
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Towards greener-by-design fine chemicals. Part 1: synthetic frontiers
Theodore A.
Gazis
a,
Jonas
Wuyts
 b,
Areti
Moutsiou
a,
Giulio
Volpin
c,
Mark J.
Ford
c,
Rodolfo I.
Teixeira
d,
Katherine M. P.
Wheelhouse
e,
Philipp
Natho
f,
Polona
Žnidaršič-Plazl
gh,
Sonja
Jost
i,
Renzo
Luisi
f,
Brahim
Benyahia
d,
Bert U. W.
Maes
b and
Gianvito
Vilé
*a
b,
Areti
Moutsiou
a,
Giulio
Volpin
c,
Mark J.
Ford
c,
Rodolfo I.
Teixeira
d,
Katherine M. P.
Wheelhouse
e,
Philipp
Natho
f,
Polona
Žnidaršič-Plazl
gh,
Sonja
Jost
i,
Renzo
Luisi
f,
Brahim
Benyahia
d,
Bert U. W.
Maes
b and
Gianvito
Vilé
*a
aDepartment of Chemistry, Materials, and Chemical Engineering “Giulio Natta”, Politecnico di Milano, Piazza Leonardo da Vinci 32, 20133 Milano, Italy. E-mail: gianvito.vile@polimi.it
bOrganic Synthesis Division, Department of Chemistry, University of Antwerp, Groenenborgerlaan 171, 2020 Antwerp, Belgium
cBayer AG, Crop Science Division, Alfred-Nobel-Straße 50, 40789 Monheim, Germany
dChemical Engineering Department, Loughborough University, Epinal Way, LE11 3TU Loughborough, Leicestershire, UK
eDrug Substance Development, GSK Medicines Research Centre, Gunnels Wood Road, SG1 2NY Stevenage, Hertfordshire, UK
fFlow Chemistry and Microreactor Technology (FLAME-Lab), Department of Pharmacy – Drug Sciences, University of Bari “Aldo Moro”, Via Edoardo Orabona 4, 70126 Bari, Italy
gFaculty of Chemistry and Chemical Technology, University of Ljubljana, Večna pot 113, 1000 Ljubljana, Slovenia
hChair of Micro Process Engineering and Technology – COMPETE, University of Ljubljana, Večna pot 113, 1000 Ljubljana, Slovenia
iDUDECHEM GmbH, Köpenicker Str. 325, 12555 Berlin, Germany
First published on 17th November 2025
Abstract
In the face of intensifying market needs and mounting environmental pressures, the pharmaceutical and agrochemical sectors must revisit core aspects of process design. This review proposes a forward-looking framework for “greener-by-design” manufacturing, emphasizing the integration of sustainability from the earliest stages of synthetic planning through to industrial implementation. We focus on four interdependent levers that collectively enable this transformation: (i) solvent choice, with an emphasis on minimization, substitution, or complete elimination; (ii) substrate sourcing, favoring renewable and biomass-derived feedstocks to reduce fossil dependency; (iii) catalyst development, exploring the use of base metals, novel heterogeneous systems, and biocatalysts; and (iv) continuous-flow processing, which enhances safety, scalability, and process control. These strategies are not meant to be applied in isolation but rather in a synergistic, end-to-end manner that accounts for the full lifecycle of chemical products. By aligning synthetic efficiency with environmental responsibility, this review outlines a practical and actionable roadmap for the sustainable production of high-value fine chemicals. The convergence of synthetic chemistry with process engineering, data science, and life cycle thinking will be critical to realizing this vision, ultimately enabling more robust, circular, and future-proof manufacturing paradigms.
1. Introduction
The fine chemical industry, broadly defined by the production of high-value (>10 USD per kg), small- to medium-volume compounds, stands as a critical economic pillar of the European Union. It sustains over 1.2 million jobs and generates a turnover of 85 billion USD, with the lion's share originating from the pharmaceutical (55 billion USD) and agrochemical (15 billion USD) sectors.1 Pharmaceutical active ingredients are typically produced at relatively low volumes, often ranging from a few kilograms to several tens of tons per annum, reflecting their high value and targeted applications. On the other hand, many agrochemical ingredients, despite sharing comparable structural complexity and stringent purity requirements, are manufactured at volumes that can exceed several thousand tons per year. This positions agrochemicals at the interface between fine and performance chemicals. Globally, the pharmaceutical and agrochemical sectors are projected to expand at nearly 6% and 4% annually through 2030, driven by growing healthcare demands for chronic and infectious disease treatments, the expansion of lifestyle and personalized medicines, aging populations, and the need to enhance agricultural productivity and ensure food security for a rising global population.2 While this growth signals economic opportunities, it also raises substantial concerns around operational costs, resource consumption, and environmental impact.Specifically, the pharmaceutical sector contributes an estimated 4–5% of worldwide carbon emissions (Fig. 1),3 with its emission intensity in 2015 registering 55% higher than that of the automotive sector.4–6 It also generates significantly more waste per unit of product than the oil refining and bulk chemical industries.7,8 In comparison, the manufacture and use of crop protection products account for about 3% of the global warming potential (GWP = contribution to global warming in CO2 equivalent) of crop production and between 1–4% of the total carbon footprint of crop cultivation.9,10 These emissions, however, must be evaluated in the broader agroecosystem context, where crop protection products boost yields, reduce land use, and improve other inputs, thus partly offsetting their direct negative environmental impact. Nevertheless, recognizing the urgency to mitigate these effects, the industry is increasingly emphasizing decarbonization efforts. For instance, the European Fine Chemical Group (EFCG) advocates for financial incentives to encourage eco-friendly manufacturing in Europe, alongside international collaboration to minimize environmental harm.11 Beyond combating climate change, such measures are viewed by the European Commission and other governing bodies as a critical lifeline to revitalize the industry's competitiveness.12
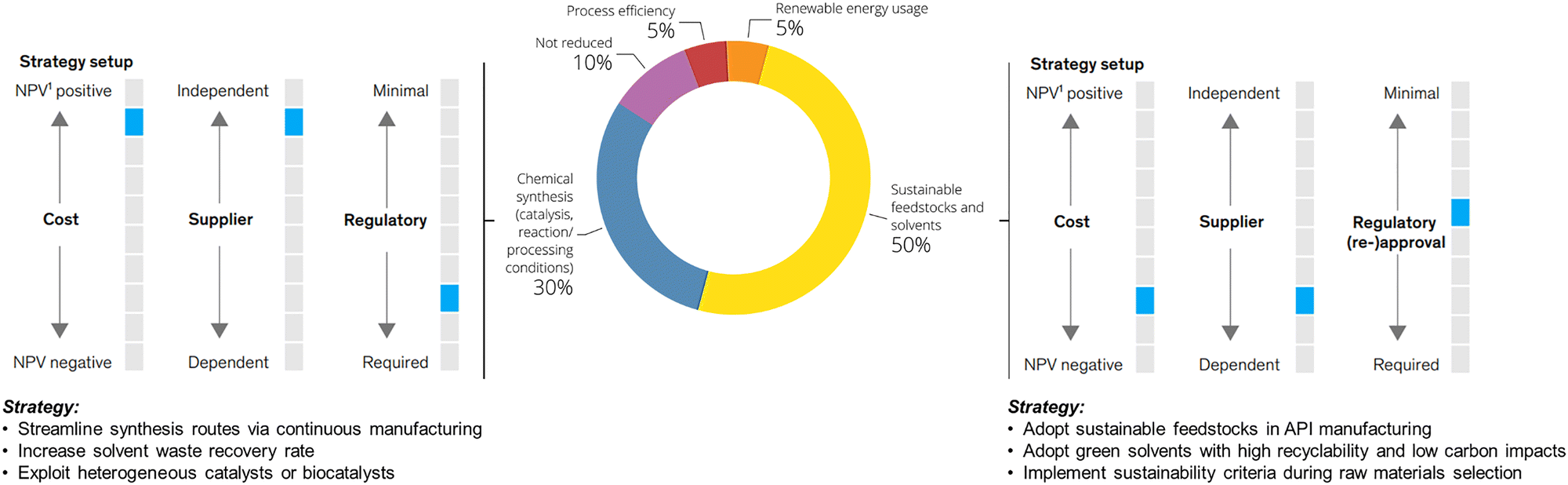 | ||
| Fig. 1 Breakdown of CO2 abatement potential in fine chemical manufacturing, as shown in the McKinsey & Company's analysis in ref. 3. The central donut chart illustrates the relative contribution of different strategies to the total achievable reduction in CO2 emissions by 2040. Approximately 80% of the abatement potential is attributed to core chemical strategies, including reaction optimization, green chemistry-based route redesign, adoption of sustainable solvents and feedstocks, and the use of heterogeneous or enzymatic catalysts. The remaining 10–20% comes from energy-related measures, including renewable electricity and low-carbon heating. Together, these levers highlight the central role of chemistry in achieving emission cuts. The diagrams on the left and right further illustrate that implementing such strategies can influence production costs, alter supplier dependencies, and introduce regulatory implications, especially when fundamental process parameters (e.g., solvent systems, reaction pathways) are modified. Nonetheless, when viewed together, these pathways show the strategic and science-driven opportunity to advance towards more sustainable fine chemical manufacturing. Adapted from ref. 3, with permission from McKinsey & Company, copyright 2025. | ||
Indeed, regulatory frameworks have progressively evolved to prioritize sustainability in pharmaceutical and agrochemical production. Central to this shift is green chemistry, a concept codified in the 12 Principles of Green Chemistry by Anastas and Warner in 1998.13 By promoting the reduction of hazardous substances and the use of safer, more sustainable alternatives, these principles align with global initiatives such as the United Nations Sustainable Development Goals.14 Notable policy milestones of this approach include the European Green Deal,15 the EU Chemicals Strategy for Sustainability toward a Toxic Free Environment,16 the Safe and Sustainable by Design (SSbD) framework,17 the US “Sustainable Chemistry Act”,18 and the Water Framework Directive,19 among others.
As a result of these intertwined economic, regulatory and environmental incentives, the fine chemical industry is undergoing a transformative shift toward more sustainable practices. A “greener-by-design” approach underpins this evolution, aiming for a circular economy through reduced chemical hazards, reduced pollution, and efficient resource utilization. However, it is important to acknowledge that reducing intrinsic chemical hazards can, in some cases, lead to unintended trade-offs, such as increased waste production, reduced atom economy, or diminished process efficiency. These potential drawbacks highlight the need for a holistic assessment of sustainability, ensuring that hazard reduction efforts do not undermine the overall environmental or economic performance of the process.
Today, according to a recent McKinsey & Company report, 50–70% of the top 20 Active Pharmaceutical Ingredients (API) manufacturers have set decarbonization targets, although fewer than 20% present detailed implementation plans encompassing abatement costs and feasibility trade-offs.3 Nevertheless, early evidence confirms the value of such initiatives. The US Environmental Protection Agency documented a 10% reduction in the emission of Toxics Release Inventory (TRI) chemicals in 2021 compared to 2012, with 89% of TRI chemical waste managed using best practices such as recycling, energy recovery, and treatment.16 Where crop protection is concerned a 40% reduction in the acute toxicity of crop protection products has been recorded since the 1960s, further illustrating a broader trend toward sustainability.20 Importantly, strong economic performance and environmental compliance need not be mutually exclusive. Between 2012 and 2015, Roche, Johnson & Johnson, and Amgen achieved a revenue growth of up to 27.2%, while reducing emissions by as much as 18.7%. Notably, Roche achieved the highest revenue increase and largest emissions reduction, suggesting that financial success and sustainable practices can go hand in hand.5 However, these environmental advancements should not be mistaken for corporate altruism; rather, they reflect growing governmental/regulatory, investor, and consumer pressure to meet increasingly stringent sustainability mandates and reduce production cost.
Beyond aggregate industry emissions, it is crucial to quantify the environmental impact of individual chemical processes. This can be achieved by using green metrics such as Process Mass Intensity (PMI) and the Environmental Factor (E-factor), which have become invaluable benchmarks due to their simplicity and broad applicability. Defined by straightforward mass-based calculations, these metrics are closely interrelated:
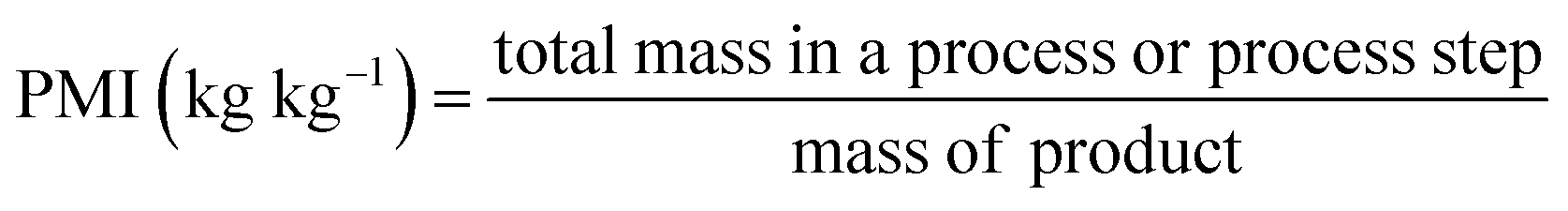 | (1) |
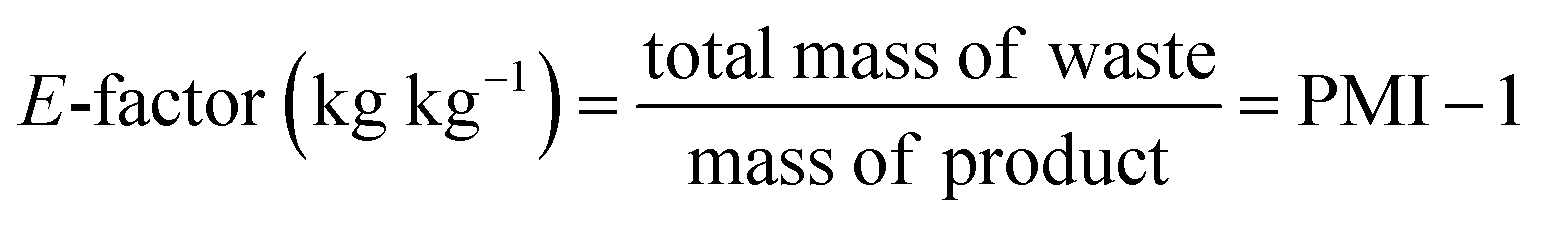 | (2) |
Variations of these metrics exist to account for by-products and recycled reactants that are not ultimately categorized as waste.21 Owing to their simplicity and accessibility, both PMI and E-factor have been instrumental in raising awareness and guiding greener practices in academic and industrial settings; as such, they will be referenced extensively throughout this review. Nonetheless, caution is required when interpreting these metrics22 as they inherently do not account for: (i) the nature (e.g., health and safety profiles) of reactants, reagents, catalysts, and solvents; (ii) energy consumption; (iii) the specifics of work-up procedures and (iv) the difference in molecular mass of products.23 Furthermore, the choice of system boundaries (i.e., the defined starting point) can significantly skew PMI and E-factor outcomes. In short, while these metrics serve as useful first approximations, they often fall short of capturing the full environmental complexity of a process. In this context, life cycle assessment delivers a far more complete picture by accounting for upstream and downstream impacts that are invisible to mass-based metrics alone,24 revealing trade-offs and blind spots in sustainability assessments. This is particularly critical in the pharmaceutical and agrochemical industry, where high solvent use, inherently complex syntheses, and energy-intensive purification steps continue to obstruct the path toward genuinely green manufacturing.
Despite the strides in implementing green chemistry principles, pharmaceutical and agrochemical industries still contribute substantially to climate change.25,26 To continue down the road to greener-by-design pharmaceuticals and agrochemicals, heightened emphasis must be placed on active ingredients, that is the bioactive components of drugs and crop chemicals which are a prime pollution hotspot. For instance, API synthesis typically accounts for about 25% of total emissions,3 but this number can rise to 60–90% for certain molecules.27
In large part, this environmental burden hinges on four core synthetic variables: the solvent, starting material, catalyst, and reaction/processing conditions. These collectively govern not only the chemical feasibility of scaling a transformation but also its sustainability profile. Targeted changes in these four pillars can drastically reduce the environmental impact of a process. For instance, a life cycle analysis conducted by GSK revealed that solvents constitute 70% of their total waste streams,28 demonstrating the potential for sweeping improvements, in line with the core principles of Green Chemistry.
A major barrier for manufacturers is the absence of a single, holistic guide outlining decarbonization solutions and their benefits, compounded by limited transparency into product-specific carbon footprints.29 This review aims to bridge that gap by consolidating recent progress in green chemistry and manufacturing technologies. In particular, the review explores how optimizing solvent selection, transitioning to renewable feedstocks, designing next-generation catalysts, and implementing continuous processing techniques could collectively drive the industry toward sustainability.
Solvent choice is the most immediate and impactful lever for improving the sustainability of chemical processes in the fine chemicals industry, making it a critical factor in reducing their environmental footprint. Solvents are the unsung enablers of synthetic transformations, by solubilizing reactants/reagents/catalysts, dictating reaction rates and selectivity, and assisting in purification, isolation, and scalability. Yet, they also account for a staggering proportion of chemical waste, often exceeding the mass of the desired product by a significant margin.30 Shifting to greener alternatives (be they lower toxicity organic solvents or solvent recycling methods, alternative media such as supercritical fluids and water-based systems or even solvent-free approaches) has the potential to radically decrease both waste generation and energy consumption. In Section 2, we will assess how innovative solvent strategies align with Green Chemistry principles to reshape the sustainability of chemical processes.
Equally transformative is the transition from fossil-based to renewable feedstocks. Petroleum and natural gas have historically provided the backbone of pharmaceutical and chemical synthesis,31 while abundant, renewable alternatives remain largely untapped. Biomass-derived starting materials (e.g., from lignocellulosic pathways), open the door to a circular chemical economy that repurposes non-edible waste streams into valuable intermediates. Hence, in Section 3, we discuss how this shift can enable defossilization while creating more resilient and diversified supply chains.
At the heart of organic synthesis lies catalysis, nature's own strategy for achieving complex reactions under mild conditions. Be it through base metals such as iron and copper, the design of highly selective heterogeneous catalysts, or tailored enzymes, next-generation catalysts are redefining synthetic methodologies and capabilities. Section 4 surveys cutting-edge catalytic development, from single-atom catalysts to enzyme-mimetic systems, and their potential implications for sustainable chemical manufacturing.
Beyond chemical inputs, one cannot forget the physical equipment within which reactions are conducted. Long has been the reign of batch reactors in the pharmaceutical and agrochemical sectors, despite their inherent constraints in process control and resource use.32 In Section 5, we illustrate how continuous-flow systems can not only address these limitations by providing on-demand rather than campaign-based manufacturing, but also allow the integration of emerging green technologies like photo- and electrocatalysis, heralding a new era of manufacturing in the pharmaceutical and agrochemical sectors.
Looking past these core synthetic frontiers, the broader landscape of fine chemicals is also transitioning to novel manufacturing technologies to streamline operations, reduce waste, and enhance product quality in real time. Innovations in smart manufacturing, process analytical technologies (PAT), and 3D printing serve as complementary tools that strengthen the “four pillars” of synthetic design. A companion perspective (Part 2: Technological Frontiers) examines these technological frontiers in greater depth, assessing their potential to revolutionize drug and agrochemical production, while upholding the principles of green chemistry and engineering.
Overall, the present perspective serves two key objectives: first, to map out existing strategies and innovations that foster greener synthetic routes across the fine chemical industry; and second, to highlight emerging research and practices that might define the future of pharmaceutical and agrochemical manufacturing. In the following sections, we take a closer look into each of the four pillars, focusing on the most promising developments, implementation challenges, and real-world opportunities. Finally, supported by insights from industry experts, we offer a critical outlook on the steps necessary to accelerate the transition toward more sustainable, efficient, and scalable manufacturing processes.
2. Solvents in fine chemical manufacturing
Solvents are an essential component of conventional chemical reactions where they dissolve reactants, facilitate reaction kinetics, and drive purification processes.33,34 Yet, their widespread use represents one of the most significant contributors to environmental burden and process inefficiency in fine chemical manufacturing. Recent analyses estimate that up to 182 kg of waste is generated per kilogram of active ingredient, largely due to solvent-intensive reactions, purification steps, and process inefficiencies.35 This corresponds to the fact that solvents can constitute between 47% and 93% of the total mass input in API synthesis, depending on the synthetic route.36 A striking example can be seen in oligonucleotide synthesis, which requires repeated wash cycles and solvent-heavy purification, resulting in exceptionally high solvent consumption.37 Because isolation and purification depend so heavily on solvents, these steps are a critical part of the sustainability equation. Herein, column chromatography is acknowledged as a particularly solvent-heavy and inefficient purification method, yet alternatives such as repeated crystallization, liquid–liquid extraction, and azeotropic drying also require substantial solvent volumes to meet stringent quality and regulatory standards.Within this context, solvent recycling is a pivotal strategy, not only in minimizing waste and emissions, but also in enhancing process efficiency. Increasing attention is therefore being directed toward selecting solvents that are both effective for the intended chemical transformation and compatible with industrial recovery and reuse methods. While many solvents are recyclable in theory, their practical suitability is governed by properties such as volatility, thermal stability, separation ease, and resistance to contamination. These attributes ultimately determine the feasibility of incorporating solvent recovery into greener-by-design manufacturing. Given the complexity of solvent recovery and its deep integration with process chemistry, a comprehensive discussion falls beyond the scope of this review. Nevertheless, selected aspects related to solvent recovery and reuse will be revisited in the Conclusion and Outlook section, to highlight their relevance in the broader framework of sustainable process chemistry.
The constraints associated with product purification alongside solvent recycling make it more practical, and in many cases more effective, to reduce or eliminate solvent use altogether, thereby circumventing the need for downstream recovery. A range of strategies are currently being explored to achieve this objective. One of the most straightforward involves replacing traditional organic solvents, such as dichloromethane (DCM), tetrahydrofuran (THF), dimethylformamide (DMF), and dimethyl sulfoxide (DMSO), with greener alternatives. These substitutions can be integrated into both established and new synthetic routes. More transformative strategies aim to eliminate organic solvents entirely by using supercritical fluids, particularly supercritical CO2, or aqueous micellar media. In this context, mechanochemistry is also gaining traction as a promising route to solvent-free synthesis, although questions remain regarding safety, scalability, and product purification. In Section 2.2, we outline solvent reduction strategies in order of increasing sustainability benefit.
2.1. Solvent selection
As environmental goals must be balanced against process efficiency and reliability, selecting an appropriate solvent minimization strategy can be both time-consuming and resource-intensive. Thus, the first step is to critically assess the environmental impact of candidate solvents. A systematic approach to rank solvent greenness has advanced significantly over the past decade, particularly within the pharmaceutical industry, where solvents can represent a large fraction of API production mass.38 Numerous organizations, including GSK,39 Pfizer,40 Sanofi,41 and ACS Green Chemistry Initiative (ACS GCI) (based on a prior AstraZeneca guide),42 have developed solvent selection guides that evaluate health, safety, and environmental impact.43 These guides commonly employ “traffic light” color-coding to highlight preferred or discouraged solvents. However, these guides often use arbitrary thresholds and criteria for color-coding and are not linked to regulatory frameworks.To address these shortcomings, the CHEM21 consortium, an industry-academic alliance,44 introduced a more flexible solvent selection methodology aligned with the Global Harmonized System (Table 1).45 By adapting insights from the existing guides, CHEM21 developed a three-tiered Environment, Health, and Safety (EHS) assessment for each solvent. Although minor discrepancies surfaced, the new guide achieved broad consensus. CHEM21's open-access tool uses a harmonized color-coding system that emphasizes a solvent's “least green” attribute rather than averaging its properties. Collectively, these attributes make CHEM21's guide the leading solvent selection reference to date.45
| Family | Solvent | BP (°C) | FP (°C) | Worst H3xx | Worst H4xx | Safety score | Health score | Env. score | Ranking by default | Ranking after discussion |
|---|---|---|---|---|---|---|---|---|---|---|
| Water | Water | 100 | n/a | None | None | 1 | 1 | 1 | Recommended | Recommended |
| Alcohols | MeOH | 65 | 11 | H301 | None | 4 | 7 | 5 | Problematic | Recommended |
| EtOH | 78 | 13 | H319 | None | 4 | 3 | 3 | Recommended | Recommended | |
| i-PrOH | 82 | 12 | H319 | None | 4 | 3 | 3 | Recommended | Recommended | |
| n-BuOH | 118 | 29 | H318 | None | 3 | 4 | 3 | Recommended | Recommended | |
| t-BuOH | 82 | 11 | H302 | None | 4 | 3 | 3 | Recommended | Recommended | |
| Benzyl alcohol | 206 | 101 | H302 | None | 1 | 2 | 7 | Problematic | Problematic | |
| Ethylene glycol | 198 | 116 | H319 | None | 1 | 2 | 5 | Recommended | Recommended | |
| Ketones | Acetone | 56 | −18 | H319 | None | 5 | 3 | 5 | Problematic | Recommended |
| MEK | 80 | −6 | H319 | None | 5 | 3 | 3 | Recommended | Recommended | |
| MIBK | 117 | 13 | H332 | None | 4 | 2 | 3 | Recommended | Recommended | |
| Cyclohexanone | 156 | 43 | H302 | None | 3 | 2 | 5 | Recommended | Problematic | |
| Esters | Methyl acetate | 57 | −10 | H319 | None | 5 | 3 | 5 | Problematic | Problematic |
| Ethyl acetate | 77 | −4 | H319 | None | 5 | 3 | 3 | Recommended | Recommended | |
| i-PrOAc | 89 | 2 | H336 | None | 4 | 2 | 3 | Recommended | Recommended | |
| n-BuOAc | 126 | 22 | H302 | None | 4 | 2 | 3 | Recommended | Recommended | |
| Ethers | Diethyl ether | 34 | −45 | H336 | None | 10 | 3 | 7 | Hazardous | HH |
| Diisopropyl ether | 69 | −28 | H315 | None | 9 | 3 | 5 | Hazardous | Hazardous | |
| MTBE | 55 | −28 | H351 | None | 8 | 3 | 5 | Hazardous | Hazardous | |
| CPME | 106 | −1 | H302 | H412 | 7 | 2 | 5 | Problematic | Problematic | |
| THF | 66 | −14 | H318 | None | 6 | 7 | 5 | Problematic | Problematic | |
| MeTHF | 80 | −11 | H351 | None | 6 | 5 | 3 | Problematic | Problematic | |
| 1,4-Dioxane | 101 | 12 | None | None | 7 | 6 | 3 | Problematic | Hazardous | |
| Anisole | 154 | 52 | H360 | None | 4 | 1 | 5 | Problematic | Recommended | |
| DME | 85 | −6 | H304 | None | 7 | 10 | 3 | Hazardous | Hazardous | |
| Hydrocarbons | Pentane | 36 | −40 | H361 | H411 | 8 | 3 | 7 | Hazardous | Hazardous |
| Hexane | 69 | −22 | H304 | H411 | 8 | 7 | 7 | Hazardous | Hazardous | |
| Heptane | 98 | −4 | H304 | H410 | 6 | 2 | 7 | Problematic | Problematic | |
| Cyclohexane | 81 | −17 | H304 | H410 | 6 | 3 | 7 | Problematic | Problematic | |
| Me-cyclohexane | 101 | −4 | H350 | H411 | 6 | 2 | 7 | Problematic | Problematic | |
| Benzene | 80 | −11 | H351 | None | 6 | 10 | 3 | Hazardous | HH | |
| Toluene | 111 | 4 | H312 | None | 5 | 6 | 3 | Problematic | Problematic | |
| Xylenes | 140 | 27 | H351 | None | 4 | 2 | 5 | Problematic | Problematic | |
| Halogenated | DCM | 40 | n/a | H351 | None | 1 | 7 | 7 | Hazardous | Hazardous |
| Chloroform | 61 | n/a | H351 | None | 2 | 7 | 5 | Problematic | HH | |
| CCl4 | 77 | n/a | H350 | H420 | 2 | 7 | 10 | Hazardous | HH | |
| DCE | 84 | 13 | H332 | None | 4 | 10 | 3 | Hazardous | HH | |
| Chlorobenzene | 132 | 29 | H319 | H411 | 3 | 2 | 7 | Problematic | Problematic | |
| Aprotic polar | Acetonitrile | 82 | 2 | H360 | None | 4 | 3 | 3 | Recommended | Problematic |
| DMF | 153 | 58 | H360 | None | 3 | 9 | 5 | Hazardous | Hazardous | |
| DMAc | 166 | 70 | H360 | None | 1 | 9 | 5 | Hazardous | Hazardous | |
| NMP | 202 | 96 | H360 | None | 1 | 9 | 7 | Hazardous | Hazardous | |
| DMPU | 246 | 121 | H361 | None | 1 | 6 | 7 | Problematic | Problematic | |
| DMSO | 189 | 95 | None | None | 1 | 1 | 5 | Recommended | Problematic | |
| Sulfolane | 287 | 177 | H360 | None | 1 | 9 | 7 | Hazardous | Hazardous | |
| HMPA | >200 | 144 | H350 | None | 1 | 9 | 7 | Hazardous | HH | |
| Nitromethane | 101 | 35 | H302 | None | 10 | 2 | 3 | Hazardous | HH | |
| Miscellaneous | Methoxy-ethanol | 125 | 42 | H360 | None | 3 | 9 | 3 | Hazardous | Hazardous |
| Carbon disulfide | 46 | −30 | H361 | H412 | 9 | 7 | 7 | Hazardous | HH | |
| Acids | Formic acid | 101 | 49 | H314 | None | 3 | 7 | 3 | Problematic | Problematic |
| Acetic acid | 118 | 39 | H314 | None | 3 | 7 | 3 | Problematic | Problematic | |
| Ac2O | 139 | 49 | H314 | None | 3 | 7 | 3 | Problematic | Problematic | |
| Amines | Pyridine | 115 | 23 | H302 | None | 4 | 2 | 3 | Recommended | Hazardous |
| NEt3 | 89 | −6 | H314 | None | 6 | 7 | 3 | Problematic | Hazardous | |
The above selection guides rank solvents according to HSE factors, regulatory compliance, physicochemical properties, and sustainability metrics. While useful for initial screening, these static rankings often fail to account for context-specific trade-offs and real-world process considerations. Dynamic tools address these limitations by enabling a more context-sensitive evaluation of solvents, integrating both environmental performance and process requirements. Herein, LCA plays a central role, offering a detailed view of environmental burdens across the entire product lifecycle, from raw material sourcing to distribution, use, and disposal.
The practical value of LCA in solvent selection is illustrated by two contrasting studies of the Suzuki–Miyaura coupling. In the first report, Sherwood et al. concluded that the reaction is too tolerant of solvent variation to serve as a meaningful screening platform, a view derived mainly from the similarities they observed in isolated yields.46 By contrast, Yamaki et al. applied a life-cycle perspective to evaluate five solvents (i.e., NMP, toluene, MEK, IPA, and EtOAc) based on production cost, CO2 emissions, and overall process efficiency.47 Their findings identified EtOAc as the optimal choice among those studied, offering a 67% reduction in production costs and a 70% decrease in CO2 emissions compared to NMP-based protocols. Crucially, the study incorporated process simulations to assess the impact of azeotropes and phase behavior on solvent recovery, achieving efficiencies of up to 85%. In summary, Yamaki's broader LCA-based analysis demonstrated that solvent choice can lead to substantial improvements in both cost and environmental impact when assessed over the full process lifecycle rather than based on yields alone.
Nevertheless, it must be emphasized that the effectiveness of LCA is often constrained by the availability of emission factors (EFs), which can differ significantly according to production methods, process efficiencies, and regional energy sources. This challenge is compounded by the lack of standardized EF values for most solvents, making carbon footprint evaluations inconsistent. For instance, reported EFs for acetonitrile range from 1.5 to 12.5 kg of CO2 per kg of solvent, depending on the synthesis route (Fig. 2). The absence of universally accepted EF databases and standardized protocols for Product Carbon Footprint (PCF) reporting further hampers efforts to establish transparent and comparable sustainability metrics across the pharmaceutical supply chain. Hence, it is hard to track and optimize the emissions associated to specific solvents hindering progress toward net-zero targets.3
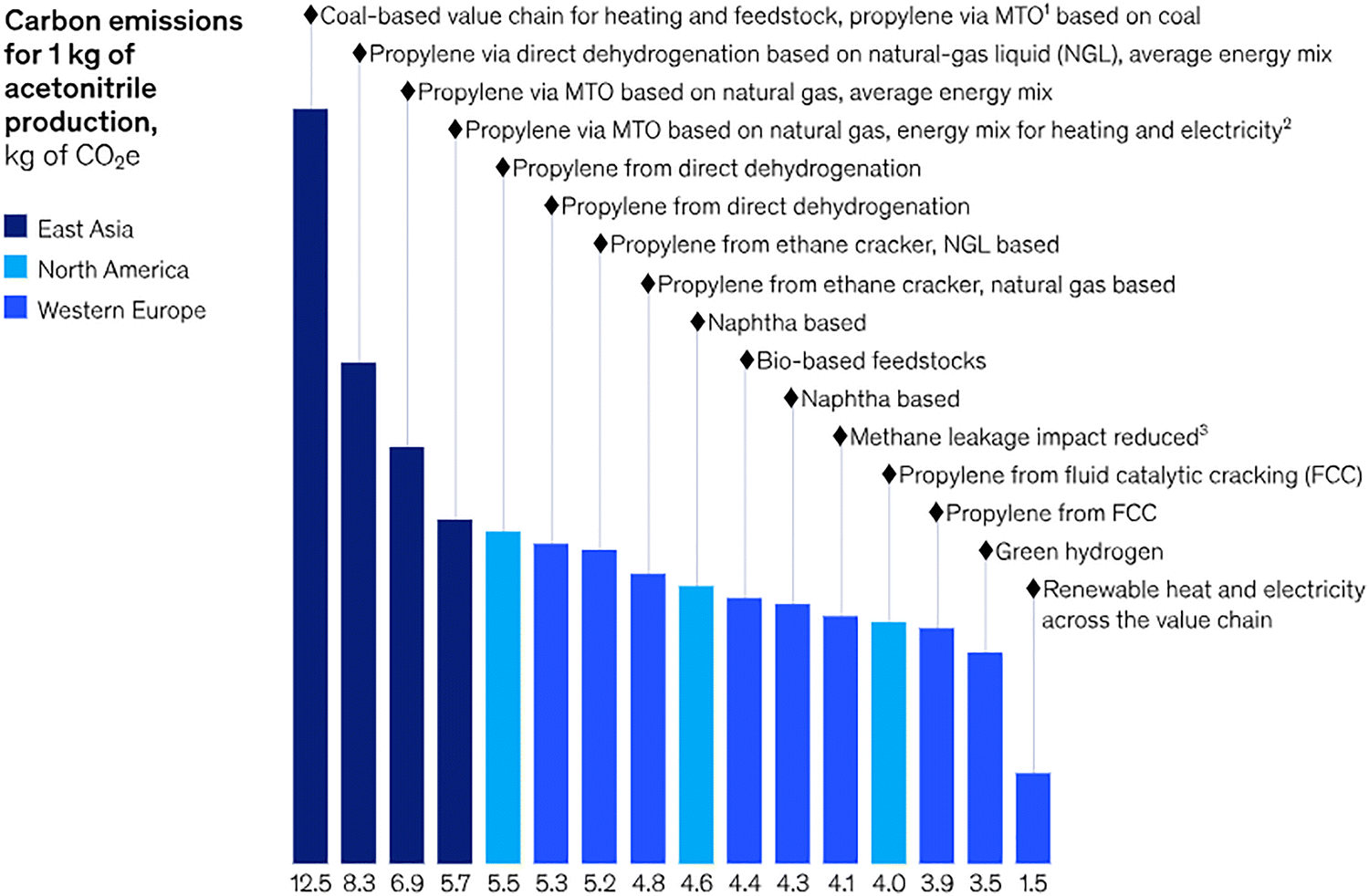 | ||
| Fig. 2 Carbon emissions associated with the use of 1 kg of acetonitrile, shown as a function of the upstream production route and feedstock source. The variability in greenhouse gas emissions, just for a single common solvent, illustrates the complexity API and fine chemical manufacturers face when conducting robust life cycle assessments: the carbon footprint of an input material is highly sensitive to regional energy mixes, reactor configurations, feedstock origins, leakages in gas supply chains, and the integration of renewable resources. Such variability complicates efforts to quantify Scope 3 emissions and to make evidence-based sustainability decisions. Reproduced from ref. 3, with permission from McKinsey & Company, copyright 2025. | ||
Beyond the limitations associated with LCA, economic and regulatory barriers further hamper the transition to greener alternatives. Although regulatory bodies like the EMA and FDA endorse the selection of eco-friendly solvents, in line with the EU's “Strategic Approach to Pharmaceuticals in the Environment”,48 any solvent change is classified as a major alteration. This designation necessitates costly new regulatory approvals and validations, deterring broader adoption of bio-based solvents. The challenge is compounded by the fact that many solvents have not been integrated into FDA classes or solvent selection guides.45 Moreover, most APIs and agrochemicals are produced via multistep synthetic sequences that require solvent mixtures or sequential solvent changes. However, selection guides such as CHEM21 are based on the properties of pure solvents. To address this gap, recent work by Peper et al. introduced separation feasibility indicators that quantify the effort involved in solvent swaps by accounting for the actual thermodynamic behavior of binary solvent systems.49 This approach enables more accurate predictions of solvent loss, energy requirements, and the number of separation steps needed.
Despite their limitations, the CHEM21 and similar guides have been undeniably successful in broadly steering both academia and industry toward more sustainable solvent choices, which has in turn spurred the development of various tools to simplify solvent impact assessment. For instance, electronic laboratory notebooks have been reported with built-in green assessments which are aligned to CHEM21 criteria.50 These systems use flashcards to provide immediate visualization and comparison of a solvent's environmental performance and physical attributes.51 While undoubtedly a step in the right direction, these tools do not recommend alternative solvents tailored to specific reactions or manufacturing processes.
To bridge this gap, additional solvent-selection tools have been introduced. One of these tools uses Principal Component Analysis (PCA), to simplify complex solvent datasets and identify greener substitutes with comparable solvation properties by highlighting key physicochemical differences.42 The Selection and Substitution Software (SUSSOL) takes this a step further by employing a neural network (Kohonen self-organizing map) to cluster solvents according to 22 physical properties (e.g., molar volume, boiling point, viscosity, surface tension). It then ranks viable replacements based on their HSE scores.52 While these tools can be valuable, simply matching physical parameters does not always capture critical factors such as solubility or solvent–solute interactions that can ultimately influence yield, selectivity, and reaction kinetics.
The rise of artificial intelligence (AI) and machine learning (ML) can take into consideration these factors to further refine solvent selection.53–55 Chung and Green, for example, constructed a framework utilizing the Conductor-like Screening Model for Real Solvents (COSMO-RS), a quantum-chemical model that predicts thermodynamic properties of molecules in solution. The framework was thus trained on over 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 neutral reactions across 295 solvents to accurately predict solvent effects on reaction kinetics.56 Their experimentally validated method facilitates rapid, reliable solvent screening and streamlined kinetic modelling. Similarly, Zhang et al. applied active learning to train machine learning potentials (MLP) for reaction mechanism modelling in explicit solvents, using a Diels–Alder reaction on water and methanol as a proof-of-concept.57 While this study highlights the potential of AI/ML to capture solute–solvent interactions in unprecedented detail, its scope is currently narrow. Limitations include the scarcity of high-quality reaction–solvent datasets, the difficulty of encoding sustainability metrics, and the high computational cost of generating training data. Broader implementation will depend on addressing these challenges and expanding curated datasets. Beyond solvent selection, AI and ML are reshaping other facets of chemical R&D, a topic examined more thoroughly in the companion review on emerging technological frontiers.
000 neutral reactions across 295 solvents to accurately predict solvent effects on reaction kinetics.56 Their experimentally validated method facilitates rapid, reliable solvent screening and streamlined kinetic modelling. Similarly, Zhang et al. applied active learning to train machine learning potentials (MLP) for reaction mechanism modelling in explicit solvents, using a Diels–Alder reaction on water and methanol as a proof-of-concept.57 While this study highlights the potential of AI/ML to capture solute–solvent interactions in unprecedented detail, its scope is currently narrow. Limitations include the scarcity of high-quality reaction–solvent datasets, the difficulty of encoding sustainability metrics, and the high computational cost of generating training data. Broader implementation will depend on addressing these challenges and expanding curated datasets. Beyond solvent selection, AI and ML are reshaping other facets of chemical R&D, a topic examined more thoroughly in the companion review on emerging technological frontiers.
2.2. Solvent substitution
Drawing on the detailed evaluations from the aforementioned guides, harmful organic solvents (e.g., DCM, THF, and DMF) can be phased out to enhance both sustainability and drug safety. Indeed, solvent choice in pharmaceutical manufacturing greatly influences drug impurity profiles, as residual solvents can generate toxic, carcinogenic, or genotoxic byproducts. For instance, widely used polar aprotic solvents such as DMF and NMP may decompose to generate amines that, in the presence of nitrosating agents, can form N-nitrosodimethylamine (NDMA) and related compounds, which are carcinogenic.58 This risk is amplified during solvent recovery and reuse, particularly when control over purification processes is inadequate.59 Factors such as insufficient cleaning, cross-contamination between solvent streams, and poor coordination with third-party recovery facilities have all been identified as contributing factors. Consequently, replacing these solvents with greener alternatives offers a dual benefit: reducing environmental impact while also minimizing the formation of harmful impurities. Among the various strategies available, substituting high-risk solvents with bio-based or non-toxic alternatives is perhaps the most straightforward. These alternatives are primarily selected to match the physical properties of existing solvents, minimizing process modification, though AI and ML are expected to broaden the selection criteria. As of June 2025, a search of the Merck Greener Alternatives website lists 60 products spanning more than 25 solvents, with continued growth.60 Among these, solvents derived from renewable biomass are particularly compelling. This section highlights a selection of such solvents as representative case studies, illustrating their ability to reduce solvent use and waste in pharmaceutical and agrochemical applications. For a more comprehensive overview, readers are directed to dedicated literature on the topic.43,61–63400 L to 24000 L per kg of drug, and served as a dehydrating agent to prevent the formation of a trans by-product.
