
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of heterobicyclo[n.1.1]alkanes
Rebecca I.
Revie†
 ,
Julia
Ragus†
and
Edward A.
Anderson
*
,
Julia
Ragus†
and
Edward A.
Anderson
*
Chemistry Research Laboratory, Department of Chemistry, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: edward.anderson@chem.ox.ac.uk
First published on 17th December 2025
Abstract
Heterocyclic bicyclo[n.1.1]alkanes have emerged as important scaffolds in contemporary drug design due to their rigid frameworks that enable the positioning of their subsituents along well defined vectors in chemical space. Offering much potential as alternative cores to traditional benzene rings, heterobicyclo[2.1.1]hexanes (HBCHexs) and heterobicyclo[3.1.1]heptanes (HBCHeps) in particular have attracted significant attention from the synthetic community. A plethora of methods have recently been developed to access these useful motifs, using both radical and polar strategies to forge the bicyclic system. This review discusses recent developments in the field, with a focus on mechanistic aspects, and those methodologies that show the most potential for general application.
 Rebecca I. Revie | Rebecca Revie obtained her MSci degree from the University of Cambridge in 2022, completing her master's research under the supervision of Prof David Spring. She has recently completed her DPhil at the University of Oxford under the supervision of Prof Ed Anderson. Her doctoral research has focused on the development of small-ring methodologies with a focus on advancing new strategies for constructing strained molecular architectures. Her research interests span organic synthesis, reaction development, and the exploration of small-ring systems as versatile building blocks in medicinal chemistry. |
 Julia Ragus | Julia Ragus received her MChem degree from the University of Oxford in 2022, with a final-year project focused on yndiamide-like molecules under the supervision of Prof Ed Anderson. She began her DPhil studies the same year at the University of Oxford, joining the research groups of Prof Veronique Gouverneur and Prof Michael Willis successively for two short research projects. In 2023, she rejoined the Anderson group to work on expanding the family of [3.1.1]propellanes as tools for the synthesis of new pharmaceutical building blocks. |
 Edward A. Anderson | Ed Anderson completed his PhD in 2001 at the University of Cambridge with Prof Andrew Holmes. After postdoctoral research periods with Profs Erik Sorensen (at the Scripps Research Institute) and Ian Paterson (again at Cambridge), Ed was appointed as an EPSRC Advanced Research Fellow at the University of Oxford in 2007. He took up a Lectureship in 2009 and was promoted to Full Professor position in 2016; he currently serves as the Head of Organic Chemistry. His research interests encompass the full range of organic chemistry, including methodology development, total synthesis, and mechanistic study. |
Introduction
Bicyclic sp3-rich scaffolds are increasingly sought after as building blocks in contemporary drug discovery. In the context of “escaping from flatland”, it has been shown that moving away from traditional planar sp2-rich structures towards sp3-rich 3D structures correlates with improved chances of clinical success.1,2 However, most drug discovery programmes are biased towards aromatic scaffolds since these are easy to construct by well-established methods, such as sp2–sp2 cross-coupling reactions. There are two main drawbacks associated with such 'flat' drug candidates: firstly, as most drugs interact with complex 3D biomacromolecules, the biasing of compound libraries towards 2D structures risks missing much of the chemical space that is relevant to their biological targets; secondly, aromatic rings can contribute to poor pharmacokinetic properties – they can be susceptible to oxidation by cytochrome P450 enzymes, leading to overly rapid metabolic clearance, while aggregation arising from π-stacking interactions can lead to poor aqueous solubility.3How can medicinal chemists overcome these challenges and escape from the problematic flatland? Instead of relying on aromatic scaffolds, new sp3-rich scaffolds are required which provide opportunities to install substituents along precise vectors in 3D space, enabling the wider exploration of chemical space in drug discovery. The properties of existing drug candidates may also be enhanced by developing arene bioisosteres consisting of sp3-rich groups that replace arenes while retaining their geometry and biological activity, but bestowing improved pharmacokinetic properties.4–6 The synthetic challenges associated with both of these goals include the construction of the scaffold, control over the precise installation of substituents at defined vectors, and the late-stage generation of a diversity of different substituents on the core scaffold.
One of the most popular types of sp3-rich scaffolds to have emerged in recent years are small-ring bridged bicyclic alkanes such as bicyclo[1.1.1]pentanes (BCPs),7 bicyclo[2.1.1]hexanes (BCHexs)8 and bicyclo[3.1.1]heptanes (BCHeps)9 (Fig. 1a). These compounds can not only act as bioisosteric candidates for ortho-, meta- and para-disubstituted benzenes, but are also novel 3D scaffolds in their own right. For example, BCPs have been successfully demonstrated to replace para-disubstituted benzenes,10 while BCHeps can serve as meta-disubstituted benzene isosteres, and BCHexs as ortho- or meta-arene analogues (Fig. 1b).7,9 Analysis of the exit vectors of the illustrated substituents reveals reasonable similarities in terms of separation of substituents and exit vector angles; however a challenge for true mimicry of an arene often lies in the reproduction of near-zero dihedral angles.
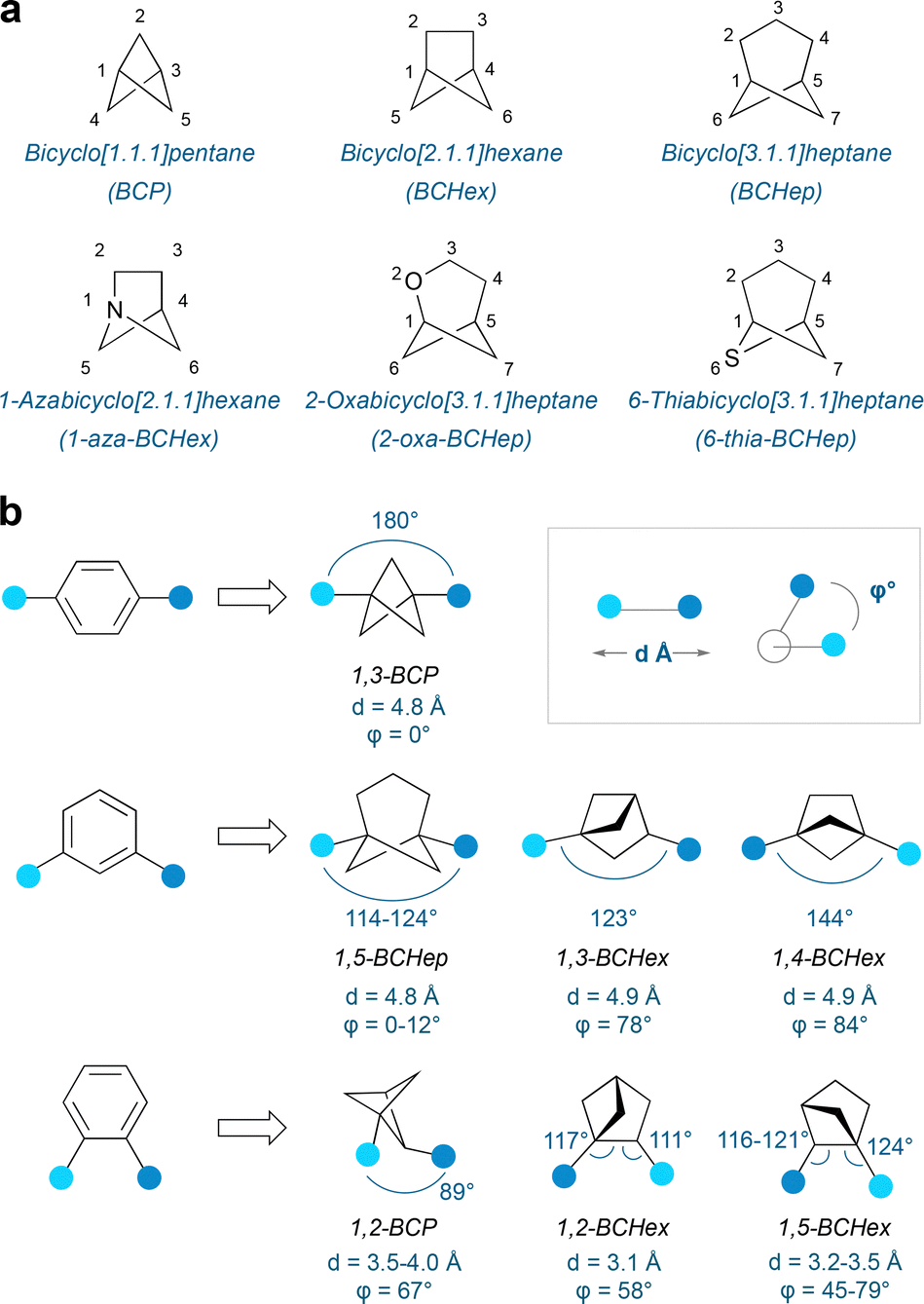 | ||
| Fig. 1 (a) Nomenclature of bicyclo[n.1.1]alkanes and heterobicyclo[n.1.1]alkanes. (b) Bicycloalkanes as bioisosteres: geometric considerations. | ||
Most early examples of small-ring bridged bicyclic systems were carbocyclic in nature, and a wealth of different methods are now available for the synthesis of such motifs;11,12 some examples are shown in Fig. 2a. These carbocyclic scaffolds provide good candidates for the bioisosteric replacement of arenes, having often been shown to improve metabolic stability, and in some cases to maintain or enhance biological activity. There are however some disadvantages to the use of bicyclic bridged carbocycles: Firstly, while BCPs improve aqueous solubility over their ‘parent’ arenes, the larger ring BCHep exhibits similar or even reduced solubility, likely due to the higher number of methylene units present;13 secondly, bicyclic carbocycles only offer opportunities to act as bioisosteres for carbocyclic aromatics; thirdly, exploration of chemical space is limited since it is challenging to introduce substituents on the bridge positions of the carbocycle at a late stage in the synthesis, and indeed there are limited opportunities for variation within the scaffold itself since it is made entirely from carbon atoms.
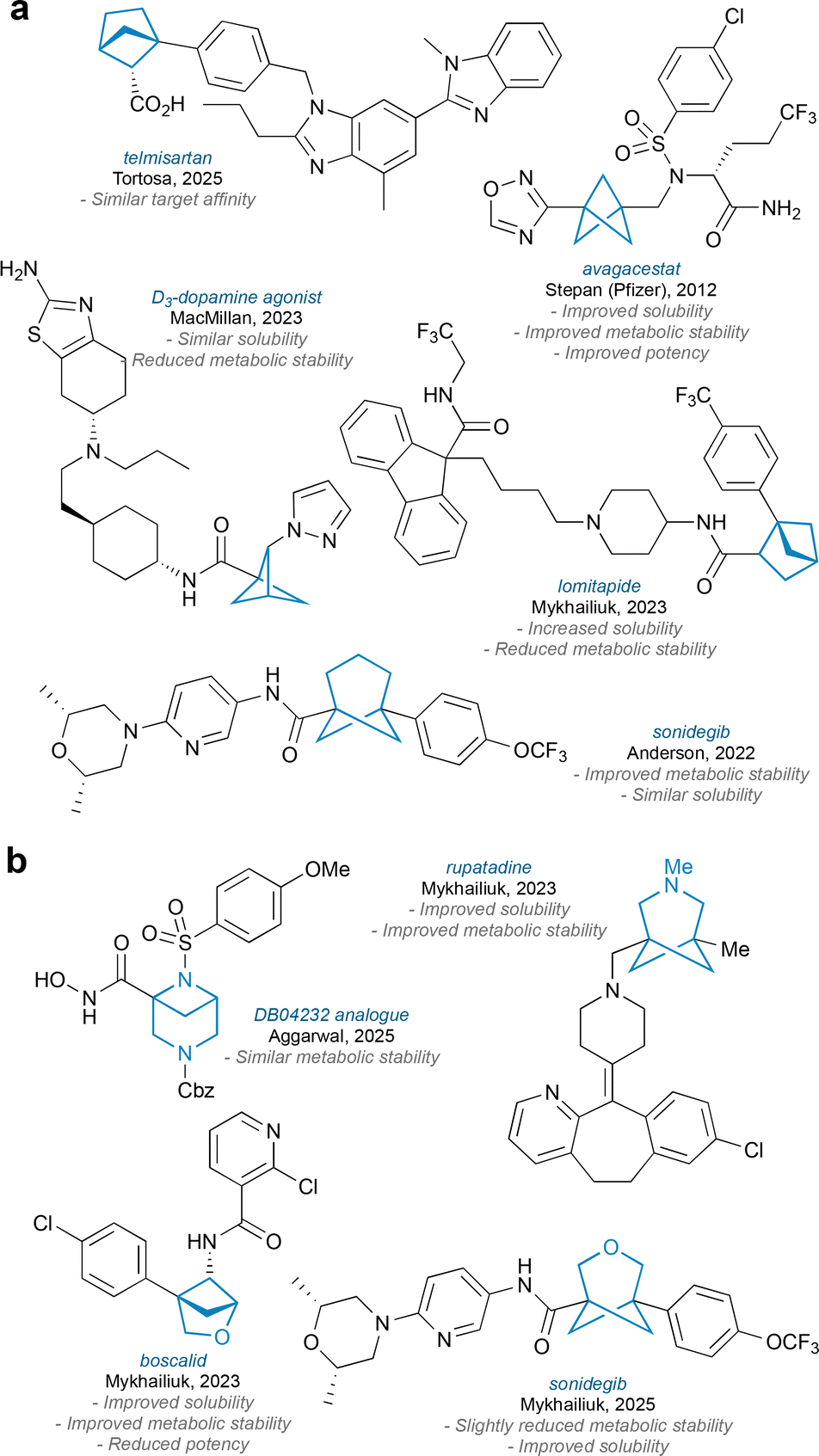 | ||
| Fig. 2 (a) Selected examples of bicyclo[n.1.1]alkane drug analogues. (b) Selected examples of heterobicyclo[n.1.1]alkane drug analogues. | ||
In light of these issues, attention has recently turned towards incorporation of heteroatoms into the bicyclic framework; some example of heterobicyclo[n.1.1]alkanes and associated nomenclature are shown in Fig. 1a. The benefits of incorporating heteroatoms include improved water solubility over their carbocyclic analogues, opportunities to act as bioisosteres for heteroarenes, opportunities for late-stage diversification on the bridge positions (when the heteroatom is nitrogen), and opportunities for diversification of the scaffold itself by incorporating heteroatoms at various positions within the bridged ring. Fig. 2b shows some examples of drug analogues that have been synthesised featuring heterobicyclo[n.1.1]alkane cores.
