
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBayesian optimization for chemical reactions
Stefan
Desimpel
 *a,
Matthieu
Dorbec
b,
Kevin M.
Van Geem
c and
Christian V.
Stevens
a
*a,
Matthieu
Dorbec
b,
Kevin M.
Van Geem
c and
Christian V.
Stevens
a
aSynBioC Research Group, Department of Green Chemistry and Technology, Faculty of Bioscience Engineering, Ghent University, Coupure Links 653, B-9000 Ghent, Belgium. E-mail: chris.stevens@ugent.be
bTechnology & Engineering, Janssen R&D, Turnhoutseweg 30, 2340 Beerse, Belgium
cLaboratory for Chemical Technology (LCT), Department of Materials, Textiles and Chemical Engineering, Faculty of Engineering & Architecture, Ghent University, B-9052 Zwijnaarde, Belgium
First published on 10th February 2026
Abstract
Bayesian optimization (BO) enables data-efficient optimization of complex chemical reactions by balancing exploration and exploitation in large, mixed-variable parameter spaces. This review provides an accessible introduction for chemists wishing to adopt BO, outlining the fundamentals of surrogate models, acquisition functions, and key mathematical concepts. Practical considerations are emphasized, including kernel design, representation of categorical variables, and strategies for multi-objective and batch optimization. Applications are comprehensively surveyed across experimental scales, from high-throughput platforms to automated flow reactors and larger-scale processes. Finally, emerging directions such as transfer learning and data reuse are discussed in the context of accelerating optimization campaigns and enabling more generalizable, data-driven strategies in chemistry.
 Stefan Desimpel | Stefan Desimpel obtained his master's degree in Bioscience Engineering from Ghent University in 2020. He completed his PhD at the same university in 2025, focusing on the development and application of practical Bayesian optimization methodologies for chemical reaction development, in close collaboration with Johnson & Johnson Innovative Medicine. His research interests include data-efficient optimization of complex reaction systems, with particular emphasis on photochemical and gas–liquid processes. |
 Matthieu Dorbec | Matthieu Dorbec is currently Scientific Associate Director in the Technology and Engineering, Chemical Process Development at Johnson & Johnson Innovative Medicine. He holds a PhD in Organic Chemistry (2006), an MSc in Chemistry (2002), and a PharmD (2003). With 20 years of R&D experience—including 14 years in pharmaceuticals—he specializes in process development and leads the process engineering group, advancing small molecule programs. His expertise includes implementing innovative technologies such as organic solvent nanofiltration, flow chemistry, flow photochemistry, and developing optimization models to support late-stage drug substance development. |
 Kevin M. Van Geem | Professor Kevin M. Van Geem is a full professor at Ghent University's Laboratory for Chemical Technology, specializing in thermochemical reaction engineering and the transition from fossil-based to renewable feedstocks. He directs pilot plants for steam cracking and chemical recycling and was formerly a Fulbright Scholar at MIT. With over 400 publications, his research spans detailed kinetic modeling, scale-up, machine learning, and advanced analytical techniques like comprehensive two-dimensional gas chromatography. He also co-founded a spin-off bridging academia and industry, contributes to sustainable chemistry initiatives, and recently secured an ERC grant for electrification of chemical processes. |
 Christian V. Stevens | Prof. Dr ir. Christian V. Stevens (°1965) is currently senior full professor of the Department of Green Chemistry and Technology at the Faculty of Bioscience Engineering (Ghent University). He graduated as bio-engineer in chemistry and agricultural industries in 1988 and obtained a PhD in 1992. He then performed postdoctoral work at the University of Florida, USA with Prof. Alan Katritzky as a NATO Research Fellow. In 2014 he was promoted to senior full professor. His research interests are focussed on microreactor technology and High Throughput Experimentation, on synthetic heterocyclic chemistry and on the use of renewable resources for the industry. |
1. Introduction: Bayesian optimization as a tool for chemical reaction optimization
Reaction condition optimization is a critical step in developing efficient and robust chemical processes. Over the past decade or so, the demand for more efficient and sustainable methodologies has driven the adoption of algorithmic optimization strategies.1,2 Among the various strategies explored, Bayesian optimization has emerged as one of the dominant approaches, due to its ability to efficiently navigate complex reaction spaces while minimizing the number of experiments needed (vide infra). This trend is evident when considering the number of publications referencing BO for chemical reactions on Web of Science, which has shown a marked increase (Fig. 1). The increasing number of publications reflects the growing recognition of the effectiveness of BO, highlighting the community's acknowledgement of its advantages.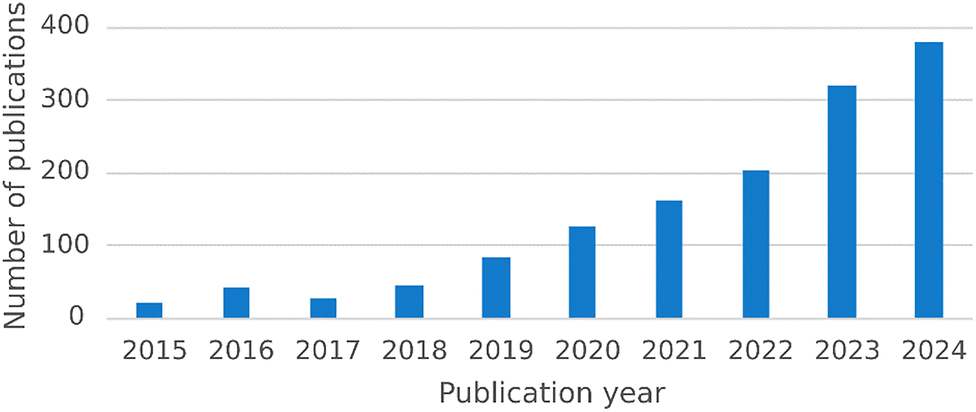 | ||
| Fig. 1 Increasing popularity of Bayesian optimization in the chemical community. Number of chemistry publications on Web of Science referencing ‘Bayesian optimization’ OR ‘Bayesian optimisation’ in the ‘Topic’ and ‘Title’ fields for each year over the last decade.3 The trend is clearly increasing, highlighting the growing adoption of BO for chemical reaction optimization. Note that papers published before 2018 are primarily computational; adoption for experimental reaction optimization gained momentum around this time (vide infra). | ||
The rise of Bayesian optimization for chemical reaction optimization is closely tied to the development of automated flow platforms. Such automated platforms, when coupled with suitable reaction analytics, allow for closed-loop optimization, making a compelling case for algorithm-guided optimization. This synergy, together with a more general overview of reactor automation, is extensively discussed in a recent review by Clayton.4
Early efforts in this field predominantly employed Simplex and SNOBFIT algorithms for reaction condition optimization.5–8 Simplex, also known as the Nelder–Mead method, is a direct search optimization technique. The algorithm progresses by modifying a set of points (a “simplex”) in the parameter space, aiming to improve reaction conditions based on function evaluations.9 Meanwhile, SNOFBIT is a derivative-free optimization algorithm designed for global optimization of noisy objective functions.10 Several studies demonstrated the effectiveness of these methods for the optimization of chemical reactions.
Notably, the group of Felpin successfully used a modified version of the Simplex algorithm to separately optimize up to three reaction parameters for four discrete synthesis steps for the synthesis of Carpanone.7 The same group used a similar algorithm to optimize temperature, residence time, catalyst loading and equivalents of a diazonium salt for a palladium-catalysed Heck–Matsuda reaction.11 Similarly, the groups of Jensen/Jamison12 and Bourne8 have demonstrated the use of SNOBFIT for chemical reaction optimization, particularly for the synthesis of pharmaceutical intermediates. However, both Simplex and SNOBFIT exhibited limitations in high-dimensional search-spaces, often struggling to reach reliable convergence and requiring significant experimental effort, making them less practical for optimizing highly complex reaction systems.13 This was confirmed by a direct comparison of a Simplex algorithm, SNOBFIT and BO by Felton et al., who found the former two approaches performed worse or only slightly better than random search for two reaction optimization case studies, while the BO algorithm showcased best-in-class performance.14 Similarly, another recent study directly compared the performance of SNOBFIT with a BO algorithm for the optimization of a mixing-sensitive coupling reaction.15 The authors reported the BO approach as a means to achieve better results with fewer experimental points. These direct comparisons further highlight the advantages of Bayesian optimization in overcoming the challenges of reaction optimization by efficiently navigating complex parameter spaces.
Around 2018, research groups began exploring Bayesian optimization as an alternative to these earlier approaches. One of the earliest applications of BO for chemical reaction optimization was reported by Schweidtmann et al. in 2018.16 The authors reported the multi-objective self-optimization of an SNAr reaction and of a N-benzylation reaction in continuous flow. This study demonstrated the ability of BO to efficiently explore chemical space, requiring a limited number of experiments to identify a Pareto front.
Over the last couple of years, Bayesian optimization has firmly established itself as a key tool for optimizing chemical reactions, as will be made clear in subsequent sections of this review. The goal is to provide an overview of some key applications of BO to chemical reaction optimization. Only wet-lab work will be covered; no computational-only studies. After an introduction to the fundamentals of Bayesian optimization, the smallest scale will be discussed, including HTE, miniaturized reactors and slug-based platforms. Next, flow-based platforms will be discussed, followed by larger-scale applications and hybrid approaches.
2. Fundamentals of Bayesian optimization
Bayesian optimization is a data-efficient, model-based framework to solve the problem of finding a global minimiser (or maximiser) of an unknown objective function.17 It is a sequential approach, particularly well-suited for expensive-to-evaluate problems, such as chemical reactions, where each experiment may involve significant time, cost, or material resources. Importantly, because BO treats the objective function as a “black-box,” it does not require a detailed mechanistic model of the reaction, making it a practical tool even when the underlying chemistry is only partially understood.In essence, the key idea is to replace the unknown, expensive objective function with an inexpensive-to-evaluate surrogate model and subsequently select new experiments based on both the predicted performance and the associated uncertainty of these surrogates via an acquisition function. When combined with a probabilistic surrogate model, Bayesian optimization aims to balance exploration of areas of the search space with high uncertainty with exploitation conditions known to yield high-performing results.18
The optimization procedure starts with an initialization phase, where initial data is collected via an experimental design such as Latin Hypercube Sampling (LHS), a Design of Experiments (DoE), or even random sampling. Alternatively, existing reaction data can be used. These data are then used to fit a surrogate model, which incorporates a prior distribution that captures initial beliefs about the behaviour of the unknown objective function, such as smoothness or expected amplitude. As the initialization data is used to train the surrogate model, the prior distribution is updated, leading to a posterior distribution that improves the accuracy of the surrogate model in reflecting the objective function.17–19
After training the surrogate model, new experiments are proposed by optimizing an acquisition function, maximizing the expected utility of candidate experiments by balancing exploration and exploitation. The objective function is then evaluated at the proposed points, the new data gets appended to the training set, and the posterior of the surrogate model is updated. This cycle continues iteratively until a termination criterion is met. In practice, this is often when a predetermined number of evaluations has been reached, until the identified optimum surpasses a certain user-specified threshold, or when the space is explored sufficiently for the uncertainty of the surrogate model to drop below a specified value.16,19,20 This process is presented schematically in Fig. 2.
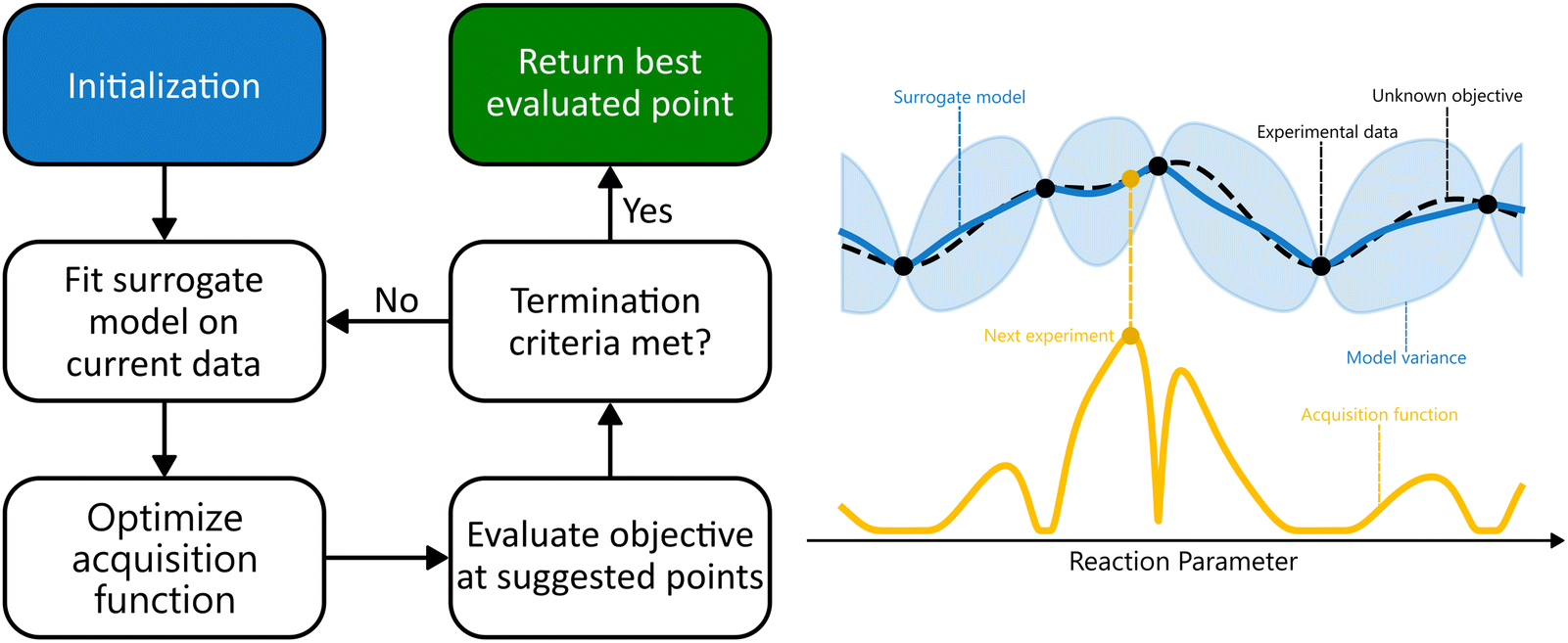 | ||
| Fig. 2 Overview of Bayesian optimization. Left: Flow chart of the optimization procedure. The algorithm starts by initialization through a small dataset, on which the surrogate model is then trained. Next, the acquisition function is optimized, and a point to evaluate next is suggested. After the objective function is evaluated at this point, the posterior of the surrogate model is updated, and the cycle continues until one of the termination criteria is met. Right: With a probabilistic surrogate model, incorporating uncertainty information, a balance between exploration and exploitation can be achieved. The acquisition function is high for parameter values where uncertainty around the value of the objective is high or for values that currently maximize the objective function. The surrogate model is plotted as the posterior mean, with the shaded area representing 2σ. | ||
Several key factors make Bayesian optimization particularly suited for chemical reaction optimization:
Data-efficiency for expensive-to-evaluate problems. The architecture of BO algorithms, using a surrogate model to quantify uncertainty, proves to be very data-efficient, reducing the number of experiments needed to locate optimal conditions.17,21
Incorporation of prior knowledge. BO frameworks allow the incorporation of prior information—such as historical data, expert intuition or knowledge of the reaction mechanism—through the choice of prior on the surrogate model's parameters. This approach can leverage previous experimental results to guide the optimization process towards more promising regions of the search space, potentially accelerating convergence.22,23
Robustness to noise. Chemical reaction data often contains a considerable amount of noise due to measurement errors, slight fluctuations in experimental conditions, or intrinsic variability of the system. Probabilistic surrogate models are well-equipped to handle this noise, allowing a separate noise term to be introduced, resulting in more robust predictions and a better estimation of uncertainty.17,24 Additionally, acquisition functions specifically designed to better handle noisy data have been developed recently.25
Global optimization. BO is inherently suited for global optimization, especially with probabilistic surrogate models that balance exploration with exploitation, as mentioned above. This is crucial for chemical reaction optimization, as it minimizes the risk of getting trapped in local optima.21,26
Flexibility. The Bayesian optimization framework is highly adaptable, capable of handling different types of variables—including both continuous and categorical—and extendable to multi-objective and constrained optimization problems. This flexibility makes BO especially valuable in real-world chemical reaction systems.24,27
In the remainder of this section, the two main components of the Bayesian Optimization framework—the surrogate model and the acquisition function—will be presented in more detail, to facilitate the discussion on various implementations and applications in chemical reaction optimization that will complete this chapter. This is by no means meant to be an exhaustive review on Bayesian optimization, but rather an introduction to its basics for people who want to start using it as a tool.
2.1. Surrogate model
As described in the previous section, a first, essential, component of Bayesian optimization is the surrogate model. In many chemical reaction optimization problems, the underlying relationship between experimental inputs (such as temperature, equivalents, solvent, and reaction time) and outputs (such as yield, selectivity, or productivity) is complex and is not analytically tractable. This ‘black-box’ nature of these systems, combined with the fact that they are expensive to evaluate (whether in terms of time, monetary cost, resource consumption, or a combination thereof), necessitates data-efficient optimization. Surrogate models address this challenge by effectively mapping these inputs to the outputs of the process, thereby approximating the unknown objective function that characterizes the reaction system.2,17Surrogate models thus provide a computationally inexpensive alternative to performing extensive experimental trials. With only a limited set of experimental data, these models learn an approximate functional relationship between the inputs and outputs of the process, enabling predictions over the entire reaction space and quantifying the uncertainty associated with those predictions. In tandem with the acquisition function, this approach enables optimization of the true objective by proxy. The uncertainty information inherent to the probabilistic surrogate models is key to balancing exploration and exploitation and to guiding further experimentation.17
Furthermore, surrogate models can, in principle, incorporate prior knowledge—be it from historical data, chemical intuition, or mechanistic insights—thereby refining their predictive capabilities and steering the optimization toward regions of interest; however, explicit demonstrations of such knowledge integration in chemical reaction optimization remain scarce. Selected examples are discussed in Section 6.
While a multitude of surrogate models exist in the broader context of Bayesian optimization, such as Random Forests (RFs) and neural networks (NN), Gaussian Processes (GPs) are by far the most routinely used surrogate model for chemical reaction optimization. This preference is largely rooted in the practical characteristics of reaction optimization problems. Chemical optimization campaigns are typically conducted in low-data regimes, where experiments are expensive and the total number of evaluations is limited. In these settings, the availability of principled predictive uncertainty is essential for guiding the exploration–exploitation trade-off central to Bayesian optimization.17 Gaussian processes natively provide principled uncertainty estimates alongside their predictions, whereas uncertainty quantification in RF- or NN-based surrogates generally relies on heuristic or computationally more complex approaches.17,28,29 Although random forests and neural networks scale favourably to higher-dimensional or larger-data problems,18,30,31 these advantages are less relevant in typical reaction optimization campaigns. On the other hand, while standard Gaussian processes scale unfavourably with increasing dataset size, this limitation is rarely encountered in practice, and scalable GP variants have been proposed for larger-data regimes and problems of higher dimensionality.32–34 For these reasons, the remainder of this section focuses on Gaussian process-based surrogate models, which dominate BO for chemical reactions.
GPs are a nonparametric, probabilistic modelling technique that defines a distribution over functions. Rather than assuming a fixed functional form, a GP models the unknown objective function as a collection of random variables such that any finite subset of these variables has a joint Gaussian distribution. This property allows GPs to predict not only the expected outcome (i.e., the mean) but also the uncertainty (i.e., the variance) associated with each prediction, a feature that is crucial when experimental data are scarce and expensive to obtain.
Put differently, a Gaussian process can be seen as a generalization of the Gaussian probability distribution. While a probability distribution describes a distribution over finite-dimensional scalars/vectors (for multivariate distributions), a stochastic process governs the properties of functions.22 Leaving mathematical rigor aside, one can loosely think of a Gaussian process as providing a “density” on the infinite-dimensional space of functions. Drawing a sample from the GP yields a function. The GP framework accomplishes this by defining how every finite set of points in the function domain is jointly distributed. By doing so consistently, it effectively captures a probability distribution over the entire function space. Fig. 3 presents an illustrative example of samples from a Gaussian process.
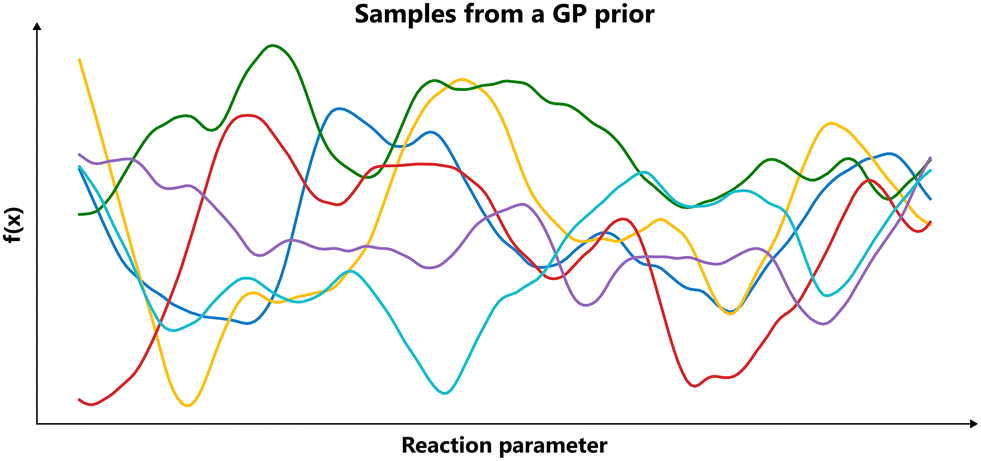 | ||
| Fig. 3 Samples drawn from a Gaussian process. The plot shows six independent samples drawn from a zero-mean Gaussian process prior with a Matérn kernel (v = 2.5, l = 1) over the interval [0, 10], illustrating how a GP represents a distribution over functions. | ||
Thus, by defining how any finite collection of function values is jointly distributed, the GP framework provides a powerful, flexible tool for modelling complex reaction systems with inherent uncertainty. The following section covers the basic mathematics about GPs. For more details, the reader is referred to the work by Rasmussen and Williams.22
| y(x) ∼ GP(m(x),k(x,x′)) | (1) |
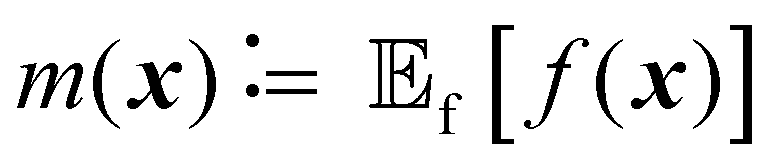 | (2) |
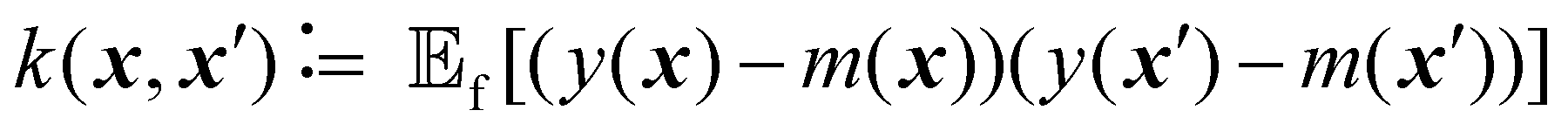 | (3) |
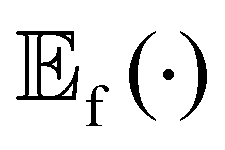 is the expected value over distribution of functions modeled by the GP. Conceptually, the mean function can be seen as the ‘average’ shape of the function—the mean value of n samples from the GP evaluated at x for n → +∞ would be m(x)—while the covariance function governs the covariance between any two function values computed at the corresponding inputs.36 In other words, the kernel characterizes similarity between data points, by mapping them to a high-dimensional feature space to better reflect the distances between data points and their similarities.37
is the expected value over distribution of functions modeled by the GP. Conceptually, the mean function can be seen as the ‘average’ shape of the function—the mean value of n samples from the GP evaluated at x for n → +∞ would be m(x)—while the covariance function governs the covariance between any two function values computed at the corresponding inputs.36 In other words, the kernel characterizes similarity between data points, by mapping them to a high-dimensional feature space to better reflect the distances between data points and their similarities.37
In GP regression, the covariance function dictates the properties of the response functions that can be fitted.17 This allows incorporating prior assumptions about the function's smoothness, periodicity and overall behaviour through a careful choice of kernel. The most used class of kernels is the Matérn class.22 The smoothness of Matérn kernels can be adjusted by the parameter ν > 0, and samples from a GP using such a kernel are differentiable [υ − 1] times.17 The general equation of the Matérn kernel is the following:22,35
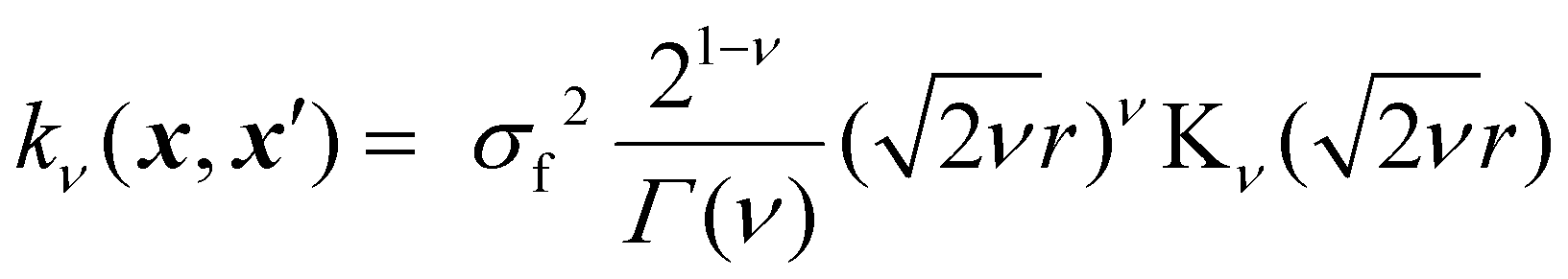 | (4) |
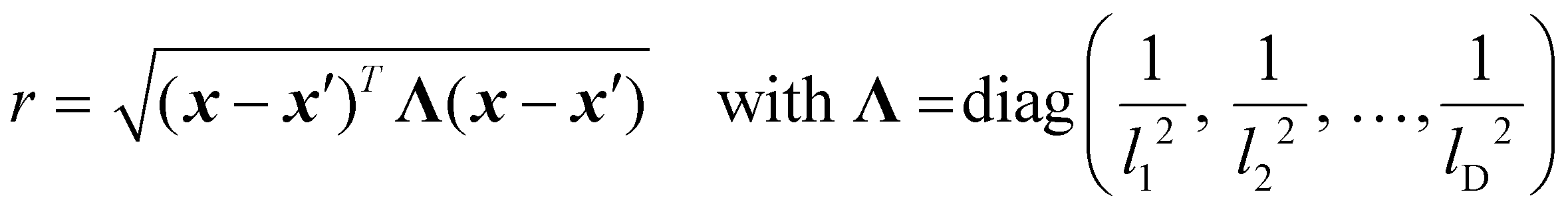 | (5) |
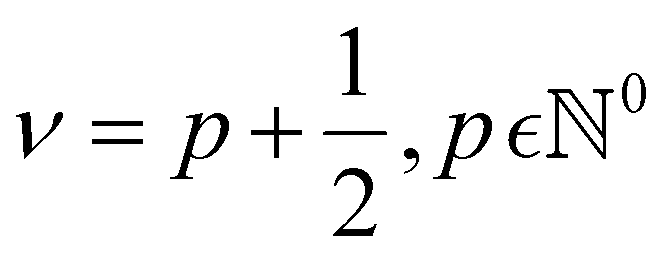 , and as ν → +∞, the Matérn kernel converges to the squared exponential kernel (SE).
, and as ν → +∞, the Matérn kernel converges to the squared exponential kernel (SE).
Kernels from this class are commonly denoted by their value for ν. The most used kernels are the Matérn 1/2, Matérn 3/2, Matérn 5/2, and squared exponential kernels:35
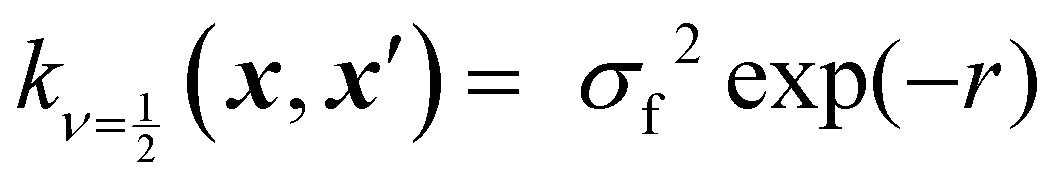 | (6) |
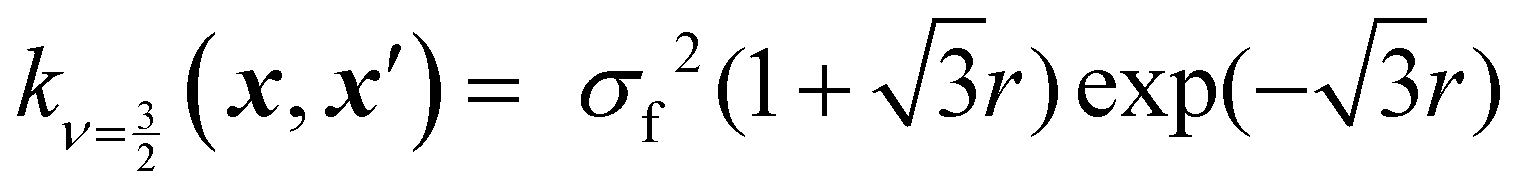 | (7) |
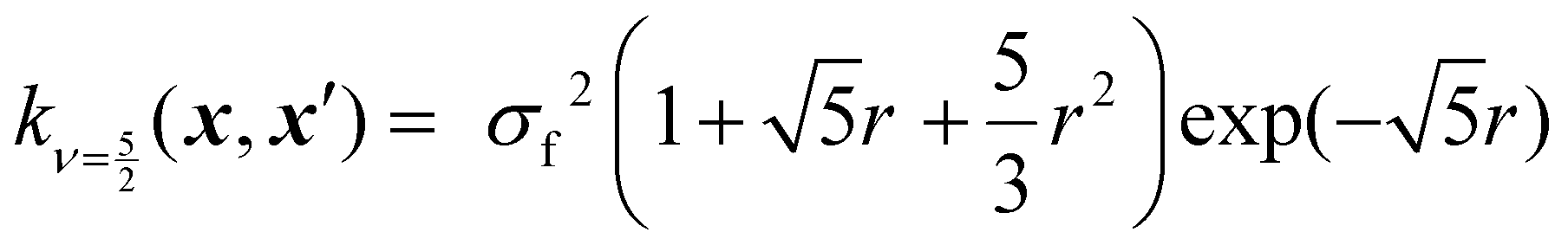 | (8) |
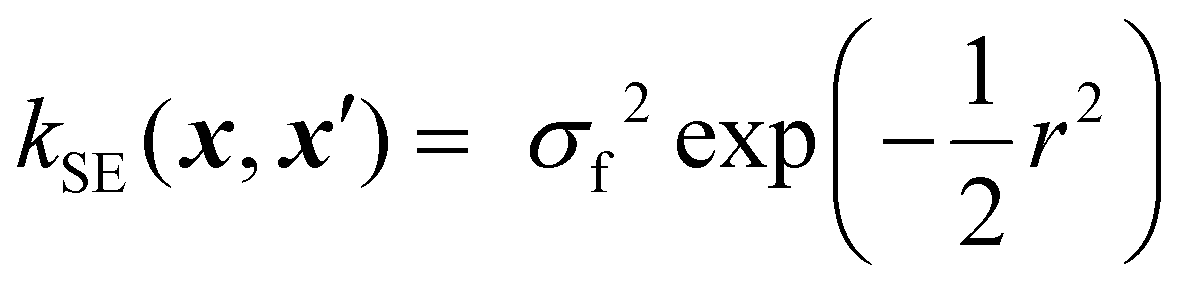 | (9) |
Beyond the Matérn family, other common kernels include the rational quadratic (RQ) and dot product kernels.
The rational quadratic kernel can be interpreted as a scale mixture (an infinite sum) of squared exponential kernels with varying length scales, making it suitable for modelling functions with non-uniform smoothness across the input space. The RQ kernel captures such behaviour by blending short- and long-range correlations in a single expression:37
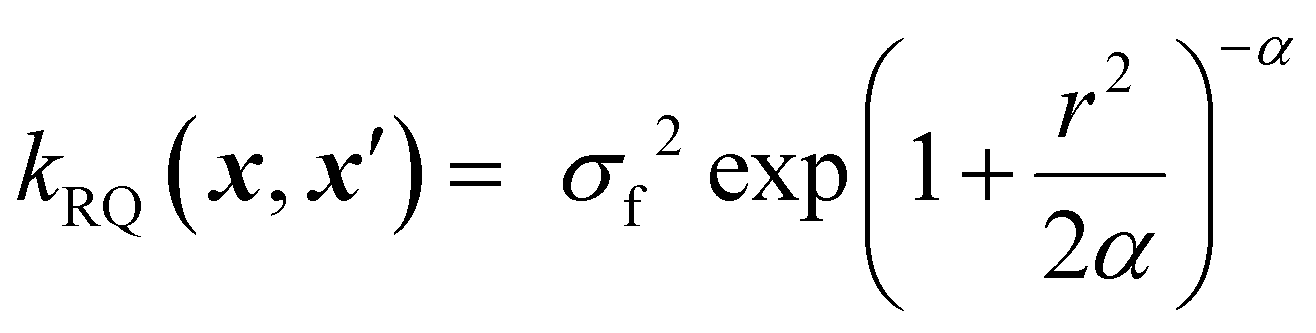 | (10) |
The dot product kernel (also known as the linear kernel) is an example of a non-stationary kernel. That is, the smoothness of the function changes depending on the location in the input space. Its mathematical representation is provided below:37
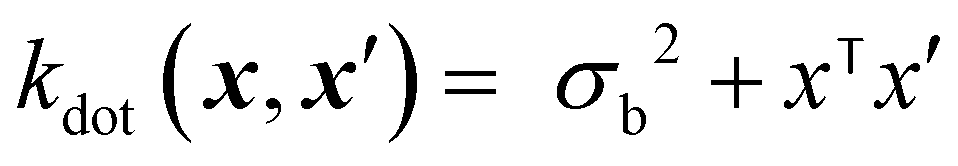 | (11) |
As shown in Fig. 4, these kernels exhibit markedly different correlation profiles.
