Madani, ProGen2: Exploring the boundaries of protein language models, Cell Syst, 2023, 14(11), 968–978 CrossRef CAS PubMed.
- M. Heinzinger, K. Weissenow, J. G. Sanchez, A. Henkel, M. Mirdita and M. Steinegger, et al., Bilingual language model for protein sequence and structure, NAR Genom. Bioinform., 2024, 6(4), lqae150 CrossRef CAS PubMed.
- X. Chen, Z. Li, M. Gao, Y. Zhang, C. Tou Leong, H. Li, et al., Protein as a Second Language for LLMs, arXiv, 2025, 2025:2510.11188 DOI:10.48550/arXiv.2510.11188.
- N. Brandes, D. Ofer, Y. Peleg, N. Rappoport and M. Linial, ProteinBERT: a universal deep-learning model of protein sequence and function, Bioinformatics, 2022, 38(8), 2102–2110 CrossRef CAS PubMed.
- J. Dauparas, I. Anishchenko, N. Bennett, H. Bai, R. J. Ragotte and L. F. Milles, et al., Robust deep learning-based protein sequence design using ProteinMPNN, Science, 2022, 378(6615), 49–56 CrossRef CAS PubMed.
- L. Wang, H. Zhang, W. Xu, Z. Xue and Y. Wang, Deciphering the protein landscape with ProtFlash, a lightweight language model, Cell Rep. Phys. Sci., 2023, 4, 101600 CrossRef CAS.
- N. Ferruz, S. Schmidt and B. Höcker, ProtGPT2 is a deep unsupervised language model for protein design, Nat. Commun., 2022, 13(1), 4348 CrossRef CAS PubMed.
- Z. Xu, J. Wu, Y. S. Song and R. Mahadevan, Enzyme Activity Prediction of Sequence Variants on Novel Substrates using Improved Substrate Encodings and Convolutional Pooling, J. Mach. Learn. Res., 2022, 165, 75–87 Search PubMed.
- M. Braun, C. C. Gruber, A. Krassnigg, A. Kummer, S. Lutz and G. Oberdorfer, et al., Accelerating Biocatalysis Discovery with Machine Learning: A Paradigm Shift in Enzyme Engineering, Discovery, and Design, ACS Catal., 2023, 13, 14454–14469 CrossRef PubMed.
- W. J. Xie and A. Warshel, Harnessing generative AI to decode enzyme catalysis and evolution for enhanced engineering, Natl. Sci. Rev., 2023, 10, nwad331 CrossRef CAS PubMed.
- J. S. Lee, O. Abdin and P. M. Kim, Language models for protein design, Curr. Opin. Struct. Biol., 2025, 92, 103027 CrossRef CAS PubMed.
- Y. G. N. Teukam, F. Zipoli, T. Laino, E. Criscuolo, F. Grisoni and M. Manica, Integrating genetic algorithms and language models for enhanced enzyme design, Brief. Bioinform., 2025, 26, bbae675 CrossRef CAS PubMed.
- J. L. Watson, D. Juergens, N. R. Bennett, B. L. Trippe, J. Yim and H. E. Eisenach, et al., De novo design of protein structure and function with RFdiffusion, Nature, 2023, 620(7976), 1089–1100 CrossRef CAS PubMed.
- W. Ahern, J. Yim, D. Tischer, S. Salike, S. M. Woodbury and D. Kim, et al., Atom-level enzyme active site scaffolding using RFdiffusion2, Nat. Methods, 2026, 23(1), 96–105 CrossRef CAS PubMed.
- M. Braun, A. Tripp, M. Chakatok, S. Kaltenbrunner, C. Fischer and D. Stoll, et al., Computational enzyme design by catalytic motif scaffolding, Nature, 2026, 649(8095), 237–245 CrossRef CAS PubMed.
- S. On, Y. Jeong and E.-S. Kim, Structure-guided sequence representation learning for generalizable protein function prediction, Bioinformatics, 2025, 41, btaf511 CrossRef CAS PubMed.
- E. C. Alley, G. Khimulya, S. Biswas, M. AlQuraishi and G. M. Church, Unified rational protein engineering with sequence-based deep representation learning, Nat. Methods, 2019, 16(12), 1315–1322 CrossRef CAS PubMed.
- S. Biswas, G. Khimulya, E. C. Alley, K. M. Esvelt and G. M. Church, Low-N protein engineering with data-efficient deep learning, Nat. Methods, 2021, 18(4), 389–396 CrossRef CAS PubMed.
- P. Carbonell, J. Wong, N. Swainston, E. Takano, N. Turner and N. Scrutton, et al., Selenzyme: Enzyme selection tool for pathway design, Bioinformatics, 2018, 34(12), 2153–2154 CrossRef PubMed.
- A. Currin, N. Swainston, P. J. Day and D. B. Kell, SpeedyGenes: a novel approach for the efficient production of error-corrected, synthetic gene libraries, Protein Eng. Des. Sel., 2014, 27, 273–280 CrossRef CAS PubMed.
- A. Currin, J. Kwok, J. C. Sadler, E. L. Bell, N. Swainston and M. Ababi, et al., GeneORator: an effective strategy for navigating protein sequence space more efficiently through Boolean OR-type DNA libraries, ACS Synth. Biol., 2019, 8, 1371–1378 CrossRef CAS PubMed.
- N. Swainston, A. Currin, P. J. Day and D. B. Kell, GeneGenie: optimised oligomer design for directed evolution, Nucleic Acids Res., 2014, 12, W395–W400 CrossRef PubMed.
- N. Swainston, A. Currin, L. Green, R. Breitling, P. J. Day and D. B. Kell, CodonGenie: optimised ambiguous codon design tools, Peer J. Comput. Sci., 2017, 3, e120 CrossRef.
- N. Swainston, M. Dunstan, A. J. Jervis, C. J. Robinson, P. Carbonell and A. R. Williams, et al., PartsGenie: an integrated tool for optimising and sharing synthetic biology parts, Bioinformatics, 2018, 34(13), 2327–2329 CrossRef CAS PubMed.
- L. Mitchener, A. Yiu, B. Chang, M. Bourdenx, T. Nadolski, A. Sulovari, et al., Kosmos: An AI Scientist for Autonomous Discovery, arXiv, 2025, 2025:2511.02824 DOI:10.48550/arXiv.2511.02824.
- R. D. King, K. E. Whelan, F. M. Jones, P. G. K. Reiser, C. H. Bryant and S. H. Muggleton, et al., Functional genomic hypothesis generation and experimentation by a robot scientist, Nature, 2004, 427, 247–252 CrossRef CAS PubMed.
- S. O'Hagan, W. B. Dunn, M. Brown, J. D. Knowles and D. B. Closed-loop Kell, multiobjective optimisation of analytical instrumentation: gas-chromatography-time-of-flight mass spectrometry of the metabolomes of human serum and of yeast fermentations, Anal. Chem., 2005, 77, 290–303 CrossRef PubMed.
- S. O'Hagan, W. B. Dunn, D. Broadhurst, R. Williams, J. A. Ashworth and M. Cameron, et al., Closed-loop, multi-objective optimisation of two-dimensional gas chromatography (GCxGC-tof-MS) for serum metabolomics, Anal. Chem., 2007, 79(2), 464–476 CrossRef PubMed.
- P. Carbonell, A. J. Jervis, C. J. Robinson, C. Yan, M. Dunstan and N. Swainston, et al., An automated Design-Build-Test-Learn pipeline for enhanced microbial production of fine chemicals, Commun. Biol., 2018, 1, 66 CrossRef PubMed.
- N. Gurdo, D. C. Volke and P. I. Nikel, Merging automation and fundamental discovery into the design-build-test-learn cycle of nontraditional microbes, Trends Biotechnol., 2022, 40, 1148–1159 CrossRef CAS PubMed.
- N. Gurdo, D. C. Volke, D. McCloskey and P. I. Nikel, Automating the design-build-test-learn cycle towards next-generation bacterial cell factories, New Biotechnol., 2023, 74, 1–15 CrossRef CAS PubMed.
- R. Matzko and S. Konur, Technologies for design-build-test-learn automation and computational modelling across the synthetic biology workflow: a review, Netw. Model Anal. Health Inf. Bioinf., 2024, 13, 22 CrossRef.
- N. Singh, S. Lane, T. Yu, J. Lu, A. Ramos and H. Cui, et al., A generalized platform for artificial intelligence-powered autonomous enzyme engineering, Nat. Commun., 2025, 16(1), 5648 CrossRef PubMed.
