
Small molecule helical emitters
Tadashi
Mori

Research Center for Environmental Preservation and Department of Applied Chemistry, Graduate School of Engineering, The University of Osaka, 2-4, Yamada-oka, Suita, Osaka 565-0871, Japan. E-mail: tmori@epc.osaka-u.ac.jp
First published on 6th January 2026
Abstract
The development of materials exhibiting circularly polarized luminescence (CPL) is a key area of research for next-generation optical technologies, including 3D displays and secure communications. The central goal in this field is to create chiral emitters with a high luminescence dissymmetry (gCPL) factor, a measure of the emission's chirality. While theoretically reaching ±2, practical values in small organic molecules have historically been much lower, on the order of 0.001 or less. This summary outlines the core strategies in molecular design focusing on helical emitters that have recently enabled significant breakthroughs, pushing g values beyond the 0.01 threshold. The magnitude of g factor is determined by the cosine of the angle between the electric (μe) and magnetic (μm) dipole transition moments, as well as their respective magnitudes. Consequently, the most successful research has moved beyond simple screening and has focused on rationally engineering molecules to optimize this relationship. One of the most direct strategies has been to design rigid, helical molecules where high symmetry forces the μe and μm to be parallel. By enforcing D2 and other symmetry in certain helicenes, helical nanographenes and related structures, researchers have minimized the angle between the moments, thus maximizing the cosine term and leading to a significant enhancement in the g factor value. A second, distinct approach targets the magnitude of the μm. In most organic chromophores, μm is inherently small, limiting the potential g factor intensity. To overcome this, researchers have designed for example belt-shaped macrocyclic molecules that function as molecular-scale solenoids. The cyclic arrangement of chromophores induces a large, circulating electric current in the excited state, which in turn generates a powerful μm along the cylinder's axis. A third innovative strategy circumvents the limitation of a small intrinsic μm by leveraging exciton coupling between two and more chromophores. In these systems, two π-conjugated units such as pyrene are held in a fixed, chiral arrangement. Upon photoexcitation, they form an intramolecular excimer, a transient excited-state complex with a well-defined helical geometry. The resulting CPL signal originates from the chiral interaction of the two strong electric transition moments, generating a large rotational strength and a high g factor without relying on the weak magnetic moment of the individual units. The progress in CPL-active materials is a testament to the power of targeted molecular engineering. As seen in the state-of-the-art examples in the review, the field has matured to a point where the fundamental photophysical principles governing CPL are being directly translated into synthetic molecular designs. While current high-performing materials are often complex and synthetically challenging, these proof-of-concept molecules validate the core design strategies.
 Tadashi Mori | Tadashi Mori is a Professor of Chemistry at The University of Osaka. He received his PhD from Kyoto University in 1997 under the supervision of Professor Hitomi Suzuki and joined the research group of Professor Yoshihisa Inoue at Osaka University in 1998. His postdoctoral and international research experiences include: a JSPS Postdoctoral Fellowship at the University of Houston in the group of Professor Jay K. Kochi (1997–1998); a Visiting Professorship at Georgetown University with Professor Richard G. Weiss (2002); an Alexander von Humboldt Fellowship at Universität Münster in the group of Professor Stefan Grimme (2005–2006); and a Fostering Joint International Research Fellowship at Technische Universität München in the group of Professor Thorsten Bach (2018–2019). His research focuses on the chiroptical properties of ground- and excited-state molecules with unique chirality—including circularly polarized luminescence (CPL)—as well as weak molecular interactions and stereoselective photoreactions. |
1. Introduction
The concept of helical systems is particularly important in the context of biological molecules, such as the secondary structures in proteins and the double helices in DNA.1,2 Related artificial systems have emerged in molecular and supramolecular foldamers and polymers, as well as in nematic and cholesteric liquid crystals.3,4 Molecularly helical systems also attract significant attention not only for the structural beauty of the molecules themselves but also for their unique physical and photophysical properties, particularly their chiroptical characteristics.5 One such property of topical interest is circularly polarized luminescence (CPL),6 a phenomenon in which the emission of light is associated with circular polarization. CPL has recently attracted growing attention in the fields of photonics and materials science, with potential applications in various advanced technologies such as light-emitting diodes, transistors, optical data storage, displays, bioimaging, and more.7–11 Among various chiral materials, small organic molecular systems are unique in that their chiral emission is exclusively dependent on their twisted molecular geometries, rather than arising from various optical effects. Organic molecules offer several advantages, including ease of molecular design, wavelength tunability, mechanical flexibility, wide-area processability, and cost competitiveness. In this context, it is noteworthy that liquid crystal assemblies have demonstrated nearly perfect dissymmetry in CPL, which is more promising for short- to medium-term real applications.12,13 However, we will not discuss such systems any further in this article.The important parameters of the CPL are characterized by luminescence dissymmetry factor (gCPL) and emission quantum yield (ΦCPL).14,15 Other features of interest may include intensity of light absorption, emission energy or color, and structure and breadth of the emission spectra. Among these various factors, the dissymmetry factor is the most significant value for assessing the performance of CPL materials, indicates the degree of chirality and is defined as follows:

![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :49. As detailed below, the gCPL value has been the most challenging parameter for many scientists in the development of superior molecularly chiral systems. Typical g factor values for small organic chiral emitters range from as low as ±0.0001 to recently improved, albeit still small, values around ±0.001. This difficulty may arise from the intrinsically weak interaction of light in the UV to visible wavelength range (hundreds of nanometres) with small molecules on the nanometre scale, as discussed likewise in relation to the fine-structure constant in quantum electrodynamics.16 Enhancing the g factor through nonconventional strategies such as supramolecular assemblies, liquid crystal materials, perovskites, and metallic materials is an active area in this field, but it is beyond the scope of this review.17
:49. As detailed below, the gCPL value has been the most challenging parameter for many scientists in the development of superior molecularly chiral systems. Typical g factor values for small organic chiral emitters range from as low as ±0.0001 to recently improved, albeit still small, values around ±0.001. This difficulty may arise from the intrinsically weak interaction of light in the UV to visible wavelength range (hundreds of nanometres) with small molecules on the nanometre scale, as discussed likewise in relation to the fine-structure constant in quantum electrodynamics.16 Enhancing the g factor through nonconventional strategies such as supramolecular assemblies, liquid crystal materials, perovskites, and metallic materials is an active area in this field, but it is beyond the scope of this review.17
There have also been several reviews on molecular CPL based on traditional and emerging chiral structures;18–22 however, due to the rapidly growing interest in this topic, considerable progress and numerous successful examples have been reported over the past couple of decades. In this article, we will illustrate selected examples of single molecule-based helical emitters with gCPL values larger than 0.01 (in absolute value) to discuss the factors and approaches for realizing state-of-the-art helical emitters; all of these outcomes have only emerged very recently. Although g factor values remain modest for practical applications, typically on the order of 0.01 at best, with only a few exceptional systems exceeding this threshold, various helical and related motifs have demonstrated significant promise. We discuss the CPL spectra of molecules in their isolated states within isotropic media, while reports based on molecular aggregates are not included, where intermolecular coupling and optical effects may play a more significant role.23,24 Finally, we will discuss the future challenges and perspectives of small molecule helical emitters.
2. Theory, formulation, and computation of CPL
In this section, we briefly introduce the theoretical background of chiroptical spectroscopies, with an emphasis on circularly polarized luminescence (CPL). We present circular dichroism (CD) and CPL in a comparative manner, as both involve transitions between the S0 and S1 states, but in different directions. For readers interested in a more detailed formulation, we refer them to the relevant literature.25,26 A quantum mechanical basis for optical rotation was introduced by Rosenfeld as early as 1928,27 and intensity of chiroptical spectroscopy of randomly oriented molecules was quantified by a rotational strength (R), analogous to the electric transition dipole strength (D). The values of R and D are experimentally evaluated by integrating CD and absorption (AB) bands over a transition frequency ν:

However, the CPL and fluorescence (FL) spectral intensities are not directly transferable due to instrumental differences. While cgs units are commonly used in (chir)optical spectroscopy (i.e., experimental assessment of R and D), Hartree atomic units are more convenient for theoretical considerations and will be used hereafter. In the CPL, therefore, g values are more frequently used to gauge the chiroptical responses. As a typical approximation in isotropic media, the following equation applies:

The R value for randomly oriented molecules in isotropic media is theoretically defined as the imaginary part of the dot product of the electric and magnetic dipole transition moment operators (μe and μm):
| R = Im(μe·μm) = |μe||μm|cosθ |
Here, θ represents the angle between μe and μm. In order to achieve a non-zero CPL and/or CD response, the electric and magnetic dipole transition moment operators must transform as the same irreducible representation. In other words, μe and μm must be non-orthogonal. This situation can occur only for Cn and Dn point groups, making only chiral molecules CPL/CD active. Neglecting quadrupole (and higher multipole) contributions, the value D is approximated as the sum squares of the lengths of μe and μm:
| D = |μe|2 + |μm|2 |
Therefore, the g factor is described as follows:

For ordinary molecules, μe is typically on the order of Debye units, while μm is measured in Bohr magnetons. Consequently, μm is around two orders of magnitude smaller than μe in atomic units. Under these circumstances, the g factor can be further approximated as:

Accordingly, when designing molecules with large g factor values in either CD or CPL, the appropriate alignment of the electric and magnetic dipole transition moments, along with their relative intensities, is crucial. To maximize the g value, for instance, μe and μm should be colinear (i.e., θ = 0 or 180°). Despite this theoretical simplicity, experimentally achieving these requirements and thus high g factor values, in particular in CPL, has proven to be quite challenging. Indeed, progress in developing highly intensified molecular CPL materials has been sluggish, with rather limited examples primarily arising through a trial-and-error approach. A theoretically sound proof-of-concept investigation or a robust theory-based design study that has been experimentally verified remains insufficient.28,29
3. Molecular and electronic structure design for CPL
As the g factor is inversely related to the electric dipole transition moment μe, a classical attempt to improve g factor values in circularly polarized luminescence (CPL) response involves using molecules with intrinsically small μe lengths. This is particularly relevant for molecules with (nearly) forbidden transitions, such as lanthanoid complexes and d- and f-block element complexes;30,31 however, these are beyond the scope of this review. In this regard, we briefly discuss the chiral ketones at the beginning of the next section. In this article, we primarily focus on small organic chiral molecules, where the probability of electronic transitions is generally high, making them more suitable for emissive materials. A second approach to improve the g factor is to increase the magnitude of the magnetic dipole transition moment μm. Within the individual helical motifs, it has been observed that increasing the inner helical cavity can enhance intensity, thereby improving g factor values, an idea analogous to that of a conventional solenoid.32 While this theoretical prediction has not been fully confirmed experimentally, an increased number of turns and/or an extension of the turns could result in a higher μm value, leading to more intense chiroptical responses.Another logical strategy to improve the g factor value is to engineer, or more specifically, appropriately align the electric and magnetic dipole transition moments, as the angle between these moments influences the chiroptical response through the cosine factor. However, identifying the direction of the dipole transition moments, particularly the magnetic one, is not a straightforward task when based solely on molecular structure. Alternatively, a common approach involves using quantum chemical computations to calculate the relevant moment vectors. In principle, since the CPL phenomenon typically corresponds to the S1 to S0 electronic transition, these vectors are obtained by calculating the optimized geometry in the S1 state, followed by property calculations. In this way, it allows for a better design of molecules with a higher CPL response. However, such computations, especially when using cost-effective methods like time-dependent density functional theory (TD-DFT), do not always yield satisfactory predictions, as the theoretical calculations of excited state species are still being actively refined.33–36
Experimentally, several efforts have been made with some success to develop effective materials.37 For instance, by introducing a donor–acceptor moiety, the charge transfer transition becomes relevant for CPL emission due to the small orbital energy separation between the donor and acceptor. Consequently, μe is aligned to some degree with this transition; however, the exact orientation of μe and μm remains ambiguous and is highly dependent on the location and nature of the donor and acceptor moieties, as well as their orbital contributions. As an alternative strategy, the extension of π systems has also proven successful. Among the naturally overlapping low-energy bands, this approach lowers the CPL-relevant π*–π transition energy, thereby isolating and aligning the transition. In this manner, the direction of the transition moments can be adjusted, allowing for some control over their relative angle. These strategies have been successfully demonstrated in various chiral systems, including helicene derivatives (vide infra).38 Molecular symmetry also plays a significant role in the alignment of dipole transition moments, although the molecular structure in the excited state is not equivalent to that of the ground state. Accordingly, highly symmetrical systems, particularly D2 symmetric molecules, have been successfully developed in helicenes and other helical compounds to enhance the CPL response.39
An alternative approach to enhancing chiroptical responses by manipulating dipole transition moments involves utilizing the Davydov splitting between two identical or similar chromophores.40–42 In this scenario of binary exciton coupling, the dipole interaction between chromophores splits the degenerate states with an energy separation of 2V (twice the correction energy), formulated as follows in zero-order approximation:


Thus, the Cotton effects for the two transitions exhibit the same and often strong intensity with opposite signs, as often observed in CD spectra of molecules with effective exciton coupling. Clearly, the rotational strength is not a function of the intrinsically small μm and can be approximated as a function of two μe alone. Here, the two μe, or more specifically, the distance and angle between the two chromophores, determines the rotational strength and, consequently, the intensity of the g factor. A similar situation is expected to hold in CPL of molecules with multiple chromophores, and some examples have indeed been reported in the literature, although the emission typically appears as a single lower-energy emission, rather than a bisignated emission. While the concept has been well explored in molecular aggregates,43,44 it has not been fully investigated or systematically studied in isolated small molecules, to the best of our knowledge; in fact, it shows particularly great promise for improving g factor values (vide infra).
Last but not least, the vibronic effect in CPL is worth mentioning.45,46 In the case of CPL from carbohelicenes, progressively structured spectra are often observed, along with occasional sign inversion of the bands. Notably, the absorption and emission bands are not fully mirror-imaged, and there are differences in the fine structures between CD and absorption bands, as well as between CPL and fluorescence bands, indicating the importance of vibronic contributions on chiroptical responses. Consequently, a reliable interpretation of CPL and the reproduction of these observations require careful consideration of vibronic contributions, including the Herzberg–Teller term.47 These aspects are significant and represent an interesting recent topic; however, they are also beyond the scope of this review.
In the following section, we showcase and summarize recent examples of state-of-the-art helical emitters reported with absolute g factor values in CPL greater than 0.01. We will discuss how to enhance the magnitude of the dissymmetric interaction between light and small organic chiral molecules through sophisticated molecular engineering efforts, as well as insights and challenges related to molecular design and the factors that control and improve dissymmetry. Finally, we will highlight advancements and future research opportunities in small molecule helical emitters. For simplicity, we provide the molecular absolute configuration primarily for the S, Sp, or M enantiomer, noting that the CPL signs are reversed if only the data for the opposite isomer are documented.
