DOI:
10.1039/D5CS01268F
(Review Article)
Chem. Soc. Rev., 2026,
55, 3687-3723
Heteroatom doping in carbon macrocycles for advanced synthesis and applications
Received
11th November 2025
First published on 27th February 2026
Abstract
Macrocycles have garnered significant research interest due to their tunable structures, unique physicochemical properties, and broad range of applications in areas such as aggregation-induced emission (AIE), molecular recognition, bioimaging, and photocatalysis. While conventional carbon-rich macrocycles depend mainly on ring-size adjustments for property modulation, the incorporation of heteroatoms (N, O, and S) allows precise control over electronic structure, band gap, and functionality. This review summarizes recent advances (2020–2025) in the synthesis and applications of heteroatom-doped (N-, O-, S-, N,O-, and N,S-doped) macrocycles. It covers metal-catalyzed (Pd, Ni, Pt, Cu, Ti, Sn, and Fe) and metal-free (acidic/basic) strategies, post-synthetic modifications, and emerging applications in host–guest systems, sensors, OLEDs, OFETs, OPVs, and (photo)catalysis, aiming to advance the field and serve as a reference for cross-disciplinary researchers.

Chao Liu
| Chao Liu obtained his MSc degree in chemistry from Soochow University in 2019. He received his PhD degree in organic chemistry from the University of Leuven (KU Leuven) in 2022, under the supervision of Professor Erik V. Van der Eycken. He finished his postdoctoral research in the same group in 2023. Currently, he is an associate professor at the Nanjing University of Posts and Telecommunications in China. He is working in the area of transition metal catalysis, heterocyclic chemistry and macrocycles. |

Ying Wei
| Ying Wei received her PhD degree from Northeast Normal University in 2014. She then joined the Key Laboratory for Organic Electronics & Information Displays and Institute of Advanced Materials (IAM), Nanjing University of Posts & Telecommunications. Currently, she is an associate professor at the Nanjing University of Posts and Telecommunications. Her research mainly focuses on the design, synthesis, and applications of organic and polymer optoelectronic materials for organic/plastic electronics. |

Linghai Xie
| Linghai Xie is a full professor at the Nanjing University of Posts and Telecommunications. He received his PhD from Fudan University in 2006. He then joined the Key Laboratory for Organic Electronics & Information Displays and Institute of Advanced Materials (IAM), Nanjing University of Posts & Telecommunications. His research mainly focuses on the design, synthesis, and applications of organic and polymer optoelectronic materials for organic/plastic electronics. He is also interested in the exploration of novel materials and processes for molecular electronics. |

Erik V. Van der Eycken
| Erik V. Van der Eycken received his PhD degree (1987) in organic chemistry from the University of Ghent, under the supervision of Professor Maurits Vandewalle. From 1988 to 1992 he worked as a scientific researcher at the R&D laboratories of AGFA-Gevaert, Belgium and moved back to the University of Ghent in 1992. In 1997 he became a Doctor Assistant at the University of Leuven (KU Leuven). He spent time as a visiting scientist at the University of Graz (C. Oliver Kappe), at The Scripps Research Institute (K. Barry Sharpless), and at Uppsala University (Mats Larhed, Anders Hallberg). He was appointed Professor at the University of Leuven (KU Leuven) in 2007 and is presently a Full Professor. |

Wei Huang
| Wei Huang received his PhD from Peking University in 1992. In 2001, he was appointed as a chair professor at Fudan University, where he founded and chaired the Institute of Advanced Materials (IAM). After that he became the vice president of Nanjing University of Posts & Telecommunications and then the president of Nanjing Tech University. He was elected as an Academician of the Chinese Academy of Sciences in 2011. His research interests include organic/plastic/flexible electronics, nanomaterials, nanotechnology, etc. |
1. Introduction
Macrocycles have attracted wide research interest owing to their versatile chemical modifications, outstanding physicochemical properties, and distinctive ring architectures.1–4 These remarkable structures have demonstrated diverse applications in aggregation-induced emission (AIE),5 molecular recognition,6 molecular machines,7 bioimaging,8 chemical sensing,9 drug delivery,10 and photocatalysis.11 Therefore, various approaches have been developed for the synthesis of new macrocycles. This has not only promoted the advancement of organic chemistry but also driven the development of materials science.
In 1970s, calixarenes were first named and characterized by Gutsche.12 Thereafter, various crown-ethers,13 pillararenes,14,15 cucurbiturils,16 and cyclodextrins (CDs),17 all exhibiting unique characteristics in host–guest chemistry and materials science, were also reported. Among these, carbon-rich macrocycles derived from benzene or polycyclic aromatic hydrocarbon (PAH) units primarily regulate their properties by adjusting the size of the macrocyclic rings.18 However, this reliance on ring-size tuning significantly limits the further application and development of these materials. Subsequently, main-group elements, including sulfur (S), boron (B), oxygen (O), and nitrogen (N), have been extensively incorporated into macrocyclic frameworks to create functional molecules and broaden applications.19 By varying the type, count, and spatial arrangement of heteroatoms, this strategy allows for fine-tuning of the electronic structure and highest occupied molecular orbital (HOMO)–lowest unoccupied molecular orbital (LUMO) band gap, thereby enabling systematic modulation of macrocycle properties.1,2 For instance, N doping can lower the LUMO energy level, while B incorporation can significantly modulate absorption and emission profiles, thereby tailoring optoelectronic properties for specific applications. Moreover, heteroatoms (e.g., N, O, and S) serve as critical handles for introducing rich non-covalent interactions. For example, N and O atoms enable directional hydrogen bonding and metal-ion coordination, while the S atom not only participates in coordination but also engages in directional chalcogen-bonding interactions (e.g., S⋯S). However, compared to all-carbon macrocycles, the synthesis of heteroatom-containing macrocycles presents several distinct challenges.3 For example, the precise control over the heteroatom type, number, and position during monomer design often leads to regioisomeric mixtures. The heteroatoms themselves can act as reactive sites that poison catalysts or promote side reactions during critical cyclization steps, significantly reducing yields. Additionally, managing the solubility and stability of sensitive intermediates requires precise condition control, and final purification often demands advanced techniques such as recycling gel-permeation chromatography (GPC). Notably, incorporating boron atoms into calixarenes, pillararenes, and crown-ether scaffolds has enabled the synthesis of various boron-doped macrocycles for diverse applications. These derivatives demonstrate distinct guest–recognition capabilities and unprecedented catalytic functions compared to their all-carbon analogues, as thoroughly summarized by Lu et al. in a recent review.20 Furthermore, as reviewed by Yang,21 borondipyrromethene (BODIPY)-based macrocycles applied in sensing, bioimaging, and photodynamic therapy show that alterations in macrocyclic linking units significantly modulate their photophysical behaviors, geometric properties, and practical applications.
This review will highlight the advances made in recent years (2020–2025) in the synthesis and applications of heteroatom-doped (e.g., N-, O-, S-, N,O-, and N,S-doped) macrocycles, while excluding less common types such as phosphorus-doped systems, because their synthetic methodologies and demonstrated applications remain limited (Fig. 1). Metal-catalyzed macrocyclization reactions using catalysts such as Pd, Ni, Pt, Cu, Ti, Sn, and Fe for constructing diverse macrocyclic scaffolds will be presented. Metal-free strategies, including acidic (Lewis and Brønsted acids) and basic conditions, for the synthesis of heteroatom-doped macrocycles, are also emphasized. Further transformations to access novel macrocycles will also be highlighted. Finally, their applications in host–guest chemistry, sensing, organic light-emitting diodes (OLEDs), organic field effect transistors (OFETs), (photo)catalysis, and organic solar cells (OSCs) will be introduced. We believe that this review will pave the way for new advancements in heteroatom-doped macrocycles and serve as a valuable resource for researchers in chemistry, materials science, biology, and related disciplines.
 |
| | Fig. 1 Synthesis and applications of heteroatom-doped macrocycles. | |
2. N-doped macrocycles
Classical N-doped macrocycles, including porphyrins, phthalocyanines, cyclens, and calixpyrroles, exhibit promising applications across multiple disciplines. Porphyrins and phthalocyanines are aromatic macrocycles with pyrrole rings, enabling metal coordination critical for biological functions and modern applications in catalysis, photodynamic therapy, and light-harvesting materials.22,23 Cyclen, a tetraazamacrocycle bearing four N-atoms for versatile functionalization, has emerged as a pivotal scaffold for constructing artificial nucleases and biological nucleotide sensors, enabling precise manipulation and detection of nucleic acids.24 Calixpyrroles, with flexible, bowl-shaped cavities, have roles as neutral/monoanionic ligands for metal coordination and anion recognition, with hybrid structures facilitating applications in catalysis and supramolecular chemistry.25 Recent advancements in synthetic methods allow precise construction of functionalized N-doped macrocycles, controlling the ring size, substitution patterns, and heteroatom integration to create complex, tailored frameworks for diverse applications.26,27
2.1. Metal catalysis
2.1.1. Palladium catalysis.
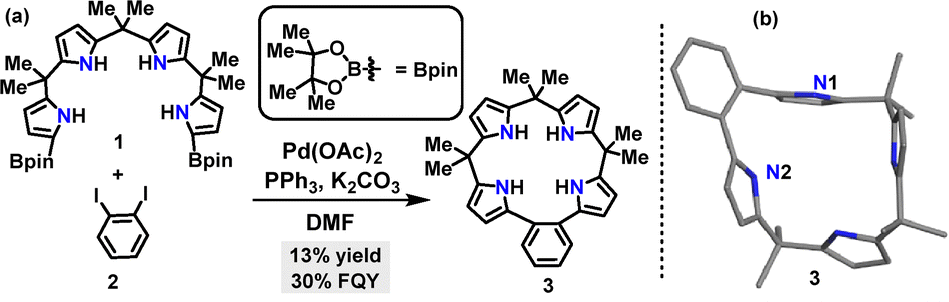
In 2021, Panda's group synthesized calix[4]pyrrole 3 in 13% yield via a palladium-catalyzed [1+1] Suzuki coupling reaction between the diborylated tetrapyrrane 1 and 2-diiodobenzene 2 (Scheme 1a).28 Crystal structure analysis revealed that macrocycle 3 adopted a slightly irregular 1,3-alternate conformation (Scheme 1b). The N1-linked pyrrole unit, connected to the o-phenylene spacer, displayed near-orthogonal geometry relative to the benzene ring plane (dihedral angle: 81°), whereas the N2-linked pyrrole unit exhibited a more coplanar arrangement (dihedral angle: 26°), suggesting potential for extended π-conjugation.
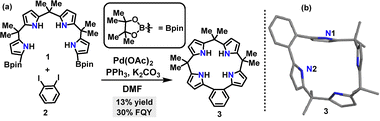 |
| | Scheme 1 (a) Synthesis and (b) crystal structure of calix[4]pyrrole 3. | |
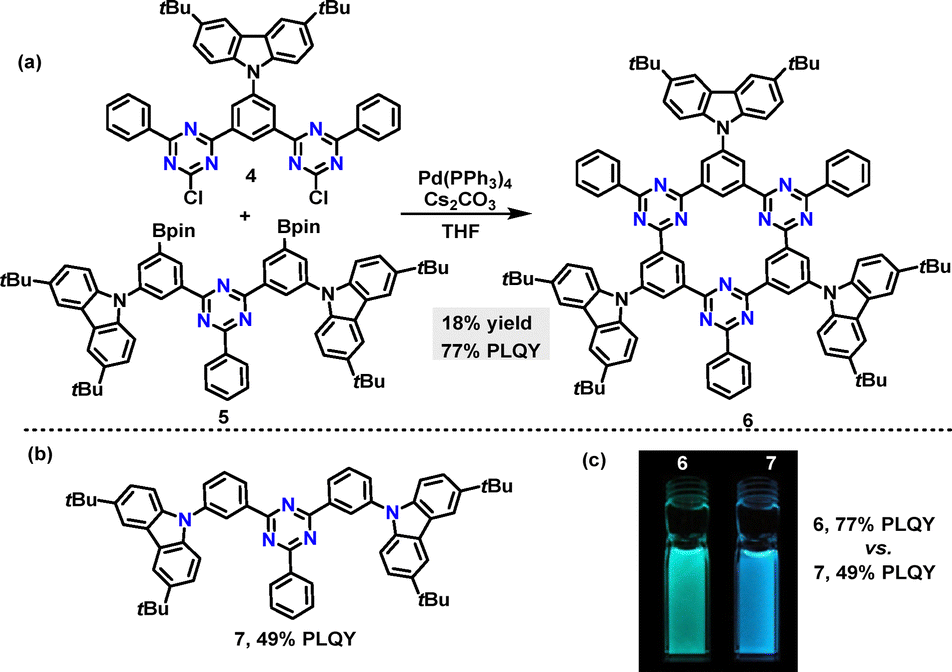
In the same year, Yasuda reported an efficient method to synthesize a thermally activated delayed fluorescence (TADF) π-conjugated macrocycle incorporating electron-donor (D) and acceptor (A) units (Scheme 2a).29 Macrocycle 6 was obtained in 18% yield via a palladium-catalyzed Suzuki–Miyaura cross-coupling reaction. The photoluminescence (PL) peak wavelengths (λPL) of macrocycle 6 exhibited a bathochromic shift in intramolecular charge transfer (ICT) absorption compared to linear 7 (Scheme 2b). Upon photoexcitation, macrocycle 6 emitted green PL at 496 nm (λPL), while 7 produced deep-blue PL at 468 nm (Scheme 2c). Owing to its rigid macrocyclic structure, macrocycle 6 displayed a smaller full width at half-maximum in its PL spectrum and lower reorganization energy than linear 7. Notably, macrocycle 6 achieved a 77% absolute photoluminescence quantum yield (PLQY) in toluene, surpassing 7's 49%, attributed to its TADF characteristics.
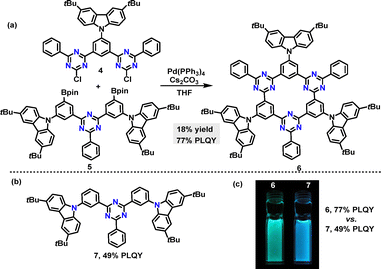 |
| | Scheme 2 (a) Synthesis of TADF π-conjugated macrocycle 6, (b) structure of linear 7, and (c) photograph of PL emissions under UV illumination at 365 nm. Adapted from ref. 29 with permission from Wiley-VCH, copyright 2021. | |
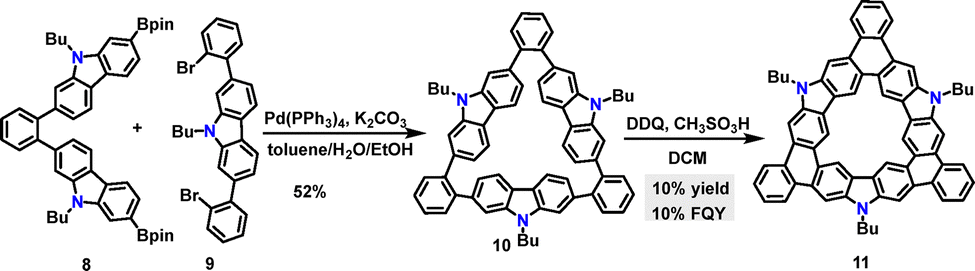
Later, Miao and co-workers reported a Suzuki coupling reaction between substrates 8 and 9, which afforded the carbazole-fused macrocycle 10 in 52% yield (Scheme 3).30 Under DDQ/CH3SO3H conditions, the fused macrocycle 11 was obtained in 10% yield via a regioselective Scholl reaction at the C-3 position of the carbazole moiety. Computational studies revealed significant bond-length alternation in the bowl structure of macrocycle 11, which was attributed to structural strain (34.7 kcal mol−1) and localized aromaticity. As an orange solid, macrocycle 11 dissolved in dichloromethane (DCM) to form a yellow solution that exhibited green fluorescence under UV irradiation, with a measured FQY of 10%.
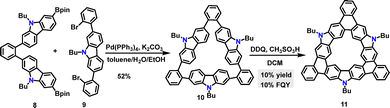 |
| | Scheme 3 Synthesis and modification of carbazole-fused macrocycle 10. | |
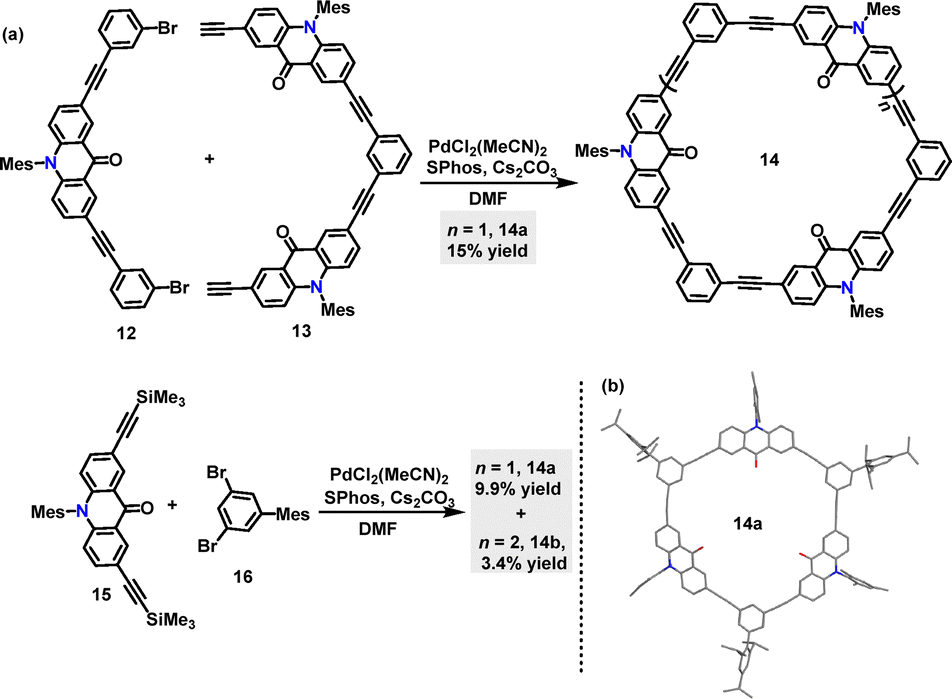
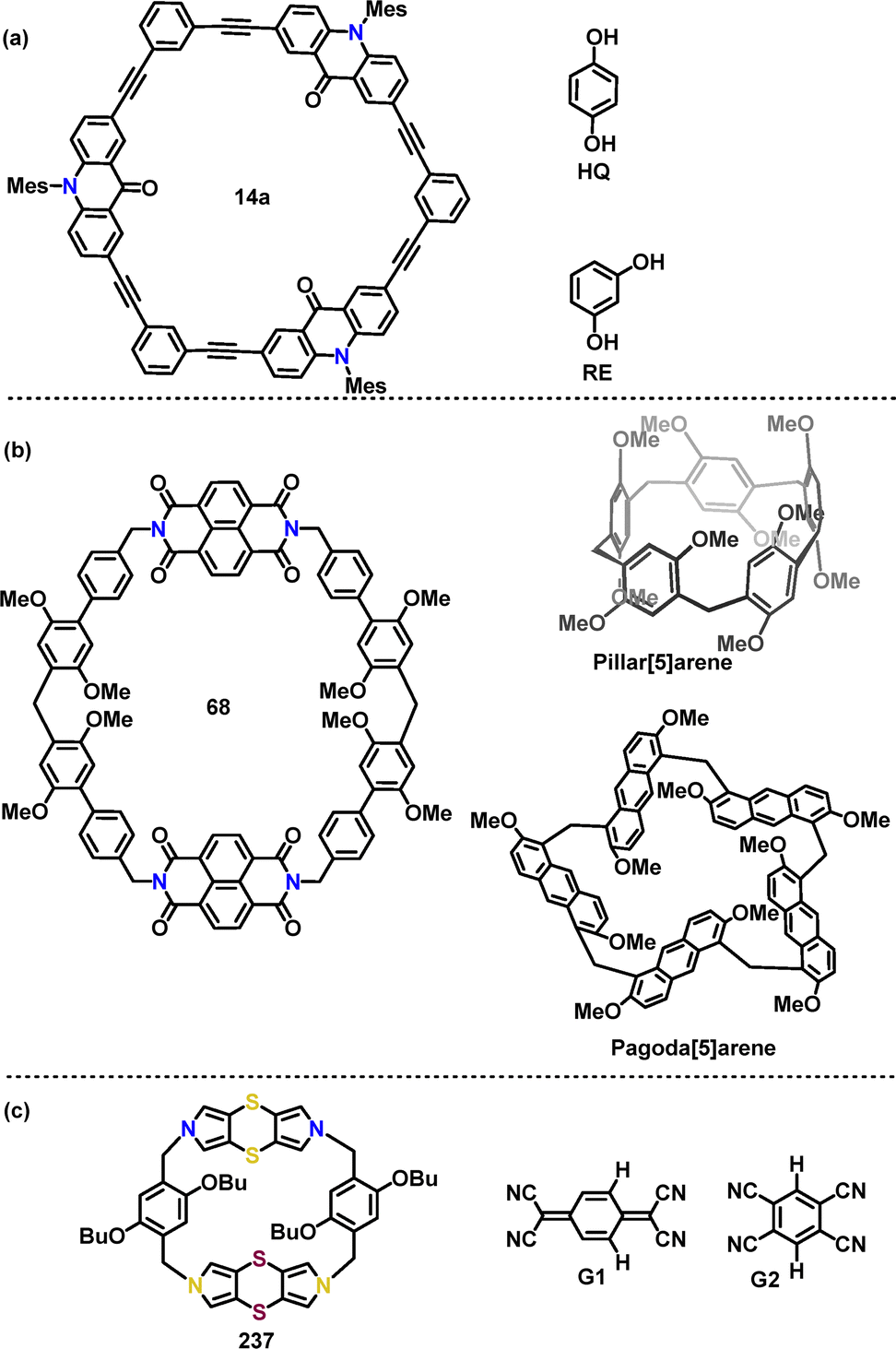
In 2023, Toyota31 reported two synthetic routes for acridone-incorporated arylene-ethynylene macrocycles 14 utilizing palladium catalysis (Scheme 4a). Macrocycle 14a, featuring three acridone-2,7-diyl units and three 1,3-phenylene units, was synthesized via Sonogashira coupling of 12 and 13 with a 15% yield. To streamline the synthesis, alternative monomeric precursors were explored, affording 14b in 9.9% and 14c in 3.4% yields. Crystal structure analysis confirmed that the trimeric macrocycle 14b adopted a near-planar framework with a cavity defined by three carbonyl groups (Scheme 4b).
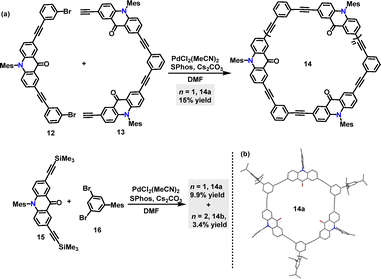 |
| | Scheme 4 Synthesis of macrocycles 14 and the crystal structure of 14a. | |
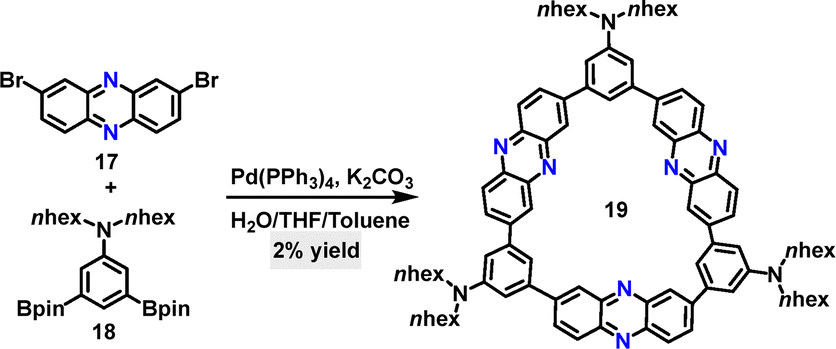
Subsequently, D–A conjugated macrocycle 19 was synthesized in 2% yield by Jiang via a one-step Suzuki coupling reaction of 2,8-dibromophenazine 17 and 3,5-diborate-N,N-dihexylaniline 18 (Scheme 5).32 The synthetic yield was relatively low due to the formation of numerous by-products, such as linear polymers and oligomers. Notably, macrocycle 19 exhibited a remarkable Stokes shift of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 422 cm−1 (361 nm) in DCM, with ultraviolet (UV) absorption at 388 nm and near-infrared (NIR) emission at 749 nm.
422 cm−1 (361 nm) in DCM, with ultraviolet (UV) absorption at 388 nm and near-infrared (NIR) emission at 749 nm.
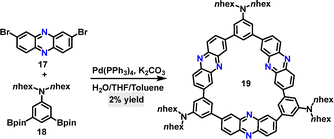 |
| | Scheme 5 Synthesis of D–A conjugated macrocycle 19. | |
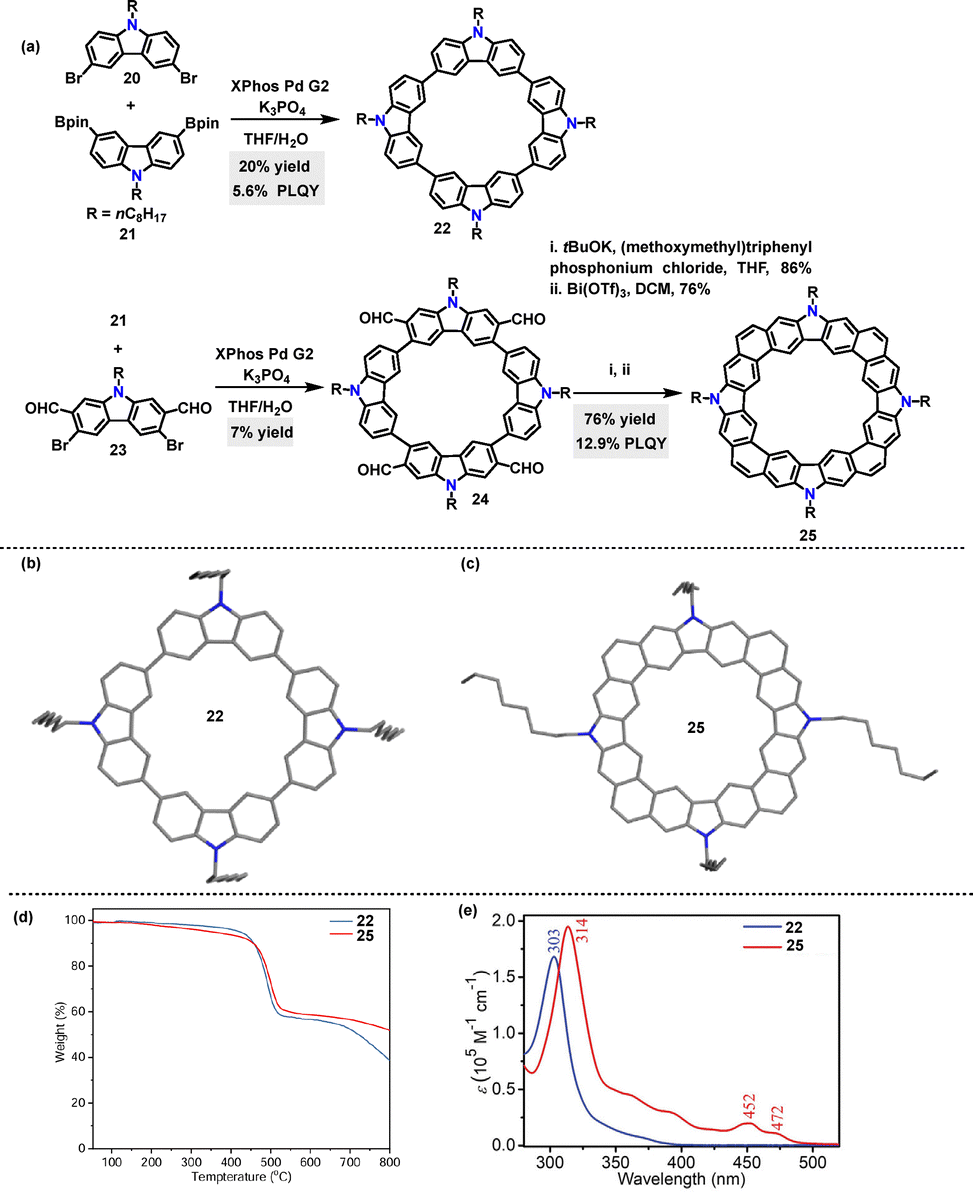
In 2023, Lu33 synthesized conjugated macrocycles 22 (20%) and 24 (7%) containing four carbazole units, via Suzuki coupling reactions. Macrocycle 24 could be further transformed into the π-conjugated species 26via a Wittig reaction and Bi(OTf)3-mediated cyclization process (Scheme 6a). Crystal structure analysis revealed that 22 adopted a herringbone packing motif without π–π overlap between adjacent molecules (Scheme 6b). In contrast, although 25 also exhibited a herringbone packing motif, it featured effective π–π stacking (Scheme 6c). Thermogravimetric analysis (TGA) revealed that both 22 and 25 exhibited high thermal stability, with 5% weight loss decomposition temperatures exceeding 350 °C (Scheme 6d). UV-vis absorption spectra indicated that 25 displayed a red-shifted absorption (314 nm) compared to 22 (303 nm), along with a narrowed optical bandgap (Scheme 6e). Additionally, 25 exhibited a higher PLQY (12.9% vs. 5.6% for 22) and a longer fluorescence lifetime (10.8 ns vs. 4.5 ns for 22), which were ascribed to reduced nonradiative decay due to its structural rigidity.
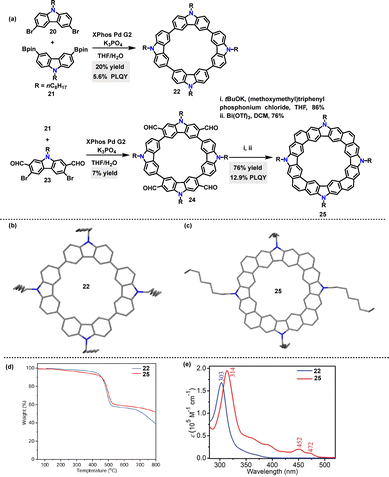 |
| | Scheme 6 (a) Synthesis of macrocycles 22, 24, and 25, (b) and (c) crystal structures of 22 and 25, (d) TGA curves of macrocycles 22 and 25, and (e) absorption spectra of macrocycles 22 and 25 in DCM. Adapted from ref. 33 with permission from Wiley-VCH, copyright 2023. | |
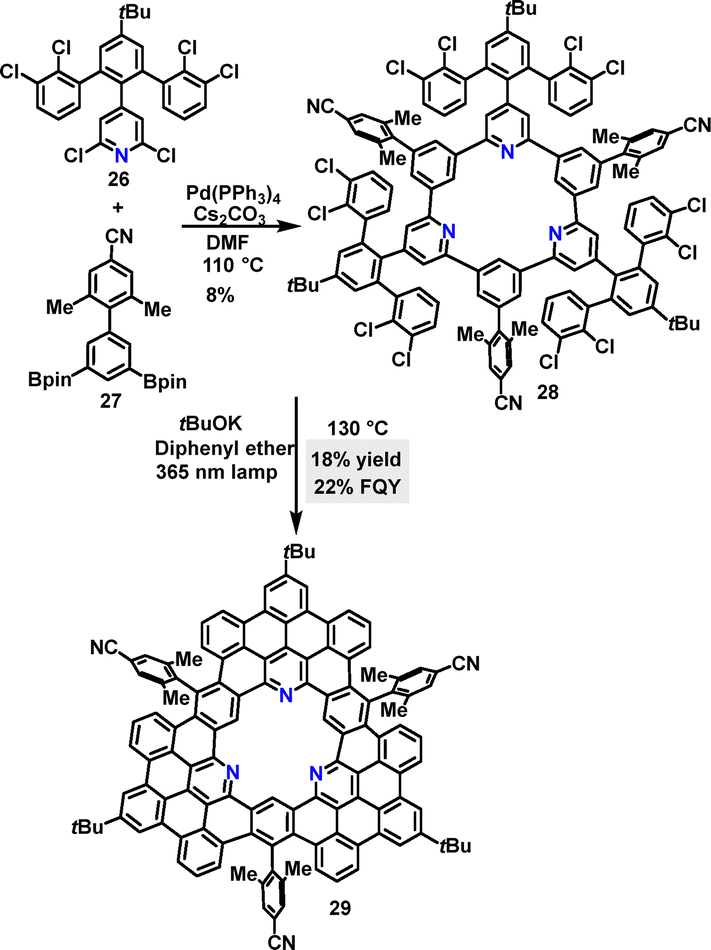
Following the same strategy, Tan synthesized the tri-N-containing macrocycle 28 in 8% yield via a palladium-catalyzed cross-coupling reaction (Scheme 7).34 Macrocycle 28 was further transformed into nanographene 29, featuring a central N-containing cavity in 18% yield under 365 nm LED irradiation. DFT simulations revealed that 29 adopted a twisted propeller-shaped molecular conformation due to the steric hindrance of the 4-cyano-2,6-dimethylphenyl group. Macrocycle 29 exhibited a FQY of 22% and a lifetime of 7.6 ns.
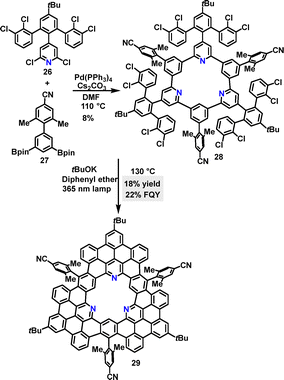 |
| | Scheme 7 Synthesis and modification of tri-N-containing macrocycle 28. | |
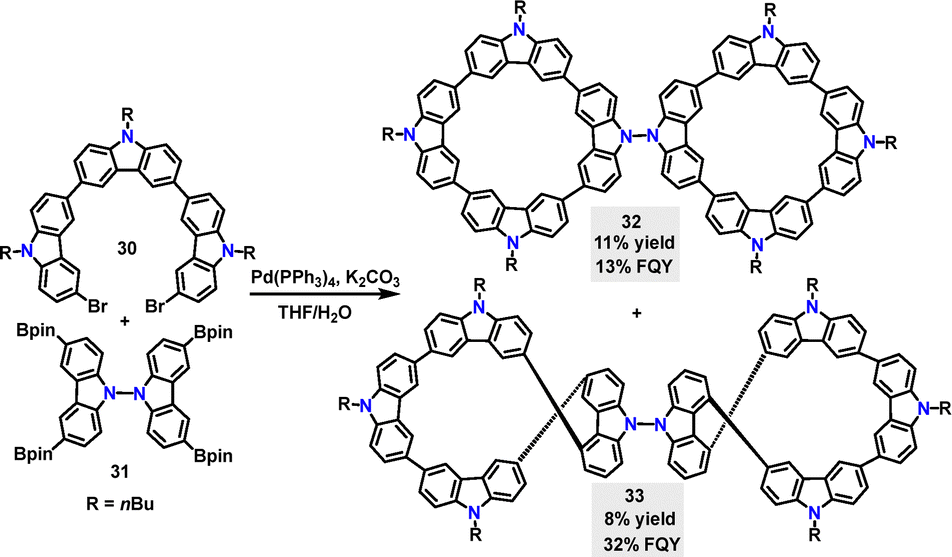
In 2024, Chen35 reported a palladium-catalyzed cross-coupling reaction, enabling the preparation of the achiral biscyclic 32 and chiral 33 in 11% and 8% yields, respectively (Scheme 8). According to DFT calculations, the two tetracyclic carbazole moieties are oriented nearly orthogonally, displaying a highly symmetric geometry. In contrast, 33, featuring a 2,2′,7,7′-substituted bicarbazole core, can’t achieve perpendicular rotation due to increased steric constraints and electronic repulsion. Instead, the molecule adopted a figure-eight conformation. Compared to 32 with an absorption maximum of 302 nm and a FQY of 13%, the figure-of-eight biscyclic 33 exhibits a red-shifted absorption maximum (311 nm) and a higher FQY (32%), which can be attributed to enhanced conjugation in the carbazole-phenyl-bridged 33 framework. Additionally, the circularly polarized luminescence (CPL) brightness of 33 was quantified as 26.0 M−1 cm−1.
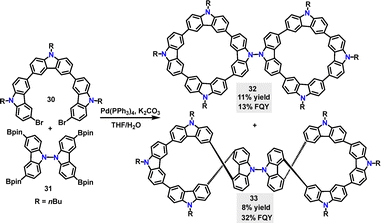 |
| | Scheme 8 Synthesis of achiral biscyclic macrocycle 32 and chiral macrocycle 33. | |
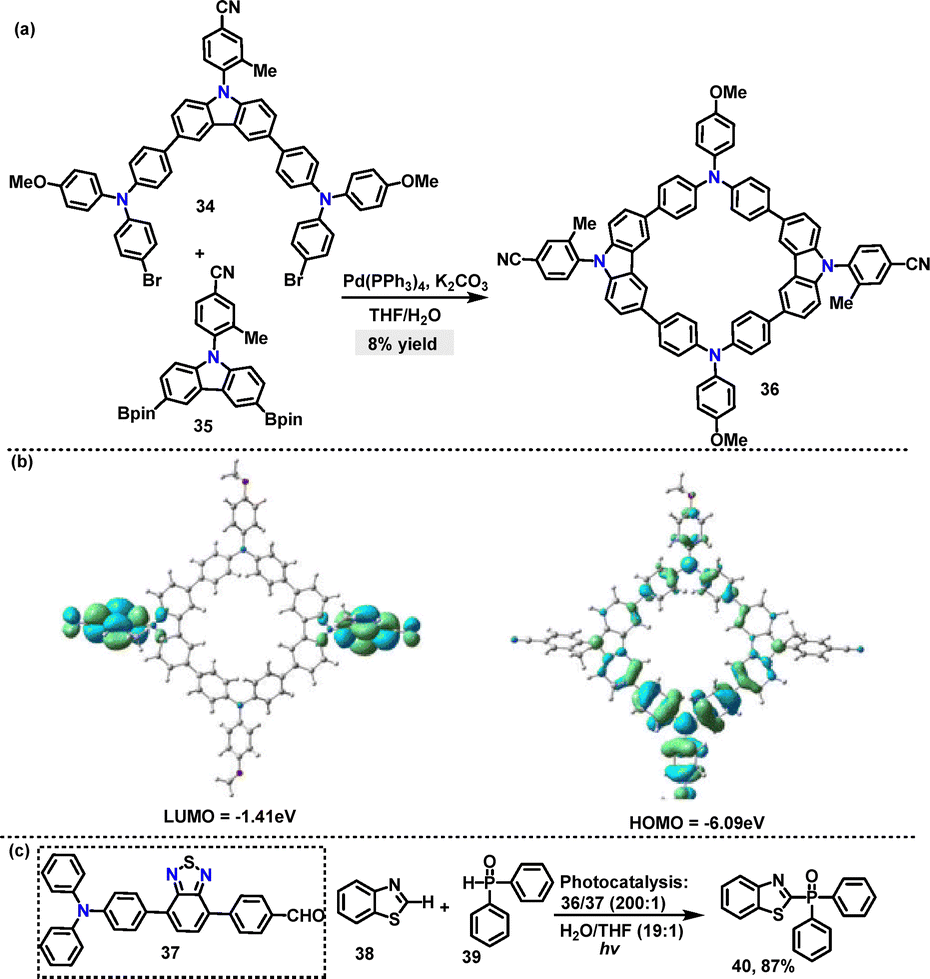
Recently, Tang36 designed and synthesized an AIE-active macrocycle 36 incorporating triphenylamine (TPA) and carbazole (Cz) units. The desired product 36 was generated in 8% yield via a palladium-catalyzed Suzuki coupling of 34 and 35 (Scheme 9a). DFT calculations revealed that 36 adopted a highly twisted conformation, where the torsion angle was significantly enhanced by the ortho-methyl group, thereby promoting AIE characteristics. The HOMO of 36 was localized on the TPA unit, whereas the LUMO was distributed over the phenylcyano group (Scheme 9b). This spatial separation ensured efficient charge separation and facilitated energy transfer processes. Moreover, both 34 and 36 exhibited AIE properties, with enhanced fluorescence intensity in THF/H2O mixtures. Superior photocatalytic performance was demonstrated by ALHS 36 in mediating cross-dehydrogenative coupling (CDC) reactions within an aqueous environment (Scheme 9b).