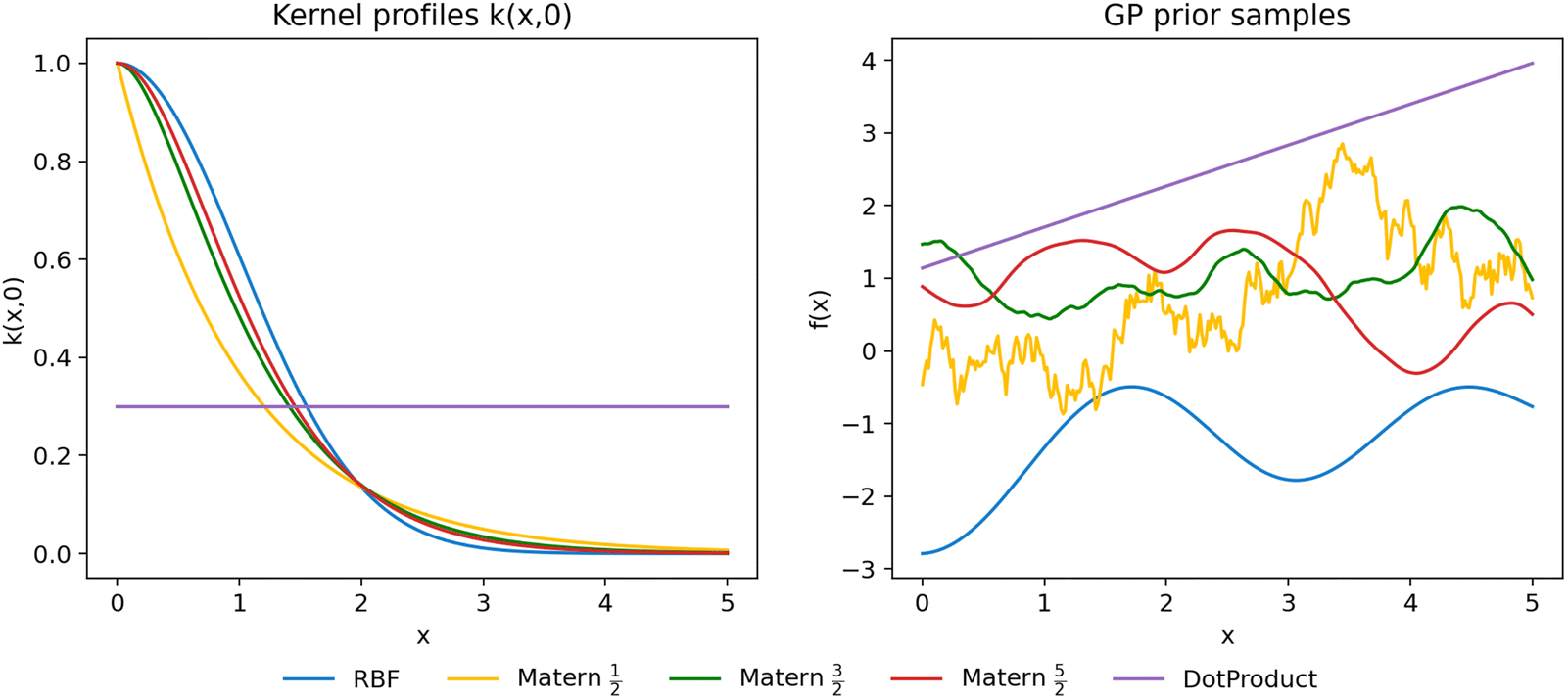 | ||
| Fig. 4 Comparison of some common kernels for GP modelling. Left: Correlation decay profiles for common GP kernels. Right: Corresponding samples drawn from a GP prior from each kernel over [0, 5], highlighting how kernel smoothness and scale influence function variability. | ||
Anisotropic kernel formulation enables Automatic Relevance Determination (ARD), where the GP learns s separate length scales ld for each input dimension. This allows the model to infer which variables most strongly influence the function output: short length scales imply a rapidly varying response and suggest importance, while long length scales imply that changes in that input have little effect.
ARD is often used as a heuristic for variable importance analysis in the chemical reaction optimization literature.16,19,20 However, using ARD has known limitations. First, it tends to overemphasize the relevance of nonlinear variables.38 Second, ARD can suffer from identifiability issues, particularly when input variables are correlated, when data is limited or noisy, or in high-dimensional parameter spaces. Alternative techniques, such as global sensitivity analysis39,40 or additive GP models41,42 may offer more robust assessments of variable importance. A detailed discussion of these techniques is outside the scope of this work, and for more information on either, the reader is referred to the corresponding references.
The kernels discussed up to this point all exclusively deal with continuous variables. However, in many practical applications—especially chemical reaction optimization—the input space contains a mix of continuous, categorical, and even structurally encoded variables. Traditional distance-based kernels, using the scaled Euclidean distance metric r, may not effectively capture the mixed nature of these inputs. Several strategies have been developed to address this issue, the most important of which will be covered here, without delving into a detailed discussion, as this is outside the scope of this literature review.
One approach is to extend existing kernels (e.g., Matérn) by replacing the standard Euclidean distance with the Gower distance, which combines scaled differences for continuous variables, and a simple mismatch indicator for categorical ones.43
The Gower distance between two input vectors x and x′ is defined as:44
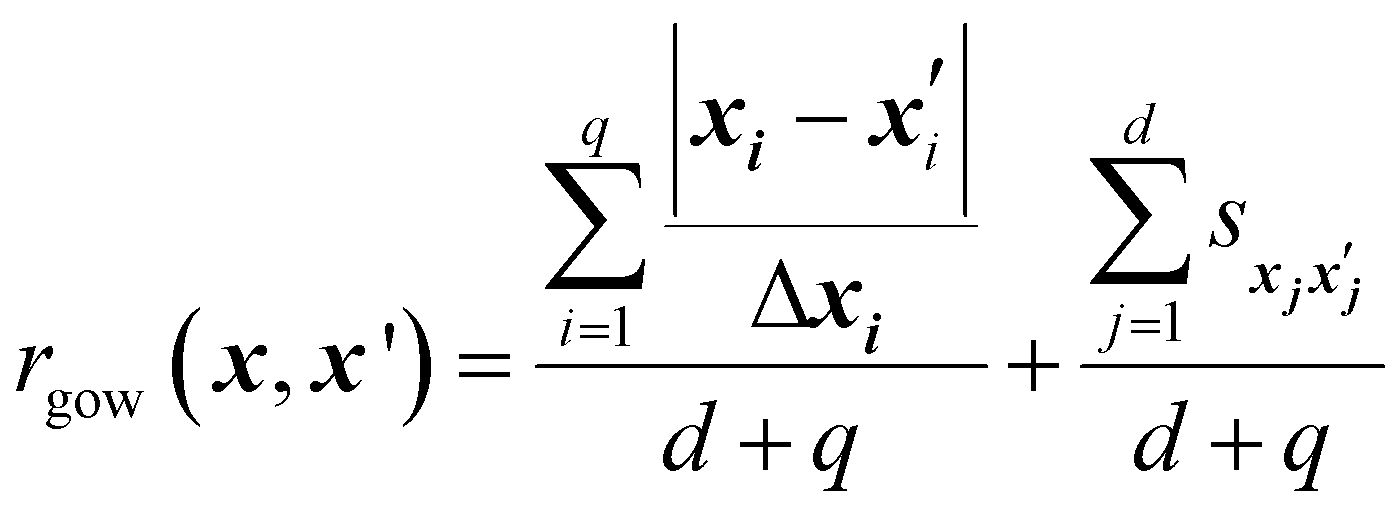 | (12) |
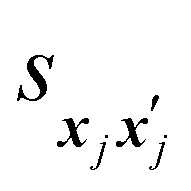 is set to 0 if the categorical variables match
is set to 0 if the categorical variables match 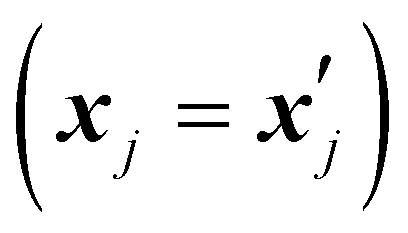 and 1 otherwise. This composite distance metric can then be put into the kernel, replacing the Euclidian distance r. For example, when applied to the Matérn kernel for the case where ν = 5/2, the mixed-variable kernel becomes:
and 1 otherwise. This composite distance metric can then be put into the kernel, replacing the Euclidian distance r. For example, when applied to the Matérn kernel for the case where ν = 5/2, the mixed-variable kernel becomes: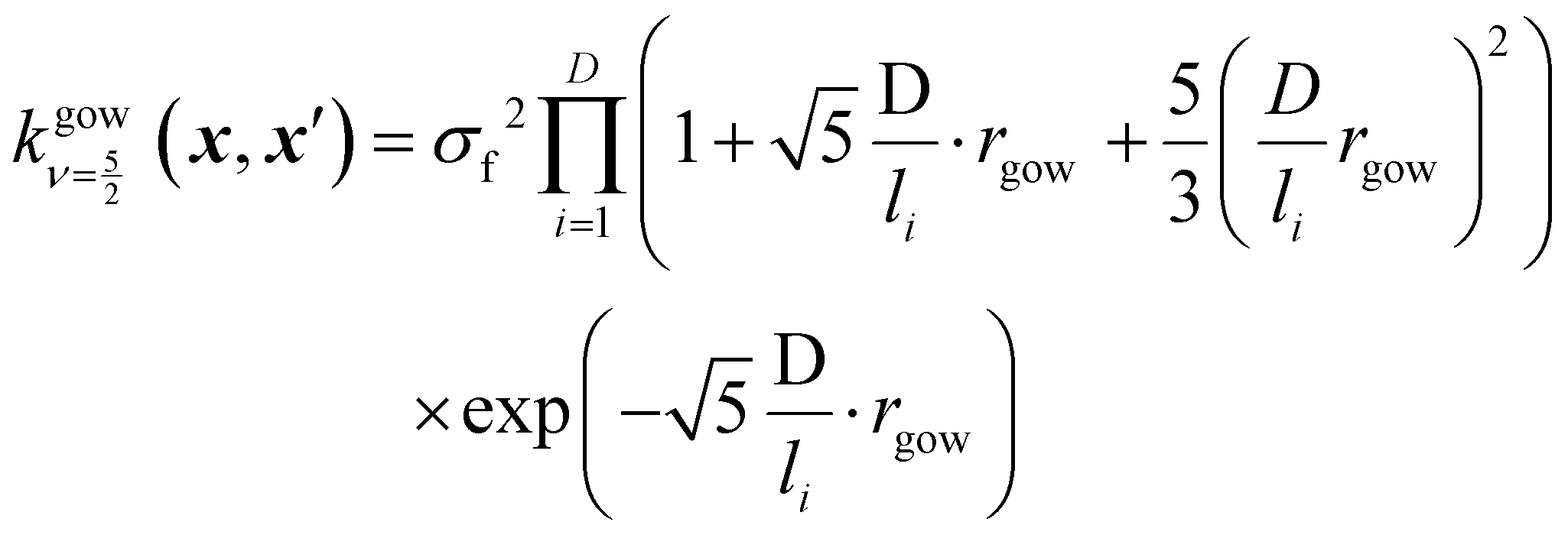 | (13) |
Another common approach in machine learning in general is to represent categorical inputs via one-hot encoding, transforming each categorical variable with k levels to a binary vector of length k. This converts categorical variables into a pseudo-continuous space, allowing them to be handled by standard kernels.45,46 This process is visualized in Fig. 5.
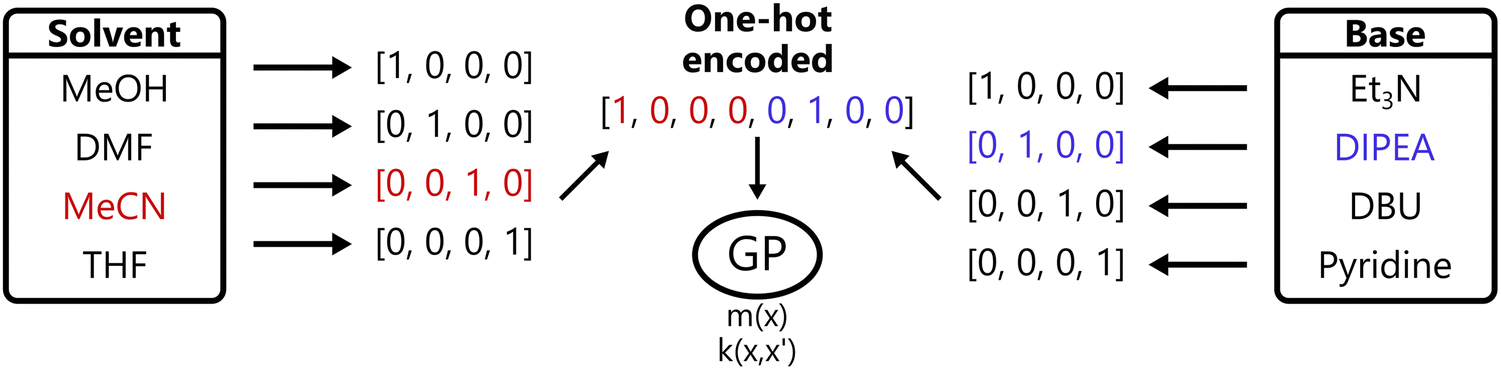 | ||
| Fig. 5 Illustration of one-hot encoding. One-hot encoding transforms each categorical choice (e.g., solvent or base) into a binary vector where exactly one position is “hot” (1) and the rest are zero, enabling GPs to handle discrete inputs via numerical feature vectors. | ||
While one-hot encoding is commonly used in BO due to its simplicity and compatibility with popular GP implementations, there is an important shortcoming. Namely, this approach does not capture structural information and thus treats all categories as equally distant, ignoring chemical similarity. This can limit the model's ability to generalize in chemically diverse spaces. Nonetheless, one-hot encoding has been found to perform well in practical applications.19,46
A more chemically informed alternative to one-hot encoding is to represent categorical inputs such as ligands or solvents via molecular descriptors. These may include:
Molecular fingerprints (e.g. Morgan fingerprints).46,47
Quantum-mechanical descriptors derived from density functional theory (DFT) calculations.46,48
These descriptor vectors can be treated as continuous inputs and used with any standard kernel. This approach embeds structural information directly into the kernel, enabling GPs to learn chemically meaningful structure–reactivity relationships. However, these descriptors can be computationally expensive to generate, especially in the case of DFT descriptors and, more importantly, in some cases, they perform only slightly better19 or worse than one-hot encoding.46
When inputs are binary vectors (e.g. molecular fingerprints or one-hot encoded input vectors), instead of using standard continuous kernels, specialized kernels can be used to more appropriately model similarity between inputs. Two examples are the Hamming and Tanimoto kernels.
The Hamming kernel measures the number of bitwise mismatches between two binary vectors:49
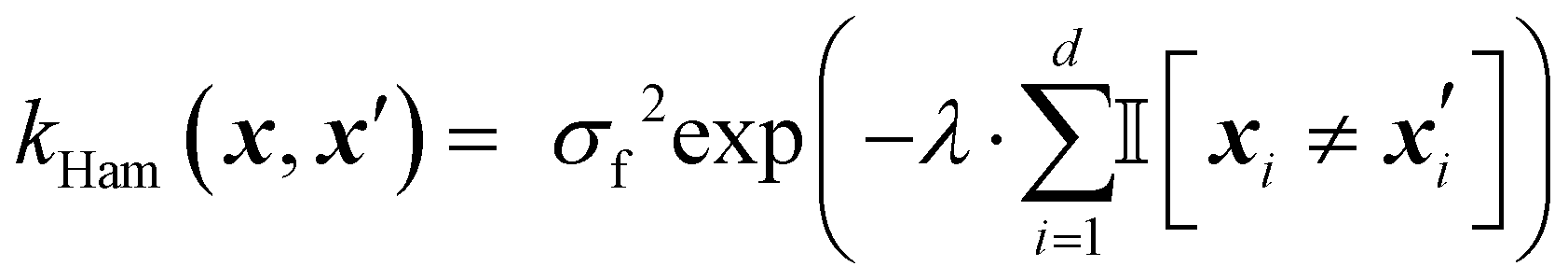 | (14) |
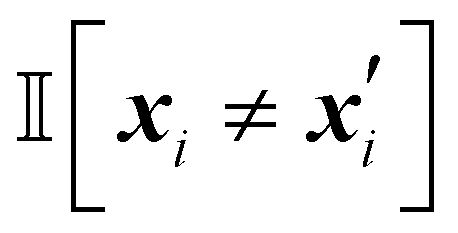 is the indicator function, which returns 1 if the inputs differ in dimension i, and 0 otherwise. It penalizes dissimilarity discretely and avoids interpolating over intermediate values.
is the indicator function, which returns 1 if the inputs differ in dimension i, and 0 otherwise. It penalizes dissimilarity discretely and avoids interpolating over intermediate values.
The Tanimoto kernel is especially well-suited for binary molecular fingerprints, and quantifies chemical similarity:50
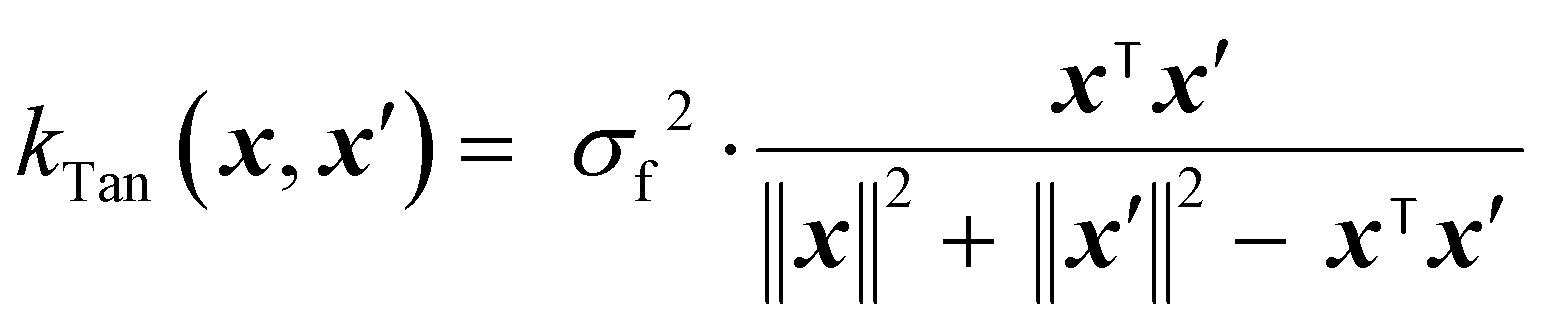 | (15) |
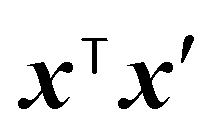 is the count of shared ‘1’ bits (common substructures), and the denominator is the union count, i.e. the total number of features present in either input. The Tanimoto kernel thus represents the overlap of molecular substructures and is commonly used in cheminformatics.51
is the count of shared ‘1’ bits (common substructures), and the denominator is the union count, i.e. the total number of features present in either input. The Tanimoto kernel thus represents the overlap of molecular substructures and is commonly used in cheminformatics.51
When inputs contain a mix of variable types and structures—such as continuous and categorical process parameters and molecular descriptors—composite kernels allow the GP to combine multiple similarity measures in a principled way.22 Two common forms are sum kernels and product kernels.
Sum kernels model independent contributions of different input groups. For example, a sum kernel combining a Matérn kernel for continuous variables and a Hamming Kernel for categorical variables might look like this:
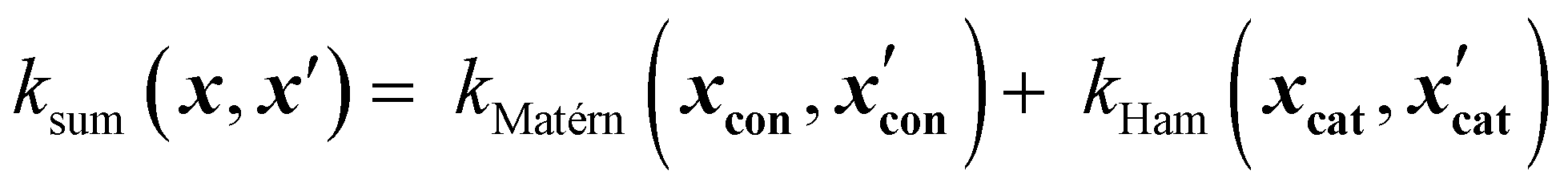 | (16) |
Similarly, product kernels assume interactions between input groups:
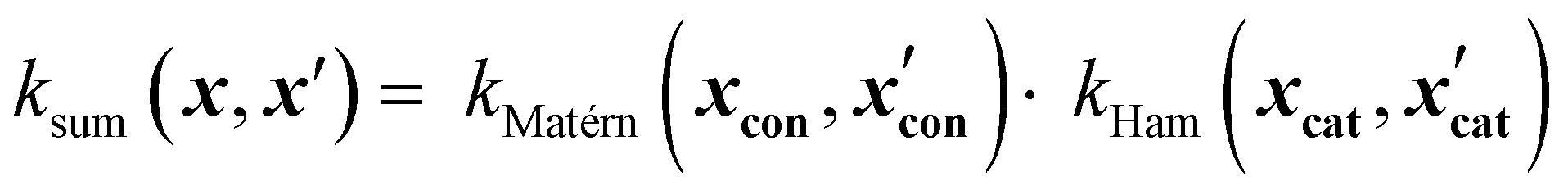 | (17) |
While the kernel determines the smoothness and amplitude of samples from a GP, the mean function represents an initial guess of the underlying objective function. In practice, this prior mean is often set to a constant μ0(x) ≡ μ0, which is commonly zero, especially when little prior information about the system is available.17 Setting the mean to zero simplifies computations and allows the kernel to capture all the structure in the data. However, the mean function can also be defined to incorporate expert knowledge, if this is available. For instance, if previous experiments or mechanistic knowledge about the reaction suggest a linear relationship with the input variable, a linear mean function could be used. Ultimately, while the choice of mean function can indeed affect the GP's predictions, the kernel typically plays the dominant role in modelling the underlying objective function, which is why the common practice is to use a zero mean when no strong prior information is available.
| y(x) ∼ GP(m(x),k(x,x′)|X,Y) | (18) |
| m(x)|X,Y = m(x) + k(x,X)(K + σn2I)−1(y − m) | (19) |
| k(x,x′)|X,Y = k(x,x′) − k(x,X)(K + σn2I)−1k(x,X)T | (20) |
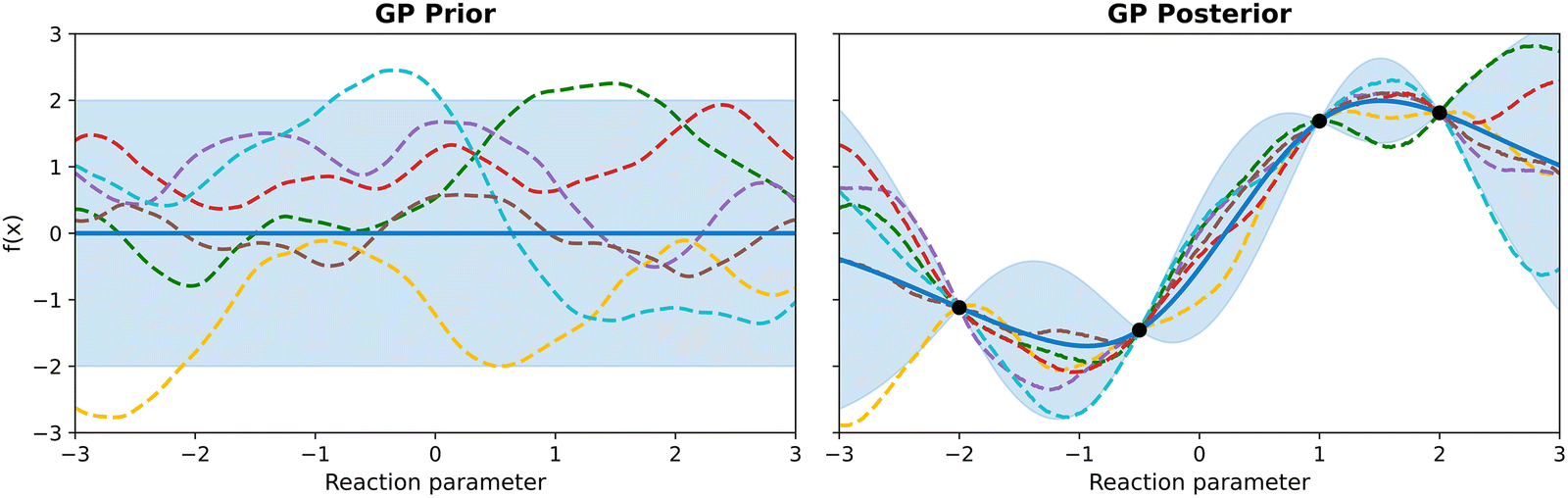 | ||
| Fig. 6 Gaussian process prior and posterior. Left: A zero-mean GP prior with a Matérn 5/2 kernel, showing the 2σ uncertainty band (shaded) and six samples from the prior (dashed). Right: The GP posterior after observing four data points (black dots), with posterior mean (solid), updated 2σ band, and six posterior samples illustrating reduced uncertainty near the observations. | ||
It should be noted that generally, appropriate hyperparameters for a given problem are unknown a priori. However, fitting these hyperparameters is essential because they tailor the GP to capture the underlying trends in the data. Therefore, the hyperparameters are fitted on the data before the posterior distribution is calculated. Typically, the hyperparameters are estimated by determining either the type II maximum marginal likelihood (MML) or the maximum a posteriori (MAP). For a more detailed overview of hyperparameter estimation, the reader is referred to.17,22
While standard Gaussian processes, as covered here, remain the dominant surrogate models for reaction optimization, various extensions—such as multi-task GPs52 and latent variable GPs53—have been developed for specific applications, but are out of scope for this work.
2.2. Acquisition function
Acquisition functions are a central component of Bayesian optimization, serving as the decision-making mechanism that guides the selection of new experimental points based on the surrogate model's predictions.17 In essence, an acquisition function quantifies the utility of evaluating the objective function at a particular input, balancing the trade-off between exploration of the search space and exploitation of current promising areas. This balance is crucial for optimizing expensive, black-box objective functions.The acquisition function leverages both the predicted mean, and the uncertainty provided by the surrogate model to calculate a scalar score for each candidate in the search space. This can be formulated as follows:17
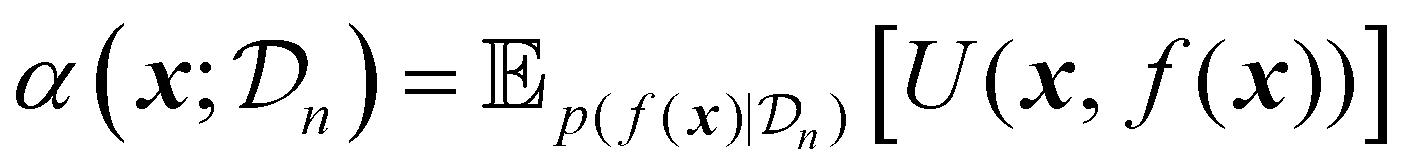 | (21) |
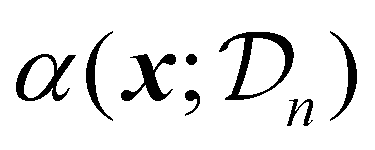 uses the surrogate model's posterior distribution
uses the surrogate model's posterior distribution 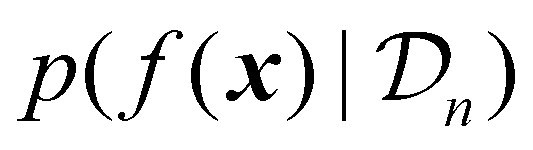 to calculate an overall utility of evaluating the objective at point x. Essentially, it takes the expected value of a utility function, U(x,f(x)), over all possible outcomes, considering both the prediction and the corresponding uncertainty around this prediction. It should be noted that in eqn (21), f(x) denotes the posterior mean prediction at x, not the surrogate model's latent function itself. By maximizing the acquisition function, the next experiment can be determined.
to calculate an overall utility of evaluating the objective at point x. Essentially, it takes the expected value of a utility function, U(x,f(x)), over all possible outcomes, considering both the prediction and the corresponding uncertainty around this prediction. It should be noted that in eqn (21), f(x) denotes the posterior mean prediction at x, not the surrogate model's latent function itself. By maximizing the acquisition function, the next experiment can be determined.
Several different acquisition functions exist.17,18 Common acquisition functions include Expected Improvement (EI), Probability of improvement (PI), Upper Confidence Bound (UCB), and Thompson Sampling (TS). Each of these functions embodies a different strategy for navigating the exploration–exploitation trade-off, yet no single acquisition strategy provides superior performance over all problem instances.17 In practice, the optimal choice of the acquisition function depends on the problem. A comparison of selected acquisition functions is provided in Fig. 7.
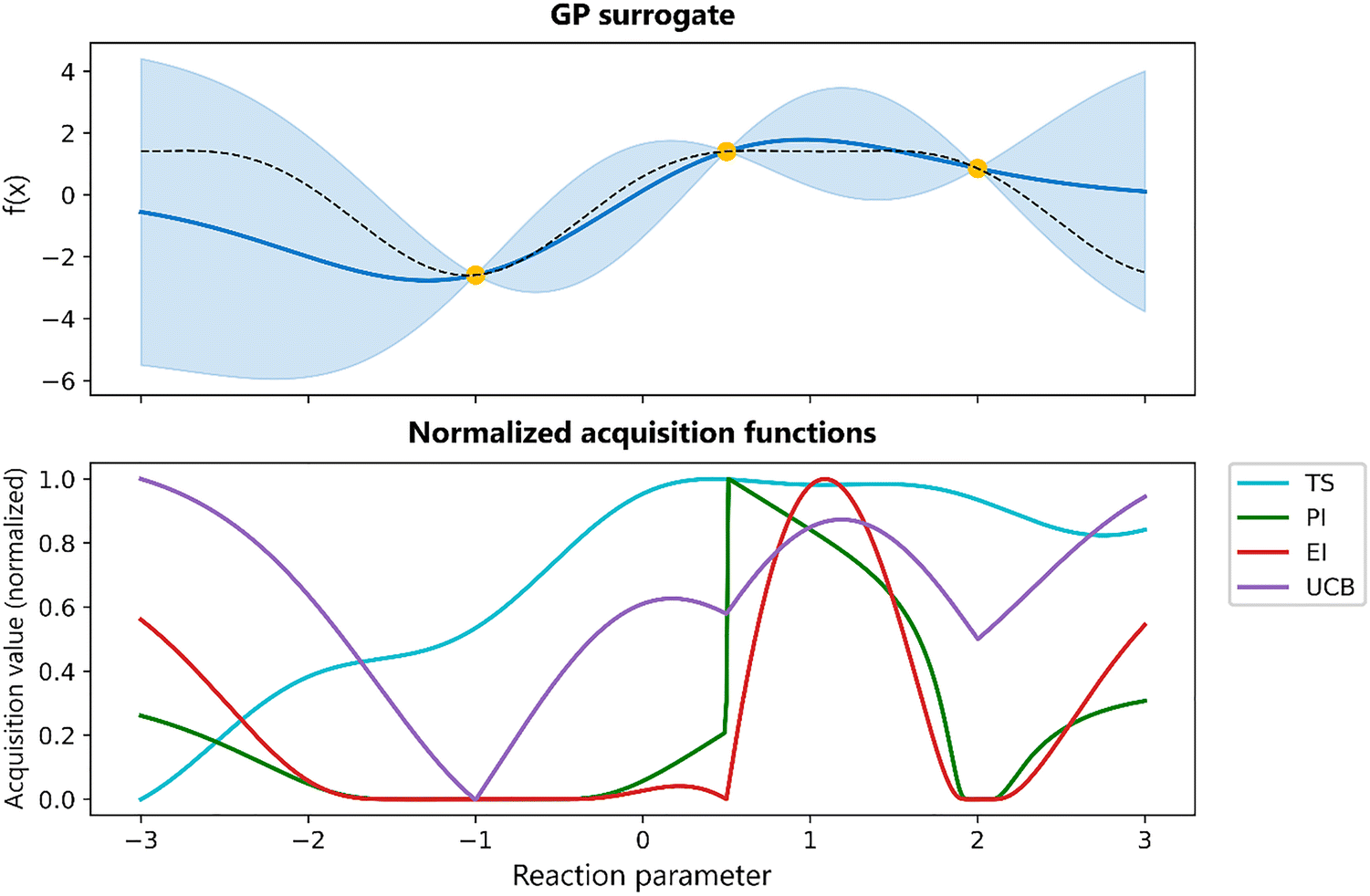 | ||
| Fig. 7 Comparison of acquisition functions derived from a shard GP surrogate. Top: The GP posterior mean (blue line), 2σ uncertainty bands (shaded), three training points (yellow), and the true function (dashed line). Bottom: Five acquisition functions, derived from the GP surrogate. TS (cyan) is a sample from the posterior and promotes broad exploration. PI (green) seeks points with high probability of improving on the current best, sharply peaking near the best observed locations. EI (red) trades off improvement magnitude and likelihood, yielding a more focused peak. UCB (purple) combines mean and uncertainty via a tuneable confidence bound. By normalizing all curves on the same scale, they can be compared more easily. | ||
Overall, acquisition functions play a critical role in directing the iterative search process. By quantifying the potential benefit of sampling at various points, they ensure that each new experiment is strategically chosen to enhance understanding of the reaction space and drive optimization towards a global optimum. However, it is essential to acknowledge that there is no one-size-fits-all acquisition function; the ultimate performance of Bayesian optimization is driven by the interplay between the surrogate model and the acquisition strategy.
As mentioned before, several acquisition functions have been developed for single-objective Bayesian optimization.18 Here, four widely used acquisition functions will be discussed: expected improvement, probability of improvement, upper confidence bound, and Thompson sampling. Each of these functions uses the predictions and uncertainty provided by the surrogate model to calculate a scalar utility value for a potential sampling point.
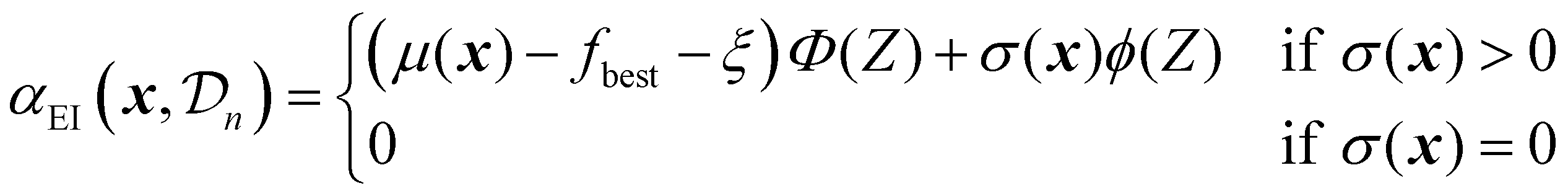 | (22) |
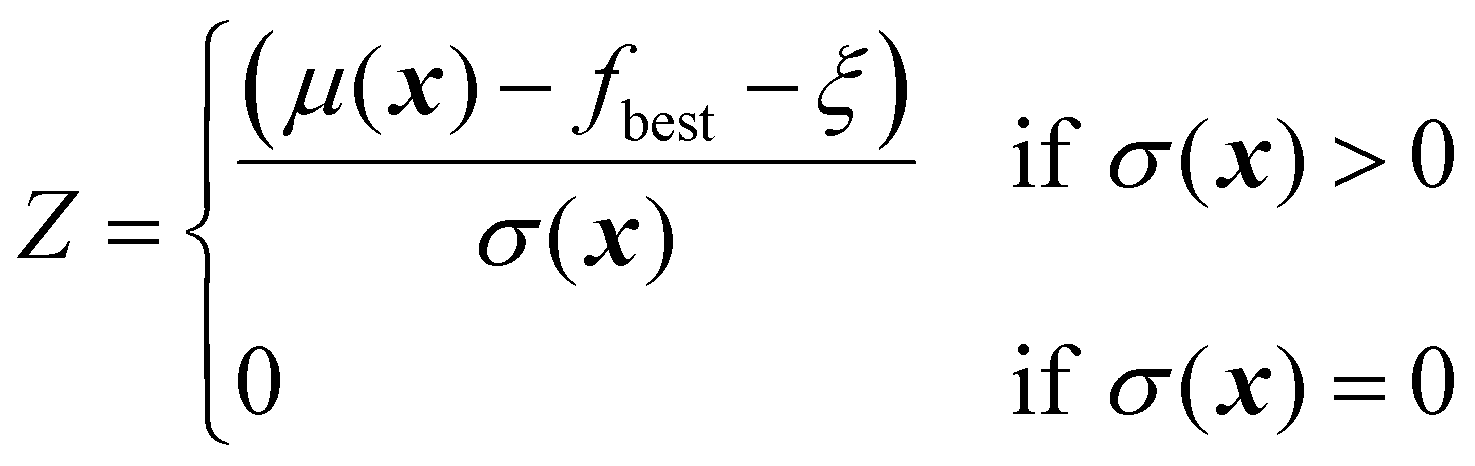 | (23) |
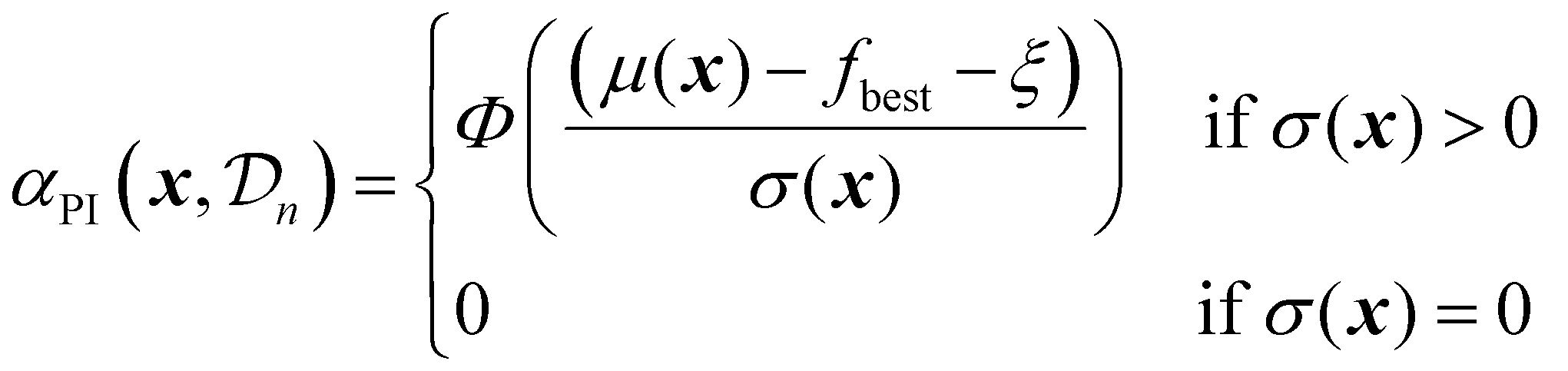 | (24) |
The parameter ξ plays an important role here, similar to its role for the EI acquisition function. In the standard implementation of PI, with ξ set to zero, the acquisition function is biased towards exploitation. This can be mitigated by adjusting the value of ξ.18 A higher ξ lowers the probability of improvement by effectively increasing the threshold that a candidate must exceed, thereby encouraging exploration of regions with higher uncertainty.