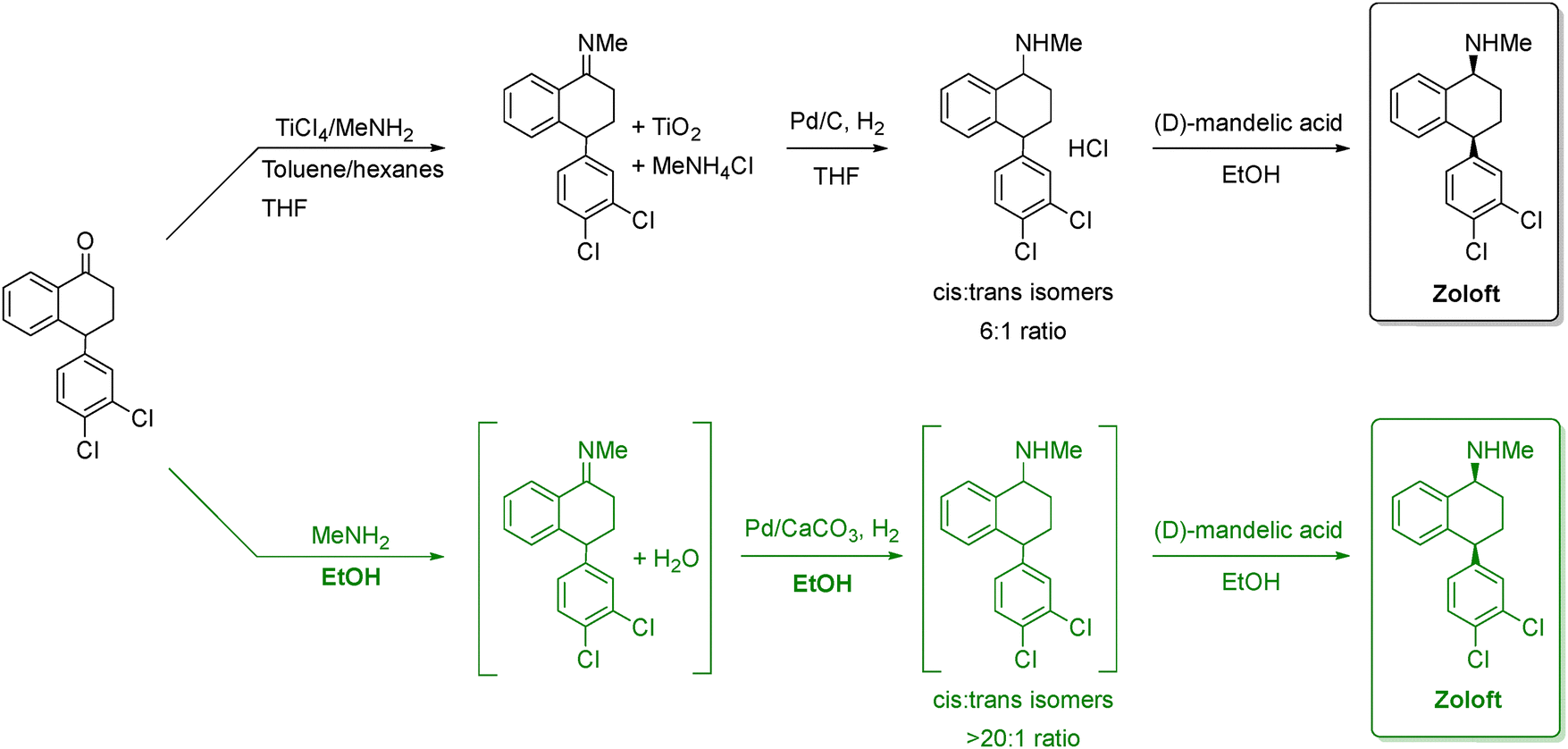 | ||
| Scheme 1 Comparison between the old and new synthetic routes to manufacture zoloft. The black route represents the traditional TiCl4-mediated reductive amination followed by Pd/C hydrogenation, which produces a modest cis:trans selectivity (6:1). The green route shows the improved process using MeNH2 in ethanol and Pd/CaCO3 hydrogenation, which avoids titanium reagents and achieves much higher stereoselectivity (>20:1). Both routes conclude with resolution using (D)-mandelic acid to give the expected drug. Adapted from ref. 66. | ||
More recently, Janssen Pharmaceuticals replaced THF with ethanol in the total synthesis of a respiratory syncytial virus drug candidate (Scheme 2, top), enabling two steps to be telescoped and eliminating an intermediate crystallization.67 Ethanol was also successfully adopted in the continuous-flow synthesis of norketamine, further showcasing its versatility as a sustainable alternative to halogenated solvents (Scheme 2, bottom).68 Importantly, LCA conducted on the production of benzophenone, a valuable API precursor, indicated that ethanol has a lower environmental impact than both conventional solvents (DCM and EtOAc) and deep eutectic alternatives, another proposed class of green solvents.69
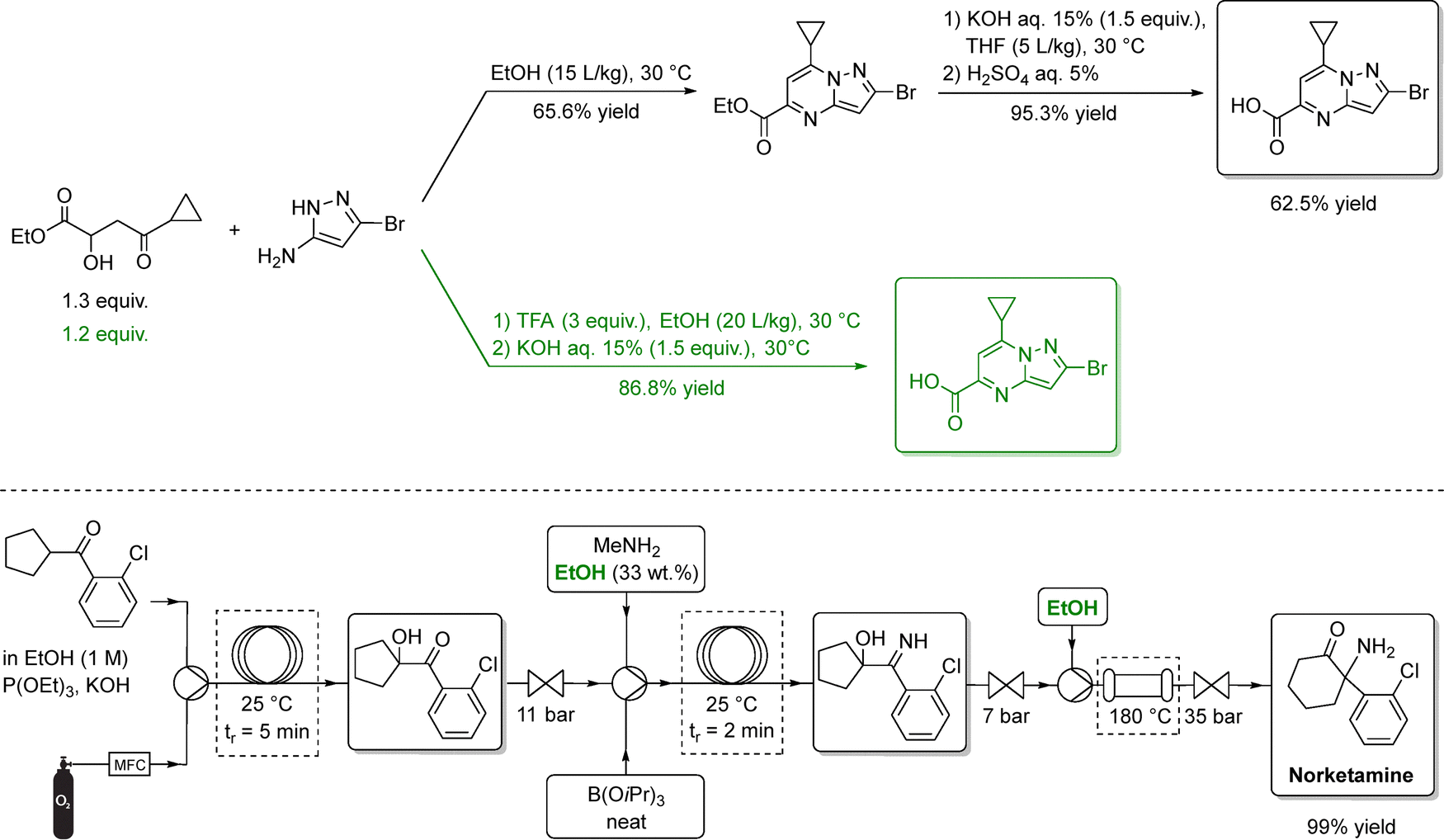 | ||
| Scheme 2 Synthesis of two selected APIs (specifically, an antiviral candidate above, and norketamine below) using ethanol as solvent. In the upper sequence, the black pathways depict the standard synthetic routes, while the green steps highlight optimized, ethanol-based transformations. In the bottom, ethanol serves as the main reaction medium for the amination step and several downstream operations. In both examples, EtOH enables cleaner reactions to afford the product in high isolated yield. Adapted from ref. 67 and 68, respectively. | ||
In addition to these environmental and process-related benefits, ethanol also illustrates how solvent selection can be shaped by regulatory constraints. For example, in Germany, the use of denatured ethanol is mandated in industrial fine chemical plants to circumvent the taxation associated with consumable-grade alcohols. This regulatory requirement necessitates continuous monitoring of distillate quality to ensure compliance with safety and purity standards. Furthermore, the denaturant itself may not be an inert additive, since some chemical agents can participate in or interfere with the reaction, potentially influencing yields, selectivity, or downstream processing. It is important at this point to clearly distinguish between denatured ethanol, which contains deliberate additives and hydrous ethanol which is simply the ∼95% ethanol–water azeotrope obtained by biomass fermentation and distillation.65 These considerations on denatured ethanol highlight the multifaceted nature of solvent selection, where regulatory, environmental, and chemical factors must be simultaneously balanced to enable truly sustainable manufacturing.
For instance, in the organometallic synthesis of tramadol (Scheme 3), substitution of conventional ethereal solvents with 2-MeTHF increased yields by roughly 10% and improved the cis/trans isomer ratio.76,77 The enhanced performance of 2-MeTHF in Grignard reactions is frequently attributed to its suppression of Wurtz couplings.78 In another study, using 2-MeTHF instead of THF enabled a scalable continuous synthesis of Grignard reagents with reduced side-product formation.79 However, it should be noted THF outperforms 2-MeTHF for certain organozinc analogues.80 More recently, Caravez et al. reported a two-step synthesis of a key intermediate for anti-HIV drug Lenacapavir using 2-MeTHF in both the Grignard reaction and the subsequent formation of an organometallic reagent.81
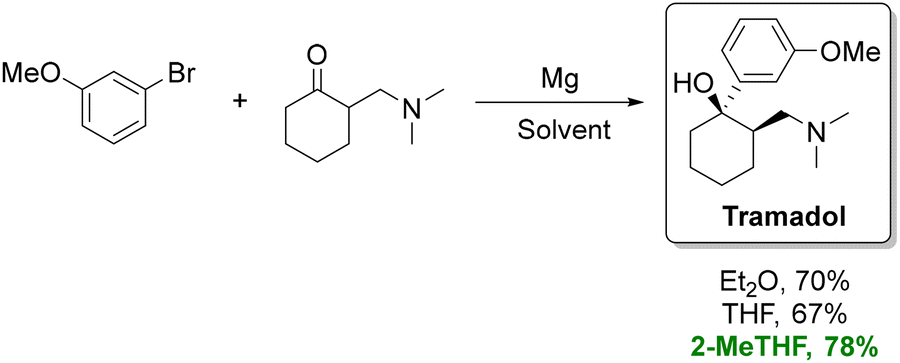 | ||
| Scheme 3 Synthesis of tramadol using Et2O, THF, and 2-MeTHF as solvents. The route performed in 2-MeTHF provides a more sustainable alternative to petroleum-derived ethers, delivering a higher isolated yield under the same reaction sequence. Adapted from ref. 77. | ||
Building on its aptitude for organometallic synthesis and high basicity, Bisz and Szostak developed an iron-catalyzed cross-coupling reaction between a Grignard reagent and aryl chlorides/tosylates. The C–C coupled products were attained in good to excellent yields within 10 min, including an intermediate for drug candidate AZD656424, a challenging target with a sterically hindered neopentyl nucleophile and sensitive ester moiety.82 Additional C–C coupling applications have been demonstrated via palladium and nickel-catalyzed Suzuki–Miyaura cross-couplings.83–85
More recently, Bhukta et al. reported that 2-MeTHF can also mediate C(sp)–H functionalization (Scheme 4).86 In their study, alkynes and anilines were combined in the presence of t-BuONO to synthesize diaryl 1,2-diketones, including a gram-scale synthesis of benzil, a precursor to the heart medication trifenagrel. Solvent screening revealed that 2-MeTHF afforded an 87% yield, whereas other solvents typically produced yields below 50%. They also reported an E-factor of 12 without considering solvent recycling, and noted that 2-MeTHF could be reused up to five times with an average 5% loss per cycle, further reducing solvent waste.
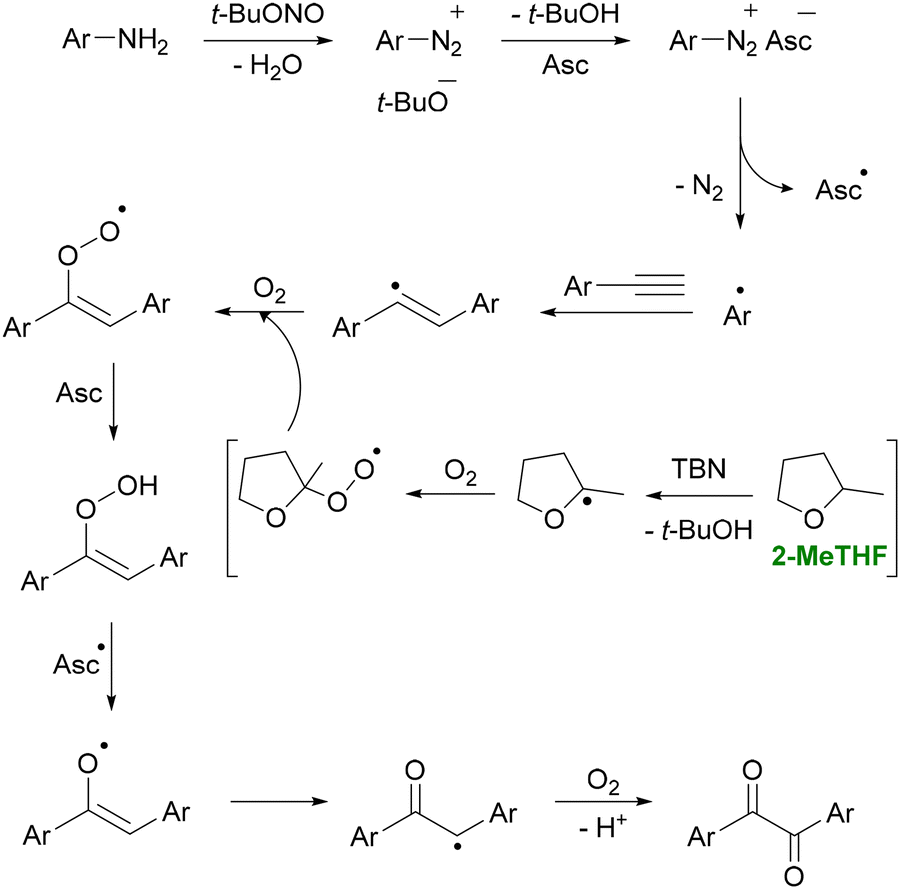 | ||
| Scheme 4 Synthesis of diaryl 1,2-diketones from anilines in 2-MeTHF. The figure shows the in situ formation of aryldiazonium species from anilines. Then, t-BuONO generates aryl radicals that add to alkynes, followed by oxygen-driven oxidation steps leading to diaryl 1,2-diketones. 2-MeTHF provides an optimal medium for this radical-oxygenation cascade, supporting efficient radical propagation and enabling the highest conversion (87% yield) among solvents screened for this transformation. Adapted from ref. 86. | ||
2-MeTHF has also been employed in the total synthesis of antihistamine dimethindene (Fenistil).87 In this case, 2-MeTHF replaced problematic toluene and hazardous diethyl ether in three out of four steps, improving the overall four-step yield from 10% to 21% and reducing the E-factor from 188 to 126.
Although continuous-flow synthesis will be discussed in more detail in Section 5, the use of 2-MeTHF in such setups can reduce hazardous waste. Bedard et al. reported a continuous-flow synthesis of diazepam through amidation followed by ammonia-induced cyclization using 2-MeTHF as the solvent (Scheme 5, top).88 2-MeTHF was selected for its suitable solubility profile, chemical inertness, and immiscibility with water, enabling efficient in-line aqueous extraction. As a result, the E-factor for diazepam synthesis dropped fourfold, from 36 to 9, with the authors noting that 2-MeTHF's importance in both reaction and extraction.
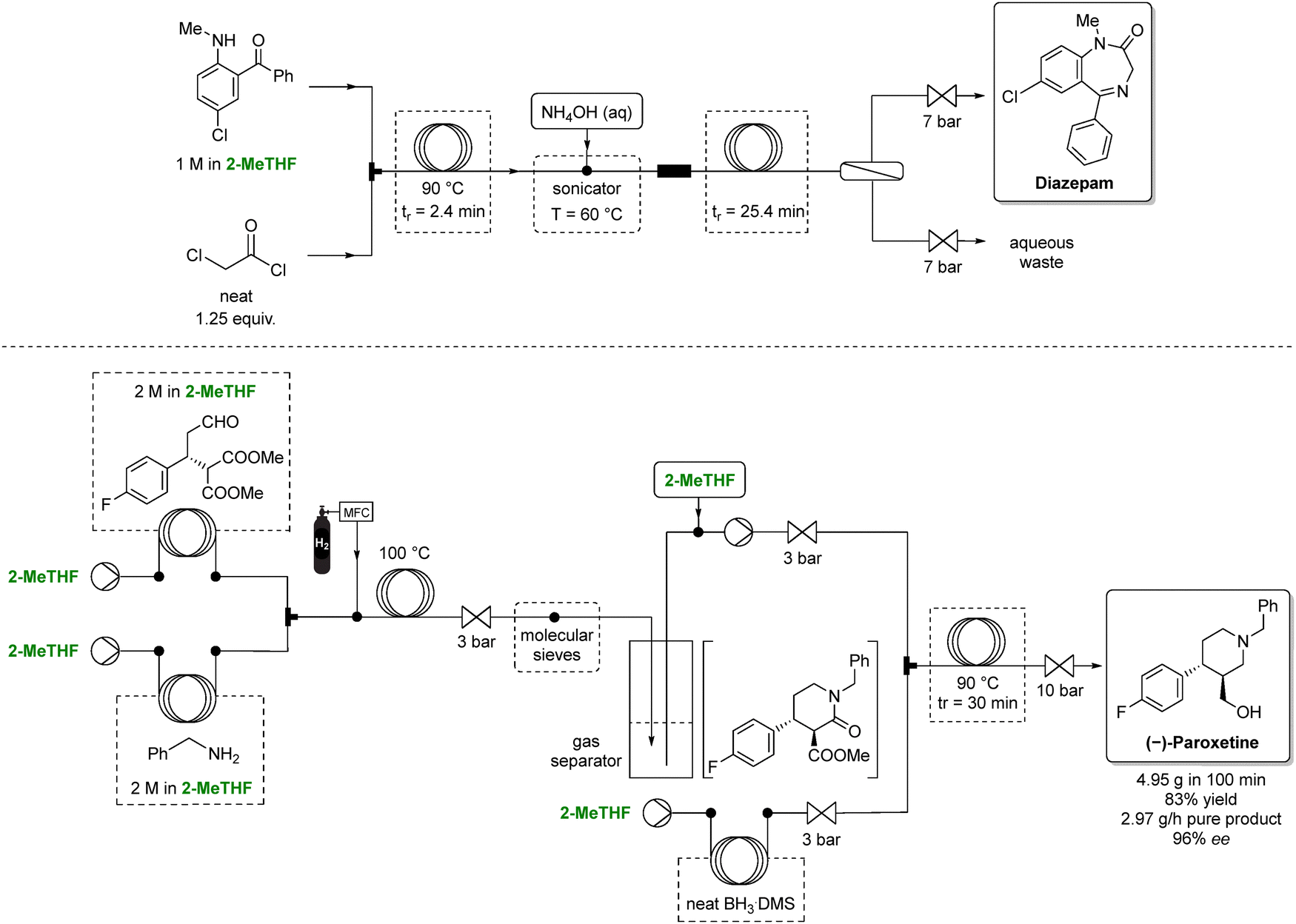 | ||
| Scheme 5 Continuous-flow strategies for the synthesis of two selected APIs (specifically, diazepam above, and (−)-paroxetine below) employing 2-MeTHF as green solvent. In the diazepam sequence, 2-MeTHF enables efficient telescoping of multiple steps under controlled heating and pressure. In the paroxetine route, 2-MeTHF supports a fully integrated multistep flow process. Overall, 2-MeTHF facilitates solvent continuity across all modules, smooth phase behavior, and robust operation under elevated temperature and pressure conditions. Adapted from ref. 88 and 89, respectively. | ||
Furthermore, Otvos et al. reported a multigram-scale flow synthesis of a chiral intermediate for (−)-paroxetine (Scheme 5, bottom).89 The phenylpiperidine intermediate was obtained in 3 steps: an asymmetric conjugate addition (conducted under solvent free conditions with excellent yield and enantioselectivity), followed by a reductive amination–lactamization, and finally a reduction of both the ester and amide. The final two steps proceeded in 2-MeTHF with >99% conversion and selectivity. The authors telescoped the 3 steps in 2-MeTHF, yielding the phenylpiperidine intermediate in 83% overall yield and 96% enantiomeric excess. Ultimately, the process delivered approximately 3 g h−1 of pure product while minimizing waste and demonstrating a cumulative E-factor of 6.
These include several pharmaceutically relevant C–C coupling reactions, such as Sonogashira100 and Baylis–Hillman coupling.101 In addition, Wilson et al. first demonstrated that cyrene could be an effective alternative to DMF in Suzuki–Miyaura cross couplings, affording good yields and broad functional group tolerance.102 The authors also reported the four-gram synthesis of 4′-methyl-2-biphenylcarbonitrile, an intermediate in the production of angiotensin II receptor antagonists (sartans) like irbesartan.103
A particularly noteworthy application was reported by Yu et al., where cyrene served not only as a solvent but also as a catalyst in the N-formylation of amines with CO2.104 In this instance, the reaction in cyrene had a tenfold higher reaction rate constant than in acetonitrile, outperforming other green solvents like γ-valerolactone (GVL). This system was further extended to the synthesis of pharmaceutically relevant benzothiazoles via the reductive conversion of CO2 with aminothiophenols.
Cyrene has also proven its utility in total synthesis. Andrew et al. replaced N-methyl pyrrolidone with cyrene in a greener route to antidepressant bupropion (wellbutrin hydrochloride).105 Their protocol, which features bromination of m-chloropropiophenone, amination with tert-butylamine, and precipitation of the resultant hydrochloride salt (Scheme 6), not only employed a greener solvent but also replaced hazardous reagents such as bromine and 12 M HCl with safer alternatives. This modification decreased the total waste from 138 kg to 46 kg per kilogram of product.
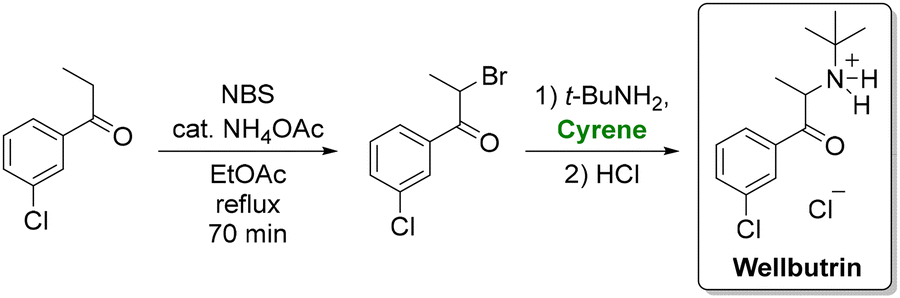 | ||
| Scheme 6 Synthesis of the antidepressant wellbutrin using cyrene as a solvent. Adapted from ref. 105. | ||
Despite its promising green credentials, cyrene presents certain practical limitations. Its high boiling point (227 °C) and complete miscibility with water complicate solvent recovery via distillation, raising concerns over its recyclability in large-scale applications. Furthermore, its ketone and acetal functionalities render it chemically labile under acidic, basic, or strongly oxidizing conditions, necessitating close monitoring of its stability throughout the process. Most concerning, however, was the recent bankruptcy of Circa Group AS, the company behind the commercial development of cyrene, casting uncertainty over the future availability of this solvent.106 This outcome can serve as a sobering reminder that the uptake and long-term success of greener alternatives hinges not only on technical performance in the academic lab or industrial plant but also on broader commercial and market dynamics. It also highlights the importance of diversifying the portfolio of green solvents under consideration, rather than relying too heavily on any single option. Fortunately, viable alternatives to NMP, DMF and other polar aprotic solvents do exist. In a comparative study, N-butylpyrrolidinone (NBP), GVL, propylene carbonate (PC) and cyrene were evaluated in Heck and Baylis-Hilman reactions, each outperforming traditional solvents in at least one context.101 NBP and GVL showed particular promise in the Heck reaction. Both solvents also benefit from simpler recovery strategies than cyrene (i.e., NBP due to its manageable boiling point and GVL due to its lower water miscibility). Moreover, GVL's production from biomass-derived levulinic acid further strengthens its profile as a circular, sustainable solvent. On the other hand, while PC demonstrated high initial reaction rates, particularly in the Baylis–Hillman reaction, challenges in product isolation due to solvent retention may limit its practicality without process modification. Nonetheless, these results illustrate that multiple bio-based solvents can effectively replace traditional polar aprotic solvents, provided recovery and practical application are adequately addressed.
In industrial settings scCO2 is commonly used to replace volatile organic compounds (VOCs) in extraction processes, famously in coffee decaffeination where it supplants chlorinated hydrocarbons. This established supercritical fluid (SCF) extraction industry minimizes technological gaps when expanding its application to fine chemistry. Nonetheless, supercritical solvents have been underutilized in pharmaceutical manufacturing, and this may be due to lack of compatible equipment already in plants.108
A notable example of scCO2 in API synthesis is found in the synthesis of antimalarial drug artemisinin. Sanofi developed a semisynthetic route to artemisinin starting from biosynthetic artemisinic acid.109 The process involved four steps: diastereoselective hydrogenation, esterification, Schenck ene oxidation via photogenerated singlet oxygen and finally acidic Hock cleavage cyclization to yield artemisinin in 55% yield with good atom economy. For the Schenck ene step, DCM was chosen both for safety reasons, as it is non-flammable, and for its ability to extend the lifetime of singlet oxygen. As scCO2 is also non-flammable and confers a similarly long singlet oxygen lifetime,110 Amara et al. envisioned a continuous-flow synthesis using scCO2 that bypassed the esterification step.111 They further simplified the process by immobilizing the photocatalyst on Amberlyst resin, eliminating the need to remove both catalyst and free acid from the final product. This approach furnished multi-gram quantities of artemisinin with yields (51%) comparable to Sanofi's report while also reducing the E-factor from 36 to 23. Additionally, singlet oxygen-mediated photooxidation in scCO2 has been explored in the synthesis of pharmaceutically relevant oxepinones,112 benzoquinones,113 and trioxanes.114
In another example, Alcalde et al. devised a flow-based route to the anticonvulsant Rufinamide (Inovelon) that integrates both synthesis and purification.115 The scCO2 platform provides a direct, streamlined method to obtain triazoles, common drug precursors, via a copper-catalyzed 1,3-dipolar azide–alkyne cycloaddition in good yields, with Rufinamide itself obtained in 52% yield at a throughput of 594 g L−1 h−1. Notably, the flow reactor was coupled to a chromatography column, enabling in-line purification and straightforward HPLC analysis of collected fractions.
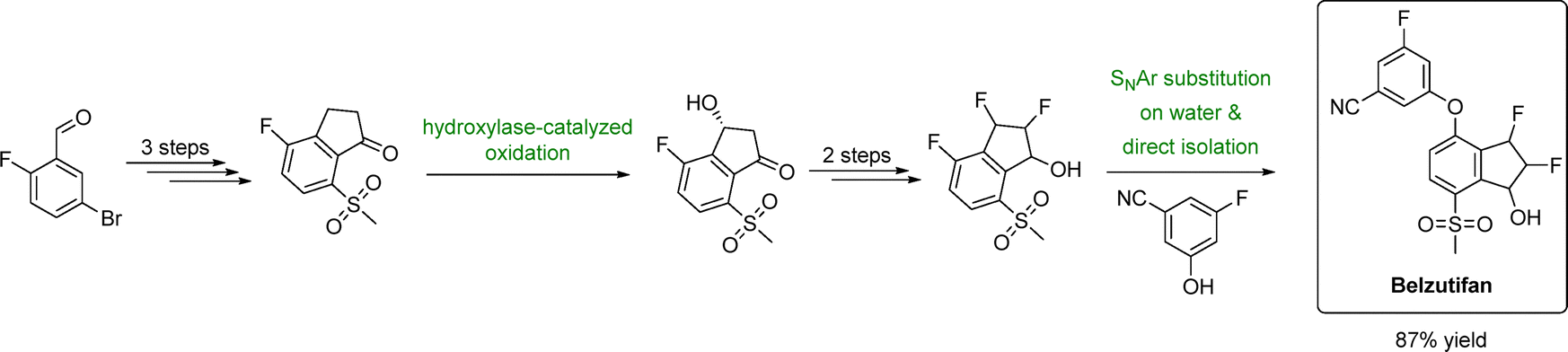 | ||
| Fig. 3 Sustainable manufacturing route to belzutifan using water as the reaction medium. Adapted from ref. 118. | ||
Where organic synthesis is concerned, many reagents and intermediates exhibit limited or negligible solubility in water. To enable reactions under aqueous conditions, micellar catalysis has been developed, wherein surfactants form a micellar environment that solubilizes organic reagents in water, enhancing both reaction efficiency and selectivity. Rather than providing an exhaustive overview, a task excellently covered elsewhere,116,117,126 this section focuses on select examples pertinent to pharmaceutical manufacturing where water use successfully minimized solvent demand and waste.
An exemplary case comes from Takeda Pharmaceuticals that devised a water-based synthetic route to felcisetrag (TAK-954), a 5-HT4 receptor agonist candidate (Scheme 7).127 In this process, aqueous media were employed in six of seven steps, with only one step carried out solely in organic solvents (acetonitrile). The first three steps (benzimidazole cyclization, amide bond formation, and reductive amidation), proceeded smoothly on water, although 15% THF was added during amide coupling to boost product solubility.
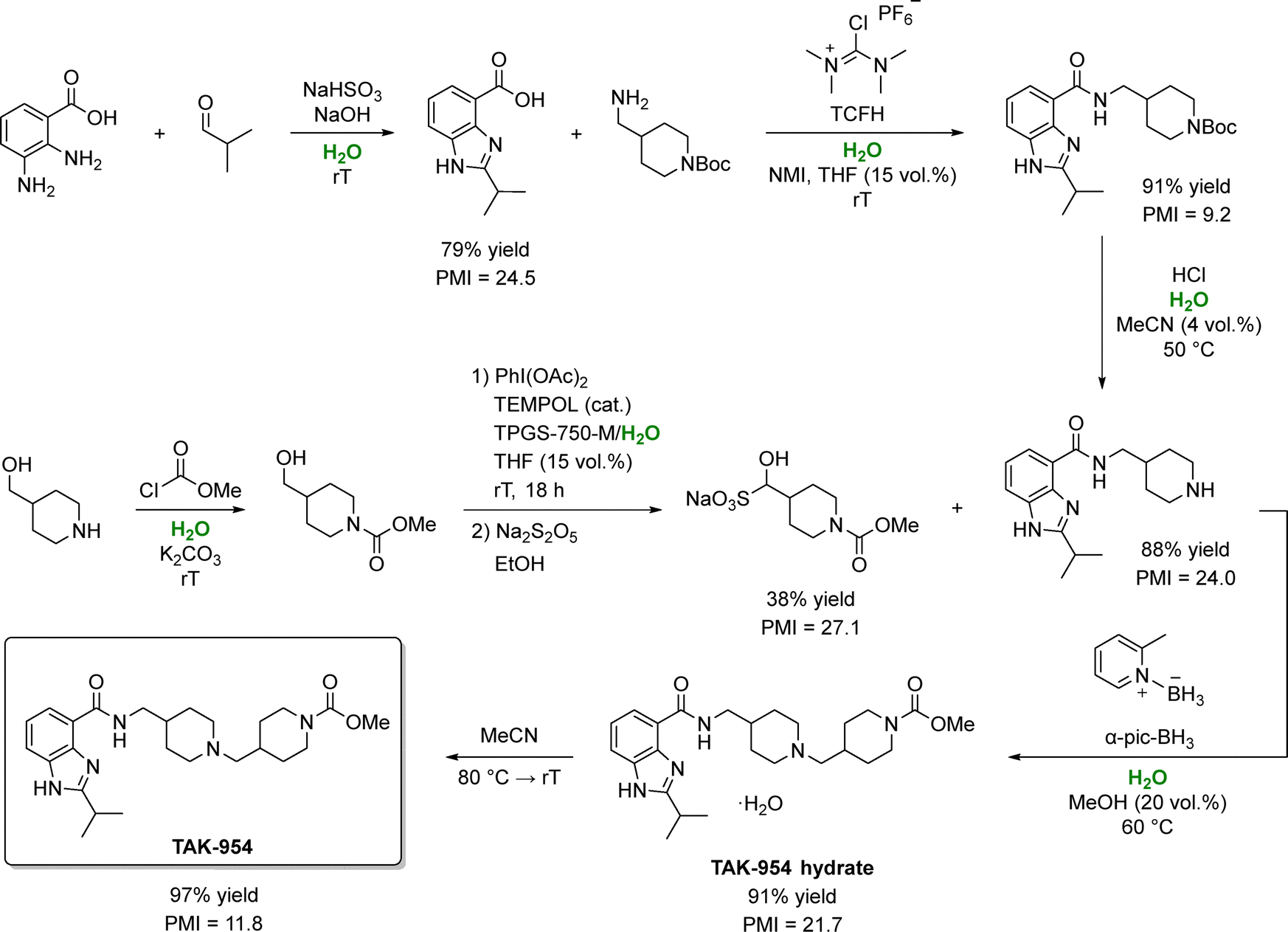 | ||
| Scheme 7 Synthesis of TAK-954 using water as the primary reaction medium. In this process, water enables selective imine formation and cleaner oxidative coupling, assists in the activation and deprotection steps, and serves as a co-solvent that reduces organic solvent demand while improving PMI. In some steps, water is used in combination with THF, MeCN, or MeOH to minimize byproduct formation. Adapted from ref. 127. | ||
While no surfactants were required in these initial steps; a small amount of TPGS-750 M (2 wt%) was necessary in step 4 (one-pot alcohol oxidation with PhI(OAc)2), owing to limited substrate solubility. Notably, water consumption was minimized by directly isolating intermediates through pH adjustments that triggered supersaturation and crystallization without organic solvents or solvent swaps. Overall, the process improved the yield from 35% to 56% and reduced the total PMI from 350 to 79, an impressive 77% reduction. In particular, switching to water led to a 94% drop in organic solvent consumption, slashing the solvent PMI from 223 to just 14. Remarkably, total water usage also fell from 106 to 55 L kgAPI−1, illustrating that using water as a reaction medium need not inflate overall water consumption. These substantial sustainability gains earned Takeda Pharmaceuticals the 2020 ACS Green Chemistry Institute's Peter J. Dunn Award.128
The low PMI of the amide bond formation (step 3) in the Takeda process is noteworthy, especially in light of the remaining inefficiencies in other steps, such as the use of PhI(OAc)2 as a terminal oxidant in step 4. This stands in contrast to traditional amide coupling strategies, which, despite their widespread use in API and agrochemical synthesis, often exhibit high PMI and rely heavily on non-renewable solvents like DCM, DMF, NMP, and DMAc.129 Recent advances have turned to flow chemistry to address these challenges. A notable example employs bioderived 2-MeTHF as solvent, di-2-pyridyldithiocarbonate (DPTDC) as a green coupling reagent and short residence times to minimize waste.130 Crucially, the by-product 2-mercaptopyridine, can be recovered (71%) and recycled into DPTDC in 91% yield (Scheme 8, top), offering a distinct advantage over conventional reagents like HATU, DCC, COMU and T3P. When applied to the synthesis of nirmatrelvir, this methodology halved the E-factor (from 108 to 54) and improved the overall yield from 48% to 70% compared to Pfizer's original route (Scheme 8, bottom).131
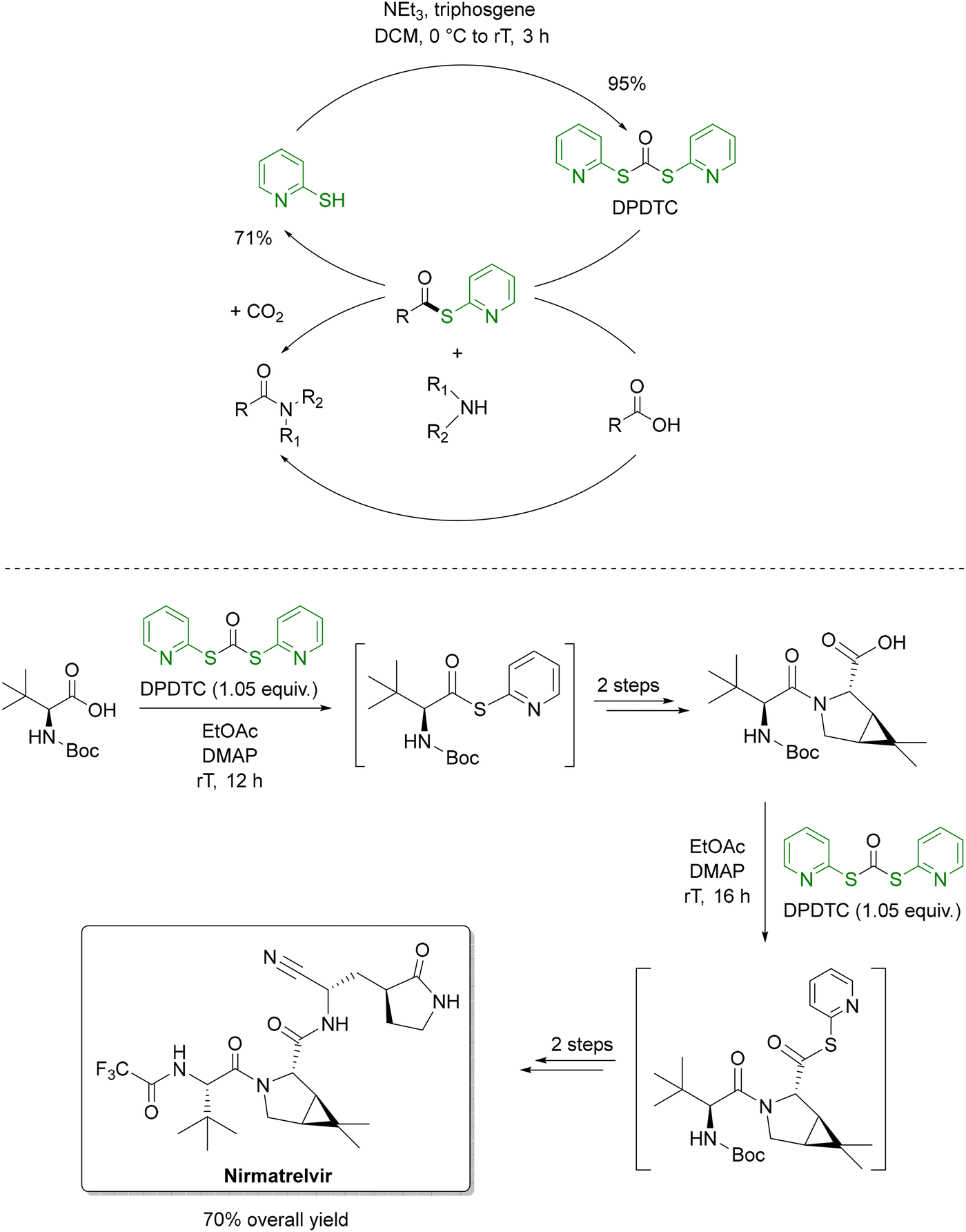 | ||
| Scheme 8 DPTDC as a recyclable amide coupling reagent (top) and its application to the synthesis of nirmatrelvir (bottom). Adapted from ref. 130 and 131. | ||
Water, though ideal from a green chemistry perspective, remains underutilized for amide couplings and peptide synthesis. Micellar techniques, however, offer a promising path forward. A seminal study from Novartis introduced a general, practical protocol for micellar amide bond formation, emphasizing the critical role of a co-solvent additive for successful reactions.132 Since then, micellar catalysis has been adopted for amide couplings in several pharmaceutical and agrochemical targets133–136 with an intriguing approach harnessing both sunlight and water for amide formation from carboxylic acids, hinting at broader possibilities for aqueous media in synthesis.137
While amide couplings remain a focal point, other organic transformations are also amenable to micellar catalysis. For instance, the Lipshutz group detailed two synthetic routes for the antimalarial drug pyronaridine in a study commissioned by the Gates foundation (Scheme 9).138 One is a linear sequence featuring a two-step, one-pot transformation in an aqueous surfactant medium, affording 87% overall yield. The alternative is a convergent, telescoped three-step sequence. It comprises of an initial solvent-free reaction followed by two aqueous micellar catalysis steps, namely a Cu(I)-catalyzed Ullmann coupling and a nitroarene reduction. In this sequence, 2 wt% aqueous TPGS-750-M surfactant enhanced the solubility and reactivity of typically water-insoluble substrates, thereby reducing reliance on hazardous organic solvents. Overall, by telescoping multiple operations, intermediate isolations were minimized, improving overall yields from 69% to 95% and achieving an impressive fivefold reduction in solvent consumption (from 46 to 9 kg kgAPI−1).
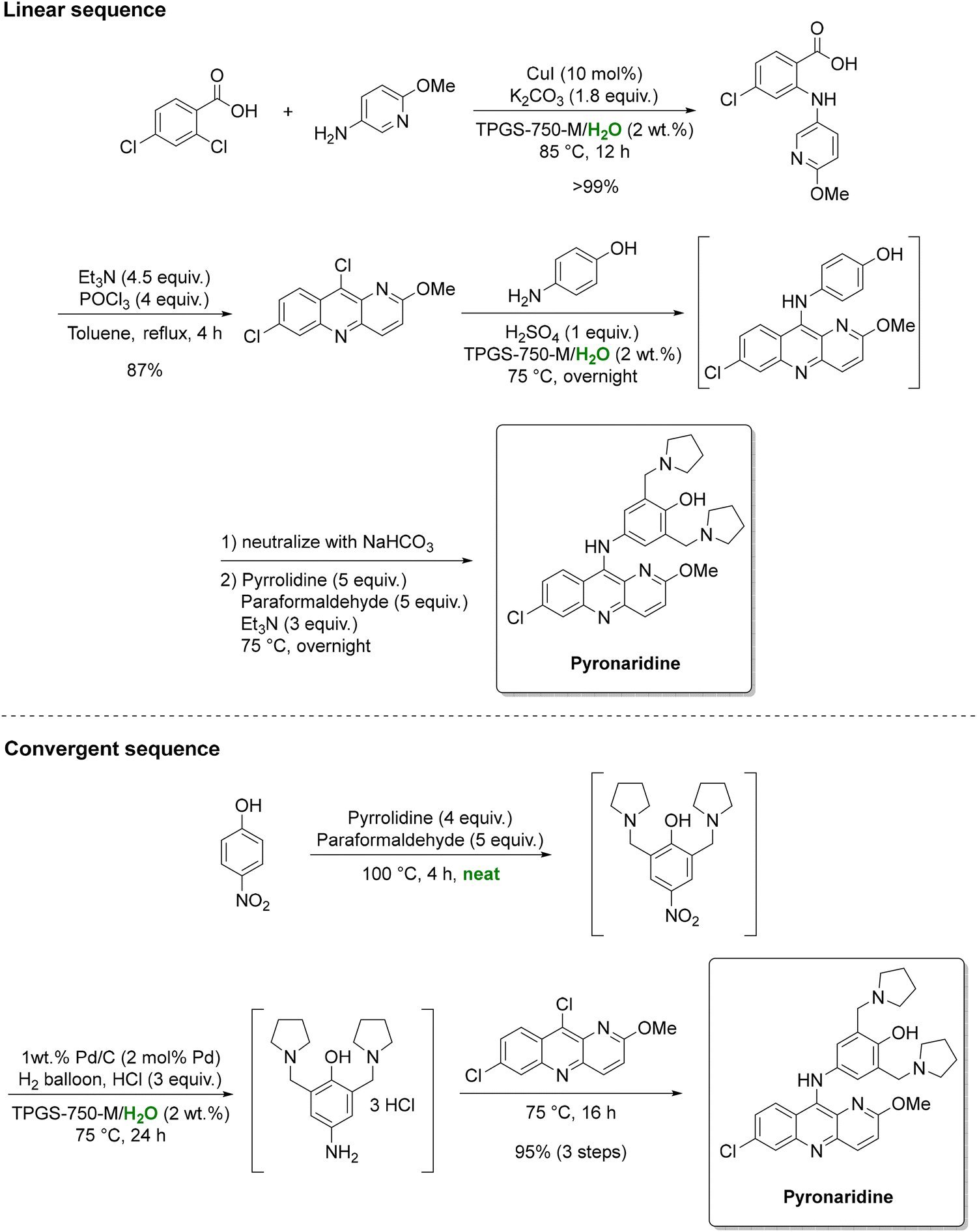 | ||
| Scheme 9 Linear and convergent strategies for the preparation of antimalarial drug pyronaridine under aqueous micellar conditions. Adapted from ref. 138. | ||
In another study, Lipshutz reported a six-step, green synthesis of MMV688533 (Scheme 10), a Medicines for Malaria Venture drug undergoing clinical evaluation.139,140 Critical transformations included two Sonogashira couplings and an amide bond formation, all under aqueous micellar conditions. Compared to Sanofi's traditional route, this approach increased the yield tenfold (67% vs. 6.4%), narrowed organic solvent use to only EtOAc and 2-MeTHF, and reduced palladium catalyst loading. The PMI also decreased by more than 50%, from 287 to 111.