- L. Pavlovic, C. Vernette, E. Pigani, L. Joigneaux, A. Labesse, J. Rigonato, et al., Deviation from Power-Law Distribution when Scaling the Distribution of Marine Plankton Folds from Genomes to Communities, bioRxiv, 2025, 2025.03.25.645231.
- V. P. Waman, N. Bordin, A. Lau, S. Kandathil, J. Wells and D. Miller, et al., CATH v4.4: major expansion of CATH by experimental and predicted structural data, Nucleic Acids Res., 2025, 53(D1), D348–D55 CrossRef CAS PubMed.
- P. Szczerbiak, L. M. Szydlowski, W. Wydmański, P. D. Renfrew, J. K. Leman and T. Kosciolek, Large protein databases reveal structural complementarity and functional locality, Nat. Commun., 2025, 16(1), 7925 CrossRef CAS PubMed.
- A. M. Lau, N. Bordin, S. M. Kandathil, I. Sillitoe, V. P. Waman and J. Wells, et al., Exploring structural diversity across the protein universe with The Encyclopedia of Domains, Science, 2024, 386(6721), eadq4946 CrossRef CAS PubMed.
- A. F. Dishman and B. F. Volkman, Design and discovery of metamorphic proteins, Curr. Opin. Struct. Biol., 2022, 74, 102380 CrossRef CAS PubMed.
- D. B. Kell, J. E. Salcedo-Sora and E. Pretorius, Amyloidogenic potential of plaque and thrombus proteomes and of fold-switching metamorphic proteins, 2025, preprints, 2025081049.
- M. Lella and R. Mahalakshmi, Metamorphic proteins: emergence of dual protein folds from one primary sequence, Biochemistry, 2017, 56(24), 2971–2984 CrossRef CAS PubMed.
- L. L. Porter, I. Artsimovitch and C. A. Ramírez-Sarmiento, Metamorphic proteins and how to find them, Curr. Opin. Struct. Biol., 2024, 86, 102807 CrossRef CAS PubMed.
- D. Chakravarty and L. L. Porter, Fold-switching Proteins, arXiv, 2025, 2025:2507.10839 DOI:10.48550/arXiv.2507.10839.
- L. L. Porter and L. L. Looger, Extant fold-switching proteins are widespread, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(23), 5968–5973 CrossRef CAS PubMed.
- I. Retamal-Farfán, J. González-Higueras, P. Galaz-Davison, M. Rivera and C. A. Ramírez-Sarmiento, Exploring the structural acrobatics of fold-switching proteins using simplified structure-based models, Biophys. Rev., 2023, 15(4), 787–799 CrossRef PubMed.
- I. Yadid, N. Kirshenbaum, M. Sharon, O. Dym and D. S. Tawfik, Metamorphic proteins mediate evolutionary transitions of structure, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(16), 7287–7292 CrossRef CAS PubMed.
- C. M. Dobson, Protein folding and misfolding, Nature, 2003, 426(6968), 884–890 CrossRef CAS PubMed.
- C. M. Dobson, Principles of protein folding, misfolding and aggregation, Semin. Cell Dev. Biol., 2004, 15(1), 3–16 CrossRef CAS PubMed.
- T. R. Jahn and S. E. Radford, Folding versus aggregation: polypeptide conformations on competing pathways, Arch. Biochem. Biophys., 2008, 469(1), 100–117 CrossRef CAS PubMed.
- M. Vendruscolo, T. P. J. Knowles and C. M. Dobson, Protein solubility and protein homeostasis: a generic view of protein misfolding disorders, Cold Spring Harb. Perspect. Biol., 2011, 3(12) CAS.
- P. Cossio, A. Trovato, F. Pietrucci, F. Seno, A. Maritan and A. Laio, Exploring the universe of protein structures beyond the Protein Data Bank, PLoS Comput. Biol., 2010, 6(11), e1000957 CrossRef PubMed.
- I. Anishchenko, S. J. Pellock, T. M. Chidyausiku, T. A. Ramelot, S. Ovchinnikov and J. Hao, et al., De novo protein design by deep network hallucination, Nature, 2021, 600(7889), 547–552 CrossRef CAS PubMed.
- L. Cao, B. Coventry, I. Goreshnik, B. Huang, J. S. Park, K. M. Jude, et al., Robust de novo design of protein binding proteins from target structural information alone, bioRxiv, 2021, 2021.09.04.459002.
- A. H.-W. Yeh, C. Norn, Y. Kipnis, D. Tischer, S. J. Pellock and D. Evans, et al., De novo design of luciferases using deep learning, Nature, 2023, 614(7949), 774–780 CrossRef CAS PubMed.
- X. Pan and T. Kortemme, De novo protein fold families expand the designable ligand binding site space, PLoS Comput. Biol., 2021, 17(11), e1009620 CrossRef CAS PubMed.
- X. Pan and T. Kortemme, Recent advances in de novo protein design: Principles, methods, and applications, J. Biol. Chem., 2021, 296, 100558 CrossRef CAS PubMed.
- L. An, D. R. Hicks, D. Zorine, J. Dauparas, B. I. M. Wicky and L. F. Milles, et al., Hallucination of closed repeat proteins containing central pockets, Nat. Struct. Mol. Biol., 2023, 30(11), 1755–1760 CrossRef CAS PubMed.
- S. Minami, N. Kobayashi, T. Sugiki, T. Nagashima, T. Fujiwara and R. Tatsumi-Koga, et al., Exploration of novel alphabeta-protein folds through de novo design, Nat. Struct. Mol. Biol., 2023, 30(8), 1132–1140 CrossRef CAS PubMed.
- R. Pearce, X. Huang, G. S. Omenn and Y. Zhang, De novo protein fold design through sequence-independent fragment assembly simulations, Proc. Natl. Acad. Sci. U. S. A., 2023, 120(4), e2208275120 CrossRef CAS PubMed.
- S. Berhanu, S. Majumder, T. Muntener, J. Whitehouse, C. Berner and A. K. Bera, et al., Sculpting conducting nanopore size and shape through de novo protein design, Science, 2024, 385(6706), 282–288 CrossRef CAS PubMed.
- N. Koga and R. Tatsumi-Koga, Inventing Novel Protein Folds, J. Mol. Biol., 2024, 436(21), 168791 CrossRef CAS PubMed.
- T. Kortemme, De novo protein design-From new structures to programmable functions, Cell, 2024, 187(3), 526–544 CrossRef CAS PubMed.
- M. A. Jendrusch, A. L. J. Yang, E. Cacace, J. Bobonis, C. G. P. Voogdt and S. Kaspar, et al., AlphaDesign: a de novo protein design framework based on AlphaFold, Mol. Syst. Biol., 2025, 21(9), 1166–1189 CrossRef PubMed.
- D. E. Kim, J. L. Watson, D. Juergens, S. Majumder, R. Sonigra and S. R. Gerben, et al., Parametrically guided design of beta barrels and transmembrane nanopores using deep learning, Proc. Natl. Acad. Sci. U. S. A., 2025, 122(38), e2425459122 CrossRef CAS PubMed.
- B. Orr, S. E. Crilly, D. Akpinaroglu, E. Zhu, M. J. Keiser and T. Kortemme, An improved model for prediction of de novo designed proteins with diverse geometries, bioRxiv, 2025.
- A. Subramanian and M. Thomson, Rapid discovery of new-to-nature protein domains by novelty-first forcing of language models, bioRxiv, 2025, 2025.10.02.679910.
- S. VázquezTorres, M. Benard Valle, S. P. Mackessy, S. K. Menzies, N. R. Casewell and S. Ahmadi, et al., De novo designed proteins neutralize lethal snake venom toxins, Nature, 2025, 639(8053), 225–231 CrossRef PubMed.
- W. J. Albery and J. R. Knowles, Evolution of enzyme function and the development of catalytic efficiency, Biochemistry, 1976, 15, 5631–5640 CrossRef CAS PubMed.
- J. R. Knowles and W. J. Albery, Perfection in enzyme catalysis - energetics of triosephosphate isomerase, Acc. Chem. Res., 1977, 10(4), 105–111 CrossRef CAS.
- M. Bazelyansky, E. Robey and J. F. Kirsch, Fractional diffusion-limited component of reactions catalyzed by acetylcholinesterase, Biochemistry, 1986, 25(1), 125–130 CrossRef CAS PubMed.
- V. Tõugu, Acetylcholinesterase: Mechanism of Catalysis and Inhibition, Curr. Med. Chem., 2001, 1, 155–170 Search PubMed.
- M. S. Kimber and E. F. Pai, The active site architecture of Pisum sativum beta-carbonic anhydrase is a mirror image of that of alpha-carbonic anhydrases, EMBO J., 2000, 19(7), 1407–1418 CrossRef CAS PubMed.
