
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Side reactions at the Zn anode: what we know and how we deal with them
Gao
Weng†
a,
Shuang
Chen†
 a,
Yang
Xiang
a,
Yufan
Xia
a,
Zhen
Luo
a,
Xuan
Zhang
b,
Muhammad Wakil
Shahzad
c,
Linhua
Zhu
d,
Ben
Bin Xu
*c,
Mi
Yan
a,
Hongge
Pan
e and
Yinzhu
Jiang
*abf
a,
Yang
Xiang
a,
Yufan
Xia
a,
Zhen
Luo
a,
Xuan
Zhang
b,
Muhammad Wakil
Shahzad
c,
Linhua
Zhu
d,
Ben
Bin Xu
*c,
Mi
Yan
a,
Hongge
Pan
e and
Yinzhu
Jiang
*abf
aSchool of Materials Science and Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: yzjiang@zju.edu.cn
bFuture Science Research Institute, ZJU-Hangzhou Global Scientific and Technological Innovation Center, Zhejiang University, Hangzhou 311215, China
cSchool of Engineering, Physics and Mathematics, Faculty of Science and Environment, Northumbria University, Newcastle upon Tyne, NE1 8ST, UK. E-mail: ben.xu@northumbria.ac.uk
dThe International Joint Research Center for Clean and Efficient Utilization of Hydrocarbon Resources in the South China Sea of Hainan Province, Engineering Research Center of Tropical Marine Functional Polymer Materials of Hainan Province, Key Laboratory of Water Pollution Treatment and Resource Reuse of Hainan Province, Key Laboratory of Functional Organic Polymers of Haikou, Hainan Normal University, Haikou, Hainan 571158, China
eInstitute of Science and Technology for New Energy, Xi'an Technological University, Xi'an 710021, China
fState Key Laboratory of Baiyunobo Rare Earth Resource Research and Comprehensive Utilization, Baotou Research Institute of Rare Earths, Baotou, 014030, China
First published on 22nd January 2026
Abstract
Aqueous Zn-ion batteries (AZIBs) have been widely regarded as promising alternatives to Li-ion batteries (LIBs) owing to their intrinsic safety and cost-effectiveness. Nonetheless, spontaneous side reactions at the Zn anode dominate the platelet packing mode of Zn plating/stripping, thus undermining the anode's reversibility. To enable the future development of AZIBs, undesirable side reactions at the Zn anode need to be fully unveiled to understand intrinsic Zn plating/stripping processes. This review offers innovative and comprehensive insights to mitigate the side reactions at the Zn anode that have not been reported to date. It commences with a profound understanding of Zn redox chemistry with side reactions involved. Subsequently, the main initiators of side reactions are discussed; the mainstream strategies used to decouple main/side reactions are considered and analyzed from the thermodynamics and kinetics viewpoints based on different mechanisms; and cutting-edge research breakthroughs are summarized. Additionally, advanced characterization technologies are expounded, equipping readers with measures to intuitively detect side reactions. Finally, current challenges related to side reactions are presented, accompanied by proposed feasible solutions to inspire readers for the future development of AZIBs.
 Gao Weng | Gao Weng received his Bachelor's degree from Soochow University in 2024. He is currently a Master's student in the School of Materials Science and Engineering, Zhejiang University, under the supervision of Prof. Yinzhu Jiang. His main research interest focuses on aqueous Zn-ion batteries. |
 Shuang Chen | Shuang Chen received her Bachelor's degree in Materials Science and Engineering from Zhejiang Sci-Tech University in 2020. She is currently working toward her Doctoral degree in Energy Storage Systems at Zhejiang University, under the supervision of Prof. Yinzhu Jiang. Her research mainly focuses on the rational design of advanced materials and interphases for energy-dense metal anodes. |
 Ben Bin Xu | Ben Bin Xu (FRSC, FIMMM, FRSA, FSCI) is a Professor (Chair) of Materials and Energy at Northumbria University, UK. His research interests cover Materials, Surface, Sustainability, Energy and Biomedical Engineering. He has published 300+ journal articles (h-73), 10 books/chapters, and 9 patents, given 100+ invited talks and won multiple awards (Stanford/Elsevier World Top 2% Scientist (2025, 2024), AIChE Excellence in Research Mid-Career Investigator Award in the ‘Composites’ area (2023), and 2016 Young Investigator award from the International Polymer Networks Group). Ben is chairing the Materials Characterization & Properties Group in the IoM3 and is a Formal Chair for the ‘Composites’ area in the AICHE (US). |
 Yinzhu Jiang | Yinzhu Jiang (FRSC, FIMMM) is a Full Professor at the School of Materials Science and Engineering, Zhejiang University. He received his PhD degree from the Department of Materials Science and Engineering, University of Science and Technology of China in 2007. He worked as a Postdoctoral Researcher at Heriot-Watt University, UK, from 2007 to 2008 and an Alexander von Humboldt Fellow at Bielefeld University, Germany, from 2008 to 2010. His research interest focuses mainly on energy-related materials and electrochemistry, including rechargeable batteries, metal anodes and solid electrolytes. He has published 200+ journal articles in top journals such as Sci. Adv., Energy Environ. Sci., Adv. Mater., J. Am. Chem. Soc., and Angew. Chem. Int. Ed. with 11000+ citations, and he has also authored 30+ invention patents. |
Key learning points(1) Our review explores the underlying principles governing the stability of the Zn anode and categorizes side reactions as essential issues associated with the Zn anode side, offering insights into the relation between side reactions and Zn2+/Zn redox chemistry.(2) Our review analyses the initiators of Zn anode-related side reactions and elaborates the corresponding targeted strategies that can decouple side reactions from the intrinsic Zn plating/stripping processes. Insightful exposition on how the optimization of different components, such as the Zn anode and electrolytes, can help to mitigate the side reactions at the Zn anode side and a critical discussion on the pros and cons of different strategies are provided. (3) Our review investigates the mainstream advanced characterization technologies for the accurate detection of side reactions. (4) Our review anticipates the challenges and future directions of research in the realm of the Zn anode, shedding light on unexplored avenues and offering opportunities to drive the development of practical aqueous Zn-ion battery-based energy storage solutions. |
1. Introduction
The depletion of fossil fuel has resulted in a pressing demand for sustainable energy sources, such as solar, wave, biomass, and wind energy.1–3 However, their intermittent and unstable supply necessitates the development of robust energy storage systems to balance electricity supply and demand, bringing battery technologies in the limelight. Li-ion batteries (LIBs), by virtue of their high energy density, have been the mainstream product to date.4 Nonetheless, their safety concerns resulting from the use of flammable organic electrolytes and high cost due to scarce Li sources are concerning. Thus, the search for alternative battery systems to replace LIBs is an ongoing mission.Aqueous batteries, featuring intrinsically safe, low-cost, and kinetically favorable aqueous electrolytes, have been considered competitive candidates to LIBs.5,6 Many aqueous secondary batteries have been developed and commercialized to date, such as lead-acid batteries, cadmium–nickel (Cd–Ni) batteries, and nickel–metal hydride (Ni–MH) batteries. Nonetheless, these aqueous battery systems can only afford a restricted energy density compared with LIBs (30–50 Wh kg−1 for lead-acid batteries, 45–80 Wh kg−1 for Ni–Cd batteries, and 60–120 Wh kg−1 for Ni–MH batteries). Furthermore, the inevitable issue of environmental contamination from their acid or alkaline electrolytes and electrode materials is concerning. Additionally, a memory effect inevitably exists in Ni–Cd batteries, while the relatively high cost and inadaptability to high-voltage fast charging conditions are intractable for Ni–MH batteries. Therefore, there is a pressing need to develop more energetic and pragmatic aqueous batteries.
Compared with the intercalation/deintercalation mode, the utilization of the plating/stripping mode with a metal anode is usually a better choice owing to its lower redox potential. Fig. 1a exhibits several corresponding parameters of reported mainstream metal anodes. It can be perceived that a high-volume specific capacity (5855 mAh cm−3) is an explicit property of the Zn anode, enabling sizing down for device development. An appropriate low electrode potential enables the Zn anode compatible with aqueous electrolytes, with the solid–liquid conversion plating/stripping mode, which cannot be achieved by other metal anodes. Consequently, benefiting from the appropriate redox potential (−0.76 V vs. standard hydrogen electrode (SHE)) and the satisfactory theoretical specific capacity (820 mAh g−1; 5855 mAh cm−3) of the Zn metal anode, aqueous Zn ion batteries (AZIBs) possess outstanding energy density merits in comparison with other aqueous batteries.7 In addition, the small ionic radius of solvated Zn2+ guarantees a rapid ion transportation rate in aqueous electrolytes, and the acceptable cost of Zn metal ensures its potential in large-scale commercialization. As explicitly exhibited in Fig. 1b, compared with LIBs, AZIBs possess the outstanding merits of safety, environmental compatibility, cost effectiveness, and abundant element (Zn) reserves. Besides, AZIBs showcase the merits of relatively higher operation voltage, superior energy density, and eco-friendliness in the realm of aqueous batteries. Thus, AZIBs with the optimized combination of intrinsic safety and satisfactory energy density are considered promising energy storage systems. They seem to be an attractive option to make up for the drawbacks of LIBs and other aqueous batteries, as demonstrated by the overwhelming research and development trends. The rapidly growing number of publication record in the last decade testifies to the flourishing development and broad prospects of AZIBs for energy storage (Fig. 1c).
 | ||
| Fig. 1 (a) Data summarizing the electrode potential, mass/volume specific capacity, and ionic radius of different metal anodes. (b) Schematic of the benefits of AZIBs over other battery systems. (c) Publications with the topic of “zinc-ion batteries”. | ||
The working mechanism of AZIBs is expounded as follows. During discharging, Zn2+ stripped from the Zn anode migrates to the cathode side and intercalates in cathode active materials, while the reverse reactions occur during charging, with Zn2+ deintercalation from the cathode and deposition on the Zn anode side. The corresponding reactions (eqn (1)–(3)) are listed below with an MnO2 cathode as an example.
| Zn anode: xZn − 2xe− ⇌ xZn2+ | (1) |
| MnO2 cathode: MnO2 + xZn2+ ⇌ ZnxMnO2 + 2xe− | (2) |
| Overall: MnO2 + xZn ⇌ ZnxMnO2 | (3) |
The development history of the Zn anode and AZIBs is summarized in Fig. 2.
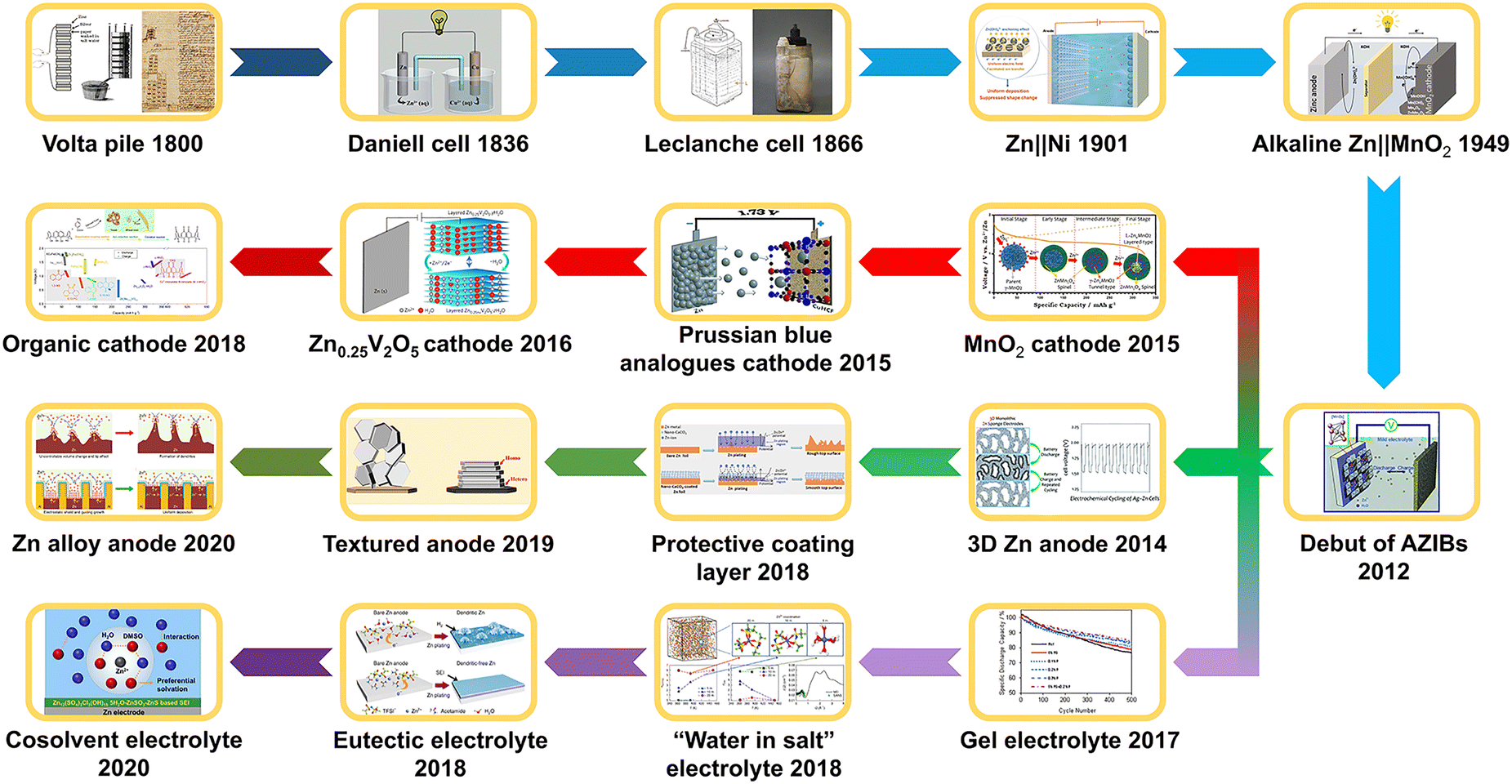 | ||
| Fig. 2 Development history of Zn anode and AZIBs. Volta pile: reproduced with permission.8 Copyright 2019, Wiley-VCH. Daniel cell: reproduced with permission.9 Copyright 2024 Wiley-VCH. Leclanche cell: reproduced with permission.10 Open Access. Zn||Ni: reproduced with permission.11 Copyright 2022, the American Chemical Society. Alkaline Zn||MnO2: reproduced with permission.12 Copyright 2022, Wiley-VCH. Debut of AZIBs: reproduced with permission.13 Copyright 2012, Wiley-VCH. MnO2 cathode: reproduced with permission.14 Copyright 2015, the American Chemical Society. Prussian blue analogue cathode: reproduced with permission.15 Copyright 2015, Wiley-VCH. Zn0.25V2O5 cathode: reproduced with permission.16 Copyright 2016, Springer Nature. Organic cathode: reproduced with permission.17 Copyright 2018, Open Access. 3D Zn anode: reproduced with permission.18 Copyright 2014, the Royal Society of Chemistry. Protective coating layer: reproduced with permission.19 Copyright 2018, Wiley-VCH. Textured anode: reproduced with permission.20 Copyright 2019, Open Access. Zn alloy anode: reproduced with permission.21 Copyright 2020, Open Access. Gel electrolyte: reproduced with permission.22 Copyright 2017, Wiley-VCH. “Water in salt” electrolyte: reproduced with permission.23 Copyright 2018, Springer Nature. Eutectic electrolyte: reproduced with permission.24 Copyright 2019, Open Access. Cosolvent electrolyte: reproduced with permission.25 Copyright 2020, the American Chemical Society. | ||
Although AZIBs are promising, the intractable issues at the Zn anode side, which are the Achilles’ heel of AZIBs, are detrimental to battery system reversibility. The main challenges encountered by the Zn anode can be classified into two, as shown in Fig. 3a, namely, dendrite growth and side reactions, including the hydrogen evolution reaction (HER), corrosion, and byproduct formation. The rampant dendrites formed due to uneven Zn deposition will pierce the separator and cause short circuits. In parallel, the thermodynamic instability of Zn in aqueous electrolytes gives rise to HER and corrosion, both chemically and electrochemically, which consume the electrolytes, generate bubbles, and roughen the Zn anode surface. Moreover, the formation of passive byproduct layers, mainly dominated by complex zinc hydroxide and originating from the increase in surficial pH, will result in large polarization during cycling and loss of active Zn.26,27 These issues are mutually exacerbated and cause battery failure ultimately.
 | ||
| Fig. 3 (a) Main challenges encountered by the Zn anodes. (b) Detrimental effects of side reactions at the Zn anode side. | ||
To alleviate these Zn anode-related problems and guarantee stable cycling, several strategies, such as artificial interphase construction, electrolyte engineering, and separator modification, have been proposed.28–31 Despite the effectiveness of these strategies, deep insights into Zn plating/stripping chemistry remain lacking. Thus far, reviews concerning the reversibility of the Zn anode can be classified into three categories. The first group focuses on modification of the Zn anode with some materials such as liquid metals and covalent organic frameworks.32,33 These reviews highlight the detailed relationships between such materials and the Zn anode with insightful views. The second group pay attention to certain strategies or systems, such as the construction of artificial interphase layers and regulation of electrocrystallization orientation.26,34 These specific strategy-based review articles furnish readers with meaningful guidance for the stabilization of the Zn anode. The last group emphasizes on the integration of physical or chemical factors in aqueous batteries, like hydrogen bonds and the formation of electric double layers (EDLs).35,36 These reviews intensively probe into the roles these factors play in Zn plating/stripping cycles and discuss the corresponding relationships.
Although these reviews provide meaningful discussions, their narratives seem to be confined to a relatively narrow range due to the thematic emphasis on certain materials, methodologies, or indicators. Hence, a holistic perspective on intrinsic Zn plating/stripping would be highly desired. Moreover, since Zn dendrites and side reactions are the major issues encountered by the Zn anode, most current reviews pay equal attention to both with ambivalent attitudes. There is a pressing need to figure out the core problem at the Zn anode and break the confusion that what governs the reversibility of Zn2+/Zn redox.
Zn anode-related issues are complex and normally occur simultaneously, which may create some difficulties in understanding Zn redox chemistry.26 Although Zn dendrites and side reactions exhibit mutual promotion effects, the fundamental factor that unbalances the original Zn plating/stripping is still vague. It should be noted that Zn metal is characterized by the close packing mode of hcp.37 Consequently, Zn platelets should be formed intrinsically during the electrodeposition processes, resulting in a dense surface morphology. Nonetheless, mossy electrodeposition structures can often be witnessed under a scanning electron microscopy (SEM), especially at small current densities, for which the occurrence of side reactions should be responsible.37,38 The redox potential differences between Zn2+/Zn and hydrogen evolution in thermodynamics indicate the spontaneity of side reactions.39 Although Zn dendrite should be the main failure mechanism if achieving the limiting current density, this situation may be far from the practical applications. Resultantly, side reactions occurrence should be laid more emphasis on given the spontaneity. The detrimental effects of side reactions can be generalized as shown in Fig. 3b. From the perspective of Zn plating/stripping, side reactions will disrupt the uniform distribution of surficial electric field, thus hindering reversibility. The occurrence of side reactions will also lead to certain electrical loss due to the active Zn loss, thereby reducing the Coulombic efficiency (CE). Regarding industrialization, the occurrence of side reactions represents the considerable consumption of the electrolyte and active Zn, and the accumulated evolved H2 will cause swelling issues in pouch cells.40 In practical applications requiring lean electrolyte conditions, rapid electrolyte consumption will lead to an insufficient capacity and incomplete electrode wetting, thus causing battery failure.41 Additionally, safety concerns regarding the evolved H2 should also be taken into account.
Herein, we categorize side reactions as an essential issue at the Zn anode, associating the HER and corrosion as the procedures initiating side reactions. As illustrated in Fig. 4, in this review, we commence with insights into Zn redox chemistry. Subsequently, we attempt to decouple main/side reactions toward intrinsic Zn plating/stripping from thermodynamic and kinetic perspectives. The thermodynamic perspective is aimed at lowering the activity of three initiators to weaken the occurrence of side reactions, while the kinetic angle is devoted to amplifying the reaction rate differences between the main/side reactions. Based on this, we provide in-depth analysis of the mainstream advanced strategies according to their mechanisms, with a summary of the latest research findings and breakthroughs. Further, a discussion on the techniques for the characterization of side reactions is presented, equipping readers with measures to intuitively detect side reactions. Finally, we offer comprehensive opinions on the challenges that still hinder the commercialization of AZIBs from the perspective of side reactions, accompanied by proposed realistic solutions and strategies.
 | ||
| Fig. 4 Schematic of the insights into side reactions based on three aspects. | ||
2. Insight into Zn redox chemistry
2.1. Thermodynamics
Zn metal is thermodynamically unstable in aqueous electrolytes. As shown in the Pourbaix diagram in Fig. 5a, there exists a redox potential difference between Zn2+/Zn and H+/H.42 As a result, Zn electrodeposition and hydrogen evolution are in direct competition. The higher redox potential of H+/H gives rise to the thermodynamic favorability for the HER, which accounts for the restricted electrochemical window of aqueous electrolytes at the Zn anode side. It should be noted that the commonly used commercial Zn foil is likely to have some impurities such as C and Ni due to the industrial processes, which result in the formation of a parasitic micro-cell that accelerates the HER.43 It was experimentally discovered that hydrogen evolution reached 3.76 mmol h−1 cm−2 in a Zn||Zn symmetric cell and 7.70 mmol h−1 cm−2 in a Zn||MnO2 full cell.44 In addition, Zn symmetric coin cells even tend to explode after resting for a period of time due to the generation of H2 gas.45 | ||
| Fig. 5 (a) Pourbaix diagram of the Zn anode in aqueous media. Reproduced with permission.42 Copyright 2021, the Royal Society of Chemistry. (b) Schematic of chemical and electrochemical side reactions. Schematic of the relationship between the occurrence of side reactions and (c) current density-induced nucleation morphology, (d) current density-induced Zn(H2O)62+ concentration gradient, (e) current density-induced textured deposition, and (f) Zn utilization ratio. Schematic of (g) H2O-rich interface formed immediately upon the application of a bias potential, (h) diluted interface formed during Zn2+ desolvation with competing HER, and (i) concentrated interface formed during Zn2+ solvation with the corrosion HER. Reproduced with permission.54 Copyright 2024, the American Chemical Society. | ||
In general, there are two types of Zn anode-related side reactions: chemical and electrochemical types, as shown in Fig. 5b. The chemical side reactions will proceed spontaneously without any connection to the external circuit, while the electrochemical side reactions are accompanied by a current flow. Chemical side reactions, mainly attributed to surficial adsorbed water, can be described by the following three equations (eqn (4)–(6)); ZnSO4 electrolyte is used as an example.46,47
| H2O → H+ + OH− | (4) |
| Zn + 2H+ → Zn2+ + H2 | (5) |
| 4Zn2+ + 6OH− + SO42− + xH2O → Zn4(OH)6SO4·xH2O (ZHS) | (6) |
Chemical side reactions proceed with a variation in surface pH. Induced by the Lewis acidity of Zn2+, the bulky electrolyte environment in AZIBs generally takes on a slightly acidic state, such as a pH value of around 4 in ZnSO4 electrolyte.48,49 Under this condition, water molecules are likely to be partly ionized into H+ and OH−. It should be noted that the oxygen coordination state endows the water molecules in the Zn2+ solvation structure with weakened H–O bonds, resulting in a deprotonation tendency, which will be discussed in detail in the following section. The hydrolysis process is also dependent on Zn salts, where the strong acid–weak base Zn salt usually causes the generation of excessive H+.50 The formed H+ induces Zn corrosion and hydrogen evolution, and the consumption of these H+ ions causes the emergence of local alkaline micro regions on the Zn anode surface. Consequently, surficial OH− will lead to the formation of complex zinc hydroxide (ZHS) passivation layers. This process from H+ to H2 includes a Volmer step (H+ + e− → Hads), Tafel step (2Hads → H2), and Heyrovsky step (Hads + H+ + e− → H2).43,51 With regard to the passivation byproducts, ZHS can be formed spontaneously, regardless of the substrate, as long as the chemical equilibrium conditions are satisfied.46 The formed ZHS byproducts, generally existing as hexagonal loose and porous structures, will aggravate polarization and intensify dendrites.52 As a result, surficial pits and byproducts can be observed if a Zn foil is directly immersed in bare ZnSO4 aqueous solution, accounting for the self-discharge phenomenon of shelved batteries.
During cycling, electrochemical side reactions that may result from the Zn(H2O)62+ species are also likely to occur at the Zn anode side, according to the following equations (eqn (7)–(9)).46,47
| 2H+ + 2e− → H2 | (7) |
| 2H2O + 2e− → 2OH− + H2 | (8) |
| 4Zn2+ + 6OH− + SO42− + xH2O → Zn4(OH)6SO4·xH2O | (9) |
The chemical side reactions described in eqn (4)–(6) will also occur with electrons from the external circuit. Similar to the situation of chemical side reactions, electrochemical side reactions proceed with a change in surface pH. Among the above-mentioned equations, H+ derived from active water dissociation possesses the lowest energy barrier for the electrochemical reduction for hydrogen formation during Zn deposition. The first electrochemical reduction of H+ will also result in the formation of local alkaline micro regions on the Zn anode surface with an increase in the pH value. Apart from the H+ contribution, during the charging process, H2O will compete with Zn2+ to gain electrons and be reduced, which will generate OH− and H2. With the formation of alkaline micro regions on Zn anode surface, this process will become more noteworthy. Concretely, the electrodeposition of water can be described as follows: H2O + e− → Hads + OH− (Volmer step); H2O + Hads + e− → H2 + OH− (Heyrovsky step); and 2Hads → H2 (Tafel step). Subsequently, the electrolyte/Zn anode surface with increased pH will result in the formation of complex zinc hydroxide (ZHS).
From the perspective of thermodynamics, the Nernst equation can describe the potential of the HER and Zn deposition, as shown in eqn (10) and (11):53
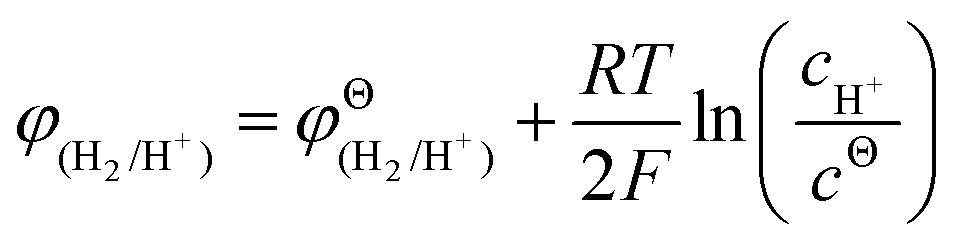 | (10) |
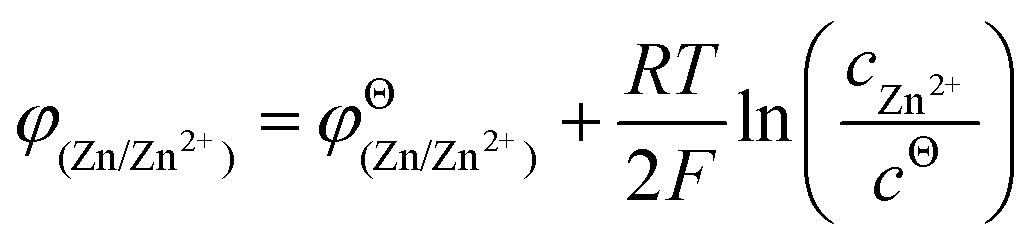 | (11) |
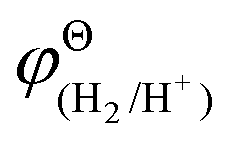 and
and 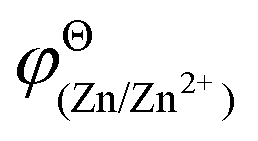 are the standard potential of the H2/H+ and Zn/Zn2+ redox, respectively. R, T, F, and cΘ represent the ideal gas constant, absolute temperature, Faraday constant, and standard molarity, respectively.
are the standard potential of the H2/H+ and Zn/Zn2+ redox, respectively. R, T, F, and cΘ represent the ideal gas constant, absolute temperature, Faraday constant, and standard molarity, respectively.