PI was the first reported acquisition function,18 and it is straightforward and computationally efficient, making it a popular choice. However, because it ignores the magnitude of potential improvement, it may lead to overly conservative exploration, and it could be argued that this makes PI a less attractive choice compared to other acquisition functions described in this section, especially when ξ is set to zero, as is often the case.
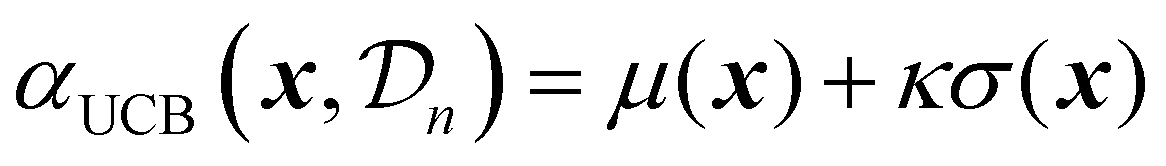 | (25) |
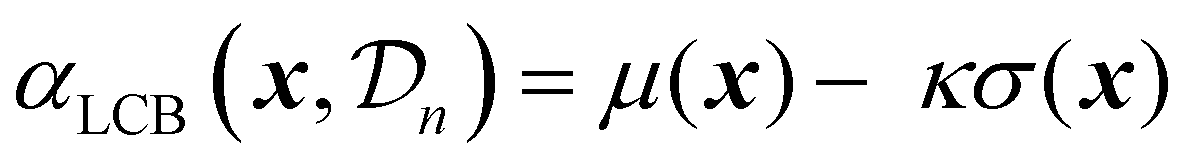 | (26) |
A theoretical guideline for setting the exploration parameter κ has been developed by Srnivas et al.56 Specifically, setting κ at iteration t as:
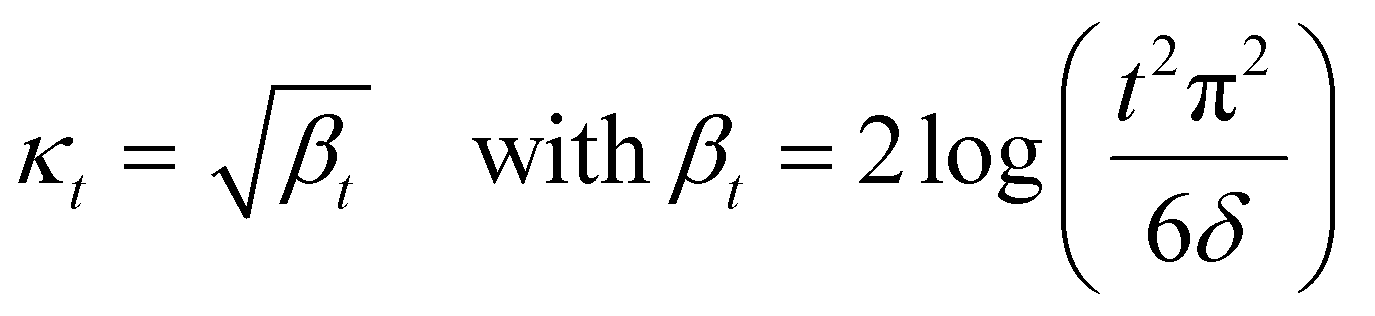 | (27) |
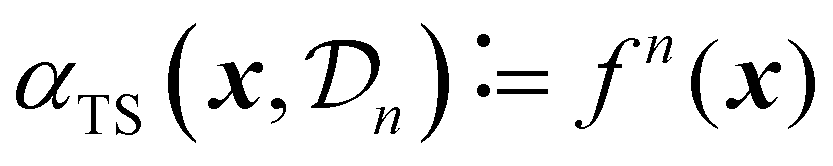 | (28) |
A critical aspect of Thompson sampling is the method used to generate samples from the surrogate model's posterior. For Gaussian processes, the most-commonly used type of surrogate model in the context of Bayesian optimization, no known methods exist to sample an exact function from the posterior. However, techniques such as spectral sampling35,58 can be used to create an approximate analytical sample from a GP, making sure that the samples accurately reflect both the central tendency and the uncertainty of the surrogate model.
2.3. Multi-objective optimization
Up until this point, the discussion has been focused on single-objective optimization. However, many real-world problems involve balancing multiple, often conflicting, objectives.1,16,60–62 During process development, for example, the importance of considering multiple performance criteria, such as yield, selectivity or productivity, becomes clear. Optimal conditions for one objective might result in sub-optimal performance on another criterium, making the process sub-optimal overall. A standard solution for this problem is the combination of multiple competing objectives into a single function.63,64 However, two major problems with linear combinations of multiple objectives have been identified: (i) quantitative a priori knowledge is necessary to determine adequate weights for the objectives, and consequently additional experiments are needed; (ii) only one optimal result is obtained, which is highly dependent on the chosen objective function and does not reveal the trade-off between multiple performance criteria, i.e., the effect they have on one another.16 A better solution to optimize a reaction towards multiple (competing) objectives is to simultaneously optimize towards all objectives at once: multi-objective optimization. In these scenarios, a single “best” solution does not exist; instead, the goal is to identify a set of trade-off solutions known as the Pareto front.65 Each solution on the Pareto front represents a state where no objective can be improved without worsening at least one other objective. This is shown for a minimization problem of two objectives in Fig. 8.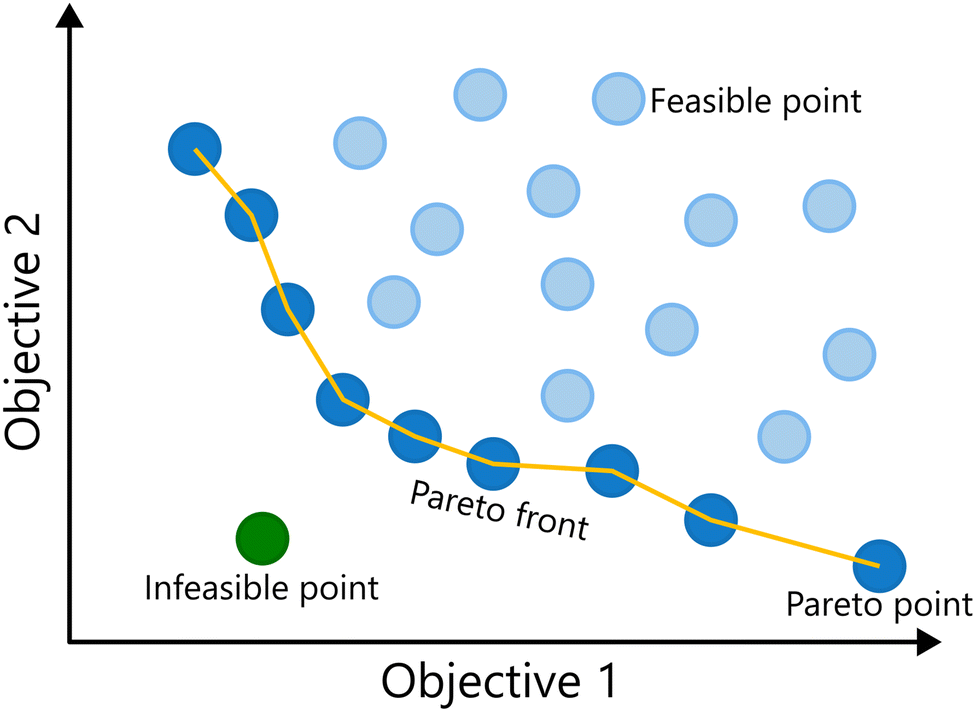 | ||
| Fig. 8 Example of a Pareto front for simultaneous minimization of two objectives. The Pareto front separates feasible and infeasible solutions. The points on the Pareto front are non-dominated solutions, as one objective cannot be improved without having a detrimental effect on the other. | ||
The core framework for multi-objective Bayesian optimization, using surrogate models and acquisition functions, is essentially unchanged from its single-objective counterpart, with some modifications to handle the added complexities introduced by optimizing multiple objectives. Typically, one surrogate model is built per objective, each providing probabilistic predictions (mean and uncertainty).35,44,59,66 Specialized multi-objective acquisition functions then leverage these models to guide the selection of new sample points that help expand and refine the Pareto front. This section introduces the key concepts of extending BO to multi-objective optimization, laying the foundation for subsequent discussion on practical implementations.
For a problem where all objectives are to be maximized, the Pareto front can be formally defined as:
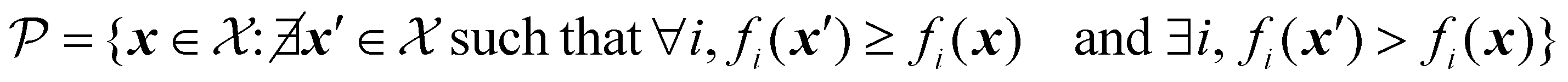 | (29) |
A key metric in evaluating the quality of a Pareto front is the hypervolume indicator.16 The hypervolume measures the size of the region in the objective space that is dominated by the Pareto front relative to a chosen reference point r (i.e., enclosed by the Pareto front on one side and the reference point on the other). A larger hypervolume corresponds to a better Pareto front, as a larger part of the objective space is enclosed. For problems with two objectives the hypervolume reduces to the area of the region dominated by the Pareto front, and for three objectives, it corresponds to the volume. The Hypervolume of a 2-d Pareto front is illustrated in Fig. 9.
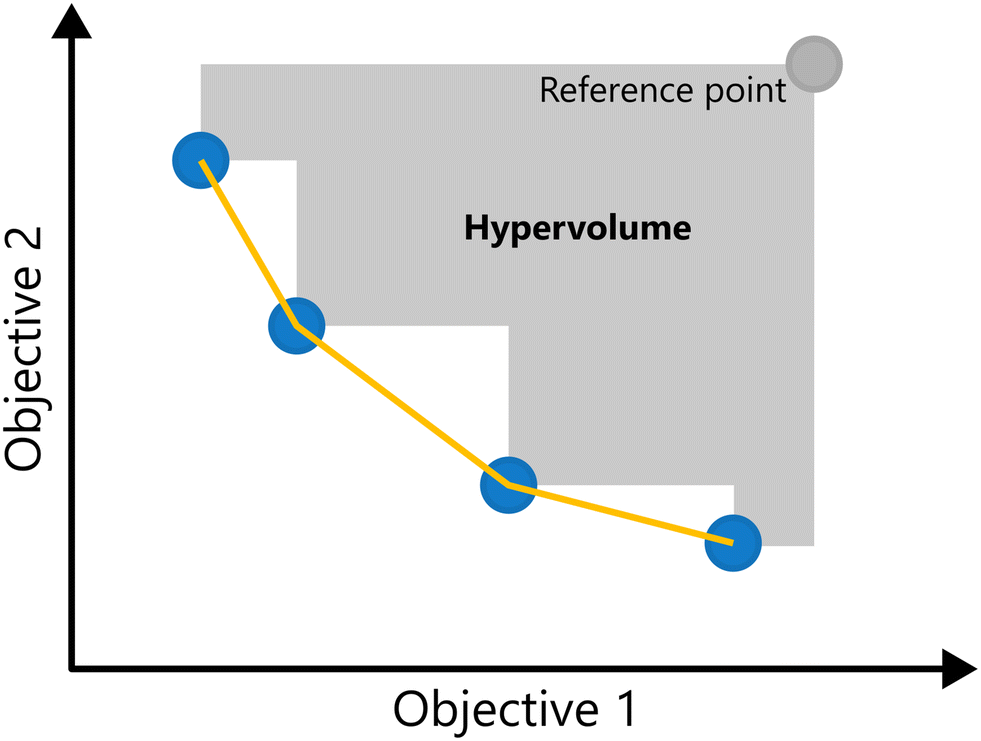 | ||
| Fig. 9 Illustration of hypervolume for a two-objective minimization problem. The blue points approximate the Pareto front (yellow line); the gray region shows the dominated space bounded by the reference point: the hypervolume. | ||
EHVI extends the idea of Expected Improvement to the multi-objective setting by measuring the expected increase in the hypervolume dominated by the Pareto front.67,68 Given the current Pareto front 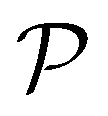 , EHVI can be expressed as:
, EHVI can be expressed as:
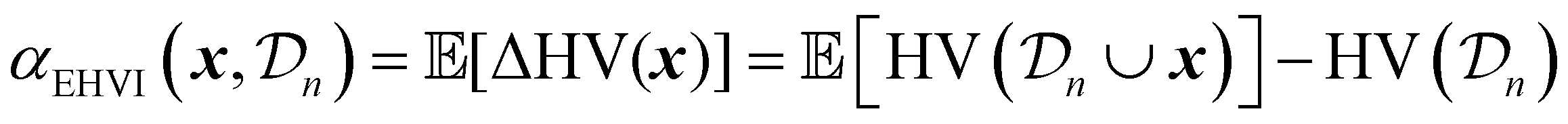 | (30) |
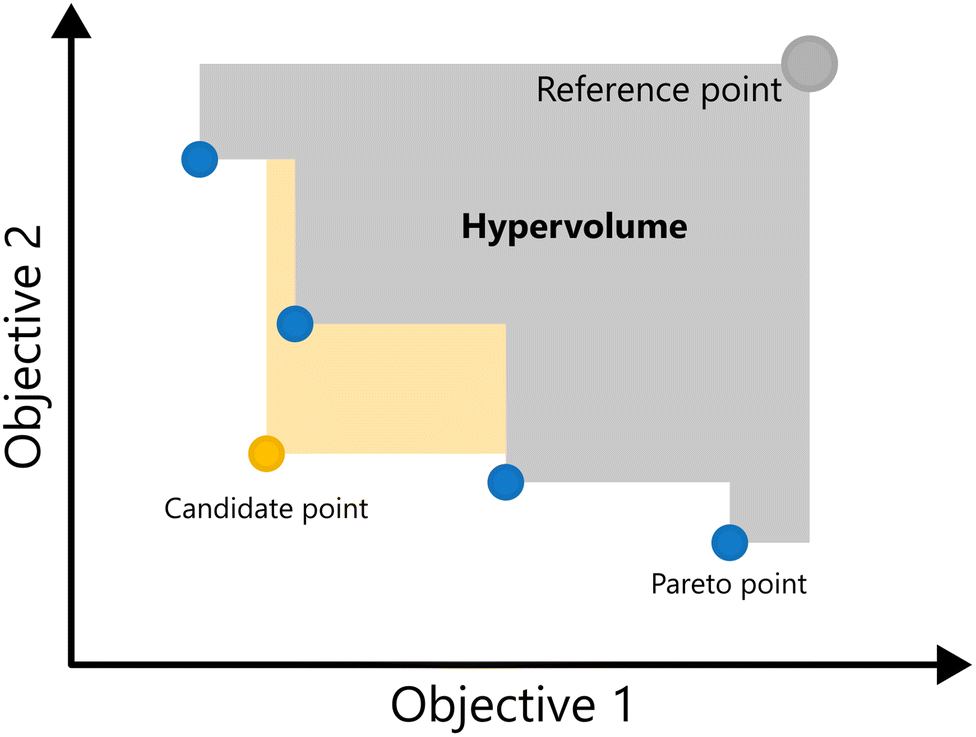 | ||
| Fig. 10 Illustration of expected hypervolume improvement for the minimization of two objective functions. The yellow area represents ΔHV(x) when adding a candidate point to the current dataset. Note that to have a non-zero hypervolume improvement, the candidate point must expand the current Pareto front, in this case dominating the blue point encompassed by the yellow area. | ||
EIM is a multi-objective adaptation of the EI acquisition function.69 In EIM, the expected improvement is expanded into a 2-d matrix. Specifically, given a Pareto set 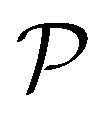 with k non-dominated points, the EIM can be represented as:69
with k non-dominated points, the EIM can be represented as:69
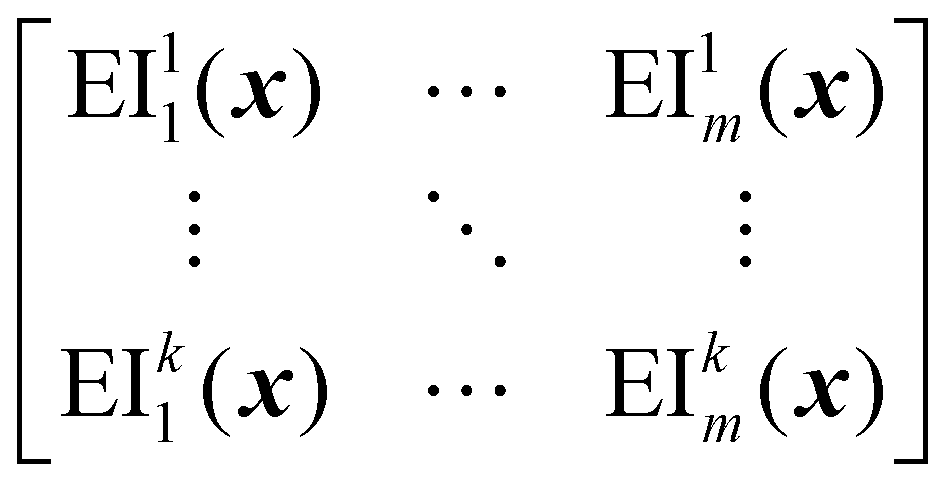 | (31) |
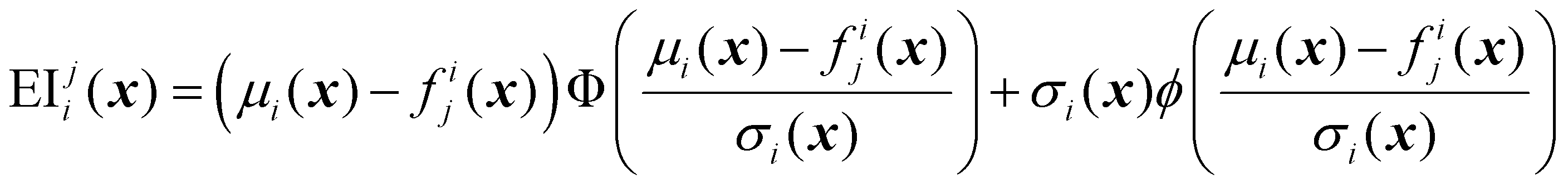 | (32) |
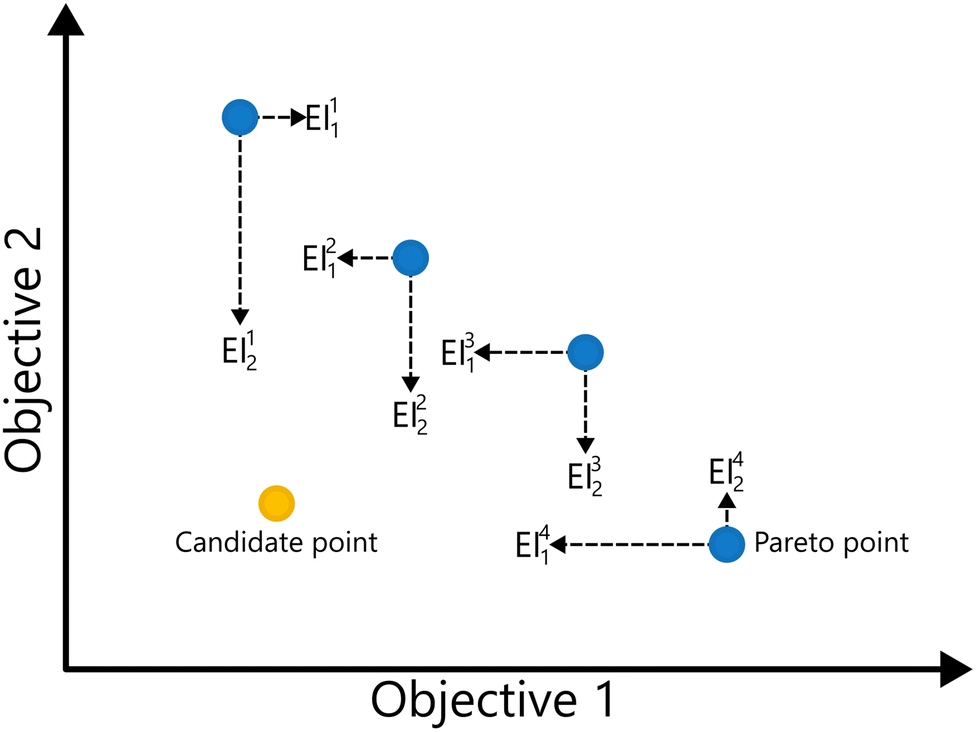 | ||
| Fig. 11 Illustration of the elements of the expected improvement matrix for the minimization of two objective functions. For a given candidate point, the surrogate's predicted means are evaluated against each Pareto point. Horizontal dashed arrows show the improvement in objective 1 (EIj1), and vertical arrows the improvement in objective 2 (EIj2) relative to the jth Pareto point, yielding the full expected improvement matrix. | ||
The matrix representation is then combined into a single scalar acquisition value using three possible transformations: Euclidean distance. Maximin distance or hypervolume improvement.69
The Euclidean distance EIM is given by:
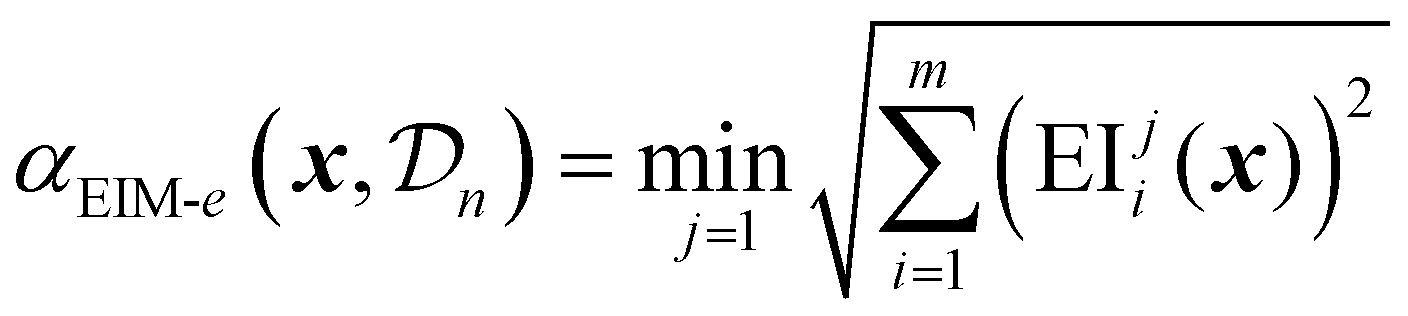 | (33) |
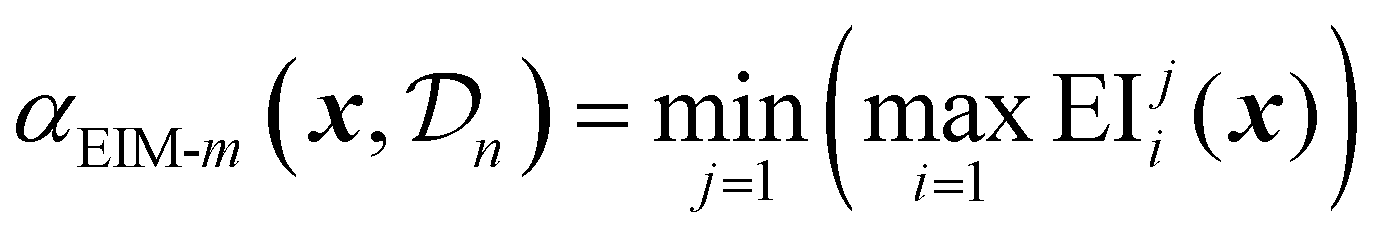 | (34) |
These aggregation strategies yield a single scalar value that reflects the overall potential for improvement across all objectives, that can be optimized to obtain the next point to evaluate.
The primary benefit of EIM over EHVI is its reduced computational cost; the formula for EIM is available in closed form, allowing for rapid calculation and optimization using classical optimization methods for acquisition functions.
2.4. Batch Bayesian optimization
Batch sampling refers to selecting multiple candidate points to evaluate in parallel during each iteration.17,70–72 This can be especially beneficial for chemical reaction optimization when the chemistry can easily be parallelized, such as in a High-Throughput Experimentation (HTE) setting,73 because although it does not reduce the total number of evaluations needed, it can significantly reduce the total time of the optimization campaign while largely preserving the data-efficiency of BO.Three common strategies for batch sampling can be distinguished:
Sequential (greedy) batch sampling . In this approach, candidate points are selected one at a time. After choosing a candidate, its impact is incorporated into the surrogate model—often using techniques such as the Constant Liar method,74 where a fixed value (e.g. the current worst value) is temporarily assigned to the selected point, and the surrogate model is updated. This update discourages subsequent selections from clustering near the already chosen point. This process repeats until the desired batch size is reached.
Simultaneous (joint) batch sampling . In simultaneous batch sampling, the entire batch of candidate points is selected simultaneously by optimizing an aggregated acquisition function. Crucially, the surrogate model remains unchanged during selection of the entire batch. Methods such as local penalization modify the acquisition function to penalize regions around already selected candidates, promoting diversity within the batch.72 Another possibility is the use of a specialized type of acquisition function that is designed to provide multiple candidate points. For example, the q-EI acquisition function extends the traditional EI criterion to jointly consider the impact of evaluating q points together.74 By capturing the interdependencies among candidates, q-EI seeks to maximize the EI of the entire batch. While this latter approach is very attractive on paper, computationally it is quite expensive.75 In general, when q is used as a prefix for an acquisition function, it implies the function is suited for batch sampling.
Batch Thompson sampling . In batch Thompson sampling, multiple samples are drawn from the surrogate model's posterior distribution, and each function is optimized. These independently derived optima together form the batch of experiments.35,76 This approach naturally promotes diversity among the batch candidates based on the model's uncertainty.
These strategies are illustrated in Fig. 12. The choice among these methods depends on the problem characteristics and available computational resources.
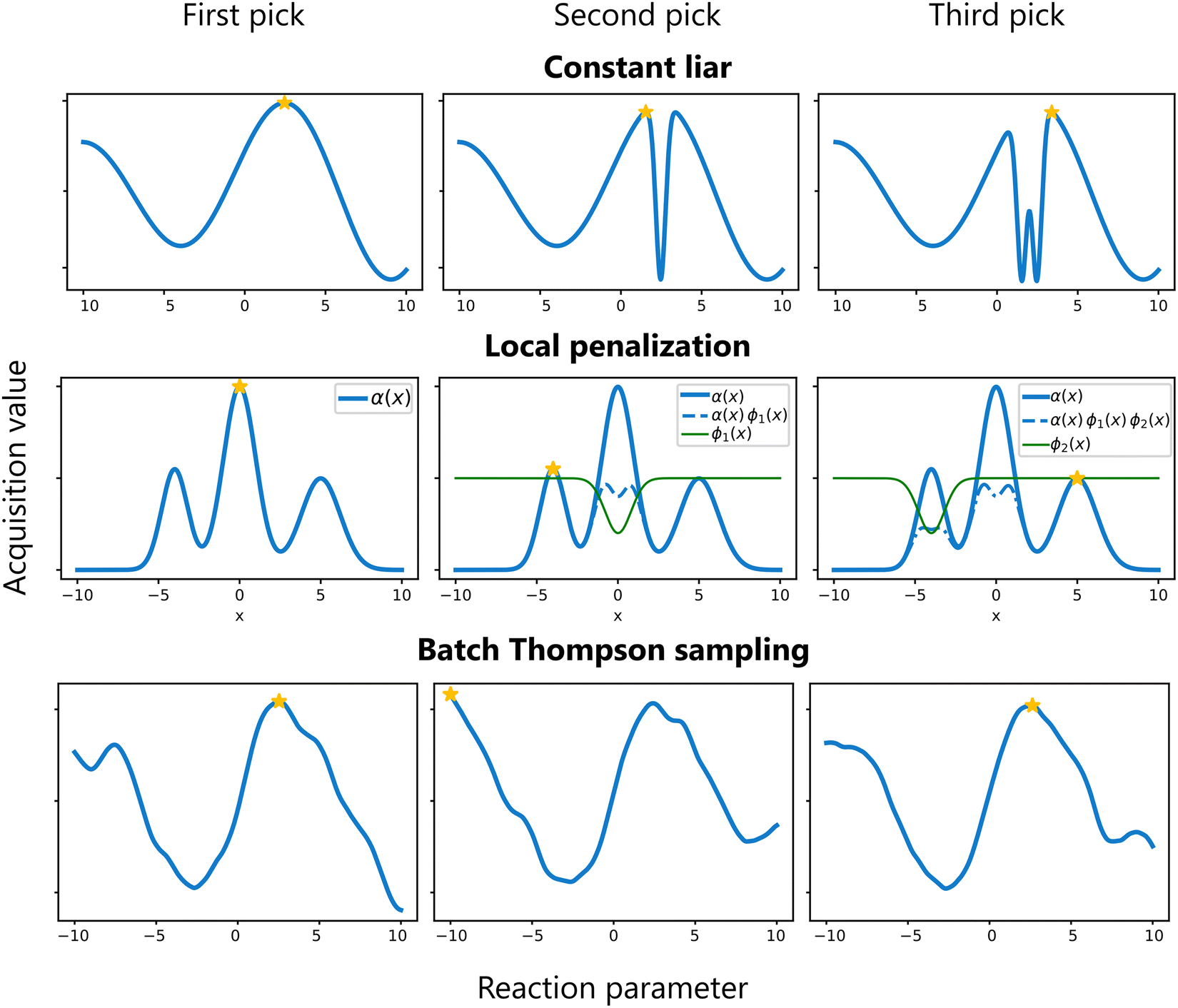 | ||
| Fig. 12 Common strategies for batch sampling in BO. Top: Constant liar with the liar value fixed at the current best. Middle: Local penalization applying sequential penalizers to discourage clustering. Bottom: Batch Thompson sampling via three independent GP posterior samples. | ||
2.5. Practical considerations
While the preceding sections introduce the core concepts and methodologies of Bayesian optimization, their practical use for chemical reaction optimization problems raises a number of recurring questions that extend beyond algorithmic details. In particular, aspiring practitioners are often faced with decisions regarding when Bayesian optimization is an appropriate tool, how to initialize a campaign, how long optimization campaigns should be continued, how reported performance can be benchmarked or assessed against other optimization methods, and how the intrinsic complexity of a given reaction system should be contextualized when interpreting and comparing studies. While these aspects are addressed implicitly throughout the literature and in the examples discussed in this review, they are rarely summarized in a single location. The following subsection therefore consolidates key practical considerations that have emerged from applications of BO in chemical reaction optimization, providing guidance for the design and interpretation of BO-driven experimental campaigns.Conversely, BO may be less advantageous when exhaustive screening is inexpensive, when broadly interpretable or uniformly accurate predictive models are required (e.g. for quality-by-design or regulatory purposes), or when optimization is dominated by hard constraints, frequent experimental failures, or other strongly discontinuous behaviour that is difficult to model explicitly.
In contrast, experimental BO studies focused on applying existing algorithms to real chemical systems rarely attempt such calibration, as their primary objective is typically to demonstrate data-efficient optimization rather than to establish algorithmic superiority. As a result, explicit experimental calibration against random or alternative optimization strategies is not performed in most reported BO applications, although notable exceptions exist, which are discussed where relevant in the following sections.
Consequently, reported experimental improvements should generally be interpreted as demonstrations of practical feasibility and efficiency, rather than as definitive performance comparisons between optimization strategies.
Early demonstrations of BO in chemical reaction optimization often focused on relatively simple, robust reactions, reflecting mostly the novelty of the approach. As the field has progressed, BO has increasingly been applied to more complex reaction systems, including higher-dimensional search spaces, and mixed-variable and multi-objective problems. Consequently, comparisons between BO studies should be interpreted in light of the underlying reaction complexity and experimental context, rather than based solely on metrics such as convergence speed or the number of experiments required.
3. Bayesian optimization in (miniaturized) batch and high-throughput reaction platforms
Bayesian optimization has been increasingly applied to small-scale reaction optimization platforms, where rapid screening and minimal reagent consumption are crucial. These (automated) platforms, including High-throughput experimentation (HTE), small-scale batch reactors (up to a few millilitres), and slug-based flow platforms, provide an efficient means to explore chemical space while reducing experimental workload. In addition, HTE platforms allow for easy parallelization of reactions,73 further accelerating reaction optimization. As will be discussed in this section, Bayesian optimization allows chemists to efficiently and systematically optimize reaction parameters, demonstrating its performance over traditional DoE or intuition-based approaches.19With their seminal paper, Shields et al. played a pivotal role in the adoption of Bayesian optimization by the broader organic chemistry community, demonstrating its effectiveness for reaction optimization.19 In this study, the authors introduced the EDBO framework, based on a GP surrogate model using DFT descriptors and a modified version of the EI acquisition function allowing for batch sampling. Designed with categorical variables in mind, the performance of this framework for a test set of one Suzuki–Miyamura reaction and five Buchwald–Hartwig couplings was found to be superior compared to that of traditional Design-of-Experiments (DoE) approaches (Scheme 1a). To further benchmark the framework's performance, the study conducted an optimization game, where 50 expert chemists competed with the EDBO algorithm to optimize a palladium-catalysed direct arylation reaction (Scheme 1b) It was observed that, while humans made significantly better initial choices than the random selection of the BO algorithm, their average performance was surpassed by that of the algorithm within three batches of five experiments. Additionally, on average BO was able to identify the global optimum more reliably and in fewer experiments. It should be noted that in this case, both BO and human experts navigated a dataset that was generated from an exhaustive screening of all parameter combinations, rather than experiments being carried out only after being proposed by the algorithm. Finally, the framework was applied to the iterative optimization of a Mitsunobu and a deoxy fluorination reaction (Scheme 1c). In both cases, the algorithm was able to quickly surpass the performance of ‘standard’ literature conditions for these reactions, even identifying quantitative conditions for the Mitsunobu case, further underscoring the robust performance of BO across diverse optimization challenges.