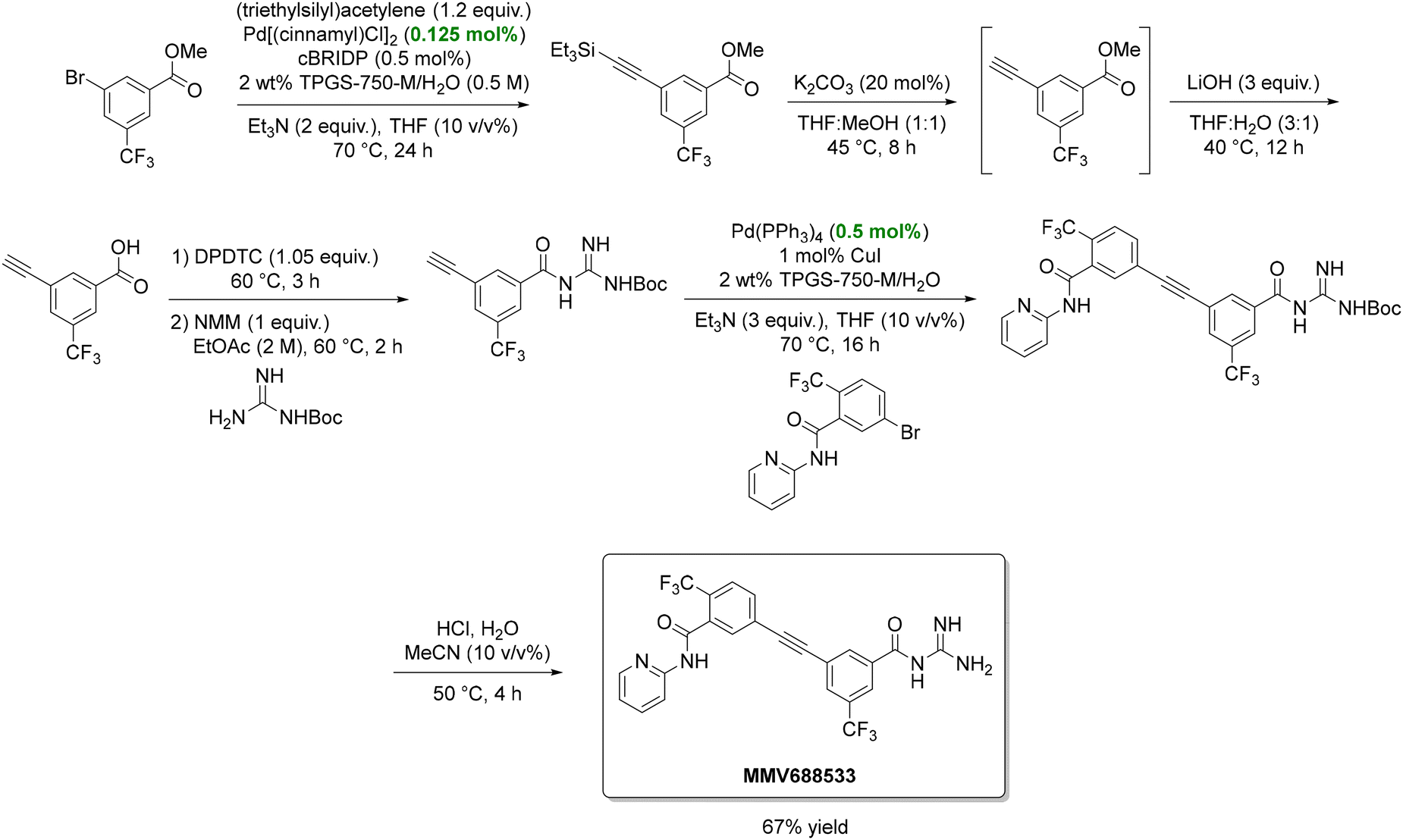 | ||
| Scheme 10 Green synthesis of the antimalarial drug candidate MMV688533. In the scheme, cBRIDP, DPDTC, and NMM are three reagents that frequently appear in amide-bond-forming reactions developed by the Lipshutz group. Specifically, cBRIDP refers to 2-chloro-N,N′-diisopropyl phosphinic chloride and serves as an activating agent for carboxylic acids. DPDTC stands for di-2-pyridyldithiocarbonate and is used to convert carboxylic acids into activated thioesters, which react readily with amines to form amides. NMM is N-methylmorpholine, and acts as a base that neutralizes the acid generated during the coupling reaction, maintaining the pH around 8–9. Adapted from ref. 139. | ||
Beyond the above examples, aqueous micellar media have been successfully employed in other key transformations including palladium catalyzed amination,141,142 borylation,143 and Suzuki–Miyaura couplings.144 A number of these transformations yielded valuable APIs including alpha blocker naftopidil (amination),145 an intermediate to lung cancer therapeutic capmatinib (borylation),143 and antitumor agent lapatinib (Suzuki–Miyaura).144 Most strikingly palladium-catalyzed aminations have recently been achieved under plug-flow conditions in recyclable water, without surfactants and using only ppm-level catalyst loadings.145 This marks a significant advance in both sustainability and scalability.
These developments in micellar catalysis are mirrored by progress in alternative water-based systems, such as those employing hydrophilic polymers. One such example is hydroxypropyl methylcellulose (HPMC) that forms hydrophobic pockets in water capable of solubilizing reactants and stabilizing catalytic nanoparticles.146 A 2017 patent by Abbvie demonstrated the use of HPMC for a range of cross-coupling and organic transformations under aqueous conditions.147 More recently, amide couplings and Buchwald–Hartwig aminations were shown to proceed within seconds to minutes using HPMC and water.148
Together, these innovations reflect a broader momentum toward aqueous methodologies in organic synthesis. Among them, micellar catalysis has shown particularly strong potential when implemented at scale, offering substantial reductions in environmental impact, solvent use, and process complexity.127,132,149,150 As the field continues to evolve, attention has turned to improving not only the reactions themselves but also the sustainability of the surfactants that enable them. For example, while the production of custom-designed surfactants such as TPGS-750-M,151 has historically relied on organic solvents, more recent alternatives, can now be prepared using recoverable solvents in more environmentally responsible processes.152 Moreover, as micellar catalysis moves from academic settings to industrial applications, process design has also shifted. Whereas laboratory-scale protocols often depend on organic co-solvents for phase separation, solubilization, or product extraction, industrial operations increasingly favor solid-phase filtration. This considerably greener approach has been demonstrated by both Takeda and Novartis.127,153 Nonetheless, replacing organic solvents with water-based micellar media is far from straightforward. One major concern is the generation of aqueous effluents containing nonionic amphiphiles, for which standard wastewater treatment technologies may not be sufficient.154 For instance, TPGS-750-M has been classified as a WGK 3 substance in Germany, indicating high water hazard.154 This necessitates specialized treatment or extraction prior to wastewater disposal, potentially adding complexity and cost to the overall process. Finally, life cycle analyses have shown that the carbon footprint of high-performance amphiphiles like TPGS-750-M can be up to 28.8 kg CO2 per kg of material, 10 to 20 times higher than for commodity surfactants such as Brij 30.154 These figures raise important questions that need to addressed for the industrial implementation of micellar processes to continue.
2.3. Solvent free synthesis and mechanochemistry
Solventless reactions represent a transformative green chemistry approach by eliminating solvents altogether to reduce environmental impact and operational hazards. Numerous reactions can proceed without solvent by exploiting either the natural liquidity of the reactants or the ability of one reactant to solubilize another.An example of a solvent-free methodology is the continuous-flow synthesis of diphenhydramine hydrochloride,155 wherein the initial alkylation between 2-dimethylaminoethanol and chlorodiphenylmethane proceeds under neat conditions at 180 °C and 1.7 MPa. Subsequent precipitation and recrystallization steps employed benign isopropanol, further reducing hazardous solvent consumption. Importantly, the end-to-end continuous system enabled inline purification that minimized solvent use further. The final product was obtained in 82% yield, with a production capacity of 4500 drug doses per day at USP standards. A similar strategy was used to synthesize an anti-HIV agent achieving 76% yield under neat conditions via a metal-free α-ketoamide synthesis.156
The success of these processes can be ascribed in part to the liquid nature of the reactants, which limits their application in producing solid APIs. To address this, mechanochemistry has been touted as a key advance in this domain. Mechanochemistry drives reactions through mechanical forces generated by techniques such as ball milling, extrusion, or resonant acoustic mixing. In addition to accelerating reaction rates and enhancing selectivity, these methods can activate otherwise inert substrates and enable reaction pathways that may be challenging or even unattainable in solution. Most importantly, the solid-state nature of these processes minimizes solvent use and reduces waste generation. However, it is worthwhile noting a more accurate description for these processes is “near-solvent-free” as they commonly require a minimal amount of solvent additive, which, in most cases, is negligible.
Paracetamol, also known as acetaminophen or Tylenol, is the most prescribed analgesic worldwide and is listed among the essential medicines of the World Health Organization (WHO).157 A key synthetic route is the catalytic transfer hydrogenation of aromatic nitro compounds, Portada et al. employed a solventless ball-milling approach in which ammonium formate served as the hydrogen donor, enabling the reaction to proceed without the need for bulk solvents (Scheme 11, top).158 Silica gel was used as a grinding auxiliary to address mixing and product recovery issues. This protocol afforded quantitative yields of paracetamol, delivering exclusively the crystalline form I and could be extended to antiarrhythmic procainamide (Scheme 11, bottom) with an 88% overall yield. Building on this concept, a related reductive milling strategy using sodium borohydride enabled the synthesis of WHO-listed antidepressant fluoxetine from an amino ketone.159 However, while the reaction conditions are highly attractive from a green chemistry standpoint, the work raised questions regarding practical aspects such as the separation of the crystalline product from the silica and Pd/C catalyst, as well as the cleaning of milling equipment. These downstream considerations are critical in assessing the true sustainability and scalability of such solventless methodologies.
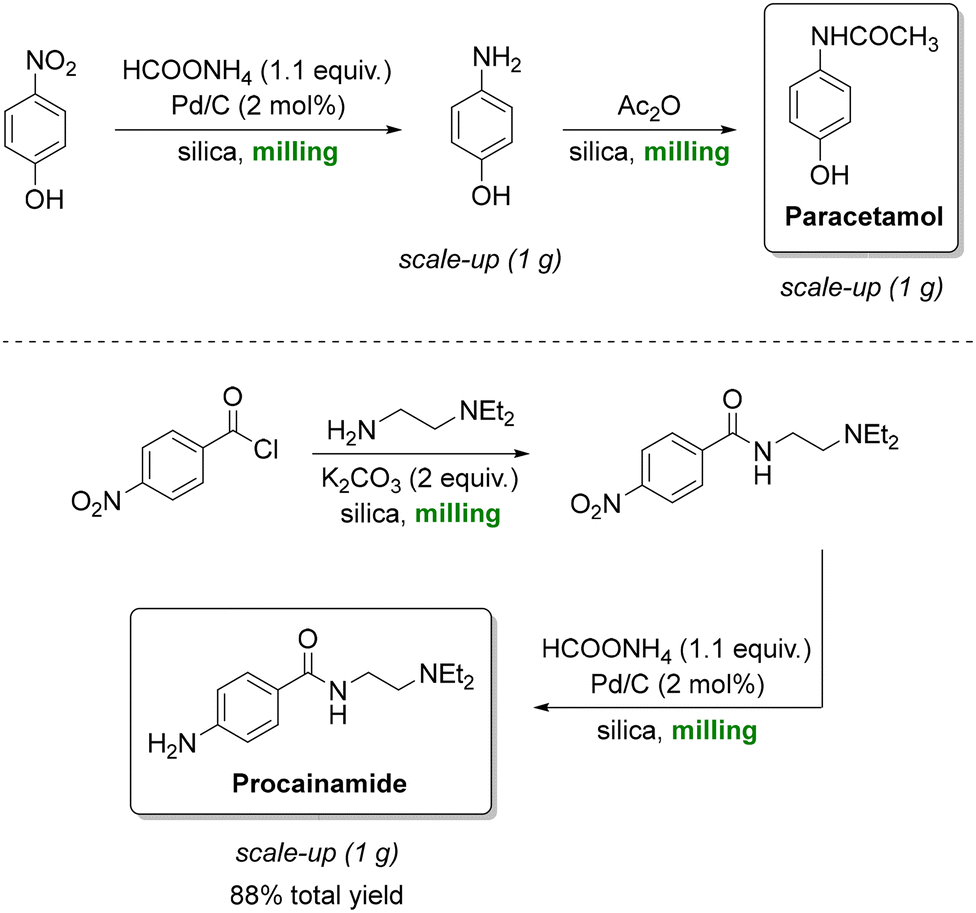 | ||
| Scheme 11 Silica-assisted mechanochemical ball milling to produce paracetamol (top) and procainamide (bottom). Adapted from ref. 158. | ||
Mechanochemistry can also be extended to amide formation reactions. For instance, Nicholson et al. described a steel jar ball-milling protocol (30 Hz) for direct amination of esters, achieving moderate to excellent yields (up to 98%) with aromatic, alkyl, and heteroaromatic esters and both primary and secondary amines (Fig. 4).160 This protocol was extended to five agrochemical and pharmaceutical agents, including antidepressant Moclobemide (92%), fungicide Fenfuram (69%), and anesthetic lidocaine (58%), and scaled successfully to multi-gram quantities without performance loss. Notably, its atom economy (55%) surpasses that of common amide coupling reagents such as phosgene (47%), HATU (28%), and EDC (41%). By eliminating solvents and stoichiometric coupling reagents, the process achieved a dramatic reduction in the PMI to 1.94, compared to 59.28 in a previously reported green synthesis using KOtBu.161 Although a striking example, numerous comparable mechanochemical PMI reductions have since been reported,162 highlighting both the progress achieved and the need for continual research in such greener methodologies. A related ball milling Mitsunobu reaction further expanded the scope to carboxylic acid–alcohol coupling, enabling multi-gram late-stage functionalizations of acid-containing APIs with a planetary ball-mill.163
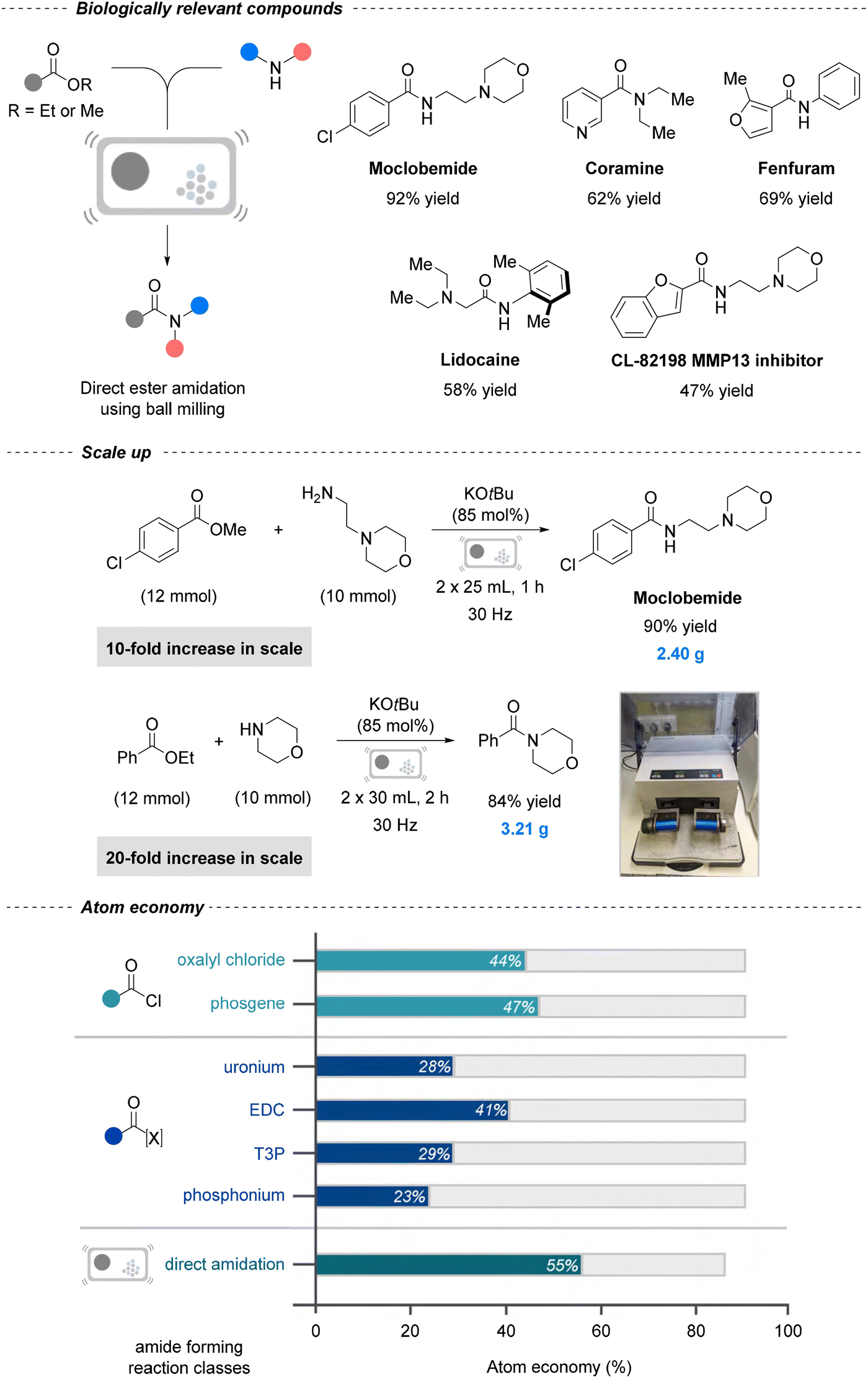 | ||
| Fig. 4 Mechanochemical strategy for solvent-free amidation of esters. General reaction scheme (top right), scale-up of API (top left), scale up experiments (middle), and atom economy calculations (bottom). Reproduced from ref. 160, with permission from Wiley, copyright 2021. | ||
Cross-coupling reactions have also found success under mechanochemical conditions. Yu et al. implemented a solventless Heck–Migita strategy (Scheme 12) to accomplish the total synthesis of anticancer drug axitinib (Inlyta).164 A key aspect of the mechanochemical process was the addition of NaBr which acted as both a grinding auxiliary and dehalogenation suppressor, minimizing by-product formation (<1%). Sequential bromination, Mizoroki–Heck coupling, and Migita cross-coupling all proceeded entirely under ball-milling conditions, culminating in a 44% overall yield of axitinib. This solvent-free process matched conventional solution-phase results while maintaining sub-2 ppm palladium contamination, thereby meeting pharmaceutical purity standards. Similar ball-milling cross-couplings have been reported for other valuable targets.165–169
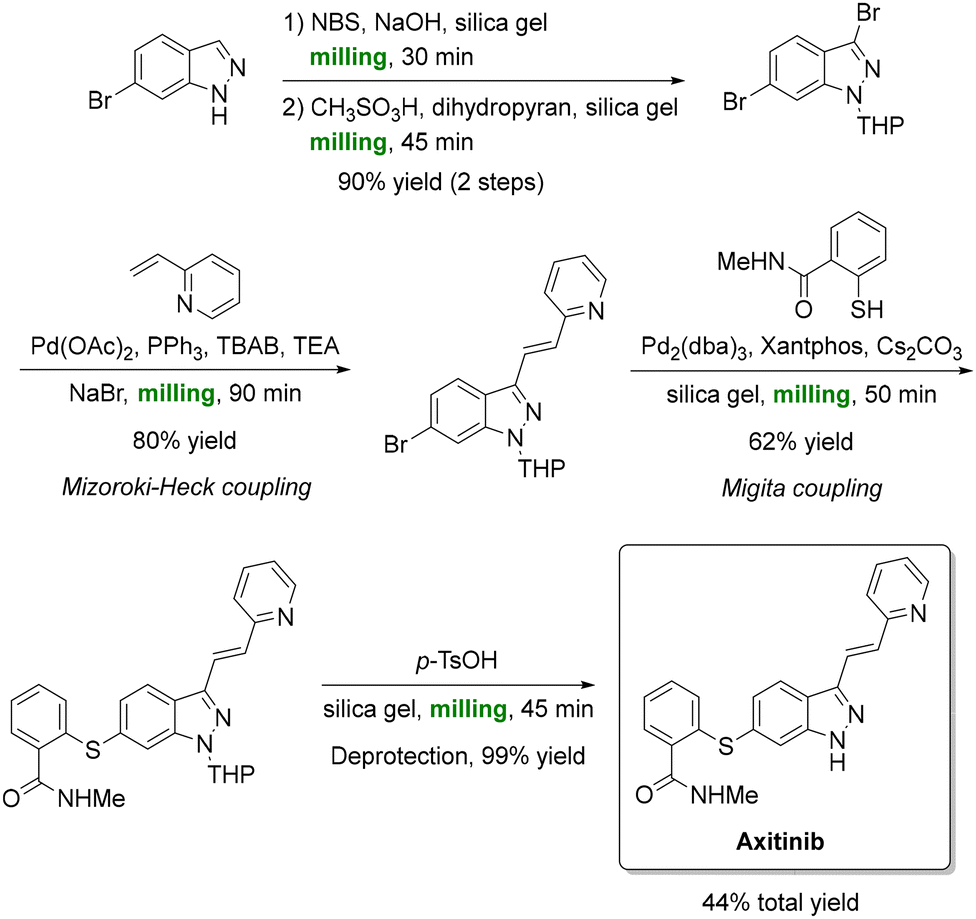 | ||
| Scheme 12 Full mechanochemical synthesis of anticancer-drug axitinib; the strategy under ball-milling conditions features Mizoroki–Heck and Migita couplings. Reproduced from ref. 164. | ||
In another study, Colacino and co-workers described a planetary ball mill synthesis of the antiepileptic drug phenytoin.170 In a one-pot, two step procedure, amino esters were ground with potassium cyanate in the presence of four equivalents of water to form a ureido intermediate in situ. This was followed by base-catalyzed cyclization to hydantoin, all without using hazardous solvents like DMF. The process afforded phenytoin in 84% yield without purification. The same group produced dantrolene (Dantrium) and nitrofurantoin (Furadantin) via ball milling at 30 Hz in a stainless-steel jar with two 5 mm steel balls, achieving over 90% yield without base, solvent, or purification, resulting in a remarkably low E-factor of 0.3 and a PMI of 1.3.171
More recently, the synthesis of dantrolene and nitrofurantoin (Fig. 5, top) was scaled up to 25 g using a continuous Twin Screw Extruder (TSE) with a two minute residence time, attaining a space-time yield of 6800 kg m−3 day−1.172 It is worth noting mechanochemistry offers a scalable solution to challenges in large-scale production of fine chemicals and agrochemicals.173 While conventional techniques like ball milling have scalability and temperature control issues, continuous techniques like TSE address these effectively by continuous processing, precise temperature control, and multi-step reactions. A life cycle assessment of nitrofurantoin synthesized by this method (Fig. 5, bottom) highlighted how continuous mechanochemistry could substantially mitigate emissions, underscoring its potential in sustainable pharmaceutical and agrochemical manufacturing.172
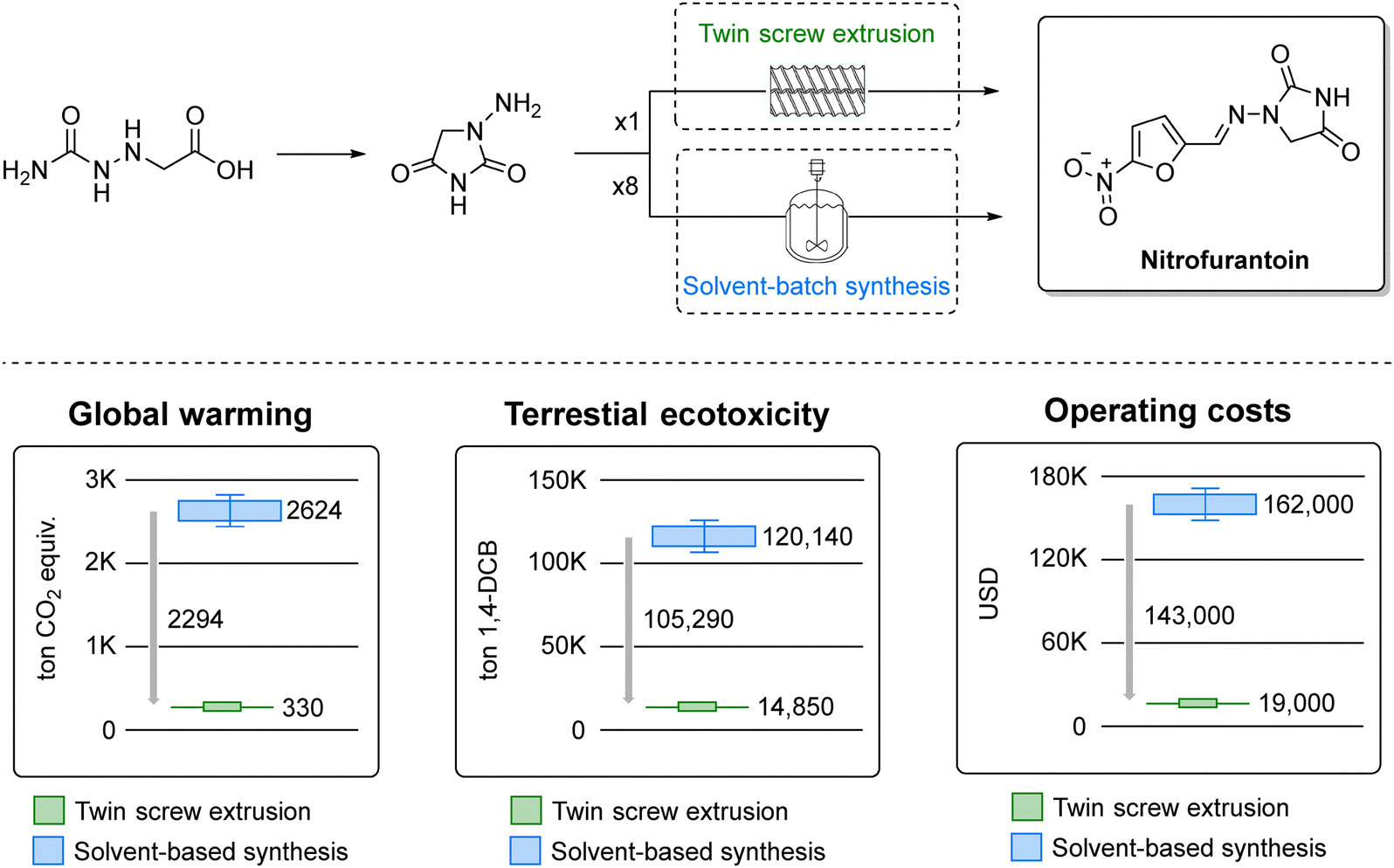 | ||
| Fig. 5 Mechanochemical synthesis of nitrofurantoin by twin screw extrusion (top) and green metrics analysis (bottom). Adapted from ref. 172. | ||
Fluorochemicals, excluding those categorized as “forever chemicals”, are pivotal in industry, yet their synthesis typically depends on energy-intensive conversions of acid-grade fluorspar into highly toxic and corrosive hydrogen fluoride, which is subsequently employed in multistep fluorination processes. By contrast, Patel et al. reported that direct milling of acid-grade fluorspar with potassium hydrogen phosphate (K2HPO4) yields a ‘Fluoromix’ reagent capable of constructing S–F and C(sp3/sp2)–F bonds without resorting to HF (Fig. 6).174 While S–F and C(sp3)–F bonds were obtained using only excess water as an additive, the construction of C(sp2)–F bonds required DMSO as the solvent. Nonetheless, the method furnished various important intermediates and pharmaceutically relevant compounds, as well as the in situ formation of other fluorinating agents. In a separate report, the degree of fluorination of 1,3-diketones with selectfluor was controlled under ball-milling conditions: liquid-assisted grinding favored monofluorination, whereas solvent-free grinding led to difluorination.175 This latter method also scaled successfully to a continuous twin-screw extruder, increasing throughput from 29 to 3395 kg m−3 day−1.176 However, it is important to consider the full process from end to end: while HF poses significant safety risks due to its intrinsic reactivity and toxicity, its atom economy and overall sustainability may be comparable to that of fluorspar plus K2HPO4, which imposes a considerable burden in terms of waste treatment and phosphate management, especially at industrial scale.
 | ||
| Fig. 6 Mechanochemical fluorination strategy using acid-grade fluorspar to obtain fluoromix. The figure illustrates the experimental setup and demonstrates the broad applicability of fluoromix across multiple classes of fluorination reactions. Specifically, fluoromix enables efficient S–F, C(sp3)–F, and C(sp2)–F bond formation, delivering a variety of fluorinated products with good-to-excellent yields. Reproduced from ref. 174, with permission from Science, copyright 2023. | ||
Beyond fluorination, mechanochemical methylation of (hetero)arenes has been conducted under solvent-free ball milling conditions (36 Hz for 1–4 h), enabling late-stage functionalization of pharmaceuticals and agrochemicals, including the crop protection product etoxazole and the anti-spasmodic papaverine.177
Qu et al. demonstrated that mechanically induced piezoelectric catalysis can effectively drive the dehalogenative deuteration of aromatic iodides, delivering good to excellent deuterium incorporation.178 This method was successfully applied to deuterate both the osteoporosis drug ipriflavone and the fungicide boscalid. Building on similar principles, Wang et al. utilized piezoelectric Li2TiO3 and molecular oxygen under mechanochemical conditions to power the triphasic hydroxysilylation of alkenes, a process often hindered by mass-transfer limitations. Their results included the late-stage functionalization of a series of drugs, achieving moderate to excellent yields.179
Beyond ball-milling techniques, alternative mechanochemical approaches have been applied to API synthesis. One promising example is a Resonant Acoustic Mixer (RAM), a non-contact mixing device that uses a forced-vibrating, high-intensity acoustic field at low frequencies (around 60 Hz) to promote efficient particle collisions. Crucially, RAM technology does not require milling media, which simplifies reaction design and scale-up while avoiding contamination from milling-element wear.162,180 Although RAM technology has successfully been used to produce cocrystals in pharmaceutical settings,181,182 Gonnet et al. pioneered its use in metal-catalyzed synthesis, specifically ring-closing metathesis, ring-closing ene–yne metathesis, and copper-catalyzed coupling to form sulfonylureas.183
Recently, Lennox et al. demonstrated a RAM-based mechanochemical synthesis of the anticonvulsant Rufinamide.184 In their process, a copper coil acted as a direct mechanocatalyst for a one-pot alkyne–azide cycloaddition. This setup avoided bulk solvents and milling media, affording an 88% isolated yield within 60 min. While the approach is promising from a green chemistry standpoint, it is important to note that the reaction likely proceeds via the formation of a copper azide intermediate. This raises an important safety concern, as copper azide is a highly sensitive and potentially explosive compound, especially under conditions of mechanical agitation typical of mechanochemical processes. It is critical that a thorough risk assessment of any new process is thus conducted as the presence of intermediates such as these pose a serious hazard during the reaction scale-up. RAM technology has also proven effective in preparing DNA and RNA fragments on a multigram scale (55–95% yields in only 15 min), while reducing solvent use by 90%.185 Moreover, high-throughput experimentation (HTE) can be combined with RAM-driven mechanochemistry to rapidly screen reaction conditions. For instance, Nanni et al. developed a solvent-free, nickel-catalyzed cross-coupling amination protocol that enabled the late-stage functionalization of drug-like and bioactive molecules.186
With various mechanochemical methods emerging across chemical synthesis, evaluating their environmental performance is critical for selecting the most sustainable process. While these techniques offer clear sustainability advantages, such as reduced solvent use and energy-efficient transformations, they also present barriers to broader adoption. First, safety remains a concern, as applying high energy to reactive materials in the absence of a solvent heat sink introduces unknown process hazards that require thorough evaluation, as shown in the example above. Second, practical aspects such as product isolation, purification, and equipment cleaning are often underreported; examples that address these issues should be clearly highlighted when available. Third, scale-up pathways remain largely undefined: transitioning from lab-scale screening to multi-kilo-per-day production requires a better understanding of key parameters, especially given the lack of established scale-up protocols. Although techniques like RAM lend themselves to high-throughput experimentation, questions remain about whether such reactors can be generalized across transformations or must be tailored specifically. Furthermore, the availability of these reactors at production-relevant scales is still limited. Finally, it is important to mention that traditional solvent-selection tools are, by nature, not directly applicable to these near-solvent-free approaches. New frameworks such as Merck MilliporeSigma's DOZNTM 2.0 offers a quantitative metric for gauging mechanochemical greenness, classifying processes under three main pillars: resource use, energy efficiency, and human/environmental hazards.187,188 Although it does not yet account for life-cycle impacts of raw material production, it takes into account their hazard profiles and resource efficiency.
3. Renewable raw materials for the synthesis of active ingredients
When developing a greener-by-design process, one cannot overlook the sourcing of raw materials. Despite the fine chemical industry's growing leadership in applying many of the 12 green chemistry principles,189 the 7th principle of using renewable feedstocks, has largely been neglected.13 The comparatively small mass fraction of raw materials relative to solvents is a common rationale for not prioritizing renewable carbon.190 Yet, raw materials can carry a disproportionate environmental impact: for example, they represent just 7% of the PMI in the synthesis of the cough suppressant gefaxipant citrate, but account for 36% of the GWP, 20% of terrestrial acidification, and 33% of freshwater eutrophication.191 Looking at the broader picture, a recent McKinsey & Company report suggests switching to sustainable feedstocks can cut API manufacturing emissions by up to 25%.3While petrochemical feedstocks are the current default for carbon raw material, there are three major sustainable raw material sources: recycled carbon, CO2-derived carbon, and biomass carbon.192 Projections suggest that to achieve net-zero emissions, the chemical industry should utilize roughly 55% recycled carbon, 25% CO2-based carbon, and 20% bio-based carbon (Fig. 7).193 Closed-loop recycling is not viable for pharmaceuticals and agrochemicals, as these molecules are metabolized or otherwise dispersed into the environment.194,195 Consequently, attention turns to CO2 or biomass feedstocks. Several C1 base chemicals like methanol and formic acid are already produced from CO2.196 Since these streams are established in today's chemical industry, they will not be discussed further. Instead, the focus falls on a broader range of biomass-derived feedstocks that can be obtained via thermal, chemocatalytic, or fermentative routes.197 These processes are, at present, at a low Technology Readiness Level (TRL).
 | ||
| Fig. 7 Projected shift in global carbon demand for chemicals from 2020 to 2050. In 2020, embedded carbon was dominated by fossil-based sources (88%), with only minor contributions from bio-based feedstocks, CO2-based processes, and recycling. By 2050, total carbon demand is expected to more than double, yet its composition is projected to change substantially: recycling is anticipated to supply over half of the demand (55%), followed by bio-based carbon (20%) and CO2-derived carbon (25%). This transformation reflects a targeted move away from fossil feedstocks towards circular and renewable carbon sources, driven by material decarbonization strategies and a compound annual growth rate of approximately 2.5% in overall carbon demand. Adapted from ref. 193. | ||
3.1. Natural product isolation
Natural products represent an early instance of bio-based feedstocks, having served as crop protection products and therapeutic agents from time immemorial.198,199 To this day, certain compounds, like quinine for antimalarial therapy, are most efficiently extracted from natural sources.200 However, the low natural abundance of these compounds necessitates large-scale biomass harvesting, inevitably yielding substantial waste.201 Consequently, while invaluable for drug discovery programs,202,203 natural product isolation rarely delivers an economically or environmentally sustainable large-scale supply of active ingredients that can fulfil market needs, nor does it significantly advance the use of biomass carbon. Instead, industrial active ingredient production relies on organic synthesis from petrochemicals, or less frequently, carbohydrate fermentation.192 In certain cases, semi-synthetic options are preferred, as in the conversion of artemisinin to artemether.204Typically, economics dictate the chosen production route. A notable example is the anticancer drug paclitaxel, whose intricate tricyclic carbon core requires approximately 40 synthetic steps, for a meagre 2% overall yield.205 Considering the bark of the most productive yew tree contains a mere 0.001–0.05 wt% of paclitaxel, direct natural product isolation does not provide a viable solution.206 To alleviate reliance on bark-harvesting, a semi-synthetic route was developed using 10-deacetylbaccatin III (10-DAB III) from European yew leaves and twigs.206 Nevertheless, challenges such as reliance on large plantations, seasonal variability, and market volatility, persisted.
The breakthrough came with bioreactor cultivation of Taxus cell lines providing biotechnological production of taxanes.207 Herein, Paclitaxel is directly extracted from cell cultures, and subjected to chromatographic purification greatly reducing biomass waste and solvent consumption.207 Notably, this method omits six intermediates compared to the semisynthetic 10-DAB III route. This achievement was recognized with a 2004 Presidential Green Chemistry Award, emphasizing the need and importance of such success stories.208 Importantly, further advances in synthetic biology hint at fully biosynthetic routes. For instance, co-expression of nine key enzymes in tobacco recently yielded (albeit at low levels) the paclitaxel precursor baccatin III, laying the foundation for a future greener and more scalable paclitaxel production route.209
3.2. Bio-based platform chemicals
In most pharmaceutical and agrochemical syntheses, commodity chemicals serve as raw materials for active ingredients. Modern biorefineries offer a renewable avenue for several C1–C6 building blocks by processing second-generation biomass (primarily side streams from the food supply chain, or agricultural and forestry residues).210 Such feedstocks consist of 50–80 wt% lignocellulose, 0–15 wt% triglycerides, and 0–30 wt% proteins.211Conventional lignocellulose processing for pulp, paper, or bioethanol prioritizes cellulose and hemicellulose extraction which leads to irreversible degradation of lignin into “technical lignin”. This by-product is relegated to low-value energy production, but can be valorized to vanillin and syringaldehyde by oxidative depolymerization.213 By contrast, “lignin-first” strategies preserve lignin's inherent functionality by depolymerizing it into methoxylated phenolics prior to treating the other fractions.214 (Scheme 13). The specific product profile depends on the biomass source (softwood, hardwood, or herbaceous) and the depolymerization method.215 For instance, during reductive catalytic fractionation, softwood primarily yields alkylguaiacols, whereas hardwood and herbaceous biomass furnish alkylguaiacols and alkylsyringols. Process conditions (catalysts, solvents) can tailor alkyl chain substituents and length.215 Oxidative depolymerization confers alternative monomers including vanillin and syringaldehyde.216 These monomers can then be funneled into drop-in commodity aromatics (e.g., phenol),217 novel platform chemicals (e.g., muconic acid),218 or complex frameworks (e.g., quinazolines).219,220 Despite this versatility, lignin valorization remains predominantly at the R&D or pilot level.221
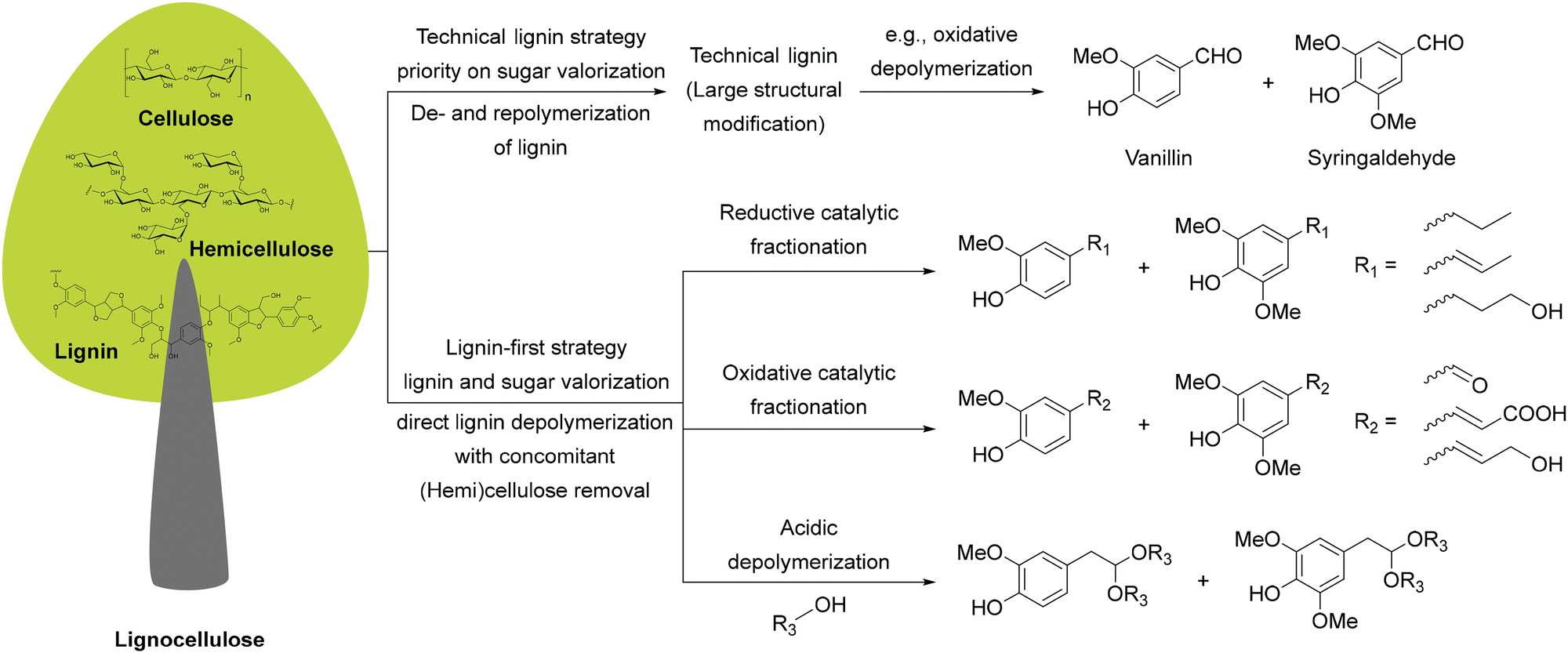 | ||
| Scheme 13 Selected lignin-derived platform chemicals accessible by depolymerization. Depending on whether sugars or lignin are prioritized, biomass can undergo technical lignin processing or lignin-first depolymerization. These approaches generate a range of aromatic platform chemicals, such as vanillin, syringaldehyde, alkylated phenols, and oxygenated aromatics, through oxidative, reductive, or acidic catalytic depolymerization routes. Adapted from ref. 217, 220 and 222. | ||
After lignin removal, the cellulose–hemicellulose solid pulp is separated for further processing.223 Acidic or enzymatic hydrolysis yields primarily glucose (from cellulose) and xylose (from hemicellulose), which can be fermented into short-chain alcohols/diols (e.g., ethanol), organic acids (e.g., acetic acid), and amino acids (e.g., L-lysine).224 Alternatively, chemical routes convert these sugars into bio-based platform chemicals. For instance, acid-catalyzed dehydration furnishes furfural or 5-hydroxymethylfurfural225 which can be valorized to bioaromatics (i.e., furanedicarboxylic acid as a terephthalic acid substitute),226 levulinic acid,227 or the bio solvent 2-MeTHF (Section 2.2) amongst others.228 Oxidation leads to sugar acids (e.g., gluconic acid),229 while hydrogenation provides sugar alcohols (e.g., mannitol), common compounds in nutraceuticals and pharmaceuticals.230
Meanwhile, glycerol, the main byproduct of biodiesel production,238 is a gateway to numerous products, notably through epichlorohydrin (hydrochlorination),239 green solvent glycerol carbonate (CO2 carbonylation),240 and various acroleins (dehydration).241
Composed of amino acids, proteins provide both carboxylic acid and amino functionalities for diverse transformations: hydrogenation yields β-amino alcohols or primary amines;244 oxidative decarboxylation forms nitriles;245 and hydrodeamination furnishes carboxylic acids.243 Side-chain modifications are also possible, e.g., glutamic acid to 2-pyrrolidone or its N-alkylated derivatives.246
3.3. Synthesis of active ingredients from bio-based platform chemicals
Two principal approaches can be pursued to produce fine chemicals from bio-based platform chemicals. The first is a “drop-in” strategy, where a bio-based intermediate simply replaces its fossil-based counterpart in the existing synthesis. For example, the expectorant guaifenesin is an ether of guaiacol and glycerol, both of which can be sourced from biomass: glycerol from triglycerides, and guaiacol from (Eucalyptus) lignin (Scheme 14).247 Similarly, L-DOPA, a Parkinson's disease therapeutic, can be prepared from vanillin produced from technical lignin.248,249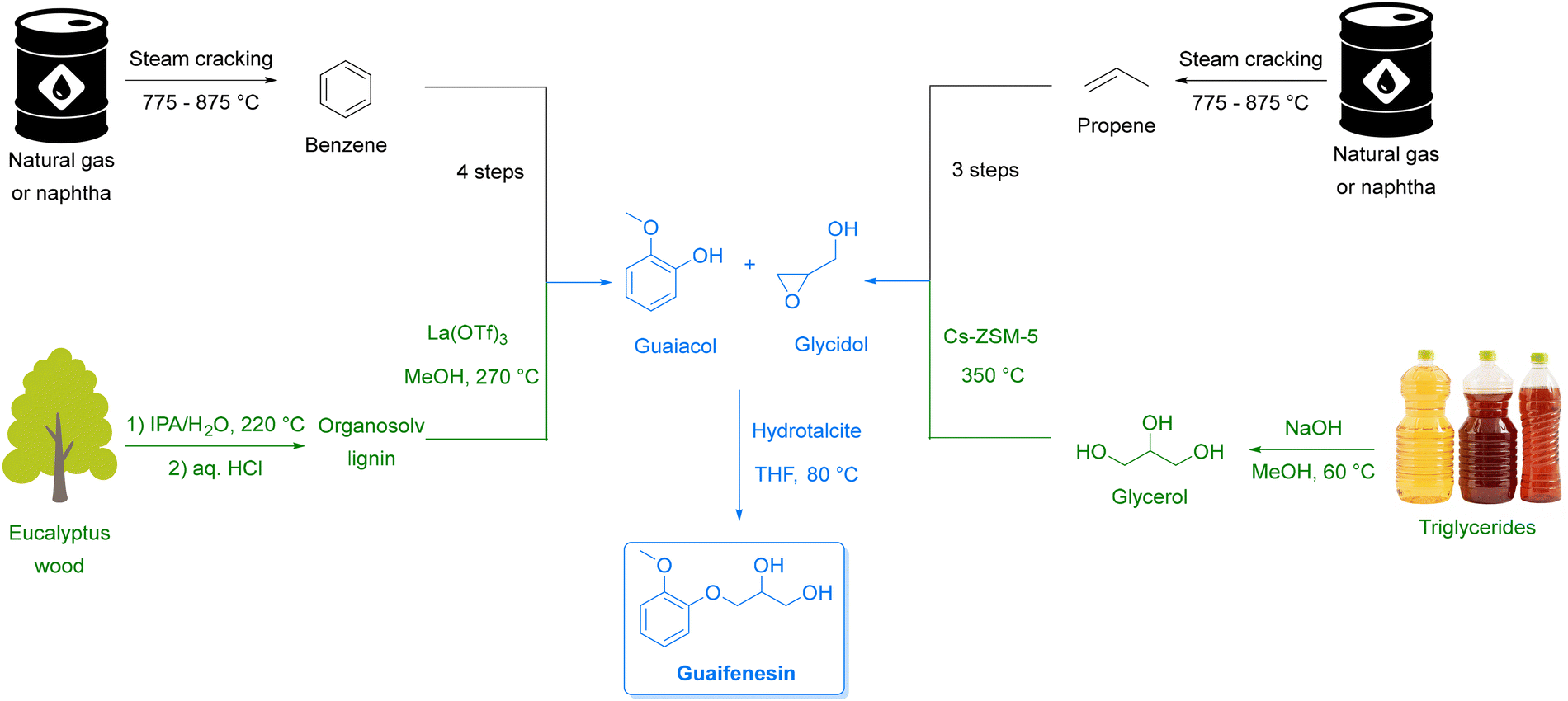 | ||
| Scheme 14 Synthesis routes to guaifenesin from biomass or fossil feedstocks. Color codes: green path = bio-based; black path = fossil-based; blue path = converged steps. Adapted from ref. 247 and 250–254. | ||
Phenol is a recurring structural motif in the pharmaceutical industry, present in at least 138 U.S. FDA-approved APIs.255 Notably paracetamol, the most prescribed analgesic worldwide,256 features a phenolic moiety. Given this prevalence, the sourcing of phenol from renewable feedstocks could yield substantial sustainability benefits. Lignin presents an appealing source in this context (cfr. Section 3.2).257 However, achieving truly sustainable API synthesis, requires not only renewable feedstocks but also greener reaction pathways. In the case of paracetamol, the conventional approach involves the nitration of phenol to yield 4-nitrophenol (Scheme 15, top). This step was also integral to the mechanochemical route discussed in Section 2.5 (Scheme 11).258 Reliance on nitration remains problematic, as it employs corrosive reagents, generates stoichiometric acetic acid waste, and results in an atom economy of just 54%.259 A more sustainable alternative involves the direct amidation of hydroquinone with ammonium acetate (Scheme 15, middle), achieving 96% conversion and 95% selectivity, with water as the by-product.259 This method eliminates salt waste, facilitates acetic acid recycling, improves atom economy to 81% and reduces the E-factor from 0.84 to 0.74. Although currently limited to batch operations and long reaction times, this route offers a compelling path forward, especially when hydroquinone is sourced from lignin-derived phenol.