Some methods for the construction of bicyclic carbocycles translate well to the synthesis of heterocycles (such as (2+2)-photocycloadditions, see below), while others appear to be limited to the carbocyclic scaffolds (especially in the case of BCP synthesis, where no heterocyclic analogues yet exist). This has driven the development of innovative new solutions for the construction of bicyclic heterocycles;14–16 this tutorial review will present an overview of current knowledge on the construction of heterobicyclo[n.1.1]alkanes, focusing on recent innovations as well as seminal discoveries. Coverage will include discussion of strategies for the synthesis of hetero-BCHexs, which can be grouped into categories of intermolecular or intramolecular cycloadditions, ring-closing reactions, and rearrangements; and similarly hetero-BCHeps, for which intermolecular and intramolecular cycloadditions, ring-closing reactions and ring-opening reactions have been developed.
Synthesis of Hetero-BCHexs
Intermolecular cycloadditions (radical)
Bicyclo[1.1.0]butanes (BCBs) are a popular precursor to hetero-BCHex (HBCHex) and hetero-BCHep (HBCHep) cores featuring a variety of bridge and bridgehead substituents, with methodologies divided into radical- and ionic-driven transformations. Radical-based approaches towards HBCHexs can be separated into two further categories: activation of the BCB component, or activation of the non-BCB component, with the latter strategy being more common in the literature.Activation of the non-BCB component usually results in formation of a biradical species via an energy transfer pathway (EnT). One end of the biradical then adds across the strained BCB central bond, leaving a radical on the opposite bridgehead position. Next, the 1,4-biradical species undergoes intersystem crossing (ISC) followed by radical recombination to lead to the hetero-BCHex structure. It is common to arrive at the triplet state by excitation of a π-bond as demonstrated by Guo et al.17 and Glorius et al.18 using cyclic sulfonyl imines and carbonyls respectively (Fig. 3a), which provides access to a range of 2-heterobicyclo[2.1.1]hexanes with the heteroatom on the side of the BCHex closest to the electron-withdrawing group (this contrasts with the analogous methodology for hetero-BCHeps, see below). In the case of the oxa-BCHep, transfer of the aryl group to one of the former BCB bridge carbon atoms was observed, which was rationalised by a secondary hydrogen atom abstraction/aryl migration process.
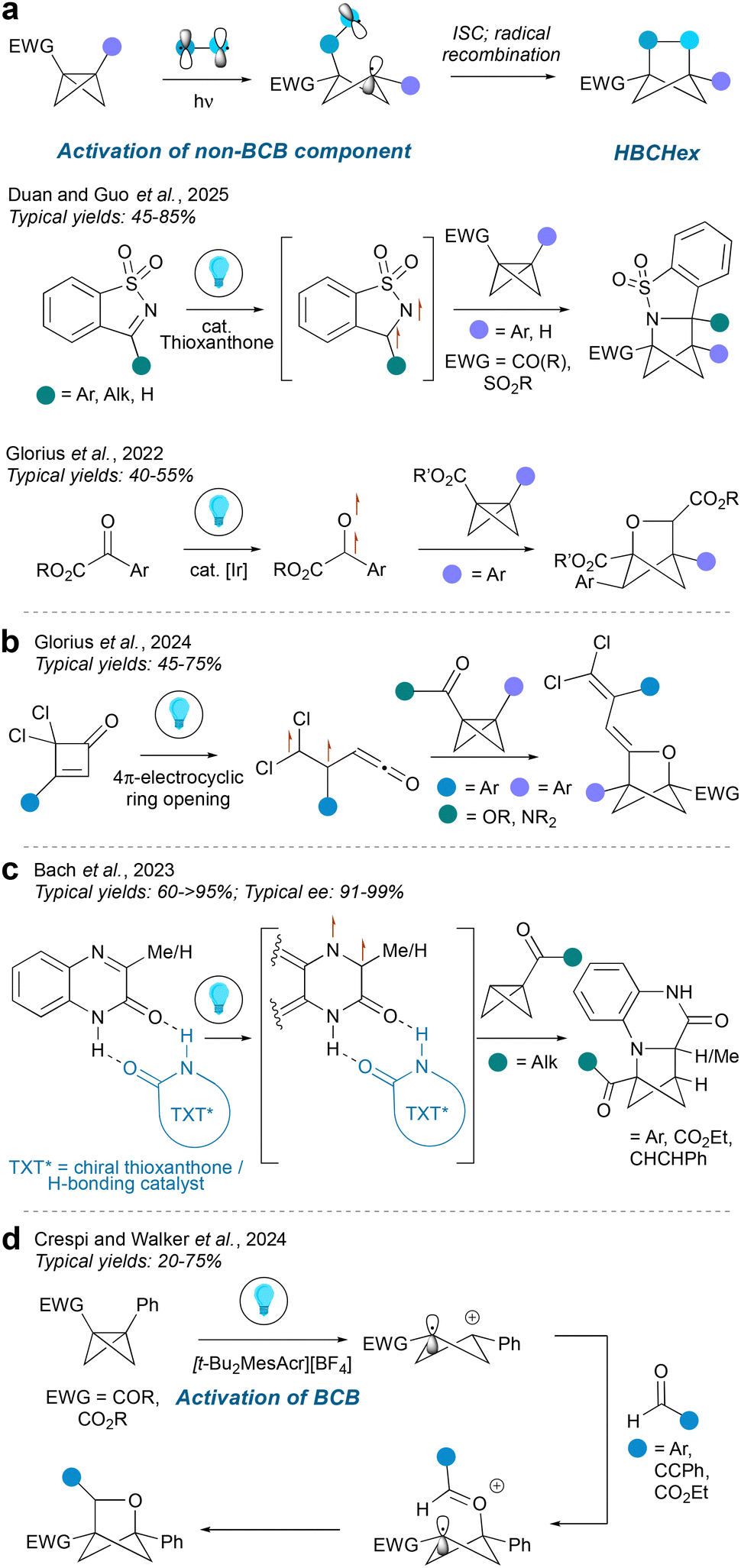 | ||
| Fig. 3 Synthesis of HBCHexs from BCBs via radical-mediated intermolecular cycloadditions. | ||
An alternative excitation pathway was reported by the Glorius group19 using a dichloroketene-derived cyclobutenone as a precursor to a triplet biradical ketene species via 4π-electrocyclic ring opening (Fig. 3b). For all these transformations, the substituents on the heteroatom-containing bridge are determined by the diradical precursors, whereas the bridgehead positions exclusively derive from the pre-installed substituents on the BCB; the latter universally features an electron-withdrawing group, and commonly an aryl ring, on the two bridgehead carbon atoms. These substituents proved particularly important in the case of the triplet ketene reaction, where it was found that the absence of an aryl group on the BCB led instead to a carbocyclic BCHex core as the major product. Enantioselective intermolecular radical insertions into BCBs were reported by Bach and co-workers, who deployed a chiral template that also served as a photosensitiser in the transformation (Fig. 3c).20 Although their attempts were mostly focused on the carbocyclic BCHex system, the method was also shown to provide access to enantioenriched aza-BCHexs.
A mechanistically distinct protocol was reported by Walker et al. (Fig. 3d),21 where the BCB is first activated via single-electron oxidation to form a cyclobutyl carbocation radical, with the carbocation located on the carbon atom distal to the EWG, and invariably featuring a cation-stabilising aryl group. This carbocation is then attacked by the carbonyl oxygen of an aldehyde; the oxocarbenium ion radical species was then proposed to undergo cyclisation followed by photoreduction.