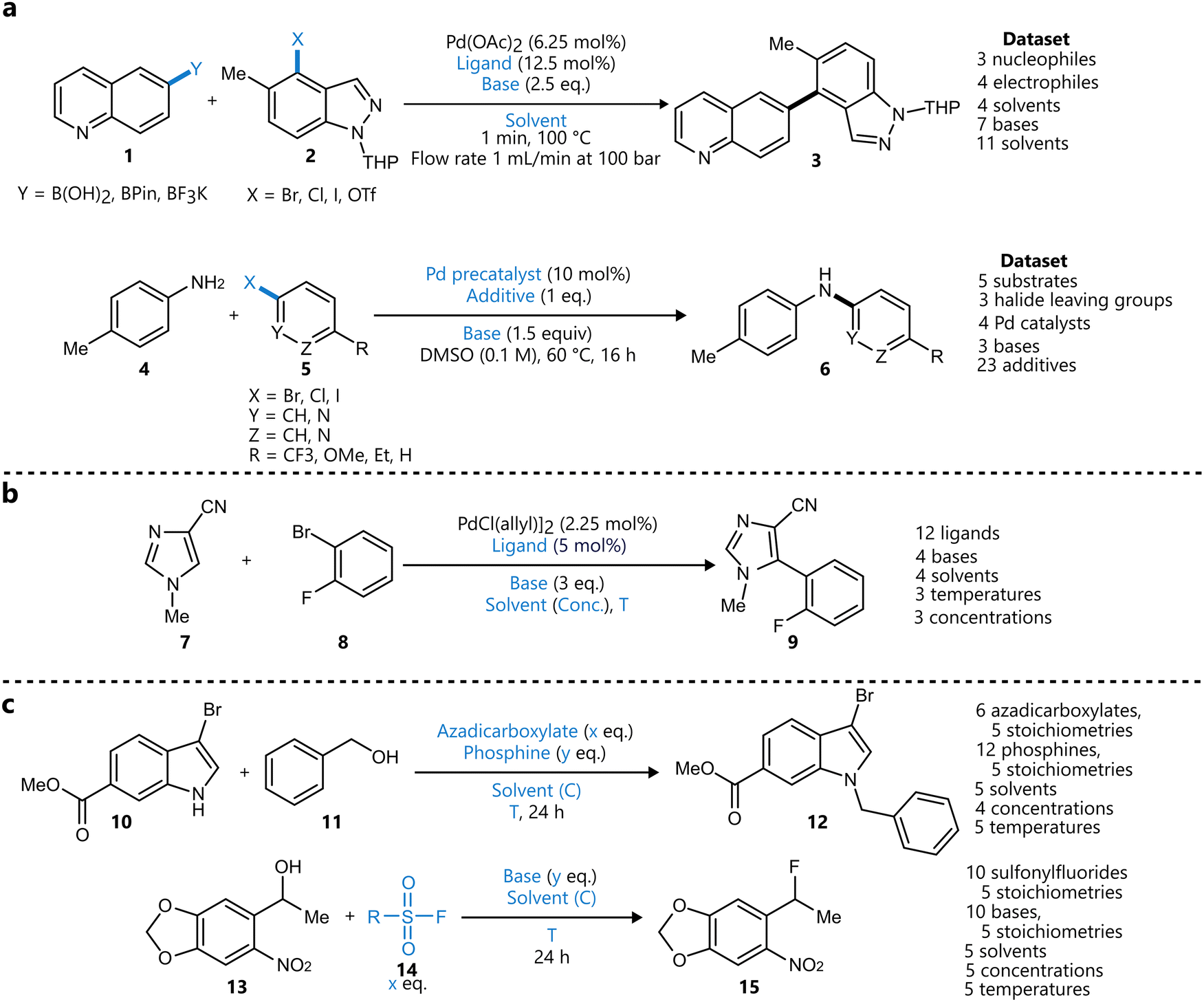 | ||
| Scheme 1 Applications of BO reported by Shields et al.19 Parameters defining the reaction space are highlighted in blue. (a) Training reactions used to test the performance of the framework and compare to a traditional DoE approach. The Suzuki reaction dataset (top) and Buchwald–Hartwig reaction dataset (bottom) were used to tune the parameters of the BO framework. (b) Direct arylation reaction used for the optimization ‘game’ to compare EDBO to the performance of expert chemists. All parameter combinations were carried out in HTE-fashion, with BO and the experts navigating the dataset to optimize the reaction. (c) Mitsunobu (top) and deoxy fluorination (bottom) reactions used to further test the EDBO framework. | ||
Building upon this foundation, Torres et al. extended the EDBO framework to multi-objective optimization, enabling simultaneous optimization of multiple reaction objectives.79 The resulting EDBO+ framework was applied to the optimization of both the yield and enantioselectivity of a Ni/photoredox-catalysed enantioselective cross-electrophile coupling of styrene oxides 16 with aryl iodides 17 and 19 (Scheme 2).
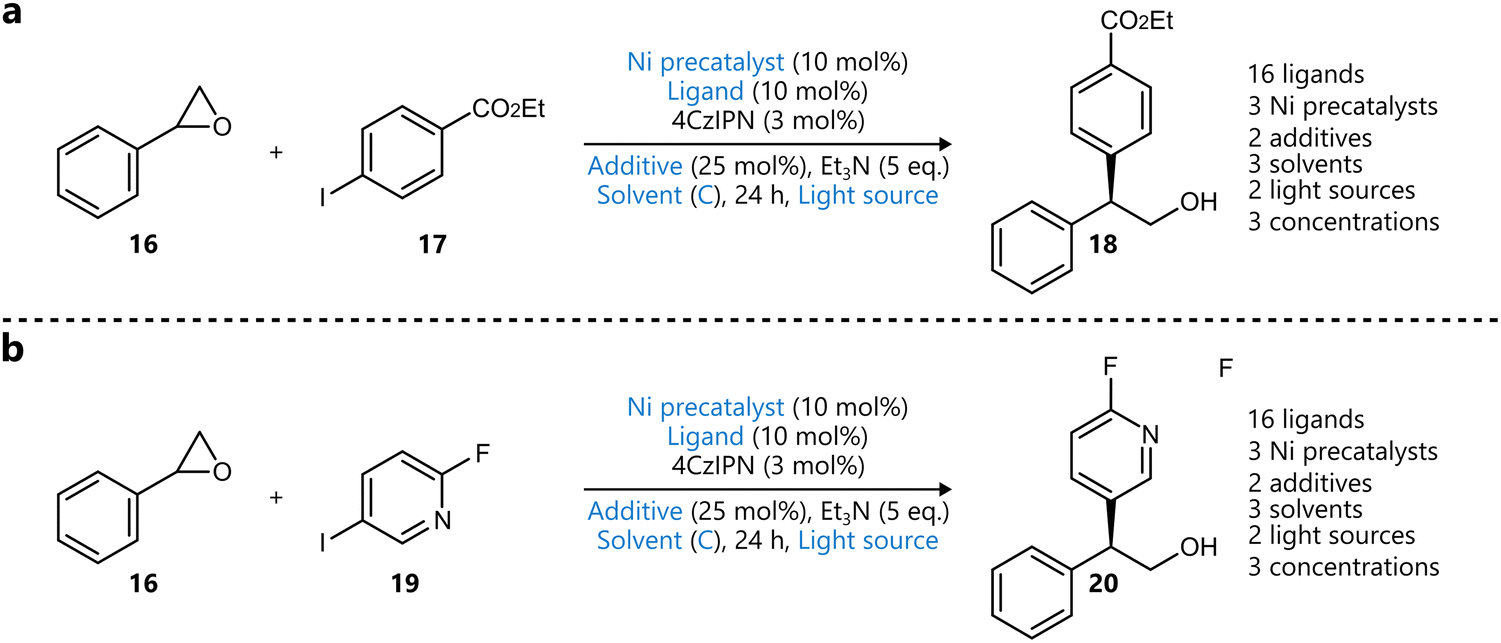 | ||
| Scheme 2 Applications of BO used to showcase optimization using EDBO+ reported by Torres et al.79 Metallaphotoredox-catalysed enantioselective cross-coupling reactions between styrene oxide 16 and two aryl iodides 17 (a) and 19 (b). Parameters defining the reaction space are highlighted in blue. Ligands were encoded using DFT descriptors, one-hot encoding was used for the rest. | ||
3.1. Advanced use of descriptors for variable selection
Also using the EDBO+ framework, Romer and colleagues reported the optimization of a nickel-catalysed reduction of enol tosylates for the stereoconvergent synthesis of trisubstituted alkenes 2280 (Scheme 3a). The authors first made a selection of phosphine ligands by successively applying principal component analysis (PCA) and clustering analysis on the kraken monophosphine ligand library81 to identify 47 clusters, with one ligand selected from each cluster for HTE screening. From this screening, eight ligands were selected to be included in the search space for the BO campaign towards the E-product, and seven for the one towards the Z-product. Other variables included were solvent, reductant, concentration, and catalyst loading, while the two objectives were yield and diastereoselectivity. Interestingly, before starting the campaigns, the GP was pretrained on both the HTE data and data from benchtop reactions, aiming to accelerate convergence. In 25 experiments across each campaign, split over five rounds of five experiments, both E-selective and Z-selective conditions were identified, affording both products in ∼90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 diastereoselectivity and >90% yield.
:10 diastereoselectivity and >90% yield.
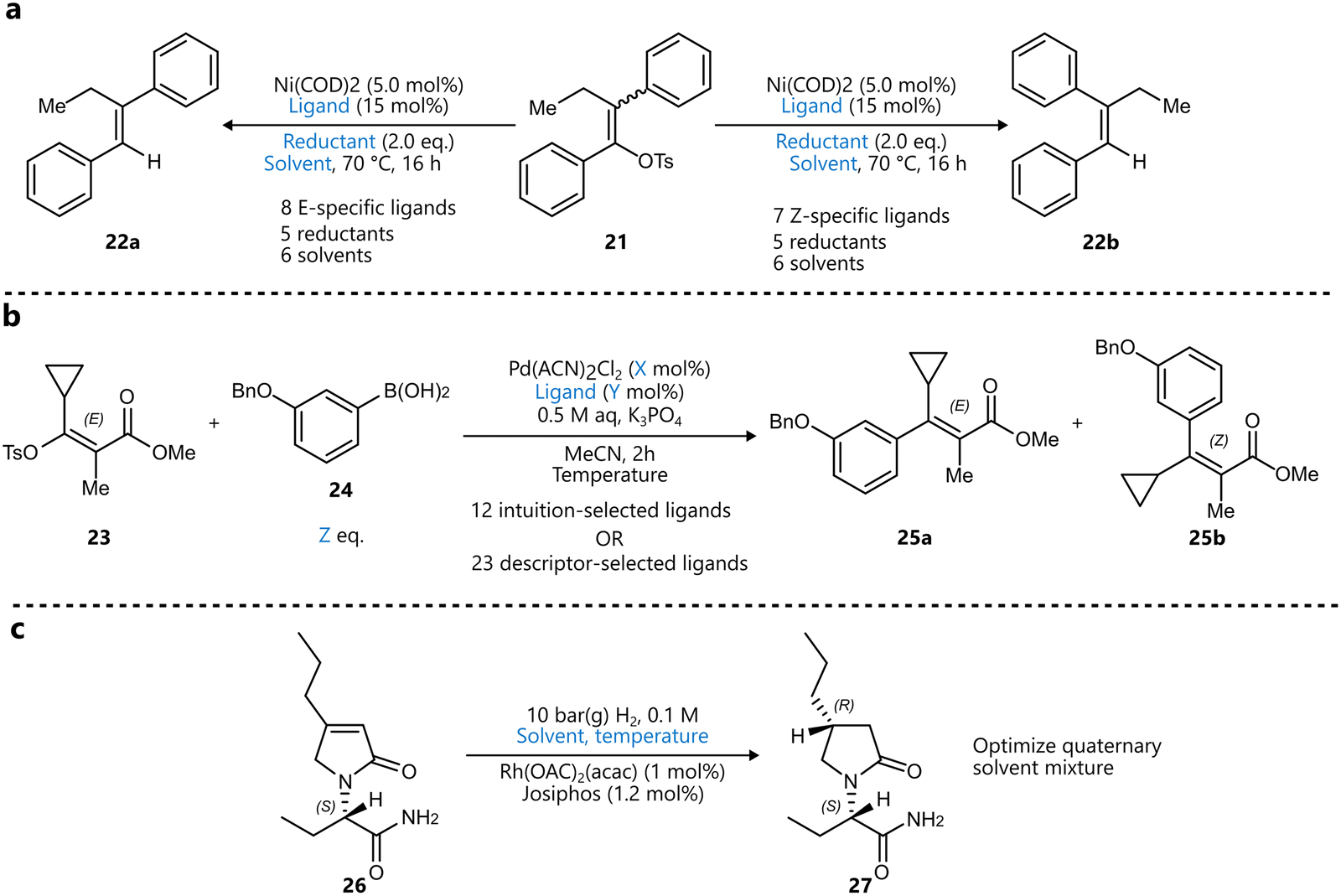 | ||
| Scheme 3 Examples of using descriptors to narrow down the variable space for BO. Parameters defining the reaction space are highlighted in blue. (a) Stereoconvergent synthesis of trisubstituted alkenes by Romer et al.80 A training set of monophosphine ligands was constructed using principal component analysis (PCA), after which a distinct set of ligands was selected for both the E- and Z-selective reaction prior to BO. (b) Pd-catalysed stereoselective Suzuki–Miyaura coupling optimized by Christensen et al. to afford maximize the yield and selectivity towards the E-product.82 The comparison between an expert-selected and a data-driven set of ligands was made, the latter outperforming the former both in terms of yield and selectivity. (c) Asymmetric hydrogenation reported by Amar et al.85 An initial descriptor-based BO campaign selected four solvents, after which a quaternary mixture of these solvents was optimized in a subsequent BO campaign. | ||
A similar study was presented by Christensen et al., who developed a fully autonomous closed-loop system for optimizing a stereoselective Suzuki–Miyaura cross-coupling reaction using standard 96-well plates.82 Their platform integrated a Chemspeed SWING robotic workstation with HPLC analytics and the ChemOS scheduling software to autonomously conduct experiments. Interestingly, the optimization was guided by a tandem of two BO algorithms: Gryffin59 and Phoenics,83 although from the paper it is not entirely clear how this was achieved. Gryffin was used for ligand selection, while Phoenics optimized the continuous parameters, including phosphine to palladium ratio, palladium loading, reaction temperature and arylboronic acid equivalents (Scheme 3b). Ligands were selected through unsupervised clustering of molecular descriptors, again from a subset of the kraken database.81 From each of the resulting 24 clusters, one ligand was selected to be included in the optimization campaign, during which no new ligands were added (e.g., from high-performing clusters). The study demonstrated that this approach outperformed intuition-based ligand selection, improving both the yield and diastereoselectivity of the desired E-isomer 25a. Interestingly, incorporating the descriptors directly into the model did not offer further benefits, which was attributed to potential bias in the selection of descriptors.
In a recent extension of this platform, the same group introduced a dynamic sampling strategy to improve the reliability of endpoint detection in time-sensitive transformations.84 By integrating a real-time plateau detection algorithm, the system could autonomously terminate experiments once product formation stabilized, rather than relying on fixed reaction times. This enhancement reduced the risk of overreaction or decomposition, improved the quality of training data for the surrogate model, and demonstrated the potential of coupling adaptive temporal control with BO-driven optimization.
Published a couple of years earlier, a study by Amar and coworkers illustrates a closely related use of molecular descriptors in conjunction with BO, applied to solvent selection for asymmetric catalysis.85 In the context of a ruthenium-catalysed asymmetric hydrogenation of a complex chiral α–β unsaturated γ-lactam 26 used for the synthesis of the anti-epileptic drug Brivaracetam, the authors aimed to simultaneously optimize yield and diastereomeric excess (Scheme 3c). The study was divided in two stages. In the first, promising solvents were selected from a database of 459 candidates. Gaussian process models were trained on several descriptor sets using an expert-curated initialization set of 25 solvents, with Thompson sampling as the acquisition strategy. Rather than optimizing the acquisition function, the sampled function was used to predict the performance of all 459 solvents, and the top candidates were selected in batches of four to five before retraining the GP. After several iterations across the different descriptor sets, the algorithm proposed 18 solvents, from which four (including two from the initial set) were carried over to the second stage. There, a BO algorithm was used to optimize the composition of a quaternary solvent mixture and the reaction temperature, yielding a Pareto front for yield and diastereomeric excess.
Further expanding the scope of Bayesian optimization beyond traditional reaction condition optimization, Li et al. reported a sequential closed-loop approach for the optimization of a metallaphotoredox reaction.47 The study applied BO to the screening of a library of 560 potential organophotoredox catalysts (OPCs) and the optimization of reaction conditions for a decarboxylative C(sp3)–C(sp2) cross-coupling reaction (Scheme 4). Using a two-stage closed-loop BO strategy with a GP surrogate model and UCB acquisition function, the authors first selected 18 promising OPCs from a library of 560 potential candidates encoded with molecular descriptors that captured a range of thermodynamic, optoelectronic, and excited-state properties. This was then followed by the optimization of three reaction parameters. In 107 total experiments, conditions resulting in an 88% yield were identified. Subsequent benchmarks showed the newly identified OPC to exhibit comparable activity to a classical Ir-catalyst, with higher yields at low nickel loadings and slightly lower yields at higher nickel loadings. Notably, BO significantly outperformed random search, where only 4.5% of the tested conditions achieved high yield, compared to 44% under BO-guided experiments. This work underscores the expanding role of BO into molecular discovery, moving beyond reaction condition optimization to include catalyst selection.
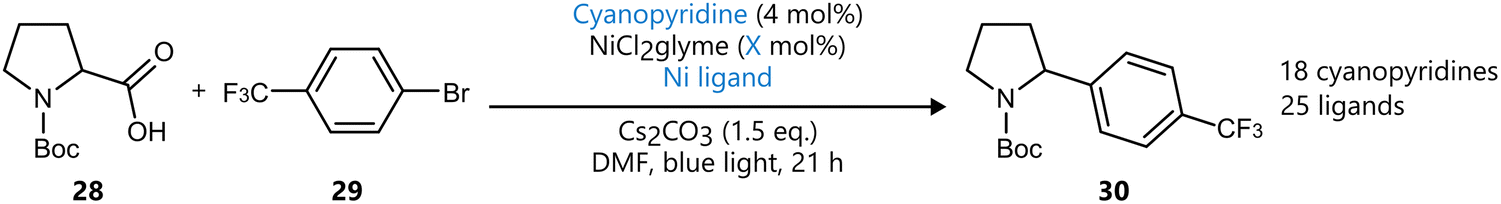 | ||
| Scheme 4 Metallaphotoredox reaction optimized using BO by Li et al.47 The two-stage BO strategy first selected 18 potential cyanopyridines from a scope of 560 candidates using descriptor-based encoding, subsequently optimizing the reaction in 107 experiments. Parameters defining the reaction space are highlighted in blue. | ||
3.2. BO for general reaction conditions
While the applications discussed so far have focused on optimizing reaction conditions for a single substrate, several recent studies have extended the use of BO to identify conditions that generalize well across a wider substrate scope. This shift towards generality reflects a practical need, mainly in medicinal and discovery chemistry, where broadly applicable conditions are highly valued. Angello et al. developed a BO framework coupled to an automated synthesis platform to identify general conditions for heteroaryl Suzuki–Miyaura couplings by optimizing across a representative panel of substrate pairs, selected by clustering analysis.86 Using a scalarized objective function to balance yields across the full set, the algorithm efficiently prioritized conditions that performed well across the entire substrate panel (Scheme 5). A comparison of three of the algorithm-identified general reactions conditions and a popular condition from the literature for this type of reaction was made for a set of 20 compounds not in the training data, with at least one of the three algorithm-identified conditions outperforming the literature conditions in 18 out of 20 cases. | ||
| Scheme 5 Optimizing a Suzuki–Miyaura coupling for generality across a substrate set, as reported by Angello et al.86 The training set included eleven target products; conditions were optimized to maximize the yield across this set. Parameters defining the reaction space are highlighted in blue. | ||
In a related approach, Wang and coworkers introduced a bandit optimization framework specifically designed to identify general reaction conditions.87 While not Bayesian optimization in the strictest sense, the method shares key features—such as probabilistic modelling and principled acquisition strategies—and can be considered part of the broader class of Bayesian-inspired optimization frameworks. Their method formulates the problem as a multi-armed bandit problem, where each ‘arm’ represents a reaction condition, having a distribution of an arbitrary reactivity metric across the entire substrate scope (Scheme 6a). Optimizing for generality then comes down to selecting the best arm, maximizing reactivity across the scope. Several optimization algorithms were benchmarked first on three reaction datasets, and the best performing one—which uses an upper confidence bound strategy—was subsequently used to identify general conditions for three reaction types (Scheme 6b–d). For the phenol alkylation (Scheme 6d), the identified conditions were compared to standard conditions on unseen substrates, with the former conditions outperforming the latter in ten out of eleven cases.
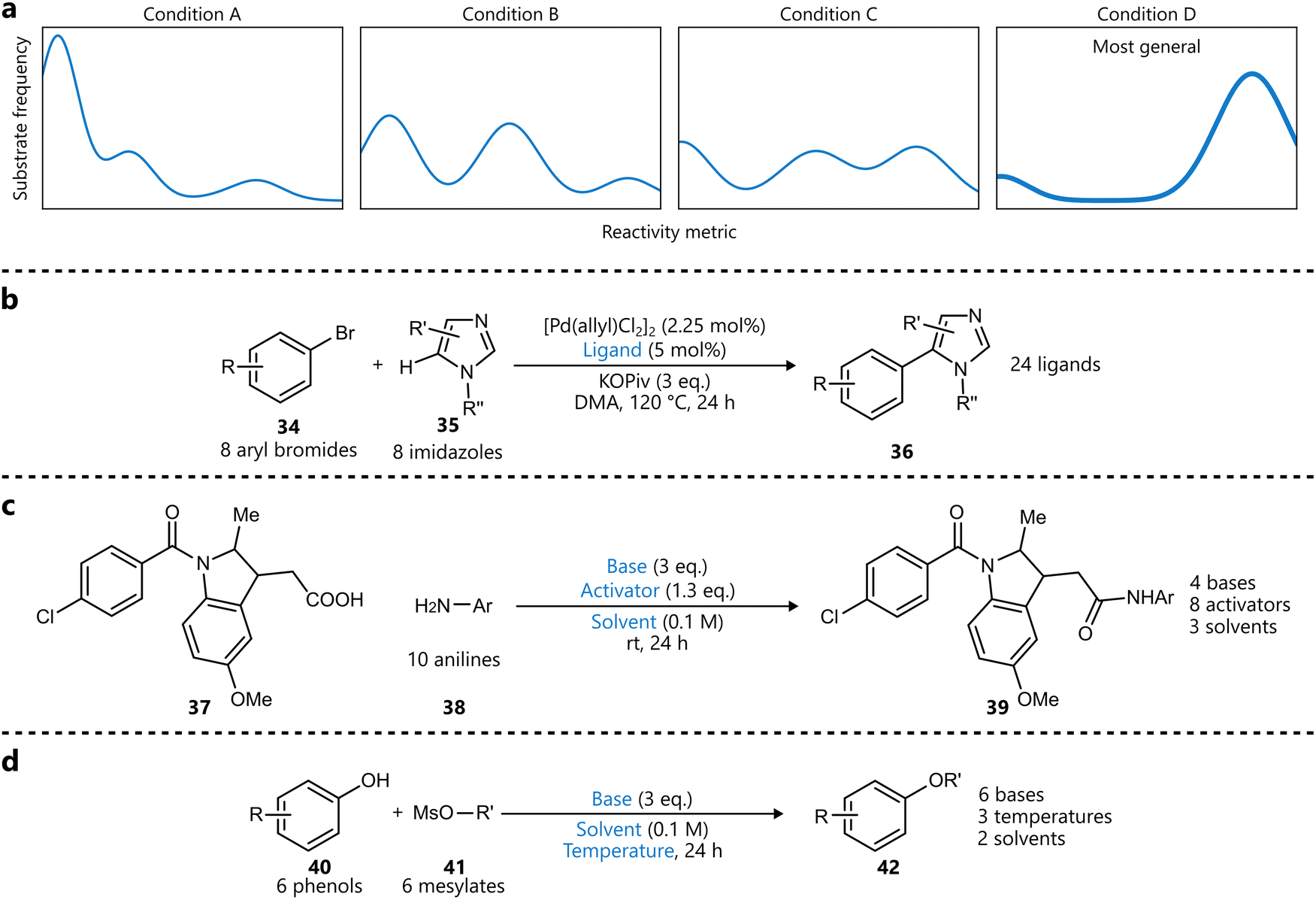 | ||
| Scheme 6 Bandit optimization for generality by Wang et al.87 Parameters defining the reaction space are highlighted in blue. (a) Illustration of how individual conditions can be seen as ‘arms’, representing a distribution of substrate reactivity. X-axis represents a reactivity metric (e.g. yield), the y-axis represents the frequency of substrates. A more general condition (like D in this example) will be skewed towards the right, representing more substrates exhibiting higher reactivity under this condition. (b) C–H arylation optimized for generality across 64 possible products. Only the ligand was optimized. (c) Amide coupling reaction optimized for generality across 10 aniline substrates. (d) Phenol alkylation reaction optimized for generality across 36 possible products. | ||
3.3. Highly automated systems
Another stream of research has explored how BO can be embedded into increasingly autonomous experimental platforms, enabling adaptive closed-loop reaction optimization with minimal human intervention.Sheng et al. developed a closed-loop system for mechanistic investigation of electrochemical reactions, combining voltammetric measurements, a deep learning model for waveform classification, and BO to refine kinetic parameters of redox-active species iteratively.88 In their work, a deep learning model was used to generate propensity values for five different common electrochemical mechanisms based on sets of voltammograms, and BO was used to guide the system towards regions of experimental space where the propensity for a certain mechanism (in this case an EC-mechanism, i.e. an electron transfer step followed by a reaction in solution) was maximized. The system was validated on the oxidative addition of 1-bromobutane to the electrogenerated CoI tetraphenylporphyrin. It's interesting to see BO being used here not for the direct optimization of reaction parameters, but rather to enhance the mechanistic understanding of a reaction by maximizing the information obtained from an experiment.
In a different context, Leonov et al. reported a “programmable synthesis engine”, capable of closed-loop optimization across a range of reaction types using Bayesian optimization with a GP surrogate and a PI acquisition function.89 The system was first demonstrated the optimization of three different transformations, including a four-component Ugi reaction, a manganese-catalysed epoxidation of styrene sulfonate, and a trifluoromethylation (Scheme 7a–d). For the latter, the authors optimized the reaction of four different starting materials in the same reactor with respect to a ‘novelty score’—a heuristic introduced based on analysis of the reaction spectrum—to explore the product space, followed by individual optimization of the yield for three identified products. Subsequently, two de novo reactions were selected from a random combinatorial search over two small chemical spaces and optimized in closed-loop fashion using BO (Scheme 7e and f). Although the platform demonstrates commendable versatility in handling multiple reaction types, the authors’ claims of generality should be interpreted in light of the system's practical constraints. All reactions were limited to a round-bottom flask. While this reactor is indeed broadly applicable, it does not encompass the full range of synthetic challenges, such as heterogeneous catalysis, gas-phase transformations, or photochemical systems.
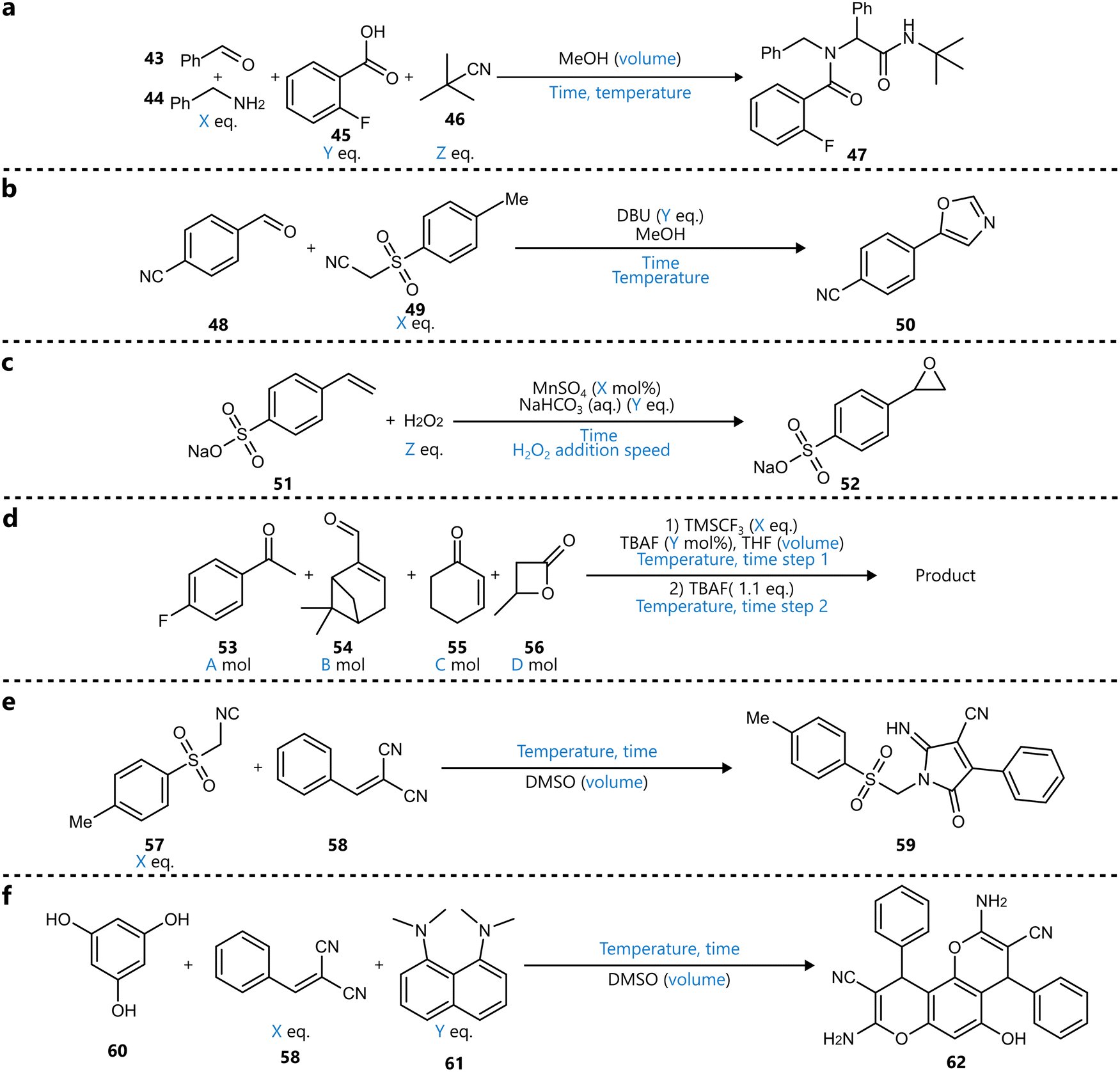 | ||
| Scheme 7 BO-optimized on the batch-based platform reported by Leonov et al.89 Parameters defining the reaction space are highlighted in blue. (a) Four-component Ugi reaction. (b) Van Leusen oxazole synthesis. (c) Manganese-catalysed epoxidation (d) explorative trifluoromethylation. Three products of this reaction were first identified and subsequently optimized in separate campaigns. (e) Reaction between toluenesulphonylmethyl isocyanide and benzylidenemalonitrile was discovered using a combinatorial search in a limited substrate set. The reaction was subsequently optimized. (f) Three-component reaction identified using a similar combinatorial search, followed by subsequent optimization. | ||
In contrast to these more centralized systems, Burger et al. developed a mobile robotic chemist that autonomously navigates a conventional laboratory environment to carry out experimental campaigns.90 The robot uses laser scanning and touch feedback to localize itself within the lab and plan its trajectory, enabling it to physically operate a range of standard and custom laboratory equipment—including solid dispensers, liquid dispensers, a capping station, photolysis station, and a headspace gas chromatography machine—and to execute protocols without human assistance. In a catalytic hydrogen evolution case study, 688 experiments (in batches of 16) were conducted over eight days, guided by Bayesian optimization to identify optimal catalyst compositions. The BO framework employed a GP surrogate and an ensemble of 16 UCB-acquisition functions, each with a different value κ to modulate the exploration–exploitation trade-off.
Each function proposed a single condition, together yielding the batch of 16 experiments. The level of spatial and operational autonomy demonstrated by this system sets it apart from fixed-deck platforms, highlighting the feasibility of integrating BO into more flexible, modular laboratory environments.
An emerging subset of highly automated small-scale (high-throughput) platforms leverages slug-based flow systems, in which small (<500 µL) slugs of reaction mixture are sent through a microreactor, significantly reducing material consumption.91–94 This can be implemented in two ways: segmented slug-flow or oscillatory flow. In the former, slugs of reaction mixture are sent through a conventional microreactor, typically separated from one another by an inert gas and carried by a solvent stream. In the latter, the reaction slug remains largely stationary in a small reaction channel while the flow is oscillated back and forth to induce mixing and temperature equilibration. Both approaches offer distinct advantages and drawbacks. Oscillatory systems are more complex to implement and cannot be parallelized within a single reactor channel, unlike slug-flow setups, limiting their throughput. However, they minimize axial dispersion and cross-contamination, making them particularly well-suited for reactions with a long residence time or those requiring high reproducibility. In both cases, these slugs can be seen as miniature batch reactors, allowing tight control over reaction conditions with minimal material consumption while retaining many of the benefits of continuous flow. Due to their small scale, these platforms are discussed here, rather than in the next section, which focuses on continuous-flow platforms.
The group of Jensen reported the use of Bayesian optimization in combination with a modified version of their previously developed oscillatory flow platform (Fig. 13a).94 To increase throughput, ten single-channel reactors were arranged in parallel. Each reactor channel was also equipped with LEDs to enable photochemical reactions. A key challenge was operating the solvent under elevated temperatures due to the oscillatory motion. To address this, the system was operated in a ‘stationary’ mode, in which each droplet was delivered to its reactor channel and held stationary for the duration of the reaction, with selector valves sealing the reactor from the rest of the system. The dragonfly95 Bayesian optimization framework, employing a GP surrogate model and an ensemble of acquisition functions (UCB, Thompson sampling, EI) was integrated into the system to guide the optimization of two Buchwald–Hartwig amination reactions (Fig. 13b and c). Both discrete (catalyst, base; two levels each) and continuous (temperature, residence time) variables were included in the optimization process, aiming for a single objective (normalized product LC area). Notably, for the first reaction, the optimization campaign was carried out twice, in different solvents, rather than including the solvent as a variable.
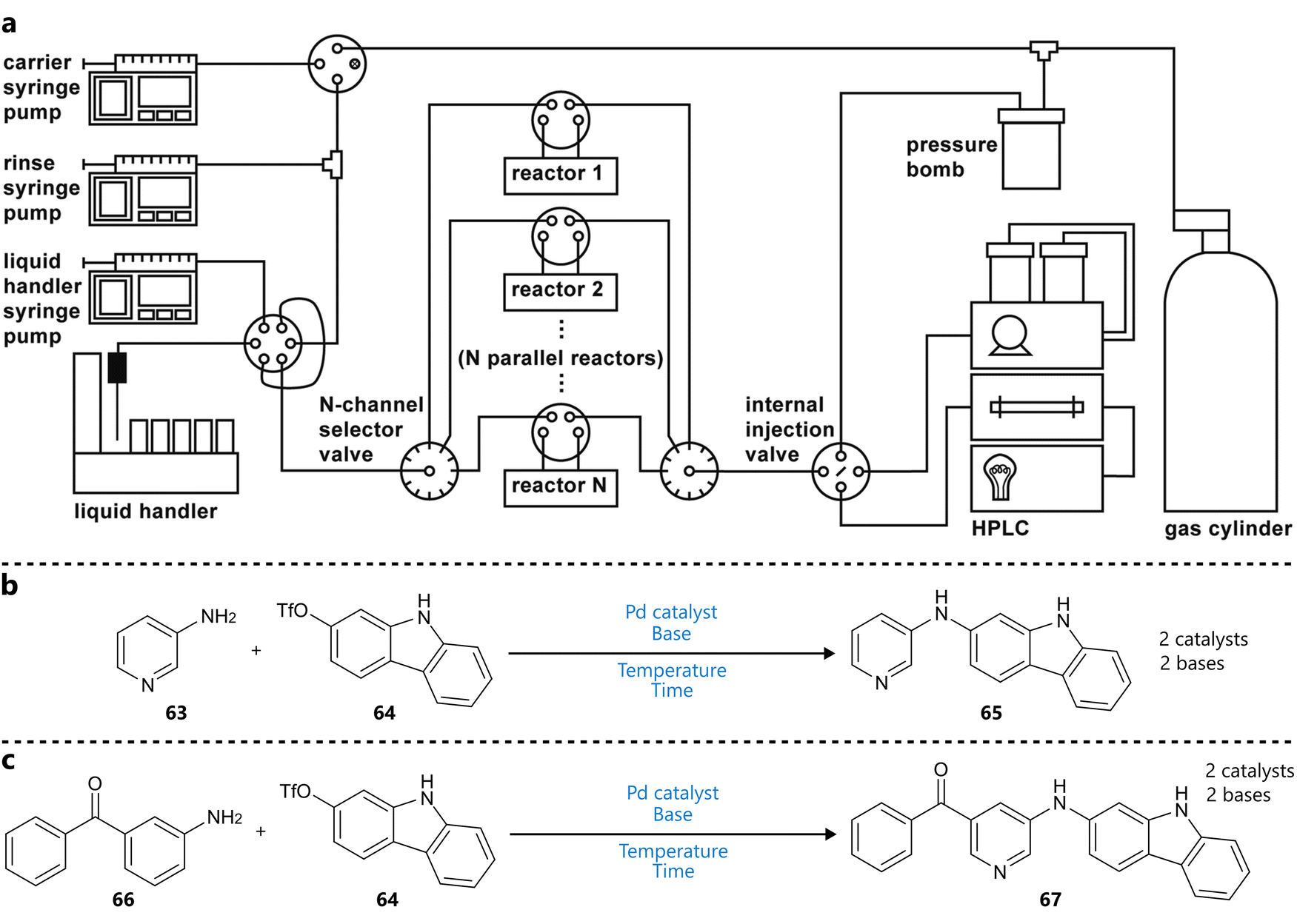 | ||
| Fig. 13 Bayesian optimization in a parallel droplet platform reported by Eyke et al.94 (a) Schematic representation of the reaction platform. Ten reactors allow parallel experimentation handled by a single reaction slug generator. Reproduced from Eyke et al.94 under CC BY 3.0 Unported.96 (b) and (c) Buchwald–Hartwig aminations used to demonstrate closed-loop optimization. Parameters defining the reaction space are highlighted in blue. | ||
Another slug-flow platform integrating BO for reaction optimization was developed by Wagner and co-workers, first introduced in a study published in Advanced science.92 Reaction slugs of 300 µL are segmented by inert gas and pushed through the heated coil reactor by a carrier solvent, after which they’re analysed by a combination of inline FT-IR and online UPLC (Fig. 14a). The platform was demonstrated through the autonomous optimization of a Buchwald–Hartwig amination (Fig. 14b), guided by Bayesian optimization using a Gaussian process surrogate and an expected improvement acquisition function. The BO campaign included six continuous variables (catalyst loading, reaction concentration, temperature, residence equivalents of base and equivalents of amine), three objectives (yield, space-time yield and cost) and ran for 60 total experiments. Interestingly, following the BO campaign, its results were used to narrow down the parameter space to a productive region, dropping residence time and concentration as parameters and reducing the ranges of the remaining ones. Next, a full-factorial Design of Experiments (DoE) was conducted on this focused design space, to construct response surface models for yield and two impurities. Finally, additional experiments were conducted to parameterize and fit a mechanistic kinetic model. This workflow illustrates the synergy of Bayesian optimization and more traditional methods, where Bayesian optimization is used to quickly identify an optimal region of a larger search space, which can then be subsequently narrowed down, enabling statistical and kinetic modelling, and mechanistic interpretation.
