
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Innovative approaches to the synthesis of five-membered heteroaromatics with emerging synthetic methodologies
Yu
Zhang†
 *ab,
Qiannan
Li†
ab,
Zhimin
Hu†
c,
Zhiyong
Leng
c,
Wei-dong
Zhang
*bc and
Shoubhik
Das
*d
*ab,
Qiannan
Li†
ab,
Zhimin
Hu†
c,
Zhiyong
Leng
c,
Wei-dong
Zhang
*bc and
Shoubhik
Das
*d
aShanghai Frontiers Science Center for Chinese Medicine Chemical Biology, Institute of Interdisciplinary Integrative Medicine Research, Shanghai University of Traditional Chinese Medicine, No. 1200, Cailun Road, Shanghai 201203, China. E-mail: yzhang@shutcm.edu.cn
bState Key Laboratory of Antiviral Drugs, Pingyuan Laboratory, Henan Normal University, Xinxiang, Henan 453007, China. E-mail: wdzhangy@hotmail.com
cSchool of Pharmacy, Second Military Medical University, Shanghai 200433, China
dDepartment of Chemistry, University of Bayreuth, Bayreuth 95447, Germany. E-mail: Shoubhik.Das@uni-bayreuth.de
First published on 18th February 2026
Abstract
According to statistics, more than 70% of drugs that are available on the market contain heterocyclic units. Among them, five-membered heteroaromatics bearing nitrogen, oxygen, and sulfur have served as the most important structural motifs found in diverse pharmaceuticals. Emerging toolboxes, such as photocatalysis, biocatalysis, electrocatalysis and skeletal editing have showcased unprecedented avenues to access them. In fact, >300 publications on the topic of synthesis of five-membered heteroaromatics by using emerging toolboxes have been recorded in the last ten years, clearly indicating the significance of this field. In this study, a conceptual overview and discussions are presented to showcase the advancement of this field. Moreover, the related drugs and bioactive compounds obtained by these approaches are highlighted.
 Yu Zhang | Yu Zhang received his MSc degree in Medicinal Chemistry from Shanghai Jiao Tong University in 2017. He obtained his PhD degree in 2020 from Georg-August-Universität Göttingen under the supervision of Prof. Dr Das and Prof. Dr Koszinowski. He then conducted postdoctoral research at the University of Antwerp with Prof. Das. In 2022, he joined Shanghai University of Traditional Chinese Medicine, where he is currently a professor. His research interests include green chemistry, medicinal chemistry, and medicinal chemistry of natural products. He has published over 50 papers in leading chemistry journals and holds five granted patents. |
 Qiannan Li | Qiannan Li received her MSc degree from Inner Mongolia University of Technology in 2022 under the supervision of Prof. Yaqi Wang. She is currently pursuing her PhD through a joint program between the Naval Medical University and Henan Normal University in the group of Prof. Weidong Zhang, with an expected completion in 2027. Her research interests focus on organic synthesis and medicinal chemistry. |
 Zhimin Hu | Zhimin Hu received her BSc and MSc degrees from the School of Pharmaceutical Sciences at Peking University in 2017 and 2019, respectively. She obtained her PhD in Traditional Chinese Pharmacy at the China Academy of Chinese Medical Sciences (2022). Since 2022, she began her research career in the research group of Prof. Wei-dong Zhang at the School of Pharmacy at Naval Medical University. Her research interests focus on the biosynthesis and chemo-enzymatic synthesis of bioactive natural products. |
 Zhiyong Leng | Zhi-Yong Leng received his Bachelor's degree from Suzhou University of Technology in 2024 under the guidance of Prof. Yi-Qing Zhou. He is currently pursuing his MS degree under Prof. Wei-Dong Zhang in a joint program between the Naval Medical University and Zhejiang Medical University. His research interests focus on medicinal chemical synthesis and chemical biology. |
 Wei-dong Zhang | Prof. Wei-Dong Zhang obtained his Bachelor's and Master's degrees in Natural Medicinal Chemistry from the Second Military Medical University in 1988 and 1991, respectively. He received his PhD in Natural Medicinal Chemistry from the Shanghai Institute of Pharmaceutical Industry (1998), under the supervision of Professor HuiTing Li. He is currently a Professor at the Shanghai University of Traditional Chinese Medicine and the Second Military Medical University. His research mainly focuses on Chinese medicine formulas, isolation, structural identification and modification, total synthesis, and structure–activity relationships in bioactive natural products. |
 Shoubhik Das | Prof. Shoubhik Das did his PhD under the guidance of Prof. Matthias Beller at LIKAT, and following this, he did postdoctoral research with Prof. Matthew Gaunt at the University of Cambridge, UK, and with Prof. Paul Dyson at the EPFL in Switzerland. He started his independent research career (habilitation) at the University of Göttingen in 2015, and after 4 years, he moved to the University of Antwerp as a tenure-track professor. Since August 2023, he has been a Chair Professor at the Department of Organic Chemistry at the University of Bayreuth, Germany. His current research interests include the development of homogeneous and heterogeneous photo-/electrocatalysts and their applications in organic synthesis as well as fuel-related molecules. |
1. Introduction
Five-membered heteroaromatics bearing nitrogen, oxygen, and sulfur (including indole, pyrazole, imidazole, benzimidazole, triazole, thiazole, indazole, tetrazole, isoxazole, and thiadiazole) have emerged as highly important structural motifs in a wide range of pharmaceuticals and natural products.1–3 The most obvious evidence is that >130 drugs containing five-membered heteroaromatics have been developed in 2013–2023, indicating their significance in the modern drug discovery.4,5 The marketed pharmaceuticals include the following: pimobendan, a first-line therapy for canine chronic heart failure in veterinary cardiology; dronedarone, used for the clinical treatment of specific types of cardiac arrhythmias; raloxifene, a selective estrogen receptor modulator that improves bone mineral density while lowering the risk of certain breast cancers; and azomycin, an agent used against anaerobic bacterial infections, amoebic dysentery, and vaginal trichomoniasis. Leveraging advanced synthetic toolkits enables the efficient assembly of key intermediates for these approved drugs, thereby streamlining and expanding their synthetic accessibility.Owing to the remarkable bioactivity of five-membered heterocycles (Fig. 1), substantial efforts have been devoted toward the development of new strategies for their synthesis.7–9 It should be noted that traditional methods have contributed tremendously to diversify the synthesis of five-membered heterocycles through thermal conditions10 and transition-metal catalysis.11–13 However, there remains a pressing need to design more efficient and convenient methods to obtain these valuable compounds and expand their structural diversity. In the last decades, emerging toolboxes such as photocatalysis, biocatalysis, electrocatalysis and skeletal editing have attracted great attention due to their numerous advantages.14–17 Photocatalysis compresses traditional multi-step synthesis into single-step light-triggered cascade reactions, providing an effective tool for constructing drug molecular skeletons under mild conditions.18 Electrocatalysis may offer high selectivity and sustainability by tuning electrode potentials to control reaction pathways, avoiding chemical oxidants or reductants and reducing byproducts.19 Biocatalysis excels in stereoselectivity and environmental benignity, enabling the efficient construction of complex five-membered aromatic heterocycles via non-natural substrates or cascade reactions.20 In this aspect, skeletal editing is an advanced method for the diversification and transformation of known active compounds via the direct modification of the core structure.21 In addition, these approaches have showcased unprecedented avenues to access five-membered heteroaromatics. Indeed, >300 recent publications on the synthesis of five-membered heteroaromatics by using emerging toolboxes have been recorded in the last ten years, which strongly indicates the significance and rapid growth of this field.22–25 Additionally, the application of these innovative tools has successfully advanced the efficient preparation of bioactive molecules and lead compounds in drug discovery.26–28
 | ||
| Fig. 1 Classification of different five-membered heteroaromatics and synthetic applications into drug molecules through emerging toolboxes. | ||
Considering the significance of this research field and our deep interest in the synthesis of heterocycles through photochemical methods,29,30 we aim to provide a critical review to mention the challenges and recent advancements in the synthesis of five-membered heteroaromatics and related drugs/bioactive molecules. This article will cover the recent advances in the application of photochemical synthesis, electrocatalysis, biocatalysis and skeletal editing to assemble five-membered heteroaromatics containing nitrogen, oxygen, sulfur, and other heteroatoms. The synthetic applications and mechanistic studies will be clearly overviewed.
2. Overview of the synthesis of five-membered heteroaromatics through emerging toolboxes
We categorized all the reported methods based on the source of heteroatoms. In the synthesis of nitrogen-containing aromatic heterocycles, a wide range of nitrogen sources such as primary amines, tertiary amines, azides and nitriles furnished the corresponding products via all the emerging strategies (Fig. 2a).31–42 Among them, tertiary amines were always suitable nitrogen sources to construct five-membered heteroaromatics via the photochemical or biocatalysis approach.43,44 Moreover, diazo compounds have been used for the synthesis of pyrazoles or triazoles in photo/electrochemical synthesis.45,46 Furthermore, nitro-compounds and aziridines have been proved as feasible nitrogen sources in photocatalysis.47,48 In addition, molecular editing has become an elegant strategy to construct five-membered heteroaromatics.49 | ||
| Fig. 2 Conceptual overview of synthesizing five-membered heteroaromatics via an emerging toolbox: (a) overall strategies for N-containing heteroarenes; (b) overall strategies for O-containing heteroarenes; (c) overall strategies for S-containing heteroarenes; (d) overall strategies for N and O-containing heteroarenes; (e) overall strategies for N and S-containing heteroarenes; (f) atom-editing synthesis of five-membered heteroarenes. | ||
For the synthesis of oxygen-containing aromatic heterocycles, phenols, alcohols, ketone derivatives and ethers have been well-established to synthesize furans and benzofurans through emerging toolboxes (Fig. 2b).50–52 In addition, spiro-isochromene derivatives have been employed into the benzofuran synthesis via a photochemical approach.53 Regarding the synthesis of sulfur/seleno-containing aromatic heterocycles, various thioethers, benzenethiol or disulfides have served as sulfur sources to obtain desirable products (Fig. 2c).54–56 Other sulfur/seleno sources such as NH4SCN/KSeCN have also been reported for the construction of benzoselenophenes and benzothiophenes.57
By designing different starting materials bearing N- and O-sources in the same molecule such as oximes, O-methyloximes, propargyl amides, phenolic amidines, and 2-aminophenols, the structural diversity of five-membered heteroarenes has been achieved (Fig. 2d).58–61 To further improve the synthetic efficiency, intermolecular synthesis from separate nitrogen and oxygen sources such as by using carboxylic acids and benzyl amines as staring materials has also been developed.62,63 Furthermore, the synthesis of N- and O-containing five-membered heteroarenes has also been developed by using starting materials bearing N and O sources such as α-azidochalcones and diazo compounds.64,65 In addition, the intermolecular synthesis by using separate nitrogen sources and sulfur sources has been reported.66–68
Five-membered heteroaromatics containing N and S have been synthesized as well by using emerging toolboxes, and structural diversity has been achieved through the rational designing of various N- and S-bearing precursors, including aminothiophenols,69 mercaptobenzonitriles,70 and thioamide derivatives,71 as well as intermolecular interactions between thiocyanic acid and diverse amine-containing compounds (Fig. 2e).72,73 The utilization of unsaturated five-membered heterocycles as starting materials has also been documented.74 Notably, innovative approaches by employing thiocyanic acid as a dual-purpose reagent to supply both N and S sources have been successfully developed for constructing high-value five-membered heteroaromatics.75
At last, skeletal editing is characterized by its ability to construct five-membered heteroaromatics via direct, atomic-level precision, eliminating the requirement of multi-step conventional synthesis.76 We also provide a concise overview for the progress of this field (Fig. 2f). It is clear that skeletal editing provided new insight for the synthesis of five-membered aromatic heterocycles via elegant atom deletion, atom transfer and atom insertion. Moreover, this capability also makes it a powerful platform for late-stage functionalization and discovering new heterocyclic compounds, which will be discussed in detail in the following sections.
3. One-nitrogen-containing aromatic heterocycles
3.1 Synthesis of pyrroles
Pyrrole, a fundamental five-membered heteroaromatic characterized by a conjugated π-electron system, occupies a central role in medicinal chemistry.77,78 Within the pharmaceutical industry, these compounds are extensively employed in the development of diverse therapeutic agents due to their multifaceted biological activities including potent antitumor efficacy via microtubule dynamics disruption, broad-spectrum antimicrobial action, and robust anti-inflammatory/analgesic properties.79 Traditional methods for synthesizing pyrrole molecules involve condensation reactions between carbonyl compounds and amines, such as the Hantzsch pyrrole synthesis, Barton–Zard pyrrole synthesis, and Paal–Knorr reaction.80–82 This section systematically explores the cutting-edge methodologies for the synthesis of pyrrole and its derivatives with a strong emphasis on innovative strategies.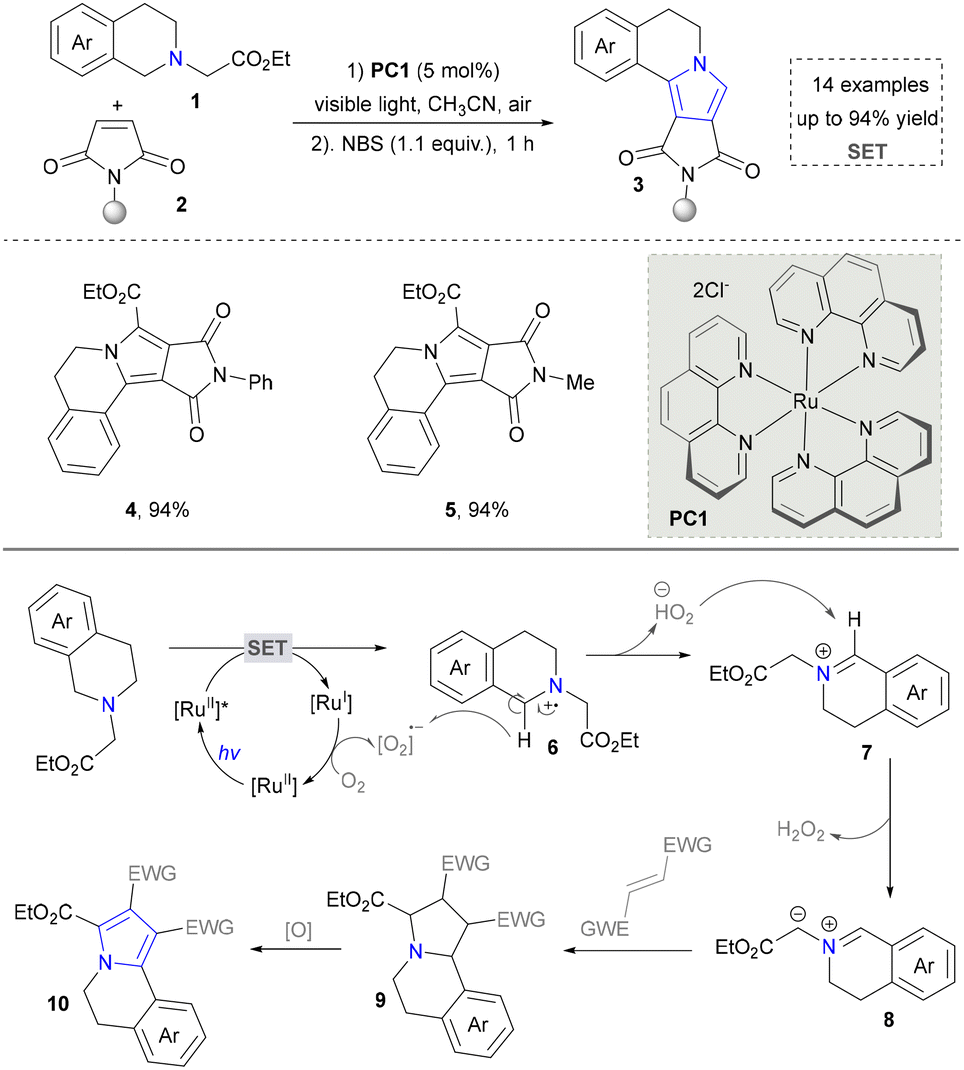 | ||
| Scheme 1 Visible-light-induced oxidation/[3+2] cycloaddition/oxidative arylation sequence to obtain pyrroles. | ||
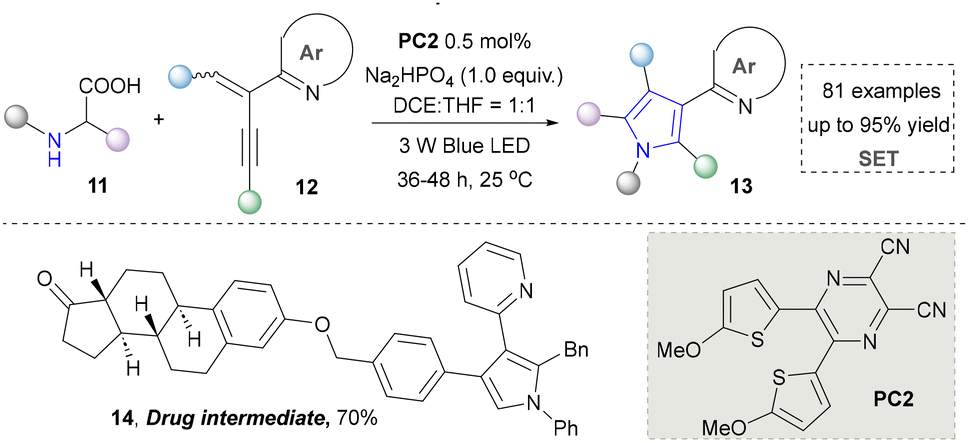
Taking advantage of the high reactivity of free radicals, a photocatalytic strategy for the construction of highly complex pyrroles was reported by Wu's group.86 In this case, α-bromoketone was activated to form the carbon radical via a single electron transfer (SET) process, which further underwent the radical addition into the enamine to afford the valuable pyrroles. Compared with the traditional Hantzsch reaction, this strategy is characterized by completion within merely 2 hours under blue-light irradiation at room temperature. Later, a similar strategy of using N-tosylhydrazones as radical precursors via iodine-mediated photo-catalysis under visible light irradiation was also reported by Adimurthy's group.87 In addition, the utilization of amino acids as nitrogen sources and the radical precursors was further developed by Jiang's group in 2021 (Scheme 2).88 This method featured easy availability of raw materials, high synthetic efficiency and good functional group tolerance. Diverse pyrroles with tri- and tetra-substituents at the carbon positions were obtained successfully in good to excellent yields through redox-neutral radical additions, cyclisation and aerobic oxidative arylation transformations. Moreover, a drug intermediate (14) was successfully obtained under standard conditions, demonstrating the utility of this method for constructing pyrrole scaffolds in bioactive molecules. Nevertheless, this approach remains constrained by its dependence on N-protected α-amino acids. Additionally, the number of substrates amenable for late-stage functionalization remained limited.
 | ||
| Scheme 2 Assembly of the azaarene-substituted pyrroles based on radical construction. | ||
In 2023, the same group extended this conceptual framework by employing TsOH as an additive to generate an electron donor–acceptor (EDA) complex with enones under visible light irradiation, thereby initiating a cascade transformation. This strategy subsequently enabled the conversion of α-amino acids into polysubstituted indole derivatives, achieving remarkable enhancement in the structural diversity of accessible indole architectures through controlled functionalization patterns.89
In an effort to further expand the structural diversity of pyrrole, a methodology for achieving highly substituted pyrroles using primary amines as the nitrogen source has been developed by Salles’ group in 2021 (Scheme 3).33 In this approach, a three-component reaction using various primary amines, carboxylic acids and ketones as synthetic synthons tremendously improved the structural diversity of pyrroles (19, 20). Moreover, the target product was obtained under solvent-free conditions, which also showed good sustainability. Although this method advanced the synthesis of highly substituted pyrroles, it required a long reaction time (72 h). In addition, the radical pathways in the proposed mechanism still need to be further verified, especially regarding the intermediate formation and conversion process.
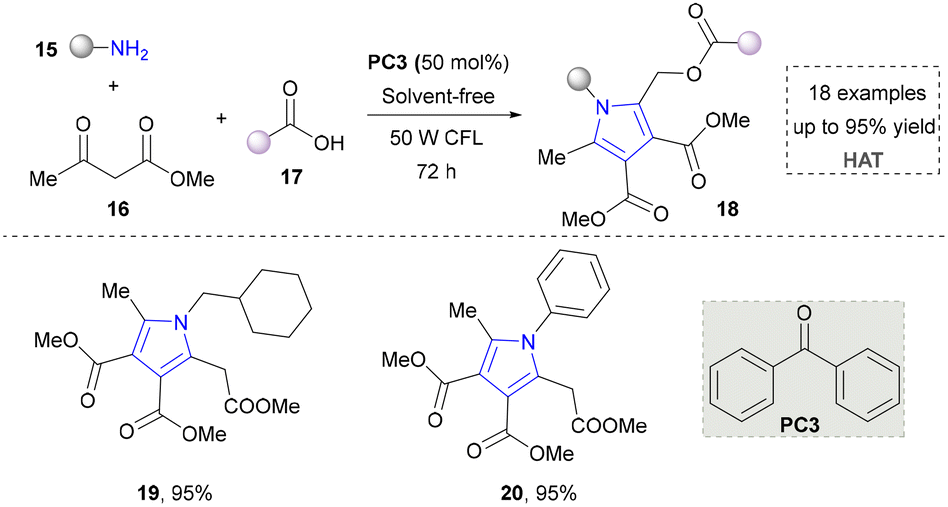 | ||
| Scheme 3 Three-component reaction for complex pyrrole synthesis. | ||
To further extend the synthesis of pyrroles through multi-component reactions, several groups persistently made significant contributions. In 2022, Tabassum's group reported a one-pot synthetic methodology for novel 6,7-bisubstituted 1H-pyrroles, employing substituted benzyl bromides, barbituric acid/Meldrum's acid, and aromatic amines under the irradiation of visible light, with fluorescein serving as the photocatalyst.90 At the same year, Maheswari's group established a another three-component synthetic strategy for the synthesis of 1,2,3,4-tetrasubstituted pyrroles at ambient temperature and under photoinduced conditions, demonstrating exceptional atom economy and operational simplicity.91 Most recently, the Heydari group devised a four-component photocatalytic assembly for functionalized pyrroles via the integration of amines, aldehydes, 1,3-dicarbonyl compounds, and nitromethane, which further diversified the synthesis of pyrroles through photochemical synthesis.92
In contrast to the most studies that focus on conventional pyrrole synthesis via the ring assembly, Park's group innovatively developed a molecular-editing strategy in 2024, achieving a furan-to-pyrrole conversion through single-atom exchange (Scheme 4).93 This strategy maintained the molecular framework while altering the heteroatom type, offering a precise protocol to investigate the impact of heteroatoms on compound properties. It also broadens the raw material sources for pyrrole synthesis, providing a new route to utilize abundant furan compounds (23, 24). Moreover, various furan derivatives and diverse nitrogen nucleophiles, including aliphatic amines, aromatic amines, and ammonia surrogates participated successfully in the system. Notably, this method achieved the late-stage functionalization of complex natural products and drug molecules containing a furan motif, efficiently converting them into pyrrole analogues under standard conditions. The substrate in this system tolerated ester and amide groups, enabling rapid derivatization of chiral precursors such as L-phemethy ester 25 and the anti-influenza drug (Tamiflu) 26. The mechanism involved the single-electron oxidation of furan by the excited state of a photocatalyst, generating a furanic cation radical. This was followed by nucleophilic addition of an amine, C–O bond homolysis, electron transfer, and proton transfer, ultimately leading to the construction of pyrroles, which was distinct from traditional pyrrole synthesis methods.
 | ||
| Scheme 4 A strategy for converting furan into pyrrole through single-atom exchange. | ||
Besides using amines/enamines as nitrogen sources, azides have emerged as a crucial alternative reagent for pyrrole synthesis due to their unique denitrification ability, photosensitising activity, and versatile intermediate generation properties.94 In 2012, Yoon and co-workers found a photocatalytic method to activate aryl and vinyl azides directly, providing a convenient method for constructing pyrroles (Scheme 5, 36–39).95 The mechanism confirmed that the vinyl azides were activated via the energy transfer to form the triplet nitrene 42. This was followed by the cyclisation of the nitrogen compartment to produce a 2H-azepane intermediate, which was subsequently rearranged to form the pyrrole. It is well known that vinyl azides decompose into nitrile or 2H-azopropylline under heat and ultraviolet light and are widely used in the synthesis of various N-heterocyclic rings.96,97
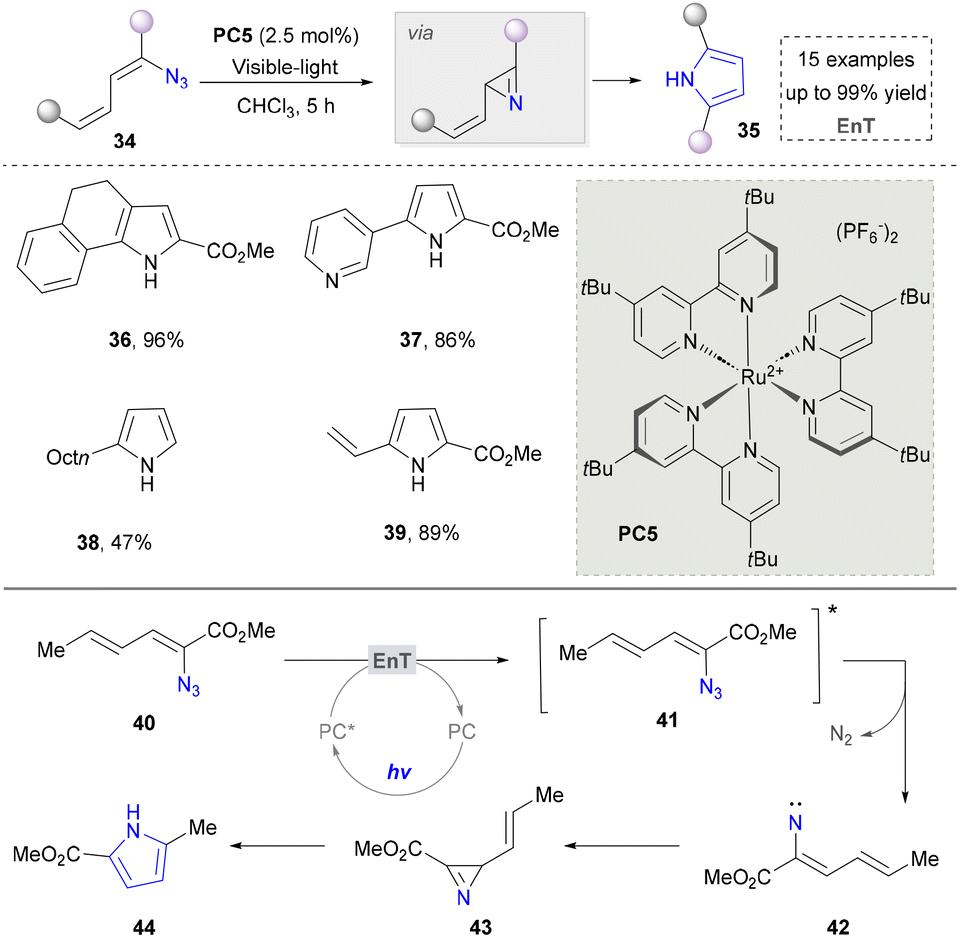 | ||
| Scheme 5 The synthesis of pyrroles via visible light activation of aryl and vinyl azides. | ||
Building on Yoon's foundational work, other groups have also explored azides as 2H-aziridine precursors for the synthesis of polysubstituted pyrroles. In 2019, the Maurya group reported a visible-light-driven protocol by utilizing α-azidochalcones coupled with 1- or 2-naphthols or 2-hydroxy-1,4-naphthoquinones in the presence of a ruthenium-based photocatalyst, achieving the construction of highly functionalized 2,3-fused pyrroles through a radical-mediated strategy.98 In 2020, they further developed a catalyst-free strategy employing α-keto-vinyl azides to access structurally diverse pyrroles. This methodology demonstrated broad applicability to vinyl azides bearing a conjugated amino group.99
In contrast to conventional intramolecular strategies by employing azides for pyrrole synthesis, the Bandini group pioneered an intermolecular approach involving the photocatalytic condensation of aryl azides with aldehydes. (Scheme 6).100 This methodology enabled the assembly of pyrrole scaffolds through cross-component coupling, thereby achieving a remarkable expansion in the structural diversity of pyrrole architectures compared to traditional ring-closing paradigms. Given that azides served as both nitrogen sources and oxidants, the addition of extra oxidants was not required. Moreover, this strategy was compatible with aryl, alkyl aldehydes, and multi-substituted azides, delivering corresponding pyrrole derivatives in good yields (up to 78%) under mild conditions (48–55).
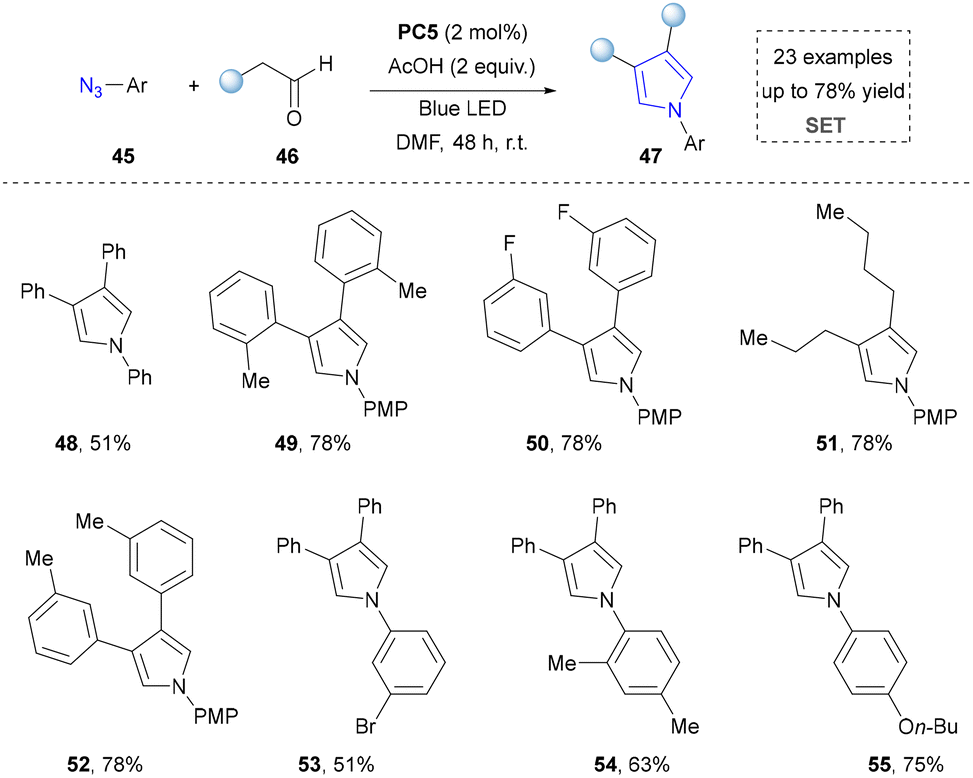 | ||
| Scheme 6 Synthesis of 1,3,4-trisubstituted pyrroles from aryl azides. | ||
Diverging from strategies by utilizing azides as precursors to 2H-aziridines, the direct employment of 2H-aziridines in pyrrole synthesis has been successfully established. In 2014, Xiao's group reported a photocatalytic strategy by employing 2H-aziridines and alkynes as starting materials to construct substituted pyrroles (59–61). Moreover, the utility of this approach was demonstrated by the efficient synthesis of pharmaceutical agents, HMG-CoA reductase inhibitor (65), showcasing its potential in drug synthesis. This strategy also featured the combination of energy transfer and redox neutral reactions, thereby facilitating the [3+2] cycloaddition of 2H-aziridine and alkyne and obtaining the desired products (Scheme 7).101 The proposed mechanism showed that the one-electron oxidation reaction of 2H-aziridine was feasible in the presence of the excited state of the photocatalyst. Then, 69 underwent a radical addition reaction with activated alkyne group to form intermediate 71, which subsequently underwent oxidation/intramolecular cyclization/aromatization to complete the reaction. This work accomplished the first metal-free photocascade catalysis that synergistically merges energy transfer and redox pathways, thereby affording novel tetra-substituted pyrroles. Based on this work, several research groups continuously worked on the photocatalytic synthesis of pyrroles by using 2H-azirines as direct building blocks. In 2019, Rastogi's group introduced nitro alkenes as new reaction partners, expanding the reaction types and product diversity of 2H-azirines.102 The same group also developed a redox-neutral 1,3-dipolar cycloaddition reaction to construct tetrasubstituted pyrroles.103 Furthermore, in 2022, Xia's team developed a single-step synthesis of pyrroles via visible-light-induced [3+2] aerobic oxidative cyclization of quinones and 2H-azirines.104
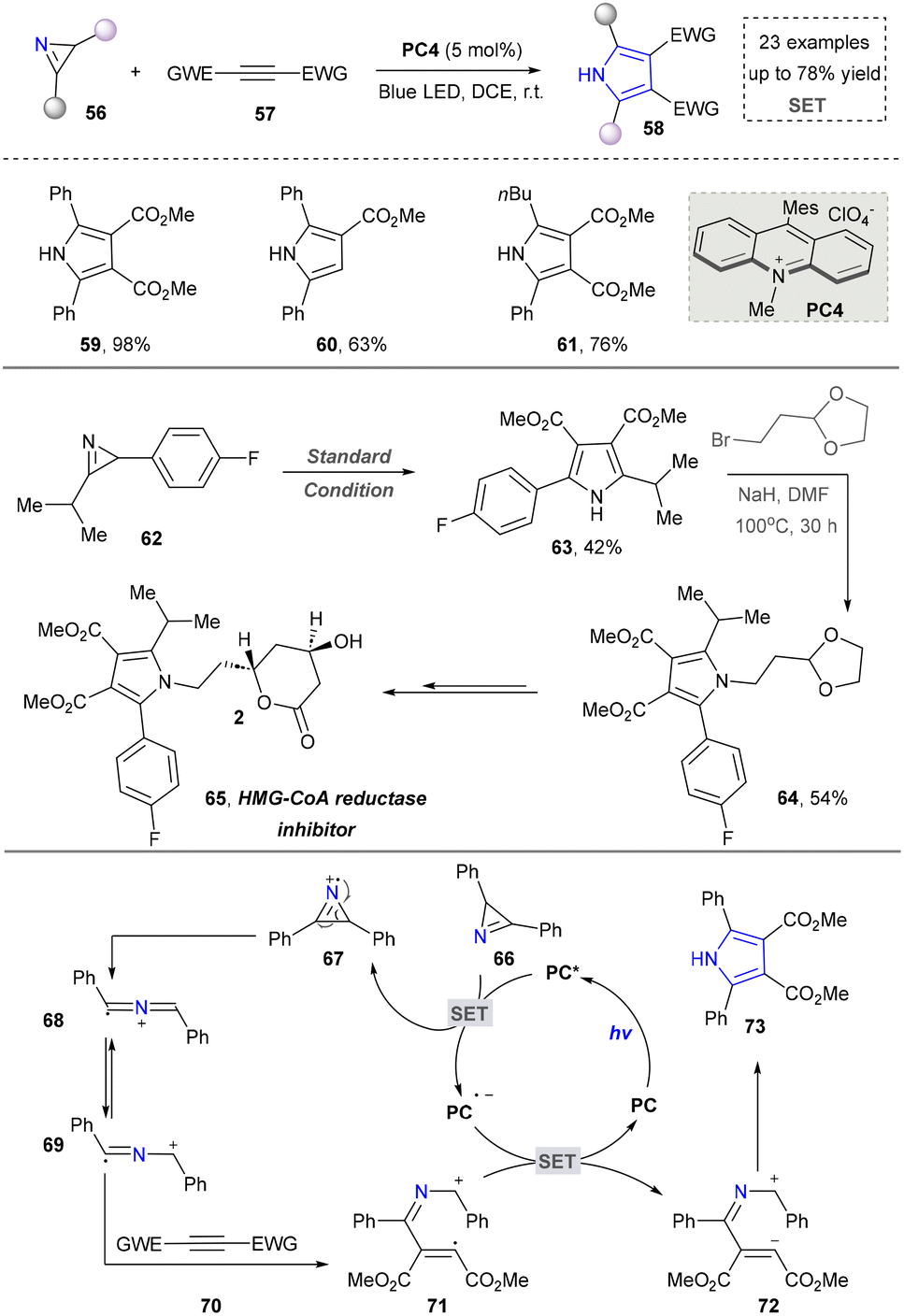 | ||
| Scheme 7 Visible-light-induced [3+2] cycloaddition for the synthesis of pyrroles. | ||
Overall, using divergent nitrogen sources, including amines, enamines, amino acids, azides and 2H-azirines, to construct pyrroles has been successfully achieved through different photochemical syntheses. Moreover, photocatalytic molecular editing has been a potential method to construct unprecedent pyrroles from other heteroarenes. However, despite these obvious advancements, there are only a few drugs and bioactive compounds that have been synthesized using these emerging approaches. A major constraint lies in the limited compatibility of sensitive functional groups, commonly present in active pharmaceutical ingredients, under photochemical conditions. Therefore, translating these strategies into practical routes for pharmaceuticals and natural product synthesis represents a compelling and underexplored frontier, holding considerable potential for future development.
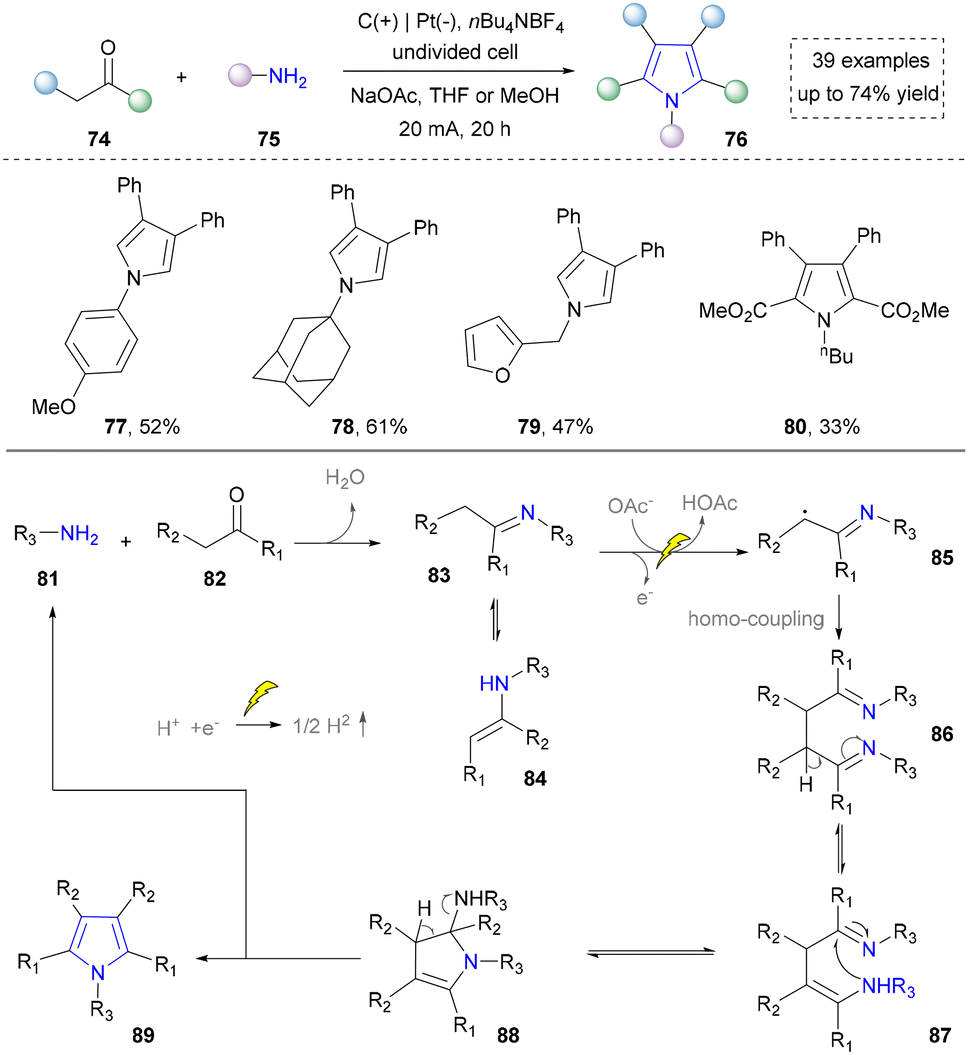 | ||
| Scheme 8 Electrochemical oxidative annulation of amines and aldehydes/ketones for the synthesis of pyrroles. | ||
Afterwards, primary amines have been consistently proposed as starting materials for the electrochemical synthesis of pyrroles. In 2021, the Chen group reported the use of aniline with dimethyl acetylenedicarboxylate to obtain the final product through consecutive condensation, single-electron-transfer (SET), ring closure and subsequent proton-eliminating aromatisation.106 Subsequently, the Yuan group disclosed that acetyl acetone was activated by iodine to form β-enamino ketone as the key intermediate. This three-component strategy significantly improved the structural diversity of pyrrole and its derivatives.107
Building upon these foundations, the Sarkar group has developed an effective electrochemical strategy for the heterocoupling of aryl- and alkyl-substituted amines to synthesize tetrasubstituted aminopyrroles in 2021 (Scheme 9).31 His approach leveraged trifluoroethanol to finely tune electrochemical parameters, achieving desired chemoselectivity through the oxidative cross-coupling of two structurally distinct enamines to construct NH-pyrroles (98–100). The mechanism involved the following challenging steps: (1) oxidation of the aryl-substituted enamine 91 to generate radical intermediate 92; (2) coupling of 92 with the alkyl-substituted enamine 90 to form radical intermediate 93. Subsequently, cyclization and oxidative aromatization yield the targeted asymmetric pyrrole product 97. This metal-free methodology expands the substrate scope for pyrrole synthesis from systems traditionally dominated by aryl groups to aryl–alkyl hybrid systems, while exhibiting broad functional group tolerance, thereby offering a highly adaptable platform for constructing complex pyrrole building blocks in medicinal chemistry. On the basis of this work, some electrochemical methods have been reported by using different enamines as building blocks, which further diversified the pyrrole synthesis.34,108,109
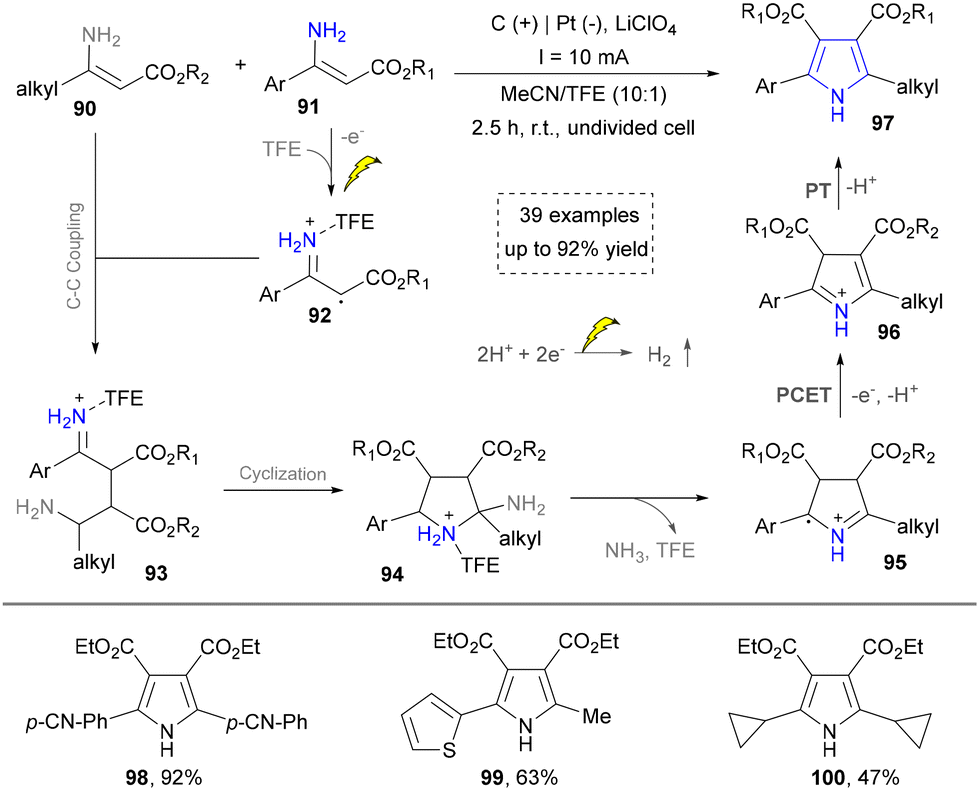 | ||
| Scheme 9 Synthesis of the asymmetrically substituted NH-pyrroles by electrooxidation. | ||
With the ongoing exploration of the nitrogen sources required for pyrrole synthesis, Liu et al. reported an innovative electrochemical ring contraction strategy by utilizing Hantzsch esters in 2021, expanding the synthetic toolbox for pyrrole preparation (Scheme 10).49 This methodology capitalized on the cathodic reduction of pyridine derivatives to generate radical anions 107, which underwent sequential intramolecular Michael addition and ring contraction processes. This transformation efficiently converted Hantzsch ester derivatives into polysubstituted pyrroles through acetate extrusion and rearomatization sequences, demonstrating exceptional atom economy and functional group diversity. In this case, estrone derivatives (103) and imidate (104) remained stable under strong reducing and Lewis acid conditions, indicating its significance in modifying steroid-based pharmaceuticals. Given that this also constitutes an atom-editing strategy through carbon deletion, we have not allocated an independent section for its discussion.
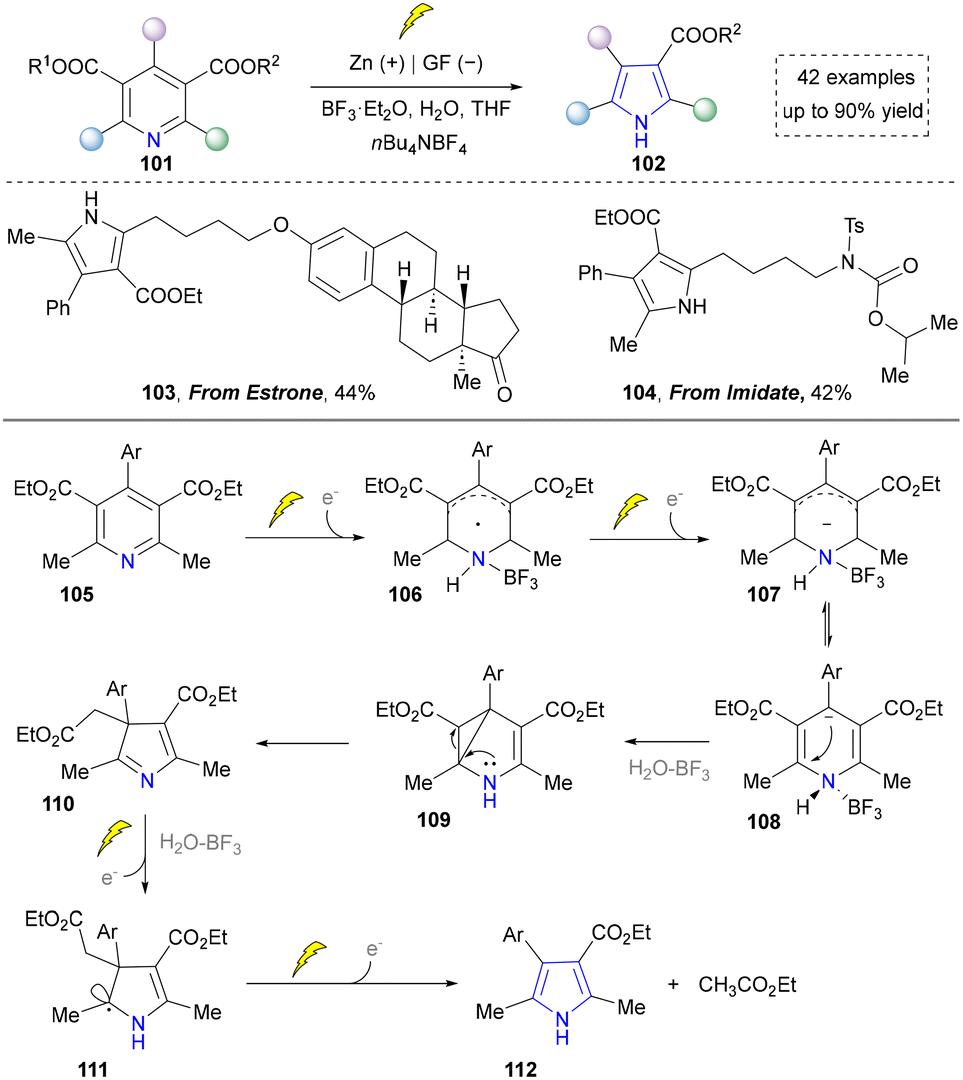 | ||
| Scheme 10 Electrochemical strategy for the generation of pyrroles by ring contraction. | ||
Overall, compared to traditional and photochemical approaches, electrochemical synthesis achieves direct functionalization of redox-sensitive groups, promotes reactions under mild and metal-free conditions, and exhibits superior compatibility and activation capability for low activity, high oxidation potential, or structurally unique alkene or amine precursors. This approach offers a distinct advantage for drug synthesis by avoiding stoichiometric oxidants and metal catalysts, thereby simplifying purification and reducing potential metal residues in active pharmaceutical ingredients. However, a notable limitation remains the challenge of scaling up these processes due to issues such as electrode passivation, limited reactor design for heterogeneous systems, and the need for precise control over potential and current density in complex molecular settings. In addition, while the use of amines and anilines as nitrogen sources in pyrrole synthesis has been extensively reported, the potential for employing other nitrogen sources through electrochemical methods remains largely unexplored, thus presenting ample opportunities for further development.
In 2014, the Lin group established a novel lipase-initiated multicomponent reaction (MCR) catalysed by Candida antarctica lipase B (CALB) for the synthesis of spirooxazino derivatives (117).113 Two rings containing a pyrrole ring were constructed in a single step, in one pot, utilizing commonly available nitrostyrene (113), aldehyde (114), cyclohexanone (115) and acetamide (116) substrates (Scheme 11). Throughout the reaction, CALB catalysed the initial three-step reaction, the aldol condensation of 120 and 121 to form the intermediate (122), the condensation of β-hydroxy ketone (122) and acetamide to afford the intermediate (123), and the hydrolysis of 123 to obtain the enamine intermediate (124). Subsequently, 124 underwent a Michael addition/intramolecular Nef reaction with nitroolefin to obtain the intermediate (125), which was eventually condensed with cyclohexanone to produce the final product spirooxazino compound (126). Building on this work, Wu and co-workers subsequently redesigned the lipase-initiated MCR to synthesize a broader range of spirooxazino derivatives and developed a two-enzymatic cascade MCR for the synthesis of chiral spirooxazinos.114 Lipase from Porcine pancreas type II (PPL) catalysed asymmetric aldol reactions to generate the optically pure chemo-aldol intermediate, which was continuously employed as a starting material of CALB along with other substrates.
 | ||
| Scheme 11 Lipase-initiated synthesis of the spirooxazino derivatives. | ||
Monoamine oxidases (MAOs) are a class of flavin-dependent enzymes that catalyse the oxygen-driven conversion of amines to their corresponding imines.115 The imines are then non-enzymatically hydrolysed to aldehydes, while the flavin adenine dinucleotide (FAD) cofactor is regenerated to oxidative state by molecular oxygen, producing hydrogen peroxide and ammonia formation.116 MAO-N from Aspergillus niger is a highly (S)-selective oxidoreductase, which has widely been used in synthetic chemistry.117,118 6-Hydroxy-D-nicotine oxidase (6-HDNO) from Arthrobacter nicotinovorans, a key enzyme involved in the metabolism of nicotine, is an enantiocomplementary (R)-selective amine oxidase.119
In 2017, the Castagnolo group reported that pyrrolines can be efficiently converted into pyrroles through the aromatization activity of MAO-N or 6-HDNO whole-cell biocatalysts under mild conditions. Among them, the variant MAO-D5 catalysed the aromatization of various N-aryl- and N-alkyl-3-pyrrolines (128) into the corresponding pyrroles (129). Afterwards, a one-pot chemoenzymatic cascade process combining ring-closing metathesis (RCM) reactions with the MAO-N enzymes was established to realize the sustainable production of pyrroles from diallyl-amines/anilines (Scheme 12).43 The MAO-Mannich chemoenzymatic methodology could be applied to synthesize the antitubercular pyrrole (132).120–122 Additionally, the pyrrole precursor (134)112 of the natural alkaloid Myrmicarin 217 (135) was obtained by the RCM-MAO chemoenzymatic cascade from the diene (133).123
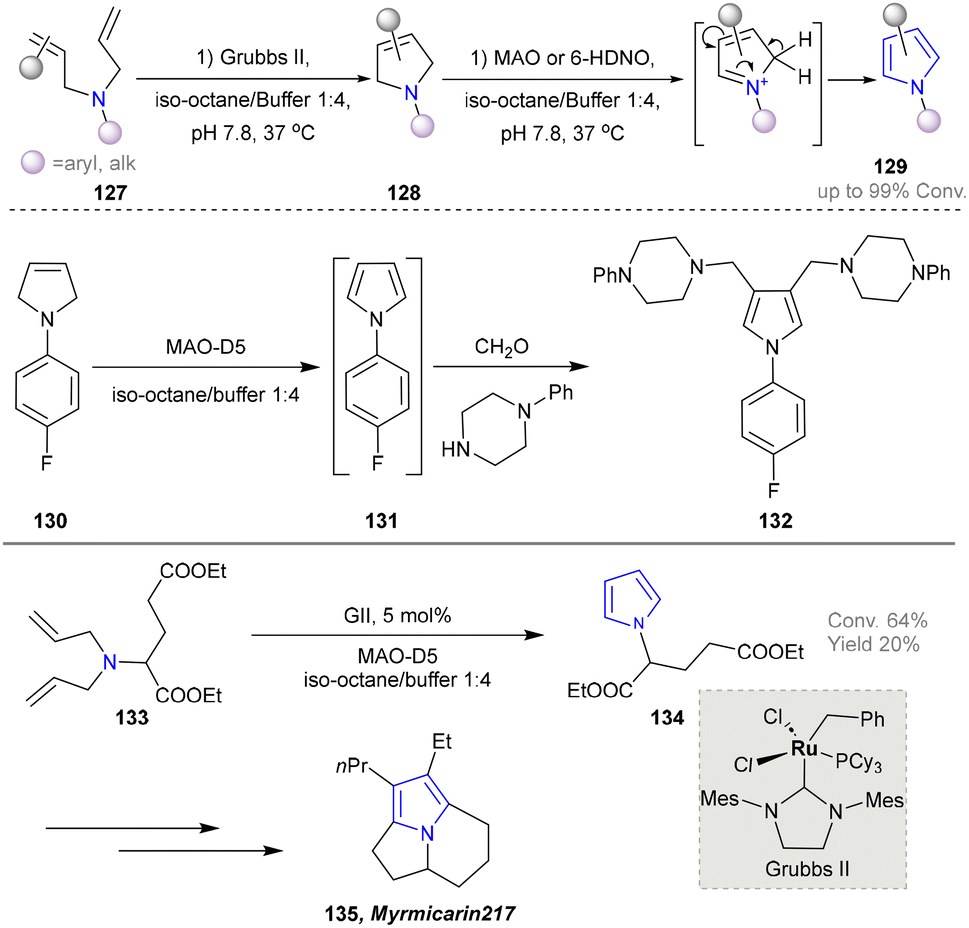 | ||
| Scheme 12 Chemoenzymatic cascade process for the synthesis of pyrroles using MAO-N. | ||
Various ketones and aldehydes can be converted into the corresponding primary amines by amine transaminases (ATAs) as biocatalysts, utilizing the reversible properties of biocatalytic amination, with a β-amino ester (141) functioning as both the substrate and the amine donor.124–126 In 2018, the Turner group designed a self-sufficient strategy for the synthesis of pyrrole without external amine donors (Scheme 13).127 The α-aminoketone (143), generated from α-diketones (140) catalysed by ATAs, condensates with β-ketone ester (142) via the Knorr pyrrole synthesis to afford substituted pyrrole (144). pH modification inhibited the undesired spontaneous oxidative dimerization of α-aminoketone (143) to produce pyrazines.