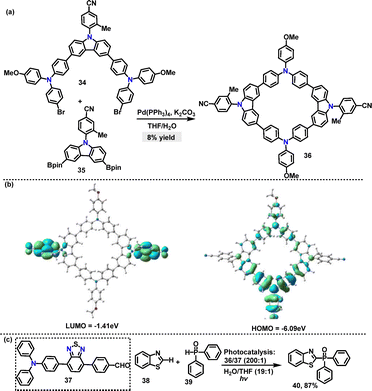 |
| | Scheme 9 (a) Synthesis, (b) molecular orbital, and (c) photocatalytic activity of AIE-active macrocycle 36. Reproduced from ref. 36 with permission from The Royal Society of Chemistry, copyright 2025. | |
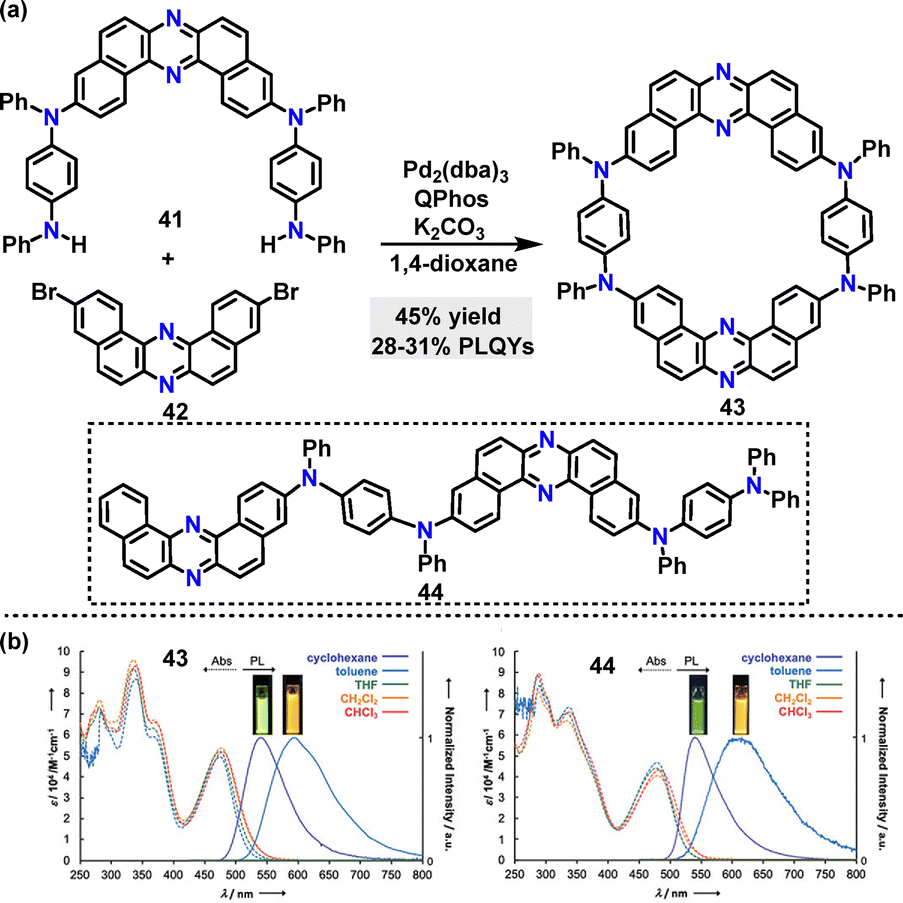
Beyond C–C coupling reactions, C–N bond formation reactions have also witnessed remarkable advancements in the synthesis of macrocycles. In 2020, Minakata37 disclosed a palladium-catalyzed C–N coupling strategy to synthesize dibenzo[a,j]phenazine-based π-conjugated macrocycle 43, in 45% yield (Scheme 10a). Macrocycle 43 incorporated two U-shaped electron-accepting cores (dibenzo[a,j]phenazine) and two electron-donating units (N,N′-diphenyl-p-phenylenediamine) and displayed emission behaviors and structural conformations influenced by polymorphism. Steady-state spectroscopy revealed that macrocycle 43 exhibited solvent-dependent emission, displaying green fluorescence in cyclohexane (Em: 540 nm, PLQY: 31%) and orange fluorescence in toluene (Em: 595 nm, PLQY: 28%). In contrast, linear 44 showed broader emission with a larger red shift (Em: 615 nm in toluene, PLQY: 20%), which was attributed to its conformational flexibility (Scheme 10b). Degassed solutions of 43 exhibited a 66% increase in emission intensity compared to aerated conditions, indicating significant delayed fluorescence (DF), whereas linear 44 showed only a 24% intensity increase.
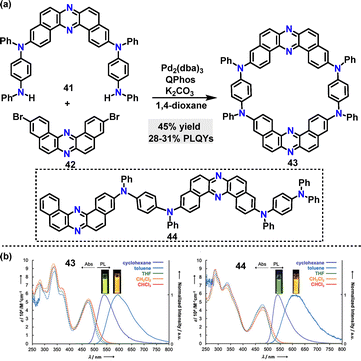 |
| | Scheme 10 (a) Synthesis of π-conjugated macrocycle 43 and (b) steady-state UV-vis absorption (Abs) and photoluminescence (PL) spectra of dilute solutions (purple, cyclohexane; sky blue, toluene; green, THF; orange, DCM; red, CHCl3) of compounds 43 and 44. Adapted from ref. 37 with permission from American Chemical Society, copyright 2020. | |
In 2021, Giuseppone38 reported a palladium-catalyzed intermolecular coupling reaction to synthesize an S6-symmetric triarylamine-based macrocycle 47 in 87% yield (Scheme 11). Upon treatment with trifluoroacetic acid (TFA), this product underwent PMB deprotection to afford macrocycle 48 in 87% yield. Optoelectronic property studies revealed that macrocycle 48 exhibited a red-shifted absorption maximum (343 nm), compared to its protected counterpart 47 (331 nm), primarily attributed to electronic inductive effects arising from the conversion of tertiary amides to secondary amides. Additionally, unbound amide side chains on macrocycle 48 enable its supramolecular polymerization, forming an axially aligned nanotubular architecture with π–π stacking interactions between phenyl rings of adjacent macrocycles along the elongation direction.
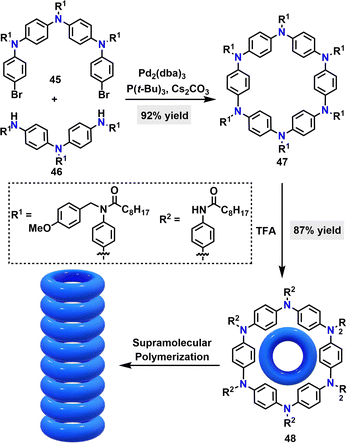 |
| | Scheme 11 Synthesis of π-conjugated macrocycles 47 and 48 and supramolecular polymerization of 48. | |
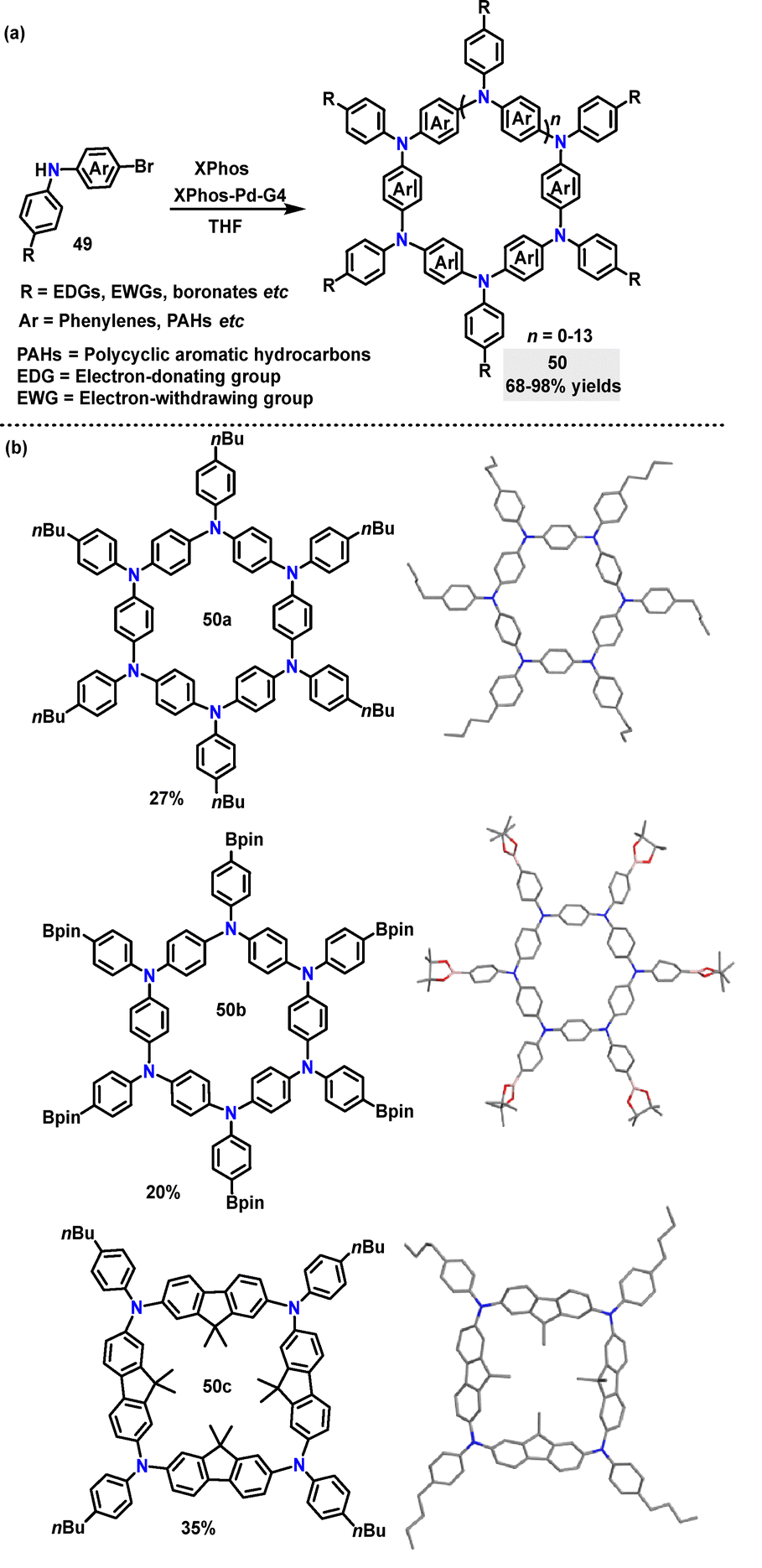
Recently, the synthesis of azaparacyclophanes 50 was developed by Bonifazi via palladium-catalyzed Buchwald–Hartwig macrocyclization.39 Diverse monomers carrying aromatic moieties, electron-donating or -withdrawing groups, and other functional groups like pinacol boronates worked well, generating the desired products 50 in 68–98% yields under mild reaction conditions (Scheme 12a). It was discovered that monomers bearing fused biphenyl endocyclic moieties like fluorene mainly yield 4-membered rings. The monomers bearing a carbazole moiety give a mixture of 4-, 5-, and 6- membered rings. Crystal structure analysis showed that 50a and 50b had a six-membered ring structure with endocyclic aryl moieties in a propeller-type conformation and coplanar N-atoms. Compound 50c displayed a four-membered square macrocycle structure with a specific conformation of the fluorene/carbazole units and coplanar N-atoms (Scheme 12b). Spectroscopic measurements showed that all compounds 50 had similar absorption ranges of 344–393 nm.
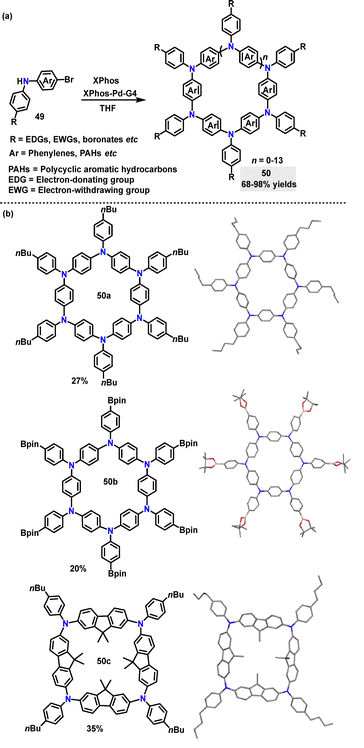 |
| | Scheme 12 (a) Synthesis of azaparacyclophane 50 and (b) crystal structures of 50a and b. | |
2.1.2. Nickel catalysis.
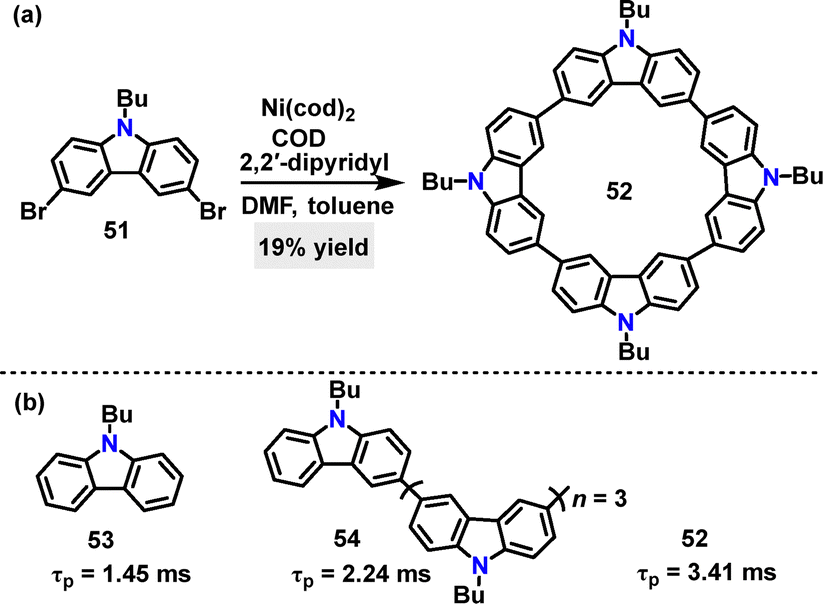
In 2021, Huang40 synthesized a cyclic tetracarbazole 52 in 19% yield via a one-pot Yamamoto coupling of a dibrominated carbazole monomer 51 (Scheme 13a). Under identical conditions, 52 exhibited more efficient ultralong phosphorescence with a phosphorescence lifetime (τp) of 3.41 ms, surpassing both monomer 53 (τp: 1.45 ms) and its linear conjugate 54 (τp: 2.24 ms) (Scheme 13b). DFT and time-dependent (TD)-DFT calculations revealed that linear conjugation increased the number of intersystem crossing (ISC) channels, whereas cyclization further reduced the energy gap between singlet and triplet states (ΔEST) and fostered a more rigid molecular structure.
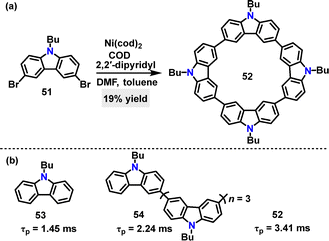 |
| | Scheme 13 (a) Synthesis of cyclic tetracarbazole 52 and (b) phosphorescence lifetime (τp) of cyclic tetracarbazole 52, monomer 53, and linear conjugate 54. | |
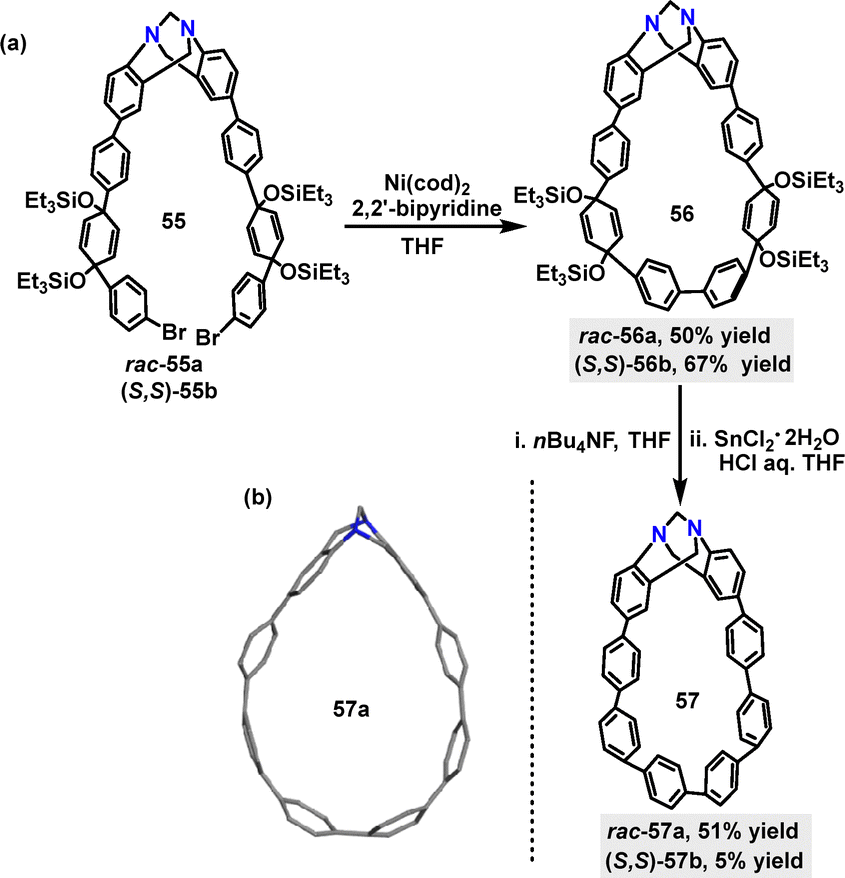
In 2024, a nanohoop incorporating a stereogenic Tröger's base skeleton was developed by Saito.41 The Ni(0)-mediated Yamamoto coupling reaction of rac-55a and (S,S)-55b monomers afforded the cyclization products rac-56a and (S,S)-56b in 50% and 67% yields, respectively (Scheme 14a). Deprotection of rac-56a and (S,S)-56b with n-BuNF, followed by reductive aromatization promoted by SnCl2 and HCl, yielded rac-57a and (S,S)-57b in 51% and 5% yields respectively. Crystal structure analysis showed that rac-57a adopted a teardrop-like structure owing to the V-shaped Tröger's base moiety, where the dihedral angle between the two phenylene rings of the Tröger's base unit measures 90° (Scheme 14b).
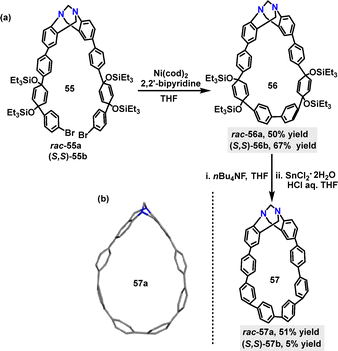 |
| | Scheme 14 (a) Synthesis of 56 and 57 and (b) crystal structure of 57a. | |
2.1.3. Platinum catalysis.
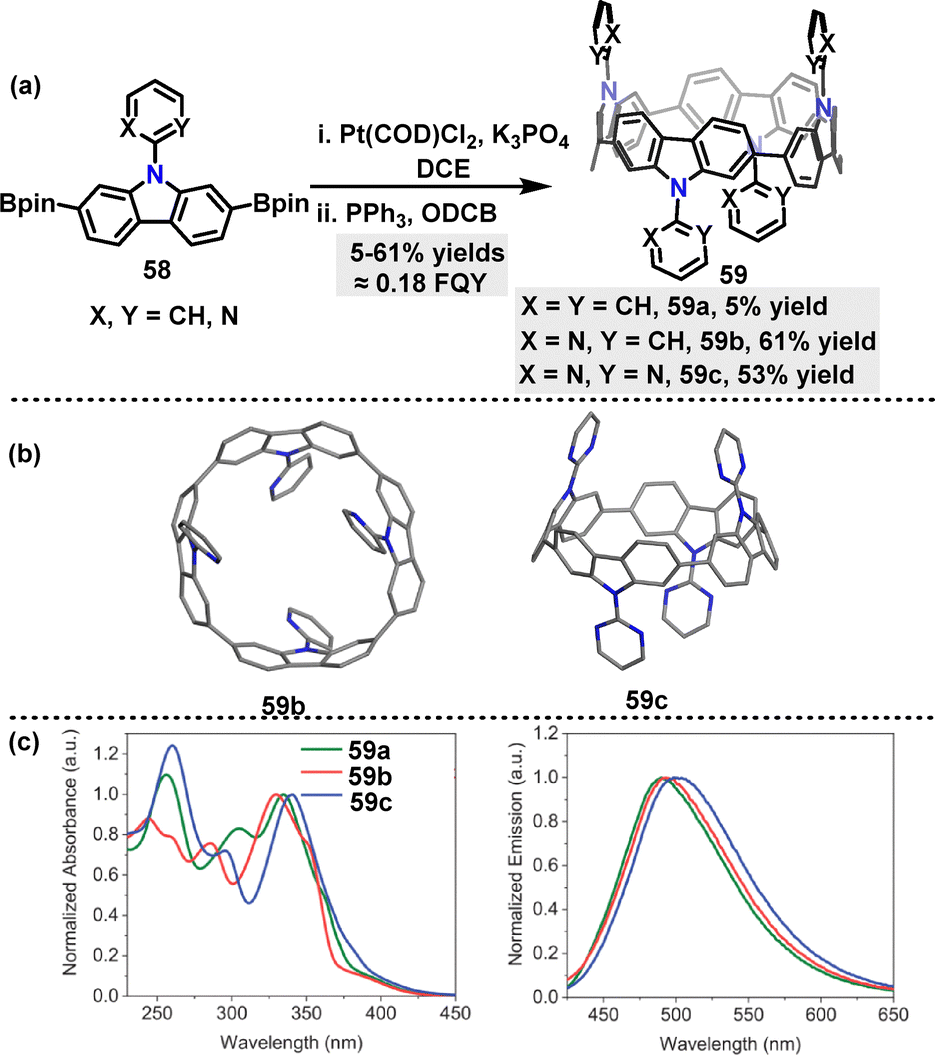
In 2024, Poriel and Quinton42 reported the synthesis of three nanohoops in 5% to 61% yields via a platinum-catalyzed cyclization, followed by reductive elimination (Scheme 15a). Crystal structures showed that the pyrimidine substituent in 59c was nearly coplanar with the carbazole moiety, due to hydrogen-bonding interactions, whereas the pyridine group in 59b exhibited reduced coplanarity (Scheme 15b). The dihedral angles between carbazole and substituents also differ significantly, affecting π-conjugation and electronic properties. UV-vis absorption spectra of the three nanohoops indicated that substituents profoundly influence absorption bands and transition characteristics (Scheme 15c). For example, the absorption maximum undergoes a blue shift from 341 nm in 59a to 335 nm and 330 nm in 59b and 59c, respectively. In emission, the broad unresolved spectra of 59a, 59b, and 59c centered at 501 nm, 490 nm, and 495 nm, respectively, demonstrated minimal substituent effects on emission wavelengths. Those nanohoops exhibited comparable radiative and non-radiative decay rates, as is evidenced by similar FQY (18%) and singlet lifetimes (τs ≈ 0.18 ns and 7 ns, respectively).
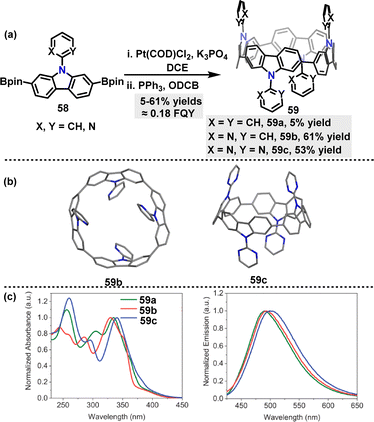 |
| | Scheme 15 (a) Synthesis of nanohoops 59 and (b) crystal structures of 59b and 59c, (c) absorption in DCM at 350 nm (left) and emission (right) in DCM at 340 nm for 59. Adapted from ref. 42 with permission from Wiley-VCH, copyright 2024. | |
2.1.4. Iron catalysis.
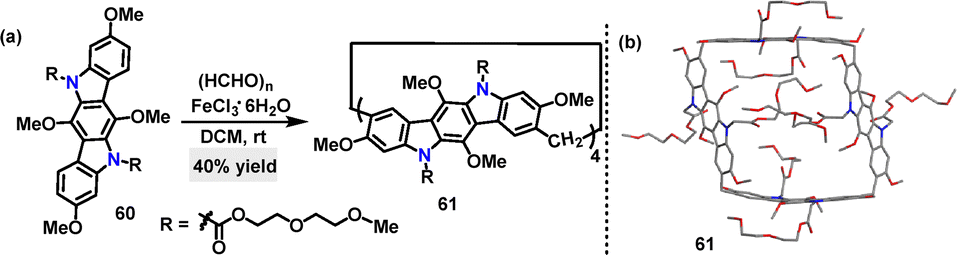
In 2020, Yang reported an FeCl3·6H2O-catalyzed cyclization reaction for the synthesis of N-embedded cubane.43 Under these conditions, the desired product 61 was obtained in 40% yield (Scheme 16a). However, when other catalysts such as trifluoroacetic acid, BF3·OEt2, and p-toluenesulfonic acid were tested, little to no product was observed. Crystal structure analysis revealed that macrocycle 61 exhibited a cubic architecture, where four indolocarbazole units were bridged by methylene groups at the 2,8-positions (Scheme 16b). The cavity measures 13.96 Å vertically and 9.01 Å horizontally, significantly larger than the cavities of pillar[5]arene (5 Å) and pagoda[4]arene (7.9 Å). DFT calculations confirmed its energy-minimized cubic conformation, with dihedral angles of 72–102° reflecting a nonplanar geometry.
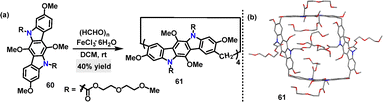 |
| | Scheme 16 (a) Synthesis and (b) crystal structure of macrocycle 61. | |
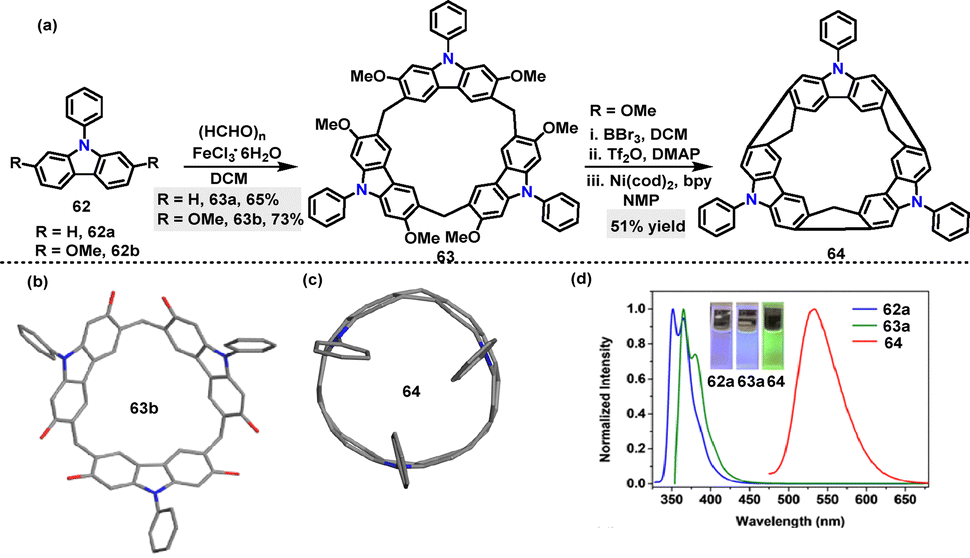
Later, Chen44 reported an FeCl3·6H2O-catalyzed one-pot condensation of substituted 9-phenylcarbazoles 62 and paraformaldehyde in DCM, affording macrocycles 63a and 63b in 65% and 73% yields, respectively (Scheme 17a). A novel N-doped aromatic belt 64 incorporating a [6]cycloparaphenylene (CPP) skeleton was then synthesized in 51% yield via the transformation of macrocycle 63a. Crystal structure analysis revealed that three carbazole subunits within macrocycle 63b were oriented on the identical side, thereby giving rise to bowl-shaped architecture (Scheme 17b). Macrocycle 64 adopted a slightly elliptical macrocyclic structure, featuring an average diameter of 7.719 Å and a deep cavity (7.948 Å in depth) (Scheme 17c). It exhibited intense green fluorescence (Em: 534 nm) with a quantum yield of 0.39 and a large Stokes shift (170 nm), which was attributed to structural relaxation in the excited state (Scheme 17d). UV-vis spectroscopy revealed a narrow HOMO–LUMO energy gap (2.02 eV), significantly smaller than those of undoped [6]CPP (3.13 eV) and methylene-bridged [6]CPP (2.66 eV).45 This observation highlighted the enhanced conjugation induced by N-doping.
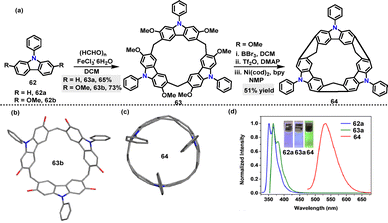 |
| | Scheme 17 (a) Synthesis of macrocycles 63 and 64 and (b) and (c) crystal structures of N-doped aromatic belts 63b and 64, and (d) fluorescence spectra of 62a, 63, and 64. Adapted from ref. 44 with permission from Wiley-VCH, copyright 2021. | |
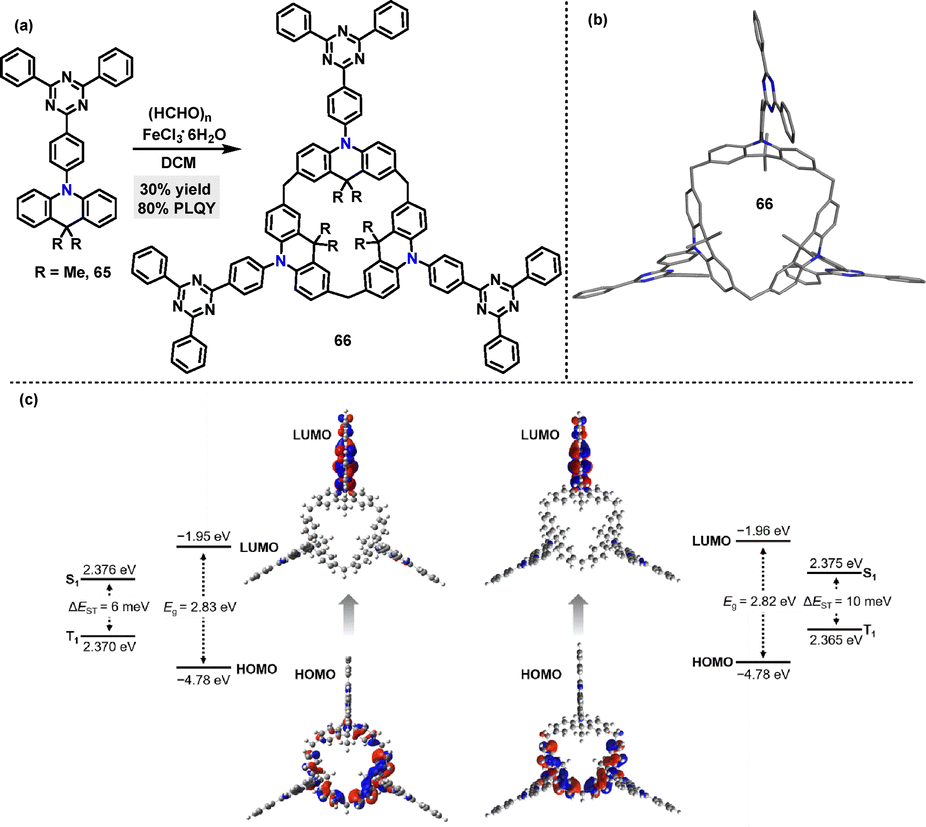
In 2022, Chen46 synthesized the luminescent macrocycle 66 in 30% yield via an FeCl3·6H2O-catalyzed one-pot Friedel–Crafts (F–C) alkylation reaction between s-triphenyltriazine-derived acridan 65 and paraformaldehyde (Scheme 18a). In the crystal structure of 66, the twisted geometry between the s-triphenyltriazine acceptor units and acridan donor units facilitates spatial separation of the HOMO and LUMO (Scheme 18b), thereby reducing the ΔEST. DFT and TD-DFT calculations revealed small ΔEST values for both cone and partial-cone conformers (Scheme 18c). The experimental ΔEST value, measured from fluorescence and phosphorescence spectra in neat film at 77 K, was 12 meV, in good agreement with theoretical analysis. Moreover, macrocycle 66 exhibited excellent TADF emission with a PLQY of 80%.
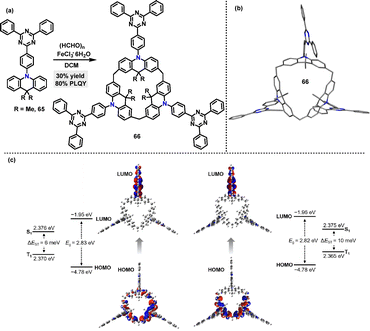 |
| | Scheme 18 (a) Synthesis and (b) crystal structures of luminescent macrocycle 66, (c) calculated energy levels of macrocycle 66: cone conformer and partial-cone conformer.46 Reproduced from ref. 46 with permission from The Royal Society of Chemistry, copyright 2022. | |
2.2. Metal-free strategies
2.2.1. Lewis acid catalysis.
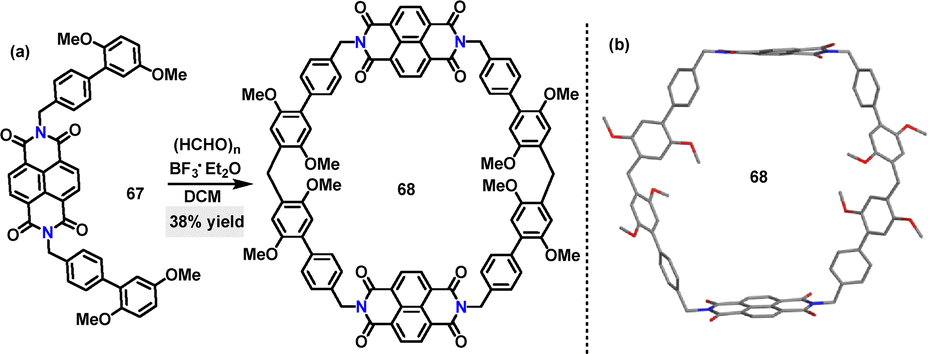
In 2023, Dessie47 reported a one-pot [2+2] condensation of substrate 67 and paraformaldehyde for the synthesis of a giant-cavity macrocycle 68 (Scheme 19a). The desired product 68 was obtained in 38% yield using CF3SO3H as the catalyst in DCM. Crystal structure analysis revealed that 68 adopted a rigid, stretched hexagonal structure (Scheme 19b). The two naphthalene diimide groups were oriented nearly face-to-face with 17.109 Å between them, while the distance between the methylene-linked carbon atoms of adjacent 1,4-dimethoxybenzene units measured 18.769 Å. These dimensions define a giant cavity with a width of 18.769 Å and a height of 17.109 Å.
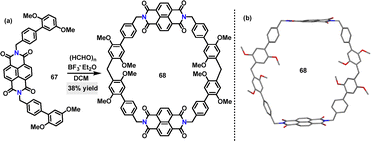 |
| | Scheme 19 (a) Synthesis and (b) crystal structures of giant-cavity macrocycle 68. | |
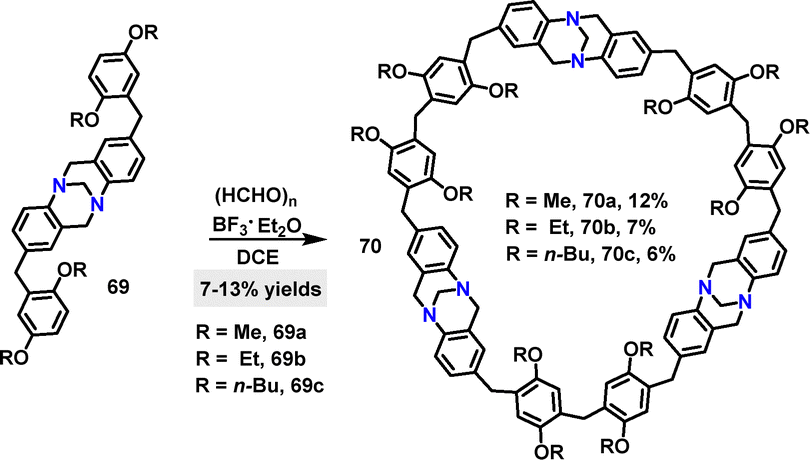
Later, Wang48 reported a BF3·Et2O-catalyzed [3+3] condensation of compound 69 with formaldehyde to construct Tröger's base-based [3]arenes 70 in 6–12% yields (Scheme 20a). TGA revealed that 70a and 70b exhibited no significant weight loss below 300 °C, indicating excellent thermal stability. Additionally, 70a and 70b demonstrated outstanding iodine-adsorption properties.49,50
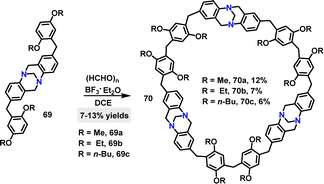 |
| | Scheme 20 Synthesis of Tröger's base-based [3]arenes 70. | |
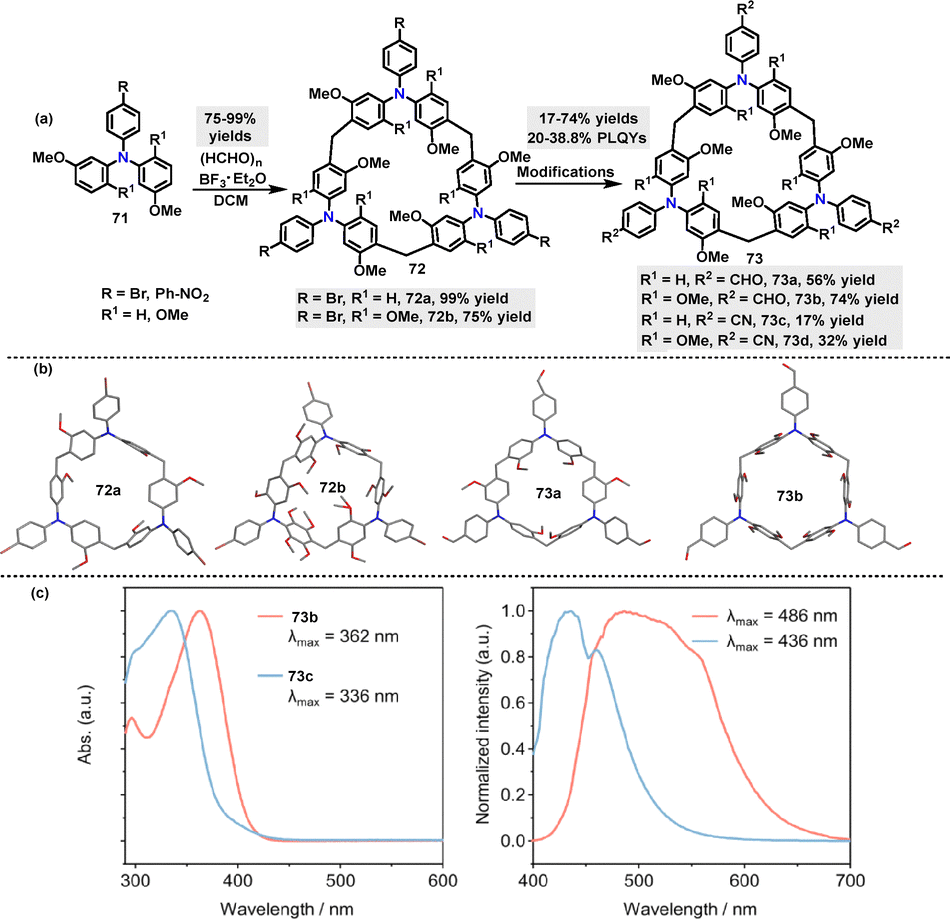
Recently, Sue51 reported a BF3·Et2O-catalyzed cyclization of triphenylamine-derivatized monomers 71 with paraformaldehyde for the synthesis of various triphenylamine[3]arenes 72. The bromine-substituent not only promoted the cyclization reaction (99% yield) but also provided opportunities for further transformations. For instance, treatment with t-BuLi in THF/MeOH converted 72 into H-terminated macrocycles 73a (56%) and 73b (74%). Cyanation reactions with CuCN yielded 73c and 73d in 17% and 32% yield, respectively (Scheme 21a). Crystal structure analysis revealed that these macrocycles adopted cylindrical propeller-like structures with approximately threefold symmetry, though not perfectly symmetric (Scheme 21b). For example, macrocycle 73b exhibited narcissistic chiral self-sorting behavior, maintaining an interlayer distance of 7.0 Å. The structure featured large one-dimensional petal-like channels (13 Å in width) and a solvent-accessible volume ratio of 56.5%. In toluene solution, 73b and 73c displayed (n–π*) transition bands at 362 nm and 336 nm, respectively, in their UV-vis absorption spectra. Their PL spectra showed strong steady-state emissions at 486 nm (73b) and 436 nm (73c) (Scheme 21c). Moreover, macrocycle 73c exhibited a PLOY of 38.8%.