- N. Muster, I. Derecho, F. Dallal, R. Alvarez, K. B. McCoy and R. Mogul, Purification, biochemical characterization, and implications of an alkali-tolerant catalase from the spacecraft-associated and oxidation-resistant Acinetobacter gyllenbergii 2P01AA, Astrobiology, 2015, 15(4), 291–300 CrossRef CAS PubMed.
- S. Srivastava, D. Singh, S. Patel and M. R. Singh, Role of enzymatic free radical scavengers in management of oxidative stress in autoimmune disorders, Int. J. Biol. Macromol., 2017, 101, 502–517 CrossRef CAS PubMed.
- J. L. Hsu, Y. Hsieh, C. Tu, D. O'Connor, H. S. Nick and D. N. Silverman, Catalytic properties of human manganese superoxide dismutase, J. Biol. Chem., 1996, 271(30), 17687–17691 CrossRef CAS PubMed.
- N. A. Popp, R. L. Powell, M. K. Wheelock, K. J. Holmes, B. D. Zapp and K. M. Sheldon, et al., Multiplex and multimodal mapping of variant effects in secreted proteins via MultiSTEP, Nat. Struct. Mol. Biol., 2025, 32(10), 2099–2111 CrossRef CAS PubMed.
- P. J. Ogden, E. D. Kelsic, S. Sinai and G. M. Church, Comprehensive AAV capsid fitness landscape reveals a viral gene and enables machine-guided design, Science, 2019, 366(6469), 1139–1143 CrossRef CAS PubMed.
- P. Carrasco, F. de la Iglesia and S. F. Elena, Distribution of fitness and virulence effects caused by single-nucleotide substitutions in Tobacco Etch virus, J. Virol., 2007, 81(23), 12979–12984 CrossRef CAS PubMed.
- F. Zanini, V. Puller, J. Brodin, J. Albert and R. A. Neher, In vivo mutation rates and the landscape of fitness costs of HIV-1, Virus Evol., 2017, 3(1), vex003 CrossRef PubMed.
- B. Yan, X. Ran, A. Gollu, Z. Cheng, X. Zhou and Y. Chen, et al., IntEnzyDB: an Integrated Structure-Kinetics Enzymology Database, J. Chem. Inf. Model., 2022, 62(22), 5841–5848 CrossRef CAS PubMed.
- M. L. Cárdenas, Michaelis and Menten and the long road to the discovery of cooperativity, FEBS Lett., 2013, 587, 2767–2771 CrossRef PubMed.
- A. Cornish-Bowden, Fundamentals of Enzyme Kinetics, Wiley, Weinheim, 4th edn, 2012 Search PubMed.
- A. Fersht, Enzyme structure and mechanism, W.H. Freeman, San Francisco, 2nd edn, 1977 Search PubMed.
- T. Keleti, Basic enzyme kinetics, Akadémiai Kiadó, Budapest, 1986 Search PubMed.
- A. Bar-Even, R. Milo, E. Noor and D. S. Tawfik, The Moderately Efficient Enzyme: Futile Encounters and Enzyme Floppiness, Biochemistry, 2015, 54(32), 4969–4977 CrossRef CAS PubMed.
- H. Kim and K. J. Shin, Diffusion influence on Michaelis–Menten kinetics: II. The low substrate concentration limit, J Phy.s: Condens Matter, 2007, 19, 065137 CrossRef.
- K. C. Chou and G. P. Zhou, Role of the protein outside active site on the diffusion-controlled reaction of enzymes, J. Am. Chem. Soc., 1982, 104, 1409–1413 CrossRef CAS.
- M. D. Truppo, Biocatalysis in the Pharmaceutical Industry: The Need for Speed, ACS Med. Chem. Lett., 2017, 8(5), 476–480 CrossRef CAS PubMed.
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Biocatalysis: Enzymatic Synthesis for Industrial Applications, Angew. Chem., Int. Ed., 2021, 60(1), 88–119 CrossRef CAS PubMed.
- B. Lin and Y. Tao, Whole-cell biocatalysts by design, Microb. Cell Fact., 2017, 16(1), 106 CrossRef PubMed.
- P. De Santis, L.-E. Meyer and S. Kara, The rise of continuous flow biocatalysis – fundamentals, very recent developments and future perspectives, React. Chem. Eng., 2020, 5, 2155–2184 RSC.
- J. M. Woodley, New frontiers in biocatalysis for sustainable synthesis, Curr. Opin. Green Sus. Chem., 2020, 21, 22–26 CAS.
- J. L. Snoep, L. P. Yomano, H. V. Westerhoff and L. O. Ingram, Protein Burden in Zymomonas mobilis - Negative Flux and Growth- Control Due to Overproduction of Glycolytic Enzymes, Microbiology, 1995, 141(Pt9), 2329–2337 CrossRef CAS.
- J. Berkhout, E. Bosdriesz, E. Nikerel, D. Molenaar, D. de Ridder and B. Teusink, et al., How biochemical constraints of cellular growth shape evolutionary adaptations in metabolism, Genetics, 2013, 194(2), 505–512 CrossRef CAS PubMed.
- Y. Chen and J. Nielsen, Energy metabolism controls phenotypes by protein efficiency and allocation, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(35), 17592–17597 CrossRef CAS PubMed.
- S. A. H. Heyde and M. H. H. Nørholm, Tailoring the evolution of BL21(DE3) uncovers a key role for RNA stability in gene expression toxicity, Commun. Biol., 2021, 4(1), 963 CrossRef CAS PubMed.
- L. La Barbera Kastberg, R. Ard, M. K. Jensen and C. T. Workman, Burden Imposed by Heterologous Protein Production in Two Major Industrial Yeast Cell Factories: Identifying Sources and Mitigation Strategies, Front. Fungal Biol., 2022, 3, 827704 CrossRef PubMed.
- G. Kudla, A. W. Murray, D. Tollervey and J. B. Plotkin, Coding-sequence determinants of gene expression in Escherichia coli, Science, 2009, 324(5924), 255–258 CrossRef CAS PubMed.
- E. Noor, A. Bar-Even, A. Flamholz, E. Reznik, W. Liebermeister and R. Milo, Pathway thermodynamics highlights kinetic obstacles in central metabolism, PLoS Comput. Biol., 2014, 10(2), e1003483 CrossRef PubMed.
- A. Sahin, D. R. Weilandt and V. Hatzimanikatis, Optimal enzyme utilization suggests that concentrations and thermodynamics determine binding mechanisms and enzyme saturations, Nat. Commun., 2023, 14(1), 2618 CrossRef CAS PubMed.
- Y. Chen and J. Nielsen, In vitro turnover numbers do not reflect in vivo activities of yeast enzymes, Proc. Natl. Acad. Sci. U S A, 2021, 118(32) Search PubMed.
- A. Bar-Even, E. Noor, Y. Savir, W. Liebermeister, D. Davidi and D. S. Tawfik, et al., The moderately efficient enzyme: evolutionary and physicochemical trends shaping enzyme parameters, Biochemistry, 2011, 50(21), 4402–4410 CrossRef CAS PubMed.
- R. Buller, S. Lutz, R. J. Kazlauskas, R. Snajdrova, J. C. Moore and U. T. Bornscheuer, From nature to industry: Harnessing enzymes for biocatalysis, Science, 2023, 382(6673), eadh8615 CrossRef CAS PubMed.
- E. J. Hossack, F. J. Hardy and A. P. Green, Building enzymes through design and evolution, ACS Catal., 2023, 13, 12436–12444 CrossRef CAS.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam and W. R. Jarvis, et al., Biocatalytic asymmetric synthesis of chiral amines from ketones applied to sitagliptin manufacture, Science, 2010, 329(5989), 305–309 CrossRef CAS PubMed.
- U. Prešern and M. Goličnik, Enzyme Databases in the Era of Omics and Artificial Intelligence, Int. J. Mol. Sci., 2023, 24(23), 16918 CrossRef PubMed.
- A. Chang, L. Jeske, S. Ulbrich, J. Hofmann, J. Koblitz and I. Schomburg, et al., BRENDA, the ELIXIR core data resource in 2021: new developments and updates, Nucleic Acids Res., 2021, 49(D1), D498–D508 CrossRef CAS PubMed.
- U. Wittig, M. Rey, A. Weidemann, R. Kania and W. Muller, SABIO-RK: an updated resource for manually curated biochemical reaction kinetics, Nucleic Acids Res., 2018, 46(D1), D656–D660 CrossRef CAS PubMed.
