
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe importance of nano–bio interfacial design in the sensing performance of nanoparticle-based affinity biosensors
Xueqian Chen
a,
Xuejie Li
a,
Richard D. Tilley
 bc and
J. Justin Gooding
*b
bc and
J. Justin Gooding
*b
aPingyuan Laboratory, College of Chemistry, Zhengzhou University, Zhengzhou 450000, China. E-mail: chenxq@zzu.edu.cn; lixuejie@gs.zzu.edu.cn
bSchool of Chemistry, Australian Centre for NanoMedicine, The University of New South Wales, Sydney, New South Wales 2052, Australia. E-mail: R.tilley@unsw.edu.au; Justin.gooding@unsw.edu.au
cMark Wainwright Analytical Centre, The University of New South Wales, Sydney, New South Wales 2052, Australia
First published on 29th April 2026
Abstract
Biosensors with good sensing performances with regards to high sensitivity, specificity, shorter response time, the ability to be multiplexed, excellent stability and reproducibility, are always in high demand. As modern biosensors are often fabricated using bioreceptors immobilized on nanoparticles to achieve efficient signal transduction or easier handling, the nanoparticle–bioreceptor (nano–bio) interface has a significant impact on the final sensing metrics. However, the role of nano–bio interfaces in sensing performance could be better understood, to facilitate the rational design of high performing nano–bio based devices. Herein, we aim to provide some basic rules and considerations to optimize nano–bio interfaces to achieve better detection performance when fabricating biosensors. The impact of the nano–bio interfaces on sensing characteristics is discussed from the perspective of bioreceptor–analyte interaction. Four interfacial parameters are included in this review: (1) the conformation of bioreceptors, (2) the coverage of the bioreceptors, (3) composition of mixed ligands, such as bioreceptors and other functional molecules and (4) spatial distribution of bioreceptors on nanoparticle surfaces. Methods to tailor these four interfacial factors are systematically investigated. In parallel, how these tailored nano–bio factors improve the sensing performances is emphasized with corresponding biosensor examples. The analytical methods for characterization of nano–bio interfaces are summarized, particularly at the single particle level. Additionally, the integration of artificial intelligence (AI) with nano–bio interfaces is discussed, highlighting how AI can improve nano–bio interfacial design. Finally, future perspectives on the role of nano–bio interfacial design in enhancing sensing capabilities are presented. This review aims to elucidate the relationship between nano–bio interfacial factors and sensing performances, as well as strategies for achieving precisely controlled nano–bio interfaces, which facilitates the rational design of high-performance biosensors.
 Xueqian Chen | Xueqian Chen received her PhD degree from the School of Chemistry, University of New South Wales. Currently, she is a lecturer at the Pingyuan Laboratory, College of Chemistry, Zhengzhou University. Her research interests include the synthesis of functional nanomaterials, investigation of their surface and interfacial properties, and the development of nanomaterial-based optical probes for biosensing and bioimaging. |
 Xuejie Li | Xuejie Li received her bachelor's degree from the College of Chemical and Materials Engineering, Fuyang Normal University. She is currently pursuing her master's degree at the College of Chemistry, Zhengzhou University. Her research focuses on the synthesis of nanomaterials and their applications in the degradation of microplastics. |
 Richard D. Tilley | Professor Richard Tilley is the Director of the Electron Microscope Unit at UNSW. His research is focused on the solution synthesis of nanoparticles and quantum dots for applications ranging from catalysis to biomedical imaging. He did his PhD in the Department of Chemistry, University of Cambridge, UK, after which he was a Postdoctoral Fellow for two years at the Toshiba basic R&D Center, Japan. A native of the UK, he graduated with a Master of Chemistry from Oxford University, UK. |
 J. Justin Gooding | J. Justin Gooding graduated with a B.Sc. (Hons) degree from Melbourne University and completed a PhD degree at the University of Oxford and postdoctoral training at the Institute of Biotechnology in Cambridge University. After starting as a Research Fellow at the University of New South Wales, he gained Professorship in 2006. He is an Arc Industry Laureate Fellow leading a research team on surface modification and nanotechnology for biosensors, biomaterials and drug delivery. |
1. Introduction
Nanoparticles have become ubiquitous in modern biosensors as nanoparticles can act as both a carrier for bioreporters and as a transducer to produce and enhance signals over the background and improve sensitivity of detection.1,2 In addition, each nanoparticle can be thought of as an independent sensing unit, which contributes to the measurement of many events in parallel. Recording the information from many individual nanoparticles in parallel creates the potential for both multiplex and high-throughput detection.3,4 Furthermore, magnetic nanoparticles (MNPs) can reduce response times due to magnetic forces facilitating mass transport.5,6 In most nanoparticle-based biosensors, bioreceptors are immobilized on the surface of nanoparticles.7 Consequently, a number of surface-related factors are involved during the process of fabricating biosensors, including the surface chemistry of nanoparticles, the self-assembled monolayer that the nanoparticle is modified with, the biofunctionalization of nanoparticles with bioreceptor molecules, and the display of the bioreceptors on nanoparticles.8–11 These surface-related factors can significantly impact the bioreceptor–analyte interaction and thereby the sensing performances.The nano–bio interface is the central component of a biosensor as it is directly involved in bioreceptor–analyte interaction, bridging the process of molecular biorecognition and signalization during biosensing (Fig. 1). The nano–bio interface determines the binding properties of nano–bio conjugates towards analytes and other biomolecules in the sample matrix. There are already some fabulous reviews about self-assembled monolayers of organic ligands on nanoparticles and biofunctionalization of nanoparticles.12–15 However, despite the important role of nano–bio interfaces in bioreceptor–analyte interaction and downstream analytical performance, there are limited reviews related to how the nano–bio interfaces affect the sensing performance of biosensors. If the nano–bio interface can be tailored and controlled precisely, we can understand the effect of nano–bio interfaces on bioreceptor–analyte interaction and how the bioreceptor–analyte interaction correlates with detection performance. This knowledge is of significance for the rational design of biosensors. Therefore, in this review, we first emphasize the important role of nano–bio interfaces in nanoparticle-based biosensors. Then, we discuss how the nanoparticle surface influences the bioreceptor–analyte interaction with particular emphasis on surface-based DNA biosensors as a case study. More deeply, the mechanisms behind the effect of nano–bio interfaces on interaction between bioreceptors and analytes are explored. Four interfacial parameters, namely conformation, coverage, spatial distribution of bioreceptors, and compositions of mixed ligands on nanoparticles, are introduced separately. Approaches to tailor these interfacial parameters and how these interfacial parameters improve the sensing capabilities of biosensors are demonstrated with corresponding examples. The sensing performances encompass sensitivity, limit of detection (LOD), specificity, selectivity, response time, multiplex detection, stability, and reproducibility. Following this, analytical approaches for characterizing nano–bio interfaces are outlined, with an emphasis on single-particle measurement. Considering the rapid progress of artificial intelligence (AI)-assisted biosensors, the integration of AI with nano–bio interfaces is explored. Finally, we conclude with perspectives on remaining challenges and future trends in this field.