2.2. Kinetics
The reaction rate, v, can be determined using the reaction rate constant, k. According to the Arrhenius equation, k and v can be obtained using eqn (12) and (13), respectively, as follows: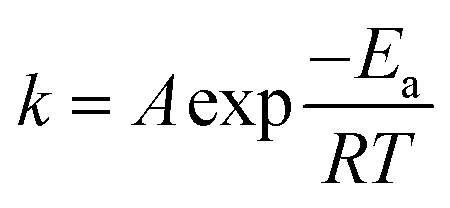 | (12) |
| v = kcn | (13) |
Although side reactions are thermodynamically favored, it is generally acknowledged that the reaction equilibrium can be regulated at the kinetic level by regulating several battery cycling-related physical factors, such as current density and Zn utilization ratio.52 Herein, we try to analyze these factors considering their correlation with Ea and c.
According to the mass transfer limitation, the current researches suggest that a small current density intensifies side reactions while a large current density inhibits them, which is attributed to the competition between Zn2+/Zn and H+/H, where Zn2+/Zn is kinetically favored due to its higher exchange current density.39 Zhou's group reported that high current densities result in a high nucleation overpotential and high Zn2+ concentration gradient at the electrolyte/Zn anode surface.55 A higher nucleation overpotential facilitates a high nuclei density, fine grains, and complete substrate electrodeposition coverage, while a high concentration gradient results in a relatively lower Zn2+ concentration near the Zn anode surface, generating less detrimental byproducts (Fig. 5c). Accordantly, Yang et al. discovered suppressed hydrogen evolution with increasing current densities, and they attributed this phenomenon to the absence of the main HER contributor Zn(H2O)62+ (discussed later) at the Zn anode surface, which was induced by the high concentration gradient (Fig. 5d).53 A similar conclusion has also been drawn by Pu's group, witnessing a change in the electrodeposition morphology from mossy to compact with an increase in current density within a certain range, and they interpreted inhibited side reactions to be kinetically unfavorable.38 They achieved a (002)-dominated, textured deposition structure at an adequate current density, which exhibited improved corrosion resistance (Fig. 5e). Thus, it can be observed that the current density affects the occurrence of side reactions kinetically via the regulation of Ea and c.
On the one hand, a large current density accelerates mass transfer and establishes an ion depletion zone, thereby reducing the concentration of hydrated Zn2+ species toward inhibited HER and byproduct formation. On the other hand, a large current density induces dense deposition or even textured surface, creating a higher energy barrier for the HER and corrosion to overcome.
Zn utilization is defined as the ratio of the cycled Zn capacity to the total Zn capacity, which is another factor. Zhi's group reported an inverse correlation between the Zn utilization ratio and Zn anode irreversibility, as illustrated in Fig. 5f.52 Batteries with a low Zn utilization usually operate for extended periods because only trace amounts of Zn participate in side reactions, and the excess available Zn compensates for the irreversible consumption. On the contrary, a high Zn utilization causes the irreversible consumption and depletion of more active Zn, which shifts the equilibrium to favor adverse side reactions. To sum up, the Zn utilization ratio is the indicator of the proportion of Zn that participates in the electrochemical redox reaction and is more related to c rather than Ea. With more proportion of Zn participating in cycling, during the electrodeposition process more hydrated Zn2+ species are involved. Consequently, more active Zn may be irreversibly consumed by side reactions and increase polarization, thus deteriorating the battery performance.
Recently, Yu et al. investigated the interfacial hydrogen evolution mechanisms and found that the establishment of an initial EDL to form an H2O-rich interface (Fig. 5g) and the subsequent dynamic evolution of hydrogen bonds are the two critical processes during Zn plating/stripping.54 The number of hydrogen bonds increases and their strength weakens in comparison with the bulk electrolyte during Zn2+ desolvation. A diluted interface is formed due to the consumption of Zn2+ and accumulation of H2O at the Zn anode surface, resulting in a strong competition between hydrogen evolution and Zn2+ reduction (Fig. 5h). By contrast, a concentrated interface (hydrogen bond number decreases and strength increases) is formed and produces a small amount of hydrogen during Zn2+ solvation. Consequently, the HER features a relatively rapid kinetics during deposition and a relatively sluggish kinetics during stripping. Corrosion HER may take place at this stage because some of the electrons originating from the oxidation of Zn will be captured by the accumulated H+ (Fig. 5i). Thus, it can be concluded that enhancing the strength of hydrogen bonds and reducing the content of active water within the initially formed EDL are essential to suppress the HER dynamics.
3. From side reactions toward intrinsic Zn plating/stripping
3.1. Side reaction initiators
To understand Zn anode-related side reactions, it is mandatory to examine the components in aqueous electrolytes and figure out the corresponding initiators. As presented in eqn (4)–(9), active water molecules and protons are mainly responsible for the occurrence of side reactions. Generally, protons are derived from the spontaneous ionization of water molecules in the presence of Zn salts. It should be noted that the active water molecules include both those in the Zn2+ solvation structure and those in the bulk electrolyte, which are termed Zn2+-coordinated water and non-coordinated water, respectively, in the following contexts.Of note, the in situ formation of a solid electrolyte interphase (SEI) stemming from the solvents and Zn salts prominently affects the occurrence of side reactions. However, to gain deeper insights into Zn deposition and side reactions, it is reasonable to exclude other possible reactions to concentrate on the trigger of side reactions. Accordingly, we choose the frequently used electrolyte systems (ZnSO4 and ZnCl2) without in situ SEI formation for discussion hereon, with the effects of SEI elaborated in subsequent sections. Yang et al. investigated the deprotonation energy of non-coordinated H2O, solvated H3O+ and Zn(H2O)62+ species and observed that their deprotonation energy followed the order of H3O+ < Zn(H2O)62+ < non-coordinated H2O (Fig. 6a).56 Nonetheless, since the concentration of H3O+ is much lower than those of the other two species, Zn2+-coordinated H2O exhibits the highest reduction activity. When H2O coordinates with ions, electron transfer occurs to weaken the H–O bond and increase the hydrolysis activity, which is responsible for the above-mentioned phenomenon.43 Based on the representative studies, we attempt to classify the Zn salt concentrations into exact ranges, such as smaller than 0.5 M as low concentration, 0.5–2.5 M as medium concentration, and larger than 2.5 M as high concentration.53,57 Different concentrations also results in variations in electrochemical stability. Yang's group reported that medium ZnSO4 concentration exhibited the narrowest electrochemical window compared with dilute or concentrated electrolytes.53 The active Zn(H2O)x2+ species is reduced in low concentrations, while at high concentrations, SO42− replaces some Zn2+-coordinated H2O in the solvation structure (Fig. 6b). These results are consistent with the high HER activity of Zn2+-coordinated H2O.
 | ||
| Fig. 6 (a) Deprotonation energy of free H2O, solvated H3O+ and Zn(H2O)62+ species. (b) Electrochemical window of high concentration, medium concentration, and low concentration ZnSO4 electrolyte. (c) Trend in the reduction activity of different existing water species. (d) Schematic of the HER-preferred and Zn deposition-preferred Zn2+ solvation structure. (e) Thermodynamic and kinetic contributions to the HER activity in electrolytes with different concentrations. (f) Schematic of three side reaction initiators and the corresponding brief reaction processes. | ||
Zhao et al. probed the thermodynamic and dynamic properties of the HER in ZnCl2 aqueous electrolytes with concentrations of 1 M and 15 M, filling the gap of the HER trends in highly concentrated electrolytes.50 From the perspective of thermodynamics, a high ZnCl2 concentration is related to a low pH with more HER activity, which is validated by the higher potential for hydrogen evolution. However, protons are not the main contributors to the HER in their case. In the battery cycling dynamics of Zn plating/stripping, Cl− will replace some Zn2+-coordinated H2O and hydrate with H2O to reduce the H2O–H2O clusters. Calculation results show that the water in Zn2+–H2O coordination structures exhibits the highest HER activity, while that in Cl−–H2O coordination structures exhibits the lowest (Fig. 6c). In the competition between Zn deposition and the HER, Zn(H2O)62+ shows greater tendency toward the HER, while the anion-involved Zn2+ solvation structure possesses greater tendency toward Zn deposition (Fig. 6d). In highly concentrated ZnCl2 electrolytes, more Zn2+-related coordination structures with decreasing strong hydrogen bonds and increasing weak hydrogen bonds exist, promoting higher charge efficiency for Zn electrodeposition instead of HER. Accordingly, the kinetics of the thermodynamically favored HER will be suppressed. Therefore, with an increase in the Zn salt concentration, the thermodynamic preference increases, while the kinetic favourability decreases, and the highest HER activity appears at medium concentrations (Fig. 6e).
Based on the existing mainstream theories, the transportation behavior of components in electrolyte systems can be classified into vehicle and hopping.58 Hopping mechanism requires for the corresponding coordination site afforded by the medium, while vehicle mechanism does not. Generally, for the bare electrolyte such as the conventional 2 M ZnSO4, Zn2+ is almost completely hydrated and Zn(H2O)62+ is characterized by vehicle mechanisms. On account of the positive charge, Zn(H2O)62+ can exhibit a rapid response under an electric field. Numerous current strategies attempt to increase Zn salt concentration and construct Zn2+ coordination sites toward facilitated Zn2+ hopping behavior due to the transformation of Zn(H2O)62+ solvation structures and inhibited side reaction trend accordingly, which will be analyzed in the following chapters. In the case of uncoordinated H2O, in aqueous solutions, H2O molecules are generally in the state of clusters and hydrogen bond networks will be formed between them. These hydrogen bond networks are dynamic, and the diffusion of uncoordinated H2O features a hopping behavior by virtue of the breakage and formation of hydrogen bonds.59 The electroneutrality and restriction of intermolecular hydrogen bonds render uncoordinated H2O transportation relatively slow. Regarding H+, it also exhibits a hopping behavior known as the Grotthuss mechanism, wherein it transports along the hydrogen bonds between the H2O molecules.60 This process consists of two steps, the translation of a proton from H3O+ to H2O by tunneling in a hydrogen bond and subsequent reorientation (rotation) of H2O to take up the next proton.61 The Grotthuss mechanism of H+ brings about a swifter transporting rate, demonstrating the nonignorable risk of H+-induced HER in spite of the low concentration of H+.
According to the discussion above, Zn2+-coordinated H2O, non-coordinated H2O, and H+ are the three initiators of the HER, which is the first side reaction. Actually, the practical situation will be more complex as other factors such as ionic solvation radius, temperature, and surface layer formation may also influence the reduction of the above-mentioned active species and H2 generation. To avoid an overly complicated discussion and focus on the intrinsic chemical properties of these initiators, we opt to primarily consider the factors of deprotonation energy, transportation behavior, and corresponding concentration, as analyzed above. Based on this, the reduction trend of different active species can be concluded as follows, which is consistent with the majority of reports in the literature. Zn2+-coordinated H2O presents the greatest reduction probability during the electrodeposition process, and the active Zn(H2O)62+ species can be regulated by manipulating the coordination environment of Zn2+. By contrast, the non-coordinated H2O in the bulk electrolyte possesses electrochemical inertness to lose electrons to some extent, while it is the main contributor to chemical side reactions due to the specific adsorption of H2O dipoles on Zn anode surface.47 With regard to H+, although its relatively lower concentration is represented by less H3O+ species, it is thermodynamically favorable for the HER and generally features a rapid transportation rate in aqueous solutions, making its contributions to side reactions nonnegligible.
Fig. 6f describes the brief behavior of these three initiators to trigger side reactions, where bulky mass transfer and interfacial electron-accepting processes are included. Resultantly, solutions to these three initiators should consider the situations of both the bulk phase and interphase, which will be described in the following section titled “thermodynamic decoupling approaches”.
3.2. Decoupling main/side reactions thermodynamically and kinetically
Following the above-mentioned logic, it is rational to decouple the side reactions and main reactions (Zn plating/stripping) from the perspective of thermodynamics and kinetics. Correspondingly, there exist two approaches. On one hand, the thermodynamic approach, from the side reactions angle, aims to regulate the occurrence tendency of side reactions toward a relatively higher Gibbs free energy difference, embodied by the restricted activity of the side reaction initiators. On the other hand, the kinetic approach is from the main reaction perspective, accelerating the main reaction of Zn plating/stripping and decelerating side reactions, thus decreasing the electricity contribution from side reactions. Strictly speaking, the kinetic approach is based on thermodynamic stability. Both the thermodynamic approach and kinetic approach can tip the balance from the side reaction-dominating state to the Zn plating/stripping main reaction-dominating state.Analyzing the mainstream strategies targeted at more Zn redox reversibility within the thermodynamic and kinetic approaches is reasonable. Taking three side reaction initiators into account, the thermodynamic approaches to tackle side reactions can be categorized into different optimization mechanisms. With respect to kinetic approaches, more emphasis will be placed on the electrochemical behavior of Zn redox, involving Zn2+ mass transfer, desolvation, electron acceptance, and surficial diffusion. From the perspectives of thermodynamics and kinetics, we will provide a profound and comprehensive interpretation of many state-of-the-art strategies applied for restraining side reactions.
3.2.1.1. Minimizing Zn2+ coordinated water. During electrodeposition, the solvated Zn2+ will transfer to the Zn anode surface and be reduced to metallic Zn. The above-mentioned processes are strongly influenced by the coordination environment of Zn2+, where the active Zn2+-coordinated water is detrimental to the reversibility of Zn anode. Based on their coordination environments, Zn2+-related solvation species can be classified into two types, i.e., solvent-separated ion pairs (SSIPs) and contact ion pairs (CIPs). Their differences lie in the fact that anions enter the primary solvation sheath of Zn2+ in CIPs but not in SSIPs. In essence, the formation of SSIPs or CIPs is dependent on the interactions between the ions and solvent molecules. Solvents with strong affinity to Zn2+ will contribute more to SSIP. Emphasis can be laid on the rational modification of the dominant SSIP species Zn(H2O)62+ and the small-proportioned CIP species (e.g., [Zn(H2O)5SO4] for ZnSO4 electrolyte). Given the strong reduction trend of the Zn2+−coordinated H2O toward undesirable side reactions, a key approach is rationalized to substituting some functional components which can coordinated with Zn2+ for the H2O in Zn2+ solvation sheath.
Bulky solvation structure optimization. Cosolvents are also proven to be effective in optimizing the undesired Zn(H2O)62+ for achieving thermodynamic-stability-favored SSIP or CIP structures. The employment of hydrophobic cosolvents can diminish the hydration of Zn2+. Zhang's team screened four typical carbonate-based solvents with different hydrophobicity, observing that diethyl carbonate (DEC) possessed the highest hydrophobicity to minimize the Zn2+-coordinated H2O and inhibit side reactions (Fig. 7a and b).62 However, it should be mentioned that the employment of hydrophobic cosolvent usually requires the utilization of amphipathic anion-based Zn salt (e.g., Zn(OTf)2) simultaneously to avert solvents delamination with the presentation of hydrophobicity meanwhile (Fig. 7c). This may increase the cost and decrease the prospect for large-scale applications. Besides, despite its thermodynamic stability, the kinetics of Zn2+ transportation may be undermined. To mitigate this, hydrophilic cosolvents have been widely investigated. Qiao's group explored in this direction using low-cost methanol featuring a simple structure and high polarity.63 It was proven that methanol would interact with H2O, hence attracting H2O out of Zn2+ solvation sheath and weakening the hydration of Zn2+ (Fig. 7d). The anti-solvation effect of methanol was conducive to the inhibit the HER and byproduct formation. Nonetheless, hydrophilic cosolvents usually suffer from unfavorable kinetics, which is ascribed to the strong-binding-induced high desolvation energy of the Zn2+ solvation sheath with organic solvents. Thus, to further address this, Mai's group investigated organic solvent molecule-free CIPs (Zn2+(CH3COO−)2(H2O)4) in an H2O and DMSO cosolvent system with Zn(CH3COO)2 salt.64 The following two observations were noted: (1) the hydrogen bonds between DMSO and H2O weakened CH3COO− hydration, which further enhanced the interaction between CH3COO− and Zn2+, and (2) the hydrogen bonds between the solvated CH3COO− and H2O prevented DMSO from replacing the Zn2+-coordinated H2O. This work achieved the construction of organic solvent-free CIPs, with the thermodynamics and kinetics simultaneously promoted (Fig. 7e). In summary, cosolvent strategies generally concentrate on the interaction between cosolvent molecules and H2O. By virtue of the higher composition ratio of cosolvent molecules compared with additives, cosolvent strategies may exhibit more conspicuous effects in solvation structure design.
 | ||
| Fig. 7 (a) Schematic of the contribution of DEC to Zn anode reversibility. (b) Molecular structures of different carbonate cosolvents. (c) Schematic of the structural evolution regarding the miscibility/immiscibility between water and hydrophobic carbonate cosolvents before (up) and after (down) the introduction of OTf− anions. Reproduced with permission.62 Copyright 2022, the American Chemical Society. (d) Changes in the Zn2+ solvent sheath with the addition of methanol. Reproduced with permission.63 Copyright 2021, Wiley-VCH. (e) SSIP solvation structure and its corresponding desolvation behavior (left), conventional CIP solvation structure and its corresponding desolvation behavior (middle), and organic-solvent-free CIP solvation structure and its corresponding desolvation behavior (right). Reproduced with permission.64 Copyright 2024, Elsevier. | ||
The concentration and type of Zn salts significantly impact the plating/stripping chemistry due to the changes in the Zn2+ coordination environments. In 2018, Wang's team developed the concept of “water in salt” electrolyte in AZIBs composed of 1 mol kg−1 Zn(TSFI)2 and 20 mol kg−1 LiTSFI, where Zn2+-coordinated water was effectively removed due to the high salt concentration and hydration competition with Li+ (Fig. 8a).23 This highly concentrated electrolyte contributed to suppressed side reactions. As mentioned above, neither low nor high concentration of Zn salt is beneficial to side reactions because a medium Zn salt concentration is characterized by the most Zn2+–H2O coordination species.53 In Fig. 8b, the Zn salt-lean electrolytes possess affordable cost and rapid kinetics merits, whereas highly concentrated electrolytes are generally equipped with wide operation temperature range. Additionally, highly concentrated electrolytes may be more effective owing to their ability to break the intermolecular hydrogen bonds between water molecules, preventing the formation of water cluster-induced HER and corrosion. Apart from concentration, the properties of anions have an effect. Chao's team developed an anionic polarity index (API) as a corrosion resistance benchmark for rational Zn salt screening: the corresponding values are presented in Fig. 8c.47 Verified by simulation, they found that low API anions not only helped to construct CIP structures but also repelled water from the inner Helmholtz plane (IHP) through characteristic adsorption, consequently suppressing electrochemical and chemical side reactions (Fig. 8d and e). On this benchmark basis, they chose a low-cost Zn(HBS)2 salt with a low API as an optimization.
 | ||
| Fig. 8 (a) Representative Zn2+ solvation structures in electrolytes with 1 mol kg−1 Zn(TFSI)2 and three different concentrations of LiTFSI (5 mol kg−1, 10 mol kg−1 and 20 mol kg−1). Reproduced with permission.23 Copyright 2018, Springer Nature. (b) Schematic of the evolution of molecules and clusters near the anode interface in aqueous electrolytes. Reproduced with permission.53 Copyright 2023, Wiley-VCH. (c) API values of different anions. Schematic of the relationships (d) between API and solvated H2O in Zn2+ solvation structure and (e) between API and interfacial water in EDL structure. Reproduced with permission.47 Copyright 2025, Wiley-VCH. (f) Schematic of the prepared hydrogel electrolyte and multiple interactions within it. (g) Zn plating behaviors and corresponding mechanisms on the Zn anode surface induced by the anionic groups. Reproduced with permission.65 Copyright 2023, Wiley-VCH. (h) Relationship between the intrinsic characteristic of molecule structure (dipole moment) and binding energy with the electrolyte components. (i) Tafel plots and (j) linear sweep voltammetry (LSV) curves of different hydrogel electrolyte samples. Reproduced with permission.66 Copyright 2024, Wiley-VCH. (k) Representative environment of active Zn species in a Zn(TFSI)2-based eutectic electrolyte. (l) Schematic of SEI-regulated uniform Zn deposition induced by eutectic electrolytes. Reproduced with permission.24 Copyright 2019, Open Access. (m) Coordination structures and corresponding average binding energies of aqueous electrolyte (left), 30% water content-hydrated deep eutectic electrolyte (middle), and deep eutectic electrolyte (right). Reproduced with permission.67 Copyright 2023, Open Access. (n) Schematic showing the frustrated coordination strategy in eutectic electrolytes. Reproduced with permission.68 Copyright 2024, Open Access. Corresponding theoretical interpretation (Sand's model) of dendritic electroplating involving (o) deep eutectic and (p) quasi-eutectic liquids. Reproduced with permission.58 Copyright 2024, Wiley-VCH. | ||
Hydrogels feature a liquid phase enveloped by 3D polymer networks and can be employed as flexible quasi-solid electrolytes.69 The application of hydrogel electrolytes in AZIBs has emerged in the application of wearable electronic devices, such as yarn batteries.70 Although pure polyacrylamide (PAM) or polyvinyl alcohol (PVA) demonstrates potential in flexible devices, their electrochemical performance remains unsatisfactory. Subsequently, emphasis has been placed on Zn2+ mass transfer and desolvation behavior. Polar groups on polymer chains can coordinate with Zn2+ to replace coordinated H2O, such as the amide group modifying the Zn2+ solvation sheath.71 Further, negatively charged groups exhibit strong affinity with Zn2+. These groups will also serve as coordination sites to facilitate Zn2+ hopping, thus improving the transportation rate and transference number. Representatively, a polyzwitterionic hydrogel electrolyte was developed by Zhang et al. containing PAM and carboxylethyl-quaternized cellulose (CEQC) double networks (Fig. 8f).65 Zn2+-coordinated water could be removed and the desolvation process could be accelerated (Fig. 8g). Compared with liquid electrolyte, their hydrogel electrolyte exhibited a 4-times lower HER exchange current density and a lower HER potential of 0.7 V. However, the underlying relationship between zwitterionic polymers and electrochemical performance was not revealed. To address this, Yang's team investigated three different polyzwitterionic hydrogel electrolytes, proposing dipole moment as a descriptor.66 Cationic groups-anions exhibited the strongest binding, anionic groups-H2O showed the moderate, and anionic groups-Zn2+ delivered the weakest. It is advisable that the cationic and anionic groups should be separated with anionic groups not located in the molecule center (Fig. 8h). With an increase in dipole moment, the monomer interacted more strongly with anions and H2O, as evidenced by the reduced percentage of uncoordinated and Zn2+-coordinated H2O. Experimentally verified, the champion sample, poly[3-dimethyl(methacryloyloxyethyl)ammonium propane sulfonate] hydrogel electrolyte, featuring the largest dipole moment, achieved the widest electrochemical window (Fig. 8i and j).
Employing eutectic electrolyte, a low-melting-point liquid mixture composed of two or more substances associated with each other through hydrogen bonds, Lewis acid–base, or van der Waals interactions, is another strategy.72 A eutectic system can be estimated based on its phase diagram, where the melting point of the mixture is lower than that of its respective pure components. Different from the conventional liquid electrolytes, eutectic electrolytes generally feature highly concentrated Zn salts with minimized water, generating compact CIP structures and Zn2+ hopping behaviors. Eutectic electrolytes for AZIBs were first reported by Cui's group, where a deep eutectic system consisting of acetamide and Zn(TFSI)2 was fabricated without extra H2O (Fig. 8k).24 The absence of H2O content and formed CIP structures prevented side reactions (Fig. 8l). However, the sluggish reaction kinetics resulted in large polarization, and most of the endeavors to date have focused on accelerating Zn2+ transportation and desolvation without sacrificing the thermodynamic stability. To improve kinetics, a small amount of H2O can be added to obtain hydrated eutectic electrolyte, where corresponding optimum equilibrium point for kinetics and thermodynamics need figuring out.73 He's group designed an H2O-deficient solvation structure ([Zn(H2O)x(EG)y(OTf)z]2−z) in a hydrated eutectic electrolyte with a precisely controlled water content (30% volume ratio), which could achieve a delicate balance between suppressed side reactions and satisfactory Zn2+ kinetics (Fig. 8m).67 Beyond H2O content, the structure and ratio of eutectic solvents need to be regulated. Wang's group utilized the asymmetry of a ligand toward frustrated cation-ligand coupling and increased CIP proportions in a eutectic electrolyte, thereby achieving improved ion mobility (Fig. 8n).68 Pan's team proposed a quasi-eutectic electrolyte model, possessing ion hopping and vehicle mechanisms in the eutectic and near low concentration regions, respectively.58 This resulted in boosted kinetics while maintaining thermodynamic stability (Fig. 8o and p).