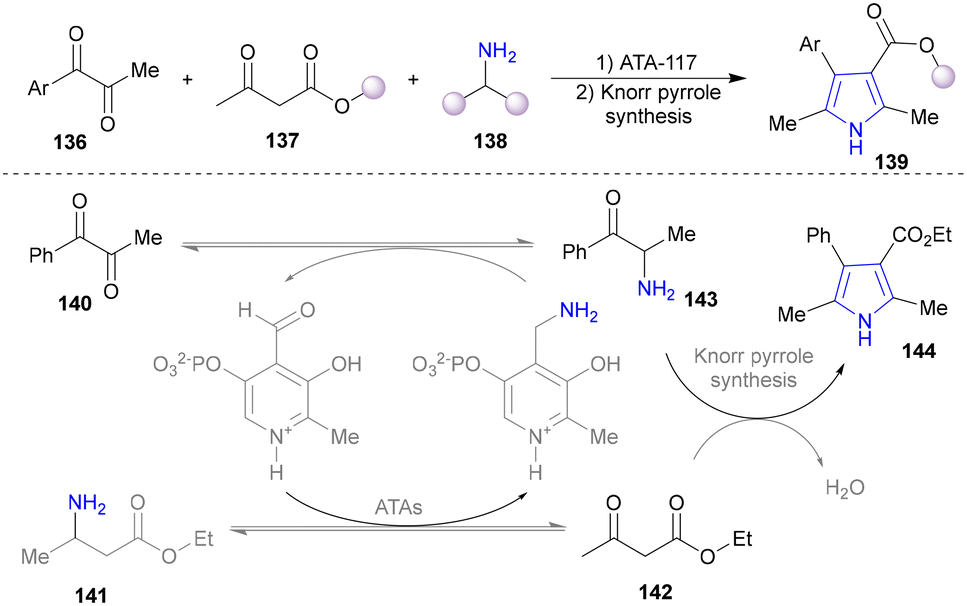 | ||
| Scheme 13 Chemo-enzymatic cascades for the synthesis of pyrroles using ATA biocatalysts. | ||
The biosynthetic pathway of violacein and indolocarbazole natural products has been fully elucidated.128–140 In 2022, Xu's group established a chemoenzymatic pathway for the biomimetic total synthesis of the spiroindimicins (Scheme 14).141 Two key enzymes, namely, tryptophan oxidase LaStaO and chromopyrrolic acid synthetase VioB, responsible for tryptophan dimerization in the biosynthetic pathway of bisindole alkaloids were utilized to catalyse the oxidative dimerization of (5-chloro-)L-tryptophan (145) in one-pot synthesis to obtain the bis-indole precursors (5,5″-dichloro-)chromopyrrolic acid (146), which were subsequently methylated and N′,N″-di-TBS-protected to synthesize 148. Afterwards, two skeletally distinct spiroindimicins with the [5,5] and [5,6] spiro-ring skeleton, including (±)-spiroindimicins D (149) and G (150), and (±)-spiroindimicins H (151) and A (152), were selectively constructed via intramolecular oxidative coupling of C3′-C4″ and C3′-C2″, respectively.
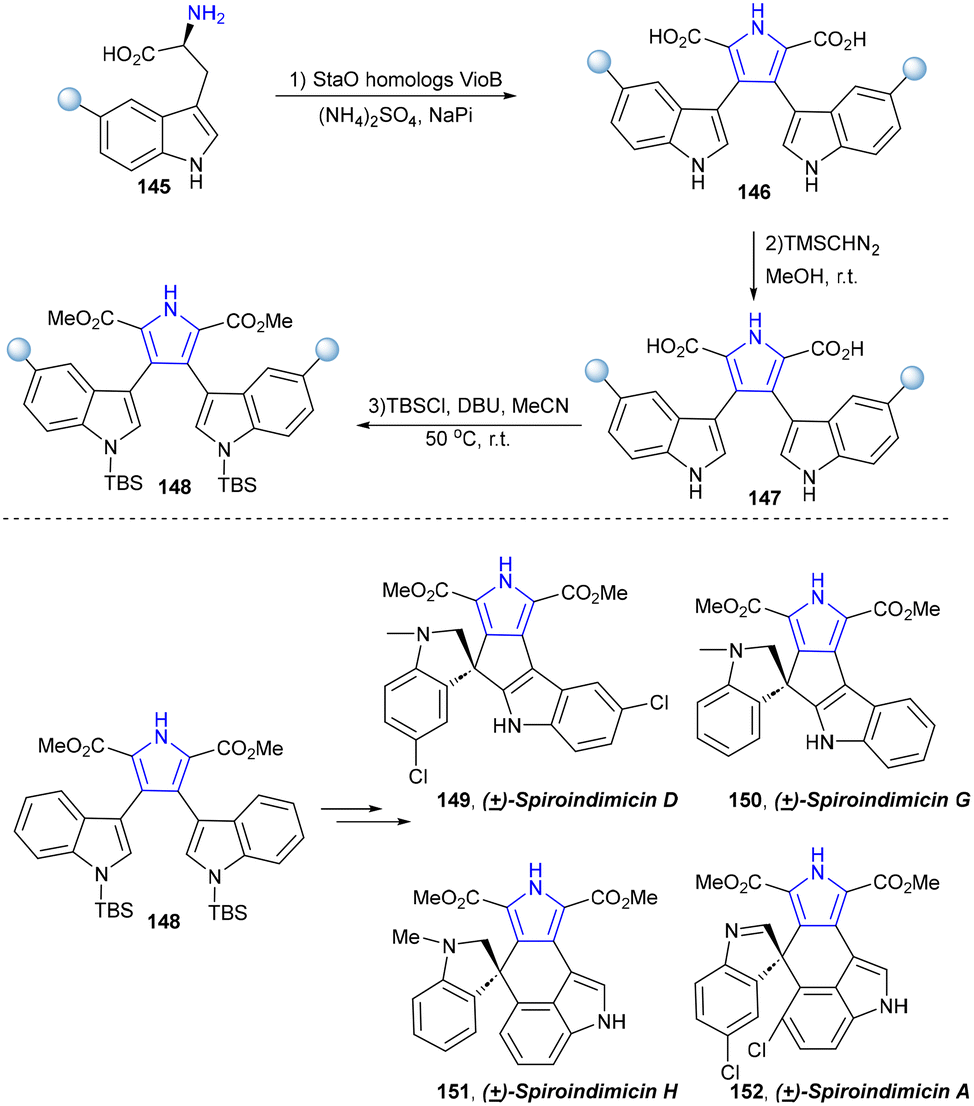 | ||
| Scheme 14 Biomimetic total synthesis of the spiroindimicin family. | ||
In 2023, an environmentally benign strategy has been developed by Wang's group to synthesize pyrrole disulfides (155), a fusion between pyrrole and disulfide functional groups, by porcine pancreatic lipase (PPL) in ethanol at 40 °C from β-ketothioamides (KTAs) 153 and ethyl cyanoacetate (Scheme 15).142 This strategy provided a significant improvement in the synthesis of pyrrole disulfides over the conventional synthesis methods by utilizing malodorous and toxic hydrogen sulfide gas or environmentally unfriendly reaction conditions,143,144 contributing to the advancement of environmentally friendly biocatalysis.
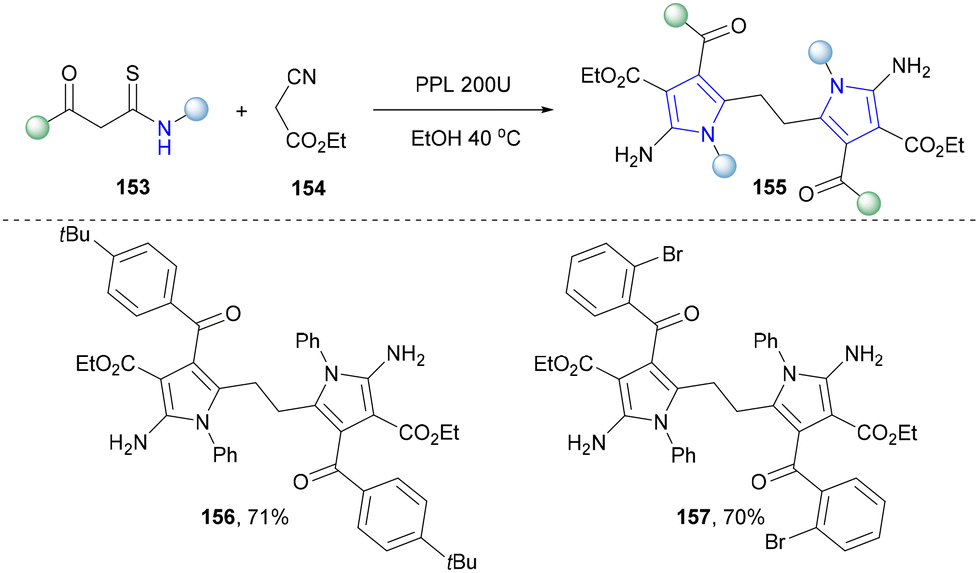 | ||
| Scheme 15 Enzymatic synthesis of pyrrole disulfides by lipase. | ||
Overall, when compared to traditional synthetic methods that may require harsh conditions and metal catalysts, the enzymatic or chemoenzymatic synthesis of pyrroles can offer a more environmentally friendly and efficient alternative. There are also some enzymes catalysing the formation of pyrrole ring involved in the biosynthetic pathway of natural products,145 such as kosinostatin146 and coumermycin A1,147 but they will not be discussed here since they have not yet been applied for enzymatic synthesis. It is expected that they will provide more possibilities for the enzymatic synthesis of pyrroles in the future.
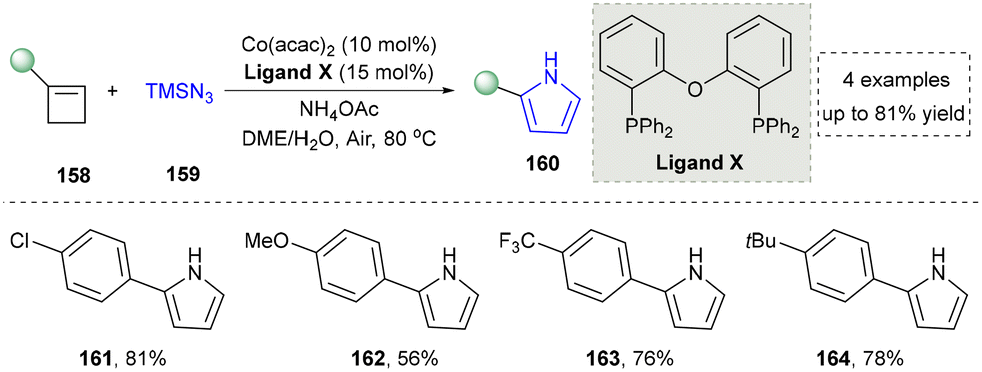 | ||
| Scheme 16 Cobalt-catalyzed nitrogen-atom insertion for the pyrrole formation. | ||
3.2 Synthesis of indoles
Indole represents a pivotal structural motif in organic synthesis,149 enabling diverse functional group transformations through a wide array of chemical reactions.150 Traditional methods include but are not limited to electrophilic halogenation, Friedel–Crafts acylation and alkylation, oxidation and reduction processes, as well as 1,2- and 1,4-addition reactions. The indole ring system is ubiquitously distributed in natural products, constituting one of the most prevalent heterocyclic frameworks in nature.151,152 Globally, >70 synthetic indole-containing drugs have been commercialized.153,154 Notably, the U.S. Food and Drug Administration (FDA) specifically approved 14 indole-derived pharmaceutical agents between 2015 and 2021, underscoring the continued importance of this heterocyclic system in modern drug discovery and development.155 In this section, the synthesis of indoles through emerging approaches will be overviewed carefully.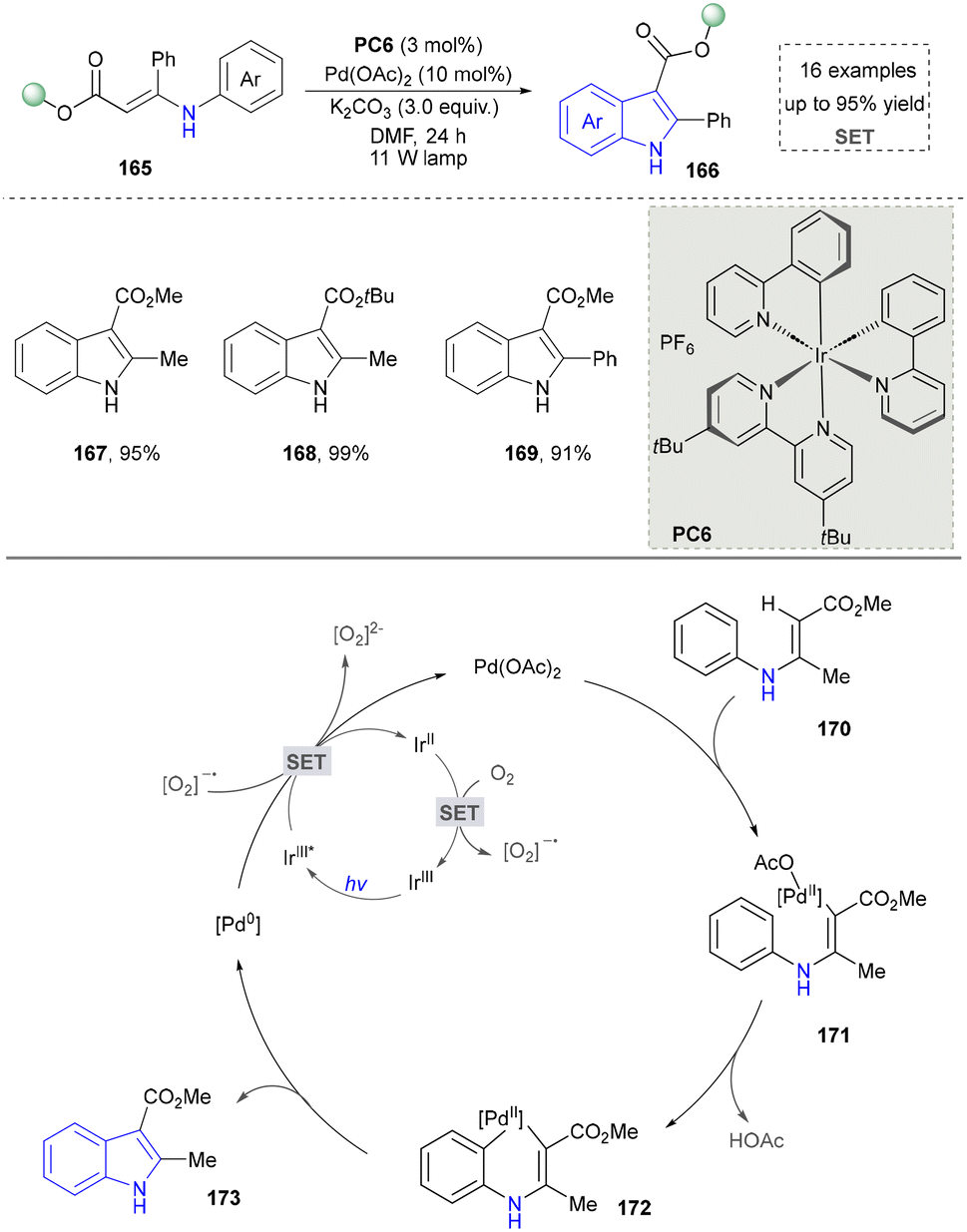 | ||
| Scheme 17 Photoredox synthesis of indoles by the palladium-catalysed C–H activation. | ||
To further improve the photocatalytic synthesis of indoles from 3-phenylamino-2-crotonates as nitrogen sources, the Wu group developed a convenient strategy for the synthesis of indole under visible light irradiation through intramolecular C–C bonds in 2016 (Scheme 18).157 By using catalytic amounts of the photosensitizer Ir(PY) and the catalyst cobalt oxime, various N-arylenamines were highly selectively converted to the corresponding indoles (176–178), with H2 being the only by-product. Compared with the previous methods, this strategy did not require the use of oxidants and additional base, so undesirable by-products can be avoided.
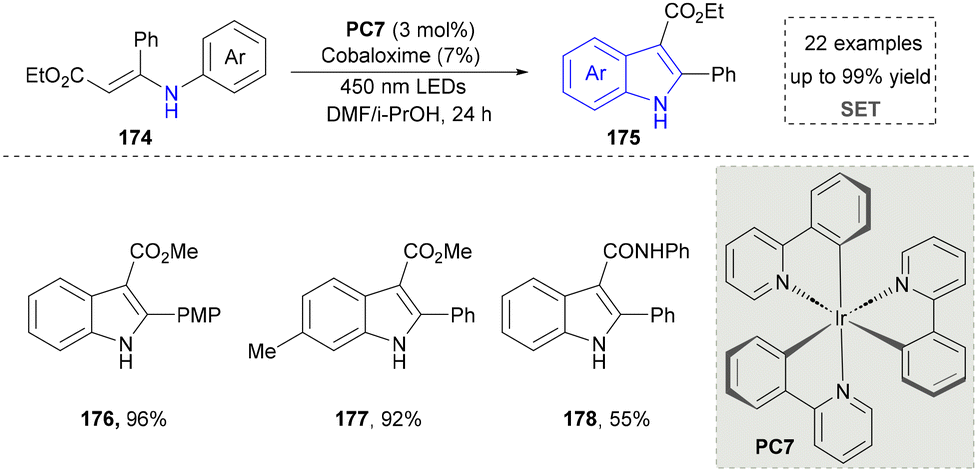 | ||
| Scheme 18 Oxidant-free strategy for the synthesis of indoles under visible light irradiation. | ||
In 2017, the Xiao group presented a light-driven radical-mediated strategy for the in situ generation of azo-o-quinone methyl esters (190) from alkenyl aniline (187) and alkyl radical precursors, followed by alkali treatment of the cyclized products for efficient and convenient synthesis of high-energy indole (Scheme 19).158 This strategy enabled efficient multicomponent reactions of alkenyl anilines, halides and thionyl groups, and showed a wide substrate range and functional group tolerance (183–186). Later, the Wang group found the similar method to afford indoles using radical-mediated strategy.59 In the same year, the Kumar group also reported the synthesis of CF3-containing indoles by using Langlois’ reagent as the radical precursor.159 This work presented a novel and practical radical Smiles rearrangement initiated by a photo-oxidation-catalysed regioselective keto-alkyne amide coupling. In 2018, Li's team developed a visible-light-induced aerobic cross-coupling of glycine derivatives with indoles for efficient BIM synthesis, featuring mild conditions and good functional group tolerance.160
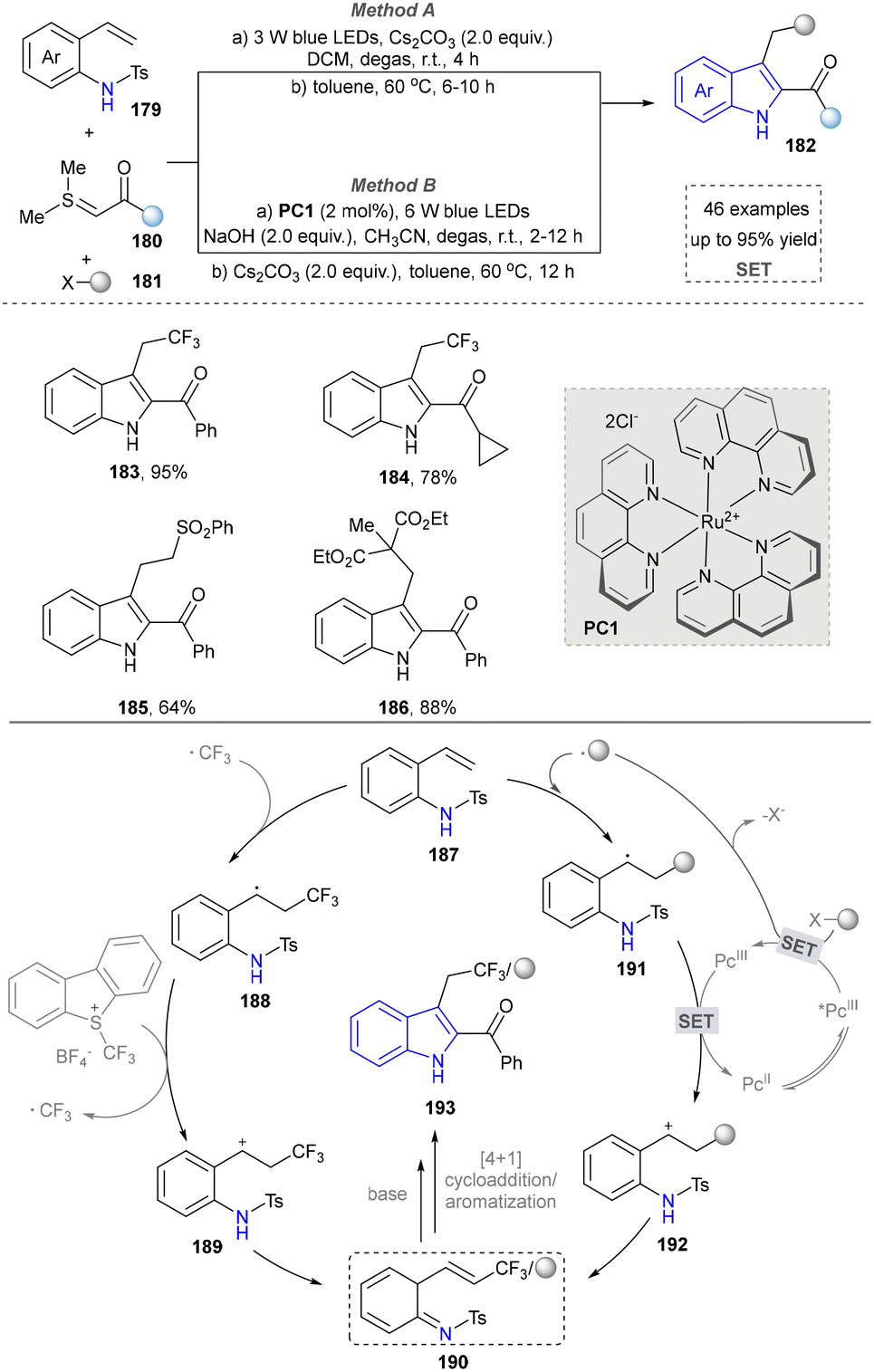 | ||
| Scheme 19 Visible-light-driven aza-ortho-quinone methide generation. | ||
In 2015, Paixão's group pioneered a visible-light-mediated intramolecular reductive cyclization reaction of substituted 2-halobenzenesulfonamides containing terminal alkynes for indole synthesis;161 however, this strategy suffered from a limited substrate scope. Subsequently, Xia's group developed an approach to access indoles via the intramolecular Mannich cyclization of N-vinyl-2-iodoaniline derivatives, which was successfully applied to natural product synthesis.162 In 2020, starting from carbonyl-acetylenamide, the Ye group successfully achieved a light-facilitated Smiles rearrangement based on acetylenamides (Scheme 20) and constructed a series of 2,3-functionalized indoles and 3,4-functionalized isoquinoline heterocyclic compounds (196, 197). The reaction showed the following characteristics: (1) the Smiles rearrangement reaction based on acetylenamide was realized for the first time; (2) two important heterocyclic skeletons of functionalized indoles and isoquinolines were synthesized with high efficiency and selectivity, and several important bioactive molecules were further synthesized by this method (200, 203). This radical Smiles rearrangement was applicable to intermolecular radical tandem reactions based on propiolamides, enabling the efficient construction of heterocyclic frameworks in a single step. The most salient feature of this strategy is its excellent compatibility with the free N–H group of indoles, allowing the product to serve directly as a building block for further functionalization.163 On this basis, the Xia group reported a visible-light-induced, copper-catalyzed oxidative cyclization of substituted ortho-aminophenylpropynes for synthesizing indole derivatives, although the substrate scope was restricted to aryl compounds.164
 | ||
| Scheme 20 Synthesis of the functionalized indoles from the visible light-mediated keto-amide coupling. | ||
In 2022, a photocatalytic method was explored by Shi's group and N-sulfonated aromatic derivatives served as starting materials, N-substituted indole products were generated efficiently through γ-fragmentation under mild conditions without any additives (Scheme 21).165 This strategy also had a wide range of substrate scope and provided a powerful strategy for the synthesis of complex indoles (206–208), which promoted the further development for the synthesis of nitrogen-containing heterocycles under neutral redox conditions.
 | ||
| Scheme 21 Photoinduced synthesis of indoles via intramolecular C–N bond formation. | ||
The proposed mechanism was depicted and the photoexcitation produced the highly oxidizing excited state of PC (Scheme 22). This was reductively quenched by the substrate (209), generating radical cationic species (210). The nitrogen atom in 210 then reacted with the alkene moiety intramolecularly, forming a cyclized radical cationic intermediate (211). A SET process between 211 and the acridine radical yielded zwitterionic intermediate (212) and regenerated an acridinium salt in the closed catalytic loop. The elimination of TsH or TsD from 212 formed another zwitterionic intermediate (213), which underwent aromatization to produce the desired indole product (214). Given the reaction's quantum yield of 1.77, a chain process occurred where 211 oxidized another molecule of 209 to produce 215, ultimately yielding the final product (214) via the same intermediate (213).
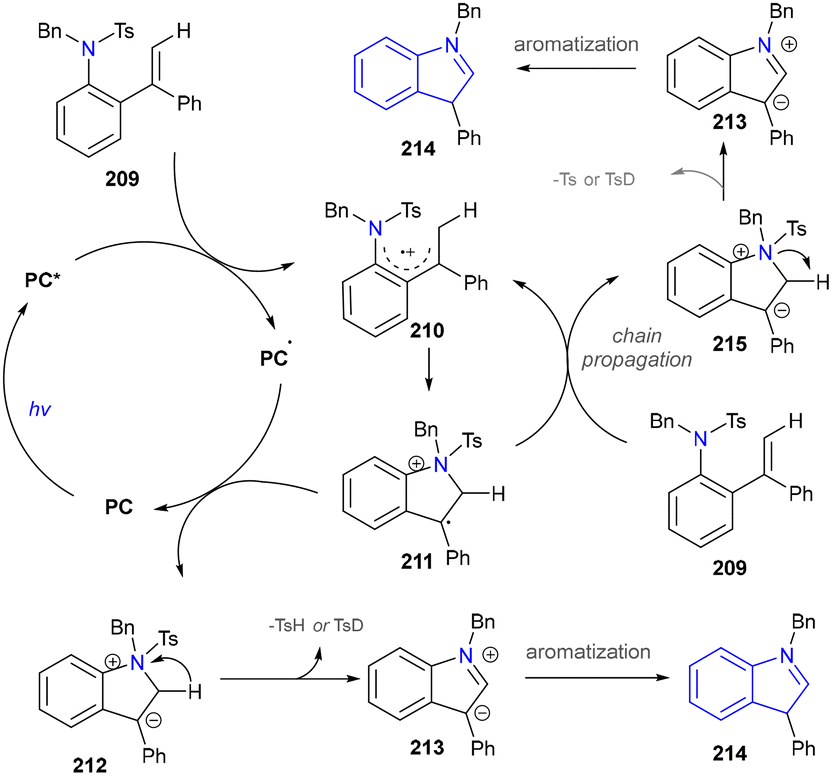 | ||
| Scheme 22 Mechanism of the visible-light-induced indole synthesis through intramolecular C–N bond formation. | ||
Cheap and commercially available nitroarenes have been ideal nitrogen sources for the construction of organic amines and N-containing heterocycles.166 The Shi group developed an environmentally friendly method for the synthesis of indoles by oxidative cleavage of olefins with nitro(hetero)aromatics in 2024 (Scheme 23).167 This strategy exploited the selective polar splitting of 1,3,2-dioxazolidine (223) to form carbonylimine intermediates (225) and N–O–C dipoles (226), and the novel reactivity of these dipoles, including the migration of carbonylimine to o-alkenyl hydroxylamine. Under the standard conditions, derivatives of bioactive molecules, such as L-menthol and S-(+)-ibuprofen (219, 220), were readily converted into indole-containing drug analogues using this photoinduced method, demonstrating the potential of this method in drug-oriented synthesis. This system exhibited notable tolerance toward ester and benzylic groups. However, the scope of late-stage functionalization of drug molecules remained relatively limited.
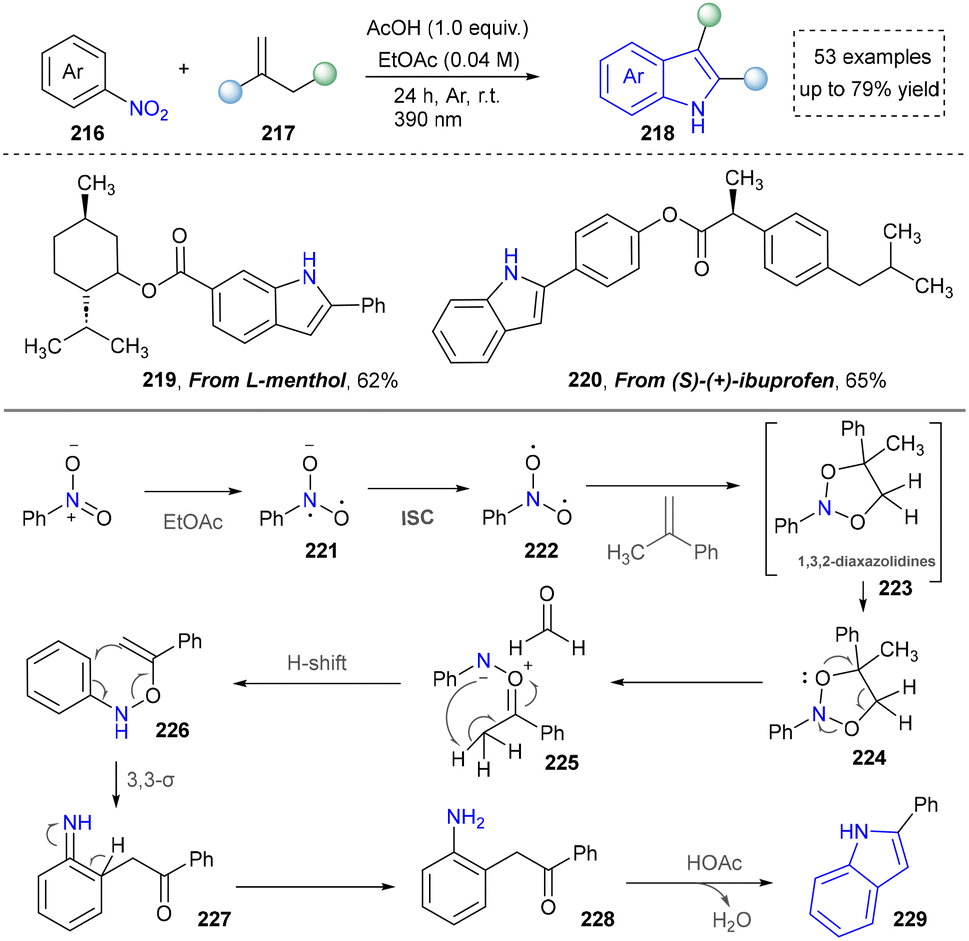 | ||
| Scheme 23 Photoinduced Bartoli indole synthesis. | ||
Similarly, the Studer group reported the cost-effective synthesis of indoles by photocatalysis. This strategy relied on a radical process for the activation of a nitro group and the annulation of nitroarenes and alkenes (Scheme 24).47 Diacetone-D-glucose, a natural product-derived compound, was readily converted into the corresponding indole derivative (232) through late-stage functionalization. This demonstrated the good tolerance of glucose compounds and acetal structures under standard conditions. Therefore, this photocatalytic system provided an efficient pathway for developing bioactive molecules containing indole skeletons. The key to success was the PPh3-mediated radical deoxygenation of nitroarenes. With the formation of nitrosoarene radical cations (234), they further reacted with various alkenes with electron-absorbing groups through the double C–H bond functionalization of nitro aromatics and alkenes at room temperature, and C3-functionalized indoles were quickly obtained. This work represents a significant shift in indole synthesis strategy, moving away from traditional reliance on alkynes and transition metals towards the use of more economical and stable alkene-based electrophiles. The method maintains high yields while demonstrating broad compatibility with a wide range of sensitive functional groups and complex bioactive scaffolds, thereby providing a new and practical approach for efficient indole construction.
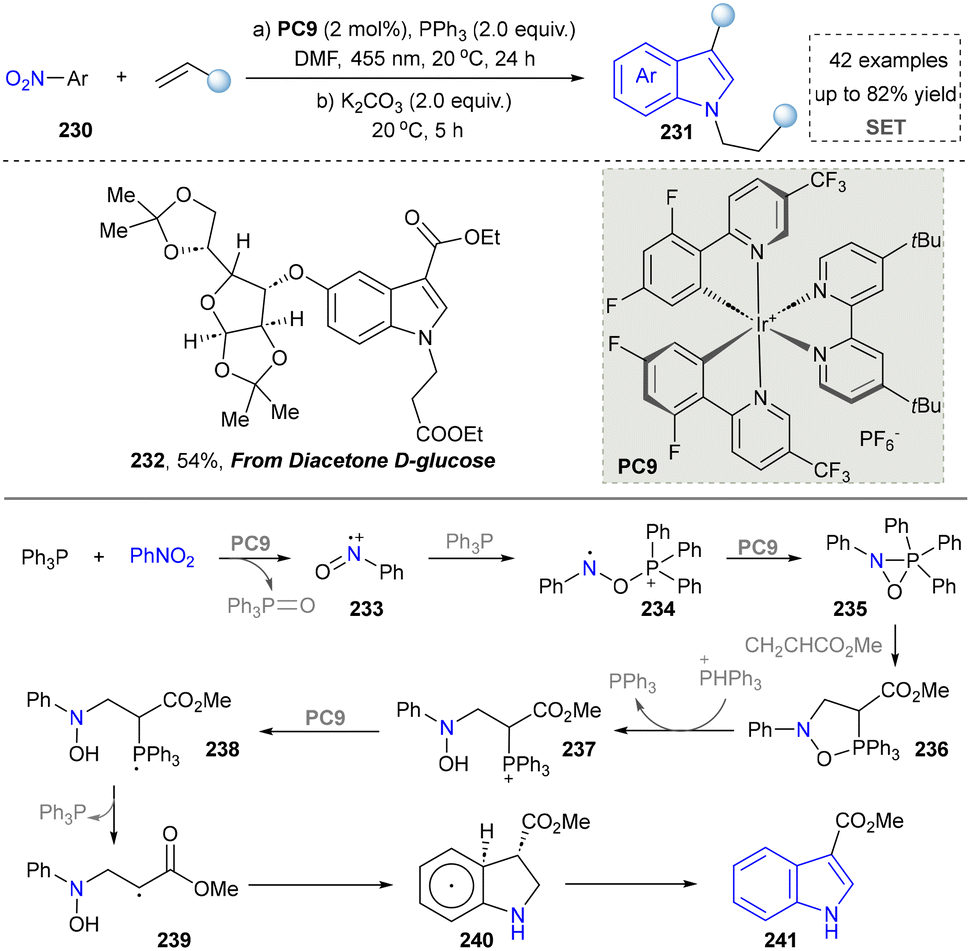 | ||
| Scheme 24 Photocatalytic PPh3-mediated synthesis of C3-functionalized indoles. | ||
Other readily available compounds have also been reported as nitrogen sources for indole synthesis. For example, in 2014, Xiao's group reported a method for efficiently constructing 2-substituted indoles from styrene azides by visible-induced photocatalysis, but the substrate scope was limited.168 In 2018, Reiser's group established a photoredox-mediated radical process for the synthesis of indoles from radical tandem cyclization of ortho-isocyano-α-bromo cinnamates.41 Recently, Wang's group has developed a strategy for the synthesis of indoles, which was driven by visible-light-mediated oxidative dehydrogenation of indolines.169
Compared to conventional methods like Fischer indole synthesis or transition-metal-catalyzed cross-coupling, photochemical synthesis revolutionizes the substrate scope for indole compounds by eliminating the need for pre-functionalization, allowing the direct modification and construction of the indole scaffold from simple, non-activated substrates under mild and neutral conditions and enabling single-step access to traditionally challenging, sterically congested or polycyclic indole derivatives, thereby providing a vastly expanded molecular scaffold library for drug discovery. Overall, the intramolecular cyclization to furnish the indoles has been well established with different photocatalytic methods. However, it is evident that the synthesis of these starting materials often involves multiple steps, which not only complicates the process but also restricts the structural diversity of the resulting indoles. It is important to highlight that intermolecular synthesis, utilizing readily available substances, holds greater promise and deserves more attention for further exploration.
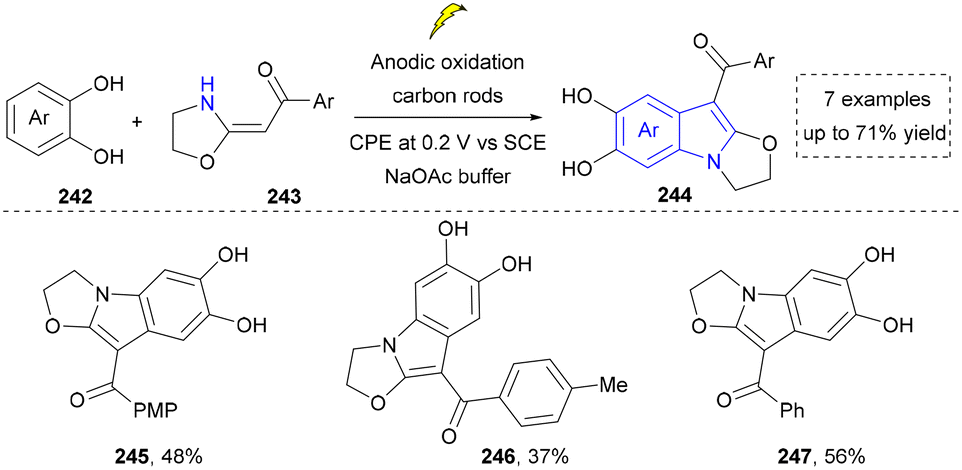 | ||
| Scheme 25 Electrochemical synthesis of the fused indole derivatives. | ||
To further obtain diverse indoles, Xu and co-workers reported the electrochemical synthesis of highly functionalized indoles and azaindoles by C–H/N–H functionalization of (hetero)-arylamines using tethered alkynes (Scheme 26).171 The broad functional-group tolerance of method was evidenced by the preparation of indoles bearing diverse substituents and the more challenging task of constructing complex azaindoles (250, 251). This strategy was successfully applied to the late-stage modification of ethinyl estradiol derivative (252) under standard conditions, demonstrating the excellent compatibility with hydroxyl groups and chiral centers, underscoring the potential for the rapid synthesis of pharmaceutically active derivatives. In addition, this method required neither precious metals nor external oxidants. Finally, the utility of this method was highlighted by the concise total synthesis of isocryptolepine, which was prepared from advanced intermediate (254), requiring only minimal further manipulations.
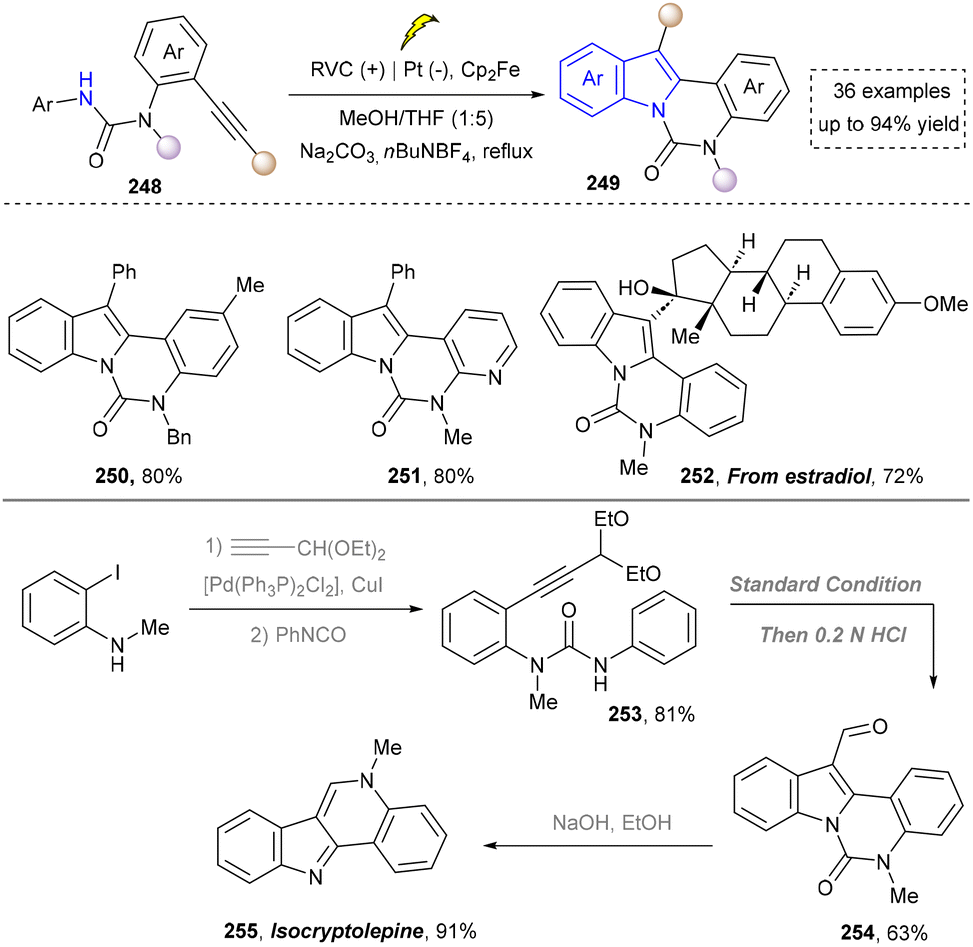 | ||
| Scheme 26 Electrochemical C–H/N–H functionalization for the synthesis of indoles. | ||
In their proposed mechanism, the electrochemical formation of (aza)indoles involved the anodic oxidation of [Cp2Fe] to [Cp2Fe+] and the cathodic reduction of methanol to methoxide (MeO−) and H2. MeO− deprotonated the substrate to form the corresponding anion (257), which underwent the single-electron transfer (SET) process with [Cp2Fe+] to generate nitrogen-centered radicals (258), which then underwent a rare 6-exo-dig cyclization reaction to form a vinyl radical (259). The further cyclization with the aryl ring produced a delocalized radical (260). Finally, the rearomatization of 260via electron and proton elimination yielded the final product (261) (Scheme 27).
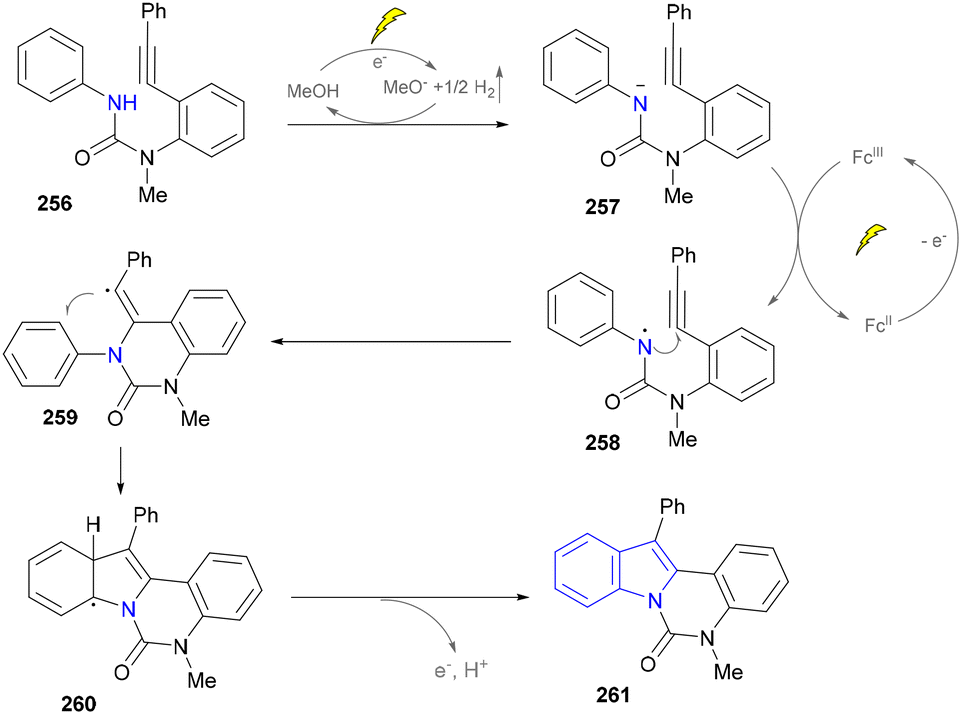 | ||
| Scheme 27 (Aza)indole synthesis by C–H/N–H functionalization. | ||
In 2018, the same team continuously developed the electrochemical dehydrogenative alkyne annulation to furnish indoles.172 This method employed an undivided cell system, where a ruthenium complex catalysed the [3+2] cyclization of aniline derivatives with available alkynes to construct indole scaffolds while releasing hydrogen gas (Scheme 28). The reaction demonstrated excellent air stability in water/alcohol mixed solvents and could be performed on a gram scale. Even though the ruthenium catalyst was required, and the substrate scope (264–266) was expanded by intermolecular synthesis. The utility of this strategy was further demonstrated by the concise synthesis of bazedoxifene (269) from the key intermediate (267), which was readily prepared under slightly modified conditions (H2O/tAmOH replace H2O/iPrOH, 3.0 F mol−1 replace 3.2 F mol−1, 2.4 h) and required further elaboration. A notable limitation, however, is the strategy's exclusive compatibility with secondary amines.
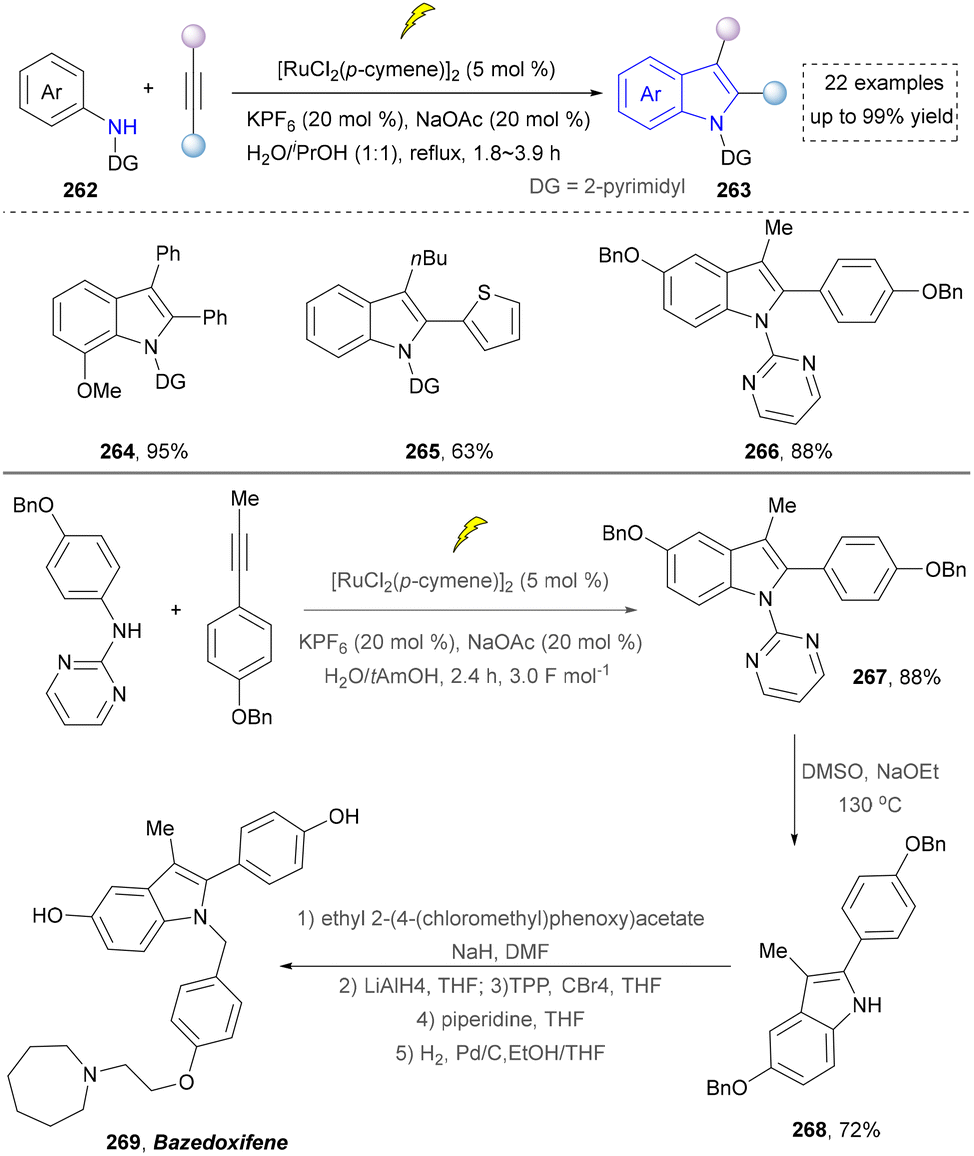 | ||
| Scheme 28 Ruthenium-catalyzed electrochemical dehydrogenative alkyne annulation. | ||
In the same year, Lei and co-workers also reported an electrochemical dehydrogenative aromatization of six- and five-membered N-heterocycles (e.g., tetrahydroquinolines and indolines) using TEMPO as an organoelectrocatalyst without relying on a transition metal catalyst (Scheme 29).173 The reaction was conducted in an undivided cell system, where anodically generated TEMPO cation mediated the stepwise dehydrogenation of N-heterocycles (with concomitant H2 evolution), achieving yields up to 82% (274–277). Earlier, the same group also successfully realized the electrocatalytic reaction of the intramolecular oxidative annulation of enamines without additional oxidants. Even though the concept was known, it improved the sustainability compared with previously photocatalytic methods.174
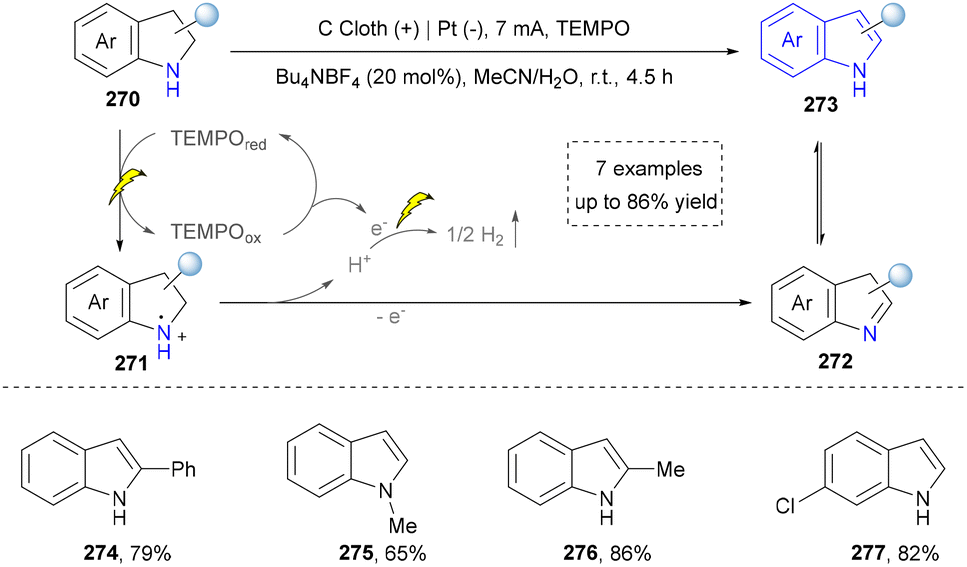 | ||
| Scheme 29 TEMPO-promoted electrochemical receptor-free dehydrogenation of N-heterocycles. | ||
In 2023, Guo and co-workers depicted a metal- and oxidant-free electrochemical protocol for the synthesis of substituted indoles. This method involved the tandem dehydration of 1-(2-aminophenyl)alcohols followed by intramolecular dehydrogenative C–N coupling to efficiently construct an indole scaffold (Scheme 30).175 Mechanistic investigations revealed that the transformation proceeded via a proton-coupled electron transfer (PCET) process to generate nitrogen-centered radicals, which subsequently underwent cyclization and oxidative aromatization. The protocol demonstrated broad functional group tolerance toward halogens, alkyl, and aryl substituents (280–283), while maintaining high efficiency even in the presence of water and atmospheric oxygen, highlighting its exceptional practicality and green chemistry features.
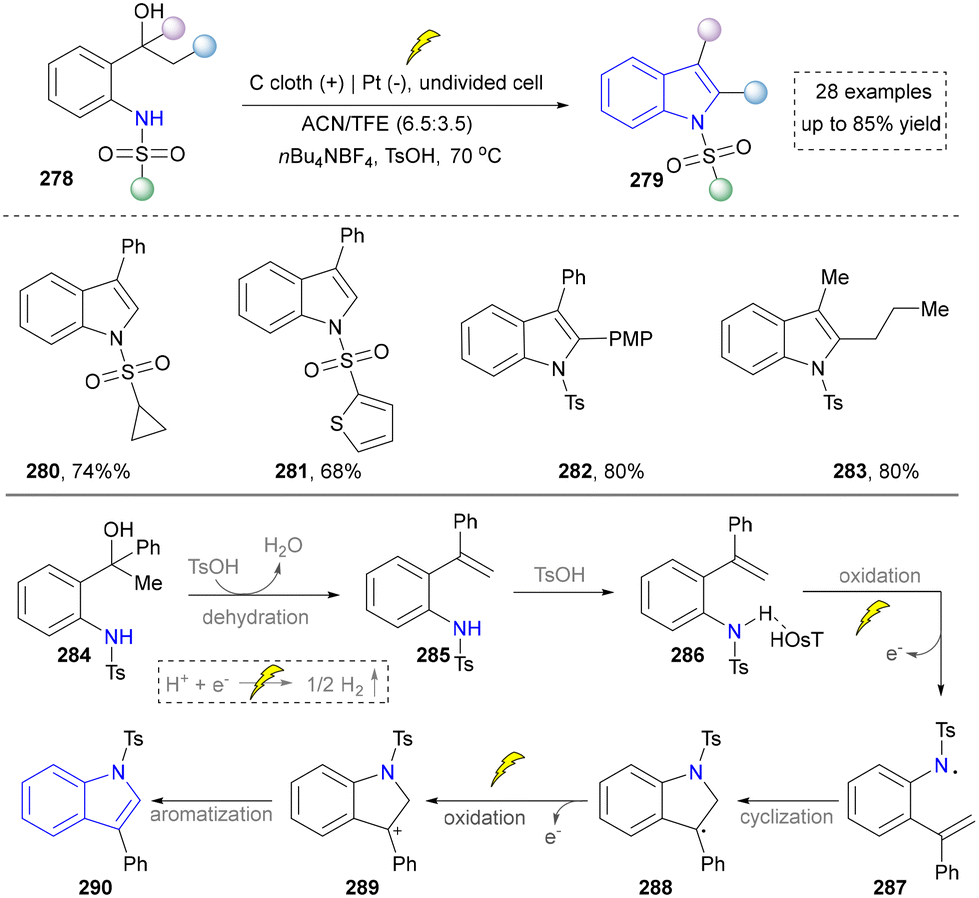 | ||
| Scheme 30 Electrosynthesis of the substituted indoles from 1-(2-aminophenyl) alcohols. | ||
Some other methods including an electrochemical [3+2] cycloaddition approach,176 pKa-regulated switchable synthesis,177 and iridium(III)-catalyzed electrochemical coupling method have been also reported based on the above-mentioned strategies.178–180 These diverse approaches have demonstrated significant potential in green synthesis and sustainable chemistry, collectively expanding the toolbox for indole construction.
Electrosynthesis demonstrates complementary advantages over photochemical methods in substrate scope, primarily due to its direct electron-transfer mechanism. It effectively activates substrates with poor light-absorption properties or those incompatible with photosensitizers. By precise potential control, electrosynthesis enables mild and selective oxidation of electron-rich arenes or reduction of electron-deficient compounds—processes that often face challenges in photocatalysis due to undesired energy transfer or overreaction. Furthermore, it exhibits better tolerance toward substrates prone to photoinduced side reactions or in high-concentration systems where light penetration is limited.