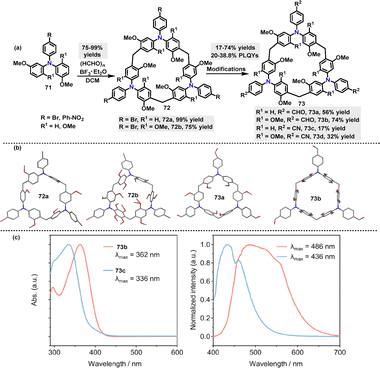 |
| | Scheme 21 (a) Synthesis of triphenylamine[3]arenes 72–73 and (b) crystal structures of 72a–b and 73a–b, (c) UV-vis and corresponding delayed fluorescence spectra of 73b–c. Reproduced from ref. 51 with permission from Wiley-VCH, copyright 2024. | |
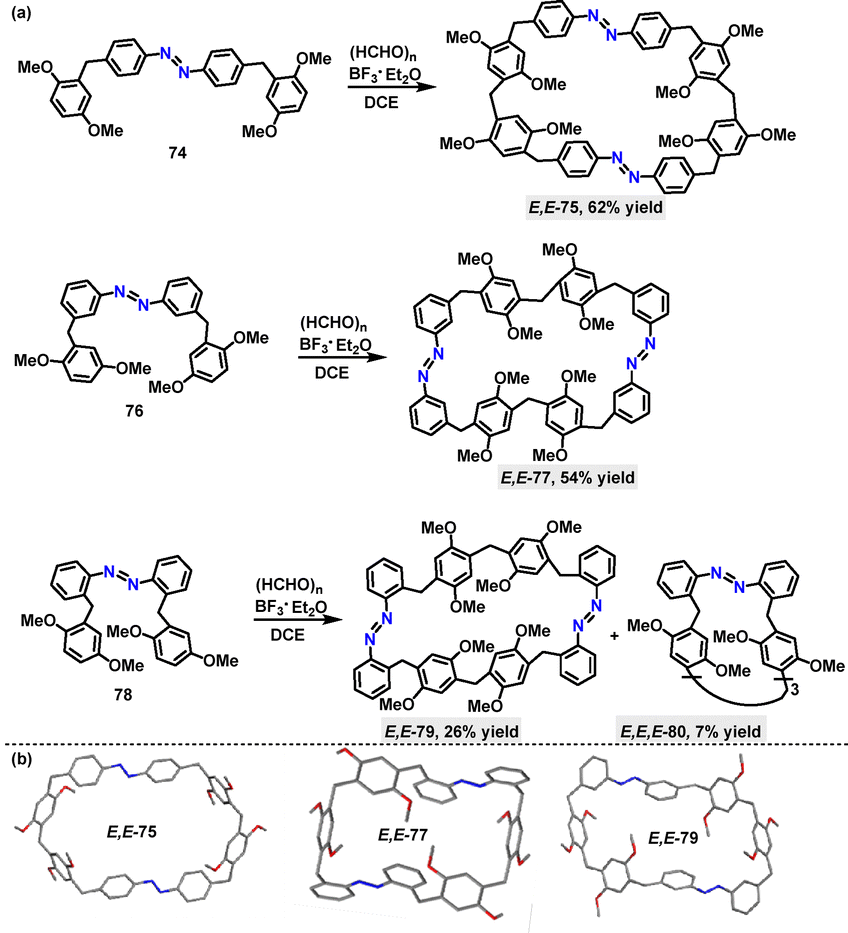
Given the crucial role of N-atoms in influencing the structures and properties of macrocycles, the azo-moiety has been employed to synthesize intriguing complex macrocyclic scaffolds. In 2020, Huang52 reported a BF3·Et2O-mediated cyclization strategy for the synthesis of azo-based macrocycles. Various derivatives with azo-groups at different substitution positions were prepared in 7–62% yields, showcasing the method's synthetic versatility (Scheme 22a). The crystal structure (Scheme 22b) analysis revealed that E,E-75 adopted an oblate hexagonal geometry with dimensions of 17.60 Å in length and 9.76 Å in width. The structure of E,E-75 is stabilized by diverse non-covalent interactions, including C–H⋯O, C–H⋯N, and C–H⋯π contacts, as well as face-to-face π–π stacking. Analogously, E,E-77 and E,E-79 exhibited distinct structural features and characteristic non-covalent interactions, highlighting the substituent-dependent structural diversity.
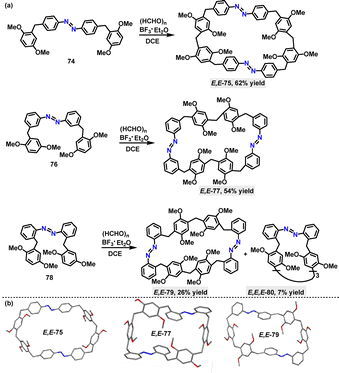 |
| | Scheme 22 (a) Synthesis of azo-macrocycles 75, 77, 79, and 80 and (b) crystal structures of 75, 77, and 79. | |
2.2.2. Brønsted acid catalysis.
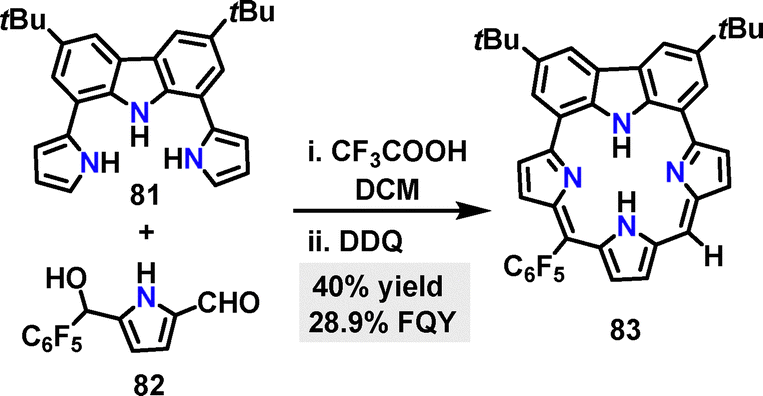
In 2020, Gokulnath53 synthesized a carbazole-based macrocycle 83 in 40% yield via a CF3COOH-catalyzed [3+1] condensation reaction followed by oxidation with DDQ (Scheme 23). The FQY of macrocycle 83 was determined to be 28.9%.
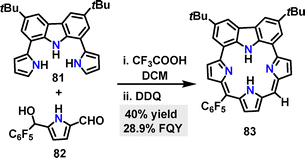 |
| | Scheme 23 Synthesis of carbazole-based macrocycle 83. | |
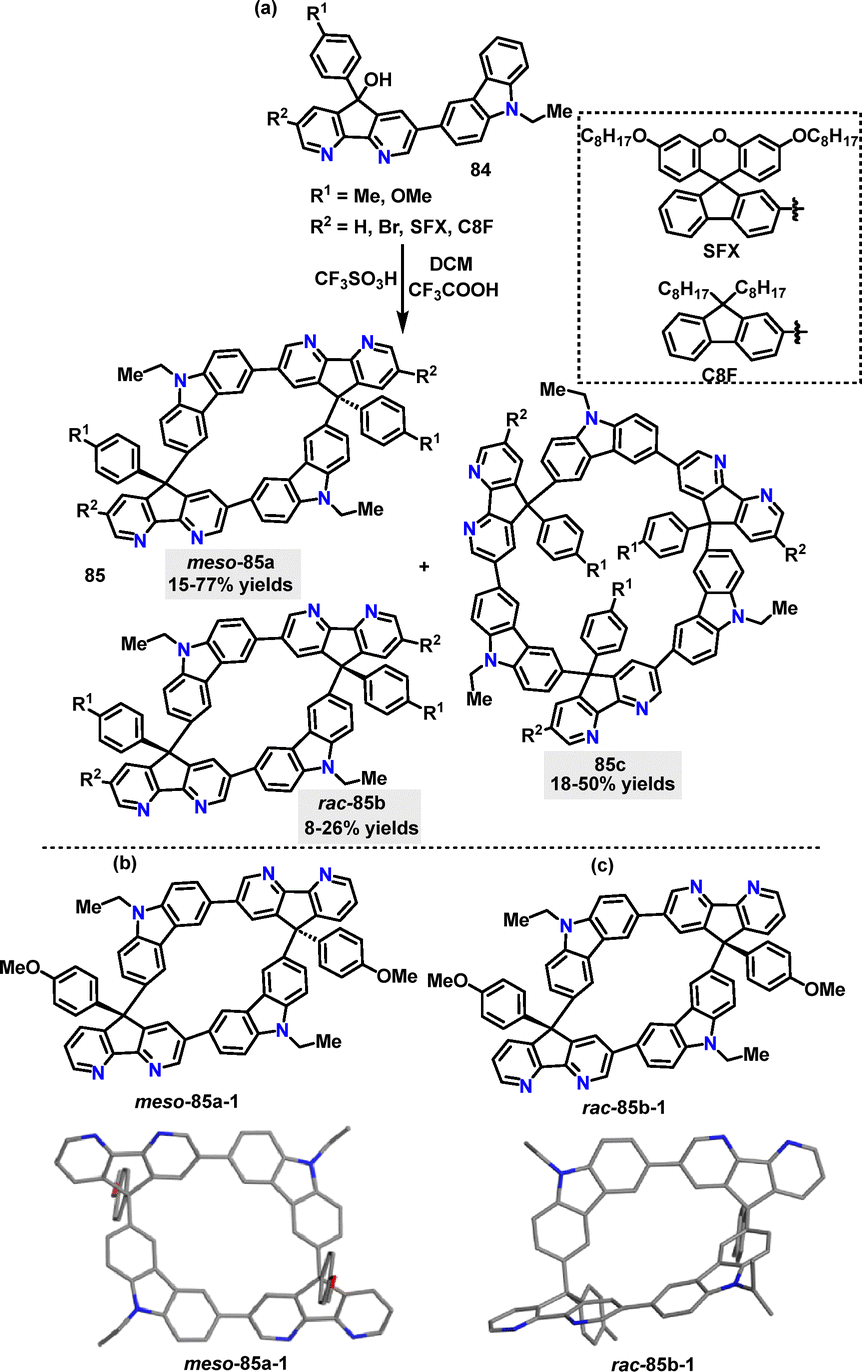
Later, our group54 reported a Brønsted acid-catalyzed F–C reaction for the stereoselective synthesis of various macrocycles 85 (Scheme 24a). Kinetic control via ultrafast quenching (within 1 min) favored meso-configurations as the dominant product, while prolonged reaction times allowed thermodynamic equilibrium to shift toward rac-isomers or oligomerization (e.g., meso-selectivity decreased from 75.6% de to 19.0% de over 54 h). The oxygen atom in methoxy substituents played a critical role in stabilizing meso-selective pathways through hydrogen-bond-enhanced aggregation, while methyl-based substrates exhibited rac-selectivity due to weaker intermolecular interactions. The crystal structures of meso- and rac-configured 85 revealed distinct centrosymmetric and asymmetric characteristics. Meso-85a-1 featured a C2-symmetric backbone, exhibiting a rigid, extended conformation with minimized intermolecular coulombic repulsion due to centrosymmetric packing (Scheme 24b). On the other hand, rac-85b-1 presented an asymmetric folded scaffold that promoted compact packing and enhanced infrared vibrational intensities for asymmetric modes (Scheme 24c).
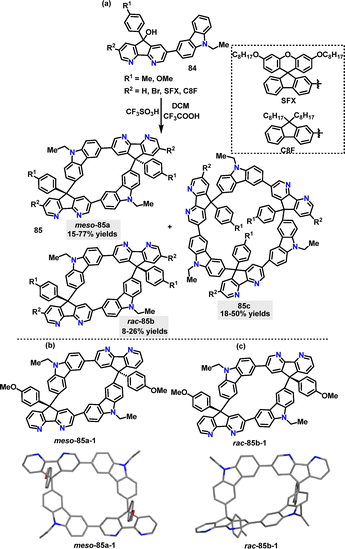 |
| | Scheme 24 (a) Synthesis of macrocycles 85 and (b) crystal structures of meso-85a-1 and rac-85b-1. | |
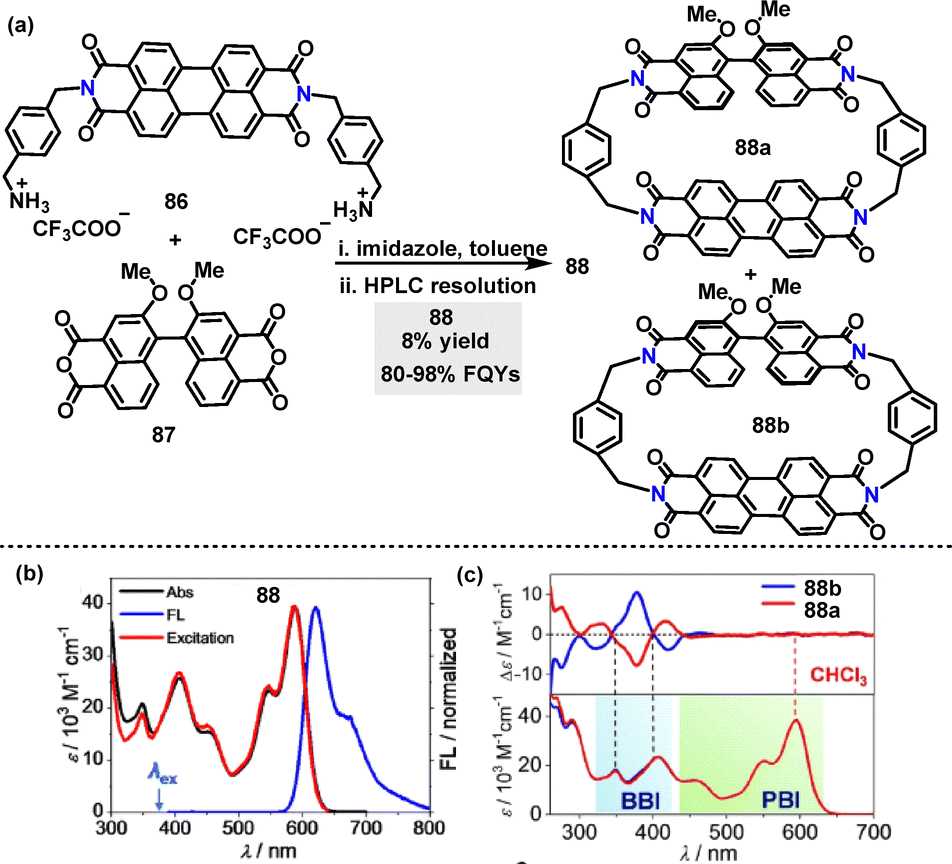
In 2022, Würthner55 reported an efficient protocol for the synthesis of chiral perylene bisimide (PBI) heterocyclophane incorporating a chiral binaphthol bisimide (BBI) moiety (Scheme 25a). Enantiomers 88 were synthesized in 8% yield via macrocyclization of perylene diimide 86 and racemic 87 under mild conditions. When 88 was excited at 380 nm, nearly quantitative FRET occurs due to the perfect spectral overlap between the BBI emission and PBI absorption bands (Scheme 25b). This led to the FQYs and lifetimes of 88 ranging from 80 to 98% and 6.9 to 7.7 ns in different solvents (e.g., CHCl3) (Scheme 25c).
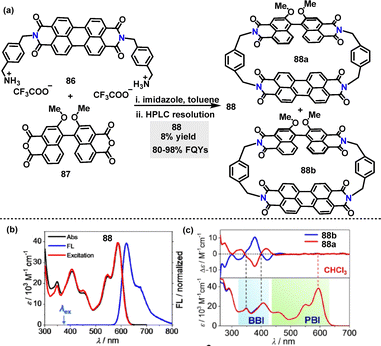 |
| | Scheme 25 (a) Synthesis of chiral perylene bisimide heterocyclophane 88, (b) UV-vis (black line), FL (blue line, λex = 380 nm) and excitation spectra (red line, λem = 650 nm) of 88, (c) CD spectra of enantiomers 88 in CHCl3. Adapted from ref. 55 with permission from Wiley-VCH, copyright 2022. | |
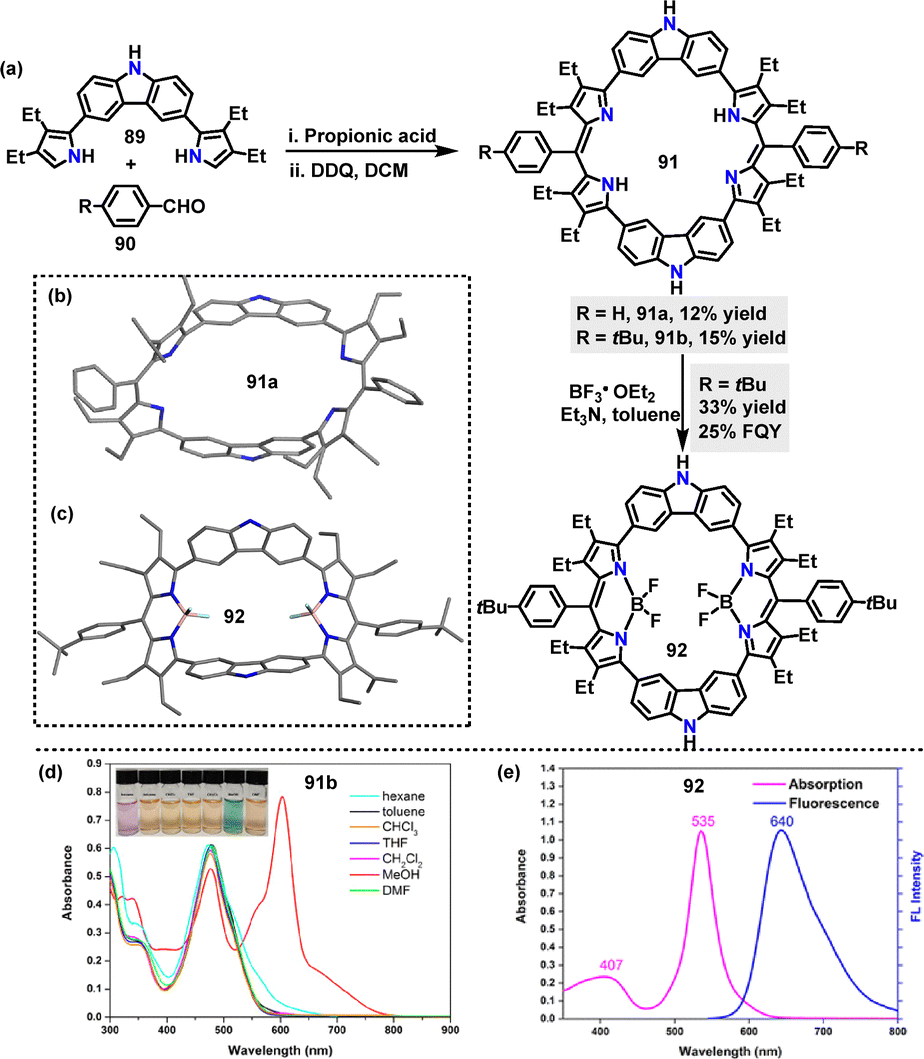
In the same year Lei56 reported a propionic acid-catalyzed condensation of the 3,6-carbazole precursor 89 with benzaldehyde, followed by oxidation with DDQ to construct macrocycle 91a in 12% yield (Scheme 26a). However, macrocycle 91a exhibited poor solubility in common organic solvents. Replacing benzaldehyde with 4-tert-butylbenzaldehyde improved solubility, affording the desired macrocycle 91b in 15% yield. Additionally, the corresponding BODIPY-like complex 92 was synthesized in 33% yield by reacting macrocycle 91b with excess boron trifluoride etherate in toluene. Crystal structure analysis revealed that macrocycle 91a adopted a nonplanar, saddle-like structure, with carbazole moieties twisted upward and dipyrrin moieties twisted downward, stabilized by intramolecular hydrogen-bonding interactions (Scheme 26b). In contrast, 92 featured a bowl-like conformation with two inverted pyrrole rings, where BF2 coordination enforced planarity around the boron-dipyrrin subunits (Scheme 26c). The UV-vis spectrum of complex 91b displayed a broad Soret-like absorption band at 477 nm (Scheme 26d). In contrast, BF2 complex 92 showed strong absorption at 535 nm and emitted red light at 640 nm with a large Stokes shift (Scheme 26e). It possessed an FQY of 25% and an excited-state lifetime of 6.7 ns, highlighting its potential for optoelectronic applications.
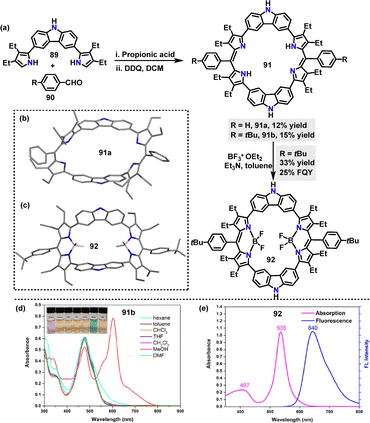 |
| | Scheme 26 (a) Synthesis of macrocycles 91 and 92, (b) and (c) crystal structures of 91a and 92, (d) UV-vis absorption spectra of 91b in different solvents, and (e) UV-vis absorption (pink line) and fluorescence (blue line) spectra of 92 in THF. Adapted from ref. 56 with permission from American Chemical Society, copyright 2022. | |
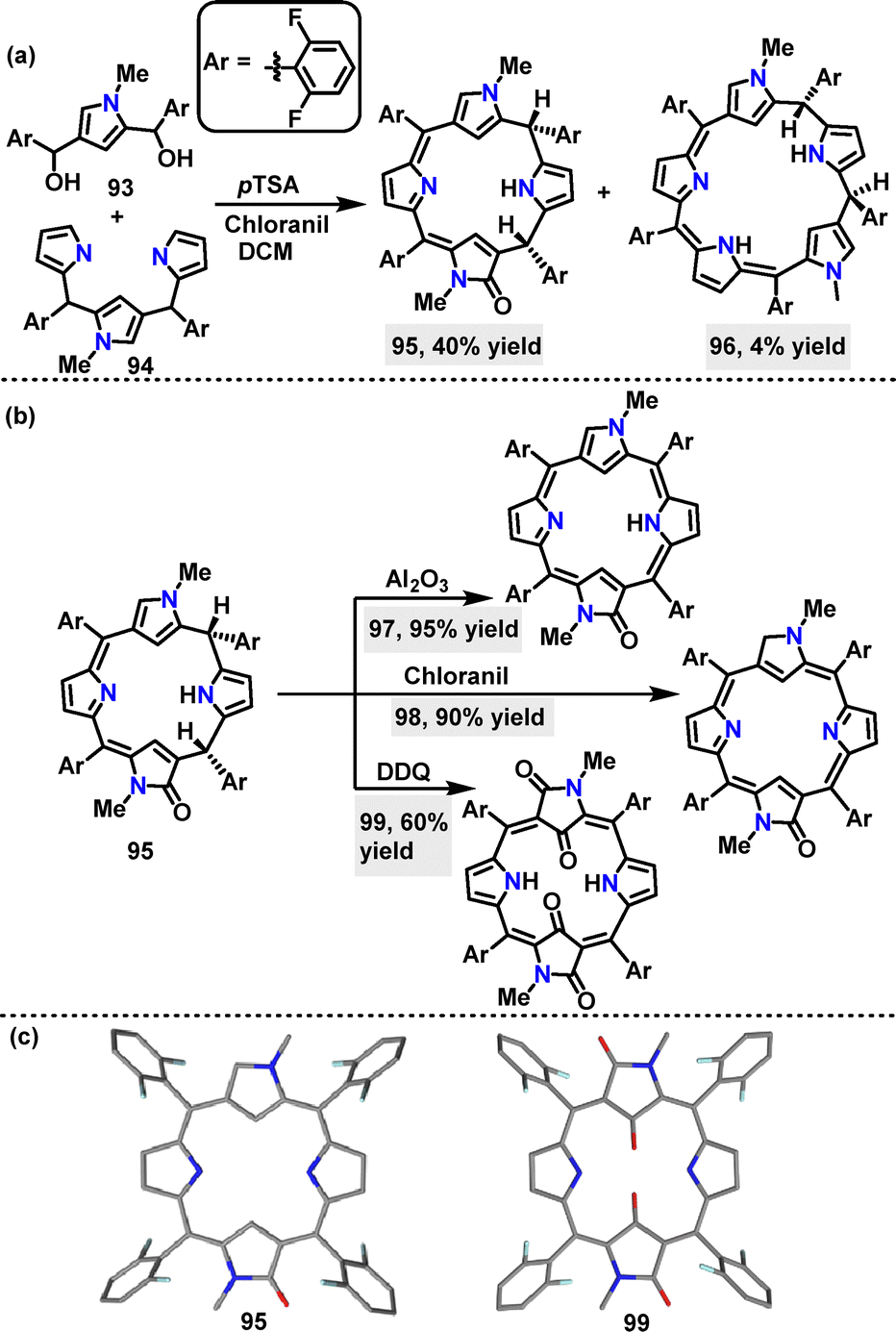
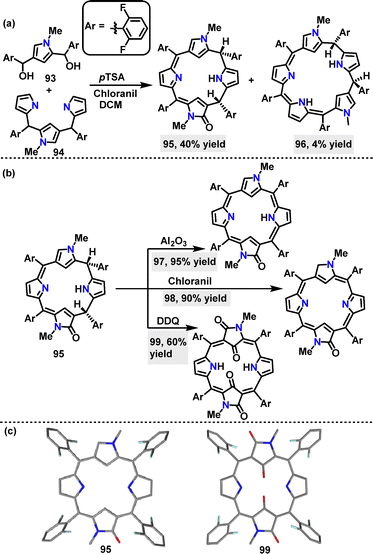
In 2022, Rath57 reported a p-toluenesulfonic acid (p-TSA)-catalyzed [3+1] oxidative condensation of N-confused N-methyl tripyrrane 93 and N-confused N-methyl dicarbinol 94, affording N-confused oxocalix[4]phyrin 95 in 40% yield, and N-confused calix[5]phyrin 96 in 4% yield, respectively (Scheme 27a). Doubly N-confused mono-oxo porphyrinoid 97 was obtained in 95% yield by dehydrogenating 95 with Al2O3. Using chloranil and DDQ as oxidants, 95 was transformed into 98 (90% yield) and 99 (60% yield), respectively (Scheme 27b). Crystal structure analysis of 95 showed a highly distorted molecular conformation, while 99 displayed a greater deviation from planarity than 95 (Scheme 27c).
 |
| | Scheme 27 (a) Synthesis and (b) modifications of macrocycles 95–99, and (c) crystal structures of 95 and 99. | |
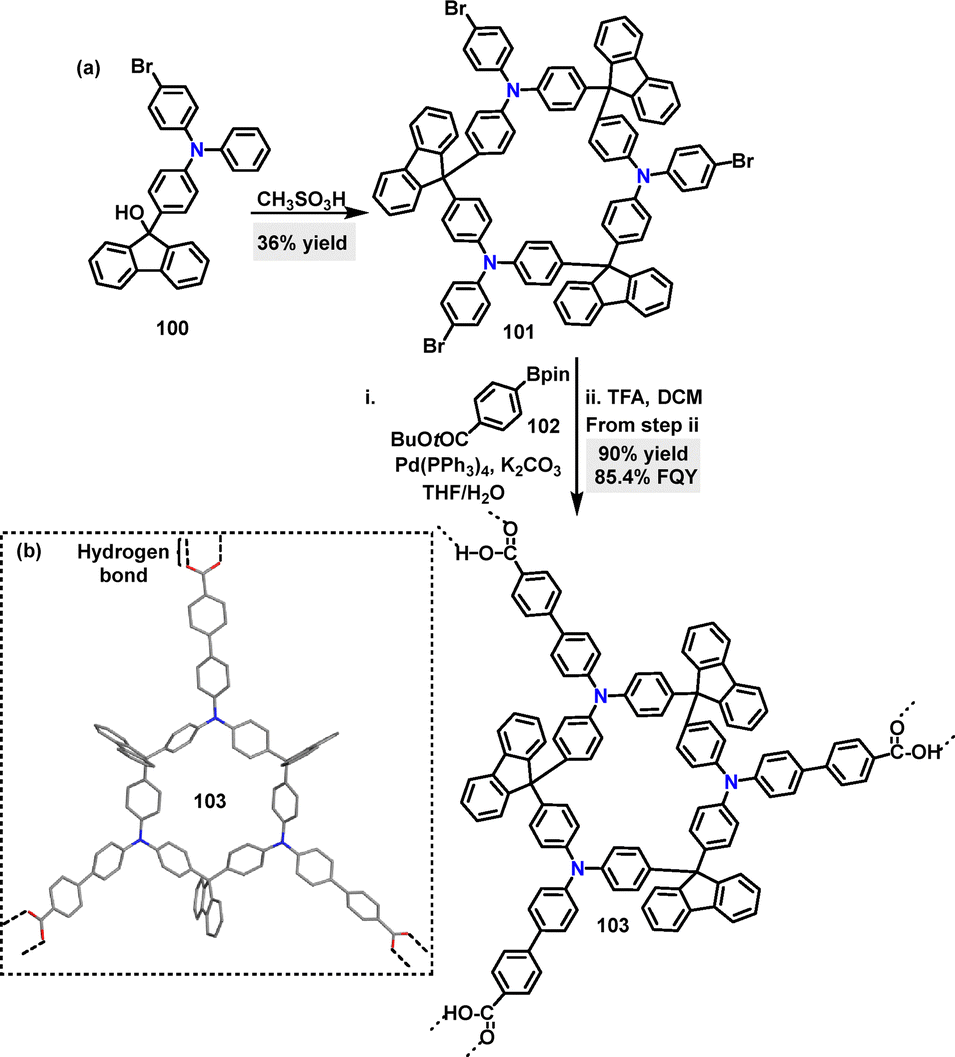
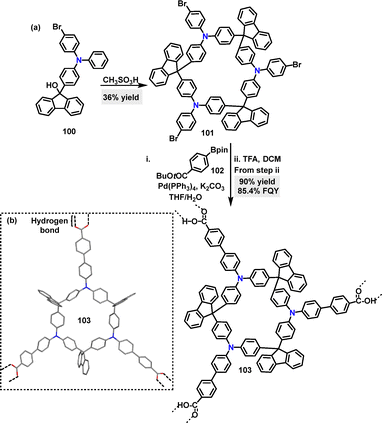
In 2023, Cong58 developed the F–C reaction for the synthesis of a 3-symmetric rigid macrocycle. Macrocycle 101 was formed in 36% yield using methanesulfonic acid catalysis. This could be further transformed into macrocycle 103via a Suzuki–Miyaura cross-coupling reaction and subsequent deprotection of tri(tert-butyl ester) using TFA as the catalyst (Scheme 28a). Crystal structure analysis of 103 showed a flattened cone-like geometry, where each macrocycle connects with three adjacent cones along radial directions through intermolecular double hydrogen-bonding interactions between carboxylic acid groups at the end of each radial arm (Scheme 28b). In THF solution, 103 exhibited strong Em at 455 nm, a high FQY of 85.4%, and dual emission lifetimes of 0.34 ns and 2.80 ns.
 |
| | Scheme 28 (a) Synthesis of macrocycles 101 and 103, and (b) crystal structure of 103. | |
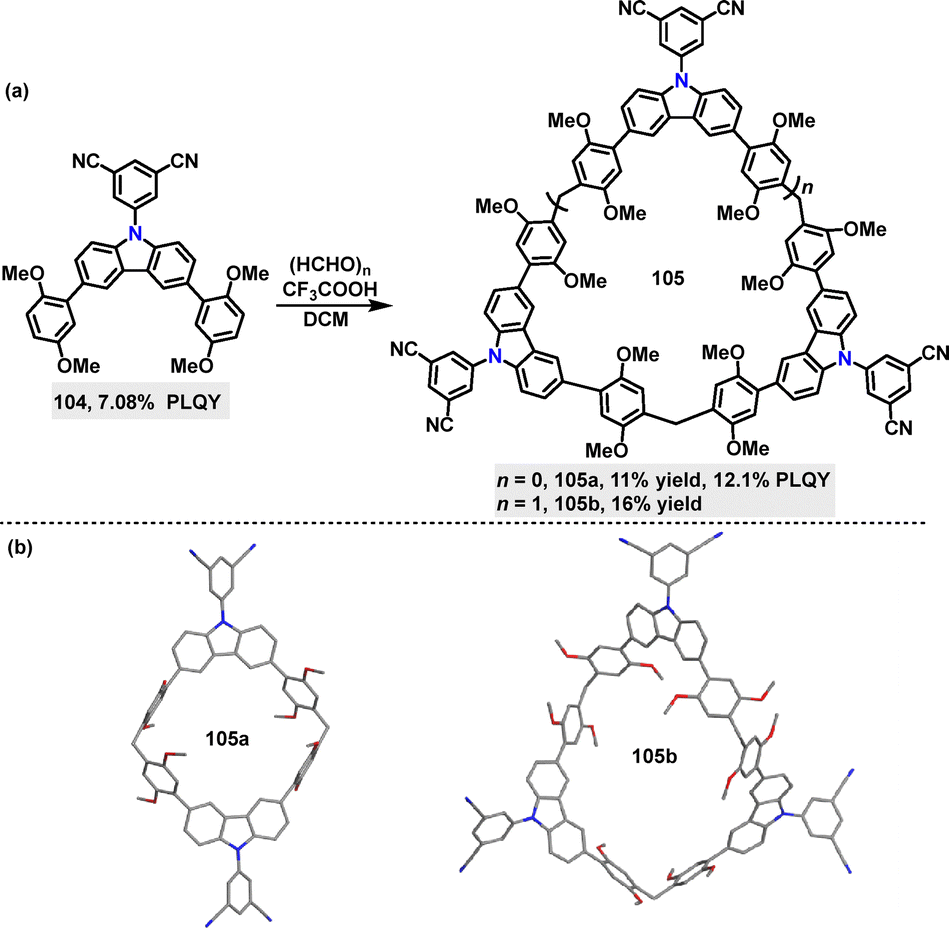
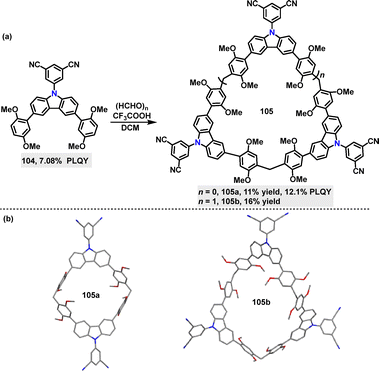
Recently, Ma59 reported the synthesis of carbazole-based macrocycles 105 (11–16% yields), comprising 5-(9H-carbazol-9-yl)isophthalonitrile (D) and 1,4-dimethoxybenzene (A) via a Suzuki–Miyaura coupling reaction (Scheme 29a). The structures of 105a and 105b were confirmed by single-crystal X-ray diffraction analysis (Scheme 29b). In various solvents, the PL spectra of 105a and 105b exhibited a bathochromic shift from toluene to DCM, indicative of ICT characteristics. The fluorescence and phosphorescence spectra recorded at 77 K in toluene solution were used to calculate the ΔEST, yielding values of 0.063 eV for 105a and 0.066 eV for 105b. DFT and TD-DFT calculations further confirmed the charge transfer nature and small ΔEST of these macrocycles, suggesting typical TADF properties. In the solid state, 105b emits at approximately 480 nm, while 105a shows a red-shifted emission peak at 510 nm, likely attributed to stronger intermolecular interactions. These intermolecular interactions restrict intramolecular motion and suppress non-radiative decay, contributing to enhanced PLQY in the solid state. For example, 105a displayed a PLQY of 12.1%, significantly higher than its monomer counterpart 104 (7.08%).
 |
| | Scheme 29 (a) Synthesis and (b) crystal structures of carbazole-based macrocycles 105. | |
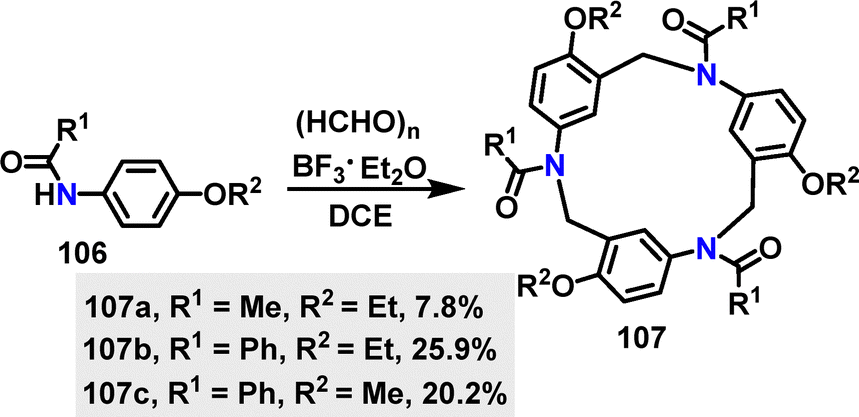
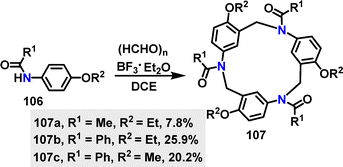
Recently, Yang, Yu, Wu, and co-workers60 developed a novel Mannich-type macrocyclization strategy for the synthesis of henacetin[3]arenes 107 (Scheme 30). The synthesis method involved a one-pot condensation under BF3·Et2O conditions, generating three substituted macrocycles in 7.8–25.9% yields. The yield improvement for 107b of 25.9% was attributed to the higher nucleophilicity of the benzamide nitrogen, which enhanced the initial imine formation and suppressed competing F–C pathways.
 |
| | Scheme 30 Synthesis of macrocycles 107. | |
2.2.3. Brønsted base catalysis.
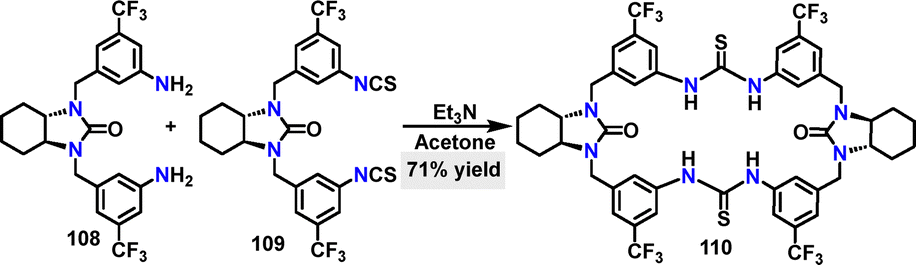
In 2020, Wang reported a chiral macrocycle through the [1+1] condensation between a bis-amine and a bis-isothiocyanate (Scheme 31).61 Employing triethylamine as the base, macrocycle 110 was generated on a multi-gram scale in 71% yield. The high yield was attributed to the preorganized geometry of the building blocks and the driving force of multiple intramolecular hydrogen bonds (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S⋯H–N), which favored cyclization over oligomerization.
S⋯H–N), which favored cyclization over oligomerization.