Intermolecular cycloadditions (polar)
Intermolecular polar cycloadditions offer an alternative entry to HBCHexs. As with radical-mediated BCB insertions, These can be promoted either by activation of the BCB, or the other reaction component. The former activation mode typically proceeds through the coordination of the BCB electron-withdrawing group (i.e., carbonyl) to a Lewis acid. The first account of such reactivity was reported by the Leitch group, who used gallium(III) triflate to promote a formal (2σ + 2π) cycloaddition in the synthesis of aza-BCHexs (Fig. 4a).22 The transformation is proposed to proceed via Ga(III)-promoted cleavage of the strained inter-bridgehead bond of the BCB to give a Ga(III) enolate which then reacts with the imine. The resulting gallium(III) amide anion then cyclises onto the cyclobutyl cation on the other side of the cyclobutane system to give the anticipated aza-BCHex (these bond formations may occur in the opposite order). A metal-free variant has been developed by Glorius et al., where boron trifluoride serves as the Lewis acid activator to produce oxa-BCHex cores on reaction with aldehydes.23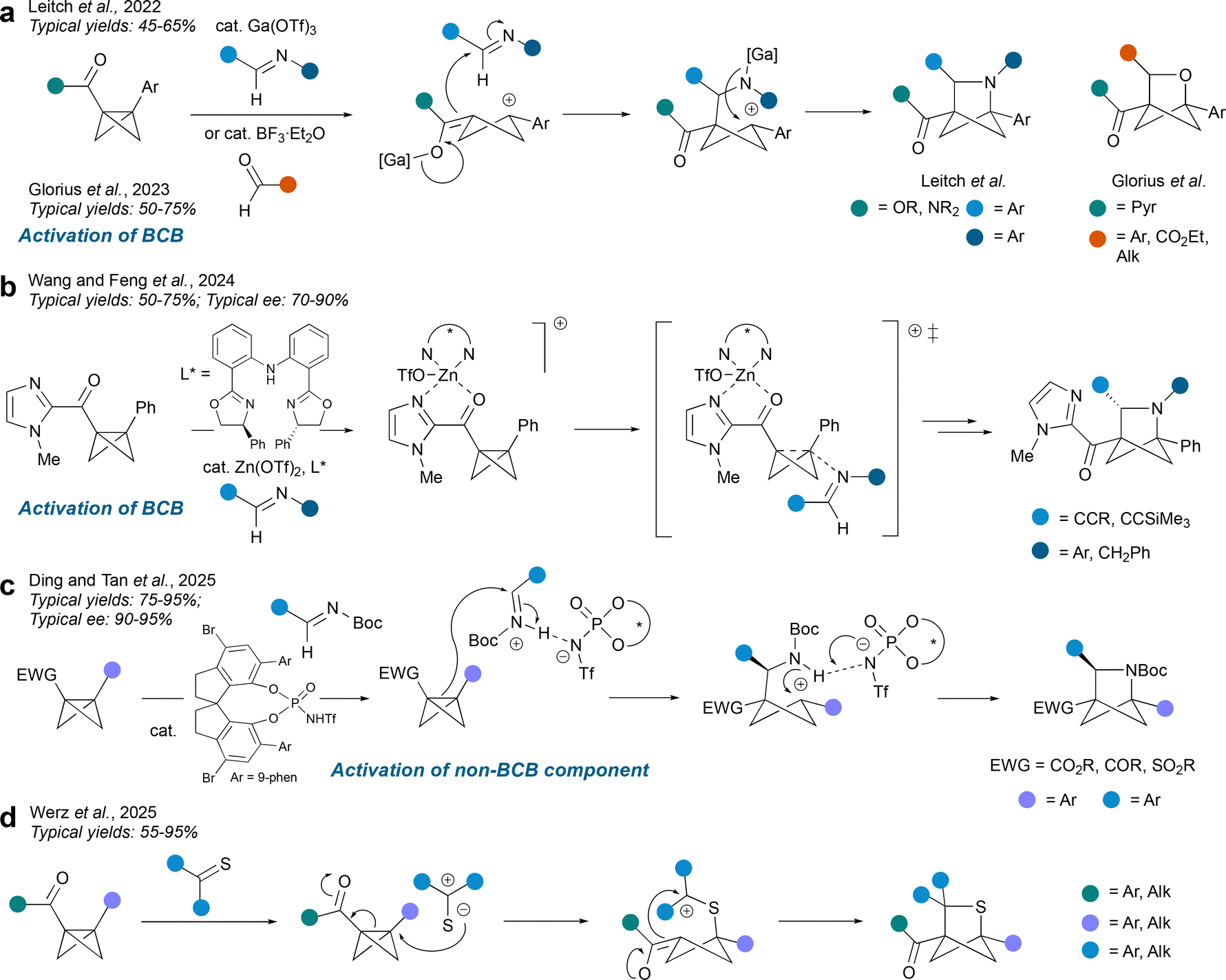 | ||
| Fig. 4 Synthesis of HBCHexs via intermolecular polar formal cycloadditions. | ||
Intermolecular polar cycloadditions can also be performed in an enantioselective manner as demonstrated by Feng et al. using a chiral zinc catalyst (Fig. 4b).24 The key step of the sequence is again the coordination of the (chiral) Lewis acid to the 2-acylimidazole group on the BCB, which then affords stereocontrol in the BCB ring opening event. In contrast to the mechanism proposed by Leitch et al., the authors propose that zinc complexation by the 2-acylimidazole promotes attack by the imine nitrogen atom on the distal bridgehead carbon, triggering BCB ring opening. Stereoselective cyclisation of the resulting zinc enolate forms the enantioenriched aza-BCHex.
Activation of the non-BCB component in polar intermolecular cycloadditions is a less explored strategy, but can be achieved by chiral Brønsted acids as demonstrated by the Tan group (Fig. 4c).25 In this work, a chiral N-triflylphosphoramide interacts with the imine electrophile via hydrogen bonding, resulting in formation of an ion-paired iminium ion that is susceptible to attack by the BCB, with the central C–C bond acting as a nucleophile. The resulting intermediate undergoes cyclisation onto the ensuing cyclobutyl cation to furnish the desired aza-BCHex.
In some cases, activation of either component is not required for a successful formal (2σ + 2π) cycloaddition (Fig. 4d). For example, the Werz group achieved the synthesis of 2-thiabicyclo[2.1.1]hexanes from thiocarbonyls in the absence of any catalyst, irradiation or heating. The reaction is proposed to proceed through a stepwise mechanism which benefits from the highly polar nature of the C–S double bond, and the consequent nucleophilicity of the sulfur atom.26
Intramolecular cycloadditions
Crossed intramolecular (2+2) cycloadditions of 1,5-dienes are a well-established strategy to form the aza-BCHex scaffold. The first such synthesis of aza-bicyclo[2.1.1]hexanes was reported independently by the Pirrung and Clardy groups in 1980,27,28 inspired by the isolation of the aza-BCHex natural product 2,4-methanoproline from the seeds of Ateleia herbert smithii. This bicyclic molecule was found to serve as a natural non-proteinogenic amino acid, and a proline analogue that has been found to be a useful tool in peptide modification.29 Since their seminal work, this photochemical strategy has been modified towards a kilogram scale30 and also used to form various HBCHex skeletons, including enantioselective examples.31,32 This reaction proceeds via initial excitation of one of the double bonds of the diene substrate (typically conjugated to an electron-withdrawing group or π-system) to a triplet biradical, mediated by a photosensitiser. This step is followed by a 5-exo-trig cyclisation to generate a 1,4-biradical, which then undergoes intersystem crossing (ISC) and internal radical recombination (cyclisation) to give the HBCHex.The non-asymmetric intramolecular (2+2) photocycloaddition can be used to access aza-BCHex,27,28 oxa-BCHex,33 and 2-thia-3-aza-BCHex 2,2-dioxide34 motifs with substitution patterns depending on the nature of the diene starting material (Fig. 5a). An enantioselective variant of the reaction was developed for aza-BCHex35 and oxa-BCHex36 cores (Fig. 5b and c). In both cases, a neighbouring heteroatom on the diene substrate coordinates to a chiral metal catalyst, with this complex then undergoing energy transfer (EnT) with the triplet state of the DPZ photosensitiser to furnish a biradical species. In the aza-BCHex case (Fig. 5b), an aza-arene is used for scandium catalyst coordination, and the stereoinduction derives from the use of chiral ligands on the Lewis acid. The synthesis of the oxa-BCHex (Fig. 5c) instead uses a chiral-at-metal Lewis acid, which forms a bidentate complex with the two oxygen atoms in the starting material.
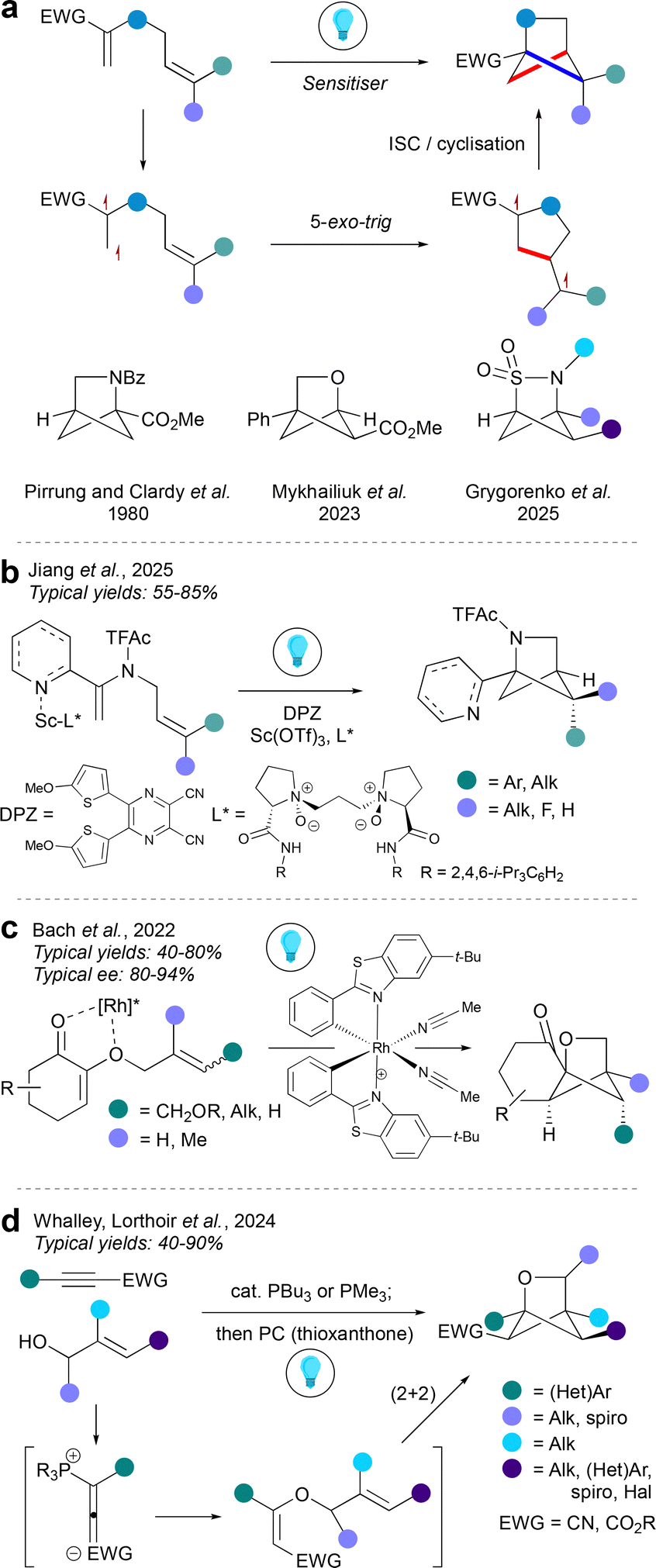 | ||
| Fig. 5 Synthesis of HBCHexs via intramolecular crossed (2+2) cycloadditions. | ||
A complementary entry to the oxa-BCHex framework by intramolecular (2+2) cycloaddition was recently reported by Whalley, Lorthoir and co-workers (Fig. 5d).37 This modular method assembles the cycloaddition substrate from an electron-deficient aryl alkyne and an allylic alcohol using a phosphine organocatalyst, which is proposed to facilitate the addition of the alcohol to the alkyne via an allenylphophonium ion intermediate. The in situ generated allyl vinyl ether then undergoes photocatalysed cycloaddition via an EnT pathway. This approach enables the installation of a wide variety of groups on several positions of the oxa-BCHex framework.