4. State-of-the-art precedents in small molecule helical emitters
Below, we present existing molecular systems that demonstrate good circularly polarized luminescence (CPL) performance, with a dissymmetry factor exceeding 0.01 in their non-aggregated state in solution. This review focuses on helical emitters, exemplified by helicenes and helicenoids, but also includes other types of molecules such as cylindrical molecules, cyclophanes, and other molecules with twisted structure. Although some of these molecules are not strictly helical, we discuss them collectively. Furthermore, we explore molecular and some supramolecular systems with multiple chromophores that exhibit excimer emission. In these systems, two chromophores are arranged in a helical manner, resulting in an intense CPL response.4.1 Ketones
As early as 1967, Emeis and Oosterhoff reported the CPL of trans-β-hydrindanone K1 (Chart 1) in isooctane at room temperature.48 Due to the very low fluorescence quantum yield, the CPL needed to be measured at relatively high concentrations; potential aggregation has not been fully ruled out, which can be tricky for some aromatic systems (vide infra). Notably, upon excitation of K1 at the carbonyl with 313 nm, an intense CPL was observed, showing a positive response for the (3aS,7aS)- or (+)-enantiomer, with dissymmetry factor values of +0.035 at 361 nm, gradually diminishing to +0.015 at 550 nm. At around the emission peak of 428 nm, a very high g value of 0.025 was found for this forbidden transition, attributed to the twisted carbonyl structure in its excited singlet state (Table 1). This phenomenon has been thoroughly discussed in relation to electric and magnetic dipole transition moments.49 Interestingly, the g factor value for the corresponding phosphorescence of the same compound was found to be nearly zero, suggesting a more or less planar carbonyl structure in the triplet state. The related thioketone also exhibited a nearly null g factor value for its fluorescence, again due to its planar excited-state structure. Subsequently, the CPL spectral band shape, width, and overall intensity of K1 were accurately reproduced by time-dependent density functional theory (TD-DFT) computations that incorporated the effects of vibronic coupling.50 In 1982, a group led by Dekkers conducted a more comprehensive study on the CPL and circular dichroism (CD) of the n–π* transition in β,γ-unsaturated cyclic ketones with varying rigidity.51 They observed marked differences in the degree of circular polarization in both absorption and emission, attributing these differences to substantial geometric changes upon electronic excitation. Among the ketones they examined, three ketones (1S,4S)-7-methylenebicyclo[2.2.1]heptan-2-one (K2), (1S,4S)-bicyco[2.2.1]hept-5-en-2-one (K3), and (5S,5′R)-adamantylideneadamantane-2-one (K4) exhibited strong negative CPL response with the absolute g factor values greater than 0.01. Table 1 summarizes the CPL parameters of the chiral ketones discussed above. All ketones exhibit high g factor intensities, compensated by their extremely low quantum yields of emission, which correspond to the forbidden n–π* transition. Accordingly, while these examples are important for molecular design and fundamental understanding related to CPL response, the materials are generally not suitable for practical applications. Recent trends have therefore shifted toward more fluorescent π–π* transitions in rigid π systems, which are detailed in the following sections. | ||
| Chart 1 | ||
| Molecule | Symmetry | λ max | Φ CPL | g CPL (λmon) | Conditions | Ref. |
|---|---|---|---|---|---|---|
| (S,S)-K1 | C 2 | ≈410 | ≈0.001 | +0.025 (428) | Isooctane, 50 mM, r.t. | 48 |
| (S,S)-K2 | C 1 | ≈420 | 0.0006 | −0.016 (—) | Heptane, 5 mM, r.t. | 51 |
| (S,S)-K3 | C 1 | ≈430 | ≈0.00005 | −0.029 (—) | Heptane, 5 mM, r.t. | 51 |
| (S,R)-K4 | C 1 | ≈420 | 0.002 | −0.012 (—) | Heptane, 5 mM, r.t. | 51 |
4.2 Cylindrical and belt-shaped macrocycles
Macrocyclic molecules have been extensively studied as chiral emitters with relatively large dissymmetry factors, and several reviews are already available on these materials from different perspectives.52–55 A significant breakthrough in this area was demonstrated by Sato and colleagues with cylinder-shaped molecules, specifically the cyclic tetramer of alkylated chrysene. It is significant to note that they structurally constitute a part of carbon nanotubes. Consequently, the isomeric [4]cyclo-2,8-chrysenylene molecules C1 and C2 (Chart 2) both exhibit strong CPL fluorescence in toluene, with exceptionally large g factor values of +0.15 and +0.10 for the (12,8)- and (11,9)-isomers, respectively, both showing positive signs for the M enantiomers (Table 2).56 The dissymmetry factor values in the CD signals were reported to be even larger, again showing slightly better values for the D4-symmetric (12,8)-isomer compared to the C2-symmetric (11,9)-isomer. These large g factor values were attributed to the oppositely oriented electric and magnetic dipole transition moments, both aligned parallel to the cylinder axis. Notably, the large g factor values in the CD spectra appear only for specific vibronic bands (with considerably low absorptivity), while the values for other vibronic and electronic transitions are at least one order of magnitude smaller. Unfortunately, this aspect of the CPL spectra has not been documented in detail. The related cyclic tetramer of pristine anthracene, [4]-cyclo-2,6-anthracene C3, was also reported to exhibit strong CPL, with g factor values of −0.10 at the CPL maximum.57 While it was not fully discussed in the text, negative CPL was obtained for the M enantiomer of this macrocycle, in contrast to the above cyclochrysenylenes, according to the absolute configuration predicted by the theoretical and experimental CD spectral comparison.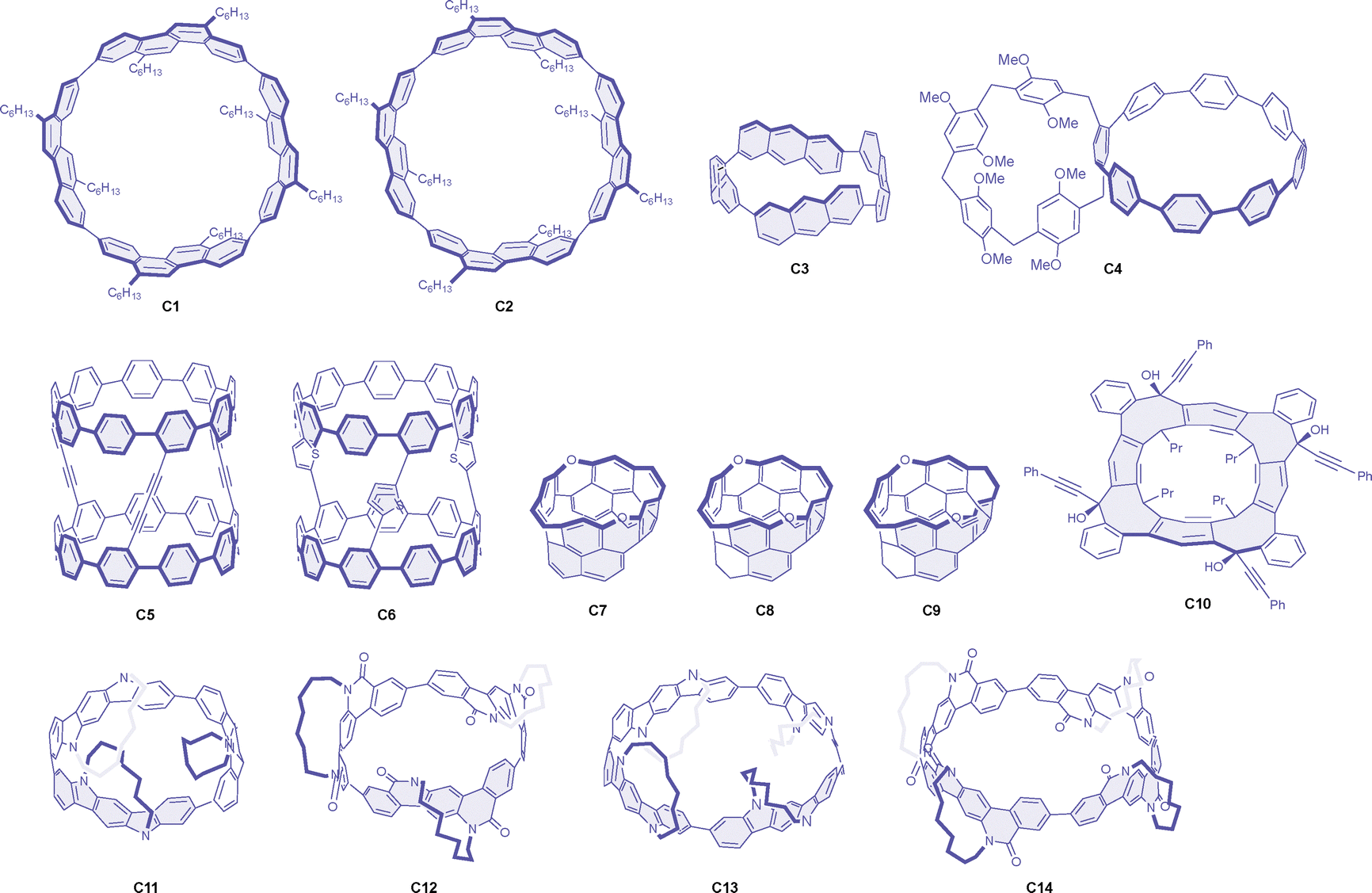 | ||
| Chart 2 | ||
| Molecule | Symmetry | λ max | Φ CPL | g CPL (λmon) | Conditions | Ref. |
|---|---|---|---|---|---|---|
| (M)-C1 | D 4 | ≈440 | 0.80 | +0.15 (443) | Toluene, 4.9 µM | 56 |
| (M)-C2 | C 2 | ≈450 | 0.74 | +0.10 (—) | Toluene, 5.1 µM | 56 |
| (M)-C3 | D 4 | 514 | 0.18 | −0.10 (536) | CH2Cl2, 10 µM | 57 |
| (Sp)-C4 | C 1 | 515 | 0.35 | −0.019 (488) | CH2Cl2 | 58 |
| (M,M,M)-C5 | C 3 | 496 | 0.68 | +0.031 (444) | CH2Cl2, 5.0 µM, 25 °C | 59 |
| (M,M,M)-C6 | C 3 | 484 | 0.45 | +0.012 (439) | CH2Cl2, 5.0 µM, 25 °C | 59 |
| (M)-C7 | C 2 | 480 | 0.20 | +0.016 (478) | Toluene, 0.10 mM, 25 °C | 60 |
| (M)-C8 | C 2 | 466 | 0.16 | +0.021 (451) | Toluene, 0.10 mM, 25 °C | 60 |
| (M)-C9 | C 2 | 477 | 0.12 | +0.015 (461) | Toluene, 0.10 mM, 25 °C | 60 |
| (M)-C10 | C 4 | ≈370 | — | +0.011 (361) | CH2Cl2, 0.10 mM, 25 °C | 61 |
| (M)-C11NR2 | D 3 | 519 | 0.25 | +0.064 (469) | Toluene, 10 µM | 62 |
| (M)-C12NR1 | D 3 | 561 | 0.42 | +0.036 (504) | Toluene, 10 µM | 62 |
| (M)-C13NR2 | D 4 | 492 | 0.43 | +0.044 (438) | Toluene, 10 µM | 62 |
| (M)-C14NR1 | D 4 | 520 | 0.80 | +0.020 (462) | Toluene, 10 µM | 62 |
A theoretical investigation revealed that larger macrocyclic molecules generally possess larger magnetic dipole transition moments and, consequently, greater rotatory strength. However, the symmetry of such molecules tends to be broken in the excited states, eventually limiting the g factor values.63 In addition to the time-consuming process of preparing cylindrical molecules, the strategy was found not to be generally applicable for achieving strong CPL. For instance, although the cyclic pyrene pentamer has been reported, it exhibited only a moderate g value of 0.001 in the CPL.64
[n]Cycloparaphenylenes have been utilized as a valuable macrocyclic backbone for the construction of emissive chiral systems. These molecules are highly conjugated and strained macrocycles with size-adjustable optical properties. In compound C4, a planar chiral pillar[5]arene unit has been incorporated into the highly emissive [8]cycloparaphenylene. The luminescence g factor value of the Sp enantiomer of this bismacrocycle was found to be −0.019, with enhanced emission efficiency compared to the parent cycloparaphenylene.58 In another example, a dyad of [9]cycloparaphenylene was employed to enhance the CPL response.59 Two tris(ethynyl)cycloparaphenylenes were oxidatively coupled to form compound C5, which exhibited strong CPL with a g value of +0.031 for the M enantiomer. The diyne linkers in C5 were subsequently converted to thiophene linkers in C6, which still exhibited intense but weaker CPL with a g factor of +0.012.
As related macrocyclic systems to the cylindrical molecules mentioned above, carbon belts are also of interest, as they also constitute a structural component of nanotubes. Starting from resorcin[4]arene, oxygen-doped chiral molecular belts have been prepared by stitching the upper fjord regions. These zigzag belts (C7–C9) are highly strained, with energies of 53–68 kcal mol−1, and exhibit strong CPL responses with dissymmetry factors ranging from +0.015 to +0.021, positive for the M enantiomers.60 It is also noteworthy that a smaller g factor value of +0.007 was reported for the half-belt shape precursor of these belts. A chiral belt containing eight-membered rings (C10) was also reported to provide comparably strong CPL, with a g factor of +0.011 for the M enantiomer.61 Very recently, D3 and D4-symmetric chiral nanorings (C11–C14) were stereoselectively synthesized and exhibited intense CPL with g factors ranging from +0.020 to +0.064.62
Due to possible symmetry breaking in the excited state, as well as orbital interactions and altered energy ordering, the above strategy using chiral and symmetrical macrocycles to enhance the CPL response does not always hold. However, cylindrical and belt-shaped macrocyclic molecules, in principle, facilitate the alignment of electric and magnetic dipole transition moments along the cylindrical axis, maximizing the rotational strength and, consequently, the dissymmetry factor values.