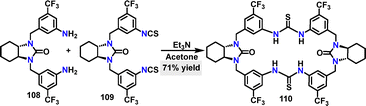 |
| | Scheme 31 Synthesis of chiral macrocycle 110. | |
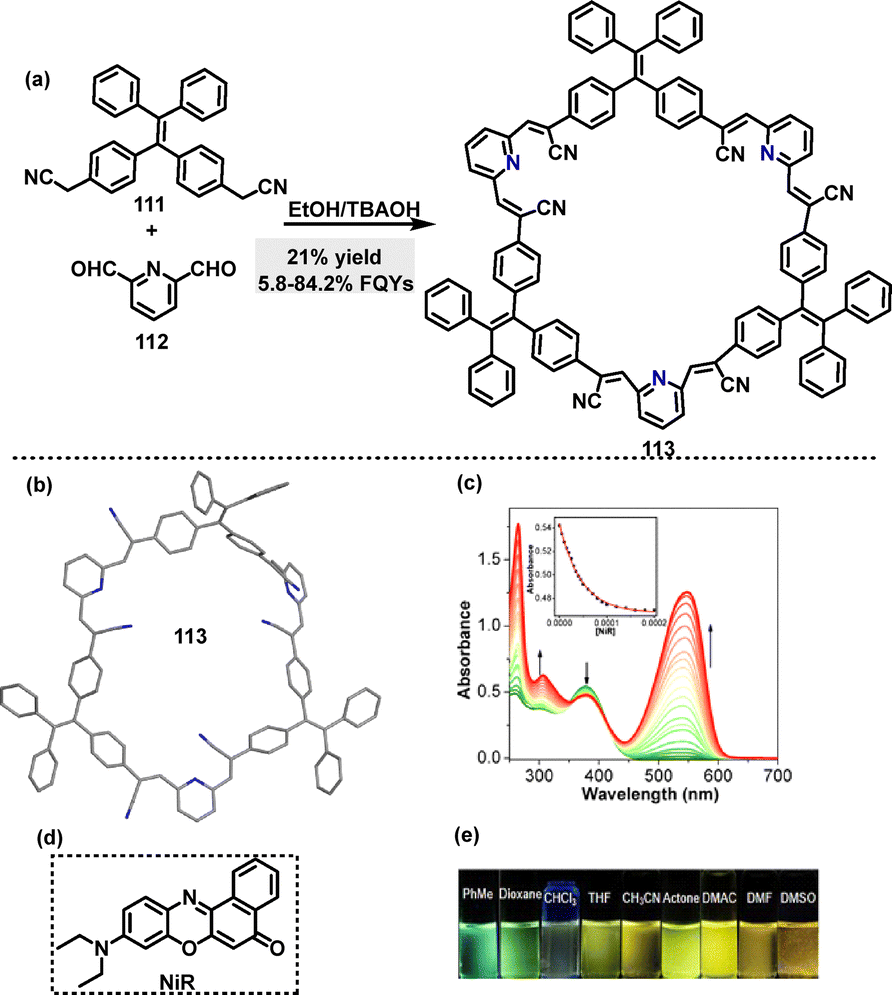
In 2021, Zang62 reported the synthesis of a fluorescent tetraphenylethene (TPEM)-based macrocycle 113 with AIE properties, enabling bright customized-color CPL. Macrocycle 113 was obtained in 21% yield via a facile Knoevenagel condensation of dibenzyl cyanide 111 with 2,6-pyridinedicarboxaldehyde 112 in ethanol (EtOH) containing tetrabutylammonium hydroxide (TBAOH) (Scheme 32a). Crystal structure analysis showed that macrocycle 113 featured a large cavity capable of encapsulating guest molecules to construct host–guest supramolecular systems (Scheme 32b). For instance, changes in absorbance were observed in the UV-vis spectra upon adding Nile red (NiR) to macrocycle 113 (Scheme 32c), indicating ground-state intermolecular interactions between TPEM and NiR (Scheme 32d). In tetrahydrofuran (THF), 113 exhibited a main absorption band at 376 nm, with its fluorescence emission color transitioning from blue to green and finally to yellow, as solvent polarity increases, indicating an ICT effect (Scheme 32e). Macrocycle 113 exhibited AIE characteristics with a low FQY of 5.8% in pure THF. This increased significantly to 84.2% in a 95% water–THF mixture.
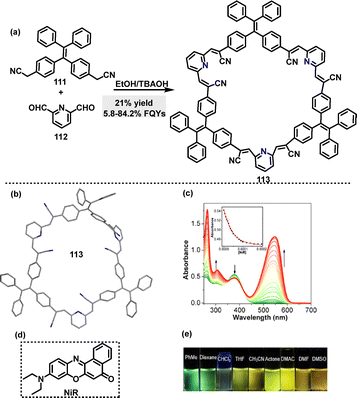 |
| | Scheme 32 (a) Synthesis and (b) crystal structure of tetraphenylethene-based 113, (c) changes in the UV-vis spectrum of TPEM in CHCl3 with the addition of NiR, (d) structure of NiR, and (e) fluorescence images. Reproduced with permission from ref. 62. American Chemical Society, copyright 2022. | |
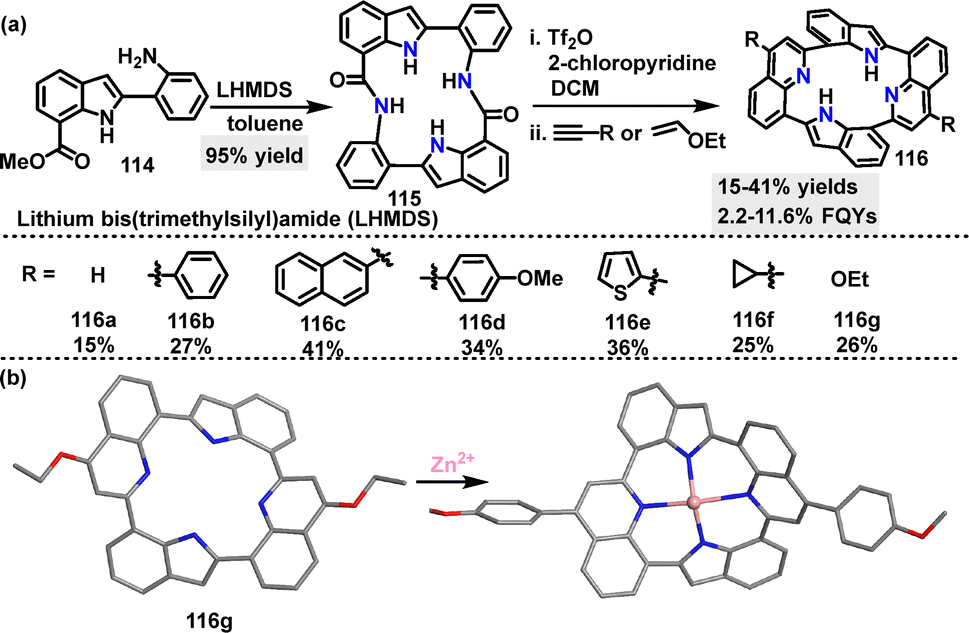
In 2024, Kumagai63 reported an efficient method for the construction of a macrocyclic diamide incorporating two indole units. Macrocycle 115 was obtained in 95% yield using Movassaghi's pyridine/quinoline-forming protocol (Scheme 33a). Further transformations of 115 yielded diverse new architecture macrocycles 116 containing two indole and two quinoline moieties in 25–42% yields. Macrocycles 116 can act as ligands due to the multiple N atoms, forming neutral Zn2+ complexes. Crystal structure analysis revealed that both 116g and the 116g/Zn2+ complex adopted saddle-shaped structures with C2 symmetry (Scheme 33b). In acetonitrile (ACN), macrocycles 116a–g exhibited absorption maxima ranging from 307 to 342 nm, with molar absorptivity values of 1.46–2.95 × 104 M−1 cm−1. These derivatives (116a–f) were emissive in both the solution (FQYs: 2.5–11.6%) and the solid state (FQYs: 2.2–5.8%). Notably, 116g bearing ethoxy substituents showed a relatively high FQY of 11.6% in ACN, likely attributed to the electron-donating nature of the ethoxy groups, which enhance intramolecular charge transfer.
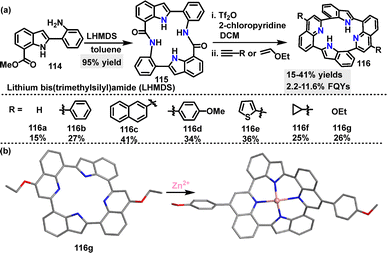 |
| | Scheme 33 (a) Synthesis and modification of macrocycle 115 and (b) crystal structures of 116g and the 116g/Zn2+ complex. | |
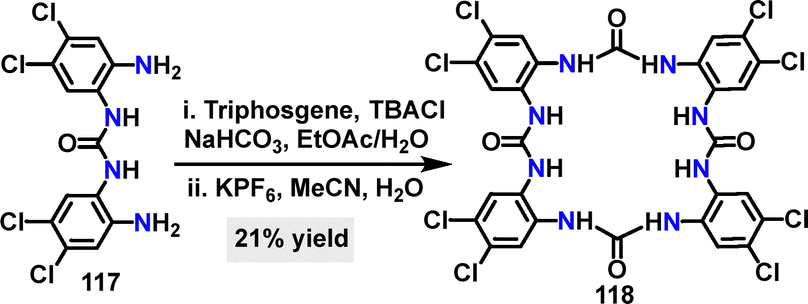
Recently, Gale64 reported an efficient and synthetically straightforward strategy for constructing tetra-urea macrocycle 118, achieving a 21% yield through a [2+2] cyclization under NaHCO3 and subsequent KPF6 conditions (Scheme 34). Macrocycle 118 had eight electron-withdrawing chlorine substituents, which enhanced its anion-binding capability.
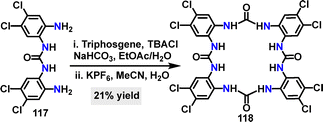 |
| | Scheme 34 Synthesis of the tetra-urea macrocycle 118. | |
2.2.4. Other conditions.
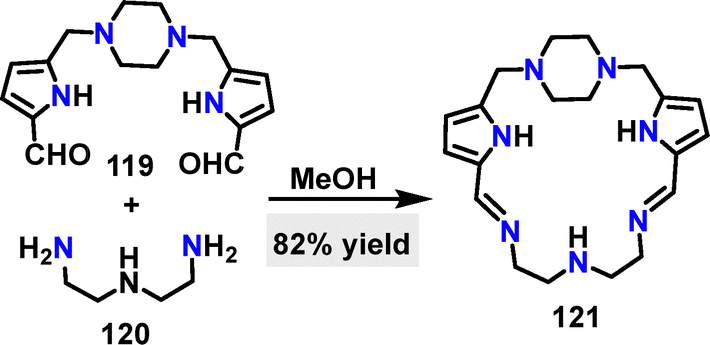
In 2024, Hamedani65 reported a one-pot condensation of an aldehyde 119 with diethylenetriamine 120 for the synthesis of a pyrrole-based macrocycle 121, achieving an 82% yield using methanol (MeOH) as the solvent (Scheme 35). The Cd2+ complex ([Cd121]) could be formed by reacting 121 with Cd(NO3)2·6H2O. Additionally, both 121 and [Cd121] demonstrated notable 2,2-diphenyl-1-picryl-hydrazyl-hydrate (DPPH) radical scavenging activity, with IC50 values of 1.03 and 1.42 mg mL−1, respectively, comparable with that of ascorbic acid.66
 |
| | Scheme 35 Synthesis of pyrrole-based macrocycle 121. | |
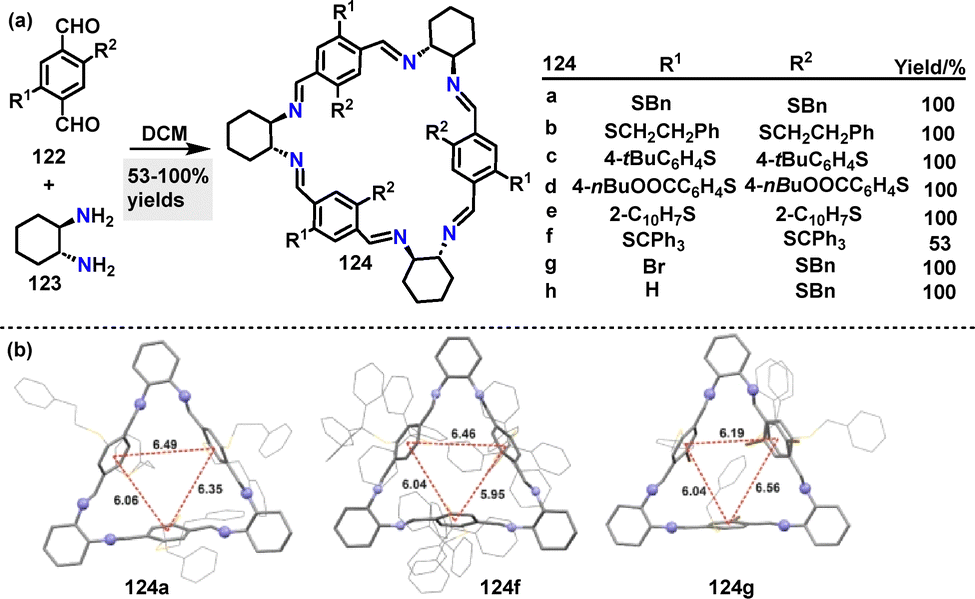
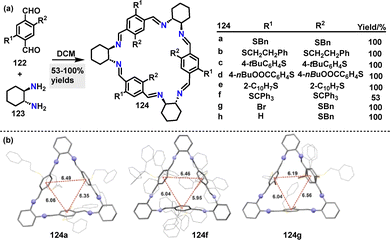
In 2025, Kwit67 reported an efficient method for the synthesis of chair-type macrocyclic compounds 124. Diverse macrocycles 124 were synthesized in 53–100% yields via [3+3] cyclocondensation reactions between equimolar dialdehydes 122 and chiral trans-1,2-diaminocyclohexane (DACH) 123 under mild conditions (Scheme 36a). Crystal structure analysis revealed that steric interactions dominate substituent arrangements, with dispersion effects playing a secondary role (Scheme 36b). The crystal structure of 124a showed columnar aggregates stabilized by π–π stacking and C–H⋯S interactions, where phenyl rings from adjacent molecules intercalate into the macrocyclic cavities. In contrast, 124f adopted a bowl-like conformation, with its trityl groups exhibiting distinct helicities influenced by combined steric and dispersive forces. Macrocycle 124g featured solvent-filled intermolecular spaces and disordered bromine substituents, resulting in variable rim diameters (4.59–8.18 Å).
 |
| | Scheme 36 (a) Synthesis of chair-type macrocycles 124 and (b) crystal structures of 124a, 124f, and 124g. Reproduced from ref. 67 with permission from Springer Nature, copyright 2025. | |
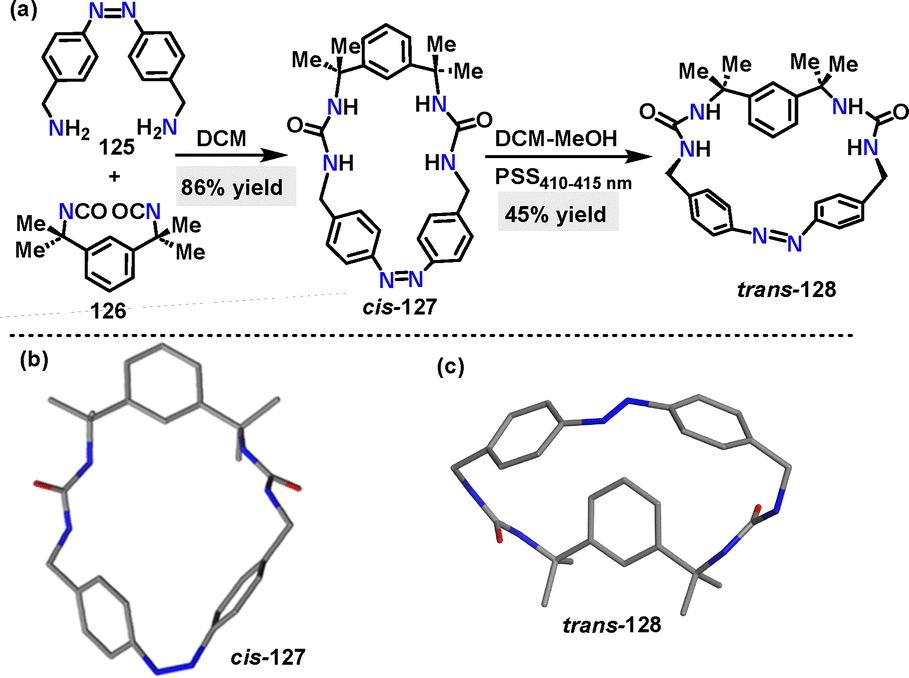
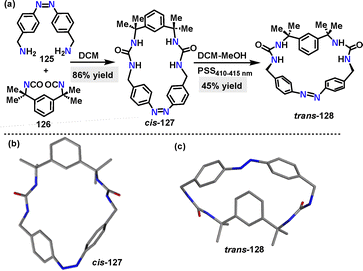
More recently, He68 and coworkers designed and synthesized an azobenzene-based macrocycle 138 through a two-step synthetic sequence. Macrocycle cis-137 was synthesized in 86% yield via the condensation of amine 135 and isocyanate 136 (Scheme 37a).
 |
| | Scheme 37 (a) Synthesis and (b) crystal structures of azobenzene-based macrocycles 127 and 128. | |
Irradiation of cis-128 with 410–415 nm light to the photostationary state (PSS) converted it to the desired trans-128 in 45% yield. Crystal structure analysis showed that cis-127 adopted a V-shaped conformation with two urea units andCsp2–H forming a preorganized binding pocket (Scheme 37b). While trans-128 exhibited a bow-shaped conformation with a bent azobenzene unit (49.19° angle between benzene rings) and a larger methylene distance (11.0 Å) (Scheme 37c).
3. O-doped macrocycles
The introduction of oxygen atoms endows macrocyclic compounds with unique capabilities in ion recognition, molecular inclusion, and dynamic response through lone pair electron coordination, hydrogen bonding, and polarity regulation.69 In 1967, Pedersen discovered crown ethers,70,71 after which oxygenated macrocycles,72 including cyclodextrins and macrocyclic polyethers, have demonstrated significant potential at the intersection of chemistry, materials science, and life sciences. Many of their derivatives, now extensively studied and reported, have opened new avenues for supramolecular chemistry. Recently, numerous synthetic methods have been developed for fascinating O-doped macrocycles, enabling their broad applications across diverse fields.
3.1. Metal catalysis
3.1.1. Palladium catalysis.
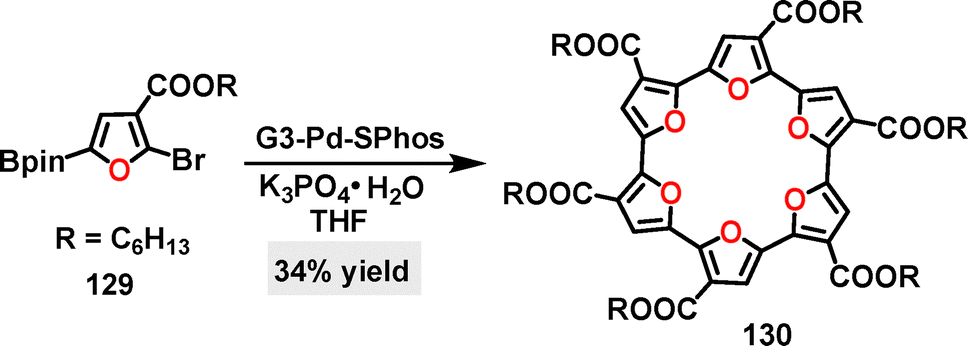
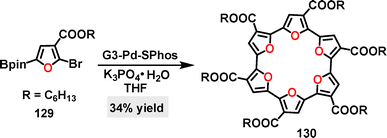
In 2021, Noonan73 reported an efficient method for the synthesis of a six-membered oligofuran macrocycle 130. Using the G3–Pd–SPhos catalyst system, macrocycle 130 was obtained in 34% yield via a cross-coupling reaction (Scheme 38). Electrochemical studies revealed two reversible reduction events at −1.49 V and −1.70 V, with no signs of instability observed after multiple reduction cycles. These potentials were cathodically shifted compared to those of the α,α′-tetramer of 2,2′-bifuran-3,3′dicarboximide74 (−1.0 V) and cyclo[6]pyrrole75 (−0.48 V).
 |
| | Scheme 38 Synthesis of six-membered oligofuran macrocycle 130. | |
3.1.2. Iron catalysis.
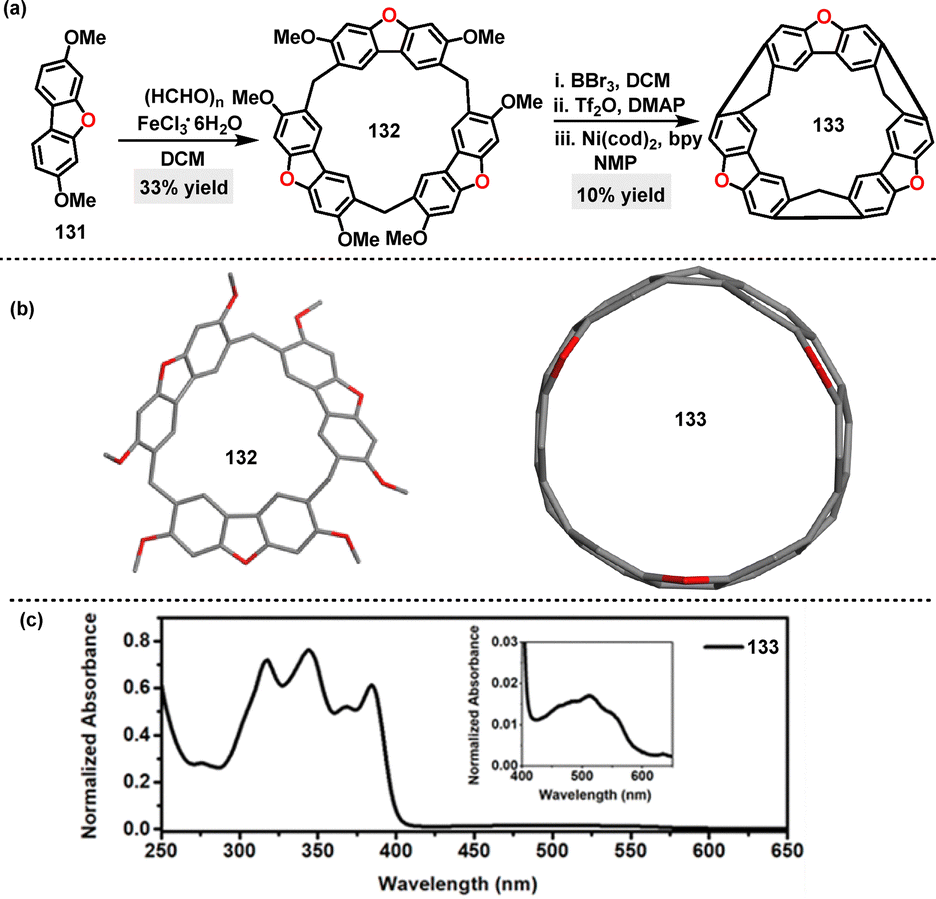
In 2024, Chen76 reported an FeCl3-catalyzed one-pot condensation for the synthesis of dibenzofuran[3]arene 132 in 33% yield. This functional macrocyclic arene 132 was further transformed into a macrocycle belt 133, featuring a rigid ring-shaped structure (Scheme 39a). Crystal structure analysis showed that macrocycle 132 exhibited a structure with a high degree of symmetry, while the macrocycle belt 133 revealed a highly symmetrical barrel-like architecture (Scheme 39b). In DCM, macrocycle belt 133 exhibited a broad absorption band with four intense peaks at 316, 344, 369, and 384 nm, accompanied by a weak, broad absorption band spanning 400–600 nm (Scheme 39c). The optical band gap derived from the UV-vis spectrum (2.16 eV) was narrower than that of methylene-bridged [6]CPP77 (2.66 eV) and slightly wider than the N-doped methylene-bridged [6]CPP78 (2.02 eV). This phenomenon can be attributed to the incorporation of O-atoms, which was designed to enhance π-conjugation.
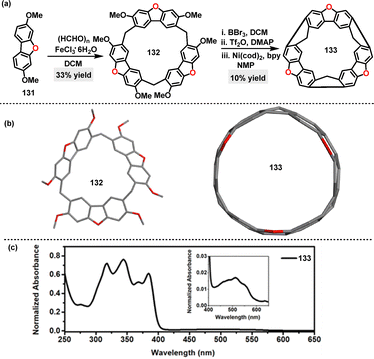 |
| | Scheme 39 (a) Synthesis and (b) crystal structures of dibenzofuran[3]arene 132 and macrocycle belt 133, and (c) UV-vis absorption spectrum of macrocycle 133. Adapted from ref. 76 with permission from The Royal Society of Chemistry, copyright 2024. | |
3.1.3. Tin catalysis.
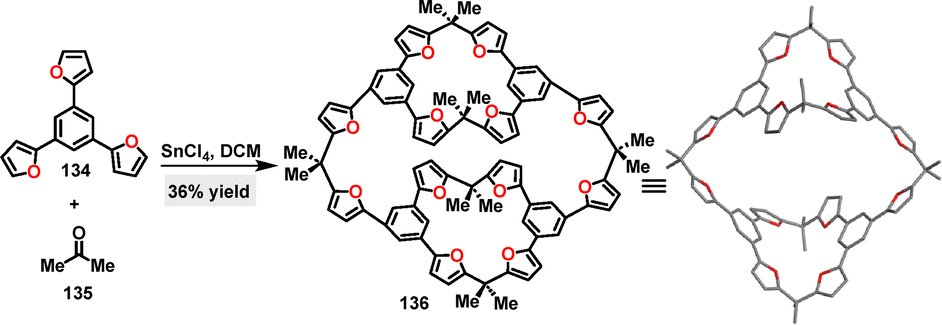
In 2021, Li79 reported a F–C cyclization between substrate 134 and acetone 135 for the synthesis of a cylindrical furan-based macrocycle 136 in 36% yield (Scheme 40). Twelve C–C bonds were formed simultaneously using SnCl4 as the Lewis acid catalyst in anhydrous DCM. In contrast, other Lewis acids (e.g., FeCl3, AlCl3, and BF3·OEt2) or Brønsted acids (e.g., TFA, HCl, and H2SO4) either failed to promote the reaction or led to polymeric byproducts. Crystal structure analysis of 136 revealed that the internuclear distance between the bridging carbon atoms connected to furans via a single acetone bridge is 16.1 Å, whereas the corresponding distance for furans linked by bis-acetone bridges was 14.4 Å.
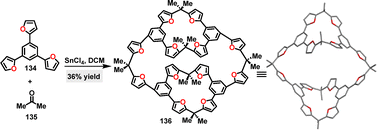 |
| | Scheme 40 Synthesis and crystal structure of furan-based macrocycle 136. | |
3.2. Metal-free strategies
3.2.1. Lewis acid catalysis.
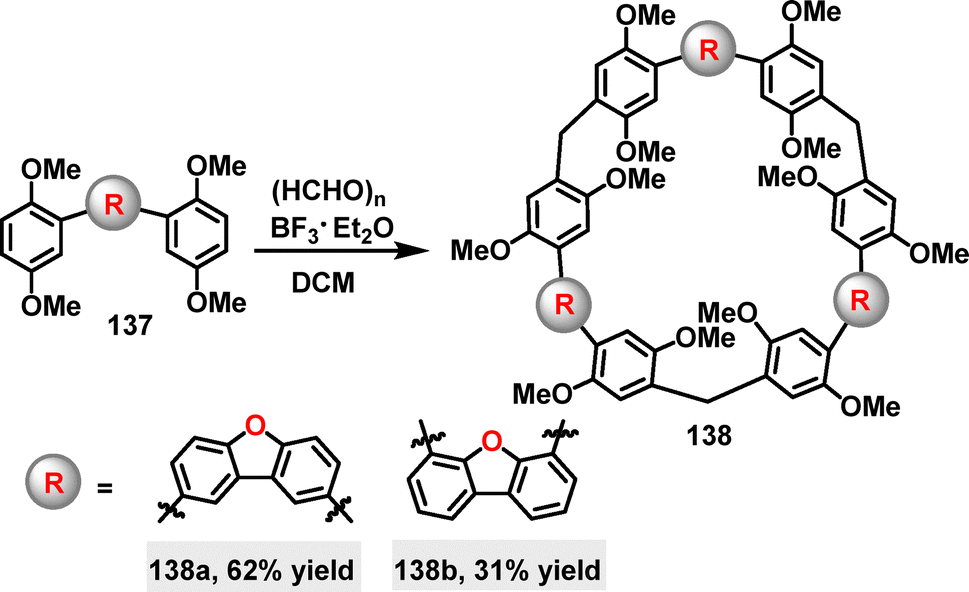
In 2022, Li, Luo, and co-workers developed a F–C reaction for the synthesis of two endo-functionalized macrocycles 138 (Scheme 41).80 Both products were demonstrated to enhance molecular recognition capabilities. Under BF3·Et2O-catalyzed condensation conditions, the target macrocycles were obtained in 62% and 31% yields, respectively. Furthermore, the benzofuranyl groups in 138a and 138b likely function as endo-binding sites of hydrogen-bonding donors, making them well-suited for binding heterocyclic guests (e.g., pyrrole and indole) through hydrogen-bonding interactions.
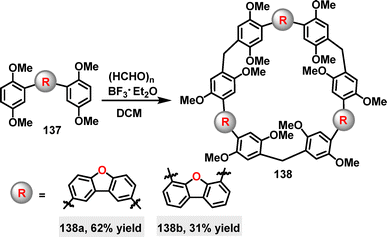 |
| | Scheme 41 Synthesis of benzofuran-based macrocycle 138. | |
3.2.2. Brønsted acid catalysis.
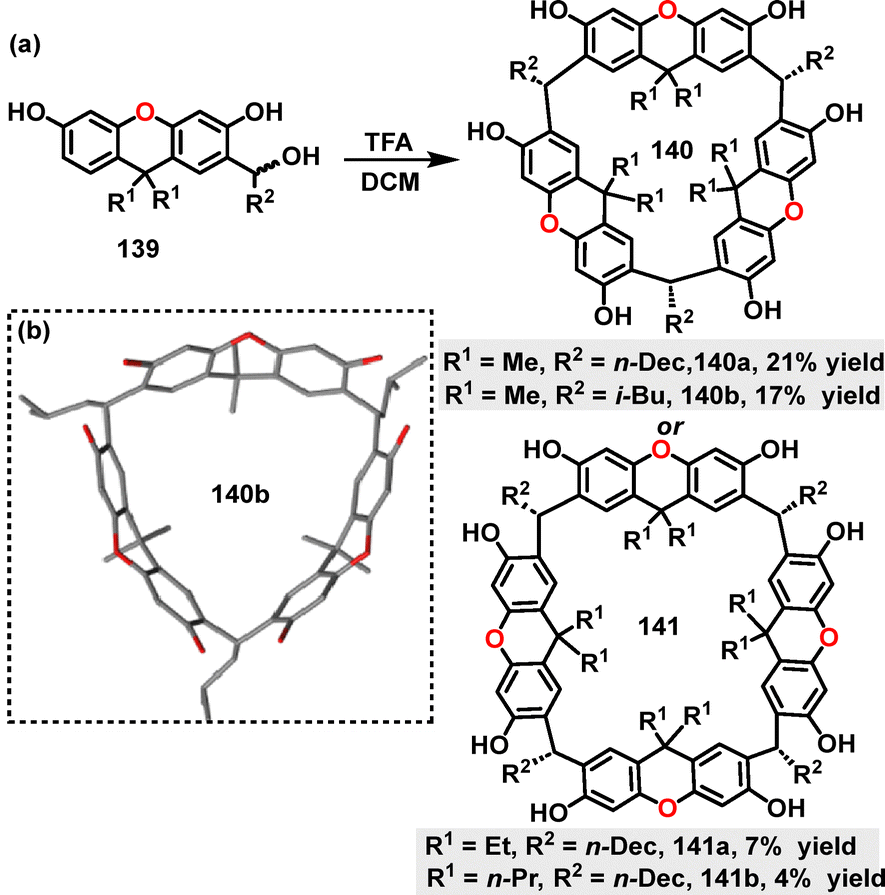
In 2021, Tiefenbacher81 reported the synthesis of a series of xanthene[n]arenes 150 and 151via a TFA-catalyzed macrocyclization of prefunctionalized xanthene monomers 149, with yields ranging from 4% to 21% (Scheme 42a). Notably, the selective formation of 150 or 151 was governed by the steric hindrance at the C9-position of monomers 149, which was modulated by different alkyl substituents (methyl, ethyl, or n-propyl). For instance, smaller substituents (e.g., methyl groups) favored the formation of 150 (17–21% yields), whereas larger substituents (e.g., ethyl or n-propyl) promoted the generation of 151 (4–7% yields). Crystal structure analysis of macrocycle 140b revealed a crown-shaped (bowl-like) conformation (Scheme 42b). Structural characterization showed tetrahedral angles of 109.4° between adjacent xanthene units and dihedral angles of 16.5° between the aromatic rings, a distortion attributed to macrocyclic strain. Additionally, the i-Bu substituents adopted an outward orientation, which minimized intramolecular steric hindrance and enhanced the compound's solubility.
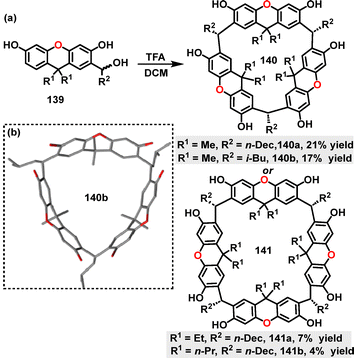 |
| | Scheme 42 (a) Synthesis of xanthene[n]arenes 140 and 141 and (b) crystal structure of 140a. | |
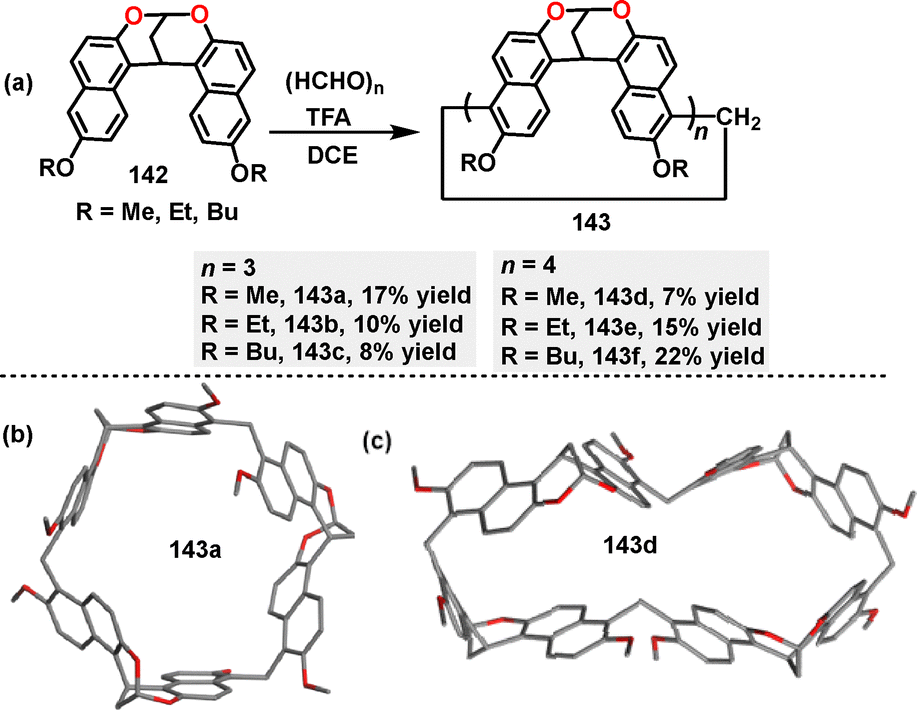
In 2022, Jiang82 reported the synthesis of a series of methylene-bridged naphthotube macrocycles 143via a F–C reaction between alkoxy-substituted bisnaphthalenes 142 and paraformaldehyde (Scheme 43a). Using trifluoroacetic acid (TFA) as a critical catalyst, various three-membered macrocycles 143a–c and four-membered macrocycles 143d–f were obtained in 7–22% yields. In contrast, the use of other acids such as CF3SO3H, p-TsOH, and BF3·OEt2 tended to induce polymer formation instead. Crystal structure analysis revealed that macrocycle 143a exclusively adopted a C3 symmetry (Scheme 43b). For 143d, a double-cavity conformation, a D2d symmetry was observed, where enantiomers underwent rapid interconversion in solution (Scheme 43c). Spectroscopic characterization highlighted distinct absorption maxima at 358 nm (143a) and 362 nm (143d). In DCE, these macrocycles displayed FQYs of 24.6% (143a) and 34.2% (143d), respectively.
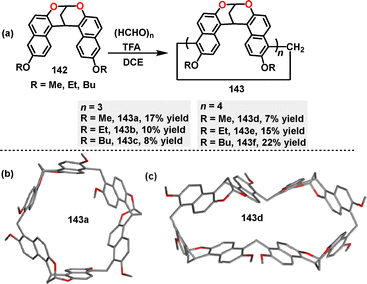 |
| | Scheme 43 (a) Synthesis of methylene-bridged naphthotube macrocycles 143 and (b) and (c) crystal structures of 143a and 143d. | |
3.2.3. Brønsted base catalysis.
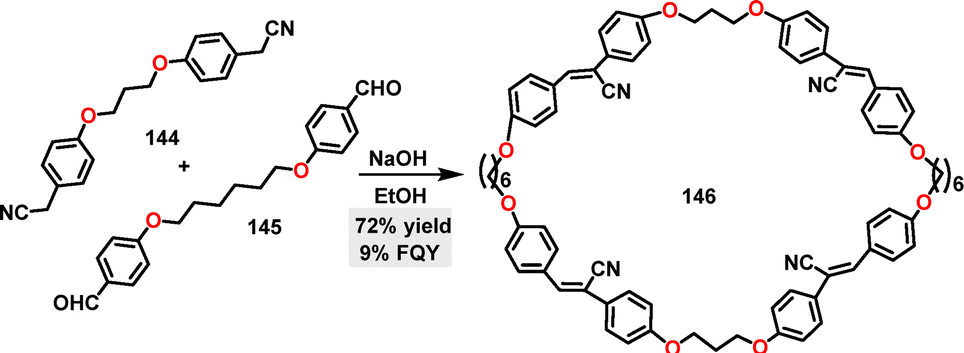
In 2021, Yang83 and co-workers reported the synthesis of a tetra-cyanostilbene macrocycle 146via a simple and rapid [2+2] condensation reaction (Scheme 44). Macrocycle 146 was obtained in 72% yield under NaOH-catalyzed conditions.
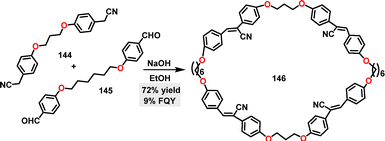 |
| | Scheme 44 Synthesis of tetra-cyanostilbene macrocycle 146. | |
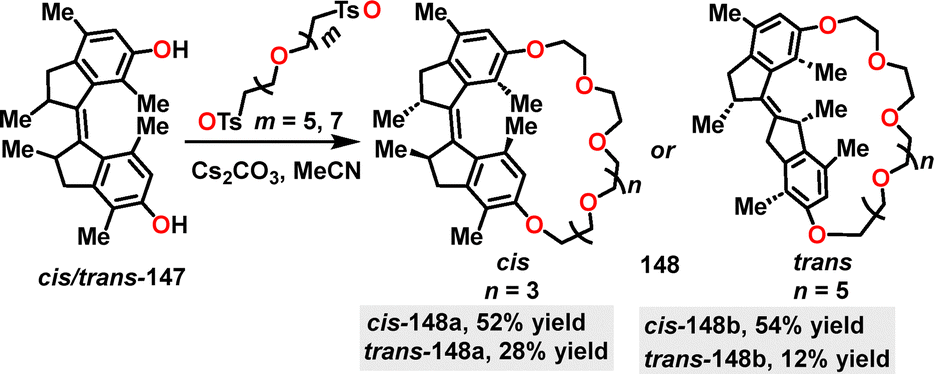
In the same year, Qu84 designed and synthesized the crown-ether-based host–guest macrocycles 148. Diverse products were obtained in 12–54% yields via a one-pot intramolecular etherification–cyclization reaction under Cs2CO3 conditions between ethylene glycol chains and diphenol motors (Scheme 45). Moreover, the observed higher yields for cis isomers, such as cis-148b (54%) compared to trans-148b (only 12%), can be attributed to the reduced strain associated with the cis configuration.
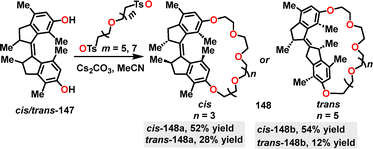 |
| | Scheme 45 Synthesis of crown-ether-based macrocycles 148. | |
3.2.4. Other conditions.
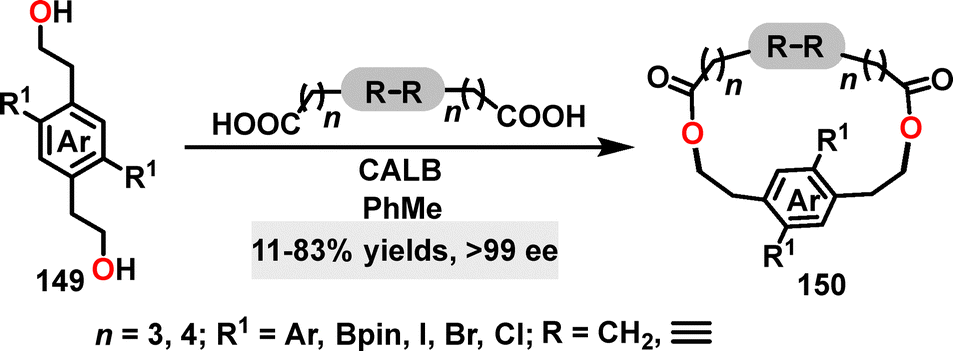
In 2020, Collins85 reported a novel methodology for the synthesis of planar chiral macrocycles 150 (Scheme 46). Using Candida antarctica lipase B (CALB) as the catalyst, this approach enabled the generation of various chiral cyclophanes via sequential acylation of diols and diacids, affording yields ranging from 35% to 83% and excellent enantioselectivities (66–99% ee). Halogen-substituted derivatives (chloro and iodo) and systems incorporating rigidified bridges (diynyl and disulfide) exhibited comparable yields (63–78%) with consistently high enantioselectivities (>99% ee in most cases). Even when diverse substituents such as alkynyl groups were introduced, the enantiomeric excesses remained exceptionally high (>99% ee), underscoring the robustness of this biocatalytic strategy.