 | ||
| Fig. 1 Scheme of the nano–bio interface, the interfacial factors, and related sensing performances. | ||
2. Importance of the nano–bio interfaces
2.1. Effect of nanoparticle surfaces on bioreceptor–analyte interaction
Bioreceptors, generally including enzymes, antibodies, DNA, aptamers, peptides and lectin, are normally immobilized on nanoparticles to give nanoparticles specificity for target analytes (Fig. 1).16–18 The sizes of nanoparticles adopted are usually smaller than 100 nm, comparable with the size of bioreceptors (2–15 nm). Consequently, when interactions between bioreceptors and analytes arise at the nanoscale surface with high curvature, interactions can be significantly different from that in solution or on a planar surface. Surface curvature, the accessibility of bioreceptor and heterogeneity of nanoparticle surfaces will all influence the bioreceptor–analyte interaction as well as the sensing performance.19,20DNA-based biosensors are used as examples to clarify how nanoparticle surfaces affect the interaction between probe DNA and target DNA. DNA probes with known sequences are immobilized on nanoparticles to capture target DNA or target RNA through DNA–DNA or DNA–RNA hybridization. The binding efficiency of probe DNA can reach 100% if equal or excess amounts of target DNA are added in the solution-phase. When probe DNA is immobilized onto a planar surface, hybridization efficiency can be nearly 100% under an intermediate surface density of 1–2 × 1012 probes per cm2. At higher or lower coverage, the efficiency of DNA hybridization can drop largely to only 20%.21 In the case of nanoparticle surfaces with higher curvature, such as 5 nm gold nanoparticles (AuNPs) with a higher DNA density of 3.43 × 1012 probes per cm2, the AuNPs–DNA conjugate diluted with 3-mercapto-1-propanol can also achieve 100% hybridization efficiency, whereas hybridization efficiency of nanoparticles without blocking is only 30–50%.22 Therefore, whether the surface is planar or spherical, the efficiency of surface DNA hybridization is comparable to that in solution, provided that neither crowding among neighbouring strands nor undesired DNA–surface interactions occur.
The Mirkin group studied the thermodynamics of hybridization between AuNPs–probe DNA and fluorophore labelled target DNA.23 Melting data of AuNPs–probe DNA (total 25 bases, including adenine (A)10 and a 15-base recognition sequence) revealed that the binding constant of the nanoparticle-based probe was 2 orders of magnitude higher than that of the 15-base molecular quencher/fluorophore DNA pair in solution under identical conditions. This increasing binding strength was due to the high surface density of DNA on AuNPs rather than the absolute amount of DNA on the AuNPs. A further study conducted by the same group confirmed that the enhancement of DNA hybridization on the AuNPs compared with DNA hybridization in free solution was enthalpically controlled rather than entropically.24 The introduction of nanoparticle surfaces mitigates against DNA from adopting enthalpically unfavorable conformations like those observed in solution.
Regarding the kinetics of DNA hybridization, the Levicky group found that hybridization rates on flat surfaces decreased by one to two orders of magnitude relative to those in solution, while dehybridization rates on flat surfaces exceeded those in solution by many orders of magnitude.25 Similar trends have been observed on the surface of nanoparticles.22 On 5 nm AuNPs, hybridization rates were slower than that in solution and dehybridization rates were accelerated on the AuNPs–target DNA conjugate, compared with target DNA in solution.26 The hybridization rate for 20-base DNA can decrease from 4.2 × 104 to 2.0 × 104 M−1 s−1, while the dehybridization rate can increase from 1.2 × 10−4 to 3.2 × 10−4 s−1.
The hybridization of surface immobilized DNA was investigated at the single molecule level with the assistance of single molecule imaging technologies. Tao's group found that the hybridization rates for individual probe strands were dominated by the local spatial arrangement of the probe DNA layer, including probe density and the degree of clustering.27,28 Recently, the Zijlstra group quantified the hybridization kinetics of AuNPs–DNA conjugates at the single particle level and investigated their dependence on DNA density. They discovered that lower DNA density could enhance hybridization kinetics by up to 15-fold, while dehybridization kinetics were almost unaffected by DNA density.29 Overall, the surface of nanoparticles can affect the thermodynamics and kinetics of probe–target DNA interaction, either promoting or inhibiting, depending on specific surface conditions. Accordingly, the next section examines the detailed mechanisms behind the impact of nanoparticle surfaces on bioreceptor–analyte interaction.
2.2. Elucidating the mechanistic basis of nanoparticle surface-mediated interactions
The surface of nanoparticles affects the bioreceptor–analyte interaction and sensing performance in several ways.Firstly, the existence of a surface brings additional interactions beyond the bioreceptor–analyte binding. These additional interactions are presented in Fig. 2a: (1) bioreceptors are likely to adsorb onto the nanoparticle surface. Such interactions may occur at ill-defined places on nanoparticles, potentially altering the conformation of bioreceptors or even causing denaturation.30,31 (2) Bioreceptors may interact with neighboring bioreceptors due to high packing density of bioreceptors on the nanoparticle. Compared with flat surfaces however, high surface curvature allows more bioreceptors to be packed onto the surface of a particle before significant neighboring interactions arise.32–34 Overly dense bioreceptor coverage causes steric hindrance for analyte binding, which reduces both efficiency and kinetics of bioreceptor–analyte interaction.21 (3) Nonspecific adsorption of interfering proteins or molecules in biofluids can occur onto the nanoparticle surface, hindering the recognition between bioreceptors and analytes. These unspecific adsorptions can not only block specific signals but also exacerbate the selectivity of biosensors.35,36
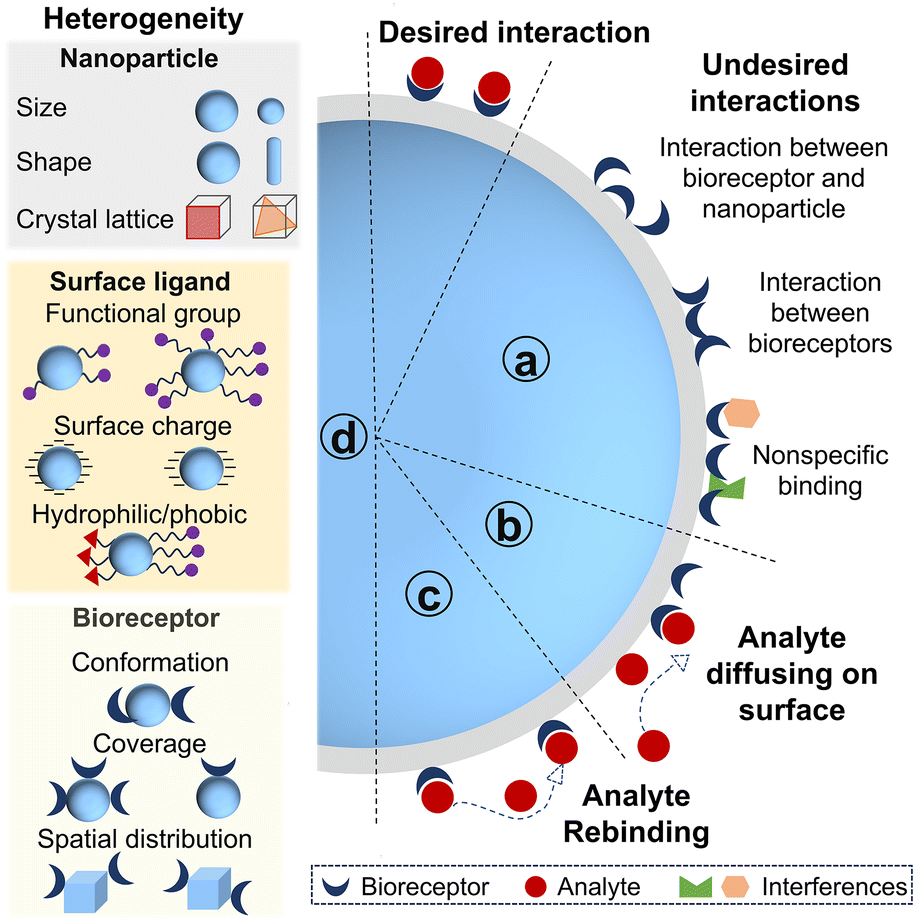 | ||