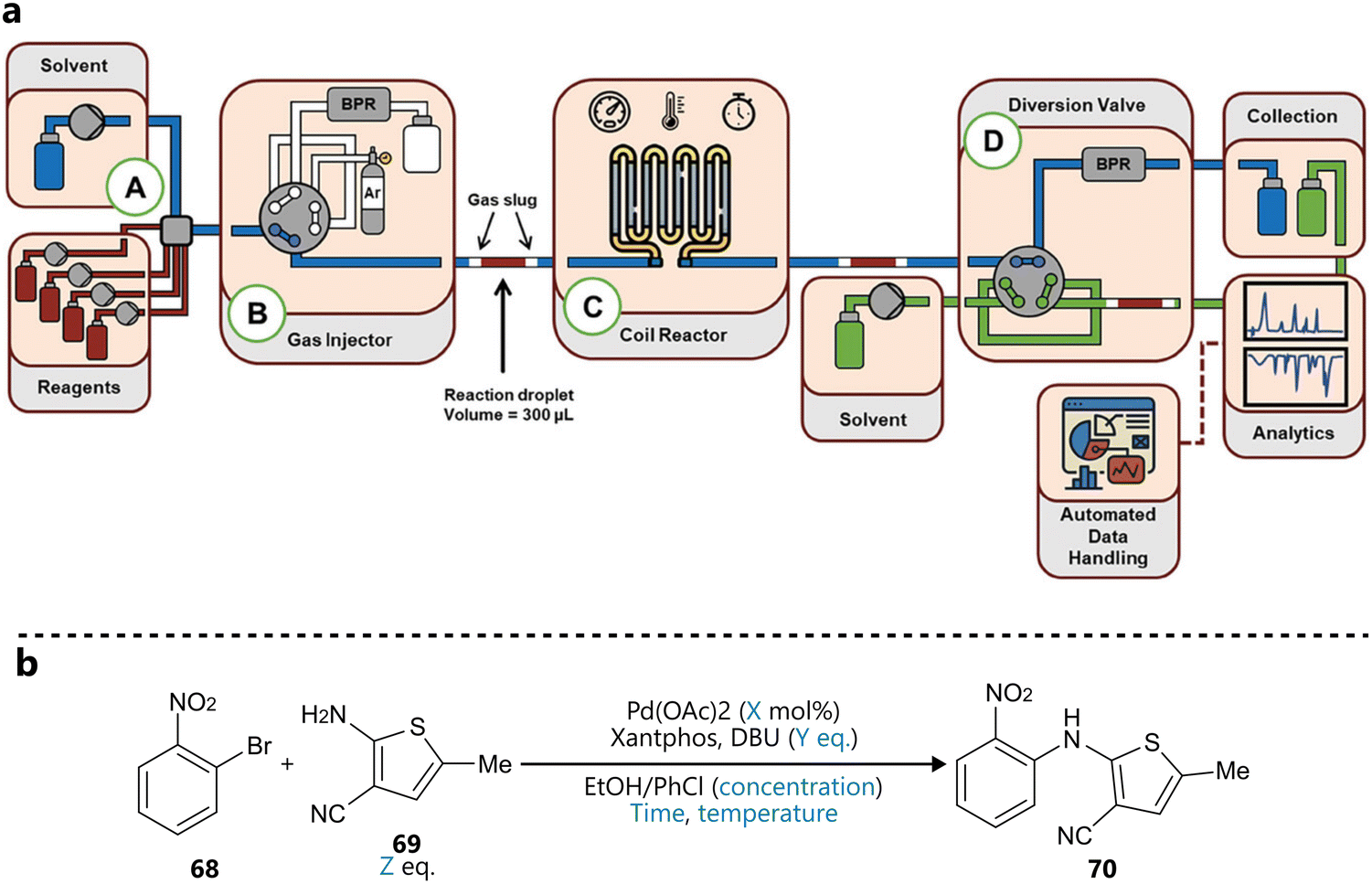 | ||
| Fig. 14 BO guided reaction optimization in a slug flow platform reported by Wagner et al.92 (a) Schematic representation of the reactor platform. Reproduced under a CC GY 4.0 license.97 (b) Buchwald–Hartwig amination used to validate the close loop optimization. Parameters defining the reaction space are highlighted in blue. | ||
In a follow-up study, Wagner et al., used the same slug-flow platform to explore several practical considerations regarding the use of Bayesian optimization.93 Using an amide coupling as a model reaction, they optimized five continuous variables: amine concentration, carboxylic acid equivalents, coupling reagent equivalents, temperature and residence time (Fig. 15a). The study revolved around three research questions around Bayesian optimization.
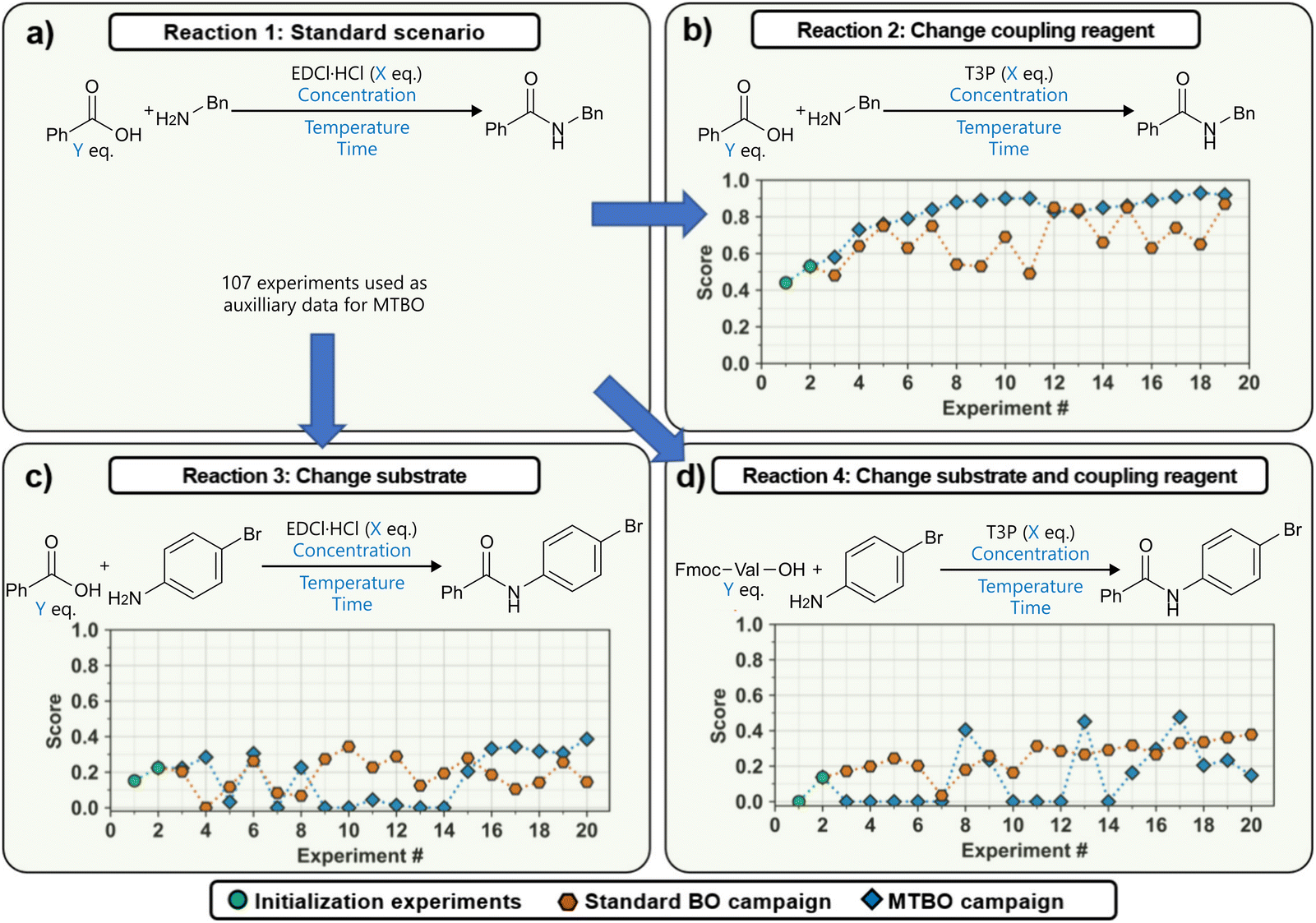 | ||
| Fig. 15 Transfer learning to accelerate BO on a slug-flow platform reported by Wagner et al.93 Adapted from the authors under CC BY 4.0.97 (a) The standard reaction scenario, from which experiments were used as auxiliary data for the transfer learning. (b) Scenario where a different coupling reagent is used. (c) Transfer to a different substrate. (d) Transfer to both a different coupling reagent and substrate. | ||
First, the authors compared two commonly used acquisition functions, EI and TS, focusing on the balance between exploration and exploitation. While TS was shown to be significantly more exploratory than EI when used with a small initialization set, this difference largely disappeared when the BO campaign was initialized using a Latin Hypercube Sampling (LHS) design, which can be considered best practice.
Second, the use of a weighted scalar objective function was examined as an alternative to formal multi-objective optimization. The aim was to prioritize high yield while penalizing long residence times and excessive reagent equivalents. While this approach is appealing due to its simplicity and ability to return a single optimal condition, it requires a priori weight assignment, and lacks a Pareto front. As such, the campaign would need to be repeated if the optimization priorities change—though it may still be suitable when a single compromise solution is desired, for example, for scale-up.
Finally, the potential for transferring knowledge across related optimization tasks was evaluated using multi-task Bayesian optimization (MTBO), a topic that will be discussed in more detail in Section 6. Three transfer scenarios were considered: (1) changing the coupling reagent, (2) changing the substrate, and (3) changing both. MTBO uses an auxiliary dataset performed on a related optimization task to try and improve optimization efficiency, in this case all 107 experiments that were previously carried out on the model reaction. While faster convergence was observed in the first case (new coupling reagent), no clear advantage was found for the more challenging substrate-related scenarios (Fig. 15b–d). Notably, these comparisons were performed with a small initialization set of only two experiments, so it could be argued that, with a proper initialization set, results may differ.
3.4. Various applications
As an alternative to a Pareto-based multi-objective optimization, Dalton et al. presented a BO-guided approach to address selectivity in the Knorr pyrazole condensation between a vinylogous amide 71 and methylhydrazine, aiming to access either N1- or N2-methyl pyrazoles 72 (Scheme 8).98 Their approach, termed ‘Utopia point Bayesian optimization’ (UPBO), defines a single, idealized outcome—or utopia point—in the objective space, and directs the optimizer to minimize the Euclidean distance to this point, thereby guiding the optimization. | ||
| Scheme 8 Knorr-pyrazole condensation to access methyl pyrazoles 72a reported by Dalton et al.98 BO was used to optimize for both N1- and N2-selective reaction conditions, targeting both yield and selectivity. Parameters defining the reaction space are highlighted in blue. | ||
The optimization was conducted over ten rounds, with 10–16 experiments per round, evenly split between conditions targeting N1 and N2 selectivity. The parameter space included temperature, equivalents of acid/base (with negative values corresponding to acid), equivalents of methylhydrazine, solvent, solvent volume, and the presence of acid catalyst, phase transfer catalyst, or molecular sieves. The latter three were implemented as Boolean variables, while the solvent was parameterized using four COSMO-RS σ-moments.
Candidate conditions were selected by ranking predicted outcomes from an ensemble of surrogate models, including Gaussian Processes, Random Forests, Extra Random Trees, Gradient Boosting, and a Bayesian Neural Network, using either Expected Improvement or Upper Confidence Bound as acquisition functions.
For the first four rounds, yield and selectivity were modelled independently, each subjected to its own acquisition function, and the resulting acquisition scores were then manually Pareto-ranked to select the next batch of experiments, rather than using a formal multi-objective acquisition function. Only from the fifth round onward was the UPBO framework adopted. While the authors reported that this approach led to more rapid convergence to high-performing regions of the search space, the comparison is weakened by the less systematic strategy employed during the initial rounds, which lacked the rigor of standard multi-objective optimization. In the end, conditions providing both high yield and high selectivity (>90:10) for both the N1 and the N2 pyrazole were identified.
Thus, while UPBO provides an intuitive and easily interpretable optimization target, it does not offer the trade-off insights inherent to Pareto-based methods, and its practical advantage over conventional scalarization remains to be systematically demonstrated.
Kondo et al. reported a BO-guided electrochemical oxidation protocol for the synthesis of α-ketiminophosphonates via oxidative dehydrogenation of α-amino phosphonates (Scheme 9a).99 After five initial experiments, a Gaussian process and EI-based BO algorithm was used to iteratively screen five parameters (current, temperature, reaction time, and concentrations of substrate and electrolyte), leading to optimal conditions in twelve experiments. The method was then applied to a broad substrate scope, and further BO-guided reoptimization improved yields across several challenging substrates.
 | ||
| Scheme 9 Various application of BO to batch-based systems. Parameters defining the reaction space are highlighted in blue. (a) Optimization of the electrochemical synthesis of α-ketiminophosphonates reported by Kondo et al.99 (b) Copper-catalysed C–N coupling optimized by BO reported by Braconi & Godineau.100 | ||
Furthermore, Da Tan et al. applied BO to polymer chemistry, where it was used for the multi-objective optimization of the synthesis of a terpolymer, balancing a high glass-transition temperature (Tg) with reduced styrene content for sustainability purposes.101 Over 89 total experiments, the authors leveraged a GP surrogate model and an EHVI-based acquisition function to efficiently refine the polymerization conditions and afford a wide range of polymers with tailored properties.
Beyond academic settings, the adoption of Bayesian reaction optimization in industrial contexts is also gaining traction. For example, Braconi and Godineau demonstrated the effectiveness of BO in an industrial setting at Syngenta, where it was used to optimize a Cu-catalysed C–N coupling of sterically hindered pyrazines as part of early-stage process development.100 The study compared multiple acquisition functions (EI, TS, UCB), each coupled to a GP surrogate, and showed that BO significantly reduced experimental workload while identifying high-yielding conditions using copper catalysts as a more sustainable alternative to palladium (Scheme 9b).
4. BO in automated flow platforms
As discussed in the previous section, a key area of research in BO is the development of closed-loop automated systems. Flow chemistry lends itself more naturally to this than batch-based platforms.102 It is thus no surprise that most automated closed-loop BO platforms are flow-based. The inherent properties of continuous flow—including greatly improved heat and mass transfer, as well as enhanced light penetration for photochemical reactions—make it particularly well-suited for integration with real-time analytics and feedback-driven optimization. These characteristics, in turn, facilitate high-frequency experimentation with high reproducibility, enabling (automated) sequential algorithmic optimization.Over the past few years, a growing number of studies have demonstrated the power of integrating BO with automated flow platforms across a wide range of chemistries and use cases. These systems generally consist of three core components: (1) an automated reactor system, capable of precise and programmable control over input parameters; (2) in-line or on-line analytical tools, providing fast and quantitative feedback on performance metrics such as yield, selectivity, or conversion; and (3) a BO optimization framework, which selects new experimental conditions based on past results. Reported applications include the optimization of single-step reactions, multistep telescoped syntheses, and complex multiphase systems—often under constraints imposed by sustainability, safety, or scalability. In the following subsections, these developments will be grouped and discussed according to their primary application domain or methodological innovation, highlighting the evolving capabilities and design principles of modern BO-guided flow systems with varying degrees of automation.
4.1. Single-step reaction optimization
In one of the first demonstrations of Bayesian optimization for chemical reaction optimization, Schweidtmann et al. reported a fully automated flow system with online HPLC analysis for multi-objective optimization.16 Two case studies were performed: a nucleophilic aromatic substitution reaction and an N-benzylation reaction, each optimized over four continuous variables (Scheme 10a and b). Using GP surrogate models and TS as acquisition function, for each of the two case studies a dense Pareto front balancing productivity (space-time yield, STY) and E-factor or % impurity was identified in under 80 experiments, showcasing the data-efficiency of multi-objective BO and its ability to reveal a comprehensive view of process trade-offs. The study was pivotal in demonstrating the feasibility of autonomous, multi-objective optimization and set the stage for the broad uptake of BO by the community. | ||
| Scheme 10 Reactions optimized by BO on the closed-loop flow platform reported by Schweidtmann et al.16 Parameters defining the reaction space are highlighted in blue. (a) SNAr reaction. (b) N-Benzylation reaction. | ||
Following this foundational study, numerous works have explored the application of Bayesian optimization for reaction optimization in flow, showcasing varying levels of automation and process complexity. These campaigns typically target—but are not limited to—a single objective, such as yield or conversion, and benefit from the reproducibility and ease of automation enabled by flow systems. A key example is the recently reported RoboChem platform, developed by the Noel research group.103 The automated platform is equipped with a photoreactor and an inline NMR. Reactions are optimized in a closed-loop fashion, using the Dragonfly95 BO algorithm, by sending slugs of reaction mixture through the photoreactor. Optimized conditions can then be scaled up by switching the operation mode to continuous flow, with isolated yields closely matching those measured with the inline NMR yield. The platform can handle both continuous and discrete reaction parameters and supports both yield and throughput as (simultaneous) objectives.
RoboChem was applied to a range of photocatalytic transformations, including C–H alkylation, C–H trifluoromethylthiolation, oxytrifluoromethylation, aryl trifluoromethylation and C(sp2)–C(sp3) cross-electrophile coupling (Scheme 11a–e). Notably, for several of these transformations, the reaction was optimized de novo for different substrates. The resulting optimal conditions were often substrate-specific, underscoring that even within a common transformation type, distinct substrates may require distinct conditions to reach optimal results. While the study did not report cross-evaluation of conditions between substrates, this result implicitly suggests that condition transferability is limited and further supports the value of closed-loop BO in efficiently tailoring conditions to specific reaction contexts.
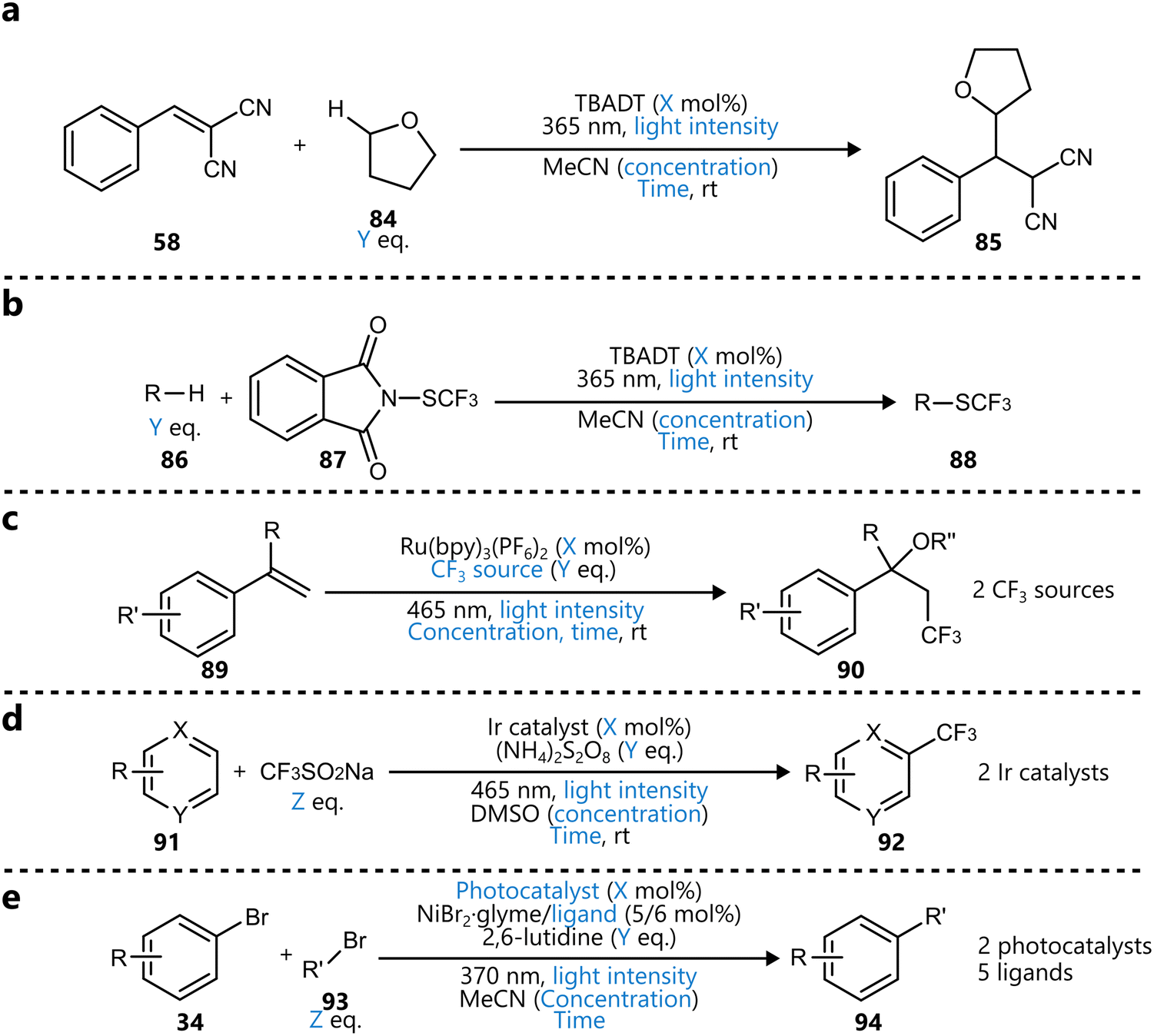 | ||
| Scheme 11 Photochemical reactions optimized on the RoboChem platform by Slattery et al.103 Parameters defining the reaction space are highlighted in blue. (a) HAT-enabled C–H alkylation. (b) HAT-enabled C–H trifluoromethylthiolation. (c) SET-enabled oxytrifluoromethylation. (d) SET-enabled aryl trifluoromethylation. (e) Metallaphotoredox C(sp2)–C(sp3) cross-electrophile coupling. | ||
A particularly illustrative example is the study by Kershaw et al., who demonstrated multi-objective Bayesian optimization for mixed-variable chemical systems.60 Instead of using molecular descriptors or one-hot encoding, the authors employed Gower similarity to enable surrogate modelling across mixed-variable spaces. Leveraging a custom, automated flow platform with online HPLC analysis, their approach was applied to the closed-loop optimization of a nucleophilic aromatic substitution reaction (Scheme 12a), targeting simultaneously the formation of both the ortho isomer 80 and the para isomer. Solvent choice was included as a discrete variable, alongside residence time, concentration, temperature and equivalents of coupling partner as continuous parameters. Five solvent candidates were selected based on their ability to provide a homogeneous reaction mixture, as well as their polarity index. After 99 experiments in total, a dense Pareto front was identified, highlighting the trade-off between formation between the ortho and para regioisomers. A second case-study involved a Sonogashira cross-coupling between an aryl bromide 95 and a terminal alkyne 96 (Scheme 12b). The choice of phosphine ligand was included as a discrete variable, alongside residence time, alkyne equivalents and temperature. The reaction was optimized simultaneously for throughput (STY) and environmental impact (reaction mass efficiency, RME). Note that catalyst loadings and base equivalents were kept constant, although these could have a profound effect on both objectives. While this choice likely reflects practical equipment constraints, such as pump count, platform complexity, minimum reliable flow rates, and solubility limits for catalyst or base solutions, it inevitably limits the number of variables that can be explored simultaneously within a closed-loop optimization framework. Nevertheless, the BO approach showed great efficiency—despite the initial LHS-set being limited to a region of low STY—in locating the Pareto front.
 | ||
| Scheme 12 Reactions optimized by Kershaw et al. to illustrate mixed-variable BO.60 Parameters defining the reaction space are highlighted in blue. (a) SNAr reaction. Only the ortho-product is displayed, but the Pareto front showing the trade-off between ortho- and para-product was identified as part of the optimization. (b) Sonogashira cross-coupling. | ||
In a related contribution, Aldulaijan et al. proposed ALaBO, a Bayesian optimization framework for mixed-variable reaction optimization that combines adaptive acquisition strategies with latent variable modelling of categorical parameters.53 Within this framework, Adaptive Expected Improvement (AEI)—an extension of the Expected Improvement acquisition function—is used to dynamically adjust the ξ parameter in response to the posterior variance of the GP surrogate model, leading to increasingly exploitative search behaviour as uncertainty diminishes. A key aspect of ALaBO is the representation of categorical variables via low-dimensional latent embeddings, which are learned jointly with the Gaussian process surrogate as optimization progresses. ALaBO was implemented as single-objective optimization algorithm, using a weighted function of yield and turnover number (TON), rather than a true multi-objective BO approach. The method was benchmarked against two other popular Bayesian optimization algorithms on five in silico case studies, where it consistently demonstrated strong performance, particularly in terms of convergence speed. Finally, the algorithm was deployed in combination with an automated flow platform for the closed-loop optimization of a Suzuki–Miyaura cross-coupling, involving four variables: ligand (five levels), temperature, residence time, and palladium loading (Scheme 13). ALaBO efficiently navigated this mixed-variable space over a campaign of 25 experiments, resulting in a yield of 93% and A TON of 95.
 | ||
| Scheme 13 Suzuki–Miyaura cross-coupling optimized to demonstrate the ALaBO optimizer by Aldulaijan et al.53 ALaBO dynamically adjusts the acquisition function during optimization to shift from exploration towards a more exploitation-focused approach. Parameters defining the reaction space are highlighted in blue. | ||
Interestingly, this optimum was achieved after only ten experiments, with the fifteen additional experiments in the full campaign not yielding any improvement upon this result. While this highlights the algorithm's efficiency, it also reflects the relatively moderate complexity of the underlying chemistry and the accompanying optimization problem, a factor that likely contributed to the rapid convergence. Nevertheless, this study underscores the practical advantage of dynamically modulated acquisition strategies for efficient reaction optimization, which could be of particular importance in systems where the underlying response surface is highly nonlinear or irregular.
At this point, it is clear that BO frameworks are highly efficient in locating optima. However, their data-sparse nature often limits the broader interpretability of models, which is a key requirement for regulatory acceptance and process scale-up. In particular, quality-by-design (QbD) principles emphasize the need for robust, data-rich models to ensure long-term process control and reproducibility.104–106 In response to this need, Knoll et al. proposed a hybrid approach that uses BO to narrow down the search space prior to formal model building.107 This was demonstrated on a photochemical benzylic bromination (Scheme 14), where six variables were to be optimized with regards to maximizing STY, conversion and selectivity: equivalents of acetic acid, temperature, concentration of starting material, light intensity, residence time, and equivalents of N-bromo succinimide (NBS) 102. The BO campaign, employing the TS-EMO algorithm, required 42 experiments to identify a high-performing region within the broader search space. Subsequently, a DoE was carried out in this region—which was defined manually from the BO campaign—to construct a local response surface model. This strategy illustrates how BO can be effectively embedded within a broader process development pipeline, combining its strength in efficient search with the modelling needed for a QbD approach.
 | ||
| Scheme 14 Photochemical bromination optimized by Knoll et al.107 After identifying an optimal region using BO, DoE was used in the narrowed-down parameter space, to construct a local response-surface model. Parameters defining the reaction space are highlighted in blue. | ||
Traditional BO relies on discrete experiments performed under steady-state conditions, which can be time- and resource-intensive. In contrast, Florit et al. developed a Bayesian optimization strategy tailored for dynamic flow experiments, where reaction parameters are varied continuously over time rather than being held constant during each run.108 Instead of selecting individual steady-state conditions, the algorithm proposes a trajectory through the design space—defined by a fixed functional form with tuneable parameters—and suggests new trajectories using a custom acquisition function based on UCB that is optimized across an entire path. Compared to traditional steady-state optimization, this approach is particularly advantageous when analysis time is relatively short compared to reaction time. Currently, the algorithm is limited to operating in Euclidean spaces, which means that only continuous variables can be used. The data acquired along the trajectories are post-processed to deconvolute the system's dynamic response and reconstruct steady-state equivalent outcomes from transient measurements, enabling direct interpretation of the results. This not only facilitates comparability with traditional approaches but also improves interpretability for chemists.
The approach was first benchmarked in silico, showing comparable sample efficiency to the Dragonfly BO algorithm, while requiring significantly less experimental time and reagent volume. The dynamic BO algorithm was initialized with steady-state experiments, after which it exclusively suggested dynamic trajectories.
As an experimental validation, the method was applied to the optimization of an ester hydrolysis using an automated flow platform (Fig. 16), varying residence time and equivalents of base. The optimal region in this simple search space was identified after only two trajectories, one initial and one suggested by the algorithm. While it should be noted, however, that the identified optima were not validated using steady-state experiments, this study does demonstrate the potential of dynamic BO to reduce experimental burden while maintaining robust optimization performance.
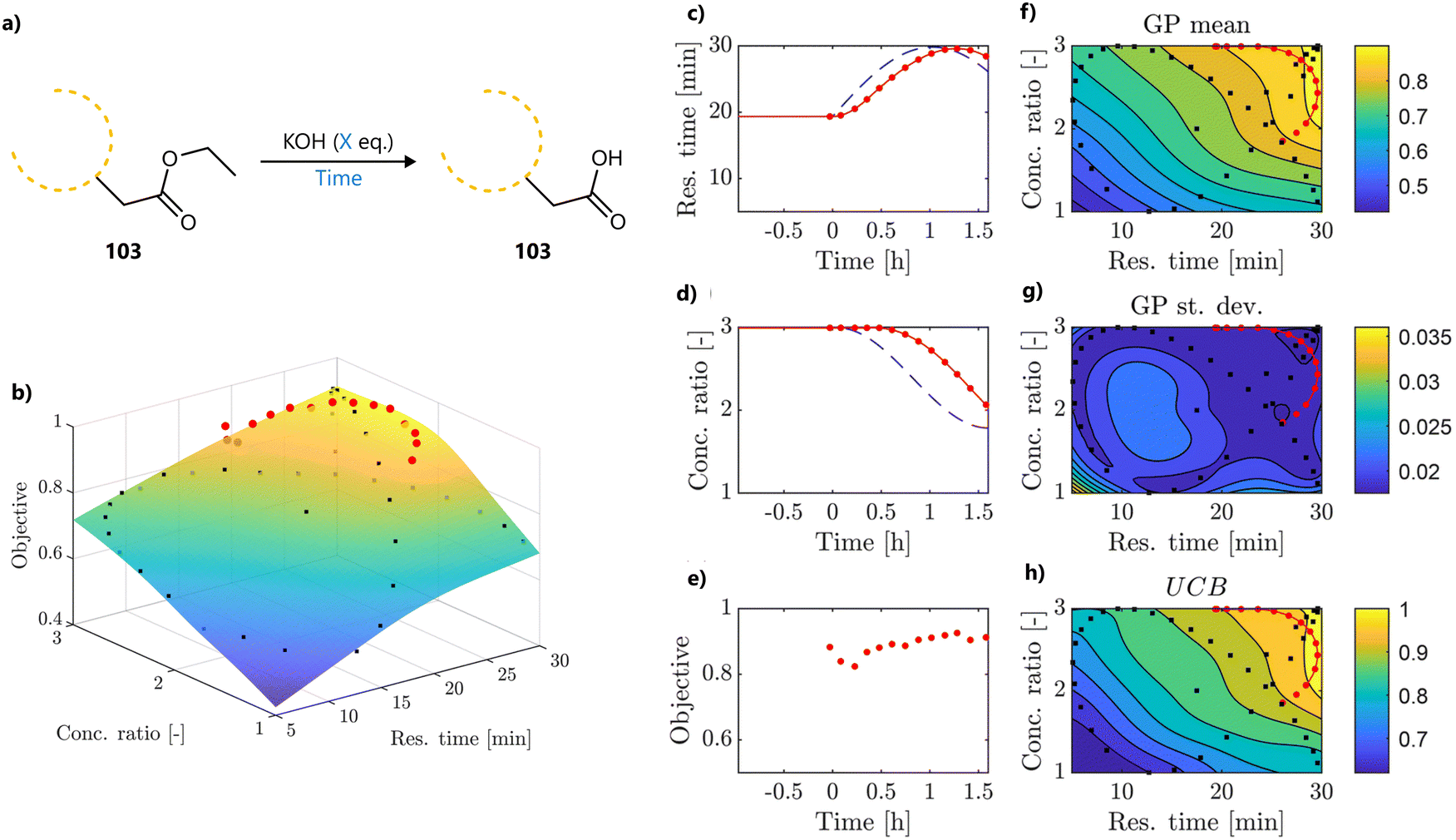 | ||
| Fig. 16 BO in combination with dynamic flow experiments, as reported by Florit et al.108 (a) Ester hydrolysis reaction that was optimized. Parameters defining the reaction space are highlighted in blue. (b) GP predictions after the second trajectory. Red points are the algorithm-suggested trajectory based on the first trajectory (black points). (c) and (d). Parameter values throughout the algorithm-suggested trajectory. Dashed lines depict the reconstructed values. (e) Value of the objective function over a trajectory. (f)–(h) Contour plots depicting the GP mean, its standard deviation and the value of the objective function. The trajectory is drawn as a continuous red line, while the red points are the sampled conditions. Points outside of the trajectory depict samples from the previous iteration. Panels c–h adapted from the authors with permission under a CC BY-NC 3.0 Unported License. | ||
As part of broader recent efforts to improve the efficiency and adaptability of Bayesian optimization, Liu et al. proposed a self-optimizing Bayesian optimization (SOBayesian) framework that departs from the conventional use of a fixed surrogate model and an acquisition function.37 Instead, multiple GP kernels (RBF, Matern, rational quadratic, and dot product) are evaluated, and their hyperparameters are optimized at each iteration, with the best-performing one selected based on cross-validation error. This adaptive surrogate modelling strategy is notable for going beyond the ‘one-size-fits-all’ kernel approach, aiming to tailor the model to the evolving structure of the problem at hand.
The method was applied to a Buchwald–Hartwig coupling between 2-bromopyridine and anisamide in flow, optimizing five continuous variables (residence time, temperature, and equivalents of anisamide, base and ligand). Notably, SOBayesian employs no formal acquisition strategy; instead, it performs brute-force global optimization over a finely discretized design space (∼250 million points in this case), selecting the global maximum of the GP mean at each round. This simplistic yet effective approach led to convergence within 21–29 experiments, depending on the initialization size, consistently identifying the same conditions.