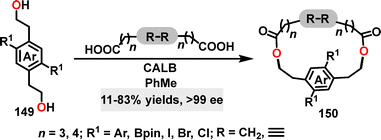 |
| | Scheme 46 Synthesis of chiral macrocycles 150. | |
4. S-doped macrocycles
The incorporation of sulfur atoms has profoundly altered the electronic properties, coordination capabilities, self-assembly behavior, and application scope of macrocyclic compounds.86 Owing to the unique electronic characteristics of sulfur (e.g., low electronegativity and high polarizability) and its distinct chemical attributes (such as the formation of polyvalent states and dynamic disulfide bonds), S-containing macrocycles exhibit exceptional functionalities. These properties are highly valuable in diverse fields including heavy metal adsorption, drug delivery systems, and optoelectronic devices.87
4.1. Metal catalysis
4.1.1. Palladium catalysis.
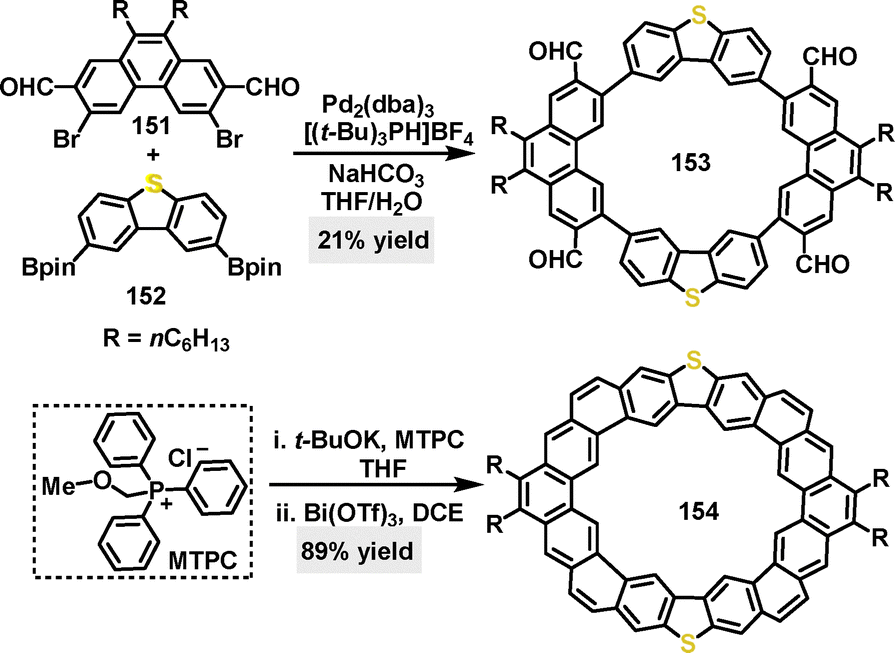
In 2020, Liu88 synthesized S-octulene 153 bearing two S-atoms via a palladium-catalyzed Suzuki coupling reaction between compounds 151 and 152. Macrocycle 154 was obtained in 21% yield and could be further transformed into the fully fused macrocycle 154 through sequential treatment with t-BuOK and subsequent Bi(OTf)3-catalyzed reactions (Scheme 47). DFT calculations revealed that S-octulene exhibited a large energy gap (3.19 eV), which implied that it may theoretically function as a solution-processable p-type organic semiconductor. Furthermore, the alkoxy-substituted heterocycloarene S-octulene demonstrated moderate solubility in common organic solvents such as DCM, chloroform, and toluene.
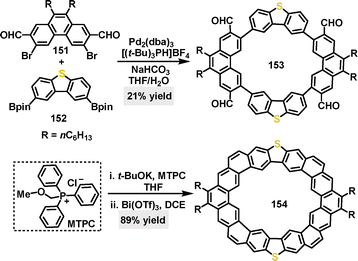 |
| | Scheme 47 Synthesis and modification of S-octulene 153. | |
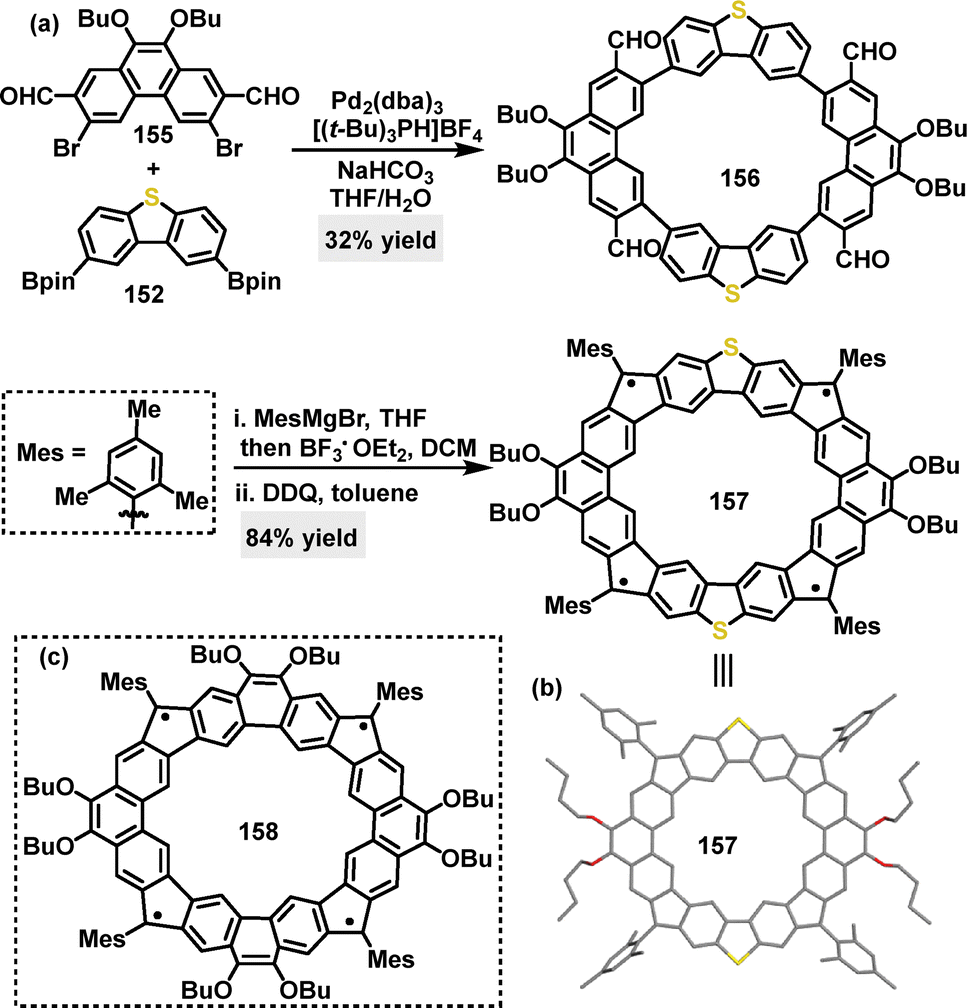
Later, the same research group reported a Suzuki coupling reaction for the synthesis of a macrocyclic structure, yielding macrocycle 156 in 32% yield (Scheme 48a).89 This intermediate could then be converted into radical macrocycle 157 (84% yield) via mesitylmagnesium bromide/BF3·Et2O-mediated F–C cyclization, followed by oxidative dehydrogenation. Crystal structure analysis revealed that radical macrocycle 157 adopted a rigid, slightly distorted coplanar backbone with a rectangle-shaped geometry (side lengths: 9.781 Å and 9.941 Å) (Scheme 48b). Compared to its all-carbon analogue 158 (Scheme 48c), radical macrocycle 157 displayed a reduced radical character (unpaired electron count 2.0 vs. 3.52 for 158) and a larger singlet–triplet energy gap (−3.47 vs. −3.04 kcal mol−1 for 158). These differences were attributed to stronger antiferromagnetic coupling through the dibenzothiophene (DBTh) units and electron donation from S-atoms.
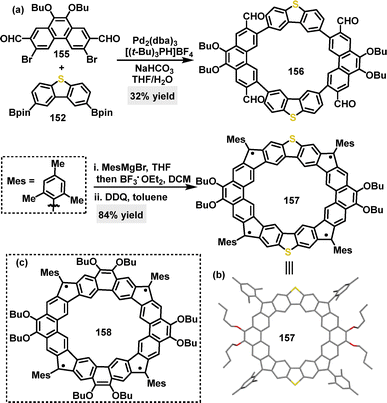 |
| | Scheme 48 (a) Synthesis and modifications of macrocycles 156, (b) crystal structure of 157, and (c) structure of 158. | |
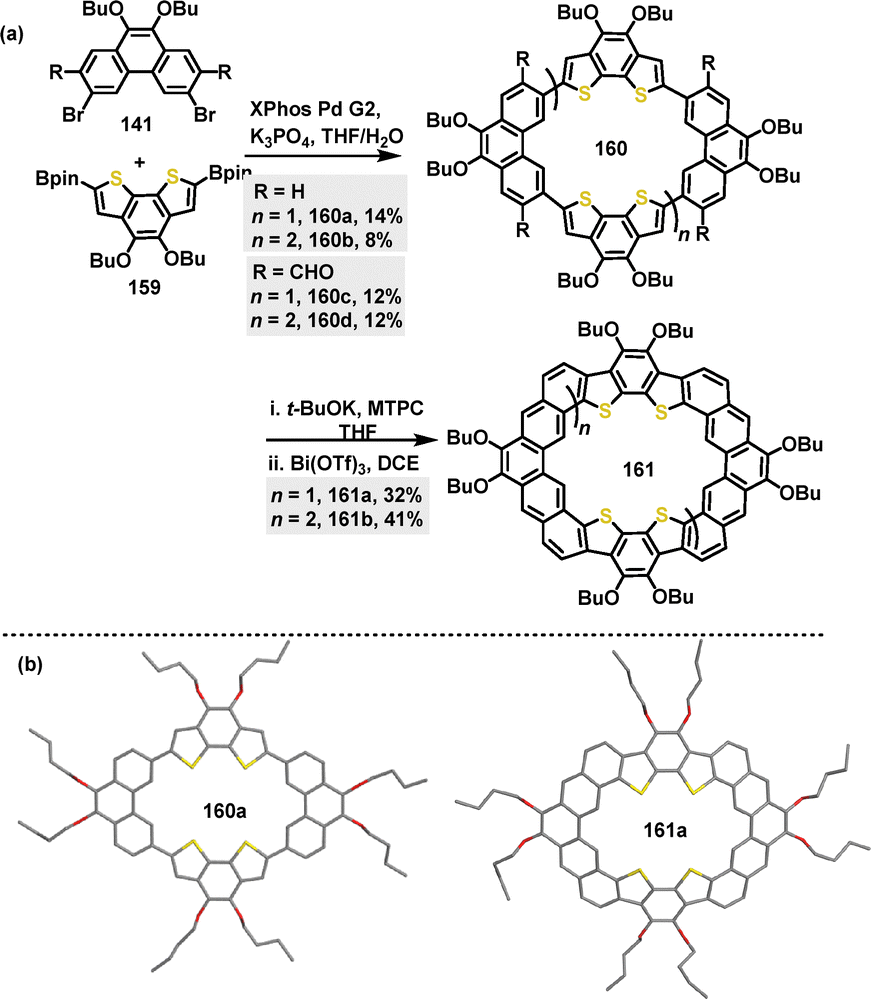
In 2024, the π-conjugated macrocycle 160,90 composed of benzo[2,1-b:3,4-b′]dithiophene (BDTh) and Phen units, was synthesized in 8–14% yield via a palladium-catalyzed Suzuki coupling reaction. Furthermore, fused macrocycles 161 were generated in 32–41% yield through a sequence of base-promoted Wittig reaction and Bi(OTf)3-catalyzed F–C reaction, starting from the aldehyde-based macrocycle 160c–d (Scheme 49a). Crystal structure analysis revealed that 160a adopted a nearly planar structure with polymorphic conformations. In contrast, 161b crystals displayed a bowl-shaped dimer with a bilayer-wavy stacking pattern (Scheme 49b). Fluorescence emission spectra revealed a notably blue-shifted peak for 161b at 470 nm compared to 161a (509 nm), indicating a significant Stokes shift for 161b.
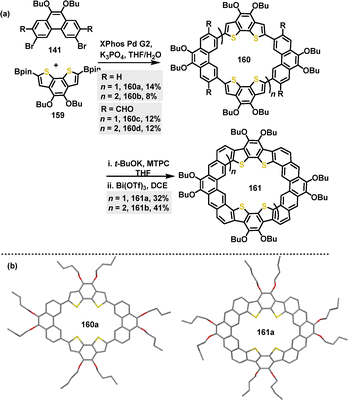 |
| | Scheme 49 (a) Synthesis and (b) crystal structures of macrocycles 160 and 161. | |
4.1.2 Nickel catalysis.
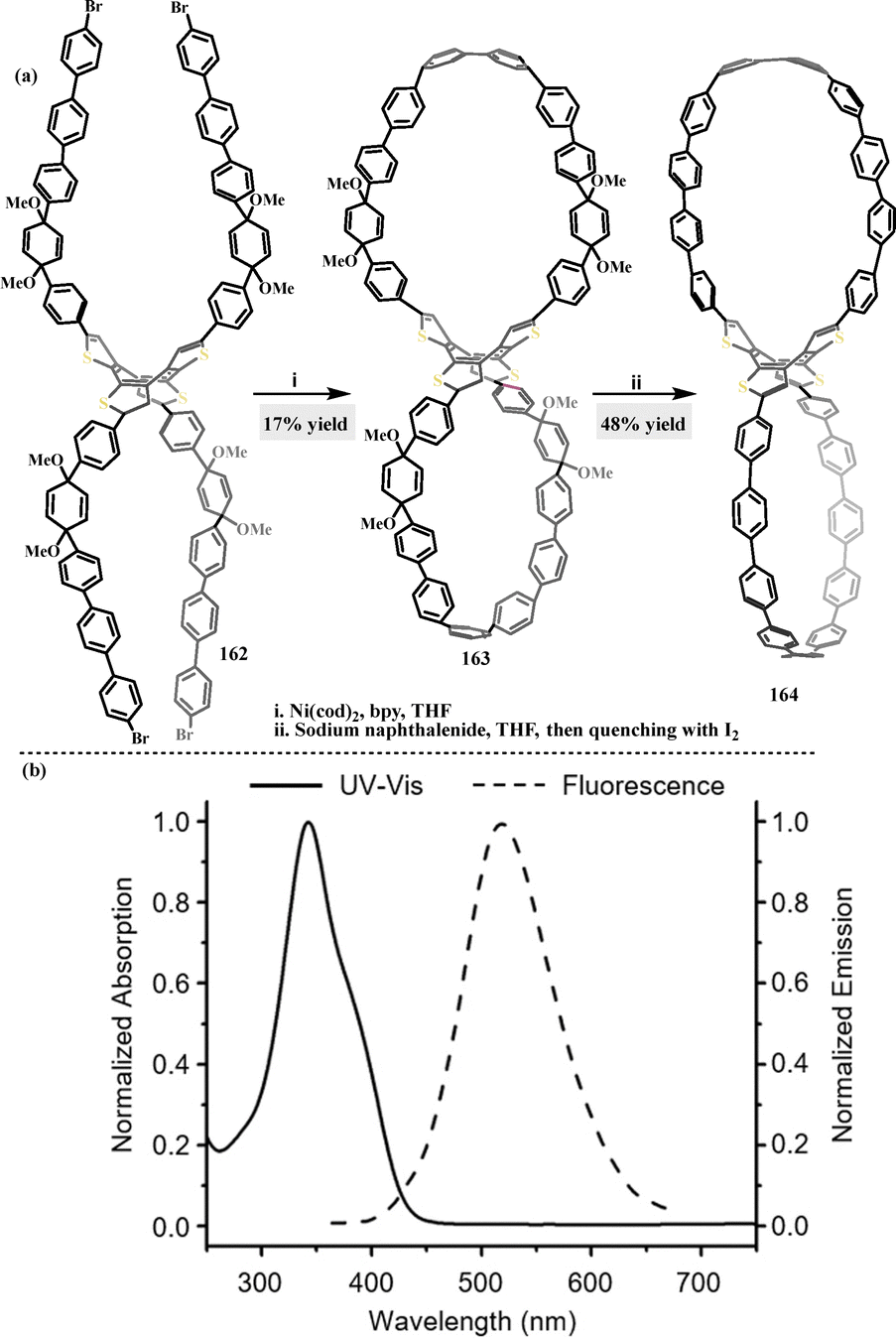
In 2021, Cong91 synthesized a fully conjugated figure-of-eight macrocycle 163, which integrates a flexible cyclooctatetrathiophene (COTh) core and two strained oligoparaphenylene loops. Macrocycle 163 was obtained in 17% yield via nickel-mediated Yamamoto homocoupling of compound 162, and 163 was further transformed into nanohoop 164 under sodium naphthalenide conditions (Scheme 50a). ICT between the electron-rich COTh core and the paraphenylene loops in 164 resulted in a red-shifted emission at 342 nm, compared to [10]CPP (338 nm).92,93 Additionally, nanohoop 164 exhibited an emission maximum at 520 nm (Stokes shift: 178 nm) (Scheme 50b), a notably shorter fluorescence lifetime (0.3 ns), and a reduced FQY (3%), indicative of charge transfer characteristics.
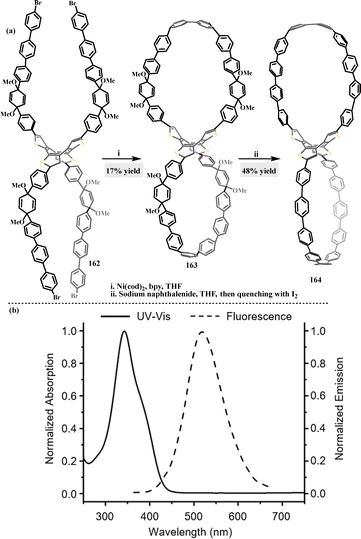 |
| | Scheme 50 (a) Synthesis of figure-of-eight macrocycles 163–164 and (b) UV-vis absorption and fluorescence of macrocycle 164 in DCM. Reproduced from ref. 91 with permission from Wiley-VCH, copyright 2021. | |
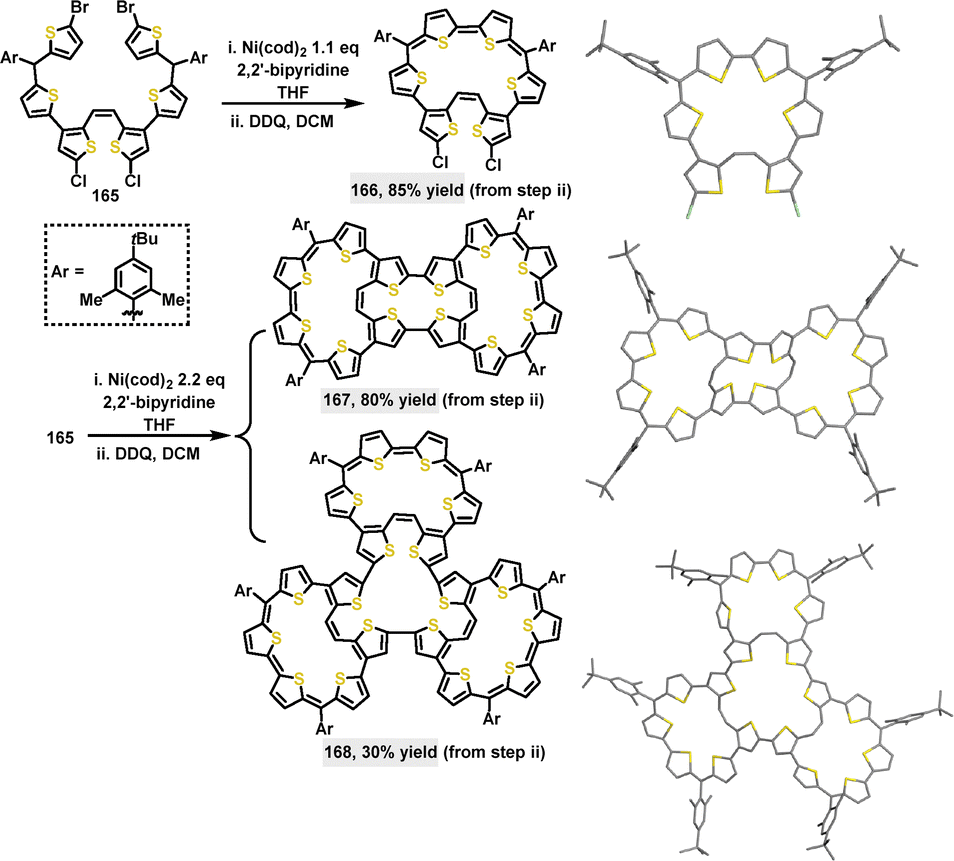
In 2023, Wu94 reported the synthesis of π-conjugated multicyclic thiophene-based macrocycles through an intermolecular Yamamoto coupling, followed by oxidative dehydrogenation. When 1.1 equivalents of Ni(cod)2 were used, a monocyclic macrocycle 166 containing four aromatic thiophene rings and a bithiophenequinodimethane moiety was obtained in an 85% yield. Notably, using 2.2 equivalents of Ni(cod)2 and DDQ led to the formation of a tricyclic macrocycle 167 (80% yield) or a three-leaf clover-like tetracyclic macrocycle 168 (30% yield) (Scheme 51). The introduction of bulky 4-tert-butyl-2,6-dimethylphenyl substituents significantly enhanced the solubility and stability of these macrocycles. Crystal structure analysis showed that macrocycle 166 adopted a twisted conformation with a dihedral angle of 28.1° and significant bond length alternation (e.g., 1.361 Å and 1.402 Å), indicative of antiaromaticity. The central ring of 167 exhibited an even more pronounced twist, with dihedral angles reaching up to 57.2°. Meanwhile, 168 displayed a bent three-leaf clover shape (length: 2.4 nm, width: 2.0 nm), characterized by dihedral angles at the crossing sites ranging from 14.8° to 70.7°.
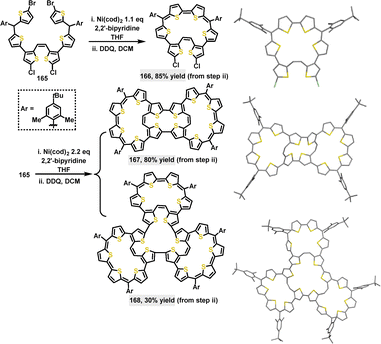 |
| | Scheme 51 Synthesis and crystal structures of thiophene-based macrocycles 166–168. | |
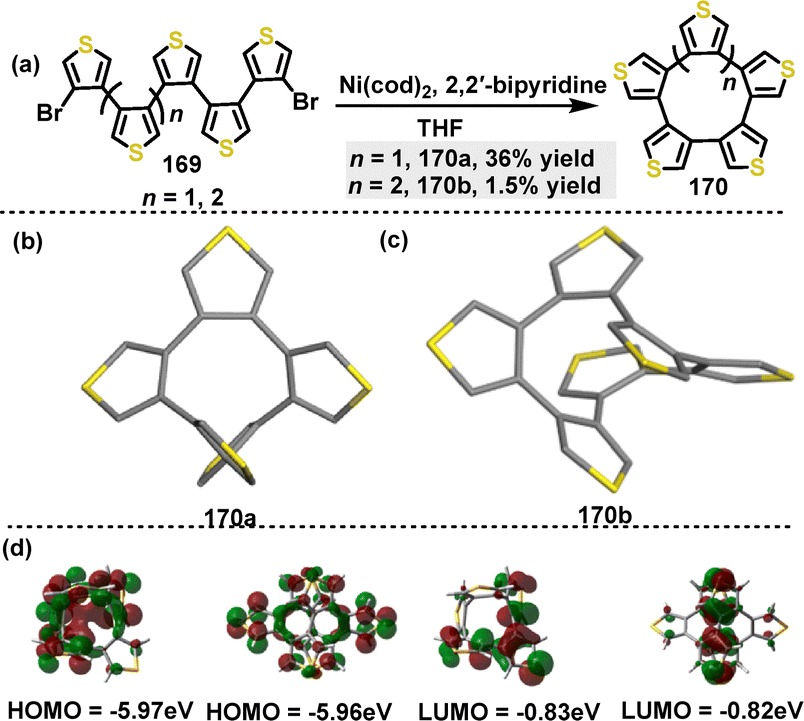
Subsequently, Segawa95 reported the synthesis of thiophene-containing polyarenes through a Ni-mediated homocoupling reaction. Under the catalytic conditions of Ni(cod)2, penta(3,4-thienylene) 170a and hexa(3,4-thienylene) 170b were synthesized with yields of 36% and 1.5%, respectively (Scheme 52a). The low yield of 170b was attributed to polymerization side reactions. Crystal structure analysis revealed that 170a adopted a C2-symmetric structure (Scheme 52b). In this structure, the thiophene moieties were connected at the 3,4-positions, forming a 10-membered ring. In contrast, 170b exhibited a D2-symmetric screw-like or helical structure, characterized by intramolecular π–π interactions, with a centroid distance of 3.4 Å (Scheme 52c). Both 170a and 170b showed size-dependent bathochromic shifts. The longest absorption maxima were observed at 273 nm for 170a and 276 nm for 170b. These shifts were ascribed to the decrease in the HOMO–LUMO gaps, as was supported by DFT calculations. The HOMO energy level decreased from −5.97 eV in 170a to −5.76 eV in 170b, while the LUMO energy level changed from −0.83 eV in 170a to −0.82 eV in 170b (Scheme 52d).
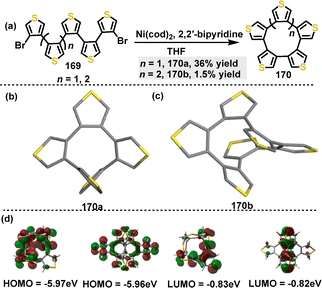 |
| | Scheme 52 (a) Synthesis and (b) and (c) crystal structures of thiophene-containing polyarenes 170, and (d) Frontier molecular orbitals and the energy levels of 170, calculated at the B3LYP-D3/6-31G(d) level. Reproduced from ref. 95 with permission from The Royal Society of Chemistry, copyright 2023. | |
4.1.3. Titanium catalysis.
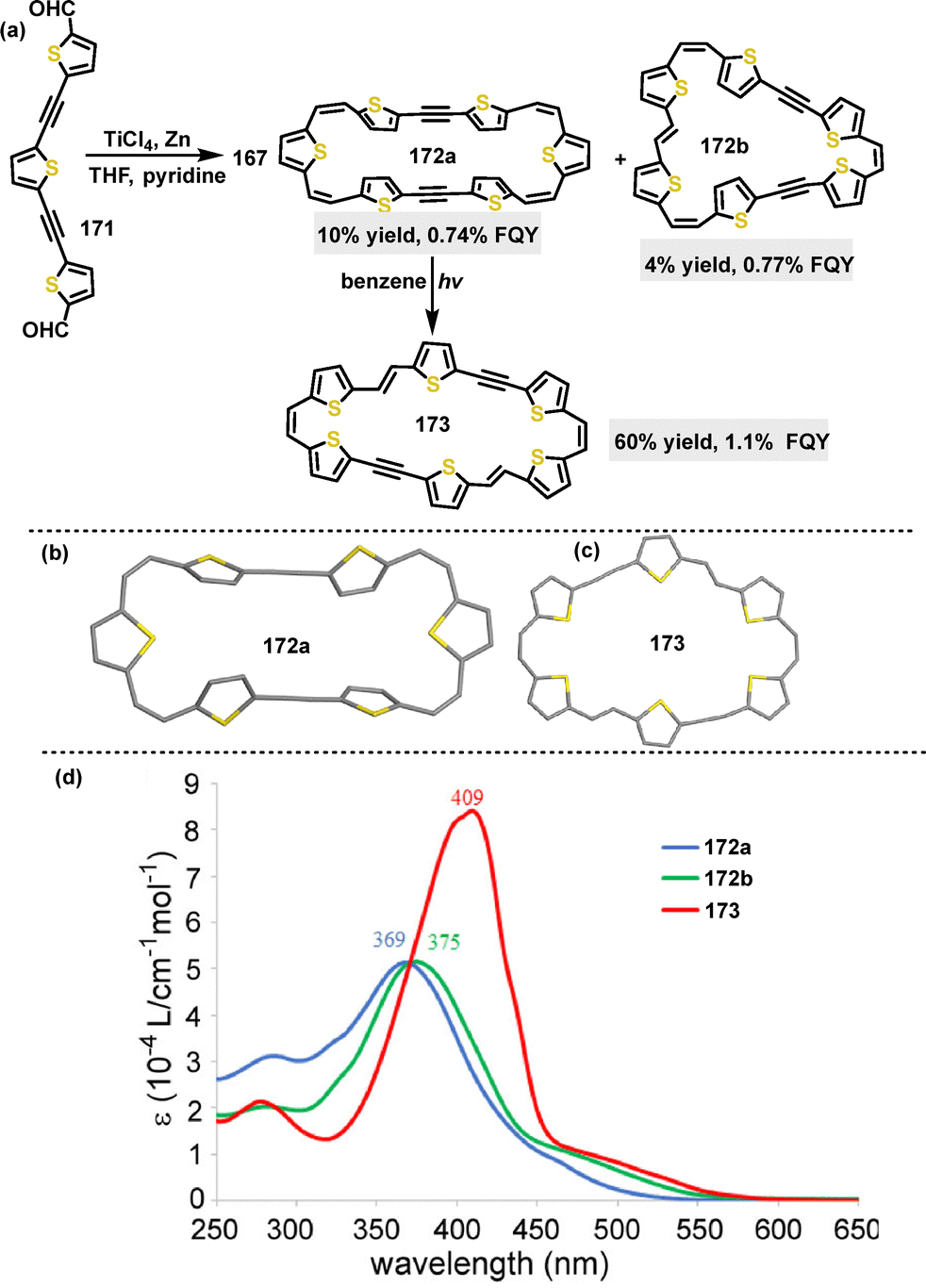
In 2020, Iyoda96 reported the synthesis of various macrocyclic π-extended thiophene skeletons through McMurry coupling of dialdehyde 171. By using TiCl4 as a catalyst, the π-conjugated macrocycles 172a and 172b were obtained with yields of 4% and 10%, respectively (Scheme 53a). Additionally, macrocycle 173 could be prepared from macrocycle 172a in a 60% yield under UV light irradiation. Crystal structure analysis of these macrocycles revealed their distinct geometries (Scheme 53b and c). UV-vis spectroscopy analysis showed that macrocycles 172a, 172b, and 713 exhibited maximum absorption wavelengths at 369 nm, 375 nm, and 409 nm, respectively (Scheme 53d). The redshift observed in macrocycle 173 implies an increased planarity. Furthermore, these compounds displayed low FQYs (172a: 0.74%; 172b: 0.77%; 173: 1.1%) due to their ring strain and conformational flexibility.
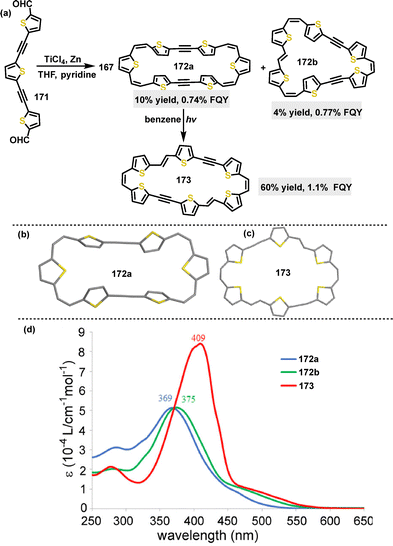 |
| | Scheme 53 (a) Synthesis of π-conjugated macrocycles 172–173, and (b) and (c) crystal structures of 172a and 173, and (d) UV-vis spectra of 172 and 173 in DCM. Adapted from ref. 96 with permission from American Chemical Society, copyright 2020. | |
4.1.4. Platinum catalysis.
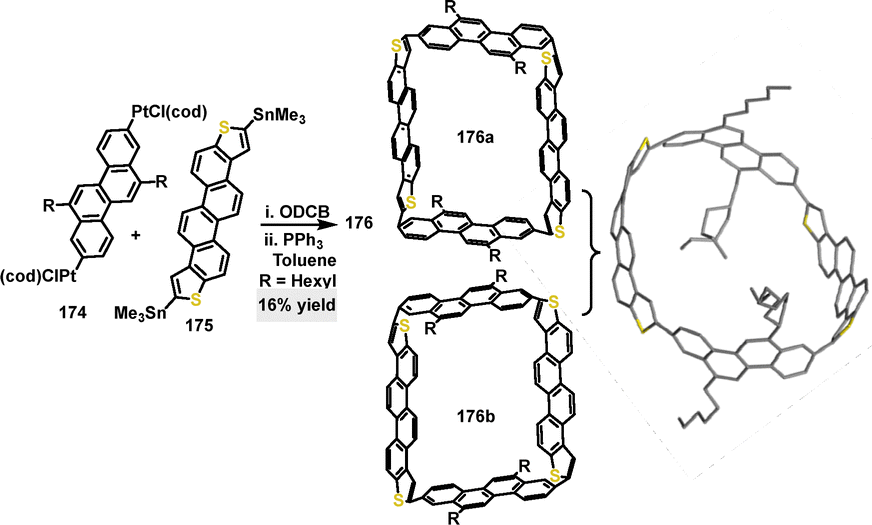
In 2021, Isobe97 reported a stereoselective cross-coupling macrocyclization. A thiophene-doped macrocycle 176 was obtained as a racemate in 16% yield through the reaction of tetrameric platinum complexes 174 and 175 in 1,2-dichlorobenzene (ODCB), involving triphenylphosphine ligand exchange and reductive elimination (Scheme 54). Crystal structure analysis of the racemate macrocycle 176 revealed an oval-shaped architecture, stemming from the pyramidalization of carbon atoms and twisted biaryl linkages with dihedral angles reaching up to 36°. CD spectroscopy revealed a strong molar ellipticity of 176a (−317 M−1 cm−1), indicative of a significant rotatory strength. Relaxed potential energy scans demonstrated that structural distortions increased the energy by less than 2 kcal mol−1, suggesting that the structure exhibits high flexibility.
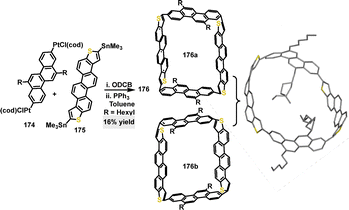 |
| | Scheme 54 Synthesis and crystal structure of thiophene-doped macrocycle 176. | |
4.2. Metal free strategies
4.2.1. Lewis acid catalysis.
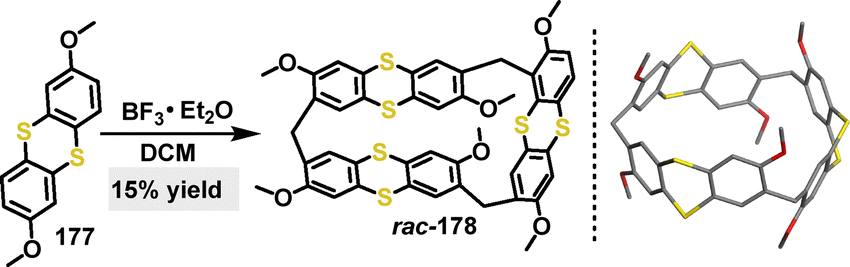
In 2023, Zhu synthesized a trimeric macrocycle rac-178 in 15% yield, via a one-pot Lewis acid-catalyzed condensation with paraformaldehyde.98 Notably, only BF3·Et2O effectively promoted cyclization, whereas other Lewis acids (e.g., AlCl3, p-TsOH, and TFA) led to polymerization or recovered the starting material, indicating a specific acid-template effect essential for this cyclization. Crystal structure analysis of rac-178 confirmed the helical belt-like structure, with a hexagonal prismatic cavity (Scheme 55).
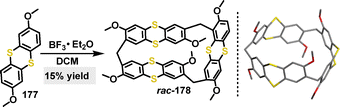 |
| | Scheme 55 Synthesis of chiral macrocycle rac-178 and crystal structure of rac-178. | |
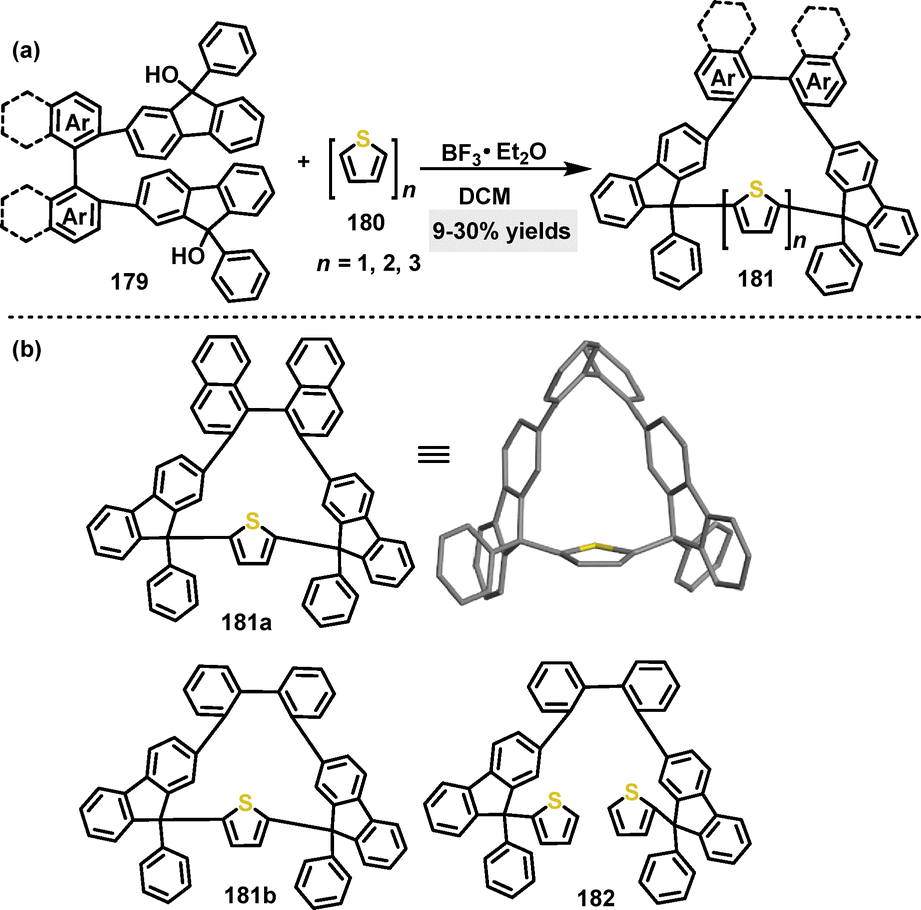
In the same year, our group developed a one-pot strategy to construct axially and centrally chiral nanogrids (AGs) via F–C gridization.99 By combining thiophene derivatives with difluorenyl biaromatic derivatives under BF3·Et2O catalysis, various macrocycles 181 were obtained in 9–30% yield (Scheme 56a). Crystal structure analysis of 181a confirmed a C2-symmetric molecular skeleton featuring waistline lengths of 1.11–1.12 nm and a biphenyl dihedral angle of 77.98° (Scheme 56b). The study compared the performance of the macrocyclic nanogrid 181b with its ungridized 182, highlighting the distinct advantage conferred by the rigid, well-defined nanogrid architecture.