Surficial solvation structure optimization. The use of an electrolyte additive is a simple strategy to modify the Zn2+ solvation structure and is considered to have great commercialization potential. Different from the above-mentioned electrolyte strategies, additives usually account for only a small proportion in electrolytes, and thus, this strategy is more dependent on the chemical environment of the Zn anode surface. Initially, Wang's group explored the coordination environment of Zn2+ and discovered the unfavourableness of the Zn(H2O)62+ species.25 Dimethyl sulfoxide (DMSO) was found to enter the solvation sheath of Zn2+ to weaken the interactions between Zn2+ and H2O, consequently suppressing the reduction trend of H2O (Fig. 9a). This work opened a new avenue, inspiring the focus on Zn(H2O)62+ SSIP in the subsequent electrolyte additive-related researches. Since then, dielectric constant and donor number have often been regarded as indexes for screening potential electrolyte additives. In 2021, our group reported the utilization of chelating chemistry to stabilize the Zn anode, shedding light on new indexes for additives toward minimized coordinated water-involved SSIP structures.74 Following this direction, in 2022, Yang's group proceeded with applying chelating ligands using the stability constant (K) as another additive screening principle.75 They chose the polydentate ligand ethylenediaminetetraacetic acid (EDTA), exhibiting a high chelating energy with Zn2+, as the side reaction inhibitor (Fig. 9b). Aside from an SSIP structure modification, an additive-containing CIP structure was developed recently using N-acetyl-ε-caprolactam (N-ac), which afforded dual solvation sites to coordinate with Zn2+.76 By coordinating with Zn2+ and aggregating Zn2+ configuration, the interaction between Zn2+ and SO42− was enhanced to engage SO42− in Zn2+ solvation structure. The minimized number of H2O molecules and the existence of SO42− in the Zn2+ solvation sheaths effectively inhibited side reactions (Fig. 9c). However, since the additive amount is generally low compared with the Zn2+ concentration, the engineering effects of solvation structures are localized and limited. Merely optimizing the Zn2+ solvation structure using additives may be not sufficient, and thus, many reported additives also function as interphase regulators in parallel.
 | ||
| Fig. 9 (a) Illustration of Zn2+ solvation structure and Zn surface passivation in ZnCl2 electrolyte without (left) and with (right) DMSO as a cosolvent. Reproduced with permission.25 Copyright 2020, the American Chemical Society. (b) Molecular structures of different chelating ligands with respective chelating energy. Reproduced with permission.75 Copyright 2022, Wiley-VCH. (c) Schematic showing the effectiveness of the N-ac additive in suppressing side reactions and Zn dendrites. Reproduced with permission.76 Copyright 2024, Wiley-VCH. (d) Role of the polyamide layer in inhibiting side reactions. Reproduced with permission.77 Copyright 2019, the Royal Society of Chemistry. (e) Zn deposition behavior at a 3D macromolecular network-modified interface. Reproduced with permission.78 Copyright 2024, Wiley-VCH. | ||
Apart from electrolyte engineering strategies, great attention has also been paid to Zn anode modification. In this regard, constructing artificial interphases can be an effective approach. As optimizers for Zn2+ dehydration and desolvation, artificial interphases generally feature Zn2+ conductivity and electronic insulation to guarantee electrodeposition beneath them. These interphases generally feature Zn2+ coordination and water bonding sites, thereby gradually destroying the hydration sheath along with Zn2+ transport. For example, Cui's group proposed a polyamide-based coating layer on the Zn anode surface.77 This coating layer not only exhibited water-/O2-resistant properties but also destroyed the Zn2+ solvation sheath via coordination with amide groups, inhibiting corrosion at the Zn anode (Fig. 9d). Nonetheless, their system exhibited poor performance under large current densities, reflecting the sluggish mass transfer issues commonly encountered by hydrophobic interphases. To address this, emphasis has also been given to hydrophilic interphases with less polarization. Recently, Chen's team developed a 3D macromolecular network interphase crosslinked by potato starch.78 The abundant hydroxyl groups in the potato starch networks facilitated Zn2+ dehydration during deposition, consequently inhibiting side reactions (Fig. 9e). Nonetheless, although the Zn2+ transportation rate is optimized, the contradiction between side reaction thermodynamics and main reaction kinetics has not been effectively averted. Moving forward, it is necessary for artificial interphases to establish ordered-Zn2+-expedited transportation channels, which will be discussed in the following “kinetic decoupling approaches” section.
3.2.1.2. Confining uncoordinated water. In the bulk phase of bare electrolyte, the abundant uncoordinated water molecules will dynamically form hydrogen bond networks. At the Zn anode surface, polar molecules will be oriented and specifically adsorbed on the IHP of EDL. These phenomena are likely to trigger water-induced side reactions.
In the case of bulky H2O, three mainstream approaches emerge accordingly, namely separating H2O–H2O clusters, minimizing H2O content, and impeding H2O diffusion. Regarding surficial H2O, efforts have also been devoted to constructing a water-poor EDL.
Separating water clusters. The hydrogen bond network is a vital factor for considering the bulk water state. Bulky H2O in the electrolyte mainly exist in a hydrogen bonding cluster state. Compared with the completely free state, the notorious H2O–H2O bonded state exhibits stronger electrochemical reduction trends.50 Given this, breaking the intermolecular hydrogen bonds in water inhibits side reactions.
Cosolvents are usually reported as hydrogen bond regulators between water molecules, restraining side reactions. Given the hydrogen bond chemistry, polar groups with electronegative atoms can be regarded as screening indexes for selecting cosolvents. These polar groups in cosolvent molecules usually coordinate with Zn2+ simultaneously. Representatively, alcohols are proven to be effective as cosolvents due to their hydroxy groups, such as ethylene glycol (EG) (Fig. 10a).79 Further, carbonyl or carboxyl groups may be more effective due to their relatively larger polarity. Huang's team reported the use of tetramethylurea featuring carbonyl groups as a cosolvent, which exhibited improved hydrogen bond regulation and Zn2+ solvation structure optimization effects (Fig. 10b).80 Nonetheless, the diversity of organic solvents accompanied by latent toxicity or flammability is still confusing, requiring corresponding evaluations based on different properties. As an organic small molecule with a large dielectric constant, high flash point and spontaneous combustion point, low viscosity, and low cost, formamide was employed as an effective cosolvent.81 Benefiting from the hydrogen bond acceptor C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O parts and donor N–H parts, formamide–H2O clusters would be formed to replace the H2O–H2O cluster, giving rise to a more stable Zn anode (Fig. 10c and d). However, there is still a gap to bridge, as an accurate indicator to evaluate the organic cosolvents from thermodynamics level is lacking. To address this, Wang's group proposed the use of Et(30), a parameter reflecting polarity and hydrogen bonding capability, as the polarity scale.82 As shown in Fig. 10e, cosolvents with a too low Et(30) interact weakly with water or Zn2+, while those with a too high Et(30) prevent the entry of anions in CIP structures. Thus, there is a volcanic correlation between cosolvent Et(30) value and battery CE performance, and the DMSO cosolvent featuring appropriate 45.1 Et(30) value harvested the highest CE (Fig. 10f).
O parts and donor N–H parts, formamide–H2O clusters would be formed to replace the H2O–H2O cluster, giving rise to a more stable Zn anode (Fig. 10c and d). However, there is still a gap to bridge, as an accurate indicator to evaluate the organic cosolvents from thermodynamics level is lacking. To address this, Wang's group proposed the use of Et(30), a parameter reflecting polarity and hydrogen bonding capability, as the polarity scale.82 As shown in Fig. 10e, cosolvents with a too low Et(30) interact weakly with water or Zn2+, while those with a too high Et(30) prevent the entry of anions in CIP structures. Thus, there is a volcanic correlation between cosolvent Et(30) value and battery CE performance, and the DMSO cosolvent featuring appropriate 45.1 Et(30) value harvested the highest CE (Fig. 10f).
 | ||
| Fig. 10 (a) Schematic of the coordination structure of Zn2+ in H2O/EG cosolvent electrolyte, presenting the effects of EG on the Zn anode reversibility. Reproduced with permission.79 Copyright 2020, Elsevier. (b) Schematic of the optimized solvation structure, bulk electrolyte, and interface chemistry with the addition of tetramethylurea. Reproduced with permission.80 Copyright 2023, Elsevier. (c) Cyclic structure of the FA–H2O complex. (d) Comparison of Zn2+ solvation sheath and the corresponding interfacial interaction between the pure aqueous electrolyte (top) and the FA–H2O cosolvent electrolyte (bottom). Reproduced with permission.81 Copyright 2023, the Royal Society of Chemistry. (e) Schematic of the variation in solvation structure with different Et(30) values. (f) Relationship between the CE of Zn anode and Et(30) values of various cosolvents. Reproduced with permission.82 Copyright 2025, Open Access. (g) Summary of the H2O/Zn2+ molar ratio of some representative related researches with the experiential and theoretical thresholds presented to comprehend “lean water”. (h) Conventional hydrogel electrolyte under much and lean-water conditions (confined Zn2+). (i) Ion transportation for hydrogels under lean-water conditions by virtue of ion transportation channels and lubrication effects. (j) Functional mechanism of zwitterion hydrogel electrolyte. (k) LSV curves of zwitterion hydrogel electrolytes with different (20, 40, and 60 wt%) water contents as well as the PAM and PAA conventional hydrogel electrolytes. Reproduced with permission.83 Copyright 2023, Open Access. (l) Digital photograph of the proposed viscoelastic soft solid electrolyte, and schematic of the corresponding composition and advantages. Reproduced with permission.84 Copyright 2024, Wiley-VCH. | ||
Minimizing the water content. Interior water content indicates uncoordinated water activity, and designing lean water electrolytes is an effective approach for inhibiting side reactions. To accurately bridge the water content and the Zn2+ coordination environments, water weight percentages may be insufficient, so we attempt to define “lean water” by the molar ratio of H2O to Zn2+ herein. As exhibited in Fig. 10g, based on many representative related researches, it seems that a H2O to Zn2+ molar ratio of about 10 is the transition point from a water-sufficient to water-poor condition.24,84–90 However, the exact definition of “lean water” is still debatable; since theoretically Zn2+ is inclined to take on hexahydrate coordination structure, below 6 molar ratio can represent the elimination of undesirable Zn(H2O)62+. Given this, we believe that a molar ratio of H2O to Zn2+ below 10 can be defined as the experiential threshold of “lean water” electrolyte, and under this condition, the bulk phase water activity is significantly inhibited with fragmented hydrogen bond networks. Meanwhile, a H2O to Zn2+ molar ratio of 6 ought to be the theoretical threshold of “lean water” electrolyte, below which value the detrimental species Zn(H2O)62+ is significantly decreased, as showcased in Fig. 10g.
Liquid phase-based lean water electrolytes have been extensively reported. Eutectic electrolyte is an example, characterized by intrinsic thermodynamic stability from redistributed hydrogen bonds and minimized water content. Typically, Cui's team reported a hydrated eutectic electrolyte composed of the hydrated Zn salt Zn(ClO4)2·H2O and succinonitrile ligand without the addition of extra water.85 Their lean water system, featuring ligand-oriented Zn2+ solvation sheaths, effectively inhibited passivation, corrosion, and hydrogen evolution. Besides, an [Emim]OTf ionic liquid-based electrolyte with only 16.7 wt% water content was reported, which could form a hydrophobic trilayer surficial adsorption interface toward a mechanically graded in situ SEI to safeguard the Zn anode.86
Another effective system is a quasi-solid-state lean water hydrogel, which can reduce the activity of water through all three approaches. Achieving a delicate H2O ratio-related balance between thermodynamics and kinetics is crucial for lean water hydrogel electrolytes (Fig. 10h). Zhi's group achieved this by proposing the lubrication effect of limited water toward rapid Zn2+ transport (Fig. 10i and j).83 In their hydrogel electrolyte, polymerized by zwitterionic monomer 3-(1-vinyl-3-imidazolio)propanesulfonate (VIPS), the 20 wt% water content sample was proven to be the optimum. The lean H2O molecules could interact with the charged groups, consequently accelerating the kinetics and achieving an outstanding ionic conductivity of 2.6 mS cm−1. Meanwhile, the limited H2O that mainly contributed to the formation of lubrication films would not allow the formation of intermolecular hydrogen bonds and Zn2+ hydration, maintaining a wide temperature window (Fig. 10k).
Soft viscoelastic materials are also promising candidates for soft solid lean water electrolytes. Recently, Li's group developed a viscoelastic soft solid electrolyte created by nano-SiO2, inducing a liquid-to-solid transition in Zn(BF4)2 salt-dissolved oligomer (glycerol polyoxyethylene-b-oxypropylene ether, GPE) and H2O liquid solution (Fig. 10l).84 The electrolyte could form a thixotropic gel structure with good fluidity under shear and stability under rest via the fracture and reconstruction of the silica network, respectively. The abundant oxygen-containing groups in the GPE matrix and nano-SiO2 filler could restrain H2O molecules through hydrogen bonds, thus reducing the activity of H2O. Notably, the lubrication effect of bound H2O and BF4− absorption by SiO2 would facilitate Zn2+ transport in parallel.
Desymmetrization of bulk phase water distribution. Although excessive water is detrimental to the Zn anode, it can accelerate the cathodic reaction kinetics by activating the lattice of the cathode materials.87 Based on lean water electrolytes, to further improve the performance of full cells with promising Zn anode reversibility, asymmetric electrolytes containing water-poor and water-rich phases have been developed. The main challenge in the mechanism is separating these two phases without spontaneous mutual diffusion. The main factor for localizing water at the cathode side is the chemical potential, and realizing a balance in the chemical potential of active water species between the two phases is necessary.
Niu's group coated a triethyl phosphate (TEP)-based electrolyte-wetted hydrophobic polypropylene separator on the Zn anode to construct a binary phase electrolyte interphase (Fig. 11a).91 The conversion of the solvation structure between [Zn(H2O)x]2+ and [Zn(TEP)n(H2O)y]2+ during Zn2+ migration and reduced water content at the anode side could effectively inhibit undesirable side reactions (Fig. 11b). Nonetheless, this method is dependent on a hydrophobic separator for physical isolation.
 | ||
| Fig. 11 (a) Schematic of the binary phase electrolyte in a cell and the corresponding mechanism of the binary phase electrolyte equilibrium in the case of hydrophobic polypropylene separator. (b) Schematic of Zn2+ solvation structure evolution and interfacial side reaction inhibition effects in the binary phase electrolyte. Reproduced with permission.91 Copyright 2023, Wiley-VCH. (c) Photographs of the miscibility of different nonaqueous cosolvents and electrolytes with water and aqueous electrolytes at ambient temperature, and the combustion tests of the methanol-based homogeneous phase and acetonitrile-/FDMA-based nonaqueous phases in the bottom panels. (d) Snapshots of the self-phase separated electrolyte at the initial chaotic state (left panel) and after 30 ns (right panel) of molecular dynamics simulation, with the nonaqueous phase region shown in purple color. (e) Cycling performance of a Zn||V2O5 pouch cell with a self-phase separated electrolyte at 0.5 A g−1 under repeated rollover. Reproduced with permission.87 Copyright 2025, the American Chemical Society. Top (f) and bottom (g) surfaces of Janus hydrogel electrolyte. (h) Schematic of the polymer composition on the top and bottom surfaces. (i) Schematic of the Zn‖NVO full cell using the Janus hydrogel electrolyte with gradient pore structures. Reproduced with permission.92 Copyright 2024, the Royal Society of Chemistry. | ||
To improve this method, Kang's group put forward a self-phase separated electrolyte, along with proposing rational screening principles.87 As shown in Fig. 11c, it was found that protic solvents generally possess small Hildebrand's solubility parameter disparities with water and spontaneously form a homogeneous phase with water. Regarding hydrophilic aprotic solvents, although phase separation occurred after adding Zn(TFSI)2 to the nonaqueous solvent and ZnSO4 to water, this stratification was thermodynamically metastable. By contrast, hydrophobic and aprotic fluorinated solvents were characterized by inherent nonflammability and could stratify with water. Consequently, a nonflammable hydrophobic and aprotic solvent, N,N-dimethyl trifluoroacetamide (FDMA), was employed to construct a nonaqueous phase. Consequently, a self-phase separated electrolyte could be realized, as verified by molecular dynamics simulation (Fig. 11d). The assembled pouch cell delivered stable cycling within 100 cycles during the rollover process (Fig. 11e).
Additionally, hydrogel-based asymmetric electrolytes have also been reported, which effectively avoid the mutual dissolution issues of liquid electrolytes. Zhu et al. proposed an asymmetrical mold method to achieve a hydrophilicity gradient in Janus hydrogel electrolytes.92 As characterized in Fig. 11f–i, the fabricated Janus hydrogel featured increasing pores and hydrophilicity from the Zn anode side to NVO cathode side, satisfying the Zn anode and cathode requirements simultaneously. However, with regard to gradient or heterogeneous hydrogel structures, there has been less exploration into the evolution of Zn2+ solvation structures and Zn2+ migration behaviors along the gradient or across the heterogeneous interphase and thus deserves more investigation.
Constructing water-poor electric double layer. Since water electron-accepting process occurs at the Zn anode/electrolyte interphase, establishing a physical obstruction to repel surficial H2O from the EDL facilitates the suppression of both chemical and electrochemical side reactions.
The construction of an artificial insulative interphase is a direct and feasible strategy. Initially, some inorganic oxide layers, as a physical barrier at the Zn anode surface to inhibit the reduction of water, have been reported such as CeO2, CaCO3, and ZnSe.19,93,94 However, these inorganic oxide interphases generally feature rigidity with a relatively higher Young's modulus, implying shedding or fracture issues during repeated cycling. Moving forward, organic interphases were investigated due to the appropriate Young's modulus and water-capture abilities of their functional groups or polymer chains. Polymer interphases are generally characterized by flexibility, which can accommodate volume changes and alleviate shedding or fracture. Hydrophobic properties can endow the polymer interphase with the ability to prevent electron acceptance of surficial water, such as (C2F4)n chains.95 The Young's modulus of metal–organic frameworks (MOFs) should fall between those of inorganic oxides and polymers, thus possessing the properties of rigidity and flexibility in parallel. The nanoscale confinement effect is a unique feature of MOFs, which is different from the hydrophobicity requirements of other materials. Water molecules will be strongly affected by the space restrictions and noncovalent interactions resulting from the massive hydrophilic groups on the inner walls of MOFs, consequently suppressing water activity (Fig. 12a).96 Our group reported a continuous amorphous MOF coating layer to safeguard the Zn anode, which could avert the sluggish kinetics caused by traditional crystal MOFs while realizing a lifespan of 7900 h in symmetric cells (Fig. 12b).28
 | ||
| Fig. 12 (a) Schematic of nanoscale confinement effect by MOFs. Reproduced with permission.96 Copyright 2023, Elsevier. (b) Schematic of the contribution of amorphous ZIF-8 coating to Zn anode reversibility. Reproduced with permission.28 Copyright 2023, the American Chemical Society. (c) Possible formation pathways of bilayer SEI. (d) Zn2+ transport behaviors and structural evolution on bilayer SEI during Zn deposition. Reproduced with permission.99 Copyright 2024, Open Access. (e) Hydrogen bond formation of SDE with H2O and SO42−, and the calculated interaction energy of hydrogen bonding between different H-bond acceptors and H-bond donors. (f) Adsorption energy and corresponding modes of SDE and H2O on the Zn(002) facet. (g) Schematic of the enhanced Zn anode stability in the SDE-added ZnSO4 electrolyte. Reproduced with permission.101 Copyright 2024, Wiley-VCH. (h) Adsorption energy and corresponding modes of triethylene glycol with horizontal and vertical adsorption on the Zn anode. (i) Schematic showing the adsorption of triethylene glycol on Zn surface and its effects. Reproduced with permission.102 Copyright 2024, Wiley-VCH. (j) Adsorption energy of various ions on Zn metal. (k) Schematic of the improved Zn anode reversibility induced by the cation–anion synergistic effect of BmBr. Reproduced with permission.103 Copyright 2025, the American Chemical Society. (l) Voltage profiles of three-electrode beaker cells at 1 mA cm−2 and 1 mA h cm−2, evidencing the markedly amplified surface overpotential. (m) Tafel curves of the bare ZnSO4 electrolyte and the cordycepin-added electrolyte. (n) Schematic of Zn plating/stripping behavior with or without cordycepin additive. Reproduced with permission.104 Copyright 2024, Open Access. | ||
The aforementioned examples are ex situ interphases that undergo pretreatment. By contrast, an in situ interphase may be more feasible, which is generally generated spontaneously when some active components in the electrolyte feature sufficiently low HOMO energy levels that exceed the electrochemical window, thereby resulting in electroreduction during cycling. Based on this, the entry of these electroreduction active species into the Zn2+ solvation structure and the ordered active species’ surficial adsorption configuration may facilitate the compactness of the in situ SEI.86,97,98 In addition, the uniformity of the microstructure and components of the in situ SEI should be exquisitely regulated owing to the complexity of Zn salts and electrolyte solutions. Chen's team proposed a robust bilayer SEI derived from 1,3-dimethyl-2-imidazolidinone (DMI) additive.99 The in situ electrochemical decomposition of DMI during cycling promoted the formation of a crystalline ZnCO3-rich outer layer and an amorphous ZnS-rich inner layer, with an overall C/N-rich organic bulk phase (Fig. 12c). The robust inorganic compositions inhibited dendrite formation, while flexible organic compositions accommodated volume changes during cycling, and the amorphous ZnS-rich inner layer homogenized Zn2+ transport (Fig. 12d). An ultra-high Zn utilization of 98% was realized. It is also worth mentioning that with regard to Zn salts, Zn(OTf)2 has been extensively investigated for in situ SEI construction. Especially, different from the aforementioned major decomposition approach, the chemistry of OTf−-induced SEI formation could be attributed to the condensation of Zn trifluorosulfonate hydroxide (Zn(OTf)x(OH)y·nH2O, ZTH). Although ZTH can be generated on the Zn anode surface during long-term stripping/plating in bare Zn(OTf)2 electrolyte, its inhomogeneity impedes the protective effects. In this case, a cathodic synthetic route toward a uniform ZTH layer was developed, mainly centering on the co-precipitation mechanism via released Zn2+ coordination with the anode surface-adsorbed OTf− and OH−.100 The optimum conditions were regulated at a current density of 0.1 mA cm−2 and duration of 24 h, and thus, the grown ZTH SEI endowed the Zn anode with over 80% DOD.
Without the construction of an artificial interphase, specific adsorption using electrolyte additives is another method to repel surficial water. According to the classic Gouy–Chapman–Stern theory, the EDL structure consists of a compact layer and diffusion layer, showing linear potential change and exponential potential change, respectively. The fixed layer is composed of IHP and an outer Helmholtz plane (OHP), and strong polarity water molecules are orientationally adsorbed on the IHP to cause Zn anode surface hydration. Consequently, the essential effect of these additives is to regulate the EDL structures with minimized adsorbed water. It is governed by the adsorption energy of the additives, with the affinity for the Zn anode afforded by polar functional groups. With the adsorption energy satisfied, other structural aspects of additive molecules, encompassing spatial arrangements and volumetric hindrance, have an influence on this. Functional groups are the primary aspect for consideration, covering the interactions not only with the Zn anode but also between two molecules. Song's group proposed a hydrogen bond interlocking mode derived from the 2,2’-sulfonyldiethano (SDE) electrolyte additive with two pairs of hydrogen bond acceptors (SO) and hydrogen bond donors (O–H) (Fig. 12e).101 These SDE additives would construct a sieving-type EDL structure by interfacial adsorption with interlocked hydrogen bonds, which repel H2O and SO42− in the EDL (Fig. 12f and g). Regarding spatial arrangement, Cao's team leveraged straight-chain molecules featuring strongly electronegative terminal hydroxyl groups for selective and horizontal adsorption on the Zn anode.102 The uniform coverage of straight-chain molecules repelled water from the Zn anode surface and guided the exposure of Zn(002) during electrodeposition (Fig. 12h and i). However, the aforementioned molecules will enter the Zn2+ solvation structure. For small additive amount, Zn2+ solvation structure may not be changed to achieve rapid bulky ion transporting, but a water poor EDL can still be guaranteed. Recently, Lu's team reported the synergistic effects of imidazole-based cations and halide anions using a small amount of ionic liquid additive 1-butyl-3-methylimidazolium bromide (BmBr).103 Halide anions would be preferentially anchored onto the Zn anode surface, facilitating the subsequent adsorption of imidazolium-based cations on the Zn anode (Fig. 12j). This anion-cation adsorption mode would effectively contribute to a water-poor Zn anode surface (Fig. 12k). Trace amount of large molecule additives, with high volumetric hindrance, are also reasonable, such as the cordycepin proposed by our group.104 Cordycepin exhibited strong affinity with the Zn anode but did not participate in the Zn2+ solvation structure. Its specific adsorption with large volumes on the Zn anode effectively constructed a water-poor EDL toward corrosion resistance. Besides, the specific adsorption of cordycepin amplified the surface overpotential, which regulated the nucleation behavior and refined the size of the Zn nuclei (Fig. 12l). The induced fine Zn grain-based plating/stripping mode with inhibited side reactions endowed the Zn anode with high reversibility (Fig. 12m and n).
3.2.1.3. Confining protons. Zn salt-based aqueous electrolytes generally take on a slightly acidic state with free moving protons. Despite their relatively low concentration, the rapid transport rate and high electron-accepting tendency of protons make them fatal to the Zn metal anode. Given their special Grotthuss mechanism-based hopping behavior, the following two aspects are valid to inhibit the mass transfer of protons. One is impairing the hydrogen bond-based transporting skeleton to restrain proton hopping, and the other is capturing protons to prevent them from combining with water and forming H3O+. In addition, to meet the cathode needs, localizing H+ in asymmetric systems has been investigated. Regarding surficial protons electron acceptance process, protons feature a small hydrated radius and good penetrability, and the same positive electrical properties shared with protons and Zn2+ makes electrostatically screening protons without influencing Zn2+ almost impracticable. Thus, a reasonable surficial perspective is ideal for enhancing the chemical energy barrier for the HER.
Destroying the hopping medium. The hydrogen bond network, as the medium for proton hopping, is the focal point. Breaking the hydrogen bond network is valid to impede the mass transfer of protons. It should be noted that the migration of both uncoordinated water and protons is correlated with the intermolecular hydrogen bonds in water, and thus, many hydrogen bond regulation strategies are effective to restrain proton mass transfer.
With regard to cosolvents, the presence of special polar groups, serving as hydrogen formation sites, can be considered a screening criterion. Recently, a Zn(OTf)2-based system containing dimethylacetamide (DMA) and benzyl alcohol (BA) was proposed.105 In this system, DMA molecules, featuring carbonyl groups as hydrogen bond acceptors, could effectively disrupt hydrogen bond networks to obstruct proton hopping. Further, with the aid of BA-assisted SEI formation, a more reversible Zn anode was realized.
The use of electrolyte additives is also an effective method for hydrogen bond regulation. Different from the above-mentioned strategies, given the low concentration of protons, the use of additives can be regarded as a bulk phase strategy here. The aspects of special polar groups should also be considered. Representatively, Tang's team proved the effectiveness of ectoine additive as a proton transport inhibitor by virtue of its high polarity with multiple sites for hydrogen bond formation.106 As shown in Fig. 13a, ectoine would form hydrogen bonds with water that are stronger than the intermolecular hydrogen bonds of water. Consequently, the ectoine additive increased the energy barrier for the dynamic reconfiguration of hydrogen bond networks, thereby impeding proton hopping. Further, a trace additive amount for confining proton transport was realized. Recently, tetrahydro-4H-pyran-4-one (THPO), featuring carbonyl and ether groups, has been reported as a proton blocker, effectively inhibiting undesirable side reactions with only 1% additive concentration.107
 | ||
| Fig. 13 (a) Schematic of proton transport based on the Grotthuss mechanism without (top) and with ectoine (bottom). Reproduced with permission.106 Copyright 2023, Wiley-VCH. (b) Mechanisms of H+ transfer in free water and PVA without salting-out, and the possible ways that hinder the transfer of H+ in the hydrogel with strong salting-out effects. Reproduced with permission.108 Copyright 2022, Wiley-VCH. (c) Schematic of Zn plating by virtue of ammonium persulfate additive. Reproduced with permission.49 Copyright 2024, Wiley-VCH. (d) pH buffering effect of the pyridinium/pyridine conversion. (e) Real-time electrolyte pH near Zn anode during discharging and charging at a current density of 1 mA cm−2. Reproduced with permission.109 Copyright 2023, Open Access. (f) Schematic of taurine's molecular structure. (g) Reaction path for in situ hybrid SEI formation. (h) Schematic of the Zn anode protection effects of the taurine additive. Reproduced with permission.48 Copyright 2023, Wiley-VCH. (i) Schematic of the Zn2+ deposition processes on bare Zn and the ZP@Zn anode. Reproduced with permission.110 Copyright 2022, Wiley-VCH. (j) Schematic of a bilayer SEI featuring a homogeneous ZnF2-rich inner layer and a mechanically stable PANI outer layer formed by the polymerization of aniline. (k) Reversible H+ holder mechanism in PANI layer. (l) Cycling stabilities of Zn anode with 70.8% DOD at 40 mA cm−2 and 40 mAh cm−2. Reproduced with permission.111 Copyright 2025, Wiley-VCH. (m) Schematic of the asymmetric electrolyte design strategy. (n) Electrochemical window of the designed asymmetric electrolyte and PAM hydrogel electrolyte measured by LSV at 5 mV s−1, with the inset showing an enlarged view of the Zn2+ deposition curves. (o) Cyclic stability of a large-capacity (∼1100 mAh) Zn||MnO2 pouch cell with an asymmetric electrolyte. Reproduced with permission.112 Copyright 2023, Open Access. | ||
In the case of polymer-based electrolytes or separators such as hydrogels, the Hofmeister effect is worth to consider, which is targeted at breaking the hydrogen bond framework. Discovered from the phenomena of protein stabilization, it has been harnessed in aqueous batteries.113 Kosmotropic ions (e.g., SO42−) facilitate intermolecular hydrogen bond formation, while chaotropic ions (e.g., ClO4−) cause opposite outcomes.113,114 Based on this, Chen's group put forward a PVA-based hydrogel separator as a proton barrier in an acidic electrolyte.108 As illustrated in Fig. 13b, the salting-out effect of the SO42− and OAc− anions resulted in the formation of disordered and discontinuous hydrogen bonds between H2O in the hydrogel networks. Therefore, H+ transport would be effectively confined to prevent the HER and corrosion.