4.3 Paracyclophanes and (phenylene)ethynylenes
Planar chiral [2.2]paracyclophane units have been used to create unique chiral systems because they position substituents in a fixed and twisted orientation (i.e., pseudo-geminal-, ortho-, meta-, and para-isomers). Among various paracyclophane derivatives, a dimesitylborylphenyl and amino groups were successfully introduced to induce intramolecular charge transfer character. This modification facilitates a significant increase in fluorescence intensity and the corresponding g factor values. Thus, the pseudo-geminal paracyclophane P1 (Chart 3) exhibited strong CPL peaking at 485 nm with a g factor of −0.013 in cyclohexane for the Sp enantiomer (Table 3).65 Due to the charge transfer character of the electronic transition, the emission was red-shifted to 542 nm in tetrahydrofuran, with reduced CPL intensity and a g factor of −0.0089. Compared to the g factor of the dimesitylboryl-substituted cyclophane, the value in P1 was nearly doubled by the incorporation of an extra phenyl ring between the boron and the cyclophane scaffold. Recently, analogous molecules featuring slightly different triarylborane-substituted cyclophanes were shown to exhibit temperature-dependent dual fluorescence from charge transfer and locally excited states, albeit with lower g factors.66 The g factor values were further improved in the pseudo-meta isomers P2, with both cyclophanes exhibiting negative CPL in cyclohexane for the Sp enantiomers, showing g factors of −0.017 at around 460 nm, along with excellent fluorescent quantum yields. More interestingly, the sign of the CPL was reversed in tetrahydrofuran and toluene, with g values of +0.0086 and +0.0069 for P2a and P2b, respectively, in the former solvent. This switching behaviour is significant for potential applications in advanced optical materials and is attributed to changes in the excited-state dynamics and the interplay between charge transfer and locally excited states. Boron difluoride complexes are well known as effective fluorescent motifs. Among the various derivatives of [2.2]paracyclophane boron difluoride complexes, biscyclophane P3 was reported to exhibit a favourable g factor of −0.01 for the Sp,Sp isomer.67 A planar chiral cyclophane core was effectively employed to induce helical twists in o-phenylenes and p-(phenylene)ethynylenes. Thus, [2.2]paracyclophane, composed of two o-quinquephenyl moieties (P4) at the pseudo-meta position, exhibited intense CPL with a g factor of −0.012 in cyclohexane.68 The Sp planar chirality in the cyclophane backbone induces complete axial chirality in the o-phenylenes, ultimately leading to a pair of M helicities and resulting in negative CPL. In more polar solvents such as chloroform and acetonitrile, the g factor intensity decreased to −0.008 and −0.006, respectively. A propeller-shaped 4,7,12,15-tetrasubstituted paracyclophane was reported as an excellent CPL emitter. A group led by Morisaki and Chujo prepared a macrocycle P5 composed of p-(phenylene)ethynylene units, which exhibited strong negative CPL with a g factor value of −0.011 for the Sp planar chirality.69 However, extending the p-(phenylene)ethynylene units further in P6 did not significantly improve the g factor; instead, a slight decrease in intensity to −0.010 was observed. Additional extension of the units further reduced the g factor intensity to −0.0075.70 Such strong CPL was attributed to the twisting between the p-(phenylene)ethynylene units. Accordingly, a unique chiral macrocycle with pairwise p-(phenylene)ethynylene units was designed using a bispyrrolidinoindoline scaffold, obtained in a stereoselective manner from tryptophan. This D2-symmetric macrocycle P7 did not require optical resolution of the racemic product and indeed afforded strong negative CPL for the D-tryptophan-based enantiomer, with a g factor value of −0.011 in cyclohexane.71 This value decreased in intensity to −0.0057 in acetonitrile due to the presence and equilibrium of varied conformers. | ||
| Chart 3 | ||
| Molecule | Symmetry | λ max | Φ CPL | g CPL (λmon) | Conditions | Ref. |
|---|---|---|---|---|---|---|
| a For an enantiomer based on D-tryptophan. | ||||||
| (Sp)-P1 | C 1 | 488 | 0.83 | −0.013 (—) | Cyclohexane, 10 µM | 65 |
| (Sp)-P2a | C 1 | 461 | 0.93 | −0.017 (—) | Cyclohexane, 10 µM | 65 |
| (Sp)-P2b | C 1 | 455 | 0.82 | −0.017 (—) | Cyclohexane, 10 µM | 65 |
| (Sp,Sp)-P3 | C 1 | 528 | 0.13 | −0.01 (—) | Tetrahydrofuran, 10 µM | 67 |
| (Sp,M,M)-P4 | C 2 | 372 | 0.18 | −0.012 (370) | Cyclohexane, 10 µM | 68 |
| (Sp)-P5 | D 2 | 460 | 0.45 | −0.011 (—) | CHCl3, 10 µM, r.t. | 69 |
| (Sp)-P6 | D 2 | 471 | 0.60 | −0.010 (471) | CHCl3, 1.0 µM | 70 |
| P7 | D 2 | 380 | 0.74 | −0.011 (384) | Cyclohexane, 10 µM | 71 |
| (M,S,S)-P8a | C 2 | ≈420 | 0.069 | −0.011 (390) | CH2Cl2 | 72 |
| (M,S,S)-P8b | C 2 | ≈420 | 0.18 | −0.012 (390) | CH2Cl2, 70 µM | 73 |
| (M,R,R)-P9 | C 2 | ≈410 | 0.029 | −0.01 (380) | Methanol, 25 µM | 74 |
| (M,R,R)-P10a | C 2 | 423 | 0.020 | −0.014 (400) | CH2Cl2, 15 µM | 75 |
| (M,R,R)-P10b | C 2 | 423 | 0.021 | −0.03 (398) | CH2Cl2, 15 µM | 75 |
| (M,R,R)-P11a | C 2 | ≈420 | 0.24 | −0.013 (407) | Methanol, 25 µM | 74 |
| (M,R,R)-P11b | C 2 | ≈420 | 0.047 | −0.013 (408) | Methanol, 25 µM | 74 |
| (M,R,R)-P11c | C 2 | ≈420 | 0.057 | −0.011 (412) | Methanol, 25 µM | 74 |
| (M,R,R)-P11d | C 2 | ≈420 | 0.16 | −0.013 (416) | Methanol, 25 µM | 74 |
| (M,R,R)-P11e | C 2 | ≈420 | 0.046 | −0.013 (414) | Methanol, 25 µM | 74 |
| (M)-(E,E)-P12 | C 2 | 395 | 0.12 | −0.034 (400) | CH2Cl2, 80 µM | 76 |
| (M)-(E,Z)-P12 | C 1 | 410 | 0.13 | −0.042 (415) | CH2Cl2, 80 µM | 76 |
Related to the aforementioned p-(phenylene)ethynylene derivatives created in the chiral scaffold, o-(phenylene)ethynylenes themselves form fluorescent helical foldamers that are used as CPL emitters. Specifically, a stapled o-(phenylene)ethynylene diol (P8a) and its pivalate ester (P8b) exclusively exhibit M helicity in the o-(phenylene)ethynylene unit induced by a chiral linker with a S,S configuration. These helical molecules, along with additional esters, all exhibited negative CPL with comparative g factors of −0.011 and −0.012 for P8a and P8b, respectively, although the quantum yield was significantly improved in the latter species.72,73 In a similar system P9, (R,R)-O-isopropylidene-D-threitol was employed to induce M helicity in the o-(phenylene)ethynylenes, which again exhibited strong negative CPL with a g factor of −0.01. In the o-(phenylene)ethynylene compound P10, arene-perfluoroarene interactions were employed to promote helical folding. The g factors of the derivatives with mono-, tri- (P10a), and penta- (P10b) fluorinated benzenes were found to be 0.009, 0.014, and 0.030, respectively, with the latter representing a three-fold increase compared to that of the non-fluorinated analogue.75 Extended helical systems (P11) have also been reported to provide comparable CPL activities.74 It is important to note that, while beyond the scope of this review, the host–guest chemistry of these helical o-(phenylene)ethynylenes, particularly in relation to Ag(I) cation binding, has been actively investigated for chiral sensing applications. Furthermore, o-(phenylene)ethynylenes were doubly stapled through sequential alkene metathesis in P12. Enhanced CPL activity was observed, with g factor values of −0.034 and −0.042 for the (E,Z)- and (E,E)-isomers, respectively, both derived from M helicity.76 The g factor value of the former isomer was found to be slightly solvent-dependent, being reported as −0.055 in hexane.
4.4 Helically twisted dimeric molecules
A successful control of the electric and magnetic dipole transition moments, and consequently the CPL emission, was also demonstrated in helically intertwined π-dimer systems described below. Accordingly, cyclooctatetraene-embedded perylene diimide dimer T1 (Chart 4) was employed by Jiang and co-workers, achieving good CPL performance with a g factor value of +0.010 for the M,M isomer, along with a moderate quantum yield of emission (Table 4). Stepwise annulation at the bay region with nitrogen atoms further improved the g factor values, reaching +0.022 for T2 and +0.030 for T3, respectively.77 Theoretical investigations revealed that nitrogen annulation gradually increases the magnitude of the magnetic dipole transition moment while slightly decreasing the electric dipole transition moment, ultimately leading to a three-fold increase in the g factor value for T3, as compared with T1. The effect of substituents on the imide nitrogen was found to be marginal; however, longitudinal π-extension to the terrylene diimide dimer resulted in a considerable decrease in the g factor to +0.002, accompanied by a significant red shift of the emission in the near-IR region. Further extension to the quarterylenediimide dimer was also conducted, but the CPL properties were not reported due to instrumental limitations.78 A different type of perylene diimide macrocycle, T4, was also reported, in which two π systems are helically twisted, a motif akin to previous paracyclophanes. A through-space interaction between the two perylene diimide moieties resulted in red-shifted emission and successfully prevented enantiomeric conversion in toluene, with a half-life of 5 days. More importantly, good CPL performance was observed, with a g factor of −0.010 at 675 nm for the M,M enantiomer.79 | ||
| Chart 4 | ||
| Molecule | Symmetry | λ max | Φ CPL | g CPL (λmon) | Conditions | Ref. |
|---|---|---|---|---|---|---|
| (M,M)-T1 | D 2 | 562 | 0.61 | +0.010 (570) | Toluene, 10 µM | 77 |
| (M,M)-T2 | C 2 | 608 | 0.50 | +0.022 (595) | Toluene, 10 µM | 77 |
| (M,M)-T3 | D 2 | 627 | 0.41 | +0.030 (609) | Toluene, 10 µM | 77 |
| (M,M)-T4 | D 2 | ≈620 | 0.48 | −0.01 (675) | Toluene, 10 µM, 25 °C | 79 |
| (S,S)-T5a | D 2 | 403 | 0.12 | +0.014 (359) | CH2Cl2, 5.7 µM, 25 °C | 80 |
| (S,S)-T5b | D 2 | 367 | 0.28 | +0.011 (345) | CH2Cl2, 9.4 µM, 25 °C | 80 |
Dimers of cyclic binaphthyls were also reported to provide good CPL responses, with g factors of −0.014 and −0.011 for the S,S enantiomer in T5a (linker = methylene) and T5b (linker = ethylene), respectively.80 The corresponding intertwined dimer with a longer propylene linker (n = 3) and the one without a linker (free methoxy groups) resulted in much lower g factors of −0.003 and −0.004, respectively.
4.5 Helicenes
Helicenes are ortho-fused conjugated aromatic systems characterized by a unique helical structure that arises from steric repulsion.81–85 This distinctive structure has been extensively investigated for its remarkable properties, particularly its chiroptical properties.86,87 CPL of helicenes has emerged as a significant research topic in recent years, positioning helicenes among the most studied molecular systems. This research spans proof-of-concept studies aimed at advancing our understanding of helical anisotropy and the development of advanced materials. As demonstrated in the following sections, these efforts have proven to be highly successful. Here, we provide an overview of the current state-of-the-art in CPL studies on molecular helicenes, focusing on the relationship between structure and chiroptical properties.The introduction of π-donor and π-acceptor moieties on the helicene has proven valuable for enhancing the CPL of [5]helicene. In this context, triarylamine and triarylborane groups were attached at the meta-positions of [5]helicene in H1 (Chart 5). This donor–acceptor helicene exhibits strong CPL performance, with a large g factor value of −0.013 (Table 5).88 Notably, symmetrically substituted [5]helicenes with either two triarylamines or two triarylboranes produced reduced CPL, with g factors of −0.0065 and −0.0040, respectively. Additionally, the position of substitution was found to be crucial for achieving this enhancement. The meta-substitution with aryl or alkynyl groups was also found to be significant for improving the CPL of [6]helicene. As such, symmetrically substituted bisethynyl[6]helicenes (H2a–f), along with the pseudo-symmetrical one (H2g), and bisaryl[6]helicenes (H3a–c) all exhibited very good g factors ranging from −0.018 to −0.028, although they displayed modest fluorescence quantum yields.89–93 Comparative g factor values were also found for other donor–acceptor [6] and [7] helicenes. Specifically, donor–acceptor moieties were introduced in the same terminal benzene rings in [6]helicene H4,94 while they were placed in different rings in [7]helicene H5.95 Both types of donor–acceptor configurations in these helicenes exhibited good CPL responses with g factor values ranging from −0.012 to −0.015. It is noteworthy that all the [5] to [7] helicene derivatives with good CPL performance mentioned above share meta-substitutions in common, highlighting the importance of orientational control of dipole transition moments by modification of electronic structure of helicenes.
 | ||
| Chart 5 | ||
| Molecule | Symmetry | λ max | Φ CPL | g CPL (λmon) | Conditions | Ref. |
|---|---|---|---|---|---|---|
|
a In toluene containing tetrahydrofuran (75:1).
b In chloroform.
|
||||||
| (M)-H1 | C 1 | 445 | 0.12 | −0.013 (446) | Hexane, 10 µM | 88 |
| (M)-H2a | C 2 | 430 | 0.16 | −0.025 (430) | CH2Cl2, 1 µM, 25 °C | 89 |
| (M)-H2b | C 2 | 426 | 0.09 | −0.027 (426) | CH2Cl2, 1 µM, 25 °C | 89 |
| (M)-H2c | C 2 | 455 | 0.06 | −0.025 (429) | CH2Cl2, 1 µM, 25 °C | 89 |
| (M)-H2d | C 2 | 426 | — | −0.028 (—) | Acetonitrile-d3, 30 µM, r.t. | 90 |
| (M)-H2e | C 2 | ≈450 | 0.06 | −0.018 (421) | CH2Cl2, 10 µM, 25 °C | 89 |
| (M)-H2f | C 2 | 453 | 0.05 | −0.018 (—) | CH2Cl2, 0.3–0.6 mM, 23 °C | 91 |
| (M)-H2g | C 2 | 449 | 0.05 | −0.025 (—) | CH2Cl2, 0.3–0.6 mM, 23 °C | 91 |
| (M)-H3a | C 2 | 425 | 0.041 | −0.02 (—) | CH2Cl2, 22.5 µM | 92 |
| (M)-H3b | C 2 | ≈430 | 0.087 | −0.024 (413) | CH2Cl2, 10 µM, r.t. | 93 |
| (M)-H3c | C 2 | 426 | 0.088 | −0.025 (418) | CH2Cl2, 10 µM, r.t. | 93 |
| (M)-H4a | C 2 | ≈430 | 0.045 | −0.015 (420) | CH2Cl2, 10 µM, 25 °C | 94 |
| (M)-H4b | C 2 | 430 | 0.05 | −0.012 (440) | CH2Cl2, 10 µM, 25 °C | 94 |
| (M)-H5 | C 2 | 477 | 0.17 | −0.013 (550) | CHCl3, 10 µM | 95 |
| (M)-H6 | C 1 | ≈490 | — | −0.012 (—) | CH2Cl2, 20 µM, 20 °C | 96 |
| (M)-H7a | C 2 | 471 | 0.15 | +0.013 (469) | CHCl3, 9.4 µM, r.t. | 97 |
| (M)-H7b | C 2 | 470 | 0.12 | +0.012 (469) | CHCl3, r.t. | 98 |
| (M)-H7c | C 2 | 467 | 0.19 | +0.012 (463) | CHCl3, r.t. | 98 |
| (M)-H8 | C 2 | 575 | 0.044 | −0.010 (574) | Toluenea, r.t. | 99 |
| (M)-H9a | C 2 | 550 | 0.089 | −0.014 (—) | CH2ClCH2Cl, 0.10 mM | 100 |
| (M)-H9b | C 2 | 554 | 0.066 | −0.021 (—) | CH2ClCH2Cl, 0.10 mM | 100 |
| (M)-H10 | C 2 | 434 | 0.17 | −0.013 (432) | CH2Cl2, 24–40 µM | 101 |
| (M)-H11 | C 2 | 492 | 0.16 | −0.020 (498) | CH2Cl2, r.t. | 102 |
| (M)-H12 | C 2 | 546 | 0.06 | −0.027 (548) | CH2Cl2, r.t. | 102 |
| (M)-H13 | C 2 | 470 | 0.035 | −0.013 (470) | CH2Cl2, r.t. | 103 |
| (M)-H14 | C 2 | 450 | 0.008 | −0.01 (480) | Toluene, 20 µM, r.t. | 104 |
| (M)-H15 | C 2 | 483 | 0.027 | −0.04 (520) | Toluene, 20 µM, r.t. | 104 |
| (M)-H16a | C 2 | 428 | 0.32 | −0.030 (428) | CHCl3, 20 µM, 25 °C | 105 |
| (M)-H16b | C 2 | 449 | 0.30 | −0.032 (449) | CHCl3, 20 µM, 25 °C | 105 |
| (M)-H17 | C 2 | 482 | 0.15 | −0.016 (482) | CHCl3, 7.8 µM, 25 °C | 106 |
| (M)-H18 | C 2 | ≈480b | — | −0.021 (—) | Methanol | 107 |
Doping with boron–nitrogen units has been a common strategy to modulate the properties of aromatic hydrocarbons. Accordingly, boron–nitrogen [6]helicene H10 is isoelectronic and isostructural to H3, and it has been found to exhibit good CPL performance, with a g value of −0.013, alongside an improved fluorescence quantum yield and molar absorptivity.101 In polyaza [7] to [9] helicenes with multiple imidazole units, blue to green emission was observed. Increasing the number of aromatic rings systematically enhanced the g factor values, with those for H11 and H12 being −0.020 and −0.027, respectively.102 Meanwhile, while the number of nitrogen atoms influenced the emission energy (and colour), it had little effect on the g value. As an additional example, sulfur atoms were incorporated into the dithia[9]helicene at the terminal rings. Interestingly, the endo-isomer H13 exhibited a better g factor than the exo-isomer, with g values of −0.013 and −0.0042, respectively.103 Notably, the exo-dithia[7]helicene displayed an oppositely signed CPL with a g value of +0.0050. These trends are explained and reproduced solely by the incorporation of vibronic contributions (i.e., Herzberg–Teller effect). Note that the vibronic effects on CPL have been discussed more thoroughly in double carbohelicenes (vide infra).45 Introducing thiadiazole units at both terminals in [7] and [9] helicenes was found to enhance the CPL anisotropy, yielding g factor values of −0.01 and −0.04 for H14 and H15, respectively.104 The theoretical investigation revealed that this enhancement is attributed to the heavy atom effect, which facilitates a magnetically allowed nature in the S1 to S0 transition. However, this also resulted in lower fluorescence quantum yields.