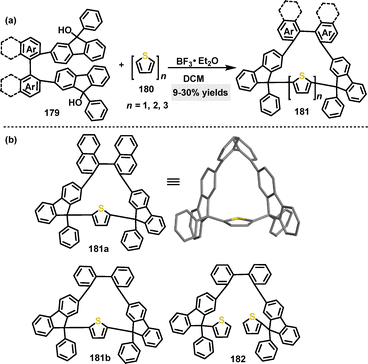 |
| | Scheme 56 (a) Synthesis of chiral nanogrids 181 and (b) crystal structure of 181a. | |
4.2.2. Other conditions.
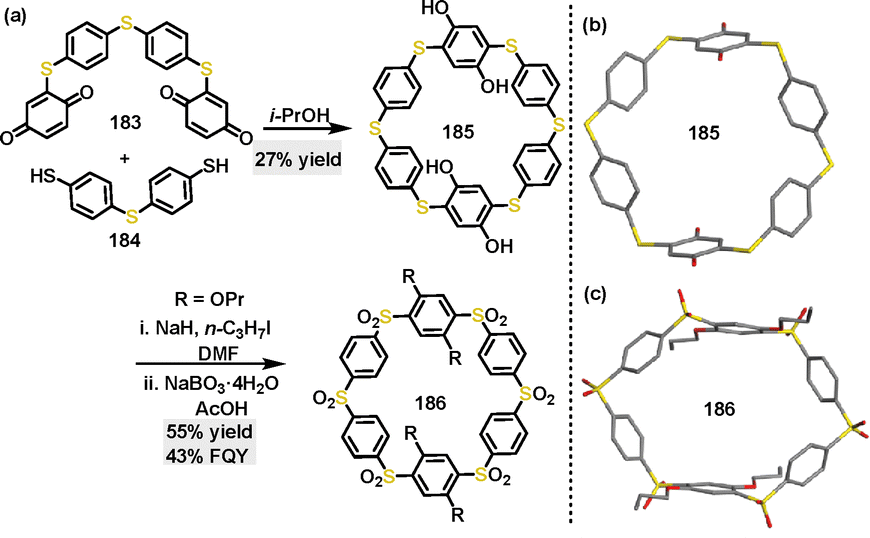
In 2022, Swager100 reported a green synthetic approach to construct thiapillar[6]arene via a thia-Michael addition of bis(cyclohexa-2,5-diene-1,4-dione) derivatives 183 and 4,4′-thiobisbenzenethiol 184. The thiapillar[6]arene macrocycle 185 was obtained in 27% yield using isopropanol (i-PrOH) as solvent, and it could be further transformed into a sulfone macrocycle 186 in 55% yield via NaH-catalyzed alkylation, followed by NaBO3·4H2O-catalyzed oxidation (Scheme 57a). Crystal structure analysis revealed that thiapillar[6]arene 185 featured a symmetrical macrocyclic cavity with a 12 Å distance between opposing sulfur atoms and 10 Å between phenyl rings, stabilized through weak intermolecular interactions (Scheme 57b). In contrast, 186 adopted a distorted “open book” conformation with reduced symmetry, characterized by alkoxy-substituted phenyl rings separated by 9 Å and a cavity width of 14 Å (Scheme 57c). In addition, 186 exhibited strong FQY (43%), attributed to D–A interactions between the appended groups and the electron-deficient sulfone cavity.
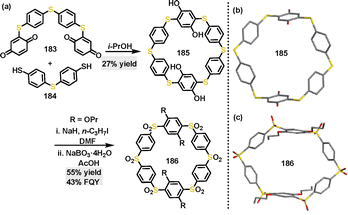 |
| | Scheme 57 (a) Synthesis and (b) and (c) crystal structures of 185 and 186. | |
5. N,O- and N,S-doped macrocycles
The synergistic incorporation of two types of heteroatoms (e.g., N,O- and N,S-pairs) can significantly tune the electronic structure, coordination selectivity, dynamic responsiveness, and functional applications of macrocyclic compounds.101,102 Interactions among heteroatoms, including electronic effects, steric hindrance, and synergistic coordination, can overcome the limitations of single-heteroatom systems, enabling the integration of more complex functionalities. For instance, aza-18-crown-6-ethers103 and diaza-18-crown-6 ethers104 exhibit remarkable versatility in heavy metal adsorption, catalysis, sensing, supramolecular assembly, and biomedicine. Their unique properties arise from N,O-synergistic coordination and distinct electronic environments, offering advantages over traditional crown ethers such as enhanced metal selectivity, dynamic responsiveness, and biocompatibility.105
5.1. Metal catalysis and N,O-doped macrocycles
5.1.1. Palladium catalysis.
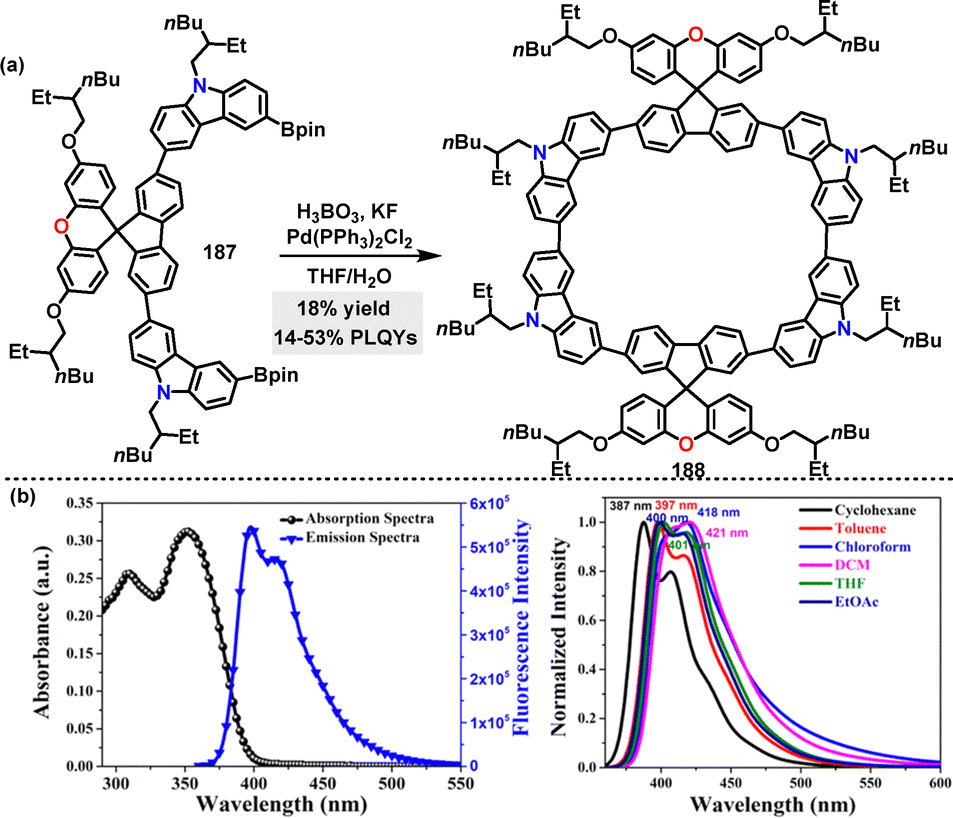
In 2024, Singh106 reported an oxidative homocoupling approach for the preparation of a D–A macrocycle in 18% yield under Pd(PPh3)2Cl2 catalysis conditions (Scheme 58a). To address solubility issues during the subsequent macrocyclization, 2-ethylhexyl alkyl chains on substrate 187 were employed for O-alkylation of the generated spiro[fluorene-9,9′-xanthene] (SFX) core. Using the space-charge-limited current method, macrocycle 188 demonstrated nearly balanced charge carrier mobilities with a hole mobility of 4.75 × 10−3 cm2 V−1 s−1 and an electron mobility of 1.0 × 10−3 cm2 V−1 s−1. In optical studies, macrocycle 188 displayed absorption peaks at 397 nm and 416 nm in nonpolar solvents, which shifted to 421 nm in polar DCM (Scheme 58b), indicative of positive solvatochromism. Additionally, macrocycle 188 exhibited strong solvent-dependent PLQY, ranging from 14% in cyclohexane, to 53% in toluene.
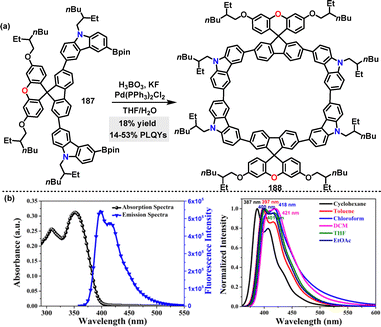 |
| | Scheme 58 (a) Synthesis of D–A macrocycle 188, (b) UV-vis and fluorescence spectra of macrocycle 188 (left) and emission spectra in solvents of increasing polarity (right). Reproduced from ref. 106 with permission from The Royal Society of Chemistry, copyright 2024. | |
5.1.2. Copper catalysis.
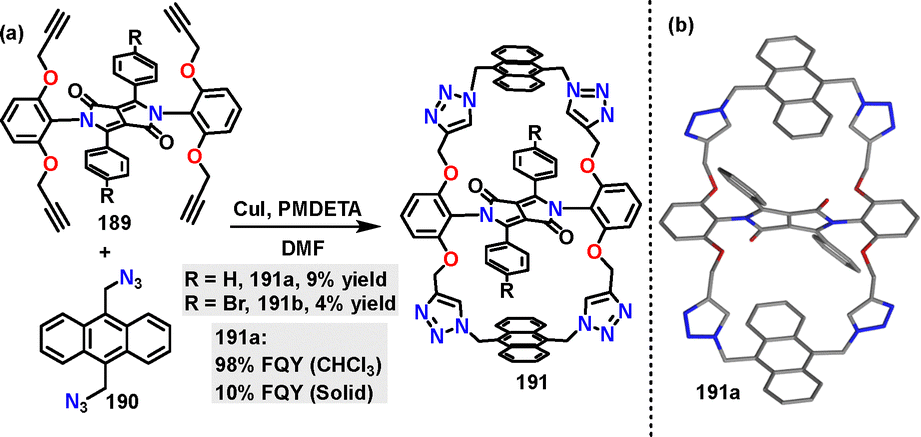
In 2024, Wang107 reported a Cu-catalyzed azide–alkyne cycloaddition (CuAAC) for constructing a diketopyrrolopyrrole (DPP)-bridged bis-anthracene macrocycle 191. The targeted doubly anthracene-strapped DPP macrocycles bearing H and Br substituents were obtained in 9% and 4% yields, respectively (Scheme 59a). Crystal structure analysis revealed that macrocycle 191a adopted a Z-shaped conformation, with anthracene straps shielding the DPP core (Scheme 59b). In CHCl3 solution, 191a exhibited a near-unity quantum yield (98%). In the solid state, thin films of 191a showed a moderate FQY (10%).
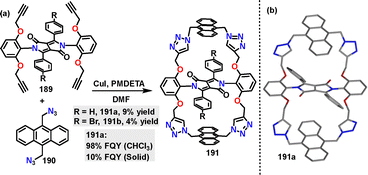 |
| | Scheme 59 (a) Synthesis and (b) crystal structure of diketopyrrolopyrrole-bridged bis-anthracene macrocycle 191. | |
5.2. Metal-free and N,O-doped macrocycles
5.2.1. Brønsted acid catalysis.
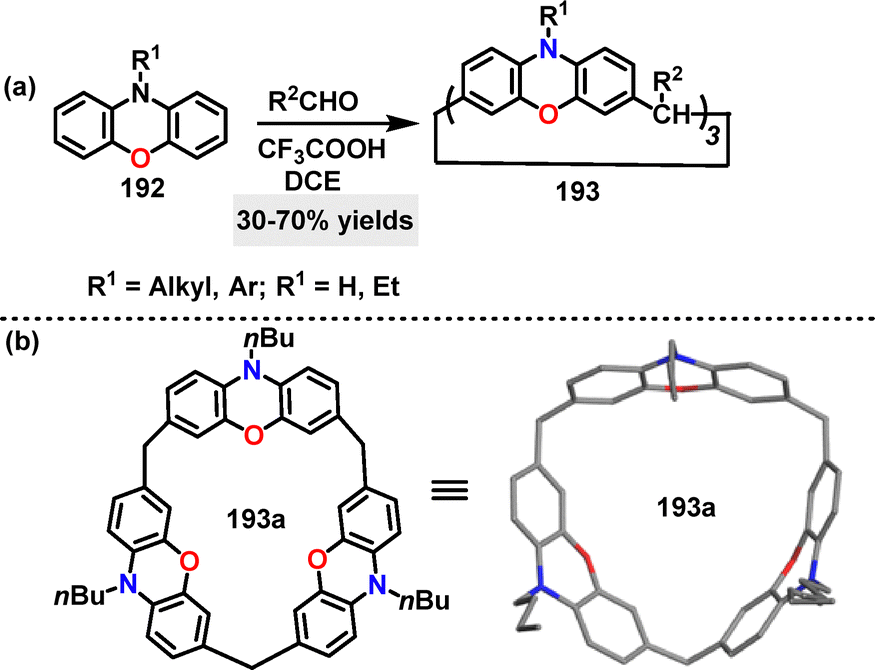
In 2023, Ma108 reported a streamlined synthesis of calix[3]phenoxazine via CF3COOH-catalyzed one-pot F–C cyclization of phenoxazine monomers 192 with substituted aldehydes. The N,O-doped macrocycles 193 bearing alkyl chains or aromatic moieties were obtained in 30–70% yield using DCE as the solvent (Scheme 60a). Crystal structure analysis showed that 193a adopted a cone-shaped conformation featuring a symmetrical cavity (7.7 Å) with phenoxazine units arranged in a butterfly configuration (Scheme 60b). The emission spectra of 193 exhibited maxima >500 nm with large Stokes shifts (100 nm), suggesting potential applications in laser materials and fluorescence-based dosimetry. Cyclic voltammetry of 193a revealed three quasi-reversible oxidation waves, each corresponding to its three phenoxazine units, highlighting its utility in optoelectronic materials.
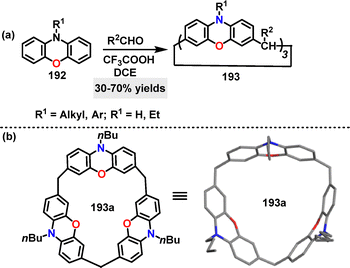 |
| | Scheme 60 (a) Synthesis of N,O-doped macrocycles 193 and (b) crystal structure of 193a. | |
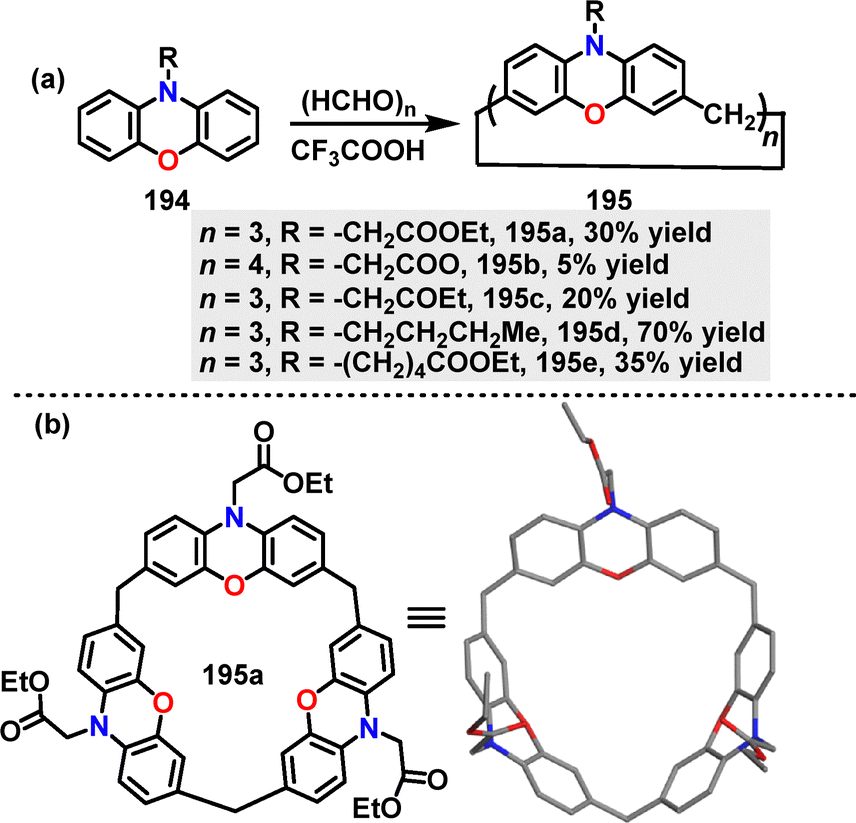
In 2025, Mao109 developed a F–C alkylation of substituted phenoxazines 194 with paraformaldehyde for the construction of calix[n]phenoxazines. Macrocycles 195 bearing different substitutions on the N-atom were generated in 5–70% yields under CF3COOH conditions (Scheme 61a). Crystal structure analysis showed that 188a adopted a symmetrical cone-shaped conformation with three ester carbonyl groups oriented toward the cavity, forming a pumpkin-shaped binding pocket (8.0 Å diameter, 4.8 Å height) (Scheme 61b).
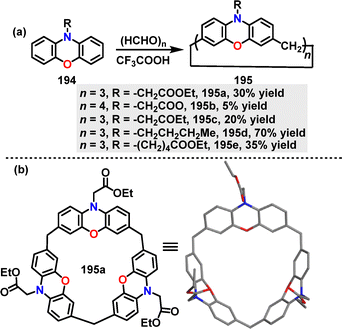 |
| | Scheme 61 (a) Synthesis of calix[n]phenoxazines 195 and (b) crystal structure of 195a. | |
5.2.2. Brønsted base catalysis.
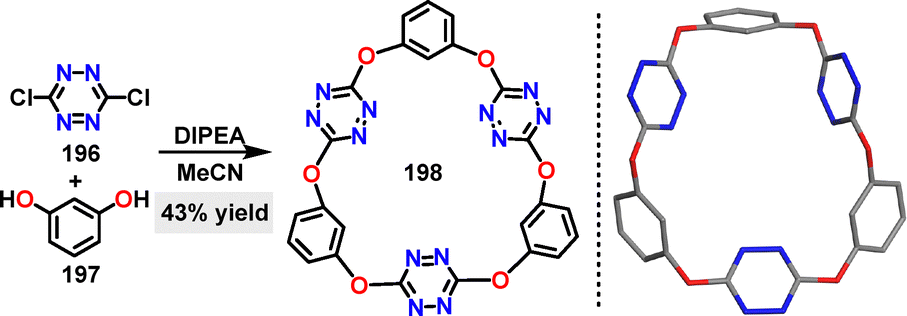
In 2020, Wang reported a novel N,O-doped macrocycle via a one-pot nucleophilic aromatic substitution reaction.110 Under N,N-diisopropylethylamine (DIPEA) conditions, the trimeric macrocycle 198 was generated in 43% yield through C–O bond formation. Crystal structure analysis revealed that 198 adopted a nearly planar hexagonal arrangement with three tetrazine centroids forming an equilateral triangle (Scheme 62).
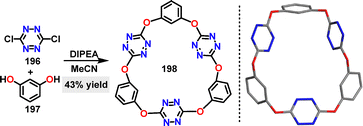 |
| | Scheme 62 Synthesis and crystal structure of macrocycle 198. | |
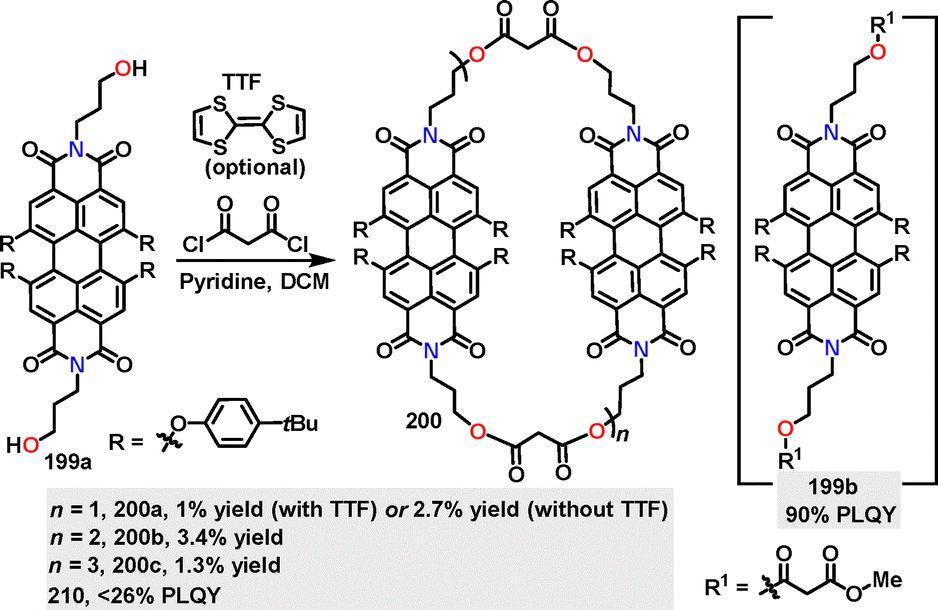
In 2021, Hirsch111 reported the synthesis of perylene bisimide cyclophane macrocycles 200via pyridine-promoted macrocyclization (n + n, n = 2, 3, 4). Macrocycles 200 with diverse ring sizes and flexible malonate linker moieties were obtained in 8.1%, 3.4%, and 1.3% yields, respectively (Scheme 63). Notably, employing electron-donating tetrathiafulvalene (TTF) as a template enhanced the yield of 200 from 2.7% to 8.1%. Monomeric 199b displayed a vibrationally resolved absorption spectrum with a maximum at 603 nm and a shoulder at 655 nm, featuring a high PLQY of 90% in toluene. This behavior is attributed to minimal π–π interactions and efficient radiative decay. In contrast, macrocycles 200 exhibited a reduced PLQY (<26%) due to π–π stacking-induced excited-state quenching, accompanied by emission maxima redshifted to 610–615 nm.
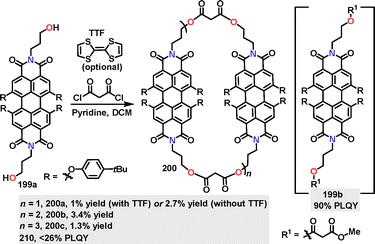 |
| | Scheme 63 Synthesis of perylene bisimide cyclophane macrocycles 200. | |
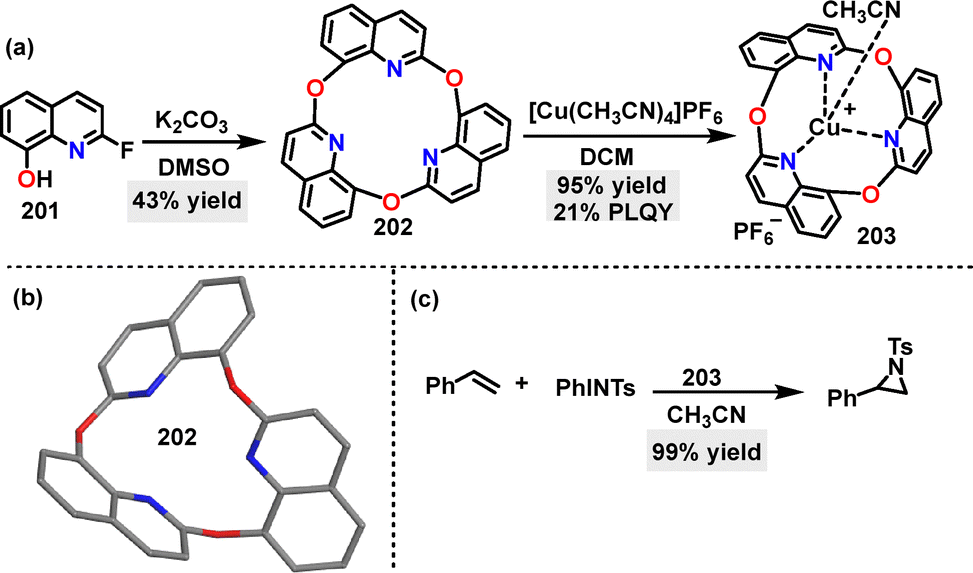
In 2023, Kumagai112 reported the synthesis of a novel oxa-triquinoline 202, a macrocycle incorporating three quinoline units and three ether O-atoms in 43% yield, via one-pot trimerization of a 2-fluoro-8-quinolinol monomer 201 under K2CO3 conditions (Scheme 64a). Crystal structure analysis revealed a bent molecular conformation of 202 (Scheme 64b). Notably, macrocycle 202 coordinated with [Cu(CH3CN)4]PF4 in DCM, inducing a bowl-shaped geometry and forming the 203 complexes in 95% yield. Moreover, this complex served as an effective catalyst, competently mediating the reaction between styrene and PhINTs (Scheme 64c).
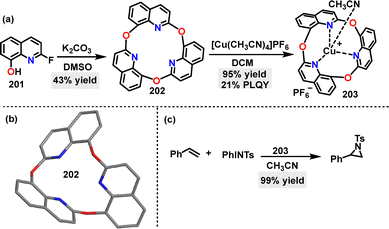 |
| | Scheme 64 (a) Synthesis of oxa-triquinoline 202 and Cu-complex 203, (b) crystal structure of 202, and (c) the catalytic activity of complex 203. | |
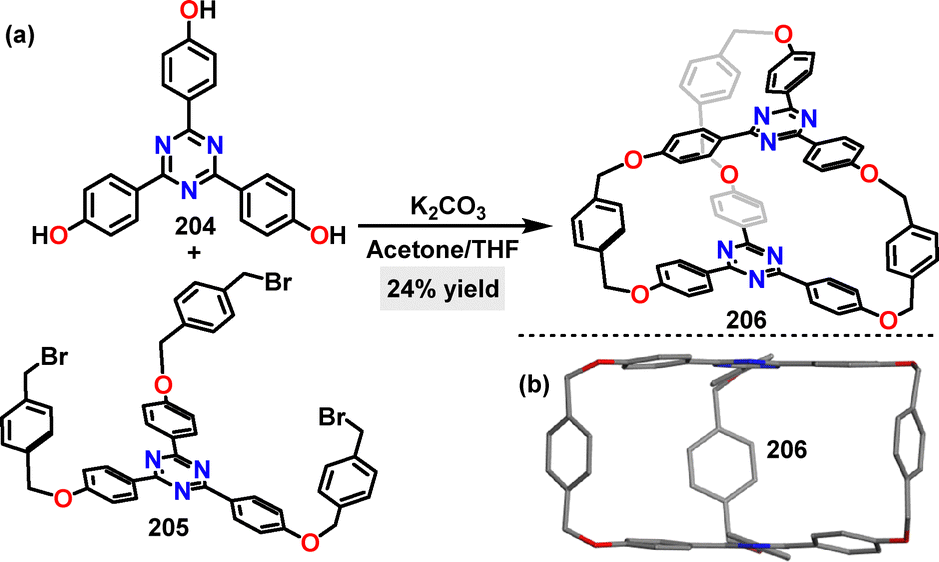
In the same year, Chou113 reported the synthesis of a macrocyclic acceptor-cage in 24% yield via K2CO3-mediated F–C alkylation (Scheme 65a). Crystal structure analysis of the cage-macrocycle 206 revealed a threefold-symmetric structure featuring two triphenyltriazine “lids” flanked by three xylylene bridges (Scheme 65b).
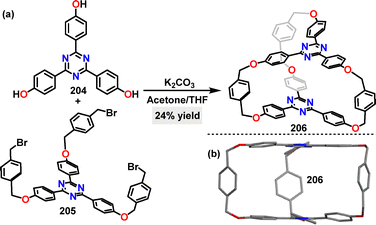 |
| | Scheme 65 (a) Synthesis and (b) crystal structure of cage-macrocycle 206. | |
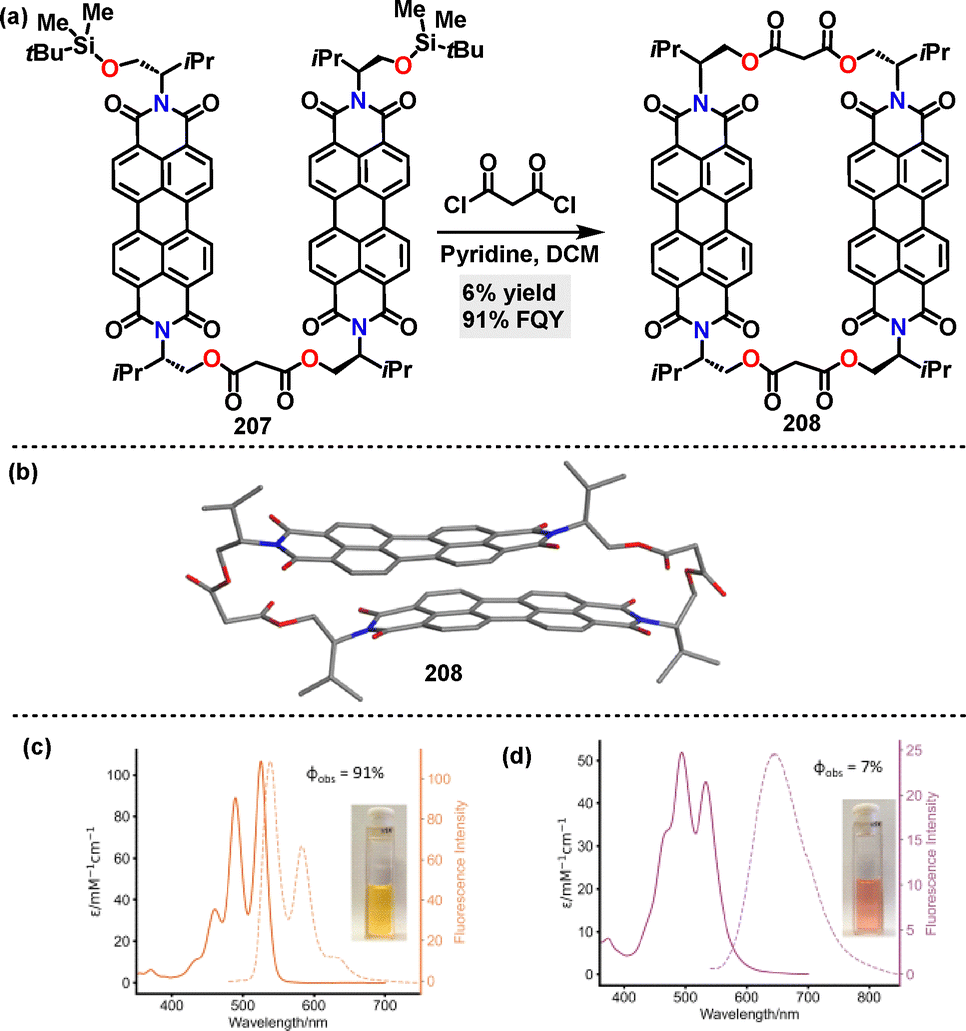
Recently, Barendt114 reported the synthesis of a chiral bis-perylene diimide macrocycle 208 in 6% yield via a [2+2] macrocyclization reaction using pyridine as the base (Scheme 66a). Crystal structure analysis of macrocycle 208 revealed a closed conformation with D2 symmetry, characterized by a π–π stacking distance of 3.3 Å (Scheme 66b). This structure was stabilized by intramolecular π–π interactions and solvent molecules encapsulated within the cavity. Fluorescence spectroscopy revealed solvent and guest-dependent emission behavior for macrocycle 208. In chloroform, the monomeric state exhibited high FQY (91%) and a small Stokes shift (13 nm) (Scheme 66c). In water, intramolecular π–π stacking induced the formation of an excimer, leading to broad emission with a large Stokes shift (130 nm) and significantly quenched emission (7%) (Scheme 66d).
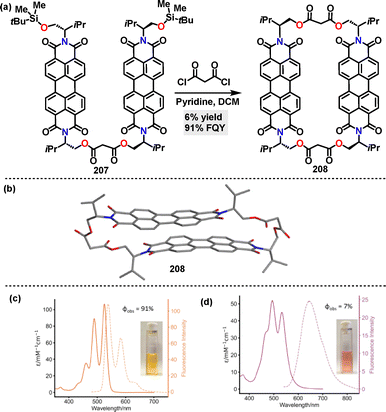 |
| | Scheme 66 (a) Synthesis and (b) crystal structure of chiral bis-perylene diimide macrocycle 208, (c) absorption and emission spectra of macrocycle 208 in CHCl3, and (d) absorption and emission spectra of macrocycle 208 in H2O. Reproduced from ref. 114 with permission from Wiley-VCH, copyright 2025. | |
5.2.3. Other conditions.
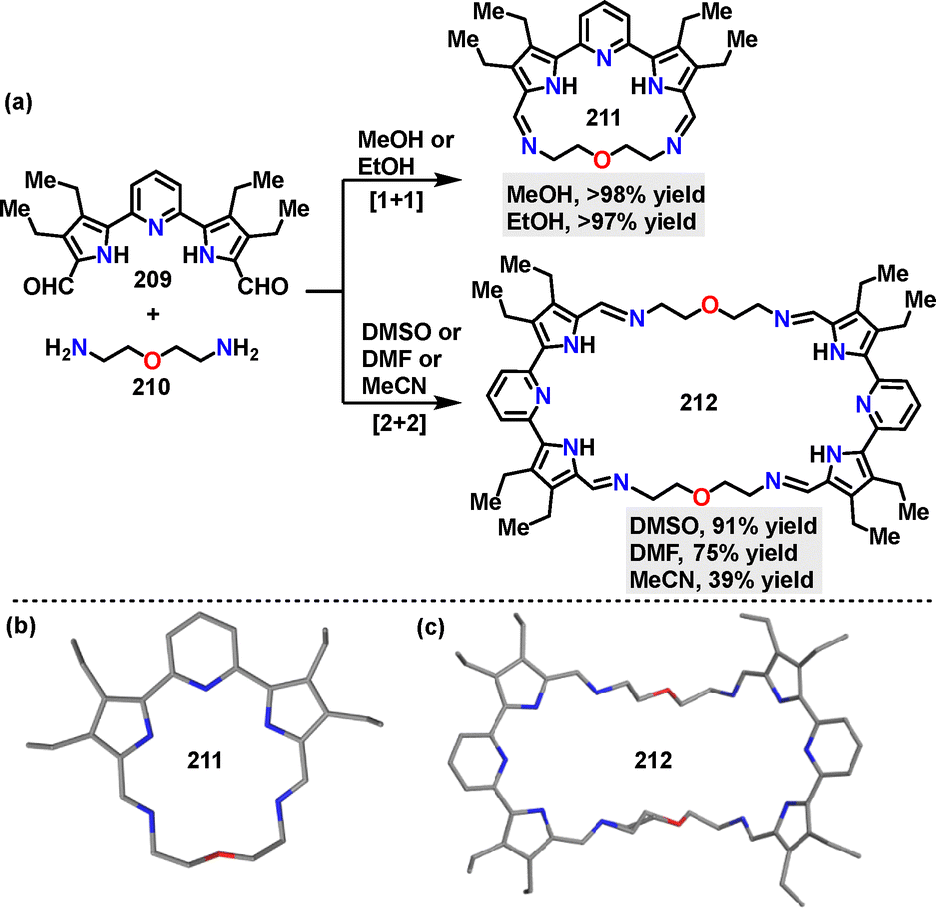
In 2023, He115 reported a one-pot strategy for constructing self-assembled macrocycles, where solvent polarity and solubility dictate the selective formation of distinct macrocycle architectures. Upon condensing dialdehyde and diamine, the desired [1+1] macrocycle 211 was obtained in 97% yield in MeOH, or in 98% yield in EtOH. Conversely, performing the same condensation in DMSO, DMF, or MeCN yielded the [2+2] macrocycle 212 in 91%, 75%, and 39% yields, respectively (Scheme 67a). Crystal structure analysis revealed that the [1+1] macrocycle 211 adopted a near-planar conformation with a bound water molecule in its cavity, stabilized by hydrogen bonds between pyrrolic NH protons and imine nitrogen atoms (Scheme 67b). In contrast, the [2+2] macrocycle 212 exhibited a chair-like geometry (Scheme 67c).
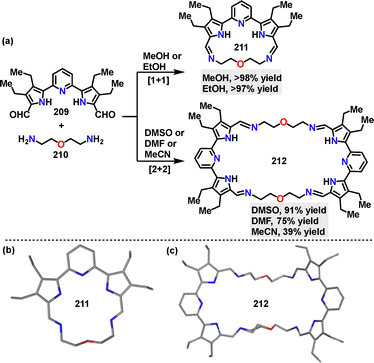 |
| | Scheme 67 (a) Synthesis and (b) and (c) crystal structures of chiral bis-perylene diimide macrocycles 211 and 212. | |
5.3. Metal catalysis and N,S-doped macrocycles
5.3.1. Palladium catalysis.
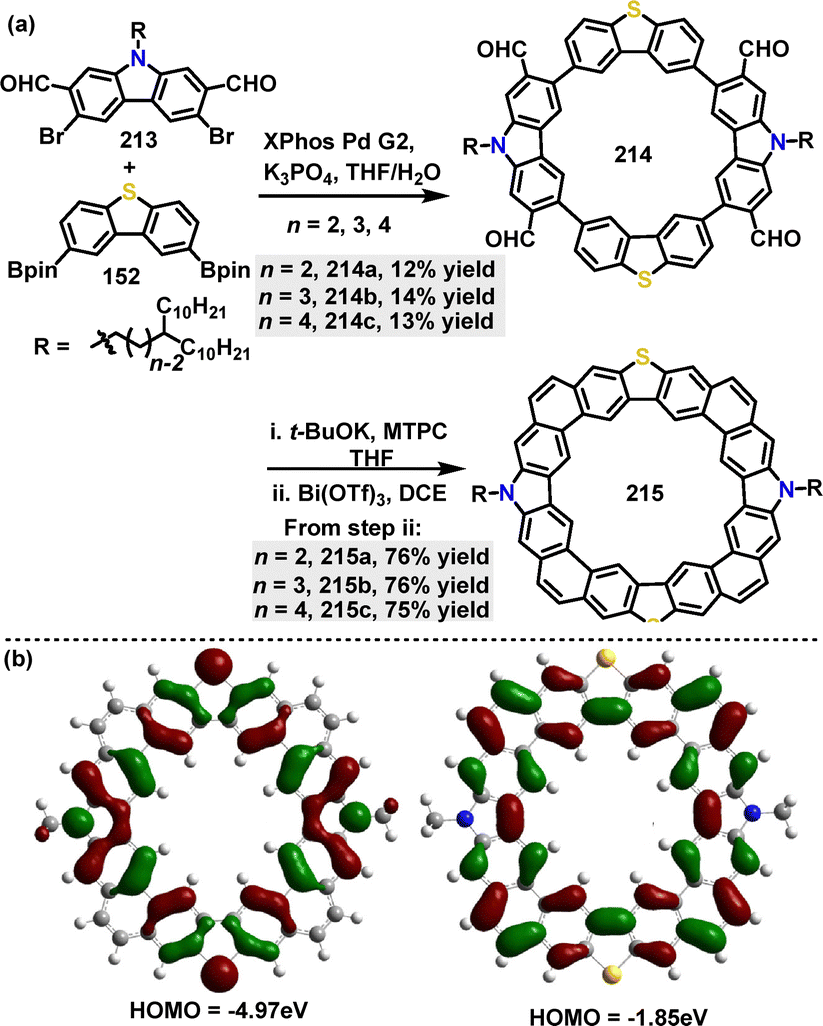
In 2022, Liu, Lu, Zhao and co-workers116 reported an efficient method for the construction of N,S-codoped macrocycles featuring branched alkyl substituents. Palladium-catalyzed Suzuki coupling reactions yielded diverse functional macrocycles 214 containing alternating dibenzo[b,d]thiophene and carbazole units in 12–14% yields. Furthermore, alkyl-substituted fully fused macrocycles 215 were obtained in 75–76% yields via sequential base-catalyzed Wittig reactions and Bi(OTf)3-catalyzed cyclization (Scheme 68a). DFT calculations revealed that the electron-rich N-atoms in these planar macrocycles elevate the energies of both the HOMO (−4.97 eV) and the LUMO (−1.85 eV), reducing the energy gap to 3.12 eV and identifying them as promising candidates for p-type semiconductors (Scheme 68b).