While the authors primarily aimed to demonstrate the feasibility of the surrogate modelling approach, the absence of an acquisition function may limit scalability to higher-dimensional problems, including mixed-variable and multi-objective problems. A benchmark against the Dragonfly algorithm on a simple two-variable system demonstrated comparable performance, although the simplicity of the task and the absence of a runtime comparison make it challenging to draw broader conclusions. Nevertheless, the study highlights the potential of adaptive surrogate modelling in Bayesian optimization (BO) and raises interesting questions about the balance between model complexity, acquisition strategies, and data efficiency.
In a related study, Mueller et al. reported the use of Bayesian optimization for the selective oxidation of phenyl sulfides in a biphasic aqueous-organic system using hydrogen peroxide and phosphotungstic acid as oxidant and catalyst, respectively.111 Conducted on a closed-loop mini-CSTR cascade platform, the system targeted optimization of reaction parameters including temperature, residence time, catalyst loading and oxidant equivalents towards maximal yield. The study evaluated four structurally distinct phenyl sulfides in separate campaigns, identifying different optimal conditions for each. This apparent substrate specificity, despite a common reaction mechanism, underscores the limitations of direct conditions transfer even in relatively well-behaved multiphasic systems, and further illustrates the strength of BO in efficiently tailoring conditions across structurally similar transformations.
Chai et al. applied BO to a heterogeneous hydrogenation in flow, targeting the synthesis of 2-amino-3-methylbenzoic acid in a packed-bed reactor, representing a typical gas–liquid–solid multiphasic system.112 Here, real-time FTIR analysis was coupled with a neural network model to provide yield and productivity (STY) data, which were then used in a multi-objective BO campaign using qNEHVI coupled to a GP surrogate.
Four continuous variables (temperature, hydrogen pressure, substrate concentration and flow rate) were optimized, identifying a Pareto front in 40 experiments. The study also compared the BO-based approach to traditional kinetic modelling. While both approaches converged to similar optima, BO required far fewer experiments, at the cost of not providing an accurate response surface model across the entire reaction space.
A complementary study by Liang et al. explored the hydrogenation of three aromatic nitro compounds under continuous flow, also using a packed-bed reactor.113 The case studies included the hydrogenation of nitrobenzene, 3,4-dichloronitrobenzene, and 5-nitroisoquinoline. The authors implemented a hybrid optimization strategy that began with the Nelder–Mead simplex algorithm to explore the local response surface. If the result met predefined performance criteria (in this case, high yield), the optimization was concluded. Otherwise, the workflow transitioned to Bayesian optimization using a GP surrogate and an expected improvement acquisition function.
This hybrid approach was benchmarked against both one-factor-at-a-time (OFAT) optimization and pure Bayesian optimization. The authors reported that their two-tiered strategy yielded superior performance over OFAT and achieved comparable or better results than full BO, often with fewer experiments. However, a significant limitation of the study lies in its benchmarking setup: each method was initialized with a different set of experimental points. Without a shared initialization set, differences in performance may stem from favourable starting conditions rather than from the relative effectiveness of the algorithms themselves. This limits the strength of the conclusions regarding comparative efficiency. Nevertheless, the study demonstrates a pragmatic and flexible framework for process development in multiphasic systems, where BO acts as a backup when local optimization alone is insufficient, a potentially resource-efficient strategy in low-complexity optimization landscapes.
This was exemplified in the work by Ahn et al., who developed an automated microreactor platform for the optimization of ultrafast organolithium chemistry.117 The synthesis of thioquinazolinone 106via the intramolecular cyclization of an in situ generated isothiocyanate-bearing aryllithium intermediate with an isocyanate was optimized using a BO framework with GP surrogate an EI acquisition function with real-time feedback from inline IR spectroscopy (Fig. 17). Unusually, reactor volume was included as an optimization variable alongside flow rate and temperature, by controlling solenoid valves that varied the point of injection of the organolithium reagent along the temperature-controlled reactor. Notably, the flow rate ratio between reagents was held constant, even though this could have been an interesting parameter given the mixing-sensitive nature of the transformation. The three-variable BO campaign was repeated five times, each converging within ten experiments and outperforming a DoE design with 80 experiments in ±80% of cases. The authors also benchmarked against random sampling, which performed significantly worse. In a subsequent campaign, the organolithium reagent (three levels) was added as a categorical variable and optimized in 15 experiments, resulting in a slight improvement in yield. These optimized conditions were then used for the combinatorial synthesis of nine S-benzylic thioquinazolinone derivatives.
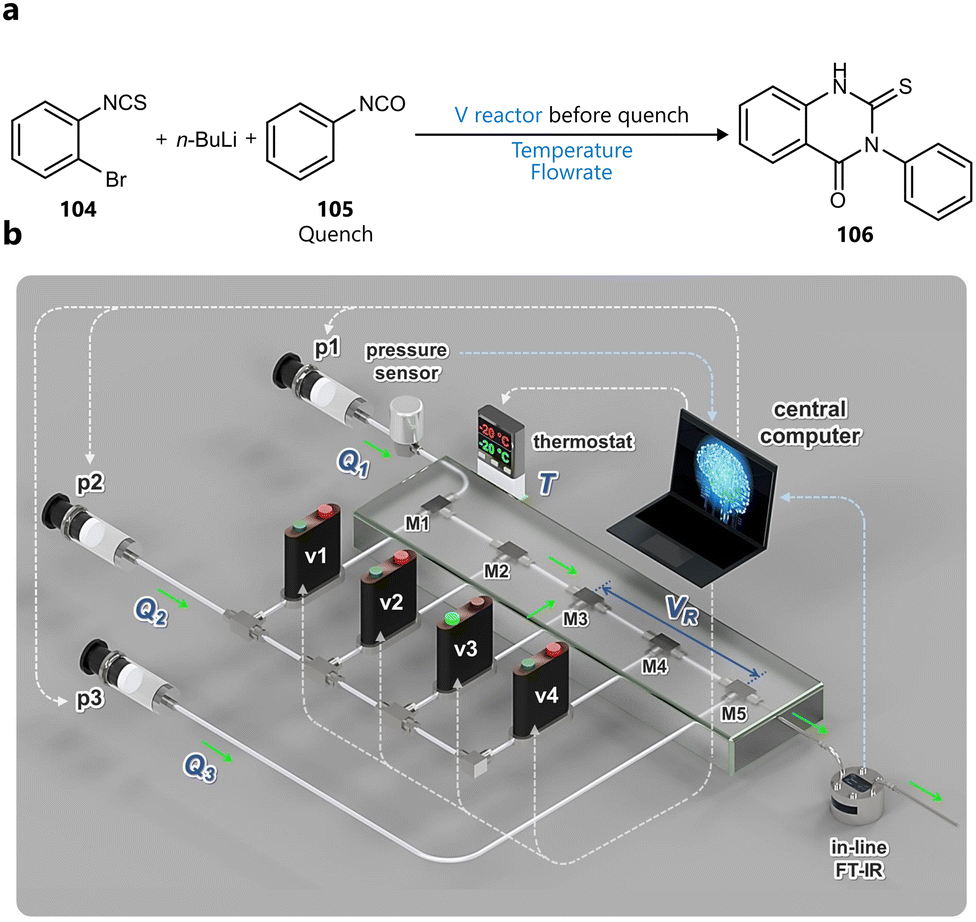 | ||
| Fig. 17 BO for optimization of ultrafast chemistry. (a) Synthesis of thioquinazolinone via an aryllithium intermediate. Parameters defining the reaction space are highlighted in blue. (b) Reaction platform developed for the study. The solenoid valves V1 to V4 can be used to change the point of injection of the organolithium reagent, changing the reactor volume. Panel b adapted from Ahn et al.117 with permission. © 2022 Elsevier B.V. | ||
In a related study, Karan and coworkers applied multi-objective Bayesian optimization to another ultrafast Br–Li exchange reaction.118 The reaction—the formation of a reactive organolithium intermediate directly followed by quenching with methanol—was optimized for both maximizing yield and minimizing impurities by varying temperature, residence time and nBuLi equivalents using the TS-EMO algorithm. Interestingly, the residence time distribution (RTD) of the reactor was modelled to estimate the time required to reach steady-state operation, ensuring reproducible experimentation while minimizing material consumption. Three optimization campaigns were performed. The first two used a capillary flow reactor with a T-piece mixer, differing only in the number of initial experiments (twelve vs. three), while the third used a microchip reactor with the same volume but superior mixing properties. Each campaign successfully identified a Pareto front of yield and impurity, with the best overall result achieved in the third campaign using the microchip reactor. The study did not show a substantial difference in convergence for the campaign with less initial experiments, although only three variables were considered, so the result might differ for more complex parameter spaces.
Although the underlying mechanism of these reactions could be viewed as comprising two steps, these processes are covered here, under single-step optimization, as it is experimentally infeasible to isolate the intermediate or to optimize the mechanistic stages independently.
Naito et al. addressed a fundamental challenge in electrochemical flow optimization by incorporating a constraint on passed charge directly into a single-objective Bayesian optimization (SOBO) framework.120 The study focused on the reductive carboxylation of imines with CO2 in a flow microreactor, where the passed charge is a function of current density, flow rate, and substrate concentration. An initial BO campaign with charge constraint (2.0–3.0 F mol−1) rapidly identified conditions delivering 90% yield, but with a limited current efficiency of 63%, reflecting the fact that SOBO, in the absence of a directly encoded efficiency target, had no incentive to minimize energy use. To steer the optimization toward more sustainable conditions, the authors re-ran the campaign under a tighter constraint (2.0–2.1 F mol−1), reusing the initial dataset. This second campaign achieved a revised optimum of 88% yield with 87% current efficiency, demonstrating that physically meaningful performance trade-offs can be enforced through constraint handling, even in single-objective settings. The study showcases constrained BO as a practical tool for balancing performance and efficiency in complex electrochemical systems without explicitly modelling mechanistic detail.
Kaven et al. extended BO to the synthesis of N-isopropylacrylamide microgels, also using the TS-EMO algorithm to simultaneously optimize mean particle size, productivity and energy efficiency.121 While the system identified a Pareto front for these three objectives within 43 experiments, a large portion of the front was constructed from GP model predictions rather than experimentally validated points, a practical decision driven by the limited number of experiments and absence of in-line or automated particle size analysis. The authors justified this approach by showing good agreement between predicted and measured values for a subset of points.
Very recently, Bayesian optimization has also been applied to biocatalytic reaction optimization in flow. Biocatalysis itself represents an important and rapidly emerging field for sustainable organic synthesis.122–124 Clayton and co-workers reported the autonomous optimization of a Novozym-435 catalysed amidation in continuous flow, employing a two-stage BO strategy.125 In a first campaign, the multi-objective TS-EMO algorithm was used to simultaneously maximize amide formation while minimizing enamine impurity formation by varying continuous reaction parameters, yielding a Pareto front. This Pareto front was subsequently used to define scalarization weights for a second, single-objective optimization campaign using the ALaBO framework, in which solvent was additionally introduced as a categorical variable. This second campaign enabled efficient exploration of the expanded mixed-variable design space, ultimately delivering reaction conditions affording 94% yield with less than 5% enamine impurity, while identifying 2-MeTHF as a more sustainable alternative to the dioxane solvent traditionally employed. Beyond optimization, the trained surrogate models were analysed using automatic relevance determination and partial dependence plots to obtain qualitative insights into the influence of individual variables and solvent choice on the biocatalytic reaction outcome.
Extending BO to hydrothermal nanoparticle synthesis in flow, Jackson et al. implemented BO to optimize the synthesis of iron oxide nanoparticles based on inline dynamic light scattering (DLS) measurements.126 They compared a BO approach with the more traditional global optimizer SNOBFIT, and found their performance—for this example, with a narrow search space and only one objective—to be similar. While convergence was achieved within 30 experiments, the optimization procedure was not described in sufficient detail to fully assess its methodology.
Kondo et al. applied Bayesian optimization to the redox-neutral TfOH-catalysed cross-coupling of both iminoquinone monoacetals (IQMAs) and quinone monoacetals (QMAs) with arenols in continuous flow.127 Two separate campaigns of 15 experiments each were performed, optimizing six variables: mixer type (categorical, using one-hot encoding), temperature, catalyst loading, equivalents of arenol, substrate concentration, and flow rate (identical for both syringes, limiting automation). A GP surrogate with a lower confidence bound (LCB) acquisition function and local penalization enabled batches of three experiments per iteration. BO efficiently identified high-yielding conditions for both substrate classes, and a limited substrate scope for each was performed to test their generality.
Dunlap and coworkers applied the EDBO+ framework described earlier to optimize the synthesis of N-butylpyrydinium bromide in flow.128 Their human-in-the-loop workflow aimed to maximize yield and productivity simultaneously, by optimizing residence time, temperature and pyridine equivalents. Interestingly, after observing that the algorithm selected the upper bound for temperature in 19 out of the first 30 experiments, the temperature range was expanded and the hypervolume of the Pareto front significantly increased, showcasing the benefit of having a human in the loop. Finally, the study also compared optimization performance between using a 60 MHz and a 400 MHz NMR instrument, finding that low-resolution data still yielded comparable outcomes, which highlights the robustness of BO and supports its suitability for real-world settings.
Two related studies applied Bayesian optimization to the synthesis and purification of N,N-di-2-ethylhexyl-isobutyramide (DEHIBA), a compound used in advanced nuclear reprocessing. In the first, Shaw et al. explored six different synthetic routes (four solvated and two solvent-free) for the synthesis of DEHIBA, optimizing productivity (STY) and reaction mass efficiency (RME) or product cost using TS-EMO.129 The optimization itself was conducted at small scale using a fully automated flow platform, but the most promising route was subsequently scaled out to provide 5L of crude product, demonstrating practical viability. In a complementary study, the same group used BO with a GP surrogate and adaptive expected improvement acquisition function to optimize the continuous purification process of crude DEHIBA.130 Using a weighted objective function balancing maximal product purity with minimal product loss, the authors leveraged multipoint sampling to enable comprehensive online analysis of the process. This second study illustrates the applicability of BO to the downstream purification of chemical reactions.
Beyond chemical synthesis, Kohl and coworkers investigated the photochemical degradation of an azo dye using a catalytic static mixer in continuous flow.131 The optimization of reaction conditions was guided by BO, converging to optimal conditions in 17 experiments. A key challenge addressed in the study was catalyst deactivation over time, which could confound the optimization algorithm by introducing temporal drift in the system response. To address this, the authors included the total amount of dye that has passed over the catalyst since regeneration as a variable, allowing the surrogate model to build an internal representation of the effect of catalyst degradation. This approach effectively decoupled performance fluctuations due to deactivation from those caused by changes in experimental parameters. Interestingly, this effect could then be extracted from the surrogate model, as is shown in Fig. 18, to provide a model without needing extra calibration experiments.
4.2. Multi-step reaction optimization
While much of the work in Bayesian reaction optimization has focused on single-step transformations, many relevant synthetic processes—particularly in the pharmaceutical and fine chemical industry—involve telescoped or multistep sequences. Optimizing such processes presents unique challenges due to the interplay between steps, accumulation of impurities, and increased experimental complexity. These challenges are compounded when integrating purification, multiphasic chemistry, or convergent synthetic strategies, where two or more separate intermediate streams are merged in a downstream reaction step. The following section illustrates how BO frameworks have been applied to tackle these complexities, highlighting both methodological innovations and practical results.One of the earliest examples of Bayesian optimization applied to a telescoped continuous flow process is the study by Sugisawa et al., who developed a mild and rapid one-flow synthesis of asymmetrical sulfamides via a two-step reaction sequence.132 The process involved initial activation of primary amines with sulfuryl chloride 107, followed by sequential introduction of a second amine, all conducted in a single, uninterrupted flow (Scheme 15). BO was employed to optimize the yield of the final product, as determined by offline 1H-NMR analysis. Considering the optimization of two steps, the search space was somewhat narrow, encompassing temperature (which was set at the same value for both steps), the residence time of the first step, the base (with six options), and the concentration of sulfuryl chloride. Reaction stoichiometry, residence time of the second step and the flow rates of all three syringes were kept constant. Residence time was varied instead by adjusting the reactor volume, and the base was parametrized by using the pKa value of the conjugate acid. Rather than treating the variables as fully continuous, the authors discretized each parameter into a set of fixed levels, effectively creating a coarse-grained optimization landscape. Nevertheless, the BO-guided optimization—leveraging a GP surrogate and UCB acquisition function—successfully identified relatively high-yielding conditions, demonstrating the feasibility of BO for telescoped syntheses.
 | ||
| Scheme 15 BO-optimized telescoped synthesis of asymmetrical sulfamides, reported by Sugisawa et al.132 Parameters defining the reaction space are highlighted in blue. | ||
Related to this, Zhu et al. reported the optimization of a two-step cryogenic flow synthesis of MAP02 113, a key intermediate in the manufacture of nemonoxacin,133 using a non-sequential, model-based Bayesian optimization workflow. The chemical sequence comprised LiHMDS-mediated hydrogen abstraction followed by stereoselective methylation with MeBr, implemented in two discrete flow modules (Scheme 16). Six variables (equivalents of reagents, residence times, and temperatures for both steps) were considered, with residence time varied via tubing length to decouple step-specific timing from overall flow rate, a recurring constraint in telescoped systems. Rather than conducting a sequential Bayesian optimization campaign with direct experimental feedback, the authors first performed 29 offline experiments and used these to train a predictive surrogate via AutoSklearn. BO (SOBO and qNEHVI) was then applied not to the physical process, but to this fixed model. In the single-objective case, three optimization strategies (random search, full factorial with three levels, and BO) were compared, with BO outperforming the others in convergence speed and predicted yield. The top-performing condition, predicted to give 89.7% yield, was subsequently validated experimentally at 89.5%. A multi-objective campaign using qNEHVI generated a Pareto front for yield and enantioselectivity, with one predicted point (81.8%, 97.85%) also confirmed closely by experiment (83.8%, 97.2%). Although the BO surrogate was not updated iteratively during experimentation, this hybrid workflow illustrates how model-driven optimization can support rational design when experimental budgets are tight and surrogate fidelity is high. However, since the surrogate model itself was a stacked ensemble trained via AutoSklearn, the decision to place a second surrogate (GP) on top—effectively running BO over an already-trained black-box model—adds conceptual redundancy. In this context, the strengths of BO as a data-efficient, adaptive experimental design tool are not leveraged. More direct global optimization methods (e.g., differential evolution, particle swarm, or even grid-based searches) might have been more appropriate for navigating a static, low-cost model landscape. As such, the study blurs the role of BO between surrogate-guided experimental planning and general-purpose black-box optimization, illustrating that methodological choices in hybrid workflows merit careful consideration, particularly when BO's core strengths are not fully exploited.
 | ||
| Scheme 16 Cryogenic telescoped synthesis of a pharmaceutical intermediate as reported by Zhu et al.133 Parameters defining the reaction space are highlighted in blue. | ||
Going a step further, Clayton et al. demonstrated how multi-point sampling within a telescoped flow system can enhance overall process understanding and guide Bayesian optimization towards more effective global optima.55 The studied reaction sequence involved a Heck coupling optionally followed by intramolecular cyclization in the first reactor, and an acid-catalysed selective deprotection in the second (Fig. 19). The deprotection could proceed from either the intermediate coupling product or the cyclized product, but multi-point analysis revealed that deprotection from the former—contrary to prior expectations—led to better overall yield and lower equivalents of acid. Four variables were included in the optimization: the residence time in the first reactor, the equivalents of ethylene glycol, the temperature of the first reactor, and the ratio of the acid flow rate to the flow rate from the first reactor. The BO algorithm consisted of a GP surrogate and the adaptive expected improvement acquisition function. Importantly, the fully automated flow platform incorporated online HPLC sampling at both reactor outlets, enabling spatially resolved data acquisition and allowing for a deeper understanding of the process. Additionally, the authors noted that had the steps been optimized sequentially, the first step would likely have been driven to maximize the yield of the cyclized product, inadvertently compromising the overall process. Instead, holistic optimization of the total system using BO afforded superior performance for the telescoped sequence.
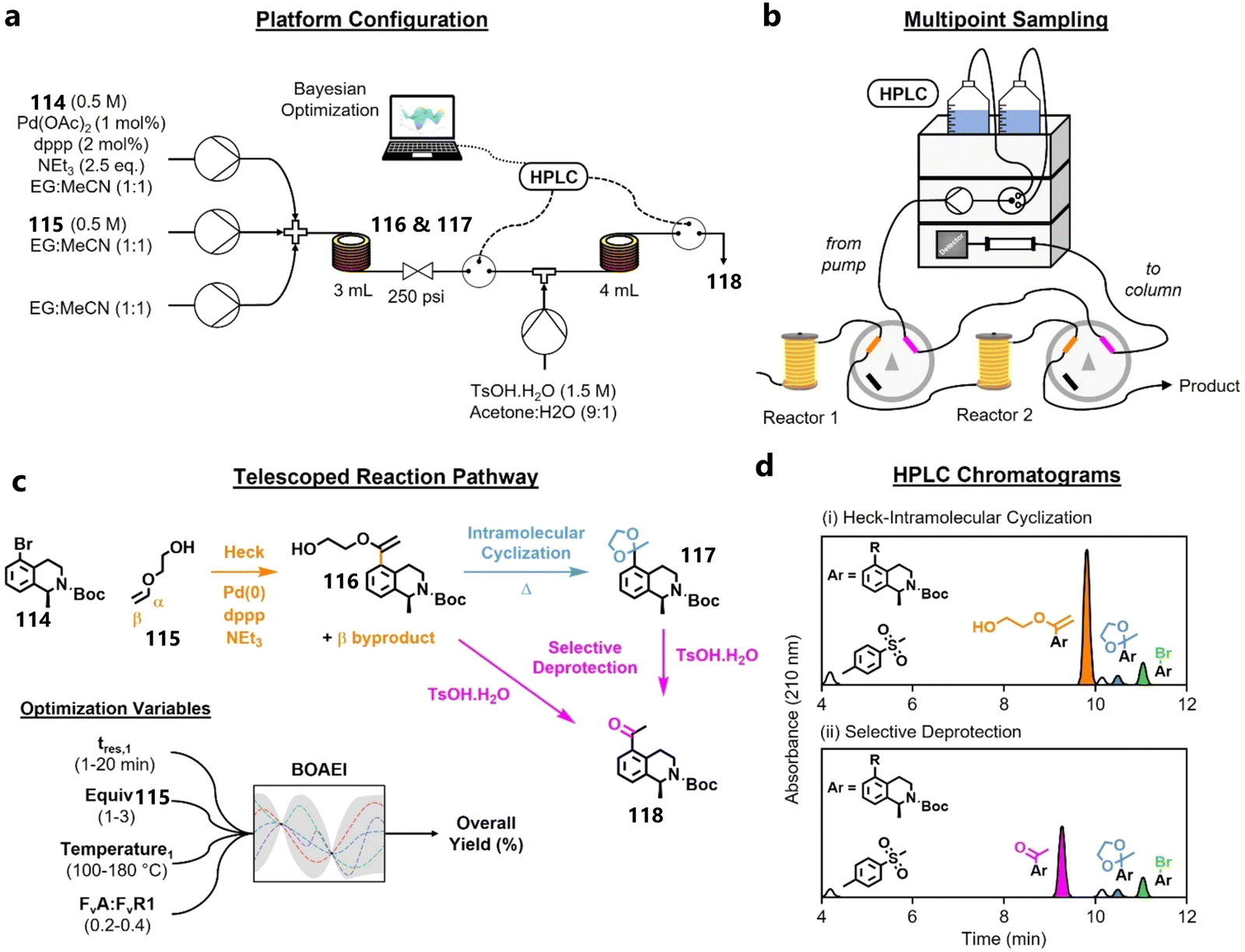 | ||
| Fig. 19 Optimization of telescoped continuous flow with multi-point sampling by Clayton et al. Adapted from the authors55 under a CC BY 4.0 license.97 (a) Platform configuration. (b) Multipoint sampling approach. (c) Reaction scheme and optimization variables. (d) Example HPLC chromatograms from both sampling points. | ||
In a conceptually related approach, Sagmeister et al. reported a fully automated flow platform for the multi-objective optimization of a two-step synthesis of edaravone, incorporating real-time process analytical technologies (PAT) and the TS-EMO BO algorithm.134 While the platform was first validated on the single-step optimization of a SNAr reaction, the focus of the study was the telescoped synthesis of edaravone, a two-step sequence involving the condensation of phenylhydrazine with ethyl acetoacetate, followed by a base-mediated cyclization (Fig. 20). Concentration profiles were obtained using inline NMR after the first step and inline FTIR after the second, with quantification based on an indirect hard model (IHM) and a partial least squares (PLS) regression model, respectively.
 | ||
| Fig. 20 Optimization of the telescoped synthesis of edaravone. Parameters defining the reaction space are highlighted in blue. Adapted from Sagmeister et al.134 under a CC BY 4.0 license.97 | ||
For the multistep optimization, three objectives were considered simultaneously: maximizing the yield of the intermediate, maximizing STY of the final product, and minimizing total reagent consumption. Seven variables were optimized, including the temperature of both reactors and the stoichiometries and residence times across both steps. The TS-EMO algorithm efficiently navigated the high-dimensional search space. Importantly, real-time process data were only collected once steady-state conditions were confirmed, and the signal was further refined using a finite impulse response (FIR) filter and time-weighted averaging. This processing step helped to reduce the impact of experimental noise, resulting in more reliable optimization performance. The optimization campaign succeeded in identifying optimal conditions over 85 experiments, achieving >95% yield of the imine intermediate and a STY of 5.42 kg L−1 h−1 for edaravone, whilst minimizing reagent excess.
This study illustrates how BO frameworks can scale to increasingly intricate multistep processes with high-dimensional search spaces, aided by the incorporation of multi-point real-time analytical feedback.
Building on this progression towards more complex, industrially relevant systems, Boyall et al. demonstrated the application of Bayesian optimization to a telescoped two-step continuous flow synthesis of paracetamol 126 (Fig. 21).20 The first step, a Pd-catalysed hydrogenation of 4-nitrophenol 123, was performed in a packed-bed reactor, with excess hydrogen subsequently removed via a tube-in-tube degasser using porous ePTFE tubing. The resulting reaction stream was directly fed into the subsequent amidation step with acetic anhydride 125. Multipoint HPLC analysis enabled tracking of intermediate and product concentrations, while catalyst stability in the packed bed reactor was monitored by repeating the same conditions every fourth experiment.
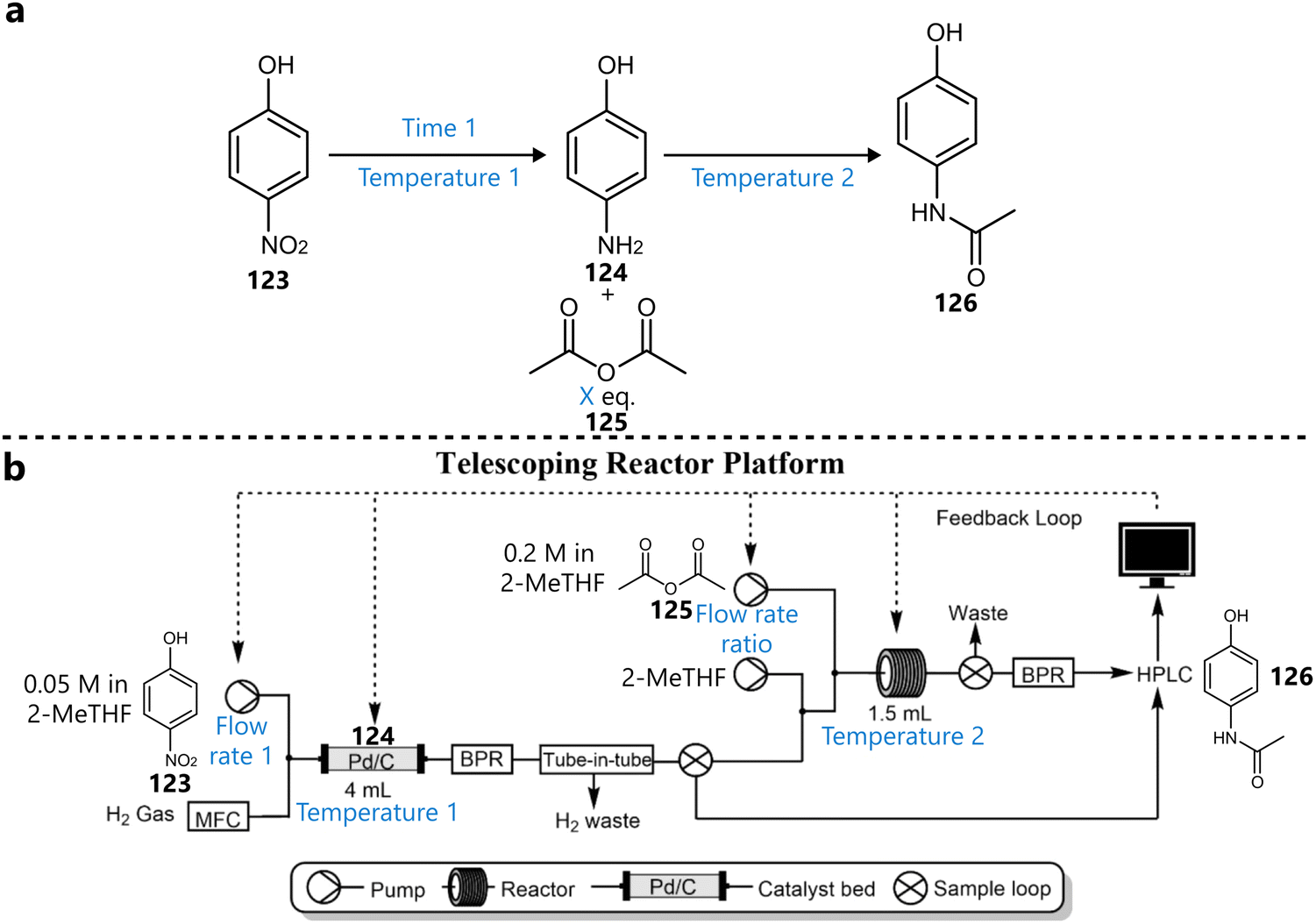 | ||
| Fig. 21 Optimization of a multi-phasic telescoped synthesis of paracetamol. Parameters defining the reaction space are highlighted in blue. (a) Reaction scheme for the two-step synthesis of paracetamol. (b) Schematic representation of the telescoped reaction platform. Adapted from Boyall et al.20 under a CC BY 4.0 license.97 | ||
Four optimization campaigns were conducted, using a GP surrogate with an adaptive expected improvement acquisition function. First, each step was optimized separately. Then, a follow-up campaign probed the second step using a reservoir of reaction mixture generated using the optimal conditions identified for the first step, allowing the authors to investigate the influence of intermediate composition on amidation performance across the design space. These latter two campaigns produced nearly identical results, suggesting that the amidation step is relatively insensitive to unreacted starting material from the first step. Finally, a telescoped campaign was run, optimizing four variables: temperature of the two reactors, flow rate of the starting material pump, and flow rate ratio of the acetic anhydride pump. In all cases, single-objective optimization was used, targeting product yield.
Importantly, the telescoped campaign required only twelve experiments to reach optimal conditions (85% overall yield), compared to twenty for the combined stepwise campaigns. Furthermore, the optimal telescoped conditions gave a significantly improved process mass intensity (PMI)—calculated a posteriori to better compare the two approaches—though this was in part due to operating the telescoped campaign at a higher starting material concentration. This study further illustrates how BO can be deployed effectively in complex multistep flow processes and again highlights the value of holistic optimization in improving overall process efficiency.
Continuing the theme of modular multistep optimization, Nambiar et al. integrated computer aided synthesis planning (CASP) with robotic flow chemistry and multi-objective Bayesian optimization for the preparation of sonidegib 132.135 The CASP-generated route, refined through human input, comprised three sequential transformations (SNAr, nitro reduction, and amide coupling) and one inline gas–liquid separation (Fig. 22a), and was implemented on a reconfigurable flow platform equipped with inline FT-IR and LC-MS. During preliminary experiments with the fully telescoped process, catalyst deactivation was observed during the reduction step, triggered by a byproduct from the upstream SNAr reaction. To avoid this, the optimization was split into two campaigns, with intermediate isolation of the SNAr product. The first campaign—the SNAr step—optimized five variables towards three objectives: yield, productivity, and cost (Fig. 22b). The Dragonfly algorithm guided the campaign, with reaction performance being monitored by both inline FTIR and LC-MS. Over 30 experiments, a clear trade-off emerged between high-yielding fluorinated substrates and lower-cost chlorinated analogues.
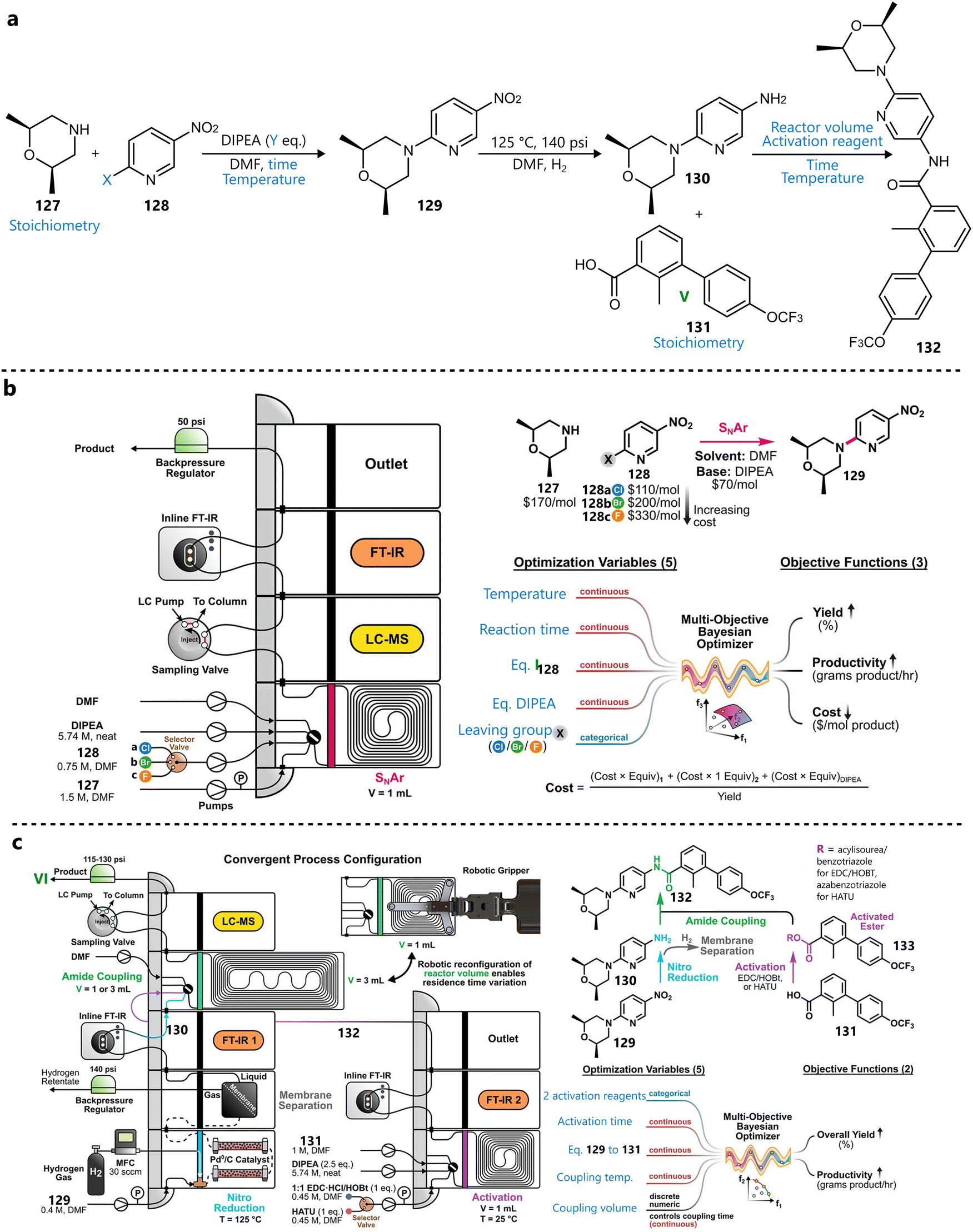 | ||
| Fig. 22 Optimization of a computer-aided synthesis pathway on an automated flow platform. Parameters defining the reaction space are highlighted in blue. (a) Synthetic pathway. (b) SNAr optimization. Left: Platform configuration. Right: Reaction scheme and optimization parameters. (c) Multistep downstream process optimization campaign. Left: Platform configuration. Right: Reaction scheme and optimization parameters. (b) and (c) Adapted from Nambiar et al.135 under a CC BY 4.0 license.97 | ||
For the multistep downstream optimization—comprising nitro reduction, gas–liquid separation, and amide coupling—the platform was reconfigured into a convergent layout, in which the carboxylic acid 131 was first pre-activated upstream before being contacted with the amine, reducing side-reactions with the coupling agent that were observed during the preliminary telescoped experiments (Fig. 22c). The BO campaign considered five variables: activation reagent (categorical), equivalents of nitro compound, activation time, coupling temperature, and reactor volume (1 or 3 mL). The latter was varied on-the-fly via robotic reconfiguration to access different residence times without altering upstream flow rates. Two objectives, yield and productivity, were optimized over fifteen experiments. Practical constraints meant that some parameters (e.g., solvent, concentration, temperature of the nitro reduction) were fixed. Inline FTIR provided real-time monitoring of reduction and activation steps, while LC-MS quantified product formation, enabling robust closed-loop optimization of the downstream sequence. The study exemplifies how BO, real-time process analytics, and modular hardware can be combined to provide efficient navigation of complex multistep chemical spaces, as well as showing the importance of sufficient understanding of the interdependencies in complex telescoped systems before starting optimization.