- D. Dudaš, U. Wittig, M. Rey, A. Weidemann and W. Müller, Improved insights into the SABIO-RK database via visualization, Database, 2023, 2023, baad011 CrossRef PubMed.
- D. Heckmann, C. J. Lloyd, N. Mih, Y. Ha, D. C. Zielinski and Z. B. Haiman, et al., Machine learning applied to enzyme turnover numbers reveals protein structural correlates and improves metabolic models, Nat. Commun., 2018, 9(1), 5252 CrossRef CAS PubMed.
- X. F. Cadet, J. C. Gelly, A. van Noord, F. Cadet and C. G. Acevedo-Rocha, Learning Strategies in Protein Directed Evolution, Methods Mol. Biol., 2022, 2461, 225–275 CrossRef CAS PubMed.
- K. Maeda, A. Hatae, Y. Sakai, F. C. Boogerd and H. Kurata, MLAGO: machine learning-aided global optimization for Michaelis constant estimation of kinetic modeling, BMC Bioinf., 2022, 23(1), 455 CrossRef CAS PubMed.
- F. Li, Y. Chen, M. Anton and J. Nielsen, GotEnzymes: an extensive database of enzyme parameter predictions, Nucleic Acids Res., 2023, 51(D1), D583–D586 CrossRef CAS PubMed.
- T. Wang, G. Xiang, S. He, L. Su, Y. Wang and X. Yan, et al., DeepEnzyme: a robust deep learning model for improved enzyme turnover number prediction by utilizing features of protein 3D-structures, Brief. Bioinform., 2024, 25(5), bbae409 CrossRef CAS PubMed.
- J. Wang, Z. Yang, C. Chen, G. Yao, X. Wan and S. Bao, et al., MPEK: a multitask deep learning framework based on pretrained language models for enzymatic reaction kinetic parameters prediction, Brief Bioinform, 2024, 25(5), bbae387 CrossRef PubMed.
- V. S. Boorla and C. D. Maranas, CatPred: a comprehensive framework for deep learning in vitro enzyme kinetic parameters, Nat. Commun., 2025, 16(1), 2072 CrossRef CAS PubMed.
- Y. Cai, W. Zhang, Z. Dou, C. Wang, W. Yu and L. Wang, PreTKcat: A pre-trained representation learning and machine learning framework for predicting enzyme turnover number, Comput. Biol. Chem., 2025, 115, 108327 CrossRef CAS PubMed.
- K. A. Sajeevan, A. B. A. Osinuga, S. Ferdous, N. Shahreen, M. S. Noor, et al., Robust Prediction of Enzyme Variant Kinetics with RealKcat, bioRxiv, 2025, 2025.02.10.637555.
- Z. Wang, D. Xie, D. Wu, X. Luo, S. Wang and Y. Li, et al., Robust enzyme discovery and engineering with deep learning using CataPro, Nat. Commun., 2025, 16(1), 2736 CrossRef CAS PubMed.
- Y. Wang, L. Cheng, Y. Zhang, Y. Cao and D. Alghazzawi, DEKP: a deep learning model for enzyme kinetic parameter prediction based on pretrained models and graph neural networks, Brief Bioinform, 2025, 26(2), bbaf187 CrossRef CAS PubMed.
- J. Wang, Y. Zhao, Z. Yang, G. Yao, P. Han and J. Liu, et al., IECata: interpretable bilinear attention network and evidential deep learning improve the catalytic efficiency prediction of enzymes, Brief. Bioinform., 2025, 26(3), bbaf283 CrossRef CAS PubMed.
- G. Wei, X. Ran, R. Ai-Abssi and Z. Yang, Finding the dark matter: Large language model-based enzyme kinetic data extractor and its validation, Protein Sci., 2025, 34(9), e70251 CrossRef CAS PubMed.
- H. Kacser and J. A. Burns, The control of flux, in Rate Control of Biological Processes Symposium of the Society for Experimental Biology, ed. D. D. Davies, Cambridge University Press, Cambridge, 1973, vol. 27, pp. 65–104 Search PubMed.
- R. Heinrich and T. A. Rapoport, A linear steady-state treatment of enzymatic chains. General properties, control and effector strength, Eur. J. Biochem., 1974, 42, 89–95 CrossRef CAS PubMed.
- D. B. Kell and H. V. Westerhoff, Metabolic control theory: its role in microbiology and biotechnology, FEMS Microbiol. Rev., 1986, 39, 305–320 CrossRef CAS.
- D. A. Fell, Understanding the control of metabolism, Portland Press, London, 1996 Search PubMed.
- D. A. Rand, Mapping global sensitivity of cellular network dynamics: sensitivity heat maps and a global summation law, J. R. Soc., Interface, 2008, 5(Suppl 1), S59–S69 CAS.
- M. K. Winson and D. B. Kell, Going places: forced and natural molecular evolution, Trends Biotechnol., 1996, 14, 323–325 CrossRef CAS PubMed.
- M. J. Oates, D. W. Corne and D. B. Kell, The bimodal feature at large population sizes and high selection pressure: implications for directed evolution, in Recent advances in simulated evolution and learning, ed. K. C. Tan, M. H. Lim, X. Yao and L. Wang, World Scientific, Singapore, 2003, pp. 215–240 Search PubMed.
- R. Y. Tsien, The green fluorescent protein, Annu. Rev. Biochem., 1998, 67, 509–544 CrossRef CAS PubMed.
- T. J. Lambert, Using FPbase: The Fluorescent Protein Database, Methods Mol. Biol., 2023, 2564, 1–45 CrossRef PubMed.
- K. S. Sarkisyan, D. A. Bolotin, M. V. Meer, D. R. Usmanova, A. S. Mishin and G. V. Sharonov, et al., Local fitness landscape of the green fluorescent protein, Nature, 2016, 533(7603), 397–401 CrossRef CAS PubMed.
- R. Gomez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hernandez-Lobato, B. Sanchez-Lengeling and D. Sheberla, et al., Automatic chemical design using a data-driven continuous representation of molecules, ACS Cent. Sci., 2018, 4(2), 268–276 CrossRef CAS PubMed.
- C. E. Gonzalez and M. Ostermeier, Pervasive Pairwise Intragenic Epistasis among Sequential Mutations in TEM-1 beta-Lactamase, J. Mol. Biol., 2019, 431(10), 1981–1992 CrossRef CAS PubMed.
- C. E. Gonzalez, P. Roberts and M. Ostermeier, Fitness Effects of Single Amino Acid Insertions and Deletions in TEM-1 beta-Lactamase, J. Mol. Biol., 2019, 431(12), 2320–2330 CrossRef CAS PubMed.
- C. B. Macdonald, D. Nedrud, P. R. Grimes, D. Trinidad, J. S. Fraser and W. Coyote-Maestas, DIMPLE: deep insertion, deletion, and missense mutation libraries for exploring protein variation in evolution, disease, and biology, Genome Biol., 2023, 24(1), 36 CrossRef CAS PubMed.
- W. R. Edwards, K. Busse, R. K. Allemann and D. D. Jones, Linking the functions of unrelated proteins using a novel directed evolution domain insertion method, Nucleic Acids Res., 2008, 36(13), e78 CrossRef PubMed.
- B. Gracia, P. Montes, A. M. Gutierrez, B. Arun and G. I. Karras, Protein-folding chaperones predict structure-function relationships and cancer risk in BRCA1 mutation carriers, Cell Rep., 2024, 43(2), 113803 CrossRef CAS PubMed.
- J. Zhou and D. M. McCandlish, Minimum epistasis interpolation for sequence-function relationships, Nat. Commun., 2020, 11(1), 1782 CrossRef CAS PubMed.
- E. E. Wrenbeck, L. R. Azouz and T. A. Whitehead, Single-mutation fitness landscapes for an enzyme on multiple substrates reveal specificity is globally encoded, Nat. Commun., 2017, 8, 15695 CrossRef CAS PubMed.
- G. R. Welch, B. Somogyi and S. Damjanovich, The role of protein fluctuations in enzyme action: a review, Prog. Biophys. Mol. Biol., 1982, 39(2), 109–146 CrossRef CAS PubMed.
- R. M. Daniel, R. V. Dunn, J. L. Finney and J. C. Smith, The role of dynamics in enzyme activity, Annu. Rev. Biophys. Biomol. Struct., 2003, 32, 69–92 CrossRef CAS PubMed.
- C. G. Acevedo-Rocha, A. Li, L. D'Amore, S. Hoebenreich, J. Sanchis and P. Lubrano, et al., Pervasive cooperative mutational effects on multiple catalytic enzyme traits emerge via long-range conformational dynamics, Nat. Commun., 2021, 12(1), 1621 CrossRef CAS PubMed.