An X-shaped double [5]helicene composed of 12H-benzo[b]phenoxazine (D1a) was reported to exhibit CPL with a g factor of −0.013 for the M enantiomer (Chart 6 and Table 6).119 Interestingly, a similar double helicene (D2) with 13H-dibenzo[b,i]phenoxazine units demonstrated even greater CPL, but with an opposite sign, featuring a g factor of +0.023 measured at the edge of the CPL band at 490 nm. A computational investigation revealed that the improved g factor values were attributed to the incorporation of nitrogen and oxygen atoms, while the sign inversion was ascribed to the reversal of the direction of electric dipole transition moments between the S0 to S1 and S1 to S0 transitions. A strong phosphorescence was observed around 600 nm for a related phenothiazine analogue (D1b) at −190 °C, along with a relatively weaker fluorescence-based CPL at 500 nm, which exhibited a positive g factor value of +0.015.120 A double aza[7]helicene fused with a Sondheimer–Wong diyne (D3) was reported to exhibit a g factor as large as +0.014 for the M isomer, although the fluorescence intensity was relatively weak.121 In this X-shaped double helicene, an octagon moiety was found to be essential for the strong chiroptical response. A structurally related saddle-shaped quadruple [7]helicenoid with a cyclooctatetrapyrrole core (D4) also demonstrated good CPL with a g value of −0.012.122 In this molecule, the M helicity in the [7]helicene structures induced P helicity in the minor helices. A lower symmetry system in double helicenes, distinct from the aforementioned D2-symmetrical examples, was also shown to be effective in enhancing chiroptical responses. Thus, an S-shaped C2-symmetric double [6]helicene (D5) and a helicenoid (D6) both exhibited intense CPL responses with g factor values of −0.011 and −0.028, respectively.123 However, these values, along with those for the helicenes H16–H18, were called into question by a detailed computational analyses.117
 | ||
| Chart 6 | ||
| Molecule | Symmetry | λ max | Φ CPL | g CPL (λmon) | Conditions | Ref. |
|---|---|---|---|---|---|---|
| a In 2-methyltetrahydrofuran at −190 °C. b In dichloromethane at room temperature. c In toluene. | ||||||
| (M,M)-D1a | D 2 | 587 | 0.035 | −0.013 (590) | CH2Cl2, 0.2 mM, r.t. | 119 |
| (M,M)-D1b | D 2 | 547b | 0.003 | +0.015 (500) | Glassa | 120 |
| (M,M)-D2 | D 2 | 569 | 0.038 | +0.023 (490) | CH2Cl2, 0.2 mM, r.t. | 119 |
| (M,M)-D3 | D 2 | 621 | <10−5 | +0.014 (693) | CH2Cl2, 10 µM, r.t. | 121 |
| (M,M,P,P)-D4 | D 2 | 575 | 0.016 | −0.012 (575) | CH2Cl2, 10 µM | 122 |
| (M,M)-D5 | C 2 | 454 | 0.094 | −0.011 (454) | CHCl3, 1.0 µM, 25 °C | 123 |
| (M,M)-D6 | C 2 | 492 | 0.190 | −0.028 (492) | CHCl3, 1.0 µM, 25 °C | 123 |
| (M,M)-D7 | D 2 | 442 | 0.08 | +0.015 (—) | CHCl3, 10 µM, r.t. | 124 |
| (M,M)-D8 | D 2 | 511c | 0.03 | −0.01 (—) | CH2Cl2 | 125 |
| (M,M)-D9 | D 2 | 480 | 0.09 | +0.025 (480) | Toluene, 10 µM, r.t. | 126 |
| (M,M)-D10 | C 2 | 424 | 0.11 | −0.02 (—) | CH2Cl2, 0.3–0.6 mM, 23 °C | 91 |
| (M,M)-D11a | C 2 | 454 | 0.25 | −0.012 (—) | CH2Cl2, 0.3–0.6 mM, 23 °C | 91 |
| (M,M)-D11b | C 2 | 455 | 0.28 | −0.012 (—) | CH2Cl2, 0.3–0.6 mM, 23 °C | 91 |
| (M,M)-D11c | C 2 | 456 | 0.28 | −0.01 (—) | CH2Cl2, 0.3–0.6 mM, 23 °C | 91 |
| (M,M)-D12 | C 2 | 420 | 0.047 | −0.011 (417) | Acetonitrile, 25 µM | 127 |
| (M,M)-D13a | C 2 | 426 | 0.021 | −0.011 (—) | CH2Cl2, 10 µM, r.t. | 128 |
| (M,M,M)-D13b | C 3 | 427 | 0.020 | −0.013 (—) | CH2Cl2, 6.7 µM, r.t. | 128 |
A helicene-derived macrocyclic framework appears to confer unique chiroptical properties, suggesting that a more robust D2-symmetric structure, maintained even in the excited state, may be achievable.129 For instance, while a directly connected figure-eight-shaped double [5]helicene exhibited only a moderate g factor value,130 a fused [5]helicene (D7) with additional p-phenylene linkers was reported to demonstrate an improved g factor of +0.015 for the M enantiomer, accomplished through appropriate manipulation of the orientation and intensity of dipole transition moments.124 In another example of a figure-eight helicene, a fused double [5]helicene (D8) was also reported, showing a similarly strong CPL with a g value of −0.010; it is noteworthy that the sign was opposite to that of D7.125 Expanding this symmetry-based molecular design to heterohelicenes is a natural progression. While free [4]helicenes suffer from racemization, the macrocylization has provided a configurationally stable figure-eight double [4]helicene (D9), which affords strong CPL with a g value of as large as +0.025.126 This molecular design also features thermally activated delayed fluorescence (TADF) property due to a relatively small ΔEST value of 0.31 eV, making D9 the first example of a red-coloured circularly polarized TADF emitter.
Dipole transition moment engineering has also been facilitated in molecular systems containing multiple helicene units, without apparent fusion between the helices. Thus, bis[6]helicenes D10–D12, which consist of alkenyl helicene moieties linked by either alkyne, arene, or o-(phenylene)ethynylene, all exhibited strong CPL responses with g factor values ranging from −0.01 to −0.02.91,127 Finally, bis and tris[6]helicene macrocycles (D13a and D13b) connected with diarylethene photoswitching moieties have been shown to exhibit strong CPL with g values of −0.011 and −0.013, respectively.128 The chiroptical responses were found to be consecutively amplified by a number of components in these series of macrocycles, and the photoswitching ability was demonstrated by the CPL silence upon photo-induced ring closure of the diarylethene moieties.
A series of helical nanographenes consisting of two hexabenzocoronenes embedded with [9], [10], and [11]helicenes have been reported. The first nanographene with a [9]helicene joint (N1a) exhibited the best chiroptical properties, with a CPL g factor value of −0.036, followed by −0.010 for the [10]helicene derivative (N1b) and −0.0089 for the [11]helicene derivative.134 It is noteworthy that the size of the helicene unit defines the overlap between the two hexabenzocoronenes, which is maximized in N1a. Enhanced chiroptical properties were supported by a computational analyses on their dipole transition moments. Larger π-extended carbo[11] and [13]helicenes (N2a and N3), consisting of layers of hexabenzocoronenes, have been prepared in an enantioselective manner via nickel-catalyzed triple [2 + 2 + 2] cycloaddition, followed by the Scholl reaction starting from the corresponding nonaynes. Along with the precursors [11]helicene (N2b) and expanded [13]helicene (N2c), all these helical nanographenes exhibited excellent CPL behaviors with moderate fluorescence intensities, showing g factors ranging from −0.010 to −0.040.135 It is noteworthy that the larger helical nanographenes tend to have slightly lower anisotropy, for reasons that are not fully understood. A unique nanographene, N4, is a π-extended pentadecabenzo[9]helicene that features four fused hexabenzocoronenes. This molecule has been shown to exhibit near-infrared emission in the range of 600–900 nm with an improved emission quantum yield and intense CPL with a high g factor value of −0.042 for the M helicity.136
As with the double helicenes mentioned in the previous section, multiple helicene units can also be incorporated into nanographenes. Consequently, geometric isomers of π-extended double [7]helicenes have been reported. Among these isomers, nanographene N5, fused at the para-position of the middle benzene ring, exhibited superior CPL performance with a g value of −0.014, followed by −0.0087 for the meta-fused derivative and −0.0027 for the ortho-fused derivative.137,138 A computational investigation revealed the importance of helicene geometry for better engineering of electric and magnetic dipole transition moments. A unique nanographene N6, exhibits formal D5 symmetry, in which five perylene diimide units are fused into a corannulene core, resulting in a quintuple [6]helicene structure. This molecule was found to exhibit greater CPL performance with a g factor value of −0.012 with all M helicities, compared to the C2-symmetrical analogue with M,M,M,M,P helicity, which has a lower g value of −0.008.139
Among various possibilities for further exploration of chiral graphene structures, the expansion of helicene-based nanographenes to heterohelicenes has indeed been realized. Of several approaches, boron–nitrogen doping has attracted significant attention due to its increased fluorescence quantum yield and tunability of frontier orbitals. Consequently, π-extended [8] and [10]heli(aminoborane)s, N7 and N8a, have been described. In these systems, carbon atoms at the inner rims of the helicenes were replaced by alternating boron and nitrogen atoms, resulting in greater ring strain and helical diameter, ultimately leading to excellent CPL performance with g factor values of −0.024 and −0.048, respectively, along with excellent emission quantum yields of 0.31 and 0.24, respectively.140 A lateral extension of the π-systems in N8a was demonstrated to induce brighter and narrowband emission in N8b, with a full width at half maximum as small as 16 nm. This modification achieved a much higher quantum yield of 0.82 while maintaining a g factor value of −0.017. However, additional fusion of benzene rings led to a decrease in both quantum yield and g factor value, with N8c displaying values of 0.67 and −0.011, and a derivative with further fusion exhibiting values of 0.072 and −0.008, respectively.141
| Molecule | Symmetry | λ max | Φ CPL | g CPL (λmon) | Conditions | Ref. |
|---|---|---|---|---|---|---|
| a 96% ee. b 92% ee. c 94% ee. | ||||||
| (M)-N1a | C 2 | 575 | 0.22 | −0.036 (580) | Tetrahydrofuran, 10 µM, r.t. | 134 |
| (M)-N1b | C 2 | 543 | 0.10 | −0.010 (540) | Tetrahydrofuran, 10 µM, r.t. | 134 |
| (M)-N2aa | C 2 | 612 | 0.31 | −0.040 (605) | CHCl3, 10 µM, 25 °C | 135 |
| (M)-N2bb | C 2 | 565 | 0.30 | −0.038 (567) | CHCl3, 10 µM, 25 °C | 135 |
| (M)-N2cc | C 2 | 461 | 0.19 | −0.010 (461) | CHCl3, 10 µM, 25 °C | 135 |
| (M)-N3 | C 2 | 607 | 0.23 | −0.034 (605) | CHCl3, 10 µM, 25 °C | 135 |
| (M)-N4 | C 2 | 684 | 0.10 | −0.042 (684) | CH2Cl2, 5 µM | 136 |
| (M,M)-N5 | C 2 | 533 | 0.44 | −0.014 (556) | CH2Cl2, 1.0 µM | 138 |
| (M,M,M,M,M)-N6 | D 5 | ≈570 | 0.10 | −0.012 (—) | Toluene, 1.0 µM | 139 |
| (M)-N7 | C 2 | 409 | 0.31 | −0.024 (409) | CH2Cl2, 10 µM | 140 |
| (M)-N8a | C 2 | 430 | 0.24 | −0.048 (430) | CH2Cl2, 10 µM | 140 |
| (M)-N8b | C 2 | 515 | 0.82 | −0.017 (515) | CH2Cl2, 10 µM | 141 |
| (M)-N8c | C 1 | 529 | 0.67 | −0.011 (529) | CH2Cl2, 10 µM | 141 |
 | ||
| Chart 7 | ||
4.6 Excimers
Another interesting molecular system that emits a strong CPL response is based on excimer emission. An excimer is a short-lived molecular dimer in its excited state, where the dimeric state is not stable in the ground state and exhibits broad and structure-less emission with a large Stokes shift.142,143 Such dynamic excimer emission has been discussed comparatively with that from the so-called static excimer, where the excited dimer is formed upon direct excitation of the ground-state dimer. Most of the relevant chiral emissions reported to date are based on intramolecular excimers, where the two interacting chromophores are closely located and spatially restricted within a single molecule, forming a twisted (helical-like) geometry. An intermolecular excimer has also been employed for chiral emission, where supramolecular chemistry plays an important role. Rigid π systems typically exhibit a linear correlation between the dissymmetry of the lowest electronic transitions in absorption (CD) and emission (CPL), as these two processes represent opposite transitions between the S0 and S1 states.144,145 However, in the excimer system, this empirical relationship does not necessarily hold, as excimer emission arises from different excited species that dynamically form from the monomer within the excited state hypersurface. As mentioned above, the exciton coupling between the chromophores in the excimer induces a much more intense electric dipole transition moment, thereby enhancing the g factor value. While such interactions become more important and significantly influence chiroptical responses upon aggregation, as well as in crystalline and film states,146,147 below we summarize the isolated molecular excimer systems that exhibit substantial CPL signals with g values larger than 0.01.A well-defined stacking pyrene dimer was successfully constructed using a planar chiral cyclophane architecture. An isomeric [2.2]pyrenophane was employed as early as 1978 by Misumi and colleagues as a model system for excimer emission.151 Very recently, the chiroptical properties of pyrenophanes (E1a–d) were reported (Chart 8).152 In E1a, the 4- and 9-positions of the two pyrene units are doubly linked by methylene linkers, while other [3.3]pyrenophanes serve as intermediates for the synthesis of E1a, containing disulfide (E1b), disulfoxide (E1c), and disulfone (E1d) linkers. A bathochromic shift in emission was observed for the [2.2]cyclophane E1a, attributed to stronger through-space interactions between the pyrene units compared to other [3.3]cyclophanes (E1b–d). X-ray structural analyses of E1a, E1c, and E1d revealed considerably different interplane distances of 3.32, 3.48, and 3.48 Å, respectively, with twist angles of 51°, 39°, and 38°, respectively. Nevertheless, all four cyclophanes exhibited strong and comparable CPL, with g values around +0.03, all positive for the Sp enantiomers (Table 8). A computational investigation provided a rationale for the strong CPL response of these planar chiral pyrenophanes, where the relative angle between the electric and magnetic dipole transition moments is markedly small, ranging from 2° to 10°. Notably, a stable and strong circularly polarized electrochemiluminescence was also observed for [2.2]paracyclophane E1a, with a g value of −0.013 at around 500 nm. Our group has also contributed to the investigation of the CPL properties of planar chiral cyclophanes. Specifically, a partially overlapped [3.3](3,9)-dicarbazolophane (E2) exhibited strong negative CPL for excimer emission at around 390 nm, with a g factor of −0.013 for the Sp isomer.153 Notably, excimer formation is well documented for carbazole units in polymers, but free carbazole does not form excimers. X-ray crystal structural and computational investigations revealed that the separation distance, twist angle, and displacement between the carbazole units are all crucial factors influencing the strong chiroptical responses.