Ring-closing reactions
Ionic cyclisations are one of the most popular and scalable methods of accessing HBCHex cores. Functionalised cyclobutanes often serve as easily accessible substrates, as demonstrated in the aza-BCHex synthesis by de Kimpe et al.38 and Kondratov et al.39 and en route to oxa-BCHex by Mykhailiuk and co-workers (Fig. 6a).40 In the second two methods, iodine serves as the promoter for cyclisation, forming an iodonium ion which triggers a 5-exo-tet cyclisation with either a nitrogen or oxygen atom acting as the nucleophile. This is a very scalable approach to the hetero-BCHex scaffold, not least as it avoids the need for preparation of BCBs. The methodology developed by the de Kimpe group uses a cyclobutane pre-functionalised with an electrophilic chloride-bearing carbon atom, where cyclization is now achieved by the addition of a nucleophile to an imine, itself derived from a cyclobutanone. Another elegant cyclisation strategy was demonstrated by Gorichko and co-workers, with a focus on the synthesis of 2,4-methanoproline, but with the potential for generalisation.41 Their strategy begins by formation of a spirooxetane which under acidic conditions opens and re-cyclises to a bicyclic lactone. Subsequent chlorination and Curtius rearrangement gives rise to an oxa-BCHep aminolactone. Treatment of this lactone with sodium hydroxide facilitates ring-opening to furnish a bicyclo[1.1.0]butane intermediate, which undergoes cyclisation of the amine onto the chloromethyl substituent to form the aza-BCHex product (Fig. 6b).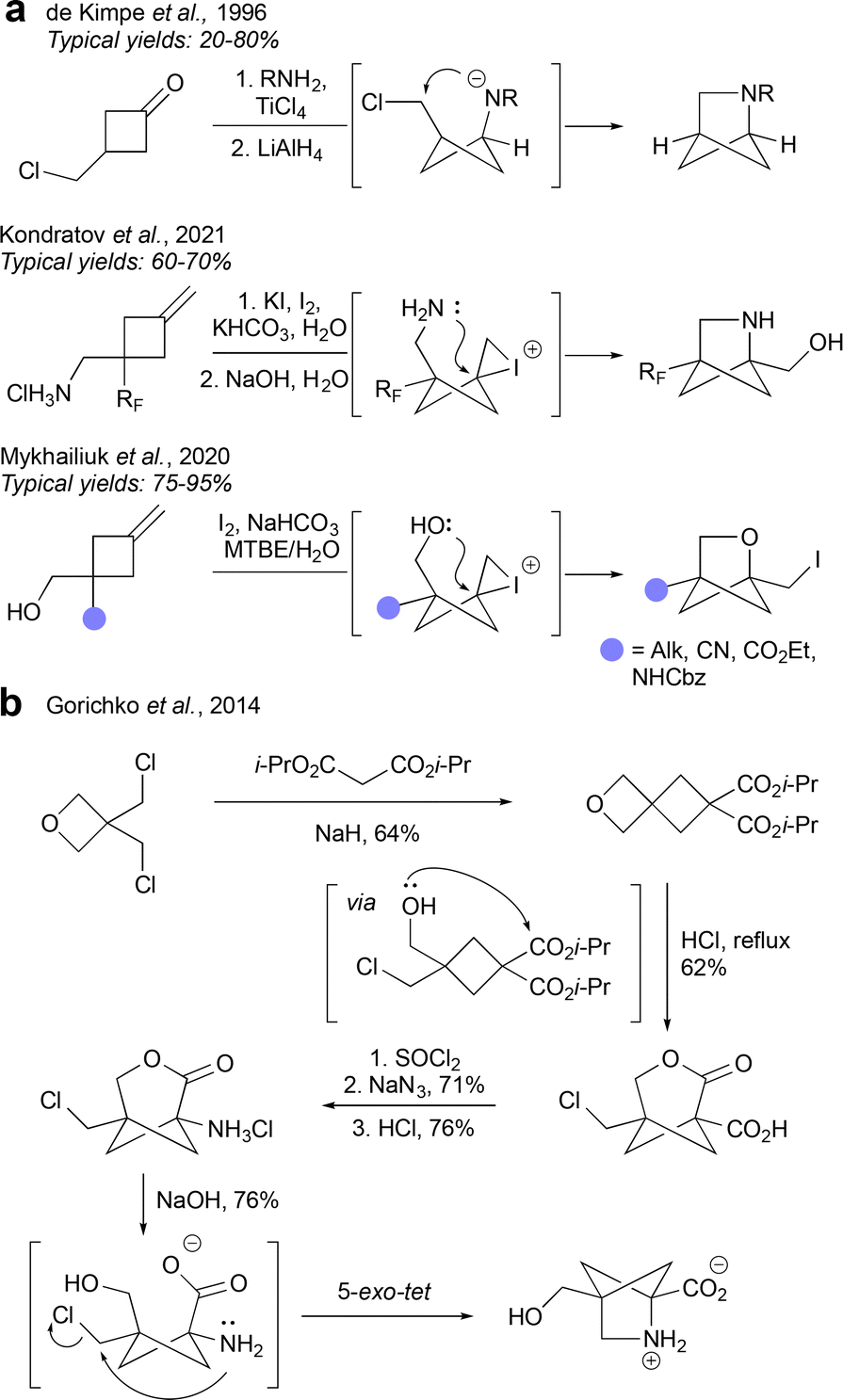 | ||
| Fig. 6 Synthesis of HBCHexs under ionic cyclisation conditions. | ||
Aside from access to 2-heterobicyclo[2.1.1]hexanes, ionic cyclisations can also be applied to the synthesis of 1-hetero-BCHexs as recently demonstrated by Aggarwal and co-workers (Fig. 7).42 The method involved is a formal (2σ + 2π) cycloaddition onto a 1-azabicyclo[1.1.0]butane (1-aza-BCB). This multistep process is initiated by acid-promoted ring-opening of the aza-BCB by a bromide nucleophile to give a 3-bromoazetidine. This is followed by an iridum-photocatalysed atom transfer radical addition reaction, where SET from the excited state photocatalyst leads to a carbon-centred radical that undergoes addition to an alkene acceptor, followed by halogen atom transfer to reform a C–Br bond. Cyclisation of the resulting bromo-amine affords the 1-aza-BCHex.
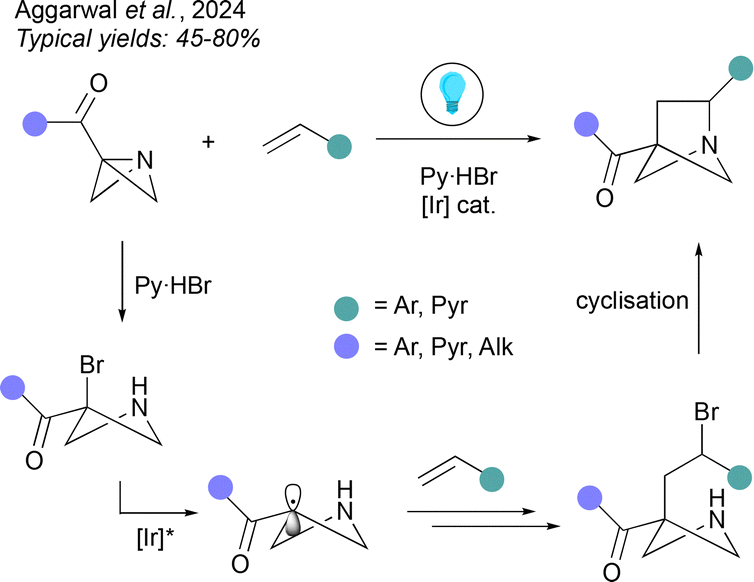 | ||
| Fig. 7 Synthesis of 1-aza-BCHexs from 1-aza-BCBs. | ||
Rearrangements
Hetero-BCHexs can also be made via rearrangement of heteroatom-containing ladderane derivatives, a strategy that does not translate to the hetero-BCHep systems described below. The Krow group have contributed multiple papers on this topic, with a particular interest in aza-BCHex structures. A good example is a 4-step approach to the aza-BCHex core, beginning with the reaction of a pyridinium ion with a Grignard reagent to form a dihydropyridine (Fig. 8).43 This dearomatised heterocycle is then directly subjected to irradiation at 300 nm to promote a photochemical 4π-electrocyclisation to an azabicyclo[2.2.0]hex-5-ene, which in the case of the example in Fig. 8 is treated with NBS as an electrophile and acetate as nucleophile. This presumably generates an intermediate tricyclic aziridinium ion that undergoes rearrangement to a highly functionalised aza-BCHex on reaction with acetate (other electrophilic activators have also been investigated).44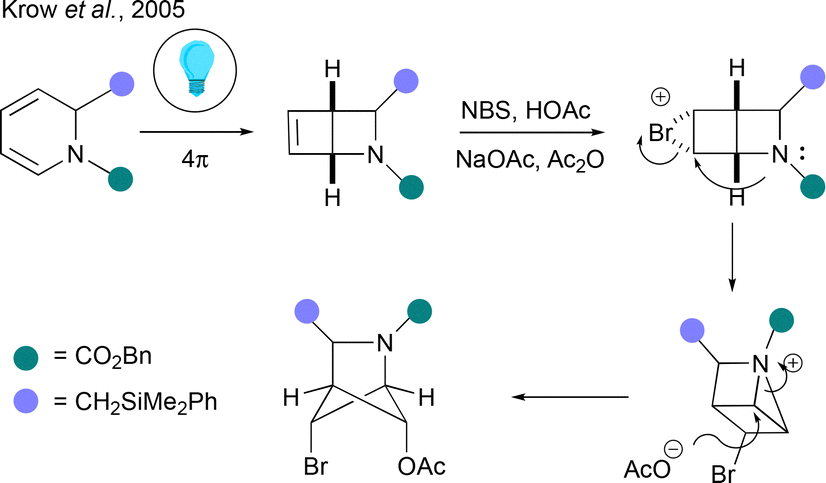 | ||
| Fig. 8 Synthesis of aza-BCHexs from dihydropyridines. | ||
Synthesis of Hetero-BCHeps
Many approaches to hetero-BCHeps (HBCHeps) are conceptually similar to HBCHex synthesis, such as insertion reactions into BCBs. However, there are some exceptions which are unique to the HBCHep scaffold, and there are also additional opportunities to vary the position of the heteroatom in the three-atom bridge.Intermolecular cycloadditions (radical)
Intermolecular formal cycloadditions involving bicyclo[1.1.0]butanes (BCBs) have emerged as one of the most popular ways to access a diverse range of hetero-BCHeps with various bridge and bridgehead substituents. Several distinct methods have been developed involving either radical or two-electron mechanisms, all of which use a catalyst to activate one or other of the cycloaddition components.Cycloadditions that proceed by radical pathways can be enabled by either generating a radical from the BCB component, or the non-BCB component. Fig. 9 shows examples of the latter strategy, which proceed by similar mechanisms. A photocatalyst (PC) is employed to firstly generate a radical from the non-BCB component by single electron transfer (SET), which then reacts with the strained central bond of the BCB to generate a cyclobutyl radical at the former bridgehead carbon atom distal from the electron-withdrawing group. This radical is then oxidised (simultaneously reducing the photocatalyst), followed by ring closure by nucleophilic capture of the carbocation. This tactic provides access to a range of 2-hetero-BCHeps, always with the nucleophilic heteroatom positioned on the side of the BCHep furthest from the bridgehead carbonyl that originated in the BCB. Using this approach, Glorius et al.45 accessed 2-oxa-BCHeps from tert-butyl α-bromoesters (Fig. 9a), with an aromatic group on the of the bridgehead positions and an ester or amide on the other, where release of the tert-butyl cation facilitates ring closure; the bridge position can feature an alkyl or benzyl substituent, or no substituent. The same group (Fig. 9b) also demonstrated the synthesis of 2-oxa-4-azaBCHeps via the addition of N-centred radicals generated from pyridinium hydrazides,46 which similarly feature an aromatic group on one of the bridgehead positions and an ester or amide on the other; various aryl groups could be installed on the bridge position. Hari et al.47 reported an elegant method to access both 2-oxa- and 2-aza-BCHeps (Fig. 9c, again with an aromatic group on one of the bridgehead positions and an ester on the other). Use of a redox-active ester enabled decarboxylative generation of an alkyl radical, with a separate β-heteroatom nucleophile undergoing cyclisation. Various alkyl and aryl groups can be installed on either of the bridge positions, including spirocyclic and fused rings. Enantioenriched HBCHeps could also be achieved using enantioenriched chiral precursors, since the stereochemistry at the nucleophile-bearing carbon atom on the backbone is maintained throughout the transformation.