| Fig. 2 Effects of nano–bio interfaces on bioreceptor–analyte interaction. Besides the desired bioreceptor–analyte interaction, the other interactions include undefined interaction between bioreceptors and nanoparticles, intermolecular interaction between bioreceptors and nonspecific binding of interferences (a); the adsorbed analyte can diffuse on the nanoparticle to bind the nearby bioreceptors (b); the analyte disassociating from the bound bioreceptor can be recaptured by the neighboring bioreceptor before diffusing away (c); the origins of heterogeneity of nano–bio interfaces (d). | ||
Secondly, nonspecific but reversible adsorption of analytes onto nanoparticles can facilitate bioreceptor–analyte interaction. This adsorption proceeds via either three-dimensional (3D) diffusion of analytes in solution to bioreceptors on nanoparticle or two-dimensional diffusion of analytes along the particle surface after transient adsorption to regions lacking bioreceptors (Fig. 2b). The Schwartz group studied the hybridization dynamics of target DNA with immobilized probe DNA at a solution–solid interface at the single molecule level using total internal reflection fluorescence microscopy.37 They found that 31% of target DNA bound to probe DNA on the fused silica and hybridized immediately, whereas 69% of target DNA adsorbed nonspecifically on the surface. Only 7% of this adsorbed population located the probe DNA via a two-dimensional surface search and successfully hybridized. The remainder desorbed and returned to solution, thereby repeating adsorption-search cycles. Meanwhile, the surface-bound DNA duplexes were more likely to melt into solution (77%) than to melt on the surface (23%). The DNA strands that melted onto the surface could either rehybridize after a brief lateral search or desorb back into solution.
Thirdly, analytes are more likely to rebind with the neighboring bioreceptors before diffusing away as the local concentration of bioreceptors confined on nanoparticles can be 105 fold higher than that in solution (Fig. 2c).38 This diffusion-limited nanointerface can therefore result in a lower apparent dissociation rate constant for nano–bio conjugate/analyte binding.38,39 The influence of bioreceptor density on rebinding at a planar surface had already been analyzed theoretically by Thompson and co-workers.40 Under analyte-limited conditions, the likelihood of prompt rebinding increases with the higher association rates and receptor density, but decreases with increasing dissociation rates and the diffusion coefficients, following an approximate square root dependence. Rebinding tends to happen when the distance a molecule can diffuse in solution during its average surface bound time is less than a length scale set by the receptor surface density relative to analyte concentration. More recently, a single molecule study investigated the impact of DNA grafting density on surface-mediated DNA transport and hybridization.41 The threshold value separating low and high grafting density regimes was ∼1012 probe per cm2. Only 1–3% of target DNA was observed to associate with probes at dilute grafting density (<1012 probe per cm2). Adsorbed target DNA frequently performed unproductive searches and flew to other distant locations via desorption-mediated diffusion, rendering rehybridization events rare. In contrast, approximately 20% of target DNA hybridized to the immobilized DNA with high density, typically in the vicinity of initial adsorption sites. Target DNA rehybridized following a dehybridization event at high density of probe DNA. As target-induced nanoparticle assembly is widely exploited in biosensing, dense bioreceptors also induce a cooperative effect to nanoparticle assembly.42 Such multivalent cooperativity between nanoparticles enhances the equilibrium binding constant and accelerates agglomerate formation kinetics.43,44 For instance, the Mirkin group found that the equilibrium binding constant for a 15 nm AuNPs–DNA conjugate (24 base strands with a 3 base pair recognition domain) is ∼3 orders of magnitude higher than that of the corresponding system in the absence of nanoparticles.45
Lastly, another important aspect is the heterogeneity introduced by nano–bio interfaces (Fig. 2d). Heterogeneity originates from multiple sources. Primarily, nanoparticles themselves are heterogeneous in size, shape, charge, roughness, surface defects and crystallographic facets.46–49 Furthermore, the immobilization of bioreceptors onto these surfaces generates additional heterogeneities.50,51 The presence of multiple attachment sites on both nanoparticles and bioreceptors makes precise control over the conformation, orientation, and density of bioreceptors on individual particle exceptionally challenging. Consequently, the surface chemistry of the nanoparticles, the immobilization strategy and the structure of bioreceptors collectively contribute to the heterogeneities of nano–bio interfaces. Additionally, bioreceptor–analyte interactions occurring at a nanoscale surface are inherently heterogeneous processes. This heterogeneity at nano–bio interfaces has attracted significant attention as advancements in high-resolution single-molecule techniques now can unravel properties hidden behind ensemble measurement.52,53 In the context of single molecule biosensing, where the detection operates at the level of individual entities, this heterogeneity becomes particularly pronounced. Any variability in the nano–bio interface can translate directly into signal fluctuation, and substantial variation will further complicate data interpretation and compromise measurement precision.54 The Prins group comprehensively studied how reactivity variability of biofunctionalized nanoparticles was determined by various independent heterogeneities.55 They classified the contributing factors into two categories: stochastic heterogeneity and non-stochastic heterogeneity. Stochastic heterogeneity arises from the discrete nature of bioreceptor immobilization, leading to a particle-to-particle variation in bioreceptor number following Poisson statistics. Non-stochastic heterogeneity refers to physical and chemical differences among nano–bio conjugates, such as the nanoparticle size, surface roughness, and the local chemical microenvironment. Their work demonstrated that reactivity variability could be stabilized by employing a large number of bioreceptors, which indicated the use of large amounts of nanoparticles, utilization of the available interaction area, suitable particle size and bioreceptor density. A key implication is that a large population of biofunctionalized nanoparticles needs to be measured to minimize variability in nanoparticle-based single molecule biosensors.