- M. A. Maria-Solano, E. Serrano-Hervás, A. Romero-Rivera, J. Iglesias-Fernandez and S. Osuna, Role of conformational dynamics in the evolution of novel enzyme function, Chem. Commun., 2018, 54(50), 6622–6634 RSC.
- S. Osuna, G. Jiménez-Osés, E. L. Noey and K. N. Houk, Molecular dynamics explorations of active site structure in designed and evolved enzymes, Acc. Chem. Res., 2015, 48(4), 1080–1089 CrossRef CAS PubMed.
- A. Romero-Rivera, M. Garcia-Borràs and S. Osuna, Role of Conformational Dynamics in the Evolution of Retro-Aldolase Activity, ACS Catal., 2017, 7(12), 8524–8532 CrossRef CAS PubMed.
- C. Acevedo-Rocha, L. Berlicki, U. T. Bornscheuer, D. J. Campopiano, P. Chaiyen and J. Čivić, et al., Enzyme evolution, engineering and design: mechanism and dynamics: general discussion, Faraday Discuss., 2024, 252, 127–156 RSC.
- G. Casadevall, J. Casadevall, C. Duran and S. Osuna, The shortest path method (SPM) webserver for computational enzyme design, Protein Eng., Des. Sel., 2024, 37, gzae005 CrossRef CAS PubMed.
- C. Duran, G. Casadevall and S. Osuna, Harnessing conformational dynamics in enzyme catalysis to achieve nature-like catalytic efficiencies: the shortest path map tool for computational enzyme redesign, Faraday Discuss., 2024, 252, 306–322 RSC.
- N. Zarifi, P. Asthana, H. Doustmohammadi, C. Klaus, J. Sanchez, S. E. Hunt, et al., Distal mutations enhance catalysis in designed enzymes by facilitating substrate binding and product release, bioRxiv, 2025, 2025.02.21.639315.
- N. Tokuriki and D. S. Tawfik, Protein dynamism and evolvability, Science, 2009, 324(5924), 203–207 CrossRef CAS PubMed.
- A. Ramanathan, J. O. Yoo and C. J. Langmead, On-the-Fly Identification of Conformational Substates from Molecular Dynamics Simulations, J. Chem. Theor. Comput., 2011, 7(3), 778–789 CrossRef CAS PubMed.
- G. Kiss, V. S. Pande and K. N. Houk, Molecular dynamics simulations for the ranking, evaluation, and refinement of computationally designed proteins, Methods Enzymol., 2013, 523, 145–170 CAS.
- A. Ramanathan, A. Savol, V. Burger, C. S. Chennubhotla and P. K. Agarwal, Protein conformational populations and functionally relevant substates, Acc. Chem. Res., 2014, 47(1), 149–156 CrossRef CAS PubMed.
- P. K. Agarwal, N. Doucet, C. Chennubhotla, A. Ramanathan and C. Narayanan, Conformational Sub-states and Populations in Enzyme Catalysis, Methods Enzymol., 2016, 578, 273–297 CAS.
- E. Campbell, M. Kaltenbach, G. J. Correy, P. D. Carr, B. T. Porebski and E. K. Livingstone, et al., The role of protein dynamics in the evolution of new enzyme function, Nat. Chem. Biol., 2016, 12(11), 944–950 CrossRef CAS PubMed.
- E. C. Campbell, G. J. Correy, P. D. Mabbitt, A. M. Buckle, N. Tokuriki and C. J. Jackson, Laboratory evolution of protein conformational dynamics, Curr. Opin. Struct. Biol., 2018, 50, 49–57 CrossRef CAS PubMed.
- H. Yu and P. A. Dalby, Exploiting correlated molecular-dynamics networks to counteract enzyme activity-stability trade-off, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(52), E12192–E12200 CrossRef CAS PubMed.
- H. Y. Cui, T. H. J. Stadtmüller, Q. J. Jiang, K. E. Jaeger, U. Schwaneberg and M. D. Davari, How to Engineer Organic Solvent Resistant Enzymes: Insights from Combined Molecular Dynamics and Directed Evolution Study, ChemCatChem, 2020, 12(16), 4073–4083 CrossRef CAS.
- R. Otten, R. A. P. Pádua, H. A. Bunzel, V. Nguyen, W. Pitsawong and M. Patterson, et al., How directed evolution reshapes the energy landscape in an enzyme to boost catalysis, Science, 2020, 370(6523), 1442–1446 CrossRef CAS PubMed.
- M. Corbella, G. P. Pinto and S. C. L. Kamerlin, Loop dynamics and the evolution of enzyme activity, Nat. Rev. Chem., 2023, 7(8), 536–547 CrossRef CAS PubMed.
- Q. Shao, Y. Jiang and Z. J. Yang, EnzyHTP Computational Directed Evolution with Adaptive Resource Allocation, J. Chem. Inf. Model., 2023, 63(17), 5650–5659 CrossRef CAS PubMed.
- V. V. Shende, N. R. Harris, J. N. Sanders, S. A. Newmister, Y. Khatri and M. Movassaghi, et al., Molecular Dynamics Simulations Guide Chimeragenesis and Engineered Control of Chemoselectivity in Diketopiperazine Dimerases, Angew. Chem. Int. Ed. Engl., 2023, 62(20), e202210254 CrossRef CAS PubMed.
- P. Wang, J. Zhang, S. Zhang, D. Lu and Y. Zhu, Using High-Throughput Molecular Dynamics Simulation to Enhance the Computational Design of Kemp Elimination Enzymes, J. Chem. Inf. Model., 2023, 63(4), 1323–1337 CrossRef CAS PubMed.
- R. M. Crean, M. Corbella, A. R. Calixto, A. C. Hengge and S. C. L. Kamerlin, Sequence - dynamics - function relationships in protein tyrosine phosphatases, QRB Discovery, 2024, 5, e4 CrossRef PubMed.
- A. Mukherjee and S. Roy, Understanding the Directed Evolution of a Natural-like Efficient Artificial Metalloenzyme, J. Phys. Chem. B, 2024, 128(49), 12122–12132 CrossRef CAS PubMed.
- R. Sun, D. Wu, P. Chen and P. Zheng, Cutting-edge computational approaches in enzyme design and activity enhancement, Biochem. R. Eng. J., 2024, 212, 109510 CrossRef CAS.
- N. A. E. Venanzi, Machine Learning for Protein Engineering Using Molecular Dynamics Simulation Data, PhD thesis, UCL, 2024.
- J. Zhou and M. Huang, Navigating the landscape of enzyme design: from molecular simulations to machine learning, Chem. Soc. Rev., 2024, 53(16), 8202–8239 RSC.
- S. Meng, Z. Li, P. Zhang, Y. Ji and U. Schwaneberg, Capturing intrinsic protein dynamics for explaining beneficial substitutions from protein engineering campaigns, Biotechnol. Adv., 2025, 83, 108660 CrossRef CAS PubMed.
- J. Planas-Iglesias, M. Majerova, D. Pluskal, M. Vasina, J. Damborsky and Z. Prokop, et al., Automated Engineering Protein Dynamics via Loop Grafting: Improving Renilla Luciferase Catalysis, ACS Catal., 2025, 15(4), 3391–3404 CrossRef CAS PubMed.
- T. Zeiske, K. A. Stafford and A. G. Palmer, Thermostability of Enzymes from Molecular Dynamics Simulations, J. Chem. Theory Comput., 2016, 12(6), 2489–2492 CrossRef CAS PubMed.
- Y. Gao, B. Wang, S. Hu, T. Zhu and J. Z. H. Zhang, An efficient method to predict protein thermostability in alanine mutation, Phys. Chem. Chem. Phys., 2022, 24, 29629 RSC.
- L. Liu, L. Cai, Y. Chu and M. Zhang, Thermostability mechanisms of β-agarase by analyzing its structure through molecular dynamics simulation, AMB Exp., 2022, 12, 50 CrossRef CAS PubMed.
- Z. Dou, Y. Sun, X. Jiang, X. Wu, Y. Li and B. Gong, et al., Data-driven strategies for the computational design of enzyme thermal stability: trends, perspectives, and prospects, Acta Biochim. Biophys. Sin., 2023, 55(3), 343–355 CrossRef CAS PubMed.
- Z. A. Rollins, T. Widatalla, A. C. Cheng and E. Metwally, AbMelt: Learning antibody thermostability from molecular dynamics, Biophys. J., 2024, 123, 2921–2933 CrossRef CAS PubMed.
- Y. Sang, X. Huang, H. Li, T. Hong, M. Zheng and Z. Li, et al., Improving the thermostability of Pseudoalteromonas porphyrae κ-carrageenase by rational design and MD simulation, AMB Exp., 2024, 14, 8 CrossRef CAS PubMed.