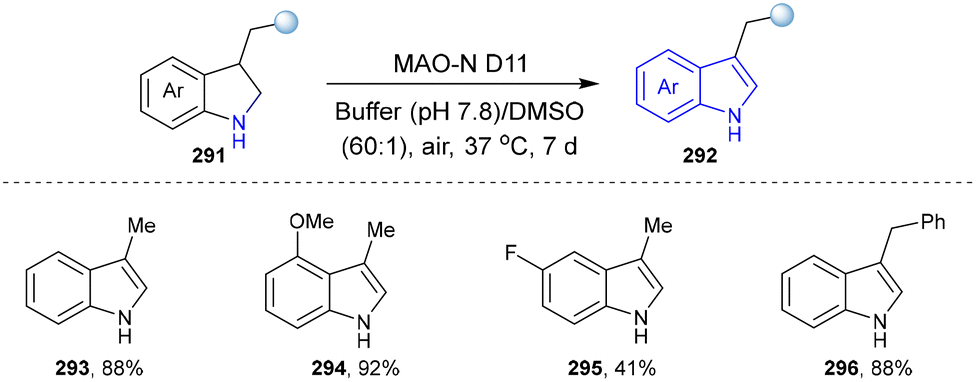 | ||
| Scheme 31 Enzymatic synthesis of the indole derivatives catalysed by MAO. | ||
In 2021, Wang and co-workers designed a green and efficient method to synthesize indoles from 1,3-diketones with fumaronitrile utilizing Candida rugosa lipase (CRL) as the catalyst with not only satisfactory regioselectivity but also high yields (Scheme 32, 300–302).40
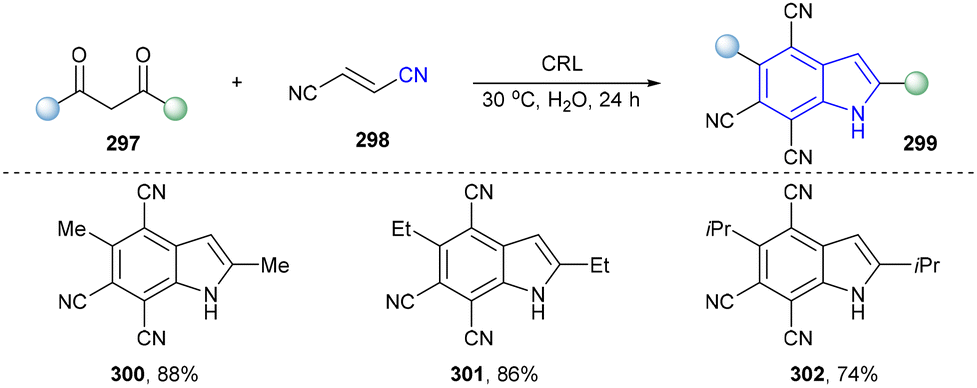 | ||
| Scheme 32 Enzymatic synthesis of indoles utilizing lipase. | ||
In 2022, Zhu's group reported a sustainable two-step, one-pot cooperative chemoenzymatic system for the efficient synthesis of indoles (Scheme 33).181 The intramolecular condensation reaction of 2-(2-aminophenyl)ethanol (303) was performed to obtain indoles (304) with 99% yield, catalysed by organocatalyst F1 and biocatalyst HLADH (horse liver alcohol dehydrogenase). Synthetic flavinium derivative F1, as a bifunctional bio-mimetic organocatalyst, participated in the bioprocess for cofactor NAD+ (nicotinamide adenine dinucleotide) regeneration and the chemoprocess for oxidative cyclodehydrogenation. The cooperative chemoenzymatic process showed excellent chemoselectivity, enhanced reactivity, more efficiency, and fewer by-products, compared with those catalysed by the above-mentioned photo-biocatalysts and metal manganese pincer complexes.182
 | ||
| Scheme 33 Cooperative chemoenzymatic synthesis of indoles. | ||
Overall, MAO, lipase and HLADH have been used to form the indole ring with chemoselectivity, regioselectivity and specificity, though the catalytic efficiency of some enzymes needs to be further improved. Indole-containing natural products such as indigo, vinblastine,183 and antidepressant psilocybin184 are derived initially from l-tryptophan, which has been de novo synthesized in a single Escherichia coli with a titre of 79.4 mg L−1 by optimizing shakeflask fermentation without any precursor feeding. To our knowledge, enzymes involved in the formation of indole-ring are limited. Therefore, Future work should focus on mining more enzymes as biocatalysts for the synthesis of indole-containing natural products.
 | ||
| Scheme 34 Construction of indoles via photochemical skeletal editing. | ||
4. Two-nitrogen-containing aromatic heterocycles: pyrazoles, imidazoles, and benzimidazoles
4.1 Synthesis of pyrazoles
Pyrazoles and their derivatives are widely studied due to their antibacterial, anti-inflammatory, and antitumor activities, and they serve as the core structural units of many important drugs such as Celecoxib.186–188 Additionally, pyrazoles are extensively applied in the development of pesticides, acting as precursors for the synthesis of highly efficient and low-toxicity fungicides, insecticides, and herbicides.189,190 In materials science, pyrazole derivatives can be used to develop fluorescent probes and organic luminescent materials.191 Their unique chemical properties also make pyrazoles important in organic synthesis, where they can be transformed into functionalized molecules via various reactions.192 Therefore, pyrazoles are not only of great significance in chemical research but also demonstrate immense potential in practical applications in pharmaceuticals, agriculture, and materials science. The cyclocondensation of hydrazine derivatives with α,β-unsaturated carbonyl compounds represents a straightforward approach to prepare polysubstituted pyrazole rings. Pyrazole formation via cyclization between ynones and hydrazine derivatives has been documented for over a century.193 In this section, the synthesis of pyrazoles via emerging protocols will be discussed carefully. | ||
| Scheme 35 Synthesis of pyrazoles from hydrazines under visible light irradiation. | ||
In 2016, Zhu's group continuously explored the photoredox aerobic annulation for the synthesis of pyrazoles. This strategy featured the concise synthesis by using different hydrazines and Michael acceptors to furnish di/-trisubstituted pyrazoles.195 The proposed mechanism suggested that the diazene was formed from the hydrazine via the SET process and oxygen worked as the electron donor. The further addition of diazene (329) into the Michael acceptor and the intramolecular condensation gave the final product. However, this strategy did not prove their applicability (Scheme 36). For instance, the gram-scale synthesis or synthesis of complicated pyrazoles was not investigated. In 2019, the same group extended their system by replacing Michael acceptors with an alkyne under photocatalytic conditions. Moreover, hydrazine hydrate and various hydrazines were successfully employed as nitrogen sources, demonstrating good substrate tolerance.196
 | ||
| Scheme 36 Photocatalytic aerobic annulation for the green synthesis of pyrazoles. | ||
Singha's group further extended the application of their strategy by using in situ formed Michael acceptors. The four-component reactions by using ethylacetoacetate, hydrazine hydrate, aromatic aldehydes and malononitrile as synthetic synthons were achieved under photoinduced conditions.197 In 2020, hydrazines as nitrogen sources to furnish pyrazoles were further explored by the same group. They presented three-component photocatalytic methods by using coumarin units, hydrazines, and aldehydes as building blocks. The advantages of this strategy were shorter reaction time and broad substrate scope.198 Moreover, using building blocks such as hydrazine, 1-fluoro-2-isothiocyanatobenzene, and malononitrile, Siddiqui's group described a method that further expanded the diversity of pyrazole synthesis via photochemical approaches.199
To further expand the structural diversity of pyrazoles via photochemical methods, the Zhu group reported a photocatalytic method to enable [4+1] annulation of hydrazones with 2-bromo-1,3-dicarbonyl compounds (Scheme 37). The key to the success was the design of three photoredox cycles, namely an oxidative quenching cycle and two reductive quenching cycles, which formed the complicated pyrazoles efficiently. However, the scope of substrate was somewhat limited due to the limitation of using 2-bromo-1,3-dicarbonyl compounds as radical precursors.200 The direct annulation of α,β-unsaturated hydrazones under photoinduced condition was also reported by Zhu's group. This method did not require any additional photocatalyst and sunlight was applied directly to promote the synthesis of pyrazoles. It was an excellent example to activate and transform the tosylhydrazones under photoinduced conditions. However, the mechanistic studies in this work was not fully investigated.201 Besides using hydrazines and hydrazones as nitrogen sources, Harrity's group described a photocatalytic method to improve the efficiency of sydnone enamine cycloaddition. In this case, the photoactivation of sydnones via energy transfer facilitate the [4+2] cycloaddition to form the key intermediate.202
 | ||
| Scheme 37 Synthesis of the pyrazole derivatives via formal [4+1] annulation and aromatization. | ||
Our group also contributed to the synthesis of pyrazoles via the photoinduced activation of N-tosylhydrazones (Scheme 38). Compared to traditional methods, this strategy facilitated the formation of diazo intermediate under the irradiation of visible light, which further reacted with alkenes to furnish diverse pyrazoles (340–342). The mechanistic studies and DFT calculations suggested the formation of a noncovalent complex, which was the key to the success. Moreover, diverse pyrazoles by using different aldehyde-derived N-tosylhdrazones and alkenes were obtained by using this strategy. However, only aryl-substituted N-tosylhdrazones were accommodated in this system.30 Nevertheless, this method enables the straightforward synthesis of pyrazoles bearing strong electron-donating groups through the use of “donor/donor” diazo intermediates, which are typically challenging to access via conventional diazo chemistry. It also efficiently constructs sterically complex pyrazolines and pyrazoles featuring spiro-quaternary carbon centers.
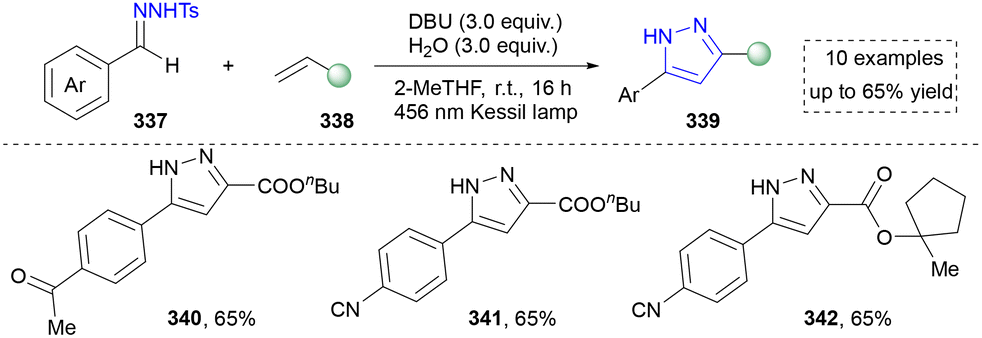 | ||
| Scheme 38 Visible-light-induced [3+2] cycloadditions to achieve pyrazoles. | ||
Overall, hydrazines and hydrzones as nitrogen sources have been well established to construct pyrroles. Other partners to furnish pyrroles have been also investigated including various alkenes and Michael acceptors. However, other nitrogen sources such as diazo compounds or amines were also possible to use in the photocatalytic synthesis of pyrroles. Using inactivated alkenes as substrates also needed to take some attentions which will tremendously expand the diversity of synthesized pyrroles. Moreover, selectivity control remained a significant challenge, especially for complex substrates with multiple reactive sites. Consequently, its application in large-scale production still requires complementation with established traditional methodologies. Moreover, the three-component reactions need to be investigated.
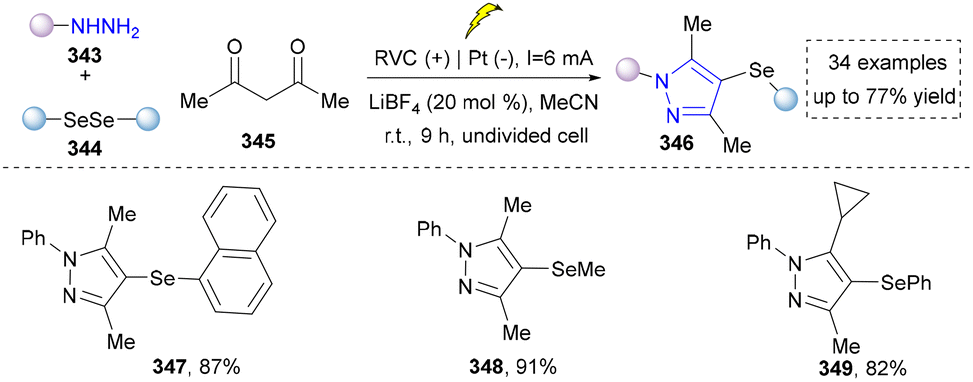 | ||
| Scheme 39 Electrochemical synthesis of 4-selanylpyrazoles. | ||
In 2022, the O. Terent’ev group disclosed the electrochemical intramolecular C–N coupling of α,β unsaturated hydrazones. The initial anodic two-electron oxidation of iodobenzene generated the hypervalent iodine species, which subsequently underwent nucleophilic substitution with N-tosylhydrazones to form the N-iodo intermediate.205 At last, the electrophilic intramolecular addition and deprotonation gave the final pyrazoles. This was a typical method to activate N-tosylhyrazones using the formed hypervalent iodine species. In 2023, the Waldvogel group continuously disclosed the electrochemical synthesis of pyrazoles via [3+2] dipolar cycloaddition. The pyrazoles were obtained directly from the hydrazones and alkenes. However, this strategy was focused on the synthesis of prazolines, and the scope and reactivities of pyrazoles were relatively limited.206 Almost at the same time, polysubstituted sulfonated pyrazoles were also obtained via electrochemical synthesis by Huang's group.207
Very recently, another nitrogen source, aryl diazonium salt, has also been reported by Tang's group (Scheme 40). In this method, aryl diazonium salts were reduced directly on the cathode, generating the aryl diazo radical intermediate, which further reacted with ketones to furnish the pyrazoles. This is the first example to from aryl radicals and aryl diazo radicals via the electroreduction of aryl diazonium salts. With this mild strategy, plenty of multi-substituted pyrazoles (353, 354) were obtained without the addition of external catalyst and oxidants. However, they did not evaluate the potential of this method in gram-scale synthesis and drug synthesis.45
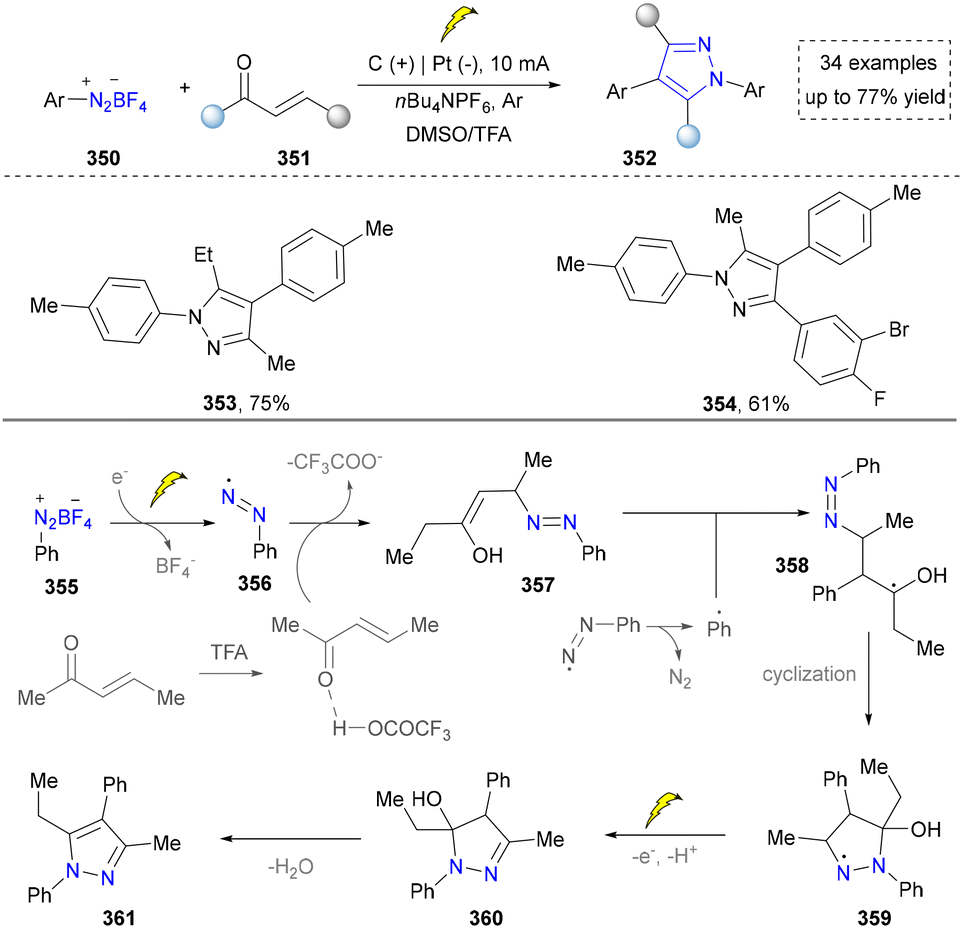 | ||
| Scheme 40 Electrochemical synthesis of pyrazoles via a radical cyclization cascade. | ||
Overall, the developments of electrochemical synthesis provided some new insights for the synthesis of pyrroles. The nitrogen sources have been expanded to the aryl diazonium salts, which was not reported in photocatalytic methods. Moreover, several three-component methods have been developed which showed good structural diversity. Furthermore, electrons serve as a clean “reagent,” reducing reagent waste and heavy metal pollution at the source, aligning with the trend toward green pharmaceutical manufacturing.
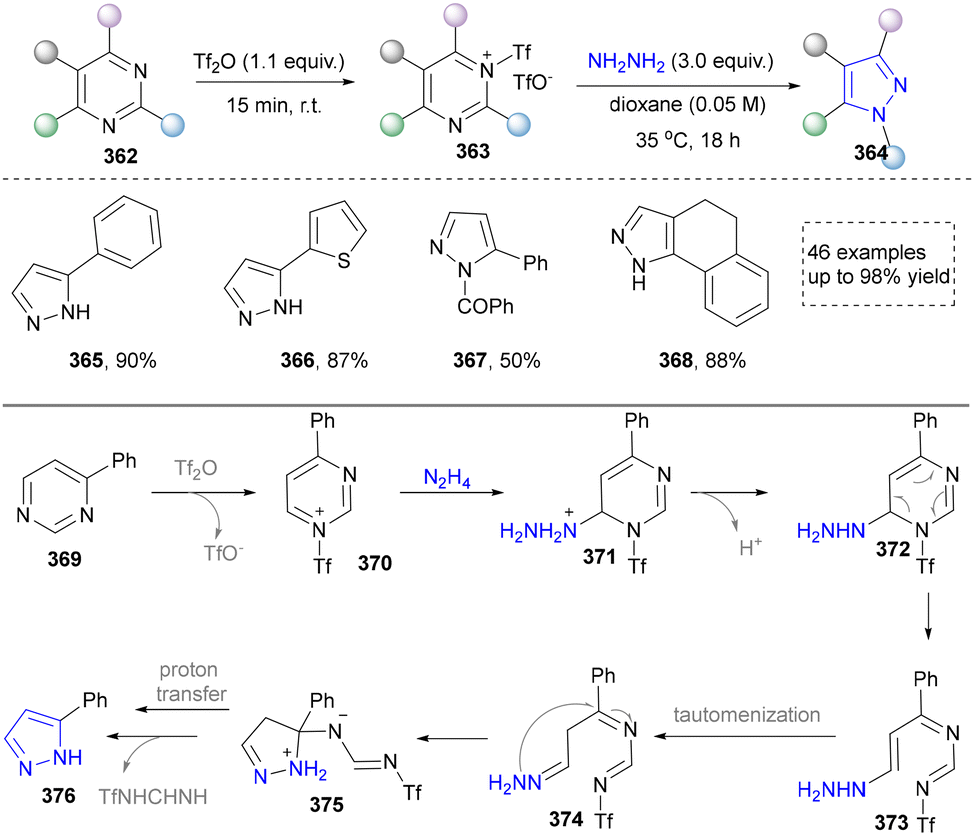 | ||
| Scheme 41 Skeletal editing of pyrimidines to pyrazoles by formal carbon deletion. | ||
In 2024, Zheng's group presented a metal- and catalyst-free method to access the skeletal editing of pyridines to pyrazoles (Scheme 42).211 With the pyridinium salt as a model substrate, two-time irradiation with different light sources accomplished cyclization to achieve pyrazolines (380–383). Subsequent photocatalytic irradiation of this intermediate with xanthone facilitated [4,5]-carbon deletion, yielding the pyrazole product. Furthermore, 1,2-diazepine, obtained via irradiation under 365 nm LEDs, was converted to pyrazoles (387–390) selectively by deleting carbon (2,3 or 5,6) at the imine position. The synthetic application of this protocol was also demonstrated by the late-stage modifications of drugs and natural products containing pyridine units. At last, the DFT studies provided the proposed mechanism for different reaction conditions. Notably, during the revision of our manuscript, Studer's group disclosed a complementary C-to-N atom swap in indoles to access indazoles, relying on oxidative cleavage and ring closure.212
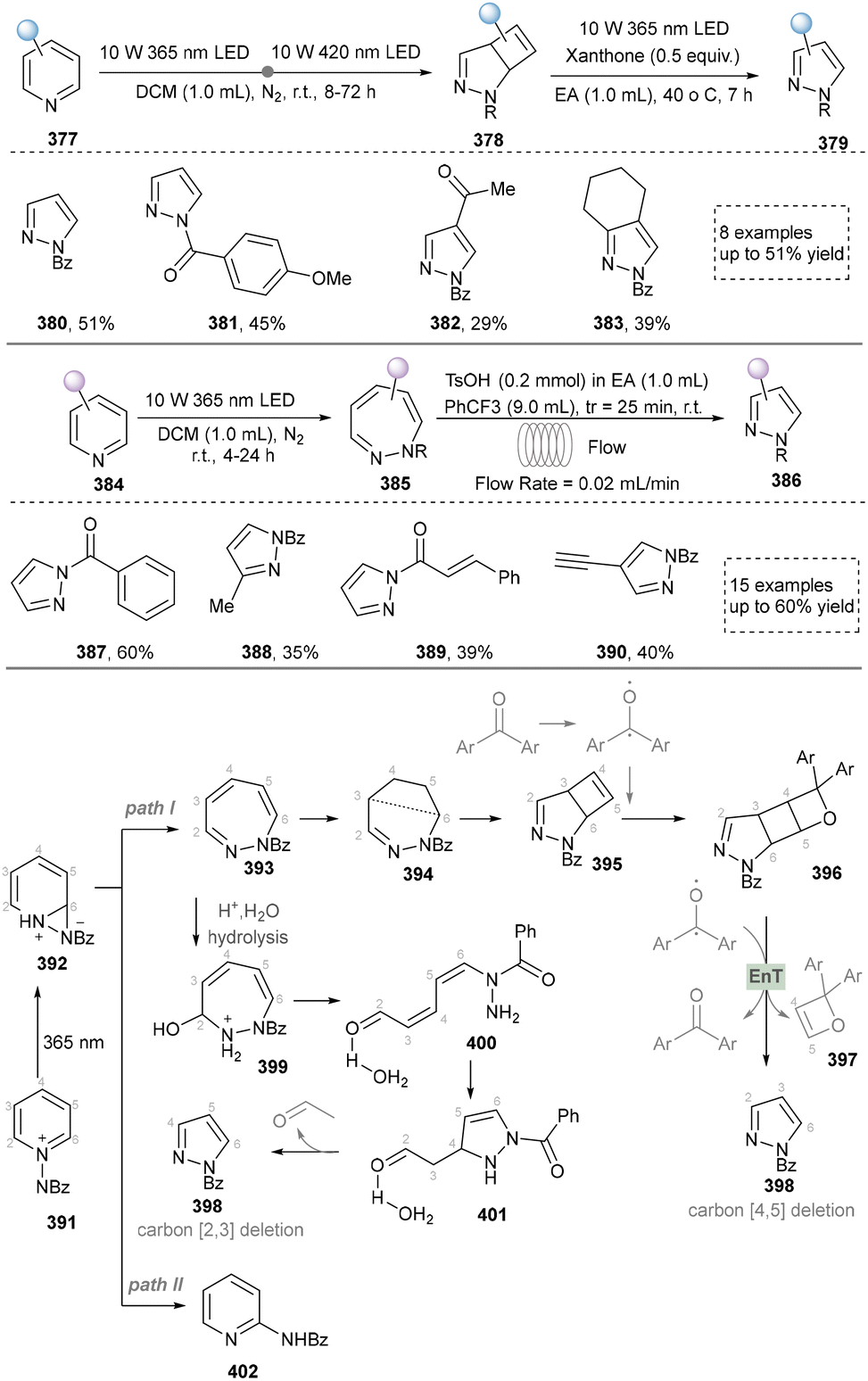 | ||
| Scheme 42 Photochemical skeletal editing of pyridines to pyrazoles. | ||
In summary, skeletal editing strategies provide a revolutionary approach for the synthesis of pyrazoles, enabling the pyrazole core by bypassing traditional lengthy de novo synthetic routes. However, this strategy currently faces challenges such as limited reaction generality, complexities in selectivity control, and dependence on specific activation modes or directing groups. Nevertheless, with a deep understanding of the mechanisms governing selective bond cleavage and reorganization, skeletal editing technology holds strong potential to evolve into a powerful tool for late-stage functionalization in medicinal chemistry. By allowing direct, “surgical” modification of the pyrazole ring within complex molecular frameworks, it could significantly accelerate lead compound optimization and structure–activity relationship studies, thereby opening a more direct and efficient pathway for the discovery of innovative drug molecules.
4.2 Synthesis of imidazoles
Imidazole is highly polar and readily soluble in water.213 In biological systems, imidazole is a key component of the side chain of histidine, an amino acid found in many proteins and enzymes.214,215 Additionally, imidazole is a common structural motif in various pharmaceuticals, including antifungal, antiprotozoal, and antihypertensive drugs.216 Industrially, it is also used in the synthesis of certain pesticides.217 The traditional methods to afford imidazoles relied on the Debus–Radziszewski reaction, Wallach reaction, Bredereck imidazole synthesis, and Kaiser–Johnson–Middleton dinitrile cyclization.218 This section will introduce the progress of synthesizing imidazoles via three emerging protocols.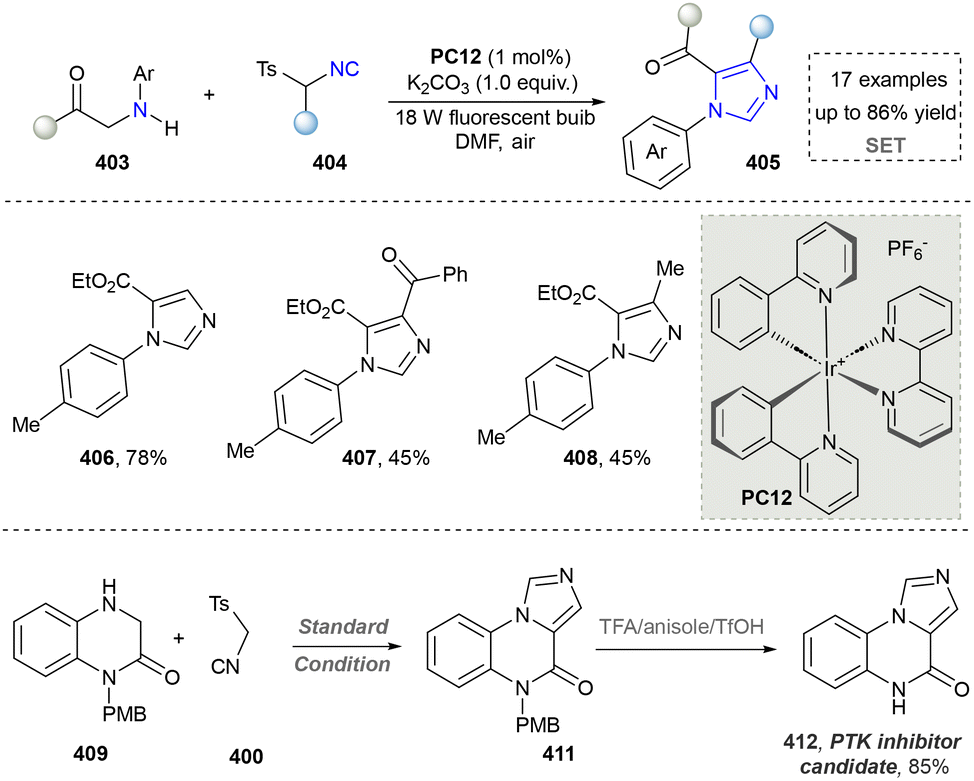 | ||
| Scheme 43 Synthesis of imidazoles by photocatalytic aerobic oxidation/[3+2] cycloaddition. | ||
Kim's group further disclosed the facile synthesis of fused imidazoles, featuring the use of readily available L-proline and α-keto vinyl azides as precursors via a photoinduced method (Scheme 44). The key to the success of this method was the formation of 2H-azirine from the α-keto vinyl azides via energy transfer. More importantly, the integration of this method with continuous-flow technology overcame a key barrier to scaling up purely photochemical synthesis. This combined photo-continuous flow strategy enabled the efficient synthesis of target compounds 416–418, reducing the reaction time dramatically from 16 hours in the batch mode to only 2 minutes. The rapid construction of imidazoles indeed was helpful in organic synthesis to save time and energy. However, further in-depth investigation in application was needed since the gram-scale synthesis was not provided, which was another obvious advantage of continuous-flow chemistry.221 Later, Maurya's group extended the photoexcitation of α-keto vinyl azides and applied into the imidazole via a three-component method.222
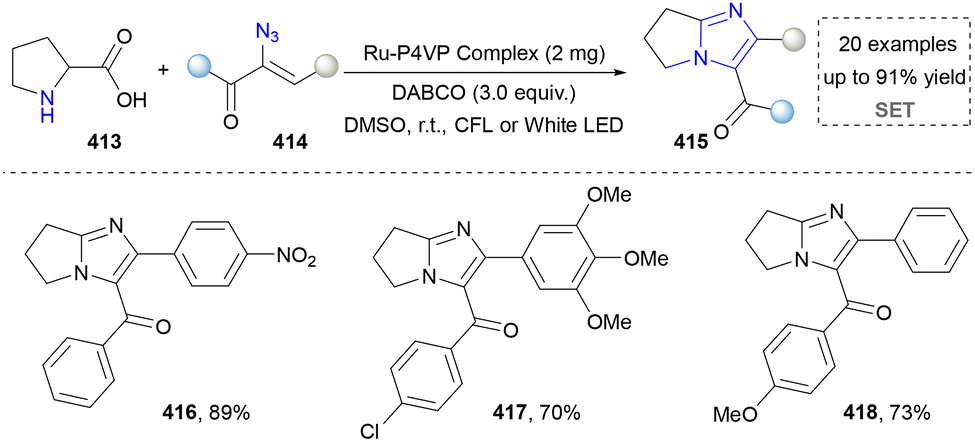 | ||
| Scheme 44 Photo-induced decarboxylated annulative achieve imidazoles. | ||
Afterwards, highly substituted imidazoles and dihydroisoquinoline-based imidazole derivatives were synthesized by Sharada's group (Scheme 45).223 The advantage of this method was that no transition metal was needed under photocatalytic conditions. Moreover, TMSN3 worked as a nitrogen source to induce nitrogen incorporation. Considering both imidazoles and dihydroisoquinolines as important drug motifs, this strategy shows potential to obtain some leading compounds via this novel skeleton assembly (421–424). However, the mechanistic investigation of this method was underexplored. We also noticed that some heterogeneous catalysts were designed and synthesized to facilitate the synthesis of imidazoles,224 but they will not be discussed here since the novelty of catalysts and their efficiency are beyond the scope of this review.
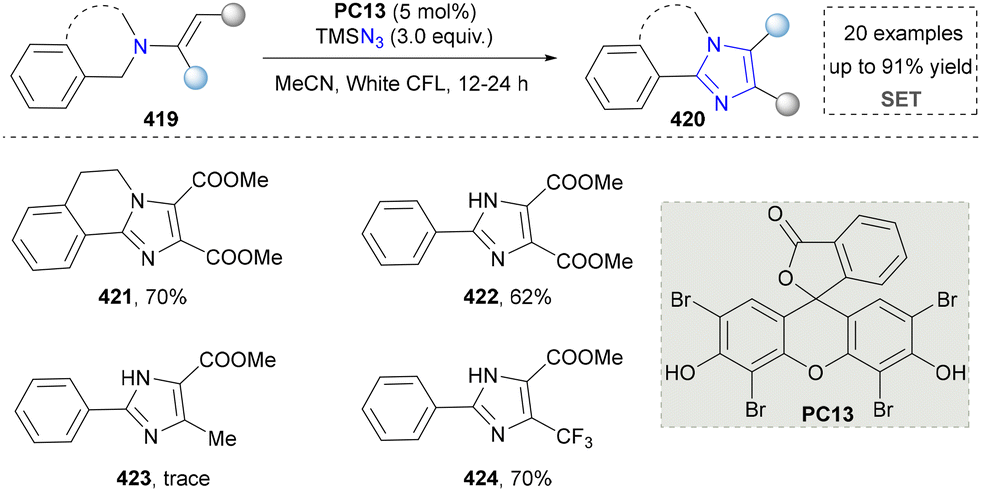 | ||
| Scheme 45 Synthesis of imidazoles via dual C(sp3)–H and C(sp2)–H bond functionalization. | ||
In conclusion, this section shows the obvious progress of imidazoles synthesis using photochemical methods. Different nitrogen sources including azides, TMSN3, and isocyanides have been investigated to form imidazoles with amines. It was anticipated that some other convenient methods such as three-component methods, or using other readily available building blocks, would be developed in the future. Moreover, the practical application into drug synthesis and gram-scale synthesis also need to be investigated in the future.
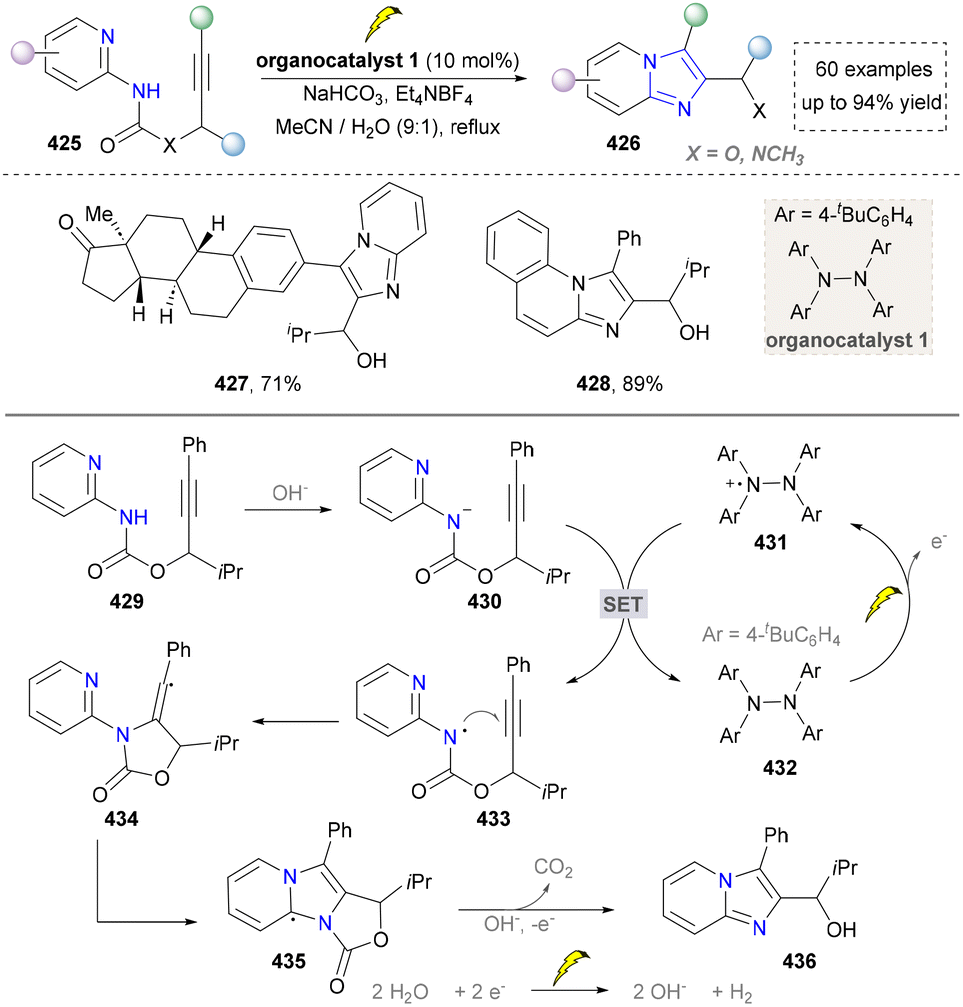 | ||
| Scheme 46 Electrochemical synthesis of imidazoles through C–N bond formation. | ||
In 2019, the synthesis of imidazo[1,5-a]quinoline was further reported by Wang's group (Scheme 47).227 They found that iodine generated from NH4I in the anode reacted with ethyl 2-(quinolin-2-yl)acetate (437) to form the iodinated intermediate (444), which was further substituted by the amine and oxidized in the presence of molecular iodine to yield an intermediate (447). At last, it underwent a tandem cyclization process to generate the final products (440–442). However, the structural diversity was limited since the synthesis of ethyl 2-(quinolin-2-yl)acetate was not convenient. Later, O. Terent’ev's group developed a three-component strategy, which applied pyridine-type aldehydes, amines and NH4SCN to construct diverse CN-substituted imidazo[1,5-a]pyridines, which showed a better substrate scope than Wang's work.228,229
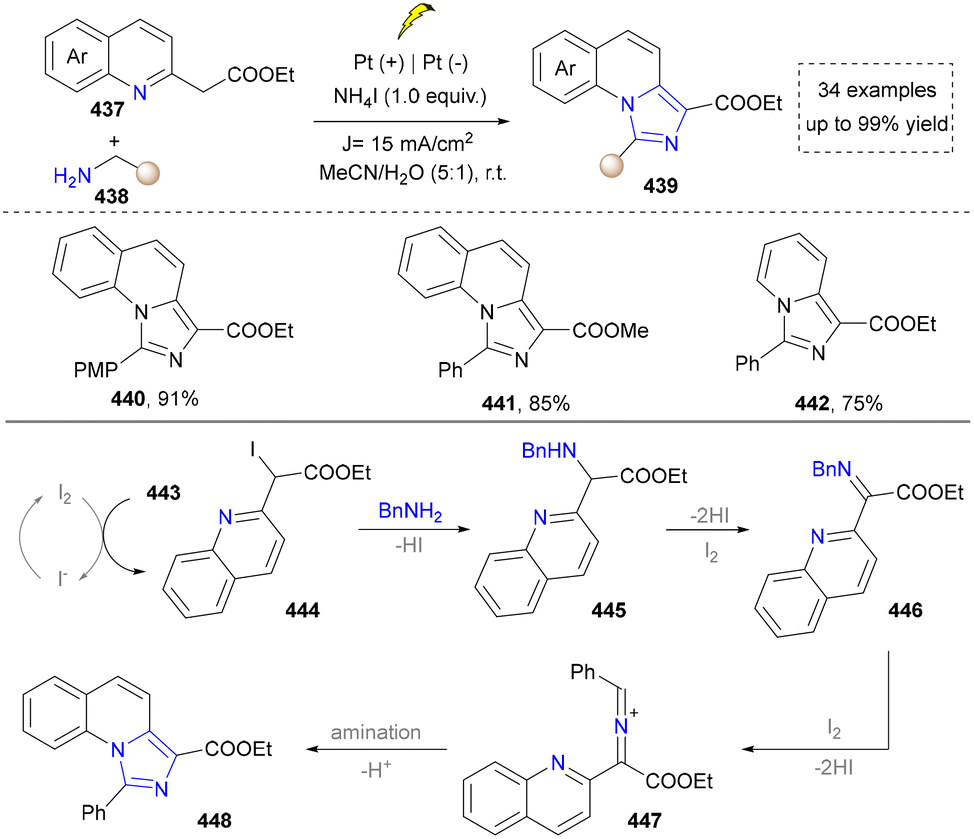 | ||
| Scheme 47 Electrocatalytic synthesis of the 1,3-disubstituted imidazo[1,5-a]quinolines. | ||
In 2020, He's group described an elegant method to construct imidazoles via electrochemical annulation of ketones and amines. In this reaction, two different amines worked as nitrogen source, one amine reacted with in situ formed α-iodo ketone to generate α-amino ketone (Scheme 48).230 The other amine was required to be a benzylic amine, as it readily condensed with the α-amino ketone to generate an imine intermediate (460), which further be oxidized to afford the imidazole. This work showed good structural diversity since the various amines could be applied to the synthesis (453–455). However, practical application such as synthesis of drug or bioactive molecules was not investigated. Later, Pan's group continuously reported the synthesis of sulfide imidazopyridines via electrochemical synthesis. In this reaction, vinyl azides, thiophenols, and pyridines reacted smoothly to form diverse imidazoles via a radical cyclization cascade, which further expanded the application of electrocatalysis in the imidazole synthesis.39 Not only are the metal- and external oxidant-free conditions noteworthy, but the [2+2+1] annulation employing readily available amines also significantly broadens the structural diversity of imidazoles.
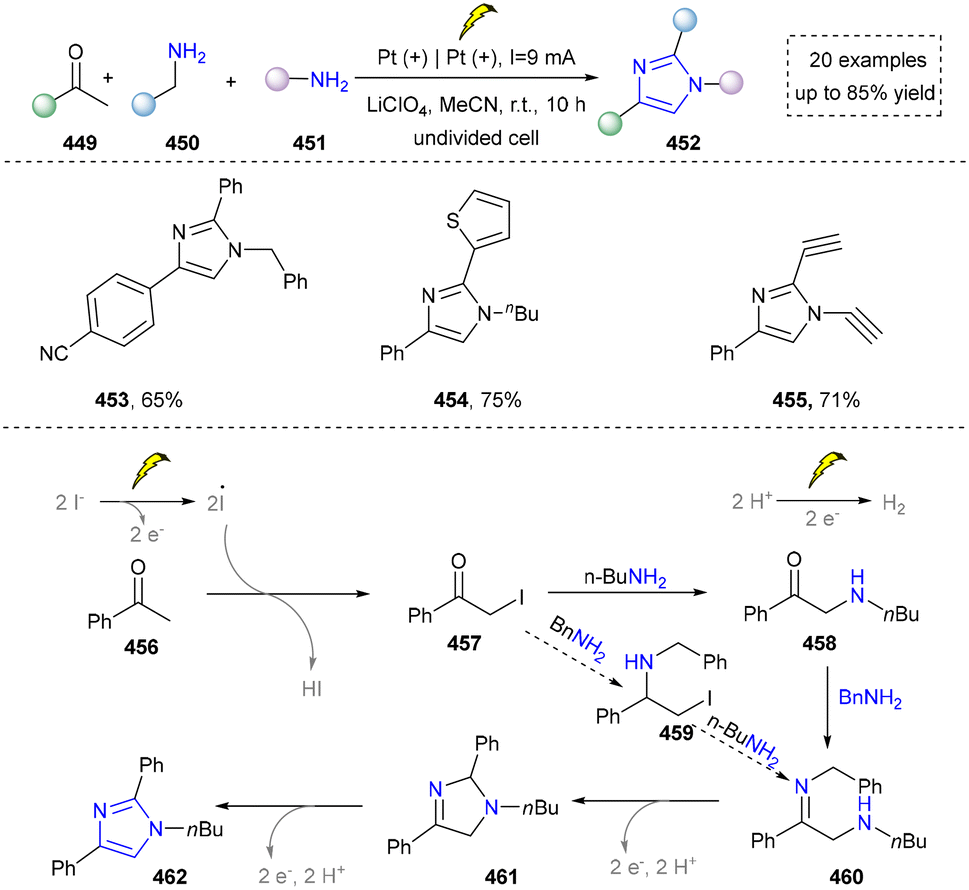 | ||
| Scheme 48 Electrochemical oxidative annulation of ketones and amines used to produce imidazoles. | ||
Inspired by their work, the Chen group developed the electrochemical synthesis of tetrasubstituted imidazoles from enamines and benzylamines. The advantage of this strategy is that the gram-scale synthesis of a drug analogue was reported.231 The Chen group also developed an electrochemical tandem Michael addition to construct imidazoles efficiently, but the concept has been developed previously.232 The Zhou group also reported the synthesis of imidazoles from ketones and amines in 2020, but this work does not explore the possibility to use two different amines.233 In 2023, the Zhou group continuously worked in this field and reported the synthesis of imidazoles via electrochemical dehydrogenative amination, this work showed some advancement such as the direct C(sp3)–H intramolecular amination.234
It has been witnessed that the electrocatalytic synthesis provided more possibility to diversify imidazoles. The key advantage of electrochemical pyrazole synthesis lies in its ability to directly activate various stable precursors through potential control, bypassing the reliance of traditional methods on diazo compounds and the dependence of photochemical approaches on photoactive substrates or catalysts. This enables broad compatibility with strongly electron-donating, high-oxidation-potential, heterocycle-modified, and oxidation-sensitive substrates, providing a more versatile and milder synthetic platform for constructing pyrazole frameworks. Moreover, it is clear that electrocatalysis could utilize some other nitrogen sources such as amides, which has been previously challenging in photochemical methods.
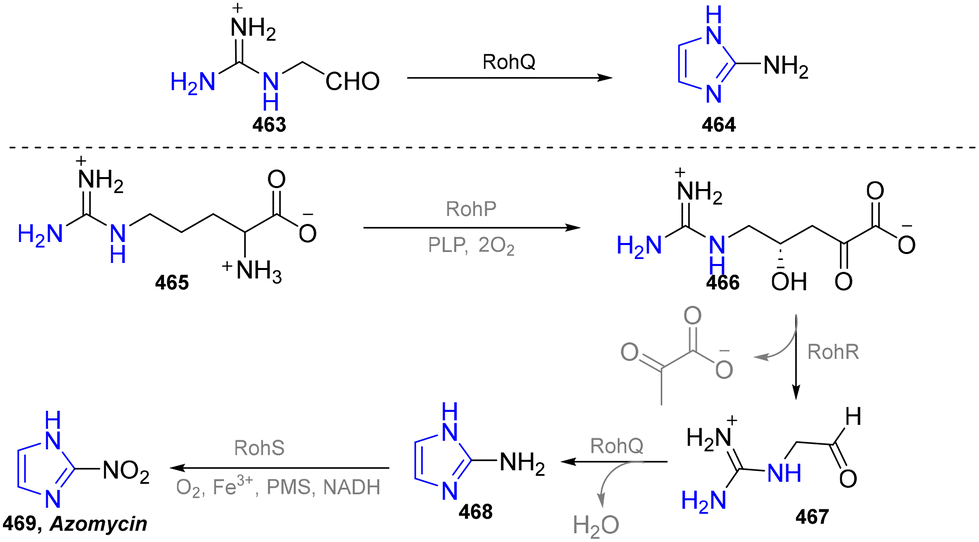 | ||
| Scheme 49 Enzymatic synthesis of imidazoles from L-arginine. | ||
Although imidazole compounds have important biological activities, enzymes that catalyse the formation of the imidazole ring need to be further explored. It is believed that the enzymatic synthesis of imidazole derivatives will gradually increase, with the analysis of the biosynthetic pathway of imidazole-containing natural products.
4.3 Synthesis of benzimidazoles
In light of its unique physicochemical properties and structural versatility, benzimidazole scaffold constitutes a privileged heterocyclic system in modern drug discovery and materials science.238,239 As a bioisostere of naturally occurring purine bases, this fused bicyclic architecture demonstrates remarkable pharmacological relevance, serving as the core structure in numerous therapeutic agents including proton pump inhibitors (e.g., omeprazole), antiparasitics (albendazole), and antiviral drugs.240,241 Benzimidazole derivatives are typically prepared through the condensation of o-phenylenediamine with carbonyl compounds (aldehydes/ketones) or carboxylic acid derivatives, or via rearrangement of quinoxaline/triazole derivatives.242,243 Consequently, the benzimidazole scaffold retains substantial synthetic values in contemporary research.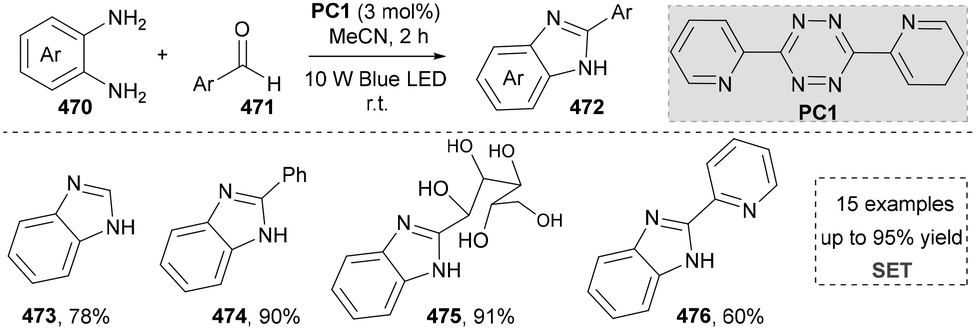 | ||
| Scheme 50 Visible-light-driven green synthesis of the 2-substituted benzimidazoles. | ||
Besides the intermolecular cyclization, another useful strategy to afford the imidazoles via photocatalysis has been reported by Wang's group in 2019 (Scheme 51).253 They used N-phenyl amidoxime esters (477) as starting materials, which formed the amidinyl radical via the SET process. The amidinyl radical reacted with the adjacent benzene ring to form a cyclized intermediate, which was subsequently oxidized by the photocatalyst, yielding the final product (479–482) after aromatization. Even though the photochemical formation of amidinyl radicals was well-established, this strategy still provided a useful method to generate 2-substituted benzimidazoles. More importantly, this method was applied to the synthesis of fungicide thiabendazole (488) under standard conditions, indicating the application potential of this protocol. This established the compatibility of this reaction with both aromatic and heteroaromatic aldehydes.
 | ||
| Scheme 51 N-Phenyl amidoxime esters to achieve the 2-substituted benzimidazoles. | ||
Benzimidazole synthesis has witnessed continuous methodological exploration. In 2016, Zhang's group established an efficient synthesis of polysubstituted benzimidazoles via a visible-light-induced intramolecular cyclization and deprotection sequence.254 Subsequent efforts prioritized sustainability, in 2020, Ke's group developed an eosin Y-catalyzed aqueous-phase reaction of benzonitrile derivatives,255 while Banerjee's group reported a catalyst- and solvent-free one-pot, four-component synthesis.256 Further advancing milder strategies, Kokotos's group demonstrated a direct photochemical synthesis from diamines and aldehydes.257 Building on this foundation, Kundu's group has recently introduced a method that merges a photocatalyst with a HAT reagent to activate ethanol for benzimidazole formation.258
The development of benzimidazole synthesis has been greatly accelerated by visible light photoredox catalysis. Initial work by Zhang's group on an intramolecular cyclization/deprotection sequence paved the way.254 Subsequent research emphasized sustainability, with Ke's group (2020) reporting an eosin Y-catalyzed aqueous protocol,255 while Banerjee's group achieved a catalyst- and solvent-free, four-component synthesis in parallel.256 Further contributions included Kokotos's257 milder synthesis from simple building blocks and, more recently, Kundu's innovative strategy that combines a photocatalyst with a HAT reagent, demonstrating the continued evolution in this field.258
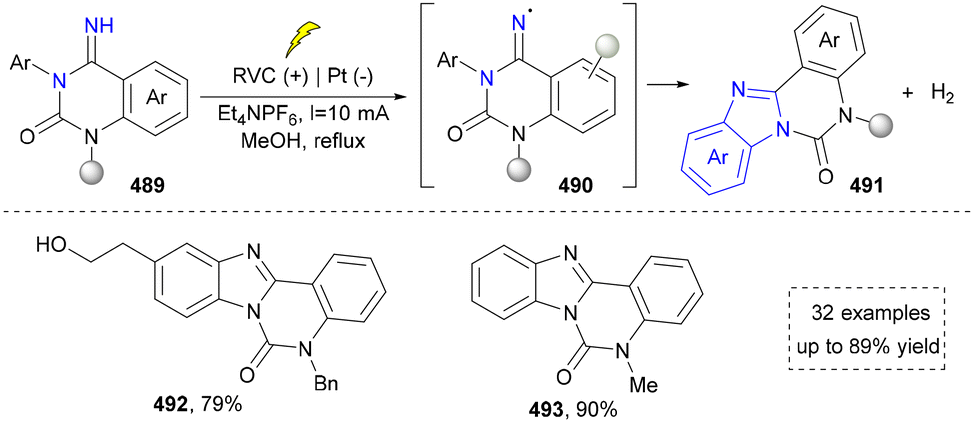 | ||
| Scheme 52 Synthesis of benzimidazoles through the anodic N–H bond cleavage. | ||
Continuously, the same group further developed an electrocatalytic method to synthesize the benzimidazole through dehydrogenative cyclization of easily available N-aryl amidines. Compared with their first work, this protocol showed a better substrate scope. In addition, they provided the detailed mechanism to show the anodic oxidation of N-aryl amidine and the subsequent formation of amidinyl radical.261 In 2023, Zhou's group also disclosed the electrocatalytic synthesis of benzimidazoles via intramolecular C(sp3)–H amination as a continuous work based on their previous reports.234
Overall, the direct electrocatalytic C–H functionalization provided some unprecedented route to construct benzimidazoles without any leading group. However, there is still some space to expand the structural diversity of benzimidazoles via intermolecular synthesis. The development of novel nitrogen sources in future electrochemical synthesis is poised for breakthroughs along the axes of more fundamental, greener, and smarter alternatives, thereby driving the advancement of sustainable synthetic chemistry.
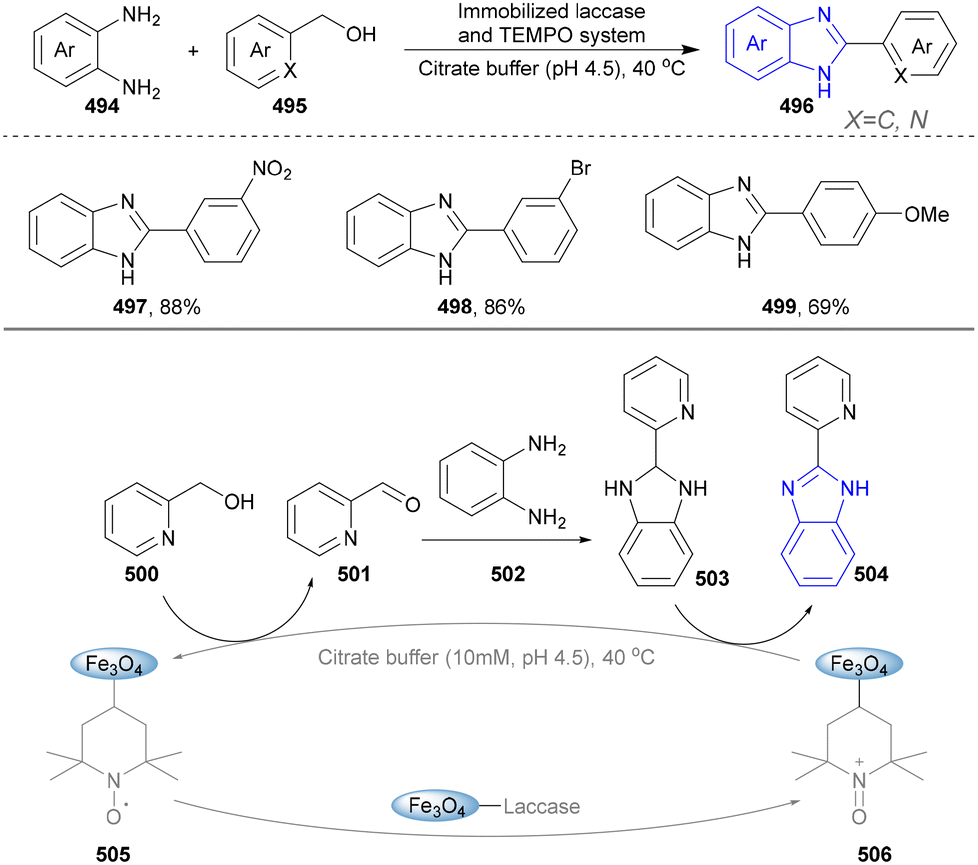 | ||
| Scheme 53 Laccase-catalysed synthesis of benzimidazoles. | ||
The synthesis of benzimidazoles via intermolecular condensation was also performed through the cooperative chemoenzymatic system catalysed by an organocatalyst (F1) and biocatalyst HLADH reported by Zhu's group (Scheme 54).181 In the bioprocess, HLADH converted alcohol (510) into aldehyde (511) at the cost of constantly consuming NAD+, which was continuously regenerated in situ by F1. The resulting F1H was subsequently reoxidized by oxygen back to F1, releasing H2O2. In the chemoprocess, the aldehyde (512) underwent spontaneous condensation with diamine (507) to generate the corresponding imine intermediate, which was cyclized into benzimidazoline (512). Finally, benzimidazoline (512) was oxidized to obtain the corresponding benzimidazole (513), while F1 was regenerated to continue the entire catalytic cycle. The transition between F1 and F1H was realized via direct hydride transfer and C10a addition. This approach offered broad applicability for the synthesis of diverse substituted benzimidazoles, with the cooperative effect of multiple catalysts proving more effective than their sequential application. Furthermore, the cooperative chemoenzymatic system was successfully employed to synthesize pimobendan (516), a calcium-sensitizing drug for the treatment of ischemic heart disease and arterial thrombotic diseases, and a moderate overall yield of 53% was obtained within 24 hours under non-optimized conditions, due to the poor aqueous solubility of the diamine in water. Furthermore, the hydroxy group was susceptible to oxidation into a carbonyl group under these reaction conditions.269,270
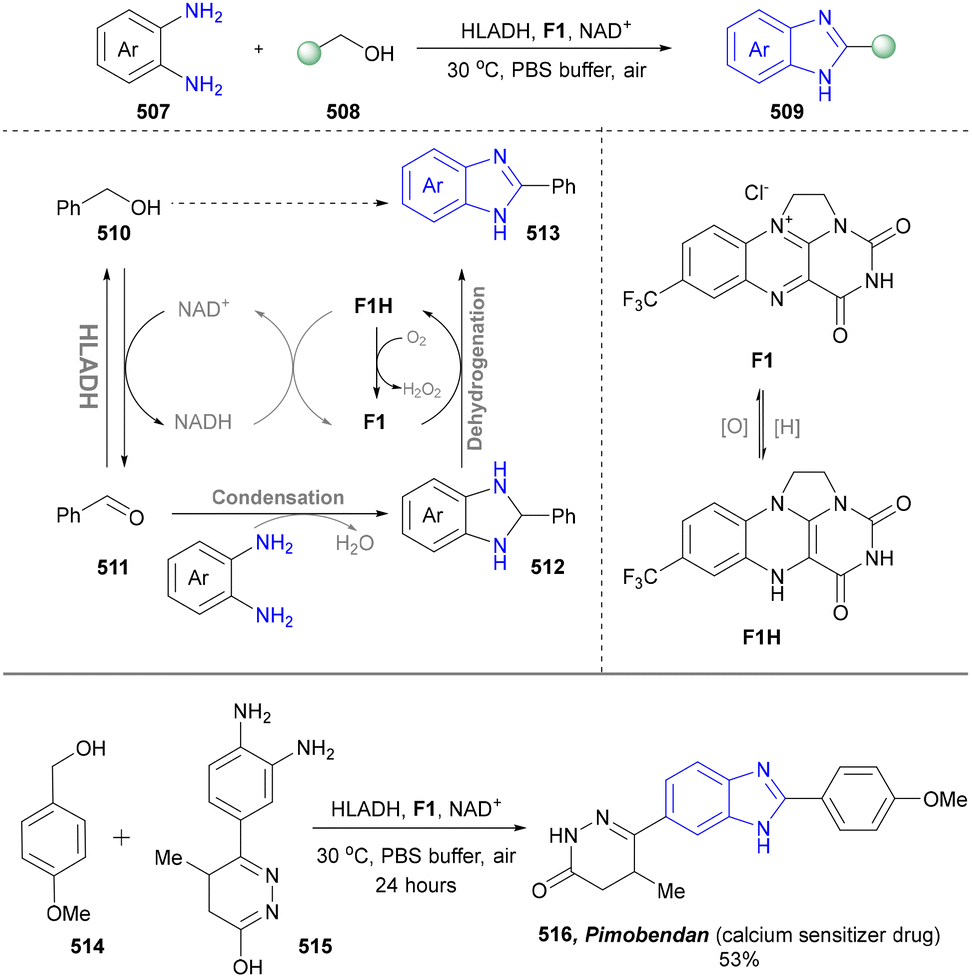 | ||
| Scheme 54 Cooperative chemoenzymatic system for the synthesis of benzimidazoles. | ||
Overall, the synthesis of benzimidazole was achieved by optimizing the reaction conditions to influence the chemoselectivity of laccase or integrating HLADH with organocatalysts. Given the increasing demand for sustainable methods, the chemoselectivity and mild reaction conditions of biocatalysis are expected to drive further research and application in the synthesis of diverse benzimidazole derivatives.