3. Interfacial parameters affecting sensing performances
There is no one-size-fits-all solution for the optimal interfacial design to enhance sensing performance, as specific applications, bioreceptors, transductions and materials employed all affect the interfacial design. Here, we focus on four critical interfacial parameters, including orientation of bioreceptors, coverage of bioreceptors, compositions of mixed ligands and spatial distribution of bioreceptors on nanoparticles. How each interfacial factor boosts the sensing performance is demonstrated with corresponding examples. Meanwhile, strategies to tailor and characterize these nano–bio interfacial parameters are summarized. To standardize the types of nanoparticles and bioreceptors across various biosensors, if applicable, AuNPs are preferentially selected as model nanoparticles and DNA serves as a model bioreceptor with priority. In many cases the same principles apply to other bioreceptors such as antibodies and peptides. Where significant differences or considerations from DNA based bioreceptors are required, these will be highlighted with an emphasis on antibodies as the representative example instead of DNA, particularly in the section on bioreceptor orientation.3.1. Orientation of bioreceptors
 | ||
| Fig. 3 (a) Schematic depiction of IgG. (b) 3D structure of IgG, consisting of the Fc region and the Fab region. Reprinted with permission,58 Copyright 2018, Springer Nature. (c) Four typical orientations of an antibody on a nanoparticle. | ||
The approaches of immobilizing antibody onto nanoparticles include direct physical adsorption, covalent/non-covalent immobilization and antibody-directed synthesis of nanoparticles.61–63 Despite its ease of immobilization, direct physical adsorption based on electrostatic forces or hydrophilicity/hydrophobicity is relatively unstable and it is difficult to control the antibody orientation. The pH of the immobilization buffer has been leveraged to regulate the orientation of antibodies during adsorption as pH can influence both charge distribution and hydrophobicity of antibodies and surface charge of nanoparticles.64–66 Driskell's group has studied the impact of pH on the orientation of the anti-horseradish peroxidase (anti-HRP) antibody adsorbed on AuNPs.67 As the specific sequence and structure of the anti-HRP antibody are unavailable, IgG1 serves as a model protein due to its fully characterized structure in the Protein Data Bank (PDB). The calculated charge distributions of IgG1 at different pH values are presented in Fig. 4a. As the pH increases from 7.5 to 8.5, the degree of protonated amino acids decreases and the number of localized positive regions of IgG1 decreases. As the electrostatic interaction between the positively charged region of the antibody and negatively charged AuNPs is the main driving force for adsorption, the surface charge distribution of the antibody can significantly affect its orientation on nanoparticles. Theoretically, the thickness of the antibody layer on 60 nm AuNPs is minimum of 9 nm for flat-on orientation or maximum of 28 nm for head-on or tail-on orientation. Correspondingly, the footprint of an antibody in tail-on/head-on orientation is 38 nm2, while the footprint in flat-on orientation is 119 nm2. Experimentally, the hydrodynamic sizes of AuNPs–anti-HRP antibody conjugates were 73.2 nm, 69.8 nm and 67.4 nm at pH 7.5, 8.0, and 8.5, respectively. The measured antibody loading increased from 171 to 240 antibodies per AuNP with increasing pH from 7.5 to 8.5. Both the decreasing hydrodynamic size and antibody loading indicated a higher proportion of antibodies oriented in tail-on or head-on orientation with decreasing pH. To further confirm the orientation of the anti-HRP antibody, its binding capacity toward HRP was tested. The antigen-binding capacities of anti-HRP antibody–AuNP conjugates prepared at pH 7.5, 8.0, and 8.5 were 33%, 23%, and 18%, respectively (Fig. 4a). Since the binding constant of the anti-HRP antibody remained unchanged across this pH range (dissociation constant (Kd) ∼ 11 nM), the decreased binding capacity suggested that more anti-HRP antibodies were preferred in tail-on orientation at lower pH. The orientation of the anti-HRP antibody was further evaluated using aggregation-based assays with anti-IgG Fab-specific antibodies. Anti-HRP antibodies adsorbed onto AuNP at pH 7.5 exposed a larger number of accessible Fab sites, indicating their tail-on orientation. Similarly, Tonigold's work found the anti-CD63 (a cell surface antigen) antibody preferably adsorbed on polymeric nanoparticles through the Fc region at pH 6.1.58 The oriented anti-CD63 antibody exhibited better capture performance to target cells. As pH varies the immobilization microenvironment and largely affects the antibody orientation, it is reasonable to optimize and control the pH of immobilization buffer precisely when preparing antibody–nanoparticle conjugates.