- K. T. Korbeld and M. J. L. J. Fürst, Enriching stabilizing mutations through automated analysis of molecular dynamics simulations using BoostMut, Prot. Sci., 2025, 34, e70334 CrossRef CAS PubMed.
- F. Peccati, C. M. Segovia, R. Núñez-Franco and G. Jiménez-Osés, Computation of Protein Thermostability and Epistasis, WIRES, 2025, 15, e70045 CAS.
- N. Zheng, Y. Cai, Z. Zhang, H. Zhou, Y. Deng and S. Du, et al., Tailoring industrial enzymes for thermostability and activity evolution by the machine learning-based iCASE strategy, Nat. Commun., 2025, 16(1), 604 CrossRef CAS PubMed.
- M. Audagnotto, W. Czechtizky, L. De Maria, H. Käck, G. Papoian and L. Tornberg, et al., Machine learning/molecular dynamic protein structure prediction approach to investigate the protein conformational ensemble, Sci. Rep., 2022, 12, 10018 CrossRef CAS PubMed.
- S. Kaptan and I. Vattulainen, Machine learning in the analysis of biomolecular simulations, Adv. Phys. X, 2022, 7, 2006080 Search PubMed.
- R. Nassar, E. Brini, S. Parui, C. Liu, G. L. Dignon and K. A. Dill, Accelerating Protein Folding Molecular Dynamics Using Inter-Residue Distances from Machine Learning Servers, J. Chem. Theor. Comput., 2022, 18, 1929–1935 CrossRef CAS PubMed.
- M. Majewski, A. Perez, P. Tholke, S. Doerr, N. E. Charron and T. Giorgino, et al., Machine learning coarse-grained potentials of protein thermodynamics, Nat. Commun., 2023, 14(1), 5739 CrossRef CAS PubMed.
- L.-E. Zheng, S. Barethiya, E. Nordquist and J. Chen, Machine Learning Generation of Dynamic Protein Conformational Ensembles, Molecules, 2023, 28, 4047 CrossRef CAS PubMed.
- E. Prašnikar, M. Ljubič, A. Perdih and J. Borišek, Machine learning heralding a new development phase in molecular dynamics simulations, Artif. Intell. Rev., 2024, 57, 102 CrossRef.
- A. Son, W. Kim, J. Park, W. Lee, Y. Lee and S. Choi, et al., Utilizing Molecular Dynamics Simulations, Machine Learning, Cryo-EM, and NMR Spectroscopy to Predict and Validate Protein Dynamics, Int. J. Mol. Sci., 2024, 25, 9725 CrossRef CAS PubMed.
- T. Wang, X. He, M. Li, Y. Li, R. Bi and Y. Wang, et al., Ab initio characterization of protein molecular dynamics with AI(2)BMD, Nature, 2024, 635(8040), 1019–1027 CrossRef CAS PubMed.
- A. R. Brownless, E. Rheaume, K. M. Kuo, S. C. L. Kamerlin and J. C. Gumbart, Using Machine Learning to Analyze Molecular Dynamics Simulations of Biomolecules, J. Phys. Chem. B, 2025, 129(22), 5375–5385 CrossRef CAS PubMed.
- G. Janson and M. Feig, Generation of protein dynamics by machine learning, Curr. Opin. Struct. Biol., 2025, 93, 103115 CrossRef CAS PubMed.
- B. J. Wittmann, K. E. Johnston, Z. Wu and F. H. Arnold, Advances in machine learning for directed evolution, Curr. Opin. Struct. Biol., 2021, 69, 11–18 CrossRef CAS PubMed.
- G. Cybenko, Approximation by Superpositions of a Sigmoidal Function, Math. Control Sign. Syst., 1989, 2(4), 303–314 CrossRef.
- K. Funahashi, On the Approximate Realization of Continuous-Mappings by Neural Networks, Neural Netw., 1989, 2(3), 183–192 CrossRef.
- K. Hornik, M. Stinchcombe and H. White, Multilayer feedforward networks are universal approximators, Neural Netw., 1989, 2(5), 359–366 CrossRef.
- K. Hornik, Approximation Capabilities of Multilayer Feedforward Networks, Neural Netw., 1991, 4(2), 251–257 CrossRef.
- A. E. Barron, Universal approximation bounds for superpositions of a sigmoidal function, IEEE Trans. Inf. Theory, 1993, 39, 930–945 Search PubMed.
- J. Park and I. W. Sandberg, Universal approximation using radial-basis-function networks, Neural Comput., 1991, 3(2), 246–257 CrossRef CAS PubMed.
- J. Park and I. W. Sandberg, Approximation and Radial-Basis-Function Networks, Neural Comput., 1993, 5(2), 305–316 CrossRef.
- Y. Liao, S. C. Fang and H. L. W. Nuttle, Relaxed conditions for radial-basis function networks to be universal approximators, Neural Netw., 2003, 16(7), 1019–1028 CrossRef PubMed.
- C. Yun, S. Bhojanapalli, A. Singh Rawat, S. J. Reddi and S. Kumar, Are Transformers universal approximators of sequence-to-sequence functions?, arXiv, 2019, 2019:1912.10077 DOI:10.48550/arXiv.1912.10077.
- S. Luo, S. Li, S. Zheng, T.-Y. Liu, L. Wang and D. He, Your Transformer May Not be as Powerful as You Expect, arXiv, 2022, 2022:2205.13401 DOI:10.48550/arXiv.2205.13401.
- T. Kajitsuka and I. Sato, Are Transformers with One Layer Self-Attention Using Low-Rank Weight Matrices Universal Approximators?, arXiv, 2023, 2023:2307.14023 DOI:10.48550/arXiv.2307.14023.
- A. Petrov, P. H. S. Torr and A. Bibi, Prompting a Pretrained Transformer Can Be a Universal Approximator, arXiv, 2024, 2024:2402.14753 DOI:10.48550/arXiv.2402.14753.
- E. Gumaan, Universal Approximation Theorem for a Single-Layer Transformer, arXiv, 2025, 2025:2507.10581 DOI:10.48550/arXiv.2507.10581.
- J. Kaplan, S. McCandlish, T. Henighan, T. B. Brown, B. Chess, R. Child, et al., Scaling Laws for Neural Language Models, arXiv, 2020:2001.08361 DOI:10.48550/arXiv.2001.08361.
- M. A. Gordon, K. Duh and J. Kaplan, Data and Parameter Scaling Laws for Neural Machine Translation, in Proc 2021 Conf Empirical Methods in Natural Language Processing, 2021, pp. 5915–5922 Search PubMed.
- Z. Chen, S. Wang, Z. Tan, X. Fu, Z. Lei, P. Wang, et al., A Survey of Scaling in Large Language Model Reasoning, arXiv, 2025, 2025:2504.02181.
- M. Li, S. Kudugunta and L. Zettlemoyer, (Mis)Fitting: A Survey of Scaling Laws, arXiv, 2025, 2025:2502.18969 DOI:10.48550/arXiv.2502.18969.
- J. Hoffmann, S. Borgeaud, A. Mensch, E. Buchatskaya, T. Cai, E. Rutherford, et al., Training Compute-Optimal Large Language Models, arXiv, 2022, 2022:2203.15556 DOI:10.48550/arXiv.2203.15556.
- T. Pearce and J. Song, Reconciling Kaplan and Chinchilla Scaling Laws, arXiv, 2024, 2024:2406.12907 DOI:10.48550/arXiv.2406.12907.
- Y. Bahri, E. Dyer, J. Kaplan, J. Lee and U. Sharma, Explaining neural scaling laws, Proc. Natl. Acad. Sci. U. S. A., 2024, 121(27), e2311878121 CrossRef CAS PubMed.
- M. Hutson, The language machines, Nature, 2021, 591(7848), 22–25 CrossRef CAS PubMed.
- G. Vernet, M. Hobisch and S. Kara, Process intensification in oxidative biocatalysis, Curr. Opin. Green Sus. Chem., 2022, 38, 100692 CAS.
- B. O. Burek, A. W. H. Dawood, F. Hollmann, A. Liese and D. Holtmann, Process Intensification as Game Changer in Enzyme Catalysis, Front. Catal., 2022, 2, 858706 CrossRef.
- J. Britton, S. Majumdar and G. A. Weiss, Continuous flow biocatalysis, Chem. Soc. Rev., 2018, 47, 5891–5918 RSC.