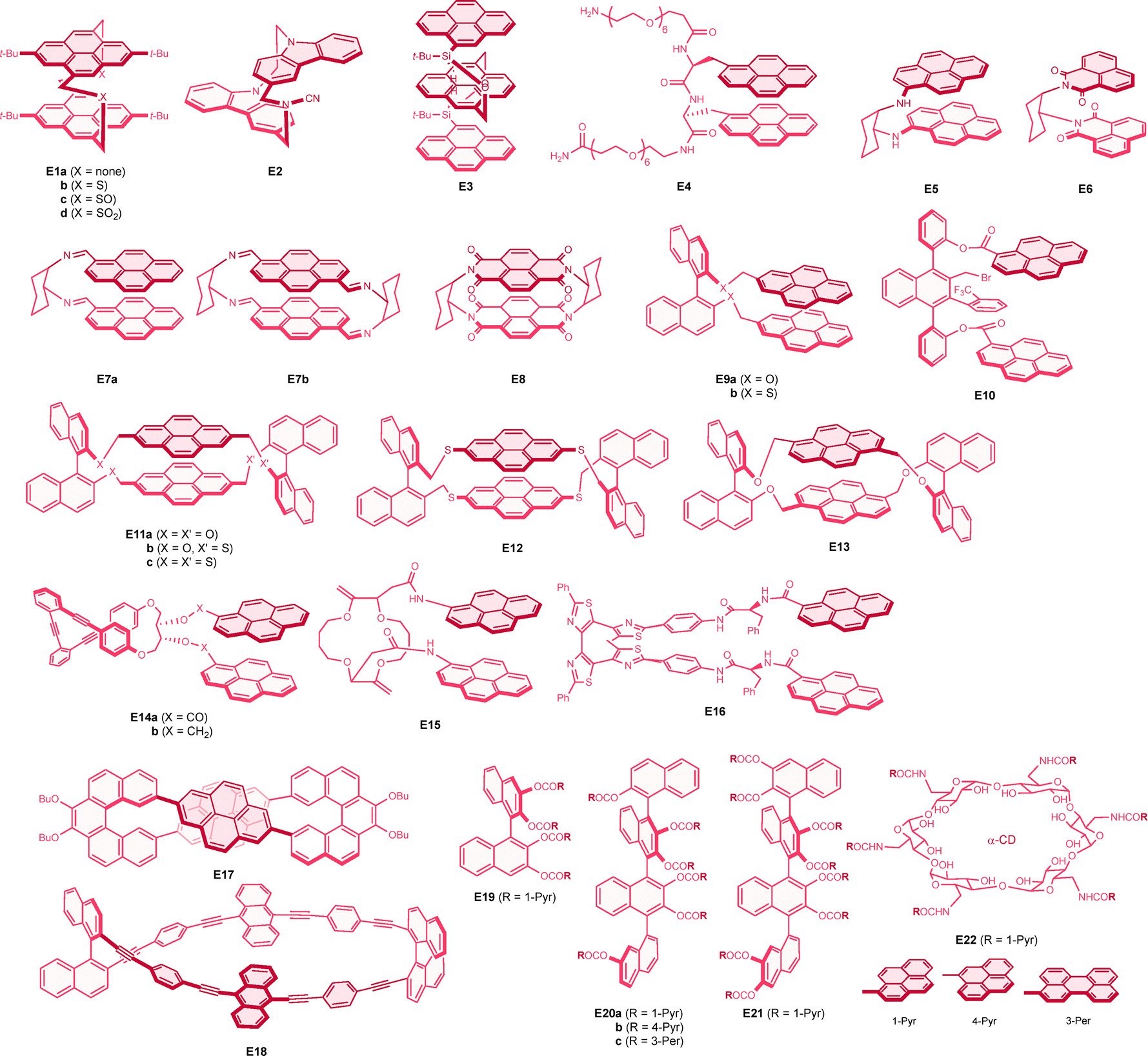 | ||
| Chart 8 | ||
| Molecule | Symmetry | λ max | Φ CPL | g CPL (λmon) | Conditions | Ref. |
|---|---|---|---|---|---|---|
| a A diastereomeric pair of enantiomers were reported, both of which provided comparable (chir)optical properties. b A value reported for the D-peptide derived enantiomer was converted. c Absolute configuration was not described. d For the 1st HPLC elute. Absolute configuration was not determined. | ||||||
| (Sp)-E1a | D 2 | 500 | 0.34 | +0.028 (—) | CH2Cl2, 10 µM, 25 °C | 152 |
| (Sp)-E1b | D 2 | 480 | 0.039 | +0.025 (—) | CH2Cl2, 10 µM, 25 °C | 152 |
| (Sp)-E1ca | D 2 | 480 | 0.017 | +0.023 (—) | CH2Cl2, 10 µM, 25 °C | 152 |
| (Sp)-E1d | D 2 | 480 | 0.074 | +0.034 (—) | CH2Cl2, 10 µM, 25 °C | 152 |
| (Sp)-E2 | C 2 | ≈390 | 0.047 | −0.013 (390) | Benzene, 25 °C | 153 |
| (S,S)-E3 | C 2 | 508 | 0.55 | −0.011 (573) | CH2Cl2, 1.0 mM | 154 |
| (S,S)-E4 | C 1 | 463 | 0.10 | −0.011 (463)b | CHCl3, 0.10 mM | 155 |
| (S,S)-E5 | C 2 | ≈460 | 0.31 | +0.016 (460) | Toluene, 50 µM | 156 |
| (S,S)-E6 | C 2 | 482 | — | +0.037 (470) | Methanol, 10 µM | 157 |
| (S,S)-E7a | C 2 | 490 | 0.19 | −0.024 (—) | CHCl3, 20 µM, r.t. | 158 |
| (S,S,S,S)-E7b | D 2 | 497 | 0.42 | −0.022 (419) | CHCl3, 20 µM, r.t. | 158 |
| (S,S,S,S)-E8 | D 2 | 515 | 0.098 | +0.040 (500) | CHCl2CHCl2, 25 µM, 25 °C | 159 |
| (S)-E9a | C 2 | 486 | 0.26 | +0.032 (—) | CH2Cl2, 20 µM, 20 °C | 160 |
| (S)-E9b | C 2 | 484 | 0.09 | −0.013 (—) | Toluene, 20 µM, 20 °C | 161 |
| E10 | C 1 | 510 | 0.29 | +0.019 (508) | CH2Cl2, 50 µM, r.t. | 162 |
| (S,S)-E11a | D 2 | 487 | 0.23 | +0.053 (—) | CH2Cl2, 20 µM, 20 °C | 160 |
| (S,S)-E11b | D 2 | 486 | 0.06 | +0.031 (—) | Toluene, 20 µM, 20 °C | 161 |
| (S,S)-E11c | D 2 | 492 | 0.07 | +0.013 (—) | Toluene, 20 µM, 20 °C | 161 |
| (S,S)-E12 | D 2 | — | 0.09 | +0.013 (—) | Toluene, 20 µM, 20 °C | 161 |
| (S,S)-E13 | D 2 | 475 | 0.27 | −0.022 (—) | CH2Cl2, 20 µM, 20 °C | 160 |
| (M,R,R)-E14a | C 2 | ≈540 | 0.44 | +0.03 (540) | CH2Cl2, 10 µM | 163 |
| (M,R,R)-E14b | C 2 | ≈420 | 0.55 | −0.012 (390) | CH2Cl2, 10 µM | 163 |
| E15 | C 2 | 491 | 0.32 | +0.017 () | CH2Cl2, 10 µM | 164 |
| (M)-E16 | C 2 | 500 | 0.04 | −0.01 (—) | CHCl3, 0.17 mM | 165 |
| (M,M)-E17 | D 2 | 487 | 0.43 | −0.038 (530) | Tetrahydrofuran, 7.7 µM, r.t. | 166 |
| (S,S)-E18 | D 2 | 668 | 0.21 | +0.020 (623) | Methanol, 1.0 µM | 167 |
| (S)-E19 | C 2 | ≈540 | 0.24 | −0.013 (—) | CH2Cl2, 10 µM, 20 °C | 168 |
| (S,S,S)-E20a | C 2 | ≈540 | 0.24 | −0.037 (—) | CH2Cl2, 6.6 µM, 20 °C | 168 |
| (S,S,S)-E20b | C 2 | ≈490 | 0.67 | −0.014 (—) | CH2Cl2, 1.0 µM, 20 °C | 169 |
| (S,S,S)-E20c | C 2 | ≈570 | 0.38 | +0.013 (—) | Tetrahydrofuran, 1.0 µM, 20 °C | 169 |
| (S,S,S)-E21 | C 2 | ≈540 | 0.25 | −0.034 (—) | CH2Cl2, 5.0 µM, 20 °C | 168 |
| E22 | C 6 | 486 | 0.39 | +0.012 (486) | CH2Cl2, 5.0 µM, r.t. | 167 |
A more direct approach to achieve chiral excimer emission is to incorporate chiral side chains into the pyrene chromophore. In E3, three pyrene groups are combined with two optically active silanes in a linear fashion. This molecule exhibited intense excimer emission without any monomer emission with a g factor value of −0.011 at around 570 nm for the S,S isomer.154 Since the CPL emission was bisignate, the absolute value of the g factor was somewhat smaller at the emission peak. A related strategy for creating chiral excimers involves appending two or more chromophores to the chiral backbone. In this context, a polypeptide backbone was used in E4 to position two pyrene groups in an appropriate proximity in a helical manner. Although the reported g factor values between the enantiomers were not fully consistent, strong excimer CPL emission on the order of ±0.01 was noted.155 A cyclohexane ring was also found to be effective as a chiral backbone for excimer CPL. Consequently, pyrene-incorporating cyclohexane 1,2-diamine (E5) was successfully demonstrated to exhibit intense CPL with a g factor value of +0.016 for the S,S isomer.156 A related cyclohexane appended with 1,8-naphthalimide groups (E6) exhibited even stronger CPL with a g factor of +0.037 in methanol.157 The corresponding 6-methoxy-naphthalimide derivative exhibited a slightly weaker CPL, with a g value of +0.0092 in tetrahydrofuran, compared to the value of +0.015 for E6 in the same solvent. Chiral 1,2-diaminocyclohexane was also employed for the construction of pyrene dimers via imine formation. Both compound E7a and a macrocyclic E7b showed comparatively intense CPL with absolute g factor of around −0.02, negative for the S,S isomers.158 Very recently, a cyclophane composed of a pair of naphthalene diimide units (E8) was reported to exhibit strong green CPL emission with a g factor value of +0.040.159 The axially chiral binaphthalene core was quite successfully utilized for chiral excimer emission. Consequently, a pyrene-appended binaphthol (E9a) was prepared by Ema and coworkers, exhibiting strong positive CPL for the S,S isomer in its excimer emission, with a g factor as large as +0.032.160 In contrast, its sulfur derivative, E9b, showed a slightly smaller g factor of −0.013, presenting an opposite sign for the same binaphthalene absolute configuration, although the details were not fully discussed in the report.161 While absolute configurations were not determined, a triaxially chiral substituted naphthalene (E10) was reported to provide a good scaffold for chiral excimer emission at 510 nm, with a g factor value of +0.019.162 With a second binaphthalene bridge, macrocyclic compounds E11a–c and related pyrenophanes E12 and E13 were also investigated by Ema's research group. Interestingly, the 2,7-linked pyrenophane E11a exhibited much more intense CPL, with a g factor value of +0.053, which was inverted in sign compared to the single binaphthol derivative E9a, despite having the same axial chirality.160 A replacement of oxygen with sulfur in the binaphthol units gradually decreased the CPL intensity, with g values of +0.031 and +0.013 for E11b and E11c, respectively.161 Notably, due to greater molecular flexibility in E11c, it showed sign inversion at elevated temperatures, exhibiting a g factor of −0.0060 at 90 °C. While the structurally similar E12 showed a comparable CPL response to that of E11c, the 1,6-linked pyrenophane E13 also exhibited sign inversion in CPL, attributed to the altered relative pyrene orientation, with a considerably intense g factor of −0.022 for the S,S enantiomer.160
Another interesting chiral scaffold involves (phenylene)ethynylenes, as discussed in Section 4.3. Accordingly, pyrene-containing o-(phenylene)ethynylenes (E14a) exhibited intense bisignate chiral emission, with g values of +0.03 (at 540 nm) and −0.007 (at 480 nm), respectively, assigned to different excimer conformations, while the o-(phenylene)ethynylenes unit exhibited no CPL emission.163 Notably, the related pyrenylmethyl ether (E14b) behaved quite differently, showing relatively small excimer CPL with a g factor of +0.005 at 515 nm, along with relatively strong CPL with a g factor of −0.012 at a transition of the o-(phenylene)ethynylenes moiety at 390 nm, comparable to those of non-pyrene o-(phenylene)ethynylenes P7 and P8. This molecule was successfully demonstrated as a ratiometric prove for silver(I) ion. The 16-crown-4 scaffold was also employed for efficient chiral emission of the excimer in E15, although other crown ethers exhibited inferior g factors.164 A chiroptical photoswitching effect was successfully demonstrated in a helical tetrathiazole containing two pyrene units.165 In the open form (E16), a strong CPL emission from the excimer was observed, with a g factor of −0.01. However, upon switching to the closed form, the emission essentially became monomeric and thus CPL-silent. A structurally unique [5]helicene-based figure-eight macrocycle (E17) exhibited very intense excimer CPL with a g factor value of −0.038 for the M,M enantiomer.166 A corresponding anthracene derivative also showed excimer CPL, albeit with a slightly smaller g factor of +0.0076 at 540 nm. In the same report, the chiroptical properties of twisted Möbius macrocycles containing pyrene and other chromophores with different symmetries were thoroughly investigated; however, the g factor values regrettably small, ranged from an order of 0.0001 to 0.001. A p-(phenylene)ethynylenes macrocycle containing two anthracene units (E18) exhibited a strong CPL response with a g factor of +0.020 for the S,S isomer,167 the value being slightly dependent on solvent and concentration. In contrast, the relevant acyclic analogue with only one binaphthyl unit exhibited a much smaller CPL, with a g factor on the order of merely 0.0001.
Multiply substituted chromophores on binaphthalene and quaternaphthalene cores have been found to serve as efficient excimer production scaffolds.168,169 Notably, tetrapyrenyl binaphthalene (E19) exhibited strong negative CPL for the excimer emission, with a g factor value of −0.013 for the S isomer. Interestingly, hexapyrenyl (E20a) and octapyrenyl (E21) quaternaphthalenes demonstrated enhanced CPL, with g factors of −0.037 and −0.034, respectively. It is noteworthy that 1-pyrenyl substitution was found to be more effective in generating chiral emission through a more twisted pyrene conformation in the excimer, compared to symmetrical 2-pyrenyl substitution. Indeed, the latter analogue of E20a exhibited much weaker CPL, even resulting in a sign inversion. In contrast, 4-pyrenyl substitution in E20b displayed a favourable g factor of −0.014, along with an enhanced fluorescence quantum yield. Meanwhile, 3-perylenyl substitution in E20c yielded an oppositely signed CPL with a g factor of +0.013. It is important to note that while excimer formation in binaphthalenes E9 (and E11–E13) is facilitated between pyrenes connected to different axially chiral naphthalene rings, the formation occurs between adjacent chromophores on the same naphthalene in the derivatives mentioned above (E19–E21). The twisting between the chromophores is restricted to a counterclockwise direction (except for E20c) due to the steric requirements from additional chromophores on the adjacent naphthalenes.