Reversibly capturing protons. The reversible capture of protons is generally based on pH buffering effects. The corresponding chemistry is based on a couple of conjugated Lewis acid–base pairs, which can combine with H+ or OH− to stabilize the pH value and inhibit side reactions.
Employing pH buffering additives has been widely reported. As we analyzed before, it can be assumed that both chemical and electrochemical side reactions are closely related to the pH value, where the acidity of the bulk electrolyte is the initial side reaction trigger, and the gradually induced Zn anode surficial alkalinity is the subsequent side reaction promoter. During plating/stripping cycling, pH increases explicitly at the Zn anode surface due to the HER-induced H+ consumption, and gradually fluctuates with the competition between the HER and ZHS formation. Three types of pH buffering additives have been reported, which all possess functions beyond restraining H+ to synergistically protect the Zn anode. The first type is ionizable NH4+ and weak acid radical-based salt, featuring pH buffering and effects. Ammonium persulfate is an example, with S2O82− stabilizing the pH and NH4+ electrostatically shielding uneven tips to improve the reversibility of the Zn anode (Fig. 13c).49 The second type is organic heterocyclic compounds featuring nitrogen-containing groups, which not only serve as donor or acceptor sites for hydrogen bonds but also induce preferential adsorption behavior to regulate deposition. In this realm, Guo's group reported pyridine and imidazole as two effective electrolyte additives, where the conversion of imidazolium/imidazole or pyridinium/pyridine achieved a stabilized pH value to inhibit side reactions (Fig. 13d and e), respectively.109 The third type is nitrogen-containing organic acid, which is also electrochemically reducible for the in situ SEI formation. Huang's team employed taurine as an effective additive toward both pH buffering and in situ SEI construction, protecting the Zn anode from active H2O or H+ (Fig. 13f–h).48
From the surficial perspective, analogous to the above-mentioned additives, reversible proton capture by pH buffering interphases can avert the contact of surficial protons with the Zn anode. Zhou's group reported the construction of a Zn3(PO4)2·4H2O-coated Zn anode (ZP@Zn) with proton reservoir properties via a mixed solution soaking method.110 The phosphate group exhibited proton self-dissociation ability, which reversibly stocked protons along with a variation in pH value (Fig. 13i). PO43− would be reversibly transformed to HPO42− and H2PO4− by trapping protons and is recovered during discharging. The pH value at the ZP@Zn anode surface remained stable, effectively inhibiting the HER process. To further avert preparation difficulties of ex situ modification, an in situ proton buffering interphase has been reported via electrolyte engineering. Recently, Huang's team proposed a tailored organic–inorganic bilayer in situ SEI strategy by employing an aniline-moderated Zn(OTF)2 electrolyte (Fig. 13j).111 During electrodeposition, Zn(OTF)2 underwent a decomposition process to in situ form a fluorine-containing inorganic interfacial inner layer. Simultaneously, the aniline monomers in the electrolyte were in situ polymerized to generate PANI, forming an organic polymer outer layer. The bilayer in situ SEI could effectively inhibit Zn dendrite growth through prolonged Sand's time and uniform surficial current density. The pH stabilization effect was derived from the PANI outer layer. As shown in Fig. 13k, the N– groups would be electrochemically reduced to –N−– during discharge, which could capture surficial H+ to form –NH–. The reverse processes occurred when charging, signifying the proton-holding properties. Consequently, the bilayer-modified Zn anode ensured cycling at large areal capacities without side reactions, achieving stable cycling at 70.8% DOD (Fig. 13l).
Desymmetrization bulk phase proton distribution. In the case of Mn-based and V-based oxides, the co-insertion of H+ and Zn2+ facilitates the full utilization of cathode materials. To achieve balance, analogous to the situations in uncoordinated water confinement, asymmetric structures have also been tasked with suppressing H+ migration to the Zn anode side and creating a partly H+-concentrated cathode side. The chemical potential of H+ between two phases, as the driving force for diffusion, is the governing quantity. However, the positive charge of protons gives them a spontaneous response under an electric field, which makes localizing protons more challenging.
The developed selective membrane can be directly employed as separators, with the virtue of polishing up the performance for H+ required MnO2 cathode in parallel, such as the Zn||Mn hybrid aqueous battery developed by Qiao's team.115 It allowed the penetration of H+ or OH− for balancing the charges and preventing the direct neutralization of anolyte and catholyte, thus optimizing the two electrodes. However, the cost of selective membranes is too high to commercialize them.
Developing (quasi-)solid asymmetric electrolytes is worthy of attempt as well. Zhi's team reported an example composed of a highly Zn2+ conductive inorganic solid-state electrolyte at the Zn anode side and a PAM hydrogel electrolyte at the MnO2 or V2O5 cathode side (Fig. 13m).112 The side reaction issues of the Zn anode were tackled with an extended electrochemical window, along with an improvement in the cathode electrochemical performance simultaneously (Fig. 13n). Their system was verified in full cells with up to ∼5.5 mAh cm−2 (Zn||MnO2) and ∼7.2 mAh cm−2 (Zn||V2O5) super high-areal capacities, which exhibited outstanding capacity retention after cycling. Their ∼1.1 Ah Zn||MnO2 pouch cell with asymmetric electrolyte retained 75.8% of its initial capacity after 280 cycles, outperforming the PAM hydrogel electrolyte sample (Fig. 13o).
Enhancing the HER theoretical overpotential. Given the Volmer step (H+ + e− → Hads), Tafel step (2Hads → H2), and Heyrovsky step (Hads + H+ + e− → H2) of proton-induced HER, Hads is an unavoidable intermediate species. Hence, regardless of kinetics factors, the corresponding theoretical overpotential of HER can be regulated by the H adsorption energy. Observing that the specific decomposition processes of water share similarities with proton-induced HER, such as the emergence of the Hads intermediate species, many related strategies can alleviate both proton and water induced HER.
To increase the HER theoretical overpotential, constructing heterogeneous anti-HER metal, such as In, or alloy interphase by simple replacement reactions has been highlighted.116 The high HER theoretical overpotential and zincophilicity of In endowed the Zn anode with a smooth electrodeposition surface and inhibited its side reaction tendency (Fig. 14a–c). Further, inspired by the lotus effect, Zhang's group described a Pb and polyvinylidene difluoride (PVDF) hybrid interphase.117 Anti-HER Pb nanoparticles were uniformly embedded on a hydrophobic PVDF matrix, which endowed the hybrid interphase with multi-channel side reaction inhibition and showcased superiorities over the bare Zn anode and single heterogeneous metal-modified Zn anode (Fig. 14d).
 | ||
| Fig. 14 (a) Schematic of Zn deposition morphology evolution on (top) bare Zn anode and (bottom) In-coated Zn anode during the plating process. Cross-sectional SEM images of Zn deposits on (b) bare Zn and (c) In-coated Zn anode. Reproduced with permission.116 Copyright 2022, the Royal Society of Chemistry. (d) Evolution of Zn metal anode with different interphase layers after plating and long-term cycling, with the heterogeneous metal-polymer hybrid interphase layer exhibiting best anti-side reaction properties. Reproduced with permission.117 Copyright 2024, the Royal Society of Chemistry. (e) Schematic of the preparation process of Pb-modified Zn anode. (f) H+ adsorption energy and (g) ΔGH* for HER of PbSO4(100), Pb(111), Zn(002), Zn(100), and Zn(101) surfaces. (h) Schematic of the effects of Pb(OAc)2 additive on Zn plating/stripping. Reproduced with permission.118 Copyright 2023, Wiley-VCH. | ||
The above-mentioned conductive artificial interphases contact the electrolyte in the shelved state, safeguarding the Zn anode from the HER. The situation changes after plating processes, given that the substrate is covered by electrodeposited Zn, where inhibiting galvanic corrosion may be the main corresponding factor.119 From a different angle, Zhou's group designed a Pb-containing interphase through the facile replacement reaction between Zn and Pb2+ (Fig. 14e).118 Subsequently, Pb(OAc)2 salt was added to ZnSO4 electrolyte for the formation of PbSO4 precipitate. The released trace Pb2+ deposited a Pb layer on the Zn plating layer, which safeguarded the newly deposited Zn from protons. Zn stripping was preferential, accompanied by the accumulated Pb, and the accumulated Pb was stripped at the end. Both Pb and PbSO4 were characterized by a low affinity for protons and a high energy barrier for the HER (Fig. 14f and g). The modified Zn anode completely covered by the Pb-containing interphase achieved a great enhancement in proton adsorption resistance, showcasing significant anti-corrosion properties (Fig. 14h).
Given the small proportion of protons in the electrolyte, simply inhibiting electron acceptance by protons may not be sufficient to realize a highly reversible Zn anode. As a result, the aforementioned artificial interphases are generally characterized by multifunctional properties like simultaneous zincophilicity and Zn2+ flux optimization effects, which will be further discussed in the following sections on kinetics approaches.
Boosting Zn2+ mass transfer. Mass transfer of Zn2+ in the electrolyte bulk phase is directly related to surficial Zn2+ supplement for Zn redox. Unsatisfactory Zn2+ mass transfer will trigger severe concentration polarization. Zn2+ mass transfer can be associated with the electrolyte ionic conductivity and Zn2+ transference number (tZn2+). Given the intrinsic ionic conductivity merit of aqueous electrolytes, tZn2+ will be mainly discussed herein.
The dielectric constant is an important factor to focus on toward decoupled cation–anion pairs. In a given system, the critical distance for ion-pair formation, q, is governed by the systematic dielectric constant, ε, according to Bjerrum's treatment, with the hypothesis that ion-pair formation occurs if the interionic distance is smaller than q:
 | (14) |
 | ||
| Fig. 15 (a) Schematic of the merits of kinetic compensation mechanism compared with other cosolvent strategies. Reproduced with permission.120 Copyright 2025, the American Chemical Society. (b) Structure and binding energy of β-CD@ClO4− complex, with blue, red, white, and green atoms representing C, O, H and Cl, respectively. (c) Independent gradient model analysis of β-CD@ClO4− complex. Reproduced with permission.121 Copyright 2022, Wiley-VCH. (d) Schematic of Zn plating in the “anions-in-colloid” system. Reproduced with permission.122 Copyright 2024, Wiley-VCH. (e) Schematic of the fabrication process and network structure of the single Zn2+ conductive hydrogel electrolyte. Reproduced with permission.123 Copyright 2023, Wiley-VCH. (f) Zn2+ hopping behaviors in the hybrid interphase layer. Reproduced with permission.124 Copyright 2020, Wiley-VCH. (g) Mechanism of tandem adsorption and desolvation of Zn (H2O)62+, originating from the bulk electrolyte, passing through the Janus mesopore channels, and reaching the Zn anode. Reproduced with permission.125 Copyright 2024, the American Chemical Society. (h) Schematic of the electrochemical configuration with the in situ formation of SAB interphase on Zn anode. (i) Schematic of the molecular mechanism of SAB for synchronous in-plane Zn/Zn2+ transfer and reaction. The exterior peptide layers contributed to coordination with Zn/Zn2+, while the hydrophobic interior forms directional desolvation/transport channels. (j) Schematic geometry at the key migration positions of Zn atoms. Reproduced with permission.126 Copyright 2024, Open Access. | ||
Localizing anion migration can be an optimization, and host–guest interaction is generally employed to attain this. β-Cyclodextrin (β-CD) is characterized by a hydrophobic cavity to trap anions, making it a potential candidate. Mai's group initiated the field of β-CD additive, which could effectively trap ClO4− anions through host–guest interactions toward achieving a tZn2+ of 0.878 (Fig. 15b and c).121 Further, Tao's group reported the “anions-in-colloid” hydrated eutectic electrolyte Zn(ClO4)2, β-cyclodextrin (β-CD), and H2O.122 In their system, β-CD, owing to its relatively larger amount, self-assembled into micelles, capturing ClO4− anions and trapping them in the colloid, resulting in a tZn2+ of 0.84 (Fig. 15d). In addition, β-CD functioned as a hydrogen bond regulator owing to the greater proportion of bound water to inhibit the HER and corrosion.
Another aspect worth noting is the transportation behavior of Zn2+. Constructing coordination-based Zn2+ hopping channels can be a strategy, which are extensively reported in polymer-based electrolytes. Typically, Sun's team developed a single Zn2+ conductive hydrogel electrolyte.123 As exhibited in Fig. 15e, it featured a pseudo-polyrotaxane (PPR) structure, composed of poly(ethylene oxide) (PEO) linear polymers and α-cyclodextrin (α-CD) ring-shaped molecules sliding along the network strands. This unique structure realized Zn2+ hopping behavior via coordination with the oxygen atoms on the PEO chains and restricted OTf− anion migration by the α-CD ring-induced spatial restriction. This dual functional effect achieved a tZn2+ of 0.923.
Facilitating Zn2+ desolvation. Solvated Zn2+ will undergo a desolvation process in EDL during deposition, which can be specifically indexed by the desolvation energy, as the corresponding barrier for the desolvation process to overcome. Thus, to facilitate desolvation, lowering the Zn2+ desolvation energy is the inevitable course. Related studies mainly focus on the interphase, harnessing surficial coordinated sites to destroy the Zn2+ hydration sheath.
The interfacial adsorption behavior of some specific structure additives has been investigated. For instance, a fructose additive featuring a circular structure, small size, quasi-planar adsorption configuration, and enhanced electron delocalization was reported.127 The oxygen-containing groups enabled multisite compact adsorption and multisite desolvation via hydrogen bonding with Zn2+ coordinated water, which could lower the Zn2+ desolvation energy from 41.51 kJ mol−1 (bare ZnSO4 electrolyte) to 21.93 kJ mol−1.
Some electron-insulative but Zn2+-conductive artificial interphases are also designed to facilitated desolvation. Aside from ensuring thermodynamics stable, kinetics perspective should further concentrate on ordered Zn2+ expedited transporting channels. For example, Pan's group established an interphase-bridged organic–inorganic layer with localized hydrophobic regions in hydrophilic matrices.124 Zn2+ was likely to hop along the organic–inorganic interphase via coordination in the form of R-SO3−[Zn(H2O)4]2+SO3−-R while the other anions and free H2O could be blocked, resulting in main/side reaction decoupling effects (Fig. 15f). Further, Chao's group decoupled and accelerated Zn(H2O)62+ capture and desolvation via a Janus mesopore accelerator.125 The hydrophilic upper layer facilitated Zn(H2O)62+ capture, while the hydrophobic bottom layer stripped active Zn2+-coordinated H2O to prevent side reactions (Fig. 15g).
Nonetheless, despite the contribution of preconstructed artificial interphases, exfoliation issues may inevitably occur. Therefore, it is of great importance to facilitate the formation of a self-assembled interphase by regulating the electrolytes. Our group previously conducted this by introducing a tiny amount of lipopeptide into ZnSO4 electrolyte, where a self-assembled supramolecular bilayer (SAB) interphase was dynamically formed toward ordered, planar Zn plating/stripping (Fig. 15h).126 As shown in Fig. 15i, the exterior peptide layers coordinated with Zn2+, and the hydrophobic interior parts formed transport channels to facilitate Zn2+ desolvation. Simultaneously, the Zn–N bindings coordinated by the peptide layer elongated surficial Zn–Zn bond lengths compared with the Zn–Zn bonds in bulky atoms, which guaranteed electrode-scaled synchronous in-plane activation (Fig. 15j). This led to rapid electrochemical reaction kinetics in the surficial Zn layer, ensuring stepwise kinetic pathways among neighboring active layers toward planar stripping. The self-assembled SAB prevented side reactions, facilitated uniform electrodeposition, and promoted the kinetics, resulting in stable cycling at an ultrahigh DOD of 90%.
Accelerating in-plane charge transfer. After transporting to the Zn anode surface, Zn2+ will undergo an electron transfer process for electroreduction, which can be evaluated by the kinetics of surficial charge transfer resistance. Accelerating surficial charge transfer will reduce the chemical barrier for Zn redox, thus averting polarization to some extent. In this regard, zincophilicity is an indispensable factor, reflecting the electron transfer trend between the substrate and Zn.
Relevantly, some graphene-based interphases have also been initially reported. Since the low zincophilicity of pure carbon is a weakness, electronegative element modification has been employed.128 Chen's group coated an ultrathin N-doped graphene oxide (GO) on a Zn anode through a one-step Langmuir–Blodgett method (Fig. 16a).129 The N-doped GO interphase facilitated Zn redox, reducing the overpotential and resulting in a dense deposition and less byproduct formation. Nonetheless, although electronegative element doping strategies afford extra zincophilicity, whether these sites will accelerate side reactions or not should be confirmed. Besides, many carbon nanomaterials are not compact microcosmically, and Zn deposition may occur at the interstice such as the interlayer spaces. This will cause more Zn anode surface area to trigger HER.
 | ||
| Fig. 16 (a) Schematic showing the fabrication of ultrathin graphene layers on Zn foil. Reproduced with permission.129 Copyright 2021, Wiley-VCH. (b) Gibbs free energy difference between H adsorption (ΔGH*) on the top site of the doping atom and the neighbor Zn hollow site. (c) Binding energy of a Zn atom on the Zn(002) plane. Reproduced with permission.130 Copyright 2023, Open Access. (d) Schematic of Zn anode reconstruction strategy, with (top) crack and surface failure in surface-modified Zn anodes and (bottom) damage-free surface in bulk-phase reconstruction after cycling. (e) Calculated binding energies of Zn atom on the Zn(001), Zn3N2(321), Zn3N2(400), and Zn3N2(222) facets. Reproduced with permission.131 Copyright 2023, Wiley-VCH. (f) ΔGH* and corresponding adsorption configurations on different crystal facets. (g) Binding energy and corresponding adsorption configurations of a Zn atom on different crystal facets. (h) Schematic of the self-construction effect. Reproduced with permission.132 Copyright 2024, Open Access. (i) Schematic of the grain boundary engineering strategy. (j) Cross-sectional SEM images of Zn anodes after 50 cycles (top) without and (bottom) with grain boundary modification. Reproduced with permission.133 Copyright 2023, Open Access. | ||
Interfacial alloying modification is another representative strategy, which could improve the zincophilicity with the guarantee of a high HER overpotential. To date, the reported alloying Zn anodes include three aspects: metal mixtures, solid solutions, or intermetallic compounds. Their differences lie in the existence of a multi-elemental intermediate phase. Metal mixtures are derived from the direct combination of two or more metals without mutual atomic dissolution and without the signal of a mutually soluble intermediate phase in X-ray diffraction (XRD) patterns. Solid solutions and intermetallic compounds consist of a blend of different metal (sometimes nonmetal for solid solutions) atoms in a certain proportion, with a specific chemical formula. To differentiate, a solid solution is based on a small amount element doping in the metal substrate, while an intermetallic compound is an alloy with a new crystal structure different from its individual components. Unconstrained by its phase diagram, a metal mixture exhibits great diversity and relatively simple synthesis processes such as electrodeposition and fusion methods.134,135 Pu's team conducted the alloy components screening and found the best candidate Bi for Zn-based binary metal mixtures benefiting from its HER-inhibiting properties and zincphilicity, lowering one order magnitude of hydrogen evolution rate than bare Zn (Fig. 16b and c).130 Ternary Zn-based metal mixtures have further been explored, such as electrodeposited Zn-Sn-Bi alloy coating layer characterized by zincophilicity and anti-HER capacity.136 However, for metal mixtures, the mutual immiscibility issue induces the respective crystallization of each component with a nonuniform distribution. This can be averted by solid solutions and intermetallic compounds.
A solid solution is made by adding slight (non)metal elements into the Zn crystal lattice. Wang's team developed a bulk phase reconstruction strategy by synergistically treating Zn foil with nitrogen plasma and thermal radiation, showing superiorities over traditional surface modification (Fig. 16d).131 Zincophilic Zn3N2 species endowed the anode with accelerated kinetics (Fig. 16e). Its corrosion resistance was also improved, featuring a more negative corrosion potential, lower corrosion current, and inconspicuous ZHS formation. Nonetheless, an understanding of solid solution-based alloy anodes remains lacking, with only a few investigations reported to date. Challenges exist in achieving significantly promoted Zn anode reversibility with the addition of a small amount of dopant. Besides, a series of practical criteria for selecting solid solution components is demanding.
For intermetallic compounds, theoretically, a self-supporting large-volume Zn-based intermetallic compounds can only be obtained from smelting, while the critical challenges in preparation direct our focus on the partial Zn anode modification by intermetallic compounds. Recently Yang's group put forward a Cu–In alloy interfacial protective layer through a potential co-replacement route.132 The Cu and In components showed complementary effects, with Cu optimizing the insufficient zincophilicity of In and In mitigating the HER catalytic activity of Cu (Fig. 16f and g). Besides, the alloy interphase underwent self-construction during cycling, which facilitated the establishment of a bottom-up anti-HER ability gradient and top-down zincophilicity gradient (Fig. 16h). Beyond the surface, the bulky grain boundary triggers side reactions due to its elevated reactivity. Zhou's team proposed a grain boundary modification strategy by constructing a Zn–Ti alloy with 0.5 wt% Ti (Fig. 16i).133 Due to the limited solid solubility of Ti in Zn, it was mainly composed of a Zn-rich solid solution and TiZn16 intermetallic compounds. The TiZn16 intermetallic compounds were thermodynamically stabilized at the grain boundaries, which effectively inhibited the occurrence of side reactions. The grain boundaries were evidenced by the Zn nucleation sites, leading to a dense deposition layer without byproducts (Fig. 16j). Nonetheless, given that the mainly reported intermetallic compounds are not free-standing, the existence of metallic Zn may affect the intrinsic properties of intermetallic compounds during cycling and undermine the energy density due to its extra weight. Further endeavors can be devoted to constructing an anode-free system with anti-corrosion and zincophilic intermetallic compounds as current collectors.
Achieving Zn epitaxy. Achieving Zn homoepitaxy with a textured Zn anode surface is an important target for a high Zn utilization ratio. Textured homoepitaxy generally results in an ordered Zn atom arrangement with less or no grain boundaries or dislocations, signifying a rapid reaction rate and fast crystal nuclei growth. In addition, a textured Zn anode can provide higher side reaction resistance than textureless surfaces due to its compact stacking mode with less grain boundaries or dislocation-induced active site exposure for side reaction initiators to attack.
Textured electron conductive substrates are a prerequisite for Zn epitaxy. In 2019, the reversible epitaxial mode of Zn(002) was proposed by Archer's team, where Zn(002)-textured electrodeposition was achieved by hexagonal graphene featuring small lattice mismatch with Zn (Fig. 17a).20 Based on this, lattice mismatch between Zn and the electrodeposition substrate has been regarded as a vital indicator for the feasibility of epitaxy, inspiring more exploration on heteroepitaxy substrates. For example, single-crystal Cu(111), featuring small lattice mismatch and zincophilicity, was also employed for heteroepitaxy later.137,138
 | ||
| Fig. 17 (a) Schematic of lattice matching-induced Zn epitaxial electrodeposition. Reproduced with permission.20 Copyright 2019, Open Access. (b) Surface energy and (c) hydrogen evolution energy barriers of the Zn(002), Zn(100), and Zn(101) facets. Reproduced with permission.37 Copyright 2025, Wiley-VCH. (d) Schematic of Zn atomic arrangements and inter-island stitching in the morphological evolution. Reproduced with permission.141 Copyright 2022, Open Access. Lattice mismatch of Zn(002) with (e) Zn(002), (f) Zn(100), and (g) Zn(101). (h) High-resolution transmission electron microscopy (HRTEM) image of the deposited Zn with corresponding nano-beam electron diffraction pattern (inset). Reproduced with permission.143 Copyright 2024, Open Access. (i) Adsorption energies of Zn atom on Zn(002) and Zn(101) planes. Ab initio molecular dynamics simulations of Zn with four layers deposited on (j) Zn(002) and (k) Zn(101), and the corresponding lattice distortion values for each layer. (l) HRTEM images of the zinc deposits with (top) Zn(002) and (bottom) Zn(101) facets. Reproduced with permission.144 Copyright 2023, Wiley-VCH. (m) Electrodeposition growth model on Zn(002) textured anode. (n) Tafel curves of different textured Zn anodes after 50 cycles with 20% DOD. (o) Electrodeposition growth model on Zn(002) textured anode. Reproduced with permission.140 Copyright 2024, Open Access. (p) SEM images of the prepared Zn foil featuring terrace with steps. (q) XRD pattern of the prepared Zn anode. (r) Adsorption energies and geometries of the Zn atom on the step edge and terrace on a Zn(002) substrate. (s) Interfacial energies of Zn(002)/Zn(002), Zn(002)/Zn(100), and Zn(002)/Zn(101). (t) Electron back-scattered diffraction inverse pole figure maps of Zn deposition on polycrystalline Zn (top) and prepared mono-oriented Zn (bottom), verifying step-edge guided Zn(002) epitaxy. Reproduced with permission.37 Copyright 2025, Wiley-VCH. | ||
Characterized by an hcp crystal structure, (100), (101), and (002) are the three main low-index facets.34 Among these facets, Zn(002) exhibits the lowest surface energy, fastest in-plane diffusion rate, and highest hydrogen evolution energy barrier (Fig. 17b and c).37,126,139,140 The Zn electrodeposition behavior on a single-crystal Zn(002) substrate was verified, proving the validity of homoepitaxy.141 Hexagonal planar Zn islands could be formed during electrodeposition, and these Zn islands would be seamlessly merged into larger single grains with the step edges flawlessly splicing subsequently (Fig. 17d). Nonetheless, the high cost of free-standing single crystal Zn is unacceptable, causing the investigation on polycrystal Zn substrate with one dominate surficial orientation. For instance, Liang's group prepared a Zn(002)-dominant anode via rolling deformation, showing enhanced Zn anode reversibility.142 In this field, one key issue is the failure mechanism of electroplating epitaxy, as investigated by Zhou's team.143 The presence of crystal grains deviating from the [001] orientation within a Zn(002) metal anode caused epitaxy failure due to lattice distortion (Fig. 17e–g). Based on this, they fabricated a [0001]-uniaxial oriented Zn metal anode featuring a Zn(002) single texture. This was achieved via electrodeposition in ZnSO4 electrolyte, where Zn(002) exhibited the lowest surface energy and biggest surface energy differences between other facets toward fast crystal growth based on the Gibbs-Wulff theory. Diminishing lattice distortion, the single [0001]-oriented Zn metal anode realized atomic “∼ABABAB∼” arrangement in Zn homoepitaxy, featuring the intrinsic properties of an hcp structure (Fig. 17h).