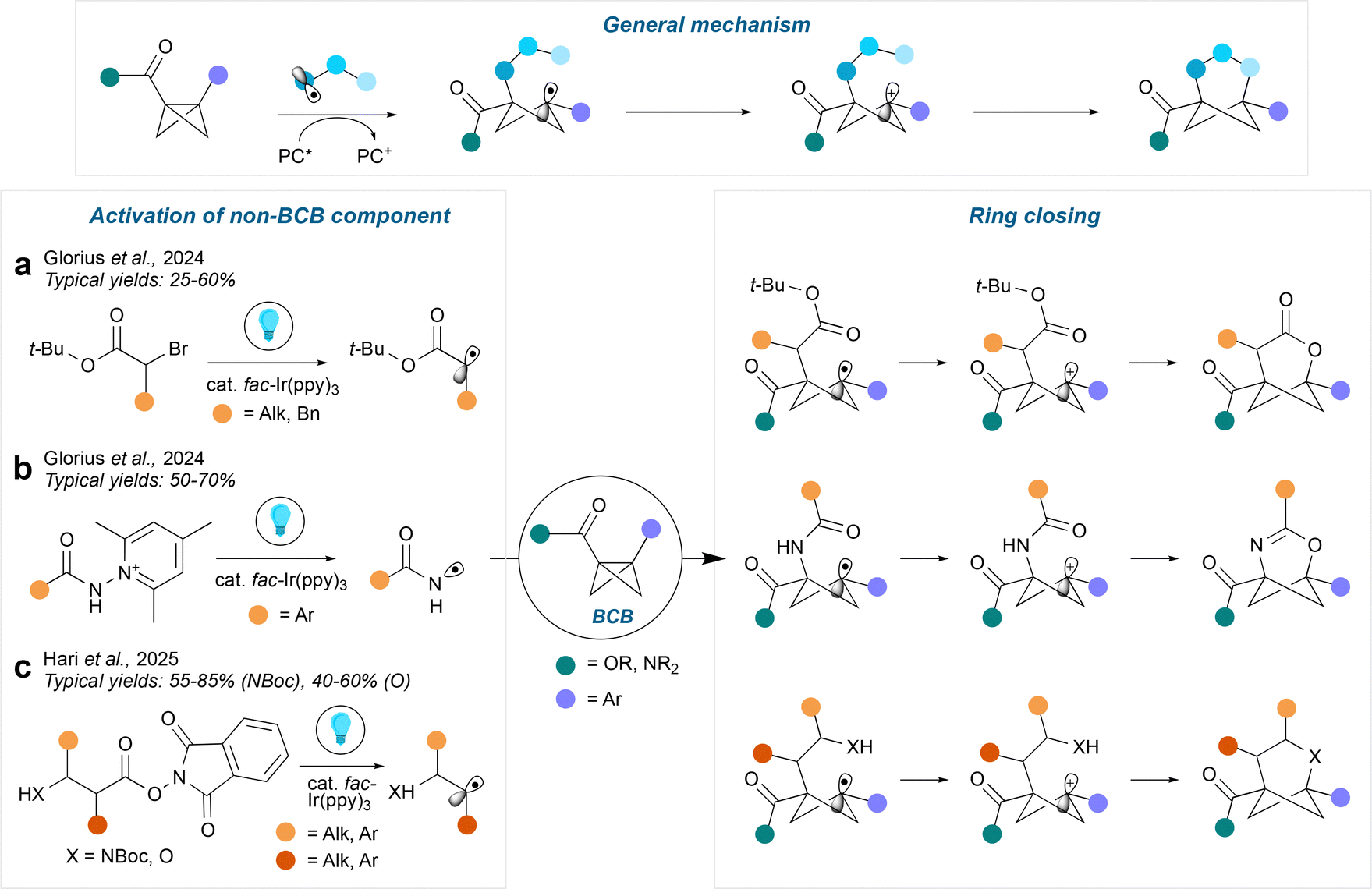 | ||
| Fig. 9 Synthesis of HBCHeps via radical-based intermolecular cycloadditions involving activation of the non-BCB component. | ||
An alternative strategy to achieve a formal (3+2) cycloaddition firstly generates a radical from the BCB, as demonstrated by Zheng et al. (Fig. 10).48 Here, a TiIII catalyst triggers reductive ring-opening of the BCB via a ketyl radical anion, leading to an enolate-cyclobutyl radical. The radical is then trapped by addition to a vinyl azide with loss of N2, and the resulting iminyl radical undergoes cyclisation by addition to the TiIV enolate, a process that ejects and regenerates the TiIII catalyst. As before, this method accommodates a range of aromatic bridge substituents, and the bridgehead substituents are again an aromatic ring and a carbonyl. However in contrast to the above methods, the heteroatom is introduced on the side of the BCHep closest to the carbonyl substituent.
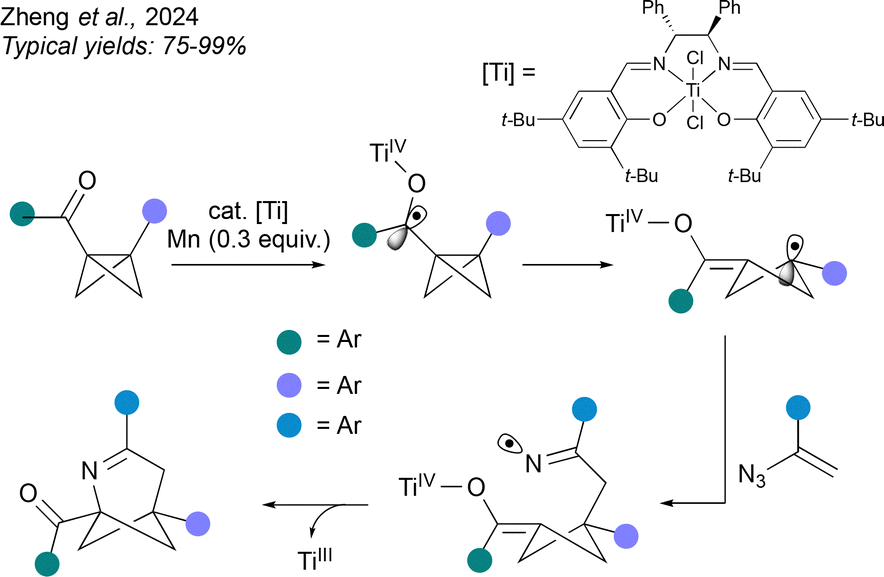 | ||
| Fig. 10 Synthesis of 2-aza-BCHeps by TiIII-catalysed radical-based intermolecular cycloaddition involving activation of the BCB component. | ||
Intermolecular cycloadditions (polar)
In a similar manner to radical-mediated formal cycloadditions, the polar pathways can be enabled by activation of either the BCB with a Lewis acid, or the non-BCB component. Examples of reactions which proceed by activating the non-BCB component are shown in Fig. 11; in most of these cases, the activated component acts as a nucleophile and attacks the BCB, causing ring-opening to form an enolate. The enolate then cyclises onto a pendent electrophile to close the ring.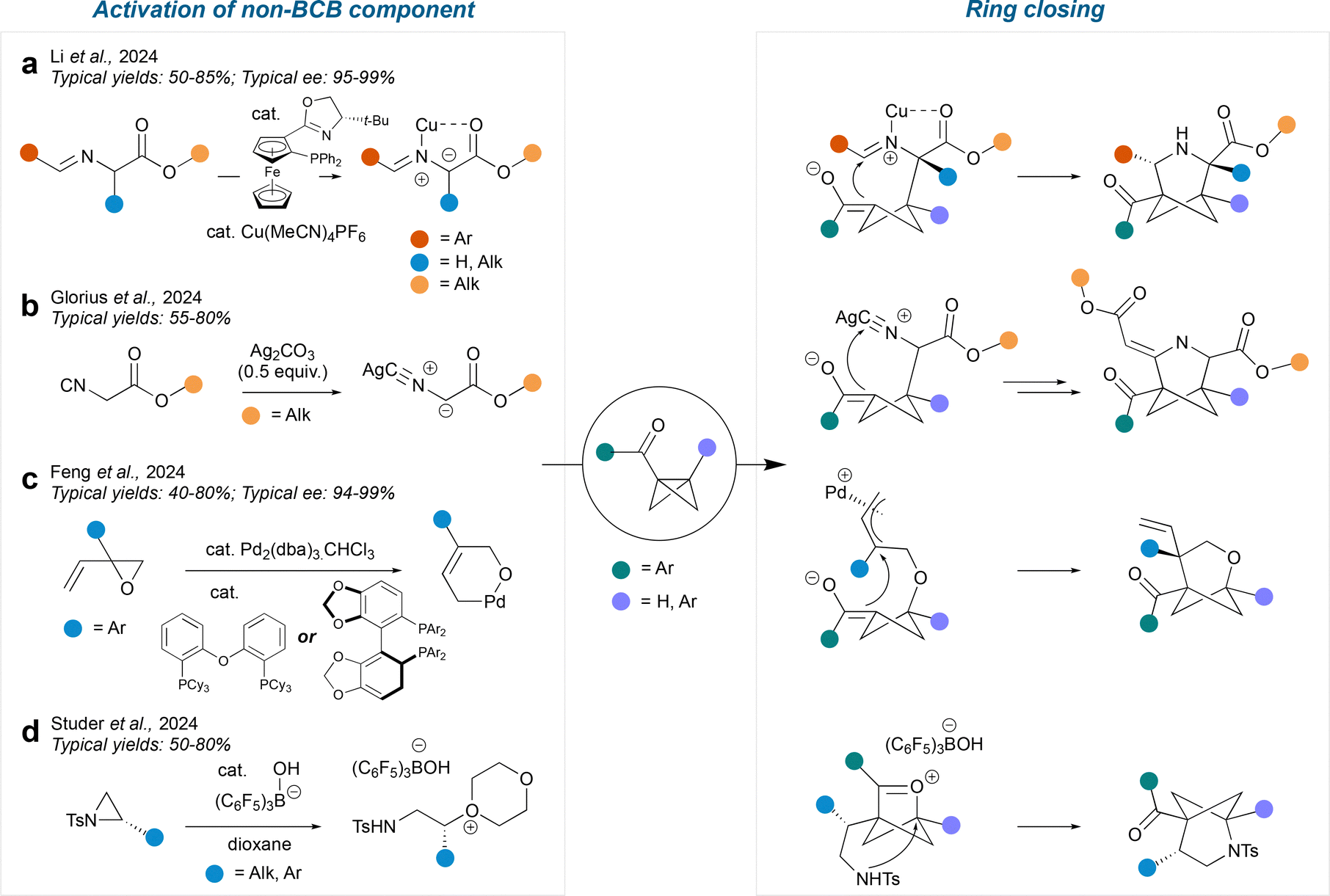 | ||
| Fig. 11 Polar intermolecular cycloadditions involving activation of non-BCB component. | ||
In a method reported by Li et al.,49 an aldimine ester is activated by a CuI catalyst to produce a Cu-azomethine ylide intermediate. This proceeds to attack the BCB, leading to a 3-azaBCHep structure. This method tolerates bridgehead substituents of aryl or hydrogen on one side and ketone on the other. The bridge substituents are most commonly aryl, ester and alkyl. If chiral ligands are used with the Cu catalyst, then enantioenriched chiral products may be obtained. The Glorius group report a related silver-enabled cycloaddition50 which uses Ag2CO3 to generate a silver–isocyanide complex. This complex acts as a nucleophile and attacks the BCB in the same way. However, after ring closure, the product undergoes a further cycloaddition reaction with another isocyanide complex to produce a 3-azaBCHep with an ester and an alkene as the bridge substituents. As before, the bridgehead substituents are mainly a combination of aryl and carbonyl.
In an alternative method by Feng et al.,51 a Pd catalyst facilitates an epoxide ring-opening by forming a π-allyl group, leaving the alkoxy group free to attack the BCB. The resulting enolate can close onto either side of the π-allyl system and the regioselectivity is controlled by the ligand chosen for the Pd catalyst. The bridge substituents are always a vinyl and aryl in the 4-position and the bridgehead substituents are always a ketone on one side and an aryl or hydrogen on the other side. The use of chiral phosphine ligands enables the synthesis of enantioenriched 2-oxaBCHeps.
Finally, Studer et al. reported a method involving the activation of an aziridine with B(C6F5)3, which results in stereospecific ring opening of the aziridine by dioxane.52 Then, in contrast to previous methods, the BCB acts as the nucleophile and attacks the dioxane-derived oxonium ion (with loss of dioxane) to form an oxocarbenium-BCHex ion intermediate. The nitrogen finally displaces the oxocarbenium ion in a transannular fashion, resulting in a 2-azaBCHep structure. The bridgehead substituents are a hydrogen on the same side as the nitrogen and an aryl ketone on the other side. Various sulfonamides can be tolerated on the nitrogen and various substituents can be introduced onto the 4-position of the bridge, including aryl, alkyl and allyl groups. The stereochemistry of the aziridine is conserved throughout the reaction, as the mechanism involved two SN2 reactions at the same site.
An alternative way to carry out a polar cycloaddition, shown in Fig. 12, is to activate the BCB with a Lewis acid, which promotes BCB fragmentation to an enolate-cyclobutyl cation. The nucleophilic part of an ambiphilic reaction partner then captures the cation, and finally the enolate reacts with the electrophilic centre of the reaction partner to close the 3-atom bridge. As these reactions rely on BCB activation, they inevitably feature a carbonyl group on one of the BCB bridgehead positions (which can coordinate to a Lewis acid) and an aryl group on the other bridgehead position (which can stabilise the intermediate carbocation). Examples include the method by Deng et al.53 which uses Eu(OTf)3 to facilitate reaction of a nitrone with the BCB, providing access to 2-oxa-3-azaBCHeps. This method has also been rendered asymmetric by Feng and co-workers using a chiral cobalt catalyst.54 In another method by the Deng group,55 In(OTf)3 catalyses the formal cycloaddition of hydrazones, leading to 2,3-diaza-BCHeps, or the stepwise addition of amine-substituted π-allylindium species, leading to 2-azaBCHeps; enantioenriched products can also be synthesised if a chiral iridium catalyst is also used. As before, these methods feature mainly aryl and carbonyl bridgehead substituents on the HBCHep product, while a range of mainly aryl substituents are available for the bridge positions. The Feng group reported a related method56 using Sc(OTf)3 as catalyst to effect the addition of 1,4-zwitterionic pyridinium thiolates, affording thiabicyclo[3.1.1]heptenes. This method produces compounds with similar aryl and carbonyl substituents on the bridgehead positions, but this time the bridge substituents are esters.