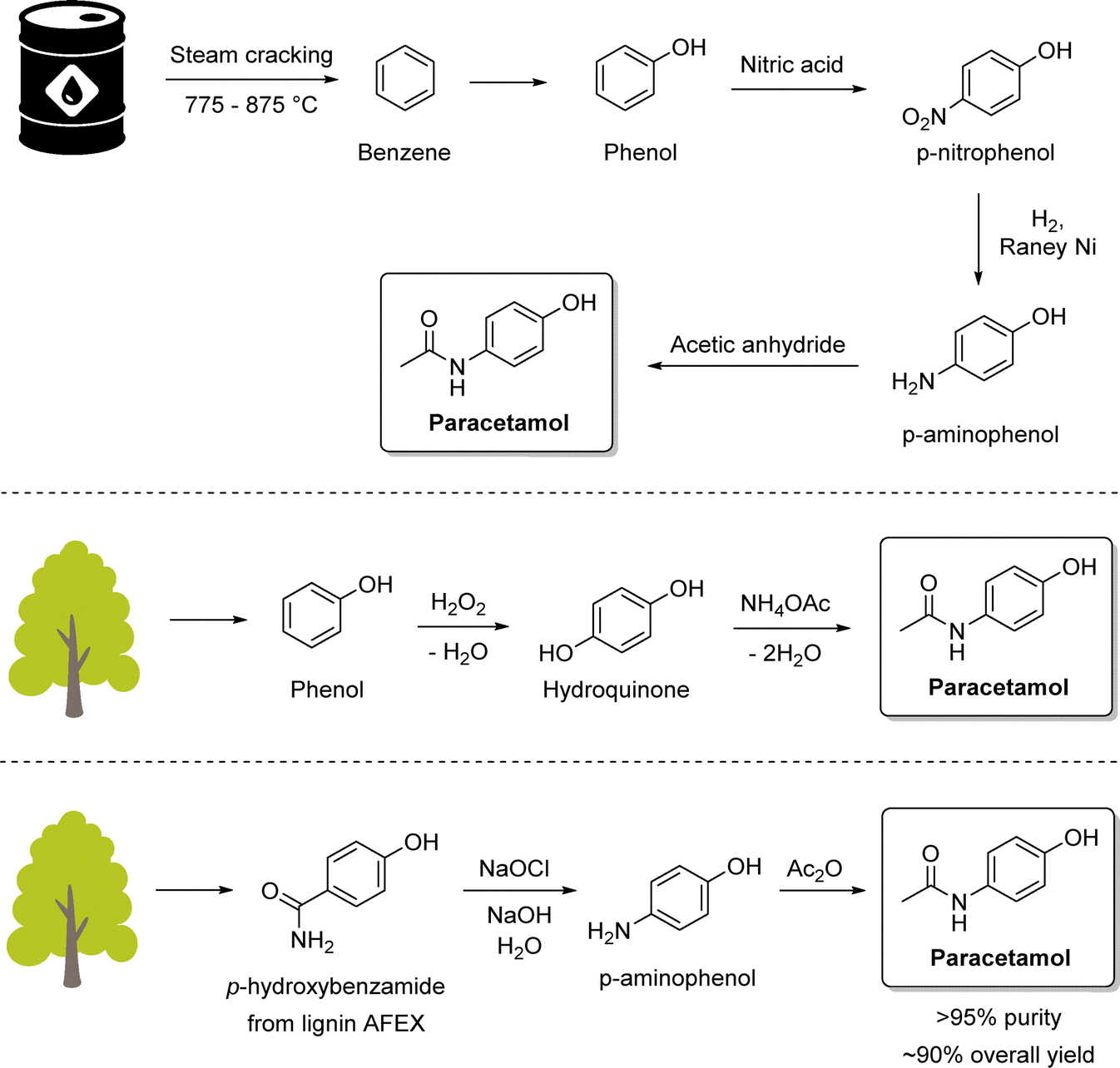 | ||
| Scheme 15 Synthesis of paracetamol from fossil fuel-derived phenol (top), lignin-derived phenol (middle), and lignin-derived p-hydroxybenzamide (bottom). Adapted from ref. 257 and 260. | ||
The second approach to produce fine chemicals from bio-based platform chemicals employs novel bio-based raw materials that lack direct petrochemical equivalents. Whereas fossil feedstocks are hydrocarbons requiring partial oxidation for functionalization, bio-based substrates come with inherent oxygen (and sometimes nitrogen) functionalities. This can bolster redox and step economy in fine chemical synthesis, but does require alternative synthetic strategies (e.g., hydrogen-borrowing).261
A striking example is a sustainable route to antihypotensive dopamine hydrochloride (Scheme 16).262 The established route utilizes vanillin derived from petrochemicals (85% of the total production) or from lignin, which is condensed with nitromethane followed by reduction (drop-in strategy).263 Alternatively, in the biomass only approach, softwood lignin is first depolymerized via acidolysis and stabilized with ethane-1,2-diol, after which 4-(hydroxyethyl)guaiacol is obtained by hydrogenation. Subsequent hydrogen-borrowing amination with ammonia followed by O-demethylation with strong acid yields dopamine hydrochloride.222 In the agrochemical realm, Maes et al. reported a softwood lignin-first route to obtain 4-propylcatechol,264 which can be converted to the fungicide diethofencarb (48% yield) over a mere 5 steps.219
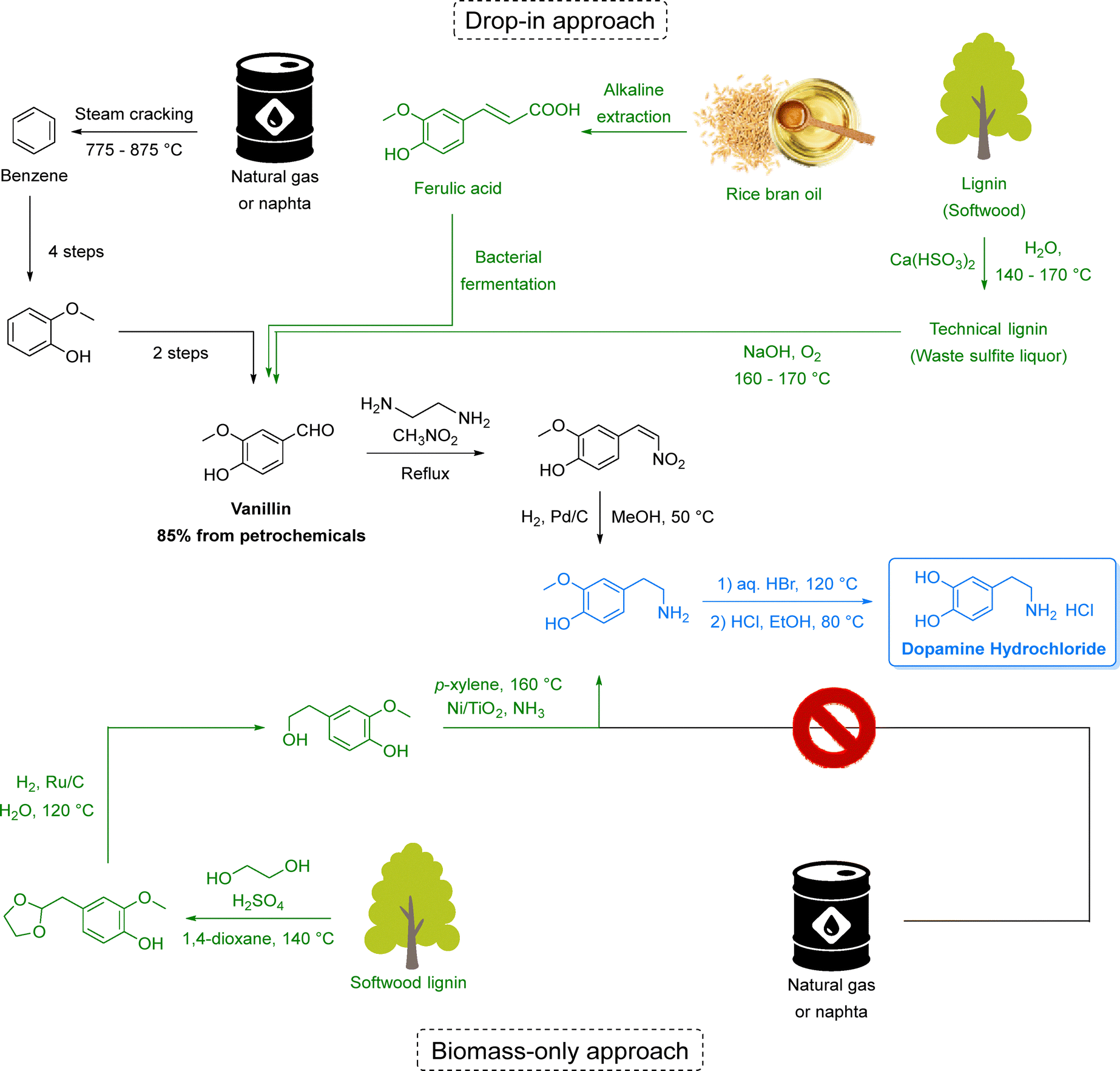 | ||
| Scheme 16 Established (drop-in approach, that is, from biomass/petrochemical-derived vanillin) and novel (biomass-only) routes to dopamine hydrochloride. Adapted from ref. 250, 262, 265 and 266. | ||
As noted earlier, while funneling lignin-derived aromatics into phenol is conceptually attractive, it requires several chemical steps.214 An elegant alternative circumvents phenol entirely by exploiting native p-hydroxybenzoate esters present in lignin derived from poplar and oil palm empty fruit bunches.260,267 Treatment of solid biomass with aqueous ammonia at 140 °C (ammonia fiber expansion or AFEX), yields p-hydroxybenzamide (pHBAm) in a single step (3.1 wt% yield). After solids separation and recovery via liquid–liquid extraction with ethyl acetate, pHBAm can be subjected to a Hofmann rearrangement under aqueous continuous-flow conditions to afford p-aminophenol. Subsequent acetylation furnishes paracetamol in >95% purity and ∼90% yield from pHBAm (Scheme 15, bottom).
Although adopting new drop-in or biomass-only routes for existing active ingredients is challenging due to altered impurity profiles, bio-based feedstocks undeniably offer substantial potential in defossilizing chemical raw materials. Indeed, literature reports indicate that bio-based pathways at pilot- or industrial-scale are already available for roughly 85% of petrochemical feedstocks.268 However, only a fraction of these has been commercialized and most still rely on first-generation feedstocks (crops), potentially competing with food supply. A large-scale example is furanedicarboxylic acid derived from fructose via HMF developed by Avantium.269 Notable examples based on second generation feedstock include certain solvents like ethanol and 2-MeTHF (Section 2). To the best of our knowledge, no current API or agrochemical is explicitly labelled as “bio-based”, though such a designation could be a compelling marketing approach.270
Several factors contribute to the slow adoption of bio-based routes. Foremost among these are economic considerations; as crude oil frequently costs under USD 100/barrel, pure bio-based synthetic routes struggle to be cost-competitive, especially since building a dedicated production facility requires significant investment.271 Additionally, the complexity inherent in synthesizing APIs and agrochemicals presents a significant challenge. These products often require multiple substrates, not all of which are currently available from bio-based sources—as exemplified by the use of nitromethane in the drop-in approach to dopamine hydrochloride (Scheme 16). A practical solution could be to adopt certification schemes based on 14C-traceable biocontent, as already practiced for commodity chemicals. This enables products such as ethyl acrylate to be marketed with a verifiable 40% 14C-traceable biocontent, since bioethanol is combined with fossil-derived acrylic acid.272 Such incremental certification strategies could facilitate a gradual, economically feasible, and smoother transition from a predominantly petroleum-based chemical industry toward a bio-based model.
Another argument is that many of the currently accessible bio-based raw materials are not necessarily more sustainable than the petroleum-based ones. This is evident from a 2023 meta-analysis by Zuiderveen et al., wherein 130 LCA's of biobased products were compared.273 They found that, while greenhouse gas emissions were on average reduced by 45%, significant environmental trade-offs were present for eutrophication (369% increase), acidification (41% increase), and land use (>5000% increase). Furthermore, large variations between individual products were observed, with some products having equal or even higher greenhouse gas emissions than their petrochemical counterpart! A relevant example is phenol, for which the greenhouse gas emissions are currently 40% higher if it is produced via a lignin-first biorefinery.274 Ultimately, it is evident that a “bio-based” label alone offers little certainty about a material's innate sustainability. An LCA-backed disclosure from suppliers is therefore vital to allow accurate comparisons across feedstocks and motivate the pharmaceutical and agrochemical industry to adopt truly sustainable feedstocks.
Finally, widespread adoption remains limited owing to elevated costs, limited supply, competition for biomass resources and reluctance of downstream customers to absorb higher prices. For instance, analysis by McKinsey & Company estimates that shifting to bio-based feedstocks (and solvents) carries an abatement cost of up to $300 per ton CO2.3 As a result, profitability is unlikely in the next 5–10 years without regulatory or financial incentives. In other words, despite technical feasibility, factors such as synthetic complexity, certification needs, sustainability trade-offs and economic constraints mean we remain closer to the beginning than the end of the transition to renewable raw materials.
4. Next generation catalysts
Although solvents and fossil-based raw materials constitute the primary source of waste and emissions in API and agrochemical production, a sizable portion of historical waste stemmed from the sector's heavy reliance on stoichiometric reagents. In earlier practices, reductions required metals and metal hydrides (such as Na, Mg, Zn, NaBH4, and LiAlH4), oxidations employed chromium(VI) compounds or permanganate, and large quantities of mineral acids (e.g., H2SO4), Lewis acids (e.g., AlCl3, ZnCl2), and bases (e.g., NaOH, KOH) were used, either as reagents or during work-up procedures.13 It was therefore not uncommon for such reactions to afford over ten times more waste than product.275 By contrast, greener-by-design manufacturing demands catalytic methods, whether homogeneous, heterogeneous, or biocatalytic, to dramatically reduce waste.Catalytic technologies were initially underutilized for several reasons. The small production volumes compared to commodity chemicals reduced the immediate pressure to minimize waste. Moreover, although less sustainable, time-tested stoichiometric methods offered broad applicability and short development times, helping companies meet tight development and market deadlines. Once these processes became established, modifying them proved challenging due to regulatory hurdles. Nevertheless, the imperative need for catalytic approaches was continuously highlighted,30,276,277 resulting in a significant shift toward catalytic methods over the past three decades. After all, the increasing complexity of the chemical structures which need to be synthesized cannot only rely on non-catalytic reactions considering the synthesis route needs to be scaled. Techniques such as hydrogenation, carbonylation, hydroformylation, and olefin metathesis, once prevalent only in commodity and material chemical production, are now widely adopted and have proven instrumental in step economy, reducing waste and enhancing selectivity under milder conditions, among other benefits.278 When catalytic methodologies are not feasible, it is essential to use inexpensive reagents that produce benign, recyclable waste streams. Reflecting their critical role, catalytic methodologies have been honored with several Noble prizes over the past three decades: for asymmetric hydrogenation and oxidation reactions in 2001;279 for olefin metathesis in 2005;280 for palladium-catalyzed cross-couplings in 2010;281 for directed enzyme evolution in 2018;282 for asymmetric organocatalysis in 2021;283 and for copper-catalyzed click (and biorthogonal) chemistry in 2022.284
Catalysis continues to evolve to meet modern demands. In homogeneous catalysis, both industry and academia are exploring how Earth-abundant catalysts compare and complement traditional precious metal systems (Section 4.1). Simultaneously, single atom catalysts and related next-generation systems target long standing issues related to the stability, selectivity, and applicability of heterogeneous catalysts for fine chemicals synthesis (Section 4.2). Finally, biocatalysis (Section 4.3) has garnered growing recognition in industry, frequently offering a more selective route than traditional catalytic protocols, particularly when introducing chiral centers. In parallel, catalytic technologies such as electrocatalysis and photocatalysis are also playing an increasingly critical role and will be discussed in greater detail in Section 5.
4.1. Homogeneous catalysis
In the fine chemical industry, the use of transition metal catalysis is particularly well established. For instance, palladium is ubiquitous in forming carbon–carbon and carbon–heteroatom (primarily C–N) bonds; rhodium, ruthenium, and iridium are critical for asymmetric hydrogenations; and ruthenium is synonymous with olefin metathesis.285 Consequently, platinum group metals (PGM) dominate most industrial catalytic transformations. In stark contrast, base metal catalysts are confined to a limited set of applications, most notably cross-coupling reactions.286,287 However, straightforward comparisons of price, natural abundance, and GWP indicate that base metals may, in principle, offer a more sustainable alternative to PGM catalysts (Table 2).| Element | Price (EUR per kg) | Price (EUR per mol)288 | Earth abundance (ppb by weight)289 | Limit API (oral use, ppm) by EMA290 | GWP (kg CO2-eq per kg)291 |
|---|---|---|---|---|---|
| Ir | 153450 |
29462 |
0.4 | 10 | 8860 |
| Pd | 35870 |
3802 | 6.3 | 10 | 3880 |
| Rh | 157580 |
16230 |
0.7 | 10 | 35100 |
| Ru | 16010 |
1616 | 1 | 10 | 12500 |
| Al | 2.37 | 0.064 | 8.2 × 107 | 8.2 | |
| Fe | 0.41 | 0.023 | 6.3 × 107 | 1300 | 1.5 |
| Ca | 3.35 | 0.134 | 5.0 × 107 | 1.0 | |
| Ti | 5.72 | 0.275 | 6.6 × 106 | 8.1 | |
| Mn | 1.65 | 0.091 | 1.1 × 106 | 250 | 1.0 |
| V | 25.94 | 1.323 | 1.9 × 105 | 10 | 33.1 |
| Ni | 15.56 | 0.918 | 9.0 × 104 | 20 | 6.5 |
| Zn | 2.81 | 0.182 | 7.9 × 104 | 1300 | 3.1 |
| Cu | 8.77 | 0.557 | 6.8 × 104 | 300 | 2.8 |
| Co | 22.46 | 1.325 | 3.0 × 104 | 5 | 8.3 |
| Mo | 62.40 | 5.990 | 1.1 × 103 | 300 | 5.7 |
A principal drawback of PGMs is their price, which is over three orders of magnitude higher than that of base metals.288 This high price stems directly from their minimal abundance in the earth's crust,289 and is further compounded by the geographic concentration of reserves with 99% of them located in South-Africa, Zimbabwe, and Russia.292 Such restricted supply chains introduce volatility in stock prices, complicating long-term planning.293 Therefore, some precious metal producers adopted chemical leasing to partially address these issues.294 PGMs also exhibit high environmental impacts.291 For instance, the GWP of palladium is 3880 kg CO2-eq per kg, compared with 6.5 kg CO2-eq per kg for nickel and just 1.5 kg CO2-eq per kg for iron. This is primarily due to the exceedingly low PGM concentrations in mineral deposits. For PGMs, extraction and refining account for as much as 90% of their total GWPs, compared to under 20% for metals like iron.295 When PGMs are recycled, their associated GWPs can decrease substantially, though not entirely eliminated due to the energy requirements of refining. For recycled palladium, estimates suggest a reduction of up to 90%,295 bringing the GWP down to around 400 kg CO2-eq per kg. Similarly, recycled nickel may have a GWP closer to 2.5 kg CO2-eq per kg, while recycled iron can drop to below 1 kg CO2-eq per kg, depending on the recycling process and energy source used.295 Nevertheless, even accounting for recyclability, noble metals such as palladium remain orders of magnitude more carbon-intensive than base metals, underscoring the importance of minimizing their use and identifying substitutes where possible.
Moreover, PGMs typically require stringent controls for trace impurities in pharmaceutical and agrochemical products. Official limits set by the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) are stricter for PGMs than for many base metals—10 ppm for PGMs, compared to 20 ppm for nickel, 300 ppm for copper, and 1300 ppm for iron and zinc.290 Consequently, manufacturers must implement rigorous purification processes and analytical testing to ensure compliance with these stringent regulatory requirements. Nevertheless, there is debate over whether their toxicity truly exceeds that of base metals,296 as it must be emphasized that some earth-abundant base metals are also far from benign. Cobalt, for instance, carries a tighter ICH limit (5 ppm for oral drugs up to 10 g day−1) and appears on REACH restriction lists due to occupational hazards including carcinogenicity. Indeed, the overall regulatory and safety landscape for base metals is far from straightforward.
In this context, although PGMs are justifiably flagged for their cost and environmental burden, and score poorly on indicators such as resource depletion, land use, and toxicity, a recent LCA by Novartis has shown that their contribution to the overall process carbon footprint can be relatively minor when employed in catalytic amounts.297 In such cases, PGMs can even indirectly reduce environmental burden by enabling shorter routes or less resource-intensive conditions. The real challenge lies in balancing catalytic efficiency with the full lifecycle impact of metal use. In this context, noteworthy achievements in reaction engineering have enabled Pd-catalyzed processes such as the Heck and Suzuki reactions to be successfully scaled up even in highly cost-sensitive, high-volume sectors like agrochemicals. A prime example is the synthesis of SDHI fungicides such as boscalid and bixafen, where innovations such as the use of boronic esters have significantly reduced boron waste, while maintaining ppm-level Pd catalyst loadings.298–300
Overall, these examples call for a more holistic view of metal sustainability, where compliance is not only a matter of meeting numerical thresholds, but also of ensuring that purification, analytical controls, and process safety are fully integrated into the design of scalable, cost-effective and responsible manufacturing routes.
Consequently, research into base metal catalysis has lagged for many years. However, the highly volatile cost of PGMs and potential supply chain issues, in addition to their huge environmental impact, has recently shifted attention toward base metals.301 Industry engagement with this topic is exemplified by the “Recent Advances in Nonprecious Metal Catalysis” review series authored by process chemists from Pfizer, AbbVie, and Boehringer Ingelheim.304 This renewed interest makes it crucial to select the best catalyst on a case-by-case basis, ideally through a predictive cradle-to-gate life cycle assessment.305 However, comprehensive LCAs, as alluded to in the introduction, hitherto remain underexploited during the R&D stage. While accelerated methods, like the fast life cycle assessment of synthetic chemistry (FLASC™) exist, they are seldom employed.28 In most instances, process decisions rest on simple mass-based metrics such as the E-factor and PMI.35,190,277 Nevertheless, as catalysts are inherently used in minute quantities, their mass contributions are negligible in these metrics, even though their environmental impact can be disproportionately high.191 Indeed, the strong correlation between metal price and GWP can serve as an indirect proxy to gauge their environmental impact (Fig. 8).
 | ||
| Fig. 8 A log–log plot correlating GWP (kg CO2-eq per kg) with price (EUR per kg) for precious and base metals using data from Table 2. The regression coefficient, being close to unity, indicates a linear relationship between price and GWP. | ||
An alternative is the C-factor, which measures CO2 emissions per kilogram of product.306 This metric allows the GWP of catalysts to be factored into the overall process impact. However, this is also not widely applied in the pharmaceutical industry due to the complexity of drug development, strict regulatory requirements, and challenges in obtaining comprehensive environmental data. In addition, pharmaceutical production involves intricate supply chains, making it difficult to standardize LCA methodologies (developed for the petrochemical sector) in and across different fine chemical product classes. The absence of industry-wide LCA frameworks complicates comparisons between different active ingredients, making it hard to establish consistent sustainability benchmarks. A similar picture emerges for complex agrochemicals. Nevertheless, regardless of the metric chosen, the use of green metrics early in the development cycle can allow us to design greener chemical processes from readily available starting materials and should be readily promoted. This will also be crucial in guiding whether, and to what extent, efforts should focus on catalyst design/substitution versus the other three core synthetic pillars discussed in this review, to ensure the greatest overall environmental benefit.297
Rather than striving for the complete replacement of PGMs with base metals, the industry is exploiting the distinct reactivities that base metals offer. These opportunities exist both for established reactions, where the substrate scope can be broadened or complemented, as well as for completely new transformations, through previously inaccessible synthesis routes not limited to the typical propensity of base metals for one electron processes.307
For example, despite palladium's dominance in cross-couplings, this metal has known drawbacks, such as slow oxidative addition with C(sp3)–electrophiles, lower reactivity toward C–O bonds, and a propensity for β-hydride elimination in alkyl–palladium intermediates.308 By contrast, nickel's heightened nucleophilicity facilitates the activation of less reactive C–O bonds, and its alkyl–nickel species undergo β-hydride elimination more slowly, making nickel superior for C(sp3)–X cross-couplings as demonstrated in recent literature.308–310 Analogously, iron can provide comparable benefits, provided more strongly activated nucleophiles such as organomagnesium or organozinc reagents are utilized.311,312 A compelling demonstration is Bristol-Myers Squibb's synthesis of a dual NK1/serotonin receptor antagonist, where an inexpensive 5–7 mol% Fe(acac)3 catalyst efficiently coupled a chloropyridine to cyclopropyl magnesium bromide, affording yields of 94–96% (Scheme 17).313
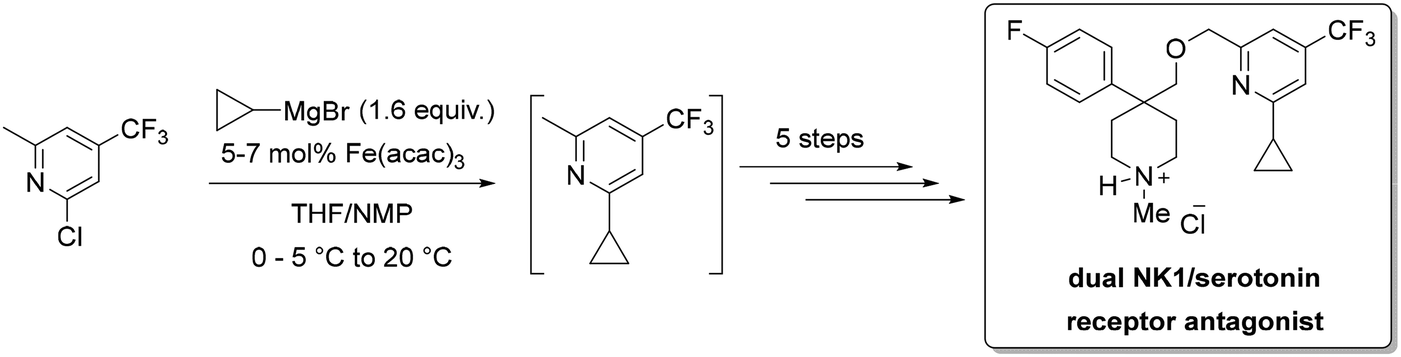 | ||
| Scheme 17 Fe(III)-catalyzed Grignard coupling in the synthesis of a dual NK1/serotonin receptor antagonist. This iron-catalyzed transformation demonstrates the effectiveness of an earth-abundant base metal as a sustainable alternative to traditional precious-metal catalysts in complex API synthesis. Adapted from ref. 313. | ||
In the field of enantioselective hydrogenations, rhodium and iridium are often unable to efficiently hydrogenate tri- and tetra-substituted double bonds or unfunctionalized substrates.314 Herein, base metals, especially cobalt, have shown significant promise.315 Highly active catalytic systems featuring bisphosphine and bisphospholane ligands have emerged,316 influenced by Chirik's seminal work on in situ reduction of bench-stable cobalt(II) chloride to cobalt(I) with zinc.317 This approach prevents deactivation caused by methanol (solvent) ligand displacement.317 One notable application is the asymmetric hydrogenation of a tri-substituted alkene to yield antiepileptic levetiracetam on a 200 g scale (Scheme 18). This route required only 0.08 mol% of cobalt catalyst, compared to 0.5 mol% rhodium in the patented route.318
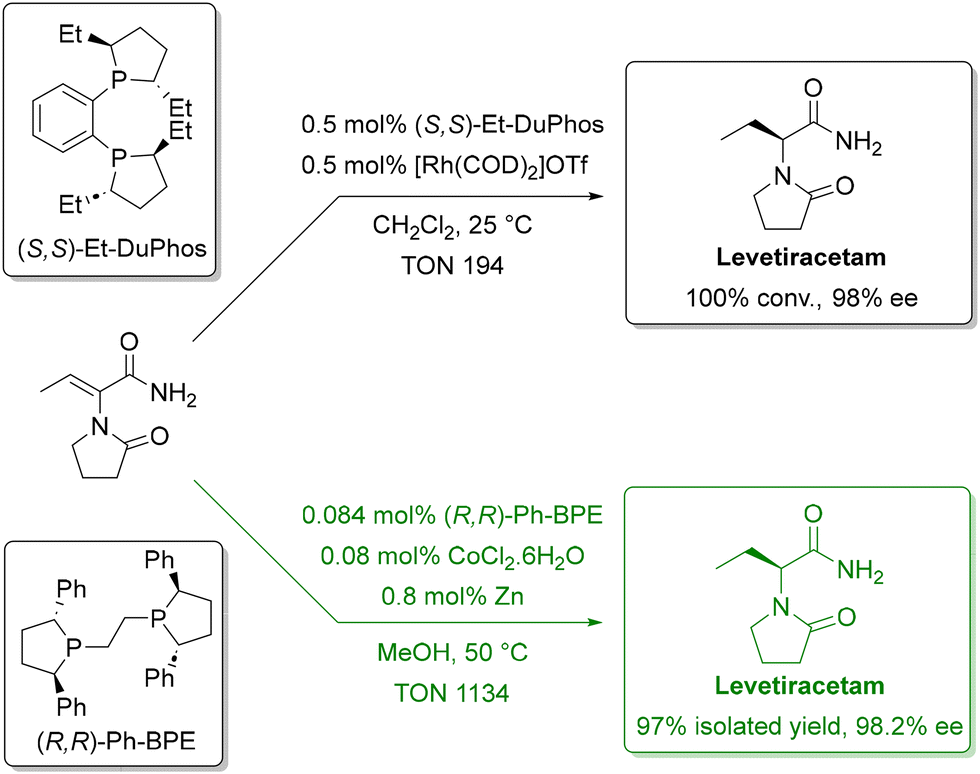 | ||
| Scheme 18 Asymmetric hydrogenation of the tri-substituted alkene in dehydro-levetiracetam with a rhodium (black) or cobalt catalyst (green). Replacing Rh with a cobalt-based catalyst paired with (R,R)-Ph-BPE enables the same key reduction in MeOH at only 0.08 mol% Co, achieving higher TON and excellent yield and ee. This example highlights how modern ligand design allows base metals such as Co to match or surpass the performance of PGM catalysts. Adapted from ref. 317. | ||
While industrial adoption of novel reactions can be slow and hindered by narrow substrate scopes, such innovations are critical when established methods prove insufficient.319 A striking illustration is Pfizer and Curia's cobalt-catalyzed cyclopropanation in the synthesis of nirmatrelvir, an antiviral used to treat SARS-CoV-2 (Scheme 19).320 Amid pressing pandemic demands, they adapted Uyeda's cobalt-based methodology to construct a bicyclic[3.1.0]proline intermediate.321 This approach effectively sidestepped supply chain bottlenecks and waste-intensive steps present in existing routes. The protocol was successfully scaled to 205 kg, furnishing the key intermediate on a multi-ton scale (across several batches) in six rather than eight to eleven steps, underscoring the value of this innovative disconnection.320 Nonetheless, the high cobalt catalyst loading (15 mol%) required for the transformation should be noted. Given the EMA's stringent 5 ppm limit for Co residues, which is stricter than for most PGM metals (Table 2), the metal will likely need to be controlled from the final product, adding to the complexity and cost of this protocol, though use of a metal-catalyzed step early on in a synthetic route is less problematic as the metal content naturally decreases in each subsequent step.
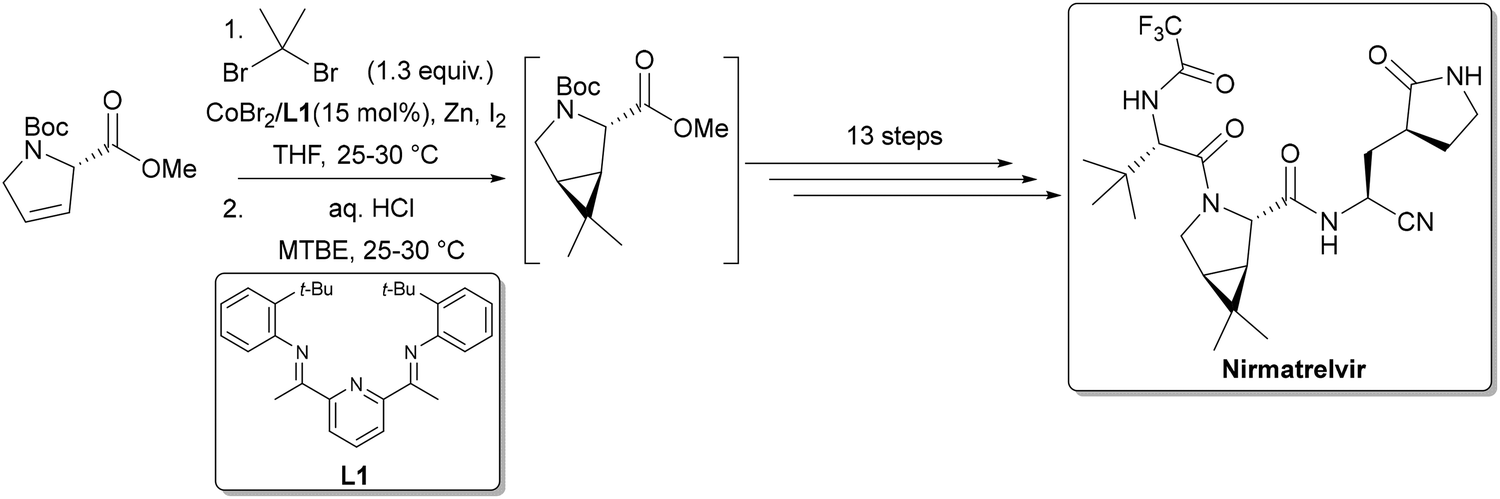 | ||
| Scheme 19 Cobalt-catalyzed cyclopropanation step in the synthesis of nirmatrelvir. Adapted from ref. 320. | ||
Another example of base metals delivering remarkable reactivity is copper-mediated Ullmann-type cross-couplings.286 Although discovered by Ullmann in 1901 and by no means novel, these transformations remain highly relevant to this day.322 Numerous industrial applications have been reported, including an Ullmann C–N coupling for the synthesis of tyclopyrazoflor, an insecticide.323 Corteva's initial [3+2] cyclization strategy raised concerns over safety, scalability, and cost.324 Three modes of conducting the reaction were explored: early-, mid-, and late-stage Ullmann coupling, ultimately selecting the mid-stage one for scale-up. In this pivotal step, a bromopyridine was coupled to a pyrazole intermediate in 70–75% yield using 3 mol% CuCl and 14 mol% N,N-dimethylethylenediamine. Demonstrating the robustness of the overall five-step route, the protocol was scaled to above 50 kg (Scheme 20). Potential room for improvement still exists for future development, as DMF, an obvious candidate for substitution, was the solvent of choice for this campaign.
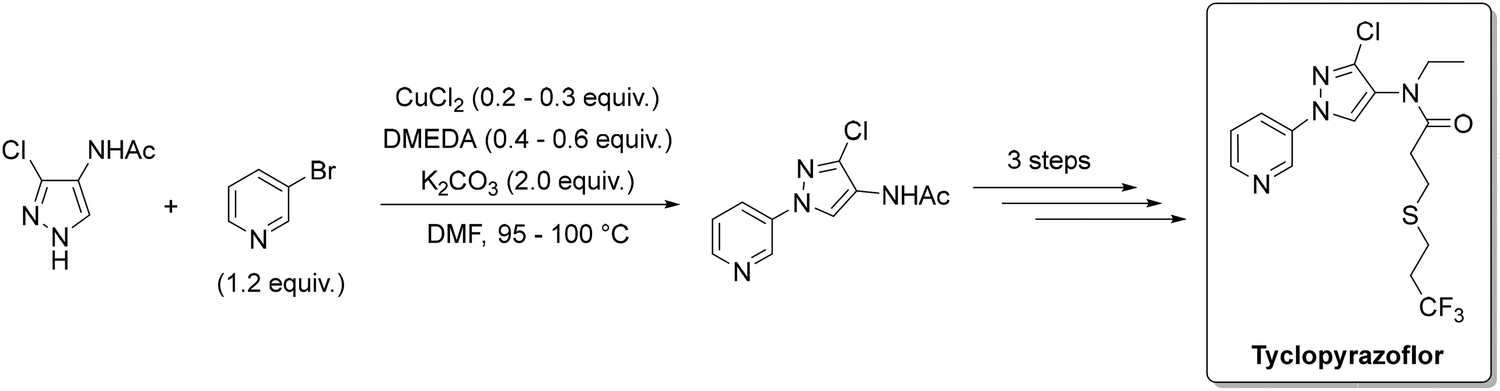 | ||
| Scheme 20 Copper-catalyzed Ullmann C–N coupling step in the synthesis of tyclopyrazoflor. Adapted from ref. 324. | ||
It must be emphasized that base metals are no panacea as they present their own distinct set of environmental and safety drawbacks, some more severe than others.293 In numerous scenarios, base-metal catalysts require higher loadings, harsher reaction conditions, more expensive ligands, less sustainable solvents, or additional additives; all factors that can negate potential benefits.325 Moreover, unlike PGMs, base metals typically lack established recycling mechanisms due to limited economic incentives.326,327 As a result, although many base-metal catalysts now operate at development and industrial scale,286,293,301,328 fully replacing PGMs is unrealistic from a holistic perspective.
To effectively move toward greener-by-design active ingredients for pharmaceuticals and agrochemicals, we must harness every available resource and methodology. In this context, heterogeneous catalysts have the potential to minimize the metal content in catalytic systems while maximizing efficiency, thus fully embodying the ‘less is more’ philosophy. Far from competing, these catalysts largely complement their homogeneous counterparts, requiring a careful catalyst selection for each individual application.
4.2. Heterogeneous catalysis
The previous subsection examined to what degree base (or even metal-free) catalysts might feasibly replace the dwindling and highly volatile supply of precious metal catalysts. However, regardless of the metal present, homogeneous catalysts are inherently limited by their nature. For instance, they generally require ligands, whose cost can be higher per mole than the metal itself, and it is exceedingly difficult to recover them.331,332 Even more critically, many homogeneous catalysts suffer from instability, a challenge that has attracted less academic attention despite its significance. This deactivation can stem from substrate or product inhibition, oxidative stress, undesired substrate reactions, inadvertent admission of air or moisture, lability of metal–ligand bonds, or ligand degradation.333At first glance, heterogeneous catalysts appear to address many of the limitations of homogeneous catalysts. For instance, straightforward recovery, reuse, and recycling are often touted, but not always realized, benefits.334 Indeed, the bulk and commodity chemical industries, where slim profit margins make catalyst recovery essential, already employ heterogeneous catalysis in up to 90% of industrial processes.335 In fine chemical production, however, the much smaller scales and higher product value diminish the immediate need to recover and reuse catalysts. Yet, this alone does not fully explain homogeneous catalysis’ dominance in APIs and agrochemicals. In homogeneous systems, the active site is a well-defined molecular complex, making it easier to understand catalyst performance through spectroscopic tools.336 The structure of the complex can then be tailored based on mechanistic insights, often by modifying the ligand or adjusting reaction conditions to optimize its performance. There exists, in essence, a rational link between the design of these catalysts and their performance. By contrast, heterogeneous catalysts typically exhibit non-uniform active sites prone to dynamic structural changes during the course of a reaction, which can lead to selectivity loss or degradation. Selectivity loss is a concern, as the formation of side products not only reduces overall efficiency but also brings additional purification steps to isolate the product, increasing waste generation, energy consumption, and process complexity. Moreover, the possibility of metal leaching is also particularly problematic in pharmaceutical manufacturing, where strict purity standards apply.337 The wide gamut of (potentially harsh) reaction conditions employed for fine chemical synthesis compared to the relatively simple transformations of bulk chemicals further magnifies these challenges.338 It is therefore essential but hugely challenging to engineer solid catalysts that afford efficient but also stable activity.
Nevertheless, several heterogeneous catalyst design strategies have been proposed in recent decades. To maintain focus on catalytic design and inherent challenges, and given that catalyst performance varies significantly with the reaction in which they are employed, this section presents examples of heterogeneous catalysts for fine chemicals primarily focusing on cross-coupling reactions (C–C and C–X bond formation). These reactions are especially important in the pharmaceutical sector, with Suzuki–Miyaura couplings being the most prevalent Pd-catalyzed process and accounting for about 19% of all reactions in drug discovery.339
Two primary categories of industrial heterogeneous catalysts are metallic and supported metal catalysts. The latter are commonly nanoparticles of metals, metal oxides, or metal sulfides dispersed on high-surface-area materials like Al2O3, SiO2, TiO2, or carbon.340 Within this class, palladium-based catalysts (e.g., Pd black, Pd/C), have become emblematic for hydrogenation protocols for both bulk and fine chemistry.341 Contrarily, their application to more complex organic transformations, including Suzuki–Miyaura has lagged behind on an industrial scale over the past decade.339 This is likely because the method typically performs well only for relatively simple systems, but introducing multiple coordinating nitrogens or significant steric hindrance often leads to poor conversion and diminished process robustness.