However, the nucleation process may be unsatisfactory for Zn(002) due to its relatively weak in-plane bonding, inducing weak nucleation driving forces with a lack of nucleation sites, especially under small current densities. Given this, epitaxy of Zn(101) was also proven to be feasible, which is characterized by stronger binding energy with Zn atoms (Fig. 17i).144 This helped the Zn(101) anode avert the accumulation of lattice distortion with an increase in the number of deposition layers (Fig. 17j–l). In addition, the corrosion resistance ability and critical epitaxy areal capacity of Zn(002) have been questioned, which may result from the relatively larger active area and horizontal crystal growth direction to form thin plates with the largest lateral size/thickness aspect ratio (Fig. 17m).140,145 In this case, Zn(100) was also investigated, where the vertically aligned deposition structures showed the smallest active surface area, which could effectively address corrosion issues (Fig. 17n).140 Besides, among different facets, the surface atom diffusion barrier of Zn(100) plane was only just higher than Zn(002), which would producing thick vertical aligned Zn plates with the smallest aspect ratio and ordered Zn(100) plane facing the separator (Fig. 17o). This is not prone to cause short-circuit under a large epitaxy capacity. However, there is still controversy about the optimal crystal orientation, and the pursuit of a higher goal that further decouple main/side reactions toward the opposite reaction rate trends is warranted.
Considering the above-mentioned issues, constructing directed and ordered nucleation sites on the textured Zn(002) anode can be an optimization strategy. Recently, our group reported a step-edge guided Zn homoepitaxy mechanism based on a melt-spinning method-derived customized Zn foil anode with the dominant Zn(002) orientation exposure (Fig. 17p).37 During the cooling processes in melt-spinning, terrace structures with steps between Zn(100) and Zn(002) could be formed, and Zn(002) exposure would be realized due to its lowest surface energy of hcp intrinsic property (Fig. 17q). The more negative adsorption energy of Zn atoms on the step edges than that on the terrace showed the nucleation favorability of the step edges, and the lowest interfacial energy between the Zn(002) facet guaranteed its homoepitaxy effects(Fig. 17r–t). The ordered distributed nucleation sites afforded by the steps would accelerate the Zn redox process and prevent local Zn accumulation toward amplified epitaxy capacity. This step-edge-guided Zn homoepitaxy strategy governed the side-reaction-free orientational planar epitaxy of Zn with rapid kinetics, achieving a highly reversible Zn anode even at 82% DOD.
3.3. Comparison of strategies
As discussed above, most studies on inhibiting side reactions focus on two parts: Zn anode and electrolyte, as exhibited in Fig. 18. Regarding the Zn anode modification part, there are two mainstream approaches: interfacial alloying and Zn anode texturing. With regard to the engineering of electrolytes, there are multiple strategies including electrolyte additives, cosolvents, Zn salt regulation, hydrogel electrolytes, eutectic electrolytes, (soft) solid lean water electrolytes, and asymmetric electrolytes. The investigation into artificial interphases can be categorized into an overlapping field of Zn anode modification and electrolyte engineering owing to the differences in the ex situ pretreatment and in situ formation mechanisms. Aside from these two parts, there is a separator-related strategy for constructing proton barriers, but its mechanisms share similarities with the strategies for electrolytes. The following Tables 1–3 summarize the performance of some representative works based on different strategies. | ||
| Fig. 18 Comprehension of strategies toward inhibited side reactions at the Zn anode. | ||
| Anode sample | Zn symmetric cell | Half cell | Full cell | Ref. | ||||
|---|---|---|---|---|---|---|---|---|
| Condition (mA cm−2, mAh cm−2) | Cycle life (h) | Contention (mA cm−2, mAh cm−2) | Cycle number and average CE | Cathode | Condition | Cycle number and capacity retention | ||
| PCu@Zn (I + III) | 10, 1 | 200 | 5, 1 | 450, 97.55% | LiMn2O4 | 1C (pouch cell) | 300, 94 mAh g−1 retained | 146 |
| ILL@Zn (I + III) | 1, 1/20, 10 | 6000/250 | 1, 1 | 120, 99.2% | Ni-doped MnO2 | 1 A g−1 | 1500, 82.6% | 147 |
| ZnBi (I + III) | 5, 5/20, 0.1 | 1000/115 | 2, 2 | 350, 99.2% | V2O5 | 5 A g−1 | 1000, 88.59% | 130 |
| CuIn@Zn (I + III) | 1, 1/10, 5 | 9100/750 | — | — | V2O5 | 2 A g−1/1 A g−1 | 1300, 3.36 mAh cm−2 in average/330, 6 mAh cm−2 in average | 132 |
| Zn0.73Al0.27@Zn (I + III) | 1, 1/2, 2 | 3060/500 | 2, 0.5/5, 1 | 400, 99.25%/480, 99.13% | MnO2 | 1.2C | 1000, 67% | 148 |
| ZnCo (I + III) | 5, 2 | 620 | 2, 1 | 2000, 99.5% | V2O5 | 5 A g−1 | 1200, 91.99% | 149 |
| ILL@Zn (I + III) | 1, 1/20, 10 | 6000/250 | 1, 1 | 120, 99.2% | Ni-doped MnO2 | 1 A g−1 | 1500, 82.6% | 147 |
| BR-Zn (I) | 5, 1/10, 3 | 1970/1100 | 5, 2 | 1000, 99.83% | Activated C | 4 A g−1/1 A g−1 | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 97%/4000, 99.95% 000, 97%/4000, 99.95% |
131 |
| DES-Zn (I) | 1, 0.5 | 1056 | — | — | α-MnO2 | 0.5 A cm−2 (pouch cell) | 1000, 83.8% | 150 |
| Zn–Ti (I) | 2, 2 | 1100 | 5, 1 | 4000, 99.85% | NH4V4O10 | 5 A g−1 | 3500, 80% | 132 |
| Zn88Al12 (I) | 0.5, — | 2000 | — | — | KxMnO2 | 0.5 A g−1 | 5000, 100% | 21 |
| Zn(002) (II) | 10, 1 | 20 | — | — | α-MnO2 | 5 A cm−2 | 250, 92% | 141 |
| Mono-Zn (II) | 1, 1/10, 5 (51% DOD) | 3000/1000 | 1, 1/8, 1 | Reservoir protocol, 99.0%/99.8% | NaV3O8·1.5H2O | 5 A g−1 | 1000, 72.6% | 37 |
| Zn(101) (II) | 4, 2 | 5350 | 15, 10 | 163, 99.3% | MnO2 | 1 A g−1 | 1000, 95.4 mAh g−1 retained | 91 |
| Zn(002) (II) | 2, 2/10, 10 (45.5% DOD) | 520/210 | 2, 2 | 100, 99.3% | K0.51V2O5 | 5 A g−1 | 2000, 161 mAh g−1 retained | 38 |
| Zn(002) (II) | 0.5, 1/5, 5 | 2100/1960 | — | — | Activated C | 5 A g−1 | 33000, 87.4% |
151 |
| Etched Zn (II) | 0.2, 0.2/1, 1/10, 5/20, 10 | 2200/1600/500/400 | 2, 1/5, 1/10, 1 | 1200, 99.81%/2000, 99.90%/4500, 99.92% | PANI-V2O5 | 5 A g−1 | 2000, 68.3% | 145 |
| Zn@CuCK (I + III) | 5, 1 | 3600 | 5, 2 | 2100, 99.6% | NaV3O8·1.5H2O | 5 A g−1 | 12000, 83.5% |
152 |
| e-TDYP@Zn (III) | 5, 5/10, 10 (28.2% DOD) | 1100/250 | 4, 1 | 800, 99.6% | MnO2 | 2 A g−1 | 6000, 90.3% | 153 |
| TiBTx-31@Zn (III) | 1, 0.5/2, 1/5, 5 | 2000/1200/1000 | 5, 1/10, 1 | 500, 98.59%/2000, 99.86% | NH4V4O10 | 5 A g−1 | 800, 85.8% | 154 |
| SA-Cu@Zn (III) | 1, 1 | 2000 | 1, 1/5, 2 | 900, 99.83%/400, 99.87% | V2O5 | 2.5 A g−1 | 2000, 50.94% | 155 |
| DP[5]-Zn (III) | 1, 1 | 5500 | 5, 2.5 | 2200, 99.76% | V2O5 | 0.5 A g−1/5 A g−1 | 500, 80.52%/6000, 92.03% | 156 |
| FPI-Zn (III) | 2, 1 | 4000 | — | — | MnO2 | 5 A g−1 | 5000, 75.21% | 157 |
| Zn@LDH@GQDs (III) | 1, 1/2, 1 | 1060/1250 | 2, 1 | 900, 99.6% | NH4V4O10 | 5 A g−1/10 A g−1 | 1000, 97.9%/2000, 98.1% | 158 |
| Pb-PVDF@Zn (III) | 5, 2.5/20, 10 (85.4% DOD) | 2000/800 | 2, 1 | 1200, 99.94% | V2O5·nH2O | 0.5 A g−1/2 A g−1 | 500, 89.3%/5000, 82.7% | 117 |
| ES-Cm@Zn (III) | 1, 1/8, 4 | 1280/530 | 1, 1 | 630, 98.5% | NH4V4O10 | 2 A g−1 | 1000, 72% | 159 |
| Zn@A-ZIF-8 (III) | 1, 1/10, 1 | 7900/1200 | 3, 3 | 1000, 99.98% | MnO2 | 1 A g−1 | 2000, without capacity attenuation | 28 |
| FCOF@Zn (III) | 5, 1/40, 1 | 1700/700 | 5, 1/40, 2 | 480, 98.4%/250, 97.3% | MnO2 | 3C | 1000, 92% | 160 |
| SEI Zn (III) | 10, 1 | 1500 | — | — | MnO2 | 1 A g−1 after 10 cycles activation at 0.2 A g−1 | 700, 96.1% | 161 |
| HB-ZnHCF@Zn (III) | 1, 0.5/5, 2.5/10, 5 | 5800/2000/1000 | 2, 1 | 3000, 99.88% | V2O5·nH2O | 0.2 A g−1/2 A g−1 | 150, 92.9%/2000, 92.6 | 162 |
| Electrolyte sample | Zn symmetric cell | Half cell | Full cell | Ref. | ||||
|---|---|---|---|---|---|---|---|---|
| Condition (mA cm−2, mAh cm−2) | Cycle life (h) | Contention (mA cm−2, mAh cm−2) | Cycle number and average CE | Cathode | Condition | Cycle number and capacity retention | ||
| ZS-26 NADS (I) | 4, 1/30, 5 | 7200/500 | — | Reservoir protocol, 96.5% | MnO2 | 5 A g−1 | 13000, 88 mAh g−1 retained |
163 |
| 0.1-LBG (I) | 1, 1/2, 2 | 1600/1160 | 4, 4 | 180, 99.6% | MnO2 | 2 A g−1 | 600, 222 mAh g−1 retained | 164 |
| IS4 (I) | 1, 1 | 1800 | 1, 0.5 | 1000, 99.2% | NaV3O8·1.5H2O | 1 A g−1 | 1200, 91.2% | 165 |
| 1Z-5H (I) | 1, 1/20, 4 | 1551/70 | — | — | V6O13 | 4 mA cm−2 | 2000, 75.97% | 166 |
| BE/5Cor (I) | 5, 5/10, 10 | 900/600 | 2, 1/5, 2.5 | 1000, 99.52%/350, 99.54% | NaV3O8·1.5H2O | 1 A g−1/5 A g−1 | 500, 70.6%/500, 70.8% | 104 |
| PZS (I) | 1, 1/10, 1 | 8688/8400 | 1, 1 | 1000, 99.2% | Zn0.25V2O5 | 1.2 A g−1 | 1000, 77.8% | 167 |
| TMAF | 4, 1 | 1400 | 10, 10 | 212, 99.78% | I2 | 1 A g−1 | 10000, 99% |
168 |
| ZS-GA (I) | 20, 1/10, 10 (85% DOD) | 1800/300 | 1, 1 | 300, 99.61% | MnO2 | 1 A g−1 | 1000, 88.2% | 169 |
| BmBr/ZSO (I) | 1, 1/1, 20 (85% DOD) | 2700/320 | 1, 1 | 2100, 99.42% | I2 | 80 mA g−1 | 400, 94.3% | 103 |
| ZSO + SPS (I) | 1, 1/5, 5 | 2100/1120 | 2, 1/5, 5 | 850, 99.41%/250, 99.36% | NaV3O8·1.35H2O | 1 A g−1 | 400, 95.5% | 170 |
| 1 wt% PS (I+ VII) | 1, 1/5, 5/10, 10 | 8060/4554/1930 | 5, 5 | 600, 99.3% | V2O5/rGO | 5 A g−1 | 3000, 81.1% | 171 |
| ZSO-SAA (I + VII) | 1, 1/10, 5/20, 20 | 3400/1700/450 | 2, 2 | 1400, 99.8% | Activated C | 10 A g−1 | 100000, 91.8% |
172 |
| Zn(HBS)2 (II) | 5, 1 | 1600 | 5, 1 | 3500, 99.8% | Mn-doped V6O13 | 10 A g−1 | 5000, 68% | 47 |
| Zn(BBI)2 (II) | 2, 2/20, 1 | 2800/700 | 5, 5 | 400, 99.7% | PANI | 1 A g−1 | 600, 91.6% | 173 |
| ZnBF/Ac-CE (II + III) | 6.26, 3.13 | 300 | 6.26, 3.13/9.39, 9.39 (60% DOD) | 250, 99.84%/150, 99.8% | PANI | 5 A cm−2 | 400, 85% | 174 |
| HMEs (I + III) | 0.5, 0.5/1, 1 | 2150/918 | 0.5, 0.5 | 690, 98% | VOPO4·2H2O | 1 A g−1 | 1500, achieving 100.1 mAh g−1 | 139 |
| BE + DEC (III + VII) | 5, 5/5, 2.5 | 750/3500 | 1, — | 400, 99.2% | V2O5·nH2O | 2 A g−1 | 5000, 72.9% | 62 |
| SFMEE (III + VII) | 1, 1 | 4000 | 1, 1 | 2864, 99% | NaV3O8·1.5H2O | 5 A g−1 | 4000, 125 mAh g−1 retained | 175 |
| W/O (III) | 1, 1 | 5500 | 5, 1 | 6000, 99.76% | V10O24·12H2O | 1 A g−1 | 1800, 83.3% | 176 |
| 2-10TP (III + VII) | 1, 1 | 2000 | — | — | V2O5 | 3 A g−1 | 3500, 71.4% | 177 |
| 50% PC-sat. (III + VII) | 2.5, 10 (68% DOD) | 100 | 1, 0.5 | 500, 99.93% | ZnMn2O4 (anode-free) | 0.35 A g−1 | 275, 80% | 178 |
| MA40 (III + VII) | 1, 1/5, 5 | 4600/1600 | 0.5, 0.5 | 2000, 99.8% | NaV3O8·1.5H2O | 1 A g−1 | 500, 77.1% | 179 |
| TSAE (III + VII) | 1, 1 | 16800 |
— | — | PANI | 5 A g−1 | 5000, 94% | 180 |
| 1 m G4 (III) | 2, 2/5, 5 | 1500/1000 | — | — | PANI | 0.5 A g−1 | 1500, 94% | 181 |
| HEDES (IV + VII) | 0.5, 0.25 | 2600 | — | — | Activated C | 0.5 A g−1 | 8000, without capacity attenuation | 182 |
| HZAH-1 (IV + VII) | 0.5, -/2, - | 3000/1650 | 1, — | 8000, 99.6% | PANI | 1 A g−1 | 10000, 95% |
183 |
| HEE-1.2-3 (IV) | 1, 0.25/2, 0.5 | 2000/400 | 0.5, 0.5 | 400, 98% | NH4V4O10 | 0.5 A g−1/1 A g−1 | 600, 91%/1000, without capacity attenuation | 184 |
| DEE-1:10 (IV + VII) |
0.1, 0.1/1, 1 | 12000/1200 |
— | — | V2O5 | 1 A g−1 | 1000, 74.5% | 185 |
| DES50 (IV) | 5, 1 | 1800 | 0.5, 0.5 | 300, 99.39% | VOX | 0.1 A g−1/1 A g−1 | 100, 80.07%/1000, 70.5% | 186 |
| HEE (IV) | 1, 1 | 2000 | 1, 0.5 | 2000 h (cycle time), 99.3% | I2 | 5C | 2000, 97.9% | 187 |
| 1 + 1 (IV) | 1, 1 | 1800 | 4, 1 | 800, 99.05% | NH4V4O10 | 0.2 A g−1 | 200, 80% | 188 |
| ACDE-3 (IV) | 0.5, 0.5 | 900 | 0.5, 0.5 | 15–200, 97.9% | Poly(1, 5-NAPD)) | 0.2 A g−1/1 A g−1 | 90, 88.2%/200, 96.6% | 122 |
| HPG-35 wt% (V) | 1, 1 | 7500 | 1, 1 | 5500 h (cycle time), 99.65% | Zn0.25V2O5·nH2O | 0.7 A g−1 | 2300, 92% | 189 |
| CHE (V) | 1, 1/5, 5 | 2850/1500 | 1, 0.5 | 600, 99.1% | PANI | 1 A g−1/5 A g−1 | 9100, 78%/1500, 80% | 190 |
| α-HP/PVA (V + VII) | 0.1, 1 | 5300 | 1, 1 | 900, 99.5% | NaV3O8·1.5H2O | 10 A g−1 | 2500, 75% | 191 |
| C18-PCHA-AAm (V) | 2, 0.1/5, 0.1/10, 0.1 | 2440/1400/840 | 1, 1 | 2464, 99.8% | MnO2 | 10C | 3000, 86% | 192 |
| PAM-1, 2-PG (V + VII) | 10, 1/100, 50 (86% DOD) | 2160/490 | 1, 0.4/5, 5 | 3700, 99.1%/300, 99.6% | VO2·0.4H2O | 1 A g−1 | 200, 73.5% | 193 |
| PVA-PAM-PAN3-Zn2 (V) | 0.5, 0.5/1, 1 | 2000/1800 | 0.5, 0.5 | Reservoir protocol, 99.56% | V2O5 | 1 A g−1 | 800, 130.6 mAh g−1 retained | 194 |
| OL-PUU/PAM (V + VII) | 1, 1/20, 1 | 2000/200 | 2, 1 | 200, 99.5% | MnO2 | 2 A g−1 | 2000, 99.82% | 195 |
| PAM/trehalose (V) | 1, 1/5, 2 | 2400/460 | 2, 0.5 | 850, 98.8% | MnO2 | 5 A g−1 | 3000, 62.7% | 196 |
| VSSE (VI) | 0.2, 0.2/0.5, 0.5 | 2000/1400 | 0.2, 0.2 | 10-500, 96.5% | rGO | 0.1 A g−1 | 500, 89.5% | 84 |
| ZIG-20 wt% (VI) | 1, 0.5 | 900 | 0.5, 0.5 | 300, 99% | MnHCF | 1C | 1500, 94% | 83 |
| LWZQE (VI) | 1, 1/1, 5 | 5500/3000 | 1, 1 | 2000, 99.2% | Al0.1V2O5·1.5H2O | 1 A g−1 | 3000, 119.8 mAh g−1 retained | 197 |
| DEE@PCN-222 (VI) | 1, 1/2, 2 | 2476/1139 | — | — | MnO2 | 1C | 517, 78.43% | 198 |
| ZP0.8@Nb2O5−x (VI) | 0.1, 0.1 | 2900 | — | Reservoir protocol, 98.9% | PANI | 1 A g−1 | 1000, 82.2% | 199 |
| WZM (VI) | 0.1, — | 360 | — | — | VS2 | 0.2 A g−1 | 250, 40% | 200 |
| d-SEI (I + VII) | 1, 1 | 5600 | 5, 1 | 800, 99.6% | I2 | 0.1C (pouch cell) | 113, 83.3% | 201 |
| Zn(OTF)2-HMPA-H2O (I + VII) | 10, 10 (85.6% DOD)/40, 10 (85.6% DOD) | 500/200 | 8, 8 (64.8% DOD)/10, 10 (85.6% DOD) | 200, 99.5%/100, 99.4% | V2O5 | 1 A cm−2 | 400, 63.63% | 202 |
| Polymer–inorganic-SEI (I + VII) | 1, 1/3, 3/10, 10/30, 30 | 4600/1500/900/200 | — | — | NH4V4O10 | 1 A g−1 | 800, 82% | 203 |
| Zn(OTF)2 + Me3EtNOTF (I + VII) | 0.5, 0.25 | 6000 | 0.5, 0.5 | 1000, 99.8% | VOPO4 | 2 A g−1 | 6000, 88.7% | 204 |
| SAB-ZnSO4 (I + VII) | 1, 1/30, 20 (90% DOD) | 3000/300 | 1, 1 | 1000, 99.6% | MnO2 | 0.5 A g−1 | 500, 64.7% | 126 |
| MMI (I + VII) | 0.5, 0.5/2, 2 | 1500/500 | 5, 1 | 400, 98.1% | MnO2 | 2 A g−1 | 500, 75.4% | 205 |
| Ternary electrolyte (III + VII) | 5, 5/20, 20 | 7200/4200 | 5, 5 | 2200, 99.8% | NaV3O8 | 1 A g−1 after 3 cycles activation at 0.2 A g−1 | 11000, 34% |
206 |
| Separator sample | Zn symmetric cell | Half cell | Full cell | Ref. | ||||
|---|---|---|---|---|---|---|---|---|
| Condition (mA cm−2, mAh cm−2) | Cycle life (h) | Contention (mA cm−2, mAh cm−2) | Cycle number and average CE | Cathode | Condition | Cycle number and capacity retention | ||
| VF | 1, 1 | 4600 | 0.5, 0.5 | 500, 99.36% | NH4V4O10 | 1 A g−1 (2VF) | 1000, 51.25% | 207 |
| BC/V | 1.169, 1.169/2.926, 2.926 (50% DOD)/2.342, 4.684 (80% DOD) | 1500/1100/500 | 1, 1 | 3500, 99.77% | Mn0.5V6O13 | 0.3 A g−1 | 500, 89.4% | 208 |
| V-NFC-CS | 10, 2/5, 25 (85.4% DOD) | 1000/300 | 1, 1 | 20–600, 99.0% | MnO2 | 1 A g−1 | 1000, 96.2% | 209 |
| ANF | 2, 1/5, 2.5 | 1470/850 | 1, 1 | 1400, 99.22% | Mn2.5V10O24·5.9H2O | 0.5 A g−1 | 300, 80.7% | 210 |
| COF-Zn | 0.2, 0.2/1, 1 | 2900/350 | 0.5, 05 | 400, 98.3% | PTO/V2O5 | 2 A g−1/2 A g−1 | 1000, 100%/500, 74.94% | 211 |
| PT@GF | 5, 2.5/10, 5 | 1600/1200 | 1, 0.5 | 500, 99.5% | I2 | 1 A g−1 | 10000, 85.8% |
212 |
| Gra-CeF3-GF | 1, 1/5, 5/10, 5 | 2500/1000/500 | 1, 1 | 320, 99.7% | V2O5 | 2 A g−1 | 2000, — | 213 |
| TCh | 1, 1/10, 10 | 2700/320 | 5, — | 600, 98.9% | MnO2 | 1.5 A g−1 | 800, 93.7% | 214 |
Nonetheless, issues, disputes, and challenges still need to be addressed for these strategies, as summarized in Fig. 18. Electrolyte additives and artificial interphases have been intensively studied with reported functions such as modifying the Zn2+ solvation structures, accelerating desolvation, and isolating active water. However, there are challenges in proposing rational and comprehensive criteria with respect to side reactions to guide the screening of additives or artificial interface design.215 In addition, deep insights are needed into the concrete processes and corresponding mechanisms of additive adsorption and in situ SEI formation at the Zn anode surface. In the case of cosolvent strategies, cost and safety issues exist because of the usage of relatively large amounts of organic components. Besides, for cosolvents, eutectics, hydrogels, and other lean water electrolytes, relatively sluggish kinetics issues are nonnegligible. In this case, the kinetic indexes, such as the electrostatic constant, for cosolvent systems might be the key.120 Especially, in the case of hydrogel electrolytes, their water content is of great importance since many hydrogels possess more than 70 wt% water content, and the excessive water may result in the majority of the bulk-phase Zn2+ adopting a hexahydrate coordination structure. Consequently, one approach is minimizing the water content with sufficient Zn2+ transporting kinetics, which deserves more investigation, such as the work by Zhi's group, and another approach is further decreasing the Zn(H2O)62+ species via coordination with Zn2+.83 In the case of electrolytes beyond the liquid state, the interfacial contact with the Zn anode should also be the focus as incomplete contact will result in huge polarization. Accordingly, some in situ treatment processes such as in situ hydrogel polymerization are preferred.216 When it comes to Zn salt regulation, it should be considered if the anion screening routes or Zn salt concentration adjustment demonstrates compatibility with other strategies. For example, the positive effects of an optimized Zn salt may be masked if some electrolyte additives are used simultaneously. Highly concentrated electrolytes may also introduce cost and safety concerns. Additionally, different Zn salts may result in different failure mechanisms, which cannot be confused together.217 In the case of asymmetric electrolytes, one main issue is the latent difficulties of preparation and assembly. Another technical issue is interfacial contact, which may not only include the electrolyte/electrode interface but also involve the interface between two electrolyte phases.
Regarding proton barrier separators, they have rarely been studied to date due to the lack of in-depth insights into hydrogen bonds and proton transportation mechanisms. The Hofmeister effect still cannot be completely understood currently, and more endeavors should be laid on the optimization of Zn2+ and proton transporting in parallel.
With respect to Zn anode texturing, various crystal facet orientations have been reported, featuring stabilized thermodynamics and accelerated kinetics, respectively. Although Zn(002) outperforms other facet orientations with anti-corrosion and fast in-plane diffusion rate enabled electrochemical properties, disputes yet exist in the optimal orientations. There is still room to explore the distinction of failure mechanisms in cycling with different textures. Distinctions between different crystal facets should also be insightfully comprehended. For the interfacial alloying strategies, it should be advisable to design the Zn anode with consistent side reaction resistance, without mechanisms failure along with repeating cycling. It will be meaningful to develop an in situ alloying/dealloying reaction route to replace the traditional Zn plating/stripping mode, which might be more effective in inhibiting side reactions.