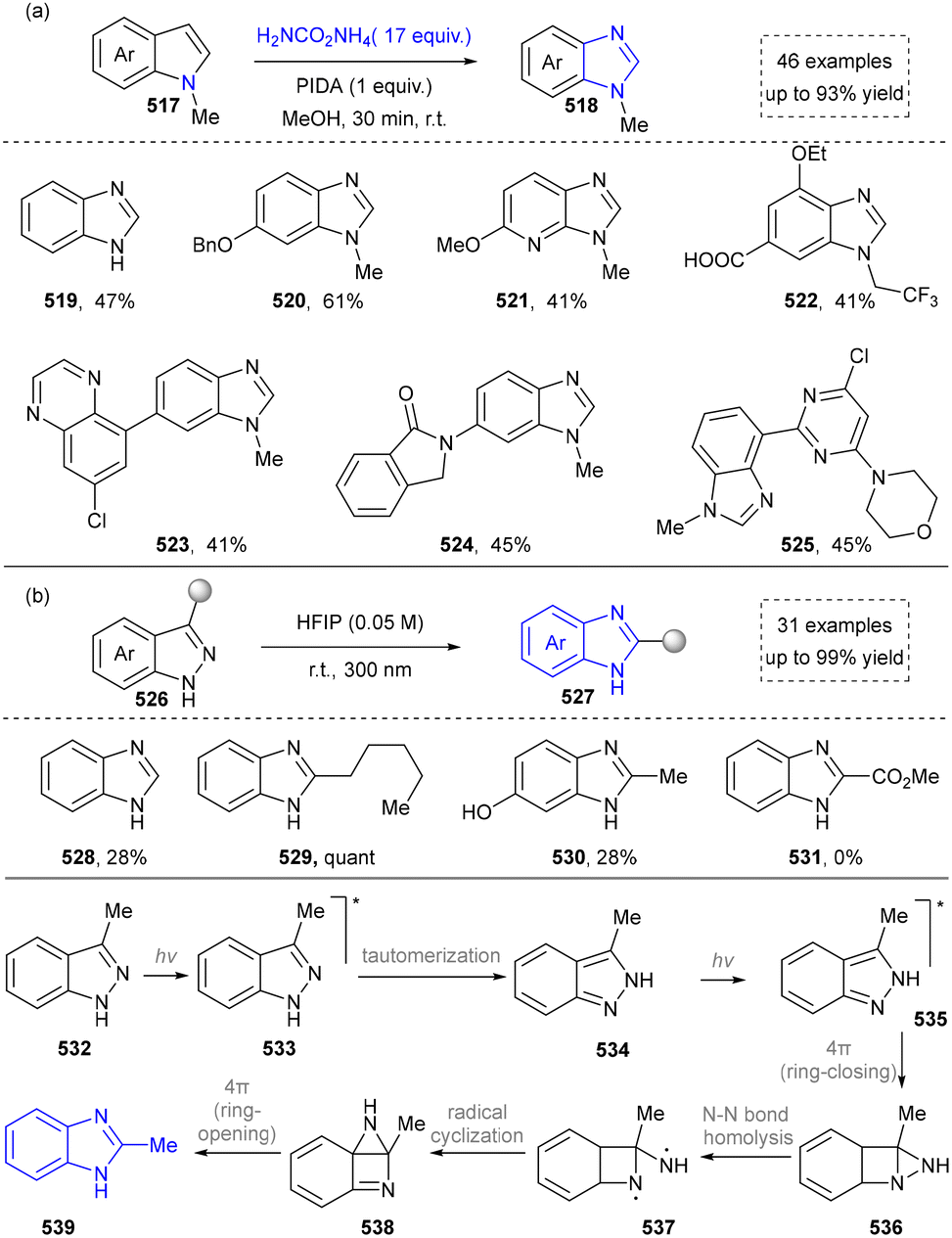 | ||
| Scheme 55 (a) Access to benzimidazole derivatives from indoles via carbon-nitrogen atom exchange. (b) Photochemical conversion of indazoles into benzimidazoles. | ||
The skeletal editing strategy for constructing benzimidazole frameworks remains in its early exploratory stages, with immense room for future development. The key challenge lies in advancing this technology from “proof-of-concept” to a “practical tool.” This transition depends on developing editing reactions with enhanced generality and selectivity, deepening the intelligent prediction of reaction mechanisms, and ultimately, integrating the approach with automated synthesis platforms. This integration will ultimately provide unprecedented efficiency for the construction and modification of molecules in drug discovery.
5. Three- and four-nitrogen-containing aromatic heterocycles: 1,2,3-triazoles, 1,2,4-triazoles, and tetrazoles
5.1 Synthesis of 1,2,3-triazoles
The synthesis of 1,2,3-triazoles holds profound importance in both fundamental and applied chemistry due to their unique structural and functional properties.274 In medicinal chemistry, 1,2,3-triazole serves as a stable bioisostere for labile functional groups (e.g., amides and esters), enhancing the metabolic stability while maintaining the key pharmacophoric interaction.274,275 This is exemplified by their incorporation into clinical agents such as the antifungal drug fluconazole and the anticancer agent cabozantinib.276 Thus, the synthesis of 1,2,3-triazole bridges fundamental research and technological innovation, driving progress across drug discovery, materials engineering, and chemical biology.277 However, the thermal cycloaddition reaction of azides to terminal alkynes remains the most prevalent method for 1,2,3-triazole synthesis.278 This section will focus on elucidating the pivotal role of emerging synthetic methodologies in the construction of 1,2,3-triazole scaffolds (including 1,2,3-triazole 1-oxides and 1,2,3-triazole 1-imines). | ||
| Scheme 56 Visible-light-assisted photocatalytic [3+2] reaction for the synthesis of 1,2,3-triazoles. | ||
Later, visible-light-mediated click chemistry for the construction of diverse 1,2,3-triazoles was further developed by several research groups.280 For instance, Pan's group developed a photocatalytic strategy under the irradiation of sunlight.281 Moreover, some novel heterogenous catalysts were designed and prepared to facilitate the synthesis of 1,2,3-triazoles under the irradiation of visible light, which further improved the efficiency and sustainability of these reactions.282–284
Besides click chemistry, the synthesis of 1,2,3-triazole by using hypervalent iodine(III) diazo reagents has recently been reported (Scheme 57).285 This unique diazo reagent has very powerful applications in organic synthesis due to their dual activities as electrophilic diazo transfer agents and oxidizing species. In this reaction, the hypervalent iodine diazo reagent (548) formed the radical intermediate (552) via the SET process, which reacted with N-Bn-quinoxalin2(1H)-one (547) and formed the final product (550, 551) after [3+2] cyclization. Gratifyingly, the obtained final products had novel structures since the triazole-fused heterocycles are highly important in drug designing.286
 | ||
| Scheme 57 Synthesis of 1,2,3-triazoles through photoredox-catalyzed [3+2] cyclization. | ||
Overall, the visible-light-promoted click chemistry to afford 1,2,3-triazoles was still the mainstream in this field. However, the other methods and nitrogen sources were applied to the synthesis of 1,2,3-triazoles, except the reported diazo compounds. The successful application of alternative nitrogen sources beyond traditional diazo compounds represents a significant and promising expansion of the methodological toolkit. This trend is expected to continue, paving the way for more sustainable, efficient, and structurally novel routes to these valuable heterocycles, ultimately broadening their potential in drug discovery.
 | ||
| Scheme 58 Electrochemical synthesis of 1,2,3-triazole 1-oxides. | ||
Inspired by this work, Fershtat's group also focused on the synthesis of 1,2,3-triazole 1-oxides through electrocatalysis.288 They improved the electrochemical conditions and enhanced the atom economy compared to previous reports. Moreover, plenty of 1,2,3-triazole 1-oxides (560–562) were afforded by this approach, using various oximinohydrazones (558, Scheme 59). To elucidate the mechanism, DFT studies and cyclic voltammetry were also performed, which provided a plausible reaction mechanism as shown below. The two-time single-electron oxidation to afford diazo species (566) was the key to this success. In addition, the subsequent cyclization and proton elimination resulted in the formation of 1,2,3-triazole 1-oxide. The same group also described the electrosynthesis of 1,2,3-triazole 1-imines via a similar strategy in 2024, expanding the structural diversity of 1,2,3-triazoles.289
 | ||
| Scheme 59 Electrochemical N–N bond forming reactions: direct access to 1,2,3-triazoles. | ||
Besides the use of oximinohydrazone, Xu's group disclosed a convenient approach to synthesize [1,2,3]triazolo[1,5-a]pyridines via the electrochemical dehydrogenative cyclization of hydrazones of acylpyridines.290 The concept was similar to the above-mentioned work. The hydrazone (569) underwent a two-electron oxidation with the help of carbon at the anode to generate a diazo intermediate (575). However, the substrate scope (571–574) was relatively limited due to the unavailability of the starting material (Scheme 60).
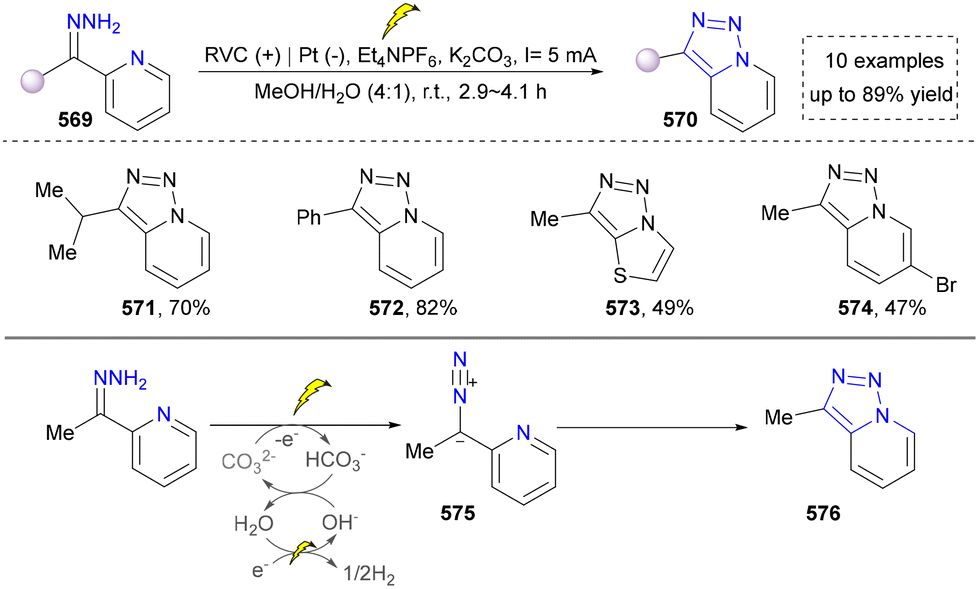 | ||
| Scheme 60 Electrochemical synthesis of 1,2,3-triazoles through dehydrogenative cyclization. | ||
Bera's group further reported that benzoyl azides could also undergo cycloaddition with propargyl alcohols to afford 4,5-disubstituted-1,2,3-triazole scaffolds (Scheme 61, 579–582). They also conducted the bioactivity screening via CLSM (confocal laser scanning microscopy) imaging, fluorescence and TCSPC (time-correlated single-photon counting) experiments, indicating the potential of these molecules in drug discovery.291
 | ||
| Scheme 61 Electrochemical synthesis of the novel N-benzoyl-1,2,3-triazole derivatives. | ||
Oximinohydrazones and benzoyl azides have been employed in the synthesis of 1,2,3-triazole, leveraging the unique redox properties and mild conditions of electrosynthesis to potentially access diverse substitution patterns. However, to date, the development remains at an early stage, and no significant practical applications in synthesizing complex functional molecules have been documented so far. Consequently, the successful implementation of these electrochemical strategies for the practical synthesis of pharmaceutical agents or their analogues is eagerly awaited, as it would mark a critical transition from conceptual demonstration to applied synthetic utility.
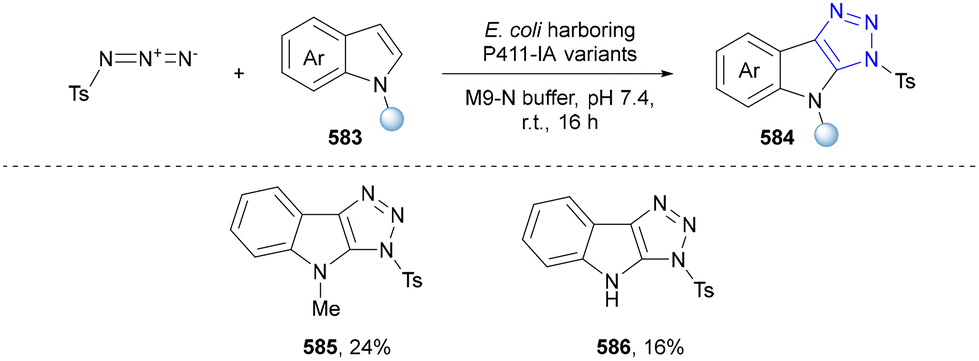 | ||
| Scheme 62 Enzymatic synthesis of triazoles via indole-azide cycloaddition. | ||
In 2020, Du's group and Huang's group successively reported the biosynthetic pathway of triazole-bearing anti-metabolite 8-azaguanine (8-AZG).292,293 The triazolopyrimidine scaffold (588) could be assembled through either a non-enzymatic or an enzymatic cascade catalysed by bacterial nitric oxide synthase (NOS) ptnF or 8-AzgA/B, with nitric oxide as a building block composing the 1,2,3-triazole moiety. The final hydrolytic cleavage of ribose 5′-monophosphate moiety from the intermediate (590) was catalysed by 8-AzgD to yield the final product 8-AZG (Scheme 63).
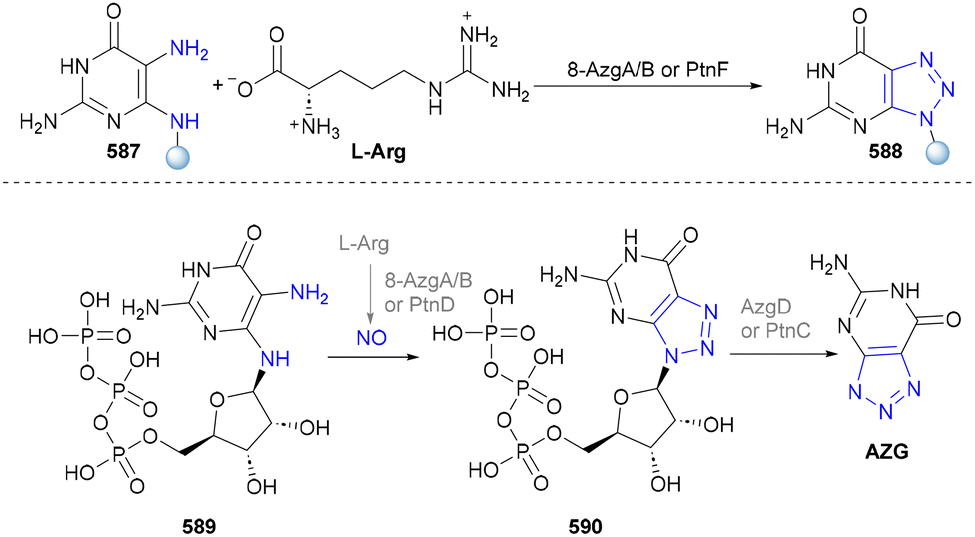 | ||
| Scheme 63 Enzymatic synthesis of the triazolopyrimidine scaffold. | ||
Enzymes were also used to afford substrates for the chemical synthesis of triazoles.293–295 In 2021, Yun and co-workers reported a multi-enzymatic cascade system for the synthesis of the sitagliptin intermediate, which was converted into sitagliptin via chemical synthesis, with benzylamine as an amino donor. The system was composed of transaminase (TA), esterase, aldehyde reductase (AHR), and formate dehydrogenase (FDH).296
Although relatively few reports have focused on the enzymatic synthesis of triazoles, the Arnold group's work demonstrated that the engineered P450 exhibits the potential to evolve indole-azide cycloaddition activity. Therefore, more enzymatic synthesis of triazoles can be realized in the future through further mining and engineering of the enzyme.
5.2 Synthesis of 1,2,4-triazoles
As a nitrogen-rich heterocyclic system, 1,2,4-triazole exhibits excellent thermal stability and the ability to form multiple hydrogen bonds, making it particularly valuable in drug designing.297–299 This scaffold is a special structure in medicinal chemistry and plays an important role in antifungal agents (such as fluconazole derivatives), anticancer drugs and central nervous system therapeutics.300 Classical strategies for 1,2,4-triazole derivatives involve cyclization of hydrazides/hydrazines, primary amine cyclization with nitrogen–oxygen exchange, and reactions employing guanidines, amidines, or ureas.301 This section comprehensively discusses the synthesis of pharmacologically significant 1,2,4-triazole scaffolds by emerging synthesis methods.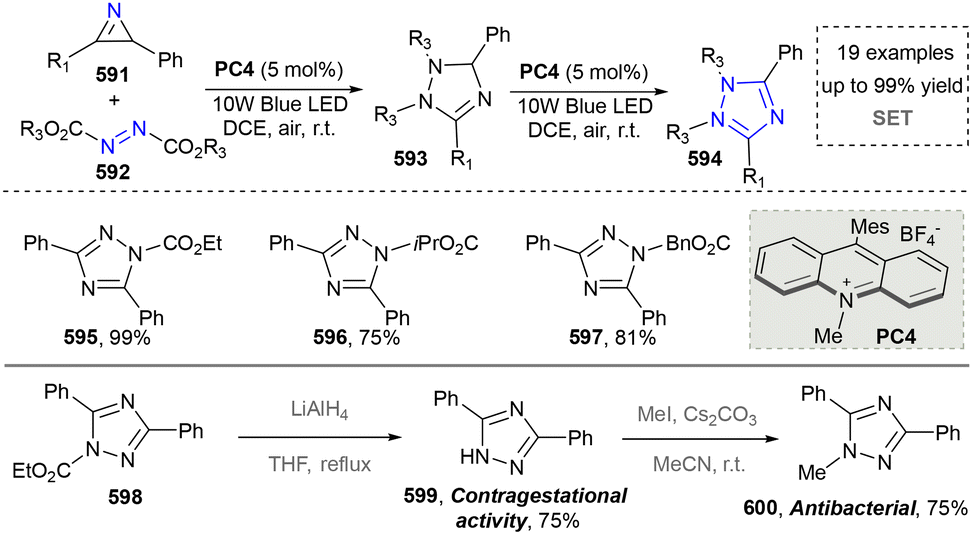 | ||
| Scheme 64 Visible-light-induced cyclization reactions for the synthesis of 1,2,4-triazoles. | ||
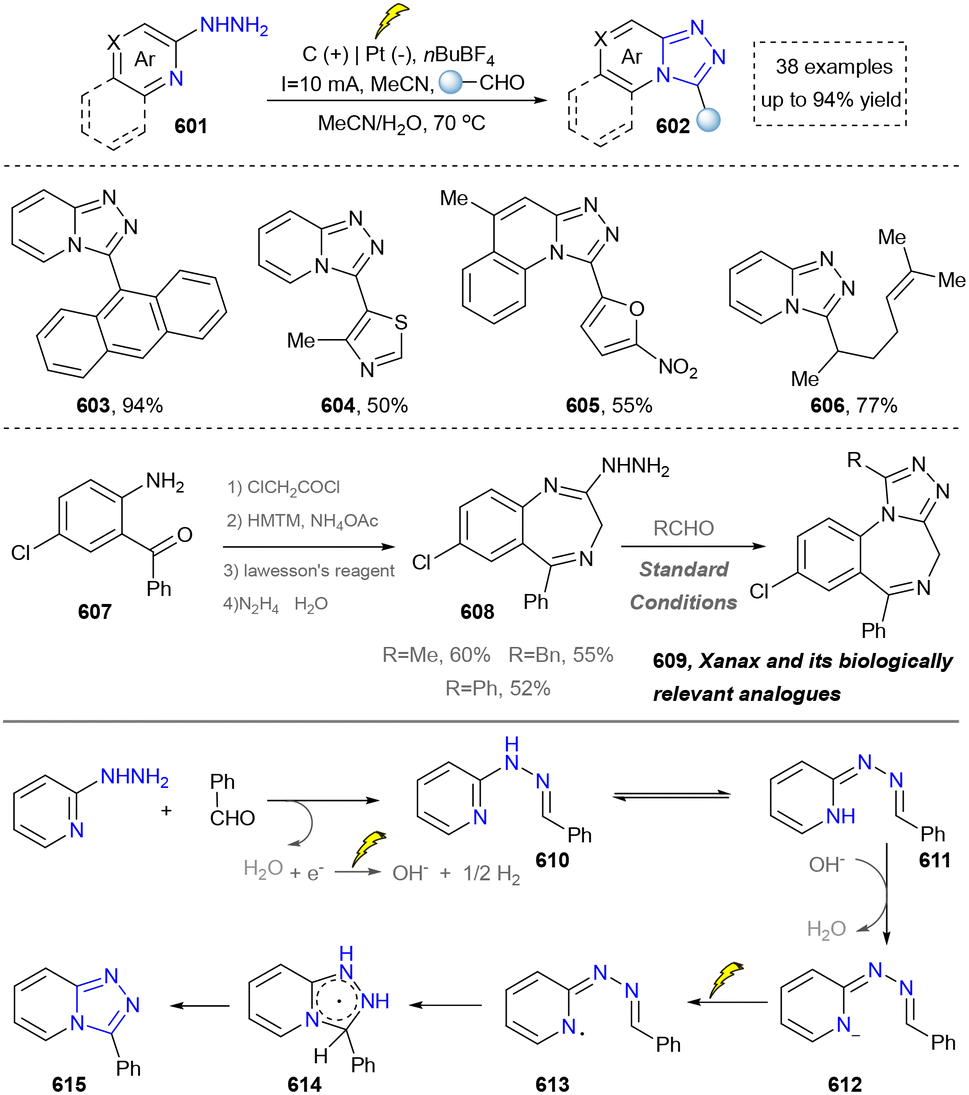 | ||
| Scheme 65 Electrochemical synthesis of 1,2,4-triazole-fused heterocycles. | ||
Later, the same group further developed their method to construct 1,2,4 triazolo[1,5-a]pyridines, the concept was similar to their previous work, and they used N-(2-pyridyl)benzamide as the starting material and the nitrogen radical was formed under the electrocatalytic condition. With this method, various 1,2,4-triazolo[1,5-a]pyridines were efficiently synthesized. More importantly, they also applied this strategy into the synthesis of several bioactive molecules (Scheme 66).303 The final bioactive compound (622) was accessible through the synthesis of their key precursor (620) under their standard conditions, followed by a streamlined conversion involving only a few simple steps. However, it should be noted that the hydroxy group was not compatible within this system.
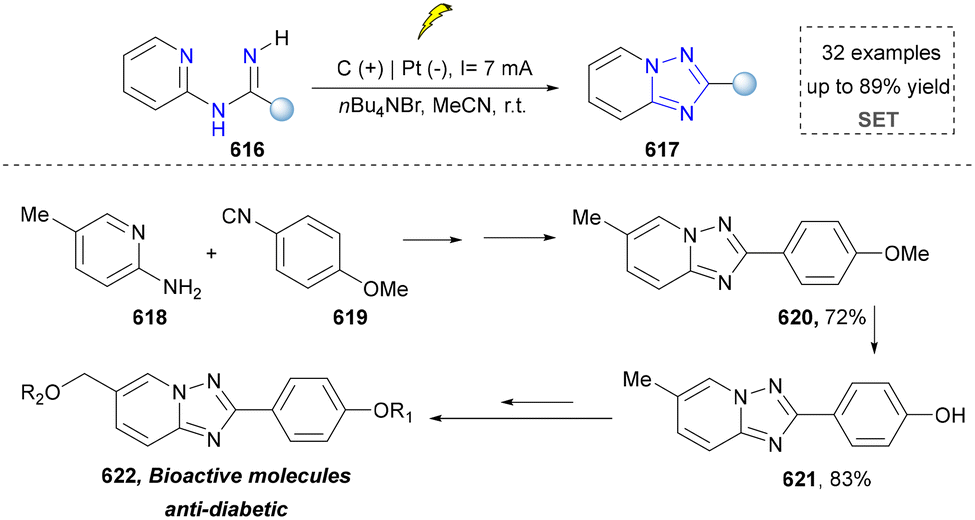 | ||
| Scheme 66 Synthesis of 1,2,4-triazoles via intramolecular electrochemical dehydrogenation. | ||
Overall, electrochemical synthesis has extended the scope of 1,2,4-triazoles, especially the construction of 1,2,4-triazole-fused heterocycles. Moreover, several bioactive compounds were afforded through electrochemical methods, such as Xanax and its related compounds, indicating their potential in drug synthesis. However, there is still some space to utilize other nitrogen sources or develop multi-component reactions to further diversify the 1,2,4-triazoles.
5.3 Synthesis of tetrazoles
As a nitrogen-rich heterocyclic ring (containing four nitrogen atoms), the tetrazole ring exhibits remarkable metabolic stability and can be used as a bioisosteric of carboxylic acids, significantly improving the pharmacokinetic profile of drug candidates.304 They play an important role in several FDA-approved drugs, including antihypertensive drugs (such as losartan) and antibiotics (such as cefazolin).305 The most common traditional approach for tetrazole synthesis involves cycloaddition reactions between azides and synthons such as nitriles, isonitriles, and imino functional groups.306 Recent advances in green synthesis methods including photocatalysis and electrochemical methods have further enhanced the sustainability of tetrazole synthesis. The structural versatility of tetrazoles, which allows functionalization at multiple locations, continues to drive innovation in medicinal chemistry, agrochemical development, and functional material design, making them indispensable materials in modern chemical research.307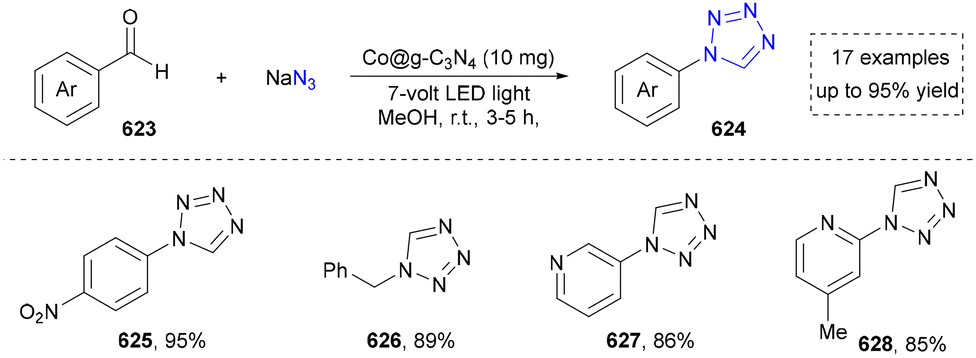 | ||
| Scheme 67 Visible-light-driven synthesis of 1H-tetrazoles through [3+2] cycloaddition. | ||
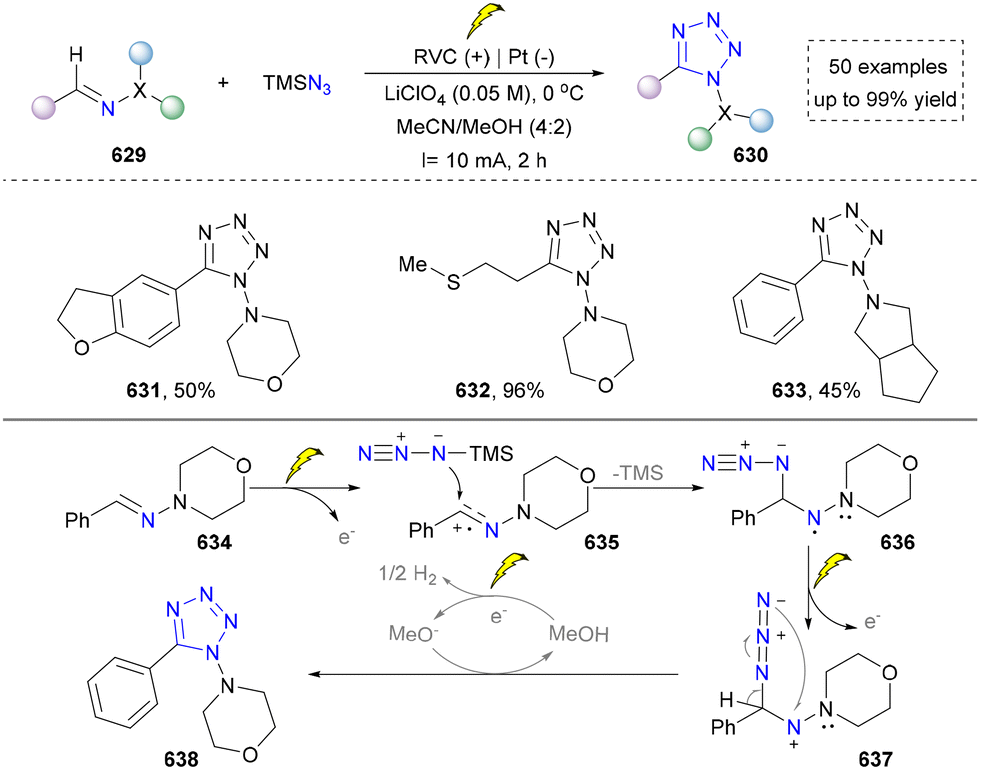 | ||
| Scheme 68 Electrochemical synthesis of tetrazoles via the [3+2] cycloaddition of azides with hydrazones. | ||
Ye's group further developed this strategy and reported a multi-component reaction (MCR) to construct tetrazoles by electrochemical synthesis (Scheme 69).42 With the advantage of the MCR, this strategy strongly expanded the structural diversity of tetrazoles by using abundant and inexpensive chemical feedstocks. Particularly, the synthesis of tetrazoles was controlled by using alkenes or alkyls as substances. Besides the synthesis of simple molecules, this strategy was applied into the late-stage modification to afford ibuprofen and fenofibric acid analogues (643, 644) under standard conditions. The electrochemical process also demonstrated excellent compatibility with a range of functional groups, including esters and ethers. These results demonstrated that this approach provided a green and sustainable synthetic methodology for the structural optimization and modification of drug molecules.
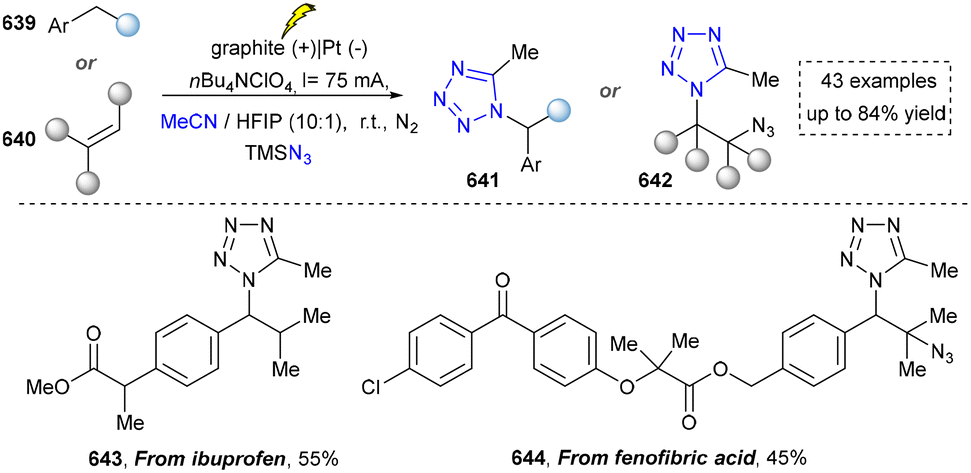 | ||
| Scheme 69 Electrochemical multicomponent tetrazolation and vicinal azidotetrazolation. | ||
Currently, prefunctionalized organic azides remained the preferred nitrogen source for both photochemical and electrochemical synthesis. In contrast, research on the precise use of stable, readily available alternative precursors such as sulfonyl hydrazides, organic amines, or nitro compounds for reactive nitrogen species remains substantially underdeveloped. Perhaps, some novel strategies using different nitrogen sources or three- or four-component reactions can be achieved in the future.
6. One-oxygen-containing aromatic heterocycles: furans and benzofurans
6.1 Synthesis of furans
As a privileged oxygen-containing heterocycle, the furan has been a versatile component in drug development, with many biologically active compounds containing this motif, including antiviral agents and anticancer drugs.310 The structural versatility of furans stems from the combination of their aromatic character (6π-electronic system) with the electronic effects of the oxoheterocyclic atoms, resulting in multiple modes of functionalisation.279 Traditional methods for the synthesis of furans include the Paal–Knorr synthesis and Feist–Bénary synthesis.311,312 This section provides a comprehensive discussion on the synthesis of furan scaffolds of pharmacological interest through emerging synthetic approaches.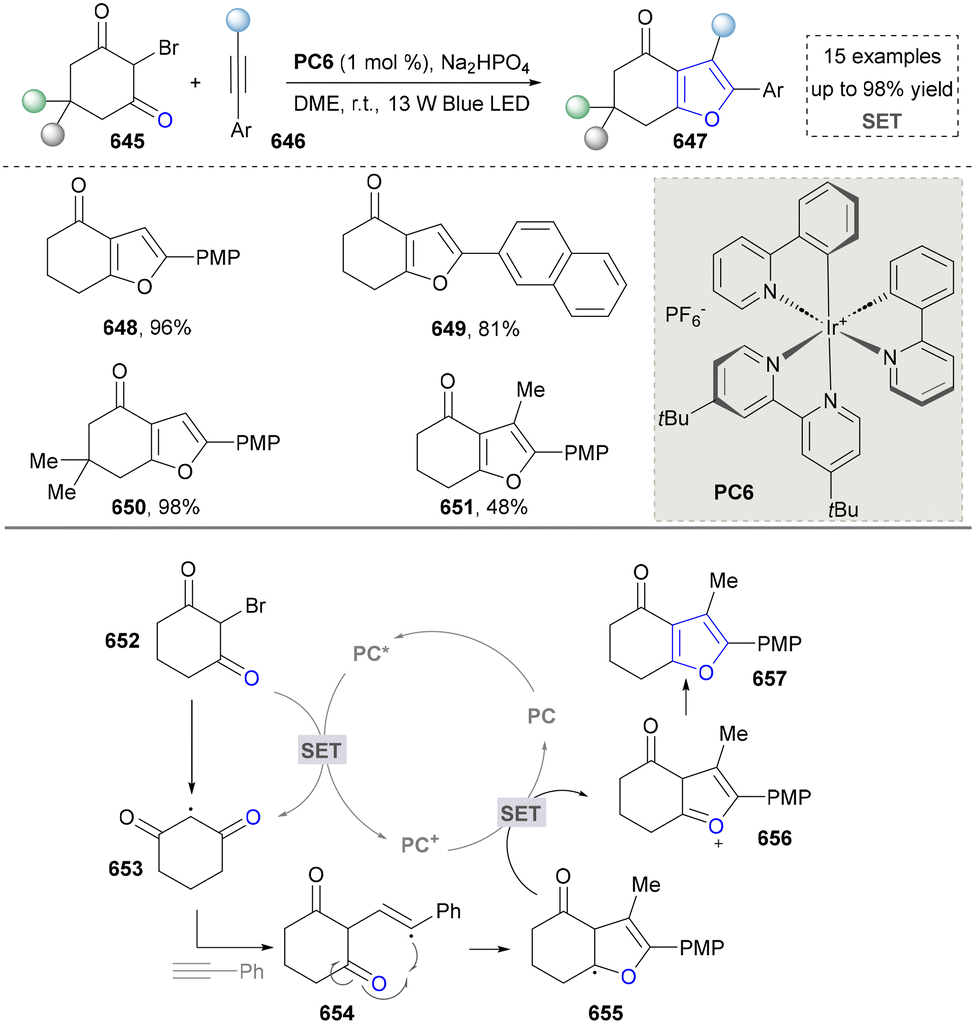 | ||
| Scheme 70 Synthesis of polysubstituted furans from cyclopropyl ketones. | ||
Inspired by this work, several methods have been reported to expand the structural diversity of furans by using various radical precursors.314–317 For instance, Wu's group disclosed that the combination of styrenes and α-alkyl ketone radicals could generate furans efficiently via photoredox catalysis.318 In 2022, a double-radical-polar crossover reaction was also described, in which an α-halo ketone was used as the radical precursor, which further reacted with various allenamides to afford functionalized 2-aminofuran.319 It should be noted that 1,3-diones without the activation of halogenation were also reported by Lei's group, and the additional persulfate salt could enable the direct hydrogen atom transfer (HAT) of the 1,3-dione.320
In addition, Xia and co-workers utilized cyclopropyl ketones as precursors for furan synthesis. Single-electron oxidation triggered ring opening to form a key unsaturated ketone intermediate (Scheme 71), followed by oxidative annulation to furnish diverse furans (660–662). However, CBr4 as a stoichiometric oxidant was not friendly to the green synthesis. Later, the direct alkene isomerization of in situ formed unsaturated ketone for the synthesis of polysubstituted furans has also been reported under the irradiation of UV light.321 In 2018, Nithyanandhan's group reported that O-acetylated (3′,5′-dimethylphenyl) as a starting material could be activated directly under the irradiation of light due to the good photophysical properties, but the structural diversity was very poor.322 Similarly, the visible-light photoredox-catalysed reaction of enynones with 2,2,2-trifluoroacetamide to access polysubstituted furfuryl trifluoroacetamide derivatives under mild reaction conditions was also reported.323 Building on previous studies, Donohoe's team utilized photoinduced alkene isomerization in 2019 to achieve efficient synthesis of polysubstituted furans under neutral conditions, thereby expanding the toolkit for the diversification of furans.324
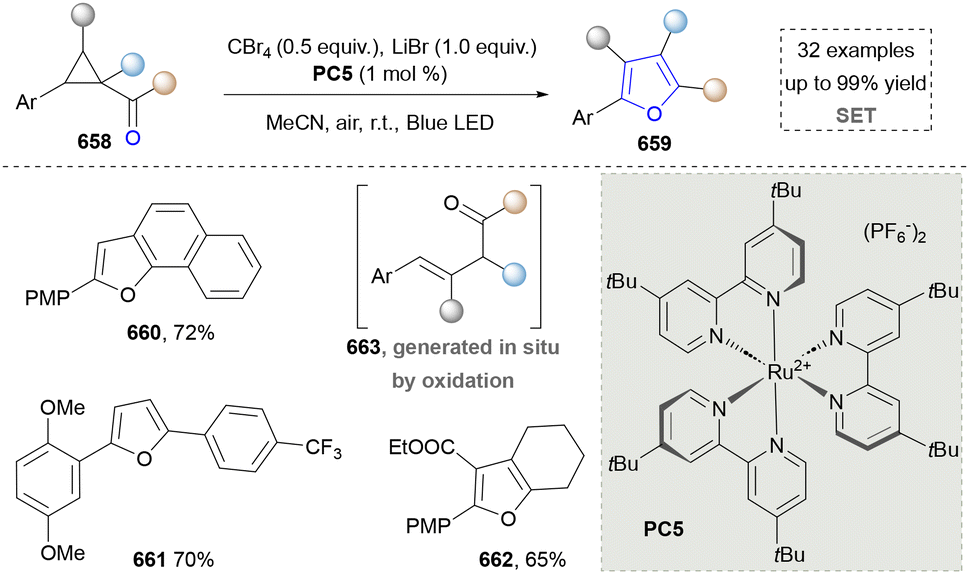 | ||
| Scheme 71 Synthesis of polysubstituted furans from cyclopropyl ketones. | ||
Later, Koenig's group significantly improved the sustainability of this strategy by the utilisation of carbon dioxide under visible light irradiation (Scheme 72). They applied 1,3-diketone as a starting material, which was deprotonated by Cs2CO3 to generate the corresponding enolate. After the SET process, it was oxidized to generate the radical species (670). 1,3-Diketone also reacted with CO2 to obtain the intermediate (671), which subsequently reacted with radical species to form acylcyclopropane (675) as a key intermediate, which further generated the final furan product through different pathways. It was also determined that CO2 was incorporated at the diketone enolic-OH position, which was key to success and facilitated the cleavage of a C–O bond during the rearrangement of acyclopropane intermediate.325 Acceptor/acceptor diazoalkanes have been successfully employed in the photocatalytic synthesis of furans through reactions with terminal alkynes. The key to this strategy lies in the energy transfer-mediated formation of triplet carbene intermediates, which facilitates the cyclization process to construct the furan heterocycle.326 Very recently, vinyl sulfoxonium ylides have also been proved as a suitable substrate to react with azides to form tri-substituted furans by Liu's group.327
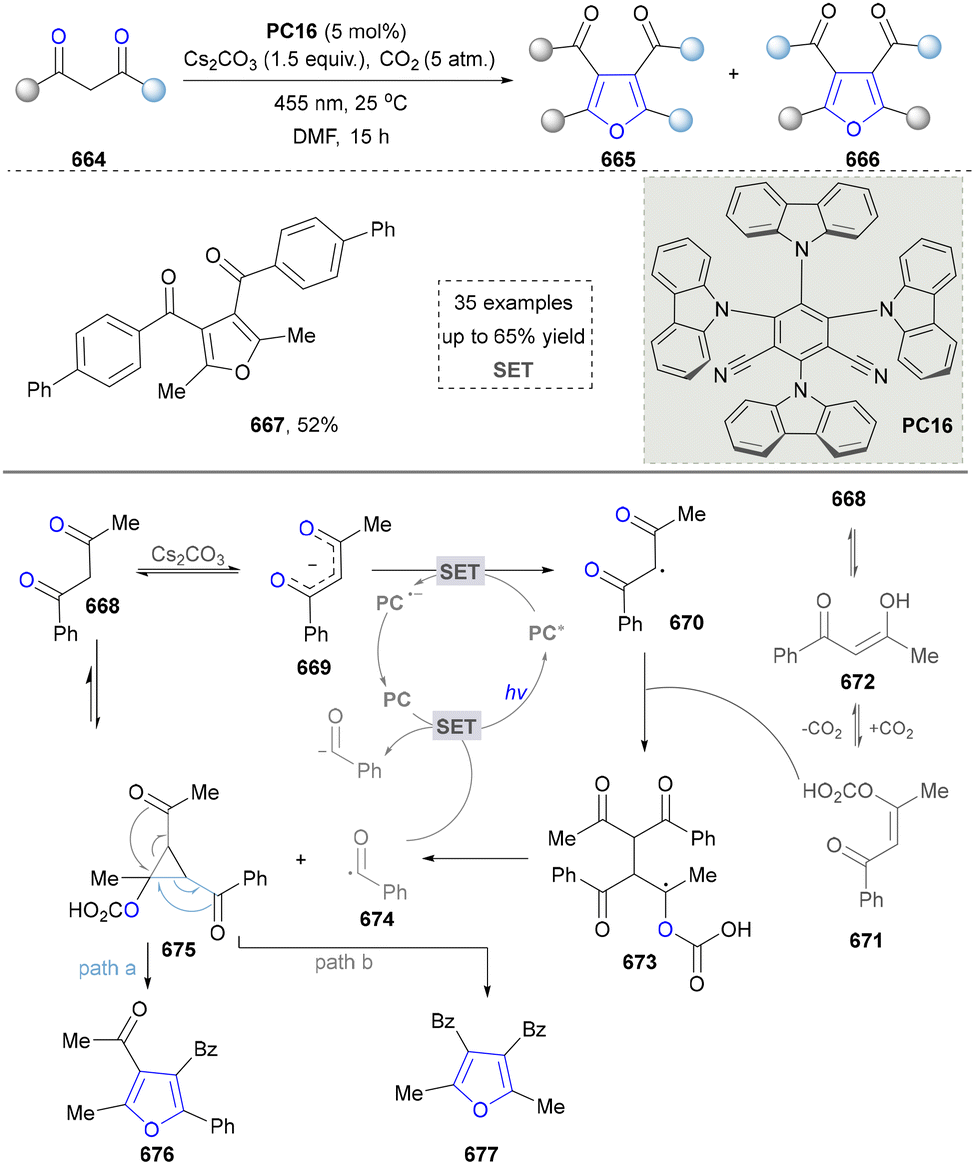 | ||
| Scheme 72 Photocatalytic synthesis of furans promoted by carbon dioxide. | ||
Afterwards, Wang's group described an elegant strategy to construct furan-fused dihydroazepines under direct photolysis without the addition of photocatalysts and additives (Scheme 73).328 By the design and synthesis of a substance containing conjugated dienes (683), the amino-substituted furan (680–682) was obtained directly by the cycloaddition with isocyanide, which showed obvious light absorption under light irradiation. This intermediate itself worked as a photosensitizer to promote the C–N bond formation through 7-endo-dig cyclization to generate the complicated furan. This strategy also featured the operational simplicity and gram-scale synthesis. The late-stage applications into complex substrates were also achieved successfully, indicating the potential of this strategy in drug synthesis.
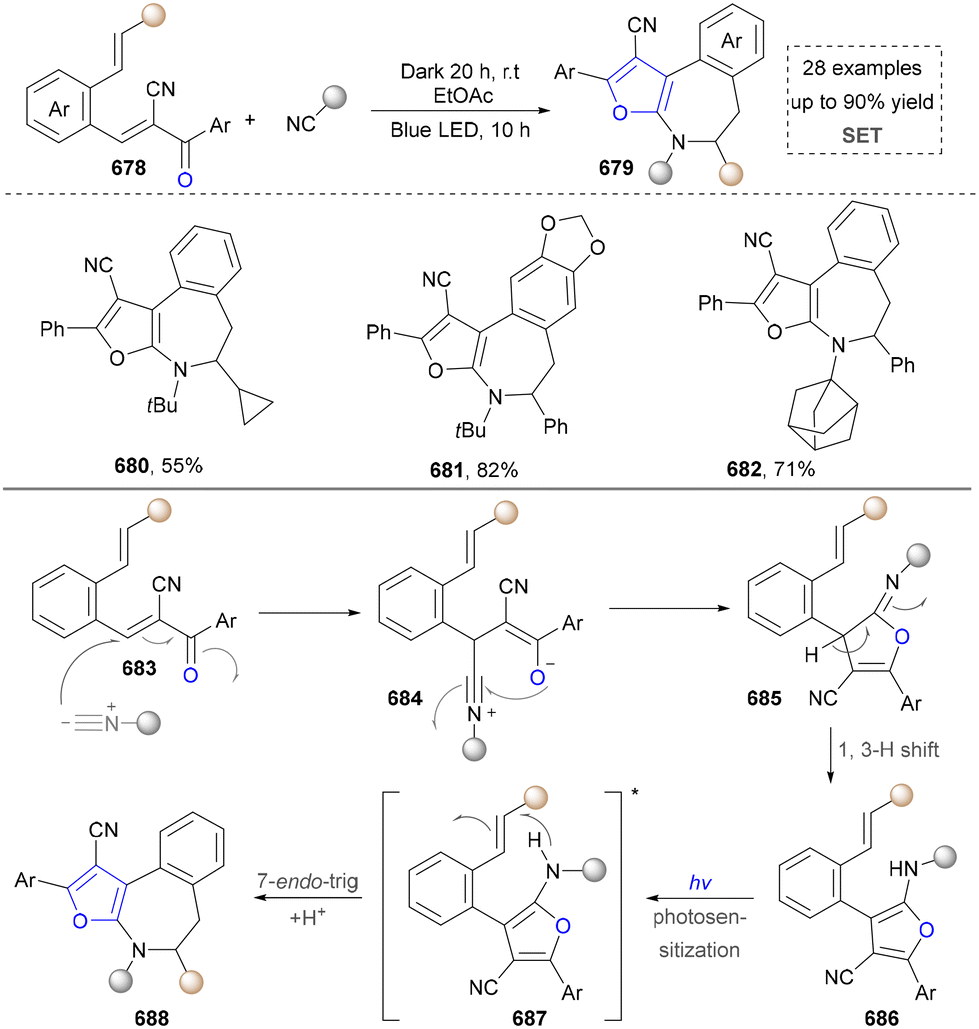 | ||
| Scheme 73 Photocatalyst-free and transition-metal-free synthesis of furans. | ||
In summary, the use of ketones as oxygen sources for the synthesis of furans has been extensively and effectively explored through various methods. It is anticipated that other oxygen sources such as alcohols and molecular oxygen could also be successfully applied in this field. Moreover, photochemical synthesis of furans circumvents the use of stoichiometric hazardous oxidants while also enabling late-stage furan cyclization of complex molecules bearing acid-, base-, or heat-sensitive functional groups, thus providing a powerful tool for the rapid diversification of leading compounds.
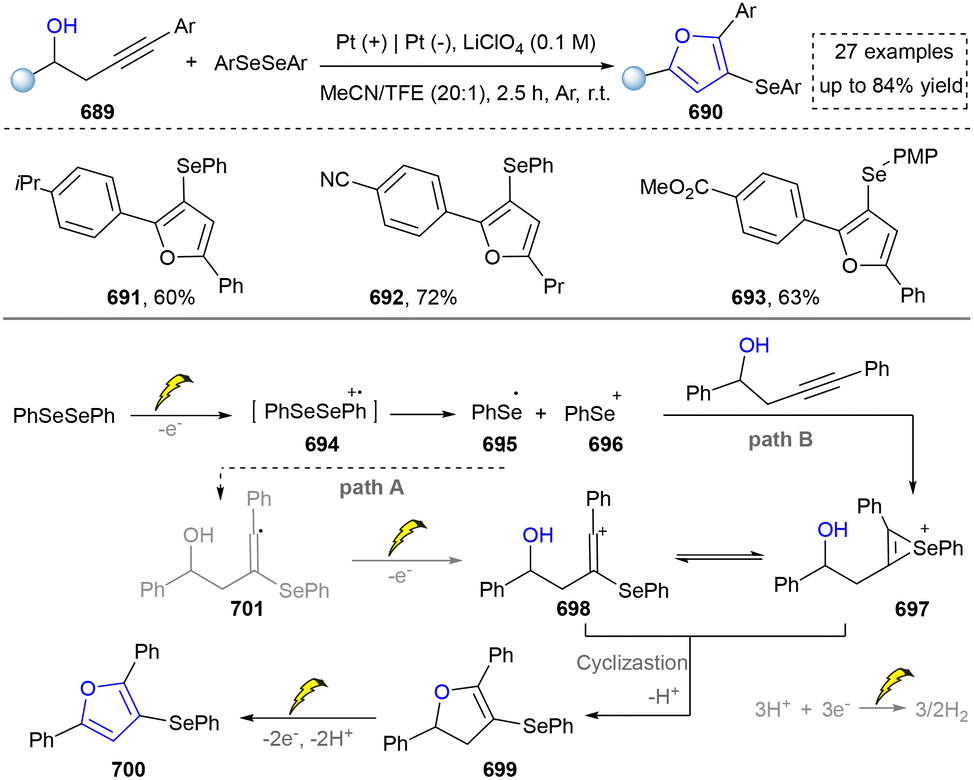 | ||
| Scheme 74 Synthesis of polysubstituted furans through electrochemical selenocyclization. | ||
Later, Tang's group also described a strategy to afford furans (705–707) via the annulation of allenes (Scheme 75). This electrochemical method avoided the traditional use of an external oxidant.51 Similarly, Zhao's group reported the electrocatalysis approach in which 1,3-dione and alkynes were used as starting materials, a known concept, but the electrocatalysis approach avoided the use of an additional oxidant.330
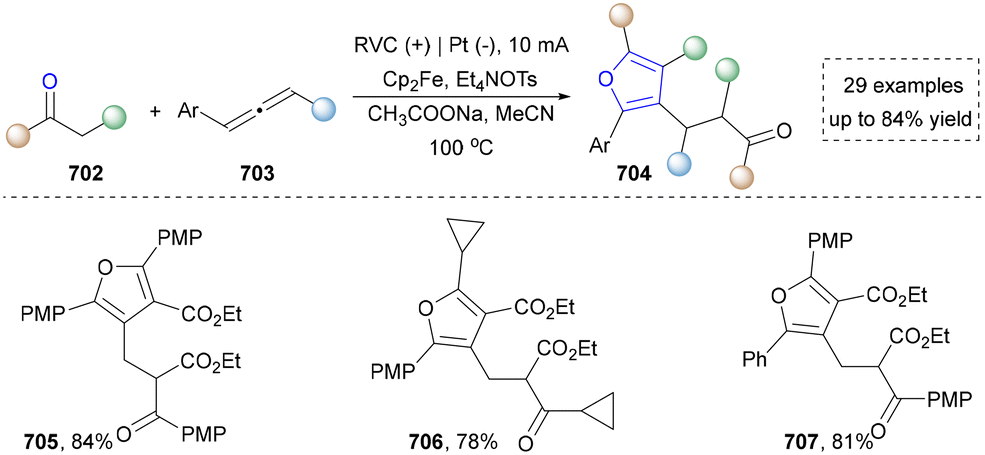 | ||
| Scheme 75 Electrochemically-mediated C–H functionalization to construct furans. | ||
Overall, electrochemical synthesis has emerged as a versatile and powerful platform for furan preparation, continuously pushing the boundaries of accessible structures. A key advancement lies in the innovative use of alcohols as green and atom-economical oxygen sources, enabling dehydrogenative cyclization reactions under mild conditions. Furthermore, the scope of cyclization partners has been significantly expanded beyond traditional substrates to include allenes, alkynes, and difunctionalized olefins. This diversification allows for the direct construction of highly substituted and functionalized furan cores, including complex benzo[b]furan scaffolds, from simple precursors, thereby greatly broadening the structural diversity and complexity achievable. Future endeavors will focus on achieving more precise oxygen-atom transfer from inert and greener oxygen sources.