Nonetheless, researchers at Eli Lilly employed a Suzuki–Miyaura coupling to synthesize the key intermediate to LY503430, a potential treatment for Parkinson's disease (Scheme 21).342 The archetypal homogeneous Pd(OAc)2/PPh3 system delivered sub-optimal yields (∼70%) and a purity below 95%. Substituting the homogeneous catalyst with Pd black afforded the single enantiomer in 88% yield on a 500 g scale. This heterogeneous approach also leached minimal (3–8 ppm) Pd amounts leading to a product purity of 99.8%. It should be noted that the researchers did not examine the recyclability of the catalytic system. Also in this case, a holistic perspective could lead to additional improvements in the future: in fact, while the use of Pd black is very efficient, the use of CDI in the subsequent step still produces 2 equiv. of imidazole waste.
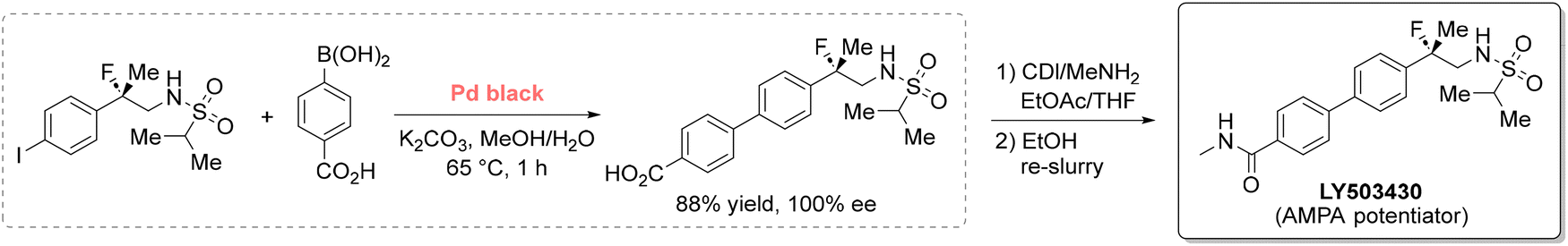 | ||
| Scheme 21 Pd black-catalyzed Suzuki–Miyaura cross-coupling toward the synthesis of LY503430, an AMPA Potentiator. Adapted from ref. 342. | ||
Meanwhile, Oril Industrie executed a multikilogram-scale synthesis of a potent Bcl-2/Bcl-xL antagonist, which promotes apoptosis in cancer cells (Scheme 22).343 Due to low purity and intellectual property concerns over the catalyst phosphine ligand, a new approach was sought. After screening catalysts with varying oxidation states, Pd loadings, and water content, Pd/C type 394 delivered the key benzyl chloride intermediate in 97% purity and boosted its yield by 30% compared to the original route. Importantly, the heterogeneous catalyst allowed the Suzuki–Miyaura coupling to be telescoped with the subsequent chlorination step, meaning mutagenic benzyl chloride did not have to be handled in powder form. In this case, the long-term stability, recyclability, or palladium leaching levels were not investigated.
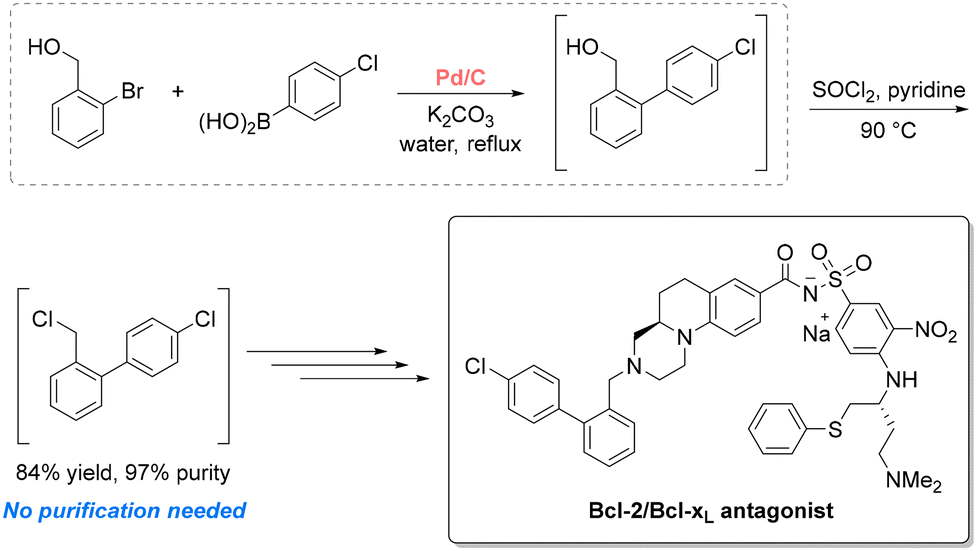 | ||
| Scheme 22 Pd/C-catalyzed Suzuki–Miyaura cross-coupling toward the synthesis of Bcl-2/Bcl-xL antagonist. Adapted from ref. 343. | ||
The select case studies above point toward the predilection of inexpensive and easily separable supported Pd-based nanoparticles in Suzuki–Miyaura couplings. However, these species show inconsistent reusability behavior and lower activity compared to the last generation of homogeneous catalysts, issues partly attributed to their uneven active site distribution.344
Several strategies exist to design solid materials with well-defined structures. One of the most established involves anchoring homogeneous complexes onto solid surfaces in the hopes of retaining their catalytic performance while enabling reuse. However, such heterogenization often introduces steric hindrance through the polymeric framework, diminishing activity.345
A compelling example comes from SiliCycle who introduced a family of organically modified silica-supported Pd catalysts (Scheme 23, top), suitable for Suzuki–Miyaura, Mizoroki–Heck, Negishi, and Sonogashira reactions.346,347 The SiliaCat catalysts can be filtered and reused up to five times with Pd and Si leaching below 1 ppm and 50 ppm, respectively. However, their stability during successive catalytic cycles is highly substrate dependent. For example, in a 100 g Suzuki–Miyaura step toward the synthesis of antihypertensive Valsartan (Scheme 23, bottom), the SiliaCat DPP-Pd catalyst initially performed excellently (100% conversion – 98% yield) but suffered significant activity loss in the second (88% conversion), third (57% conversion), and fourth (30% conversion) cycle.348
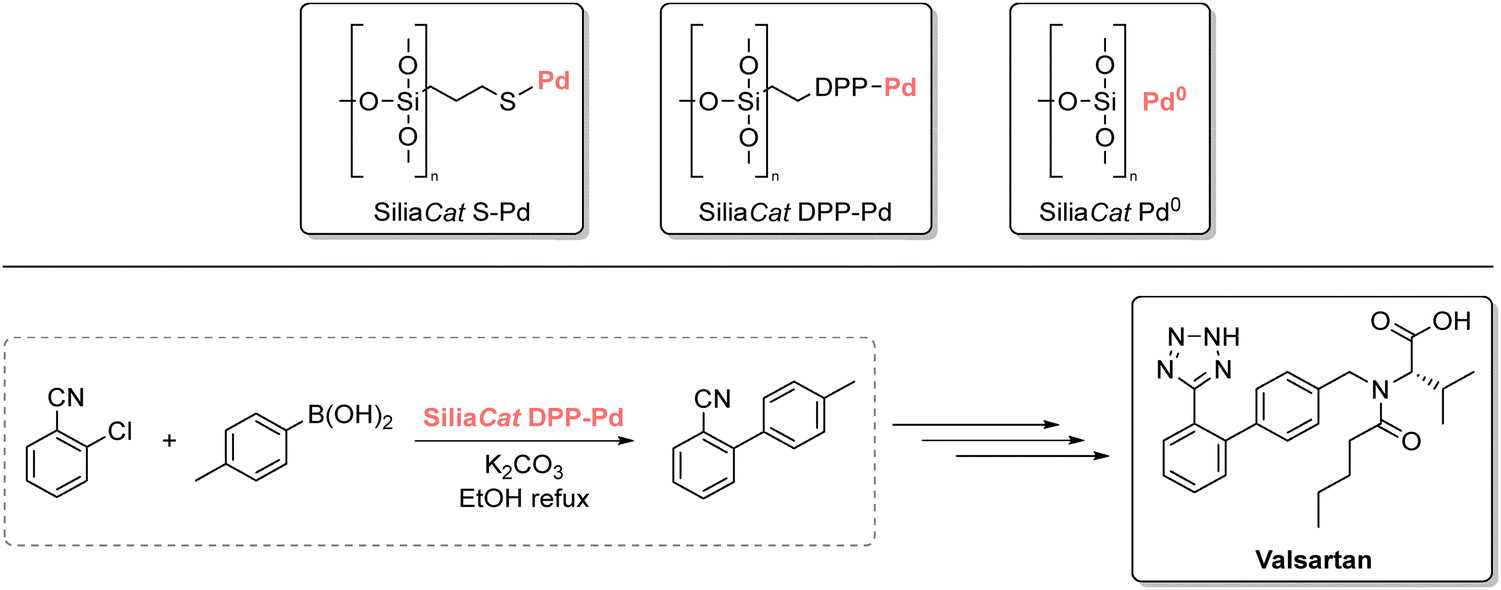 | ||
| Scheme 23 Structures of SiliaCat palladium-based catalysts (top) and SiliaCat DPP-Pd-catalyzed Suzuki–Miyaura cross-coupling towards the synthesis of valsartan (bottom). The top panel shows a series of SiliaCat palladium-based catalysts, in which Pd is immobilized within an organosilica matrix to maintain a well-defined, active form. This controlled environment enables cross-coupling reactivity using solid catalysts. Although this design aims to limit Pd release, these materials can still suffer from measurable leaching and generally offer limited recyclability. The bottom panel exemplifies this aspect through a SiliaCat DPP-Pd-catalyzed Suzuki–Miyaura coupling reaction, illustrating how these materials are applied in practice despite the persistent challenge of achieving truly leach-free, reusable heterogeneous systems. Adapted from ref. 346–348. | ||
These examples represent only a fraction of the extensive research on heterogeneous catalyst design strategies, a subject expertly reviewed elsewhere.349–351 Still, they reveal a common pattern: whether as supported nanoparticles or immobilized complexes, heterogeneous catalysts often undergo dynamic structural changes that hinder reusability—the very benefit championed as a reason to switch from homogeneous catalysis. This is especially problematic given that several industrial techniques exist for metal recovery from homogeneous catalysts.352,353 For this reason, in a well-known opinion paper, de Vries and Farina succinctly observed that “almost all methods of heterogenization of homogeneous catalysts that have been invented over the years will never be used in industry, for the simple reason that these methods do not reduce catalyst-related costs but rather increase them”.345 Consequently, to commercialize heterogeneous catalysts, it is essential to consider long-term stability rather than solely high activity. A range of academic innovations are poised to make this shift possible.
In this context, single-atom catalysts (SACs) are attracting particular attention as they confine isolated metal atoms on a solid support (Fig. 9a).355,356 This maximizes atom utilization efficiency and confers high selectivity, since uniform, single-atom sites favor specific reaction pathways and products.357 Moreover, SACs often exhibit unique electronic and geometric properties, that improve selectivity and minimize side reactions compared to conventional heterogeneous catalysts. Over the last decade, rational catalyst design has created SACs with performance rivaling homogeneous systems,358 utilizing strong electronic or covalent metal–support interactions for added stability.359 For instance, SACs featuring iron, cobalt, and nickel atoms isolated on nitrogen-doped carbon supports, have shown exceptional catalytic activity in oxygen reduction, hydrogen evolution, and carbon dioxide reduction.360 Although, their application in more complex pharmaceutically-relevant transformations is still emerging, several Pd-based SACs have already shown promise in Suzuki–Miyaura coupling.361,362
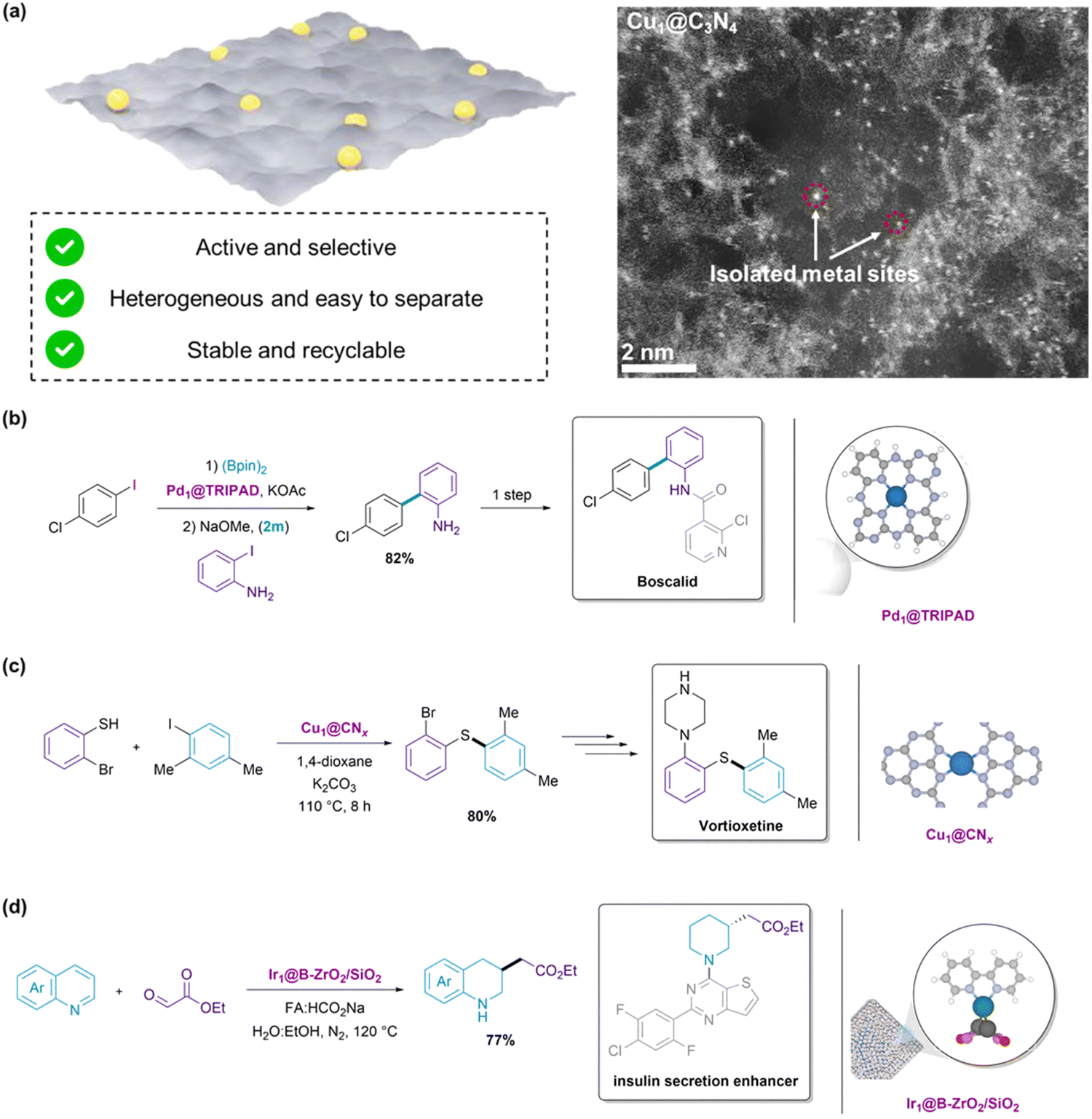 | ||
| Fig. 9 Representative SACs and their application in organic synthesis (a) structural model and HAADF-STEM image of a Cu SAC supported on carbon nitride. (b) Pd SAC anchored on a covalent organic framework used in one-pot cross borylative coupling. (c) Cu SAC supported on mesoporous carbon nitride for thiocoupling reactions. (d) Ir SAC immobilized on a metal organic framework for the synthesis of γ-amino acids, esters and ketones. Reproduced from ref. 363–365, with permission from Wiley and the American Chemical Society, copyright 2025. | ||
Chen et al. impregnated Pd single atoms on exfoliated graphitic carbon nitride via microwave-assisted wet-impregnation.362 Here, Boudart's turnover frequency (TOF) is a key metric that highlights how effectively each active site drives the reaction.366 The catalyst was tested at Idorsia Pharmaceuticals and achieved a TOF of 549 h−1, outperforming commercially available homogeneous and heterogeneous Pd systems for a broad range of substrates. Prolonged operation (12 h) resulted in no detectable metal leaching demonstrating the catalyst's stability. Similarly, Liu et al. employed porphyrin precursors to construct a two dimensional Pd catalyst in a M–N3C1 coordination environment.367 Although the performance of this catalyst (3.8 h−1 TOF) paled in comparison to the previous one, it was applied toward the late-stage functionalization of indomethacin, an anti-inflammatory drug.
Single atoms need not be restricted to two-dimensional surfaces. Ji et al., for example, anchored Pd single atoms on UiO-66-NH2, a Zr-based metal–organic framework (MOF) enabling mild Suzuki–Miyaura cross-coupling of a diverse range of substrates.368 Notably an exceptional TOF of 13000 h−1 was achieved, a result attributed to optimized electronic metal–support interactions. The catalyst was successfully recycled at least eight times with no discernible loss of activity, and scaled to 5 g. Similarly, Jin et al. exploited the electronic properties of Pd sites to anchor it on defective titanium oxide through charge transfer.369 The catalyst achieved TOF of up to 11000 h−1 at room temperature, surpassing benchmark homogeneous catalysts. It retained its catalytic activity over five cycles.
Collectively, these studies highlight the promise of SACs for agrochemical and pharmaceutical applications. Beyond Suzuki–Miyaura reactions, SACs continue to expand their footprint in organic synthesis with recent examples in borylative cross-coupling (Fig. 9b),363 thiocouplings (Fig. 9c),364 and γ-amino acid, ester, and ketone synthesis (Fig. 9d)365 among others.361
In developing next-generation catalysts, researchers have revisited the stability and recyclability challenges of earlier systems to design active sites at the atomic level. These advances have yielded catalysts that demonstrate superior activity and selectivity to established homogeneous analogues. A recent techno-economic analysis and life cycle assessment indicates that recyclable single-atom catalysts (SACs) based on earth-abundant metals can substantially reduce greenhouse gas emissions and costs in fine chemical synthesis compared to current industrial precious-metal catalysts.370 However, significant challenges remain. Their efficacy relies heavily on metal–support interactions, which can deteriorate under harsh reaction environments, resulting in metal leaching, an extensively studied and ongoing concern. Additionally, the intrinsic spatial isolation of active sites in SACs can hinder the simultaneous activation of multiple or challenging substrates.371 Among the various strategies proposed, dual-atom catalysts (DACs) offer a compelling approach by positioning one single-atom site in close proximity to another, enabling synergistic regulation of catalytic activity. Although primarily deployed in simple electrocatalytic transformations,372 such as the oxygen reduction reaction and the oxygen evolution reaction,373 recent studies have explored their potential in cross-coupling protocols.374
Key remaining hurdles include developing scalable synthesis techniques that consistently produce SACs with uniform dispersion and robust long-term stability. Creating advanced supports capable of enhancing both catalyst durability and activity is equally vital. Advancements in characterization tools, computational modeling, and the design of specialized ligands and supports will be essential for overcoming these issues. Ultimately, whether SACs and related next-generation catalysts will surpass their predecessors and gain broad industrial application remains an open question.
4.3. Biocatalysis
Biocatalysis is the final pillar of catalysis considered in this review. Over the years, biocatalysis has emerged as a key sustainable technology in the pharmaceutical industry, prized for its high regio- and stereoselectivity and for the biodegradability of its catalysts. Crucially, it typically operates under milder, more eco-friendly conditions and generates less waste than chemical catalysis.375 These benefits largely stem from the intrinsic compatibility of enzymes and microbes with aqueous environments rather than organic solvents, inherently reducing waste generation.376A primary requirement for industrial-scale pharmaceutical and agrochemical production is a high total turnover number, expressed as the mass of product per mass of (bio)catalyst. Beyond anchoring individual enzymes, recent strategies focus on optimizing enzyme structure with precise spatial arrangements377 and on multi-enzyme assemblies affixed to protein scaffolds.378 Advanced cofactor recycling approaches have also emerged, such as co-immobilized enzyme assemblies featuring a movable cofactor “swing arm”379 and the co-encapsulation of enzymes and cofactors within microgels using nanoparticles.380 However, in many industrial settings, simpler and more practical approaches are often preferred. A common alternative involves the use of lyophilized enzyme powders at low loadings (typically <1%), which offer ease of use and avoid some of the challenges associated with immobilization, such as variability in performance, activity loss, and increased process complexity. While this can occasionally complicate downstream separation due to enzyme denaturation and emulsion formation, it remains a widely adopted and cost-effective strategy in commercial-scale biocatalysis.
While these state-of-the-art synthetic enzymes are broadening the application of biocatalysts, it bears noting that naturally occurring enzymes or whole cells have, in fact, been employed as bio-based catalysts for centuries, predominantly in food production. Following World War II, their use expanded to industrial conversion of complex substrates (e.g., steroids and penicillin) requiring site-specific or enantioselective transformations.381 However, limited insights into enzyme structures, mechanisms, and the challenges in improving their performance initially hampered wider uptake. Moreover, enzymes often perform poorly under the high substrate concentrations and conditions that characterize industrial processes, especially when organic media are required to dissolve hydrophobic substrates.382,383
At the turn of the millennium, breakthroughs in rational design, directed evolution, and other enzyme engineering techniques unlocked unprecedented opportunities for biocatalysis, notably in biomass valorization and the circular economy.375 Acknowledging these advances, Frances Arnold received the 2018 Nobel Prize for her work on directed enzyme evolution.384 One prominent application is the amine transaminase that supplanted a rhodium catalyst in industrial sitagliptin production (Fig. 10).385 Through substrate walking, modeling, and directed evolution, the resultant transaminase was capable of industrial-grade performance at high substrate concentrations and in the presence of a cosolvent. Crucially, it delivered chiral amines previously attainable only through resolution methods.
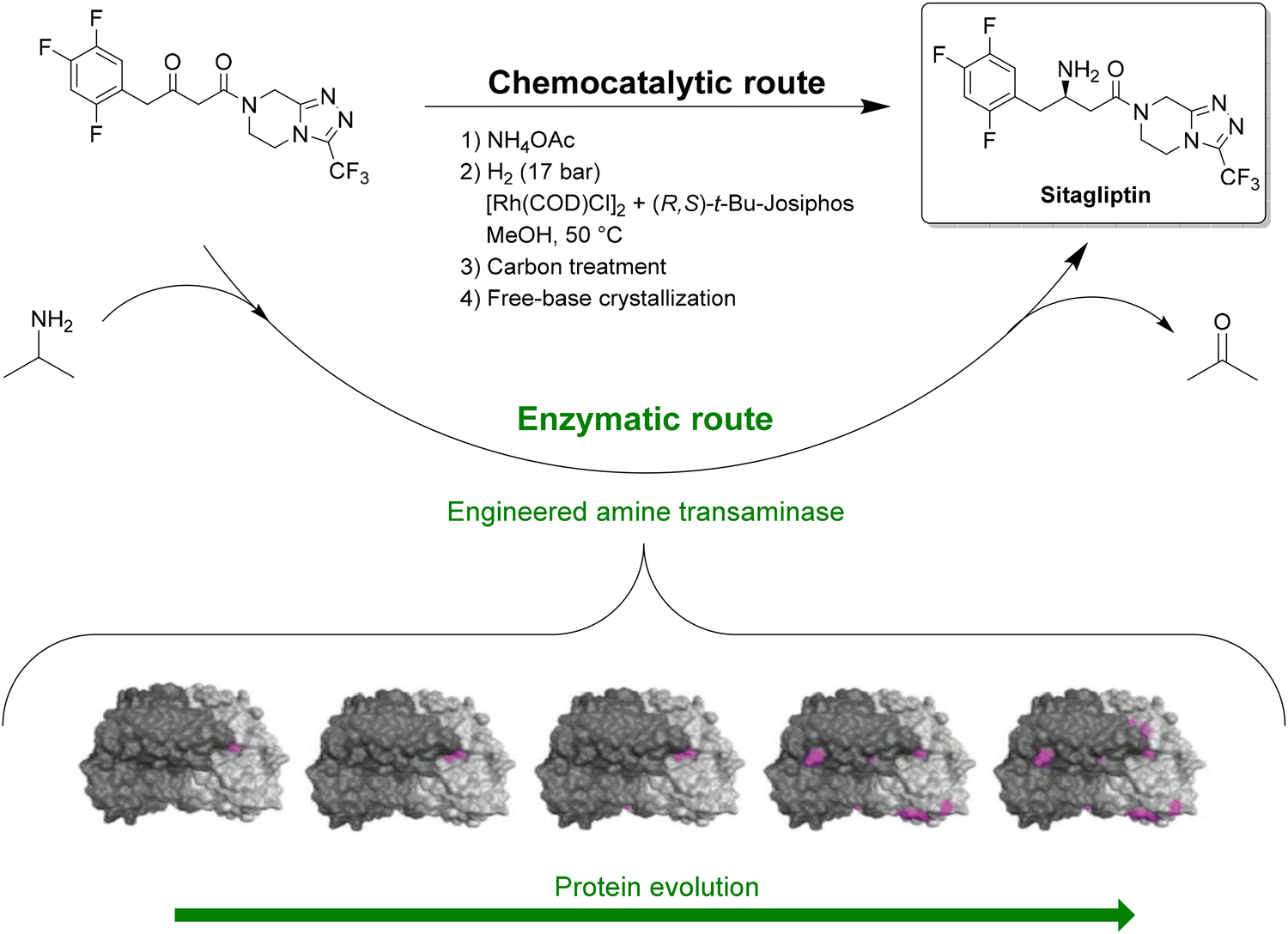 | ||
| Fig. 10 Synthesis of sitagliptin. The upper route relies on a multi-step sequence including high-pressure hydrogenation with a Rh/Josiphos catalyst. In contrast, the enzymatic route achieves the same C–N bond formation using a transaminase evolved through iterative protein-engineering cycles (structures shown at bottom), ultimately affording sitagliptin under milder, greener conditions. Reproduced from ref. 385, with permission from Science, copyright 2010. | ||
A similar strategy was devised for montelukast sodium (Singulair), a widely prescribed treatment for allergies and asthma.386 By means of directed evolution, a keto reductase (KRED) was developed to asymmetrically reduce the ketone precursor into its (S)-alcohol, the key intermediate of Singulair. The engineered KRED tolerated elevated temperatures (40–45 °C) and up to 70% v/v water-miscible organic cosolvent (2-propanol), conditions necessary to dissolve the hydrophobic precursor. Relative to the traditional moisture-sensitive, corrosive, and atom-inefficient diisopinocampheylborane approach, the KRED route lowered PMI and organic solvent consumption by approximately 30% and 25%, respectively.
In the past decade, further insights into enzyme mechanisms and the exploitation of enzyme promiscuity have triggered renewed interest in biocatalysis,387 aided by experimental and computational tools. These innovations enabled researchers to design enzymes for reactions absent in nature.388 Concurrently, the combination of microfluidics with machine learning has made target screening at over 1 kHz in picolitre-scale emulsion droplets possible.389 As a result, libraries of over 10 million enzyme variants can be assessed in a single day with minimal material and time usage compared to microtiter plate methods. These ultrahigh-throughput screening methods create a data-rich environment on enzyme activity, kinetics, stability, and structure, thereby deepening our understanding of enzymatic mechanisms and informing optimal protein engineering.389,390 Simultaneously, digital enzyme evolution consolidates AI-driven protein design, precision DNA editing, and robotics to shrink previously lengthy workflows into mere weeks.391 Beyond enzyme engineering, AI is reshaping retrosynthetic analysis for chemoenzymatic pathways,392,393 and refined biocatalyst informatics is accelerating enzyme selection and process development.394,395
The next logical step is to integrate biocatalysts into automated synthesis platforms that unify route design and execution, reflecting trends already emerging in organic synthesis.396 Consequently, numerous chemoenzymatic and multi-enzymatic pathways have been devised for producing natural products, pharmaceuticals, and food supplements.375,388,397–400
The recent synthesis of molnupiravir, a COVID-19 antiviral401 is a striking example of the potential of chemoenzymatic integration.402 To enable rapid, large-scale production, researchers replaced the original ten-step process (Scheme 24, top) with a concise three-step route starting from ribose (Scheme 24, middle). The central sequence featured a biocatalytic cascade using two engineered enzymes—ribosyl-1-kinase and uridine phosphorylase—with phosphate recycling via pyruvate oxidase and acetate kinase. This setup eliminated the need for stoichiometric phosphate donors and required isolation of only one intermediate. The biocatalytic steps operated at substrate loadings exceeding 80 g L−1 and delivered >99.5% purity after simple extraction and crystallization. ATP regeneration from pyruvate and inorganic phosphate addressed inorganic phosphate waste and cofactor cost.
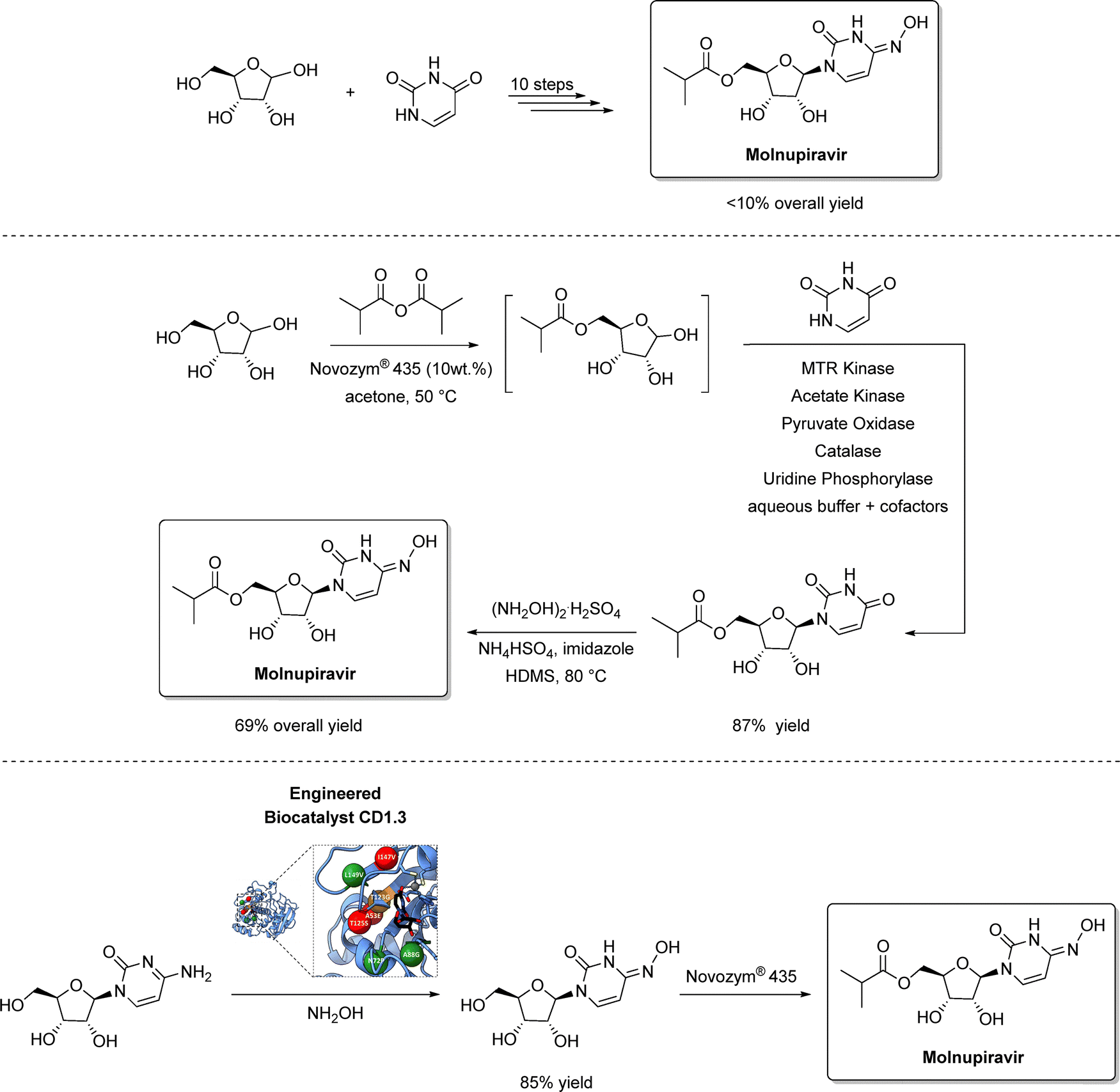 | ||
| Scheme 24 Synthesis of the COVID-19 antiviral molnupiravir via original route (top), biocatalytic cascade (middle), and engineered cytidine deaminase (bottom). Comparison of three synthetic approaches to COVID-19 antiviral molnupiravir. The original chemical route (top) required 10 steps and delivered the API in <10% overall yield. A subsequent chemoenzymatic strategy (middle) dramatically improved efficiency by combining Novozym® 435–mediated esterification with an enzymatic cascade involving MTR kinase, acetate kinase, pyruvate oxidase, catalase, and uridine phosphorylase, enabling concise access to the key intermediate and raising the overall yield to 69%. The most advanced version (bottom) employs an engineered biocatalyst, CD1.3, to achieve direct and high-yielding conversion of the nucleoside precursor with hydroxylamine, providing molnupiravir in 85% yield. Adapted from ref. 402 and 403, respectively. | ||
The final chemocatalytic conversion of uracil carbonyl to an oxime was redesigned for sustainability using hexamethyldisilazane (HMDS) as both solvent and reagent, with catalytic imidazole as a mild dehydrating agent. This avoided toxic activators like diethyl chlorophosphate and enabled in situ silylation with aqueous workup, simplifying purification.
In contrast to this hybrid approach, Burke et al. developed a fully enzymatic route by placing multiple engineered enzymes in a cascade to generate novel biochemical pathways. (Scheme 24, bottom).403 Starting from cytidine, they selectively formed N-hydroxy-cytidine using an engineered cytidine aminotransferase, followed by 5′-acylation via immobilized Candida antarctica lipase B (Novozym® 435). Conducted at high substrate concentrations, this route achieved 90% conversion to the target nucleoside analogue within 24 h.403
Similarly, the antiretroviral islatravir was synthesized via a multienzymatic cascade involving five engineered enzymes and four auxiliary enzymes (Scheme 25).404 The process starts with 2-ethynylglycerol oxidation by a galactose oxidase variant, with the resulting hydrogen peroxide byproduct decomposed by catalase and horseradish peroxidase. Next, a pantothenate kinase variant regioselectively phosphorylates the sensitive aldehyde intermediate using ATP, which is recycled from acetyl phosphate through acetate kinase. This phosphorylated aldehyde undergoes an aldol reaction with acetaldehyde, catalyzed by a deoxyribose 5-phosphate aldolase variant. A phosphopentomutase variant shifts the phosphate group from position 5 to 1. Finally, a purine nucleoside phosphorylase variant couples 2-fluoroadenine to the deoxyribose derivative. To drive product formation, sucrose phosphorylase removes phosphate from the last three steps. This aqueous approach achieved a 51% yield with no intermediate isolation, while improving atom economy and halving the total number of steps.399,404
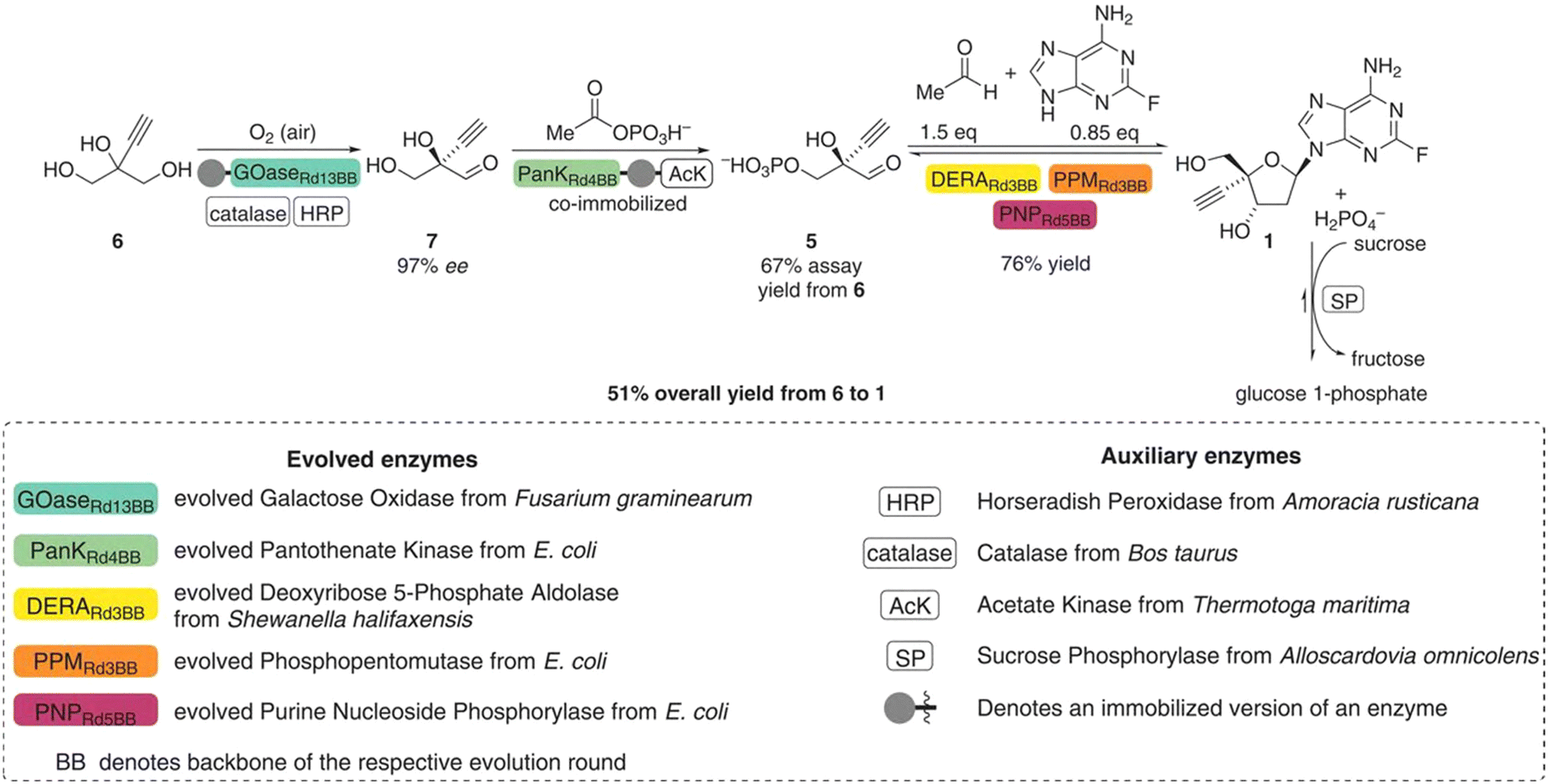 | ||
| Scheme 25 Synthesis of antiretroviral islatravir via a multienzymatic cascade. Auxiliary enzymes such as HRP, catalase, acetate kinase, and sucrose phosphorylase regenerate key cofactors and drive thermodynamic efficiency. The figure highlights the specific evolved enzymes employed at each stage and the immobilized variants used to streamline handling and enhance stability. Reproduced from ref. 404, with permission from Science, copyright 2019. | ||
Typically, chemo- or multi-enzymatic cascades are conducted in a one-pot format, wherein multiple catalysts operate within a single vessel. Increasingly, a single enzyme, whether multifunctional or not, can carry out these multi-step reactions alone.397 Advances in computational tools have further expanded these possibilities, as seen in PluriZymes, genetically engineered proteins with dual active sites.405,406
Alternatively, flow biocatalysis situates each catalyst in sequential chambers within a flow-through reactor, offering high productivity when integrated with downstream processing (continuous manufacturing). Miniaturized flow-through systems enable rigorous process control, efficient mass transport and easy scale-up by numbering-up strategies. However, harnessing flow biocatalysis to its fullest necessitates model-based reactor design.383,407
Despite considerable academic enthusiasm and select industrial cases, a number of which were highlighted above, biocatalysis is still not widely adopted in the fine chemical and pharmaceutical industries. A recent industry-wide survey attributed this to a combination of technical, cultural, educational, and organizational hurdles.408 A major barrier is the high complexity and entry cost associated with enzyme screening and directed evolution, as well as the need for interdisciplinary collaboration across biologists, chemists, data scientists, enzymologists, and engineers, which many companies find difficult to coordinate. Moreover, the adoption of biocatalysis is hindered by challenges in enzyme robustness, post-reaction workup, analytical demands, high dilution requirements, sensitivity to impurities, limited commercial enzyme availability, and a general reluctance to invest in the specialized infrastructure required to overcome these barriers. Finally, biocatalysis remains underrepresented in academic curricula, meaning most chemists enter industry with limited exposure to enzymatic methods, leading to reluctance and cultural resistance to adopt unfamiliar tools. This is compounded by the conservative mindset prevalent in highly regulated sectors such as pharmaceuticals, where risk aversion and a focus on short-term business priorities often take precedence over innovation, significantly slowing the adoption of more sustainable catalytic processes despite their clear long-term benefits.