 | ||
| Fig. 4 (a) Charge distribution calculated for the IgG1 antibody and corresponding hydrodynamic diameters of AuNPs–antibody conjugates and Fab accessibility at different pH values. Reprinted with permission,67 Copyright 2019, American Chemical Society. (b) Schematic illustration of MPBA-oriented antibody conjugation and sensing performance for MMP-9 detection. Reprinted with permission,75 Copyright 2025, American Chemical Society. (c) Oriented immobilization of antibody on nanoparticles based on affinity tags, including protein A/protein G, biotin–streptavidin, SpyTag-SpyCatcher and Fc-targeting monobody. Reprinted with permission,88 Copyright 2022, American Chemical Society. (d) Pictures of LFA strips and standard curves of protein A oriented and non-oriented probes. Reprinted with permission,78 Copyright 2023, Elsevier. (e) Oriented antibody conjugates adopting SpyTag-SpyCatcher and their application in cellphone-based portable detection. Reprinted with permission,87 Copyright 2022, Elsevier. | ||
Covalent immobilization based on the abundant functional moieties of antibodies, such as amino, carboxyl, and sulfhydryl groups, is relatively simple to achieve but typically results in random orientation. Detailed reviews on the covalent immobilization of antibody are covered elsewhere.15,68,69 To achieve orientation-controlled covalent immobilization of antibodies, functional groups located specifically outside the Fab region are typically used, such as polysaccharide chains in the Fc region and disulfide bonds between two heavy chains (Fig. 3a).70,71 For example, intact antibodies can be reduced into half antibodies to orientally immobilize on gold surfaces via their two native thiol groups, exhibiting 2-fold improved sensitivity without loss of selectivity.72 Wu et al. further introduced half antibody fragments as bioreceptors in a microcantilever-based immunosensor for ginsenoside Re detection.73,74 Compared with intact antibodies immobilized via thiolated secondary antibodies, immunosensors utilizing half antibodies as bioreceptors exhibited a 1500–4000-fold increase in sensitivity. Meanwhile, the LOD of immunosensors based on half-antibodies was 1 pg mL−1, whereas the LOD of the immunosensor adopting an intact antibody was approximately 0.5 ng mL−1. In addition to disulfide bonds, Sun and coworkers developed lateral flow immunoassay (LFIA) using plasmonic yolk–shell-satellite nanostructures (YSSNs) functionalized with 4-mercaptophenylboronic acid (MPBA).75 The boronic acid group of MPBA forms stable cyclic esters with cis-diol groups on the carbohydrate moieties in the Fc region of antibodies, orienting the Fab region outward from the nanoparticle surface, maximizing their accessibility of Fab regions for the target antigen (Fig. 4b). The YSSN–MPBA conjugate achieved an antibody conjugation efficiency of 72%, which was 1.79-fold and 1.06-fold higher than those of YSSNs (67%) and AuNPs (40%), respectively. Additionally, the antibody orientation efficiency of the YSSN-MPBA conjugate reached 77%, significantly exceeding 36% observed for randomly conjugated antibodies on YSSNs, directly resulting in a higher number of accessible Fab sites. Furthermore, the test results of YSSN-MPBA-LFIA and AuNP-LFIA were obtained using a handheld optical densitometer. Based on the optical density (OD) values of the test line, the LODs were 0.041 ng mL−1 for YSSN-MPBA-LFIA and 1.71 ng mL−1 for AuNP-LFIA, respectively. Owing to the strong Raman scattering signal generated by the MPBA–YSSN structure, this platform was also suited for SERS sensing. The SERS-based LFIA achieved a 305-fold improvement in LOD (0.0056 ng mL−1) compared to the conventional AuNP-LFIA (1.71 ng mL−1). Meanwhile, this platform demonstrated robustness against the matrix effect under Raman detection mode, maintaining high sensitivity and accuracy in serum and tear, with excellent recovery rates ranging from 86.4 to 98.2%.
As for non-covalent immobilization, affinity tags are adopted to modulate the orientation of antibodies.76 Protein A and protein G are frequently employed to regulate antibody orientation through specifically binding the Fc region of IgG (Fig. 4c).77 Hu and coworkers conjugated the anti-enrofloxacin antibody onto carboxyl-functionalized microspheres in an oriented manner utilizing cysteine-tagged recombinant protein A (Fig. 4d).78 The carboxyl on microspheres was activated by EDC (1-ethyl-3-(3-dimethylaminopropyl) carbodiimide) and 2-chloroacetohydrazide. Next, the recombinant protein A was conjugated onto the activated microspheres by a substitution reaction between chloroacetamide and sulfhydryl. The anti-enrofloxacin antibody was then bound to recombinant protein A on microspheres, resulting in the formation of oriented antibodies. The proportion of tail-on orientation was significantly higher for the oriented antibody-microspheres (89.1%) than that for the non-oriented counterparts (62.1%), indicating more accessible antigen-binding sites. Consequently, the LODs for enrofloxacin were 0.035 ng mL−1 for the oriented probe and 0.079 ng mL−1 for the non-oriented probe. Moreover, the linear range for the oriented probe was 0.25–10 ng mL−1, which was wider than that of the non-oriented probe (0.1–2.5 ng mL−1). However, protein A and protein G themselves are typically immobilized on nanoparticles via EDC-NHS (N-hydroxysuccinimide) crosslinking in a random orientation, which inevitably leads to partially random orientation of the capture antibodies. Biotin–streptavidin is another affinity tag to orientally immobilize antibodies, requiring site-specific biotin modification of antibodies (Fig. 4c). Sulfhydryl groups located at the hinge region of antibodies are commonly harnessed for site-selective biotinylation.79 A site-selectively biotinylated Fab fragment of an antibody demonstrated 20-fold higher recognition capacity than random biotinylation.80
Except for natural antibodies, engineered antibodies have been utilized to achieve oriented immobilization. Specific amino acids can be recombinantly introduced at particular positions within an antibody, thereby offering distinct anchoring sites for tethering antibodies onto nanoparticles.81 In parallel, genetic fusion of proteins with binding modules can also provide active-site accessibility for immobilization.82,83 SpyTag and SpyCatcher are a pair of peptide partners that can form irreversible isopeptides within minutes.84 A nanobody-SpyTag fusion protein can selectively couple to SpyCatcher functionalized surfaces with controlled orientation.85 Compared with a random-oriented nanobody dimer, the signal/background ratio of detecting Dengue virus nonstructural protein 1 (DENV NS1) increased by at least a factor of 5 using the oriented nanobody dimer built on SpyTag-SpyCatcher.86 In a recent study, Jongnam Park orientally conjugated an antibody onto polystyrene-co-poly(acrylic acid)-coated quantum beads utilizing SpyCatcher and a SpyTag-fused Fc-specific nanobody (Fig. 4e).87 This oriented conjugate resulted in an ∼91% accessibility of the Fab region, a significant increase compared to randomly conjugated antibodies using EDC-NHS chemistry (∼23.4% accessibility). The number of accessible antigen-binding sites per bead increased dramatically from 389 to 1978. The LFA utilizing oriented conjugates demonstrated an LOD of 5.1 pg mL−1 for human chorionic gonadotropin, an ∼6-fold improvement in sensitivity compared to that of the LFA using randomly oriented antibodies. While effective, antibody engineering is of high-cost and labor-intensive, requiring screening and engineering distinct antibodies according to specific antigens. To overcome the above limitations, a more adaptable approach was creatively developed.88 An engineered Fc-binding monobody, including a cysteine to enable thiol-based coupling onto poly(lactic acid)–poly(ethylene glycol) (PLA–PEG) nanoparticles, acts as an adapter between nanoparticles and antibodies (Fig. 4c). The conjugation efficiency of antibodies with a monobody adapter was substantially higher than that coupled by EDC-NHS (47 vs. 23 mg of Ab per mg of NPs). This novel immobilization strategy resulted in a significant improvement in binding efficiency (>1000-fold) relative to the standard EDC-NHS coupling. In addition to the above adapters, DNA, aptamers and peptides have also been harnessed as affinity tags to mediate the orientation of antibodies.89–92
Besides conjugating antibodies on the pre-synthesized nanoparticles, antibody-directed synthesis of nanoparticles has emerged as a novel approach to regulate antibody orientation. Metal–organic frameworks (MOFs) have emerged as a suitable scaffold for antibody and enzyme immobilization because of their well-defined pore structure, excellent chemico-thermal stability and high surface area. One-step synthesis of an oriented antibody-decorated MOF was presented by Falcaro's group.93 A Zn-based zeolitic imidazolate framework (ZIF), (Zn2(mIM)2(CO3)), produced from Zn2+ and 2-methylimidazole (mIM), was triggered by the Fc region of the antibody (Fig. 5a). As the negatively charged carboxyl and histidine groups were mostly located in the Fc region of the antibody, Zn2+ accumulated in the Fc region and triggered the formation of ZIFs. The Fc region of antibodies was partially inserted within ZIFs while the Fab region protruded from ZIFs, yielding oriented antibody-ZIF conjugates. Quantum dots (QDs) were then encapsulated in the antibody–ZIF conjugates to generate a fluorescence signal for diagnostic applications. This ordered configuration preserved target binding capacity compared to the antibodies randomly conjugated on QDs. Similarly, Zhang et al. coated various nanoparticles including gold nanorods (AuNRs), layered double hydroxide, superparamagnetic iron oxide nanoparticles, and ultrasmall superparamagnetic iron oxide using an orientated antibody-ZIF-8 composite via a generalizable film-coating approach (Fig. 5b).94 The antibodies selectively bound to ZIF-8 through histidine-rich Fc regions, thereby exposing the functional Fab regions to targets. The generated ZIF8-antibody@NPs exhibited high antibody loading and targeting efficiencies. Notably, the MOF-orientated antibodies in both studies exhibited the enhanced capture efficiency in targeting cancer cells. Recently Tian et al. applied the oriented antibody–MOF to wearable, label-free and persistent sweat cortisol detection (Fig. 5c).95 This full integrated system exhibited satisfactory on-body biosensing performance with a good linear detection range from 1 pg mL−1 to 1 µg mL−1 and a LOD of 0.26 pg mL−1. Meanwhile, this wearable sensor demonstrated good persistence in detecting cortisol, with only 4.1% decay after 9 days of storage.