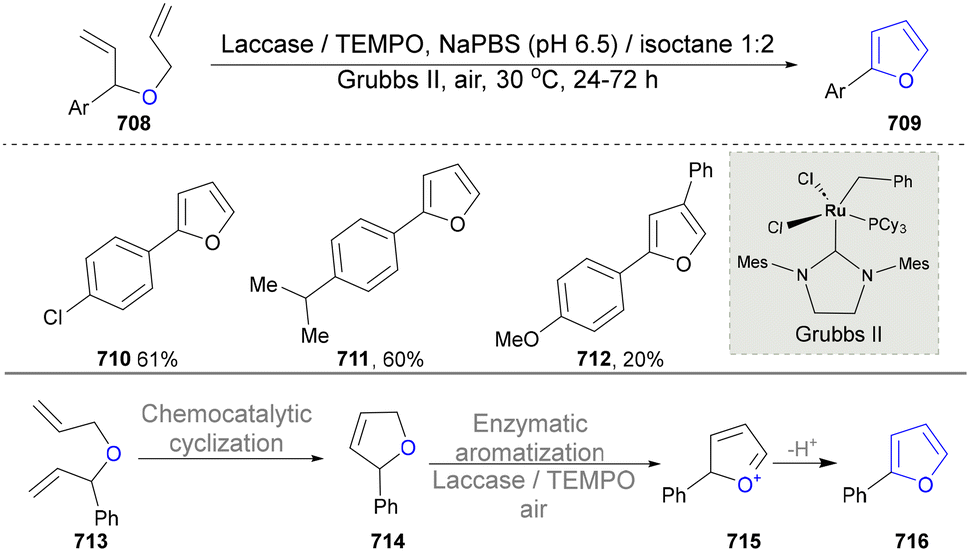 | ||
| Scheme 76 Enzymatic synthesis of furans by T. versicolor laccase. | ||
In 2023, Ouyang's group reported an environmentally sustainable chemoenzymatic strategy to generate n-butyl-5-formyl-2-furancarboxylate (nBu-FFCA), the precursor of furan-2,5-dicarboxylic acid (FDCA) from commercial sodium gluconate (GA).332 GA was catalysed by gluconate 5-dehydrogenase from Gluconobacter oxydans (Ga5DH) coupled with the short-chain reductase from Pichia stipitis (PsCR) to form 5-keto-D-gluconic acid (5KGA), which subsequently reacted to produce nBu-FFC via dehydration and esterification.
It is attractive to convert cheap biomass into chemicals directly through furanic platform molecules. The chemo- and bio-catalytic synthesis of 2,5-bis(hydroxymethyl)furan (BHMF), a crucial building block for biobased polymers and fine chemicals, has made great progress in past decades.333–336 In 2024, Liao et al.337 established an efficient chemobiocatalytic one-pot process for converting glucose into BHMF by combining CaCl2 with engineered Saccharomyces cerevisiae harboring MgADH1 (an HMF-resistant ADH from Meyerozyma guilliermondii SC1103), without the requirement of intensive 5-hydroxymethylfurfural (HMF) purification335,338 or expensive fructose and sugar syrup.339–343
Although only laccase-catalysed furan ring formation has been reported, there are various enzymes involved in the formation of furan ring in natural products. For example, AteafoF from Aspergillus terreus catalysed the formation of furan ring in asperfuranone, an azaphilone first isolated from A. nidulans.344 In the biosynthetic pathway of furanocoumarin, the formation of the furan ring involved one prenyltransferase (PT) and two cytochrome P450s.345–347 In Salvia miltiorrhiza Bunge, CYP71Ds catalysed the formation of the characteristic furan ring of tanshinones,348 then a 2-oxoglutarate-dependent dioxygenase converted dihydrofuran to furan.349 We will not provide a detailed discussion of these examples here; however, they have a great potential for enzymatic synthesis of furan, like laccase.
6.2 Synthesis of benzofurans
Considering that benzofurans have exceptional structural and pharmacological properties, the synthesis of benzofuran derivatives represents a critical research area in organic synthesis and medicinal chemistry.350 Moreover, as a privileged heterocyclic scaffold, benzofuran combines the aromatic stability of benzene with the electronic diversity of the furan ring, creating unique opportunities for drug designing and material development.351 In medicinal chemistry, benzofuran-containing compounds demonstrate broad bioactivity,352 featuring prominently in FDA-approved drugs such as the anticoagulant dronedarone and the antipsychotic amiodarone.353 Traditional benzofuran synthesis relies on Lewis acid-catalysed or transition metal-catalysed oxidative cyclization of substituted benzene derivatives, though the requirement for metal catalysts, ligands, and additives complicates these processes for industrial applications.354,355 This section focuses on highlighting the groundbreaking contributions of emerging synthetic methodologies in the construction of benzofurans and related drug analogues.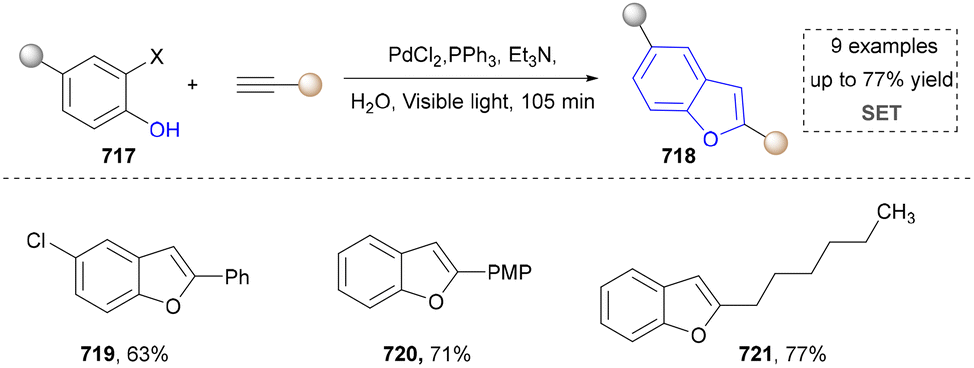 | ||
| Scheme 77 The formation of benzofurans by the cyclisation of ortho-halophenols and terminal alkynes. | ||
To further achieve the photochemical synthesis of benzofurans, Kumar's group described an efficient method to construct benzofurans by using trifluoromethylating Langlois' reagent as a radical precursor to release the –CF3 radical after single electron transfer event (Scheme 78).159 The trifluoromethyl radical further reacted with a vinylic double bond to form the radical species (728) which further underwent 5-exo-dig cyclization and oxidation to form the carbocation (729). At last, the subsequent addition of water to the vinylic cation (731) with the elimination of hydrogen gas provided the final products (724–726). It should be noted that this strategy was very powerful to synthesize diverse five-membered heteroarenes including benzofurans and indoles indicating the potential of this strategy in the synthesis of drugs. In addition, various radical precursors such as arylsulfinic acids, thiosulfonates and aryldiazonium salts were applied in the synthesis of diverse benzofurans by this similar strategy.50,358–362 α-Azidochalcones was also designed to achieve the activation of an alkene group, which further underwent the cyclization to furnish the benzofuran-2-amines.363
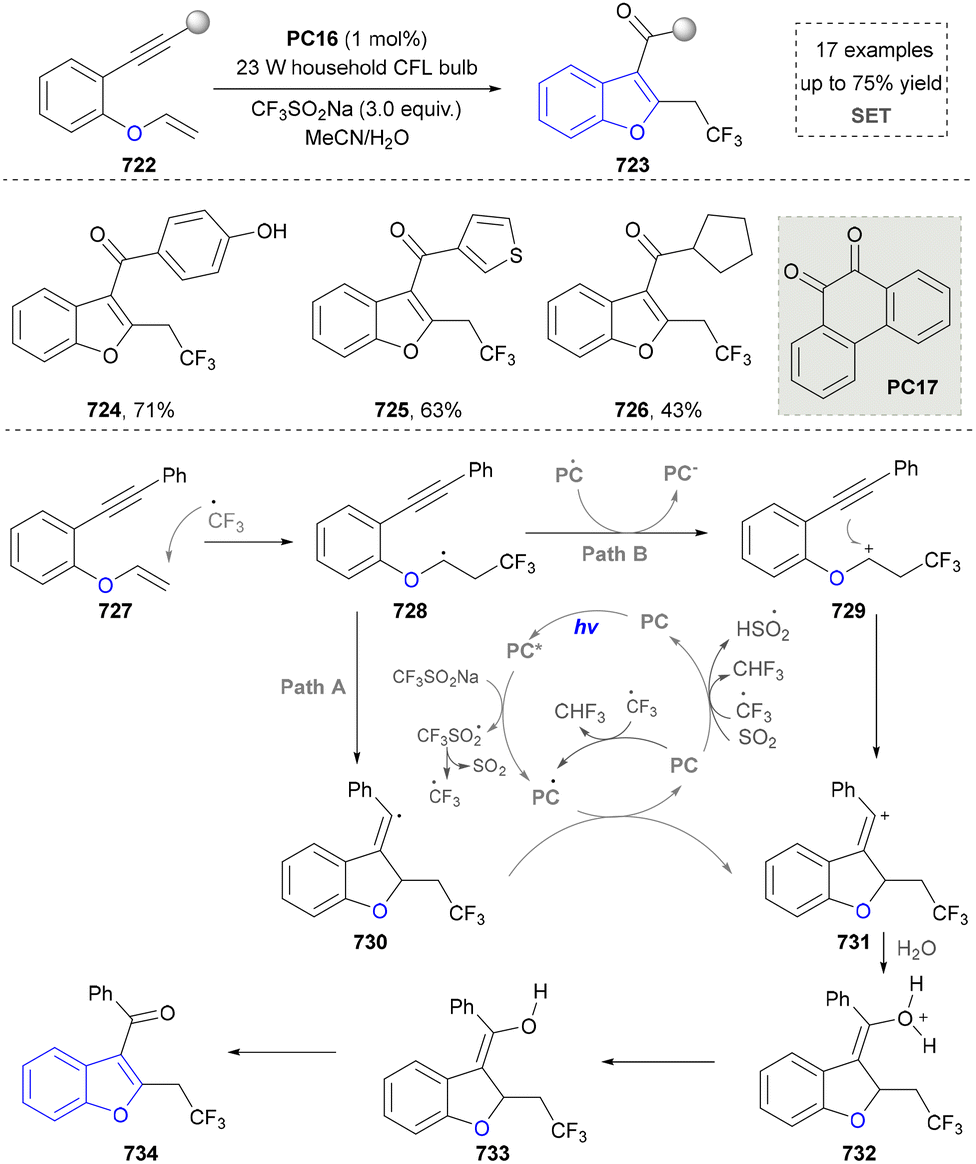 | ||
| Scheme 78 Dehydrogenative cascade trifluoromethylation and oxidation. | ||
In 2019, Zhou's group designed benzofuranone-based spiroisochromenes to investigate the oxa-6π electrocyclic reaction of cis,cis-1,8-dioxatetraene. The targeted spiroisochromene derivatives were activated under the irradiation of 390 nm light and were transformed into the 5H-benzofuro[3,2-c]isochromene derivatives, a class of scaffolds with pronounced bioactive potential (Scheme 79, 737–739). Interestingly, the formed product was easily turned back to the original starting material under heating conditions.53 The direct intramolecular synthesis of dibenzofuran derivative was also reported by using 2-(2′-aminophenyl) phenol as a starting material.364
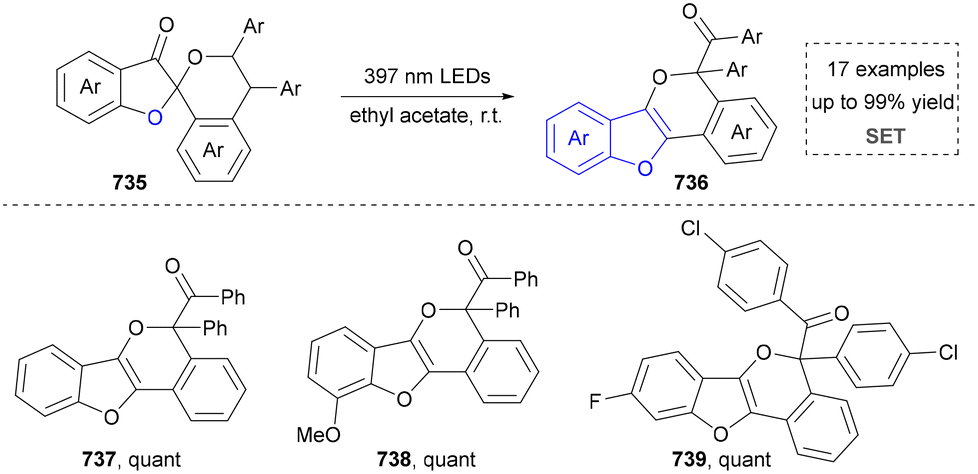 | ||
| Scheme 79 Electrocyclic reactions of 1,8-dioxatetraene. | ||
Hashmi's group also reported a novel method to access the diborylated benzofurans, indoles, and benzothiophenes by using photocatalysis (Scheme 80).365 Since the geminal diboronates have attracted great attention due to their unique bioactivities, the synthesis of heteroarenes bearing five-membered rings with diboronates is highly important in medicinal chemistry. The mechanistic experiments in this work proved the generation of new photosensitive gold complexes from dinuclear gold catalysts with Na2CO3 under blue LED illumination. With the activation of alkyne via energy transfer, the product (742–745) was formed via subsequent cyclization. Moreover, 10 gram-scale synthesis was also achieved, indicating the practicability of this method. This strategy pioneers the gem-diborylation of three core heteroaromatic scaffolds, namely benzofurans, indoles, and benzothiophenes, effectively addressing a critical synthetic gap for these biologically essential building blocks.
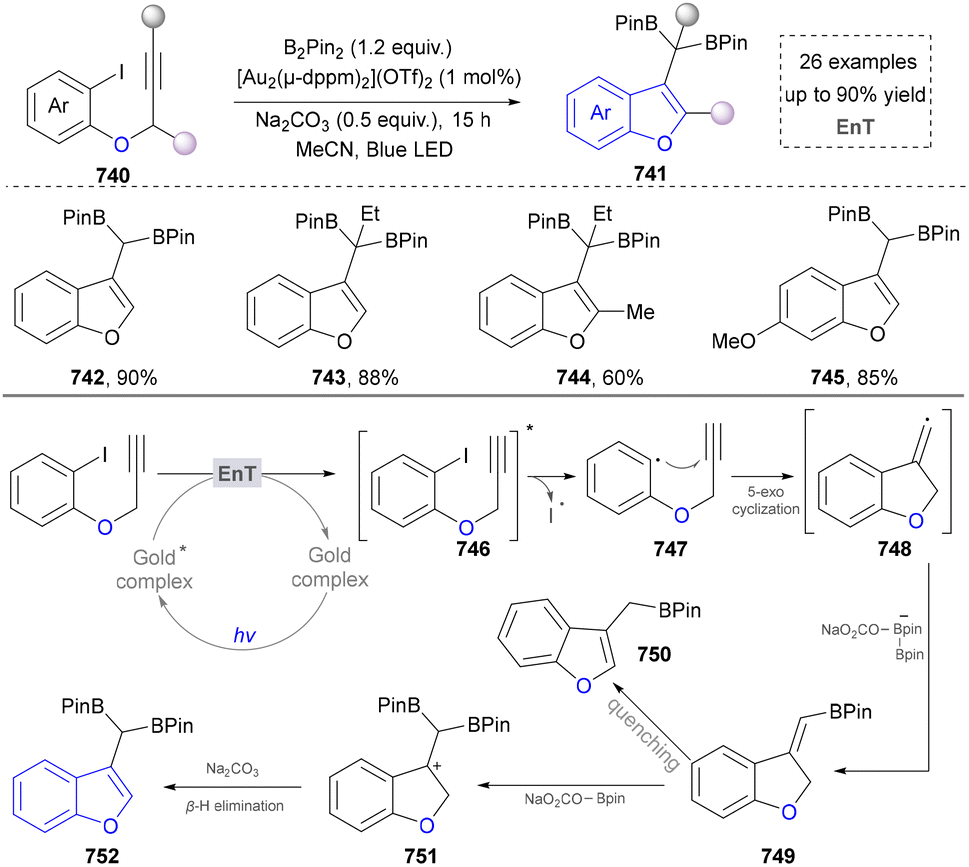 | ||
| Scheme 80 Visible-light-induced gem-diborylation for the synthesis of benzofurans. | ||
In summary, divergent methods through photochemical synthesis have been developed in this field. Most of the approaches focused on the radical addition of vinylic double bond and subsequent oxidation. However, some other novel protocols via energy transfer have also reported recently to broaden the reaction patterns. It is also promising to utilize three-component methods to further expand the photochemical synthesis of benzofurans.
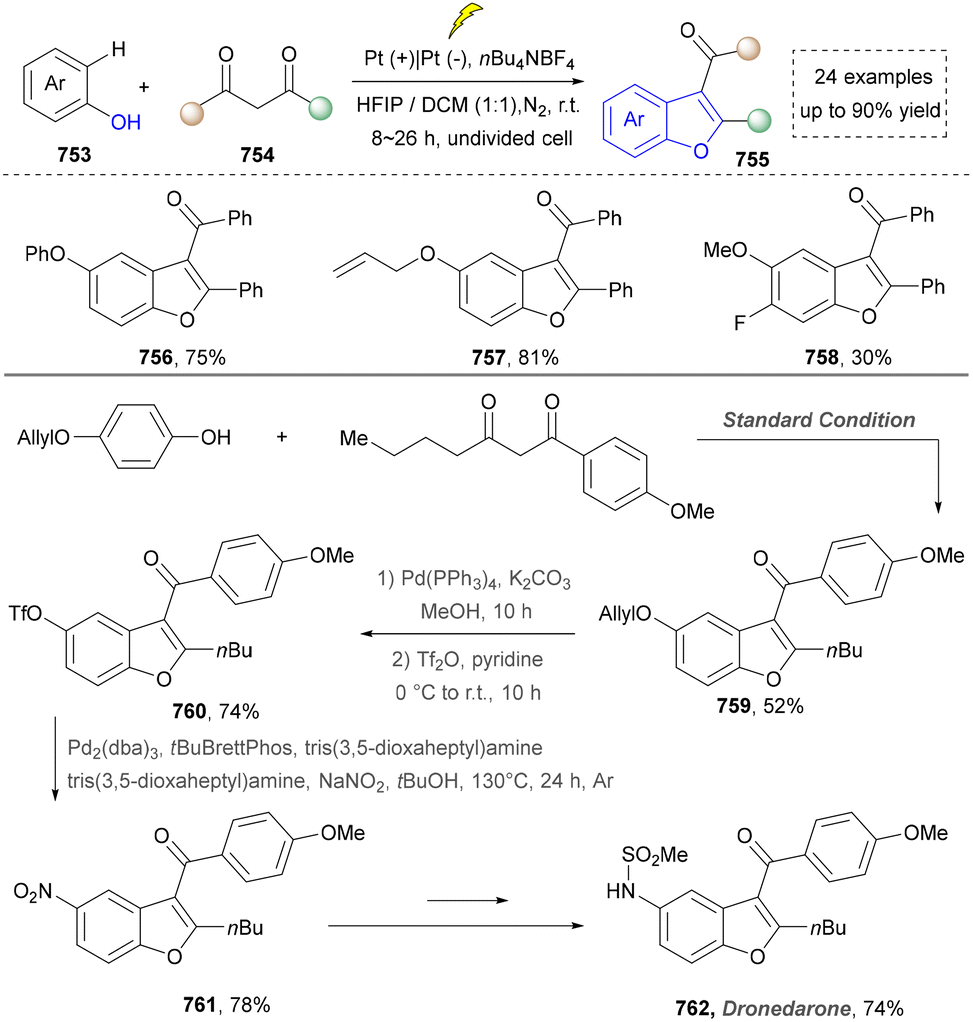 | ||
| Scheme 81 Electrochemical cross-dehydrogenative coupling between phenols and β-dicarbonyl compounds. | ||
Recently, Sun's group disclosed an elegant method to achieve the selective synthesis of benzofurans from propargylic aryl ethers in the presence of dialkyl(aryl) diselenides (Scheme 82).368 The key to success is the single-electron oxidation of propargylic aryl ethers to generate the radical species (769), which further reacted with a phenyl selenium radical via radical-radical cross coupling to access the intermediate (770). The subsequent [3,3]-sigmatropic rearrangement, anodic oxidation and cyclization furnished the final benzofurans (765–767) under simple reaction conditions. At the same time, Cai's group also reported the synthesis of naphtho[1,2-b]furan-2-carbaldehyde and related derivatives via a similar strategy, indicating the potential of electrocatalysis upon the synthesis of benzofurans.369
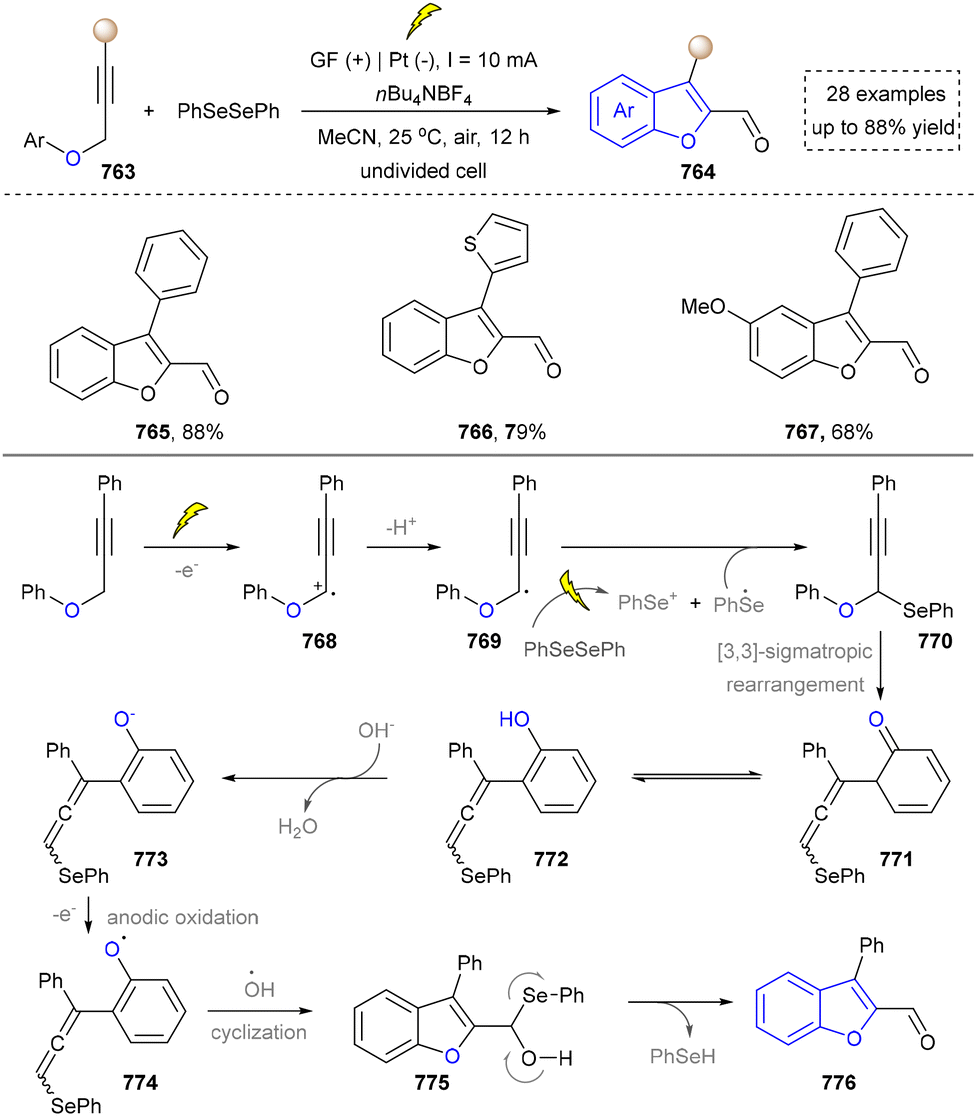 | ||
| Scheme 82 Synthesis of benzofurans via an electrochemical cyclization reaction. | ||
Overall, the electrochemical synthesis of benzofurans significantly broadens the scope of applicable starting materials, which were previously challenging to use in the synthesis of benzofurans, even though current methods still predominantly rely on the built-in oxygen sources within pre-functionalized substrates. Additionally, the successful synthesis of certain drugs and intermediates through electrochemical methods highlights its promising potential in the field of pharmaceutical synthesis.
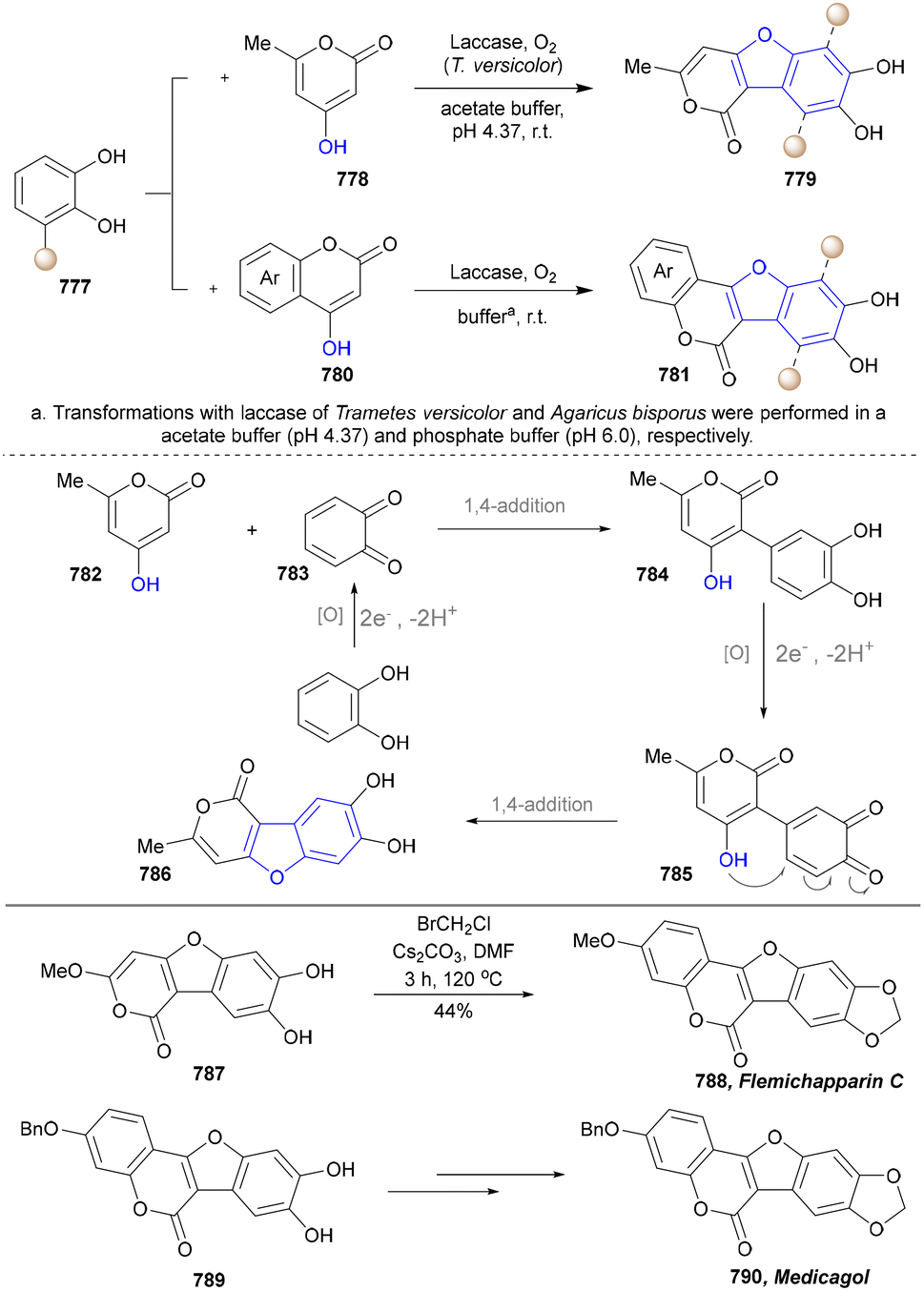 | ||
| Scheme 83 Enzymatic synthesis of benzofurans by laccase. | ||
More benzofuran derivatives such as coumestans had been obtained via the oxidation–Michael addition reactions between catechols and various 1,3-dicarbonyl compounds in the presence of Pycnoporus cinnabarinus laccase372 and Myceliophthora thermophila laccase Suberase®, which exhibited non-stereoselectivity but high regioselectivity (Scheme 84).373,374
 | ||
| Scheme 84 Enzymatic synthesis of the benzofuran derivatives by laccase. | ||
Overall, laccase-catalysed domino reactions exhibited great potential for the synthesis of benzofurans from catechols and various substrates. These studies highlighted the versatility of the laccase-initiated cascade reactions as a green pathway for the synthesis of organic compounds. As described above, the selectivity of natural laccases was not ideal; therefore, further research should focus on laccase engineering and optimization of reaction conditions to improve the selectivity.
7. One-sulfur-containing aromatic heterocycles: thiophenes and benzothiophenes
7.1 Synthesis of thiophenes
Thiophene is a key heterocyclic scaffold, which is widely found in pharmaceutical preparations, dyes and natural products, and is an important intermediate in pharmaceuticals.375,376 Representative examples include cephalosporin antibiotics (e.g., cefalotin, cefaloridine, and cefoxitin), exhibiting broad-spectrum antibacterial activity.377 Emerging strategies including light-mediated and electrochemical annulation are attracting significant attention for their ability to streamline the construction of complex thiophene-containing architectures while enhancing structural diversity and pharmacological potential. Conventional approaches for thiophene synthesis typically utilize the Gewald and Fiesselmann methods.378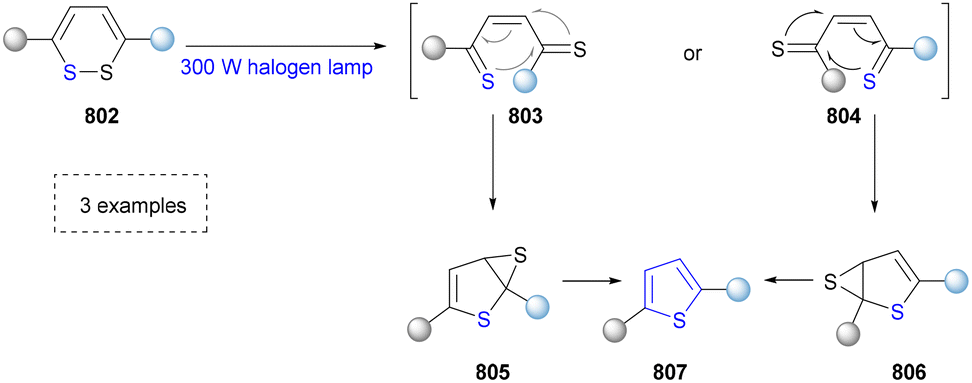 | ||
| Scheme 85 Photoinduced synthesis of thiophenes. | ||
In 2012, Wu and co-workers reported a facile visible-light-driven protocol for synthesizing 3,4-diarylthiophenes from 3,4-diaryl-2,5-dihydrothiophenes, utilizing a platinum(II) terpyridyl complex as a photocatalyst and acetonitrile as the solvent.380 This strategy achieved yields, which were comparable to conventional thermal synthesis under visible light irradiation, demonstrating the viability of photoredox catalysis in thiophene synthesis.
Advancing this field, Luo's group disclosed a relay catalytic approach in 2020, integrating chemical catalysis and photocatalysis to streamline the synthesis of complex thiophenes (813–815) from simple precursors (Scheme 86).56 Their one-pot protocol first employed chemical catalysis to assemble dihydrothiophene intermediates (811), followed by visible-light-mediated iodocyclization/deiodinative transformation of alkyne thioether intermediates using substoichiometric iodine.
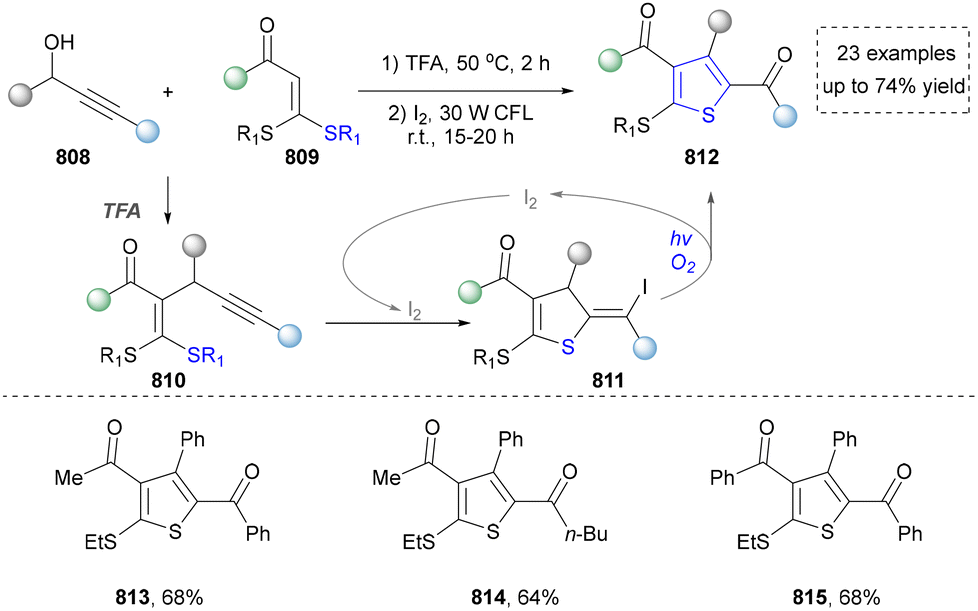 | ||
| Scheme 86 Synthesis of 2,4-diacyl thiophenes from α-oxo-ketene dithioacetals and propargylic alcohols. | ||
Afterwards, Pan's group developed a similar method for constructing polysubstituted thiophenes through [3+2] cyclization of alkynes with easily available ketene dithioacetals, further expanding the synthetic toolbox for modular thiophene functionalization.381
Overall, light-driven technologies in thiophene synthesis remain in their infancy. Complicated starting materials are required to prepare to furnish the thiophene synthesis. Furthermore, systematic investigations into key scientific questions such as photosensitizer design and energy transfer mechanisms remain underexplored. Notably, electrochemical synthesis routes of thiophenes have not been reported to date. This gap may stem from challenges such as redox potential matching and electrode interface modulation. The development of novel photo-/electro-responsive catalysts to overcome the energy barrier limitations of conventional pathways remains a valuable research direction for providing innovative strategies toward the green synthesis of thiophene derivatives.
7.2 Synthesis of benzothiophenes (benzoselenophenes)
The benzothiophene scaffold represents a privileged heterocyclic system. Within medicinal chemistry, benzothiophene-containing compounds demonstrate remarkable pharmacological diversity, as exemplified by clinically significant agents such as raloxifene (a selective estrogen receptor modulator with anticancer applications) and numerous other therapeutic candidates, exhibiting potent antimicrobial and anti-inflammatory activities.382,383 Benzothiophenes are conventionally synthesized via intramolecular electrophilic cyclization of O-alkynyl aryl thioethers or transition metal (Pd/Cu/Au)-catalysed intramolecular C–S bond formation. Alternative approaches include Cu/Pd-catalysed intermolecular C–S coupling and intramolecular Heck reactions.384 However, the green and efficient synthesis of benzothiophene remains a focus of interest.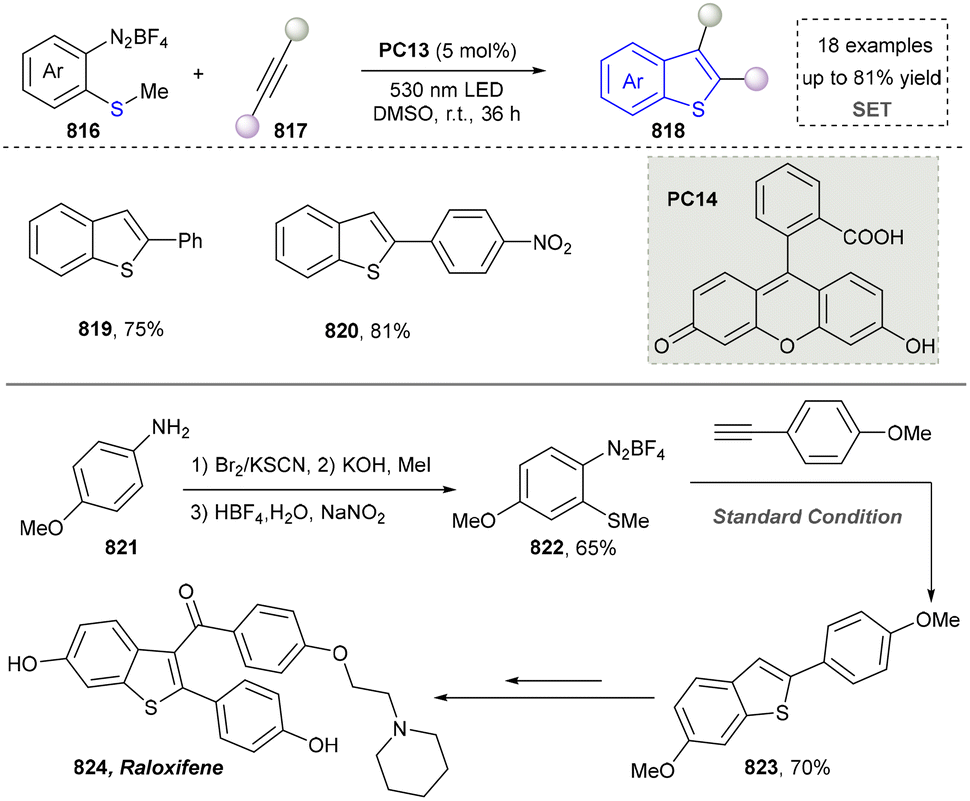 | ||
| Scheme 87 Photocatalytic synthesis of benzothiophenes. | ||
Inspired by this work, Luna's group also reported a similar method to construct the benzothiophenes from diazonium salts and alkynes, but this method relied on the precious gold catalysis.386 Moreover, several continuous reports focused on the photocatalytic synthesis of benzothiophenes from alkynes and various radical precursors.387–393 For example, Zhu's group reported the synthesis of benzothiophenes and benzoselenophenes through the photocatalytic annulation of available thiophenols or 1,2-diphenyldiselane with alkynes.394 Wu's group reported the synthesis of benzothiophenes using sodium metabisulfite as a radical precursor, which further reacted with 2-alkynylthioanisoles.395 With all of these methods, the divergent synthesis of benzothiophenes has been achieved. In addition, Kitamura's group disclosed the irradiation of the iodinated benzo[b]thiophenes with a Hg lamp (>290 nm) and afforded [1]benzothieno[3,2-b][1]benzothiophene derivatives (BTBTs) successfully.396
In 2024, Li's group further developed this method. The synthesis of 3-(alkyl/arylsulfonyl)benzothiophenes and benzoselenophenes was achieved via the photoinduced tandem cyclization of 2-alkynylthioanisoles or-selenoanisoles with DABSO as radical precursors (Scheme 88, 827–830). Moreover, this reaction proceeded smoothly without any external photocatalyst, improving the sustainability of benzothiophene synthesis.397 Benzenethiol-linked 1,6-enynes were also applied to the synthesis of benzothiophenes, whereas this strategy has been well-established in the synthesis of benzofurans.398
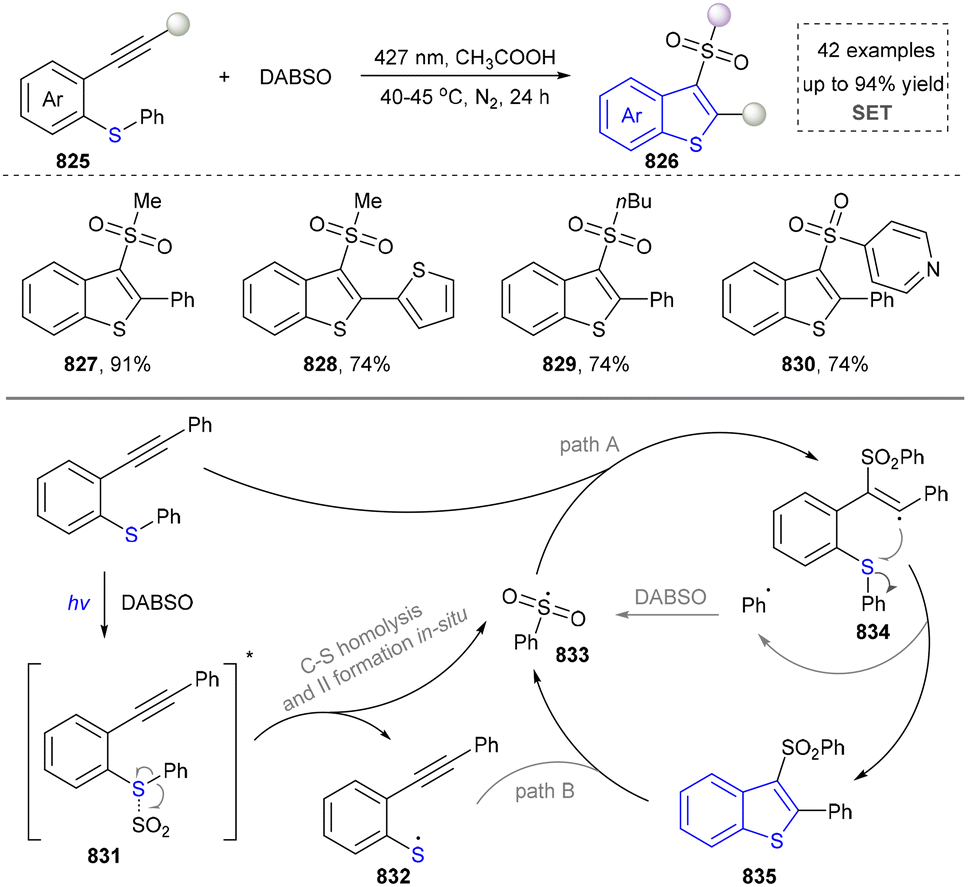 | ||
| Scheme 88 Photo-induced synthesis of 3-(alkyl/arylsulfonyl) benzothiophenes. | ||
Overall, the synthesis of benzothiophenes from photochemical approaches has been well explored. The thiol source was mostly from thioethers, which reacted with different alkynes to obtain benzofurans through the photoredox catalysis. However, the starting material in these cases was relatively difficult to prepare. The radical precursors had to be introduced into the starting material in most of the cases, which limited the practical use of this protocol.
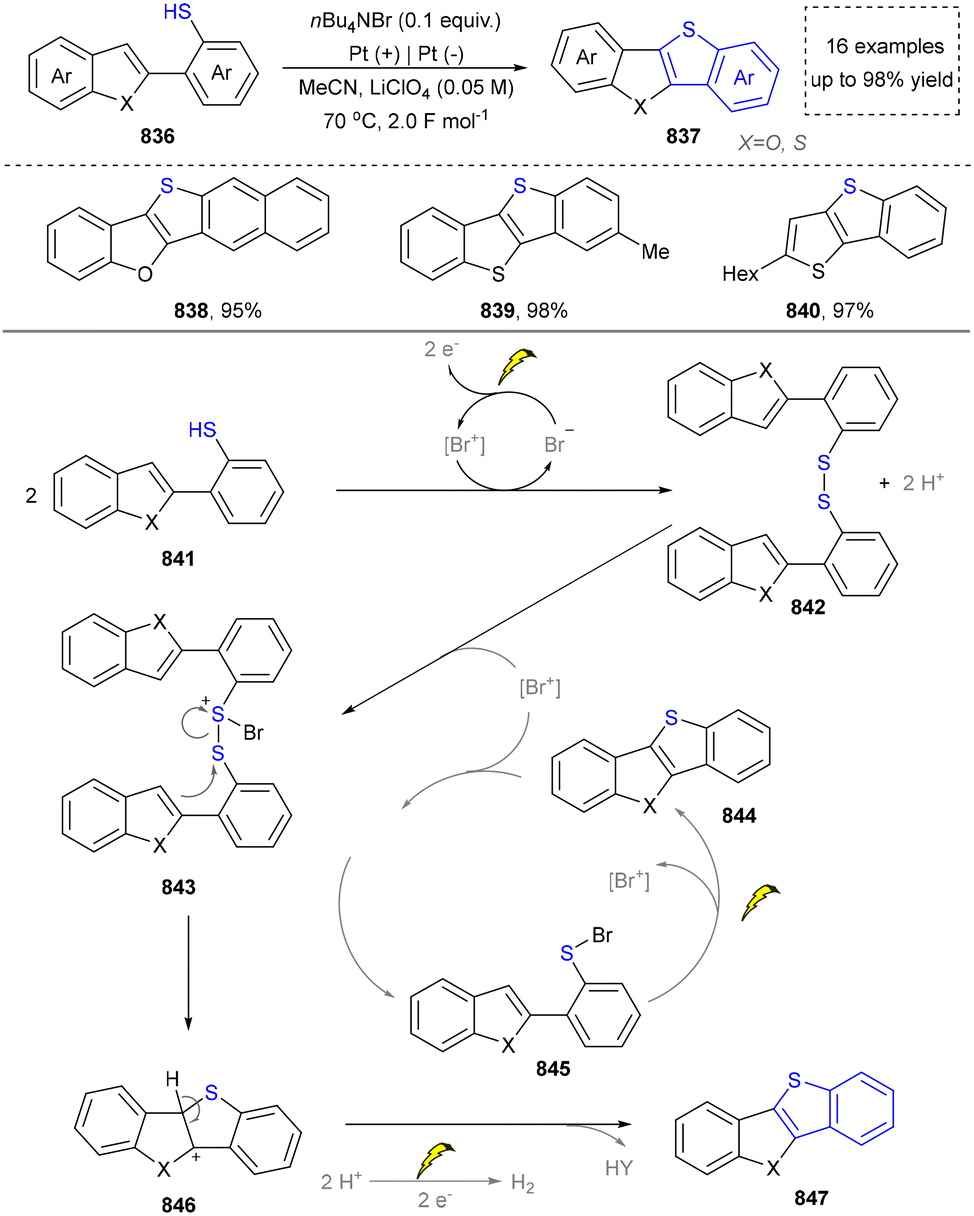 | ||
| Scheme 89 Electrochemical synthesis of the thienoacene derivatives. | ||
The synthesis of benzoselenophenes and benzothiophenes via electrochemical oxidative method has also been disclosed by Zeng's group (Scheme 90). The enamide underwent oxidative electrochemical selenocyanation with KSeCN/NH4SCN to afford radical species as the key intermediate, which further underwent cyclization to furnish the final products (851–854).401
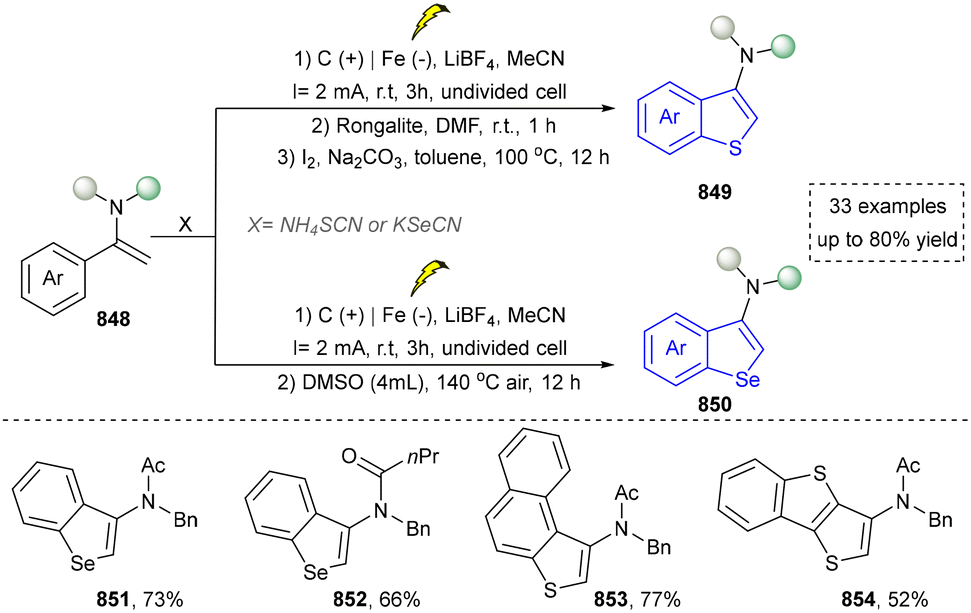 | ||
| Scheme 90 Electrocatalytic synthesis of benzoselenophenes and benzothiophenes. | ||
Overall, the electrochemical synthesis of benzothiophenes has contributed to the utilization of different starting materials such as thiols and disulfides. There are also some methods, which are very similar to photochemical methods; however, concise reactions by using simple synthetic synthons should be the future target. Another future integrated photo-electro system holds promise. Here, photocatalysis would generate key sulfur radicals, while electrochemical control of electrode potential would facilitate the cyclization and aromatization. This synergy could offer unprecedented command over reaction pathways and selectivity, presenting a new approach to overcome current selectivity challenges.
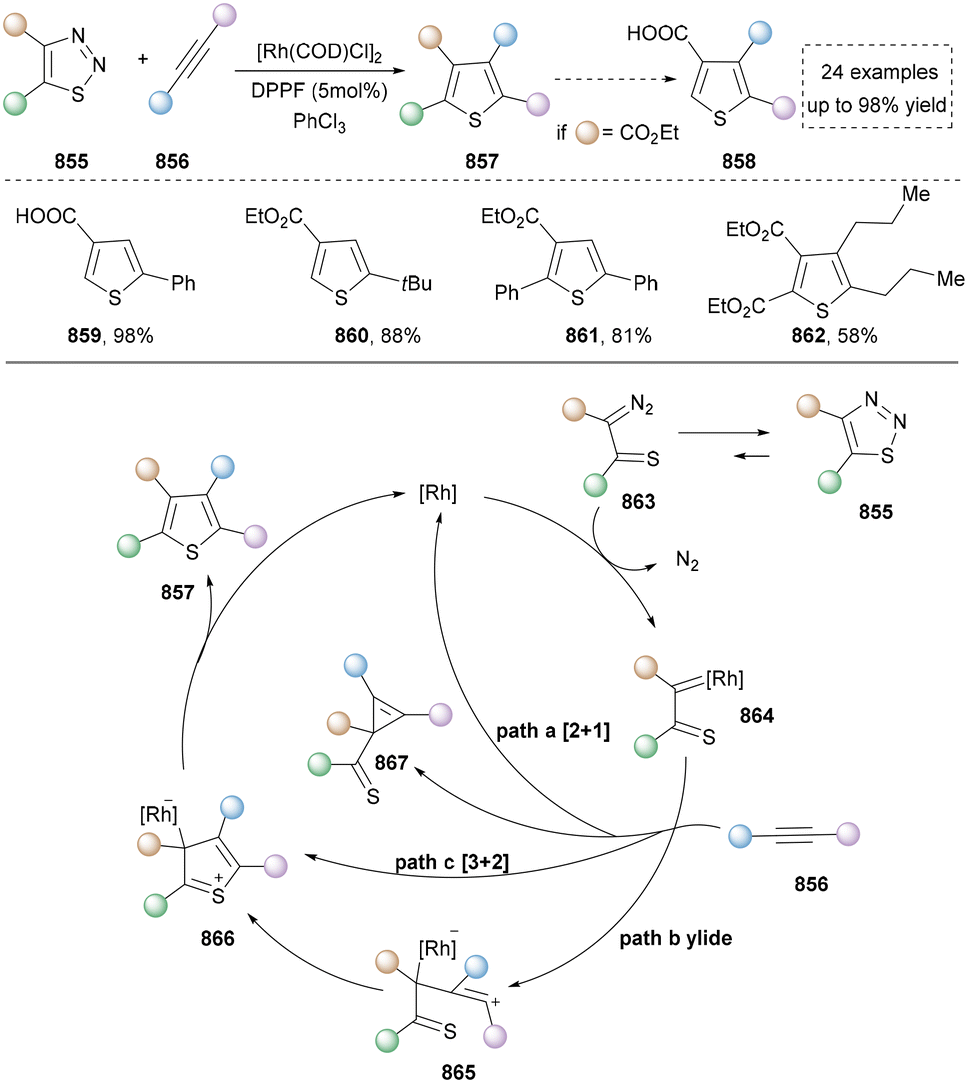 | ||
| Scheme 91 Modular synthesis of polysubstituted thiophenes from 1,2,3-thiadiazoles. | ||
8. Aromatic heterocycles containing hybrid heteroatoms: isoxazoles, oxazoles, benzoxazoles, isothiazoles, thiazoles, and benzothiazoles
8.1 Synthesis of isoxazoles
As a privileged heterocyclic scaffold containing adjacent nitrogen and oxygen atoms, the isoxazole ring exhibits remarkable metabolic stability and serves as a bioisostere for various carboxylic acid derivatives and other heterocycles, which significantly enhance the pharmacokinetic profiles of bioactive molecules.403,404 This structural motif features prominently in numerous pharmaceuticals, including COX-2 inhibitors (e.g., valdecoxib), antibacterial agents, and CNS-active compounds.405,406 Isoxazole rings are traditionally constructed through [3+2] cycloaddition of N-chlorosuccinimide-chlorinated hydroxylamines with alkynes, or via 1,3-dipolar cycloaddition of oxidized nitriles with terminal alkynes.407 This section provides a comprehensive discussion on emerging eco-friendly methodologies that have significantly enhanced the efficient synthesis of isoxazoles. | ||
| Scheme 92 Visible light promoted the synthesis of isoxazoles. | ||
Recently, our group has also disclosed the synthesis of isoxazoles via a photoinduced three-component strategy. In this case, the N-tosylhydrazones (879) were activated under the irradiation of light to generate in situ diazo compounds (880), which further reacted with tert-butyl nitrite to form the intermediate nitronate 881.29 The nitronate underwent the [3+2] cycloadditions with different alkynes to furnish the final isoxazoles (876–878). The isoxazolines was also afforded by using alkene as a starting material. More importantly, two antitrypanosomal agents were synthesized efficiently by using this strategy (Scheme 93).
 | ||
| Scheme 93 Visible-light-mediated strategy for the synthesis of Isoxazoles. | ||
Overall, the photochemical synthesis of isoxazoles was underexplored, and the reported cases demonstrated certain advantages over traditional thermal conditions or transition metal catalysis. Although the concept is not entirely novel, there is still significant room for further exploration. In particular, it is worth noting that the investigation of different nitrogen sources, beyond the commonly studied oximes and hydrazones, is also highly warranted.
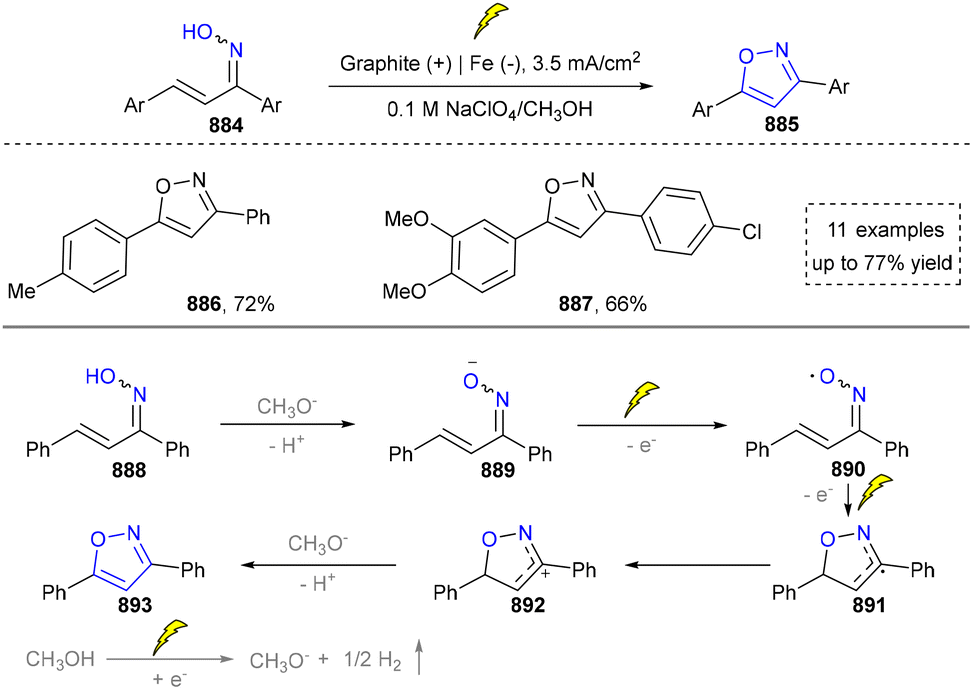 | ||
| Scheme 94 Electrochemical synthesis of the 3,5-disubstituted isoxazoles. | ||
To further expand the structural diversity of isoxazoles, the synthesis of organoselenyl isoxazoles has been disclosed by Hu's group in 2024 (Scheme 95).412 In this method, diverse organoselenyl isoxazoles were afforded under electrocatalytic conditions. In their proposed mechanism, the phenyl selenium radical generated from diorganyl diselenides reacted with 2-alkyn-1-one O-methyloximes to generate the vinyl radical (904), which further underwent an intramolecular radical cyclization to form the cyclized intermediate (905). Follow-up oxidation at the anode afforded the corresponding pentacyclic cation intermediate (906), which further dropped the methyl cation to form the product.