5. Streamlined synthesis with flow chemistry
Making pharmaceutical and agrochemical synthesis greener involves not just altering what goes into them, but also how they are run. This section thus explores how continuous-flow reactors can tie these elements together to deliver major gains in energy efficiency and waste reduction. Continuous-flow reactors are extensively employed in the petrochemical and bulk-chemical industries, where large production volumes justify the significant capital investment and development work required. By contrast, pharmaceutical and agrochemical production typically occurs on smaller, more varied scales, making batch reactors the dominant choice in these sectors.409 Indeed, batch reactors are inherently flexible and multipurpose, allowing manufacturers to handle a wide range of chemistries within existing infrastructure. This flexibility is especially valuable when the same equipment must accommodate different products over time, reducing the incentive to invest in dedicated flow systems unless there is a clear performance or economic benefit. Nonetheless, the disadvantages of batch reactors become apparent when examinig a conventional manufacturing setup. In a batch workflow, raw materials are charged and processed in a reactor, followed by separate work-up (e.g., extraction, filtration, etc.) and purification (e.g., crystallization, drying, etc.) steps, often within the same facility or modular plant setup. While starting materials or intermediates may be sourced globally as part of a distributed supply chain, most unit operations typically occur within the same site, not across different geographic locations. Scaling up entails repeating these cycles until the desired product quantity is attained, reducing overall productivity due to prolonged processing times. Delays are further compounded by necessary hold times and interim storage needs, coupled with stringent quality-control measures implemented after each step to detect any batch-to-batch variations (Fig. 11, top).32,410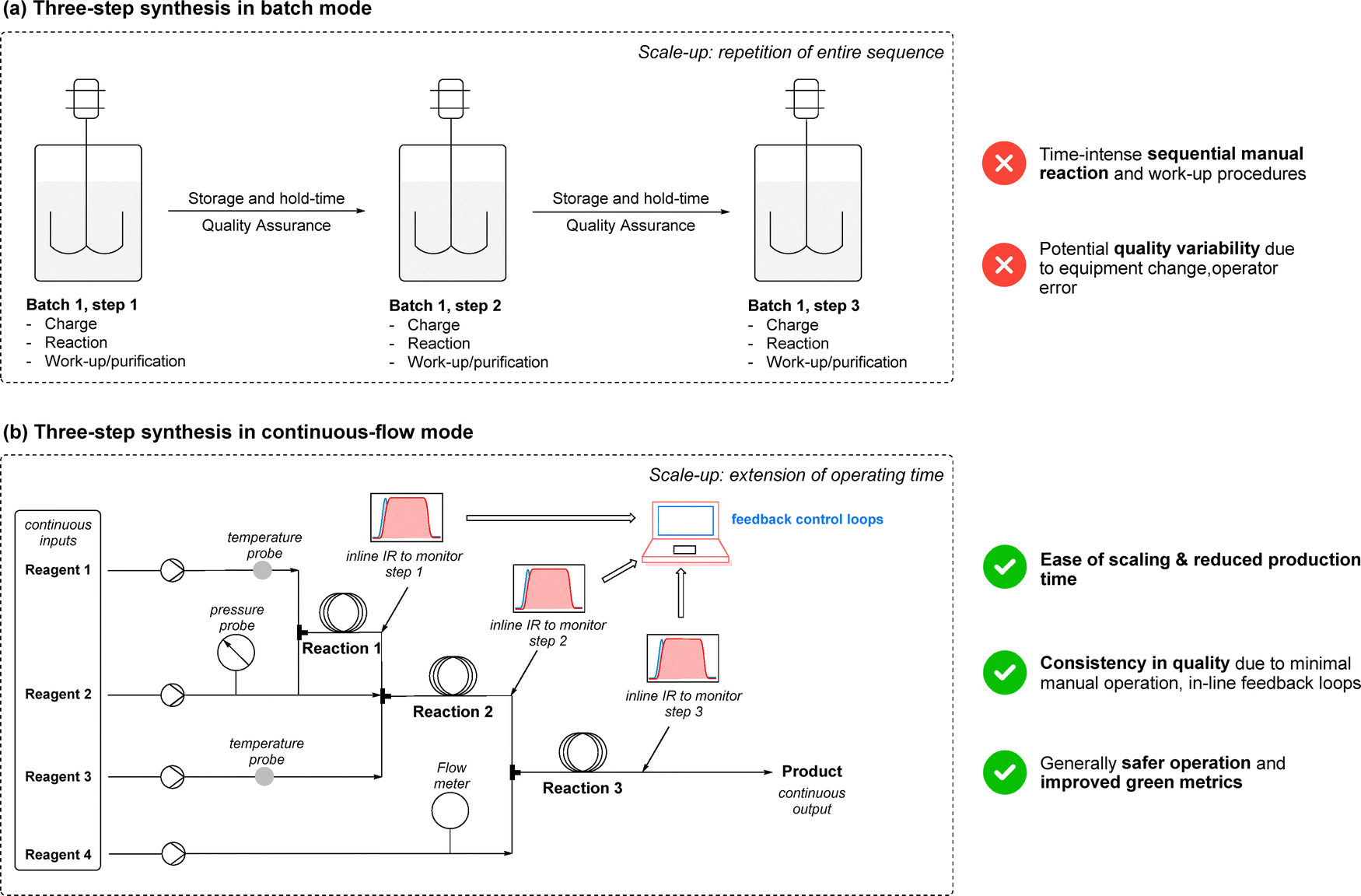 | ||
| Fig. 11 Comparison of a typical batch (i) and continuous-flow (ii) approach for the synthesis of a general API. Comparison of a three-step synthesis run as sequential batch operations versus an integrated multi-step continuous-flow process. In the batch approach (a), each step requires separate charging, reaction, work-up, and quality-assurance activities, leading to long cycle times, intermediate storage, and accumulated variability from equipment changes and manual handling. Scale-up simply multiplies these steps, compounding time, cost, and risk. The continuous-flow alternative (b) links all three reactions into a unified process equipped with inline temperature, pressure, and IR monitoring as well as feedback-control loops. Scaling is achieved by extending runtime rather than enlarging reactor volume, enabling stable operation, shorter production timelines, and consistent product quality. Adapted from ref. 32 and 410. | ||
To address these inefficiencies, continuous-flow processing is emerging as a transformative approach for the fine chemical sector. Continuous-flow processes involve continuously pumping reagents into a plug flow reactor (PFR) or mixed reactors [e.g., continuous-stirred tank reactor (CSTR)], from which product is continuously discharged. This transforms productivity metrics from measurements of unit time (temporally) to ones of unit space (spatially) as product amounts can be simply scaled up by prolonged operation.32,410 Beyond minimizing hold and processing times, flow reactors enable multiple synthetic steps to be consolidated within a single continuous run—a strategy known as multistep flow synthesis (Fig. 11, bottom).32,410 Conducting reactions in miniaturized continuous-flow reactors confers additional advantages.411–414 From a safety perspective, flow reactors offer steady-state operations, which can reduce the frequency of start-up and shutdown cycles. This feature can help limit the immediate consequences of process deviations. However, it is important to note that hazardous or toxic reagents are still present in the overall system, often in similar or greater quantities than in batch, and while their instantaneous inventory within the reactor is typically lower than in batch setups, appropriate precautions remain essential during reagent handling, charging, and storage. In addition, superior mixing and heat transfer mitigate the risk of thermal runaway, particularly in miniaturized flow systems. In some cases, reactive intermediates that are unstable in batch can be generated and used in situ in flow, without the need for isolation. Furthermore, greater control over key reaction parameters (temperature, pressure, and residence time) is one of the defining advantages of flow systems. This control can allow access to more extreme or highly precise conditions that may be challenging to achieve safely in batch, enabling reactions that require elevated temperatures, pressures, or rapid quenching.415–417 Flow methods are also increasingly integrated with advanced digitization and in-line Process Analytical Technology (PAT), such as real-time IR, NMR, UV-VIS, or UHPLC.418 These technologies support reaction monitoring, kinetic analysis, self-optimization, and dynamic experimentation, contributing to greater process understanding and control.419–422 That said, the implementation of flow methods does not automatically guarantee a greener process. In some cases, flow systems may require more dilute conditions to maintain solubility, which can negatively impact the PMI unless effective solvent recovery strategies are in place. On the other hand, the ability to heat or cool smaller reaction volumes more efficiently can offer energy savings and tighter thermal control, particularly for reactions that benefit from either precise or extreme conditions.
To date, the adoption of continuous-flow methods in the agrochemical and pharmaceutical sectors has been met with caution. One key reason is economic: investing in new equipment demands significant upfront capital and research.410 As product patents only provide market exclusivity for a short period, generic competitors quickly enter the market, reducing the financial incentive to invest in costly new equipment when existing batch infrastructure remains serviceable. However, it must be emphasized that the landscape has been shifting in recent years. Contract Development and Manufacturing Organizations (CDMOs) are increasingly investing in advanced capabilities, such as continuous processing, automation, and specialized catalytic technologies, that many legacy companies may not have prioritized developing in-house. As a result, strategic partnerships with CDMOs are becoming important, allowing companies to tap into external expertise, accelerate development timelines, and share the risk and cost associated with technological upgrades.
Scientific and technical challenges also hinder the implementation of flow methods. These include whether the underlying chemistry is compatible across sequential steps, particularly in telescoped processes, where maintaining a single solvent system and ensuring tolerance to byproducts or side products is essential. This constraint applies equally in batch, where telescoping is only feasible when reaction intermediates and impurities do not interfere with downstream steps. Nonetheless, as illustrated by the example herein, there are clearly defined cases where flow chemistry offers distinct and transformative advantages over batch processing. For instance, drug discovery is often conducted via well-established batch methods to rapidly deliver target libraries. When it comes to scaling up production, converting these batch synthetic processes to flow may necessitate re-evaluating synthetic sequences, retraining personnel, adopting new PATs, and recruiting interdisciplinary teams (e.g., chemical engineers, mechanical engineers, analytical specialists, and AI experts).413 The challenges posed by these changes can thus slow the shift to continuous approaches.410 Additionally, flow development faces both practical (e.g., clogging due to solid formation) and physical (e.g., solids as starting materials, products, and or by-products) limitations absent in batch settings.423 Finally, regulatory factors must be considered. Since API production is subject to strict oversight, companies can be reluctant to risk prolonged approval timelines or complex post-approval modifications.424–426 With the FDA, EMA and ICH endorsing continuous manufacturing, barriers to continuous-flow R&D have been lowered, advancing sustainable API and agrochemical development.427–429 The examples below focus on the benefits of flow over batch reactors in three specific areas: metalated intermediates, photochemistry, and electrochemistry.
Continuous-flow technology has gained recognition for the safe and efficient generation of hazardous and highly reactive metalated intermediates.415,416 A noteworthy example was reported in 2024 by Genentech and Hoffmann-La Roche, who developed a continuous-flow synthesis of quinazoline organozinc 3.430 This species is a late-stage intermediate to divarasib (Scheme 26), a cancer therapeutic at advanced clinical trials. The primary synthetic challenge lies in establishing an atropisomeric axis between the aminopyridine moiety and quinazoline core through a Negishi reaction. In the second-generation synthetic route, the traditional batch process involves metal–halogen exchange of bromoquinazoline 1 with i-PrMgCl·LiCl at −70 °C, followed by transmetalation with ZnCl2 at 10 °C. Although this method enabled production of up to 125 kg of product 2, it suffered several limitations: firstly, not only is the exchange highly exothermic, but the resultant Grignard intermediate also decomposes above −60 °C, posing quality risks from potential thermal hotspots; secondly, further scale-up would require specialized low-capacity cryogenic reactors and large volumes of liquid nitrogen, an environmental and logistical challenge.
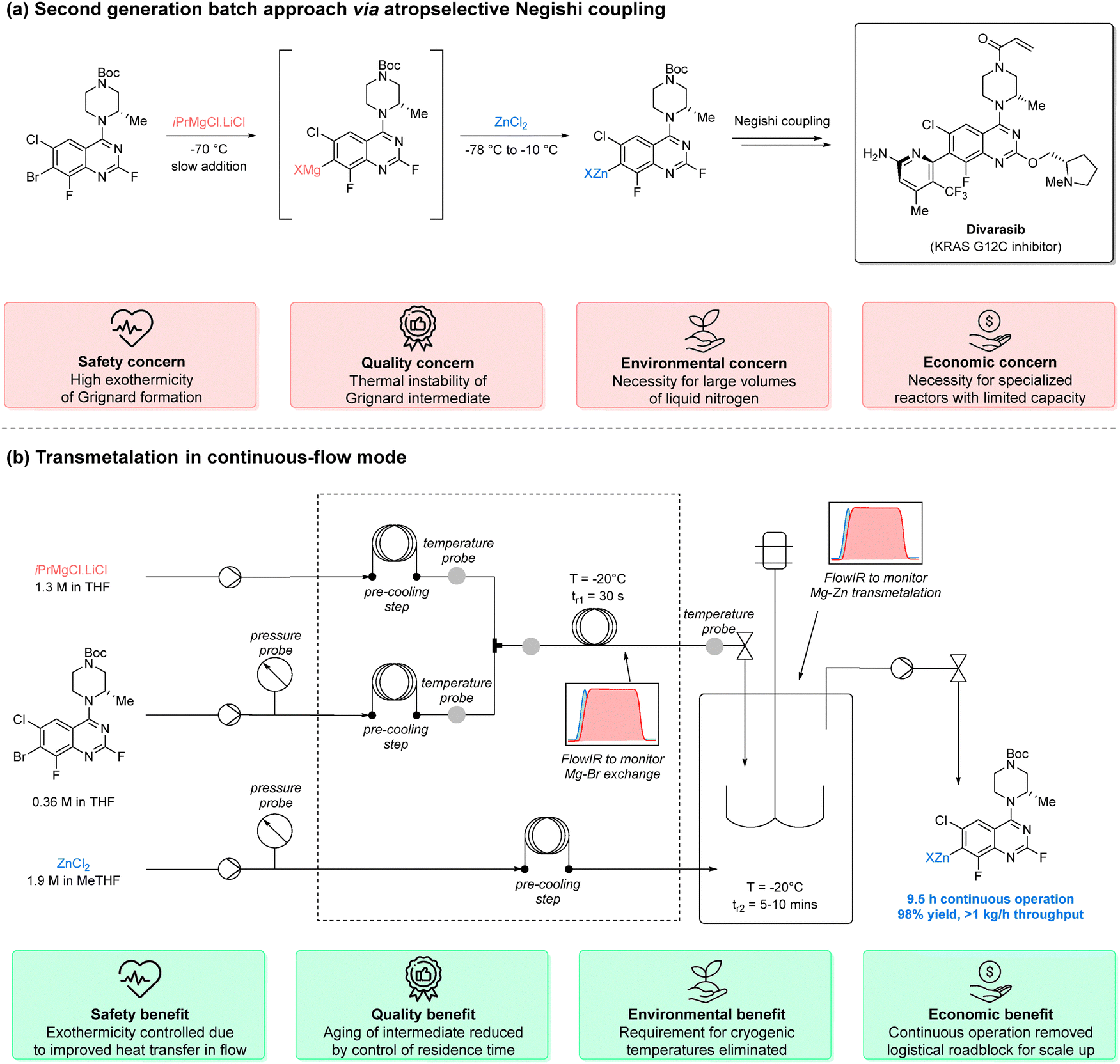 | ||
| Scheme 26 Batch vs. continuous-flow approach to obtain a key intermediate of divarasib. The batch route (a) relies on highly exothermic Grignard formation, thermally unstable intermediates, and deep-cryogenic conditions, creating safety, quality, environmental, and economic limitations. In contrast, the continuous-flow transmetalation (b) precisely controls exotherms, minimizes intermediate aging, and operates at significantly higher, more manageable temperatures. Inline FlowIR monitoring ensures tight control of both the Mg–Br exchange and Zn transmetalation steps, enabling long continuous operation with high assay yield and high throughput. Adapted from ref. 430. | ||
Switching to a continuous-flow setup eliminated these hurdles. Conducting the initial metal–halogen exchange in a high surface–area-to-volume plug-flow reactor allowed the temperature to rise to −20 °C, thanks to superior heat transfer. In-line process analytical technology supported safe operation (temperature and pressure probes) and facilitated real-time quality checks (FTIR monitoring at each step). By setting a strict 30-second residence time, the temperature-dependent decomposition of intermediate 2 was minimized. The subsequent transmetalation to aryl zinc halide 3 proceeded in a continuous-stirred tank reactor at −10 °C, with a five- to ten-minute residence time. Over 9.5 h of steady-state operation, the pilot plant delivered comparable yields and product quality to the batch process while maintaining stable reaction parameters. This example highlights a growing trend in industry: the strategic use of flow reactors or CSTRs as intermediate solutions between traditional batch reactors. In such cases, continuous methods would enable transformations that would be extremely difficult, if not unfeasible, in batch, offering real, tangible benefits in terms of safety, control, and scalability.
Comparable benefits emerged in the scalable preparation of a canagliflozin intermediate (Scheme 27).431 Canagliflozin, a treatment for type 2 diabetes mellitus, is traditionally produced in batch via a lithium–halogen exchange on aryl bromide 4, followed by addition to gluconolactone 6. Subsequent steps (methoxylation, desilylation, and reduction) yield the final API. However, the lithium–halogen exchange and addition sequence require temperatures below −70 °C; if the temperature rises to −40 °C the yields drop precipitously (50% vs. 8%). Even under cryogenic conditions, suboptimal mixing and heat removal lead to the formation of impurities from side-reactions involving the unstable aryllithium. Translating this process into a large-scale flow setup remediated these issues. Solutions of aryl bromide 4 and n-butyllithium were fed into a plug-flow reactor at −5 °C (jacket temperature −36 °C), achieving the metal–halogen exchange in just five seconds. The unstable aryllithium intermediate 5 was subsequently quenched in a second plug-flow reactor by adding gluconolactone 6, also within a five-second residence time. Operating at a total flow rate of 44 L h−1, this setup produced the desired intermediate in 88% yield, at a productivity of 3.4 kg h−1. Notably, no clogging from lithium salts occurred, and side reactions were markedly reduced, emphasizing the advantages of flow-based metalation strategies.
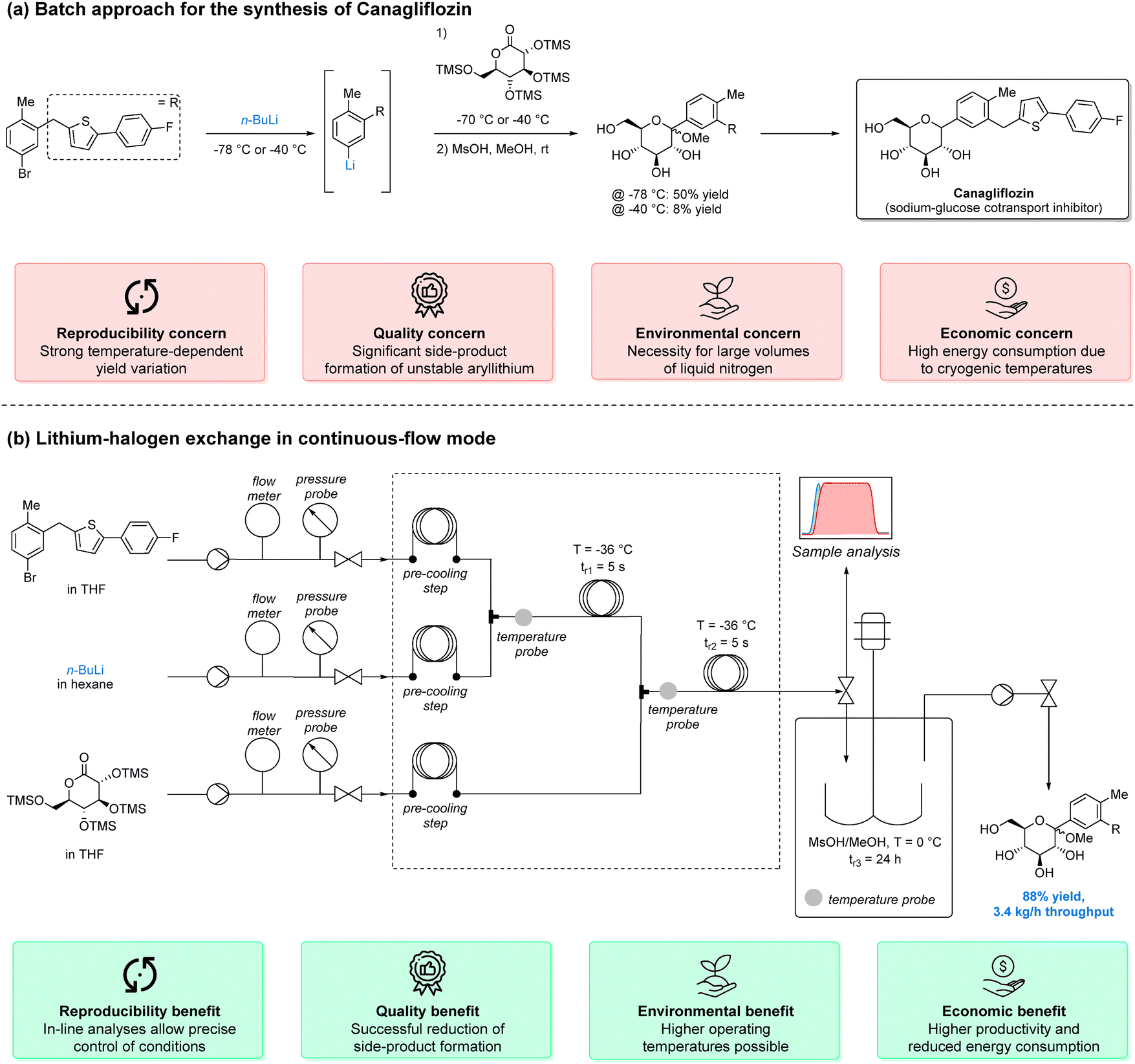 | ||
| Scheme 27 Batch vs. continuous-flow approach to obtain a key intermediate of canagliflozin. The batch approach (a) requires deep cryogenic temperatures, suffers from strong temperature-dependent variability, and produces unstable arylium intermediates, leading to reproducibility, quality, environmental, and economic concerns. The continuous-flow alternative (b) enables precise control of residence time and temperature, ensuring reproducible organolithium formation and clean downstream coupling. Adapted from ref. 431. | ||
Although continuous-flow technology has been successfully applied to industrial flash/metalation processes, its use in photochemical reactions beyond the laboratory remains limited.432 Indeed, the scarcity of standardized large-scale photoreactors, coupled with physical constraints imposed by the Lambert–Bouguer–Beer law, complicates scale-up. Additional challenges include achieving adequate photon flux and mitigating solvent vapor hazards. Nonetheless, several recent studies illustrate the promise of continuous-flow photochemistry for industrial applications.433–439
Merck recently reported a visible light-induced benzylic bromination process for manufacturing belzutifan (Scheme 28, top).440 Brominated indane 9 represents a key intermediate in the synthesis of this API. In the initial synthesis route developed for clinical trials, bromination of compound 8 occurred via a thermal radical mechanism using 1,3-dibromo-5,5-dimethylhydantoin (DBDMH) and azobisisobutyronitrile (AIBN). While this method was suitable for smaller scales, it presented several issues for mass production. For instance, the elevated temperatures required to initiate the reaction led to partial product decomposition, and the use of explosive AIBN together with hazardous dichloromethane raised significant safety concerns. Additionally, the unstable bromo-intermediate 9 had to be isolated before subsequent steps, and the non-selective radical process often generated over-brominated and deketalized impurities, leading to quality challenges.
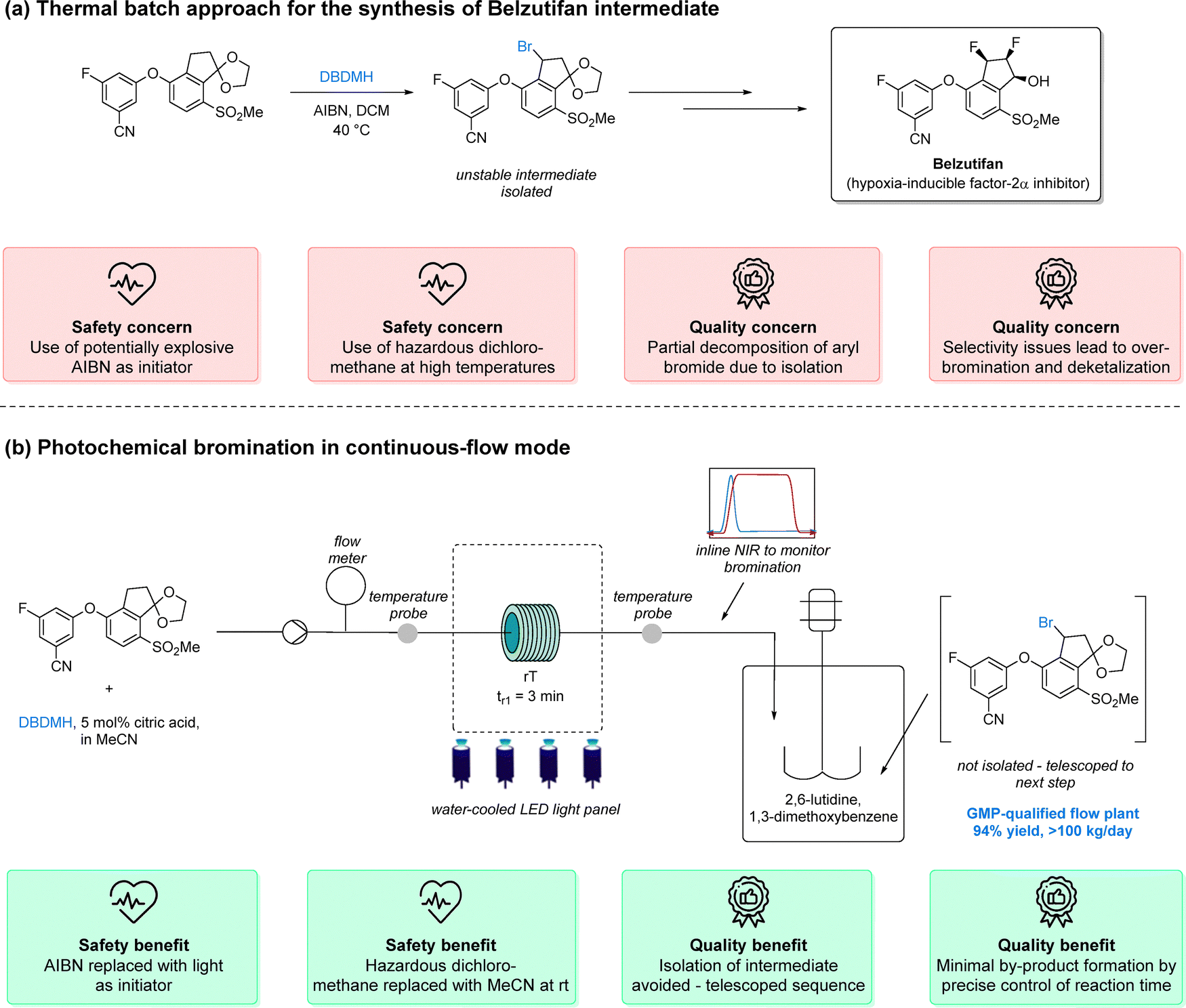 | ||
| Scheme 28 Comparison of batch vs. continuous-flow approach to obtain a key intermediate of belzutifan. The traditional batch process (a) requires azobisisobutyronitrile (AIBN) as a potentially explosive radical initiator and uses dichloromethane at elevated temperature, leading to safety risks, intermediate instability, and quality issues such as overbromination and debromination. The photochemical flow process (b) replaces AIBN with light, eliminates hazardous DCM in favor of acetonitrile (MeCN) at room temperature, and integrates real-time NIR monitoring to precisely control the bromination. This also enables telescoped processing, and delivers a GMP-ready intermediate with high assay yield and scalable productivity. In the figure, DBDMH stands for 1,3-dibromo-5,5-dimethylhydantoin. Adapted from ref. 440. | ||
To address these concerns, Merck developed a continuous-flow benzylic photo-bromination that operates under ambient conditions (Scheme 28, bottom).440 By retaining DBDMH as the bromine source and replacing AIBN with visible light as a radical initiator, the reaction avoided both elevated temperatures and potentially explosive materials. Additionally, switching the solvent from dichloromethane to acetonitrile not only enhanced safety but also enabled a telescoped process that directly fed to the next synthetic step, thereby eliminating the isolation of intermediate 9. Precise control of the product attributes and process parameters in the flow system ensured consistent conversion rates and minimized the formation of over-brominated by-products. To match the productivity of the thermal batch process, a large-scale tubular plug-flow reactor (0.83 L, 7.1 mm i.d.) was deployed. The reactor was arranged in a spiral coil, enclosed between two glass plates for cooling-fluid circulation, and surrounded by high-intensity LED chips. In-line near-infrared spectroscopy monitored the bromination reaction in real time for precise quality assurance. A reactor train consisting of several such units in sequence achieved a productivity of >100 kg day−1 and an assay yield of 94%, an impressive demonstration of industrial-scale continuous photochemistry.
Beyond photochemistry, synthetic electrochemistry has experienced a renaissance over the past two decades, owing to attractive features like the ability to control reaction rates by adjusting current, tailor selectivity through potential, and substitute stoichiometric reagents with more sustainable electron donors or acceptors.441,442 Yet, its application in large-scale industrial settings remains somewhat limited.443–448 Multi-kilogram processes present safety concerns, including the use of high currents in flammable solvents, potential generation of hydrogen or oxygen, and risks associated with high cell voltages and electrical arcing.449 These issues are further compounded by a lack of established scale-up workflows, especially for continuous-flow operations.
In response, Merck recently devised a strategy for scaling electrochemical continuous-flow route to pharmaceutically relevant kilogram scales. They illustrated this approach by oxidizing thioether 10 to sulfone 11 (Scheme 29).450 Sulfones are prevalent in APIs, yet their synthesis typically depends on stoichiometric oxidants that pose safety risks during scale-up (e.g., oxygen in organic solvents, potential formation of explosive peroxides or peracids). Consequently, a scalable, greener, and safer alternative is highly desirable. After establishing baseline conditions in batch, the process was transferred to a small-scale continuous-flow setup and scaled to kilogram quantities using a recirculating Electro Syn Cell parallel plate reactor. This reactor featured five electrode frames, each with a RuO2/Ti electrode, providing four active anodic surfaces of 400 cm2 each (a total of 1600 cm2), and was coupled to a 20 L jacketed reaction vessel. The flow rate was maintained at 5 L min−1 to achieve optimal linear velocity along the electrodes, and a current density of 30 mA cm−2 (48 A total, 4.5 F mol−1) delivered complete conversion in under 18 h, yielding over 1 kg of sulfone 11.
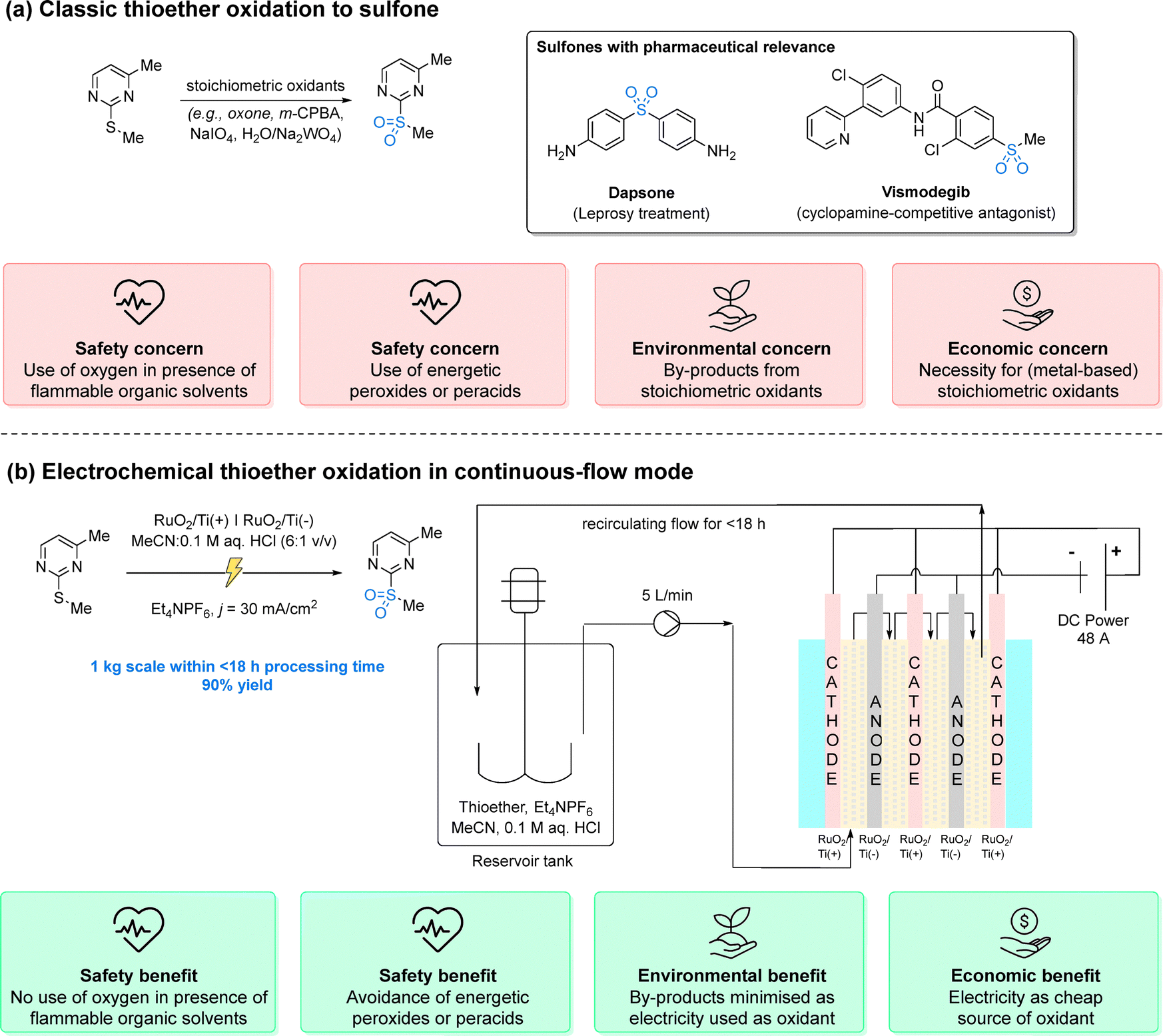 | ||
| Scheme 29 Batch vs. continuous-flow approach for thioether oxidation. The traditional methods (a) rely on oxygen, peroxides, or metal-based oxidants to obtain the sulfone, leading to safety risks, hazardous byproducts, and higher costs. The continuous-flow electrochemical approach (b) replaces these reagents with electricity as the oxidant, eliminating energetic peroxides, minimizing waste, and avoiding flammable oxygen–solvent mixtures. Adapted from ref. 450. | ||
A similar workflow was recently adopted at Pfizer as a safer and more scalable route to ketal 14 (Scheme 30).451 In the original linear batch route, a telescoped Birch reduction/ketalization of anisole 12 afforded ketal 14 in 74% yield in batches exceeding 50 kg. However, this approach entailed using flammable lithium metal (7.2 kg per batch) and large quantities of liquid ammonia (210 kg per batch) raising concerns about overpressure hazards as well as the economic and environmental costs of maintaining cryogenic conditions (−50 °C to −60 °C). Furthermore, the process was labor-intensive, necessitating multiple lithium additions to control exotherms, meticulous drying to prevent moisture-sensitive decomposition of ketone 13, and the use of several vessels for reaction and work-up.
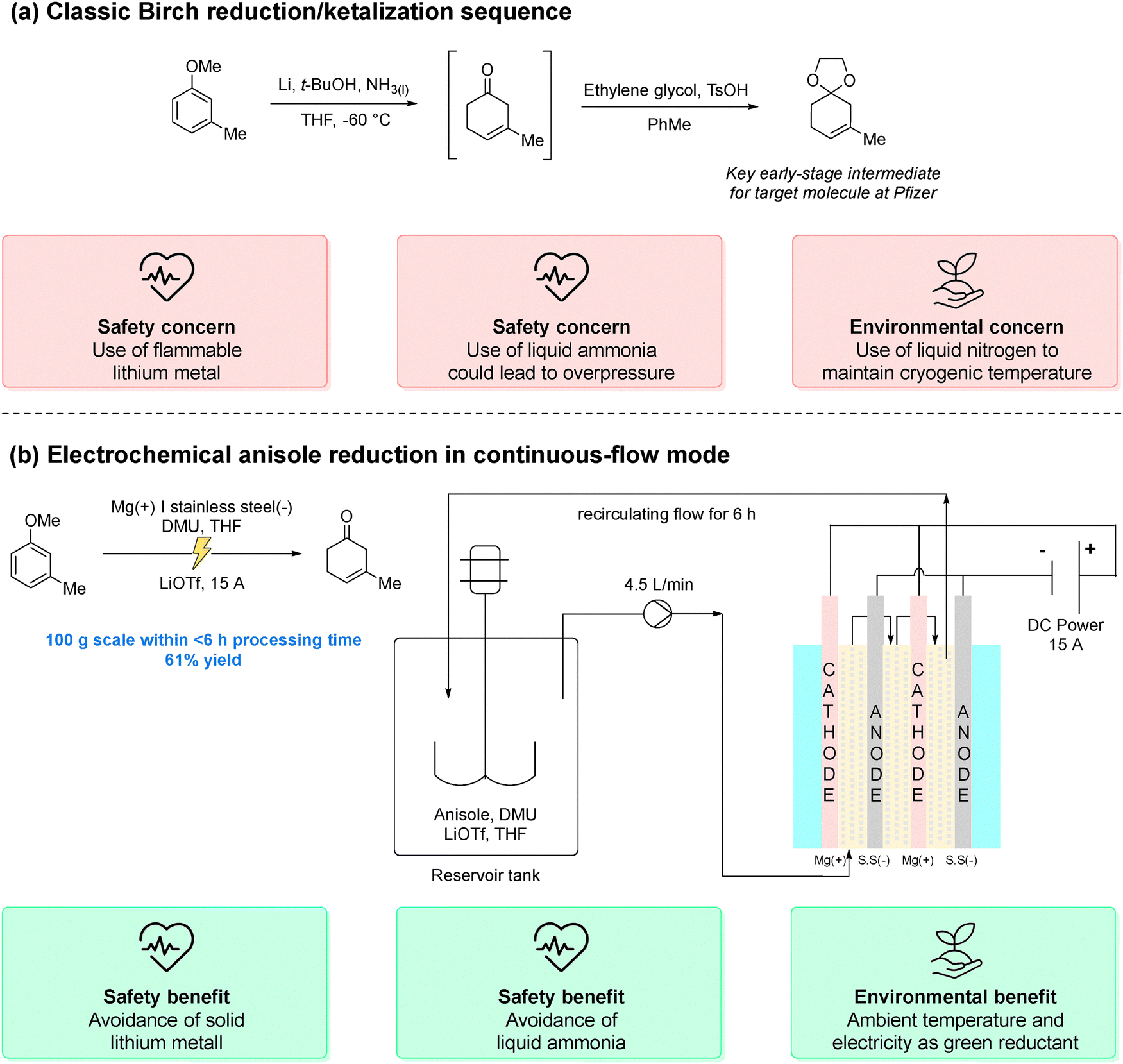 | ||
| Scheme 30 Batch vs. continuous-flow approach for Birch-type reduction. The traditional Birch reduction/ketalization sequence (a) relies on lithium metal, liquid ammonia, and cryogenic temperatures, introducing major safety and environmental hazards. In contrast, the continuous-flow electrochemical reduction (b) avoids both lithium and liquid ammonia, operates at ambient temperature, and uses electricity as a greener reductant. Adapted from ref. 451. | ||
To mitigate these risks, a continuous-flow electrochemical reduction was explored. Conditions first optimized in batch experiments were successfully transferred to a parallel plate reactor operating in recirculation mode. Employing a Mg(+)/stainless steel(−) electrode system, DMU as the proton donor, LiOTf as the electrolyte, and a flow rate of 4.5 L min−1 (15 A current, 4 F mol−1), 100 grams of the desired ketone 13 was produced at a 61% assay yield in a six-hour run. Although the yield obtained was slightly lower than for the Birch reduction, and the voltage continuously increased during the demonstration run, this approach effectively addressed the previous safety and handling issues. These achievements not only demonstrate the feasibility of electrochemical synthesis on a large scale in continuous mode, but also demonstrate its potential impact on both safety and sustainability.
Collectively, these recent examples demonstrate the tangible benefits of continuous-flow methods over conventional batch processes. It must be emphasized, however, that while flash flow chemistry for highly reactive metalated intermediates has matured into a widely accepted option that can be scaled up to multi-kilogram quantities, photochemical and electrochemical flow processes are just beginning to transition into industrial settings. Unquestionably, the benefits of flow reactors extend beyond these reaction classes to more established API syntheses. For instance, a recent techno-economic analysis comparing seven APIs (amitriptyline hydrochloride, tamoxifen, zolpidem, rufinamide, artesunate, ibuprofen, and phenibut), showed that continuous-flow manufacturing is markedly more energy-efficient, resource-saving, and cost-effective, reducing energy use and CO2 emissions by up to an order of magnitude.452 To fully realize their potential, however, further development of specialized equipment and best-practice workflows is required, with the scalable demonstrations discussed in this section serving as a promising springboard for sustainable pharmaceutical and agrochemical production.
6. Conclusion and future perspectives
Today, both the pharmaceutical and agrochemical industries are at a pivotal moment. Integrating sustainability into the very core of reaction and process design is no longer optional: it is essential for long-term viability. Four pillars (i.e., solvent choice, substrate sourcing, catalyst development, and continuous processing) are central to achieving this vision. However, these must be pursued not in isolation, but within a holistic, end-to-end framework. A greener solvent or a novel catalyst cannot compensate for a fundamentally flawed route design and improvements in one synthetic pillar must not undermine another. Several of the examples included in this review are not in accordance with this principle. For instance the synthesis of tyclopyrazoflor in Section 4.1 (Scheme 20),323 employs an earth-abundant catalyst but relies on dimethylformamide, a solvent classified as hazardous by the CHEM21 guide.45 Unfortunately, this reflects the current reality of the field, where many high-performance systems, such as single-atom catalysts or complex photocatalysts, have been optimized in conventional and widely applied solvents due to solubility, stability, or reactivity constraints. The integration of greener solvents into such advanced catalytic systems remains a significant and largely unmet challenge to date. Thus, advancing green chemistry requires consistency across all dimensions of process design and sustainability must be built into the architecture of a process from the outset. This means not only evaluating how reactions are conducted but also reconsidering which synthetic variable should be prioritized in the first place. Among the many decisions that expose this tension, solvent choice is perhaps the most immediate and illustrative given the large quantities used in a process.For example, bio-based solvents, while intuitively attractive, often present recovery and reusability challenges that undermine their green appeal. Terpene-based solvents, despite being renewable, are typically more viscous and heavier than traditional options, making separation and recycling more difficult and energy intensive. As a result, conventional solvents like toluene, with their well-understood profiles and simpler recovery routes, continue to play a dominant role in industry despite their fossil origin. Choosing a solvent, therefore, must strike a balance between theoretical sustainability and practical performance, particularly at scale. A biobased alternative solvent is therefore not necessarily beneficial, unless it is a drop-in. Inherent to any sustainable and circular process is the necessity of reagent and solvent recovery. While such recovery efforts may be minor considerations during initial pilot stages, at multi-hundred-ton-per-year production scales they become a non-negotiable cornerstone of economic and environmental performance. The choice of a solvent must be driven not only by its reaction compatibility but also by its process integration potential, i.e., how easily it can be recovered and reused.
In catalysis, biocatalysis has emerged as a potent enabler of greener chemical transformations. High-performance enzymes are now increasingly used for complex cascade reactions and specific oxygenation processes, which can dramatically improve process efficiency and selectivity. What once required lengthy and resource-intensive development efforts can now often be accessed via collaborations with specialist partners. When applied appropriately, biocatalysis offers an unparalleled potential for sustainable synthesis. Nonetheless, as noted previously, industrial implementation of biocatalysis hitherto remains limited, hampered by high development costs particularly in downstream processes, regulatory complexity, limited internal expertise, and cultural inertia, suggesting that much work is still needed to convert potential into practice. While base metal catalysis may seem to offer a greener alternative, this approach often shifts the burden to other parts of the process, demanding more energy, complex ligands, or unsustainable solvents that offset the environmental gains. Heterogeneous single-atom catalysts are also gaining traction, though they require deeper mechanistic understanding and rigorous process optimization to match the performance and selectivity of established homogeneous catalysts. Moreover, their applicability in transformations of more immediate interest in active ingredient synthesis still has to be shown.
Among advanced processing technologies, continuous-flow chemistry (and increasingly mechanochemistry and reactive extrusion) offer transformative potential versus the established batch approaches commonly applied to scaling for fine chemicals synthesis. While initial capital investment remains a barrier, leveraging external flow chemistry providers for pilot-scale development is an increasingly common and effective solution. These collaborations can reduce the adoption threshold and help overcome the infrastructural gap in the critical early stages of scale-up, where internal resources may be insufficient due to competing priorities. Over time, successful pilots can justify internalization of these platforms, unlocking long-term process efficiencies and sustainability gains.
Beyond individual synthetic tools, collaboration is essential. Trust-based partnerships between businesses with complementary expertise and academia are central to creating actionable insights. These collaborations can foster the development of shared platforms, talent exchange programs, and open innovation models that can transform theoretical academic knowledge into real-world sustainable processes. In this context, as pharmaceuticals and agrochemicals evolve toward greater structural and functional sophistication, they inherently demand increasingly advanced tools and competences. Finding and establishing the best possible technical production synthesis for such complex molecules not only requires designing the most sustainable synthesis routes possible but also to work within the constraints of cost-effectiveness. Doing so often involves pushing the boundaries of nearly every chemical technology available. However, no single industrial team, regardless of expertise, can master all emerging innovations in the chemical sciences indicating the necessity for specialized CRO's and open innovation programs.
One cannot overstate the importance of holistic route design in achieving sustainability goals. Choosing the wrong synthetic tools can preclude sustainability, no matter how green the individual steps may be. Take, for instance, multiply substituted benzoic acid.453 Classical linear chemical transformation sequences, such as nitration, reduction to aniline, and substitution of nitrogen via, for example, Sandmeyer chemistry, would undoubtably enable the compound to be produced on a large scale. However, the overall synthesis would be inefficient and waste intensive. The less intuitive, but more convergent, single step construction of the phenyl ring (via Michael reaction of a β-ketoester and an α,β-unsaturated ketone followed by intramolecular aldol condensation and elimination) may offer a path to a more sustainable outcome.453 As part of this holistic view, the degree of convergence in a synthesis can be a good measure for the potential sustainability of a production process.454 To achieve this, chemists and chemical engineers must adopt a holistic, systems-level mindset in process design. Rather than optimizing individual steps in isolation, prioritizing streamlined and well-integrated workflows can reduce complexity, energy consumption (e.g., from unnecessary solvent changes), and waste generation, ultimately leading to more efficient and sustainable manufacturing.
LCA remains a critical tool for a comprehensive and accurate comparison of sustainability across routes and processes.30,455 However, LCA is data-intensive in early discovery phases. Many chemical intermediates and routes are missing from standard databases, and proprietary process data remains inaccessible, complicating efforts to produce accurate and actionable assessments. A cohesive and harmonized LCA framework is essential here, and industry-academia collaboration is needed to unlock predictive, comparable, and scalable LCA models. Only through this alignment can the pharmaceutical and agrochemical sectors meaningfully and objectively measure and reduce their environmental impact.
As synthesis and technology continue to converge, the boundary between chemistry and chemical/process engineering is becoming increasingly blurred. In this context, the field of computational modeling (from first-principles quantum chemistry to large-scale numeric and mechanistic process simulations) has the potential to completely reshape what is considered the optimal synthetic route. A synthesis once deemed superior based on laboratory findings may be eclipsed by an alternative set of conditions when computational models explore operating conditions beyond the constraints of classical physical experimentation. Such in silico experimentation can simulate vast parameter spaces and unveil global optima that are otherwise inaccessible. Advanced retrosynthesis software (e.g., SYNTHIATM, ICSYNTH, IBM RXN) can also facilitate the identification of optimal synthetic routes early in development, thereby also embracing less obvious disconnections. Meanwhile, real-time, self-optimizing experimental platforms, powered by sequential design of experiments (DoE) and machine learning algorithms, can dynamically adjust process parameters to maintain product quality and maximize efficiency. The full integration of AI, ML, and computational modeling into chemical process development is vast, and will be addressed in greater detail in a separate review.