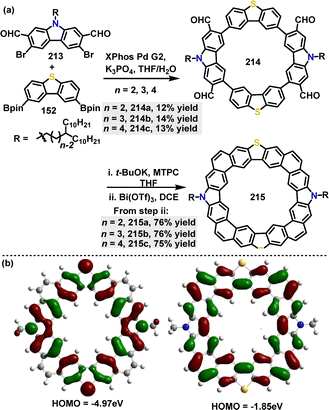 |
| | Scheme 68 (a) Synthesis and modification of N,S-codoped macrocycle 214 and (b) Frontier orbitals and energy levels of this series of N,S-embedded macrocycles 225. Reproduced from ref. 116 with permission from American Chemical Society, copyright 2022. | |
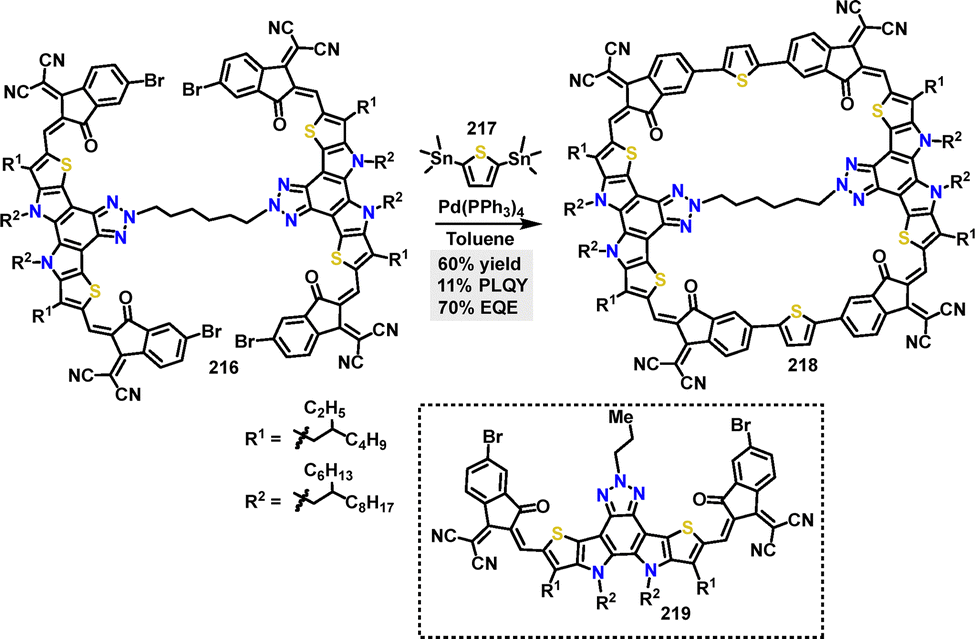
In 2023, Yuan117 reported the synthesis of a cyclic conjugated macrocycle 218 featuring a multicomponent D–A architecture. The π-conjugated macrocycle 218 was efficiently synthesized in 60% yield via a palladium-catalyzed Stille coupling reaction (Scheme 69). Macrocycle 218 demonstrated a high PLQY of 11% in solution and 4% in the solid state, significantly higher than its linear analogue 219 (2%). This enhancement was attributed to reduced molecular vibrations enabled by the rigid macrocyclic structure, which suppressed nonradiative decay. TGA revealed superior thermal stability for macrocycle 218, with 5% weight loss occurring at 393.8 °C (vs. 330.3 °C for 219).
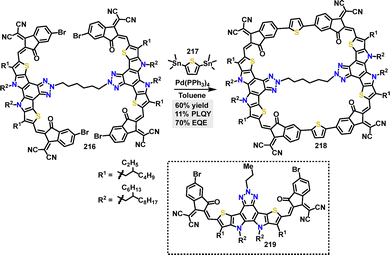 |
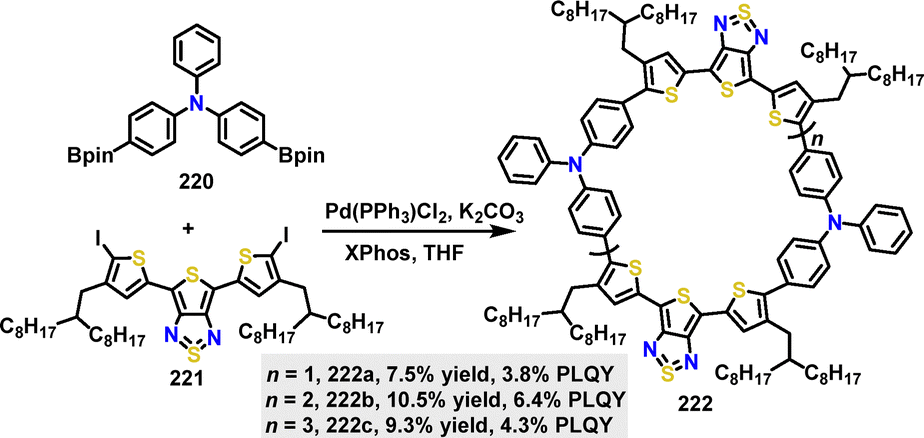
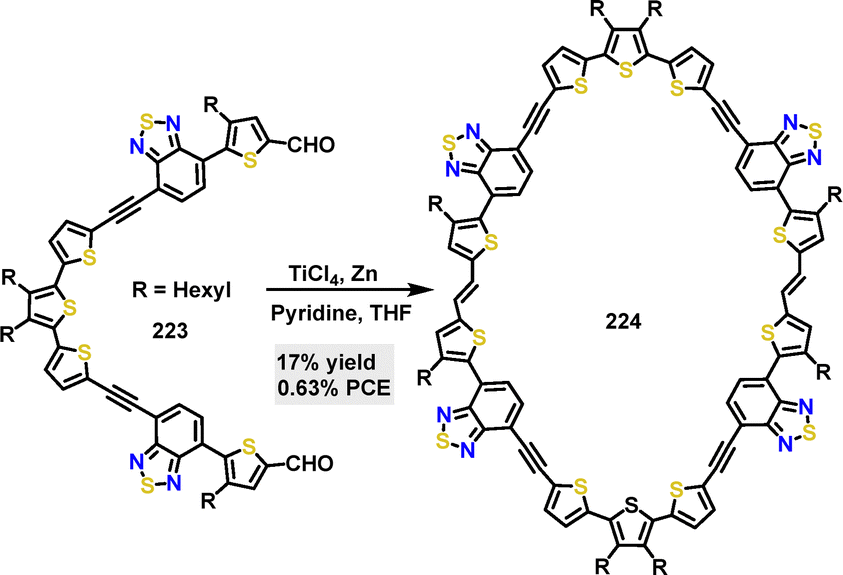
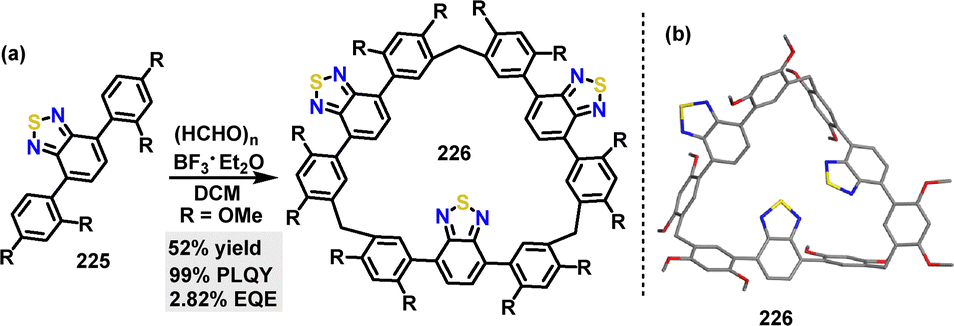
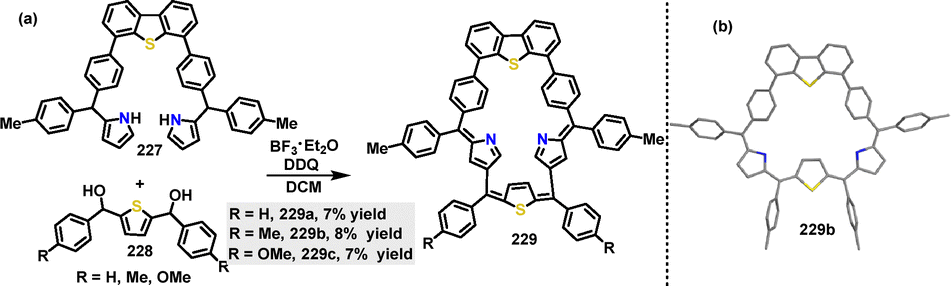
| | Scheme 69 Synthesis of cyclic conjugated macrocycle 218. | |
In 2024, Wan118 and co-workers reported a series of D–A macrocycles 222via a one-pot Suzuki cross-coupling reaction. Under palladium catalysis, D–A dimeric macrocycle 222a, trimeric macrocycle 222b, and tetrameric macrocycle 222c were selectively formed in [2+2], [3+3], and [4+4] cyclization pathways with yields of 7.5%, 10.5%, and 9.3%, respectively (Scheme 70). The trimeric macrocycle 222b demonstrated an optimal balance between ring strain and conformational flexibility, achieving the highest PLQY of 6.4% due to mixed conformations and allowed electronic transitions. Smaller macrocycle 222a suffered from high ring strain, resulting in lower PLQY (3.8%), while larger tetramers 222c showed moderate PLQY (4.3%) attributed to the stability of transoid conformations.
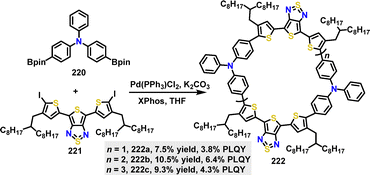 |
| | Scheme 70 Synthesis of D–A macrocycle 222. | |
5.3.2. Titanium catalysis.
In 2021, Cooke119 reported the synthesis of a thiophene-based conjugated macrocycle via a one-pot Sonogashira reaction. Using a TiCl4/Zn reagent system, the D–A macrocycle 224 featuring electron-deficient benzothiadiazole (BT) units was obtained in 17% yield (Scheme 71).
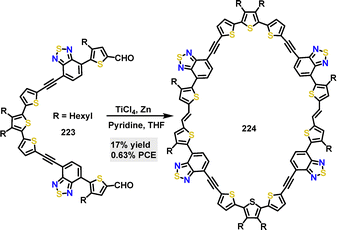 |
| | Scheme 71 Synthesis of D–A macrocycle 224. | |
5.4. Metal-free and N,S-doped macrocycles
5.4.1. Lewis acid catalysis.
In 2022, Li120 reported an efficient synthetic method for constructing a benzothiadiazole(BT)-based macrocycle. The target macrocycle 226, incorporating a dimethoxyphenyl donor and 2,1,3-benzothiadiazole acceptor, was synthesized in 52% yield via a one-pot condensation of BT precursors 225 with paraformaldehyde, using BF3·Et2O as a Lewis acid catalyst (Scheme 72a). Crystal structure analysis revealed that macrocycle 226 adopted a rigid triangular conformation, where C(sp3) bridges restrict the rotation of benzothiadiazole and phenyl groups, thereby reducing conformational flexibility (Scheme 72b). Macrocycle 226 demonstrated an exceptionally high PLQY of 99%, significantly exceeding that of monomer 225 (65%). Additionally, macrocycle 226 showed a longer fluorescence lifetime (11.25 ns) than monomer 225 (8.45 ns), indicating reduced nonradiative relaxation in the macrocycle.
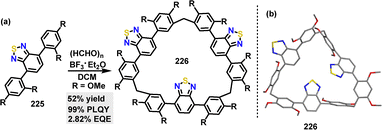 |
| | Scheme 72 (a) Synthesis and (b) crystal structure of benzothiadiazole-based macrocycle 226. | |
In 2023, Ravikanth121 reported the synthesis of DBTh-containing thiacarbaporphyrinoids under acidic conditions. Macrocycles 229 were obtained in 7–8% yields via the condensation of pentapyrranes 227 with thiophene diols 228 in the presence of BF3·OEt2, followed by DDQ-mediated oxidation (Scheme 73a). Crystal structure analysis of compound 229b revealed a nonplanar macrocyclic structure (Scheme 73b).
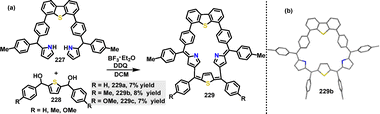 |
| | Scheme 73 (a) Synthesis of thiacarbaporphyrinoids 229 and (b) crystal structure of 229b. | |
In 2024, Lin122 reported the synthesis of a calix[3]phenothiazine macrocycle 231 featuring rigid cavitand structures for guest encapsulation. Under BF3·OEt2 catalysis, this macrocyclic donor molecule was obtained in 47.1% yield via a one-pot cyclization reaction (Scheme 74).
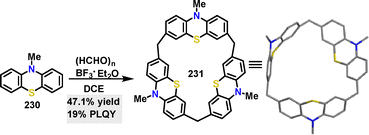 |
| | Scheme 74 Synthesis and crystal structure of macrocycle 241. | |
In 2025, Gokulnath123 designed and synthesized two rigid dithienopyrrole (DTP)-based macrocycles 233 and 234via a macrocyclization reaction. Under BF3·OEt2/DDQ conditions, the cyclotrimer 233 and cyclotetramer 234 were obtained in 10% and 4% yields, respectively (Scheme 75a). Crystal structure analysis of 233 revealed a fully planar conformation, with pyrrolic N-atoms and DTP S-atoms oriented toward the macrocyclic core (Scheme 75b).
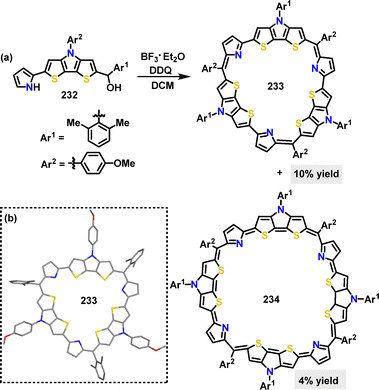 |
| | Scheme 75 (a) Synthesis of dithienopyrrole-based macrocycles 233 and 234, and (b) crystal structure of 233. | |
5.4.2. Brønsted base catalysis.
In 2023, Zhu124 reported an efficient one-pot cyclization of 235 with 1,4-dibutoxy-2,5-dibromomethylbenzene 236 for the synthesis of a pyrrolodithiin-derived box-like cyclophane 237. Using NaH as the base, macrocycle 237 was obtained in 21% yield (Scheme 76). Crystal structure analysis revealed that macrocycle 237 adopted a hex-nut-shaped geometry featuring a central cavity (width: 8.95 Å), which is capable of guest encapsulation.
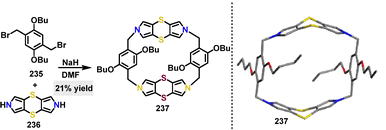 |
| | Scheme 76 Synthesis and crystal structure of 237. | |
5.4.3. Other conditions.
In 2021, Würthner125 reported a cross-coupling strategy for constructing macrocyclic architectures incorporating perylene bisimide (PBI) units. Macrocycles 240a and 240b were synthesized via a one-pot protocol in 30% and 4% yields, respectively (Scheme 77a). The lower yield of 240b was attributed to its higher strain energy (30.6 kJ mol−1) compared to 240a (13.9 kJ mol−1). Crystal structure analysis of 240b revealed oligothiophene bridges adopting an all-syn, twisted S-shape that encloses a planar PBI core (Scheme 77b). This geometry exhibited moderate strain (30.6 kJ mol−1), originating from thiophene torsion angles ranging from 68° to 158°, and featured symmetric and slightly deformed molecular motifs. S-atoms in the thiophene units point toward the PBI core, with close S–S distances of 3.37 Å. In UV-vis spectroscopy, the strong, low-energy absorption bands of 240a and 240b at 530–536 nm (54000–64800 M−1 cm−1; Scheme 77c), originating from their PBI core and absent in 241 and 242, collectively demonstrate the pivotal role of ring strain in both the synthesis and the resulting structure–absorption relationship of PBI-based oligothiophene macrocycles.
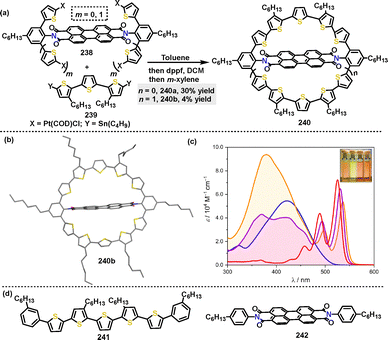 |
| | Scheme 77 (a) Synthesis of perylene bisimide-based macrocycle 240, (b) crystal structure of 240b, (c) UV-vis absorption spectra of 240a (purple line), 240b (orange line), 241 (blue line) and 242 (red line) in DCM, and (d) structures of compounds 241 and 242. Reproduced from ref. 125 with permission from Wiley-VCH, copyright 2022. | |
6. Applications of heteroatom-doped macrocycles
Benefiting from their heteroatom-doped architectures, novel macrocycles exhibit intriguing host–guest properties, enabling the selective recognition of diverse neutral, cationic, and anion molecules. Furthermore, their tunable photophysical characteristics allow them to serve as highly selective chemosensors, operating via distinct fluorescence turn-on or turn-off mechanisms. Beyond molecular recognition and sensing, these functional macrocycles have also found broad range of applications in organic electronic devices (e.g., OLEDs, OFETs, and OSCs), catalysis, and molecular adsorption and separation applications.
6.1. Molecular recognition
6.1.1. Recognition of neutral guests.
The design of synthetic macrocyclic hosts capable of selective molecular recognition is a central theme in supramolecular chemistry. In 2022, Komori reported the host–guest properties of acridone-incorporated arylene-ethynylene macrocycles 14a, with a particular focus on their ability to bind dihydric phenolic guests (G) via hydrogen bonding (Fig. 2a).31 The cyclic trimer 14a, which featured a planar hexagonal cavity lined with three carbonyl groups, was found to form 1:1 host–guest complexes with phenolic species such as hydroquinone (HQ) of binding affinity (Ka) (1.00 ± 0.09) × 103) and resorcinol (RE) of Ka (0.99 ± 0.14) × 103).
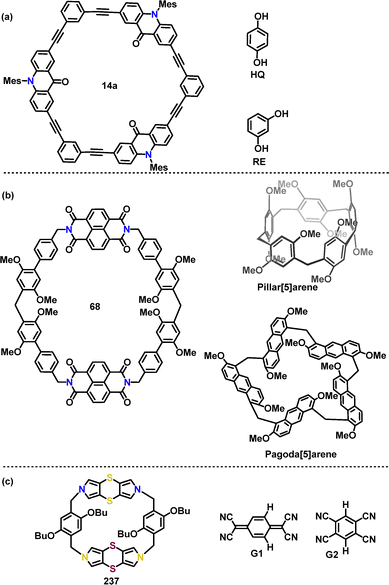 |
| | Fig. 2 Structures of (a) host 14a and guests HQ, RE, (b) host 68 and guests pillar[5]arene, pagoda[5]arene, (c) host 237 and guests G1 and G2. | |
In 2023, Zeng's group designed a giant electron-deficient macrocycle, naphthalene diimide-extended-pillar[6]arene 68. In contrast to its negligible interaction with pillar[5]arene, macrocycle 68 displayed exceptional selectivity for pagoda[5]arene, forming a 1:1 inclusion complex with a Ka of (1.77 ± 0.04) × 105 M−1 (Fig. 2b). The complexation process was marked by a significant color change and the rise of a new broad absorption feature at 600–800 nm, underscoring the formation of a charge-transfer complex stabilized by strong intermolecular interactions.47
Later, Zhu demonstrated the host–guest binding behavior of macrocycle 237, exhibiting remarkable adaptability toward electron-deficient guests G1 and G2 with varying sizes and geometries (Fig. 2c), with Ka values of 1.2 × 104 M−1 and 1.6 × 104 M−1, respectively (Scheme 72c).124 In the presence of G2, the cyclic voltammetry (CV) profile of 237 changed significantly (from −0.50 to 0.60 V), indicating a two-electron oxidation process that led to the release of the guest due to charge repulsion. This redox-triggered dissociation demonstrated the potential of macrocycle 237 as a switchable molecular host for controllable supramolecular systems.
In 2024, Chen, Lin, and coworkers introduced a pioneering supramolecular strategy to construct efficient organic scintillators, moving beyond traditional covalent synthesis by leveraging programmable host–guest interactions.122 When complexed with guests G3 and G4 (Fig. 3a), macrocycle 231 formed supramolecular co-crystals G3@231 (Ka: 540 M−1) and G4@231 (Ka: 2.9 × 104 M−1) that exhibited TADF properties (Fig. 3c). G3@231 emitted green light at 541 nm with CIE coordinates (0.36, 0.56), whereas G4@231 displayed orange emission at 633 nm (CIE: 0.39, 0.58). The red-shifted emission in G4@231 was attributed to the heavy-atom effect of bromine in the guest molecule (Fig. 3b). Notably, under N2, the PLQY values of G3@231 (73%) and G4@231 (34%) were significantly higher than that of macrocycle 231 (19%). This enhancement arose from molecular structure rigidification and suppression of nonradiative decay mediated by strong noncovalent host–guest interactions.
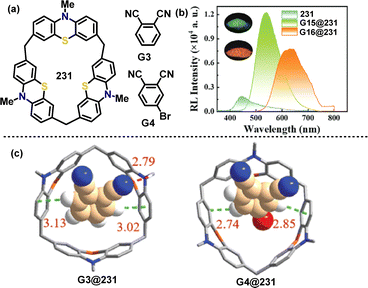 |
| | Fig. 3 (a) Structures of host 231 and neutral guests G3 and G4, (b) radioluminescence spectra of 231, G3@231, and G4@231, and (c) diagram of crystal structures of G3@231 and G4@231. Adapted from ref. 122 with permission from Wiley-VCH, copyright 2024. | |
Recently, Wong and Chou presented a detailed thermodynamic analysis of inclusion complexes between a macrocyclic, triazine-based electron-accepting host 206 and various electron-donating guests, with Ka ranging from 852 M−1 to 1.5 × 103 M−1 (Fig. 4a). Moreover, G5@206 was found to be an endothermic and entropy-driven process, which was attributed to the release of solvent molecules (e.g., DCM) from the host cavity upon guest inclusion.113
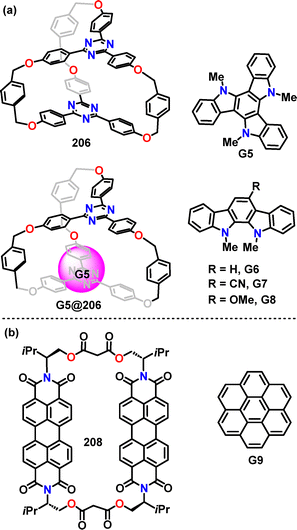 |
| | Fig. 4 Structures of (a) 206, G5@206, and guests G5–G8, (b) 208, and guest G9. | |
In 2025, Barendt presented the intrinsic cavity and conformational flexibility of the chiral bis-perylene diimide (PDI) macrocycle 208, making it an exceptional host for studying tunable host–guest interactions.114 Macrocycle 206 demonstrated strong binding to G9 (Ka: 3.8 × 104 M−1) through cavity-based π–π stacking, forming 1:1 host–guest complexes (Fig. 4b). These complexes exhibited charge-transfer exciplex emission with a significant emission shift (116 nm), highlighting the macrocycle's ability to modulate photophysical properties through guest inclusion.
The recognition of fullerenes such as C60 and C70 by macrocyclic hosts is well-established. In 2020, Yang and coworkers reported the saddle-shaped, S-containing heterocycloarene S-octulene 154.88 Its distinctive three-dimensional geometry, which possessed a vertical extent of 4.6 Å, provided a complementary cavity for the curved surfaces of fullerenes. This structural complementarity was quantitatively confirmed by binding studies in toluene, which revealed a higher association constant for C60 (Ka: 1.25 × 106 M−1) than that for C70 (Ka: 9.49 × 106 M−1), indicating a discernible preference and stronger host–guest interaction with C60 (Fig. 5a).
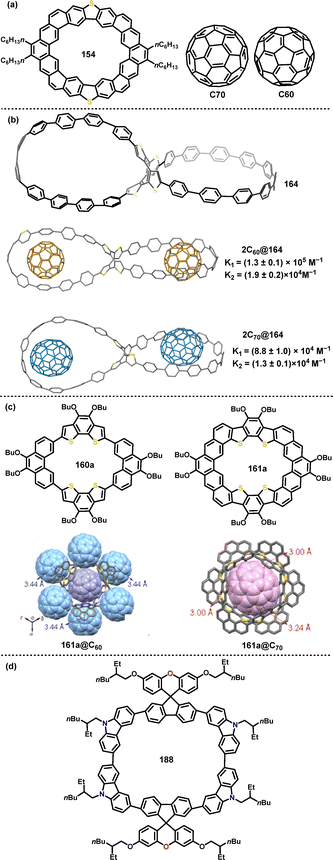 |
| | Fig. 5 Structure of (a) 154 and guests C60, C70, (b) 164, and co-crystals 2C60@164, 2C70@164, (c) 160a, 161a, and co-crystals 161a@C60, 161a@C70, (d) 188. Reproduced from ref. 90 with permission from The Royal Society of Chemistry, copyright 2024. | |
Later, Zhan introduced the fully conjugated nanohoop 164, which featured a unique architecture combining strained oligoparaphenylene loops and a flexible COTh core.91 This synergy endowed it with guest-adaptive cavities, a property spectacularly demonstrated by X-ray structures of its 2C60@164 and 2C70@164 complexes (Fig. 5b). These peanut-like assemblies constituted the first isolable 1:2 complexes of their kind, wherein the host's cavities underwent distinct unsymmetrical distortions to encapsulate the different fullerenes, stabilized by significant π–π interactions between the complementary curved surfaces.
In 2024, Liu, Lu and coworkers investigated the host–guest properties of π-conjugated macrocycle 160a and its fully fused heterocycloarene analogue 161a, revealing pronounced size- and geometry-dependent binding preferences toward C60 and C70.90 For instance, 160a showed stronger binding to C70 (Ka: 2.44 × 105 M−1) than to C60, attributed to better geometric complementarity. Conversely, 161a exhibited higher affinity for C70 (Ka: 1.62 × 105 M−1) due to its bowl-shaped cavity, which accommodated the ellipsoidal C70 guest (Fig. 5c). In the same year, Singh and co-workers reported macrocycle 188, an ambipolar system constructed from alternating spirofluorene-9,9′-xanthene (SFX) and carbazole units.106 This nanoring possessed a well-defined cavity with internal dimensions suitable for guest encapsulation, and it exhibited strong host–guest binding with C60 (Ka: 9.95 × 104 M−1), which was primarily mediated by noncovalent interactions (Fig. 5d).
6.1.2. Recognition of cationic guests.
In 2021, Qu and Feringa introduced two macrocyclic hosts by integrating a first-generation molecular motor into a crown-ether-like structure.84 The stable-cis-148b, adopting a folded conformation, exhibited strong and selective 1:1 binding with dialkylammonium guests (e.g., Ka: 219.0 M−1 for G10, Ka: 162.4 M−1 for G11) via hydrogen bonds and ion–dipole interactions (Fig. 6a). In stark contrast, the stable-trans-148b, which adopted a more linear and stretched conformation, showed negligible binding affinity (Ka < 1 M−1) for the same guests. In the same year, Li reported that macrocycle 136 exhibited selective binding for cationic guests (Fig. 6b),79 driven by cation–dipole interactions (between cationic centers and electron-rich furan O-atoms) and CH–π interactions (between alkyl protons of the guests and π-electron clouds of the furan rings). For instance, G12 binds most strongly due to its optimal size, which fitted well within the macrocycle's cylindrical cavity of (4.1 ± 1.4) × 103 M−1 (Ka). In contrast, G13 was too large, leading to steric repulsion and a weaker binding Ka of (1.2 ± 0.2) × 103 M−1.
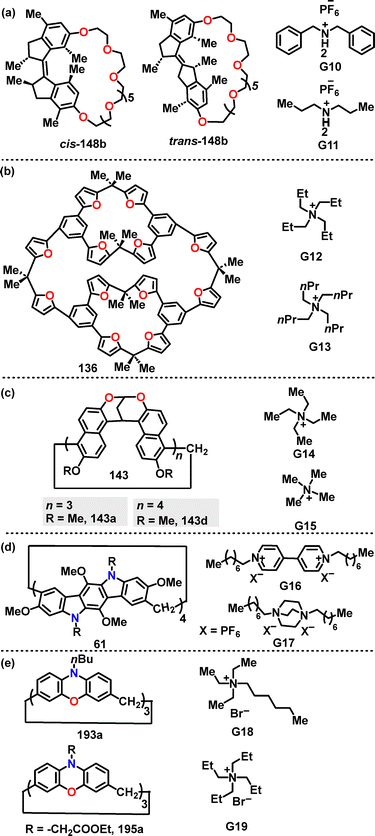 |
| | Fig. 6 Structures of cis-148b, trans-148b, 136, 143a, 143d, 61, 193a, and 195a and cationic guests G10–G19. | |
Later, the group of Jiang systematically investigated the host–guest complexation behaviour of methylene-bridged naphthotubes (Fig. 6c).82 For example, 143a exhibited strong binding affinity toward organic cations G14 and G15 (e.g., Ka: (1.7 ± 0.2) × 105 M−1 for G14; (5.1 ± 0.2) × 103 M−1 for G15), attributed to its rigid, electron-rich cavity (Scheme 41d). In contrast, 143d showed weaker binding (Ka: 102–103 M−1 for G14) due to double-cavity distortion and charge repulsion.
In 2023, Yang systematically investigated the host–guest chemistry of N-embedded cubarene 61 (Fig. 6d).43 The macrocycle was found to form stable, intramolecular complexes with a series of quaternary ammonium salts. Notably, high binding affinities were quantified for specific guests, with Ka values reaching (5.33 ± 0.70) × 105 M−1 for G16 and (5.31 ± 0.50) × 105 M−1 for G17.
Recently, Ma and colleagues demonstrated that the cone-shaped calix[3]phenoxazine 193a formed 1:1 host–guest complexes with quaternary ammonium salts, leveraging its electron-rich, well-defined cavity (Fig. 6e).108 The binding affinity was critically dependent on guest size and structure, with the Ka for ammonium salt G18 (featuring a trimethylammonium head and a hexyl chain) being (1.66 ± 0.15) × 103 M−1. In sharp contrast, the significantly weaker binding for the smaller guest G19 (14.6 ± 1.1) M−1) underscored this pronounced size selectivity. Building on this, the same group engineered an ester-functionalized analogue 195a, in which the pendant carbonyl groups served as auxiliary binding sites.109 This strategic modification yielded a superior binding affinity (2.61 ± 0.22) × 103 M−1) with guest G19 compared to the non-ester parent macrocycle 193a, underscoring the efficacy of side-chain functionalization in enhancing molecular recognition.
6.1.3. Recognition of anion guests.
In 2020, Wang reported a novel N,O-doped macrocycle 198.110 It showed strong selectivity for nitrate (NO3−) (Ka ≈ 2.2 × 105 M−1) through a combination of anion–π interactions and hydrogen bonding, forming an [NO3·198]− complex (Fig. 7a). Later, He and colleagues demonstrated that cis-127 exhibited high selectivity and a Ka of 1.93 × 103 M−1 for the hydrogen pyrophosphate (HPPi) anion (Fig. 7b), whereas trans-128 showed considerably weaker Ka (2.82 × 102 M−1) for HPPi.68 DFT calculations revealed that V-shaped cis-127 perfectly complemented the HPPi anion by multiple cooperative hydrogen bonds, engaging all available hydrogen bond donors in the macrocycle. In contrast, the bow-shaped conformation of trans-128 was far less complementary, resulting in fewer hydrogen bonds and a significantly weaker interaction with the guest.
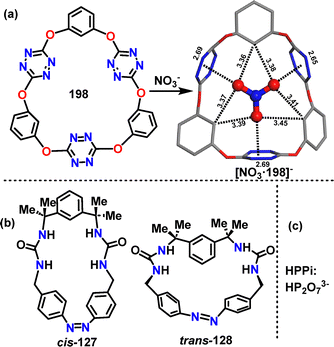 |
| | Fig. 7 (a) Single-crystal X-ray molecular structures of 198 complexes with nitrate, (b) structures of cis-127 and trans-128, and (c) anion guest HPPi. | |
6.2. Molecular sensing
6.2.1. Fluorescence turn-on sensing.
In 2021, the Panda group demonstrated that macrocycle 7 served as a highly selective fluorescent sensor for the fluoride ion (F−) in acetonitrile (Fig. 8).28 Upon complexation with F−, the host exhibited a distinct “turn-on” fluorescence response with an emission maximum at 421 nm. This signal enhancement was accompanied by a measured FQY of 30%. Furthermore, the sensor displayed excellent selectivity for F− over a range of competing anions, including Cl−, Br−, I−, and AcO−. Fluorimetric titration experiments and Job's plot analysis revealed that the host–guest complexation follows a 1:1 stoichiometric ratio.
 |
| | Fig. 8 (a) Structure of macrocycle 3, and (b) emission spectra of its fluoride complex in acetonitrile. Adapted from ref. 28 with permission from American Chemical Society, copyright 2021. | |
In 2023, Gong reported a novel tetra-cyanostilbene-based macrocycle 146 that functioned as a “turn-on” blue fluorescence response for sensing oxalic acid (OA) in aqueous media (Fig. 9).83 This behavior was attributed to restricted intramolecular rotation (RIR), a characteristic of the AIE effect. The absolute FQY increased from 9% (free 146) to 55% upon binding with OA. DFT calculations further clarified that the HOMO–LUMO energy gap decreased from 3.50 eV (free 146) to 3.33 eV (146-OA complex), accounting for the observed fluorescence red shift.
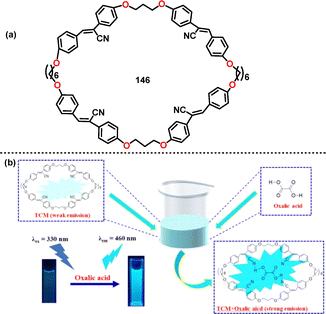 |
| | Fig. 9 (a) Structure of macrocycle 146 and (b) the proposed detecting mechanism of 146 for oxalic acid. Reproduced from ref. 83 with permission from Elsevier, copyright 2023. | |
In 2024, Keypour and co-workers developed a novel pyrrole-based macrocyclic Schiff base 121 that served as a highly selective fluorescent sensor for Cd2+ ions (Fig. 10a).65 Coordination with Cd2+ triggered a distinct “turn-on” response, marked by a 3-fold enhancement in emission intensity at 430 nm, with excellent selectivity over 18 competing metal ions. Later, the group of Yang and Jiang developed a novel D–A phenazine-based macrocycle that served as a dual-channel fluorescent sensor for transition metal ions.32 While most tested ions quenched its near-infrared (NIR) emission, the addition of Fe3+ triggered a distinct turn-on response, marked by a strong, blue-shifted emission band at 545 nm (Fig. 10b).
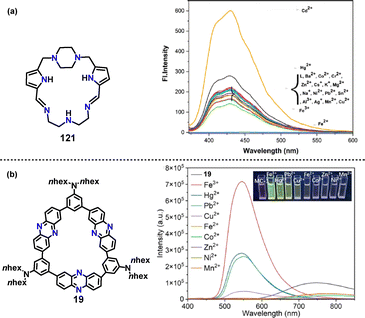 |
| | Fig. 10 Structures and fluorescence responses of macrocycles (a) 121 and (b) 19 upon interaction with different metal ions. Reproduced from ref. 65 with permission from Elsevier, copyright 2024. | |
6.2.2. Fluorescence turn-off sensing.
In 2025, Sessler reported a highly selective fluorescence turn-off sensor for Cu2+ based on a unique diketopyrrolopyrrole (DPP)-centered macrocycle 191a (Fig. 11a).107 The addition of 100 equivalents of other metal ions, including Na+, K+, Ca2+, Mg2+, Zn2+, Fe2+, Ni2+, Cd2+, and Ag+, resulted in negligible changes to its fluorescence (Fig. 11b). In stark contrast, the introduction of only 5 equivalents of Cu2+ led to an almost complete and rapid quenching of the strong green fluorescence (Fig. 11c).
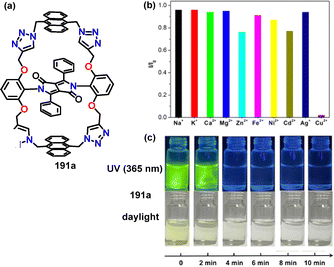 |
| | Fig. 11 (a) Structure of macrocycle 191a, (b) relative fluorescence intensities of 191a measured upon addition of various metal ions, and (c) the addition of 5 equivalents of Cu2+ to acetonitrile solutions of 191a induced a clear color change. Reproduced from ref. 107 with permission from The Royal Society of Chemistry, copyright 2025. | |
6.3. Organic electronic devices
6.3.1. OLEDs.
In 2020, Minakata developed the novel TADF D–A–D–A π-conjugated macrocycle 43.37 An OLED device using macrocycle 43 as the emitter achieved a high EQE of 11.6%, significantly exceeding the 6.9% EQE of devices employing the linear analogue 44 as the fluorescent emitter. This indicated that macrocycle 43 exhibited more efficient TADF than its flexible linear counterpart (Fig. 12a). Later, the group of Yasuda reported the successful implementation of a novel D–A π-conjugated macrocycle 6 as an efficient emitter in OLEDs (Fig. 12b).29 OLEDs incorporating macrocycle 6 achieved an external quantum efficiency (EQE) of up to 15.7%, i.e. four times higher than that of devices using linear 7. This enhanced EQE arises from macrocycle 6's strong TADF capability and favorable molecular orientation in the film, which improves light out-coupling efficiency.
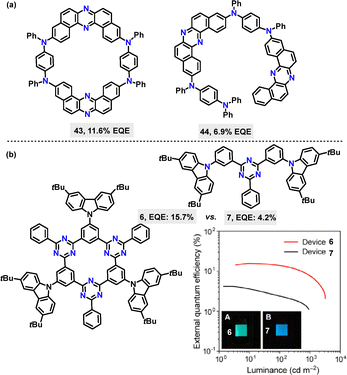 |
| | Fig. 12 (a) Structures of macrocycle 43 and linear analogue 44, (b) structures of macrocycle 6 and linear analogue 7 with their spectra of external EQE. Reproduced from ref. 29 with permission from Wiley-VCH, copyright 2021. | |
In 2022, Zheng, Zang, and co-workers reported a breakthrough in chiral light-harvesting systems that held significant promise for the development of circularly polarized (CP)-OLEDs.62 In the chiral 1,1′-bi-2-naphthol-di-octadecylamide BDA/113/NiR system (Fig. 13), the circularly polarized luminescence (CPL) signal was transferred to NiR, enabling the generation of bright white-light-emitting CPL with CIE coordinates of (0.33, 0.34), a QY of 37.3% and a dissymmetrical factor (glum) of 0.025. Moreover, a prototype white-light-emitting diode (WLED) was constructed by depositing the BDA/TPEM/NiR composite onto a commercial ultraviolet B (UVB) LED chip (λex ≈ 300 nm), which subsequently emitted bright white light upon operation at 5.5 V.
 |
| | Fig. 13 (a) Structures of macrocycle 113, (b) compound NiR and (c) photographs of the WLED assembly: a 300 nm UVB LED is coated with BDA/TPEM/NiR (1 and 2: the coated UVB LEDs are turned off and turned on). Reproduced with permission. Reproduced from ref. 62 with permission from American Chemical Society, copyright 2021. | |
Later, Li, Bai, and Cui introduced a novel macrocyclization-induced emission enhancement (MIEE) to significantly boost the solid-state luminescence efficiency of organic chromophores.120 Owing to its D–A structure, macrocycle 226 exhibited a red-shifted emission at 562 nm (orange-red), compared to 225, which emitted at 491 nm (green) (Fig. 14). An OLED device employing the macrocycle 226 emitter demonstrated superior electroluminescence performance compared to monomer 225, achieving a 47% enhancement in maximum EQE (2.82% for 226vs. 1.92% for 225) and 84% higher peak brightness (4355 cd m−2 for 226vs. 2369 cd m−2 for 225).
 |
| | Fig. 14 Structures of (a) compound 225 and macrocycle 226, and (b) their PL spectra in a solid state. Adapted from ref. 120 with permission from Springer Nature, copyright 2022. | |
In 2023, Wong and Chou reported a groundbreaking strategy for achieving highly efficient TADF in OLEDs through entropy-driven charge-transfer complexation.113 The OLED device incorporating the G10@206 complex displayed bright green electroluminescence (EL) with a main peak at 522 nm (Fig. 15a) and a full-width at half-maximum of 91 nm (at 4 V). It featured a low turn-on voltage (2.6 V) and a maximum EQE of 15.2% (at 4.2 V).