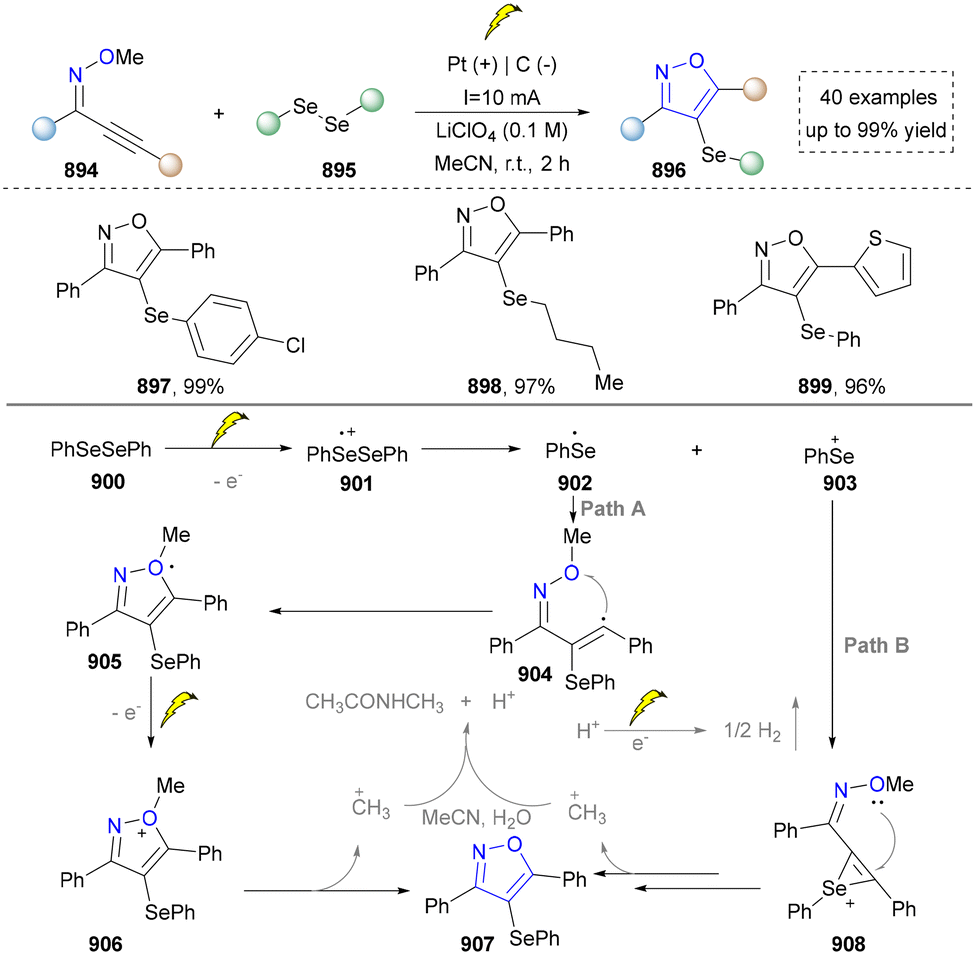 | ||
| Scheme 95 Electrochemically promoted synthesis of organoselenyl isoxazoles. | ||
In summary, the synthesis of isoxazoles via electrochemical methods has been well established, especially the application of oximes as a nitrogen source. Moreover, nitrogen sources were extended to the nitro substance, which further diversified the synthesis of isoxazoles. Therefore, the substrate scope of isoxazole synthesis was strongly extended compared with traditional or photochemical methods such as the generation of organoselenyl isoxazoles. Therefore, there is still some space to explore other nitrogen sources and reaction reagents to construct novel isoxazoles.
8.2 Synthesis of oxazoles
Oxazole, a distinctive heterocyclic scaffold incorporating both nitrogen and oxygen atoms represents a privileged structural motif, which is extensively distributed in natural products and pharmaceutical agents.413 As evidenced by its presence in FDA-approved therapeutics such as the antibacterial drug linezolid and the antiviral agent pleconaril, this scaffold maintains persistent research interest and occupies a pivotal position in both organic synthesis and medicinal chemistry.414–416 Oxazole synthesis methodologies include the Robinson–Gabriel synthesis, Fischer oxazole synthesis (using cyanohydrins and aldehydes), and Bredereck reaction.417 This section will critically examine the transformative role of environmentally benign strategies in constructing this pharmacologically significant heterocyclic framework.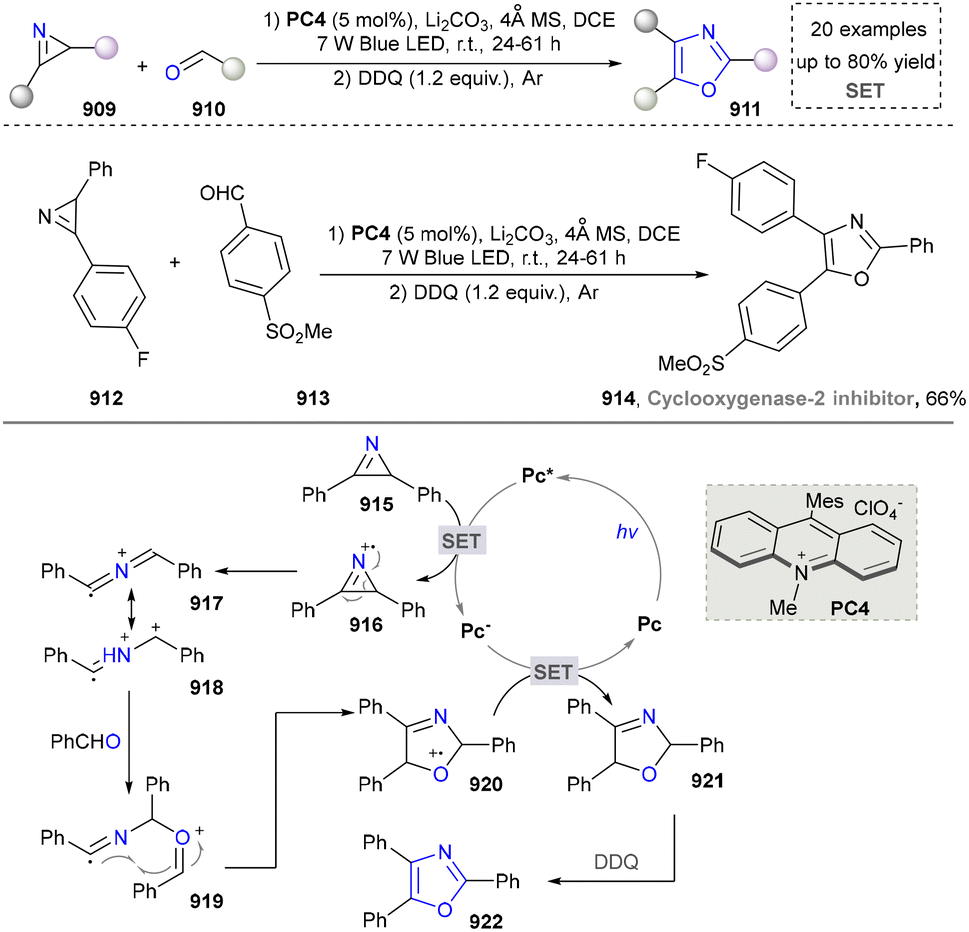 | ||
| Scheme 96 [3+2] cycloaddition/oxidative aromatization sequence to afford oxazoles. | ||
α-Bromoketones were also applied to the synthesis of oxazoles by Cho's group (Scheme 97).419 In this case, α-bromoketones reacted with benzylamines to give diverse oxazoles (926–928) under photocatalytic conditions. Furthermore, the methodology enabled the efficient synthesis of the natural product Texaline (931), which was obtained in 71% yield from 929 and 930, under optimized conditions (PC 1 mol%, CCl3Br 1.5 equiv., K3PO4 3.0 equiv., DMF 0.1 M, Blue LEDs 7 W, 16 h). The successful incorporation of a pyridine-based substrate (930) as the starting material confirmed the compatibility of the pyridine moiety with the reaction conditions. Moreover, this natural product was produced on a gram scale, highlighting the applicability of this method. Later, CO2-pomoted cyclization of α—bromo ketones and amines to construct substituted oxazoles has also been reported by Li's group, which improved the sustainability of this method.420 In 2023, Srivastava's group found that using benzil and benzyl amine could also form the oxazoles via photocatalysis.63 The concept was similar to the above work and both of them relied on the synthesis of imine as an intermediate.
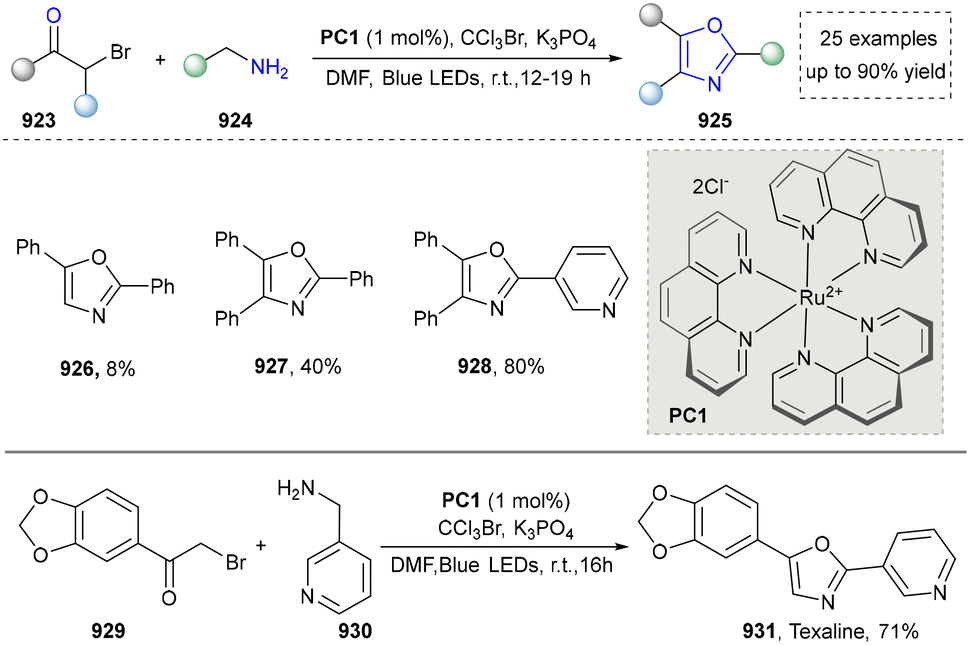 | ||
| Scheme 97 Synthesis of substituted oxazoles by photocatalysis. | ||
To further expand the structural diversity of oxazoles via photochemical synthesis, Wang's group employed the readily available propargyl amide as a starting material, which underwent cyclization to generate a valuable oxazole (Scheme 98).59 In the mechanistic studies, they proved that linked I2/visible light activated the C–C triple bond efficiently, which was followed by nucleophilic addition by electron-donating amide groups, providing the final products (934–936). Inspired by this pioneering work, several groups continuously utilized the propargyl amides to synthesize oxazoles under photocatalytic conditions.421,422 For example, Mal's group further developed this method by using charge-transfer complexes as semi-heterogeneous photocatalysts.423 Selenium-π-acid catalysis has also been reported by Liu's group to facilitate the activation of propargyl amide, thereby forming the oxazoles under photocatalytic conditions.424
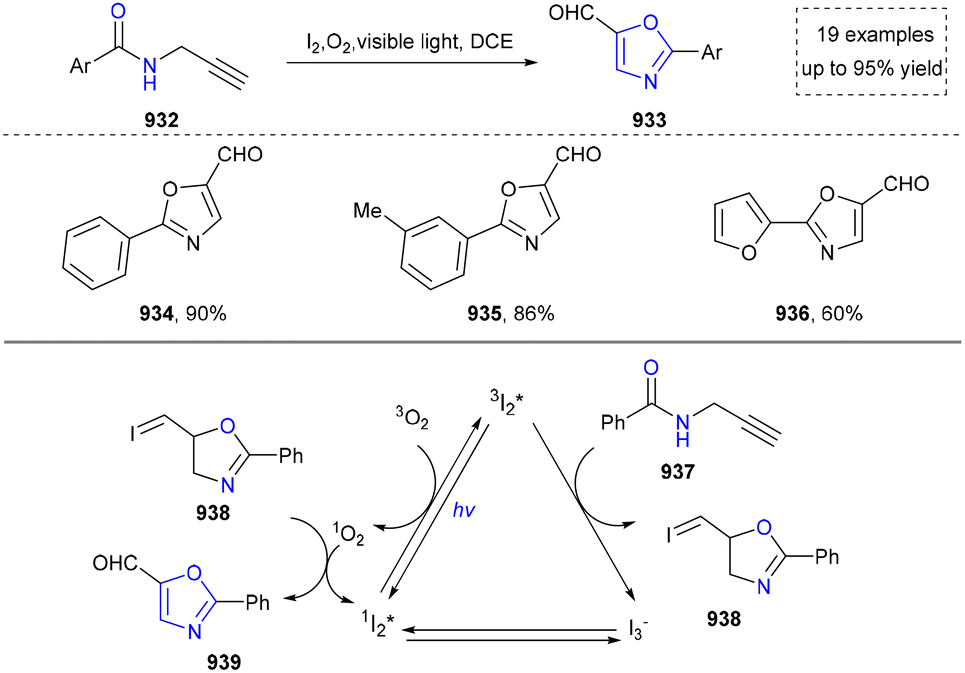 | ||
| Scheme 98 Photocatalytic activation of alkynes for cyclization reactions. | ||
Later, the photoannulation of α-azidochalcones into 2,5-diaryloxazoles was reported by Maurya's group in 2022 (Scheme 99). They also proposed the mechanism as shown below. The α-azidochalcones (942) were activated via energy transfer to form the excited-state of the substrate (943), which further released N2 to afford 2H-azirine (944) as a key intermediate. 2H-Azirine either further reacted with 2,4-dinitrophenol to form 2,5-diaryloxazoles (946) via the photoexcitation again or formed indole directly after the rearrangement by replacing the photocatalyst to Ru(bpy)3(PF6)2.65 Enaminone was also reported as a nitrogen source to afford oxazoles by Baell and Liu's group, repectively.67,425
 | ||
| Scheme 99 Visible light promoted the conversion of α-azidochalcones into 2,5-diaryloxazoles. | ||
Besides the use of azide as the nitrogen source, the diazo compounds were applied for the synthesis of oxazoles in 2023. Maiti's group disclosed the initial photolysis of the diazo compound to afford singlet carbenes, which were tapped by nitriles and underwent [3+2] cycloaddition to form substituted oxazoles (Scheme 100). Compatible with diverse diazo compounds and nitriles, it efficiently builds complex oxazole scaffolds, including drug intermediates and isotope-labelled derivatives. Its integration with continuous-flow technology successfully addresses the scalability challenge in photochemistry, offering a practical and sustainable synthetic route. This method further enabled a concise two- to three-step synthesis of bioactive oxazoles from 953 and 955, providing efficient access to compounds such as hinduchelins B (954) and picomolar inhibitors of cytosolic human carbonic anhydrase II (956).68 This group continuously unveiled that the combination of photoelectro-chemical (PEC) reaction to facilitate the synthesis of oxazoles and imide-fused pyrroles via the formation of a carbene radical anion.64 Moreover, arylazo sulfones were applied as an arylating agent to furnish the oxazoles via the formation of diazenyl radicals.426 Xia's group also described a similar strategy to afford oxazoles at almost the same time.427
 | ||
| Scheme 100 Photoinduced [3+2] cycloaddition for the synthesis of oxazoles. | ||
Overall, various nitrogen sources have been applied to the synthesis of oxazoles through photochemical methods. Moreover, photochemical strategies for synthesizing oxazoles demonstrate excellent compatibility with heterocyclic substituents. They also enable the retention of sensitive functional groups such as aldehyde groups on the skeleton that conventional methods seldom achieve. In addition, several drugs and natural products were synthesized successfully from these approaches. Therefore, it is highly possible that other heteroarenes also have possibilities to be afforded by photochemical methods.
 | ||
| Scheme 101 Electrochemically promoted synthesis of polysubstituted oxazoles. | ||
Xia's group also described an elegant method to afford oxazoles by using abundant and inexpensive carboxylic acids and benzyl amines as starting materials (Scheme 102). Besides typical advantages of electrochemical synthesis such as free of oxidants, this method was successfully employed for the late-stage modification of drugs, and several drug analogues bearing oxazoles were efficiently afforded under standard conditions (972–974).62 The system demonstrated excellent functional group tolerance, accommodating stable unsaturated bonds, benzylic group and other heteroaromatics. Furthermore, its applicability was underscored by a successful gram-scale synthesis.
 | ||
| Scheme 102 Electrochemical synthesis of oxazoles via [3+2] cycloaddition. | ||
Almost at the same time, MeCN worked simultaneously as a solvent and as a nitrogen source, which has been achieved by several groups.430–433 With this method, polysubstituted oxazoles were synthesized from readily available ketones/alkynes and acetonitrile under electrocatalytic conditions. Inspired by this work, Li's group also reported a convenient method to construct the oxazole by using MeCN as a nitrogen source. This four-component reaction by using terminal alkynes, (thio)xanthenes, nitriles, and water as starting materials has been developed in the absence of any catalyst and external oxidant with excellent structural diversity.434 The N,N,N,N-tetramethyl ethylenediamine (TMEDA) has also been applied as a nitrogen source for the synthesis of oxazoles by Cho's group.435 Moreover, TMEDA played the role of a mediator in this system to facilitate the reaction. In addition, intramolecular electro-oxidative addition of N-propargyl benzamides has been reported.436,437
Compared to the photochemical synthesis of oxazoles, electrochemical synthesis provided novel routes to expand the diversity of the oxazoles. For instance, new nitrogen sources including MeCN to construct oxazoles have been reported, which showed unique advantage of the electrosynthesis.
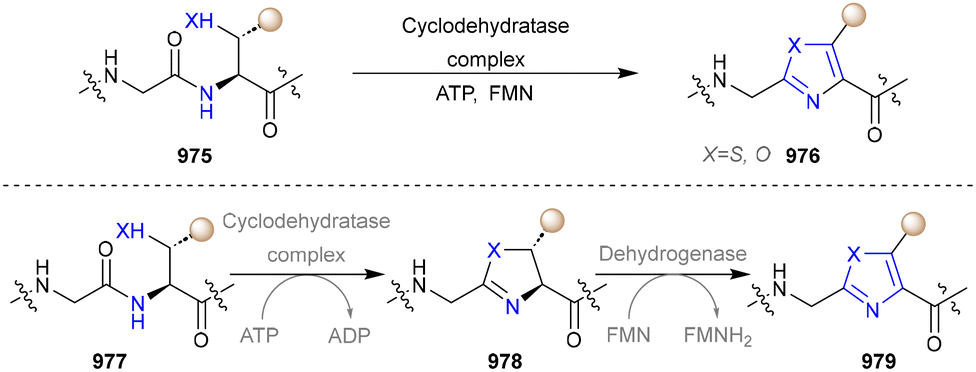 | ||
| Scheme 103 Enzymatic synthesis of thiazole/oxazole-modified microcin. | ||
The studies remain on the discovery of the enzyme, responsible for azoline biogenesis in thiazole/oxazole-modified microcin (TOMM) natural products. Therefore, further research should focus on the application of enzymes in the synthesis of thiazoles and oxazoles.
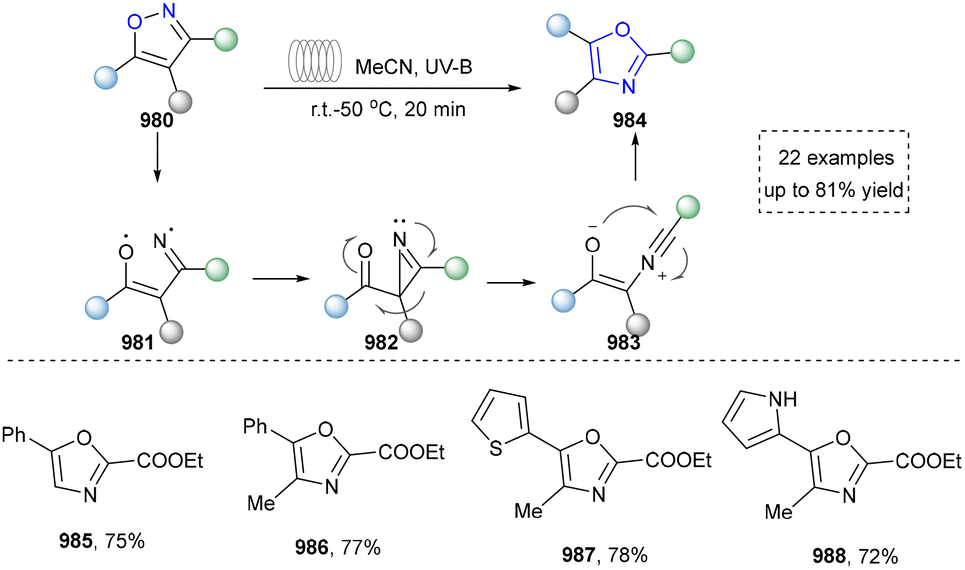 | ||
| Scheme 104 Access to diverse oxazoles from isoxazoles enabled by continuous-flow photoisomerization. | ||
8.3 Synthesis of benzoxazoles
The synthesis of benzoxazoles is particularly valuable as it combines synthetic accessibility with exceptional drug-like properties.439,440 Numerous benzoxazole derivatives have demonstrated broad-spectrum pharmacological potential, encompassing antifungal, anti-inflammatory, antimalarial, anti-trypanosomal, and anticancer properties, as well as ectoparasitic, antipneumococcal, and antituberculosis activities, among other pharmacological effects.441–444 Benzoxazoles are classically prepared by reacting o-aminophenols with carboxylic acid derivatives, aldehydes, or alcohols.445 This section provides a comprehensive discussion on emerging synthetic strategies for benzoxazole derivatives, highlighting their significance in contemporary medicinal chemistry. | ||
| Scheme 105 Visible-light-triggered oxidation for the synthesis of 2-aminobenzoxazoles. | ||
Overall, the synthesis of imines has been a crucial step in constructing benzoxazoles, using directly either as a starting material or as an intermediate. However, considering the potential drawbacks and limitations associated with the use of imines, such as their instability or the complexity of their synthesis, it will be highly innovative and valuable to explore alternative synthetic strategies that bypass the requirement of imines.
 | ||
| Scheme 106 Electrochemical synthesis of the 2-substituted benzoxazoles. | ||
To avoid the preparation of Schiff bases from aminophenols and aldehydes, direct use of aminophenols became an ideal protocol to access benzoxazoles, which have been developed recently.456–459 Among the related studies, Wacharasindhu's work developed an electrochemical method, which achieved the direct synthesis of 2-aminobenzoxazoles from 2-aminophenols and isothiocyanates (Scheme 107).460 This method showcased a decent substrate scope (1011–1013) and demonstrated good compatibility with various starting materials. Furthermore, Wang's group developed an electrochemical approach by using simple nitrogen sources such as NH4+ and formamide, which avoided the synthesis of aminophenols and further expanded the diversity of synthesized benzoxazoles.461
 | ||
| Scheme 107 Electrochemical NaI/NaCl-mediated synthesis of 2-aminobenzoxazoles. | ||
Besides using aminophenols or related analogues to synthesize benzoxazoles, electrochemical synthesis of benzoxazoles from anilides has been disclosed by Waldvogel's group (Scheme 108).462 The key to success was the direct N–H activation to an amidyl radical through the anodic oxidation, which was stabilized by the adjacent aromatic ring. The subsequent cyclization and oxidation provided the final products (1021–1023). To further gain insights into the mechanism, the same group further conducted a series of mechanistic studies including potential dependence in CV data, substituent effect-induced changes in product selectivity, and solvent trapping experiments supporting the presence of cations to validate their proposed mechanism.463
 | ||
| Scheme 108 Electrochemical synthesis of benzoxazoles from anilides. | ||
Therefore, it is clear that the electrosynthesis approaches have already overcome some limitations of photocatalysis methods such as the feasibility of other nitrogen sources besides imines. Electrosynthesis has also expanded the scope of nitrogen sources to include available amines and anilides, which not only broadens the synthetic pathways but also provides valuable clues for further development. However, although the diversity of starting materials and sustainability has increased, there is no significant improvement in the structural diversity of the final products compared to traditional methods.
 | ||
| Scheme 109 Enzymatic synthesis of 2-substituted benzoxazoles via the GOX-CPO system. | ||
The above-mentioned immobilized laccase and TEMPO system could also be utilized to synthesize 2-substituted benzoxazoles.268 The one-pot enzymatic cascade included the generation of salicylaldehyde derivatives, the condensation of salicylaldehyde derivatives with 2-hydroxy-anilines and the biocatalytic dehydrogenation.
In 2019, Wang's group employed hemoglobin from Vitreoscilla (VHb) as a biocatalyst for the oxidative cyclization to synthesize 2-substituted benzoxazoles with satisfactory yields (84–97%) (Scheme 110).465
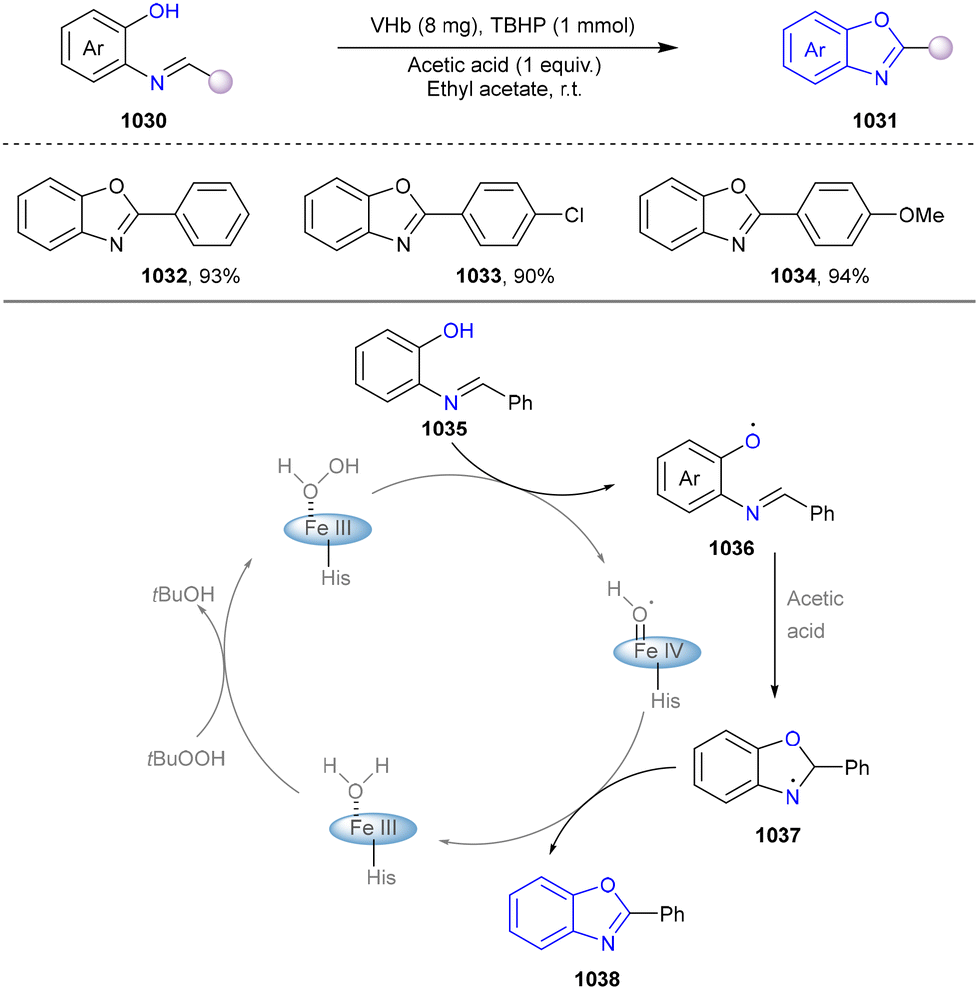 | ||
| Scheme 110 Enzymatic synthesis of 2-substituted benzoxazoles via oxidative cyclization. | ||
In this subsection, three examples of enzymatic syntheses of benzoxazoles were described. Nevertheless, the biosynthetic pathway of bioactive benzoxazoles with meta- and ortho-substituted heterocycles has been gradually elucidated, such as cytotoxic benzoxazole nataxazole,466 antibiotics A33853,467 antibiotic caboxamycin,468 and closoxazole A and B,469 providing new insight for further enzymatic synthesis of benzoxazole moiety.
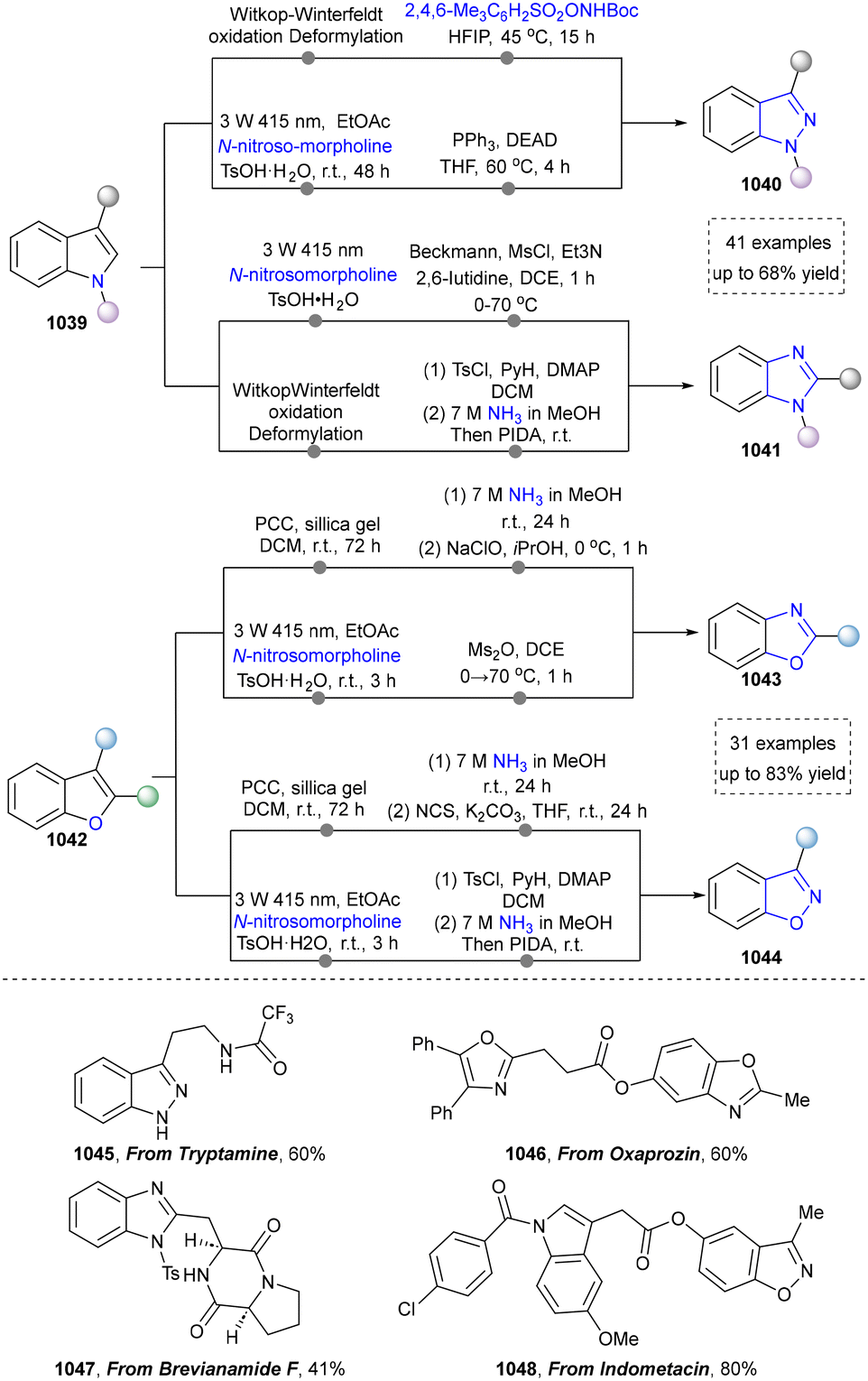 | ||
| Scheme 111 Carbon-to-nitrogen atom exchange of indoles and benzofurans to form aromatic heterocycles. | ||
In summary, although the atom editing synthesis of benzoxazoles is still in its early stages, the direct-editing paradigm it represents is providing medicinal chemists with a powerful and unprecedented tool, holding significant potential to greatly accelerate the processes of lead discovery and optimization. Future efforts will probably expand directly toward developing traceless editing strategies that avoid the introduction of or need for additional protecting groups.
8.4 Synthesis of thiazoles and isothiazoles
The isothiazole scaffold represents a structurally unique heterocyclic system of growing importance in medicinal chemistry.470 Isoxazole derivatives offer diverse bioactive scaffolds that remain at the forefront of modern drug discovery, exemplified by marketed antiviral (denotivir), antibacterial (sulfafurazole), and antipsychotic (ziprasidone) agents, along with numerous investigational agents in anticancer, antidiabetic, antifungal, and neuroprotective categories.471,472 Conventional isothiazole synthesis depends on condensation reactions followed by coupling/alkylations.470 However, the green and efficient synthesis of isoxazoles continues to represent a high-value research endeavour in contemporary medicinal chemistry.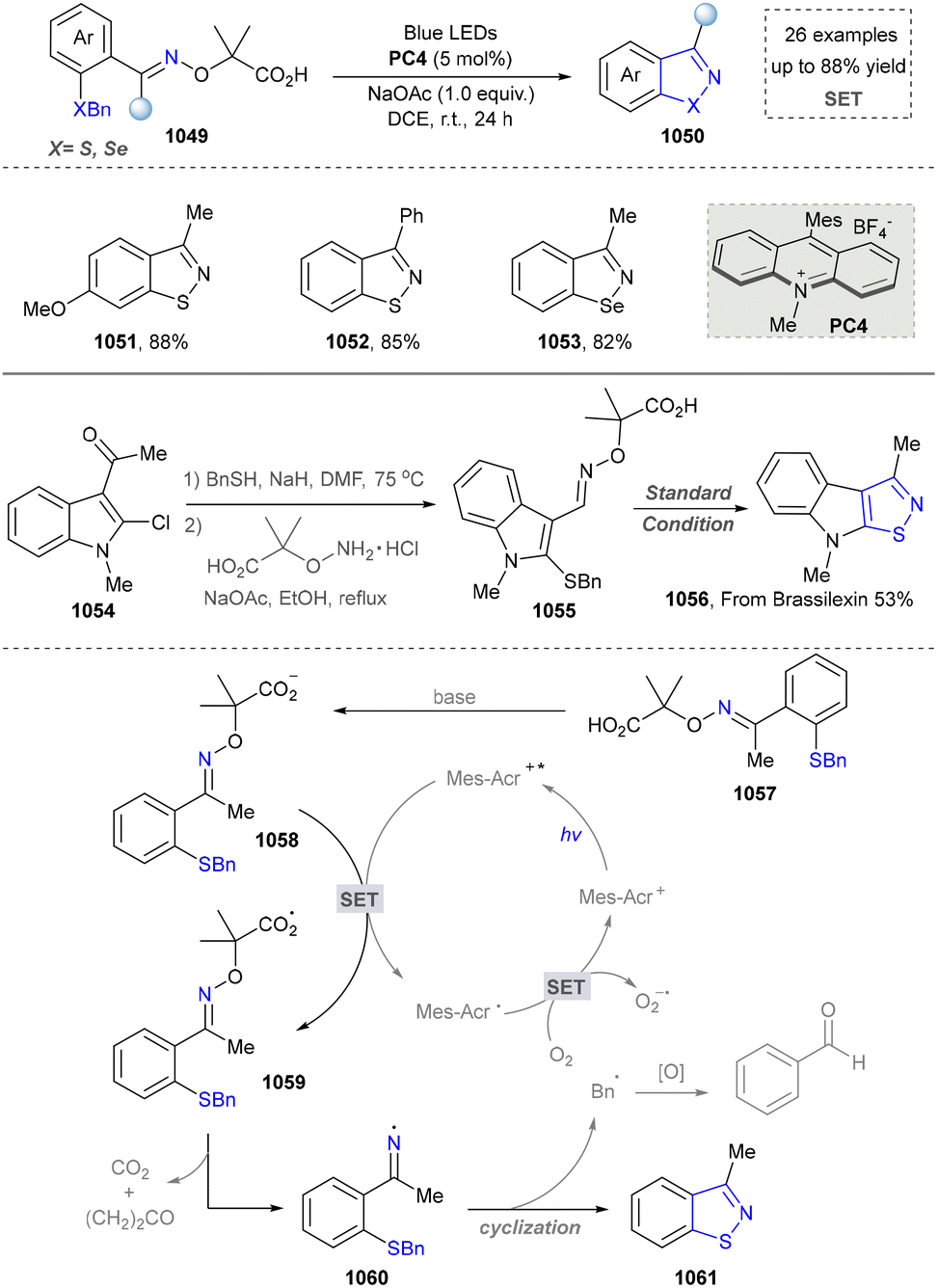 | ||
| Scheme 112 Visible light-promoted N–S bond formation for the synthesis of isothiazoles. | ||
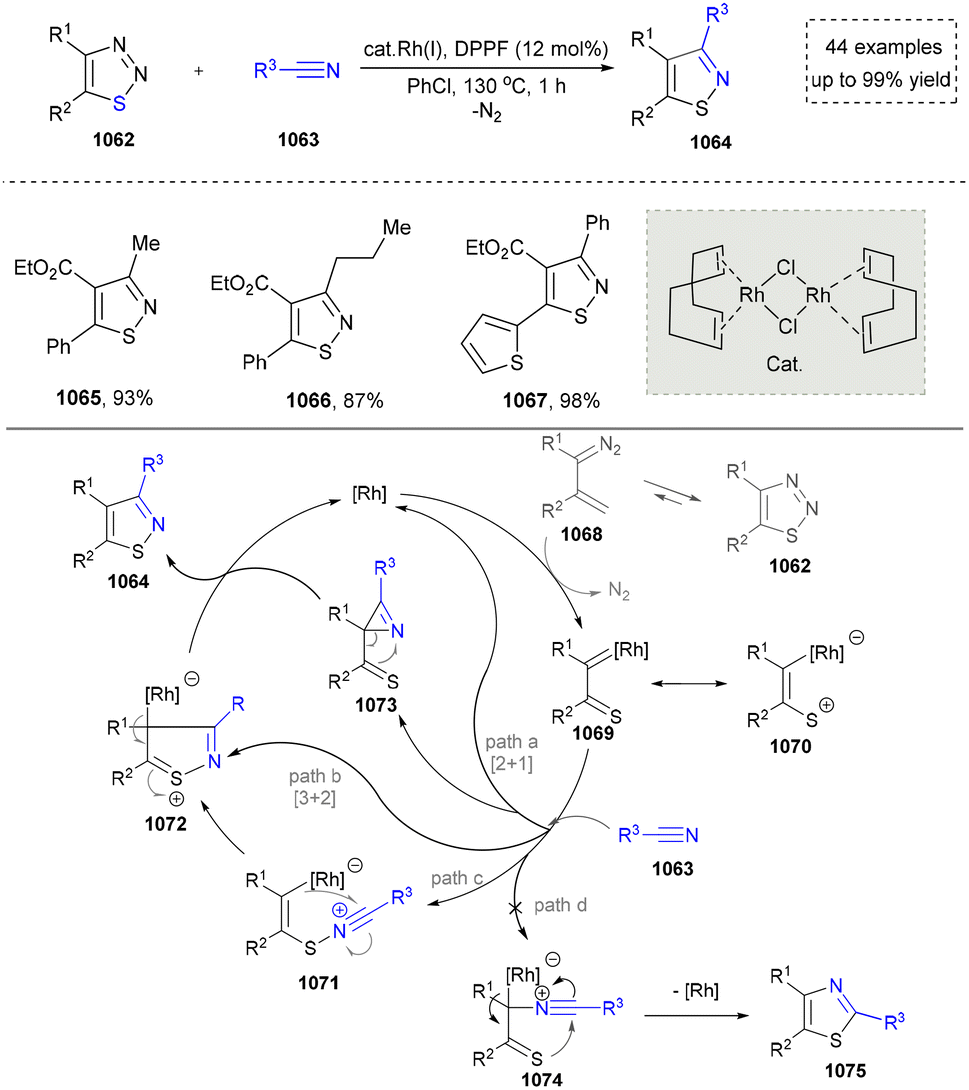 | ||
| Scheme 113 Synthesis of isothiazoles via ring transformation of 1,2,3-thiadiazoles using nitriles. | ||
In 2025, Sarpong's group reported a cheminformatics platform designed to identify which skeletal editing maximized the exploration of new chemical space.476 This work introduced a framework for quantifying the impact of core-atom transformations in heteroaromatic compounds and highlighted specific skeletal editing that most significantly enabled accessing unexplored chemical regions. Furthermore, it called for the development of new synthetic methodologies to achieve these prioritized core-atom modifications.
In the context of drug discovery, photochemical, electrochemical and enzymatic synthesis holds a distinct advantage for the rapid exploration of a new chemical substrate scope from scratch. Conversely, when the objective shifts to the precise skeletal modification of existing complex drug molecules, atom editing emerges as a more promising and cutting-edge direction.
8.5 Synthesis of thiazoles
The thiazole motif is a pharmacologically indispensable heterocycle, valued for its unique electronic properties and broad-spectrum of bioactivity.477 It is also crucial in medicinal chemistry, in fact, thiazole derivatives exhibit versatile bioactivities through interactions with multiple enzymatic and receptor targets in biological systems, demonstrating therapeutic relevance across nearly all medical domains.478 Thiazole derivatives exhibited promising therapeutic potential across antibacterial, antifungal, antitubercular, anticancer, antiviral, anti-inflammatory/analgesic, hypoglycemic, antiepileptic, antiparasitic, and antioxidant applications.479–482 Thiazole derivatives are typically synthesized through multi-step functional group transformations, exemplified by the Cook–Heilbron thiazole synthesis and Hantzsch method.483,484 It remains a highly active area of research, and this section will provide a comprehensive overview of the implementation of emerging green technologies in the synthesis of thiazole compounds. | ||
| Scheme 114 Visible-light-induced synthesis of the 2-(2-hydrazinyl)thiazole derivatives. | ||
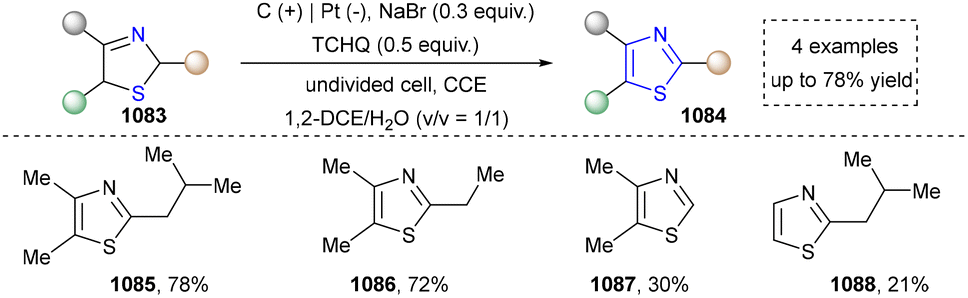 | ||
| Scheme 115 Electrochemical synthesis of thiazoles. | ||
In 2022, an electrochemical approach for the construction of 2-aminothiazoles by using readily accessible enaminones and thioureas has been reported by Wan's group (Scheme 116), which thoroughly explored the chemical space of aminothiazoles (1092, 1093) by using diverse starting materials. Furthermore, this strategy was successfully employed in the synthesis of chiral steroids, including a progesterone derivative (1094) under slightly modified conditions (stirred for 3 h, 3.7 F mol−1), which confirmed that the cyclic enone was tolerable in this system. Moreover, detail mechanistic studies have been performed which showed that cascade enamine α-C–H thiolation (1110) and C–N bond amination (1102) were the key to success. Moreover, hydrochloric acid worked as both the electrolyte and the acid promoter in this reaction.490
 | ||
| Scheme 116 Electrochemical C–H thiolation/C–N amination for thiazole synthesis. | ||
In 2024, Wan's group further developed their method and the electrochemical oxidative cyclization of enaminones with thioamides to achieve the thiazoles without using any catalyst and oxidants, improving the sustainability of this method.71
To further expand the structural diversity of thiazoles by using enaminones as the nitrogen source, Wen's group developed the cascade three-component cyclization of enaminones with potassium thiocyanate and alcohols (Scheme 117).75 The reaction proceeded smoothly via the oxidative C–H thiolation and nucleophilic tandem cyclization to construct the C–O, C–S, and C–N bonds, and the subsequent cleavage of C–N bond generated various 2-alkoxythiazoles (1107–1109).
 | ||
| Scheme 117 An electrochemical-enabled cascaded cyclization to obtain 2-alkoxythiazoles. | ||
Overall, electrochemical synthesis offers an efficient route to thiazole skeletons with varying degrees of substitution, bypassing the need for elaborate synthetic sequences. Moreover, the successful use of enaminones as a nitrogen source demonstrates its promising potential for expanding the scope of suitable starting materials. This progress not only highlighted the versatility of electrochemical methods but also suggested that numerous other possibilities awaited exploration in the future.
8.6 Synthesis of benzothiazoles
The benzothiazole scaffold represents a privileged heterocyclic system in medicinal chemistry due to its versatile pharmacological profile and structural adaptability.491 Its synthesis is pivotal for developing bioactive molecules, as the core structure confers enhanced binding affinity and metabolic stability.492 In fact, benzothiazole derivatives exhibit diverse therapeutic properties, including anticancer (e.g., tasisulam), antimicrobial (e.g., ethoxzolamide), and neuroprotective (e.g., riluzole) activities.6,493 The advancement of benzothiazole-based medicinal chemistry has emerged as a rapidly evolving and highly dynamic research frontier.494 In clinical practice, numerous benzothiazole-derived compounds have been successfully implemented as therapeutic agents, demonstrating high therapeutic efficacy against diverse disease pathologies.495 Benzothiazoles are traditionally prepared via the Phillips method or Jacobson synthesis.496 Emerging methods of benzothiazole synthesis are discussed in this section. | ||
| Scheme 118 Photochemical synthesis of 2-methylbenzothiazoles. | ||
To avoid halogen-substituted thioamides, Li's group further developed a photocatalytic method to construct the 2-substituted benzothiazoles in 2012 (Scheme 119).499 With the assistance of photocatalyst, this reaction was finalized under the irradiation of visible light with diverse thioamides as substrates. The mechanism suggested that the thioamide was first deprotonated to generate the anion intermediate (1126), which was oxidized by the excited state of photocatalyst to generate radical species (1127). The radical intermediate further reacted with the benzene ring, which further underwent the oxidation to provide the final products (1123, 1124). In 2015, Lei's group further found that a dual catalytic system via photoredox cobalt-catalysis could obviate the use of the oxidant, providing a novel strategy to construct the C–S bonds.500 A study by Gustafson and others also expanded the application of this method.501,502 Some heterogeneous photocatalysts have also been applied to the synthesis of benzothiazoles from thioamides.503
 | ||
| Scheme 119 Visible light photoredox radicals for the synthesis of the 2-substituted benzothiazoles. | ||
Besides the use of thioamides, 2-aminothiophenols and various aldehydes as synthetic synthons have been disclosed by Cho's group (Scheme 120). Considering the ready availability of aldehydes and the convenience to prepare the 2-aminothiophenols, this strategy was highly convenient and provided diverse benzothiazoles (1133, 1134). In this reaction, benzothiazoline (1136) was the key intermediate from the starting materials, which was oxidized by the excited state of the photocatalyst through an SET process. Afterwards, oxygen was involved in facilitating the deprotonation of radical species and finally completed the reaction.504 The same group further reported that in situ-generated photosensitizing disulfides from 2-aminothiophenols could also promote the synthesis of benzothiazoles.505 Biswas’ group also found a similar strategy to construct the benzothiazoles with different photocatalysts.244 Some heterogeneous photocatalysts have also been applied to the synthesis of benzothiazoles from aminothiophenols.506–508
 | ||
| Scheme 120 Synthesis of the 2-substituted benzothiazoles via visible light-driven photoredox. | ||
In 2019, Zhang's group also reported a photocatalytic method by using the 2-isocyanoaryl thioethers as the starting material (Scheme 121). With ethyl 2-bromo-2,2-difluoroacetate and 5-(trifluoromethyl)-5H dibenzo[b,d]thiophen-5-ium trifluoromethanesulfonate (1146) as radical precursors, 2-CF2/CF3-containing benzothiazoles (1143, 1144) have been successfully obtained in good to excellent yields via radical cascade cyclization.509
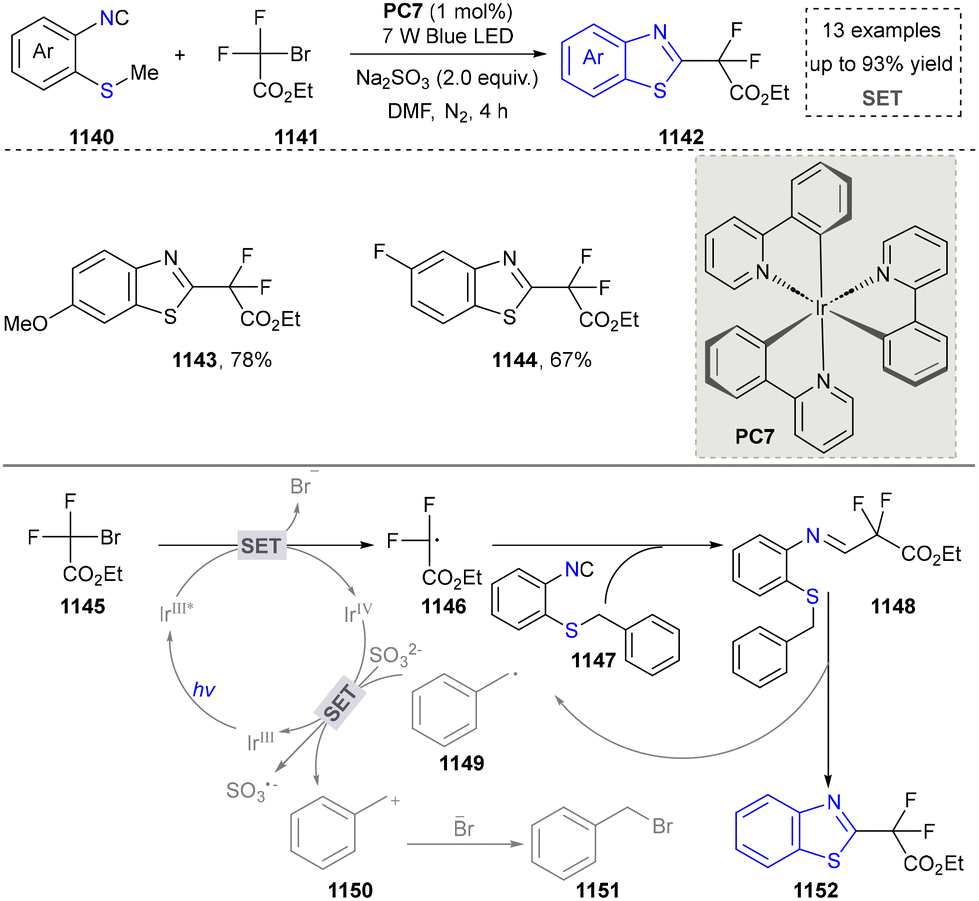 | ||
| Scheme 121 Photocatalytic cyclization for the synthesis of 2-CF2/CF3-containing benzothiazoles. | ||
Overall, the development of photochemical synthesis has significantly advanced the construction of benzothiazoles. In addition, it is possible to use simple starting materials such as 2-aminothiophenols to construct benzothiazoles. The intermolecular cyclization also improved the structural diversity. Hopefully, more synthetic synthons and convenient methods, such as multicomponent reactions (MCRs), can be developed to further expand the scope and efficiency of benzothiazole synthesis.
 | ||
| Scheme 122 Electrochemical synthesis of benzothiazole derivatives. | ||
In 2017, Lei's group found that 2-aminobenzothiazoles could be synthesized via electrochemical synthesis (Scheme 123). Compared to traditional methods, this work utilized aryl isothio cyanates and various amines as starting materials, which strongly expanded the structural diversity of benzothiazoles (1161–1163). The thiourea generated from nucleophilic addition of amine to isothiocyanates (1066) was the key intermediate, which underwent anodic oxidation twice to complete the synthesis of benzothiazoles.70 This approach allows for the efficient preparation of benzothiazole derivatives that are challenging to obtain through conventional oxidative methods due to the presence of sensitive functional groups, notably halogenated compounds and those bearing strong electron-withdrawing substituents. It thus offers a robust tool for the optimization of drug lead candidates. Wirth's group further expanded the electrocatalytic activation of thioamides and afforded valuable thiazolopyridines.511 Liu's group also developed a three-component method, but the concept was similar to Lei's work.512
 | ||
| Scheme 123 Electrochemical dehydrogenation for the synthesis of benzothiazoles. | ||
2-Aminothiophenols as nitrogen sources to construct benzothiazoles via electrochemical synthesis have also been established. In 2022, Luo's group reported an efficient Fe-catalysed electrochemical approach to synthesize benzothiazoles by using different 2-aminothiophenols and ethers as starting materials. In this case, in situ formation of aldehydes from ethers was the key to success.457 To further expand the use of electrochemical synthesis, Du's group disclosed a three-component strategy to construct benzothiazoles (Scheme 124). The structural diversity was strongly expanded by using various 2-aminothiophenols, aldehydes and malononitrile. In their proposed mechanism, a Knoevenagel condensation between malononitrile and benzaldehyde generated the intermediate (1179), the thiol radical from Cu-catalysis reacted with 1179 to give the radical species (1180), which further underwent 1,6-HAT and intramolecular cyclization to afford 1182. The subsequent release of -NH2 radicals completed the reaction.69 The intermolecular cyclization of isocyanides with thiols or diselenides has also been reported recently by He's group.513
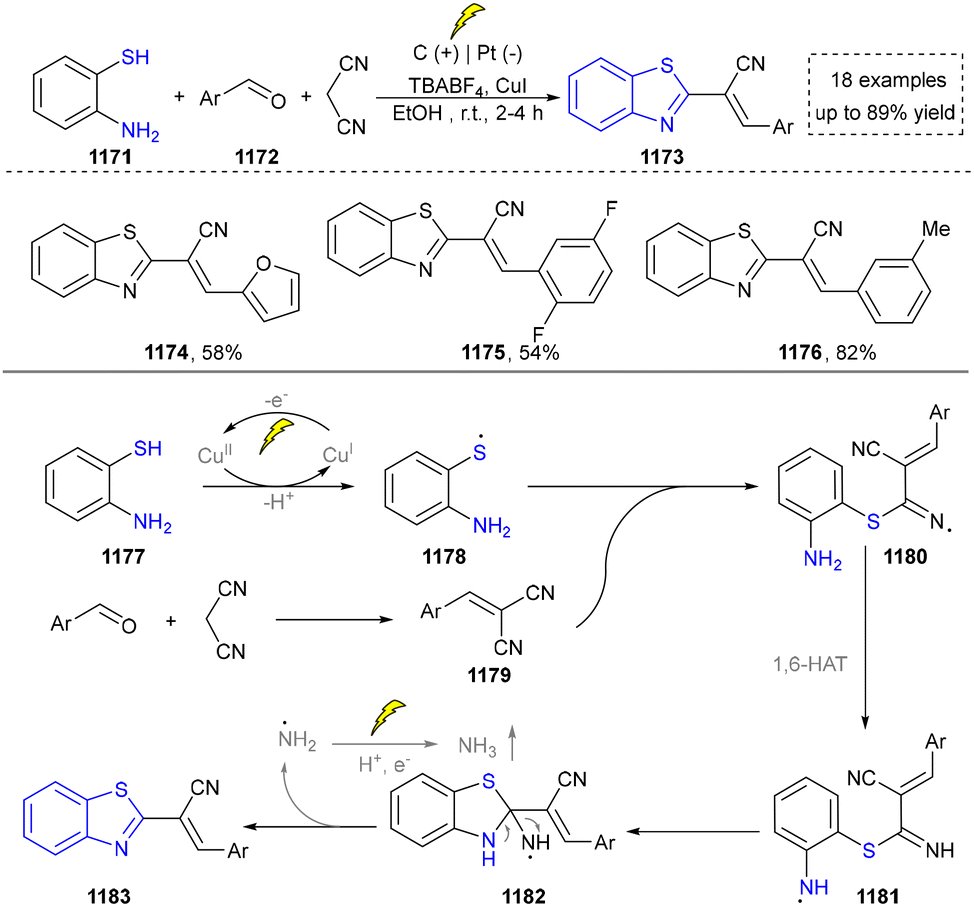 | ||
| Scheme 124 Cu-catalyzed electron-relayed synthesis of 2-alkenylbenzothiazoles. | ||
In summary, the synthesis of benzothiazoles by using electrochemical methods was extended to other nitrogen sources including isothiocyanates, which have been previously challenging to use. Moreover, three-component reactions including simple aldehydes and different 2-aminothiophenols as starting materials have also been developed, which strongly extended the structural diversity of obtained benzothiazoles such as the construction of 2-alkenylbenzothiazoles. However, the practical application of these methods is still in its early stage and warrants further exploration.