4. Technologies for the characterization of side reactions
For the characterization of side reactions, the three main reported categories are microscopy technologies, spectrometer analysis and electrochemical methods (Fig. 19). Herein, we summarize the extensively adopted characterization based on the aforementioned classifications. | ||
| Fig. 19 Mainstream characterization methodologies for side reactions at the Zn anode and their respective contributions and restrictions. | ||
4.1. Microscopy technologies
The mechanisms of microscopy are dependent on the interactions between specific microscopic particles and observed samples. For optical microscopy, images can be obtained by the reflected light through the amplification of eyepieces and objective lens. Optical microscopy is generally employed as an in situ characterization method, observing dendrite growth and generation of bubbles from a side view. For example, as shown in Fig. 20a and b, obvious Zn dendrites and H2 bubbles are observed in bare ZnSO4 electrolyte while dense deposition emerged using an effective N-acetyl-D-glucosamine additive.218 However, a restriction is the relatively large wavelength of visible light. Using a laser with a smaller wavelength effectively improves the resolution ratio according to the Rayleigh criterion, such as in the case of confocal laser scanning microscopy (CLSM). The mechanism of CLSM is derived from surficial morphology-induced focus differences, and this method offers insights into smoothness-related surficial morphology fluctuations or 3D structures on the Zn anode surface. An uneven deposition surface with obvious color contrast manifests poor reversibility in the bare ZnSO4 electrolyte, while a uniform surface smoothness signifies a stable Zn anode with cordycepin additive (Fig. 20c). | ||
| Fig. 20 In situ optical microscopy images of Zn deposition on Zn substrates at 1 mA cm−2 in ZnSO4 electrolyte (a) with and (b) without additives. Reproduced with permission.218 Copyright 2025, Wiley-VCH. (c) CLSM images of 1 mAh cm−2 deposited Zn on a Cu substrate (left) without and (right) with cordycepin additive. Reproduced with permission.104 Copyright 2024, Open Access. SEM images of Zn plates after soaking in ZnSO4 electrolyte (d) without and (e) with additives. Reproduced with permission.219 Copyright 2024, Open Access. (f) SEM images of Zn deposition on (top) polycrystalline Zn anode and (down) mono-oriented Zn(002) anode with different plating times at 10 mA cm−2. Reproduced with permission.37 Copyright 2025, Wiley-VCH. (g) In situ AFM images of initial nucleation and early growth on (top) Ag-modified Zn anode and (down) bare Zn anode. Reproduced with permission.220 Copyright 2024, Wiley-VCH. | ||
Focused electron beam accelerated by an applied voltage can feature a wavelength on the picometer scale to acquire more microscopic information. SEM is conducted by collecting electron feedback information after electron beam bombardment. Backscattered electron images and secondary electron images are mainly employed, featuring surficial morphology sensitivity and component sensitivity, respectively. For the characterization of chemical side reactions, a commonly used routine is to immerse the Zn anode in the electrolyte solution for a period of time, simulating the Zn anode stability in the battery-shelved state. After soaking, SEM can be carried out to determine the uneven surface caused by corrosion and byproduct formation. For instance, our group reported an effective electrolyte additive 3-hydroxy-4-(trimethylammonio)butyrate, where the SEM images showed that it safeguarded the Zn anode from chemical side reactions (Fig. 20d and e).219 In electrochemical systems, such as coin cells and electrolytic tanks, the situations on the Zn anode surface can also be investigated by SEM after plating, stripping, or cycling. For example, in the mono-oriented Zn(002) anode prepared by our group, an optimized Zn deposition morphology was achieved compared with polycrystalline Zn anode (Fig. 20f).37 Additionally, the transmission and diffraction electrons can be collected to obtain some texture-related information based on high-resolution transmission electron microscope (HRTEM) and selected area electron diffraction (SAED) technologies.37
The image formation principle of scanning probe microscopy is based on the interaction between the atom probe and sample surface. Given that the interatomic interaction is sensitive to distance, these methods can precisely respond to surface morphologies. In this field, atomic force microscopy (AFM), harnessing laser feedback to detect the change in probe position toward surficial smoothness reflection, is an effective method. It is capable of evaluating the surficial smoothness after deposition or cycling more microscopically owing to its greater sensitivity to surface topography. In situ AFM can further probe the timely information of nucleation and deposition. An example was reported by Li's group, where the Zn nucleation behavior on different substrates was investigated.220 The corresponding in situ AFM images showed that the plated Zn adopted highly oriented planes with Ag modification while uneven morphologies appeared on the bare Zn anode (Fig. 20g).
Among the aforementioned microscopy technologies, only the in situ optical microscopy can directly observe of hydrogen gas generation. Nonetheless, occasionality also exists for the hydrogen bubbles observation, which depends on the selection of characterization area. Besides, the generated bubbles derived from micro-region hydrogen evolution corrosion may not be captured by an optical microscopy due to the limitation of its characterization scale. In the case of SEM, it enables the observation of byproduct formation due to the recognizable morphology of Zn salt-based hydroxide and its appropriate characterization scales. However, this is not generally an in situ approach, and the detection of byproduct formation processes during cycling is lacking. Overall, the majority of microscopy technologies are more suitable for surface morphology evolution analysis, which serves as an indirect indicator of side reactions as the occurrence of side reactions and dendrite growth are strongly related. Acquiring timely in situ information about side reactions may be difficult using the current conventional microscopy technologies.
4.2. Spectrometry analysis
Spectrometry methods are established by distinguishing different feedback in continuous signals based on a certain parameter. Depending on the variation in the information on dipole moment and polarizability due to light absorption/scattering, in situ Fourier transform infrared (FTIR) spectroscopy and Raman spectroscopy have been tasked with probing the real-time Zn anode/electrolyte interphase evaluation during Zn plating/stripping, providing information on corresponding indicators such as the states of hydrogen bonds and anions.54,221,222 For instance, recently a D-valine-filled all-in-one hydrogel electrolyte was reported, where the adsorption of D-valine anion at IHP of the Zn anode could construct a water-deficient surface with weakened hydrogen bonds.221 As proven in Fig. 21a–d, with the adsorption of D-valine anion, both the νO–H signals in FTIR spectroscopy and the ν-HOH stretching signals in Raman spectroscopy exhibited reduced intensity during the Zn plating/stripping processes. | ||
| Fig. 21 (a) Contour plots of in situ FTIR at the interface under different Zn plating/stripping time in a bare ZnSO4-based hydrogel electrolyte. (b) In situ Raman spectra of water signals at the Zn anode/electrolyte interface in the bare ZnSO4-based hydrogel electrolyte. (c) Contour plots of in situ FTIR at the interface under different Zn plating/stripping times in D-valine-filled hydrogel electrolyte. (d) In situ Raman spectra of water signals at the Zn anode/electrolyte interface in D-valine-filled hydrogel electrolyte. Reproduced with permission.221 Copyright 2025, Wiley-VCH. (e) XRD patterns of Zn anodes cycled in different electrolytes after 50 cycles. (f) In situ XRD patterns of Zn(100) plane in Zn||Zn symmetric cells with (left) ZnSO4 and (right) PAM/trehalose hydrogel electrolytes. Reproduced with permission.196 Copyright 2024, Wiley-VCH. High-resolution XPS spectra of (left) Zn 2p and (right) O 1s from Zn anodes in (g) bare electrolyte and (h) Al2O3-modified electrolyte. (i) SEM image of the cycled Zn anode in a full cell with bare electrolyte and (j) corresponding surface EDS mapping. (k) SEM image of the cycled Zn anode in the full cell with Al2O3-modified electrolyte and (l) corresponding surface EDS mapping. Reproduced with permission.225 Copyright 2025, Wiley-VCH. (m) Schematic of the ECMS instrument. QMS is short for quadrupole mass spectrometer. (n) Quantification of evolved hydrogen as a function of time, during (0 to 600 s) and after discontinuing (600 to 4000 s) the application of current (–1.5 mA cm−2). Reproduced with permission.226 Copyright 2024, Open Access. In situ EIS of Zn||Zn symmetric cells during resting in (o) Zn(HBS)2, (p) ZnSO4, and (q) Zn(OTf)2 electrolyte. Reproduced with permission.47 Copyright 2025, Wiley-VCH. | ||
The aforementioned in situ FTIR and Raman spectroscopies mainly characterize the occurrence trends of side reactions, while the following discussed approaches, from another angle, reflect the outcome caused by side reactions. In the case of XRD, it shows signal strength peaks at specific angles as scattered X-ray emerging coherent diffraction. Strictly, XRD cannot be categorized as a spectrometry method, given that 2θ is a parameter on the spatial scale, but it is discussed here owing to its similarities with spectrometry in terms of the data provided.223 XRD is generally tasked with identifying byproduct formation and Zn anode texture after chemical soaking, electrochemical deposition or cycling. In addition, in situ XRD technology has been applied, testifying the texture evolution during plating/stripping cycles. In the work by Chen's group, they developed a PAM/trehalose hydrogel electrolyte and representatively utilized XRD to investigate the Zn anode chemistry.196Ex situ XRD patterns evidenced inhibited byproduct formation and more Zn(002) exposure (Fig. 21e). In addition, in situ XRD was carried out, as shown in Fig. 21f. With a prolonged plating time, the diffraction intensity of Zn(100) in ZnSO4 electrolyte remained extremely high while that of a PAM/trehalose hydrogel electrolyte remained relatively low and constant, indicating the inhibition of unwanted Zn(100) exposure and the facilitation of dense Zn deposition. X-rays can also simulate the photoelectrons from surficial atoms, possessing the characteristic binding energy of elements. X-ray photoelectron spectroscopy (XPS) is based on collecting photoelectrons according to their different binding energies, acquiring information on surficial element components and bonding. XPS provides an approach to characterize the passivation byproducts or SEI composition. Upon electron beam irradiation, characteristic X-rays may also be stimulated due to the energy level transition of inner electrons. Because of the energy differences, the characteristic X-rays are collected by energy dispersive spectroscopy (EDS) to identify the element information, further affirming the formation of byproducts. A corresponding example was given by Wu's team, who proposed Al2O3 nanoparticles as a bifunctional additive for Zn||LiCoO2 batteries. To investigate the Zn anode interphase chemistry, both XPS and EDS were carried out. According to the XPS result, as shown in Fig. 21g, the Zn anode cycled in the bare electrolyte displayed two peaks at 1021.9 and 1023 eV, corresponding to the Zn 2p3/2 spectra of metallic Zn and ZnO byproduct, respectively. This result aligned with the peak at 531.6 eV in the O 1s XPS spectra. By contrast, the Zn anode cycled in Al2O3-added electrolyte exhibited only one peak at 1021.9 eV, with the metal oxide peak at 531.6 eV in the O 1s XPS spectra stemming from surficial residual Al2O3, signifying less byproduct formation (Fig. 21h). The surface SEM images with the corresponding EDS mapping indicated that in the bare electrolyte, uneven byproduct formation occurred, while a uniform deposition surface with no byproducts was realized with Al2O3 additives (Fig. 21i–l).
With regard to spectrometry methods, it can be assumed that they are mainly targeted at detecting the formation of byproducts, with less focus on the information on hydrogen gas production. Among them, in situ FTIR and Raman spectroscopies detect the zone of electrolytes near the Zn anode surface, mainly affording real-time information of the EDL. In the case of other electrode characterizations, XPS possesses a micrometer-level characterization scale, showing great surface element sensitivity. In comparison, XRD and EDS tend to be bulk phase analysis methods, which can obtain chemical component information on the micrometer scale. To bridge the gap between surface and bulk phase signals and obtain the element depth distribution profiles of byproducts and formed SEI, different sputtering depth-based XPS and time-of-flight secondary ion mass spectrometry (ToF-SIMS) analyses may be effective.24,224 It should be mentioned that although spectrometry methods are restricted in the characterization of hydrogen gas formation at the Zn anode, it is undeniable that many spectrometry technologies (e.g., FTIR spectroscopy and Raman spectroscopy) contribute greatly to the understanding of the bulk phase properties of electrolyte such as Zn2+ solvation structure and hydrogen bond networks.82 These parameters are strongly related to the stability of the Zn anode/electrolyte interphase and help us forecast the occurrence trends of hydrogen evolution and corrosion.
4.3. Electrochemical methods
Electrochemical methods rely on the assembly of electrochemical systems such as coin cells and electrolytic tanks. To date, the mainstream electrochemical measurements for side reactions are LSV and Tafel testing, with many relevant examples covered in previous texts. LSV measurement can be carried out by imposing a negatively directed linearly changing voltage to the Zn anode side. Once side reactions occur with interfacial electron transfer, an obvious change in the current signal responses can be observed. LSV is able to detect the initial potential and the corresponding current contribution of hydrogen evolution but the electricity contribution of Zn2+/Zn redox cannot be effectively differentiated. To exclude the distraction of Zn2+/Zn redox, it is advisable to employ Na2SO4-based electrolyte in a three-electrode system.With regard to the Tafel analysis, its mechanism is relevant to the Butler–Volmer equation according to eqn (15), as follows:
 | (15) |
In situ electrochemical mass spectrometry (ECMS), as a combination of spectrometry and electrochemical method, is usually employed to detect hydrogen generation.46 It is able to detect the formation of different gas products, with the produced substance ionized, separated, and collected based on the identified charge/mass ratio. Different from the LSV and Tafel tests that give qualitative analysis, in situ ECMS can provide real-time quantitative analysis about hydrogen evolution, thus affording more rational information of side reactions. For example, Roy et al. quantified hydrogen evolution by means of in situ ECMS, with the schematic of the system presented in Fig. 21m.226 The hydrogen evolution data harvested in Fig. 21n exhibited the HER response and different corresponding sources with(out) an applied current. Assuming that charge to HER is completely balanced by Zn corrosion, they calculated a corrosion rate of 96 pg s−1.
With regard to byproduct formation, the results from electrochemical impedance spectroscopy (EIS) may be helpful. Surficial passivation byproduct formation results in a significant increase in impedance, which can be employed to further support the characterization by microscopy and spectrometry methods. Further, in situ EIS provides an approach for impedance dynamic monitoring toward surficial component evolution, tracing the processes for the formation of byproducts.47 For example, in the study by Chao's group, the Zn(HBS)2 salt with low API anion featured improved anti-corrosion ability compared with the generally used ZnSO4 and Zn(OTf)2, as verified by the small increase in ohmic and charge transfer resistances (Fig. 21o-q).47
Different from spectrometry analysis, electrochemical methods further contribute to the characterization of hydrogen evolution and corrosion. Notably, in situ ECMS is a useful quantitative analysis for hydrogen gas formation. The byproduct-related results from EIS may be relatively indirect, which requires the coordination of other characterization techniques. Given that in the series of side reactions, the HER is the initial step, electrochemical characterization is generally indispensable. Owing to this reason, under many circumstances, electrochemical characterization results for the occurrence of side reactions are regarded as rational screening indexes for materials component/ratio regulation in concrete Zn anode optimization strategies.
4.4. Corresponding restrictions
Although the characterizations discussed above can effectively detect the occurrence of side reactions, there still exist some corresponding challenges, as listed in Fig. 19. With regard to microscopy technologies, the main issue is the restricted characterization area. For instance, the scale bar of SEM with regard to the Zn anode surface observation is generally in the order of micrometers or even smaller, and the characterization area is much smaller compared with the total Zn anode. Given this, discrepancy may exist in the characterization results. Moreover, the significance of focusing on a small region should be rethinked, for the area of commercial Zn anode may be at least in the order of square centimeter. A more reasonable larger characterization area or more rational comprehensive evaluation methods for the Zn anode should be taken into account. Besides, many microscopy technologies are not capable of detecting side reactions in a straightforward manner since the majority techniques only indirectly characterize the deposition or cycling morphology of the Zn anode surface. In the realm of spectrometry analysis, one main issue is the difficulty to distinguish the effective information of the interphase and bulk phase from the mixed results. Besides, the component analysis feasibility of spectrometry is restricted to some extent, such as the relatively subjective peak separation results of XPS. When it comes to electrochemical methods, although LSV and Tafel analysis can effectively reflect the trends of hydrogen evolution and corrosion, they cannot analyze side reactions quantitatively. Under this condition, in situ ECMS should be conducted for auxiliary analysis. Besides, the accuracy of electrochemical methods should be given particular attention and repeated measurements are required. Even in the case of the same sample, such as 2 M ZnSO4 electrolyte, different results have been obtained in the LSV and Tafel tests in different reports in the literature. In addition, in different electrolyte systems, the corrosion reaction may differ, and thus, the corrosion current/potential in Tafel tests should be determined exactly.5. Conclusions and perspectives
To summarize, side reactions at the Zn anode induce a deviation from the hcp structure featuring the intrinsic plating/stripping mode. Consequently, we innovatively proposed the viewpoint that side reactions are the primary issues for the Zn anode and need to be addressed. In our review, we commenced by elaborating the significance of focusing on side reactions. Subsequently, we gave profound insights into the Zn2+/Zn redox chemistry, expounding the thermodynamic and kinetic properties of side reactions at the Zn anode. Afterward we lay emphasis on decoupling side reactions toward intrinsic Zn plating/stripping behavior based on different mechanisms, with side reactions initiators constraint and Zn plating/stripping facilitation. Numerous state-of-the-art studies were considered and analyzed in detail. Moreover, the reported mainstream characterization techniques to detect the occurrence side reactions were discussed. However, there are some challenges related to the side reactions at the Zn anode to be tackled, necessitating a follow-up discussion. Given these facts, we attempt to propose our realistic solutions and strategies and the future outlook in the corresponding fields.Firstly, we should note that even with side reactions excluded, Zn plating/stripping will not always take on the intrinsic state, especially in high DOD situations, given that the plating/stripping processes of polycrystalline Zn are non-uniform. Accumulated polycrystalline Zn hexagonal platelets cause reaction rate differences at the edges or in different crystal planes. Although intrinsic Zn hexagonal platelets emerge, they are not desirable because the uneven accumulation will deteriorate the Zn anode surface continuously, resulting in electrode perforation. For commercialization, an appropriately high Zn utilization is required, for which simply getting rid of side reactions is not sufficient. Moving a step further, herein we tentatively propose the standpoint that the ultimate goal of the Zn anode is to realize the side reaction-free planar plating/stripping mode, with the roadmap presented in Fig. 22a. These two demands are also closely related, for eliminating side reactions facilitates Zn intrinsic plating/stripping, and the planar electrochemical redox results in an anti-corrosion compact surface. In this field, the previous work of our group concentrating on the step-edge-guided Zn homoepitaxy can be considered an example.37 This research achieved record-high lifespans of over 1000 h and 480 h at high DOD of 51% and 82%, respectively. However, great endeavors are still needed in many mechanism-related aspects. For instance, compared with the nucleation theory extensively employed in the plating process, there is a lack of theoretical direction for the stripping process. More endeavors are needed in constructing the electrochemical stripping mode, covering electron transfer, surficial diffusion, and dissolution (solid–liquid transformation) processes supported by in situ characterization and calculations. Based on this, the corresponding tunable factors like stripping overpotential can be investigated in depth.228
 | ||
| Fig. 22 Schematic of the challenges and future perspective of side reactions and Zn anode in the realm of (a) Zn redox chemistry, (b) mechanisms, (c) deviceization, and (d) applications. | ||
Secondly, the characterization technologies for side reactions are still deficient, making the mechanisms of some detailed aspects unclear, as shown in Fig. 22b. Although it has been extensively reported that active Zn2+ coordinated water has a great contribution to side reactions, the specific Zn2+ desolvation process is ambiguous. Consequently, the root cause of the above-mentioned phenomena may be still unclear, and there are less specific indicators bridging the Zn2+ solvation structure and occurrence of side reactions. In this field, many in situ characterization techniques such as in situ FTIR spectroscopy, Raman spectroscopy, and extended X-ray absorption fine structure (EXAFS) are advisable.229,230 Regarding the occurrence of side reactions, some detailed procedures about how these reactions proceed are still lacking. Knowledge about bond breaking and formation is required to get further understanding of the inhibition of side reactions, and in situ spectroscopy and density functional theory (DFT) calculations may be effective to detect some intermediate state substances. In the case of side reaction initiators, a study also shed light on a previously neglected factor, dissolved oxygen.231 Besides, further investigation is demanded from a kinetic perspective of side reactions such as the reaction order of hydrogen evolution. This may be more beneficial for side reaction suppression such as developing the HER anti-catalysis mechanisms. Employing the ECMS to further quantitatively analyze hydrogen generation kinetically along with the reaction time may be the key. Moreover, the development of in situ characterization techniques to gain insights into the Zn anode/electrolyte interphase is urgently needed. Since both additive-specific adsorption and in situ SEI formation occur at the interphases, the concrete corresponding behaviors should be ascertained by effective characterization beyond theoretical calculations. In this direction, we can refer to the researches in other fields. For example, magic angle spinning nuclear magnetic resonance (NMR) spectroscopy was employed to probe the local structural order of carbon and its impact on ion adsorption capacitance, and operando scanning transmission electron microscopy (STEM)-electron energy-loss spectroscopy (EELS) was developed to in situ characterize SEI formation dynamically in LIBs.232,233
Furthermore, deviceization beyond coin cells is an inevitable course. The device configuration should be standardized with regard to large-scale commercialization. To accelerate deviceization, herein, we propose some viewpoints for reference, as exhibited in Fig. 22c. Above all, in an amplified electrochemical system, although side reactions are more serious, their total elimination may be impractical. Hydrogen evolution leads to swelling issues with an increase in the inner pressure of the device, and safety hazards may also be introduced owing to the flammability of hydrogen. Therefore, we consider the rationality of installing convenient gas exhaust units on devices. Challenges exist in the requirements for constructing an open system without liquid leakage, which may be achieved by (quasi-)solid electrolytes. For example, Yang's team reported an open pouch cell system based on a well-designed hydrogel electrolyte, which could not only release hydrogen but also enable electrolyte replenishing to compensate for irreversible consumption.234 Based on this, we further assume the possibility of effectively and portably collecting the evolved hydrogen for other energy-related applications, such as hydrogen fuel cells. Besides, with regard to deviceization, the form of the Zn anode, including Zn foil, Zn powder, and anode-free system, is worth considering. In the case of Zn foil with the merits of tractability and low cost, its shortcomings include restricted Zn utilization and uneven Zn plating/stripping-induced perforation at a high DOD. Beyond Zn foil, other systems have higher demands for the inhibition of side reactions at the anode side. In the case of Zn powder with a larger specific surface area, although it features a uniform surficial electric field distribution to inhibit dendrite growth and theoretically higher Zn utilization, the more severe side reactions caused by the larger contact area with aqueous electrolytes as well as the relatively high cost and safety issues should be overcome.235 Designing active water repelling binders or anti-galvanic corrosion current collectors has been reported to alleviate side reactions at the Zn powder anode.119,236 To realize an anode-free system targeted at high energy density, it is still challenging to optimize the CE loss resulting from side reactions and dead Zn to a record level. Constructing zincophilic and anti-HER current collectors may be necessary.237 Moreover, given that the ultimate practical system is a full battery, simply assessing the reversibility of the Zn anode may be meaningless. Given that cathode is an indispensable part, the significance of the performance of full cells should outweigh that of Zn symmetric cells and half cells. It should be noted that the dissolution of cathode materials may also affect the side reactions on Zn anode side, which is mainly related to transition metal oxide and halogen cathodes. In the case of a transition metal oxide cathode, the dissolved transition metal ions will cause crosstalk effects, with the competitive solvation between the transition metal ions and Zn2+ enhancing the Zn2+ desolvation energy barrier toward non-uniform deposition and facilitated side reactions.224 Regarding the halogen cathode, the dissolution of polyhalides is undesirable, which will shuttle across the separator and result in self-discharge and soft-short-circuit when chemically reduced by Zn metal.238 Although the root cause of aforementioned phenomena lies on the cathode side, the impartibility of anode and cathode in full battery system render inevitability with these issues for Zn anode, resulting in inferior performance. Therefore, on one hand, strategies for inhibiting side reactions at the Zn anode side should be further taken into consideration in terms of compatibility with different cathodes. On the other hand, exploration into the simultaneous improvement of both the Zn anode and cathode reversibility can be a solution. In this realm, asymmetric electrolyte is one strategy such as the recently reported work composed of a Zn anode stability favored and Mn2+ shuttling unfavored nonaqueous-phase gel anolyte and a reaction kinetics favored aqueous phase catholyte toward crosstalk-free Zn||MnO2 batteries.224 Besides, coupled strategies also deserve consideration such as employing alloying Zn anode accompanied by cosolvent electrolyte can stabilize Zn anode and cathode simultaneously.239 For full cells performance consideration, with regard to Zn||halogen batteries, recently, Qiao's group put forward an electroactive redox coupling strategy by integrating ferrocene into I2 cathodes, which enables shuttle-free and capacity-boosted Ah-level Zn||I2 pouch cells.240 Finally, we should also focus on a systematization approach from cells to batteries, which may demand rigorous assembly and regulation technologies. The commercial battery module accompanied by some corresponding standard requirements should be considered.
Furthermore, the development of AZIBs is still in its primary stage, signifying that there is a long way to go for commercialization. Taking the intrinsic properties of AZIBs into consideration, herein we put forward four main potential application aspects, as presented in Fig. 22d. The high safety and the volume specific capacity of the Zn anode enable its application in grid-connected energy storage stations and portable chargers. These situations require strict safety for inflammability and ought to minimize the volume toward effective energy storage. With regard to electric vehicles, fire disasters induced by flammable LIBs are frequently reported, which makes the application of safety-characterized AZIBs necessary in this field. However, noting that the power density of AZIBs cannot reach the same level as that of LIBs, AZIBs can only cater to low-speed situations. In addition, AZIBs can actuate some low-power consumption wearable devices functioning as intelligent displays and healthcare monitoring devices, and these electronics require high safety and low toxicity to be compatible with the human body.241
Author contributions
G. W., S. C., and Y. J. conceived the idea. G. W., S. C., Y. X., Y. X., Z. L., B. B. X., and Y. J. wrote and revised the manuscript. X. Z., M. Y., L. H. Z., M. W. S. and H. P. reviewed the manuscript and provided revisions. G. W., S. C., B. B. X., and Y. J. processed the figures and visualizations. The work was supervised by B. B. X. and Y. J. G. W. and S. C. contributed equally to this work.Conflicts of interest
The authors declare no conflicts of interest. The authors have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Data availability
No primary research results, software or code have been included, and no new data were generated or analyzed as part of this review.Acknowledgements
This work was supported by the National Natural Science Foundation of China (52471240), Zhejiang Provincial Natural Science Foundation of China (LZ23B030003), and the Fundamental Research Funds for the Central Universities (226-2024-00075). B. B. X. is grateful for the support from the Engineering and Physical Sciences Research Council (EPSRC, UK) RiR grant - RIR18221018-1 and EU COST CA23155 for the conference funding support.References
- D. Larcher and J. M. Tarascon, Nat. Chem., 2015, 7, 19–29 Search PubMed.
- N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Nature, 2015, 517, 476–480 Search PubMed.
- J. K. Kirkegaard, D. P. Rudolph, S. Nyborg, H. Solman, E. Gill, T. Cronin and M. Hallisey, Nat. Energy, 2023, 8, 655–664 Search PubMed.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 Search PubMed.