- S. Patti, I. M. Alunno, S. Pedroni, S. Riva, E. E. Ferrandi and D. Monti, Advances and Challenges in the Development of Immobilized Enzymes for Batch and Flow Biocatalyzed Processes, ChemSusChem, 2024, 18, e202402007 CrossRef PubMed.
- R. A. Rocha, R. E. Speight and C. Scott, Engineering Enzyme Properties for Improved Biocatalytic Processes in Batch and Continuous Flow. Org Proc, Res Dev, 2022, 26, 1914–1924 CAS.
- L.-E. Meyer, M. Hobisch and S. Kara, Process intensification in continuous flow biocatalysis by up and downstream processing strategies, Curr. Opin. Biotechnol, 2022, 78, 102835 CrossRef CAS PubMed.
- Y. Chalopin, The physical origin of rate promoting vibrations in enzymes revealed by structural rigidity, Sci. Rep., 2020, 10(1), 17465 CrossRef CAS PubMed.
- S. R. Miller, An appraisal of the enzyme stability-activity trade-off, Evolution, 2017, 71(7), 1876–1887 CrossRef CAS PubMed.
- G. Feller, Protein stability and enzyme activity at extreme biological temperatures, J. Phys.: Condens. Matter, 2010, 22(32), 323101 CrossRef PubMed.
- N. Tokuriki, C. J. Jackson, L. Afriat-Jurnou, K. T. Wyganowski, R. Tang and D. S. Tawfik, Diminishing returns and tradeoffs constrain the laboratory optimization of an enzyme, Nat. Commun., 2012, 3, 1257 CrossRef PubMed.
- R. A. Studer, P. A. Christin, M. A. Williams and C. A. Orengo, Stability-activity tradeoffs constrain the adaptive evolution of RubisCO, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(6), 2223–2228 CrossRef CAS PubMed.
- R. Kurahashi, S. I. Tanaka and K. Takano, Activity-stability trade-off in random mutant proteins, J. Biosci. Bioeng., 2019, 128(4), 405–409 CrossRef CAS PubMed.
- V. L. Arcus and A. J. Mulholland, Temperature, Dynamics, and Enzyme-Catalyzed Reaction Rates, Annu. Rev. Biophys., 2020, 49, 163–180 CrossRef CAS PubMed.
- M. Teufl, C. U. Zajc and M. W. Traxlmayr, Engineering Strategies to Overcome the Stability-Function Trade-Off in Proteins, ACS Synth. Biol., 2022, 11(3), 1030–1039 CrossRef CAS PubMed.
- Q. Hou, M. Rooman and F. Pucci, Enzyme Stability-Activity Trade-Off: New Insights from Protein Stability Weaknesses and Evolutionary Conservation, J. Chem. Theory Comput., 2023, 19(12), 3664–3671 CrossRef CAS PubMed.
- J. R. Klesmith, J. P. Bacik, E. E. Wrenbeck, R. Michalczyk and T. A. Whitehead, Trade-offs between enzyme fitness and solubility illuminated by deep mutational scanning, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(9), 2265–2270 CrossRef CAS PubMed.
- J. Knowles, ParEGO: A hybrid algorithm with on-line landscape approximation for expensive multiobjective optimization problems, IEEE Trans Evol Comput, 2006, 10(1), 50–66 Search PubMed.
- E. Zitzler, M. Laumanns and L. Thiele, SPEA2: Improving the Strength Pareto Evolutionary Algorithm for multiobjective optimization, in EUROGEN 2001, Evolutionary Methods for Design, Optimization and Control with Applications to Industrial Problems, ed K. Giannakoglou, 2001, pp. 95–100 Search PubMed.
- C. A. Coello Coello, D. A. van Veldhuizen and G. B. Lamont, Evolutionary algorithms for solving multi-objective problems, Kluwer Academic Publishers, New York, 2002 Search PubMed.
- K. Deb, A. Pratap, S. Agarwal and T. Meyarivan, A fast and elitist multiobjective genetic algorithm: NSGA-II, IEEE Trans. Evol. Comput., 2002, 6(2), 182–197 CrossRef.
- E. Zitzler, L. Thiele, M. Laumanns, C. M. Fonseca and V. G. da Fonseca, Performance assessment of multiobjective optimizers: An analysis and review, IEEE Trans. Evol. Comput., 2003, 7(2), 117–132 CrossRef.
- M. Kim, T. Hiroyasu, M. Miki and S. Watanabe, SPEA2+: Improving the Performance of the Strength Pareto Evolutionary Algorithm 2, LNCS, 2004, 4242, 742–751 Search PubMed.
- W. Sheng, Y. Liu, X. Meng and T. Zhang, An Improved Strength Pareto Evolutionary Algorithm 2 with application to the optimization of distributed generations, Comput Maths Appl, 2012, 64, 944–955 CrossRef.
- C. Qian, Y. Yu and Z.-H. Zhou, An analysis on recombination in multi-objective evolutionary optimization, Artif. Intell., 2013, 204, 99–119 CrossRef.
- K. Deb and H. Jain, An Evolutionary Many-Objective Optimization Algorithm Using Reference-Point-Based Nondominated Sorting Approach, Part I: Solving Problems With Box Constraints, IEEE Trans. Evol. Comput., 2014, 18(4), 577–601 Search PubMed.
- J. Blank and K. Deb, Pymoo: Multi-Objective Optimization in Python, IEEE Access, 2020, 8, 89497–89509 Search PubMed.
- Z. Wang, Y. Pei and J. Li, A Survey on Search Strategy of Evolutionary Multi-Objective Optimization Algorithms, Appl. Sci., 2023, 13, 4643 CrossRef CAS.
- Y. Xu, H. Zhang, L. Huang, R. Qu and Y. Nojima, A Pareto Front grid guided multi-objective evolutionary algorithm, Appl. Soft Comput., 2023, 136, 110095 CrossRef.
- D.-C. Dang, A. Opris and D. Sudholt, Crossover can guarantee exponential speed-ups in evolutionary multi-objective optimisation, Artif. Intell., 2024, 330 Search PubMed.
- S. Kang, K. Li and R. Wang, A survey on pareto front learning for multi-objective optimization, J. Membr. Comput., 2025, 7, 128–134 CrossRef.
- J. Z. Salazar, D. Hadka, P. Reed, H. Seada and K. Deb, Diagnostic benchmarking of many-objective evolutionary algorithms for real-world problems, Eng. Opt., 2025, 57, 287–308 CrossRef.
- M. Garza-Fabre, S. M. Kandathil, J. Handl, J. Knowles and S. C. Lovell, Generating, Maintaining, and Exploiting Diversity in a Memetic Algorithm for Protein Structure Prediction, Evol. Comput., 2016, 24(4), 577–607 CrossRef PubMed.
- J. D. Knowles and D. W. Corne, M-PAES: A memetic algorithm for multiobjective optimization, Proc 2000 Congr Evol Computation, Vols 1 and 2, 2000, pp. 325–332 Search PubMed.
- N. Krasnogor, A. Aragon and J. Pacheco, Memetic Algorithms. Metaheuristic Procedures for Training, Neutral Netw., 2006, 36, 225–248 Search PubMed.
- P. Merz and B. Freisleben, Fitness landscapes and memetic algorithm design, in New ideas in optimization, ed. D. Corne, M. Dorigo and F. Glover, McGraw-Hill, London, 1999, pp. 245–260 Search PubMed.
- P. Moscato, Memetic algorithms: a short introduction, in New Ideas in Optimisation, ed. D. Corne, M. Dorigo and F. Glover, McGraw-Hill, London, 1999, pp. 219–243 Search PubMed.
- Handbook of Memetic Algorithms, ed. F. Neri, C. Cotta and P. Moscato, Springer, Berlin, 2012 Search PubMed.
- F. Neri and C. Cotta, Memetic algorithms and memetic computing optimization: A literature review, Swarm Evolution. Comput., 2012, 2, 1–14 CrossRef.
- D. Varela, V. Karlin and I. Andre, A memetic algorithm enables efficient local and global all-atom protein-protein docking with backbone and side-chain flexibility, Structure, 2022, 30(11), 1550–1558 CrossRef CAS PubMed.
- A. J. Wilson, D. R. Pallavi, M. Ramachandran, S. Chinnasamy and S. Sowmiya, A Review On Memetic Algorithms and Its Developments, Electr. Automat. Eng., 2022, 1, 7–12 Search PubMed.
- W. Zhang and Y. Lan, A Novel Memetic Algorithm Based on Multiparent Evolution and Adaptive Local Search for Large-Scale Global Optimization, Comput. Intell. Neurosci., 2022, 2022, 3558385 Search PubMed.