While the Nambiar platform demonstrated how BO can manage cross-step dependencies in multistep synthesis, downstream purification was not considered. Clayton et al. addressed this gap directly, applying multi-objective Bayesian optimization to both reaction and work-up stages within a unified, closed-loop framework.136 Using the TS-EMO algorithm, they optimized a Sonogashira coupling, and a Claisen–Schmidt condensation with in-line phase separation. The first case targeted mono-coupled product formation (as a model for lanabecestat synthesis) and optimized three continuous variables (residence time, temperature, and alkyne equivalents) toward two objectives: space–time yield (STY) and residual starting material, as the latter has a significant impact on the downstream work-up. The parameter space was relatively simple, and although 80 total experiments (20 LHC + 60 BO-guided) may be considered high, the resulting Pareto front was densely populated and offered a well-resolved trade-off between amount of starting material remaining and throughput. In the second case study, a biphasic Claisen–Schmidt condensation and its aqueous work-up were optimized holistically: sampling occurred only after the in-line liquid–liquid separator, ensuring that the optimization fully captured interactions between the upstream reaction and downstream partitioning. Four continuous variables (flow rates, aqueous-to-organic ratio, temperature) were tuned toward three objectives: STY, purity, and reaction mass efficiency. Across 109 total experiments, the algorithm identified eighteen non-dominated points spanning a broad Pareto front. While the platform was treated as a black-box, lacking intermediate analytics, the study provided an early and important demonstration of BO applied to purification-inclusive multistep optimization.
Finaly, Luo et al. applied multi-objective Bayesian optimization to a telescoped two-step flow synthesis of hexafluoroisopropanol (HFIP), combining the catalytic gas-phase rearrangement of hexafluoropropylene oxide (HFPO) to hexafluoroacetone (HFA), followed by Pd/C-catalysed hydrogenation of HFA to HFIP in a sequential gas–liquid and gas–liquid–solid heterogeneous setup.137 Four process variables (gas flow rate and temperature in each step) were optimized with respect to conversion and E-factor, using qNEHVI in closed loop. Sampling was performed only at the outlet of the full telescoped sequence, with no intermediate analytics. Prior to experimental application, the authors benchmarked four acquisition functions (qNEHVI, qEHVI, qNParEGO, and TS-EMO) using kinetic models of unrelated reactions, artificially perturbed with varying levels of Gaussian noise. qNEHVI demonstrated the most robust performance under noise and outpaced the other methods in computational efficiency. The final wet-lab campaign began with ten Latin hypercube samples and proceeded through ten BO-guided iterations, yielding a best condition of 76.2% conversion and an E-factor of 0.3744. As the final example in this section, the study highlights the continued expansion of BO beyond idealized systems into telescoped, heterogeneous, and noise-prone processes, where both experimental and algorithmic resilience are equally critical to success.
5. BO in industrial and large-scale processes
Despite the growing adoption of Bayesian optimization, its application to larger-scale or manufacturing-relevant processes remains rare. Most reported studies remain conceptual or are at an early stage, often limited to digital optimization workflows or lab-scale analogues of industrial problems. One reason for this limited uptake is that BO is naturally suited to exploratory optimization—where objectives are poorly defined, prior knowledge is limited, and experimental budgets are tight. In contrast, process development at larger scales tends to be guided by regulatory frameworks such as Quality by Design (QbD), which emphasize robustness, interpretability, and traceability—features not inherently built into most BO workflows. As a result, while BO may not be directly applicable during late-stage scale-up, it holds significant promise for early-phase development, particularly in rapidly identifying viable process windows or operating regions that can subsequently be refined through conventional DoE or mechanistic modelling. This role has been highlighted in hybrid workflows, such as those reported by the Kappe group,107 where BO is used to accelerate model building. The examples discussed in this section represent early efforts to bring BO closer to manufacturing contexts, whether by applying it to scale-relevant batch or flow reactions, or by embedding it into computational workflows involving process dynamics and cost models. While diverse in scope, they collectively underline that BO for industrial optimization is still in its infancy, with clear opportunities for further methodological and infrastructural development.Kumar et al. presented a data-driven workflow for optimizing methanol synthesis from syngas, using Gaussian process regression (GPR) models trained on laboratory-scale data to predict methanol selectivity and CO conversion.138 Ten kernel configurations were benchmarked for each objective, and SHAP values were used to assess feature influence. This approach assigns feature importance scores by averaging each variable's marginal contribution to the model output over all possible feature combinations. A weighted-sum objective was then constructed and optimized using Bayesian optimization, though no details were provided on the structure or updating of the BO surrogate. As in the Zhu et al. study described earlier, BO appears to have been applied post hoc to a static surrogate, introducing an additional surrogate layer whose role is conceptually ambiguous. While the modeling strategy is rigorous and interpretable, the use of BO remains secondary and structurally disconnected from the data acquisition process. The study is best viewed as an interpretable ML workflow with BO appended for global optimization, rather than as a focused, sequential BO campaign.
Jorayev et al. reported multi-objective Bayesian optimization for the valorization of crude sulfate turpentine (CST), targeting the two-step synthesis of p-cymene from a controlled CST surrogate mixture composed of α-pinene, β-pinene, 3-carene, limonene, and dimethyl sulfide.61 The isomerization was performed in batch and the hydrogenation in flow. Each step was optimized independently using TS-EMO, targeting p-cymene yield and conversion. In the first campaign, temperature, reaction time, sulfuric acid concentration and ratio of aqueous to organic phase were varied; in the second, temperature, gas and liquid flow rates, and residence time were optimized. Although the system was not fully continuous and the optimizations were decoupled, the workflow integrates batch and flow elements under a common, surrogate model-based framework. The use of chemically representative feedstocks and larger reactor volumes places the study closer to early-stage process development than discovery-scale screening. It demonstrates how BO can be extended to semi-continuous, multi-variable systems involving industrially relevant input mixtures.
Whereas the study by Jorayev et al. addressed process-relevant feedstock complexity through surrogate-based optimization of batch–flow integration, Kondo et al. focused more directly on experimental scale-up. They applied Bayesian optimization to improve the sulfurization-based synthesis of 2,3,7,8-tetrathiaspiro[4.4]nonane, a key intermediate in the preparation of polyvalent mercaptans used in industrial materials.139 The study targeted a second-generation reaction protocol involving pentaerythrityl tetrachloride and NaSH in toluene, and optimized four variables: NaSH equivalents, sulfur equivalents, NaSH concentration in water, and substrate concentration. Optimization was carried out using Gaussian process regression and the expected improvement acquisition function. Two BO campaigns were conducted, one with random initialization, the other using Latin hypercube sampling (LHS). The LHS-guided campaign reached an 89% GC yield in just eight experiments while also reducing sulfur and NaSH loadings. Importantly, the optimized conditions were scaled directly from 2 g to 100 g without reoptimization, maintaining isolated yields of 81–87%. While the optimization was conducted in batch and in a non-automated manner, the study stands out in this section for demonstrating that BO-guided condition refinement can be effectively transferred to decagram-scale synthesis. It underscores BO's potential as a practical tool for scaling early reaction optimization into preparative workflows with minimal experimental overhead.
Liu et al. applied Bayesian optimization to the design of a batch solvent removal process, specifically the concentration of a pharmaceutical intermediate via evaporation, using a dynamic simulation model.140 The objective was to minimize a customized total cost function that combined operational costs (labor, equipment, utilities) with the cost of lost toluene, including downstream treatment. Three process variables—evaporation pressure, temperature difference (ΔT), and outlet temperature—were optimized using GP surrogate and expected improvement as the acquisition function. The BO framework was benchmarked against four alternative optimization strategies: tree-structured Parzen estimators (TPE), genetic algorithms (GA), particle swarm optimization (PSO), and simulated annealing (SA). BO achieved the lowest final cost and fastest convergence, identifying a process configuration that reduced the total cost by nearly 50% in around 20 simulation calls. Although the study remained purely in silico, it demonstrates how BO can be integrated with mechanistic process simulations to optimize cost-relevant unit operations under dynamic conditions.
Collectively, the studies reviewed in this section illustrate the emerging role of Bayesian optimization in process-relevant and scale-conscious chemical applications. While none of the workflows reviewed here are fully automated or implemented at manufacturing scale, they reflect a growing interest in using BO to support condition refinement, early-phase scale-up, and unit operation design. One case study demonstrates effective transfer of BO-derived conditions to gram or 100 g scale, while others integrate BO with mechanistic models to explore cost-sensitive or constrained design spaces. Nevertheless, the limited number of examples and the reliance on surrogate-based or offline workflows underscore that BO in industrial process optimization remains in its early stages. Future development will likely depend on deeper integration with automation platforms, hybrid modelling strategies, and continued efforts to balance data efficiency with robustness and interpretability.
6. Hybrid Bayesian optimization
While standard Bayesian optimization has proven effective for low-throughput reaction optimization under data scarcity, recent work has begun exploring how BO can be adapted or extended to overcome domain-specific limitations. These efforts fall broadly under what might be termed “hybrid” BO strategies, approaches that seek to incorporate mechanistic knowledge, pre-trained models, auxiliary data, or experimental constraints into the BO framework. Many remain at a conceptual or simulated stage. Still, they highlight important methodological directions: how to accelerate early-stage discovery using historical or multitask data, how to incorporate cost or feasibility constraints, and how to develop chemically informed surrogates that encode structure beyond black-box regression. This section surveys a representative selection of such approaches, organized by theme rather than implementation, and assesses their potential impact in reaction optimization contexts.6.1. Chemically informed and physics-aware Bayesian optimization
Frey et al. proposed a hybrid surrogate strategy that leverages physics-based modeling to improve early-stage Bayesian optimization for chemical reactions.141 Termed Chemically-Informed Data-Driven Optimization (ChIDDO), the method augments each BO iteration by adding synthetic data from a mechanistic model—in this case, a mass transport-based electrochemical model—into the experimental dataset prior to Gaussian process surrogate fitting (Fig. 23). A simple accounting rule determines the number of such insertions: at each iteration, the difference between a fixed total budget and the number of available experimental points is filled with low-fidelity model predictions. Importantly, the parameters of the physics model are updated iteratively by minimizing the root mean square error (RMSE) between model outputs and experimental data, thereby improving model fidelity over time. The MRB (Model and Region Balancing) acquisition function is then applied to the fused dataset to select the next experimental query. The method was benchmarked on synthetic electrochemical landscapes and showed improved sample efficiency over standard BO, but only in cases with more than three optimization variables. Moreover, the benefit of the approach depended strongly on the fidelity of the underlying physical model, potentially limiting its practical applicability in domains where mechanistic models are unavailable or poorly predictive. Nevertheless, the framework provides a structured route for embedding mechanistic priors into BO workflows, particularly in high-dimensional or data-scarce chemical optimization problems.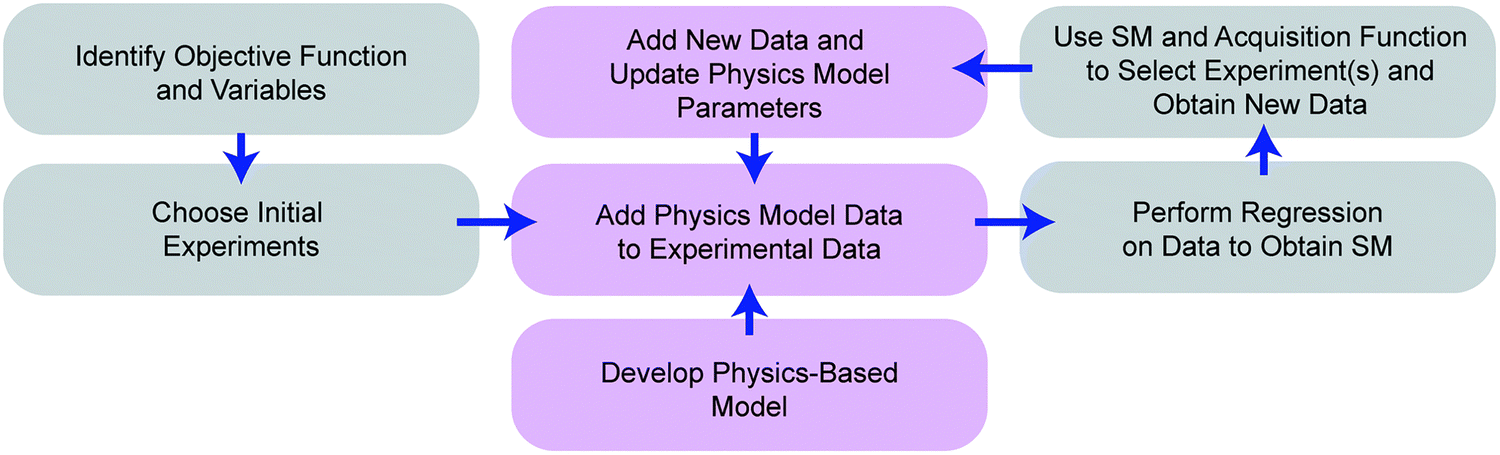 | ||
| Fig. 23 Schematic representation of the ChiDDO algorithm. Gray blocks correspond to steps related to acquisition of experimental data. Purple blocks represent steps to incorporate the physics model. Reproduced from ref. 141 with permission from The Royal Society of Chemistry, copyright 2022. | ||
Hickman et al. developed a Bayesian optimization framework that incorporates known design and experimental constraints directly into the acquisition function optimization step.142 Constraints are modeled as independent functions and used to restrict the domain over which the acquisition function is optimized. During acquisition optimization, feasibility is checked explicitly to ensure that proposed experiments do not violate the predefined constraints, thus eliminating infeasible regions a priori. Two chemically motivated benchmarks were presented. In the first, the goal was to maximize yield and minimize cost for the synthesis of fullerene derivatives in flow, with constraints expressed as linear combinations of flow rates to reflect hardware limitations. In the second, a multi-objective molecular design task was considered, targeting redox-active materials with desirable redox potentials, solvation free energies, and absorption wavelengths. Here, the constraint was a synthetic accessibility threshold, limiting the search to candidates with plausible retrosynthetic routes. Notably, the advantage of constraint-aware BO proved highly objective-specific: improved convergence and optima were observed for cost in the first benchmark, and for redox potential and solvation free energy in the second, but no gain was seen for optimizing yield or spectral properties. While the benchmarking was performed entirely in silico, the methodology offers a principled way to encode known feasibility restrictions—such as instrument limits or synthetic access—into BO campaigns and could be directly transferable to constrained experimental domains.
While ChIDDO and constraint-aware BO focus on embedding physical feasibility and domain boundaries, other forms of chemical realism—such as reagent cost—can also be directly encoded into the BO loop. The Cost-Informed Bayesian Optimization (CIBO) framework, as reported by Schoepfer et al., is one such example.143 In their work, the authors modified the expected improvement acquisition function by subtracting a term proportional to the reagent cost of each candidate, thereby reducing the acquisition value of high-cost experiments (Fig. 24). This encourages the selection of lower-cost conditions without explicitly modelling cost as an optimization objective. The framework was tested on two literature-derived reaction datasets: a direct arylation and a Buchwald–Hartwig cross-coupling. In both cases, the comparison between standard BO and CIBO was based on total campaign cost rather than per-experiment cost. CIBO demonstrated a clear advantage in the first case, where it achieved optimal conditions at a reduced cost. In the second, its benefit was conditional: it offered lower campaign cost only if the optimization was halted early, for example, after reaching a 70% yield threshold. When allowed to continue, both CIBO and standard BO ultimately converged on the same total cost, as all relevant reagents were eventually sampled. This raises a broader point: because standard BO had no access to cost information, the comparison may be skewed. A more transparent and flexible alternative would be to treat cost as a second objective within a multi-objective BO framework, enabling explicit trade-off modelling and Pareto front visualization. Moreover, the assumed cost model—where a reagent incurs a one-time penalty upon first use—may be less relevant in practical settings where reagent availability is pre-constrained. Still, CIBO demonstrates how modifying the acquisition function can introduce chemically meaningful prioritization into BO, particularly in early-phase screening campaigns where resource constraints are prevalent.
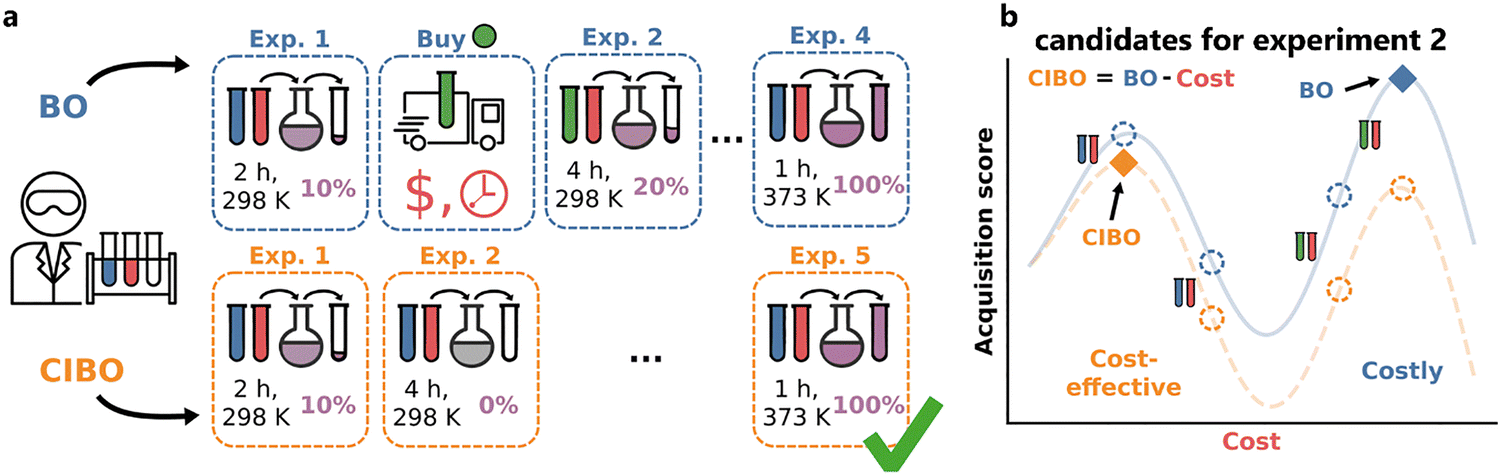 | ||
| Fig. 24 Comparison between standard and cost-informed BO (CIBO). (a and b) CIBO balances the expected improvement of the experiment with its cost, suggesting more cost-effective experiments. Adapted from Schoepfer et al.143 under a CC BY 3.0 Unported license.96 | ||
6.2. Transfer learning and generalization strategies in Bayesian optimization
Beyond modifying acquisition functions, another line of research focuses on the use of pretrained or multitask surrogate models to accelerate BO convergence, either by reusing historical data or embedding generalizable chemical knowledge into the model architecture. These approaches seek to bypass the data inefficiency of BO's cold start, especially in scenarios with well-structured prior data or repeated tasks across chemical series.Taylor et al. developed a multi-task Bayesian optimization (MTBO) framework aimed at accelerating chemical reaction optimization by transferring information from related, previously optimized tasks.52 The method employs a multi-task Gaussian process (MTGP) surrogate that models both the current and auxiliary reactions jointly, with a shared kernel structure capturing cross-task correlations (Fig. 25). The framework was experimentally validated using a closed-loop flow platform to optimize a C–H activation reaction, with five parameters—residence time, temperature, catalyst concentration, solvent, and ligand—varied.
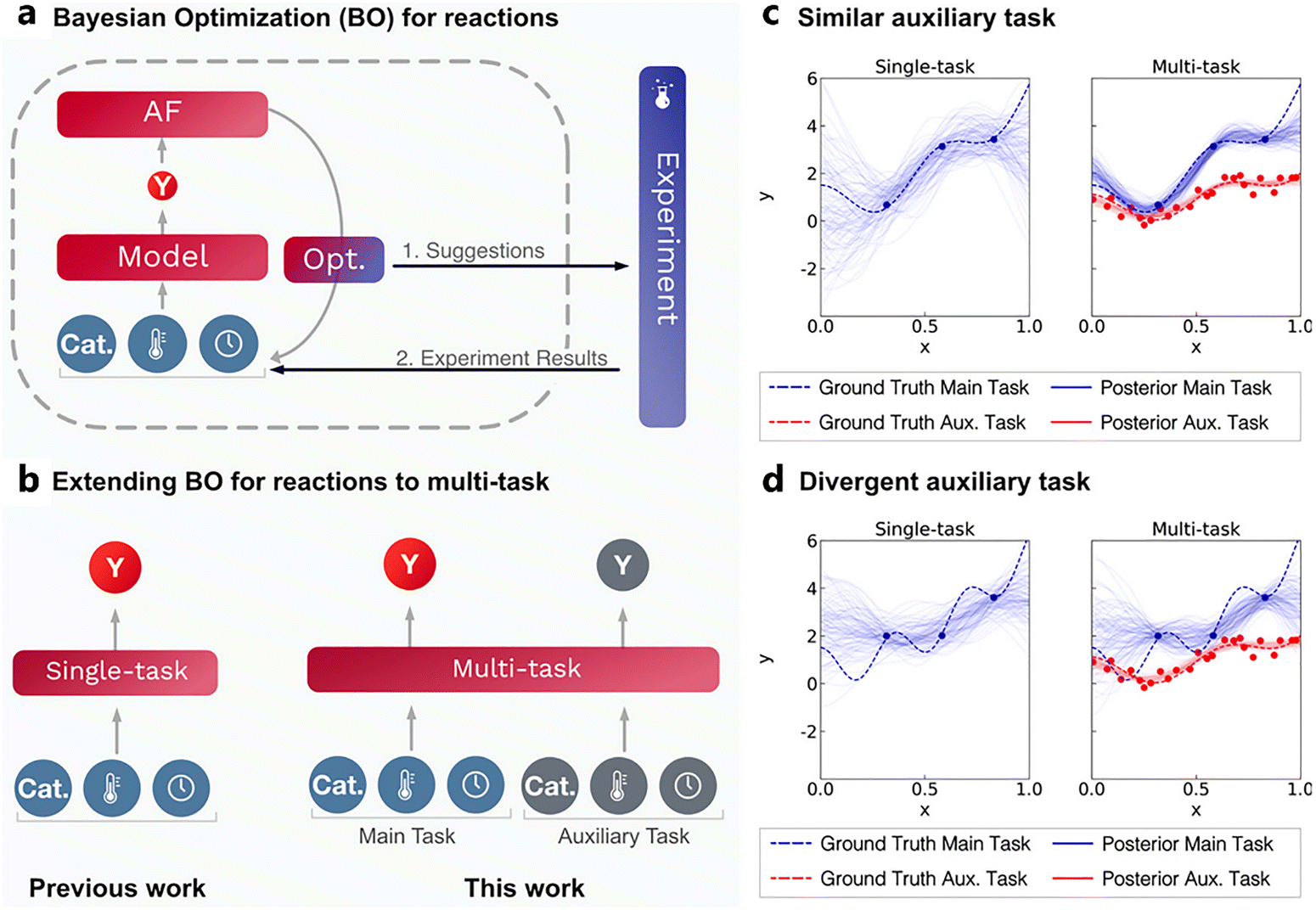 | ||
| Fig. 25 Multitask learning (MTBO) to accelerate Bayesian reaction optimization. (a) Standard BO. (b) Multitask BO replaces the GP with a multitask GP trained simultaneously on an auxiliary task (e.g. a similar chemical reaction). (c) When the auxiliary task is similar, predictions on the main task are greatly improved. (d) When the auxiliary task is divergent, performance is comparable to single-task BO. Adapted from Taylor et al.52 under a CC BY 4.0 license.97 | ||
MTBO consistently accelerated convergence when auxiliary tasks displayed similar reactivity to the target, and performance improved further as more auxiliary tasks were included. However, when auxiliary reactions were poorly correlated—exemplified by low-yielding systems—the surrogate model could become biased, slowing convergence or misdirecting early iterations. The authors showed that this effect could be mitigated by using a broader set of auxiliary tasks to balance individual influences. This approach is particularly well-suited to discovery workflows, where a single transformation is often applied across a substrate scope, and offers an alternative to optimizing for general conditions as discussed in Section 3.2. The results highlight both the potential of MTBO in data-scarce optimization campaigns and the importance of curating auxiliary tasks based on chemical relevance.
Taking another approach to knowledge transfer between reactions, Faurschou et al. implemented a closed-loop Bayesian optimization workflow for substrate-specific condition refinement in carbohydrate protective group chemistry, focusing on enabling practical transfer learning across structurally related substrates.144 The authors employed a single GP surrogate with an additional categorical variable to encode the identity of each substrate, enabling data from previously optimized substrates to inform the model's predictions for new ones. While simple in formulation, this approach effectively enabled transfer learning without the need for a more complex multitask architecture. Optimization was carried out over eight reaction parameters: four discrete including the substrate, and four continuous. The platform operated in closed loop, with reaction outcomes automatically evaluated and fed back into the optimization. The authors observed that including prior data improved early-stage performance for new substrates, though the benefit plateaued as more data was added. While the method assumes consistent data structure and reaction format across substrates, it demonstrates that even minimal structural adaptation can yield effective knowledge transfer in substrate scope optimization.
Hickman et al. introduced SeMOpt, a transfer learning framework for reaction optimization that incorporates prior experimental knowledge via a neural process (NP) model.145 Neural processes are a class of probabilistic models that learn a distribution over functions conditioned on data from previous tasks. In this context, the NP is trained on multiple completed optimization campaigns and used to predict likely high-performing regions of the design space for a new task. Rather than replacing the standard BO loop, SeMOpt blends the NP's prediction with that of a task-specific Gaussian process using a compound acquisition function—effectively biasing early search toward regions historically associated with high yields. As more task-specific data accumulate, the influence of the NP is gradually diminished, allowing the GP to take over. The framework was validated in silico using both a synthetic reaction family and a dataset of 15 Buchwald–Hartwig coupling campaigns derived from Ahneman et al.,146 each comprising 276 experiments spanning all possible parameter combinations for a given product (Fig. 26). While SeMOpt significantly reduced the number of experiments required to identify high-performing conditions, the authors note several limitations—such as the need for consistently encoded historical data, the sensitivity of transfer performance to task similarity, and challenges in representing categorical variables—which are not specific to SeMOpt but reflect broader obstacles in applying BO to chemical reaction optimization. Among the approaches surveyed in this section, SeMOpt stands out as the most formalized and modular transfer learning framework—offering explicit task conditioning, dynamic influence balancing, and a principled fallback to standard BO when prior data prove uninformative.
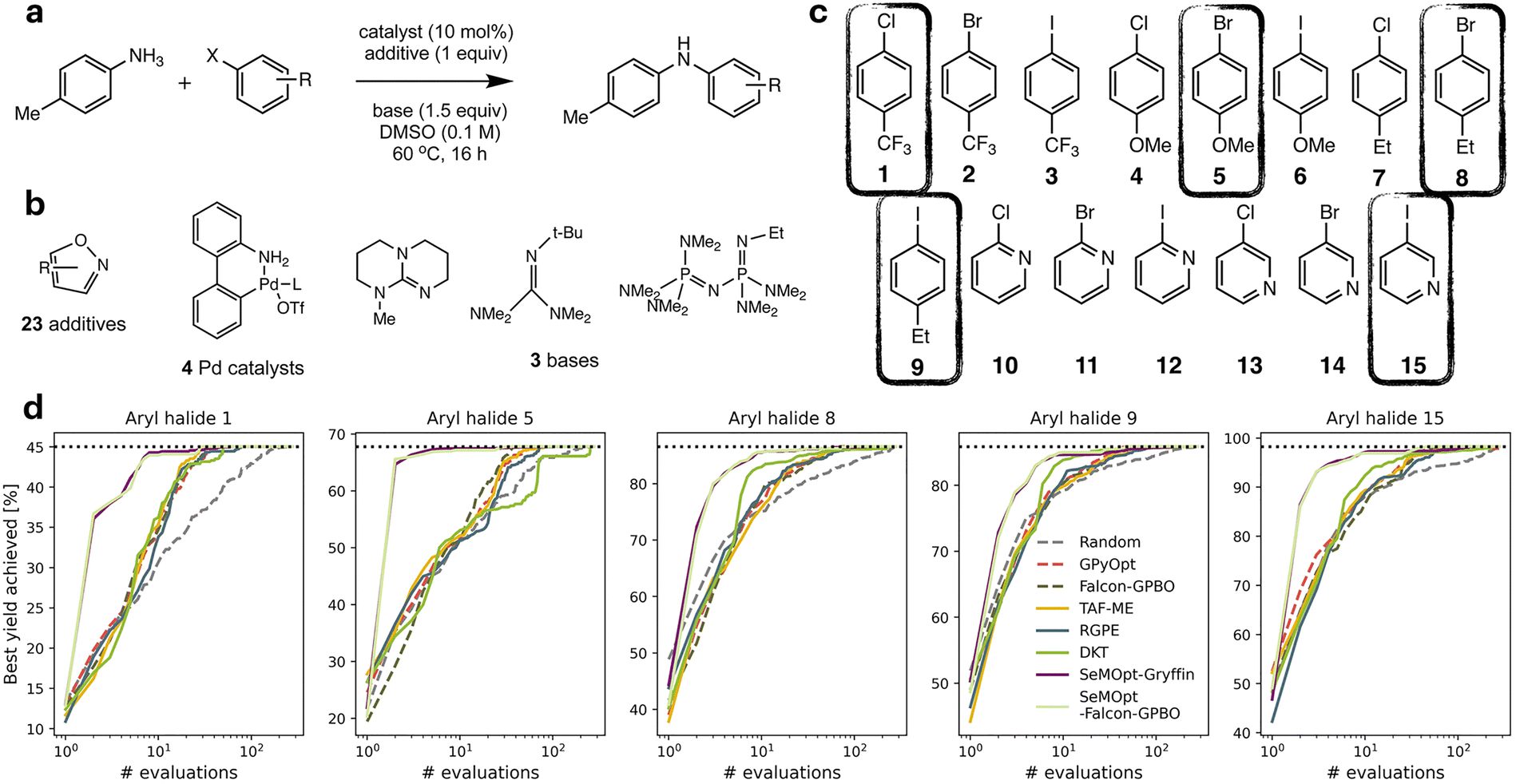 | ||
| Fig. 26 Example use of SeMOpt for transfer learning during the BO-guided optimization of a Buchwald–Hartwig cross-coupling. (a) Reaction scheme. All data comes from a real-world dataset. (b) Parameter space, comprised of only categorical variables. (c) Structure of the aryl halide substrates. The ones circled in black are selected as targets for five optimization campaigns. (d) Results of the optimization campaigns. Dashed lines indicate single-task strategies, where no prior information is fed to the algorithm. In comparison, the solid lines indicate transfer learning. For each of the five target substrates, the algorithm was pretrained on the other 14 substrates. The horizontal dotted line represents the highest yield reported. The transfer-learning approach clearly accelerates convergence. Reproduced from ref. 145 with permission from The Royal Society of Chemistry, copyright 2023. | ||
Attempting to optimize reaction conditions for generality across a substrate scope—like the Angello86 and Wang87 papers discussed earlier—Schmid et al. introduced a formulation of generality-aware reaction optimization based on curried functions.147 While the paper presents a mathematically formal treatment of the problem, the discussion here is limited to its high-level conceptual contributions and implications for chemical reaction optimization. In the framework, the reaction outcome is modelled as a function f(x,s), where x denotes the reaction conditions and s the substrate. Currying reinterprets this as f(x)(s), meaning that for fixed reaction conditions x, the resulting function maps each substrate s to its predicted performance. This allows the optimization to focus on selecting a globally effective condition set x* that performs well across substrates, rather than optimizing a separate x for each task. The authors frame this as a min-max objective: minimize over x the maximum loss across all substrates. Practically, this means seeking the condition set with the best worst-case performance, a formulation well-suited to discovery settings where generality is often prioritized over substrate-specific fine-tuning. Importantly, the paper also addresses the partial observation problem: in a real-world setting, it is impossible to try every condition for every substrate. To mitigate this, the authors design acquisition strategies—such as max-loss, average-loss, and a sequential one-step lookahead—that operate robustly in this incomplete setting. These were then benchmarked on real-world datasets, demonstrating their effectiveness.
Finally, Kwon et al. proposed a BO workflow guided by a graph neural network (GNN) pretrained on a large reaction database to prioritize candidate conditions for initial sampling.148 First, the GNN was used to rank the full set of conditions, restricting the search space to a smaller subset, after which a Bayesian optimization algorithm was initialized on the selected set of best predicted conditions. A hybrid acquisition function was proposed to blend predictions from the BO with those from the GNN, with the influence of the latter diminishing as the number of iterations increases. To mitigate the GNN imposing a too limited parameter space, a heuristic for expanding the search space was implemented upon stagnation of the BO. This warm-start approach led to an ±8.5% improvement in convergence speed over random initialization across a set of benchmark reactions. While the concept illustrates the potential of pretrained models to inform early-stage optimization, the modest performance gain must be weighed against the need for a substantial, labelled dataset to train the GNN, limiting its practical applicability in typical low-data scenarios.
7. State of the art in Bayesian optimization
Most BO campaigns still rely on a standard Gaussian process with a Matérn or squared-exponential kernel and a basic acquisition function such as expected improvement or probability of improvement. Discrete variables (catalysts, ligands, solvents, etc.) are usually treated as categorical or via one-hot encoding; chemically or physically informed descriptor-based encodings remain relatively rare. Descriptors can, in principle, accelerate convergence by allowing the surrogate to extrapolate beyond previously tested options, an advantage for large search spaces where hundreds of ligands or additives are under consideration. Yet a persistent hurdle is descriptor selection: which numerical features truly capture the steric, electronic or other effects that govern reactivity? The previous sections surveyed the few systematic comparisons to date and finds only modest, or even negligible, benefits for elaborate, high-dimensional descriptors relative to simple one-hot encoding. These data suggest that effort might be better spent on simple, chemically transparent descriptors—such as pKa, solvent polarity indices, σ-donor/π-acceptor numbers—than on elaborate features that may not generalize or even improve performance at all. It can be argued that standardized descriptor libraries, such as the Kraken dataset for organophosphorus ligands,81 which provide curated steric and electronic descriptors, could add value without resorting to ad hoc feature engineering.149A second area of interest concerning algorithms is transfer learning. Most optimization campaigns still begin ‘cold,’ without leveraging any prior data. Although the multi-task and neural-process-based approaches covered here hold great promise, a method that generalizes across broad reagent and substrate spaces has yet to emerge.
Even partial success here would shorten initialization phases and further accelerate optimization. Similarly, extending the chemically informed Bayesian optimization by embedding first-principles relations, such as the Bouguer–Lambert–Beer law or gas–liquid mass transfer correlations, as priors in the surrogate could further reduce the experimental burden.
Another theme is robust optimization, in which the acquisition function not only considers high performance, but also low local sensitivity, which is critical for process development. Initial efforts have been made but have not yet been applied to chemical reactions.150
For very high-dimensional problems, recent work on nested-subspace151,152 and trust-region BO153 could provide better scalability, although their relevance to chemical reaction optimization remains to be demonstrated.
A closely related approach is multi-fidelity Bayesian optimization (MF-BO), where inexpensive, lower-accuracy data sources (e.g., semi-empirical calculations, coarse HTE plates, or simplified kinetic models) are modelled alongside higher-fidelity data sources (e.g., flow experiments) within a single probabilistic surrogate.154 By allowing the optimizer to query low-cost information first, and reserve expensive laboratory experiments for the most promising points. Although unproven for chemistry, MF-BO promises to accelerate convergence significantly.