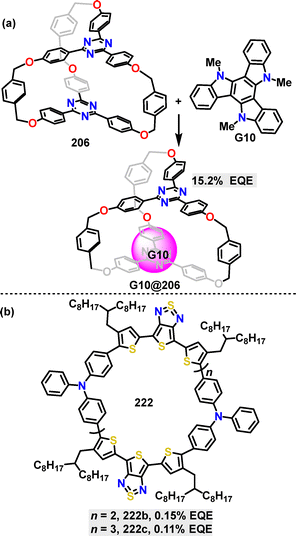 |
| | Fig. 15 (a) Structures of macrocycle 206, G10, and the G10@206 complex and (b) structures of macrocycles 222b–c. | |
Recently, Yi, Wu and Wan reported the fabrication and characterization of near-infrared (NiR) OLEDs based on a series of D–A cyclic oligomers 222 (Fig. 15b).118 Notably, OLEDs incorporating macrocycle 222b achieved efficient NIR electroluminescence exceeding 900 nm, with an EQE of 0.15% and a high radiance of 2897 mW sr−1 m−2. The 222c device showed a slight red-shift at 907 nm and a higher maximum radiance of 2897 mW sr−1 m−2, though with a lower EQE of 0.11%.
6.3.2. OFETs.
In 2020, Lu, Wu, and Liu synthesized a novel saddle-shaped S-containing heterocycloarene 154.88 Consequently, the bottom-gate, bottom-contact (BGBC) OFET device of 154 (Fig. 16a) exhibited a hole mobility of 1.08 × 10−3 cm2 V−1 s−1 and an on/off ratio of 5.6 × 103.
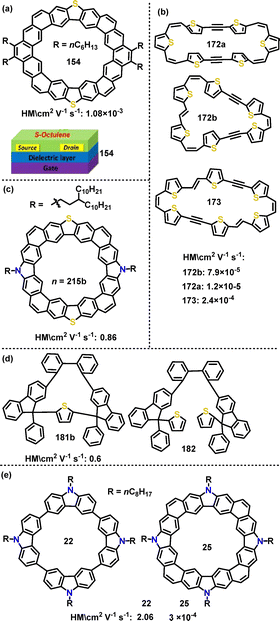 |
| | Fig. 16 Structures of macrocycles (a) 154, (b) 172a, 172b, and 173, (c) 215b, (d) 181b and 182, (e) 22 and 25. Reproduced from ref. 88 with permission from The Royal Society of Chemistry, copyright 2020. | |
Later, Iyoda studied the OFET behavior of macrocyclic π-extended thiophene hexamers.96 The OFET properties of these macrocycles showed a strong dependence on their structural planarity (Fig. 16b). For instance, macrocycle 172a with a twisted geometry had a hole mobility of 1.2 × 10−5 cm2 V s−1, and macrocycle 172b with an E,Z,Z,Z configuration had a hole mobility of 7.9 × 10−5 cm2 V−1 s−1. In contrast, 173, featuring a rigid planar E,Z,E,Z structure, exhibited the highest mobility of 2.4 × 10−4 cm2 V−1 s−1, which can be attributed to its inherent columnar stacking. In 2022, Liu, Lu and Zhao designed and synthesized a novel series of N,S-embedded heterocycloarenes (Fig. 16c),116 which featured a fully coplanar aromatic backbone. Notably, OFET devices incorporating 215b exhibited an on/off current ratio of 107 and thermal stability up to 100 °C. Additionally, the charge carrier mobility of the N,S-embedded macrocycle 215b (0.86 cm2 V−1 s−1) was significantly enhanced by two orders of magnitude compared to its S-embedded macrocycle 154 (1.08 × 10−3 cm2 V−1 s−1).113 This stark contrast underscores how molecular geometry engineering effectively boosted charge carrier mobilities in organic semiconductors.
In 2023, Xie and Huang explored the application of a novel class of axially and centrally chiral A-type nanogrids in OFET memory devices (Fig. 16d).99 OFET memory device measurements of 181b revealed a significantly larger memory window (28.3 V) compared to 182 (12.7 V) and an on/off ratio over 104. This performance disparity was attributed to the rigid nanogrid architecture of 181b, which enabled efficient electron/hole trapping through its well-defined chiral framework. In the same year, Zhao, Lu, and co-workers demonstrated a periphery fusion strategy to significantly enhance charge transport in OFET devices.33 The device based on 25 displayed typical p-type characteristics with a maximum hole mobility (HM) of 2.06 cm2 V−1 s−1 (Fig. 16e), which is four orders of magnitude higher than that of the device based on 22 (3 × 10−4 cm2 V−1 s−1). DFT calculations further indicated that both 22 and 25 possessed delocalized HOMO/LUMO distributions, highlighting their potential as p-type semiconductor materials.
6.3.3. OSCs.
In 2021, Skabara, Samuel, and Cooke investigated the performance of thiophene-based conjugated macrocycle 224 in organic electronic devices (Fig. 17a), including bulk heterojunction (BHJ) OSCs.119 Macrocycle 224 was employed as an electron donor in BHJ OSCs, which demonstrated a maximum power conversion efficiency (PCE) of 0.63%, highlighting the macrocycle's potential in low-bandgap photovoltaic materials. Later, Yuan presented a significant advancement in the use of cyclic thiophene-based conjugated molecules for photovoltaics (Fig. 17b).117 Notably, OSCs fabricated with macrocycle 218 achieved a record-breaking (PCE) of 14.2%. The devices exhibited an EQE of up to 70% across the 400–800 nm spectral range, highlighting the macrocycle's exceptional optoelectronic performance and potential for advanced photovoltaic applications.
 |
| | Fig. 17 Structures of macrocycles (a) 224 and (b) 218. | |
6.4. Catalysis
In 2023, Kobayashi and Kumagai demonstrated the catalytic efficacy of complex 203,112 which exhibited high activity in the aziridination of styrene with (N-tosylimino)phenyliodinane (PhINTs), affording the corresponding aziridine in 99% yield (Fig. 18c). Although complex 203 showed negligible emission in solution, adding H2O to its methanol solutions enhanced PL intensity with orange emission, indicative of AIE properties. In the solid state, the complex displayed strong emission (Em: 592 nm, PLQY: 21%) and long PL lifetimes (τ: 10–30 µs), suggesting phosphorescence via ISC facilitated by the Cu(I) cation.
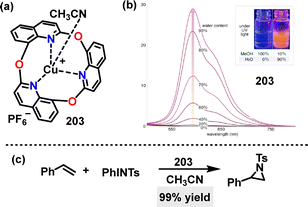 |
| | Fig. 18 (a) Structure of macrocycle 203, (b) PL emission spectra of the 203 complexes in MeOH/H2O, and (c) the catalytic activity of Cu-complex 203. Adapted from ref. 112 with permission from Wiley-VCH, copyright 2023. | |
Recently, Tang reported the construction of a novel supramolecular artificial light-harvesting system (ALHS) employing an AIE-active macrocycle 36 for applications in photocatalysis.36 The emission spectra of 34 (460 nm) and 36 (450 nm) showed good overlap with the absorption profile of the orange-emitting acceptor 37, enabling efficient Förster resonance energy transfer (FRET). In the 36/37 system, a one-step FRET process achieved an energy transfer efficiency (ETE) of 82.6% at a D/A ratio of 1000:40, outperforming the 34/PBTB system with ETE 77.9%. Rigid macrocyclic 36-based ALHSs exhibited significantly higher ETE (82.6%) compared to 34-based counterparts. Notably, under white light irradiation, the 36-based ALHSs demonstrated superior photocatalytic activity toward the cross-dehydrogenative coupling reaction in water, generating product 40 in 87% yield (Fig. 19b). In contrast, 34-based systems showed substantially lower catalytic activity (36%), underscoring the structural advantage of macrocyclic architecture.
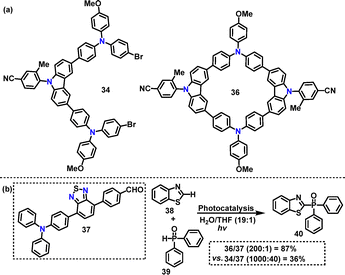 |
| | Fig. 19 (a) Structures of macrocycles 34, 36, and 37 and (b) a comparison of the catalytic performance for systems 34/37 and 36/37. | |
6.5. Molecular adsorption and separation
6.5.1. Molecular adsorption.
In 2023, Wang, Jiang, and co-workers developed a novel class of Tröger Base-derived macrocycles 70a–b and demonstrated their exceptional performance as adsorbents for iodine vapor capture.48 In vapor-phase adsorption tests (Fig. 20), 70a adsorbed up to 4.02 g g−1 (402 wt%), while 70b achieved an adsorption capacity of 3.65 g g−1 (365 wt%). These values were significantly higher than those of many previously reported N-free macrocyclic adsorbents,50,51 such as cyclotrixylohydroquinoylene derivatives, terphen[n]arenes, and quaterphen[n]arenes.
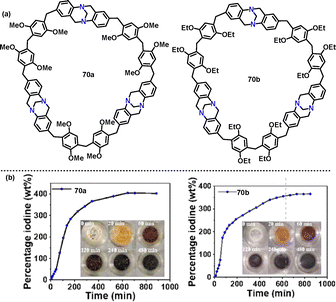 |
| | Fig. 20 (a) Structures of macrocycles 70a and 70b, and (b) their time-dependent iodine capture behaviours. Adapted from ref. 48 with permission from The Royal Society of Chemistry, copyright 2023. | |
In 2024, Zhang, Zhao, Sue, and co-workers established triphenylamine[3]arene macrocycles 73a and b as a highly efficient and versatile platform for iodine adsorption,49 with their superior performance originating from the strong electron-donating character of the intrinsic triphenylamine (TPA) moieties. For example, at 343 K under ambient pressure, these materials achieved iodine uptake capacities of 3.74 and 4.44 g g−1 after 24 h, peaking at 3.85 and 4.56 g g−1 after 34 h (Fig. 21b). Over five days under ambient conditions, they showed gradual weight loss, indicating stable yet reversible interactions with iodine.
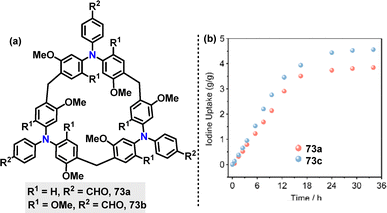 |
| | Fig. 21 (a) Structures of macrocycles 73a and 73b, and (b) their static iodine adsorption. Reprinted (naming of compounds adapted) with permission. Reproduced from ref. 49 with permission from Wiley-VCH, copyright 2024. | |
6.5.2. Molecular separation.
In 2025, Gale, Wu, Félix and co-workers developed a novel supramolecular strategy for the molecular separation of toxic anions from water, using an ion-pair assembly 118–G20 composed of macrocycle 118 and G20 (Fig. 22a).64 The 118–G20 system exhibited pronounced binding affinity for anions such as CrO42−, SO42−, and Cl−, with particularly strong interactions toward multicharged oxoanions. Notably, the assembly enabled the removal of CrO42− from aqueous solutions at concentrations ranging from 2.5 to 20 µM, achieving efficiencies between 78% and 32% through simple syringe filtration (Fig. 22b).
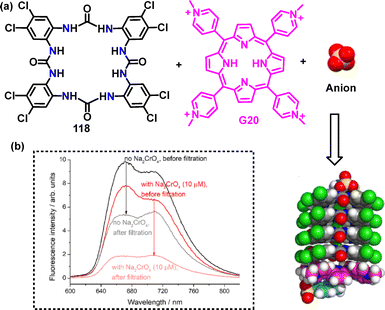 |
| | Fig. 22 (a) MD simulations of 118 co-assemblies with G20, and (b) fluorescence spectra of the solutions containing 118 and G20 in the absence and presence of CrO42−. Reproduced from ref. 64 with permission from American Chemical Society, copyright 2025. | |
Conclusion
In recent years, numerous innovative methods have emerged for the synthesis of heteroatom-doped macrocycles, which play a crucial role in organic chemistry, materials science, catalysis, and medicine. Techniques such as acid- or base-catalyzed cycloaddition and condensation reactions are commonly employed to construct macrocyclic frameworks. Transition metal-catalyzed cyclization reactions further facilitate the incorporation of heteroatoms into macrocycles, thereby improving selectivity and yields. For example, Pd-catalyzed cross-coupling, Ni-catalyzed Yamamoto polymerization, Cu-mediated azide–alkyne cycloaddition, Fe-catalyzed cyclization, and other cyclization reactions have proven effective in synthesizing diverse complex heteroatom-doped macrocyclic compounds. These macrocycles exhibit unique physical and chemical properties due to heteroatom incorporation, which alters the electronic distribution within the macrocycle, thereby influencing its reactivity and stability. For instance, N-doped macrocycles are widely used as catalysts in organic synthesis, while the ability of nitrogen to interact with specific ions or small molecules enables the development of selective sensors. O-doped macrocycles can detect specific ions or small molecules (e.g., heavy metals) through selective binding, making them effective tools for environmental monitoring. S-doped macrocycles have been employed in the development of advanced materials, such as OFETs and host–guest materials. Moreover, the presence of polar heteroatoms often enhances solubility in various solvents, increasing their versatility in applications such as the construction of metal–organic frameworks (MOFs)/covalent organic frameworks (COFs), optoelectronic devices, sensing, and drug delivery.
To propel the field of heteroatom-doped macrocycles forward, the development of more efficient, selective, and general synthetic strategies is of critical importance. This necessitates a concerted effort to overcome three primary challenges, including enantioselective synthesis, generality and structural diversity, and scalability and efficiency. (1) The construction of well-defined chiral macrocycles remains a significant bottleneck, limiting applications in asymmetric catalysis, enantioselective sensing, and chiroptical materials. Pioneering catalytic asymmetric macrocyclization strategies, such as synergistic systems combining transition metals (e.g., Pd, Cu, and Ni) with chiral ligands or organocatalysts, are essential to achieve precise stereochemical control. (2) Achieving greater structural diversity and expanding substrate scope will rely on transformative methods, such as photoredox-mediated annulations for novel bond formation, and the strategic use of multicomponent reactions (e.g., A3-coupling and Ugi reaction) in macrocyclization. (3) To truly transition these molecules from the laboratory stage to industrially relevant materials, synthetic routes must be fundamentally optimized for yield and scalability. Integrating robust catalytic methods, continuous-flow processes, and other process intensification techniques will be vital for enabling large-scale production and unlocking their full practical application potential.
The advancement of innovative synthetic methodologies will be the key to unlocking a new generation of applications that fully leverage the unique properties of heteroatom-doped macrocycles. Their well-defined, functionalized cavities position them as ideal platforms for supramolecular catalysis and biomimetics, enabling the design of artificial enzymes and catalysts for high-selectivity transformations under mild conditions. Concurrently, these macrocycles are emerging as essential components in advanced functional materials, such as those for molecular separation, organic electronics, and stimuli-responsive membranes, where their tunable host–guest chemistry and porosity can be precisely exploited. Furthermore, their tailored biocompatibility, molecular recognition capabilities, and unique spectroscopic signatures open significant avenues in biomedical translation, including targeted drug delivery, biosensing, and bioimaging, thereby creating a vital bridge between supramolecular chemistry and biomedical science.
In conclusion, developing new synthetic strategies for heteroatom-doped macrocycles, paired with rigorous property and application studies, will drive progress in supramolecular chemistry, catalysis, biology, and materials science. We anticipate that this review will offer valuable insights and directions for researchers navigating these interdisciplinary Frontiers.
Author contributions
Writing (original draft): CL, SL, XC and TY; writing (reviewing and editing): YW, LX, EVVdE and WH; funding acquisition: CL, YW, LX, EVVdE and WH; the final manuscript has been read and approved by all authors.
Conflicts of interest
There are no conflicts to declare.
Data availability
Data sharing is not applicable to this article as no new data were generated or analyzed in this review.
Acknowledgements
This work was supported by the Natural Science Research Start-up Foundation of Recruiting Talents of Nanjing University of Posts and Telecommunications (Grant No. NY224034; recipient CL, writing), the National Key R&D Program of China (Grant No. 2024YFB361260; recipient LX, writing), the National Natural Science Foundation of China (22275098, 62288102 and 22071112; recipients YW, LX, and WH, writing), and the Basic Research Program of Jiangsu (BK20243057; recipient LX, writing). The authors acknowledge the FWO [Fund for Scientific Research-Flanders (Belgium)] for financial support (recipient EVVdE, writing) and the Research Council of the KU Leuven (recipient EVVdE, writing). This paper has been prepared with the support of the “RUDN University Strategic Academic Leadership Program” (recipient EVVdE, writing).
Notes and references
- S. Zhang, S. Fan, H. He, J. Zhu, L. Murray, G. Liang, S. Ran, Y. Zhu, M. J. Cryle, H.-Y. He and Y. Zhang, Chem. Soc. Rev., 2025, 54, 396–464 RSC.
- X. Q. Ma, H. P. Xiao, Y. Chen, Q. S. Lai, X. X. Li and S. T. Zheng, Coord. Chem. Rev., 2024, 510, 215818 CrossRef CAS.
- K. T. Mortensen, T. J. Osberger, T. A. King, H. F. Sore and D. R. Spring, Chem. Rev., 2019, 119, 10288–10317 CrossRef CAS PubMed.
- X. Y. Lou, S. Zhang, Y. Wang and Y. W. Yang, Chem. Soc. Rev., 2023, 52, 6644–6663 RSC.
- H. T. Feng, Y. X. Yuan, J. B. Xiong, Y. S. Zheng and B. Z. Tang, Chem. Soc. Rev., 2018, 47, 7452–7476 RSC.
- C. D. Zhao, H. Yao, S. Y. Li, F. Du, L. L. Wang and L. P. Yang, Chin. Chem. Lett., 2024, 35, 108879 CrossRef CAS.
- Q. Feng, S. Zhu, B. Wang, F. Yu, H. Li, M. Yu, M. Xu and L. Xie, Adv. Funct. Mater., 2024, 34, 2312622 CrossRef CAS.
- X. N. Han, Y. Han and C. F. Chen, Chem. Soc. Rev., 2023, 52, 3265–3298 RSC.
- R. Paolesse, S. Nardis, D. Monti, M. Stefanelli and C. Di Natale, Chem. Rev., 2017, 117, 2517–2583 CrossRef CAS PubMed.
- R. Kumar, A. Sharma, H. Singh, P. Suating, H. S. Kim, K. Sunwoo, I. Shim, B. C. Gibb and J. S. Kim, Chem. Rev., 2019, 119, 9657–9721 CrossRef CAS PubMed.
- S. Bleus and W. Dehaen, Coord. Chem. Rev., 2024, 509, 215762 CrossRef CAS.
-
C. D. Gutsche, Calixarenes: An Introduction, Royal Society of Chemistry, Cambridge, UK, 1989 Search PubMed.
- G. W. Gokel, W. M. Leevy and M. E. Weber, Chem. Rev., 2004, 104, 2723–2750 CrossRef CAS PubMed.
- Z. Q. Wang, X. Wang and Y. W. Yang, Adv. Mater., 2024, 36, 2301721 CrossRef CAS PubMed.
- J. R. Wu, G. Wu and Y. W. Yang, Acc. Chem. Res., 2022, 55, 3191–3204 CrossRef CAS PubMed.
- Z. Liu and Y. Liu, Chem. Soc. Rev., 2022, 51, 4786–4827 RSC.
- M. Votava and B. J. Ravoo, Chem. Soc. Rev., 2021, 50, 10009–10024 RSC.
- Y. Li, H. Kono, T. Maekawa, Y. Segawa, A. Yagi and K. Itami, Acc. Mater. Res., 2021, 2, 681–691 CrossRef CAS.
- T. M. Fukunaga, C. Sawabe, T. Matsuno, J. Takeya, T. Okamoto and H. Isobe, Angew. Chem., Int. Ed., 2021, 60, 19097–19101 CrossRef CAS PubMed.
- Y. Wu, S. Li, H. Li, R. Ye and Z. Lu, J. Mater. Chem. C, 2023, 11, 7144–7158 RSC.
- Y. Qin, X. Liu, P. P. Jia, L. Xu and H. B. Yang, Chem. Soc. Rev., 2020, 49, 5678–5703 RSC.
- M. Toganoh and H. Furuta, Chem. Rev., 2022, 122, 8313–8437 CrossRef CAS PubMed.
- H. Lu and N. Kobayashi, Chem. Rev., 2016, 116, 6184–6261 CrossRef CAS PubMed.
- M. D. Tomczyk, N. Kuźnik and K. Walczak, Coord. Chem. Rev., 2023, 481, 215047 CrossRef CAS.
- Y. Matano and H. Imahori, Acc. Chem. Res., 2009, 42, 1193–1204 CrossRef CAS PubMed.
- R. Sengupta, M. Ravikanth and T. K. Chandrashekar, Chem. Soc. Rev., 2021, 50, 13268–13320 RSC.
- P. Li, D. Shimoyama, N. Zhang, Y. Jia, G. Hu, C. Li, X. Yin, N. Wang, F. Jäkle and P. Chen, Angew. Chem., Int. Ed., 2022, 61, e202200612 CrossRef CAS PubMed.
- B. S. Kumar, B. Chandra, K. V. Jovan Jose and P. K. Panda, J. Org. Chem., 2021, 86, 10536–10543 CrossRef CAS PubMed.
- S. Shikita, G. Watanabe, D. Kanouchi, J. Saito and T. Yasuda, Adv. Photonics Res., 2021, 2, 2100021 CrossRef CAS.
- M. Zhao, S. H. Pun, Q. Gong and Q. Miao, Angew. Chem., Int. Ed., 2021, 60, 24124–24130 CrossRef CAS PubMed.
- T. Komori, E. Tsurumaki and S. Toyota, Chem. – Asian J., 2023, 18, e202201003 CrossRef CAS PubMed.
- H. Li, X. Zhang, J. Peng, S. Yang, R. Hu and X. Jiang, J. Mater. Chem. C, 2024, 12, 10127–10134 RSC.
- N. Zhang, W. Li, J. Zhu, T. Wang, R. Zhang, K. Chi, Y. Liu, Y. Zhao and X. Lu, Adv. Mater., 2023, 35, 2300094 CrossRef CAS PubMed.
- F. F. Wang, Y. X. Wang, Q. Wu, L. Chai, X. W. Chen and Y. Z. Tan, Angew. Chem., Int. Ed., 2024, 136, e202315302 CrossRef.
- Y. Shi, X. Li, J. Di, Y. Xue, N. Zhang, T. Jin, C.-F. Chen and P. Chen, CCS Chem., 2025, 7, 1–17 Search PubMed.
- J. C. Yang, K. Chen, G. L. Zhang, C. Qi, H. T. Feng and B. Z. Tang, Chem. Sci., 2025, 16, 4741–4748 RSC.
- S. Izumi, H. F. Higginbotham, A. Nyga, P. Stachelek, N. Tohnai, P. D. Silva, P. Data, Y. Takeda and S. Minakata, J. Am. Chem. Soc., 2020, 142, 1482–1491 CrossRef CAS PubMed.
- F. Picini, S. Schneider, O. Gavat, A. Vargas Jentzsch, J. Tan, M. Maaloum, J.-M. Strub, S. Tokunaga, J.-M. Lehn, E. Moulin and N. Giuseppone, J. Am. Chem. Soc., 2021, 143, 6498–6504 CrossRef CAS PubMed.
- J. Ayuso-Carrillo, F. Fina, E. C. Galleposo, R. R. Ferreira, P. K. D. Mondal, B. D. Ward and D. Bonifazi, J. Am. Chem. Soc., 2024, 146, 16440–16457 CrossRef CAS PubMed.
- H. Zhu, I. Badía-Domínguez, B. Shi, Q. Li, P. Wei, H. Xing, M. C. R. Delgado and F. Huang, J. Am. Chem. Soc., 2021, 143, 2164–2169 CrossRef CAS PubMed.
- Y. Yoshigoe, H. Shimada, T. Takaki, Y. Imai and S. Saito, Chem. – Eur. J., 2024, 30, e202304059 CrossRef CAS PubMed.
- C. Brouillac, N. McIntosh, B. Heinrich, O. Jeannin, O. De Sagazan, N. Coulon, J. Rault-Berthelot, J. Cornil, E. Jacques, C. Quinton and C. Poriel, Adv. Sci., 2024, 11, 2309115 CrossRef CAS PubMed.
- H. Zhang, H. Li, S. Sun, L. Tan, H. Shen, B. Lin and P. Yang, Org. Lett., 2023, 25, 2078–2083 CrossRef CAS PubMed.
- F. Zhang, X. S. Du, D. W. Zhang, Y. F. Wang, H. Y. Lu and C. F. Chen, Angew. Chem., Int. Ed., 2021, 60, 15291–15295 CrossRef CAS PubMed.
- Y. Li, Y. Segawa, A. Yagi and K. Itami, J. Am. Chem. Soc., 2020, 142, 12850–12856 CrossRef CAS PubMed.
- H. Y. Zhou, D. W. Zhang, X. N. Han, Y. Han and C. F. Chen, Chem. Commun., 2022, 58, 12180–12183 RSC.
- F. Zeng, L. L. Tang, M. H. Ding and W. Dessie, Org. Lett., 2023, 25, 6290–6294 CrossRef CAS PubMed.
- P. Niu, C. Shi, J. Jiao, W. Xie, H. Qiu, Z. Yang, J. Jiang and L. Wang, Chem. Commun., 2023, 59, 10960–10963 RSC.
- J. Jiao, W. Xie, Y. Li, C. Lin, J. Jiang and L. Wang, Chem. – Eur. J., 2022, 28, e202201933 CrossRef CAS PubMed.
- B. Li, B. Wang, X. Huang, L. Dai, L. Cui, J. Li, X. Jia and C. Li, Angew. Chem., Int. Ed., 2019, 131, 3925–3929 CrossRef.
- W. Fang, J. Zhang, M. Guo, Y. Zhao and A. C. H. Sue, Angew. Chem., Int. Ed., 2024, 63, e202409120 CrossRef CAS PubMed.
- Y. Liu, H. Wang, P. Liu, H. Zhu, B. Shi, X. Hong and F. Huang, Angew. Chem., Int. Ed., 2021, 133, 5830–5834 CrossRef.
- A. Kalaiselvan, I. S. Vamsi Krishna, A. P. Nambiar, A. Edwin, V. S. Reddy and S. Gokulnath, Org. Lett., 2020, 22, 4494–4499 CrossRef CAS PubMed.
- D. Lin, Y. Wei, A. Peng, H. Zhang, C. Zhong, D. Lu, H. Zhang, X. Zheng, L. Yang, Q. Feng, L. H. Xie and W. Huang, Nat. Commun., 2020, 11, 1756 CrossRef CAS PubMed.
- G. Ouyang, J. Rühe, Y. Zhang, M. J. Lin, M. Liu and F. Würthner, Angew. Chem., Int. Ed., 2022, 134, e202206706 Search PubMed.
- H. Chen, X. Shi, Y. Lun, Y. Xu, T. Lu, Z. Duan, M. Shao, J. L. Sessler, C. Yu and H. Lei, J. Am. Chem. Soc., 2022, 144, 8194–8203 Search PubMed.
- N. Halder, R. Naryanasamy, D. Usharani and H. Rath, Org. Chem. Front., 2022, 9, 2333–2342 RSC.
- J. Q. Zhao, L. L. Mao, G. H. Zhang, S. Z. Zhan, H. Xiao, S. Zhang, C. H. Tung, L. Z. Wu and H. Cong, J. Mater. Chem. A, 2023, 11, 4672–4678 RSC.
- S. Li, C. Zhu, L. Mao, X. Zhang, Z. Ye, C. Li and D. Ma, Sci. China: Chem., 2025, 68, 3116–3122 CrossRef CAS.
- Y. Shen, X. Liang, T. Ma, D. Zhou, W. Li, J. Ma, J. Ma, W. Wu, Z. Yu and C. Yang, Angew. Chem., Int. Ed., 2025, 137, e202504211 CrossRef.
- S. X. Nie, H. Guo, T. Y. Huang, Y. F. Ao, D. X. Wang and Q. Q. Wang, Nat. Commun., 2020, 11, 6257 CrossRef CAS PubMed.
- Y. X. Yuan, J. H. Jia, Y. P. Song, F. Y. Ye, Y. S. Zheng and S. Q. Zang, J. Am. Chem. Soc., 2022, 144, 5389–5399 CrossRef CAS PubMed.
- K. Kihara, T. Kobayashi, W. Xu and N. Kumagai, Chem. – Eur. J., 2024, 30, e202304176 CrossRef CAS PubMed.
- J. Li, O. Catal, I. Marques, D. A. McNaughton, R. M. Maklad, W. G. Ryder, M. J. S. Hill, A. Seddo, W. Lewis, D. J. Adams, V. Félix, X. Wu and P. A. Gale, J. Am. Chem. Soc., 2025, 147, 3392–3401 Search PubMed.
- H. Keypour, M. Abdollahi-Moghadam, H. Zeynali, R. Karamian and N. B. Hamedani, J. Mol. Struct., 2024, 1295, 136803 CrossRef CAS.
- R. Singla, A. Ganguli and M. Ghosh, Int. J. Food Prop., 2010, 13, 1290–1299 CrossRef CAS.
- N. Prusinowska, A. Czapik, J. Szymkowiak and M. Kwit, Sci. Rep., 2025, 15, 890 CrossRef CAS PubMed.
- S. Xiong, Y. Zhang, Y. Jiang, F. Wang, W. Zhou, A. Li, Q. Zhang, Q. Wang and Q. He, Chem. Commun., 2023, 59, 12994–12997 RSC.
- S. K. M. Nalluri and J. F. Stoddart, Chem. Soc. Rev., 2017, 46, 2459–2478 RSC.
- C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 2495–2496 CrossRef CAS.
- C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 7017–7036 CrossRef CAS.
- G. W. Gokel, L. J. Barbour, L. Stephen and E. S. Meadows, Coord. Chem. Rev., 2001, 222, 127–154 CrossRef CAS.
- A. J. Varni, M. Kawakami, S. A. Tristram-Nagle, D. Yaron, T. Kowalewski and K. J. Noonan, Org. Chem. Front., 2021, 8, 1775–1782 RSC.
- S. V. Mulay, O. Dishi, Y. Fang, M. R. Niazi, L. J. W. Shimon, D. F. Perepichka and O. Gidron, Chem. Sci., 2019, 10, 8527–8532 RSC.
- T. Köhler, D. Seidel, V. Lynch, F. O. Arp, Z. Ou, K. M. Kadish and J. L. Sessler, J. Am. Chem. Soc., 2003, 125, 6872–6873 CrossRef PubMed.
- Y. Han, W. C. Guo, X. S. Du and C. F. Chen, Chem. Commun., 2024, 60, 5719–5722 RSC.
- X. S. Du, D. W. Zhang, Y. Guo, J. Li, Y. Han and C. F. Chen, Angew. Chem., Int. Ed., 2021, 60, 13021–13028 CrossRef CAS PubMed.
- F. Zhang, X. S. Du, D. W. Zhang, Y. F. Wang, H. Y. Lu and C. F. Chen, Angew. Chem., Int. Ed., 2021, 60, 15291–15295 CrossRef CAS PubMed.
- D. Zhu, S. Fang, L. Tong, Y. Lei, G. Wu, T. Chudhary and H. Li, Chem. Commun., 2021, 57, 4440–4443 RSC.
- K. Xu, B. Li, S. Yao, Z. Li, Y. Lu, M. Dong, J. Qiu, L. Luo and C. Li, Angew. Chem., Int. Ed., 2022, 134, e202203016 CrossRef.
- J. Pfeuffer-Rooschuz, L. Schmid, A. Prescimone and K. Tiefenbacher, JACS Au, 2021, 1, 1885–1891 CrossRef CAS PubMed.
- Y. F. Wang, H. Yao, L. P. Yang, M. Quan and W. Jiang, Angew. Chem., Int. Ed., 2022, 134, e202211853 CrossRef.
- Y. Gong, S. Fang, Y. Zheng, H. Guo and F. Yang, J. Photochem. Photobiol., A, 2023, 435, 114307 CrossRef CAS.
- Y. Liu, Q. Zhang, S. Crespi, S. Chen, X. K. Zhang, T. Y. Xu, C. S. Ma, S. W. Zhou, Z. T. Shi, H. Tian, B. L. Feringa and D. H. Qu, Angew. Chem., Int. Ed., 2021, 133, 16265–16274 CrossRef.
- C. Gagnon, É. Godin, C. Minozzi, J. Sosoe, C. Pochet and S. K. Collins, Science, 2020, 367, 917–921 CrossRef CAS PubMed.
- T. Yang, T. Wang, F. Deng, S. Ma, F. Yang, Y. Wei, C. Zhong and L. Xie, Sci. Bull., 2025, 70, 1902–1906 CrossRef CAS PubMed.
- A. Jana, S. Bähring, M. Ishida, S. Goeb, D. Canevet, M. Sallé, J. O. Jeppesen and J. L. Sessler, Chem. Soc. Rev., 2018, 47, 5614–5645 RSC.
- L. Yang, N. Zhang, Y. Han, Y. Zou, Y. Qiao, D. Chang, Y. Zhao, X. Lu, J. Wu and Y. Liu, Chem. Commun., 2020, 56, 9990–9993 RSC.
- X. Lu, D. An, Y. Han, Y. Zou, Y. Qiao, N. Zhang, D. Chang, J. Wu and Y. Liu, Chem. Sci., 2021, 12, 3952–3957 RSC.
- D. An, R. Zhang, J. Zhu, T. Wang, Y. Zhao, X. Lu and Y. Liu, Chem. Sci., 2024, 15, 4590–4601 RSC.
- L. Zhan, C. Dai, G. Zhang, J. Zhu, S. Zhang, H. Wang, Y. Zeng, C. H. Tung, L. Z. Wu and H. Cong, Angew. Chem., Int. Ed., 2022, 61, e202113334 CrossRef CAS PubMed.
- T. C. Lovell, Z. R. Garrison and R. Jasti, Angew. Chem., Int. Ed., 2020, 59, 14363–14367 CrossRef CAS PubMed.
- C. Zhao, F. Liu, L. Feng, M. Nie, Y. Lu, J. Zhang, C. Wang and T. Wang, Nanoscale, 2021, 13, 4880–4886 RSC.
- L. Ren, Y. Han, X. Hou, Y. Ni, Y. Zou, T. Jiao and J. Wu, J. Am. Chem. Soc., 2023, 145, 12398–12406 Search PubMed.
- M. Nagase, S. Nakano and Y. Segawa, Chem. Commun., 2023, 59, 11129–11132 RSC.
- K. Shirahata, M. Takashika, K. Hirabayashi, M. Hasegawa, H. Otani, K. Yamamoto, Y. Le, T. Shimizu, A. Shinobu and M. Iyoda, J. Org. Chem., 2020, 86, 302–309 CrossRef PubMed.
- T. M. Fukunaga, C. Sawabe, T. Matsuno, J. Takeya, T. Okamoto and H. Isobe, Angew. Chem., Int. Ed., 2021, 60, 19097–19101 CrossRef CAS PubMed.
- M. Lin, L. Bian, Q. Chen, H. Xu, Z. Liu and K. Zhu, Angew. Chem., Int. Ed., 2023, 62, e202303035 CrossRef CAS PubMed.
- Y. Wei, X. Du, W. X. Huang, M. Y. Fu, Z. Zhang, C. X. Zhong, Y. Wang, H. F. Ling, L. H. Xie and W. Huang, Chin. J. Chem., 2023, 41, 2969–2974 CrossRef CAS.
- S. I. Etkind, S. Ichii, N. A. Romero and T. M. Swager, Synlett, 2022, 1532–1538 CAS.
- A. Ovsyannikov, S. Solovieva, I. Antipin and S. Ferlay, Coord. Chem. Rev., 2017, 352, 151–186 CrossRef CAS.
- X. Sun, J. Bai, X. Y. Wang and H. Y. Gong, Coord. Chem. Rev., 2024, 518, 216063 CrossRef CAS.
- A. M. Camp, M. R. Kita, P. T. Blackburn, H. M. Dodge, C. H. Chen and A. J. Miller, J. Am. Chem. Soc., 2021, 143, 2792–2800 CrossRef CAS PubMed.
- C. H. Wei, S. Dong, Z. Xu, M. Li, T. Zhang, Z. Xu, M. Li, T. Zhang, Z. Xu, S. Lan, S. Wang and L. Mao, Angew. Chem., Int. Ed., 2024, 63, e202412253 CrossRef CAS PubMed.
- J. F. Stoddart, A Platform for Change, Supramol. Chem., 2015, 27, 567–570 CrossRef CAS.
- P. K. Kodali, S. Choppella, D. Kumar, U. K. Pandey, M. K. Ravva and S. P. Singh, Chem. Commun., 2024, 60, 11726–11729 RSC.
- H. Zhou, Y. Zhang, Z. Zheng, J. Wan, H. Zhang, K. Lin, J. L. Sessler and H. Wang, Chem. Sci., 2025, 16, 910–919 RSC.
- L. Mao, F. Li, L. Huang, X. Qu, K. Wang, Y. Zhang and D. Ma, Org. Lett., 2023, 25, 5597–5601 CrossRef CAS PubMed.
- L. Wang, Y. Zhang, J. Chen, S. Jongaksorn, Z. Lu, X. Zhang, S. Li, C. Zhu, D. Ma and L. Mao, J. Org. Chem., 2025, 90, 1671–1677 CrossRef CAS PubMed.
- S. Y. Zhuang, Y. Cheng, Q. Zhang, S. Tong and M. X. Wang, Angew. Chem., Int. Ed., 2020, 132, 23924–23931 CrossRef.
- I. Solymosi, S. Krishna, E. Nuin, H. Maid, B. Scholz, D. M. Guldi, M. E. Pérez-Ojeda and A. Hirsch, Chem. Sci., 2021, 12, 15491–15502 RSC.
- T. Kobayashi and N. Kumagai, Angew. Chem., Int. Ed., 2023, 135, e202307896 CrossRef.
- C. Y. Lin, C. H. Hsu, C. M. Hung, C. C. Wu, Y. H. Liu, E. H. C. Shi, T. H. Lin, Y. C. Hu, W. Y. Hung, K. T. Wong and P. T. Chou, Nat. Chem., 2024, 16, 98–106 CrossRef CAS PubMed.
- D. Hartmann, S. E. Penty, M. A. Zwijnenburg, R. Pal and T. A. Barendt, Angew. Chem., Int. Ed., 2025, 64, e202501122 CrossRef CAS PubMed.
- F. Wang, X. Shi, Y. Zhang, W. Zhou, A. Li, Y. Liu, J. L. Sessler and Q. He, J. Am. Chem. Soc., 2023, 145, 10943–10947 CrossRef CAS PubMed.
- N. Zhang, L. Yang, W. Li, J. Zhu, K. Chi, D. Chang, Y. Qiao, T. Wang, Y. Zhao, X. Lu and Y. Liu, J. Am. Chem. Soc., 2022, 144, 21521–21529 CrossRef CAS PubMed.
- W. Liu, H. Zhang, S. Liang, T. Wang, S. He, Y. Hu, R. Zhang, H. Ning, J. Ren, A. Bakulin, F. Gao, Y. Jun and Y. Zou, Angew. Chem., Int. Ed., 2023, 62, e202311645 CrossRef CAS PubMed.
- W. Zhang, W. Liu, Z. Xu, K. An, Q. H. Li, Z. Wang, P. Lei, Q. Zeng, N. Li, Y. P. Yi, H. Wu and J. H. Wan, Adv. Funct. Mater., 2024, 34, 2411745 CrossRef CAS.
- J. M. dos Santos, L. K. Jagadamma, J. Cameron, A. A. Wiles, C. Wilson, P. J. Skabara, I. D. W. Samuel and G. Cooke, J. Mater. Chem. C, 2021, 9, 16257–16271 RSC.
- S. Li, K. Liu, X. C. Feng, Z. X. Li, Z. Y. Zhang, B. Wang, M. Li, Y. L. Bai, L. Cui and C. Li, Nat. Commun., 2022, 13, 2850 CrossRef CAS PubMed.
- G. Kaur, N. Rawat and M. Ravikanth, J. Org. Chem., 2023, 88, 14989–14997 CrossRef CAS PubMed.
- G. Zhang, F. Chen, Y. Di, S. Yuan, Y. Zhang, X. Quan, Y. Chen, H. Chen and M. Lin, Adv. Funct. Mater., 2024, 34, 2404123 CrossRef CAS.
- J. Ajay, T. Sulfikarali, A. Edwin, M. Sasi, S. Mori and S. Gokulnath, Org. Lett., 2025, 27, 2128–2132 Search PubMed.
- Z. Lu, W. Lv, H. Liu, Y. Liu, S. Liao, X. Wang and K. Zhu, Org. Lett., 2023, 25, 3508–3511 Search PubMed.
- K. Bold, M. Stolte, K. Shoyama, M. Holzapfel, A. Schmiedel, C. Lambert and F. Würthner, Angew. Chem., Int. Ed., 2022, 61, e202113598 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Shuangyi
Li
a,
Xin
Chen
a,
Tonglin
Yang
a,
Ying
Wei
a,
Shuangyi
Li
a,
Xin
Chen
a,
Tonglin
Yang
a,
Ying
Wei