- Z. Jakšić, S. Devi, O. Jakšić and K. Guha, A Comprehensive Review of Bio-Inspired Optimization Algorithms Including Applications in Microelectronics and Nanophotonics, Biomimetics., 2023, 8, 278 CrossRef PubMed.
- K. G. Reddy and D. Mishra, Advances in Feature Selection Using Memetic Algorithms: A Comprehensive Review, WiRES, 2025, 15, e70026 Search PubMed.
- 662–678.
- S. Gault, P. M. Higgins, C. S. Cockell and K. Gillies, A meta-analysis of the activity, stability, and mutational characteristics of temperature-adapted enzymes, Biosci. Rep., 2021, 41(4) CrossRef CAS PubMed.
- B. T. Porebski, P. J. Conroy, N. Drinkwater, P. Schofield, R. Vazquez-Lombardi and M. R. Hunter, et al., Circumventing the stability-function trade-off in an engineered FN3 domain, Protein Eng., Des. Sel., 2016, 29(11), 541–550 CrossRef CAS PubMed.
- K. S. Siddiqui, Defying the activity-stability trade-off in enzymes: taking advantage of entropy to enhance activity and thermostability, Crit. Rev. Biotechnol., 2017, 37(3), 309–322 CrossRef PubMed.
- S. Akanuma, M. Bessho, H. Kimura, R. Furukawa, S. I. Yokobori and A. Yamagishi, Establishment of mesophilic-like catalytic properties in a thermophilic enzyme without affecting its thermal stability, Sci. Rep., 2019, 9(1), 9346 CrossRef PubMed.
- M. A. Siddiq and J. W. Thornton, Fitness effects but no temperature-mediated balancing selection at the polymorphic Adh gene of Drosophila melanogaster, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(43), 21634–21640 CrossRef CAS PubMed.
- R. Furukawa, W. Toma, K. Yamazaki and S. Akanuma, Ancestral sequence reconstruction produces thermally stable enzymes with mesophilic enzyme-like catalytic properties, Sci. Rep., 2020, 10(1), 15493 CrossRef CAS PubMed.
- S. D. Stimple, M. D. Smith and P. M. Tessier, Directed evolution methods for overcoming trade-offs between protein activity and stability, AIChE J., 2020, 66(3) CrossRef CAS PubMed.
- M. L. Romero, H. Garcia Seisdedos and B. Ibarra-Molero, Active site center redesign increases protein stability preserving catalysis in thioredoxin, Protein Sci., 2022, 31(9), e4417 CrossRef CAS PubMed.
- K. Nguyen and C. Lee, Catalytic His-loop flexibility drives high activity in hyperthermophilic esterase EstE1 while preserving structural stability, Microbiol. Spectrosc., 2025, 13(10), e0100325 CrossRef PubMed.
- S. E. Calvo, K. R. Clauser and V. K. Mootha, MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins, Nucleic Acids Res., 2016, 44(D1), D1251–D1257 CrossRef CAS PubMed.
- P. Thagard and K. Verbeurgt, Coherence as constraint satisfaction, Cogn. Sci., 1998, 22(1), 1–24 CrossRef PubMed.
- P. Thagard, Coherence, truth, and the development of scientific knowledge, Philos. Sci., 2007, 74(1), 28–47 CrossRef.
- P. Thagard, Explanatory Coherence. Reasoning: Studies of Human Inference and Its Foundations, 2008, pp. 471–513 Search PubMed.
- The cognitive science of science: explanation, discovery, and conceptual change, ed. P. Thagard, MIT Press, Cambridge, MA, 2012 Search PubMed.
- J. Spidlen, W. Moore, D. Parks, M. Goldberg, K. Blenman and J. S. Cavenaugh, et al., Data File Standard for Flow Cytometry, Version FCS 3.2, Cytometry, A, 2021, 99(1), 100–102 CrossRef PubMed.
- M. Hucka, A. Finney, H. M. Sauro, H. Bolouri, J. C. Doyle and H. Kitano, et al., The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models, Bioinformatics, 2003, 19(4), 524–531 CrossRef CAS PubMed.
- M. Galdzicki, K. P. Clancy, E. Oberortner, M. Pocock, J. Y. Quinn and C. A. Rodriguez, et al., The Synthetic Biology Open Language (SBOL) provides a community standard for communicating designs in synthetic biology, Nat. Biotechnol., 2014, 32(6), 545–550 CrossRef CAS PubMed.
- S. M. Keating, D. Waltemath, M. Konig, F. Zhang, A. Drager and C. Chaouiya, et al., SBML Level 3: an extensible format for the exchange and reuse of biological models, Mol. Syst. Biol., 2020, 16(8), e9110 CrossRef PubMed.
- J. M. Burel, S. Besson, C. Blackburn, M. Carroll, R. K. Ferguson and H. Flynn, et al., Publishing and sharing multi-dimensional image data with OMERO, Mamm. Genome, 2015, 26(9–10), 441–447 CrossRef PubMed.
- J. Range, C. Halupczok, J. Lohmann, N. Swainston, C. Kettner and F. T. Bergmann, et al., EnzymeML-a data exchange format for biocatalysis and enzymology, FEBS J., 2022, 289(19), 5864–5874 CrossRef CAS PubMed.
- Y. Long, F. Abbasinejad, F. Z. Li, P. Reinprecht, B. Wittmann and J. L. Kennemur, et al., Enzyme Engineering Database (EnzEngDB): a platform for sharing and interpreting sequence-function relationships across protein engineering campaigns, Nucleic Acids Res., 2026, 54(D1), D564–D571 CrossRef PubMed.
- D. B. Kell and S. G. Oliver, The metabolome 18 years on: a concept comes of age, Metabolomics, 2016, 12(9), 148 CrossRef PubMed.
- O. Yurekten, T. Payne, N. Tejera, F. X. Amaladoss, C. Martin and M. Williams, et al., MetaboLights: open data repository for metabolomics, Nucleic Acids Res., 2024, 52(D1), D640–D646 CrossRef CAS PubMed.
- M. Sud, E. Fahy, D. Cotter, K. Azam, I. Vadivelu and C. Burant, et al., Metabolomics Workbench: An international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools, Nucleic Acids Res., 2016, 44(D1), D463–D470 CrossRef CAS PubMed.
- A. Smelter and H. N. B. Moseley, A Python library for FAIRer access and deposition to the Metabolomics Workbench Data Repository, Metabolomics, 2018, 14(5), 64 CrossRef PubMed.
- S. Leo, M. R. Crusoe, L. Rodriguez-Navas, R. Sirvent, A. Kanitz and P. De Geest, et al., Recording provenance of workflow runs with RO-Crate, PLoS One, 2024, 19(9), e0309210 CrossRef CAS PubMed.
- B. A. Schäfer, D. Poetz and G. W. Kramer, Documenting Laboratory Workflows Using the Analytical Information Markup Language, J. Assoc. Lab. Autom., 2004, 9, 375–381 CrossRef.
- S. M. Kearnes, M. R. Maser, M. Wleklinski, A. Kast, A. G. Doyle and S. D. Dreher, et al., The Open Reaction Database, J. Am. Chem. Soc., 2021, 143(45), 18820–18826 CrossRef CAS PubMed.
- W. Finnigan, L. J. Hepworth, S. L. Flitsch and N. J. Turner, RetroBioCat as a computer-aided synthesis planning tool for biocatalytic reactions and cascades, Nat. Catal., 2021, 4(2), 98–104 CrossRef CAS PubMed.
- The UniProt Consortium, UniProt: the Universal Protein Knowledgebase in 2025, Nucl. Acids Res., 2025, 63, D609–D617 CrossRef PubMed.
- H. M. Berman and S. K. Burley, Protein Data Bank (PDB): Fifty-three years young and having a transformative impact on science and society, Q. Rev. Biophys., 2025, 58, e9 CrossRef CAS PubMed.
- H. Derouiche, Z. Brahmi and H. Mazeni, Agentic AI Frameworks: Architectures, Protocols, and Design Challenges, arXiv, 2025, 2025:2508.10146 DOI:10.48550/arXiv.2508.10146.
- B. Ni and M. J. Buehler, Agentic End-to-End De Novo Protein Design for Tailored Dynamics Using a Language Diffusion Model, arXiv, 2025, 2025:2502.10173 DOI:10.48550/arXiv.2502.10173.
- Y. Yamada, R. Tjarko Lange, C. Lu, S. Hu, C. Lu, J. Foerster, et al., The AI Scientist-v2: Workshop-Level Automated Scientific Discovery via Agentic Tree Search, arXiv, 2025, 2025:2504.08066 DOI:10.48550/arXiv.2504.08066.
| This journal is © The Royal Society of Chemistry 2026 |