Finally, there is growing interest in coupling BO with large language models (LLMs). A recent pre-print from the Cooper group demonstrates an LLM contextualizing optimizer suggestions with expert knowledge, suggesting new, better-performing areas of the search space when the BO algorithm reaches a plateau.155 While promising, this approach should be taken with caution, as model explainability is crucial.
8. Conclusions and outlook
Bayesian optimization (BO) has emerged as a powerful methodology for chemical reaction optimization, particularly beneficial in contexts where experimental data are expensive, scarce, or otherwise challenging to obtain. Fundamentally, BO combines surrogate models to approximate reaction behaviour with acquisition functions that strategically balance exploration of uncertain parameter spaces with exploitation of promising reaction conditions. Throughout diverse chemical domains—including electrochemistry, photoredox catalysis, polymerization, and multiphase reactions—BO consistently demonstrates versatility, efficiency, and significant practical utility. Notable examples include the optimization of metallaphotoredox-catalysed cross-coupling reactions by Torres et al. and Li et al., and the systematic exploration of photochemical transformations using advanced automated flow platforms such as RoboChem or the modular telescoped flow platform reported by Nambiar and coworkers.The primary strength of BO lies in its ability to navigate experimental uncertainty and achieve remarkable data efficiency intelligently. Gaussian Processes (GPs) remain the preferred surrogate model due to their inherent flexibility and reliable quantification of uncertainty. Recent methodological advancements have substantially improved BO's practical performance and applicability. Adaptive acquisition functions like Adaptive Expected Improvement (AEI), specialized acquisition functions designed explicitly for noisy data environments (such as qNEHVI), and batch-oriented strategies (e.g., qEI) have been particularly influential. Additionally, mixed-variable kernels employing methods like the Gower distance and chemically informed descriptors have enabled BO to tackle complex, realistic chemical reaction spaces more effectively.
Despite these methodological successes, several important limitations persist. Much of the published literature still focuses on relatively simple chemical reactions, such as Suzuki couplings and Buchwald–Hartwig aminations, which typically yield robust outcomes across broad parameter ranges.
These reactions serve as valuable benchmarks due to their well-characterized nature, but tackling more challenging reactions remains an important challenge for future research. Low initial reactivity, complex reaction mechanisms, or multiphase scenarios are underrepresented in current studies, largely due to practical constraints, including experimental complexity and the challenges associated with accurate surrogate modelling.
Another critical issue is the current reliance on substantial manual intervention in narrowing down the parameter space prior to optimization. Optimization campaigns often consider only a limited number of variables, a scenario not truly representative of complex real-world optimization tasks where BO could excel. Furthermore, different experimental platforms present distinct strengths and weaknesses. High-throughput experimentation (HTE) setups effectively handle categorical variables but often struggle with continuously varying parameters, whereas flow chemistry platforms typically face opposite constraints. Consequently, developing integrated systems that combine the advantages of batch and flow platforms and designing more flexible automated experimental setups represent a valuable direction for further advancement.
Interpretability and robustness also represent recurring challenges in BO-driven optimizations. Although BO excels at rapidly identifying promising experimental conditions, its reliance on sparse sampling can result in surrogate models lacking comprehensive, data-rich response surfaces, which are required for robust process scale-up. Recent hybrid strategies, such as those described by Knoll et al., which employ detailed Design of Experiments (DoE) or mechanistic kinetic modelling within BO-identified regions, demonstrate promising solutions to enhance interpretability and reliability. Additionally, Automatic Relevance Determination (ARD) is widely used for identifying parameter importance. Yet, its interpretation can be misleading, particularly in high-dimensional or correlated parameter spaces, suggesting caution in relying solely on ARD for robust insights.
In practice, BO should thus be regarded as an integral component of broader experimental and process optimization strategies rather than a standalone solution. It is particularly valuable in early-stage exploratory research, where rapidly pinpointing promising reaction conditions is critical. Recent innovations in flow chemistry platforms, such as the dynamic trajectories proposed by Florit et al. and multi-point sampling methodologies, have further enhanced the utility of BO by maximizing the information extracted per experiment, thereby enabling deeper mechanistic insights at minimal experimental cost.
Several promising future directions have emerged clearly from the literature. Hybrid BO methodologies that integrate chemical and physical insights directly into the optimization process, such as chemically informed surrogates (e.g., ChIDDO by Frey et al.), show considerable promise. Additionally, transfer learning and multi-task BO strategies, exemplified by MTBO (Taylor et al.) and SeMOpt (Hickman et al.), offer valuable avenues for accelerating optimization across related reaction series or extensive substrate scopes.
The growing interest in BO in industrial settings is also encouraging, although substantial opportunities remain for further integration into regulatory frameworks and large-scale manufacturing environments.
In summary, Bayesian optimization has matured into a crucial and highly effective tool for optimizing chemical reactions. Demonstrated successes across numerous chemical domains justify its rapid development and increasing sophistication. Nevertheless, researchers and practitioners must remain critically aware of its limitations, applicability, and practical integration with traditional optimization strategies. Continued methodological innovation, thoughtful integration with established experimental frameworks, and focused exploration of more challenging chemical contexts will ensure that BO continues to support efficient, sustainable, and robust chemical reaction development.
Author contributions
Stefan Desimpel: conceptualization, methodology, investigation, writing – original draft, visualization, writing – review and editing. Matthieu Dorbec: writing – review and editing, supervision. Kevin M. Van Geem: writing – review and editing, supervision Christian V. Stevens: writing – review and editing, supervision.Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included, and no new data were generated or analysed as part of this review.Acknowledgements
Stefan Desimpel acknowledges financial support from Janssen Pharmaceutica NV and the Flemish government through a Baekeland mandate under grant agreement HBC.2021.0182.Notes and references
- C. J. Taylor, A. Pomberger, K. C. Felton, R. Grainger, M. Barecka, T. W. Chamberlain, R. A. Bourne, C. N. Johnson and A. A. Lapkin, Chem. Rev., 2023, 123, 3089–3126 CrossRef CAS PubMed.
- A. D. Clayton, J. A. Manson, C. J. Taylor, T. W. Chamberlain, B. A. Taylor, G. Clemens and R. A. Bourne, React. Chem. Eng., 2019, 4, 1545–1554 RSC.
- Bayesian optimization (All Fields) OR Bayesian optimisation (All Fields) – 1615 – Web of Science Core Collection, https://www.webofscience.com/wos/woscc/summary/b67dc2dc-3cc2-426d-aec9-84e65e97f78c-014c9a478f/times-cited-descending/1, (accessed 27 February 2025).
- A. D. Clayton, Chem. Methods, 2023, 3, e202300021 CrossRef CAS.
- A. D. Clayton, L. A. Power, W. R. Reynolds, C. Ainsworth, D. R. J. Hose, M. F. Jones, T. W. Chamberlain, A. J. Blacker and R. A. Bourne, J. Flow Chem., 2020, 10, 199–206 CrossRef.
- E. Wimmer, D. Cortés-Borda, S. Brochard, E. Barré, C. Truchet and F.-X. Felpin, React. Chem. Eng., 2019, 4, 1608–1615 Search PubMed.
- D. Cortés-Borda, E. Wimmer, B. Gouilleux, E. Barré, N. Oger, L. Goulamaly, L. Peault, B. Charrier, C. Truchet, P. Giraudeau, M. Rodriguez-Zubiri, E. Le Grognec and F. X. Felpin, J. Org. Chem., 2018, 83, 14286–14289 CrossRef.
- N. Holmes, G. R. Akien, A. J. Blacker, R. L. Woodward, R. E. Meadows and R. A. Bourne, React. Chem. Eng., 2016, 1, 366–371 RSC.
- J. C. Lagarias, J. A. Reeds, M. H. Wright and P. E. Wright, SIAM J. Optim., 1998, 9, 112–147 Search PubMed.
- W. Huyer and A. Neumaier, ACM Trans. Math. Softw., 2008, 35, 1–25 CrossRef.
- D. Cortés-Borda, K. V. Kutonova, C. Jamet, M. E. Trusova, F. Zammattio, C. Truchet, M. Rodriguez-Zubiri and F.-X. Felpin, Org. Process Res. Dev., 2016, 20, 1979–1987 CrossRef.
- A.-C. Bédard, A. Adamo, K. C. Aroh, M. G. Russell, A. A. Bedermann, J. Torosian, B. Yue, K. F. Jensen and T. F. Jamison, Science, 2018, 361, 1220–1225 CrossRef PubMed.
- S. Soritz, D. Moser and H. Gruber-Wölfler, Chem. Methods, 2022, 2, e202100091 CrossRef CAS.
- K. C. Felton, J. G. Rittig and A. A. Lapkin, Chem. Methods, 2021, 1, 116–122 Search PubMed.
- C. Lehmann, K. Eckey, M. Viehoff, C. Greve and T. Röder, Org. Process Res. Dev., 2024, 28, 3108–3118 Search PubMed.
- A. M. Schweidtmann, A. D. Clayton, N. Holmes, E. Bradford, R. A. Bourne and A. A. Lapkin, Chem. Eng. J., 2018, 352, 277–282 CrossRef CAS.
- B. Shahriari, K. Swersky, Z. Wang, R. P. Adams and N. de Freitas, Proc. IEEE, 2016, 104, 148–175 Search PubMed.
- F. Archetti and A. Candelieri, Bayesian Optimization and Data Science, Springer, Cham, 2019 Search PubMed.
- B. J. Shields, J. Stevens, J. Li, M. Parasram, F. Damani, J. I. M. Alvarado, J. M. Janey, R. P. Adams and A. G. Doyle, Nature, 2021, 590, 89–96 Search PubMed.
- S. L. Boyall, H. Clarke, T. Dixon, R. W. M. Davidson, K. Leslie, G. Clemens, A. D. Muller Frans, L. Clayton, R. A. Bourne and T. W. Chamberlain, ACS Sustainable Chem. Eng., 2024, 12, 15125–15133 CrossRef CAS PubMed.
- D. R. Jones, M. Schonlau and W. J. Welch, J. Glob. Optim., 1998, 13, 455–492 CrossRef.
- C. E. Rasmussen and C. K. I. Williams, Gaussian Processes for Machine Learning, The MIT Press, Cambridge, 2005 Search PubMed.
- K. Swersky, J. Snoek and R. P. Adams, in Advances in Neural Information Processing Systems, ed. C. J. Burges, L. Bottou, M. Welling, Z. Ghahramani and K. Q. Weinberger, Curran Associates Inc., Red Hook NY, 2013, vol. 26 Search PubMed.
- J. Snoek, H. Larochelle and R. P. Adams, arXiv, 2012, preprint, arxiv:1206.2944v2 DOI:10.48550/arXiv.1206.2944.
- H. Zhou, X. Ma and M. B. Blaschko, arXiv, 2023, preprint, arxiv:2310.05166v3 DOI:10.48550/arXiv.2310.05166.
- J. Mockus, Bayesian Approach to Global Optimization, Springer, Netherlands, Dordrecht, 1989 Search PubMed.
- P. I. Frazier, arXiv, 2018, preprint, arxiv:1807.02811 DOI:10.48550/arXiv.1807.02811.
- F. Hutter, H. H. Hoos and K. Leyton-Brown, Sequential Model-Based Optimization for General Algorithm Configuration, Springer Berlin Heidelberg, 2011 Search PubMed.
- C. M. Bishop, Pattern Recognition and Machine Learning, Springer, New York, 2006 Search PubMed.
- A. Criminisi, J. Shotton and E. Konukoglu, Found. Trends Comput. Graph. Vis., 2012, 7, 81–227 CrossRef.
- N. S. Eyke, W. H. Green and K. F. Jensen, React. Chem. Eng., 2020, 5, 1963–1972 Search PubMed.
- X. Wang, Y. Jin, S. Schmitt and M. Olhofer, ACM Comput. Surv., 2023, 55, 1–36 Search PubMed.
- H. Liu, J. Cai, Y. S. Ong and Y. Wang, Knowl. Based Syst., 2018, 164, 324–335 CrossRef.
- R. Moriconi, M. P. Deisenroth and K. S. Sesh Kumar, Mach. Learn., 2020, 109, 1925–1943 Search PubMed.
- E. Bradford, A. M. Schweidtmann and A. Lapkin, J. Glob. Optim., 2018, 71, 407–438 CrossRef.
- J. M. Maciejowski and X. Yang, in 2013 Conference on Control and Fault-Tolerant Systems (SysTol), IEEE, 2013, pp. 1–12.
- R. Liu, Z. Wang, W. Yang, J. Cao and S. Tao, Digital Discovery, 2024, 3, 1958–1966 RSC.
- J. Piironen and A. Vehtari, in 2016 IEEE 26th International Workshop on Machine Learning for Signal Processing (MLSP), IEEE, 2016, pp. 1–6.
- A. Saltelli, M. Ratto, T. Andres, F. Campolongo, J. Cariboni, D. Gatelli, M. Saisana and S. Tarantola, Global Sensitivity Analysis. The Primer, Wiley, Hoboken, NJ, 2007 Search PubMed.
- A. Marrel, B. Iooss, B. Laurent and O. Roustant, Reliab. Eng. Syst. Saf., 2009, 94, 742–751 CrossRef.
- K. Kandasamy, J. Schneider and B. Poczos, 32nd International Conference on Machine Learning, 2015, 1, 295–304.
- J. Bergstra, R. Bardenet, Y. Bengio and B. Kégl, Algorithms for Hyper-Parameter Optimization, Curran Associates Inc., Red Hook NY, 2011 Search PubMed.
- M. Halstrup, PhD Thesis, Technical University Dortmund, 2016.
- J. A. Manson, T. W. Chamberlain and R. A. Bourne, J. Glob. Optim., 2021, 80, 865–886 CrossRef.
- B. Ranković, R.-R. Griffiths, H. B. Moss and P. Schwaller, Digital Discovery, 2024, 3, 654–666 RSC.
- A. Pomberger, A. A. Pedrina McCarthy, A. Khan, S. Sung, C. J. Taylor, M. J. Gaunt, L. Colwell, D. Walz and A. A. Lapkin, React. Chem. Eng., 2022, 7, 1368–1379 RSC.
- X. Li, Y. Che, L. Chen, T. Liu, K. Wang, L. Liu, H. Yang, E. O. Pyzer-Knapp and A. I. Cooper, Nat. Chem., 2024, 16, 1286–1294 CrossRef CAS PubMed.
- M. Nouman, R. B. Canty, B. A. Koscher, M. A. McDonald and K. F. Jensen, J. Chem. Inf. Model., 2025, 65, 6499–6512 CrossRef CAS.
- F. Hutter, L. Xu, H. H. Hoos and K. Leyton-Brown, Artif. Intell., 2014, 206, 79–111 Search PubMed.
- A. Tripp, S. Bacallado, S. Singh and J. M. Hernández-Lobato, arXiv, preprint, 2023, arxiv:2306.14809 DOI:10.48550/arXiv.2306.14809.
- L. Ralaivola, S. J. Swamidass, H. Saigo and P. Baldi, Neural Networks, 2005, 18, 1093–1110 CrossRef PubMed.
- C. J. Taylor, K. C. Felton, D. Wigh, M. I. Jeraal, R. Grainger, G. Chessari, C. N. Johnson and A. A. Lapkin, ACS Cent. Sci., 2023, 9, 957–968 CrossRef CAS.
- N. Aldulaijan, J. A. Marsden, J. A. Manson and A. D. Clayton, React. Chem. Eng., 2024, 9, 308–316 Search PubMed.
- D. Jasrasaria and E. O. Pyzer-Knapp, Dynamic Control of Explore/Exploit Trade-Off in Bayesian Optimization, Springer Verlag, Cham, 2019, vol. 858 Search PubMed.
- A. D. Clayton, E. O. Pyzer-Knapp, M. Purdie, M. F. Jones, A. Barthelme, J. Pavey, N. Kapur, T. W. Chamberlain, A. J. Blacker and R. A. Bourne, Angew. Chem., Int. Ed., 2023, 62, e202214511 CrossRef CAS PubMed.
- N. Srinivas, A. Krause, S. M. Kakade and M. Seeger, IEEE Trans. Inf. Theory, 2009, 58, 3250–3265 Search PubMed.
- B. Do, T. Adebiyi and R. Zhang, J. Comput. Inf. Sci. Eng., 2024, 24, 121006 Search PubMed.
- J. M. Hernández-Lobato, M. W. Hoffman and Z. Ghahramani, Adv. Neural Inf. Process. Syst., 2014, 1, 918–926 Search PubMed.
- F. Häse, M. Aldeghi, R. J. Hickman, L. M. Roch and A. Aspuru-Guzik, Appl. Phys. Rev., 2021, 8, 031406, DOI:10.1063/5.0048164.
- O. J. Kershaw, A. D. Clayton, J. A. Manson, A. Barthelme, J. Pavey, P. Peach, J. Mustakis, R. M. Howard, T. W. Chamberlain, N. J. Warren and R. A. Bourne, Chem. Eng. J., 2023, 451, 138443 Search PubMed.
- P. Jorayev, D. Russo, J. D. Tibbetts, A. M. Schweidtmann, P. Deutsch, S. D. Bull and A. A. Lapkin, Chem. Eng. Sci., 2022, 247, 116938 Search PubMed.
- J. C. Fromer, D. E. Graff and C. W. Coley, Digital Discovery, 2024, 3, 467–481 Search PubMed.
- D. E. Fitzpatrick, C. Battilocchio and S. V. Ley, Org. Process Res. Dev., 2016, 20, 386–394 Search PubMed.
- C. Houben, N. Peremezhney, A. Zubov, J. Kosek and A. A. Lapkin, Org. Process Res. Dev., 2015, 19, 1049–1053 CrossRef CAS.
- K. Deb, Multi-objective optimization using evolutionary algorithms, John Wiley & Sons, Ltd, Hoboken, NJ, 2004 Search PubMed.
- F. Häse, L. M. Roch and A. Aspuru-Guzik, Chem. Sci., 2018, 9, 7642–7655 RSC.
- M. T. M. Emmerich, PhD Thesis, Technische Universität Dortmund, 2005.
- K. Yang, M. Emmerich, A. Deutz and T. Bäck, J. Glob. Optim., 2019, 75, 3–34 CrossRef.
- D. Zhan, Y. Cheng and J. Liu, IEEE Trans. Evol. Comput., 2017, 21, 956–975 Search PubMed.
- S. R. Chowdhury and A. Gopalan, arXiv, preprint, 2019, arXiv:1911.01032 DOI:10.48550/arXiv.1911.01032.
- J. Azimi, A. Fern and X. Z. Fern, Batch Bayesian Optimization via Simulation Matching, NIPS, 2010, vol. 23 Search PubMed.
- J. González, Z. Dai, P. Hennig and N. Lawrence, Proceedings of the 19th International Conference on Artificial Intelligence and Statistics, 2016, 648–657.
- S. M. Mennen, C. Alhambra, C. L. Allen, M. Barberis, S. Berritt, T. A. Brandt, A. D. Campbell, J. Castañón, A. H. Cherney, M. Christensen, D. B. Damon, J. Eugenio de Diego, S. García-Cerrada, P. García-Losada, R. Haro, J. Janey, D. C. Leitch, L. Li, F. Liu, P. C. Lobben, D. W. C. MacMillan, J. Magano, E. McInturff, S. Monfette, R. J. Post, D. Schultz, B. J. Sitter, J. M. Stevens, I. I. Strambeanu, J. Twilton, K. Wang and M. A. Zajac, Org. Process Res. Dev., 2019, 23, 1213–1242 Search PubMed.
- D. Ginsbourger, R. Le Riche and L. Carraro, A Multi-points Criterion for Deterministic Parallel Global Optimization based on Gaussian Processes, 2008 Search PubMed.
- J. Wang, S. C. Clark, E. Liu and P. I. Frazier, Oper. Res., 2016, 68, 1850–1865 Search PubMed.
- K. Kandasamy, A. Krishnamurthy, J. Schneider and B. Poczos, in Proceedings of the Twenty-First International Conference on Artificial Intelligence and Statistics, ed. A. Storkey and F. Perez-Cruz, PMLR, 2018, vol. 84, pp. 133–142 Search PubMed.
- A. Senthil Vel, D. Cortés-Borda and F.-X. Felpin, React. Chem. Eng., 2024, 9, 2882–2891 Search PubMed.
- P. Müller, A. D. Clayton, J. Manson, S. Riley, O. S. May, N. Govan, S. Notman, S. V. Ley, T. W. Chamberlain and R. A. Bourne, React. Chem. Eng., 2022, 7, 987–993 RSC.
- J. A. G. Torres, S. H. Lau, P. Anchuri, J. M. Stevens, J. E. Tabora, J. Li, A. Borovika, R. P. Adams and A. G. Doyle, J. Am. Chem. Soc., 2022, 144, 19999–20007 Search PubMed.
- N. P. Romer, D. S. Min, J. Y. Wang, R. C. Walroth, K. A. Mack, L. E. Sirois, F. Gosselin, D. Zell, A. G. Doyle and M. S. Sigman, ACS Catal., 2024, 14, 4699–4708 Search PubMed.
- T. Gensch, G. dos Passos Gomes, P. Friederich, E. Peters, T. Gaudin, R. Pollice, K. Jorner, A. Nigam, M. Lindner-D’Addario, M. S. Sigman and A. Aspuru-Guzik, J. Am. Chem. Soc., 2022, 144, 1205–1217 CrossRef CAS PubMed.
- M. Christensen, L. P. E. Yunker, F. Adedeji, F. Häse, L. M. Roch, T. Gensch, G. dos Passos Gomes, T. Zepel, M. S. Sigman, A. Aspuru-Guzik and J. E. Hein, Commun. Chem., 2021, 4, 112 CrossRef.
- F. Häse, L. M. Roch, C. Kreisbeck and A. Aspuru-Guzik, ACS Cent. Sci., 2018, 4, 1134–1145 Search PubMed.
- M. Christensen, Y. Xu, E. E. Kwan, M. J. Di Maso, Y. Ji, M. Reibarkh, A. C. Sun, A. Liaw, P. S. Fier, S. Grosser and J. E. Hein, Chem. Sci., 2024, 15, 7160–7169 Search PubMed.
- Y. Amar, A. M. Schweidtmann, P. Deutsch, L. Cao and A. Lapkin, Chem. Sci., 2019, 10, 6697–6706 RSC.
- N. H. Angello, V. Rathore, W. Beker, A. Wołos, E. R. Jira, R. Roszak, T. C. Wu, C. M. Schroeder, A. Aspuru-Guzik, B. A. Grzybowski and M. D. Burke, Science, 2022, 378, 399–405 Search PubMed.
- J. Y. Wang, J. M. Stevens, S. K. Kariofillis, M.-J. Tom, D. L. Golden, J. Li, J. E. Tabora, M. Parasram, B. J. Shields, D. N. Primer, B. Hao, D. Del Valle, S. DiSomma, A. Furman, G. G. Zipp, S. Melnikov, J. Paulson and A. G. Doyle, Nature, 2024, 626, 1025–1033 Search PubMed.
- H. Sheng, J. Sun, O. Rodríguez, B. B. Hoar, W. Zhang, D. Xiang, T. Tang, A. Hazra, D. S. Min, A. G. Doyle, M. S. Sigman, C. Costentin, Q. Gu, J. Rodríguez-López and C. Liu, Nat. Commun., 2024, 15, 2781 CrossRef CAS.
- A. I. Leonov, A. J. S. Hammer, S. Lach, S. H. M. Mehr, D. Caramelli, D. Angelone, A. Khan, S. O’Sullivan, M. Craven, L. Wilbraham and L. Cronin, Nat. Commun., 2024, 15, 1240 CrossRef CAS.
- B. Burger, P. M. Maffettone, V. V. Gusev, C. M. Aitchison, Y. Bai, X. Wang, X. Li, B. M. Alston, B. Li, R. Clowes, N. Rankin, B. Harris, R. S. Sprick and A. I. Cooper, Nature, 2020, 583, 237–241 CrossRef.
- Y. J. Hwang, C. W. Coley, M. Abolhasani, A. L. Marzinzik, G. Koch, C. Spanka, H. Lehmann and K. F. Jensen, Chem. Commun., 2017, 53, 6649–6652 RSC.
- F. Wagner, P. Sagmeister, C. E. Jusner, T. G. Tampone, V. Manee, F. G. Buono, J. D. Williams and C. O. Kappe, Adv. Sci., 2024, 11, 2308034 Search PubMed.
- F. L. Wagner, P. Sagmeister, T. G. Tampone, V. Manee, D. Yerkozhanov, F. G. Buono, J. D. Williams and C. O. Kappe, ACS Sustainable Chem. Eng., 2024, 12, 10002–10010 CrossRef.
- N. S. Eyke, T. N. Schneider, B. Jin, T. Hart, S. Monfette, J. M. Hawkins, P. D. Morse, R. M. Howard, D. M. Pfisterer, K. Y. Nandiwale and K. F. Jensen, Chem. Sci., 2023, 14, 8798–8809 RSC.
- K. Kandasamy, K. R. Vysyaraju, W. Neiswanger, B. Paria, C. R. Collins, J. Schneider, B. Poczos and E. P. Xing, arXiv, 2020, preprint, arxiv:1903.06694 DOI:10.48550/arXiv.1903.06694.
- Deed – Attribution 3.0 Unported – Creative Commons, https://creativecommons.org/licenses/by/3.0/, (accessed 27 April 2025).
- Deed – Attribution 4.0 International – Creative Commons, https://creativecommons.org/licenses/by/4.0/, (accessed 27 April 2025).
- D. M. Dalton, R. C. Walroth, C. Rouget-Virbel, K. A. Mack and F. D. Toste, J. Am. Chem. Soc., 2024, 146, 15779–15786 Search PubMed.
- M. Kondo, A. Sugizaki, M. I. Khalid, H. D. P. Wathsala, K. Ishikawa, S. Hara, T. Takaai, T. Washio, S. Takizawa and H. Sasai, Green Chem., 2021, 23, 5825–5831 RSC.
- E. Braconi and E. Godineau, ACS Sustainable Chem. Eng., 2023, 11, 10545–10554 CrossRef.
- J. Da Tan, A. K. Y. Low, S. T. Rui Ying, S. Y. Tan, W. Zhao, Y.-F. Lim, Q. Li, S. A. Khan, B. Ramalingam and K. Hippalgaonkar, Digital Discovery, 2024, 3, 2628–2636 Search PubMed.
- C. P. Breen, A. M. K. Nambiar, T. F. Jamison and K. F. Jensen, Trends Chem., 2021, 3, 373–386 Search PubMed.
- A. Slattery, Z. Wen, P. Tenblad, J. Sanjosé-Orduna, D. Pintossi, T. den Hartog and T. Noël, Science, 2024, 383, eadj1817 Search PubMed.
- A. A. Zlota, Org. Process Res. Dev., 2022, 26, 899–914 Search PubMed.
- A. Q. Mohammed, P. K. Sunkari, P. Srinivas and A. K. Roy, Org. Process Res. Dev., 2015, 19, 1634–1644 CrossRef.
- A. Q. Mohammed, P. K. Sunkari, A. B. Mohammed, P. Srinivas and A. K. Roy, Org. Process Res. Dev., 2015, 19, 1645–1654 CrossRef.
- S. Knoll, C. E. Jusner, P. Sagmeister, J. D. Williams, C. A. Hone, M. Horn and C. O. Kappe, React. Chem. Eng., 2022, 7, 2375–2384 RSC.
- F. Florit, K. Y. Nandiwale, C. T. Armstrong, K. Grohowalski, A. R. Diaz, J. Mustakis, S. M. Guinness and K. F. Jensen, React. Chem. Eng., 2025, 10, 656–666 RSC.
- J. Zhang, N. Sugisawa, K. C. Felton, S. Fuse and A. A. Lapkin, React. Chem. Eng., 2024, 9, 706–712 Search PubMed.
- Z. Wang, in Comprehensive Organic Name Reactions and Reagents, John Wiley & Sons, Ltd, 2010, pp. 2536–2539 Search PubMed.
- P. Mueller, A. Vriza, A. D. Clayton, O. S. May, N. Govan, S. Notman, S. V. Ley, T. W. Chamberlain and R. A. Bourne, React. Chem. Eng., 2023, 8, 538–542 RSC.
- K. Chai, W. Xia, R. Shen, G. Luo, Y. Cheng, W. Su and A. Su, Chem. Eng. Sci., 2025, 302, 120901 CrossRef.
- R. Liang, X. Duan, J. Zhang and Z. Yuan, React. Chem. Eng., 2022, 7, 590–598 RSC.
- J. Yoshida, A. Nagaki and T. Yamada, Chem. – Eur. J., 2008, 14, 7450–7459 CrossRef PubMed.
- J. Yoshida, Y. Takahashi and A. Nagaki, Chem. Commun., 2013, 49, 9896–9904 RSC.
- T. Wirth, Angew. Chem., Int. Ed., 2017, 56, 682–684 Search PubMed.
- G.-N. Ahn, J.-H. Kang, H.-J. Lee, B. E. Park, M. Kwon, G.-S. Na, H. Kim, D.-H. Seo and D.-P. Kim, Chem. Eng. J., 2023, 453, 139707 CrossRef.
- D. Karan, G. Chen, N. Jose, J. Bai, P. Mcdaid and A. Lapkin, React. Chem. Eng., 2024, 9, 619–629 RSC.
- M. Purwa, G. Chandrakanth, A. Rana, A. Mottafegh, S. Kumar, D. Kim and A. K. Singh, Chem. – Asian J., 2024, 19, e202400438 CrossRef PubMed.
- Y. Naito, M. Kondo, Y. Nakamura, N. Shida, K. Ishikawa, T. Washio, S. Takizawa and M. Atobe, Chem. Commun., 2022, 58, 3893–3896 RSC.
- L. F. Kaven, A. M. Schweidtmann, J. Keil, J. Israel, N. Wolter and A. Mitsos, Chem. Eng. J., 2024, 479, 147567 CrossRef.
- E. L. Bell, W. Finnigan, S. P. France, A. P. Green, M. A. Hayes, L. J. Hepworth, S. L. Lovelock, H. Niikura, S. Osuna, E. Romero, K. S. Ryan, N. J. Turner and S. L. Flitsch, Nat. Rev. Methods Primers, 2021, 1, 46 CrossRef.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef PubMed.
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 88–119 CrossRef.
- M. J. Takle, S. C. Cosgrove and A. D. Clayton, Chem. Sci., 2025, 16, 18783–18790 RSC.
- C. Jackson, K. Robertson, V. Sechenyh, T. W. Chamberlain, R. A. Bourne and E. Lester, React. Chem. Eng., 2025, 10, 511–514 RSC.
- M. Kondo, H. D. P. Wathsala, M. S. H. Salem, K. Ishikawa, S. Hara, T. Takaai, T. Washio, H. Sasai and S. Takizawa, Commun. Chem., 2022, 5, 148 CrossRef CAS.
- J. H. H. Dunlap, J. G. G. Ethier, A. A. A. Putnam-Neeb, S. Iyer, S.-X. L. Luo, H. Feng, J. A. G. Torres, A. G. G. Doyle, T. M. M. Swager, R. A. A. Vaia, P. Mirau, C. A. A. Crouse and L. A. A. Baldwin, Chem. Sci., 2023, 14, 8061–8069 RSC.
- T. Shaw, A. D. Clayton, R. Labes, T. M. Dixon, S. Boyall, O. J. Kershaw, R. A. Bourne and B. C. Hanson, React. Chem. Eng., 2024, 9, 426–438 RSC.
- T. Shaw, A. D. Clayton, J. A. Houghton, N. Kapur, R. A. Bourne and B. C. Hanson, Sep. Purif. Technol., 2025, 361, 131288 CrossRef CAS.
- T. M. Kohl, Y. Zuo, B. W. Muir, C. H. Hornung, A. Polyzos, Y. Zhu, X. D. Wang and D. L. J. Alexander, React. Chem. Eng., 2024, 9, 872–882 RSC.
- N. Sugisawa, H. Sugisawa, Y. Otake, H. Krems Roman, V. Nakamura and S. Fuse, Chem. Methods, 2021, 1, 484–490 CrossRef CAS.
- J. Zhu, C. Zhao, L. Sheng, D. Shen, G. Fan, X. Wu, L. Yu and K. Du, J. Flow Chem., 2024, 14, 539–546 CrossRef CAS.
- P. Sagmeister, F. F. Ort, C. E. Jusner, D. Hebrault, T. Tampone, F. G. Buono, J. D. Williams and C. O. Kappe, Adv. Sci., 2022, 9, 2105547 Search PubMed.
- A. M. K. Nambiar, C. P. Breen, T. Hart, T. Kulesza, T. F. Jamison and K. F. Jensen, ACS Cent. Sci., 2022, 8, 825–836 Search PubMed.
- A. D. Clayton, A. M. Schweidtmann, G. Clemens, J. A. Manson, C. J. Taylor, C. G. Niño, T. W. Chamberlain, N. Kapur, A. J. Blacker, A. A. Lapkin and R. A. Bourne, Chem. Eng. J., 2020, 384, 123340 CrossRef CAS.
- G. Luo, X. Yang, W. Su, T. Qi, Q. Xu and A. Su, Chem. Eng. Sci., 2024, 298, 120434 CrossRef CAS.
- A. Kumar, K. K. Pant, S. Upadhyayula and H. Kodamana, ACS Omega, 2023, 8, 410–421 CrossRef CAS PubMed.
- M. Kondo, H. D. P. Wathsala, K. Ishikawa, D. Yamashita, T. Miyazaki, Y. Ohno, H. Sasai, T. Washio and S. Takizawa, Molecules, 2023, 28, 5180 CrossRef CAS.
- C. Liu, C. Han, C. Gu, W. Sun, J. Wang and X. Tang, Comput. Chem. Eng., 2024, 189, 108813 Search PubMed.
- D. Frey, J. H. Shin, C. Musco and M. A. Modestino, React. Chem. Eng., 2022, 7, 855–865 RSC.
- R. J. Hickman, M. Aldeghi, F. Häse and A. Aspuru-Guzik, Digital Discovery, 2022, 1, 732–744 RSC.
- A. A. Schoepfer, J. Weinreich, R. Laplaza, J. Waser and C. Corminboeuf, Digital Discovery, 2024, 3, 2289–2297 RSC.
- N. V. Faurschou, R. H. Taaning and C. M. Pedersen, Chem. Sci., 2023, 14, 6319–6329 Search PubMed.
- R. J. Hickman, J. Ruža, H. Tribukait, L. M. Roch and A. García-Durán, React. Chem. Eng., 2023, 8, 2284–2296 Search PubMed.
- D. T. Ahneman, J. G. Estrada, S. Lin, S. D. Dreher and A. G. Doyle, Science, 2018, 360, 186–190 CrossRef CAS PubMed.
- S. P. Schmid, E. M. Rajaonson, C. T. Ser, M. Haddadnia, S. X. Leong, A. Aspuru-Guzik, A. Kristiadi, K. Jorner and F. Strieth-Kalthoff, arXiv, preprint, 2025, arxiv:2502.18966 DOI:10.48550/arXiv.2502.18966.
- Y. Kwon, D. Lee, J. W. Kim, Y.-S. Choi and S. Kim, ACS Omega, 2022, 7, 44939–44950 CrossRef CAS.
- T. Morishita and H. Kaneko, ACS Omega, 2023, 8, 33032–33038 CrossRef CAS.
- L. Yang, J. Lyu, W. Lyu and Z. Chen, arXiv, preprint, 2023, arxiv: 2310.20145 DOI:10.48550/arXiv.2310.20145.
- N. Jaquier and L. Rozo, High-dimensional Bayesian Optimization via Nested Riemannian Manifolds, in Neural Information Processing Systems (NeurIPS), 2020 Search PubMed.
- L. Papenmeier, L. Nardi and M. Poloczek, arXiv, preprint, 2022, arxiv: 2304.11468 DOI:10.48550/arXiv.2304.11468.
- N. Namura and S. Takemori, Proc. AAAI Conf. Artif. Intell., 2025, 39, 19624–19632 Search PubMed.
- B. Do and R. Zhang, AIAA J., 2025, 63, 2286–2322 CrossRef.
- A. Cissé, X. Evangelopoulos, V. V. Gusev and A. I. Cooper, in Proceedings of the Thirty-Fourth International Joint Conference on Artificial Intelligence, International Joint Conferences on Artificial Intelligence Organization, California, 2025, pp. 4967–4975.
| This journal is © The Royal Society of Chemistry 2026 |