Finally, we successfully demonstrated that the chiral sugar in α-cyclodextrin serves as an effective platform for chiral excimer emission.170 In this study, hexapyrenyl cyclodextrin derivative (E22) was prepared to exhibit strong CPL with a g factor of +0.012 and a fluorescence quantum yield of 0.39. It was observed that the removal of one pyrene moiety slightly decreased the g factor value to +0.0090. Additionally, increasing the connecting chain length, and thus the flexibility between the chromophores, considerably reduced the g factor values. Consequently, it is postulated that twisted excimer formation is facilitated between neighbouring pyrenes, as effective spatial restriction is provided by the cumulative interactions with surrounding pyrene rings. A related γ-cyclodextrin derivative with two separated pyrene units (i.e., AC and AD types) exhibited excimer emission in solution but showed pronounced excimer CPL only upon aggregation.171
 | ||
| Chart 9 | ||
| Molecule | λ max | Φ CPL | g CPL (λmon) | Conditions | Ref. |
|---|---|---|---|---|---|
| a In water containing a small amount of ammonia (pH ≈ 9.5). b In 0.1 M NaCl and NaH2PO4 (pH = 7). | |||||
| [(S,S)-S1]2 | 538 | — | +0.016 (—) | Toluene, 4.0 mM, 0 °C | 172 |
| (Pyrene)2⊂[γ-CD]2 | ≈470 | — | +0.012 (—) | Water, 2 mM | 173 |
| [S2]2⊂[γ-CD]2 | ≈530 | 0.37 | −0.015 (480) | Watera, 45 µM, 25 °C | 174 |
| {S3a⊂[CB6]2}2⊂[γ-CD]2 | 448 | 0.47 | −0.011 (—) | Watera, 10 µM, 25 °C | 175 |
| {S3b⊂[CB6]2}2⊂[γ-CD]2 | 452 | 0.45 | −0.019 (—) | Watera, 10 µM, 25 °C | 175 |
| {S3c⊂[CB6]2}2⊂[γ-CD]2 | 454 | 0.40 | −0.015 (—) | Watera, 10 µM, 25 °C | 175 |
| [S4]2⊂[γ-CD]2 | ≈570 | 0.15 | −0.021 (573) | Watera, 45 µM, 25 °C | 176 |
| (Smt)-[S5]2 | 480 | 0.05 | −0.012 (—) | CH2Cl2, 10 µM | 177 |
| [(Sp)-S6]2 | 463 | 0.038 | +0.010 (463) | CH2Cl2, 10 µM, 25 °C | 178 |
| TO2⊂(S,S,S,S)-S7 | ≈610 | — | −0.011 (—) | PBS buffer, 0 µM | 179 |
| [ATP⊂S8]2 | 550 | 0.442 | +0.01 (550) | Water, 25 µM | 180 |
| RNA duplex | 490 | 0.05 ~ 0.09 | +0.020 ~ +0.035 | Bufferb, 2.4 µM, r.t. | 181 |
Controlling the relative chromophore geometry in intermolecular excimers is indeed challenging. Therefore, supramolecular interactions have been effectively employed to organize these geometries, enhancing CPL responses. In a classical study by Kano and colleagues, pristine pyrene was dissolved in a 10 mM aqueous γ-cyclodextrin solution, forming a 2:2 complex. Although details such as the degree of complexation were not fully characterized, this solution exhibited intense CPL with a g factor value of +0.012 at around 460 nm.173 A similar 2:2 complex of N-(pyren-1-yl)adamantane-1-carboxamide with γ-cyclodextrin was also studied, yielding slightly weaker CPL with a g factor of −0.0078 at a peak of 495 nm,182 with the sign opposite to that of the above pyrene-cyclodextrin complex. Recently, alkynylpyrenes were threaded into the γ-cyclodextrin cavity to form a discrete [4]rotaxane ([S2]2⊂[γ-CD]2). This rotaxane showed strong excimer emission with a fluorescence quantum yield of 0.37, along with intense negative CPL with a g factor of −0.015.174 The sign of the CPL was consistent with the corresponding CD band at the longest wavelength, suggesting that the chiral twisting of the complex is preserved within the cyclodextrin cavity in the excited state. Recently, related [8]rotaxanes consisting of a pair of dialkynylpyerene threaders of varying length (S3a–c), capped with two cucurbit[6]uril [CB6] units and two γ-cyclodextrin molecules, were developed, all exhibiting excellent CPL responses.175 An analogous [4]rotaxane featuring perylene chromophores ([S4]2⊂[γ-CD]2) was also reported, affording a slightly improved g factor value of −0.021.176
A topologically chiral [2]catenane was also utilized as a platform for efficient excimer CPL and switching functions. Thus, the pyrene-functionalized [2]catenane, S5 dimer, exhibited strong CPL with a g factor of −0.012 for the Smt topology.177 Upon protonation with trifluoroacetic acid, the fluorescence quantum yield increased to 0.17, along with a 1.8-fold amplification of the CPL response, accompanied by slight bathochromic shifts in emission. In contrast, the addition of sodium ions induced the open form, resulting in a loss of excimer emission and CPL. A pyrene-functionalized chiral molecular muscle, the [c2]daisy chain ([S6]2), was also reported to exhibit strong CPL with a g factor of +0.010.178 This interlocked molecule demonstrated multiple switching behaviors upon the addition of anions and solvents, leading to reduced CPL for the former and sign-inverted, 1.5-fold enhanced CPL for the latter.
Recently, a 1:2 complex formed between the chiral corral[4] binol macrocycle (S7) and thiazole orange (TO) was shown to exhibit strong CPL in the relatively long-wavelength region with a g factor of −0.011, while excimer emission was not explicitly confirmed.179 As a supplementary example, a tetracationic achiral nanotube S8 with two anthracene units underwent 1:1 complexation with adenosine 5′-triphosphate in water, adopting an M-twisted conformation. This complex further engaged in second-level supramolecular interactions, leading to strong excimer CPL with a positive g factor of +0.010 at 550 nm, resulting from P-twisting dimerization between the anthracene units of each complex.180 Lastly, three RNA duplexes consisting of complementary 21-mer RNA strands with five 2′-O-pyrene modified nucleosides were reported to exhibit CPL excimer emission with g factors up to +0.035.181
5. Summary and perspectives
The field of small-molecule helical emitters has successfully transitioned from exploratory synthesis to the era of rational design. Recent breakthroughs in achieving high luminescence dissymmetry, or gCPL, factor values through engineering molecular symmetry, transition moment alignment, and exciton coupling validate our fundamental understanding. The most successful helical emitters reported to date—spanning diverse molecular architectures—achieve luminescence dissymmetry factors |gCPL| exceeding 0.04, corresponding to an enantiomeric emission ratio greater than 51:49. These high-performance systems include cylindrical macrocycles (C1–C3, and C11–C13), phenylene(ethynylene) oligomer (P12), helicene and helical nanographenes (H15, N2a, N4, and N8a), and excimer-based systems (E8 and E13a). While these successes provide a robust foundation, the next phase of innovation will hinge on overcoming critical challenges to translate these molecular triumphs into functional technologies. The foremost challenge in circularly polarized luminescence (CPL) molecular design is to resolve the persistent trade-off between high dissymmetry and high photoluminescence quantum yield. Future molecular approaches must optimize both parameters simultaneously. Integrating principles from highly efficient emitters, such as designing chiral frameworks that support thermally activated delayed fluorescence (TADF) or phosphorescence, represents a crucial and promising direction for achieving this synergy. Emitters capable of harvesting triplet excitons while maintaining a rigid helical structure will be key to developing materials suitable for high-performance circularly polarized organic light-emitting diodes.183
Beyond the single molecule, controlling chiroptical properties in the solid state is also paramount. The elegant properties designed into a molecule can be negated by aggregation-caused quenching or unpredictable packing in films or crystals. Therefore, a greater focus on supramolecular and materials chemistry is essential. Strategies that utilize chiral liquid crystal matrices, engineer CPL-active polymers, or develop self-assembling systems will be vital for amplifying chirality from the molecular to the macroscopic scale. It is crucial to ensure that the exceptional properties of individual emitters are preserved and enhanced in the bulk material. As these fundamental hurdles are overcome, the application landscape for CPL materials will expand significantly beyond displays and anti-counterfeiting. The unique sensitivity of CPL to the chiral environment positions it as a powerful tool for high-contrast bioimaging and real-time sensing of biological processes. Furthermore, the intrinsic link between molecular chirality and electron spin opens exciting possibilities at the nexus of CPL and spintronics, enabling novel opto-spintronic devices. To accelerate progress across these fronts, integrating computational chemistry with machine learning will be indispensable, facilitating the inverse design of molecules with tailored properties and rapidly identifying the most promising candidates for synthesis.184–186
In essence, while foundational design rules for high-g molecular emitters are now established to a certain extent, the future lies in holistic, multi-parameter optimization and the hierarchical control of chirality across scales. By bridging molecular design with materials engineering and advanced applications, the field is poised to deliver on the promise of CPL, harnessing the chirality of light to drive next-generation technologies. We hope this summary on helical emitters provides a solid foundation for a more detailed and sophisticated discussion aimed at elucidating the underlying factors contributing to the enhancement of the emission dissymmetry factor and the further advancement of CPL materials design.
Author contributions
Tadashi Mori fulfils all applicable contributor roles defined by CRediT taxonomy for this manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results have been included in this article, and no new datasets were generated or analysed as part of this review.Acknowledgements
Financial support from Grant-in-Aids for Scientific Research, JSPS (Grant Number JP25K01752), and from CREST, JST, Japan (Grant Number JPMJCR2001) is greatly acknowledged.Notes and references
- L. Pauling and R. B. Corey, Proc. Natl. Acad. Sci. U. S. A., 1951, 37, 729–740, DOI:10.1073/pnas.37.11.729.
- J. D. Watson and F. H. C. Crick, Nature, 1953, 171, 737–738, DOI:10.1038/171737a0.
- E. Yashima, K. Maeda, H. Iida, Y. Furusho and K. Nagai, Chem. Rev., 2009, 109, 6102–6211, DOI:10.1021/cr900162q.
- H. K. Bisoyi and Q. Li, Chem. Rev., 2022, 122, 4887–4926, DOI:10.1021/acs.chemrev.1c00761.
- J. R. Brandt, F. Salerno and M. J. Fuchter, Nat. Rev. Chem., 2017, 1, 0045, DOI:10.1038/s41570-017-0045.
- J. P. Riehl and F. S. Richardson, Chem. Rev., 1986, 86, 1–16, DOI:10.1021/cr00071a001.
- F. Furlan, J. M. Moreno-Naranjo, N. Gasparini, S. Feldmann, J. Wade and M. J. Fuchter, Nat. Photonics, 2024, 18, 658–668, DOI:10.1038/s41566-024-01408-z.
- J. Crassous, M. J. Fuchter, D. E. Freedman, N. A. Kotov, J. Moon, M. C. Beard and S. Feldmann, Nat. Rev. Mater., 2023, 8, 365–371, DOI:10.1038/s41578-023-00543-3.
- B. H. Jhun, S. Y. Park and Y. You, Dyes Pigm., 2023, 208, 110786, DOI:10.1016/j.dyepig.2022.110786.
- D.-W. Zhang, M. Li and C.-F. Chen, Chem. Soc. Rev., 2020, 49, 1331–1343, 10.1039/C9CS00680J.
- J. Han, S. Guo, H. Lu, S. Liu, Q. Zhao and W. Huang, Adv. Opt. Mater., 2018, 6, 1800538, DOI:10.1002/adom.201800538.
- Y. Wu, M. Li, Z.-G. Zheng, Z.-Q. Yu and W.-H. Zhu, J. Am. Chem. Soc., 2023, 145, 12951–12966, DOI:10.1021/jacs.3c01122.
- J. Ahn, S. H. Lee, I. Song, P. Chidchob, Y. Kwon and J. H. Oh, Device, 2023, 1, 100176, DOI:10.1016/j.device.2023.100176.
- Y. Nagata and T. Mori, Front. Chem., 2020, 8, 448, DOI:10.3389/fchem.2020.00448.
- H. Lu, L. Di Bari and L. Favereau, Nat. Photonics, 2025, 19, 1041–1047, DOI:10.1038/s41566-025-01729-7.
- E. Borie, Metrologia, 1986, 22, 140–145, DOI:10.1088/0026-1394/22/3/004.
- Q. Xu, J. Fu, M. Tang, H. Yao and J. Lin, Adv. Opt. Mater., 2025, 13, e01569, DOI:10.1002/adom.202501569.
- E. M. Sánchez-Carnerero, A. R. Agarrabeitia, F. Moreno, B. L. Maroto, G. Muller, M. J. Ortiz and S. de la Moya, Chem. – Eur. J., 2015, 21, 13488–13500, DOI:10.1002/chem.201501178.
- J. Kumar, T. Nakashima and T. Kawai, J. Phys. Chem. Lett., 2015, 6, 3445–3452, DOI:10.1021/acs.jpclett.5b01452.
- J.-L. Ma, Q. Peng and C.-H. Zhao, Chem. – Eur. J., 2019, 25, 15441–15454, DOI:10.1002/chem.201903252.
- Y. Zhang, S. Yu, B. Han, Y. Zhou, X. Zhang, X. Gao and Z. Tang, Matter, 2022, 5, 837–875, DOI:10.1016/j.matt.2022.01.001.
- T. Zhang, Y. Zhang, Z. He, T. Yang, X. Hu, T. Zhu, Y. Zhang, Y. Tang and J. Jiao, Chem. – Asian J., 2024, 19, e202400049, DOI:10.1002/asia.202400049.
- W. R. Kitzmann, J. Freudenthal, A.-P. M. Reponen, Z. A. VanOrman and S. Feldmann, Adv. Mater., 2023, 35, 2302279, DOI:10.1002/adma.202302279.
- C. Zhang, S. Li, X.-Y. Dong and S.-Q. Zang, Aggregate, 2021, 2, e48, DOI:10.1002/agt2.48.
- S. S. Andrews and J. Tretton, J. Chem. Educ., 2020, 97, 4370–4376, DOI:10.1021/acs.jchemed.0c01061.
- M. Caricato, J. Phys. Chem. A, 2024, 128, 1197–1206, DOI:10.1021/acs.jpca.3c08095.
- L. Rosenfeld, Z. Phys., 1929, 52, 161–174, DOI:10.1007/BF01342393.
- Z. Chen, M. Huang, C. Zhong, S. Gong, V. Coropceanu, J.-L. Brédas and C. Yang, Chem. Sci., 2023, 14, 6022–6031, 10.1039/D3SC01809A.
- G. Sini, Q. Sun, E. Cho, J.-L. Brédas and V. Coropceanu, J. Phys. Chem. Lett., 2025, 16, 3715–3720, DOI:10.1021/acs.jpclett.5c00690.
- D. Parker, J. D. Fradgley and K.-L. Wong, Chem. Soc. Rev., 2021, 50, 8193–8213, 10.1039/D1CS00310K.
- B. Doistau, J.-R. Jiménez and C. Piguet, Front. Chem., 2020, 8, 555, DOI:10.3389/fchem.2020.00555.
- R. G. Uceda, C. M. Cruz, S. Míguez-Lago, L. Á. de Cienfuegos, G. Longhi, D. A. Pelta, P. Novoa, A. J. Mota, J. M. Cuerva and D. Miguel, Angew. Chem., Int. Ed., 2024, 63, e202316696, DOI:10.1002/anie.202316696.
- C. A. Guido, F. Zinna and G. Pescitelli, Chem. Rev., 2025, 125, 10492–10656, DOI:10.1021/acs.chemrev.5c00359.
- G. Longhi, E. Castiglioni, J. Koshoubu, G. Mazzeo and S. Abbate, Chirality, 2016, 28, 696–707, DOI:10.1002/chir.22647.
- I. Warnke and F. Furche, Wiley Interdiscip. Rev.:Comput. Mol. Sci., 2012, 2, 150–166, DOI:10.1002/wcms.55.
- J. Autschbach, Chirality, 2009, 21, E116–E152, DOI:10.1002/chir.20789.
- J. L. Greenfield, J. Wade, J. R. Brandt, X. Shi, T. J. Penfold and M. J. Fuchter, Chem. Sci., 2021, 12, 8589–8602, 10.1039/D1SC02335G.
- W.-L. Zhao, M. Li, H.-Y. Lu and C.-F. Chen, Chem. Commun., 2019, 55, 13793–13803, 10.1039/C9CC06861A.
- T. Mori, Chem. Rev., 2021, 121, 2373–2412, DOI:10.1021/acs.chemrev.0c01017.
- I. Gómez-Oya, J. Portela-Pino, L. M. Salonen, Á. Peña-Gallego and J. L. Alonso-Gómez, Chem. – Eur. J., 2025, 31, e202501266, DOI:10.1002/chem.202501266.
- G. Pescitelli, Chirality, 2022, 34, 333–363, DOI:10.1002/chir.23393.
- S. Castro-Fernández, Á. Peña-Gallego, R. A. Mosquera and J. L. Alonso-Gómez, Molecules, 2019, 24, 141, DOI:10.3390/molecules24010141.
- M. Li, S. Nizar, S. Saha, A. Thomas, S. Azzini, T. W. Ebbesen and C. Genet, Angew. Chem., Int. Ed., 2023, 62, e202212724, DOI:10.1002/anie.202212724.
- G. Pescitelli, L. Di Bari and N. Berova, Chem. Soc. Rev., 2014, 43, 5211–5233, 10.1039/C4CS00104D.
- T. Mori, Angew. Chem., Int. Ed., 2024, 63, e202319702, DOI:10.1002/anie.202319702.
- Y. Liu, J. Cerezo, G. Mazzeo, N. Lin, X. Zhao, G. Longhi, S. Abbate and F. Santoro, J. Chem. Theory Comput., 2016, 12, 2799–2819, DOI:10.1021/acs.jctc.6b00109.