 | ||
| Fig. 5 (a) The nucleation of ZIFs was triggered by the Fc region of the antibody. Reprinted with permission, Copyright 2022, Wiley-VCH.93 (b) Illustration of the site-specific antibody coating on nanoparticles based on the interaction between the histidine-rich Fc region of antibodies and ZIF-8. Reprinted with permission, Copyright 2023, Wiley-VCH.94 (c) Scheme of the working electrode for cortisol sensing. The ZIF-8@antibody was drop-coated after the electrodeposition of Prussian blue. PEDOT:PSS stands for poly(3,4-ethylenedioxythiophene):polystyrene sulfonate. Reprinted with permission, Copyright 2023, American Chemical Society.95 (d) Illustration of the dot-blot immunoassay using IgG-AuNC conjugates. Reprinted with permission, Copyright 2019, American Chemical Society.96 (e) ECL platform for detecting HER2 in breast cancer patients using herceptin-functionalized AuNCs. Reprinted with permission, Copyright 2025, American Chemical Society.98 | ||
In addition to MOFs, Chen's group prepared IgG-encapsulated gold nanoclusters (IgG-AuNCs) with high photoluminescence quantum yield and satisfactory bioactivity via a facile biomineralization process.96 By contrast, IgG/AuNC conjugates produced by coupling IgG to pre-synthesized AuNCs via EDC-NHS chemistry exhibited poor stability, leading to the pronounced nonspecific adsorption and elevated background signals. Consequently, under identical concentration of anti-human IgG, the signal change obtained in IgG/AuNC conjugate-based assay was markedly lower than that achieved with IgG-AuNCs in dot-blot immunoassay (Fig. 5d). Later, IgG-AuNCs were adopted as a “two in one” probe in electrochemiluminescence immunoassay, offering facile preparation, rapid detection, and good analytical performance in spike-and-recovery tests using 0.1% diluted serum.97 More recently, his group developed human epidermal growth factor receptor 2 (HER2)-specific monoclonal antibody (herceptin)-functionalized AuNCs for the quantitative detection of HER2 expression levels in breast cancer patients using electrochemiluminescence immunoassay (Fig. 5e). This assay showed high consistency with the standard and commercial ELISA kit with the Pearson correlation coefficient of 0.993. Serum HER2 extracellular domain levels were positively correlated with tissue HER2 expression determined by immunohistochemistry and fluorescence in situ hybridization, with particularly pronounced differences among the immunohistochemistry 3+, 2+, 1+, and 0 groups (p < 0.001).98
Strategies for achieving an upright and extended conformation of probe DNA on AuNPs include adding spacers, backfilling after bioconjugation, freezing-directed stretching, and utilizing DNA frameworks. Spacers, including alkyl, short DNA sequences or polyethylene glycol (PEG) units, are inserted in the end of probe DNA to avoid undesired interaction between the recognition region of DNA and nanoparticle surface (Fig. 6a).105 As the recognition region of the probe DNA is spatially isolated from the AuNPs, the probe DNA maintains a higher binding capacity for the target. The affinity of nucleobases toward gold ranks as: thymine (T) < cytosine (C) < guanine (G) < adenine (A).106 The Fan group creatively adopted a polyadenine (polyA) tail as an effective anchoring block and systematically investigated its effect on DNA hybridization (Fig. 6b). The hybridization efficiency of AuNPs–thiolated DNA conjugates without polyA as a spacer was only 5–10% at a loading density of 50 strands per AuNP.107 In contrast, once introducing polyA as an anchoring block instead of thiol, the hybridization efficiency increased with the length of polyA, from 42% for polyA5 to 90% for polyA30, even though the number of DNA per AuNP decreased from 70 to 10. Furthermore, how the polyA feature affected the hybridization kinetics and response time was investigated using a plasmonic sensor. The working principle is that target DNA would bring two AuNPs–probe DNA conjugates together, resulting in red shift in the plasmonic resonance peak. This plasmonic sensor exhibited a visualizable color change within 10 min for AuNPs–DNA conjugated with polyA. However, it took more than 12 h to display a color change for AuNPs–DNA conjugates without polyA. Quantitative analysis of kinetics was performed by fitting response curves using the Avrami law (inset of Fig. 6b). In the equation, t0 is the reaction onset time, τ is the characteristic time that depends on the reaction rate and aggregation geometry, and n is the Avrami exponent related to the physical mechanism of aggregate growth. The τ and n for AuNPs–DNA conjugates with polyA were 112 min and 0.77, while τ and n for AuNPs–DNA conjugates without polyA were 595 min and 0.99, respectively. Later on, the Fan group further quantitatively studied the hybridization thermodynamics and kinetics of AuNPs–DNA conjugates with polyA.108 Regarding the thermodynamics of DNA hybridization, the free energy of the DNA duplex in solution (−31.96 kcal mol−1) is higher than that of the DNA duplex on AuNPs immobilized via the Au–S bond (−22.09 kcal mol−1). The free energy cost between the DNA duplex in solution and the DNA duplex on AuNPs is 9.87 kcal mol−1, indicating that DNA duplexes are less stable on AuNPs. When the length of polyA increases from 5 to 40 nucleotides, the free energy cost decreases from 11.02 to 4.5 kcal mol−1, suggesting the improved DNA duplex stability on AuNPs. Consistent with this trend, the apparent binding constant enhances over 4 orders of magnitude with the increasing length of polyA, from 2.3 × 1015 to 1.4 × 1020 M−1 cm−1. Meanwhile, the rate constants of hybridization increase from 0.003 to 0.021 s−1 when the polyA tail is extended from 5 to 40 nucleotides.