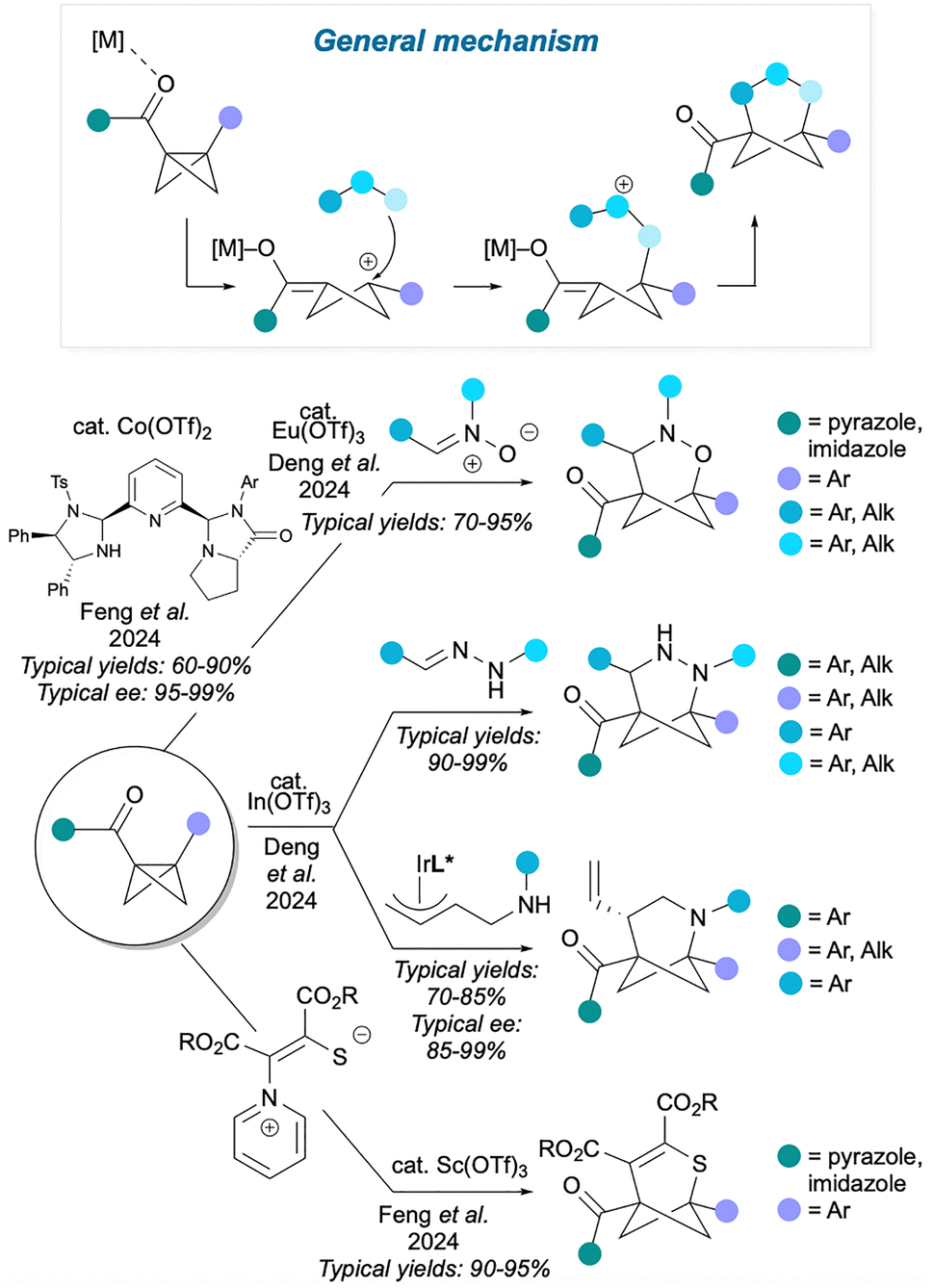 | ||
| Fig. 12 Polar intermolecular cycloadditions involving activation of BCB component. | ||
To summarise this section, it is possible to synthesise a wide variety of hetero-BCHeps by intermolecular cycloadditions, with heteroatoms in various positions in the ring, via a wide selection of different radical and polar catalytic methods. However, all methods share the common feature of a strain-release reaction involving the BCB central bond, followed by ring closure to form the BCHep; this arguably limits the diversity of bridgehead substituents, which must be amenable to the activation process, and are therefore most commonly an aryl group on one side and a carbonyl on the other. These methods offer wider variation in the pattern of bridge substitution, however most of them are limited by the requirement to have at least one bridge substituent of a certain type at a particular site on the ring. Such methods could therefore become more broadly applicable if it were possible to precisely introduce any desired substituent at any position on the ring, including with control of absolute stereochemistry, or indeed to prepare BCHeps with no bridge substituents at all, if so desired.
Intramolecular cycloadditions
Despite a longer history, intramolecular cycloadditions are relatively underexplored as an approach to hetero-BCHeps compared to the intermolecular variant. (2+2) Cycloadditions of 1,5 dienes usually favour the “cross product” bicyclo[2.1.1]hexane over the “straight product” bicyclo[2.2.0]hexane (Fig. 13), since the “cross product” is formed via a kinetically favoured 5-exo-trig cyclisation. However, cycloadditions of 1,6 dienes usually favour the “straight product” bicyclo[3.2.0]heptane, as this is also formed via a kinetically favoured 5-exo-trig cyclisation, over the “cross product” bicyclo[3.1.1]heptane,57 although there are a small number of reports where the “cross product” has been achieved.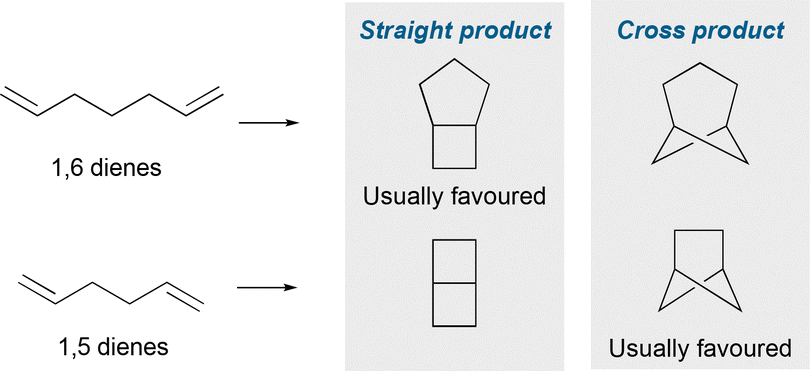 | ||
| Fig. 13 Straight product vs. cross product in intramolecular cycloadditions towards bicycloalkanes. | ||
In one of the earliest syntheses of aza-BCHeps, Schieweck et al. described a thermal (2+2) cycloaddition of N-acryloyl acrylimides, decorated with various substituents, to generate 3-aza-BCHep diones (Fig. 14a).58 In this reaction, one of the bridgehead substituents is an aryl group, but the other can be aryl, alkyl or a hydrogen atom, while the nitrogen substituent can be aryl, alkyl or H. This method could therefore potentially provide access to a wide range of medicinally-relevant aza-BCHeps with varying substitution patterns, albeit with the limitation that carbonyls must be included on the bridge, which introduces potential issues with downstream imide stability.
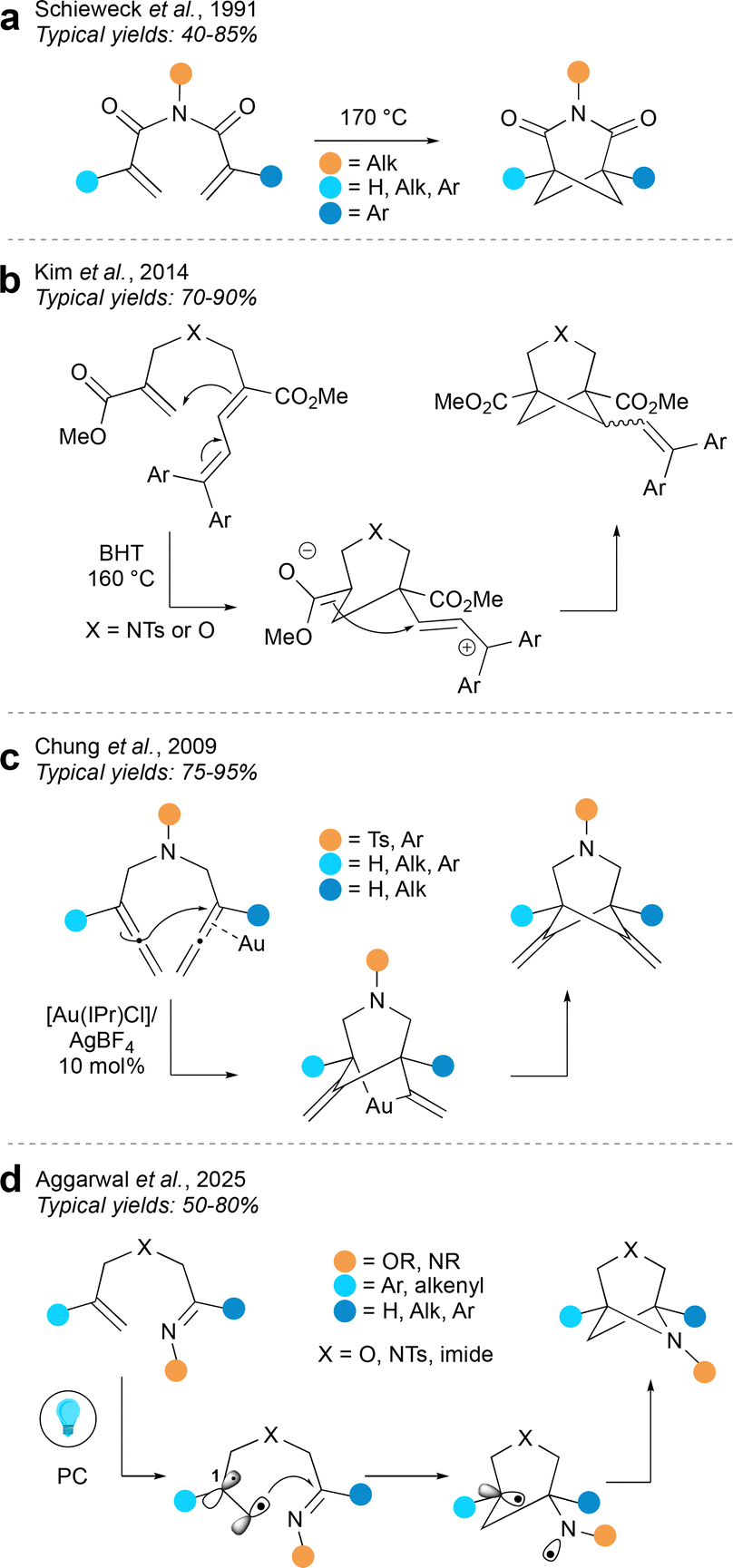 | ||
| Fig. 14 Synthesis of HBCHeps via intramolecular cycloadditions. | ||
A complementary method described by Kim et al.59 (Fig. 14b) circumvents the need for carbonyls on the three-atom bridge by including a sidechain on the smaller bridge that can stabilise a positive charge, allowing the reaction to proceed in a stepwise manner via Prins-type cyclisation. This produces BCHeps with no substituents on the larger bridge and with esters on the bridgehead positions, which would be useful for further functionalisation. However the diarylvinyl group, which is necessary for reactivity, is retained on the cyclobutane bridge. This method could certainly become more applicable should the diaryl part prove amenable to further functionalisation, or if other, more medicinally relevant groups could be installed at this position.
An alternative method developed by Chung et al. (Fig. 14c) uses an N-heterocyclic carbene/Au(I) catalyst system to mediate a formal cycloaddition between two allenes.60 This reaction is proposed to begin with nucleophilic attack of one allene component onto the Au-bound allene. This gives a gold-bridged intermediate which then undergoes reductive elimination to form the product. A limited selection of aryl and alkyl groups may be installed at the bridgehead positions, and the substituent on the nitrogen atom can be either an aryl or sulfonyl group. An advantage of this method is that it does not introduce unnecessary substituents on the larger bridge, however it does result in alkenes on both of the smaller bridges; its utility could be expanded by further functionalisation of these alkenes, and by extending the scope of the bridgehead substituents.