In 2017, Beifuss's group utilized a commercial laccase from A. bisporus to catalyse the synthesis of novel pyrimidobenzothiazoles without regio-selectivity from 1185 and catechol 1184 (Scheme 125).514 2-Phenylbenzothiazole was also obtained by the condensation of aldehydes and 2-aminothiophenol, which was catalysed by Novoprime Base 268 or the laccase (T. versicolor)/DDQ (2,3-dichloro-5,6-dicyano-1,4-benzoquinone) catalytic system.32,515
 | ||
| Scheme 125 Laccase-catalysed synthesis of pyrimidobenzothiazoles. | ||
As described above, the enzymatic synthesis of benzothiazole and benzoxazoles was catalysed by laccase and the GOX-CPO system. Therefore, further engineering of these enzymes, which can be applied to the synthesis of both benzothiazoles and benzoxazoles, is highly desirable.
8.7 Synthesis of 1,2,3-thiadiazoles
1,2,3-Thiadiazoles represent crucial nitrogen–sulfur heterocyclic scaffolds extensively present in natural products and pharmaceutical molecules.516 Numerous bioactive compounds containing this framework exhibit significant therapeutic effects including antiviral, anticancer, and antifungal activities.517 Additionally, 1,2,3-thiadiazoles serve as versatile synthetic intermediates in organic synthesis.518 While traditional synthetic methodologies such as the Hurd-Mori,519 Wolff,520,521 and Pechmann syntheses522 have been established, this section will emphasize the emerging synthetic strategies that demonstrate enhanced values in constructing 1,2,3-thiadiazole derivatives.In 2019, Rao's group reported a photocatalytic method to afford 1,2,3-thiadiazoles efficiently (Scheme 126).72 This strategy relied on the [4+1] annulations of α-bromo-N-benzoyl-hydrazone 1203 and potassiumthiocyanate 1198 to furnish the key intermediate 1203, which was further oxidized by singlet oxygen to give the final products (1200–1202). However, the preparation of the starting material was complicated, which limited the application of this method. Moreover, no progress has been made toward significantly expanding the structural diversity of 1,2,3-thiadiazoles.
 | ||
| Scheme 126 Photocatalytic method to afford the 1,2,3-thiadiazole derivatives. | ||
 | ||
| Scheme 127 Electrosynthesis of 1,2,3-thiadiazoles. | ||
Overall, the synthesis of 1,2,3-thiadiazoles via emerging methods remains underexplored. Only the hydrazones and related derivatives were reported as the nitrogen sources to construct 1,2,3-thiadiazoles. Compared to privileged scaffolds like benzothiazole and benzoxazole, which are prevalent in pharmaceuticals, the 1,2,3-thiadiazole core is relatively uncommon in marketed drugs. This has, to some extent, reduced the urgency for intensive exploration of new synthetic methods targeting it. Despite the challenges, the successful development of an electrochemical synthesis would represent the precise electrochemical construction of N–N and C–S bonds under mild conditions. Its underlying mechanism could potentially inspire the synthesis of other unstable heterocycles.
 | ||
| Scheme 128 Benzo[1,2,3]thiadiazole synthesis from benzothiazol-2(3H)-ones and 2-halobenzothiazoles by single-step skeletal editing. | ||
8.8 Synthesis of 1,2,4-thiadiazoles
1,2,4-Thiadiazoles constitute another important class of heterocyclic compounds with distinctive biological properties, frequently incorporated into bioactive molecules.525,526 Notably, the commercial antibiotic cefozopran, containing this scaffold, exhibits remarkable antibacterial activity.527 Conventional synthetic routes typically require stoichiometric oxidants,528 whereas this section will highlight the advantages of modern synthetic approaches in constructing 1,2,4-thiadiazoles systems. | ||
| Scheme 129 Visible-light-driven aerobic oxidative cyclization to afford 1,2,4-thiadiazoles. | ||
To further improve the synthetic efficiency, the He group disclosed that WS2 served as the semiconductor photocatalyst to achieve the synthesis of 1,2,4-thiadiazoles (1235–1237) efficiently with TEMPO and O2 (in air) as the redox catalysts. This strategy featured the more structural diversity using phenyl isothiocyanate and benzimidamide hydrochloride as substrates. Importantly, the large-scale synthesis was also applied to prove its applicability. The catalyst was also recyclable up to five times, which further improved the sustainability of this strategy (Scheme 130).530 Photochemical synthesis provides an effective method for preparing aliphatic 1,2,4-thiadiazoles, which are challenging to obtain through conventional approaches and allow for lipophilicity adjustment. The retained N–H bond in these high-yield products facilitates subsequent functionalization.
 | ||
| Scheme 130 Semi-heterogeneous photosynthesis of 5-amino-1,2,4-thiadiazoles. | ||
 | ||
| Scheme 131 Electrochemical synthesis of 3,5-disubstituted-1,2,4-thiadiazoles. | ||
Later, the Liu group also extended the electrocatalytic synthesis of 1,2,4-thiadiazoles from phenyl isothiocyanates and 2-aminopyridines.73 In 2020, the amino 1,2,4-thiadiazoles were also achieved by using imidoyl thiourea as the starting material under electrocatalytic conditions.532 Recently, the Wan group has also found that the electrocatalytic protocol furnished the oxidative cyclization of enaminones with thioamides.71
Overall, the synthesis of 1,2,4-thiadiazoles via an electrocatalytic method gives some new insights to broaden the substrate scope. The structural diversity was further improved using some other substances such as 2-aminopyridines and enaminones, which were previously challenging for photochemical synthesis. Hopefully other reagents can be further explored in this field.
 | ||
| Scheme 132 1,2,4-Thiadiazole synthesis by vanadium-dependent haloperoxidases. | ||
8.9 Synthesis of 1,3,4-thiadiazoles
As a significant member of azole heterocycles, thiadiazoles demonstrate diverse biological and pharmacological activities.534 Their unique aromatic structure containing two nitrogen atoms and one sulfur atom enables chemical modifications to develop highly bioactive derivatives, making the synthesis and bioactivity exploration of thiadiazole derivatives a research focus. Representative drugs include sulfamethizole (a sulfonamide antibiotic with a 1,3,4-thiadiazole moiety for bacterial infections) and acetazolamide (a carbonic anhydrase inhibitor for treating edema, epilepsy, and glaucoma).535 Although dehydration cyclization and direct cyclization remain common methods for 1,3,4-thiadiazole synthesis,536–539 this section will discuss the substantial contributions of innovative synthetic methodologies in this field. | ||
| Scheme 133 Visible-light-promoted synthesis of 1,3,4-thiadiazoles. | ||
Thiadiazoles (1277–1279) and selenadiazoles have also been achieved using the light-assisted method. In this case, benzoylhydrazines 1285 and 2-methylquinolines 1280 worked as starting materials and the products were formed through subsequent photocatalysis-induced iodine substitution, oxydic aldehyde formation, and nucleophilic addition. Importantly, the synthesized products showed attractive fluorescence properties for cell imaging, indicating their potential (Scheme 134).541 Therefore, intermolecular synthesis of 1,3,4-thiadiazoles using different nitrogen sources and sulfur sources provided an efficient method, which also showed good structural diversity.
 | ||
| Scheme 134 Photoinduced construction of thiadiazole derivatives. | ||
 | ||
| Scheme 135 Electrochemical oxidative synthesis of 1,3,4-thiadiazoles. | ||
Electrochemical synthesis of 1,3,4-thiadiazoles overcomes the inherent limitations of traditional methods in handling sensitive functional groups such as naked N–H bonds, eliminating the need for multi-step protection/deprotection procedures. This strategy enables the direct utilization of inexpensive starting materials containing sensitive motifs, allowing for the straightforward and efficient modification of complex molecular frameworks. Concurrently, applying this powerful tool to the direct modification of complex bioactive molecules represents a promising frontier.
Conclusions
Over the past decade, the advent of emerging approaches such as photochemical, electrochemical, biocatalysis and atom editing has revolutionized the construction of five-membered heteroarenes. These innovative approaches have not only provided new methods but also made the synthesis of previously challenging five-membered ring heteroarenes more feasible and efficient. Compared to traditional methods, photochemistry and electrochemistry always offer mild, eco-friendly conditions to avoid using additional additives or stoichiometric oxidants. More importantly, they could also create some unique pathways to activate inert bonds and precisely functionalize substrates, significantly broadening accessible substrate scopes. Enzymatic catalysis operates with biocompatible media and high selectivity, ideal for modifying complex bio-derived molecules and expanding natural substrate libraries. Atomic editing performs atom-precise modifications directly constructing novel heterocycles beyond conventional reach, always providing novel structural motifs. It is also important to acknowledge that these strategies come with their own inherent limitations. For example, photochemistry is constrained by limited light penetration depth and challenges in scale-up, with strategies employing photosensitizers in conjunction with continuous-flow systems being adopted to enhance the photon utilization efficiency.544,545 Furthermore, electrochemistry suffers from limitations such as mass transport constraints at the electrode interface and competing side reactions, which can be effectively mitigated through the modification of the electrode interface.546 The limitations of enzyme chemistry primarily stem from its narrow substrate specificity and instability under non-physiological conditions, making rational design and directed evolution via protein engineering key to expanding its application scope, even though such technologies have not yet been applied to the synthesis of five-membered aromatic heterocycles.547 Meanwhile, molecular editing, an emerging strategy, is progressively evolving towards greater refinement.In addition to integrating emerging technologies with continuous-flow synthesis to address scalability challenges, the development of integrated and hybrid catalytic systems has emerged as a pivotal strategic direction. For instance, continuous flow reactors are promising platforms for electrochemical synthesis, as they allow for the rapid refreshment of reagents at the electrode surface.548 The incorporation of artificial intelligence and machine learning is poised to enable the predictive modelling and rational design of complex hybrid catalytic networks, shifting the discovery paradigm from empirical screening to informed design.549,550 Another highly significant trend is the convergence and synergy among these four strategies. For example, the combination of enzymatic stereocontrol with photoredox-mediated bond activation within catalytic cascades promises to transcend the boundaries of singular methodologies, creating novel and sustainable pathways to complex heterocycles from basic feedstocks.551 In addition, photoenzyme catalysis and electroenzyme catalysis have emerged as cutting-edge areas in chiral biocatalysis.552,553 There is no doubt that the future impact of these hybrid technologies on the synthesis of five-membered heteroarenes will be impossible to ignore.
The application to natural product and drug synthesis has also been achieved, even though the related findings are still very poorly compared with the quantities of marked drugs and natural products baring these scaffolds. Furthermore, these methods enabled the late-stage modification of natural products and drugs. Moving forward, it is also necessary to develop synthetic methods that utilize more drug candidates without active group protection, with the long-term goal of applying these strategies to the total synthesis of natural products. We have also noticed that the direct application of these emerging toolboxes in drug discovery and lead modification remains largely underexplored. Hence, it is highly encouraging for medicinal chemists to apply these approaches more widely.
It is also worth noting that the synthesis of some of the five-membered heteroarenes using these emerging toolboxes remains underexplored, thus leaving ample room for further investigation. For instance, syntheses of thiophenes and isothiazoles by electrochemical synthesis have remained unexplored. The synthesis of pyrazoles, 1,2,4-triazoles, tetrazoles, thiophenes, benzothiophenes, isoxazoles, and isothiazoles via biocatalysis was also unexplored. Moreover, some five-membered heteroarenes have only a few reports, indicating that more concerted efforts are needed to fully explore their potential. The growing field of atom editing has also attracted considerable interest for its ability to diversify five-membered heteroarenes from other core structures. Overall, we are convinced that these emerging toolboxes will continue to yield more significant findings and applications in this field. Looking ahead, medicinal chemistry is also poised to greatly benefit from these innovative approaches.
In summary, each of these methods has its own distinct advantages in the synthesis of five-membered heteroaromatics. It is precisely through their complementary strengths that they have collectively driven the field forward at a remarkable pace.
Author contributions
Y. Z., W. D. Z, and S. D. conceived the project. Z. H. and Q. L. conducted the literature survey. Y. Z., Z. H., Z. L and Q. L. wrote the manuscript. W. D. Z and S. D. guided and reviewed the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included, and no new data were generated or analysed as part of this review.Acknowledgements
This work was supported by the National Natural Science Foundation of China (82404460, Y. Z. and 82304667, Z. H.), the General Project of Shanghai Natural Science Foundation (24ZR1466700), the Open Research Fund of the School of Chemistry and Chemical Engineering, Henan Normal University (2024Y08, Y. Z.), the CAMS Innovation Fund for Medical Sciences (CIFMS) (2023-I2M-3-009, W. Z.), and the Key Project at Central Government Level: The Ability Establishment of Sustainable Use for Valuable Chinese Medicine Resources (2060302-2305-02, W. Z.). We are grateful to Dingding Xia and Weijie Wang for their valuable contributions to the preliminary literature research.Notes and references
- F. Zhao, D. Masci, E. Tomarelli and D. Castagnolo, Synthesis, 2020, 2948–2961 CAS.
- V. Srivastava, P. K. Singh, S. Tivari and P. P. Singh, Org. Chem. Front., 2022, 9, 1485–1507 RSC.
- S. Devi, D. Wadhwa and J. Sindhu, Org. Biomol. Chem., 2022, 20, 5163–5229 RSC.
- P. Bhutani, G. Joshi, N. Raja, N. Bachhav, P. K. Rajanna, H. Bhutani, A. T. Paul and R. Kumar, J. Med. Chem., 2021, 64, 2339–2381 CrossRef CAS PubMed.
- P. Das, M. D. Delost, M. H. Qureshi, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2018, 62, 4265–4311 CrossRef PubMed.
- C. M. Marshall, J. G. Federice, C. N. Bell, P. B. Cox and J. T. Njardarson, J. Med. Chem., 2024, 67, 11622–11655 CrossRef CAS PubMed.
- K. W. Wellington, T. Qwebani-Ogunleye, N. I. Kolesnikova, D. Brady and C. B. de Koning, Arch. Pharm., 2013, 346, 266–277 CrossRef CAS PubMed.
- R. A. Craig, 2nd and B. M. Stoltz, Chem. Rev., 2017, 117, 7878–7909 CrossRef PubMed.
- Z. Meng, J. Yan, C. Ning, M. Shi and Y. Wei, Chem. Sci., 2023, 14, 7648–7655 RSC.
- J. C. van Meel, A. B. Mauz, W. Wienen and W. Diederen, J. Cardiovasc. Pharmacol., 1989, 13, 508–509 CrossRef CAS PubMed.
- A. H. Romero., Top. Curr. Chem., 2019, 377, 21–90 CrossRef PubMed.
- M. Gao, C. He, H. Chen, R. Bai, B. Cheng and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 6958–6961 Search PubMed.
- H. Jin and A. Furstner, Angew. Chem., Int. Ed., 2020, 59, 13618–13622 CrossRef CAS PubMed.
- J. Xuan, L. Q. Lu, J. R. Chen and W. J. Xiao, Eur. J. Org. Chem., 2013, 6755–6770 Search PubMed.
- S. C. Cosgrove and G. J. Miller, Expert Opin. Drug Discovery, 2022, 17, 355–364 Search PubMed.
- Y. Zhao, M. Cai, J. Xian, Y. Sun and G. Li, J. Mater. Chem. A, 2021, 9, 20164–20183 RSC.
- L. Ding, Y. Fan and H. Lu, Chem. Soc. Rev., 2025, 54, 8145–8169 RSC.
- S. Han, Z. Chen, Y. Guo, J. Chen, Z. Wang and Y.-F. Zeng, Org. Chem. Front., 2024, 11, 6694–6699 RSC.
- K. Kaur, R. Mandal, J. R. Walensky, F. Gallou and S. Handa, Angew. Chem., Int. Ed., 2025, 64, e202416132 CrossRef CAS PubMed.
- S. Jaiswal, A. Bhardwaj, J. Dwivedi and S. Sharma, Catal. Surv. Asia, 2025, 29, 1–23 Search PubMed.
- Q.-Q. Yang, J.-X. Li, G. Zhan, C. Peng, J.-L. Li and B. Han, J. Chem. Educ., 2025, 102, 2372–2377 CrossRef CAS.
- S. Slagman and W.-D. Fessner, Chem. Soc. Rev., 2021, 50, 1968–2009 RSC.
- M. K. Yadav and S. Chowdhury, Org. Biomol. Chem., 2025, 23, 506–545 RSC.
- S. Ghara, P. Barik, S. Ghosh, S. Ghosh, A. Mandal, C. Pramanik, M. Ikbal, S. Dhara and S. Samanta, Org. Chem. Front., 2025, 12, 2790–2837 Search PubMed.
- R. Sharma, M. Arisawa, S. Takizawa and M. S. Salem, Org. Chem. Front., 2025, 12, 1633–1670 Search PubMed.
- E. K. Taskinen and B. König, J. Nat. Prod., 2025, 88, 2822–2848 Search PubMed.
- C. E. Hatch and W. J. Chain, ChemElectroChem, 2023, 10, e202300140 CrossRef CAS PubMed.
- T. Manna, A. De, K. Nurjamal and S. M. Husain, Org. Biomol. Chem, 2022, 20, 7410–7414 RSC.
- Y. Li, Y. Zhang, J. Wang, D. Xia, M. Zhuo, L. Zhu, D. Li, S.-F. Ni, Y. Zhu and W.-D. Zhang, Org. Lett., 2024, 26, 3130–3134 CrossRef CAS PubMed.
- Y. Zhang, Y. Li, S.-F. Ni, J.-P. Li, D. Xia, X. Han, J. Lin, J. Wang, S. Das and W.-D. Zhang, Chem. Sci., 2023, 14, 10411–10419 RSC.
- M. Baidya, D. Maiti, L. Roy and S. De Sarkar, Angew. Chem., Int. Ed., 2022, 134, e202111679 CrossRef.
- M. Maphupha, W. P. Juma, C. B. de Koning and D. Brady, RSC Adv., 2018, 8, 39496–39510 RSC.
- T. W. von Zuben, G. Cariello Silva and A. G. Salles, Green Chem., 2021, 23, 6361–6365 RSC.
- Z. Chen, G. Shi, W. Tang, J. Sun and W. Wang, Eur. J. Org. Chem., 2021, 951–955 CrossRef CAS.
- W.-Q. Liu, T. Lei, Z.-Q. Song, X.-L. Yang, C.-J. Wu, X. Jiang, B. Chen, C.-H. Tung and L.-Z. Wu, Org. Lett., 2017, 19, 3251–3254 Search PubMed.
- F. Zhao, D. Masci, S. Ferla, C. Varricchio, A. Brancale, S. Colonna, G. W. Black, N. J. Turner and D. Castagnolo, ACS Catal., 2020, 10, 6414–6421 CrossRef CAS.
- O. F. Brandenberg, D. C. Miller, U. Markel, A. Ouald Chaib and F. H. Arnold, ACS Catal., 2019, 9, 8271–8275 Search PubMed.
- F. Verma, A. Sahu, P. K. Singh, A. Rai, M. Singh and V. K. Rai, Green Chem., 2018, 20, 3783–3789 Search PubMed.
- P.-F. Zhong, H.-M. Lin, L.-W. Wang, Z.-Y. Mo, X.-J. Meng, H.-T. Tang and Y.-M. Pan, Green Chem., 2020, 22, 6334–6339 RSC.
- F. Li, Y. Xu, C. Wang, C. Wang, R. Zhao and L. Wang, Bioorg. Chem., 2021, 107, 104583 CrossRef CAS PubMed.
- A. Vidyasagar, J. Shi, P. Kreitmeier and O. Reiser, Org. Lett., 2018, 20, 6984–6989 CrossRef CAS PubMed.
- Y. Yu, X.-B. Zhu, Y. Yuan and K.-Y. Ye, Chem. Sci., 2022, 13, 13851–13856 RSC.
- N. Scalacci, G. W. Black, G. Mattedi, N. L. Brown, N. J. Turner and D. Castagnolo, ACS Catal., 2017, 7, 1295–1300 Search PubMed.
- P. Zhang, T. Xiao, S. Xiong, X. Dong and L. Zhou, Org. Lett., 2014, 16, 3264–3267 CrossRef CAS PubMed.
- W.-J. Wei, Y.-Q. Zeng, X.-F. Liang, F.-H. Cui, M.-R. Wang, Y.-M. Pan, W.-G. Duan and H.-T. Tang, Green Chem., 2025, 27, 1006–1012 RSC.
- J. Wen, W. Zhao, X. Gao, X. Ren, C. Dong, C. Wang, L. Liu and J. Li, J. Org. Chem., 2022, 87, 4415–4423 CrossRef CAS PubMed.
- J. Zhang, C. Mück-Lichtenfeld, M. A. Wiethoff and A. Studer, Angew. Chem., Int. Ed., 2024, 63, e202416726 CrossRef CAS PubMed.
- H. Wang, Y. Ren, K. Wang, Y. Man, Y. Xiang, N. Li and B. Tang, Chem. Commun., 2017, 53, 9644–9647 RSC.
- X. Liu, C. Liu and X. Cheng, Green Chem., 2021, 23, 3468–3473 RSC.
- H. Chen, Y. Yan, N. Zhang, Z. Mo, Y. Xu and Y. Chen, Org. Lett., 2020, 23, 376–381 CrossRef PubMed.
- M.-X. He, Y. Yao, C.-Z. Ai, Z.-Y. Mo, Y.-Z. Wu, Q. Zhou, Y.-M. Pan and H.-T. Tang, Org. Chem. Front., 2022, 9, 781–787 RSC.
- Y. Wang, B. Tian, M. Ding and Z. Shi, Chem. – Eur. J., 2020, 26, 4297–4303 CrossRef CAS PubMed.
- X. Si, Y. Jia, X. Luan, L. Yang, Y. Pei and W. Zhou, Angew. Chem., Int. Ed., 2019, 58, 2660–2664 CrossRef CAS PubMed.
- J. Lee, E. Yu and C.-M. Park, Org. Biomol. Chem., 2022, 20, 7499–7502 RSC.
- K. Mitsudo, R. Matsuo, T. Yonezawa, H. Inoue, H. Mandai and S. Suga, Angew. Chem., Int. Ed., 2020, 59, 7803–7807 CrossRef CAS PubMed.
- J. Xue, L.-G. Bai, L. Zhang, Y. Zhou, X.-L. Lin, N.-J. Mou, D.-R. Xiao and Q.-L. Luo, J. Org. Chem., 2020, 85, 9761–9775 CrossRef CAS PubMed.
- Z. Cheng, Q. Gu and X. Zeng, Asian J. Org. Chem., 2023, 12, e202300461 CrossRef CAS.
- K. L. Dunbar, J. R. Chekan, C. L. Cox, B. J. Burkhart, S. K. Nair and D. A. Mitchell, Nat. Chem. Bioly., 2014, 10, 823–829 CrossRef CAS PubMed.
- Y. Liu, B. Wang, X. Qiao, C.-H. Tung and Y. Wang, ACS Catal., 2017, 7, 4093–4099 CrossRef CAS.
- H. Jiang, C. Zang, H. Cheng, B. Sun and X. Gao, Catal. Sci. Technol., 2021, 11, 7955–7962 RSC.
- T. D. Svejstrup, W. Zawodny, J. J. Douglas, D. Bidgeli, N. S. Sheikh and D. Leonori, Chem. Commun., 2016, 52, 12302–12305 RSC.
- X. Zhang, Q. Yuan, H. Zhang, Z.-J. Shen, L. Zhao, C. Yang, L. Guo and W. Xia, Green Chem., 2023, 25, 1435–1441 RSC.
- M. Z. Beg, S. Tivari, A. Kashyap, P. K. Singh, P. P. Singh, P. Nainwal and V. Srivastava, J. Heterocycl. Chem., 2024, 61, 458–465 CrossRef CAS.
- D. Maiti, A. Saha, S. Guin, D. Maiti and S. Sen, Chem. Sci., 2023, 14, 6216–6225 RSC.
- U. D. Newar, S. Borra and R. A. Maurya, Org. Lett., 2022, 24, 4454–4458 CrossRef CAS PubMed.
- T.-T. Zeng, J. Xuan, W. Ding, K. Wang, L.-Q. Lu and W.-J. Xiao, Org. Lett., 2015, 17, 4070–4073 CrossRef CAS PubMed.
- M. Li, Z. He, W. Zhao, Y. Yu, F. Huang and J. B. Baell, J. Org. Chem., 2023, 88, 8257–8267 CrossRef CAS PubMed.
- A. Saha, C. Sen, S. Guin, C. Das, D. Maiti, S. Sen and D. Maiti, Angew. Chem., Int. Ed., 2023, 62, e202308916 CrossRef CAS PubMed.
- C. Hu, D. Wang, L. Wang, Y. Fu and Z. Du, Green Chem., 2024, 26, 312–316 RSC.
- P. Wang, S. Tang and A. Lei, Green Chem., 2017, 19, 2092–2095 Search PubMed.
- Q. Huang, J. Liu and J.-P. Wan, Org. Lett., 2024, 26, 5263–5268 CrossRef PubMed.
- Y. Zhang, Y. Cao, L. Lu, S. Zhang, W. Bao, S. Huang and Y. Rao, J. Org. Chem., 2019, 84, 7711–7721 CrossRef CAS PubMed.
- J. S. Li, X. Y. Xie, P. P. Yang, S. Jiang, L. Tao, Z. W. Li, C. H. Lu and W. D. Liu, Adv. Synth. Catal., 2019, 362, 771–775 CrossRef.
- Y. Li, C.-C. Sun and C.-C. Zeng, J. Electroanal. Chem., 2020, 861, 113941 CrossRef CAS.
- D. Li, L. Chen, Y. Jin, X. Wang, L. Liu, Y. Li, G. Chen, G. Wu, Y. Qin, L. Yang, M. Wang, L. Zhao, Z. Xu and J. Wen, Green Chem., 2023, 25, 4656–4661 RSC.
- A. R. White, R. A. Kozlowski, S.-C. Tsai and C. D. Vanderwal, Angew. Chem., Int. Ed., 2017, 56, 10525–10529 CrossRef CAS PubMed.
- T. Shi, G. Yin, X. Wang, Y. Xiong, Y. Peng, S. Li, Y. Zeng and Z. Wang, Green. Synth. Catal., 2023, 4, 20–34 CAS.
- J. Pu, G. Liu, L. Wu and Z. Zhang, Sci. Sin. Chim., 2022, 52, 1357–1370 CrossRef.
- S. C. Philkhana, F. O. Badmus, I. C. Dos Reis and R. Kartika, Synthesis, 2021, 1531–1555 CrossRef CAS PubMed.
- A. Balakrishna, A. Aguiar, P. J. M. Sobral, M. Y. Wani, J. Almeida e Silva and A. J. F. N. Sobral, Catal. Rev., 2018, 61, 84–110 CrossRef.
- D. H. R. Barton and S. Z. Zard, J. Chem. Soc., Chem. Commun., 1985, 1098–1100 RSC.
- D. H. R. Barton, J. Kervagoret and S. Z. Zard, Tetrahedron Lett., 1990, 46, 7587–7598 CrossRef CAS.
- Y. Q. Zou, L. Q. Lu, L. Fu, N. J. Chang, J. Rong, J. R. Chen and W. J. Xiao, Angew. Chem., Int. Ed., 2011, 50, 7171–7175 CrossRef CAS PubMed.
- L. Huang and J. Zhao, Chem. Commun., 2013, 49, 3751–3753 RSC.
- B. Kurpil, K. Otte, A. Mishchenko, P. Lamagni, W. Lipiński, N. Lock, M. Antonietti and A. Savateev, Nat. Commun., 2019, 10, 945–954 CrossRef PubMed.
- T. Lei, W. Q. Liu, J. Li, M. Y. Huang, B. Yang, Q. Y. Meng, B. Chen, C. H. Tung and L. Z. Wu, Org. Lett., 2016, 18, 2479–2482 CrossRef CAS PubMed.
- N. N. Kumar Reddy, D. Rawat and S. Adimurthy, J. Org. Chem., 2018, 83, 9412–9421 CrossRef CAS PubMed.
- W. Hu, Q. Zhan, H. Zhou, S. Cao and Z. Jiang, Chem. Sci., 2021, 12, 6543–6550 RSC.
- S. Yao, X. Zhao, X. Ban, T. Shao, Y. Yin, W. Hu and Z. Jiang, Org. Chem. Front., 2023, 10, 4779–4785 RSC.
- S. Govindaraju and S. Tabassum, Top. Catal., 2022, 1–13 Search PubMed.
- T. Pavithra, D. B. Rajkumar, K. Gnanaoli, S. N. Sunil Gowda, N. Devipriya and C. U. Maheswari, ChemistrySelect, 2023, 8, e202204564 CrossRef CAS.
- F. Ahmadi, M. Shariatipour, M. Jadidi Nejad and A. Heydari, J. Photochem. Photobiol., A, 2024, 457, 115863 CrossRef CAS.
- D. Kim, J. You, D. H. Lee, H. Hong, D. Kim and Y. Park, Science, 2024, 386, 99–105 CrossRef CAS PubMed.
- A. G. O'Brien, F. Levesque and P. H. Seeberger, Chem. Commun., 2011, 47, 2688–2690 RSC.
- E. P. Farney and T. P. Yoon, Angew. Chem., Int. Ed., 2013, 53, 793–797 CrossRef PubMed.
- A. De and A. Majee, J. Heterocycl. Chem., 2022, 59, 422–448 CrossRef CAS.
- M. B. Harisha, P. Dhanalakshmi, R. Suresh, R. R. Kumar, S. Muthusubramanian and N. Bhuvanesh, ChemistrySelect, 2019, 4, 2954–2958 CrossRef CAS.
- S. Borra, D. Chandrasekhar, U. D. Newar and R. A. Maurya, J. Org. Chem., 2018, 84, 1042–1052 CrossRef PubMed.
- S. Borra, L. Borkotoky, U. D. Newar, B. Das and R. A. Maurya, Adv. Synth. Catal., 2020, 362, 3364–3368 CrossRef CAS.
- Y. Liu, A. Parodi, S. Battaglioli, M. Monari, S. Protti and M. Bandini, Org. Lett., 2019, 21, 7782–7786 CrossRef CAS PubMed.
- J. Xuan, X. D. Xia, T. T. Zeng, Z. J. Feng, J. R. Chen, L. Q. Lu and W. J. Xiao, Angew. Chem., Int. Ed., 2014, 53, 5653–5656 CrossRef CAS PubMed.
- B. S. Karki, L. Devi, A. Pokhriyal, R. Kant and N. Rastogi, Chem. – Asian J., 2019, 14, 4793–4797 CrossRef CAS PubMed.
- A. Pokhriyal, B. Singh Karki, R. Kant and N. Rastogi, J. Org. Chem., 2021, 86, 4661–4670 CrossRef CAS PubMed.
- L. Wang, C. Liu, L. Li, X. Wang, R. Sun, M. D. Zhou and H. Wang, Chin. J. Chem., 2022, 40, 719–724 CrossRef CAS.
- X. Gao, P. Wang, Q. Wang, J. Chen and A. Lei, Green Chem., 2019, 21, 4941–4945 RSC.
- G. Shi, J. Sun and Z. Chen, ChemistrySelect, 2021, 6, 12342–12345 CrossRef CAS.
- W.-J. Chen and G.-Q. Yuan, Tetrahedron Lett., 2022, 90, 153615 CrossRef CAS.
- M. Baidya, J. Dutta and S. De Sarkar, Org. Lett., 2023, 25, 3812–3817 CrossRef CAS PubMed.
- Y. Zhao, Y. Fan, X. Meng, X. Kang, Z. Ji, S. Yan and L. Tian, J. Org. Chem., 2022, 87, 11131–11140 CrossRef CAS PubMed.
- E. K. Jaffe, Prog. Mol. Biol. Transl., 2019, 169, 85–104 Search PubMed.
- R. M. Lüönd and R. Neier, Biochim. Biophys. Acta, Gen. Subj., 1996, 1289, 83–86 CrossRef PubMed.
- Q. Kang, H. Fang, M. Xiang, K. Xiao, P. Jiang, C. You, S. Y. Lee and D. Zhang, Nat. Commun., 2023, 14, 5177–5191 CrossRef CAS PubMed.
- J. L. Wang, X. Y. Chen, Q. Wu and X. F. Lin, Adv. Synth. Catal., 2014, 356, 999–1005 CrossRef CAS.
- X.-Y. Chen, J.-L. Wang, X.-F. Lin and Q. Wu, Tetrahedron, 2016, 72, 3318–3323 CrossRef CAS.
- H. Gaweska and P. F. Fitzpatrick, Biomol. Concepts, 2011, 2, 365–377 CrossRef CAS PubMed.
- R. R. Ramsay and A. Albreht, J. Neural Transm., 2018, 125, 1659–1683 CrossRef CAS PubMed.
- K. E. Atkin, R. Reiss, V. Koehler, K. R. Bailey, S. Hart, J. P. Turkenburg, N. J. Turner, A. M. Brzozowski and G. Grogan, J. Mol. Biol., 2008, 384, 1218–1231 CrossRef CAS PubMed.
- D. Ghislieri, A. P. Green, M. Pontini, S. C. Willies, I. Rowles, A. Frank, G. Grogan and N. J. Turner, J. Am. Chem. Soc., 2013, 135, 10863–10869 CrossRef CAS PubMed.
- R. S. Heath, M. Pontini, B. Bechi and N. J. Turner, ChemCatChem, 2014, 6, 996–1002 CrossRef CAS.
- H. Chachignon, N. Scalacci, E. Petricci and D. Castagnolo, J. Org. Chem., 2015, 80, 5287–5295 CrossRef CAS PubMed.
- S. Bhakta, N. Scalacci, A. Maitra, A. K. Brown, S. Dasugari, D. Eyangelopoulos, T. D. McHugh, P. N. Mortazavi, A. Twist, E. Petricci, F. Manetti and D. Castagnolo, J. Med. Chem., 2016, 59, 2780–2793 CrossRef CAS PubMed.
- M. Arend, B. Westermann, N. Risch, N. L. Brown, N. J. Turner and D Castagnolo, Angew. Chem., Int. Ed., 1998, 37, 1044–1070 CrossRef PubMed.
- A. E. Ondrus and M. Movassaghi, Chem. Commun., 2009, 4151–4165 RSC.
- A. Gomm and E. O’Reilly, Curr. Opin. Chem. Biol., 2018, 43, 106–112 CrossRef CAS PubMed.
- F. Guo and P. Berglund, Green Chem., 2017, 19, 333–360 RSC.
- S. A. Kelly, S. Pohle, S. Wharry, S. Mix, C. C. R. Allen, T. S. Moody and B. F. Gilmore, Chem. Rev., 2018, 118, 349–367 CrossRef CAS PubMed.
- J. Xu, A. P. Green and N. J. Turner, Angew. Chem., Int. Ed., 2018, 57, 16760–16763 CrossRef CAS PubMed.
- P. R. August, T. H. Grossman, C. Minor, M. P. Draper, I. A. MacNeil, J. M. Pemberton, K. M. Call, D. Holt and M. S. Osburne, J. Mol. Microbiol. Biotechnol., 2000, 2, 513–519 CAS.
- H.-E. Lai, A. M. C. Obled, S. M. Chee, R. M. Morgan, R. Lynch, S. V. Sharma, S. J. Moore, K. M. Polizzi, R. J. M. Goss and P. S. Freemont, ACS Chem. Biol., 2021, 16, 2116–2123 CrossRef CAS PubMed.
- A. Ahmed, A. Ahmad, R. Li, W. Al-Ansi, M. Fatima, B. S. Mushtaq, S. Basharat, Y. Li and Z. Bai, Microbiol. Biotechnol., 2021, 31, 1465–1480 CrossRef CAS PubMed.
- T. Nishizawa, S. Grüschow, D. H. E. Jayamaha, C. Nishizawa-Harada and D. H. Sherman, J. Am. Chem. Soc., 2006, 128, 724–725 CrossRef CAS PubMed.
- A. R. Howard-Jones and C. T. Walsh, Biochemistry, 2005, 44, 15652–15663 CrossRef CAS PubMed.
- T. Spolitak and D. P. Ballou, Arch. Biochem. Biophys., 2015, 573, 111–119 CrossRef CAS PubMed.
- H. Onaka, S.-i Taniguchi, Y. Igarashi and T. Furumai, J. Antibiot., 2002, 55, 1063–1071 CrossRef CAS PubMed.
- Q. Gao, C. Zhang, S. Blanchard and J. S. Thorson, Chem. Biol., 2006, 13, 733–743 CrossRef CAS PubMed.
- S.-Y. Kim, J.-S. Park, C.-S. Chae, C.-G. Hyun, B. W. Choi, J. Shin and K.-B. Oh, Appl. Microbiol. Biotechnol., 2007, 75, 1119–1126 CrossRef CAS PubMed.
- H.-T. Chiu, Y.-L. Chen, C.-Y. Chen, C. Jin, M.-N. Lee and Y.-C. Lin, Mol. Biosyst., 2009, 5, 1180–1191 CrossRef CAS PubMed.
- H.-T. Chiu, Y.-C. Lin, M.-N. Lee, Y.-L. Chen, M.-S. Wang and C.-C. Lai, Mol. Biosyst., 2009, 5, 1192–1203 CrossRef CAS PubMed.
- F.-Y. Chang, M. A. Ternei, P. Y. Calle and S. F. Brady, J. Am. Chem. Soc., 2013, 135, 17906–17912 CrossRef CAS PubMed.
- C. L. Yang, B. Zhang, W. W. Xue, W. Li, Z. F. Xu, J. Shi, Y. Shen, R. H. Jiao, R. X. Tan and H. M. Ge, Org. Lett., 2020, 22, 4665–4669 CrossRef CAS PubMed.
- X. Zheng, Y. Li, M. Guan, L. Wang, S. Wei, Y. C. Li, C. Y. Chang and Z. Xu, Angew. Chem., Int. Ed., 2022, 61, e202208802 CrossRef CAS PubMed.
- F. Wen, Y. Xu, F. Li, J. Ma, Z. Wang, H. Zhang and L. Wang, Catalysts, 2023, 13, 1493–1504 CrossRef CAS.
- W. A. A. Arafa and M. F. Hussein, Chin. J. Chem., 2020, 38, 501–508 CrossRef CAS.
- A. Abdelhamid, K. S. M. Salama, A. M. Elsayed, M. A. Gad and M. El-Remaily, Acs. Omega, 2022, 7, 3990–4000 CrossRef CAS PubMed.
- C. T. Walsh, S. Garneau-Tsodikova and A. R. Howard-Jones, Nat. Prod. Rep, 2006, 23, 517–531 RSC.
- Y. Hu, Q. Zhou, Z. Zhang, H. X. Pan, Y. Ilina, M. Metsä-Ketelä, Y. Igarashi and G. L. Tang, Chin. J. Chem., 2021, 39, 3329–3333 CrossRef CAS.
- S. Siebenberg, N. Burkard, A. Knuplesch, B. Gust, S. Grond and L. Heide, ChemBioChem, 2011, 12, 2677–2685 CrossRef CAS PubMed.
- J. Wang, H. Lu, Y. He, C. Jing and H. Wei, J. Am. Chem. Soc, 2022, 144, 22433–22439 CrossRef CAS PubMed.
- W. Wei, Q. Xie, X. Li, Y. Xie and H. Zhou, Org. Biomol. Chem., 2025, 23, 3937–3941 RSC.
- S. Liu, F. Zhao, X. Chen, G. J. Deng and H. Huang, Adv. Synth. Catal., 2020, 362, 3795–3823 CrossRef CAS.
- O. G. Julia, C. Reisenbauer, A. Franchino, P. Finkelstein and B. Morandi, Science, 2022, 377, 1104–1109 CrossRef PubMed.
- S. M. Umer, M. Solangi, K. M. Khan and R. S. Z. Saleem, Molecules, 2022, 27, 7586–7630 CrossRef CAS PubMed.
- M. Chauhan, A. Saxena and B. Saha, Eur. J. Med. Chem., 2021, 218, 113400 CrossRef CAS PubMed.
- M. J. Nieto and H. K. Lupton, Curr. Med. Chem., 2021, 28, 4828–4844 CrossRef CAS PubMed.
- W. Zeng, C. Han, S. Mohammed, S. Li, Y. Song, F. Sun and Y. Du, RSC Med. Chem., 2024, 15, 788–808 RSC.
- J. Zoller, D. C. Fabry, M. A. Ronge and M. Rueping, Angew. Chem., Int. Ed., 2014, 53, 13264–13268 CrossRef CAS PubMed.
- C.-J. Wu, Q.-Y. Meng, T. Lei, J.-J. Zhong, W.-Q. Liu, L.-M. Zhao, Z.-J. Li, B. Chen, C.-H. Tung and L.-Z. Wu, ACS Catal., 2016, 6, 4635–4639 CrossRef CAS.
- Y. Y. Liu, X. Y. Yu, J. R. Chen, M. M. Qiao, X. Qi, D. Q. Shi and W. J. Xiao, Angew. Chem., Int. Ed., 2017, 56, 9527–9531 CrossRef CAS PubMed.
- S. Jana, A. Verma, R. Kadu and S. Kumar, Chem. Sci., 2017, 8, 6633–6644 RSC.
- Y. Zhang, X. Yang, H. Zhou, S. Li, Y. Zhu and Y. Li, Org. Chem. Front., 2018, 5, 2120–2125 RSC.
- G. P. da Silva, A. Ali, R. C. da Silva, H. Jiang and M. W. Paixão, Chem. Commun., 2015, 51, 15110–15113 RSC.
- J. Tang, L. Ren, J. Li, Y. Wang, D. Hu, X. Tong and C. Xia, Org. Lett., 2022, 24, 3582–3587 CrossRef CAS PubMed.
- Z. S. Wang, Y. B. Chen, H. W. Zhang, Z. Sun, C. Zhu and L. W. Ye, J. Am. Chem. Soc., 2020, 142, 3636–3644 CrossRef CAS PubMed.
- Q. Huang, M. Zhao, Y. Yang, Y. N. Niu and X. F. Xia, Org. Chem. Front., 2021, 8, 5988–5993 RSC.
- Q. Li, X. Gu, Y. Wei and M. Shi, Chem. Sci., 2022, 13, 11623–11632 RSC.
- X.-X. Du, Q.-X. Zi, Y.-M. Wu, Y. Jin, J. Lin and S.-J. Yan, Green Chem., 2019, 21, 1505–1516 RSC.
- H. Qin, R. Liu, Z. Wang, F. Xu, X. Li, C. Shi, J. Chen, W. Shan, C. Liu, P. Xing, J. Zhu, X. Li and D. Shi, Angew. Chem., Int. Ed., 2024, 137, e202416923 CrossRef.
- X. D. Xia, J. Xuan, Q. Wang, L. Q. Lu, J. R. Chen and W. J. Xiao, Adv. Synth. Catal., 2014, 356, 2807–2812 CrossRef CAS.
- Z. Wang, G. Fei, S. Shi, M. Liu, P. Li and P. Xie, Mol. Catal., 2024, 564, 114334 CAS.
- C.-C. Zeng, F.-J. Liu, D.-W. Ping, L.-M. Hu, Y.-L. Cai and R.-G. Zhong, J. Org. Chem., 2009, 74, 6386–6389 CrossRef CAS PubMed.
- Z. W. Hou, Z. Y. Mao, H. B. Zhao, Y. Y. Melcamu, X. Lu, J. Song and H. C. Xu, Angew. Chem., Int. Ed., 2016, 55, 9168–9172 CrossRef CAS PubMed.
- F. Xu, Y.-J. Li, C. Huang and H.-C. Xu, ACS Catal., 2018, 8, 3820–3824 Search PubMed.
- Y. Wu, H. Yi and A. Lei, ACS Catal., 2018, 8, 1192–1196 CrossRef CAS.
- S. Tang, X. Gao and A. Lei, Chem. Commun., 2017, 53, 3354–3356 RSC.
- C. Yuan, X. Huang, Y. Lu, Z. Fang, C. Liu, B. Chen and K. Guo, Green. Synth. Catal., 2023, 4, 311–315 CAS.
- X. Chang, X. Chen, S. Lu, Y. Zhao, Y. Ma, D. Zhang, L. Yang and P. Sun, Adv. Synth. Catal., 2022, 364, 2865–2871 Search PubMed.
- S. Mallick, T. Mandal, N. Kumari, L. Roy and S. De Sarkar, Chem. – Eur. J., 2024, 30, e202304002 Search PubMed.
- P. Du, J. L. Brosmer and D. G. Peters, Org. Lett., 2011, 13, 4072–4075 CrossRef CAS PubMed.
- B. Huang, G. Chen, H. Zhang, X. Tang, J. Yuan, C. Lu and J. Wang, Org. Chem. Front., 2023, 10, 3515–3521 RSC.
- Y. Kim, D. Kim and S. Chang, Chem. Commun., 2021, 57, 12309–12312 RSC.
- Z. Tan, X. Zhang, M. Xu, Y. Fu, W. Zhuang, M. Li and C. Zhu, Sci. Adv., 2022, 8, eadd1912 Search PubMed.
- S. Elangovan, J. Neumann, J.-B. Sortais, K. Junge, C. Darcel and M. Beller, Nat. Commun., 2016, 7, 12641–12648 CrossRef PubMed.
- L. Ferrer, M. Mindt, V. F. Wendisch and K. Cankar, Syst. Microbiol. Biomanuf., 2023, 4, 511–527 CrossRef.
- Z. Huang, Y. Yao, R. Di, J. Zhang, Y. Pan and G. Liu, Microb. Biotechnol., 2025, 18, e70135 CrossRef CAS PubMed.
- J. Woo, A. H. Christian, S. A. Burgess, Y. Jiang, U. F. Mansoor and M. D. Levin, Science, 2022, 376, 527–532 Search PubMed.
- I. Ameziane El Hassani, K. Rouzi, H. Assila, K. Karrouchi and M. H. Ansar, Reactions, 2023, 4, 478–504 Search PubMed.
- K. Karrouchi, S. Radi, Y. Ramli, J. Taoufik, Y. N. Mabkhot, F. A. Al-aizari and M. H. Ansar, Molecules, 2018, 23, 134–219 Search PubMed.
- M. Sadeghpour and A. Olyaei, Res. Chem. Intermed., 2021, 47, 4399–4441 Search PubMed.
- D. Liu, Q. Bi, J. Zhang, Y. Gao, C. Luo, H. Tian, J. He and L. Zhang, Bioorg. Chem., 2024, 143, 107024 CrossRef CAS PubMed.
- S. P. Chandrasekharan, A. Dhami, S. Kumar and K. Mohanan, Org. Biomol. Chem., 2022, 20, 8787–8817 RSC.
- Y. Duan, Q. Zhao, Y. Yang, J. Zhang, X. Tao and Y. Shen, J. Heterocycl. Chem., 2019, 56, 1464–1471 CrossRef CAS.
- M. K. Hunjan, S. Panday, A. Gupta, J. Bhaumik, P. Das and J. K. Laha, Chem. Rec., 2021, 21, 715–780 CrossRef CAS PubMed.
- B. Bishop, K. Brands, A. Gibb and D. Kennedy, Synthesis, 2003, 43–52 CrossRef.
- S. Yadav, P. Rai, M. Srivastava, J. Singh, K. P. Tiwari and J. Singh, Tetrahedron Lett., 2015, 56, 5831–5835 CrossRef CAS.
- Y. Ding, T. Zhang, Q.-Y. Chen and C. Zhu, Org. Lett., 2016, 18, 4206–4209 CrossRef CAS PubMed.
- Y. Meng, T. Zhang, X. Gong, M. Zhang and C. Zhu, Tetrahedron Lett., 2019, 60, 171–174 CrossRef CAS.
- B. Pati Tripathi, A. Mishra, P. Rai, Y. Kumar Pandey, M. Srivastava, S. Yadav, J. Singh and J. Singh, New J. Chem., 2017, 41, 11148–11154 RSC.
- A. K. Sharma, A. Jaiswal, A. Mishra, J. Tiwari, D. Jaiswal, S. Singh, J. Singh and J. Singh, New J. Chem., 2020, 44, 13350–13356 RSC.
- H. Sagir, P. Rai, A. Ibad, F. Ibad and I. R. Siddiqui, Catal. Commun., 2017, 100, 153–156 Search PubMed.
- J. Cheng, W. Li, Y. Duan, Y. Cheng, S. Yu and C. Zhu, Org. Lett., 2016, 19, 214–217 Search PubMed.
- T. Zhang, Y. Meng, J. Lu, Y. Yang, G. Q. Li and C. Zhu, Adv. Synth. Catal., 2017, 360, 3063–3068 Search PubMed.
- C. P. Lakeland, D. W. Watson and J. P. A. Harrity, Chem. – Eur. J., 2019, 26, 155–159 CrossRef PubMed.
- Y. Wu, J.-Y. Chen, J. Ning, X. Jiang, J. Deng, Y. Deng, R. Xu and W.-M. He, Green Chem., 2021, 23, 3950–3954 RSC.
- W.-B. He, S.-J. Zhao, J.-Y. Chen, J. Jiang, X. Chen, X. Xu and W.-M. He, Chin. Chem. Lett., 2023, 34, 107640 CrossRef CAS.
- S. A. Paveliev, O. O. Segida, O. V. Bityukov, H. T. Tang, Y. M. Pan, G. I. Nikishin and A. O. Terent'ev, Adv. Synth. Catal., 2022, 364, 3910–3916 CrossRef CAS.
- M. Linden, S. Hofmann, A. Herman, N. Ehler, R. M. Bär and S. R. Waldvogel, Angew. Chem., Int. Ed., 2023, 62, e202214820 CrossRef CAS PubMed.
- J. Feng, Y. Wang, L. Gao, Y. Yu, J. B. Baell and F. Huang, J. Org. Chem., 2022, 87, 13138–13153 CrossRef CAS PubMed.
- G. L. Bartholomew, F. Carpaneto and R. Sarpong, J. Am. Chem. Soc., 2022, 144, 22309–22315 CrossRef CAS PubMed.
- H. C. Van der Plas and H. Jongejan, Recl. Trav. Chim. Pays-Bas, 1968, 87, 1065–1072 CrossRef CAS.
- H. C. Van der Plas and H. Jongejan, Tetrahedron Lett., 1967, 8, 4385–4388 CrossRef.
- J. Luo, Q. Zhou, Z. Xu, K. N. Houk and K. Zheng, J. Am. Chem. Soc., 2024, 146, 21389–21400 CrossRef PubMed.
- Z. Wang, P. Xu, S. M. Guo, C. G. Daniliuc and A. Studer, Nature, 2025, 642, 92–98 CrossRef CAS PubMed.
- A. Siwach and P. K. Verma, BMC Chem., 2021, 15, 1–69 Search PubMed.
- J.-C. Tsou, C.-J. Tsou, C.-H. Wang, A.-L. A. Ko, Y.-H. Wang, H.-H. Liang, J.-C. Sun, K.-F. Huang, T.-P. Ko, S.-Y. Lin and Y.-S. Wang, J. Am. Chem. Soc., 2024, 146, 33309–33315 CrossRef CAS PubMed.
- H. Huang, T. Yan, C. Liu, Y. Lu, Z. Wu, X. Wang and J. Wang, Nat. Commun., 2024, 15, 5714–5724 CrossRef CAS PubMed.
- L. Zhang, X.-M. Peng, G. L. V. Damu, R.-X. Geng and C.-H. Zhou, Med. Res. Rev., 2014, 34, 340–437 Search PubMed.
- F. K. Keter and J. Darkwa, BioMetals, 2011, 25, 9–21 CrossRef PubMed.
- N. Rani, R. Singh and P. Kumar, Curr. Org. Synth., 2023, 20, 630–662 CrossRef CAS PubMed.
- Q. H. Deng, Y. Q. Zou, L. Q. Lu, Z. L. Tang, J. R. Chen and W. J. Xiao, Chem – Asian J., 2014, 9, 2432–2435 CrossRef CAS PubMed.
- S. Park, J. Jung and E. J. Cho, Eur. J. Org. Chem., 2014, 4148–4154 CrossRef CAS.
- P. R. Adiyala, S. Jang, N. K. Vishwakarma, Y.-H. Hwang and D.-P. Kim, Green Chem., 2020, 22, 1565–1571 RSC.
- L. Borkotoky, K. Bora and R. A. Maurya, Asian J. Org. Chem., 2023, 12, e202300146 CrossRef CAS.
- S. M. Patel, E. P. Prasad, M. Bakthadoss and D. S. Sharada, Org. Lett., 2021, 23, 257–261 CrossRef CAS PubMed.
- A. R. Patel, G. Patel and S. Banerjee, ACS Omega, 2019, 4, 22445–22455 CrossRef CAS PubMed.
- N. Sbei, A. V. Listratova, A. A. Titov and L. G. Voskressensky, Synthesis, 2019, 2455–2473 CAS.
- Z. W. Hou, Z. Y. Mao, Y. Y. Melcamu, X. Lu and H. C. Xu, Angew. Chem., Int. Ed., 2018, 57, 1636–1639 Search PubMed.
- P. Qian, Z. Yan, Z. Zhou, K. Hu, J. Wang, Z. Li, Z. Zha and Z. Wang, J. Org. Chem., 2019, 84, 3148–3157 CrossRef CAS PubMed.
- S. S. Grishin, O. M. Mulina, V. A. Vil’ and A. O. Terent'ev, Org. Chem. Front., 2024, 11, 327–335 RSC.
- V. A. Vil’, S. S. Grishin and A. O. Terent’ev, Molecules, 2022, 27, 7721–7735 CrossRef PubMed.
- L. Zeng, J. Li, J. Gao, X. Huang, W. Wang, X. Zheng, L. Gu, G. Li, S. Zhang and Y. He, Green Chem., 2020, 22, 3416–3420 Search PubMed.
- W. Wang, S. Zhang, G. Shi and Z. Chen, Org. Biomol. Chem., 2021, 19, 6682–6686 RSC.
- K. Zhou, S. Xia, Y. Liu and Z. Chen, Org. Biomol. Chem., 2022, 20, 7840–7844 Search PubMed.
- Z. Yang, J. Zhang, L. Hu, A. Li, L. Li, K. Liu, T. Yang and C. Zhou, J. Org. Chem., 2020, 85, 5952–5958 CrossRef CAS PubMed.
- A. Li, C. Li, T. Yang, Z. Yang, Y. Liu, L. Li, K. Tang and C. Zhou, J. Org. Chem., 2023, 88, 1928–1935 Search PubMed.
- C. Thomas and C. D. Gwenin, Biology, 2021, 10, 388–403 CrossRef CAS PubMed.
- R. J. Knox, R. C. Knight and D. I. Edwards, Biochem. Pharmacol., 1983, 32, 2149–2156 CrossRef CAS PubMed.
- J. B. Hedges and K. S. Ryan, Angew. Chem., Int. Ed., 2019, 58, 11647–11651 Search PubMed.
- N. D. Mahurkar, N. D. Gawhale, M. N. Lokhande, S. J. Uke and M. M. Kodape, Results Chem., 2023, 6, 101139 CrossRef CAS.
- N. T. Chung, V. C. Dung and D. X. Duc, RSC Adv., 2023, 13, 32734–32771 RSC.