 | ||
| Fig. 6 (a) Different spacers for probe DNA. (b) Detection principle of plasmonic biosensors and kinetic plots of AuNPs–DNA nanoconjugates. Reprinted with permission,107 Copyright 2013, American Chemical Society. (c) Regulate the conformation of DNA through backfilling. (d) Schematic illustration of nanoflare probes and their response curves. Reprinted with permission,114 Copyright 2018, American Chemical Society. (e) Freezing-directed conjugation of DNA onto AuNPs and other nanomaterials. Reprinted with permission,116,117 Copyright 2019, American Chemical Society and Copyright 2019, Wiley-VCH. (f) Schematic representation of the signal transduction mechanism of an ssDNA-tetrahedral DNA framework complex and its sensing performances. Reprinted with permission,121 Copyright 2020, American Chemical Society. (g) Sensing characterization of recognition interfaces assembled by a tetrahedral DNA framework. Reprinted with permission,122 Copyright 2025, American Chemical Society. | ||
Backfilling using alkanethiols and thiolated PEG also contributed to the upright orientation of DNA (Fig. 6c).109–111 The alkanethiol 6-mercaptohexanol (MCH) is commonly used to backfill the prepared AuNPs–DNA conjugates. The hybridization efficiency can reach 100% for a mixed layer of ssDNA and MCH on planar gold.112 Nevertheless, the suitable concentration range for MCH backfilling is narrow (around 100 mM). If the added MCH concentration is too high, AuNPs would undergo aggregation as less DNA strands to maintain stability of AuNPs.113 Bromide was reported as an alternative backfiller with higher tolerable concentration to 300 mM by the Liu group.114 The hybridization efficiencies of AuNPs–DNA conjugates without backfilling and backfilling with MCH or Br− were investigated. Fluorescein (FAM)-labeled complementary DNA was added to hybridize with probe DNA on AuNPs. Each AuNP hybridized with only ∼7 cDNA strands without backfilling. The number of hybridized complementary DNA per AuNP increased to 22 and 20 after backfilling with Br− (200 mM) and MCH (100 µM), respectively. A nanoflare probe was constructed based on AuNPs–DNA conjugates and FAM–complementary DNA for preliminary biosensing applications. Nanoflares are spherical nucleic acids (SNA) assembled on AuNPs that release a fluorescent reporter upon target binding.115 The fluorescence of the FAM fluorophore on complementary DNA was quenched by AuNPs in the absence of target DNA. Upon target DNA binding and displacing the FAM-complementary DNA from AuNPs, the fluorescence intensity significantly increased (Fig. 6d). The saturated signal of the nanoflare backfilled with Br− was 50% higher than that with MCH. Also, the sensitivity of nanoflares was 70% higher for Br− backfilling than that for MCH, indicating superior sensing performance of Br− in this DNA biosensor. Additionally, the Liu group also found that freezing can stretch and align the probe DNA based on lateral DNA–DNA interactions (Fig. 6e). DNA oligonucleotides can be readily adsorbed onto a range of nanomaterials, including AuNPs, graphene oxide (GO), iron oxide, and WS2 with a stretched conformation upon freezing.116,117 The freezing-directed assembly of DNA on gold electrodes was used to enable robust electrochemical sensing in whole blood.118
Similarly, DNA frameworks, regarded as organic nanoparticles, are also utilized to tailor the conformation of probe DNA on selective sites.119 The Song group employed a tetrahedral DNA framework to keep capture DNA extended from the surface of screen-printed carbon electrodes.120 The rough surface of screen-printed carbon electrodes results in the disorder of capture DNA and the nonspecific adsorption of signaling probes, which sterically hinders the target binding. The capture DNA hanging on tetrahedral DNA frameworks possesses enhanced capturing efficiency, leading to hundred times increase in both the signal-to-noise ratio and sensitivity compared with the surface directedly decorated with a capture probe. This sensing platform can be extended to a variety of target molecules as the recognition DNA sequences on tetrahedral DNA frameworks can be tailored to match different targets. Three representative targets, including miRNA-141, thrombin, and ATP, have been successfully detected. Beyond single tetrahedral DNA frameworks, Fan and his coworkers creatively utilized two tetrahedral DNA frameworks to extend probe DNA, shown in Fig. 6f.121 A tetrahedral DNA framework-based electrochemical biosensor was built to investigate the stretching effect on DNA hybridization. Probe DNA–tetrahedral DNA frameworks were immobilized on a gold electrode, one end of target DNA was hybridized to the tetrahedral DNA framework, and the other end of target DNA was hybridized with biotin labeled signaling DNA. Subsequently, avidin-HRP bound to a biotin labelled signaling strand and catalyzed the oxidization of tetramethylbenzidine, generating an electrochemical signal proportional to the concentration of target DNA. The LOD of this stretching DNA-enabled electrochemical biosensor can be as low as 1 fM. This response time was within 30 min, substantially faster than that of many previously reported biosensors. Both the hybridization kinetics and hybridization efficiency of the probe DNA were significantly improved by stretching DNA with two tetrahedral DNA frameworks. More recently, the same group further utilized tetrahedral DNA frameworks to engineer a biosensing interface with densely monodispersed DNA probes.122 Tetrahedral DNA frameworks serve as rigid and programmable frameworks to precisely control the conformation and spatial organization of pendant ssDNA probes on the electrode (Fig. 6g). By effectively suppressing the nonspecific adsorption and inter-probe entanglement, the tetrahedral DNA framework-programmed interface exhibited superior performance: the hybridization kinetics of a tetrahedral DNA framework immobilized DNA probe exhibited a 16-fold higher rate constant due to the improved probe accessibility (0.16 min−1 for tetrahedral DNA framework immobilized ssDNA vs. 0.01 min−1 for ssDNA). The Kd of tetrahedral DNA framework immobilized ssDNA was 18.12 nM, 3-fold lower compared with that of ssDNA (55.11 nM). A dramatically higher signal-to-noise ratio of 214 (vs. 17 for ssDNA) was achieved by maximizing the specific signal while minimizing nonspecific background noise, enabling reliable detection of target DNA down to 200 fM. Collectively, all these works highlight tetrahedral DNA frameworks as a powerful tool for interfacial engineering. Tetrahedral DNA frameworks overcome the fundamental limitation of conventional probe immobilization by programming DNA conformation at the nanoscale, providing a general and effective framework for constructing biosensors with exceptional speed, specificity, and sensitivity.