- D. Chao, W. Zhou, F. Xie, C. Ye, H. Li, M. Jaroniec and S.-Z. Qiao, Sci. Adv., 2020, 6, eaba4098 Search PubMed.
- H. Lu, X. Zhang, M. Luo, K. Cao, Y. Lu, B. B. Xu, H. Pan, K. Tao and Y. Jiang, Adv. Funct. Mater., 2021, 31, 2103514 Search PubMed.
- Z. Luo, Y. Xia, S. Chen, X. Wu, R. Zeng, X. Zhang, H. Pan, M. Yan, T. Shi, K. Tao, B. B. Xu and Y. Jiang, Nano-Micro Lett., 2023, 15, 205 Search PubMed.
- L. Fabbrizzi, Angew. Chem., Int. Ed., 2019, 58, 5810–5822 Search PubMed.
- H. Yuan, J. Luan, J. Liu and C. Zhong, Adv. Funct. Mater., 2024, 34, 2400289 Search PubMed.
- G. Cadeco, P. Costa, B. B. Campos, M. Algarra, M. V. Martinez de Yuso, P. R. Moreira, P. Silva and E. Vieira, Conserv. Patrim., 2021, 38, 35–43 Search PubMed.
- X. Liu, Q. Wang, H. Wang, J. Liu, X. Fan, C. Zhong and W. Hu, ACS Appl. Mater. Interfaces, 2022, 14, 56856–56866 Search PubMed.
- S. Debnath, A. Maiti, P. Naskar and A. Banerjee, ChemNanoMat, 2022, 8, e202200261 CrossRef CAS.
- C. Xu, B. Li, H. Du and F. Kang, Angew. Chem., Int. Ed., 2012, 51, 933–935 CrossRef CAS PubMed.
- M. H. Alfaruqi, V. Mathew, J. Gim, S. Kim, J. Song, J. P. Baboo, S. H. Choi and J. Kim, Chem. Mater., 2015, 27, 3609–3620 Search PubMed.
- R. Trocoli and F. La Mantia, ChemSusChem, 2015, 8, 481–485 CrossRef CAS PubMed.
- D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, Nat. Energy, 2016, 1, 16119 Search PubMed.
- Q. Zhao, W. Huang, Z. Luo, L. Liu, Y. Lu, Y. Li, L. Li, J. Hu, H. Ma and J. Chen, Sci. Adv., 2018, 4, eaao1761 Search PubMed.
- J. F. Parker, C. N. Chervin, E. S. Nelson, D. R. Rolison and J. W. Long, Energy Environ. Sci., 2014, 7, 1117–1124 Search PubMed.
- L. Kang, M. Cui, F. Jiang, Y. Gao, H. Luo, J. Liu, W. Liang and C. Zhi, Adv. Energy Mater., 2018, 8, 1801090 CrossRef.
- J. Zheng, Q. Zhao, T. Tang, J. Yin, C. D. Quilty, G. D. Renderos, X. Liu, Y. Deng, L. Wang, D. C. Bock, C. Jaye, D. Zhang, E. S. Takeuchi, K. J. Takeuchi, A. C. Marschilok and L. A. Archer, Science, 2019, 366, 645–648 CrossRef CAS PubMed.
- S.-B. Wang, Q. Ran, R.-Q. Yao, H. Shi, Z. Wen, M. Zhao, X.-Y. Lang and Q. Jiang, Nat. Commun., 2020, 11, 1634 CrossRef CAS PubMed.
- T. K. A. Hoang, D. T. Nam Long, J. H. Cho, J. Y. J. Su, C. Lee, C. Lu and P. Chen, ChemSusChem, 2017, 10, 2816–2822 CrossRef CAS PubMed.
- F. Wang, O. Borodin, T. Gao, X. Fan, W. Sun, F. Han, A. Faraone, J. A. Dura, K. Xu and C. Wang, Nat. Mater., 2018, 17, 543–549 Search PubMed.
- H. Qiu, X. Du, J. Zhao, Y. Wang, J. Ju, Z. Chen, Z. Hu, D. Yan, X. Zhou and G. Cui, Nat. Commun., 2019, 10, 5374 CrossRef PubMed.
- L. Cao, D. Li, E. Hu, J. Xu, T. Deng, L. Ma, Y. Wang, X.-Q. Yang and C. Wang, J. Am. Chem. Soc., 2020, 142, 21404–21409 CrossRef CAS PubMed.
- J. Yang, R. Zhao, Y. Wang, Z. Hu, Y. Wang, A. Zhang, C. Wu and Y. Bai, Adv. Funct. Mater., 2023, 33, 2213510 Search PubMed.
- Z. Dong, C. Zhong, H. Chai, G. Weng, J. Chen, X. Guo, Y. Wang, J. Jiang, X. Huang, J. Fan, C. Wei, C. Wu, H. Liu, S. Dou and X. Yang, Adv. Funct. Mater., 2025, 35, 2503502 CrossRef CAS.
- Y. Xiang, L. Zhou, P. Tan, S. Dai, Y. Wang, S. Bao, Y. Lu, Y. Jiang, M. Xu and X. Zhang, ACS Nano, 2023, 17, 19275–19287 CrossRef CAS PubMed.
- Y. Xia, Z. Hong, L. Zhou, S. Chen, Z. Luo, S. Jin, Y. Huang, Y. Jiang and Y. Wu, J. Energy Chem., 2023, 87, 153–161 Search PubMed.
- X. Wu, Y. Xia, S. Chen, Z. Luo, X. Zhang, Y. Lu, H. Pan, B. B. Xu, M. Yan and Y. Jiang, Small, 2024, 20, 2306739 Search PubMed.
- J. Cao, D. Zhang, C. Gu, X. Wang, S. Wang, X. Zhang, J. Qin and Z.-S. Wu, Adv. Energy Mater., 2021, 11, 2101299 CrossRef CAS.
- S. Hong, Z. Choi, B. Hwang and A. Matic, ACS Energy Lett., 2024, 9, 5421–5433 CrossRef CAS.
- C. Wei, L. Tan, Y. Zhang, K. Zhang, B. Xi, S. Xiong, J. Feng and Y. Qian, ACS Nano, 2021, 15, 12741–12767 CrossRef CAS PubMed.
- X. Yang, Z. Dong, G. Weng, Y. Su, J. Huang, H. Chai, Y. Zhang, K. Wu, J.-B. Baek, J. Sun, D. Chao, H. Liu, S. Dou and C. Wu, Adv. Energy Mater., 2024, 14, 2401293 Search PubMed.
- M. Li, X. Wang, J. Meng, C. Zuo, B. Wu, C. Li, W. Sun and L. Mai, Adv. Mater., 2024, 36, 2308628 Search PubMed.
- L. Jiang, D. Li, X. Xie, D. Ji, L. Li, L. Li, Z. He, B. Lu, S. Liang and J. Zhou, Energy Storage Mater., 2023, 62, 102932 Search PubMed.
- S. Chen, Z. Luo, Y. Xia, X. Wu, K. Wang, M. Yan, H. Pan, B. Bin Xu and Y. Jiang, Angew. Chem., Int. Ed., 2025, 64, e202501176 CrossRef CAS PubMed.
- J. Zhang, W. Huang, L. Li, C. Chang, K. Yang, L. Gao and X. Pu, Adv. Mater., 2023, 35, 2300073 Search PubMed.
- J. Zhang, L. N. Y. Cao, R. Li, J. Yang, L. Li, K. Yang, Z. L. Wang and X. Pu, Adv. Mater., 2025, 37, 2410244 CrossRef CAS PubMed.
- Z. Cai, Y. Ou, B. Zhang, J. Wang, L. Fu, M. Wan, G. Li, W. Wang, L. Wang, J. Jiang, Z. W. Seh, E. Hu, X.-Q. Yang, Y. Cui and Y. Sun, J. Am. Chem. Soc., 2021, 143, 3143–3152 CrossRef CAS PubMed.
- Z. Wu, Q. Li, Z. Chen and C. Zhi, Matter, 2025, 8, 102137 CrossRef CAS.
- L. Yuan, J. Hao, C.-C. Kao, C. Wu, H.-K. Liu, S.-X. Dou and S.-Z. Qiao, Energy Environ. Sci., 2021, 14, 5669–5689 RSC.
- J. Chen, W. Zhao, J. Jiang, X. Zhao, S. Zheng, Z. Pan and X. Yang, Energy Storage Mater., 2023, 59, 102767 CrossRef.
- L. Ma, Q. Li, Y. Ying, F. Ma, S. Chen, Y. Li, H. Huang and C. Zhi, Adv. Mater., 2021, 33, 2007406 CrossRef CAS PubMed.
- Z. Cai, Y. Ou, J. Wang, R. Xiao, L. Fu, Z. Yuan, R. Zhan and Y. Sun, Energy Storage Mater., 2020, 27, 205–211 Search PubMed.
- X. Yu, Z. Li, X. Wu, H. Zhang, Q. Zhao, H. Liang, H. Wang, D. Chao, F. Wang, Y. Qiao, H. Zhou and S. G. Sun, Joule, 2023, 7, 1145–1175 Search PubMed.
- X. Wang, W. Zhou, L. Wang, Y. Zhang, S. Li, X. Li, Z. Zhao, T. Zhang, H. Jin, X. Song, P. Liang, B. Zhang, D. Zhao and D. Chao, Adv. Mater., 2025, 37, 2501049 Search PubMed.
- K. Ouyang, S. Chen, W. Ling, M. Cui, Q. Ma, K. Zhang, P. Zhang and Y. Huang, Angew. Chem., Int. Ed., 2023, 62, e202311988 Search PubMed.
- H. Li, L. Yang, S. Zhou, J. Li, Y. Chen, X. Meng, D. Xu, C. Han, H. Duan and A. Pan, Adv. Funct. Mater., 2024, 34, 2313859 Search PubMed.
- Q. Zhao, X. Yu, J. Xue, M. Zhang, Z. Li, J. Wang, Y. Yang, Y. Zou, Y. Qiao and S.-G. Sun, ACS Energy Lett., 2024, 9, 4102–4110 Search PubMed.
- C. Liu, X. Xie, B. Lu, J. Zhou and S. Liang, ACS Energy Lett., 2021, 6, 1015–1033 Search PubMed.
- Z. Wu, Y. Wang and C. Zhi, Joule, 2024, 8, 2442–2448 CrossRef.
- L. Qian, H. Zhu, T. Qin, R. Yao, J. Zhao, F. Kang and C. Yang, Adv. Funct. Mater., 2023, 33, 2301118 CrossRef CAS.
- X. Yu, M. Chen, Z. Li, X. Tan, H. Zhang, J. Wang, Y. Tang, J. Xu, W. Yin, Y. Yang, D. Chao, F. Wang, Y. Zou, G. Feng, Y. Qiao, H. Zhou and S.-G. Sun, J. Am. Chem. Soc., 2024, 146, 17103–17113 Search PubMed.
- Y. Yang, H. Yang, R. Zhu and H. Zhou, Energy Environ. Sci., 2023, 16, 2723–2731 Search PubMed.
- H. Yang, Y. Yang, W. Yang, G. Wu and R. Zhu, Energy Environ. Sci., 2024, 17, 1975–1983 Search PubMed.
- J. Zheng, J. Yin, D. Zhang, G. Li, D. C. Bock, T. Tang, Q. Zhao, X. Liu, A. Warren, Y. Deng, S. Jin, A. C. Marschilok, E. S. Takeuchi, K. J. Takeuchi, C. D. Rahn and L. A. Archer, Sci. Adv., 2020, 6, eabb1122 Search PubMed.
- J. Li, Y. Lou, S. Zhou, Y. Chen, X. Zhao, A. Azizi, S. Lin, L. Fu, C. Han, Z. Su and A. Pan, Angew. Chem., Int. Ed., 2024, 63, e202406906 CrossRef CAS PubMed.
- M. F. Kropman and H. J. Bakker, Science, 2001, 291, 2118–2120 CrossRef CAS.
- G. Zhou, W. Ding, Y. Guan, T. Wang, C. Zhao, C. Liu, L. Zhang and J. Zhu, Nano Energy, 2024, 132, 110398 Search PubMed.
- K.-D. Kreuer, A. Rabenau and W. Weppner, Angew. Chem., Int. Ed. Engl., 1982, 21, 208–209 CrossRef.
- L. Miao, R. Wang, S. Di, Z. Qian, L. Zhang, W. Xin, M. Liu, Z. Zhu, S. Chu, Y. Du and N. Zhang, ACS Nano, 2022, 16, 9667–9678 Search PubMed.
- J. Hao, L. Yuan, C. Ye, D. Chao, K. Davey, Z. Guo and S.-Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 7366–7375 Search PubMed.
- L. Geng, J. Meng, X. Wang, W. Wu, K. Han, M. Huang, C. Han, L. Wu, J. Li, L. Zhou and L. Mai, Chem, 2025, 11, 102302–102315 Search PubMed.
- H. Zhang, X. Gan, Z. Song and J. Zhou, Angew. Chem., Int. Ed., 2023, 62, e202217833 CrossRef CAS.
- S. Zhang, H. Ao, J. Dong, D. Wang, C. Wang, X. Xu, Z. Hou and J. Yang, Angew. Chem., Int. Ed., 2025, 64, e202414702 CrossRef CAS.
- R. Chen, C. Zhang, J. Li, Z. Du, F. Guo, W. Zhang, Y. Dai, W. Zong, X. Gao, J. Zhu, Y. Zhao, X. Wang and G. He, Energy Environ. Sci., 2023, 16, 2540–2549 Search PubMed.
- W. Deng, Z. Deng, X. Zhang, Y. Chen, R. Feng, G. Li and X. Wang, Angew. Chem., Int. Ed., 2025, 64, e202416482 Search PubMed.
- G. Weng, X. Yang, Z. Wang, Y. Xu and R. Liu, Small, 2023, 19, 2303949 CrossRef CAS.
- H. Li, Z. Liu, G. Liang, Y. Huang, Y. Huan, M. Zhu, Z. Pei, Q. Xue, Z. Tang, Y. Wang, B. Li and C. Zhi, ACS Nano, 2018, 12, 3140–3148 CrossRef CAS.
- P. Lin, J. Cong, J. Li, M. Zhang, P. Lai, J. Zeng, Y. Yang and J. Zhao, Energy Storage Mater., 2022, 49, 172–180 Search PubMed.
- L. Geng, X. Wang, K. Han, P. Hu, L. Zhou, Y. Zhao, W. Luo and L. Mai, ACS Energy Lett., 2022, 7, 247–260 Search PubMed.
- M. Qiu, Y. Liang, J. Hong, J. Li, P. Sun and W. Mai, Angew. Chem., Int. Ed., 2024, 63, e202407012 Search PubMed.
- M. Luo, C. Wang, H. Lu, Y. Lu, B. Bin Xu, W. Sun, H. Pan, M. Yan and Y. Jiang, Energy Storage Mater., 2021, 41, 515–521 CrossRef.
- R. Meng, H. Li, Z. Lu, C. Zhang, Z. Wang, Y. Liu, W. Wang, G. Ling, F. Kang and Q.-H. Yang, Adv. Mater., 2022, 34, 2200677 Search PubMed.
- D. Xu, X. Ren, H. Li, Y. Zhou, S. Chai, Y. Chen, H. Li, L. Bai, Z. Chang, A. Pan and H. Zhou, Angew. Chem., Int. Ed., 2024, 63, e202402833 CrossRef CAS PubMed.
- Z. Zhao, J. Zhao, Z. Hu, J. Li, J. Li, Y. Zhang, C. Wang and G. Cui, Energy Environ. Sci., 2019, 12, 1938–1949 Search PubMed.
- Y.-F. Qu, J.-W. Qian, F. Zhang, Z. Zhu, Y. Zhu, Z. Hou, Q. Meng, K. Chen, S. X. Dou and L.-F. Chen, Adv. Mater., 2025, 37, 2413370 CrossRef CAS PubMed.
- R. Qin, Y. Wang, M. Zhang, Y. Wang, S. Ding, A. Song, H. Yi, L. Yang, Y. Song, Y. Cui, J. Liu, Z. Wang, S. Li, Q. Zhao and F. Pan, Nano Energy, 2021, 80, 105478 CrossRef CAS.
- Z. Li, Y. Liao, Y. Wang, J. Cong, H. Ji, Z. Huang and Y. Huang, Energy Storage Mater., 2023, 56, 174–182 CrossRef.
- C. You, R. Wu, X. Yuan, L. Liu, J. Ye, L. Fu, P. Han and Y. Wu, Energy Environ. Sci., 2023, 16, 5096–5107 Search PubMed.
- J. Heo, D. Dong, Z. Wang, F. Chen and C. Wang, Joule, 2025, 9, 101844 CrossRef CAS.
- Y. Wang, Q. Li, H. Hong, S. Yang, R. Zhang, X. Wang, X. Jin, B. Xiong, S. Bai and C. Zhi, Nat. Commun., 2023, 14, 3890 CrossRef CAS PubMed.
- W. Lin, K. Zhou, L. Xing, S. Huang, M. Ye, Y. Zhang, Y. Tang, X. Liu, Z. Wen, W. Du and C. C. Li, Adv. Energy Mater., 2024, 14, 2404545 Search PubMed.
- W. Yang, X. Du, J. Zhao, Z. Chen, J. Li, J. Xie, Y. Zhang, Z. Cui, Q. Kong, Z. Zhao, C. Wang, Q. Zhang and G. Cui, Joule, 2020, 4, 1557–1574 CrossRef CAS.
- C. Liu, W. Xu, L. Zhang, D. Zhang, W. Xu, X. Liao, W. Chen, Y. Cao, M.-C. Li, C. Mei and K. Zhao, Angew. Chem., Int. Ed., 2024, 63, e202318063 CrossRef CAS PubMed.
- X. Zhao, J. Fu, M. Chen, Y. Wang, C. Huang, K. Qian, G. Feng, B. Li, D. Zhou and F. Kang, J. Am. Chem. Soc., 2025, 147, 2714–2725 Search PubMed.
- Z. Liu, Y. Zhang, M. Li, H. Li, J. Zhang, Y. Zhao, G. Xu, J. Hu, T. Lin and N. Zhang, Angew. Chem., Int. Ed., 2025, 64, e202511520 CrossRef CAS PubMed.
- L. Lin, Z. Shao, S. Liu, P. Yang, K. Zhu, W. Zhuang, C. Li, G. Guo, W. Wang, G. Hong, B. Wu, Q. Zhang and Y. Yao, Angew. Chem., Int. Ed., 2025, 64, e202425008 CrossRef CAS PubMed.
- D. Han, T. Sun, R. Zhang, W. Zhang, T. Ma, H. Du, Q. Wang, D. He, S. Zheng and Z. Tao, Adv. Funct. Mater., 2022, 32, 2209065 Search PubMed.
- J. Zhu, M. Yang, Y. Hu, M. Yao, J. Chen and Z. Niu, Adv. Mater., 2024, 36, 2304426 CrossRef CAS PubMed.
- K. Zhu, X. Niu, W. Xie, H. Yang, W. Jiang, M. Ma and W. Yang, Energy Environ. Sci., 2024, 17, 4126–4136 Search PubMed.
- H. Liu, J.-G. Wang, W. Hua, H. Sun, Y. Huyan, S. Tian, Z. Hou, J. Yang, C. Wei and F. Kang, Adv. Sci., 2021, 8, 2102612 Search PubMed.
- X. Yang, C. Li, Z. Sun, S. Yang, Z. Shi, R. Huang, B. Liu, S. Li, Y. Wu, M. Wang, Y. Su, S. Dou and J. Sun, Adv. Mater., 2021, 33, 2105951 Search PubMed.
- Q. Li, H. Wang, H. Yu, M. Fu, W. Liu, Q. Zhao, S. Huang, L. Zhou, W. Wei, X. Ji, Y. Chen and L. Chen, Adv. Funct. Mater., 2023, 33, 2303466 Search PubMed.
- Y. Xiang, H. Pan, Y. Jiang, S. Xie, M. Xu and X. Zhang, Nano Energy, 2023, 117, 108873 Search PubMed.
- R. Zhang, T. Shui, A. Li, H. Xia, G. Xu, L. Ji, C. Lu, W. Zhang and Z. Sun, Energy Environ. Sci., 2025, 18, 1011–1026 Search PubMed.
- H. Wu, H.-T. Yin, J.-L. Yang and R. Liu, Adv. Energy Mater., 2025, 15, 2501359 Search PubMed.
- Y. Meng, M. Wang, J. Wang, X. Huang, X. Zhou, M. Sajid, Z. Xie, R. Luo, Z. Zhu, Z. Zhang, N. A. Khan, Y. Wang, Z. Li and W. Chen, Nat. Commun., 2024, 15, 8431 Search PubMed.
- H. Yao, Y. Li, Z. Chen, J. Chen, C.-F. Du, Y. Chen, J. Chen, M. W. Wong, J. Zhao and D. Yuan, Angew. Chem., Int. Ed., 2024, 63, e202411056 Search PubMed.
- T. Yan, B. Wu, S. Liu, M. Tao, J. Liang, M. Li, C. Xiang, Z. Cui, L. Du, Z. Liang and H. Song, Angew. Chem., Int. Ed., 2024, 63, e202411470 Search PubMed.
- F. Wu, J. Zhang, L. Ma, P. Ruan, Y. Chen, S. Meng, R. Yin, W. Shi, W. Liu, J. Zhou and X. Cao, Angew. Chem., Int. Ed., 2024, 63, e202421787 Search PubMed.
- Y. Lv, C. Huang, M. Zhao, M. Fang, Q. Dong, W. Tang, J. Yang, X. Zhu, X. Qiao, H. Zheng, C. Sun, L. Zheng, M. Zheng, Y. Xu and J. Lu, J. Am. Chem. Soc., 2025, 147, 8523–8533 Search PubMed.
- Z. Luo, Y. Xia, S. Chen, X. Wu, E. Akinlabi, B. B. Xu, H. Pan, M. Yan and Y. Jiang, Energy Environ. Sci., 2024, 17, 6787–6798 Search PubMed.
- Y. Gao, Y. Liu, Y. Jia, B. Feng, L. Ma and M. Zhu, Adv. Funct. Mater., 2025, 35, 2514985 Search PubMed.
- Q. Meng, Q. Bai, R. Zhao, P. Cao, G. Zhang, J. Wang, F. Su, X. Zhou, J. Yang and J. Tang, Adv. Energy Mater., 2023, 13, 2302828 Search PubMed.
- C. Xiang, T. Yan, B. Wu, S. Liu, L. Du, Z. Cui, H. Song and Z. Liang, Adv. Funct. Mater., 2025, 35, e16431 CrossRef.
- Y. Yuan, J. Yang, Z. Liu, R. Tan, M. Chuai, J. Sun, Y. Xu, X. Zheng, M. Wang, T. Ahmad, N. Chen, Z. Zhu, K. Li and W. Chen, Adv. Energy Mater., 2022, 12, 2103705 Search PubMed.
- Y. Lyu, J. A. Yuwono, P. Wang, Y. Wang, F. Yang, S. Liu, S. Zhang, B. Wang, K. Davey, J. Mao and Z. Guo, Angew. Chem., Int. Ed., 2023, 62, e202303011 Search PubMed.
- Z. Xing, Y. Sun, X. Xie, Y. Tang, G. Xu, J. Han, B. Lu, S. Liang, G. Chen and J. Zhou, Angew. Chem., Int. Ed., 2023, 62, e202215324 Search PubMed.
- M. Cui, L. Yu, J. Hu, S. He, C. Zhi and Y. Huang, Angew. Chem., Int. Ed., 2025, 64, e202423531 CrossRef CAS PubMed.
- S. Chen, Y. Ying, L. Ma, D. Zhu, H. Huang, L. Song and C. Zhi, Nat. Commun., 2023, 14, 2925 CrossRef CAS.
- M. Qiu, P. Sun, K. Han, Z. Pang, J. Du, J. Li, J. Chen, Z. L. Wang and W. Mai, Nat. Commun., 2023, 14, 601 Search PubMed.
- I. Aguilar, J. Brown, L. Godeffroy, F. Dorchies, V. Balland, F. Kanoufi and J.-M. Tarascon, Joule, 2025, 9, 101784 Search PubMed.
- D. Chao, C. Ye, F. Xie, W. Zhou, Q. Zhang, Q. Gu, K. Davey, L. Gu and S.-Z. Qiao, Adv. Mater., 2020, 32, 2001894 CrossRef CAS PubMed.
- P. Xiao, H. Li, J. Fu, C. Zeng, Y. Zhao, T. Zhai and H. Li, Energy Environ. Sci., 2022, 15, 1638–1646 RSC.
- M. Liu, W. Yuan, X. Qu, X. Ru, X. Li, T. Wang, X. Wang, Y. Wang, Y. Liu and N. Zhang, Energy Environ. Sci., 2024, 17, 9611–9622 Search PubMed.
- P. Ruan, X. Chen, L. Qin, Y. Tang, B. Lu, Z. Zeng, S. Liang and J. Zhou, Adv. Mater., 2023, 35, 2300577 CrossRef CAS PubMed.
- Q. Li, Y. Wang, F. Mo, D. Wang, G. Liang, Y. Zhao, Q. Yang, Z. Huang and C. Zhi, Adv. Energy Mater., 2021, 11, 2003931 CrossRef CAS.
- J. Cong, Y. Wang, X. Lin, Z. Huang, H. Wang, J. Li, L. Hu, H. Hua, J. Huang, Y.-C. Lin, H. Xu, Z. Li and Y. Huang, J. Am. Chem. Soc., 2025, 147, 8607–8617 CrossRef CAS PubMed.
- M. Qiu, P. Sun, Y. Wang, L. Ma, C. Zhi and W. Mai, Angew. Chem., Int. Ed., 2022, 61, e202210979 CrossRef CAS PubMed.
- M. Cheng, D. Li, J. Cao, T. Sun, Q. Sun, W. Zhang, Z. Zha, M. Shi, K. Zhang and Z. Tao, Angew. Chem., Int. Ed., 2024, 63, e202410210 CAS.
- H. Xia, G. Xu, X. Cao, C. Miao, H. Zhang, P. Chen, Y. Zhou, W. Zhang and Z. Sun, Adv. Mater., 2023, 35, 2301996 CrossRef CAS PubMed.
- Y. Cui, Q. Zhao, X. Wu, X. Chen, J. Yang, Y. Wang, R. Qin, S. Ding, Y. Song, J. Wu, K. Yang, Z. Wang, Z. Mei, Z. Song, H. Wu, Z. Jiang, G. Qian, L. Yang and F. Pan, Angew. Chem., Int. Ed., 2020, 59, 16594–16601 CrossRef CAS PubMed.
- L. Wang, B. Zhang, W. Zhou, Z. Zhao, X. Liu, R. Zhao, Z. Sun, H. Li, X. Wang, T. Zhang, H. Jin, W. Li, A. Elzatahry, Y. Hassan, H. J. Fan, D. Zhao and D. Chao, J. Am. Chem. Soc., 2024, 146, 6199–6208 CrossRef CAS PubMed.
- S. Chen, Y. Xia, R. Zeng, Z. Luo, X. Wu, X. Hu, J. Lu, E. Gazit, H. Pan, Z. Hong, M. Yan, K. Tao and Y. Jiang, Sci. Adv., 2024, 10, eadn2265 CrossRef CAS.