The intramolecular cycloaddition tactic has recently been exploited by the Aggarwal group for the synthesis of bis-hetero-BCHeps (Fig. 14d).61 This method proceeds by a radical mechanism, which is typically challenging because the “straight product” is generally favoured over the “cross product” due to the kinetic preference of a 5-exo-trig over a 6-exo-trig cyclisation. This issue was overcome by including a radical-stabilising group at the C1 position to favour the 6-exo-trig pathway, leading to the desired “cross product”. The group X in the larger bridge may be oxygen, carbon or nitrogen, while the bridgehead substituents can be a range of aryl, allyl or carbonyl groups. Additional carbonyls may also be attached to the bridge positions but these are not necessary. Aside from the last example, these intramolecular cycloadditions were reported before the current trends in using (hetero)bicycloalkanes as bioisosteres in drug scaffolds. As such, they represent a relatively underexplored area which could be profitable to revisit; if the scope of substituents could be expanded and further functionalisations developed, these methods could become useful tools for the synthesis of hetero-BCHeps.
Ring-closing reactions
Aside from cycloadditions, the main way to construct hetero-BCHeps is by cyclisation reactions. There are several varieties of ring-closing strategies to choose from, which provide access to a range of HBCHeps with heteroatoms in various positions on the ring.Perhaps the most prominent cyclisation method in the recent literature is that reported by Mykhailiuk et al. (Fig. 15a), where a spirocyclic cyclobutane–oxetane is used as a starting material. Reduction of either a nitrile62 or ester13 on the cyclobutane precedes Lewis acid-promoted cyclisation onto the oxetane, resulting in 3-hetero-BCHeps with no bridge substituents. A restriction of this strategy is that one of the bridgehead substituents is inevitably a (primary) alcohol, while the other (cyclobutyl) substituent can be one of a wide range of aromatic, alkyl, ester or nitrile groups. Importantly, this method is scalable and provides opportunities for further facile functionalisation to access a wide range of pharmaceutically relevant compounds. Subsequent to Mykhailiuk's work, Johnson et al. described a related strategy using a bromonium ion as electrophilic trigger for 2-oxa-4-azaBCHep synthesis.63 Possible substituents for the bridge position in this chemistry include a wide range of aryl and alkyl groups, while the alkyl bromide on the bridgehead position offers opportunities for further functionalisation. It is also possible to have a limited number of additional substituents already installed on the cyclobutane before the ring closure.
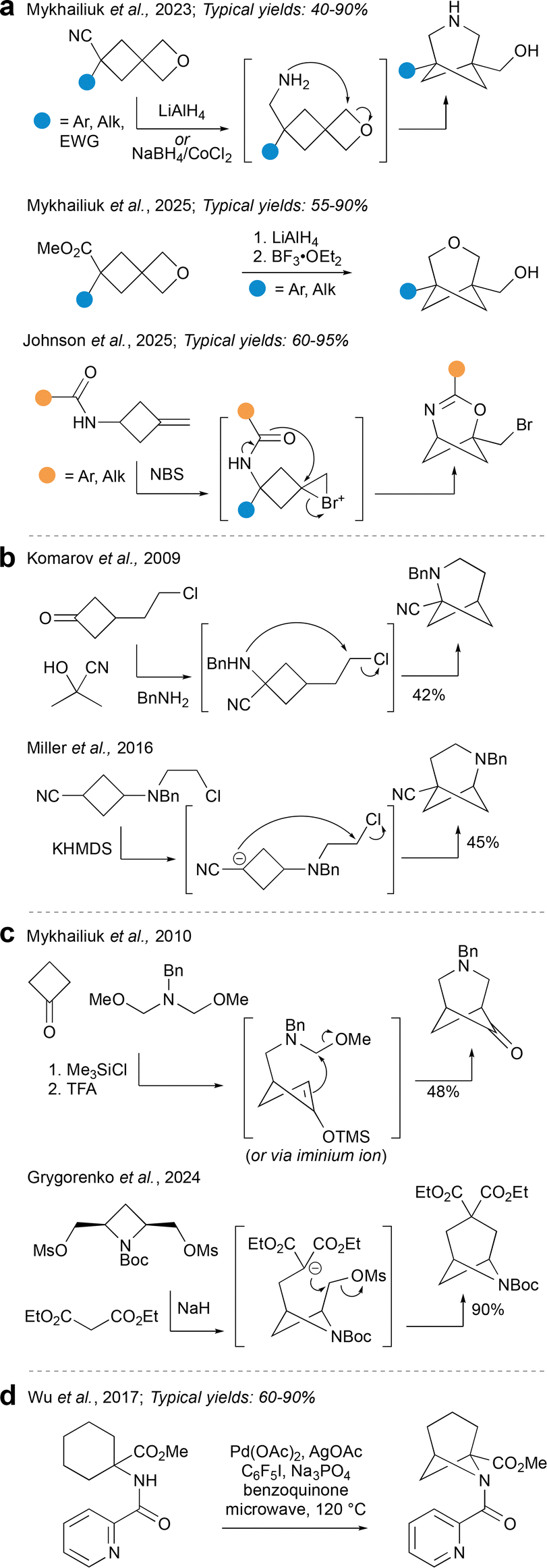 | ||
| Fig. 15 Synthesis of HBCHeps via cyclisation reactions. | ||
The next group of methods (shown in Fig. 15b) also involves cyclobutane-containing starting materials, however these are not spirocyclic and the mode of ring-closure varies between methods; all furnish HBCHeps with the heteroatom in the 2-position. Komarov et al.64 report a Strecker-type cyclisation which forms a C–N bond to make 2-azaBCHeps. The cyclisation was only attempted with the substituents shown, however there are opportunities for further functionalisation on the nitrogen or nitrile. A similar method was reported by Miller et al.,65 but this time a C–C bond is formed last by deprotonation adjacent to the nitrile, then attack of the aza-enolate on the pendent chloride. This results in 2-aza-BCHeps with the bridgehead nitrile group at the other side, which similarly present opportunities for further functionalisation.
Another variety of cyclisation reaction, shown in Fig. 15c, involves a one-pot tandem reaction between two symmetrical components. Mykhailiuk et al.66 report such a method which employs a double Mannich reaction to afford 3-aza-BCHeps, installing a ketone at the 6-position (Fig. 15c). More recently, Grygorenko et al.67 reported a double alkylation of diethyl malonate with a symmetric 1,3-disubstituted azetidine to generate 6-aza-BCHeps. Similarly, the transformation was performed with only one set of substituents, but was scalable and a variety of further functionalisations were possible. Both of these symmetrical additions furnish building blocks that can be prepared on multi-decagram scale, and can be easily modified to provide access to medicinally relevant compounds.
A final and rather unique mode of cyclisation is a C–H amination reported by Wu et al. (Fig. 15d).68 In this process, Pd-catalysed directed C–H activation occurs at the γ-position of the aminocyclohexane substrate, followed by formation of the 6-aza-BCHep by reductive elimination. The bridgehead substituents explored included a hydrogen atom, and a limited number of esters, while the nitrogen atom was substituted only with the pyridyl ketone directing group shown. C–H activation at the δ-position could also be achieved, so it seems possible that through careful choice of starting materials, this method could be expanded to access aza-BCHeps with the nitrogen in alternative positions on the ring. Cyclisation by C–H activation is certainly an underdeveloped area when it comes to bridged small ring chemistry, and could merit further exploration to unearth its full potential.
Ring-opening reactions
To complete the survey of methods for the synthesis of HBCHeps, a final category covers ring opening reactions of hetero[3.1.1]propellanes, developed by our group (Fig. 16).69 A unified synthetic strategy was employed to access the heteropropellane substrates, which proved capable of undergoing a range of radical-based ring opening reactions across the strained central bond to afford disubstituted 3-hetero-BCHeps. Bridgehead substituents included alkyl, aryl, halide, heteroatom and hydrogen groups. Variation of the substituent on the nitrogen atom, and further functionalisation at the bridgehead positions, are also possible. The yields for the synthesis of 3-oxa-BCHeps were generally the highest, followed by the 3-aza-BCHeps, while the scope of ring-opening reactions to form 3-thia-BCHeps was limited by competing fragmentation reactions.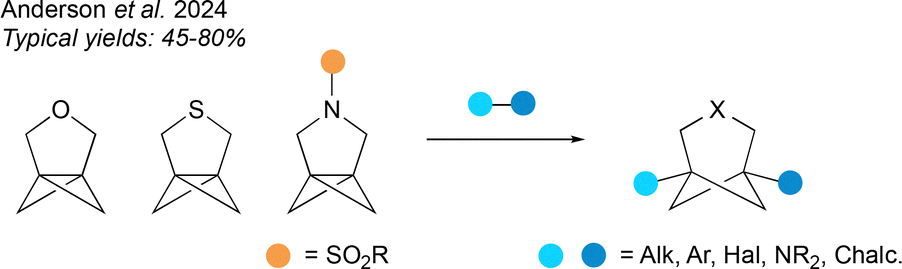 | ||
| Fig. 16 Ring-opening reactions of hetero[3.1.1]propellanes. | ||
The advantages of this method are the ability to introduce a diverse range of bridgehead substituents at a late stage in the BCHep synthesis, however further work is needed to develop a greater range of ring opening reactions for these heteropropellanes, since the ring-opening scope is not yet as extensive as that of their well-established carbocyclic cousins.
Conclusions
The growing interest within the medicinal chemistry community in heterobicyclo[n.1.1]alkanes has prompted the discovery of a variety of methods for their synthesis. A range of complementary and mechanistically distinct methods are now available to add to the medicinal chemist's toolbox. The most common strategies, applicable to both hetero-BCHeps and hetero-BCHexs, are intermolecular cycloadditions (both radical and polar), intramolecular cycloadditions, and ring-closing reactions. Hetero-BCHeps can additionally be prepared from ring-opening reactions of hetero[3.1.1]propellanes; this route is not yet possible for hetero-BCHexs since hetero[2.1.1]propellanes are currently unknown. Hetero-BCHexs can additionally be prepared via some unique rearrangement reactions.While these methods addressed the synthetic challenge of constructing these important scaffolds, there is still room for development regarding the need for precise installation of substituents at defined angles, and the late-stage generation of a diversity of different substituents on the scaffold. Many of the methods described within require specific groups to be present in the starting materials for their stability and reactivity, which can limit the diversity of accessible products and also result in the introduction of unnecessary substituents on the scaffolds. Many of the methods also have limitations in substrate scope, which, if overcome, could allow access to a wider diversity of substituted scaffolds. Conquering these limitations in current methods could greatly expand the applicability of this chemistry in drug discovery programmes.
Opportunities for further development may also lie in targeting new compound classes and transformations that have not yet been achieved. For example, despite the abundance of carbocyclic bicyclo[1.1.1]pentanes in the literature, there are still no examples of heterobicyclo[1.1.1]pentanes. Additionally, despite the heavy reliance on propellanes for the synthesis of carbocyclic BCPs and BCHeps, this chemistry remains underdeveloped for heterocyclic systems. Another unexplored area that could be worth investigating is the late-stage functionalisation of heterobicyclo[n.1.1]alkanes by C–H activation; the asymmetric synthesis or functionalisation of these scaffolds is a further goal that to date has only been partially met.
We hope that this review will provide a useful overview of the current state of the art at an exciting time in a field which is rapidly growing and developing. While excellent progress has recently been made, room undoubtedly remains for new innovations to address outstanding challenges with current methods, and to target new scaffolds, transformations and applications.
Conflicts of interest
There are no conflicts to declare.Data availability
No data is associated with this Review.Acknowledgements
RIR thanks AbbVie for a studentship. JR thanks the Centre for Doctoral Training in Synthesis for Biology and Medicine for a studentship (EP/L015838/1), generously supported by GlaxoSmithKline, MSD, Syngenta and Vertex, and to the Oxford Radcliffe Scholarship for funding. EA thanks the EPSRC (EP/S013172/1) for additional support.Notes and references
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed.
- F. Lovering, MedChemComm, 2013, 4, 515–519 RSC.
- J. R. Timothy and J. F. M. Simon, Drug Discovery Today, 2009, 14, 1011–1020 CrossRef PubMed.
- J. Tsien, C. Hu, R. R. Merchant and T. Qin, Nat. Rev. Chem., 2024, 8, 605–627 CrossRef CAS PubMed.
- P. K. Mykhailiuk, Org. Biomol. Chem., 2019, 17, 2839–2849 RSC.