We are at the beginning of a new chapter. Having long relied on petrochemical foundations, the fine chemical industry is now opening the door to a more circular, resource efficient, and environmentally responsible way of operating, where sustainability is a central pillar of innovation and strategic development. This shift towards a greener-by-design manufacturing philosophy is by no means an expression of corporate altruism, and cannot be viewed in isolation. Rather it represents one element of a complex interplay of regulatory, societal, political, and economic forces – sometimes aligned, sometimes conflicting, and often uneven across geographic regions and sectors.456 Collectively, these pressures are (broadly) steering companies toward the pursuit of the ‘best chemistry’, irrespective of motive or ultimate goal. But as Paul Anastas reminds us: “we have no desire to do the best ‘green’ chemistry; we will do the best chemistry, and it will happen to be green.”457
Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Acknowledgements
The authors gratefully acknowledge financial support from the European Commission through the Marie Skłodowska-Curie Fellowships under the “SOLCAT” project (T. A. G., grant agreement no. 101152890) and the “Expand Flow” project (P. N., grant agreement no. 101106497); the Horizon Europe's “Global Challenges and European Industrial Competitiveness” programme for the “SusPharma” project (A. M., R. I. T., R. L., and B. B., grant agreement no. 101057430); the UKRI Horizon Europe Guarantee Scheme supporting the “SusPharma” project at Loughborough University (R.I.T., B.B., grant agreement 10038378); the European Research Council for the ERC Starting Grant “SAC_2.0” (G. V., grant agreement no. 101075832); the Horizon Europe's “Twinning Bottom-Up” programme for the “FLOWCAT” project (P. Ž. P., grant agreement no. 101160108); VLAIO (Flanders Innovation & Entrepreneurship, Belgium) through the “AC2GEN” project (Flanders Industry Innovation Moonshot—Strategic Basic Research for Clusters) (B. U. W. M.); Special Research Fund UAntwerpen (BOF Center of Excellence “CASCH” and SEP) (B. U. W. M.); and the Slovenian Research and Innovation Agency (P. Ž. P., grant agreement no. P2-0191).References
- European Chemical Industry Council, https://efcg.cefic.org/about-us/, (accessed 21 February 2025).
- Active Pharmaceutical Ingredients Market Size Report, https://www.grandviewresearch.com/industry-analysis/active-pharmaceutical-ingredients-market, (accessed 21 February 2025).
- API manufacturing: The cost of decarbonization | McKinsey & Company, https://www.mckinsey.com/industries/life-sciences/our-insights/decarbonizing-api-manufacturing-unpacking-the-cost-and-regulatory-requirements#/, (accessed 21 February 2025).
- 2022 Carbon Impact of Biotech & Pharma Report, https://www.mygreenlab.org/2022-carbon-impact-of-biotech--pharma-report.html, (accessed 5 June 2025).
- L. Belkhir and A. Elmeligi, J. Cleaner Prod., 2019, 214, 185–194 CrossRef CAS.
- The Carbon Impact of Biotech and Pharma, https://shcoalition.org/the-carbon-impact-of-biotech-and-pharma-a-roadmap-to-1-5c/, (accessed 17 June 2025).
- B. W. Cue and J. Zhang, Green Chem. Lett. Rev., 2009, 2, 193–211 CrossRef CAS.
- G. Wernet, S. Conradt, H. P. Isenring, C. Jiménez-González and K. Hungerbühler, Int. J. Life Cycle Assess., 2010, 15, 294–303 CrossRef CAS.
- Pesticides and climate change | Pesticide Action & Agroecology Network (PAN), https://www.panna.org/news/pesticides-and-climate-change/, (accessed 14 April 2025).
- E. Audsley, K. Stacey, D. J. Parsons and A. G. Williams, Estimation of the Greenhouse Gas Emissions from Agricultural Pesticide Manufacture and Use, Cranfield University, 2009 Search PubMed.
- A strong European API industry can achieve strategic autonomy of the EU health system, https://efcg.cefic.org/mediaroom/a-strong-european-api-industry-can-achieve-strategic-autonomy-of-the-eu-health-system/, (accessed 21 February 2025).
- The Draghi report on EU competitiveness, https://commission.europa.eu/topics/eu-competitiveness/draghi-report_en, (accessed 21 February 2025).
- P. Anastas and J. Warner, Green Chemistry: Theory and Practice, Oxford University Press, Oxford, New York, 2000 Search PubMed.
- The 17 Goals | Sustainable Development, https://sdgs.un.org/goals, (accessed 21 February 2025).
- The European Green Deal – European Commission, https://commission.europa.eu/strategy-and-policy/priorities-2019-2024/european-green-deal_en, (accessed 21 February 2025).
- Chemicals strategy – European Commission, https://environment.ec.europa.eu/strategy/chemicals-strategy_en, (accessed 21 February 2025).
- Safe and sustainable by design – European Commission, https://research-and-innovation.ec.europa.eu/research-area/industrial-research-and-innovation/chemicals-and-advanced-materials/safe-and-sustainable-design_en, (accessed 21 February 2025).
- Sustainable Chemistry Research and Development Act of 2019, https://www.congress.gov/bill/116th-congress/house-bill/2051/text, (accessed 14 April 2025).
- Water Framework Directive – European Commission, https://environment.ec.europa.eu/topics/water/water-framework-directive_en, (accessed 21 February 2025).
- A. Shattuck, J. Peasant Stud., 2021, 48, 231–253 CrossRef.
- K. Lokesh, A. S. Matharu, I. K. Kookos, D. Ladakis, A. Koutinas, P. Morone and J. Clark, Green Chem., 2020, 22, 803–813 RSC.
- E. R. Monteith, P. Mampuys, L. Summerton, J. H. Clark, B. U. W. Maes and C. R. McElroy, Green Chem., 2020, 22, 123–135 RSC.
- R. A. Sheldon, M. L. Bode and S. G. Akakios, Curr. Opin. Green Sustainable Chem., 2022, 33, 100569 CrossRef CAS.
- Z. Chen, J. Z. Lian, H. Zhu, J. Zhang, Y. Zhang, X. Xiang, D. Huang, K. Tjokro, V. Barbarossa, S. Cucurachi and B. Dong, J. Cleaner Prod., 2024, 459, 142550 CrossRef CAS.
- A. Booth, A. Jager, S. D. Faulkner, C. C. Winchester and S. E. Shaw, Int. J. Environ. Res. Public Health, 2023, 20, 3206 CrossRef PubMed.
- Y. Yang, D. Tilman, Z. Jin, P. Smith, C. B. Barrett, Y.-G. Zhu, J. Burney, P. D’Odorico, P. Fantke, J. Fargione, J. C. Finlay, M. C. Rulli, L. Sloat, K. Jan van Groenigen, P. C. West, L. Ziska, A. M. Michalak, the Clim-Ag Team and D. B. Lobell, Science, 2024, 385, eadn3747 CrossRef CAS PubMed.
- Y. Tao, S. Zhu, J. Smith, N. Lakhani and F. You, ACS Sustainable Chem. Eng., 2023, 11, 6510–6522 CrossRef CAS.
- A. D. Curzons, C. Jiménez-González, A. L. Duncan, D. J. C. Constable and V. L. Cunningham, Int. J. Life Cycle Assess., 2007, 12, 272–280 CrossRef CAS.
- Green Chemistry Challenge Eligibility and Scope, https://www.epa.gov/greenchemistry/green-chemistry-challenge-eligibility-and-scope, (accessed 11 June 2025).
- R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC.
- L. Wollensack, K. Budzinski and J. Backmann, Curr. Opin. Green Sustainable Chem., 2022, 33, 100586 CrossRef CAS.
- S. L. Lee, T. F. O’Connor, X. Yang, C. N. Cruz, S. Chatterjee, R. D. Madurawe, C. M. V. Moore, L. X. Yu and J. Woodcock, J. Pharm. Innov., 2015, 10, 191–199 CrossRef.
- D. J. C. Constable, C. Jimenez-Gonzalez and R. K. Henderson, Org. Process Res. Dev., 2007, 11, 133–137 CrossRef CAS.
- S. Kar, H. Sanderson, K. Roy, E. Benfenati and J. Leszczynski, Chem. Rev., 2022, 122, 3637–3710 CrossRef CAS PubMed.
- F. Roschangar, J. Li, Y. Zhou, W. Aelterman, A. Borovika, J. Colberg, D. P. Dickson, F. Gallou, J. D. Hayler, S. G. Koenig, M. E. Kopach, B. Kosjek, D. K. Leahy, E. O’Brien, A. G. Smith, M. Henry, J. Cook and R. A. Sheldon, ACS Sustainable Chem. Eng., 2022, 10, 5148–5162 CrossRef CAS.
- S. Gina Akakios, M. Leanne Bode and R. Arthur Sheldon, Green Chem., 2021, 23, 3334–3347 RSC.
- B. I. Andrews, F. D. Antia, S. B. Brueggemeier, L. J. Diorazio, S. G. Koenig, M. E. Kopach, H. Lee, M. Olbrich and A. L. Watson, J. Org. Chem., 2021, 86, 49–61 CrossRef CAS.
- B. Benyahia, R. Lakerveld and P. I. Barton, Ind. Eng. Chem. Res., 2012, 51, 15393–15412 CrossRef CAS.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC.
- K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31–36 RSC.
- D. Prat, O. Pardigon, H.-W. Flemming, S. Letestu, V. Ducandas, P. Isnard, E. Guntrum, T. Senac, S. Ruisseau, P. Cruciani and P. Hosek, Org. Process Res. Dev., 2013, 17, 1517–1525 CrossRef CAS.
- L. J. Diorazio, D. R. J. Hose and N. K. Adlington, Org. Process Res. Dev., 2016, 20, 760–773 CrossRef CAS.
- C. J. Clarke, W.-C. Tu, O. Levers, A. Bröhl and J. P. Hallett, Chem. Rev., 2018, 118, 747–800 CrossRef CAS.
- M. J. B. Brown, in Green and Sustainable Medicinal Chemistry: Methods, Tools and Strategies for the 21st Century Pharmaceutical Industry, ed. L. Summerton, H. F. Sneddon, L. C. Jones and J. H. Clark, The Royal Society of Chemistry, 2016 Search PubMed.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2015, 18, 288–296 RSC.
- J. Sherwood, Beilstein J. Org. Chem., 2020, 16, 1001–1005 CrossRef CAS PubMed.
- T. Yamaki, T. T. H. Nguyen, N. Hara, S. Taniguchi and S. Kataoka, Green Chem., 2024, 26, 3758–3766 RSC.
- Strategic approach to pharmaceuticals in the environment, https://www.europarl.europa.eu/doceo/document/TA-9-2020-0226_EN.html, (accessed 21 February 2025).
- S. Peper, A. González de Castilla, T. Kochenburger, J. Hillenbrand, J. Gries and D. Förtsch, ACS Sustainable Chem. Eng., 2024, 12, 18004–18017 CrossRef CAS.
- S. Boobier, J. C. Davies, I. N. Derbenev, C. M. Handley and J. D. Hirst, J. Chem. Inf. Model., 2023, 63, 2895–2901 CrossRef CAS PubMed.
- J. Heeley, S. Boobier and J. D. Hirst, J. Cheminf., 2024, 16, 60 CAS.
- H. Sels, H. De Smet and J. Geuens, Molecules, 2020, 25, 3037 CrossRef CAS PubMed.
- Y. Amar, A. M. Schweidtmann, P. Deutsch, L. Cao and A. Lapkin, Chem. Sci., 2019, 10, 6697–6706 RSC.
- M. Meuwly, Chem. Rev., 2021, 121, 10218–10239 CrossRef CAS.
- M. Gastegger, K. T. Schütt and K.-R. Müller, Chem. Sci., 2021, 12, 11473–11483 RSC.
- Y. Chung and W. H. Green, Chem. Sci., 2024, 15, 2410–2424 RSC.
- H. Zhang, V. Juraskova and F. Duarte, Nat. Commun., 2024, 15, 6114 CrossRef CAS.
- Nitrosamine impurities, https://www.ema.europa.eu/en/human-regulatory-overview/post-authorisation/pharmacovigilance-post-authorisation/referral-procedures-human-medicines/nitrosamine-impurities/nitrosamine-impurities-guidance-marketing-authorisation-holders, (accessed 21 February 2025).
- Control of Nitrosamine Impurities in Human Drugs, https://www.fda.gov/regulatory-information/search-fda-guidance-documents/control-nitrosamine-impurities-human-drugs, (accessed 16 June 2025).
- Greener Alternative Products, https://www.sigmaaldrich.com/IT/en/products/chemistry-and-biochemicals/greener-alternative-products?country=IT&language=en&cmsRoute=products&cmsRoute=chemistry-and-biochemicals&cmsRoute=greener-alternative-products&facets=facet_product_category%3Asolvents&page=3, (accessed 6 June 2025).
- N. Winterton, Clean Technol. Environ. Policy, 2021, 23, 2499–2522 CrossRef.
- J. Sperry and J. García-Álvarez, Molecules, 2016, 21, 1527 CrossRef PubMed.
- in Green Solvents in Organic Synthesis, ed. X. Wu, F. Wang, Z. Yin and L. He, Wiley, 1st edn, 2024 Search PubMed.
- B. M. Jenkins, in Encyclopedia of Agriculture and Food Systems, ed. N. K. Van Alfen, Academic Press, Oxford, 2014, pp. 461–498 Search PubMed.
- B. Amigun, R. Sigamoney and H. Von Blottnitz, Renewable Sustainable Energy Rev., 2008, 12, 690–711 CrossRef.
- G. P. Taber, D. M. Pfisterer and J. C. Colberg, Org. Process Res. Dev., 2004, 8, 385–388 CrossRef CAS.
- X. Jusseau, E. Cleator, W. M. Maton, Q. Chen, R. Geertman, Y. Yuan, X. Wang, H. Wei, F. Medina and M. Giannerini, Org. Process Res. Dev., 2022, 26, 976–986 CrossRef CAS.
- V.-E. H. Kassin, R. Gérardy, T. Toupy, D. Collin, E. Salvadeo, F. Toussaint, K. Van Hecke and J.-C. M. Monbaliu, Green Chem., 2019, 21, 2952–2966 RSC.
- Q. Zaib, M. J. Eckelman, Y. Yang and D. Kyung, Green Chem., 2022, 24, 7924–7930 RSC.
- V. Antonucci, J. Coleman, J. B. Ferry, N. Johnson, M. Mathe, J. P. Scott and J. Xu, Org. Process Res. Dev., 2011, 15, 939–941 CrossRef CAS.
- C. S. Slater, M. J. Savelski, D. Hitchcock and E. J. Cavanagh, J. Environ. Sci. Health, Part A, 2016, 51, 487–494 CrossRef CAS.
- V. Pace, P. Hoyos, L. Castoldi, P. Domínguez de María and A. R. Alcántara, ChemSusChem, 2012, 5, 1369–1379 CrossRef CAS PubMed.
- M. Miele, V. Pillari, V. Pace, A. R. Alcántara and G. de Gonzalo, Molecules, 2022, 27, 6701 CrossRef CAS.
- A. Kamal, H. K. Singh, S. K. Maury, A. K. Kushwaha, V. Srivastava and S. Singh, Asian J. Org. Chem., 2023, 12, e202200632 CrossRef CAS.
- E. Skolia and C. G. Kokotos, Eur. J. Org. Chem., 2024, e202400105 CrossRef CAS.
- K. Flick and E. Frankus, US3652589A, 1972.
- Z. Wang, Z. Ge, Z. Ye, L. Shu, L. Zeng, M. Fang, Y. Sun and K. Zhang, CN101265201B, 2011.
- A. Kadam, M. Nguyen, M. Kopach, P. Richardson, F. Gallou, Z.-K. Wan and W. Zhang, Green Chem., 2013, 15, 1880–1888 RSC.
- G. Menges-Flanagan, E. Deitmann, L. Gössl, C. Hofmann and P. Löb, Org. Process Res. Dev., 2020, 24, 315–321 CrossRef CAS.
- L. Gössl, K. Dahms, G. Menges-Flanagan and M. Maskos, Org. Process Res. Dev., 2024, 28, 2028–2040 CrossRef.
- J. C. Caravez, Y. Hu, E. Oftadeh, K. T. Mamo and B. H. Lipshutz, J. Org. Chem., 2024, 89, 3995–4000 CrossRef CAS.
- L. Cheng, D. Pettersen, B. Ohlsson, P. Schell, M. Karle, E. Evertsson, S. Pahlén, M. Jonforsen, A. T. Plowright, J. Boström, T. Fex, A. Thelin, C. Hilgendorf, Y. Xue, G. Wahlund, W. Lindberg, L.-O. Larsson and D. Gustafsson, ACS Med. Chem. Lett., 2014, 5, 538–543 CrossRef CAS PubMed.
- S. D. Ramgren, L. Hie, Y. Ye and N. K. Garg, Org. Lett., 2013, 15, 3950–3953 CrossRef CAS PubMed.
- P. Lei, Y. Mu, Y. Wang, Y. Wang, Z. Ma, J. Feng, X. Liu and M. Szostak, ACS Sustainable Chem. Eng., 2021, 9, 552–559 CrossRef CAS.
- A. E. Lubaev, C. C. Marvin, A. W. Dombrowski and Z. Qureshi, RSC Adv., 2024, 14, 29184–29188 RSC.
- S. Bhukta, R. Chatterjee and R. Dandela, Green Chem., 2023, 25, 3034–3039 RSC.
- A. F. Quivelli, F. V. Rossi, C. Alario, F. Sannicolò, P. Vitale, J. García-Álvarez, F. M. Perna and V. Capriati, Mol. Basel Switz., 2022, 27, 7594 CAS.
- A.-C. Bédard, A. R. Longstreet, J. Britton, Y. Wang, H. Moriguchi, R. W. Hicklin, W. H. Green and T. F. Jamison, Bioorg. Med. Chem., 2017, 25, 6233–6241 CrossRef.
- S. B. Ötvös, M. A. Pericàs and C. O. Kappe, Chem. Sci., 2019, 10, 11141–11146 RSC.
- Toxicity screening of potential bio-based Polar Aprotic Solvents|RIVM, https://www.rivm.nl/toxicity-screening-of-potential-bio-based-polar-aprotic-solvents, (accessed 21 February 2025).
- J. Sherwood, M. D. Bruyn, A. Constantinou, L. Moity, C. R. McElroy, T. J. Farmer, T. Duncan, W. Raverty, A. J. Hunt and J. H. Clark, Chem. Commun., 2014, 50, 9650–9652 RSC.
- R. J. I. Tamargo, P. Y. M. Rubio, S. Mohandoss, J.-J. Shim and Y. R. Lee, ChemSusChem, 2021, 14, 2133–2140 CrossRef CAS PubMed.
- A. Citarella, M. Cavinato, A. Amenta, M. Nardini, A. Silvani, D. Passarella and V. Fasano, Eur. J. Org. Chem., 2024, e202301305 CrossRef CAS.
- R. Nickisch, P. Conen, S. M. Gabrielsen and M. A. R. Meier, RSC Adv., 2021, 11, 3134–3142 RSC.
- L. Mistry, K. Mapesa, T. W. Bousfield and J. E. Camp, Green Chem., 2017, 19, 2123–2128 RSC.
- U. Veerabagu, G. Jaikumar, F. Lu and F. Quero, React. Chem. Eng., 2021, 6, 1900–1910 RSC.
- T. W. Bousfield, K. P. R. Pearce, S. B. Nyamini, A. Angelis-Dimakis and J. E. Camp, Green Chem., 2019, 21, 3675–3681 RSC.
- K. L. Wilson, J. Murray, C. Jamieson and A. J. B. Watson, Org. Biomol. Chem., 2018, 16, 2851–2854 RSC.
- J. M. Ramos-Villaseñor, J. Sotelo-Gil, S. E. Rodil and B. A. Frontana-Uribe, Faraday Discuss., 2023, 247, 182–194 RSC.
- K. L. Wilson, A. R. Kennedy, J. Murray, B. Greatrex, C. Jamieson and A. J. B. Watson, Beilstein J. Org. Chem., 2016, 12, 2005–2011 CrossRef CAS PubMed.
- S. Sangon, N. Supanchaiyamat, J. Sherwood, C. R. McElroy and A. J. Hunt, React. Chem. Eng., 2020, 5, 1798–1804 RSC.
- K. L. Wilson, J. Murray, C. Jamieson and A. J. B. Watson, Synlett, 2018, 650–654 CAS.
- I. A. Darwish, H. W. Darwish, A. H. Bakheit, H. M. Al-Kahtani and Z. Alanazi, in Profiles of Drug Substances, Excipients and Related Methodology, ed. A. A. Al-Majed, Academic Press, 2021, vol. 46, pp. 185–272 Search PubMed.
- H. Yu, D. Yu, Z. Xue, B. Zhang and T. Mu, Chem. Eng. J., 2022, 431, 133397 CrossRef CAS.
- O. B. Andrew, J. Sherwood and G. A. Hurst, J. Chem. Educ., 2022, 99, 3277–3282 CrossRef CAS.
- Circa Group AS – Decision to file petition for bankruptcy, https://live.euronext.com/en/products/equities/company-news/2024-10-07-circa-group-decision-file-petition-bankruptcy, (accessed 22 August 2025).
- K. A. Fayemiwo, N. Chiarasumran, S. A. Nabavi, V. Manović, B. Benyahia and G. T. Vladisavljević, J. Environ. Chem. Eng., 2020, 8, 103536 CrossRef CAS.
- P. Franco and I. De Marco, Processes, 2020, 8, 938 CrossRef CAS.
- J. Turconi, F. Griolet, R. Guevel, G. Oddon, R. Villa, A. Geatti, M. Hvala, K. Rossen, R. Göller and A. Burgard, Org. Process Res. Dev., 2014, 18, 417–422 CrossRef CAS.
- M. Okamoto, T. Takagi and F. Tanaka, Chem. Lett., 2000, 1396–1397 CrossRef CAS.
- Z. Amara, J. F. B. Bellamy, R. Horvath, S. J. Miller, A. Beeby, A. Burgard, K. Rossen, M. Poliakoff and M. W. George, Nat. Chem., 2015, 7, 489–495 CrossRef CAS.
- L. Wu, D. S. Lee, H. Boufroura, M. Poliakoff and M. W. George, ChemPhotoChem, 2018, 2, 580–585 CrossRef CAS.
- B. L. Abreu, H. Boufroura, J. C. Moore, M. Poliakoff and M. W. George, Synthesis, 2022, 3651–3657 CAS.
- L. Wu, B. L. Abreu, A. J. Blake, L. J. Taylor, W. Lewis, S. P. Argent, M. Poliakoff, H. Boufroura and M. W. George, Org. Process Res. Dev., 2021, 25, 1873–1881 CrossRef CAS.
- S. Alcalde, R. Porcar, M. L. De La Puente, G. R. Cumming, C. Mateos, P. García-Losada, C. Anta, J. A. Rincón and E. García-Verdugo, Org. Process Res. Dev., 2023, 27, 276–285 CrossRef CAS.
- M. Cortes-Clerget, J. Yu, J. R. A. Kincaid, P. Walde, F. Gallou and B. H. Lipshutz, Chem. Sci., 2021, 12, 4237–4266 RSC.
- B. H. Lipshutz and S. Ghorai, Green Chem., 2014, 16, 3660–3679 RSC.
- D. A. DiRocco, Y.-L. Zhong, D. N. Le, S. D. McCann, J. C. Hethcox, J. Kim, J. N. Kolev, B. Kosjek, S. M. Dalby, J. P. McMullen, R. Gangam and W. J. Morris, Org. Process Res. Dev., 2024, 28, 404–412 CrossRef CAS.
- J. Kim, V. Zhang, K. Abe, Y. Qin, D. A. DiRocco, J. P. McMullen, A. Sun, R. Gangam, M. Chow, A. Pitts-McCoy and A. S. Malkani, Org. Process Res. Dev., 2024, 28, 422–431 CrossRef CAS.
- J. C. Hethcox, J. Kim, H. C. Johnson, Y. Ji, M. Chow, J. A. Newman, D. A. DiRocco and J. P. McMullen, Org. Process Res. Dev., 2024, 28, 413–421 CrossRef CAS.
- Y. Qin, K. A. Mattern, V. Zhang, K. Abe, J. Kim, M. Zheng, R. Gangam, A. Kalinin, J. N. Kolev, S. Axnanda, Z. E. X. Dance, U. Ayesa, Y. Ji, S. T. Grosser, E. Appiah-Amponsah and J. P. McMullen, Org. Process Res. Dev., 2024, 28, 432–440 CrossRef CAS.
- D. N. Le, T. J. Wright, E. Alwedi, C. Hartmanshenn, H. Li, J. P. McMullen and D. A. DiRocco, Org. Process Res. Dev., 2024, 28, 451–459 CrossRef CAS.
- H. Gröger, F. Gallou and B. H. Lipshutz, Chem. Rev., 2023, 123, 5262–5296 CrossRef.
- E. O. Romero, A. T. Saucedo, J. R. Hernández-Meléndez, D. Yang, S. Chakrabarty and A. R. H. Narayan, JACS Au, 2023, 3, 2073–2085 CrossRef CAS.
- E. L. Bell, W. Finnigan, S. P. France, A. P. Green, M. A. Hayes, L. J. Hepworth, S. L. Lovelock, H. Niikura, S. Osuna, E. Romero, K. S. Ryan, N. J. Turner and S. L. Flitsch, Nat. Rev. Methods Primer, 2021, 1, 1–21 CrossRef.
- N. Das and C. Maity, Commun. Chem., 2022, 5, 1–23 CrossRef PubMed.
- J. D. Bailey, E. Helbling, A. Mankar, M. Stirling, F. Hicks and D. K. Leahy, Green Chem., 2021, 23, 788–795 RSC.
- Daniel Bailey wins Peter J. Dunn Award, https://cen.acs.org/people/awards/Daniel-Bailey-wins-Peter-J/98/i20, (accessed 3 June 2025).
- M. T. Sabatini, L. T. Boulton, H. F. Sneddon and T. D. Sheppard, Nat. Catal., 2019, 2, 10–17 CrossRef CAS.
- J. M. Saunders, E. O. Nava, J. Li, M. J. Wong, K. M. Freiberg and B. H. Lipshutz, ACS Sustainable Chem. Eng., 2025, 13, 6646–6655 CrossRef CAS.
- J. R. A. Kincaid, J. C. Caravez, K. S. Iyer, R. D. Kavthe, N. Fleck, D. H. Aue and B. H. Lipshutz, Commun. Chem., 2022, 5, 156 CrossRef CAS.
- F. Gallou, P. Guo, M. Parmentier and J. Zhou, Org. Process Res. Dev., 2016, 20, 1388–1391 CrossRef CAS.
- S. Hazra, F. Gallou and S. Handa, ACS Sustainable Chem. Eng., 2022, 10, 5299–5306 CrossRef CAS.
- K. M. Freiberg, R. D. Kavthe, R. M. Thomas, D. M. Fialho, P. Dee, M. Scurria and B. H. Lipshutz, Chem. Sci., 2023, 14, 3462–3469 RSC.
- C. M. Gabriel, M. Keener, F. Gallou and B. H. Lipshutz, Org. Lett., 2015, 17, 3968–3971 CrossRef CAS PubMed.
- F. Bordignon, A. Scarso and A. Angelini, ChemBioChem, 2025, 2500099 Search PubMed.
- A. Bourboula, O. G. Mountanea, G. Krasakis, C. Mantzourani, M. G. Kokotou, C. G. Kokotos and G. Kokotos, Eur. J. Org. Chem., 2023, e202300008 CrossRef CAS.
- J. R. A. Kincaid, R. D. Kavthe, J. C. Caravez, B. S. Takale, R. R. Thakore and B. H. Lipshutz, Org. Lett., 2022, 24, 3342–3346 CrossRef CAS.
- R. D. Kavthe, K. S. Iyer, J. C. Caravez and B. H. Lipshutz, Chem. Sci., 2023, 14, 6399–6407 RSC.
- Antimalarial drugs to save lives, https://www.mmv.org/our-work/antimalarial-to-save-lives, (accessed 4 June 2025).
- K. S. Iyer, R. D. Kavthe, R. M. Lammert, J. R. Yirak and B. H. Lipshutz, JACS Au, 2024, 4, 680–689 CrossRef CAS.
- K. S. Iyer, K. B. Dismuke Rodriguez, R. M. Lammert, J. R. Yirak, J. M. Saunders, R. D. Kavthe, D. H. Aue and B. H. Lipshutz, Angew. Chem., Int. Ed., 2024, 63, e202411295 CAS.
- N. Compagno, N. Lucchetti, A. Palmisano, R. Profeta and A. Scarso, J. Org. Chem., 2024, 89, 12452–12461 CrossRef CAS.
- C. Palladino, T. Fantoni, L. Ferrazzano, A. Tolomelli and W. Cabri, ACS Sustainable Chem. Eng., 2023, 11, 15994–16004 CrossRef CAS.
- M. J. Wong, E. Oftadeh, J. M. Saunders, A. B. Wood and B. H. Lipshutz, ACS Catal., 2024, 14, 1545–1552 CrossRef CAS.
- S. Sharma, T. N. Ansari and S. Handa, ACS Sustainable Chem. Eng., 2021, 9, 12719–12728 CrossRef CAS.
- W. Braje, K. Britze, J. D. Dietrich, A. Jolit, J. Kasch, J. Klee and T. Lindner, WO2017129796A1, 2017.
- D. Petkova, N. Borlinghaus, S. Sharma, J. Kaschel, T. Lindner, J. Klee, A. Jolit, V. Haller, S. Heitz, K. Britze, J. Dietrich, W. M. Braje and S. Handa, ACS Sustainable Chem. Eng., 2020, 8, 12612–12617 CrossRef CAS.
- F. Gallou, N. A. Isley, A. Ganic, U. Onken and M. Parmentier, Green Chem., 2016, 18, 14–19 RSC.
- D. J. Lippincott, E. Landstrom, M. Cortes-Clerget, B. H. Lipshutz, K. Buescher, R. Schreiber, C. Durano, M. Parmentier, N. Ye, B. Wu, M. Shi, H. Yang, M. Andersson and F. Gallou, Org. Process Res. Dev., 2020, 24, 841–849 CrossRef CAS.
- B. H. Lipshutz, S. Ghorai, A. R. Abela, R. Moser, T. Nishikata, C. Duplais, A. Krasovskiy, R. D. Gaston and R. C. Gadwood, J. Org. Chem., 2011, 76, 4379–4391 CrossRef CAS.
- N. R. Lee, M. Cortes-Clerget, A. B. Wood, D. J. Lippincott, H. Pang, F. A. Moghadam, F. Gallou and B. H. Lipshutz, ChemSusChem, 2019, 12, 3159–3165 CrossRef CAS.
- C. Krell, R. Schreiber, L. Hueber, L. Sciascera, X. Zheng, A. Clarke, R. Haenggi, M. Parmentier, H. Baguia, S. Rodde and F. Gallou, Org. Process Res. Dev., 2021, 25, 900–915 CrossRef CAS.
- T. N. Ansari, G. Xu, A. Preston and P. Gao, Org. Process Res. Dev., 2024, 28, 816–830 CrossRef CAS.
- A. Adamo, R. L. Beingessner, M. Behnam, J. Chen, T. F. Jamison, K. F. Jensen, J.-C. M. Monbaliu, A. S. Myerson, E. M. Revalor, D. R. Snead, T. Stelzer, N. Weeranoppanant, S. Y. Wong and P. Zhang, Science, 2016, 352, 61–67 CrossRef CAS PubMed.
- S. R. Bankar, S. S. Ghadge and V. H. Jadhav, New J. Chem., 2023, 47, 19679–19687 RSC.
- Z. N. Ennis, D. Dideriksen, H. B. Vægter, G. Handberg and A. Pottegård, Basic Clin. Pharmacol. Toxicol., 2016, 118, 184–189 CrossRef CAS PubMed.
- T. Portada, D. Margetić and V. Štrukil, Molecules, 2018, 23, 3163 CrossRef.
- R. Solà, O. B. Sutcliffe, C. E. Banks and B. Maciá, Sustainable Chem. Pharm., 2017, 5, 14–21 CrossRef.
- W. I. Nicholson, F. Barreteau, J. A. Leitch, R. Payne, I. Priestley, E. Godineau, C. Battilocchio and D. L. Browne, Angew. Chem., Int. Ed., 2021, 60, 21868–21874 CrossRef CAS.
- B. R. Kim, H.-G. Lee, S.-B. Kang, G. H. Sung, J.-J. Kim, J. K. Park, S.-G. Lee and Y.-J. Yoon, Synthesis, 2012, 42–50 CAS.
- N. Fantozzi, J.-N. Volle, A. Porcheddu, D. Virieux, F. García and E. Colacino, Chem. Soc. Rev., 2023, 52, 6680–6714 RSC.
- M. T. J. Williams, L. Adarve Cardona and C. Bolm, Adv. Synth. Catal., 2024, 366, 2220–2225 CrossRef CAS.
- J. Yu, Z. Hong, X. Yang, Y. Jiang, Z. Jiang and W. Su, Beilstein J. Org. Chem., 2018, 14, 786–795 CrossRef CAS.
- E. P. T. Leitão, RSC Sustainability, 2024, 2, 3655–3668 RSC.
- X. Yang, C. Wu, W. Su and J. Yu, Eur. J. Org. Chem., 2022, e202101440 CrossRef CAS.
- O. Bento, F. Luttringer, T. Mohy El Dine, N. Pétry, X. Bantreil and F. Lamaty, Eur. J. Org. Chem., 2022, e202101516 CrossRef CAS.
- J. Yu, Y. Zhang, Z. Zheng and W. Su, RSC Mechanochem., 2024, 1, 367–374 RSC.
- T. Nikonovich, T. Jarg, J. Martõnova, A. Kudrjašov, D. Merzhyievskyi, M. Kudrjašova, F. Gallou, R. Aav and D. Kananovich, RSC Mechanochem., 2024, 1, 189–195 RSC.
- L. Konnert, B. Reneaud, R. M. de Figueiredo, J.-M. Campagne, F. Lamaty, J. Martinez and E. Colacino, J. Org. Chem., 2014, 79, 10132–10142 CrossRef CAS.
- E. Colacino, A. Porcheddu, I. Halasz, C. Charnay, F. Delogu, R. Guerra and J. Fullenwarth, Green Chem., 2018, 20, 2973–2977 RSC.
- O. Galant, G. Cerfeda, A. S. McCalmont, S. L. James, A. Porcheddu, F. Delogu, D. E. Crawford, E. Colacino and S. Spatari, ACS Sustainable Chem. Eng., 2022, 10, 1430–1439 CrossRef CAS.
- E. Colacino, V. Isoni, D. Crawford and F. García, Trends Chem., 2021, 3, 335–339 CrossRef CAS.
- C. Patel, E. André-Joyaux, J. A. Leitch, X. M. de Irujo-Labalde, F. Ibba, J. Struijs, M. A. Ellwanger, R. Paton, D. L. Browne, G. Pupo, S. Aldridge, M. A. Hayward and V. Gouverneur, Science, 2023, 381, 302–306 CrossRef CAS PubMed.
- J. L. Howard, Y. Sagatov, L. Repusseau, C. Schotten and D. L. Browne, Green Chem., 2017, 19, 2798–2802 RSC.
- Q. Cao, J. L. Howard, D. E. Crawford, S. L. James and D. L. Browne, Green Chem., 2018, 20, 4443–4447 RSC.
- S. Ni, M. Hribersek, S. K. Baddigam, F. J. L. Ingner, A. Orthaber, P. J. Gates and L. T. Pilarski, Angew. Chem., Int. Ed., 2021, 60, 6660–6666 CrossRef CAS PubMed.
- R. Qu, S. Wan, X. Zhang, X. Wang, L. Xue, Q. Wang, G.-J. Cheng, L. Dai and Z. Lian, Angew. Chem., Int. Ed., 2024, 63, e202400645 CrossRef CAS.
- X. Wang, X. Zhang, X. He, G. Guo, Q. Huang, F. You, Q. Wang, R. Qu, F. Zhou and Z. Lian, Angew. Chem., Int. Ed., 2024, 63, e202410334 CrossRef CAS.
- J. F. Reynes, V. Isoni and F. García, Angew. Chem., Int. Ed., 2023, 62, e202300819 CrossRef CAS PubMed.
- D. J. am Ende, S. R. Anderson and J. S. Salan, Org. Process Res. Dev., 2014, 18, 331–341 CrossRef CAS.
- A. A. L. Michalchuk, K. S. Hope, S. R. Kennedy, M. V. Blanco, E. V. Boldyreva and C. R. Pulham, Chem. Commun., 2018, 54, 4033–4036 RSC.
- L. Gonnet, C. B. Lennox, J.-L. Do, I. Malvestiti, S. G. Koenig, K. Nagapudi and T. Friščić, Angew. Chem., Int. Ed., 2022, 61, e202115030 CrossRef CAS.
- C. B. Lennox, T. H. Borchers, L. Gonnet, C. J. Barrett, S. G. Koenig, K. Nagapudi and T. Friščić, Chem. Sci., 2023, 14, 7475–7481 RSC.
- J. D. Thorpe, J. Marlyn, S. G. Koenig and M. J. Damha, RSC Mechanochem., 2024, 1, 244–249 RSC.
- A. Nanni, D. Kong, C. Zhu and M. Rueping, Green Chem., 2024, 26, 8341–8347 RSC.
- P. Sharma, E. Ponnusamy, S. Ghorai and T. J. Colacot, J. Organomet. Chem., 2022, 970–971, 122367 CrossRef CAS.
- P. Sharma, C. Vetter, E. Ponnusamy and E. Colacino, ACS Sustainable Chem. Eng., 2022, 10, 5110–5116 CrossRef CAS.
- R. A. Sheldon, Green Chem., 2016, 18, 3180–3183 RSC.
- C. Jimenez-Gonzalez, C. S. Ponder, Q. B. Broxterman and J. B. Manley, Org. Process Res. Dev., 2011, 15, 912–917 CrossRef CAS.
- H. B. Rose, B. Kosjek, B. M. Armstrong and S. A. Robaire, Curr. Res. Green Sustainable Chem., 2022, 5, 100324 CrossRef CAS.
- J. Spekreijse, K. Vikla, M. Vis, K. Boysen-Urban, G. Philippidis and R. M’Barek, Bio-based value chains for chemicals, plastics and pharmaceuticals: a comparison of bio based and fossil based value chains, Publications Office of the European Union, 2021 Search PubMed.
- Turning off the Tap for Fossil Carbon – Future Prospects for a Global Chemical and Derived Material Sector Based on Renewable Carbon, https://renewable-carbon.eu/publications/product/turning-off-the-tap-for-fossil-carbon-future-prospects-for-a-global-chemical-and-derived-material-sector-based-on-renewable-carbon/, (accessed 27 February 2025).
- T. J. Dijkman, M. Birkved and M. Z. Hauschild, Int. J. Life Cycle Assess., 2012, 17, 973–986 CrossRef CAS.
- M.-W. Siegert, A. Lehmann, Y. Emara and M. Finkbeiner, Int. J. Life Cycle Assess., 2020, 25, 1436–1454 CrossRef CAS.
- W. Gao, S. Liang, R. Wang, Q. Jiang, Y. Zhang, Q. Zheng, B. Xie, C. Y. Toe, X. Zhu, J. Wang, L. Huang, Y. Gao, Z. Wang, C. Jo, Q. Wang, L. Wang, Y. Liu, B. Louis, J. Scott, A.-C. Roger, R. Amal, H. He and S.-E. Park, Chem. Soc. Rev., 2020, 49, 8584–8686 RSC.
- R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC.
- F. E. Dayan, C. L. Cantrell and S. O. Duke, Bioorg. Med. Chem., 2009, 17, 4022–4034 CrossRef CAS PubMed.
- A. G. Atanasov, B. Waltenberger, E.-M. Pferschy-Wenzig, T. Linder, C. Wawrosch, P. Uhrin, V. Temml, L. Wang, S. Schwaiger, E. H. Heiss, J. M. Rollinger, D. Schuster, J. M. Breuss, V. Bochkov, M. D. Mihovilovic, B. Kopp, R. Bauer, V. M. Dirsch and H. Stuppner, Biotechnol. Adv., 2015, 33, 1582–1614 CrossRef CAS PubMed.
- K. M. Kacprzak, in Natural Products: Phytochemistry, Botany and Metabolism of Alkaloids, Phenolics and Terpenes, ed. K. G. Ramawat and J.-M. Mérillon, Springer, Berlin, Heidelberg, 2013, pp. 605–641 Search PubMed.
- J. Gallego-Jara, G. Lozano-Terol, R. A. Sola-Martínez, M. Cánovas-Díaz and T. de Diego Puente, Molecules, 2020, 25, 5986 CrossRef CAS.
- P. Zhang, C.-B. Duan, B. Jin, A. S. Ali, X. Han, H. Zhang, M.-Z. Zhang, W.-H. Zhang and Y.-C. Gu, Adv. Agrochem, 2023, 2, 324–339 CrossRef CAS.
- A. G. Atanasov, S. B. Zotchev, V. M. Dirsch and C. T. Supuran, Nat. Rev. Drug Discovery, 2021, 20, 200–216 CrossRef CAS PubMed.
- M. Boehm, P. C. Fuenfschilling, M. Krieger, E. Kuesters and F. Struber, Org. Process Res. Dev., 2007, 11, 336–340 CrossRef CAS.
- L. Min, J.-C. Han, W. Zhang, C.-C. Gu, Y.-P. Zou and C.-C. Li, Chem. Rev., 2023, 123, 4934–4971 CrossRef CAS PubMed.
- W. C. Liu, T. Gong and P. Zhu, RSC Adv., 2016, 6, 48800–48809 RSC.
- S. Malik, R. M. Cusidó, M. H. Mirjalili, E. Moyano, J. Palazón and M. Bonfill, Process Biochem., 2011, 46, 23–34 CrossRef CAS.
- Presidential Green Chemistry Challenge, https://www.epa.gov/greenchemistry/presidential-green-chemistry-challenge-2004-greener-synthetic-pathways-award, (accessed 27 February 2025).
- B. Jiang, L. Gao, H. Wang, Y. Sun, X. Zhang, H. Ke, S. Liu, P. Ma, Q. Liao, Y. Wang, H. Wang, Y. Liu, R. Du, T. Rogge, W. Li, Y. Shang, K. N. Houk, X. Xiong, D. Xie, S. Huang, X. Lei and J. Yan, Science, 2024, 383, 622–629 CrossRef CAS.
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695–699 CrossRef CAS PubMed.
- K. Wang and J. W. Tester, Green Energy Resour., 2023, 1, 100005 CrossRef.
- C. S. K. Lin, L. A. Pfaltzgraff, L. Herrero-Davila, E. B. Mubofu, S. Abderrahim, J. H. Clark, A. A. Koutinas, N. Kopsahelis, K. Stamatelatou, F. Dickson, S. Thankappan, Z. Mohamed, R. Brocklesby and R. Luque, Energy Environ. Sci., 2013, 6, 426–464 RSC.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- Y. Liao, S.-F. Koelewijn, G. Van den Bossche, J. Van Aelst, S. Van den Bosch, T. Renders, K. Navare, T. Nicolaï, K. Van Aelst, M. Maesen, H. Matsushima, J. M. Thevelein, K. Van Acker, B. Lagrain, D. Verboekend and B. F. Sels, Science, 2020, 367, 1385–1390 CrossRef CAS.
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- C. Xu, R. A. D. Arancon, J. Labidi and R. Luque, Chem. Soc. Rev., 2014, 43, 7485–7500 RSC.
- S. Zheng, Z. Zhang, S. He, H. Yang, H. Atia, A. M. Abdel-Mageed, S. Wohlrab, E. Baráth, S. Tin, H. J. Heeres, P. J. Deuss and J. G. de Vries, Chem. Rev., 2024, 124, 10701–10876 CrossRef CAS PubMed.
- C. Ver Elst, R. Vroemans, M. Bal, S. Sergeyev, C. Mensch and B. U. W. Maes, Angew. Chem., Int. Ed., 2023, 62, e202309597 CrossRef CAS.
- E. Blondiaux, J. Bomon, M. Smoleń, N. Kaval, F. Lemière, S. Sergeyev, L. Diels, B. Sels and B. U. W. Maes, ACS Sustainable Chem. Eng., 2019, 7, 6906–6916 CrossRef CAS.
- A. Afanasenko and K. Barta, iScience, 2021, 24, 102211 CrossRef CAS.
- S. Sethupathy, G. Murillo Morales, L. Gao, H. Wang, B. Yang, J. Jiang, J. Sun and D. Zhu, Bioresour. Technol., 2022, 347, 126696 CrossRef CAS PubMed.
- X. Wu, E. Smet, F. Brandi, D. Raikwar, Z. Zhang, B. U. W. Maes and B. F. Sels, Angew. Chem., Int. Ed., 2024, 63, e202317257 CrossRef CAS.
- E. Paone, T. Tabanelli and F. Mauriello, Curr. Opin. Green Sustainable Chem., 2020, 24, 1–6 CrossRef.