- J. Yan, H. Dou, M. Su, M. Xu, S. Liu, S. Qin, B. Zhang, K. Zong, L. Tan, X. Wang and Z. Chen, Angew. Chem., Int. Ed., 2025, 64, e202505372 CrossRef CAS PubMed.
- G. Weng, Z. Dong, P. Xiang, Y. Zhu, C. Wu, X. Yang, H. Liu and S. Dou, Adv. Funct. Mater., 2024, 34, 2400839 CrossRef CAS.
- J. Zhou, M. Xie, F. Wu, Y. Mei, Y. Hao, R. Huang, G. Wei, A. Liu, L. Li and R. Chen, Adv. Mater., 2021, 33, 2101649 CrossRef CAS.
- L. Wang, S. Zhou, K. Yang, W. Huang, S. Ogata, L. Gao and X. Pu, Adv. Sci., 2024, 11, 2307667 CrossRef CAS PubMed.
- Z. Yang, C. Hu, Q. Zhang, T. Wu, C. Xie, H. Wang, Y. Tang, X. Ji and H. Wang, Angew. Chem., Int. Ed., 2023, 62, e202308017 CrossRef CAS PubMed.
- M. Zhang, C. Sun, G. Chen, Y. Kang, Z. Lv, J. Yang, S. Li, P. Lin, R. Tang, Z. Wen, C. C. Li, J. Zhao and Y. Yang, Nat. Commun., 2024, 15, 9455 Search PubMed.
- Y. Zhao, S. Guo, M. Chen, B. Lu, X. Zhang, S. Liang and J. Zhou, Nat. Commun., 2023, 14, 7080 CrossRef CAS PubMed.
- L. Wang, W. Huang, W. Guo, Z. H. Guo, C. Chang, L. Gao and X. Pu, Adv. Funct. Mater., 2022, 32, 2108533 CrossRef CAS.
- Y. Du, Y. Feng, R. Li, Z. Peng, X. Yao, S. Duan, S. Liu, S. C. Jun, J. Zhu, L. Dai, Q. Yang, L. Wang and Z. He, Small, 2024, 20, 2108533 Search PubMed.
- Y. Xin, J. Qi, H. Xie, Y. Ge, Z. Wang, F. Zhang, B. He, S. Wang and H. Tian, Adv. Funct. Mater., 2024, 34 CAS.
- Z. Hao, Y. Zhang, Z. Hao, G. Li, Y. Lu, S. Jin, G. Yang, S. Zhang, Z. Yan, Q. Zhao and J. Chen, Adv. Mater., 2023, 35, 2209985 Search PubMed.
- X. Xiao, L. C. Greenburg, Y. Li, M. Yang, Y.-K. Tzeng, C. Sui, Y. Peng, Y. Wu, Z. Zhang, X. Gao, R. Xu, Y. Ye, P. Zhang, Y. Yang, A. Vailionis, P.-C. Hsu, J. Qin and Y. Cui, Nano Lett., 2025, 25, 1305–1313 Search PubMed.
- G. Liu, Y. Tang, Y. Wei, H. Li, J. Yan, Z. Feng, W. Du, Q. Yang, M. Ye, Y. Zhang, Z. Wen, X. Liu and C. C. Li, Angew. Chem., Int. Ed., 2024, 63, e202407639 Search PubMed.
- L. Ren, Z. Hu, C. Peng, L. Zhang, N. Wang, F. Wang, Y. Xia, S. Zhang, E. Hu and J. Luo, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2309981121 Search PubMed.
- S. D. Pu, C. Gong, Y. T. Tang, Z. Ning, J. Liu, S. Zhang, Y. Yuan, D. Melvin, S. Yang, L. Pi, J.-J. Marie, B. Hu, M. Jenkins, Z. Li, B. Liu, S. C. E. Tsang, T. J. Marrow, R. C. Reed, X. Gao, P. G. Bruce and A. W. Robertson, Adv. Mater., 2022, 34, 2202552 Search PubMed.
- M. Zhou, S. Guo, J. Li, X. Luo, Z. Liu, T. Zhang, X. Cao, M. Long, B. Lu, A. Pan, G. Fang, J. Zhou and S. Liang, Adv. Mater., 2021, 33, 2100187 CrossRef CAS PubMed.
- X. Zhang, J. Li, Y. Liu, B. Lu, S. Liang and J. Zhou, Nat. Commun., 2024, 15, 2735 CrossRef CAS PubMed.
- Z. Liu, Z. Guo, L. Fan, C. Zhao, A. Chen, M. Wang, M. Li, X. Lu, J. Zhang, Y. Zhang and N. Zhang, Adv. Mater., 2024, 36, 2305988 CrossRef CAS PubMed.
- D. Xu, B. Chen, X. Ren, C. Han, Z. Chang, A. Pan and H. Zhou, Energy Environ. Sci., 2024, 17, 642–654 Search PubMed.
- J. Zhou, F. Wu, Y. Mei, Y. Hao, L. Li, M. Xie and R. Chen, Adv. Mater., 2022, 34, 2200782 CrossRef CAS PubMed.
- J. Ji, Z. Zhu, H. Du, X. Qi, J. Yao, H. Wan, H. Wang, L. Qie and Y. Huang, Adv. Mater., 2023, 35, 2211961 Search PubMed.
- J. Zheng, Z. Huang, Y. Zeng, W. Liu, B. Wei, Z. Qi, Z. Wang, C. Xia and H. Liang, Nano Lett., 2022, 22, 1017–1023 Search PubMed.
- T. Wang, Y. Tang, M. Yu, B. Lu, X. Zhang and J. Zhou, Adv. Funct. Mater., 2023, 33, 2306101 CrossRef CAS.
- M. Kwon, J. Lee, S. Ko, G. Lim, S.-H. Yu, J. Hong and M. Lee, Energy Environ. Sci., 2022, 15, 2889–2899 RSC.
- Y. Li, X. Ma, X. Zhang, F. Zhang, Q. Wang, Q. Guo, J. Liu, Y. Wang, J. Huang and Y. Xia, Energy Environ. Sci., 2024, 17, 9205–9214 RSC.
- X. Liu, J.-W. Qian, J.-W. Chen, Y.-K. Xu, W.-Y. Wang, W.-X. Dong, W. Hu, G.-R. Cai, J. Lu, S.-H. Yu and L.-F. Chen, Angew. Chem., Int. Ed., 2025, 64, e202504613 CrossRef CAS PubMed.
- X. Liu, W. Zhang, Y. Liu, X. Li, D. Zhang, K. Wang, L. Liu and C. Huang, Energy Environ. Sci., 2024, 17, 9538–9547 Search PubMed.
- Q. Shen, J. Wang, Z. He, W. Fu, E. H. Ang and J. Wang, Angew. Chem., Int. Ed., 2025, 64, e202507504 CrossRef CAS PubMed.
- L. Han, Y. Guo, F. Ning, X. Liu, J. Yi, Q. Luo, B. Qu, J. Yue, Y. Lu and Q. Li, Adv. Mater., 2024, 36, 2308086 Search PubMed.
- J. Tao, X. Cai, Y. Li, L. Huang, X. Zhang, H. Zhang, D. Ma, L. Ran and W. Song, Adv. Energy Mater., 2025, 15, 2403662 CrossRef CAS.
- Y. Wang, W. Chen, F. Wang, X. Li, Z. Zhang, W. Li and F. Wang, Adv. Mater., 2025, 37, 2500596 Search PubMed.
- Y.-Q. Liu, Y.-H. Song, G.-D. Yang, J.-Y. Zhang, X.-Y. Shen, X.-L. Wu and H.-Z. Sun, Energy Storage Mater., 2024, 72, 103761 CrossRef.
- Z. Jiang, Z. Du, R. Pan, F. Cui, G. Zhang, S. Lei, G. He, K. Yin and L. Sun, Adv. Energy Mater., 2024, 14, 2402150 CrossRef CAS.
- Z. Zhao, R. Wang, C. Peng, W. Chen, T. Wu, B. Hu, W. Weng, Y. Yao, J. Zeng, Z. Chen, P. Liu, Y. Liu, G. Li, J. Guo, H. Lu and Z. Guo, Nat. Commun., 2021, 12, 6606 CrossRef CAS PubMed.
- D. Ma, F. Li, K. Ouyang, Q. Chen, J. Zhao, M. Chen, M. Yang, Y. Wang, J. Chen, H. Mi, C. He and P. Zhang, Nat. Commun., 2025, 16, 4817 CrossRef CAS PubMed.
- M. Liu, W. Yuan, G. Ma, K. Qiu, X. Nie, Y. Liu, S. Shen and N. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202304444 CrossRef CAS PubMed.
- H. Lin, L. Zeng, C. Lin, J. Wu, H. He, C. Huang, W. Lai, P. Xiong, F. Xiao, Q. Qian, Q. Chen and J. Lu, Energy Environ. Sci., 2025, 18, 1282–1293 Search PubMed.
- K. Wang, H. Zhan, W. Su, X.-X. Liu and X. Sun, Energy Environ. Sci., 2025, 18, 1398–1407 RSC.
- L. Liu, X. Wang, Z. Hu, X. Wang, Q. Zheng, C. Han, J. Xu, X. Xu, H.-K. Liu, S.-X. Dou and W. Li, Angew. Chem., Int. Ed., 2024, 63, e202405209 CrossRef CAS PubMed.
- M. Kim, J. Lee, Y. Kim, Y. Park, H. Kim and J. W. Choi, J. Am. Chem. Soc., 2023, 145, 15776–15787 CrossRef CAS PubMed.
- S. Wang, S. Wang, Z. Wei, Y. Wang, D. Zhang, Z. Chen and C. Zhi, Nat. Commun., 2025, 16, 1800 Search PubMed.
- W. Zhong, H. Cheng, S. Zhang, L. Li, C. Tan, W. Chen and Y. Lu, Nat. Commun., 2025, 16, 4586 Search PubMed.
- Y. Ouyang, W. Zong, X. Gao, S. X. Leong, J. R. T. Chen, Y. Dai, H. Dong, I. Y. Phang, P. R. Shearing, G. He, Y.-E. Miao, T. Liu and X. Y. Ling, Angew. Chem., Int. Ed., 2025, 64, e202504965 CrossRef CAS PubMed.
- T. Li, A. Naveed, J. Zheng, B. Chen, M. Jiang, B. Liu, Y. Zhou, X. Li, M. Su, R. Guo, J. Sumner, C. C. Li and Y. Liu, Angew. Chem., Int. Ed., 2025, 64, e202424095 Search PubMed.
- Z. Peng, S. Li, L. Tang, J. Zheng, L. Tan and Y. Chen, Nat. Commun., 2025, 16, 4490 CrossRef CAS PubMed.
- K. Liu, M. Sun, Y. Wu, T. Zhang, A. Zhu, S. Bu, C. Luan, K. Liu, Y. Zhou, D. Lin, S. Wu, C. S. Lee, B. Huang, G. Hong and W. Zhang, Adv. Mater., 2025, 37, 2420079 Search PubMed.
- H. Du, Y. Dong, Q.-J. Li, R. Zhao, X. Qi, W.-H. Kan, L. Suo, L. Qie, J. Li and Y. Huang, Adv. Mater., 2023, 35, 2210055 Search PubMed.
- K. Guan, W. Chen, Y. Yang, F. Ye, Y. Hong, J. Zhang, Q. Gu, Y. Wu and L. Hu, Adv. Mater., 2024, 36, 2405889 CrossRef CAS PubMed.
- Y. Qiu, Y. Lin, D. Shi, H. Zhang, J. Luo, J. Chen, Z. Liu, Y. Yu, D. Lin, W. Zhang, Y. Li and C. Yang, Adv. Mater., 2025, 37, 2415373 CrossRef CAS PubMed.
- Y. Ding, D. Huang, Y. Wang, X. Zhao, R. Han, M. Tang, B. Li, D. Zhou and F. Kang, Adv. Mater., 2025, 37, 2419221 Search PubMed.
- T. Zhang, J. Yang, H. Wang, H. Yu, Q. Li, L. Chen, Y. Chen and T. Wang, Energy Storage Mater., 2024, 65, 103085 CrossRef.
- F. Ming, Y. Zhu, G. Huang, A.-H. Emwas, H. Liang, Y. Cui and H. N. Alshareef, J. Am. Chem. Soc., 2022, 144, 7160–7170 CrossRef CAS PubMed.
- L. Liu, H. Lu, C. Han, X. Chen, S. Liu, J. Zhang, X. Chen, X. Wang, R. Wang, J. Xu, H. K. Liu, S. X. Dou and W. Li, ACS Nano, 2023, 17, 23065–23078 CrossRef CAS PubMed.
- G. Qu, H. Wei, S. Zhao, Y. Yang, X. Zhang, G. Chen, Z. Liu, H. Li and C. Han, Adv. Mater., 2024, 36, 2400370 CrossRef CAS PubMed.
- L. Miao, R. Wang, W. Xin, L. Zhang, Y. Geng, H. Peng, Z. Yan, D. Jiang, Z. Qian and Z. Zhu, Energy Storage Mater., 2022, 49, 445–453 Search PubMed.
- N. Wang, M. Zhang, X. Ren, M. Sun, T. Pian, P. Wang, Y.-F. Cui, X. Yao, C. Jia and H. Y. Yang, Angew. Chem., Int. Ed., 2025, 64, e202502761 CrossRef CAS PubMed.
- W. Deng, Z. Deng, Y. Chen, R. Feng and X. Wang, Angew. Chem., Int. Ed., 2024, 63, e202316499 CrossRef CAS PubMed.
- M. Han, J. Huang, X. Xie, T. C. Li, J. Huang, S. Liang, J. Zhou and H. J. Fan, Adv. Funct. Mater., 2022, 32, 2110957 CrossRef CAS.
- X. Bai, M. Sun, J. Yang, B. Deng, K. Yang, B. Huang, W. Hu and X. Pu, Energy Environ. Sci., 2024, 17, 7330–7341 Search PubMed.
- W. Lv, Y. Tan, C. Guo, X. He, L. Zeng, J. Zhu, L. Yang, Z. Chen, X. Yin, J. Xu and H. He, Adv. Energy Mater., 2025, 15, 2403689 CrossRef CAS.
- J. Hao, L. Yuan, Y. Zhu, X. Bai, C. Ye, Y. Jiao and S.-Z. Qiao, Angew. Chem., Int. Ed., 2023, 62, e202310284 CrossRef CAS PubMed.
- Y. Zhong, X. Xie, Z. Zeng, B. Lu, G. Chen and J. Zhou, Angew. Chem., Int. Ed., 2023, 62, e202310577 CrossRef CAS PubMed.
- Y. Wang, B. Liang, D. Li, Y. Wang, C. Li, H. Cui, R. Zhang, S. Yang, Z. Chen, Q. Li, F. Mo, J. Fan and C. Zhi, Joule, 2025, 9, 101944 CrossRef CAS.
- Z. Du, S. Shen, X. Su, Y. Zhuang, M. Chen, X. Zhang, Z. Lin, L. Yu, P. Zhou, M. Wu, X. Lyu and Z. Zou, Adv. Mater., 2025, 37, 2502328 CrossRef CAS PubMed.
- X. Xu, S. Li, S. Yang and B. Li, Energy Environ. Sci., 2024, 17, 7919–7931 RSC.
- X. Wang, B. Wang, P. Lei, X. Wang, L. Zhou, J. Zhang, J. Zhang and J. Cheng, Energy Environ. Sci., 2024, 17, 6640–6655 Search PubMed.
- S.-J. Guo, M.-Y. Yan, D.-M. Xu, P. He, K.-J. Yan, J.-X. Zhu, Y.-K. Yu, Z.-Y. Peng, Y.-Z. Luo and F.-F. Cao, Energy Environ. Sci., 2025, 18, 418–429 Search PubMed.
- Z.-J. Chen, T.-Y. Shen, X. Xiao, X.-C. He, Y.-L. Luo, Z. Jin and C.-H. Li, Adv. Mater., 2024, 36, 2413268 Search PubMed.
- K. Zhu, J. Luo, D. Zhang, N. Wang, S. Pan, S. Zhou, Z. Zhang, G. Guo, P. Yang, Y. Fan, S. Hou, Z. Shao, S. Liu, L. Lin, P. Xue, G. Hong, Y. Yang and Y. Yao, Adv. Mater., 2024, 36, 2311082 Search PubMed.
- S. Yang, Q. Wu, Y. Li, F. Luo, J. Zhang, K. Chen, Y. You, J. Huang, H. Xie and Y. Chen, Angew. Chem., Int. Ed., 2024, 63, e202409160 Search PubMed.
- Y. Zhang, Y. Wang, X. Yang, H. Liu, K. Wu, L. Zhang, D. Chao, L. Lin, S.-X. Dou and C. Wu, Energy Storage Mater., 2025, 78, 104246 CrossRef.
- C.-L. Miao, X.-X. Wang, D.-H. Guan, J.-X. Li, J.-Y. Li and J.-J. Xu, Angew. Chem., Int. Ed., 2024, 63, e202410208 CrossRef CAS PubMed.
- X. Shi, Y. Zhong, Y. Yang, J. Zhou, X. Cao and S. Liang, Angew. Chem., Int. Ed., 2025, 64, e202414777 Search PubMed.
- Z. Wang, J. Hu, L. Han, Z. Wang, H. Wang, Q. Zhao, J. Liu and F. Pan, Nano Energy, 2019, 56, 92–99 CrossRef CAS.
- M. Zhao, Y. Lv, Y. Xu, H. Yang, Z. Bo and J. Lu, Nat. Commun., 2025, 16, 2843 CrossRef CAS PubMed.
- D. Wang, D. Lv, H. Peng, C. Wang, H. Liu, J. Yang and Y. Qian, Angew. Chem., Int. Ed., 2023, 62, e202310290 CrossRef CAS PubMed.
- R. Chen, W. Zhang, C. Guan, Y. Zhou, I. Gilmore, H. Tang, Z. Zhang, H. Dong, Y. Dai, Z. Du, X. Gao, W. Zong, Y. Xu, P. Jiang, J. Liu, F. Zhao, J. Li, X. Wang and G. He, Angew. Chem., Int. Ed., 2024, 63, e202401987 CrossRef CAS PubMed.
- L. Cao, D. Li, T. Pollard, T. Deng, B. Zhang, C. Yang, L. Chen, J. Vatamanu, E. Hu, M. J. Hourwitz, L. Ma, M. Ding, Q. Li, S. Hou, K. Gaskell, J. T. Fourkas, X.-Q. Yang, K. Xu, O. Borodin and C. Wang, Nat. Nanotechnol., 2021, 16, 902–910 CrossRef CAS PubMed.
- T. Chen, Y. Wang, X. Li, T. Yuan, Y. Pang and S. Zheng, Angew. Chem., Int. Ed., 2025, 64, e202424642 CrossRef CAS PubMed.
- C. Chang, S. Hu, T. Li, F. Zeng, D. Wang, S. Guo, M. Xu, G. Liang, Y. Tang, H. Li, C. Han and H.-M. Cheng, Energy Environ. Sci., 2024, 17, 680–694 RSC.
- S. Ou, J. Zheng, X. Chen, R. Li, Z. Yuan, S. Liu, Y. Niu, M. An, G. Zhou, Y. Yamauchi and X. Zhang, Energy Environ. Sci., 2025, 18, 5457–5469 Search PubMed.
- T. Li, Y. Gong, H. Yang, Y. Liu, X. Dong, Y. Xu, H. Ma, C. Wei, S. Zhang, F. Huang, M. Yang and T. Lin, Energy Storage Mater., 2025, 79, 104311 CrossRef.
- H. Ma, H. Chen, M. Chen, A. Li, X. Han, D. Ma, P. Zhang and J. Chen, Nat. Commun., 2025, 16, 1014 Search PubMed.
- L. Yang, Y.-J. Zhu, H.-P. Yu, Z.-Y. Wang, L. Cheng, D.-D. Li, J. Tao, G. He and H. Li, Adv. Energy Mater., 2024, 14, 2401858 Search PubMed.
- J. Xu, Y. Yang, Q. Dai, Z. Zheng, Y. Cao, Y. Cheng, B. Peng, L. Ma and Y. Wang, Angew. Chem., Int. Ed., 2025, 64, e202423118 Search PubMed.
- W. Yuan, X. Qu, Y. Wang, X. Li, X. Ru, D. Jia, L. Zhao, Y. Hou, J. Shen, Z. Shen and N. Zhang, Energy Storage Mater., 2025, 76, 104130 Search PubMed.
- Z. Zhao, Y. Zhang, H. Zhang, X. Shi, H. Zhao, J. Liu, J. Liu and L. Li, Nano Lett., 2025, 25, 7483–7490 Search PubMed.
- Q. Wang, J. Zhao, J. Zhang, M. Li, F. Tan, X. Xue, Z. Sui, Y. Zou, X. Zhang, W. Zhang and C. Lu, Adv. Funct. Mater., 2024, 34, 2405957 Search PubMed.
- X. Wang, X. Li, H. Fan and L. Ma, Nano-Micro Lett., 2022, 14, 205 CrossRef CAS PubMed.
- Y. Qin, H. Li, C. Han, F. Mo and X. Wang, Adv. Mater., 2022, 34, 2207118 Search PubMed.
- X. Shi, J. Zeng, A. Yi, F. Wang, X. Liu and X. Lu, J. Am. Chem. Soc., 2024, 146, 20508–20517 Search PubMed.
- X. Yang, X. Tang, J. Lei, X. Zeng, J. Wen, A. Liu, S. Xia, Q. Luo, J. Liu, A. Xue, D. Han and G. Zhou, Angew. Chem., Int. Ed., 2025, 64, e202504003 Search PubMed.
- X. Wu, Y. Xia, S. Chen, Z. Luo, X. Zhang, M. W. Shahzad, B. B. Xu, H. Pan, M. Yan and Y. Jiang, EcoMat, 2024, 6, e12438 CrossRef CAS.
- X. Zhou, B. Wen, Y. Cai, X. Chen, L. Li, Q. Zhao, S.-L. Chou and F. Li, Angew. Chem., Int. Ed., 2024, 63, e202402342 Search PubMed.
- J. Lin, C. Ji, G. Guo, Y. Luo, P. Huang, F. Xu, L. Sun, W. Pfleging and K. S. Novoselov, Angew. Chem., Int. Ed., 2025, 64, e202501721 CrossRef CAS PubMed.
- G. Duan, K. Zhang, Y. Wang, L. Sun, B. Luo, S. Zheng, Z. Bao, M. Zhou, H. Hu, D. Chen, L. Gong, Z. Ye and J. Huang, Energy Storage Mater., 2025, 83, 104619 CrossRef.
- X. Li, P. Liu, C. Han, T. Cai, Y. Cui, W. Xing and C. Zhi, Energy Environ. Sci., 2025, 18, 2050–2094 Search PubMed.
- M. Tang, X. Zhao, R. Han, Y. Wang, Y. Ding, Z. Si, B. Li, D. Zhou and F. Kang, Angew. Chem., Int. Ed., 2025, 64, e202421574 CrossRef CAS PubMed.
- R. Deng, A. S. Menon, M. Walker, L. F. J. Piper, A. W. Robertson and F. Wu, Adv. Funct. Mater., 2025, 35, 2424954 Search PubMed.
- K. Roy, A. Rana, J. N. Heil, B. M. Tackett and J. E. Dick, Angew. Chem., Int. Ed., 2024, 63, e202319010 Search PubMed.
- Z. Wen, W. Fang, F. Wang, H. Kang, S. Zhao, S. Guo and G. Chen, Angew. Chem., Int. Ed., 2024, 63, e202314876 Search PubMed.
- Z. Liu, G. Xu, Y. Zhang, M. Li, H. Li, J. Zhang, J. Hu, T. Lin and N. Zhang, Angew. Chem., Int. Ed., 2025, 64, e202501960 CrossRef CAS PubMed.
- X. Yu, M. Chen, J. Wang, S. Li, H. Zhang, Q. Zhao, H. Luo, Y. Deng, H. Liang, J. Zhou, F. Wang, D. Chao, Y. Zou, G. Feng, Y. Qiao and S.-G. Sun, Nat. Commun., 2025, 16, 3820 CrossRef CAS PubMed.
- T. Wu, C. Hu, Q. Zhang, Z. Yang, G. Jin, Y. Li, Y. Tang, H. Li and H. Wang, Adv. Funct. Mater., 2024, 34, 2315716 CrossRef CAS.
- R. Sun, D. Han, C. Cui, Z. Han, X. Guo, B. Zhang, Y. Guo, Y. Liu, Z. Weng and Q.-H. Yang, Angew. Chem., Int. Ed., 2023, 62, e202303557 Search PubMed.
- X. Liu, D. Lyu, C. Merlet, M. J. A. Leesmith, X. Hua, Z. Xu, C. P. Grey and A. C. Forse, Science, 2024, 384, 321–325 CrossRef CAS PubMed.
- J. J. Lodico, M. Mecklenburg, H. L. Chan, Y. Chen, X. Y. Ling and B. C. Regan, Sci. Adv., 2023, 9, eadg5135 CrossRef CAS PubMed.
- F. Wang, J. Zhang, H. Lu, H. Zhu, Z. Chen, L. Wang, J. Yu, C. You, W. Li, J. Song, Z. Weng, C. Yang and Q.-H. Yang, Nat. Commun., 2023, 14, 4211 CrossRef CAS PubMed.
- Z. Cai, J. Wang and Y. Sun, eScience, 2023, 3, 100093 Search PubMed.
- Y. Wang, X. Ma, X. Yang, R. Zhang, H. Hong, S. Wang, Q. Li, Z. Chen, Z. Huang, H. Lv and C. Zhi, Adv. Mater., 2025, 37, 2419702 CrossRef CAS PubMed.
- X. Zheng, Z. Liu, J. Sun, R. Luo, K. Xu, M. Si, J. Kang, Y. Yuan, S. Liu, T. Ahmad, T. Jiang, N. Chen, M. Wang, Y. Xu, M. Chuai, Z. Zhu, Q. Peng, Y. Meng, K. Zhang, W. Wang and W. Chen, Nat. Commun., 2023, 14, 76 CrossRef CAS PubMed.
- H. Zhao, L. Zhao, D. Yin, N. Gao, Y. Zhang, J. Feng, X. Chen, X. Liu, J. Wei, C. Fan, G. Gao, C. Xiao and S. Ding, Adv. Mater., 2025, 37, e15759 Search PubMed.
- H. Meng, Q. Ran, T.-Y. Dai, J.-H. Jia, J. Liu, H. Shi, G.-F. Han, T.-H. Wang, Z. Wen, X.-Y. Lang and Q. Jiang, Adv. Mater., 2024, 36, 2403803 Search PubMed.
- S.-J. Zhang, J. Hao, H. Wu, Y. Hu, Q. Chen, B. Johannessen, Q. Ma, D. Luo and S.-Z. Qiao, Nat. Chem., 2025 DOI:10.1038/s41557-025-01986-7.
- J. Li, P. Ruan, X. Chen, S. Lei, B. Lu, Z. Chen and J. Zhou, ACS Energy Lett., 2023, 8, 2904–2918 CrossRef CAS.
Footnote |
| † G. Weng and S. Chen contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2026 |