- P. Hernández-Lladó, N. A. Meanwell and A. J. Russell, J. Med. Chem., 2025, 68, 16921–16939 CrossRef PubMed.
- B. R. Shire and E. A. Anderson, JACS Au, 2023, 3, 1539–1553 CrossRef CAS PubMed.
- A. Denisenko, P. Garbuz, S. V. Shishkina, N. M. Voloshchuk and P. K. Mykhailiuk, Angew. Chem., Int. Ed., 2020, 59, 20515–20521 CrossRef CAS PubMed.
- N. Frank, J. Nugent, B. R. Shire, H. D. Pickford, P. Rabe, A. J. Sterling, T. Zarganes-Tzitzikas, T. Grimes, A. L. Thompson, R. C. Smith, C. J. Schofield, P. E. Brennan, F. Duarte and E. A. Anderson, Nature, 2022, 611, 721–726 CrossRef CAS PubMed.
- Z. Fang, Q. Xu, X. Lu, N. Wan and W.-L. Yang, Synthesis, 2025, 1171–1179 CrossRef CAS.
- H. E. Diepers and J. C. L. Walker, Beilstein J. Org. Chem., 2024, 20, 859–890 CrossRef CAS PubMed.
- S. Nagasawa and Y. Iwabuchi, Synthesis, 2024, 1153–1170 Search PubMed.
- P. K. Mykhailiuk and D. Dibchak, Angew. Chem., Int. Ed., 2025, 64, e202505519 CrossRef PubMed.
- J. Dong, H. Liao and D. Xue, Synthesis, 2025, 722–731 CrossRef CAS.
- J.-J. Feng, Synlett, 2025, 621–629 CrossRef CAS.
- D. M. Whalley, O. Lorthioir, S. C. Coote and N. A. Anderson, Org. Biomol. Chem., 2025, 23, 7538–7565 RSC.
- S. Tian, R. Liu, K. Zhang, Y. Xia, Y. Liu, P. Li, X.-H. Duan and L.-N. Guo, Org. Lett., 2025, 27, 3818–3824 CrossRef CAS PubMed.
- Y. Liang, R. Kleinmans, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2022, 144, 20207–20213 CrossRef CAS PubMed.
- S. Dutta, Y.-L. Lu, J. E. Erchinger, H. Shao, E. Studer, F. Schäfer, H. Wang, D. Rana, C. G. Daniliuc, K. N. Houk and F. Glorius, J. Am. Chem. Soc., 2024, 146, 5232–5241 CrossRef CAS PubMed.
- M. de Robichon, T. Kratz, F. Beyer, J. Zuber, C. Merten and T. Bach, J. Am. Chem. Soc., 2023, 145, 24466–24470 CrossRef CAS PubMed.
- M. Golfmann, M. Reinhold, J. D. Steen, M. S. Deike, B. Rodemann, C. Golz, S. Crespi and J. C. L. Walker, ACS Catal., 2024, 14, 13987–13998 CrossRef CAS.
- K. Dhake, K. J. Woelk, J. Becica, A. Un, S. E. Jenny and D. C. Leitch, Angew. Chem., Int. Ed., 2022, 61, e202204719 CrossRef CAS PubMed.
- Y. Liang, F. Paulus, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2023, 62, e202305043 CrossRef CAS PubMed.
- F. Wu, W.-B. Wu, Y. Xiao, Z. Li, L. Tang, H.-X. He, X.-C. Yang, J.-J. Wang, Y. Cai, T.-T. Xu, J.-H. Tao, G. Wang and J.-J. Feng, Angew. Chem., Int. Ed., 2024, 63, e202406548 CrossRef CAS PubMed.
- J.-T. Che, W.-Y. Ding, H.-B. Zhang, Y.-B. Wang, S.-H. Xiang and B. Tan, Nat. Chem., 2025, 17, 393–402 CrossRef CAS PubMed.
- D. B. Werz, M. George and D. A. Knyazev, Chem. Sci., 2025, 16, 8588–8593 RSC.
- M. C. Pirrung, Tetrahedron Lett., 1980, 21, 4577–4578 CrossRef CAS.
- P. Hughes, M. Martin and J. Clardy, Tetrahedron Lett., 1980, 21, 4579–4580 CrossRef CAS.
- V. V. Levterov, O. Michurin, P. O. Borysko, S. Zozulya, I. V. Sadkova, A. A. Tolmachev and P. K. Mykhailiuk, J. Org. Chem., 2018, 83, 14350–14361 CrossRef CAS PubMed.
- L. D. Elliott, M. Berry, B. Harji, D. Klauber, J. Leonard and K. I. Booker-Milburn, Org. Proc. Res. Dev., 2016, 20, 1806–1811 CrossRef CAS.
- D. Tian, Y. Pan, X. Zhao, Y. Yin and Z. Jiang, J. Am. Chem. Soc., 2025, 147, 12410–12417 CrossRef CAS PubMed.
- T. Rigotti, D. P. Schwinger, R. Graßl, C. Jandl and T. Bach, Chem. Sci., 2022, 13, 2378–2384 RSC.
- A. Denisenko, P. Garbuz, N. M. Voloshchuk, Y. Holota, G. Al-Maali, P. Borysko and P. K. Mykhailiuk, Nat. Chem., 2023, 15, 1155–1163 CrossRef CAS PubMed.
- Y. O. Zaika, I. O. Borodin, H. O. Olekh, M. V. Kovalov, O. D. Diachenko, V. S. Brovarets, B. V. Vashchenko and O. O. Grygorenko, Org. Lett., 2025, 27, 2858–2862 CrossRef CAS PubMed.
- D. Tian, Y. Pan, X. Zhao, Y. Yin and Z. Jiang, J. Am. Chem. Soc., 2025, 147, 12410–12417 CrossRef CAS PubMed.
- T. Rigotti, D. P. Schwinger, R. Graßl, C. Jandl and T. Bach, Chem. Sci., 2022, 13, 2378–2384 RSC.
- D. M. Whalley, L. Carlino, O. D. Putra, N. A. Anderson, S. C. Coote and O. Lorthioir, Chem. Sci., 2024, 15, 19564–19570 RSC.
- C. Stevens and N. De Kimpe, J. Org. Chem., 1996, 61, 2174–2178 CrossRef CAS.
- A. A. Homon, O. V. Hryshchuk, O. V. Mykhailenko, B. V. Vashchenko, K. P. Melnykov, O. M. Michurin, C. G. Daniliuc, I. I. Gerus, V. O. Kovtunenko, I. S. Kondratov and O. O. Grygorenko, Eur. J. Org. Chem., 2021, 6580–6590 CrossRef CAS.
- V. V. Levterov, Y. Panasyuk, V. O. Pivnytska and P. K. Mykhailiuk, Angew. Chem., Int. Ed., 2020, 59, 7161–7167 CrossRef CAS PubMed.
- R. I. Vasiuta and M. V. Gorichko, Tetrahedron Lett., 2014, 55, 466–468 CrossRef CAS.
- M. Zanini, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2024, 63, e202410207 CrossRef CAS PubMed.
- G. R. Krow, G. Lin and F. Yu, J. Org. Chem., 2005, 70, 590–595 CrossRef PubMed.
- G. R. Krow, R. Edupuganti, D. Gandla, A. Choudhary, G. Lin, P. E. Sonnet, C. DeBrosse, C. W. Ross III, K. C. Cannon and R. T. Raines, J. Org. Chem., 2009, 74, 8232–8242 CrossRef CAS PubMed and references therein.
- F. Zhang, S. Dutta, A. Petti, D. Rana, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2025, 64, e202418239 CrossRef CAS PubMed.
- C. C. Chintawar, R. Laskar, D. Rana, F. Schäfer, N. Van Wyngaerden, S. Dutta, C. G. Daniliuc and F. Glorius, Nat. Catal., 2024, 7, 1232–1242 CrossRef CAS.
- B. Ghorai, B. Sahana and D. P. Hari, ChemRxiv, 2025, preprint DOI:10.26434/chemrxiv-2024-px1f3-v2.
- Z. Lin, H. Ren, X. Lin, X. Yu and J. Zheng, J. Am. Chem. Soc., 2024, 146, 18565–18575 CrossRef CAS PubMed.
- X. Wang, R. Gao and X. Li, J. Am. Chem. Soc., 2024, 146, 21069–21077 CrossRef CAS PubMed.
- Y. Liang, R. Nematswerani, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2024, 63, e202402730 CrossRef CAS PubMed.
- J.-L. Zhou, Y. Xiao, L. He, X.-Y. Gao, X.-C. Yang, W.-B. Wu, G. Wang, J. Zhang and J.-J. Feng, J. Am. Chem. Soc., 2024, 146, 19621–19628 CrossRef CAS PubMed.
- S. Dutta, C. G. Daniliuc, C. Mück-Lichtenfeld and A. Studer, J. Am. Chem. Soc., 2024, 146, 27204–27212 CrossRef CAS PubMed.
- J. Zhang, J. Y. Su, H. Zheng, H. Li and W. P. Deng, Angew. Chem., Int. Ed., 2024, 63, e202318476 CrossRef CAS PubMed.
- W.-B. Wu, B. Xu, X.-C. Yang, F. Wu, H.-X. He, X. Zhang and J.-J. Feng, Nat. Commun., 2024, 15, 8005 CrossRef CAS PubMed.
- J. Zhang, J.-Y. Su, H. Zheng, H. Li and W.-P. Deng, ACS Catal., 2024, 14, 17837–17849 CrossRef CAS.
- Y. Xiao, F. Wu, L. Tang, X. Zhang, M. Wei, G. Wang and J. J. Feng, Angew. Chem., Int. Ed., 2024, 63, e202408578 CrossRef CAS PubMed.
- R. Srinivasan and K. H. Carlough, J. Am. Chem. Soc., 1967, 89, 4932–4936 CrossRef CAS.
- J. Stanek, A. Alder, D. Bellus, A. S. Bhatnagar, A. Haeusler and K. Schieweck, J. Med. Chem., 1991, 34, 1329–1334 CrossRef CAS PubMed.
- K. H. Kim, J. W. Lim, J. Lee, M. J. Go and J. N. Kim, Adv. Synth. Catal., 2014, 356, 3363–3369 CrossRef CAS.
- S. M. Kim, J. H. Park, Y. K. Kang and Y. K. Chung, Angew. Chem., Int. Ed., 2009, 48, 4532–4535 CrossRef CAS PubMed.
- V. Aggarwal, Z.-X. Zhang, K. Shu, M. Mandigma, M. Tilby, Y. Guo and A. Noble, ChemRxiv, 2025, preprint DOI:10.26434/chemrxiv-2025-285mh.
- D. Dibchak, M. Snisarenko, A. Mishuk, O. Shablykin, L. Bortnichuk, O. Klymenko-Ulianov, Y. Kheylik, I. V. Sadkova, H. S. Rzepa and P. K. Mykhailiuk, Angew. Chem., Int. Ed., 2023, 62, e202304246 CrossRef CAS PubMed.
- D. Wood, N. Hernandez, J. Newman and R. Johnson, Org. Lett., 2025, 27, 11890–11895 CrossRef CAS PubMed.
- D. S. Radchenko, N. Kopylova, O. O. Grygorenko and I. V. Komarov, J. Org. Chem., 2009, 74, 5541–5544 CrossRef CAS PubMed.
- H. Shuwen, H. Jinsong, R. A. Eric, R. C. Harry, P. Barbara, L. C. Steven and M. Michael, Tetrahedron Lett., 2016, 57, 1268–1269 CrossRef.
- A. V. Denisenko, A. P. Mityuk, O. O. Grygorenko, D. M. Volochnyuk, O. V. Shishkin, A. A. Tolmachev and P. K. Mykhailiuk, Org. Lett., 2010, 12, 4372–4375 CrossRef CAS PubMed.
- A. V. Chernykh, B. V. Vashchenko, S. V. Shishkina, D. M. Volochnyuk and O. O. Grygorenko, J. Org. Chem., 2024, 89, 10440–10450 CrossRef CAS PubMed.
- J. Zhao, X.-J. Zhao, P. Cao, J.-K. Liu and B. Wu, Org. Lett., 2017, 19, 4880–4883 CrossRef CAS PubMed.
- R. Revie, A. Dasgupta, Y. Biddick, K. Christensen, R. Smith and E. Anderson, ChemRxiv, 2024, preprint DOI:10.26434/chemrxiv-2024-tnx0l.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2026 |