- S. H. Lin and H. Eyring, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 3802–3804, DOI:10.1073/pnas.71.10.3802.
- C. A. Emeis and L. J. Oosterhoff, Chem. Phys. Lett., 1967, 1, 129–132, DOI:10.1016/0009-2614(67)85007-3.
- C. A. Emeis and L. J. Oosterhoff, J. Chem. Phys., 1971, 54, 4809–4819, DOI:10.1063/1.1674756.
- B. Pritchard and J. Autschbach, ChemPhysChem, 2010, 11, 2409–2415, DOI:10.1002/cphc.201000054.
- P. H. Schippers, J. P. M. van der Ploeg and H. P. J. M. Dekkers, J. Am. Chem. Soc., 1983, 105, 84–89, DOI:10.1021/ja00339a015.
- Z. Sun, H. Tang, L. Wang and D. Cao, Chem. – Eur. J., 2025, 31, e202404217, DOI:10.1002/chem.202404217.
- Y. Xue, Y. Shi and P. Chen, Adv. Opt. Mater., 2024, 12, 2303322, DOI:10.1002/adom.202303322.
- M. Hasegawa and Y. Mazaki, Synlett, 2024, 1361–1374, DOI:10.1055/a-2158-8820.
- M. Hasegawa, Y. Nojima and Y. Mazaki, ChemPhotoChem, 2021, 5, 1042–1058, DOI:10.1002/cptc.202100162.
- S. Sato, A. Yoshii, S. Takahashi, S. Furumi, M. Takeuchi and H. Isobe, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 13097–13101, DOI:10.1073/pnas.1717524114 ; Correction: S. Sato, A. Yoshii, S. Takahashi, S. Furumi, M. Takeuchi and H. Isobe, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 5194–5195, DOI:10.1073/pnas.1902266116.
- J. Wang, G. Zhuang, M. Chen, D. Lu, Z. Li, Q. Huang, H. Jia, S. Cui, X. Shao, S. Yang and P. Du, Angew. Chem., Int. Ed., 2020, 59, 1619–1626, DOI:10.1002/anie.201909401.
- Y. Xu, F. Steudel, M.-Y. Leung, B. Xia, M. von Delius and V. W. W. Yam, Angew. Chem., Int. Ed., 2023, 62, e202302978, DOI:10.1002/anie.202302978.
- G. Li, L.-L. Mao, J.-N. Gao, X. Shi, Z.-Y. Huo, J. Yang, W. Zhou, H. Li, H.-B. Yang, C.-H. Tung, L.-Z. Wu and H. Cong, Angew. Chem., Int. Ed., 2025, 64, e202419435, DOI:10.1002/anie.202419435.
- J.-H. Chen, Z.-Y. Jiang, H. Xiao, S. Tong, T.-H. Shi, J. Zhu and M.-X. Wang, Angew. Chem., Int. Ed., 2023, 62, e202301782, DOI:10.1002/anie.202301782.
- Y. Peng, S. Tong, Y. Zhang and M.-X. Wang, Angew. Chem., Int. Ed., 2023, 62, e202302646, DOI:10.1002/anie.202302646.
- T. An, J. Yao, Z. Xiao, Q. Yu, Y. Wang, Y. Gao, Y. Song, Z. Huang, Z. Ding, X. Zhang, Y. Xie, M. Lv, C. Zuo, J. Ding and L. Ding, Chem. Sci., 2025, 16, 21404–21415, 10.1039/D5SC06445G.
- G. Sini, Q. Sun, E. Cho, J.-L. Brédas and V. Coropceanu, J. Phys. Chem. Lett., 2025, 16, 3715–3720, DOI:10.1021/acs.jpclett.5c00690.
- R. Kurosaki, M. Suzuki, H. Hayashi, M. Fujiki, N. Aratani and H. Yamada, Chem. Commun., 2019, 55, 9618–9621, 10.1039/C9CC03123E.
- M.-Y. Zhang, X. Liang, D.-N. Ni, D.-H. Liu, Q. Peng and C.-H. Zhao, Org. Lett., 2021, 23, 2–7, DOI:10.1021/acs.orglett.0c02463.
- M.-Y. Zhang, C.-L. Xin, S.-F. Hou and M.-J. Song, Org. Lett., 2025, 27, 9235–9240, DOI:10.1021/acs.orglett.5c02800.
- C.-H. Chen and W.-H. Zheng, Org. Chem. Front., 2021, 8, 6622–6627, 10.1039/D1QO01202A.
- A. Yanagawa, R. Inoue and Y. Morisaki, Chem. Commun., 2024, 60, 1468–1471, 10.1039/D3CC05000A.
- Y. Morisaki, M. Gon, T. Sasamori, N. Tokitoh and Y. Chujo, J. Am. Chem. Soc., 2014, 136, 3350–3353, DOI:10.1021/ja412197j.
- M. Gon, Y. Morisaki and Y. Chujo, J. Mater. Chem. C, 2015, 3, 521–529, 10.1039/C4TC02339K.
- T. Honda, D. Ogata, M. Tsurui, S. Yoshida, S. Sato, T. Muraoka, Y. Kitagawa, Y. Hasegawa, J. Yuasa and H. Oguri, Angew. Chem., Int. Ed., 2024, 63, e202318548, DOI:10.1002/anie.202318548.
- S. P. Morcillo, D. Miguel, L. Álvarez de Cienfuegos, J. Justicia, S. Abbate, E. Castiglioni, C. Bour, M. Ribagorda, D. J. Cárdenas, J. M. Paredes, L. Crovetto, D. Choquesillo-Lazarte, A. J. Mota, M. C. Carreño, G. Longhi and J. M. Cuerva, Chem. Sci., 2016, 7, 5663–5670, 10.1039/C6SC01808D.
- P. Reiné, J. Justicia, S. P. Morcillo, G. Mazzeo, E. García-Fernández, A. Rodríguez-Diéguez, L. Álvarez de Cienfuegos, S. Abbate, J. M. Cuerva, G. Longhi and D. Miguel, Chirality, 2018, 30, 43–54, DOI:10.1002/chir.22774.
- A. M. Ortuño, P. Reiné, S. Resa, L. Álvarez de Cienfuegos, V. Blanco, J. M. Paredes, A. J. Mota, G. Mazzeo, S. Abbate, J. M. Ugalde, V. Mujica, G. Longhi, D. Miguel and J. M. Cuerva, Org. Chem. Front., 2021, 8, 5071–5086, 10.1039/D1QO00822F.
- D. Otero, Á. Martínez-Pinel, A. M. Ortuño, L. Álvarez de Cienfuegos, J. M. Cuerva, G. Longhi, A. Millán and D. Miguel, Org. Lett., 2025, 27, 8459–8463, DOI:10.1021/acs.orglett.5c02277.
- P. Reine, A. G. Campaña, L. Alvarez de Cienfuegos, V. Blanco, S. Abbate, A. J. Mota, G. Longhi, D. Miguel and J. M. Cuerva, Chem. Commun., 2019, 55, 10685–10688, 10.1039/C9CC04885E.
- Y. Liu, Z. Ma, Z. Wang and W. Jiang, J. Am. Chem. Soc., 2022, 144, 11397–11404, DOI:10.1021/jacs.2c04012.
- K. Chen, Y. Liu, Z. Wang, S. Hu, Y. Zhao, W. Wang, G. Liu, Z. Wang and W. Jiang, J. Am. Chem. Soc., 2024, 146, 13499–13508, DOI:10.1021/jacs.4c02914.
- S. E. Penty, M. A. Zwijnenburg, G. R. F. Orton, P. Stachelek, R. Pal, Y. Xie, S. L. Griffin and T. A. Barendt, J. Am. Chem. Soc., 2022, 144, 12290–12298, DOI:10.1021/jacs.2c03531.
- Y. Nojima, M. Hasegawa, N. Hara, Y. Imai and Y. Mazaki, Chem. – Eur. J., 2021, 27, 5923–5929, DOI:10.1002/chem.202005320.
- Y. Shen and C.-F. Chen, Chem. Rev., 2012, 112, 1463–1535, DOI:10.1021/cr200087r.
- M. Gingras, Chem. Soc. Rev., 2013, 42, 968–1006, 10.1039/c2cs35154d.
- M. Gingras, G. Felix and R. Peresutti, Chem. Soc. Rev., 2013, 42, 1007–1050, 10.1039/c2cs35111k.
- M. Gingras, Chem. Soc. Rev., 2013, 42, 1051–1095, 10.1039/c2cs35134j.
- N. Hoffmann, J. Photochem. Photobiol., C, 2014, 19, 1–19, DOI:10.1016/j.jphotochemrev.2013.11.001.
- Y. Nakai, T. Mori and Y. Inoue, J. Phys. Chem. A, 2012, 116, 7372–7385, DOI:10.1021/jp304576g.
- M. Vanzan, S. Bertuletti, G. Becatti, B. Bazan, M. T. Rispens, S. I. C. Wan, M. Leeman, W. L. Noorduin and F. Baletto, J. Phys. Chem. A, 2025, 129, 9537–9547, DOI:10.1021/acs.jpca.5c04360.
- F. Zhao, J. Zhao, Y. Wang, H.-T. Liu, Q. Shang, N. Wang, X. Yin, X. Zheng and P. Chen, Dalton Trans., 2022, 51, 6226–6234, 10.1039/d2dt00677d.
- K. Dhbaibi, L. Abella, S. Meunier-Della-Gatta, T. Roisnel, N. Vanthuyne, B. Jamoussi, G. Pieters, B. Racine, E. Quesnel, J. Autschbach, J. Crassous and L. Favereau, Chem. Sci., 2021, 12, 5522–5533, 10.1039/d0sc06895k.
- K. Wu, J. Tessarolo, A. Baksi and G. H. Clever, Angew. Chem., Int. Ed., 2022, 61, e202205725, DOI:10.1002/anie.202205725.
- C. Schaack, L. Arrico, E. Sidler, M. Górecki, L. Di Bari and F. Diederich, Chem. – Eur. J., 2019, 25, 8003–8007, DOI:10.1002/chem.201901248.
- J. Malinčík, S. Gaikwad, J. P. Mora-Fuentes, M.-A. Boillat, A. Prescimone, D. Häussinger, A. G. Campaña and T. Šolomek, Angew. Chem., Int. Ed., 2022, 61, e202208591, DOI:10.1002/anie.202208591.
- D. Gobel, S. Miguez-Lago, M. J. Ruedas-Rama, A. Orte, A. G. Campana and M. Juricek, Helv. Chim. Acta, 2022, 105, e202100221, DOI:10.1002/hlca.202100221.
- S. Cadeddu, R. Chowdhury, C. Delpiano Cordeiro, S. Parmar, A. Kramer, M. Cordier, A. Pensel, N. Vanthuyne, R. Sessoli, M. Chiesa, Y.-K. Liao, R. H. Friend, E. Salvadori, J. Autschbach and J. Crassous, J. Am. Chem. Soc., 2025, 147, 23643–23653, DOI:10.1021/jacs.5c04887.
- H. Kubo, T. Hirose, T. Nakashima, T. Kawai, J.-Y. Hasegawa and K. Matsuda, J. Phys. Chem. Lett., 2021, 12, 686–695, DOI:10.1021/acs.jpclett.0c03174 ; Correction: H. Kubo, T. Hirose, T. Nakashima, T. Kawai, J.-Y. Hasegawa and K. Matsuda, J. Phys. Chem. Lett., 2021, 12, 1124, DOI:10.1021/acs.jpclett.1c00143.
- C. Shen, G. Zhang, Y. Ding, N. Yang, F. Gan, J. Crassous and H. Qiu, Nat. Commun., 2021, 12, 2786, DOI:10.1038/s41467-021-22992-6.
- K. Fujise, E. Tsurumaki, G. Fukuhara, N. Hara, Y. Imai and S. Toyota, Chem. – Asian J., 2020, 15, 2456–2461, DOI:10.1002/asia.202000394.
- K. Suzuki, H. Fukuda, H. Toda, Y. Imai, Y. Nojima, M. Hasegawa, E. Tsurumaki and S. Toyota, Tetrahedron, 2023, 132, 133243, DOI:10.1016/j.tet.2022.133243.
- H. Fukuda, M. Kobayashi, E. Tsurumaki, M. Yamashina, M. Hasegawa, K. Wakamatsu and S. Toyota, Chem. – Eur. J., 2025, 31, e202404348, DOI:10.1002/chem.202404348.
- G.-F. Huo, W.-T. Xu, Y. Han, J. Zhu, X. Hou, W. Fan, Y. Ni, S. Wu, H.-B. Yang and J. Wu, Angew. Chem., Int. Ed., 2024, 63, e202403149, DOI:10.1002/anie.202403149.
- Y. Appiarius, S. Míguez-Lago, P. Puylaert, N. Wolf, S. Kumar, M. Molkenthin, D. Miguel, T. Neudecker, M. Juríček, A. G. Campaña and A. Staubitz, Chem. Sci., 2024, 15, 466–476, 10.1039/D3SC02685J.
- T. Otani, T. Sasayama, C. Iwashimizu, K. S. Kanyiva, H. Kawai and T. Shibata, Chem. Commun., 2020, 56, 4484–4487, 10.1039/d0cc01194k.
- B. C. Baciu, P. J. Bronk, T. de Ara, R. Rodriguez, P. Morgante, N. Vanthuyne, C. Sabater, C. Untiedt, J. Autschbach, J. Crassous and A. Guijarro, J. Mater. Chem. C, 2022, 10, 14306–14318, 10.1039/D2TC02910C.
- Z. Zhang, Y. Murata and T. Hirose, Tetrahedron, 2023, 142, 133514, DOI:10.1016/j.tet.2023.133514.
- Y. Sawada, S. Furumi, A. Takai, M. Takeuchi, K. Noguchi and K. Tanaka, J. Am. Chem. Soc., 2012, 134, 4080–4083, DOI:10.1021/ja300278e.
- K. Murayama, Y. Oike, S. Furumi, M. Takeuchi, K. Noguchi and K. Tanaka, Eur. J. Org. Chem., 2015, 1409–1414, DOI:10.1002/ejoc.201403565.
- T. Kaseyama, S. Furumi, X. Zhang, K. Tanaka and M. Takeuchi, Angew. Chem., Int. Ed., 2011, 50, 3684–3687, DOI:10.1002/anie.201007849.
- K. Mori, T. Murase and M. Fujita, Angew. Chem., Int. Ed., 2015, 54, 6847–6851, DOI:10.1002/anie.201502436.
- G. R. Kiel, H. M. Bergman, A. E. Samkian, N. J. Schuster, R. C. Handford, A. J. Rothenberger, R. Gomez-Bombarelli, C. Nuckolls and T. D. Tilley, J. Am. Chem. Soc., 2022, 144, 23421–23427, DOI:10.1021/jacs.2c09555.
- J. B. Birks, D. J. S. Birch, E. Cordemans and E. Vander Donckt, Chem. Phys. Lett., 1976, 43, 33–36, DOI:10.1016/0009-2614(76)80750-6.
- M. Sapir and E. V. Donckt, Chem. Phys. Lett., 1975, 36, 108–110, DOI:10.1016/0009-2614(75)85698-3.
- S. Toyota, Chem. – Eur. J., 2025, 31, e02193, DOI:10.1002/chem.202502193.
- Y. Nakakuki, T. Hirose and K. Matsuda, J. Am. Chem. Soc., 2018, 140, 15461–15469, DOI:10.1021/jacs.8b09825.
- M. Qiu, J. Du, N.-T. Yao, X.-Y. Wang and H.-Y. Gong, Beilstein J. Org. Chem., 2025, 21, 1422–1453, DOI:10.3762/bjoc.21.106.
- C. Maeda and T. Ema, Chem. Commun., 2025, 61, 4757–4773, 10.1039/d4cc06307d.
- K. Dhbaibi, L. Favereau and J. Crassous, Chem. Rev., 2019, 119, 8846–8953, DOI:10.1021/acs.chemrev.9b00033.
- C. A. Guido, F. Zinna and G. Pescitelli, J. Mater. Chem. C, 2023, 11, 10474–10482, 10.1039/D3TC01532G.