- T. A. Ewing, N. Nouse, M. van Lint, J. van Haveren, J. Hugenholtz and D. S. van Es, Green Chem., 2022, 24, 6373–6405 RSC.
- N. Li and M.-H. Zong, ACS Catal., 2022, 12, 10080–10114 CrossRef CAS.
- R. C. Cioc, M. Crockatt, J. C. van der Waal and P. C. A. Bruijnincx, Angew. Chem., Int. Ed., 2022, 61, e202114720 CrossRef CAS.
- J. C. van der Waal and E. de Jong, Industrial Biorenewables, John Wiley & Sons, Ltd, 2016, pp. 97–120 Search PubMed.
- B. Yang, P. Chen, T. Zhu, G. Dong, H. Wang, Y. Liao, X. Zhang and L. Ma, Energy Fuels, 2024, 38, 5303–5313 CrossRef CAS.
- T. Mehtiö, M. Toivari, M. G. Wiebe, A. Harlin, M. Penttilä and A. Koivula, Crit. Rev. Biotechnol., 2016, 36, 904–916 CrossRef.
- H. Schiweck, A. Bär, R. Vogel, E. Schwarz, M. Kunz, C. Dusautois, A. Clement, C. Lefranc, B. Lüssem, M. Moser and S. Peters, Ullmann's Encyclopedia of Industrial Chemistry, John Wiley & Sons, Ltd, 2012 Search PubMed.
- A. Thomas, B. Matthäus and H.-J. Fiebig, Ullmann's Encyclopedia of Industrial Chemistry, John Wiley & Sons, Ltd, 2015, pp. 1–84 Search PubMed.
- U. Biermann, U. T. Bornscheuer, I. Feussner, M. A. R. Meier and J. O. Metzger, Angew. Chem., Int. Ed., 2021, 60, 20144–20165 CrossRef CAS.
- J. O. Metzger and U. Bornscheuer, Appl. Microbiol. Biotechnol., 2006, 71, 13–22 CrossRef CAS.
- A. Suhara, K. Karyadi, S. G. Herawan, A. Tirta, M. Idris, M. F. Roslan, N. R. Putra, A. L. Hananto and I. Veza, Clean Technol., 2024, 6, 886–906 CrossRef.
- A. Soutelo-Maria, J.-L. Dubois, J.-L. Couturier and G. Cravotto, Catalysts, 2018, 8, 464 CrossRef.
- V. Yelchuri, K. Srikanth, R. B. N. Prasad and M. S. L. Karuna, J. Chem. Sci., 2019, 131, 39 CrossRef.
- E. B. Mubofu, Sustainable Chem. Processes, 2016, 4, 11 CrossRef.
- Y. Liu, B. Zhong and A. Lawal, RSC Adv., 2022, 12, 27997–28008 RSC.
- Epinity – AGC Vinythai, https://epinitychem.com/, (accessed 27 February 2025).
- S. Sahani, S. N. Upadhyay and Y. C. Sharma, Ind. Eng. Chem. Res., 2021, 60, 67–88 Search PubMed.
- A. Abdullah, A. Zuhairi Abdullah, M. Ahmed, J. Khan, M. Shahadat, K. Umar and M. A. Alim, J. Cleaner Prod., 2022, 341, 130876 Search PubMed.
- T. M. Lammens, M. C. R. Franssen, E. L. Scott and J. P. M. Sanders, Biomass Bioenergy, 2012, 44, 168–181 CrossRef CAS.
- F. De Schouwer, L. Claes, A. Vandekerkhove, J. Verduyckt and D. E. De Vos, ChemSusChem, 2019, 12, 1272–1303 CrossRef CAS PubMed.
- J. Verduyckt, R. Coeck and D. E. De Vos, ACS Sustainable Chem. Eng., 2017, 5, 3290–3295 CrossRef CAS.
- L. Claes, J. Verduyckt, I. Stassen, B. Lagrain and D. E. De Vos, Chem. Commun., 2015, 51, 6528–6531 RSC.
- F. D. Schouwer, S. Adriaansen, L. Claes and D. E. De Vos, Green Chem., 2017, 19, 4919–4929 RSC.
- S. L. Bhanawase and G. D. Yadav, Catal. Today, 2017, 291, 213–222 CrossRef CAS.
- W. S. Knowles, Asymmetric Catalysis on Industrial Scale, John Wiley & Sons, Ltd, 2003, pp. 21–38 Search PubMed.
- M. Fache, E. Darroman, V. Besse, R. Auvergne, S. Caillol and B. Boutevin, Green Chem., 2014, 16, 1987–1998 RSC.
- H. Fiege, H. Voges, T. Hamamoto, S. Umemura, T. Iwata, H. Miki, Y. Fujita, H. Buysch, D. Garbe and W. Paulus, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Wiley, 1st edn, 2000 Search PubMed.
- R. Christoph, B. Schmidt, U. Steinberner, W. Dilla and R. Karinen, Ullmann's Encyclopedia of Industrial Chemistry, John Wiley & Sons, Ltd, 2006 Search PubMed.
- X. Shen, Q. Meng, Q. Mei, H. Liu, J. Yan, J. Song, D. Tan, B. Chen, Z. Zhang, G. Yang and B. Han, Chem. Sci., 2020, 11, 1347–1352 RSC.
- D. Singh, D. Sharma, S. L. Soni, C. S. Inda, S. Sharma, P. K. Sharma and A. Jhalani, J. Cleaner Prod., 2021, 307, 127299 CrossRef CAS.
- A. Kostyniuk, D. Bajec, P. Djinović and B. Likozar, Chem. Eng. J., 2020, 394, 124945 CrossRef CAS.
- K. A. Scott, P. B. Cox and J. T. Njardarson, J. Med. Chem., 2022, 65, 7044–7072 CrossRef CAS PubMed.
- Paracetamol API Market Size & Share [2033], https://www.marketreportsworld.com/market-reports/paracetamol-api-market-14714703, (accessed 9 June 2025).
- J. Park, M. A. Kelly, J. X. Kang, S. S. Seemakurti, J. L. Ramirez, M. C. Hatzell, C. Sievers and A. S. Bommarius, Green Chem., 2021, 23, 7488–7498 RSC.
- ed. A. Kleemann, J. Engel, B. Kutscher and D. Reichert, Pharmaceutical Substances: Syntheses, Patents and Applications of the most relevant APIs, Georg Thieme Verlag, Stuttgart, 5th edn, 2009 Search PubMed.
- R. Joncour, N. Duguet, E. Métay, A. Ferreira and M. Lemaire, Green Chem., 2014, 16, 2997 Search PubMed.
- S. D. Karlen, V. I. Timokhin, C. Sener, J. K. Mobley, T. Runge and J. Ralph, ChemSusChem, 2024, 17, e202400234 CrossRef CAS.
- N. Z. Burns, P. S. Baran and R. W. Hoffmann, Angew. Chem., Int. Ed., 2009, 48, 2854–2867 CrossRef CAS.
- L. Dong, Y. Wang, Y. Dong, Y. Zhang, M. Pan, X. Liu, X. Gu, M. Antonietti and Z. Chen, Nat. Commun., 2023, 14, 4996 CrossRef CAS PubMed.
- P. D’Arrigo, L. A. M. Rossato, A. Strini and S. Serra, Molecules, 2024, 29, 442 CrossRef PubMed.
- G. De Smet, X. Bai, C. Mensch, S. Sergeyev, G. Evano and B. U. W. Maes, Angew. Chem., Int. Ed., 2022, 61, e202201751 CrossRef CAS PubMed.
- J. Lora, Monomers, Polymers and Composites from Renewable Resources, ed. M. N. Belgacem and A. Gandini, Elsevier, Amsterdam, 2008, pp. 225–241 Search PubMed.
- Z. Sheng, C. Deng, C. Tian, F. Liu, G. Li, H. Wang, F. Yin, F. Ding and R. Yang, CN111892507A, 2020.
- J. Ralph, S. Karlen and J. Mobley, US10286504B2, 2019.
- RoadToBio | Publications, https://roadtobio.eu/index.php?page=publications, (accessed 18 March 2025).
- Avantium Starts Unloading of High Fructose Syrup at FDCA Flagship Plant, https://newsroom.avantium.com/avantium-starts-unloading-of-high-fructose-syrup-at-fdca-flagship-plant/, (accessed 22 August 2025).
- J. Ruf, A. Emberger-Klein and K. Menrad, Sustainable Prod. Consum., 2022, 34, 353–370 CrossRef.
- The Mass Balance Approach, https://www.basf.com/basf/www/global/en/who-we-are/sustainability/we-drive-sustainable-solutions/circular-economy/mass-balance-approach, (accessed 27 February 2025).
- Acrylic Monomers, https://chemicals.basf.com/basf/chemicals/global/en/Petrochemicals/acrylic-monomers, (accessed 18 March 2025).
- E. A. R. Zuiderveen, K. J. J. Kuipers, C. Caldeira, S. V. Hanssen, M. K. Van Der Hulst, M. M. J. De Jonge, A. Vlysidis, R. Van Zelm, S. Sala and M. A. J. Huijbregts, Nat. Commun., 2023, 14, 8521 Search PubMed.
- M. Tschulkow, M. Pizzol, T. Compernolle, S. Van Den Bosch, B. Sels and S. Van Passel, Resour., Conserv. Recycl., 2024, 204, 107466 CrossRef CAS.
- J.-P. Lange, Nat. Catal., 2021, 4, 186–192 CrossRef CAS.
- R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC.
- R. Arthur Sheldon, Green Chem., 2023, 25, 1704–1728 RSC.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2009, 39, 301–312 Search PubMed.
- W. S. Knowles, Adv. Synth. Catal., 2003, 345, 3–13 CrossRef CAS.
- Y. Chauvin, Angew. Chem., Int. Ed., 2006, 45, 3740–3747 CrossRef.
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS.
- K. Voskarides, J. Mol. Evol., 2021, 89, 189–191 CrossRef CAS.
- T. Ooi and C. Crudden, ACS Catal., 2021, 11, 15234 CrossRef CAS.
- Nat. Mater., 2023, 22, 147 Search PubMed.
- C. A. Busacca, D. R. Fandrick, J. J. Song and C. H. Senanayake, Applications of Transition Metal Catalysis in Drug Discovery and Development, John Wiley & Sons, Ltd, 2012, pp. 1–24 Search PubMed.
- Q. Yang, Y. Zhao and D. Ma, Org. Process Res. Dev., 2022, 26, 1690–1750 CrossRef CAS.
- B. A. Baviskar, P. V. Ajmire, D. S. Chumbhale, M. S. Khan, V. G. Kuchake, M. Singupuram and P. R. Laddha, Sustainable Chem. Pharm., 2023, 32, 100953 CrossRef CAS.
- Steel, Aluminum, Nickel, Rare earth,Copper Prices Charts and News-Shanghai Metals Market, https://www.metal.com/, (accessed 24 February 2025).
- WebElements Periodic Table “Periodicity” Abundance in Earth's crust (by weight) Periodic table gallery, https://www.webelements.com/periodicity/abund_crust/, (accessed 25 February 2025).
- ICH Q3D Elemental impurities – Scientific guideline | European Medicines Agency (EMA), https://www.ema.europa.eu/en/ich-q3d-elemental-impurities-scientific-guideline, (accessed 24 February 2025).
- P. Nuss and M. J. Eckelman, PLoS One, 2014, 9, e101298 CrossRef.
- Mineral commodity summaries 2023|U.S. Geological Survey, https://www.usgs.gov/publications/mineral-commodity-summaries-2023, (accessed 25 February 2025).
- J. D. Hayler, D. K. Leahy and E. M. Simmons, Organometallics, 2019, 38, 36–46 CrossRef CAS.
- Precious metals leasing transaction, https://www.marsh.com/content/marsh2/en_us/services/trade-credit/insights/how-credit-insurance-can-help-leasing-companies.html, (accessed 24 February 2025).
- C. Harpprecht, B. Miranda Xicotencatl, S. van Nielen, M. van der Meide, C. Li, Z. Li, A. Tukker and B. Steubing, Resour., Conserv. Recycl., 2024, 205, 107572 CrossRef CAS.
- K. S. Egorova and V. P. Ananikov, Organometallics, 2017, 36, 4071–4090 CrossRef CAS.
- M. U. Luescher and F. Gallou, Green Chem., 2024, 26, 5239–5252 RSC.
- S. Smidt, J. Dietz, M. Keil and T. Grote, WO2007138089, 2007.
- A. Straub, N. Lui, J. Wieschemeyer, U. Klöttschen and E. Damen, WO2009003650, 2009.
- M. Dockner, L. Rodefeld and J. Heinrich, WO2015011032, 2015.
- H. Zhao, A. K. Ravn, M. C. Haibach, K. M. Engle and C. C. C. Johansson Seechurn, ACS Catal., 2024, 14, 9708–9733 CrossRef CAS PubMed.
- M. P. Watson and D. J. Weix, Acc. Chem. Res., 2024, 57, 2451–2452 CrossRef CAS.
- O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440–1459 RSC.
- R. A. Singer, S. Monfette, D. J. Bernhardson, S. Tcyrulnikov and E. C. Hansen, Org. Process Res. Dev., 2020, 24, 909–915 CrossRef CAS.
- R. A. Sheldon, ACS Sustainable Chem. Eng., 2018, 6, 32–48 CrossRef CAS.
- B. Voss, S. I. Andersen, E. Taarning and C. H. Christensen, ChemSusChem, 2009, 2, 1152–1162 CrossRef CAS PubMed.
- M. L. Clapson, C. S. Durfy, D. Facchinato and M. W. Drover, Cell Rep. Phys. Sci., 2023, 4, 101548 CrossRef CAS.
- R. Kranthikumar, Organometallics, 2022, 41, 667–679 CrossRef CAS.
- L.-M. Chen and S. E. Reisman, Acc. Chem. Res., 2024, 57, 751–762 CrossRef CAS.
- V. M. Chernyshev and V. P. Ananikov, ACS Catal., 2022, 12, 1180–1200 CrossRef CAS.
- J. D. Sears, P. G. N. Neate and M. L. Neidig, J. Am. Chem. Soc., 2018, 140, 11872–11883 CrossRef CAS.
- R. B. Bedford and P. B. Brenner, in Iron Catalysis II, ed. E. Bauer, Springer International Publishing, Cham, 2015, pp. 19–46 Search PubMed.
- C. Risatti, K. J. Jr. Natalie, Z. Shi and D. A. Conlon, Org. Process Res. Dev., 2013, 17, 257–264 CrossRef CAS.
- C. Margarita and P. G. Andersson, J. Am. Chem. Soc., 2017, 139, 1346–1356 CrossRef CAS PubMed.
- J. Wen, F. Wang and X. Zhang, Chem. Soc. Rev., 2021, 50, 3211–3237 RSC.
- S. Chakrabortty, B. de Bruin and J. G. de Vries, Angew. Chem., Int. Ed., 2024, 63, e202315773 CrossRef CAS.
- M. R. Friedfeld, H. Zhong, R. T. Ruck, M. Shevlin and P. J. Chirik, Science, 2018, 360, 888–893 CrossRef CAS.
- J. Surtees, V. Marmon, E. Differding and V. Zimmermann, WO2001064637A1, 2001.
- P. W. Sutton, J. P. Adams, C. Wade and K. Wheelhouse, Early Drug Development, John Wiley & Sons, Ltd, 2018, pp. 73–124 Search PubMed.
- R. F. Algera, C. Allais, A. F. Baldwin, T. Busch, F. Colombo, M. Colombo, C. Depretz, Y. R. Dumond, A. R. Faria Quintero, M. Heredia, J. Jung, A. Lall, T. Lee, Y. Liu, S. Mandelli, M. Mantel, R. Morris, J. Mustakis, B. Nguyen, R. Pearson, J. L. Piper, J. A. Ragan, B. Ruffin, C. Talicska, S. Tcyrulnikov, C. Uyeda, R. M. Weekly and M. Zeng, Org. Process Res. Dev., 2023, 27, 2260–2270 CrossRef CAS.
- J. Werth and C. Uyeda, Angew. Chem., Int. Ed., 2018, 57, 13902–13906 CrossRef CAS PubMed.
- F. Ullmann and J. Bielecki, Berichte Dtsch. Chem. Ges., 1901, 34, 2174–2185 CrossRef CAS.
- X. Li, Q. Yang, B. A. Lorsbach, A. Buysse, N. Niyaz, L. Cui and R. Jr. Ross, Org. Process Res. Dev., 2022, 26, 3290–3302 CrossRef CAS.
- Q. Yang, X. Li, B. A. Lorsbach, G. Roth, D. E. Podhorez, R. Jr. Ross, N. Niyaz, A. Buysse, D. Knueppel and J. Nissen, Org. Process Res. Dev., 2019, 23, 2122–2132 CrossRef CAS.
- M. U. Luescher, F. Gallou and B. H. Lipshutz, Chem. Sci., 2024, 15, 9016–9025 RSC.
- T. Chiarini, I. Ciabatti, G. Rossi, C. Tavanti and C. Zambardi, The role and the recovery of Platinum-Group Metal catalysts in the pharmaceutical industry, https://www.tcaspa.com/en/news/the-role-and-the-recovery-of-platinum-group-metal-catalysts-in-the-pharmaceutical-industry, (accessed 24 February 2025).
- X. Liang, J. Tang, L. Li, Y. Wu and Y. Sun, J. Cleaner Prod., 2022, 376, 134108 CrossRef CAS.
- P. Devendar, R.-Y. Qu, W.-M. Kang, B. He and G.-F. Yang, J. Agric. Food Chem., 2018, 66, 8914–8934 CrossRef CAS.
- O. García Mancheño and M. Waser, Eur. J. Org. Chem., 2023, e202200950 CrossRef.
- A. Antenucci, S. Dughera and P. Renzi, ChemSusChem, 2021, 14, 2785–2853 CrossRef CAS.
- J. Maes, E. A. Mitchell and B. U. W. Maes, in Green Chemistry Series, ed. L. Summerton, H. F. Sneddon, L. C. Jones and J. H. Clark, Royal Society of Chemistry, Cambridge, 2016, pp. 192–202 Search PubMed.
- H. Xiao, W. R. F. Goundry, R. Griffiths, Y. Feng and S. Karlsson, Green Chem., 2025, 27, 3186–3196 RSC.
- R. H. Crabtree, Chem. Rev., 2015, 115, 127–150 CrossRef CAS.
- C. W. Jones, Top. Catal., 2010, 53, 942–952 CrossRef CAS.
- S. Wacławek, V. V. T. Padil and M. Černík, Ecol. Chem. Eng. Sci., 2018, 25, 9–34 Search PubMed.
- S. Bhaduri and M. Doble, Homogeneous catalysis: mechanisms and industrial applications, Wiley, Hoboken, New Jersey, 2nd edn, 2014 Search PubMed.
- J. M. Thomas, Principles and Practice of Heterogeneous Catalysis, John Wiley & Sons, Incorporated, Newark, 1st edn, 2015 Search PubMed.
- R. A. Sheldon, I. Arends and U. Hanefeld, Green chemistry and catalysis, Wiley-VCH, Weinheim, 2007 Search PubMed.
- S. Vásquez-Céspedes, R. C. Betori, M. A. Cismesia, J. K. Kirsch and Q. Yang, Org. Process Res. Dev., 2021, 25, 740–753 CrossRef.
- R. A. Sheldon and H. van Bekkum, Fine Chemicals through Heterogeneous Catalysis, John Wiley & Sons, 2008 Search PubMed.
- C. W. A. Chan, A. H. Mahadi, M. M.-J. Li, E. C. Corbos, C. Tang, G. Jones, W. C. H. Kuo, J. Cookson, C. M. Brown, P. T. Bishop and S. C. E. Tsang, Nat. Commun., 2014, 5, 5787 CrossRef CAS PubMed.
- N. A. Magnus, J. A. Aikins, J. S. Cronin, W. D. Diseroad, A. D. Hargis, M. E. LeTourneau, B. E. Parker, S. M. Reutzel-Edens, J. P. Schafer, M. A. Staszak, G. A. Stephenson, S. L. Tameze and L. M. H. Zollars, Org. Process Res. Dev., 2005, 9, 621–628 CrossRef CAS.
- C. Hardouin, S. Baillard, F. Barière, C. Copin, A. Craquelin, S. Janvier, S. Lemaitre, S. Le Roux, O. Russo and S. Samson, Org. Process Res. Dev., 2020, 24, 652–669 CrossRef CAS.
- F.-X. Felpin, T. Ayad and S. Mitra, Eur. J. Org. Chem., 2006, 2679–2690 CrossRef CAS.
- S. Hübner, J. G. de Vries and V. Farina, Adv. Synth. Catal., 2016, 358, 3–25 CrossRef.
- V. Pandarus, G. Gingras, F. Béland, R. Ciriminna and M. Pagliaro, Org. Process Res. Dev., 2012, 16, 117–122 CrossRef CAS.
- R. Ciriminna, V. Pandarus, A. Fidalgo, L. M. Ilharco, F. Béland and M. Pagliaro, Org. Process Res. Dev., 2015, 19, 755–768 CrossRef CAS.
- V. Pandarus, D. Desplantier-Giscard, G. Gingras, F. Béland, R. Ciriminna and M. Pagliaro, Org. Process Res. Dev., 2013, 17, 1492–1497 CrossRef CAS.
- in Design of Heterogeneous Catalysts, ed. U. S. Ozkan, Wiley, 1st edn, 2009 Search PubMed.
- S. Bhat, Y. J. Pagán-Torres and E. Nikolla, Top. Catal., 2023, 66, 1217–1243 CrossRef CAS.
- J. S. Bates, Nat. Chem., 2025, 1–7 Search PubMed.
- T. A. Fassbach, J.-M. Ji, A. J. Vorholt and W. Leitner, ACS Catal., 2024, 14, 7289–7298 CrossRef CAS.
- J. Hagen, Industrial Catalysis: A Practical Approach, Wiley, 1st edn, 2015 Search PubMed.
- M. C. Bryan, P. J. Dunn, D. Entwistle, F. Gallou, S. G. Koenig, J. D. Hayler, M. R. Hickey, S. Hughes, M. E. Kopach, G. Moine, P. Richardson, F. Roschangar, A. Steven and F. J. Weiberth, Green Chem., 2018, 20, 5082–5103 RSC.
- J. M. Thomas, Nature, 2015, 525, 325–326 CrossRef CAS.
- G. Vilé, D. Albani, M. Nachtegaal, Z. Chen, D. Dontsova, M. Antonietti, N. López and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2015, 54, 11265–11269 CrossRef.
- M. A. Bajada, J. Sanjosé-Orduna, G. D. Liberto, S. Tosoni, G. Pacchioni, T. Noël and G. Vilé, Chem. Soc. Rev., 2022, 51, 3898–3925 RSC.
- A. C. M. Loy, S. Y. Teng, B. S. How, X. Zhang, K. W. Cheah, V. Butera, W. D. Leong, B. L. F. Chin, C. L. Yiin, M. J. Taylor and G. Kyriakou, Prog. Energy Combust. Sci., 2023, 96, 101074 CrossRef.
- H. Zhang, G. Liu, L. Shi and J. Ye, Adv. Energy Mater., 2018, 8, 1701343 CrossRef.
- S. Mitchell and J. Pérez-Ramírez, Nat. Commun., 2020, 11, 4302 CrossRef CAS PubMed.
- V. B. Saptal, V. Ruta, M. A. Bajada and G. Vilé, Angew. Chem., Int. Ed., 2023, 62, e202219306 CrossRef CAS.
- Z. Chen, E. Vorobyeva, S. Mitchell, E. Fako, M. A. Ortuño, N. López, S. M. Collins, P. A. Midgley, S. Richard, G. Vilé and J. Pérez-Ramírez, Nat. Nanotechnol., 2018, 13, 702–707 CrossRef CAS PubMed.
- V. B. Saptal, C. Saetta, A. Laufenböck, M. Sterrer, I. S. Kwon, A. Lucotti, M. Tommasini, O. Tomanec, A. Bakandritsos, G. Di Liberto, G. Pacchioni and G. Vilé, J. Am. Chem. Soc., 2025, 147, 18524–18540 CrossRef CAS PubMed.
- T. A. Gazis, S. Palit, L. A. Cipriano, N. Allasia, S. M. Collins, Q. M. Ramasse, I. S. Kwon, M. Sterrer, G. Di Liberto and G. Vilé, Angew. Chem., Int. Ed., 2025, 64, e202510632 CrossRef CAS.
- H. Jia, Q. Liao, W. Liu, L. A. Cipriano, H. Jiang, P. H. Dixneuf, G. Vilé and M. Zhang, J. Am. Chem. Soc., 2024, 146, 31647–31655 CrossRef CAS.
- M. Boudart, Chem. Rev., 1995, 95, 661–666 CrossRef CAS.
- J. Liu, Z. Chen, C. Liu, B. Zhang, Y. Du, C.-F. Liu, L. Ma, S. Xi, R. Li, X. Zhao, J. Song, X. Z. Sui, W. Yu, L. Miao, J. Jiang, M. J. Koh and K. P. Loh, J. Mater. Chem. A, 2021, 9, 11427–11432 RSC.
- S. Ji, X. Lu, M. Zhang, L. Leng, H. Liu, K. Yin, C. Xu, C. He, J. H. Horton, J. Zhang and Z. Li, Chem. Eng. J., 2023, 452, 139205 CrossRef CAS.
- Y. Jin, F. Lu, D. Yi, J. Li, F. Zhang, T. Sheng, F. Zhan, Y. Duan, G. Huang, J. Dong, B. Zhou, X. Wang and J. Yao, CCS Chem., 2020, 3, 1453–1462 CrossRef.
- M. A. Bajada, M. Tschulkow and G. Vilé, Cell Rep. Sustain., 2025, 2, 100286 Search PubMed.
- H. Yan, C. Su, J. He and W. Chen, J. Mater. Chem. A, 2018, 6, 8793–8814 RSC.
- T. A. Gazis, V. Ruta and G. Vilé, ACS Catal., 2025, 15, 6852–6873 CrossRef CAS.
- S. Liu, Y. Chen, C. Chen, Y. Wu, J. Du, X. Feng, Q. Wu, P. Qi, H. Wang, N. Ren and W.-Q. Guo, Sep. Purif. Technol., 2024, 351, 127989 CrossRef CAS.
- X. Hai, Y. Zheng, Q. Yu, N. Guo, S. Xi, X. Zhao, S. Mitchell, X. Luo, V. Tulus, M. Wang, X. Sheng, L. Ren, X. Long, J. Li, P. He, H. Lin, Y. Cui, X. Peng, J. Shi, J. Wu, C. Zhang, R. Zou, G. Guillén-Gosálbez, J. Pérez-Ramírez, M. J. Koh, Y. Zhu, J. Li and J. Lu, Nature, 2023, 622, 754–760 CrossRef CAS.
- A. R. Alcántara, P. Domínguez de María, J. A. Littlechild, M. Schürmann, R. A. Sheldon and R. Wohlgemuth, ChemSusChem, 2022, 15, e202102709 CrossRef.
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef CAS PubMed.
- J. Santiago-Arcos, S. Velasco-Lozano, E. Diamanti, A. I. Benítez-Mateos, D. Grajales-Hernández, F. Paradisi and F. López-Gallego, ACS Sustainable Chem. Eng., 2024, 12, 9474–9489 CrossRef CAS PubMed.
- A. Ledesma-Fernandez, S. Velasco-Lozano, P. Campos-Muelas, R. Madrid, F. López-Gallego and A. L. Cortajarena, Protein Sci., 2024, 33, e4984 CrossRef CAS.
- H. Bak, J. Cha and I. Kwon, Chem. Eng. J., 2024, 497, 154463 CrossRef CAS.
- S. Liu, X. Wu, X. Dong, Q. Shi and Y. Sun, ACS Sustainable Chem. Eng., 2024, 12, 6366–6375 CrossRef CAS.
- C. M. Heckmann and F. Paradisi, ChemCatChem, 2020, 12, 6082–6102 CrossRef CAS PubMed.
- R. A. Sheldon and P. C. Pereira, Chem. Soc. Rev., 2017, 46, 2678–2691 RSC.
- P. Žnidaršič-Plazl, Curr. Opin. Green Sustainable Chem., 2021, 32, 100546 CrossRef.
- ‘Revolution based on evolution’ honored with chemistry Nobel | Science | AAAS, https://www.science.org/content/article/revolution-based-evolution-honored-chemistry-nobel, (accessed 25 February 2025).
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef CAS PubMed.
- J. Liang, J. Lalonde, B. Borup, V. Mitchell, E. Mundorff, N. Trinh, D. A. Kochrekar, R. Nair Cherat and G. G. Pai, Org. Process Res. Dev., 2010, 14, 193–198 CrossRef CAS.
- N. J. Turner and R. Kumar, Curr. Opin. Chem. Biol., 2018, 43, A1–A3 CrossRef CAS.
- R. Buller, S. Lutz, R. J. Kazlauskas, R. Snajdrova, J. C. Moore and U. T. Bornscheuer, Science, 2023, 382, eadh8615 CrossRef CAS.
- M. Gantz, S. V. Mathis, F. E. H. Nintzel, P. Lio and F. Hollfelder, Faraday Discuss., 2024, 252, 89–114 RSC.
- M. Vasina, D. Kovar, J. Damborsky, Y. Ding, T. Yang, A. deMello, S. Mazurenko, S. Stavrakis and Z. Prokop, Biotechnol. Adv., 2023, 66, 108171 CrossRef CAS PubMed.
- Imperagen – Reinventing Enzyme Engineering, https://imperagen.com/, (accessed 25 February 2025).
- I. Levin, M. Liu, C. A. Voigt and C. W. Coley, Nat. Commun., 2022, 13, 7747 CrossRef CAS PubMed.
- D. Kreutter and J.-L. Reymond, Chem. Sci., 2024, 15, 18031–18047 RSC.
- W. Finnigan, M. Lubberink, L. J. Hepworth, J. Citoler, A. P. Mattey, G. J. Ford, J. Sangster, S. C. Cosgrove, B. Z. da Costa, R. S. Heath, T. W. Thorpe, Y. Yu, S. L. Flitsch and N. J. Turner, ACS Catal., 2023, 13, 11771–11780 CrossRef CAS.
- S. P. France, R. D. Lewis and C. A. Martinez, JACS Au, 2023, 3, 715–735 CrossRef CAS.
- C. W. Coley, D. A. Thomas, J. A. M. Lummiss, J. N. Jaworski, C. P. Breen, V. Schultz, T. Hart, J. S. Fishman, L. Rogers, H. Gao, R. W. Hicklin, P. P. Plehiers, J. Byington, J. S. Piotti, W. H. Green, A. J. Hart, T. F. Jamison and K. F. Jensen, Science, 2019, 365, eaax1566 CrossRef CAS.
- D. Wu and X. Lei, Tetrahedron, 2022, 127, 133099 CrossRef CAS.
- F. Kaspar and A. Schallmey, Curr. Opin. Biotechnol, 2022, 77, 102759 CrossRef CAS.
- S. Simić, E. Zukić, L. Schmermund, K. Faber, C. K. Winkler and W. Kroutil, Chem. Rev., 2022, 122, 1052–1126 CrossRef.
- R. Tanifuji and H. Oguri, Beilstein J. Org. Chem., 2024, 20, 1693–1712 CrossRef CAS.
- G. Cruz, J. Acosta, J. Del Arco, V. J. Clemente-Suarez, V. Deroncele and J. Fernández-Lucas, ChemCatChem, 2022, 14, e202200140 CrossRef CAS.
- J. A. McIntosh, T. Benkovics, S. M. Silverman, M. A. Huffman, J. Kong, P. E. Maligres, T. Itoh, H. Yang, D. Verma, W. Pan, H.-I. Ho, J. Vroom, A. M. Knight, J. A. Hurtak, A. Klapars, A. Fryszkowska, W. J. Morris, N. A. Strotman, G. S. Murphy, K. M. Maloney and P. S. Fier, ACS Cent. Sci., 2021, 7, 1980–1985 CrossRef CAS.
- A. J. Burke, W. R. Birmingham, Y. Zhuo, T. W. Thorpe, B. Zucoloto da Costa, R. Crawshaw, I. Rowles, J. D. Finnigan, C. Young, G. M. Holgate, M. P. Muldowney, S. J. Charnock, S. L. Lovelock, N. J. Turner and A. P. Green, J. Am. Chem. Soc., 2022, 144, 3761–3765 CrossRef CAS.
- M. A. Huffman, A. Fryszkowska, O. Alvizo, M. Borra-Garske, K. R. Campos, K. A. Canada, P. N. Devine, D. Duan, J. H. Forstater, S. T. Grosser, H. M. Halsey, G. J. Hughes, J. Jo, L. A. Joyce, J. N. Kolev, J. Liang, K. M. Maloney, B. F. Mann, N. M. Marshall, M. McLaughlin, J. C. Moore, G. S. Murphy, C. C. Nawrat, J. Nazor, S. Novick, N. R. Patel, A. Rodriguez-Granillo, S. A. Robaire, E. C. Sherer, M. D. Truppo, A. M. Whittaker, D. Verma, L. Xiao, Y. Xu and H. Yang, Science, 2019, 366, 1255–1259 CrossRef CAS PubMed.
- S. Alonso, G. Santiago, I. Cea-Rama, L. Fernandez-Lopez, C. Coscolín, J. Modregger, A. K. Ressmann, M. Martínez-Martínez, H. Marrero, R. Bargiela, M. Pita, J. L. Gonzalez-Alfonso, M. L. Briand, D. Rojo, C. Barbas, F. J. Plou, P. N. Golyshin, P. Shahgaldian, J. Sanz-Aparicio, V. Guallar and M. Ferrer, Nat. Catal., 2020, 3, 319–328 CrossRef CAS.
- S. Roda, L. Fernandez-Lopez, M. Benedens, A. Bollinger, S. Thies, J. Schumacher, C. Coscolín, M. Kazemi, G. Santiago, C. G. W. Gertzen, J. L. Gonzalez-Alfonso, F. J. Plou, K.-E. Jaeger, S. H. J. Smits, M. Ferrer and V. Guallar, Angew. Chem., Int. Ed., 2022, 61, e202207344 CrossRef CAS.
- T. Menegatti, I. Plazl and P. Žnidaršič-Plazl, Chem. Eng. J., 2024, 483, 149317 CrossRef CAS.
- F. Gallou, H. Gröger and B. H. Lipshutz, Green Chem., 2023, 25, 6092–6107 RSC.
- S. V. Ley, Y. Chen, A. Robinson, B. Otter, E. Godineau and C. Battilocchio, Org. Process Res. Dev., 2021, 25, 713–720 CrossRef CAS.
- K. P. Cole and M. D. Johnson, Expert Rev. Clin. Pharmacol., 2018, 11, 5–13 CrossRef CAS PubMed.
- M. Baumann, T. S. Moody, M. Smyth and S. Wharry, Eur. J. Org. Chem., 2020, 7398–7406 CrossRef CAS.
- A. S. Burange, S. M. Osman and R. Luque, iScience, 2022, 25, 103892 CrossRef CAS.
- A. Domokos, B. Nagy, B. Szilágyi, G. Marosi and Z. K. Nagy, Org. Process Res. Dev., 2021, 25, 721–739 CrossRef CAS.
- M. Colella, A. Nagaki and R. Luisi, Chem. – Eur. J., 2020, 26, 19–32 CrossRef CAS.
- P. Natho and R. Luisi, Tetrahedron Green Chem., 2023, 2, 100015 CrossRef.
- M. Spennacchio, P. Natho, M. Andresini and M. Colella, J. Flow Chem., 2024, 14, 43–83 CrossRef CAS.
- P. Natho, M. Colella, M. Andresini, L. Degennaro and R. Luisi, Org. Lett., 2024, 26, 3032–3036 CrossRef CAS PubMed.
- L. J. Nolan, S. J. King, S. Wharry, T. S. Moody and M. Smyth, Curr. Opin. Green Sustainable Chem., 2024, 46, 100886 CrossRef CAS.
- P. Sagmeister, R. Lebl, I. Castillo, J. Rehrl, J. Kruisz, M. Sipek, M. Horn, S. Sacher, D. Cantillo, J. D. Williams and C. O. Kappe, Angew. Chem., Int. Ed., 2021, 60, 8139–8148 CrossRef CAS PubMed.
- A. M. Kearney, S. G. Collins and A. R. Maguire, React. Chem. Eng., 2024, 9, 990–1013 RSC.
- M. Fonteyne, J. Vercruysse, F. De Leersnyder, B. Van Snick, C. Vervaet, J. P. Remon and T. De Beer, TrAC, Trends Anal. Chem., 2015, 67, 159–166 CrossRef CAS.
- P. Liu, H. Jin, Y. Chen, D. Wang, H. Yan, M. Wu, F. Zhao and W. Zhu, Chin. Chem. Lett., 2024, 35, 108877 CrossRef CAS.
- F. M. Akwi and P. Watts, Chem. Commun., 2018, 54, 13894–13928 RSC.
- M. M. Nasr, M. Krumme, Y. Matsuda, B. L. Trout, C. Badman, S. Mascia, C. L. Cooney, K. D. Jensen, A. Florence, C. Johnston, K. Konstantinov and S. L. Lee, J. Pharm. Sci., 2017, 106, 3199–3206 CrossRef CAS.
- C. Badman, C. L. Cooney, A. Florence, K. Konstantinov, M. Krumme, S. Mascia, M. Nasr and B. L. Trout, J. Pharm. Sci., 2019, 108, 3521–3523 CrossRef CAS.
- A. C. Fisher, W. Liu, A. Schick, M. Ramanadham, S. Chatterjee, R. Brykman, S. L. Lee, S. Kozlowski, A. B. Boam, S. C. Tsinontides and M. Kopcha, Int. J. Pharm., 2022, 622, 121778 CrossRef CAS.
- ICH guideline Q13 on continuous manufacturing of drug substances and drug products – Scientific guideline | European Medicines Agency (EMA), https://www.ema.europa.eu/en/ich-guideline-q13-continuous-manufacturing-drug-substances-and-drug-products-scientific-guideline, (accessed 25 February 2025).
- ICH Q13 Continuous Manufacturing of Drug Substances and Drug Products, https://www.fda.gov/regulatory-information/search-fda-guidance-documents/q13-continuous-manufacturing-drug-substances-and-drug-products, (accessed 25 February 2025).
- ICH Official web site: ICH, https://ich.org/page/quality-guidelines#13-2, (accessed 25 February 2025).
- S. M. Kelly, R. Lebl, T. C. Malig, T. M. Bass, D. Kummli, D. Kaldre, U. Orcel, L. Tröndlin, D. Linder, J. Sedelmeier, S. Bachmann, C. Han, H. Zhang and F. Gosselin, Org. Process Res. Dev., 2024, 28, 1546–1555 CrossRef CAS.
- G. Yano, M. Hyakumura, K. Nakano, H. Yasukouchi, H. Kawachi, M. Funabashi, T. Ohishi, Y. Ogawa and A. Nishiyama, Org. Process Res. Dev., 2024, 28, 1814–1821 CrossRef CAS.
- E. G. Moschetta, G. C. Cook, L. J. Edwards, M. A. Ischay, Z. Lei, F. Buono, F. Lévesque, J. A. O. Garber, M. MacTaggart, M. Sezen-Edmonds, K. P. Cole, M. G. Beaver, J. Doerfler, S. M. Opalka, W. Liang, P. D. Morse and N. Miyake, Org. Process Res. Dev., 2024, 28, 831–846 CrossRef CAS.
- M. G. Beaver, E. Zhang, Z. Liu, S. Zheng, B. Wang, J. Lu, J. Tao, M. Gonzalez, S. Jones and J. S. Tedrow, Org. Process Res. Dev., 2020, 24, 2139–2146 CrossRef CAS.
- A. Robinson, M. Dieckmann, J.-P. Krieger, T. Vent-Schmidt, D. Marantelli, R. Kohlbrenner, D. Gribkov, L. L. Simon, D. Austrup, A. Rod and C. G. Bochet, Org. Process Res. Dev., 2021, 25, 2205–2220 CrossRef CAS.
- C. Pratley, Y. Shaalan, L. Boulton, C. Jamieson, J. A. Murphy and L. J. Edwards, Org. Process Res. Dev., 2024, 28, 1725–1733 CrossRef CAS.
- A. Steiner, P. M. C. Roth, F. J. Strauss, G. Gauron, G. Tekautz, M. Winter, J. D. Williams and C. O. Kappe, Org. Process Res. Dev., 2020, 24, 2208–2216 CrossRef CAS.
- A. Chaudhuri, W. F. C. de Groot, J. H. A. Schuurmans, S. D. A. Zondag, A. Bianchi, K. P. L. Kuijpers, R. Broersma, A. Delparish, M. Dorbec, J. van der Schaaf and T. Noël, Org. Process Res. Dev., 2025, 29, 460–471 CrossRef CAS.
- R. I. Teixeira, T. H. Waldron Clarke, A. Love, X.-Z. Sun, S. Kayal and M. W. George, Org. Process Res. Dev., 2025, 29, 34–47 CrossRef CAS PubMed.
- K. C. Harper, E.-X. Zhang, Z.-Q. Liu, T. Grieme, T. B. Towne, D. J. Mack, J. Griffin, S.-Y. Zheng, N.-N. Zhang, S. Gangula, J.-L. Yuan, R. Miller, P.-Z. Huang, J. Gage, M. Diwan and Y.-Y. Ku, Org. Process Res. Dev., 2022, 26, 404–412 CrossRef CAS.
- C. Bottecchia, F. Lévesque, J. P. McMullen, Y. Ji, M. Reibarkh, F. Peng, L. Tan, G. Spencer, J. Nappi, D. Lehnherr, K. Narsimhan, M. K. Wismer, L. Chen, Y. Lin and S. M. Dalby, Org. Process Res. Dev., 2022, 26, 516–524 CrossRef CAS.
- N. Tanbouza, T. Ollevier and K. Lam, iScience, 2020, 23, 101720 CrossRef CAS.
- M. C. Leech, A. D. Garcia, A. Petti, A. P. Dobbs and K. Lam, React. Chem. Eng., 2020, 5, 977–990 RSC.
- N. Petrović, B. K. Malviya, C. O. Kappe and D. Cantillo, Org. Process Res. Dev., 2023, 27, 2072–2081 CrossRef.
- X. Zhong, M. A. Hoque, M. D. Graaf, K. C. Harper, F. Wang, J. D. Genders and S. S. Stahl, Org. Process Res. Dev., 2021, 25, 2601–2607 CrossRef CAS.
- J. D. Griffin, K. C. Harper, S. Velasquez Morales, W. H. Morrill, W. I. Thornton, D. Sutherland and B. A. Greiner, Org. Process Res. Dev., 2024, 28, 1877–1885 CrossRef CAS.
- A. G. Fink, F. Navarro-Pardo, J. R. Tavares and U. Legrand, ChemCatChem, 2024, 16, e202300977 CrossRef CAS.
- A. Love, D. S. Lee, G. Gennari, R. Jefferson-Loveday, S. J. Pickering, M. Poliakoff and M. George, Org. Process Res. Dev., 2021, 25, 1619–1627 CrossRef CAS.
- F. Xu, L. Chen, D. Lehnherr and F. Lévesque, Org. Process Res. Dev., 2025, 29, 872–880 CrossRef CAS.
- Y. Yuan and A. Lei, Nat. Commun., 2020, 11, 802 CrossRef CAS PubMed.
- C. Bottecchia, D. Lehnherr, F. Lévesque, M. Reibarkh, Y. Ji, V. L. Rodrigues, H. Wang, Y. Lam, T. P. Vickery, B. M. Armstrong, K. A. Mattern, K. Stone, M. K. Wismer, A. N. Singh, E. L. Regalado, K. M. Maloney and N. A. Strotman, Org. Process Res. Dev., 2022, 26, 2423–2437 CrossRef CAS.
- A. E. Goetz, J. Arcari, S. W. Bagley, R. Hicklin, L. Jin, Z. Jin, X. Li, Z. Liu, J. C. McWilliams, M. D. Parikh, T. Potter, W. J. Smith, L. Sun, J. Trujillo, P. Wang and X. Zhao, Org. Process Res. Dev., 2024, 28, 2168–2176 CrossRef CAS.
- M. C. Ince, B. Benyahia and G. Vilé, ACS Sustainable Chem. Eng., 2025, 13, 2864–2874 CrossRef CAS PubMed.
- M. J. Ford and G. Karig, US9440917B2, 2016.
- V. Farina, Org. Process Res. Dev., 2025, 29, 979–985 CrossRef CAS.
- C. R. McElroy, A. Constantinou, L. C. Jones, L. Summerton and J. H. Clark, Green Chem., 2015, 17, 3111–3121 RSC.
- Stockholm Declaration on Chemistry for the Future, https://www.stockholm-declaration.org/ (accessed 15 July 2025).
- C. Sansom, Solvents and sustainability, https://www.chemistryworld.com/features/solvents-and-sustainability/3008751.article, (accessed 24 August 2025).
| This journal is © The Royal Society of Chemistry 2026 |