- X. Li, M. D. Romero, S. Tcaturian, K. Kurpiewska and A. Dömling, J. Org. Chem., 2023, 88, 9823–9834 CrossRef CAS PubMed.
- O. Ebenezer, F. Oyetunde-Joshua, O. D. Omotoso and M. Shapi, Results Chem., 2023, 5, 100925 CrossRef CAS.
- V. A. Mamedov and N. A. Zhukova, Synthesis, 2021, 1849–1878 CrossRef CAS.
- G. K. Padhy, J. Panda, C. Sekhar Patr, S. K. Raul and A. Kumar Behera, Rasayan J. Chem., 2023, 16, 2009–2018 CrossRef CAS.
- S. Samanta, S. Das and P. Biswas, J. Org. Chem., 2013, 78, 11184–11193 CrossRef CAS PubMed.
- Z. Li, H. Song, R. Guo, M. Zuo, C. Hou, S. Sun, X. He, Z. Sun and W. Chu, Green Chem., 2019, 21, 3602–3605 Search PubMed.
- W.-K. An, S.-J. Zheng, H.-X. Zhang, T.-T. Shang, H.-R. Wang, X.-J. Xu, Q. Jin, Y. Qin, Y. Ren, S. Jiang, C.-L. Xu, M.-S. Hou and Z. Pan, Green Chem., 2021, 23, 1292–1299 RSC.
- Y. Shiraishi, Y. Sugano, S. Tanaka and T. Hirai, Angew. Chem., Int. Ed., 2010, 49, 1656–1660 CrossRef CAS PubMed.
- M. H. Li, Z. Yang, H. Hui, B. Yang, Y. Wang and Y. W. Yang, Angew. Chem., Int. Ed., 2023, 62, e202313358 CrossRef CAS PubMed.
- Y. F. Wang, M. Y. Qi, M. Conte, Z. R. Tang and Y. J. Xu, Angew. Chem., Int. Ed., 2023, 62, e202304306 CrossRef CAS PubMed.
- M. Hosseini-Sarvari and H. Sheikh, React. Chem. Eng., 2022, 7, 2202–2210 RSC.
- T. Montini, V. Gombac, J. J. Delgado, A. M. Venezia, G. Adami and P. Fornasiero, Inorg. Chim. Acta, 2021, 520, 120289 Search PubMed.
- G. Kumaraswamy, G. Sadanandam, K. Ledwaba and R. Maroju, J. Photochem. Photobiol., A, 2022, 429, 113888 Search PubMed.
- G. Li, R. He, Q. Liu, Z. Wang, Y. Liu and Q. Wang, J. Org. Chem., 2019, 84, 8646–8660 CrossRef CAS PubMed.
- B. Hu, W. Dong, Z. Feng, X. Gao, H. Gao, X. Xie and Z. Zhang, Asian J. Org. Chem, 2016, 5, 1467–1470 CrossRef CAS.
- M. Lin, F. Wu, T. Liu, Z. Chen, X. Xu and F. Ke, Chin. J. Org. Chem., 2020, 40, 2563–2569 CrossRef CAS.
- G. Patel, A. R. Patel and S. Banerjee, New J. Chem., 2020, 44, 13295–13300 RSC.
- E. Skolia, M. K. Apostolopoulou, N. F. Nikitas and C. G. Kokotos, Eur. J. Org. Chem., 2021, 422–428 CrossRef CAS.
- S. Kumari, A. Joshi, I. Borthakur and S. Kundu, J. Org. Chem., 2023, 88, 11523–11533 CrossRef CAS PubMed.
- M. Laćan, V. Rogić, I. Tabaković, D. Galijaš and T. Solomun, Electrochim. Acta, 1983, 28, 199–207 CrossRef.
- H.-B. Zhao, Z.-W. Hou, Z.-J. Liu, Z.-F. Zhou, J. Song and H. Xu, Angew. Chem., Int. Ed., 2016, 56, 587–590 CrossRef PubMed.
- H. B. Zhao, J. L. Zhuang and H. C. Xu, ChemSusChem, 2021, 14, 1692–1695 CrossRef CAS PubMed.
- K. Sun, S. Li, Y. Si and Q. Huang, Crit. Rev. Biotechnol., 2021, 41, 969–993 CrossRef CAS PubMed.
- T. Kudanga, G. S. Nyanhongo, G. M. Guebitz and S. Burton, Enzyme Microb. Technol., 2011, 48, 195–208 CrossRef CAS PubMed.
- A. Díaz-Rodríguez, I. Lavandera, S. Kanbak-Aksu, R. A. Sheldon, V. Gotor and V. Gotor-Fernández, Adv. Synth. Catal., 2012, 354, 3405–3408 CrossRef.
- K. Kędziora, A. Díaz-Rodríguez, I. Lavandera, V. Gotor-Fernández and V. Gotor, Green Chem., 2014, 16, 2448–2453 RSC.
- H. Leutbecher, M.-A. Constantin, S. Mika, J. Conrad and U. Beifuss, Tetrahedron Lett., 2011, 52, 605–608 CrossRef CAS.
- M. Maphupha, W. P. Juma, C. B. de Koning and D. Brady, RSC Adv., 2018, 8, 39496–39510 RSC.
- M. Mogharabi-Manzari, M. Kiani, S. Aryanejad, S. Imanparast, M. Amini and M. A. Faramarzi, Adv. Synth. Catal., 2018, 360, 3563–3571 CrossRef CAS.
- J. C. van Meel, A. B. Mauz, W. Wienen and W. Diederen, J. Cardiovasc. Pharmacol., 1989, 13, 508–509 CrossRef CAS PubMed.
- K. L. Boyle and E. Leech, J. Vet. Emerg. Crit. Car., 2012, 22, 398–408 CrossRef PubMed.
- H. Li, J. Liu, Y. Huo, X. Li, Q. Chen and Y. Gao, Green Chem., 2025, 27, 12580–12585 Search PubMed.
- A. S. Paschke, Y. Brägger, B. Botlik, E. Staudinger, O. Green and B. Morandi, Nat. Chem., 2025, 17, 1750–1756 CrossRef CAS PubMed.
- C. S. dos Santos, C. S. Buettner, D. B. Yildiz, M. Mamone, A. Ruffoni and D. Leonori, Angew. Chem., Int. Ed., 2025, 64, e202423804 CrossRef PubMed.
- D. Lengerli, K. Ibis, Y. Nural and E. Banoglu, Expert Opin. Drug Discovery, 2022, 17, 1209–1236 CrossRef CAS PubMed.
- H. Hernández-López, S. Leyva-Ramos, C. F. Azael Gómez-Durán, A. Pedraza-Alvarez, I. R. Rodríguez-Gutiérrez, M. A. Leyva-Peralta and R. S. Razo-Hernández, ACS Omega, 2020, 5, 14061–14068 CrossRef PubMed.
- J. Dai, S. Tian, X. Yang and Z. Liu, Front. Chem., 2022, 10, 891484 Search PubMed.
- M. Cui, C. Su, R. Wang, Q. Yang and C. Kuang, RSC Adv., 2021, 11, 38933–38937 RSC.
- R. Khandelwal, M. Vasava, R. B. Abhirami and M. Karsharma, Bioorg. Med. Chem. Lett., 2024, 112, 129927 Search PubMed.
- P. Kumar, C. Joshi, A. K. Srivastava, P. Gupta, R. Boukherroub and S. L. Jain, ACS Sustainable Chem. Eng, 2015, 4, 69–75 Search PubMed.
- W. D. Castro-Godoy, A. A. Heredia, L. C. Schmidt and J. E. Argüello, RSC Adv., 2017, 7, 33967–33973 Search PubMed.
- Z. G. Wu, X. J. Liao, L. Yuan, Y. Wang, Y. X. Zheng, J. L. Zuo and Y. Pan, Chem. – Eur. J., 2020, 26, 5694–5700 Search PubMed.
- P. Gogoi, T. Saikia, R. Hazarika, A. Garg, K. Deori and D. Sarma, ACS Sustainable Chem. Eng., 2023, 11, 15207–15217 Search PubMed.
- X. Guo, L. Zeng, Z. Wang, T. Zhang, C. He and C. Duan, RSC Adv., 2017, 7, 52907–52913 RSC.
- B. Wang, J. Durantini, J. Nie, A. E. Lanterna and J. C. Scaiano, J. Am. Chem. Soc., 2016, 138, 13127–13130 CrossRef CAS PubMed.
- J. Wen, W. Zhao, X. Gao, X. Ren, C. Dong, C. Wang and J. Li, J. Org. Chem., 2022, 87, 4415–4423 CrossRef CAS PubMed.
- Q. Guan, S. Xing, L. Wang, J. Zhu, C. Guo, C. Xu, Q. Zhao, Y. Wu, Y. Chen and H. Sun, J. Med. Chem., 2024, 67, 7788–7824 CrossRef CAS PubMed.
- N. Henning, T. Daßler and W. Jugelt, Z. Chem., 1982, 22, 25–26 CrossRef CAS.
- K. Titenkova, A. D. Shuvaev, F. E. Teslenko, E. S. Zhilin and L. L. Fershtat, Green Chem., 2023, 25, 6686–6693 RSC.
- A. D. Shuvaev, M. A. Feoktistov, F. E. Teslenko and L. L. Fershtat, Adv. Synth. Catal., 2024, 366, 5050–5060 CrossRef CAS.
- P. Xu and H. C. Xu, ChemElectroChem, 2019, 6, 4177–4179 CrossRef CAS.
- M. Bandyopadhyay, U. Goswami, S. Ghorai, S. Pathak, D. Ganguly, J. Escorihuela, J. Ganguly and M. K. Bera, Asian J. Org. Chem., 2024, 14, e202400415 CrossRef.
- G. Zhao, Y.-Y. Guo, S. Yao, X. Shi, L. Lv and Y.-L. Du, Nat. Commun., 2020, 11, 1614–1623 CrossRef CAS PubMed.
- F. Hou, Y. Wan, Q. Gan, M. Xian and W. Huang, Front. Bioeng. Biotechnol., 2020, 8, 603514 CrossRef PubMed.
- L. Kiss, E. Forró, R. Sillanpää and F. Fülöp, Tetrahedron, 2008, 19, 2856–2860 CrossRef CAS.
- F. Rutjes, S. Groothuys, B. Kuijpers, P. Quaedflieg, H. Roelen, R. Wiertz, R. Blaauw and F. van Delft, Synthesis, 2006, 3146–3152 Search PubMed.
- T. P. Khobragade, S. Sarak, A. D. Pagar, H. Jeon, P. Giri and H. Yun, Front. Bioeng. Biotechnol., 2021, 9, 757062 CrossRef PubMed.
- B.-L. Li, B. Li, R.-L. Zhang, J.-J. Zhao, X.-F. Wang, Y.-M. Liu, Y.-P. Shi, J.-B. Liu and B.-Q. Chen, Bioorg. Med. Chem. Lett., 2016, 26, 1279–1281 CrossRef CAS PubMed.
- N. Liu, J. Tu, G. Dong, Y. Wang and C. Sheng, J. Med. Chem., 2018, 61, 5484–5511 CrossRef CAS PubMed.
- A. Marwaha, J. White, F. El_Mazouni, S. A. Creason, S. Kokkonda, F. S. Buckner, S. A. Charman, M. A. Phillips and P. K. Rathod, J. Med. Chem., 2012, 55, 7425–7436 CrossRef CAS PubMed.
- U. Salma, S. Ahmad, M. Zafer Alam and S. A. Khan, J. Mol. Struct., 2024, 1301, 137240 CrossRef CAS.
- S. Couto Rodrigues, R. Silva Moratório de Moraes, G. Tavares de Almeida Pinto, M. T. Miranda Martins, P. Antunes do Nascimento, D. L. Alves Soares, A. B. Mestre Botelho, C. Cardoso Cruz and A. C. Cunha, Chem. Rec., 2024, 25, e202400190 CrossRef PubMed.
- Z. Ye, M. Ding, Y. Wu, Y. Li, W. Hua and F. Zhang, Green Chem., 2018, 20, 1732–1737 RSC.
- Y. Li, Z. Ye, N. Chen, Z. Chen and F. Zhang, Green Chem., 2019, 21, 4035–4039 RSC.
- H. Huang, M. P. Winters, S. K. Meegalla, E. Arnoult, S. Paul Lee, S. Zhao, T. Martin, B. Rady, J. Liu, M. Towers, M. Otieno, F. Xu, H. K. Lim, J. Silva, A. Pocai and M. R. Player, Bioorg. Med. Chem. Lett., 2018, 28, 429–436 CrossRef CAS PubMed.
- L. Dai, S. Cui, G. L. V. Damu and C. Zhou, Chin. J. Org. Chem., 2013, 33, 224–244 CrossRef CAS.
- S. Leyva-Ramos and J. Cardoso-Ortiz, Cur. Org. Chem., 2021, 25, 388–403 CrossRef CAS.
- S.-Q. Wang, Y.-F. Wang and Z. Xu, Eur. J. Med. Chem., 2019, 170, 225–234 CrossRef CAS PubMed.
- Y. Uozumi and Y. Sugiyama, Synfacts, 2018, 1199 Search PubMed.
- Z. Ye, F. Wang, Y. Li and F. Zhang, Green Chem., 2018, 20, 5271–5275 RSC.
- K. Chand, Rajeshwari, A. Hiremathad, M. Singh, M. A. Santos and R. S. Keri, Pharmacol. Rep., 2017, 69, 281–295 CrossRef CAS PubMed.
- M. Abu Yousef and R. Matsubara, RSC Adv., 2023, 13, 5228–5248 RSC.
- W. Zhang, W. Xu, F. Zhang, C. Ma, K. Ma and Y. Li, Chin. J. Org. Chem., 2020, 40, 2338 CrossRef CAS.
- Y. C. H. Jiang, Y. Zhang and S. Yu, Org. Lett., 2013, 15, 4884–4887 CrossRef PubMed.
- Y. Y. Han, Y. Y. Jiao, D. Ren, Z. Hu, S. Shen and S. Yu, Asian J. Org. Chem., 2016, 6, 414–417 CrossRef.
- H. Jiang, Y. Cheng, Y. Zhang and S. Yu, Org. Lett., 2013, 15, 4884–4887 CrossRef CAS PubMed.
- Y. Li, H. Liu, Z. Huang, Y. He, B.-H. Xu, H. Wang and Z. Yu, J. Org. Chem., 2021, 86, 8402–8413 CrossRef CAS PubMed.
- Y. Liu, W. Luo, T. Xia, Y. Fang, C. Du, X. Jin, Y. Li, L. Zhang, W. Lei and H. Wu, Org. Chem. Front., 2021, 8, 1732–1738 RSC.
- S. Wang, W. L. Jia, L. Wang, Q. Liu and L. Z. Wu, Chem. – Eur. J., 2016, 22, 13794–13798 CrossRef CAS PubMed.
- A. Quintavalla, R. Veronesi, L. Speziali, A. Martinelli, N. Zaccheroni, L. Mummolo and M. Lombardo, Adv. Synth. Catal., 2022, 364, 362–371 CrossRef CAS.
- A. Shao, X. Luo, C. W. Chiang, M. Gao and A. Lei, Chem. – Eur. J., 2017, 23, 17874–17878 CrossRef CAS PubMed.
- L. Feng, H. Yan, C. Yang, D. Chen and W. Xia, J. Org. Chem., 2016, 81, 7008–7022 CrossRef CAS PubMed.
- R. Bisht, S. Singh, K. Krishnamoorthy and J. Nithyanandhan, Photochem. Photobiol. Sci., 2018, 17, 835–845 CrossRef CAS PubMed.
- T. Chen, W. Wu and Z. Weng, Tetrahedron, 2019, 75, 130751 CrossRef CAS.
- J. C. L. Walker, S. Werrel and T. J. Donohoe, Chem. – Eur. J., 2019, 25, 13114–13118 CrossRef CAS PubMed.
- Y.-M. Tian, H. Wang, Ritu and B. König, Chem. Sci., 2022, 13, 241–246 RSC.
- S. Zhu, F. Li, C. Empel, S. Jana, C. Pei and R. M. Koenigs, Adv. Synth. Catal., 2022, 364, 3149–3154 CrossRef CAS.
- J.-H. Cui, M. Tan, J. Zhang, Y. He, X. Li and P.-N. Liu, Org. Chem. Front., 2025, 12, 1905–1910 RSC.
- B. S. Gore, C.-Y. Kuo and J.-J. Wang, Green Chem., 2023, 25, 8074–8081 RSC.
- D. Maiti, A. Halder, A. Sasidharan Pillai and S. De Sarkar, J. Org. Chem., 2021, 86, 16084–16094 CrossRef CAS PubMed.
- M. Chen, J. Wang, Y. Kan, X. Jia, B. Huang, T. Li and X. Zhao, Org. Lett., 2023, 25, 4540–4545 CrossRef CAS PubMed.
- C. Risi, F. Zhao and D. Castagnolo, ACS Catal., 2019, 9, 7264–7269 CrossRef CAS.
- J. Chen, J. Cai, F. Sha, W. Sun, X. Lyu, Y. Chang, F. Cao, L. Zhao, H. Wu and P. Ouyang, Green Chem., 2023, 25, 7126–7140 RSC.
- Z. Du, D. Yang, Q. Cao, J. Dai, R. Yang, X. Gu and F. Li, Bioresour. Bioprocess., 2023, 10, 52–68 CrossRef PubMed.
- Z. Huang, J. Wang, J. Lei, W. Zhao, H. Chen, Y. Yang, Q. Xu and X. Liu, Front. Chem., 2022, 10, 925603 CrossRef CAS PubMed.
- N. Li and M.-H. Zong, ACS Catal., 2022, 12, 10080–10114 CrossRef CAS.
- W. Zhao, F. Wang, K. Zhao, X. Liu, X. Zhu, L. Yan, Y. Yin, Q. Xu and D. Yin, Carbon Resour. Convers., 2023, 6, 116–131 CAS.
- X.-P. Liao, Q. Wu, M.-H. Zong and N. Li, Bioresour. Bioprocess., 2024, 11, 38–47 CrossRef.
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- B. Liu and Z. Zhang, ChemSusChem, 2016, 9, 2015–2036 CrossRef CAS PubMed.
- P. Wrigstedt, J. Keskiväli, J. E. Perea-Buceta and T. Repo, ChemCatChem, 2017, 9, 4244–4255 CrossRef CAS.
- Y. Peng, L. Jiang, J. Di, C. Ma, Q. Li and Y. He, ACS Sustainable Chem. Eng., 2022, 10, 12165–12176 CrossRef CAS.
- Q. Li, C.-L. Ma and Y.-C. He, Bioresour. Technol., 2023, 378, 128965 CrossRef CAS PubMed.
- Q. Wu, M.-H. Zong and N. Li, ACS Catal., 2023, 13, 9404–9414 CrossRef CAS.
- Y.-M. Chiang, C. E. Oakley, M. Ahuja, R. Entwistle, A. Schultz, S.-L. Chang, C. T. Sung, C. C. C. Wang and B. R. Oakley, J. Am. Chem. Soc., 2013, 135, 7720–7731 CrossRef CAS PubMed.
- Y. Zhao, Y. He, L. Han, L. Zhang, Y. Xia, F. Yin, X. Wang, D. Zhao, S. Xu, F. Qiao, Y. Xiao and L. Kong, Acta Pharm. Sin. B, 2024, 14, 869–880 CrossRef CAS PubMed.
- C. Villard, R. Munakata, S. Kitajima, R. van Velzen, M. E. Schranz, R. Larbat and A. Hehn, New Phytol., 2021, 231, 1923–1939 CrossRef CAS PubMed.
- A. Bouillé, R. Larbat, R. Kumari, A. Olry, C. Charles, D. R. Nelson, J. Thornton, C. Villard and A. Hehn, New Phytol., 2025, 245, 2085–2102 CrossRef.
- Y. Ma, G. Cui, T. Chen, X. Ma, R. Wang, B. Jin, J. Yang, L. Kang, J. Tang, C. Lai, Y. Wang, Y. Zhao, Y. Shen, W. Zeng, R. J. Peters, X. Qi, J. Guo and L. Huang, Nat. Commun., 2021, 12, 685–695 CrossRef CAS PubMed.
- J. J. Song, X. Fang, C. Y. Li, Y. Jiang, J. X. Li, S. Wu, J. Guo, Y. Liu, H. Fan, Y. B. Huang, Y. K. Wei, Y. Kong, Q. Zhao, J. J. Xu, Y. H. Hu, X. Y. Chen and L. Yang, Plant Physiol., 2022, 188, 1496–1506 Search PubMed.
- A. Mushtaq, A. F. Zahoor, S. Ahmad, M. J. Saif, A. ul Haq, S. G. Khan, A. A. Al-Mutairi, A. Irfan, S. A. Al-Hussain and M. E. A. Zaki, ACS Omega, 2024, 9, 20728–20752 CrossRef CAS PubMed.
- P. Patel, R. Shakya, Vishakha, V. Asati, B. D. Kurmi, S. K. Verma, G. D. Gupta and H. Rajak, J. Mol. Struct., 2024, 1299, 137098 Search PubMed.
- S. Bhargava and D. Rathore, Lett. Org. Chem., 2017, 14, 381–402 CrossRef CAS.
- D. Goyal, A. Kaur and B. Goyal, ChemMedChem, 2018, 13, 1275–1299 CrossRef CAS PubMed.
- D. X. Duc, Curr. Org. Synth., 2020, 17, 498–517 CrossRef CAS PubMed.
- A. S. G. E. A. Chugunova, A. R. Burilov, L. M. Yusupova, M. A. Pudovik and O. G. Sinyashin, Russ. Chem. Bull., 2019, 68, 887–910 CrossRef.
- S. Ghosh, J. Das and F. Saikh, Tetrahedron Lett., 2012, 53, 5883–5886 CrossRef CAS.
- Y. Suzuki, Y. Okita, T. Morita and Y. Yoshimi, Tetrahedron Lett., 2014, 55, 3355–3357 CrossRef CAS.
- X. Li, Z. Xu, L. Wang, F. Wang, J. Yang and P. Li, ChemPhotoChem, 2020, 5, 142–148 CrossRef.
- J. Liu, S. Tang, S. Wang, M. Cao, J. Zhao, P. Zhang and P. Li, J. Org. Chem., 2022, 87, 9250–9258 Search PubMed.
- H. Wang, Y. Huang, Q. Wu, J. Lu, Y.-L. Xu and Y.-Y. Chen, J. Org. Chem, 2022, 87, 13288–13299 Search PubMed.
- L. Wang, M. Zhang, Y. Zhang, Q. Liu, X. Zhao, J.-S. Li, Z. Luo and W. Wei, Chin. Chem. Lett., 2020, 31, 67–70 CrossRef CAS.
- Z. Xia, O. Khaled, V. Mouriès-Mansuy, C. Ollivier and L. Fensterbank, J. Org. Chem., 2016, 81, 7182–7190 CrossRef CAS PubMed.
- S. Borra, D. Chandrasekhar, S. Khound and R. A. Maurya, Org. Lett., 2017, 19, 5364–5367 Search PubMed.
- J. Y. Cho, G.-b Roh and E. J. Cho, J. Org. Chem., 2018, 83, 805–811 Search PubMed.
- L. Zhang, X. Si, F. Rominger and A. S. K. Hashmi, J. Am. Chem. Soc., 2020, 142, 10485–10493 Search PubMed.
- X.-S. Ji, H.-D. Zuo, Y.-T. Shen, W.-J. Hao, S.-J. Tu and B. Jiang, Chem. Commun., 2022, 58, 10420–10423 Search PubMed.
- Y. Wang, B. Tian, M. Ding and Z. Shi, Chem. – Eur. J., 2019, 26, 4297–4303 Search PubMed.
- Z. Feng, X. Guan, H. Ma, Y. Fan, P. Liu and P. Sun, Green Chem., 2024, 26, 11216–11221 Search PubMed.
- K. Cen, M. Ouyang, G. He, Z. Zeng, Q. Wang, X. Yu, F. Zhao and J. Cai, Green. Synth. Catal., 2024, 6, 444–448 Search PubMed.
- U. Beifuss, H. Leutbecher, J. Conrad and I. Klaiber, Synlett, 2005, 3126–3130 Search PubMed.
- S. Hajdok, H. Leutbecher, G. Greiner, J. Conrad and U. Beifuss, Tetrahedron Lett., 2007, 48, 5073–5076 CrossRef CAS.
- M. Kidwai, A. Jain, A. Sharma and R. C. Kuhad, C. R. Chim., 2013, 16, 728–735 Search PubMed.
- K. W. Wellington, T. Qwebani-Ogunleye, N. I. Kolesnikova, D. Brady and C. B. de Koning, Arch. Pharm., 2013, 346, 266–277 Search PubMed.
- T. Qwebani-Ogunleye, N. I. Kolesnikova, P. Steenkamp, C. B. de Koning, D. Brady and K. W. Wellington, Bioorgan. Med. Chem., 2017, 25, 1172–1182 Search PubMed.
- X. Peng, G. Yin, K. Wu, G. Wu, J. Chen and Z. Wang, Asian J. Org. Chem, 2024, 13, e202300631 Search PubMed.
- K. A. Scott and J. T. Njardarson, Sulfur Chem., 2019, 1–34 Search PubMed.
- R. Shah and P. K. Verma, Chem. Cent. J., 2018, 12, 1–22 Search PubMed.
- D. D. Xuan, Mini-Rev. Org. Chem., 2021, 18, 110–134 Search PubMed.
- J. E. Page, E. Block and G. H. N. Towers, Photochem. Photobiol., 1999, 70, 159–165 Search PubMed.
- Y.-Z. Chen, D.-H. Wang, B. Chen, J.-J. Zhong, C.-H. Tung and L.-Z. Wu, J. Org. Chem., 2012, 77, 6773–6777 Search PubMed.
- B. Zheng, X. Li, Y. Song, S. Meng, Y. Li, Q. Liu and L. Pan, Org. Lett., 2021, 23, 3453–3459 Search PubMed.
- D. Clemett and C. M. Spencer, Adis Drug Eval., 2000, 60, 379–411 Search PubMed.
- J. D. Croxtall and G. L. Plosker, Adis Drug Eval., 2009, 69, 339–359 CAS.
- S. K. Nandana, P. Rahul, S. Ann Babu, J. John and H. Hopf, Chem. Rec., 2024, 24, e202400019 CrossRef PubMed.
- D. P. Hari, T. Hering and B. König, Org. Lett., 2012, 14, 5334–5337 CrossRef CAS PubMed.
- B. Alcaide, P. Almendros, E. Busto, F. Herrera, C. Lázaro-Milla and A. Luna, Adv. Synth. Catal., 2017, 359, 2640–2652 CrossRef CAS.
- F. Gao, J.-T. Wang, L.-L. Liu, N. Ma, C. Yang, Y. Gao and W. Xia, Chem. Commun., 2017, 53, 8533–8536 RSC.
- L. Gao, B. Chang, W. Qiu, L. Wang, X. Fu and R. Yuan, Adv. Synth. Catal., 2016, 358, 1202–1207 CrossRef CAS.
- K. P. Sujith and A. Lee, Eur. J. Org. Chem., 2023, e202300257 Search PubMed.
- Z. Wang, J.-L. Li, S.-P. Zhang and W.-C. Yang, Mol. Catal., 2023, 549, 113469 CAS.
- J. Yan, J. Xu, Y. Zhou, J. Chen and Q. Song, Org. Chem. Front., 2018, 5, 1483–1487 RSC.
- X.-Y. Yuan, F.-L. Zeng, H.-L. Zhu, Y. Liu, Q.-Y. Lv, X.-L. Chen, L. Peng and B. Yu, Org. Chem. Front., 2020, 7, 1884–1889 RSC.
- D. Zhang, Y. Sun, G. Wang, Y. Liu, C. Ni, Q. Ji, X. Xu and Z. Fang, J. Org. Chem, 2024, 89, 13367–13372 Search PubMed.
- X.-F. Xia, G.-W. Zhang and S.-L. Zhu, Tetrahedron, 2017, 73, 2727–2730 Search PubMed.
- X. Gong, M. Wang, S. Ye and J. Wu, Org. Lett., 2019, 21, 1156–1160 Search PubMed.
- T. Kitamura, K. Morita, H. Nakamori and J. Oyamada, J. Org. Chem., 2019, 84, 4191–4199 Search PubMed.
- T. Xu, F.-D. Wang, W.-C. Yang, T. Lu, M. Wang and P. Li, Green Chem., 2025, 27, 2386–2391 RSC.
- S. Rajput, Rajat, Nitesh, P. Gupta, H. Singh and N. Jain, J. Org. Chem., 2025, 90, 493–502 CrossRef CAS PubMed.
- K. Mitsudo, Y. Tachibana, E. Sato and S. Suga, Org. Lett., 2022, 24, 8547–8552 CrossRef CAS PubMed.
- M.-M. Zhang, Y. Sun, W.-W. Wang, K.-K. Chen, W.-C. Yang and L. Wang, Org. Biomol. Chem., 2021, 19, 3844–3849 RSC.
- Z. Cheng, Q. Gu and X. Zeng, Asian J. Org. Chem., 2023, 12, e202300461 Search PubMed.
- D. Kurandina and V. Gevorgyan, Org. Lett., 2016, 18, 1804–1807 CrossRef CAS PubMed.
- J. Zhu, J. Mo, H.-Z. Lin, Y. Chen and H.-P. Sun, Bioorgan. Med. Chem., 2018, 26, 3065–3075 Search PubMed.
- Y. Shinde, B. Khairnar and S. Bangale, ChemistrySelect, 2024, 9, e202401423 Search PubMed.
- A. Arzine, H. Hadni, K. Boujdi, K. Chebbac, N. Barghady, Y. Rhazi and M. El Yazidi, Molecules, 2024, 29, 3366–3384 CrossRef CAS PubMed.
- G. C. Arya, K. Kaur and V. Jaitak, Eur. J. Med. Chem., 2021, 221, 113511 CrossRef CAS PubMed.
- Y. Walunj, P. Mhaske and P. Kulkarni, Mini-Rev. Org. Chem., 2021, 18, 55–77 CAS.
- H.-L. Xiao, C.-C. Zeng, H.-Y. Tian, L.-M. Hu and R. D. Little, J. Electroanal. Chem., 2014, 727, 120–124 CrossRef CAS.
- S. Hofmann, J. Winter, T. Prenzel, M. de Jesús Gálvez-Vázquez and S. R. Waldvogel, ChemElectroChem, 2023, 10, e202300434 CrossRef CAS.
- S. Hosseini, S. A. Bawel, M. S. Mubarak and D. G. Peters, ChemElectroChem, 2019, 6, 4318–4324 Search PubMed.
- E. Rodrigo, H. Baunis, E. Suna and S. R. Waldvogel, Chem. Commun., 2019, 55, 12255–12258 Search PubMed.
- N. Sun, Z. Qiao, J. Li, J. Gu, L. Jin and X. Hu, Green Chem., 2024, 26, 10240–10246 RSC.
- H.-Z. Zhang, Z.-L. Zhao and C.-H. Zhou, Eur. J. Med. Chem., 2017, 144, 444–492 CrossRef PubMed.
- Z. Jin, Nat. Prod. Rep., 2013, 30, 869–915 Search PubMed.
- M. C. Wenlock, P. Barton and T. Luker, Bioorg. Med. Chem. Lett., 2011, 21, 3550–3556 Search PubMed.
- C. M. Marshall, J. G. Federice, C. N. Bell, P. B. Cox and J. T. Njardarson, J. Med. Chem., 2024, 67, 11622–11655 CrossRef CAS PubMed.
- A. Saito, Curr. Org. Chem., 2020, 24, 2048–2069 Search PubMed.
- L. Chen, H. Li, P. Li and L. Wang, Org. Lett., 2016, 18, 3646–3649 CrossRef CAS PubMed.
- T. Chatterjee, J. Y. Cho and E. J. Cho, J. Org. Chem., 2016, 81, 6995–7000 CrossRef CAS PubMed.
- X. Zhang, Y. He, J. Li, R. Wang, L. Gu and G. Li, J. Org. Chem., 2019, 84, 8225–8231 CrossRef CAS PubMed.
- Y. Liu, Y. Shi, L. Wei, K. Zhao, J. Zhao, P. Zhang, X. Xu and P. Li, Chem – Asian J., 2021, 16, 2417–2420 CrossRef CAS PubMed.
- X. Y. Wang, Q. B. Zhang, X. L. Jin, L. Z. Wu and Q. Liu, ChemPhotoChem, 2021, 5, 240–244 CrossRef CAS.
- S. Sahoo, T. K. Dinda and P. Mal, Chem. – Eur. J., 2024, 30, e202402192 Search PubMed.
- X. Y. Wang, Q. B. Zhang, X. L. Jin, L. Z. Wu and Q. Liu, ChemPhotoChem, 2021, 5, 240–244 CrossRef CAS.
- Y. H. Wang, Y. Q. Jiang, Y. Q. Zhang, Y. Ling, L. Ming and G. Q. Liu, Chem. – Eur. J., 2023, 29, e202300530 CrossRef CAS PubMed.
- E. Delalande, L. di Terlizzi, C. Russo, C. Volpe, S. Protti and M. Giustiniano, Adv. Synth. Catal., 2025, 367, e202401079 CrossRef CAS.
- J. Bai, D. Qi, Z. Song, B. Li, L. Guo, C. Yang and W. Xia, Org. Biomol. Chem., 2023, 21, 5511–5515 RSC.
- G. Yuan, Z. Zhu, X. Gao and H. Jiang, RSC Adv., 2014, 4, 24300–24303 RSC.
- L. Wei and G. Yuan, Tetrahedron, 2023, 132, 133246 CrossRef CAS.
- Y. Wang, X. J. Zhao, X. Wu, L. Zhang, G. Li and Y. He, ChemElectroChem, 2022, 9, e202200378 CrossRef CAS.
- L. Bao, C. Liu, W. Li, J. Yu, M. Wang and Y. Zhang, Org. Lett., 2022, 24, 5762–5766 CrossRef CAS PubMed.
- L. E. Sattler and G. Hilt, Chem. – Eur. J., 2020, 27, 605–608 CrossRef PubMed.
- J.-Q. Zhang, C. Shen, S. Shuai, L. Fang, D. Hu, J. Wang, Y. Zhou, B. Ni and H. Ren, Org. Lett., 2022, 24, 9419–9424 Search PubMed.
- N. Yang, A. Li, H. Gao, L.-M. Liao, Y.-P. Yang, P.-L. Wang and H. Li, Green Chem., 2023, 25, 5128–5133 RSC.
- J. Jang and E. J. Cho, Adv. Synth. Catal., 2024, 366, 3450–3454 Search PubMed.
- J. D. Herszman, M. Berger and S. R. Waldvogel, Org. Lett., 2019, 21, 7893–7896 CrossRef CAS PubMed.
- H. Zhang, Y. Xiong, M.-J. Luo, R. Yang, J. Bai, X.-R. Song and Q. Xiao, Org. Chem. Front., 2023, 10, 3786–3791 Search PubMed.
- C. Bracken and M. Baumann, J. Org. Chem., 2020, 85, 2607–2617 CrossRef CAS PubMed.
- X. K. Wong and K. Y. Yeong, ChemMedChem, 2021, 16, 3237–3262 CrossRef CAS PubMed.
- T. Horch, E. M. Molloy, F. Bredy, V. G. Haensch, K. Scherlach, K. L. Dunbar, J. Franke and C. Hertweck, Angew. Chem., Int. Ed., 2022, 61, e202205409 CrossRef CAS PubMed.
- H. Razavi, S. K. Palaninathan, E. T. Powers, R. L. Wiseman, H. E. Purkey, N. N. Mohamedmohaideen, S. Deechongkit, K. P. Chiang, M. T. A. Dendle, J. C. Sacchettini and J. W. Kelly, Angew. Chem., Int. Ed., 2003, 115, 2864–2867 Search PubMed.
- V. Šlachtová and L. Brulíková, ChemistrySelect, 2018, 3, 4653–4662 CrossRef.
- T. Coelho, L. F. Maia, A. M. da Silva, M. W. Cruz, V. Planté-Bordeneuve, O. B. Suhr, I. Conceiçao, H. H. J. Schmidt, P. Trigo, J. W. Kelly, R. Labaudinière, J. Chan, J. Packman and D. R. Grogan, J. Neurol., 2013, 260, 2802–2814 Search PubMed.
- Y. Ando, Y. Sekijima, K. Obayashi, T. Yamashita, M. Ueda, Y. Misumi, H. Morita, K. Machii, M. Ohta, A. Takata and S.-I. Ikeda, J. Neurol. Sci., 2016, 362, 266–271 Search PubMed.
- S. Soni, N. Sahiba, S. Teli, P. Teli, L. K. Agarwal and S. Agarwal, RSC Adv., 2023, 13, 24093–24111 Search PubMed.
- L. Yadav and V. Srivastava, Synlett, 2013, 2758–2762 Search PubMed.
- L. Wang, Z.-G. Ma, X.-J. Wei, Q.-Y. Meng, D.-T. Yang, S.-F. Du, Z.-F. Chen, L.-Z. Wu and Q. Liu, Green Chem., 2014, 16, 3752–3757 Search PubMed.
- H. A. N. Le, L. H. Nguyen, Q. N. B. Nguyen, H. T. Nguyen, K. Q. Nguyen and P. H. Tran, Catal. Commun., 2020, 145, 106120 Search PubMed.
- Z. Q. Zhu, S. Liu, Z. Y. Hu, Z. B. Xie, J. Tang and Z. G. Le, Adv. Synth. Catal., 2021, 363, 2568–2572 CrossRef CAS.
- V. R. M. Laćan, Electrochim. Acta, 1983, 28, 199–207 Search PubMed.
- W. C. Li, C. C. Zeng, L. M. Hu, H. Y. Tian and R. D. Little, Adv. Synth. Catal., 2013, 355, 2884–2890 CrossRef CAS.
- L.-S. Kang, H.-l Xiao, C.-C. Zeng, L.-M. Hu and R. D. Little, J. Electroanal. Chem., 2016, 767, 13–17 Search PubMed.
- F. Ferlin, F. Valentini, F. Campana and L. Vaccaro, Green Chem., 2024, 26, 6625–6633 RSC.
- L. Liu, Z. Xu, T. Liu, C. Xu, W. Zhang, X. Hua, F. Ling and W. Zhong, J. Org. Chem., 2022, 87, 11379–11386 CrossRef CAS PubMed.
- T. Morofuji, A. Shimizu and J. I. Yoshida, Chem. – Eur. J., 2015, 21, 3211–3214 CrossRef CAS PubMed.
- T. Ghoshal, T. M. Patel and S. Kotturi, ChemistrySelect, 2021, 6, 8080–8084 CrossRef CAS.
- Y. L. Lai, X. Yang, S. C. Wu, X. Y. Li, Y. X. Gong, S. L. Zhang, R. M. Zhong, J. H. Liao and J. M. Luo, ChemElectroChem, 2022, 9, e202200787 CrossRef CAS.
- Y. L. Lai, J. S. Ye and J. M. Huang, Chem. – Eur. J., 2016, 22, 5425–5429 CrossRef CAS PubMed.
- A. Ziarati, A. Sobhani-Nasab, M. Rahimi-Nasrabadi, M. R. Ganjali and A. Badiei, J. Rare Earths, 2017, 35, 374–381 CrossRef CAS.
- T. N. T. Huynh, T. Tankam, S. Koguchi, T. Rerkrachaneekorn, M. Sukwattanasinitt and S. Wacharasindhu, Green Chem., 2021, 23, 5189–5194 Search PubMed.
- T. Li, K. Li, J. Yu, Q. Sun and Z. Wang, Org. Lett., 2024, 26, 10809–10815 CrossRef CAS PubMed.
- T. Gieshoff, A. Kehl, D. Schollmeyer, K. D. Moeller and S. R. Waldvogel, Chem. Commun., 2017, 53, 2974–2977 RSC.
- T. Gieshoff, A. Kehl, D. Schollmeyer, K. D. Moeller and S. R. Waldvogel, J. Am. Chem. Soc., 2017, 139, 12317–12324 CrossRef CAS PubMed.
- A. Kumar, S. Sharma and R. A. Maurya, Tetrahedron Lett., 2010, 51, 6224–6226 CrossRef CAS.
- F. Li, Z. Li, X. Tang, X. Cao, C. Wang, J. Li and L. Wang, ChemCatChem, 2019, 11, 1192–1195 CrossRef CAS.
- C. Cano-Prieto, R. García-Salcedo, M. Sánchez-Hidalgo, A. F. Braña, H. P. Fiedler, C. Méndez, J. A. Salas and C. Olano, ChemBioChem, 2015, 16, 1461–1473 CrossRef CAS PubMed.
- M. Lv, J. Zhao, Z. Deng and Y. Yu, Chem. Bio., 2015, 22, 1313–1324 CrossRef CAS PubMed.
- A. A. Losada, C. Cano-Prieto, R. García-Salcedo, A. F. Braña, C. Méndez, J. A. Salas and C. Olano, Microb. Biotechnol., 2017, 10, 873–885 CrossRef PubMed.
- A. De Oliveira Silva, J. McQuade and M. Szostak, Adv. Synth. Catal., 2019, 361, 3050–3067 CrossRef CAS.
- J. Wang, A. Liao, R. J. Guo, X. Ma and J. Wu, J. Agric. Food. Chem., 2024, 73, 30–46 Search PubMed.
- X. Zhang, C. Cai, Z. Sui, M. Macielag, Y. Wang, W. Yan, A. Suckow, H. Hua, A. Bell, P. Haug, W. Clapper, C. Jenkinson, J. Gunnet, J. Leonard and W. V. Murray, ACS Med. Chem. Lett., 2017, 8, 947–952 CrossRef CAS PubMed.
- C. A. Lipinski, o Franc0 I, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
- M. J. Cabrera-Afonso, S. Cembellín, A. Halima-Salem, M. Berton, L. Marzo, A. Miloudi, M. C. Maestro and J. Alemán, Green Chem., 2020, 22, 6792–6797 Search PubMed.
- B. Seo, Y. G. Kim and P. H. Lee, Org. Lett., 2016, 18, 5050–5053 CrossRef CAS PubMed.
- B. Roure, M. Alonso, G. Lonardi, D. B. Yildiz, C. S. Buettner, T. Dos Santos and D. Leonori, Nature, 2025, 637, 860–867 Search PubMed.
- G. L. Bartholomew, L. J. Karas, R. M. Eason, C. S. Yeung, M. S. Sigman and R. Sarpong, J. Med. Chem., 2025, 68, 6027–6040 CrossRef CAS PubMed.
- A. Singh, D. Malhotra, K. Singh, R. Chadha and P. M. S. Bedi, J. Mol. Struct., 2022, 1266, 133479 Search PubMed.
- S. J. Kashyap, V. K. Garg, P. K. Sharma, N. Kumar, R. Dudhe and J. K. Gupta, Med. Chem. Res., 2011, 21, 2123–2132 Search PubMed.
- M. S. Moghadam, S. Maleki, E. Darabpour, H. Motamedi and S. M. S. Nejad, Asian Pac. J. Trop. Biomed., 2010, 3, 262–265 Search PubMed.
- P. C. Sharma, K. K. Bansal, A. Sharma, D. Sharma and A. Deep, Eur. J. Med. Chem., 2020, 188, 112016 CrossRef CAS PubMed.
- D. Teoh, T. A. Ayeni, J. M. Rubatt, D. J. Adams, L. Grace, M. D. Starr, W. T. Barry, A. Berchuck, S. K. Murphy and A. A. Secord, Gynecol. Oncol., 2011, 121, 187–192 CrossRef CAS PubMed.
- C. Zhou, S. Cui, J. Lv, G. L. V. Damu and Y. Wang, Sci. Sin. Chim., 2012, 42, 1105–1131 CrossRef.
- K. O. M. I. G. Shahin, A. T. Taher, A. S. Mayhoub and A. E. Kassab, Mini-Rev. Org. Chem., 2022, 20, 270–284 Search PubMed.
- D. X. Duc and N. T. Chung, Curr. Org. Synth., 2022, 19, 702–730 CrossRef CAS PubMed.
- S. Dey, A. Das, R. N. Yadav, P. J. Boruah, P. Bakli, T. Baishya, K. Sarkar, A. Barman, R. Sahu, B. Maji, A. K. Paul and M. F. Hossain, Org. Biomol. Chem., 2023, 21, 1771–1779 RSC.
- Y. Li, C.-C. Sun and C.-C. Zeng, J. Electroanal. Chem., 2020, 861, 113941 CrossRef CAS.
- P. Li, S.-F. Yang, Z.-L. Fang, H.-R. Cui, S. Liang, H.-Y. Tian, B.-G. Sun and C.-C. Zeng, J. Environ. Chem. Eng., 2022, 10, 107847 CrossRef.
- S.-F. Yang, P. Li, Z.-L. Fang, S. Liang, H.-Y. Tian, B.-G. Sun, K. Xu and C.-C. Zeng, Beilstein J. Org. Chem, 2022, 18, 1249–1255 CrossRef CAS PubMed.
- C. Zhang and G. Yuan, Chem. Commun., 2023, 59, 12188–12191 RSC.
- H. Guo, Y. Liu, C. Wen and J.-P. Wan, Green Chem., 2022, 24, 5058–5063 RSC.
- K. Haider, N. Shrivastava, A. Pathak, R. Prasad Dewangan, S. Yahya and M. Shahar Yar, Results Chem., 2022, 4, 100258 CrossRef CAS.
- R. S. Keri, M. R. Patil, S. A. Patil and S. Budagumpi, Eur. J. Med. Chem., 2015, 89, 207–251 CrossRef CAS PubMed.
- P. Chander Sharma, D. Sharma, A. Sharma, K. K. Bansal, H. Rajak, S. Sharma and V. K. Thakur, Appl. Mater. Today, 2020, 20, 100783 CrossRef.
- M. Bhat and S. L. Belagali, Mini-Rev. in Org. Chem., 2020, 17, 323–350 CrossRef CAS.
- V. V. K. V. Juthiga, S. N. M. Boddapati, M. Balha and R. Tamminana, Results Chem., 2025, 15, 102212 CrossRef.
- S. Dhadda, A. K. Raigar, K. Saini, Manju and A. Guleria, Sustainable Chem. Pharm., 2021, 24, 100521 CrossRef CAS.
- R. Paramasivam, R. Palaniappan and V. T. Ramakrishnan, J. Chem. Soc., Chem. Commun., 1979, 260–261 Search PubMed.
- H. Wang, Q. Wu, J.-D. Zhang, H.-Y. Li and H.-X. Li, Org. Lett., 2021, 23, 2078–2083 Search PubMed.
- Y. Cheng, J. Yang, Y. Qu and P. Li, Org. Lett., 2012, 14, 98–101 CrossRef CAS PubMed.
- G. Zhang, C. Liu, H. Yi, Q. Meng, C. Bian, H. Chen, J.-X. Jian, L.-Z. Wu and A. Lei, J. Am. Chem. Soc., 2015, 137, 9273–9280 CrossRef CAS PubMed.
- A. N. Dinh, A. D. Nguyen, E. M. Aceves, S. T. Albright, M. R. Cedano, D. K. Smith and J. L. Gustafson, Synlett, 2019, 1648–1655 CAS.
- A. S. Kostyuchenko, E. B. Uliankin, A. J. Stasyuk, A. L. Samsonenko, T. Y. Zheleznova, A. L. Shatsauskas and A. S. Fisyuk, J. Org. Chem., 2022, 87, 6657–6667 Search PubMed.
- J. Bai, S. Yan, Z. Zhang, Z. Guo and C.-Y. Zhou, Org. Lett., 2021, 23, 4843–4848 Search PubMed.
- C. Yu, K. Lee, Y. You and E. J. Cho, Adv. Synth. Catal., 2013, 355, 1471–1476 CrossRef CAS.
- H. S. Hwang, S. Lee, S. S. Han, Y. K. Moon, Y. You and E. J. Cho, J. Org. Chem., 2020, 85, 11835–11843 Search PubMed.
- S. Li, J. Yin, H. Zhang and K. A. I. Zhang, ACS Appl. Mater. Interfaces, 2023, 15, 2825–2831 CrossRef CAS PubMed.
- H. Liu, Q. Li, P. Pan, L. Zhou, B. Deng, S. Zhao, P. Liu, Y. Wang and J. Li, Chin. Chem. Lett., 2023, 34, 108562 CrossRef CAS.
- H. Liu, W.-W. Yi, Q.-Q. Li and S.-Y. Zhao, Inorg. Chem. Front., 2024, 11, 5973–5978 RSC.
- Y. Yuan, W. Dong, X. Gao, X. Xie and Z. Zhang, Org. Lett., 2019, 21, 469–472 Search PubMed.
- I. Tabaković, M. Trkovnik, M. Batušić and K. Tabaković, Synthesis, 1979, 590–592 CrossRef.
- A. A. Folgueiras-Amador, X. Y. Qian, H. C. Xu and T. Wirth, Chem. – Eur. J., 2018, 24, 487–491 CrossRef CAS PubMed.
- P.-F. Huang, J.-L. Fu, Y. Peng, J.-H. Fan, L.-J. Zhong, K.-W. Tang and Y. Liu, Org. Biomol. Chem., 2024, 22, 3752–3760 RSC.
- Y. Zhang, Z. Y. Mo, X. Y. Tang, H. T. Tang, Y. M. Pan, K. D. Yang and M. X. He, Adv. Synth. Catal., 2024, 366, 3808–3814 Search PubMed.
- H. T. Abdel-Mohsen, J. Conrad, K. Harms, D. Nohr and U. Beifuss, RSC Adv., 2017, 7, 17427–17441 Search PubMed.
- N. Ghorashi, Z. Shokri, R. Moradi, A. Abdelrasoul and A. Rostami, RSC Adv., 2020, 10, 14254–14261 RSC.
- C. Wang, X. Geng, P. Zhao, Y. Zhou, Y.-D. Wu, Y.-F. Cui and A.-X. Wu, Chem. Commun., 2019, 55, 8134–8137 Search PubMed.
- H.-W. Cui, S. Peng, X.-Z. Gu, H. Chen, Y. He, W. Gao, F. Lv, J.-H. Wang, Y. Wang, J. Xie, M.-Y. Liu, Z. Yi and W.-W. Qiu, Eur. J. Med. Chem., 2016, 111, 126–137 CrossRef CAS PubMed.
- A. G. Lyapunova, D. A. Androsov and M. L. Petrov, Tetrahedron Lett., 2013, 54, 3427–3430 CrossRef CAS.
- S. B. Yonghan (Fred) Hu, O. W. Gooding, J. W. Labadie, W. Miller and J. A. Porco, Jr., J. Org. Chem, 1998, 64, 1049–1051 Search PubMed.
- M. Caron, J. Org. Chem., 1986, 51, 4077–4078 CrossRef.
- S. M. S. Atta, D. S. Farrag, A. M. K. Sweed and A. H. Abdel-Rahman, Eur. J. Med. Chem., 2010, 45, 4920–4927 CrossRef CAS PubMed.
- P. T. I. John and C. Sheehan, J. Am. Chem. Soc., 1949, 72, 4059–4062 Search PubMed.
- S. K. Mo, Q. H. Teng, Y. M. Pan and H. T. Tang, Adv. Synth. Catal., 2019, 361, 1756–1760 Search PubMed.
- J. Nociarová, A. Purkait, R. Gyepes and P. Hrobárik, Org. Lett., 2024, 26, 619–624 Search PubMed.
- M. Madhu Sekhar, U. Nagarjuna, V. Padmavathi, A. Padmaja, N. V. Reddy and T. Vijaya, Eur. J. Med. Chem, 2018, 145, 1–10 Search PubMed.
- D. G. Anstis, A. C. Lindsay, T. Söhnel and J. Sperry, J. Nat. Prod., 2020, 83, 1721–1724 Search PubMed.
- S.-Y. Liu, D.-S. Zhang and C.-Q. Hu, Eur. J. Med. Chem., 2010, 45, 5808–5816 Search PubMed.
- L. M. T. Frija, A. J. L. Pombeiro and M. N. Kopylovich, Eur. J. Org. Chem., 2017, 2670–2682 Search PubMed.
- L. Yadav, V. Srivastava and A. Yadav, Synlett, 2013, 465–470 Search PubMed.
- J.-C. Hou, H.-T. Ji, Y.-H. Lu, J.-S. Wang, Y.-D. Xu, Y.-Y. Zeng and W.-M. He, Chin. Chem. Lett., 2024, 35, 109514 CrossRef CAS.
- Z. Q. Wang, X. J. Meng, Q. Y. Li, H. T. Tang, H. S. Wang and Y. M. Pan, Adv. Synth. Catal., 2018, 360, 4043–4048 CrossRef CAS.
- Z. Yang, J. Zhang, L. Hu, L. Li, K. Liu, T. Yang and C. Zhou, J. Org. Chem., 2020, 85, 3358–3363 CrossRef CAS PubMed.
- M. Sharma, C. A. Pascoe, S. K. Jones, S. G. Barthel, K. M. Davis and K. F. Biegasiewicz, J. Am. Chem. Soc., 2025, 147, 10698–10705 CrossRef CAS PubMed.
- Y. Hu, C.-Y. Li, X.-M. Wang, Y.-H. Yang and H.-L. Zhu, Chem. Rev., 2014, 114, 5572–5610 CrossRef CAS PubMed.
- M. Bala, P. Piplani, A. Ankalgi, A. Jain and L. Chandel, Med. Chem., 2023, 19, 730–756 CrossRef CAS PubMed.
- H.-M. Kuo, S.-Y. Li, H.-S. Sheu and C. K. Lai, Tetrahedron, 2012, 68, 7331–7337 CrossRef CAS.
- M. K. P. J. M. Farrar and P. Kaszynski, J. Org. Chem., 2000, 65, 930–941 Search PubMed.
- J. K. Augustine, V. Vairaperumal, S. Narasimhan, P. Alagarsamy and A. Radhakrishnan, Tetrahedron, 2009, 65, 9989–9996 CrossRef CAS.
- G. L. Almajan, S.-F. Barbuceanu, G. Bancescu, I. Saramet, G. Saramet and C. Draghici, Eur. J. Med. Chem., 2010, 45, 6139–6146 Search PubMed.
- V. Srivastava, P. K. Singh and P. P. Singh, Croat. Chem. Acta, 2015, 88, 59–65 Search PubMed.
- C. Wen, G. Sun, L. Liu, J. Zhang, M. She, Z. Yang, P. Liu, S. Zhang and J. Li, Green Chem., 2024, 26, 7944–7950 RSC.
- Z. Ma, X. Hu, Y. Li, D. Liang, Y. Dong, B. Wang and W. Li, Org. Chem. Front., 2021, 8, 2208–2214 RSC.
- Q. Wang, X. Wang, Q. Liu, G. Xie, S. Ding, X. Wang and H. Fan, Org. Chem. Front., 2020, 7, 3912–3917 RSC.
- F. Sacchelli, E. Quadri, L. Raineri, A. Jorea, M. Pessina, A. Lo Presti and L. Capaldo, ChemSusChem, 2025, 18, e202501012 CrossRef CAS PubMed.
- Z. Haider, R. Archana and H. Ju, Molecules, 2025, 30, 3490 Search PubMed.
- J. C. Bui, E. W. Lees, L. M. Pant, I. V. Zenyuk, A. T. Bell and A. Z. Weber, Chem. Rev., 2022, 122, 11022–11084 CrossRef CAS PubMed.
- S. Hecko, A. Schiefer, C. P. Badenhorst, M. J. Fink, M. D. Mihovilovic, U. T. Bornscheuer and F. Rudroff, Chem. Rev., 2023, 123, 2832–2901 Search PubMed.
- W. N. Klement, E. Savino, S. Rooijmans, P. P. Mulder, N. S. Lynn Jr, W. R. Browne and E. Verpoorte, ACS Electrochem., 2025, 1, 504–515 CrossRef CAS PubMed.
- Y. Zhang, Y. Han, S. Chen, R. Yu, X. Zhao, X. Liu and Y. Xu, Nat. Mach. Intell., 2025, 7, 1010–1022 Search PubMed.
- B. A. Grzybowski, A. Żądło-Dobrowolska, N. Onishchenko and E. S. Larsen, Nat. Rev. Bioeng., 2025, 3, 791–803 CrossRef CAS.
- J. Rui, X. Mu, J. Soler, J. C. Paris, Y. Guo, M. Garcia-Borràs and X. Huang, Nat. Catal., 2024, 7, 1394–1403 CrossRef CAS.
- A. Choi, D. Kim, D. Yim, J. Park, A. Sharma, W. Kim and H. Kim, J. Am. Chem. Soc, 2025, 147, 30897–30906 Search PubMed.
- B. Zhao, Y. Xu, Q. Zhu, A. Liu, X. Peng, T. Zhang and X. Huang, Nature, 2025, 643, 699–704 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2026 |