- H. Zhuo, X. Shen, W. Zhao, Z. Li, K. Gao, Z. Wang, W. Wu, J. Bai, G. Chang, Y. Wu, W. Ma, M. Zhang, G. Long, R. Li, V. Coropceanu, F. Gao and X. Shang, Matter, 2025, 8, 102371, DOI:10.1016/j.matt.2025.102371.
- D. Sakamaki, S. Tanaka, K. Tanaka, M. Takino, M. Gon, K. Tanaka, T. Hirose, D. Hirobe, H. M. Yamamoto and H. Fujiwara, J. Phys. Chem. Lett., 2021, 12, 9283–9292, DOI:10.1021/acs.jpclett.1c02896.
- S. Tanaka, D. Sakamaki, N. Haruta, T. Sato, M. Gon, K. Tanaka and H. Fujiwara, J. Mater. Chem. C, 2023, 11, 4846–4854, 10.1039/D3TC00871A.
- G. Zhu, T. Ding, H. Chen, J. Liang, W. Wang, W. Cai and G. Zhang, Org. Lett., 2025, 27, 6688–6694, DOI:10.1021/acs.orglett.5c01719.
- X. L. Chen, S. Q. Yu, Z. Y. Cheng, Z. Y. Zheng, A. N. Chen, J. Bai, J. Q. Liang, C. Zheng, X. Huang and H. Y. Gong, Org. Lett., 2025, 27, 3511–3516, DOI:10.1021/acs.orglett.5c00294.
- K. Nakamura, S. Furumi, M. Takeuchi, T. Shibuya and K. Tanaka, J. Am. Chem. Soc., 2014, 136, 5555–5558, DOI:10.1021/ja500841f.
- H. Kubo, D. Shimizu, T. Hirose and K. Matsuda, Org. Lett., 2020, 22, 9276–9281, DOI:10.1021/acs.orglett.0c03506.
- Q. Zhou, X. Hou, J. Wang, Y. Ni, W. Fan, Z. Li, X. Wei, K. Li, W. Yuan, Z. Xu, M. Zhu, Y. Zhao, Z. Sun and J. Wu, Angew. Chem., Int. Ed., 2023, 62, e202302266, DOI:10.1002/anie.202302266.
- C. Qu, Y. Xu, Y. Wang, Y. Nie, K. Ye, H. Zhang and Z. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202400661, DOI:10.1002/anie.202400661.
- R. G. Uceda, A. M. Ortuño, L. Álvarez de Cienfuegos, F. Movilla, A. J. Mota, D. Miguel and J. M. Cuerva, Org. Lett., 2025, 27, 7967–7972, DOI:10.1021/acs.orglett.5c02299.
- L. Ning, C.-L. Sun, X. Zhang, Y. Wang, Y. Song, J. Feng, F. Liu, Q. Gong, Y. Mai, Q. Zhang and Y. Huang, Angew. Chem., Int. Ed., 2025, 64, e202513070, DOI:10.1002/anie.202513070.
- X. Zhang, D. An, R. Zhang, Y. Liu and X. Lu, CCS Chem., 2025, 7 DOI:10.31635/ccschem.025.202506153 , Just Published.
- A. Robert, G. Naulet, H. Bock, N. Vanthuyne, M. Jean, M. Giorgi, Y. Carissan, C. Aroulanda, A. Scalabre, E. Pouget, F. Durola and Y. Coquerel, Chem. – Eur. J., 2019, 25, 14364–14369, DOI:10.1002/chem.201902637.
- R. J. Malone and W. A. Chalifoux, Acc. Chem. Res., 2025, 58, 2379–2393, DOI:10.1021/acs.accounts.5c00211.
- I. G. Stará and I. Starý, Acc. Chem. Res., 2020, 53, 144–158, DOI:10.1021/acs.accounts.9b00364.
- H. V. Anderson, N. D. Gois and W. A. Chalifoux, Org. Chem. Front., 2023, 10, 4167–4197, 10.1039/D3QO00517H.
- P. Izquierdo-García, J. M. Fernández-García, S. Medina Rivero, M. Šámal, J. Rybáček, L. Bednárová, S. Ramírez-Barroso, F. J. Ramírez, R. Rodríguez, J. Perles, D. García-Fresnadillo, J. Crassous, J. Casado, I. G. Stará and N. Martín, J. Am. Chem. Soc., 2023, 145, 11599–11610, DOI:10.1021/jacs.3c01088.
- F. Morita, Y. Kishida, Y. Sato, H. Sugiyama, M. Abekura, J. Nogami, N. Toriumi, Y. Nagashima, T. Kinoshita, G. Fukuhara, M. Uchiyama, H. Uekusa and K. Tanaka, Nat. Synth., 2024, 3, 774–786, DOI:10.1038/s44160-024-00527-3.
- Y.-J. Shen, N.-T. Yao, L.-N. Diao, Y. Yang, X.-L. Chen and H.-Y. Gong, Angew. Chem., Int. Ed., 2023, 62, e202300840, DOI:10.1002/anie.202300840.
- W. Niu, Y. Fu, Z.-L. Qiu, C. J. Schürmann, S. Obermann, F. Liu, A. A. Popov, H. Komber, J. Ma and X. Feng, J. Am. Chem. Soc., 2023, 145, 26824–26832, DOI:10.1021/jacs.3c09350.
- W. Niu, Y. Fu, Q. Deng, Z.-L. Qiu, F. Liu, A. A. Popov, H. Komber, J. Ma and X. Feng, Angew. Chem., Int. Ed., 2024, 63, e202319874, DOI:10.1002/anie.202319874.
- Y. Wu, Y. Liu, W. Jiang and Z. Wang, Sci. China Chem., 2023, 66, 2400–2407, DOI:10.1007/s11426-023-1686-4.
- Y. Yu, C. Wang, F.-F. Hung, C. Chen, D. Pan, C.-M. Che and J. Liu, J. Am. Chem. Soc., 2024, 146, 22600–22611, DOI:10.1021/jacs.4c06997.
- Y. Yu, C. Wang, F.-F. Hung, L. Jiang, C.-M. Che and J. Liu, Angew. Chem., Int. Ed., 2025, 64, e202501645, DOI:10.1002/anie.202501645.
- B. Stevens, Adv. Photochem., 1971, 8, 161–226, DOI:10.1002/9780470133385.
- J. B. Birks, Rep. Prog. Phys., 1975, 38, 903–974, DOI:10.1088/0034-4885/38/8/001.
- H. Tanaka, Y. Inoue and T. Mori, ChemPhotoChem, 2018, 2, 386–402, DOI:10.1002/cptc.201800015.
- M. Cei, L. Di Bari and F. Zinna, Chirality, 2023, 35, 192–210, DOI:10.1002/chir.23535.
- G. Albano, G. Pescitelli and L. Di Bari, Chem. Rev., 2020, 120, 10145–10243, DOI:10.1021/acs.chemrev.0c00195.
- X. Feng, X. Wang, C. Redshaw and B. Z. Tang, Chem. Soc. Rev., 2023, 52, 6715–6753, 10.1039/D3CS00251A.
- J. Zhao, S. Shi, X. Zhang, D. Liu, F. Song, Y. Cheng and F. Li, Coord. Chem. Rev., 2026, 548, 217173, DOI:10.1016/j.ccr.2025.217173.
- B. Manna and D. K. Palit, J. Phys. Chem. C, 2020, 124, 24470–24487, DOI:10.1021/acs.jpcc.0c07977.
- D. Chakraborty, H. Lischka and W. L. Hase, J. Phys. Chem. A, 2020, 124, 8907–8917 CrossRef CAS.
- T. Kawashima, T. Otsubo, Y. Sakata and S. Misumi, Tetrahedron Lett., 1978, 19, 5115–5118, DOI:10.1016/S0040-4039(01)85826-3.
- L.-T. Bao, R.-H. Zhang, X. Yuan, X. Wang, P. Wu, X.-Q. Wang, J. Chen, A. Zhu, H.-B. Yang and W. Wang, Angew. Chem., Int. Ed., 2025, 64, e202500472, DOI:10.1002/anie.202500472.
- K. Tani, R. Imafuku, K. Miyanaga, M. E. Masaki, H. Kato, K. Hori, K. Kubono, M. Taneda, T. Harada, K. Goto, F. Tani and T. Mori, J. Phys. Chem. A, 2020, 124, 2057–2063, DOI:10.1021/acs.jpca.0c00286.
- J. Zhu, S. Chen and C. He, J. Am. Chem. Soc., 2021, 143, 5301–5307, DOI:10.1021/jacs.1c01106.
- T. Nishikawa, N. Tajima, M. Kitamatsu, M. Fujiki and Y. Imai, Org. Biomol. Chem., 2015, 13, 11426–11431, 10.1039/C5OB01710F.
- M. J. Álvaro-Martins, C. Billiaux, P. Godard, R. Oda, G. Raffy and D. M. Bassani, Chem. Commun., 2023, 59, 7963–7966, 10.1039/D3CC01802D.
- Y. Sheng, J. Ma, S. Liu, Y. Wang, C. Zhu and Y. Cheng, Chem. – Eur. J., 2016, 22, 9519–9522, DOI:10.1002/chem.201600891.
- Q.-Y. Meng, X.-F. Hou, J.-Q. Zhao, J.-Q. Luo, W.-K. Chen, Z. Sun, J.-M. Cheng, X.-M. Chen and H.-L. Sun, Chin. J. Chem., 2025, 43, 2655–2660, DOI:10.1002/cjoc.70156.
- D. Deb, M. Parashar, S. Sarkar, S. Ghosh, S. Balasubramanian and S. J. George, Angew. Chem., Int. Ed., 2025, 64, e20385, DOI:10.1002/anie.202520385.
- K. Takaishi, S. Murakami, F. Yoshinami and T. Ema, Angew. Chem., Int. Ed., 2022, 61, e202204609, DOI:10.1002/anie.202204609.
- K. Takaishi, F. Yoshinami, Y. Sato and T. Ema, Chem. – Eur. J., 2024, 30, e202400866, DOI:10.1002/chem.202400866.
- Y.-B. Du, Q.-T. Lu, Y.-S. Cui, K.-W. Wu, Y. Wang, Y.-Z. Zhang, Z. Zhao, J.-L. Hou and Q. Cai, Angew. Chem., Int. Ed., 2025, 64, e202421060, DOI:10.1002/anie.202421060.
- P. Reiné, J. Justicia, S. P. Morcillo, S. Abbate, B. Vaz, M. Ribagorda, Á. Orte, L. Álvarez de Cienfuegos, G. Longhi, A. G. Campaña, D. Miguel and J. M. Cuerva, J. Org. Chem., 2018, 83, 4455–4463, DOI:10.1021/acs.joc.8b00162.
- A. Homberg, E. Brun, F. Zinna, S. Pascal, M. Górecki, L. Monnier, C. Besnard, G. Pescitelli, L. Di Bari and J. Lacour, Chem. Sci., 2018, 9, 7043–7052, 10.1039/C8SC02935K.
- Y. Hashimoto, T. Nakashima, D. Shimizu and T. Kawai, Chem. Commun., 2016, 52, 5171–5174, 10.1039/C6CC01277A.
- Q. Zhou, W. Yuan, Y. Li, Y. Han, L. Bao, W. Fan, L. Jiao, Y. Zhao, Y. Ni, Y. Zou, H.-B. Yang and J. Wu, Angew. Chem., Int. Ed., 2024, 63, e202417749, DOI:10.1002/anie.202417749.
- K. Miki, T. Noda, M. Gon, K. Tanaka, Y. Chujo, Y. Mizuhata, N. Tokitoh and K. Ohe, Chem. – Eur. J., 2019, 25, 9211–9216, DOI:10.1002/chem.201901467.
- K. Takaishi, R. Takehana and T. Ema, Chem. Commun., 2018, 54, 1449–1452, 10.1039/C7CC09187G.
- K. Takaishi, K. Iwachido, R. Takehana, M. Uchiyama and T. Ema, J. Am. Chem. Soc., 2019, 141, 6185–6190, DOI:10.1021/jacs.9b02582.
- H. Shigemitsu, K. Kawakami, Y. Nagata, R. Kajiwara, S. Yamada, T. Mori and T. Kida, Angew. Chem., Int. Ed., 2022, 61, e202114700, DOI:10.1002/anie.202114700.
- C. Tu, W. Wu, W. Liang, D. Zhang, W. Xu, S. Wan, W. Lu and C. Yang, Angew. Chem., Int. Ed., 2022, 61, e202203541, DOI:10.1002/anie202203541.
- K. Takaishi, K. Iwachido and T. Ema, J. Am. Chem. Soc., 2020, 142, 1774–1779, DOI:10.1021/jacs.9b13184.
- K. Kano, H. Matsumoto, S. Hashimoto, M. Sisido and Y. Imanishi, J. Am. Chem. Soc., 1985, 107, 6117–6118, DOI:10.1021/ja00307a055.
- M. Inouye, K. Hayashi, Y. Yonenaga, T. Itou, K. Fujimoto, T.-A. Uchida, M. Iwamura and K. Nozaki, Angew. Chem., Int. Ed., 2014, 53, 14392–14396, DOI:10.1002/anie.201408193.
- K. Nishioki, A. Kimura, Y. Ohishi, J. Yamashita, M. Kitamoto, M. Iwamura, K. Nozaki, J. Chiba and M. Inouye, Chem. Sci., 2025, 16, 20414–20421, 10.1039/D5SC06304C.
- K. Hayashi, Y. Miyaoka, Y. Ohishi, T.-A. Uchida, M. Iwamura, K. Nozaki and M. Inouye, Chem. – Eur. J., 2018, 24, 14613–14616, DOI:10.1002/chem.201803215.
- Y. Wang, J. Gong, X. Wang, W.-J. Li, X.-Q. Wang, X. He, W. Wang and H.-B. Yang, Angew. Chem., Int. Ed., 2022, 61, e202210542, DOI:10.1002/anie.202210542.
- X. Li, W.-T. Xu, X.-Q. Xu, Y. Wang, X.-Q. Wang, H.-B. Yang and W. Wang, Angew. Chem., Int. Ed., 2025, 64, e202412548, DOI:10.1002/anie.202412548.
- R. Fu, F.-Y. Chen, L. Dai, D.-Y. Li, Q.-Y. Zhao, S.-D. Guo, Z.-H. Song, L.-B. Jing, H. Wang, X. Li, D.-S. Guo and K. Cai, Nat. Commun., 2025, 16, 9718, DOI:10.1038/s41467-025-64739-7.
- H. Nian, L. Cheng, L. Wang, H. Zhang, P. Wang, Y. Li and L. Cao, Angew. Chem., Int. Ed., 2021, 60, 15354–15358, DOI:10.1002/anie.202105593.
- M. Nakamura, J. Suzuki, F. Ota, T. Takada, K. Akagi and K. Yamana, Chem. – Eur. J., 2016, 22, 9121–9124, DOI:10.1002/chem.201602043.
- C. Yang, W. Chen, X. Zhu, X. Song and M. Liu, J. Phys. Chem. Lett., 2021, 12, 7491–7496, DOI:10.1021/acs.jpclett.1c02013.
- S. Veglianti, A. Michieletti and D. Padula, ACS Mater. Lett., 2025, 7, 3220–3226, DOI:10.1021/acsmaterialslett.5c00872.
- Y. Zhou, H. Zhu, Y. Yuan, Z. Song and B. C. Mort, J. Chem. Inf. Model., 2025, 65, 4281–4292, DOI:10.1021/acs.jcim.4c02374.
- R. G. Uceda, A. Gijón, S. Míguez-Lago, C. M. Cruz, L. Álvarez de Cienfuegos, A. J. Mota, D. Miguel and J. M. Cuerva, ChemPhotoChem, 2025, 9, e202500079, DOI:10.1002/cptc.202500079.
- R. G. Uceda, A. Gijón, S. Míguez-Lago, C. M. Cruz, V. Blanco, F. Fernández-Álvarez, L. Álvarez de Cienfuegos, M. Molina-Solana, J. Gómez-Romero, D. Miguel, A. J. Mota and J. M. Cuerva, Angew. Chem., Int. Ed., 2024, 63, e202409998, DOI:10.1002/anie.202409998.
| This journal is © The Royal Society of Chemistry 2026 |