Collectively, the orientation of bioreceptors on nanoparticles is critical to accessibility of analytes to bioreceptors, which directly determines the sensitivity, LOD, signal-to-noise ratio and response time of biosensors. A range of approaches have been illustrated to regulate the orientation of bioreceptors on nanoparticles for superior sensing performance, listed in Table 1. The approaches to modulate the orientation of antibody are classified into pH regulation, affinity tags, genetically engineered antibody and antibody-directed synthesis of nanomaterials. pH regulation is straightforward to perform; however, it may not be applicable for all antibodies as it depends on the protonation/deprotonation of amino acid residues. Meanwhile, pH can affect both antibody activity and colloidal stability of nanoparticles.123 Genetic modification of antibodies based on protein engineering requires detailed knowledge of protein structure, high cost and long preparation time.124 The strategy of antibody-directed synthesis remains nascent with limited types of nanomaterials. Compared with antibodies, DNA is much easier to synthesize chemically with tailored functional groups. Strategies to orientate DNA in upright and extended conformation include incorporating spacers, backfilling, freezing or tetrahedral DNA framework-direct alignment. Except for approaches described above, applying electric field is also a promising approach to modulate the orientation of bioreceptors.125 Kim's group applied an electric field to align the antibody orientation on planar surfaces.126 The impedance change was maximized when antibodies were directionally aligned. The sensitivity enhancements from aligning the Fab and Fc regions were reported to be 1.31- and 1.60-fold in phosphate-buffered saline (PBS) and 1.41- and 2.18-fold in plasma, respectively. Similarly, Ye et al. found that the DNA conformation was sensitive to the applied electric field on planar gold. DNA tends to coil around the edge of large defects under positive potential, whereas it is lifted out of defects under negative potentials.127 However, there is no study of tailoring orientation of antibodies or DNA on nanoparticle surfaces via electric field yet. Given its cost-effectiveness, scalability, and ease of integration with chemical and biological modification strategies, substantial opportunities remain for exploiting electric fields to control the orientation of bioreceptors on nanoparticles.
| Surface-bioreceptor | Tailoring approach | Analyte | Signal strategy | Enhanced sensing performance | Ref. |
|---|---|---|---|---|---|
| Notes: FL represents the fluorescence signal. | |||||
| AuNPs–antibody | pH | HRP | Abs. | 2-fold increase in the percentage of active antibody | 67 |
| Gold–antibody | Thiol groups | GRe | Optical | 1500–4000-fold improved sensitivity; LOD: decrease from 0.5 ng mL−1 to 1 pg mL−1 | 71, 73 and 74 |
| YSSNs–antibody | Carbohydrate moiety | MMP-9 | OD | Linear range: 0.1–20 ng mL−1 (OD mode) and 0.01–1000 ng mL−1 (SERS mode) for YSSN-MPBA-LFIA; 5–200 ng mL−1 for AuNP-LFIA (OD mode) | 75 |
| SERS | LOD: 0.041 ng mL−1 (OD mode) and 0.012 ng mL−1 (SERS mode) for YSSN-MPBA-LFIA; 1.71 ng mL−1 for AuNP-LFIA (OD mode) | ||||
| Microsphere-antibody | Protein A | Enrofloxacin | FL | Linear range: 0.25–10 ng mL−1 for an oriented probe; 0.1–2.5 ng mL−1 for a non-oriented probe | 78 |
| LOD: 0.035 ng mL−1 for an oriented probe and 0.079 ng mL−1 for a non-oriented probe | |||||
| Polystyrene surface-antibody | Avidin–biotin | cTnI | Abs. | 20-fold higher detection capability; LOD: decrease from 2.65 ng mL−1 to 0.13 ng mL−1 | 80 |
| Polystyrene sphere-antibody | SpyTag-SpyCatcher | DENV NS1 | FL | Signal-to-noise ratio: 5-fold increase; LOD: decrease from 0.32 ng mL−1 to 0.064 ng mL−1 | 86 |
| Quantum bead-nanobody | SpyTag-SpyCatcher | Human chorionic gonadotropin | FL | LOD: 5.1 pg mL−1 | 87 |
| Sensitivity: 6-fold improvement compared to the randomly oriented antibodies | |||||
| PLA–PEG NPs–antibody | Monobody adopter | Vascular endothelial cells | FL | >1000-fold improvement in binding efficiency | 88 |
| MOF-antibody containing QDs | Ab-directed synthesis | SKOV-3 cells | FL | Fully preserved targeting efficiency | 93 |
| ZIF-8–antibody | Ab-directed synthesis | Cortisol | Current | Linear range:1 pg mL−1 to 1 µg mL−1 | 95 |
| LOD: 0.26 pg mL−1 and good storage stability | |||||
| AuNCs–antibody | Ab-directed synthesis | Goat anti-human IgG | FL | Excellent sensitivity | 96 |
| Good storage stability | |||||
| AuNPs–DNA | Spacer | DNA | Abs. | Hybridization efficiency increased from 5% to 90%; 5-fold faster in aggregate time | 107 |
| AuNPs–DNA | Backfilling | DNA | FL | Sensitivity: 2-fold improvement | 114 |
| DNA framework-DNA | DNA framework | miRNA-141, thrombin, ATP | Current | Hundred times higher sensitivity and signal-to-noise ratio | 120 |
| DNA framework-DNA | DNA framework | DNA | Current | Higher hybridization efficiency and LOD:1 fM; response time: 2 times faster than other sensors | 121 |
| DNA framework-DNA | DNA framework | DNA | Current | Rate constant: 10.16 min−1 for tetrahedral DNA framework-ssDNA vs. 0.01 min−1 for ssDNA | 122 |
| Kd: 18.12 nM for tetrahedral DNA framework-ssDNA vs. 55.11 nM for ssDNA | |||||
| Signal-to-noise ratio: 214 for tetrahedral DNA framework immobilized ssDNA vs. 17 for ssDNA | |||||
3.2. Coverage of bioreceptors
 | ||
| Fig. 7 (a) Detection principle of the optomagnetic immunoassay. (b) Response performances of MNPs with different antibody coverages. Reprinted with permission,130 Copyright 2014, American Chemical Society. (c) Schematic illustration of the strong and weakly ionized AuNPs for ultrasensitive LFIAs. (d) The snapshots of IgG on Cit-Au and AA-Au surfaces at different simulation times. (e) Testing strips of AA-AuNPs LFIAs responding to α-fetoprotein with different concentrations and the correlated linear fitting curve. Reprinted with permission,132 Copyright 2024, American Association for the Advancement of Science. | ||
Regarding the optimal coverage of bioreceptors, other groups investigated the effect of bioreceptor coverage on the enhanced sensing performance and obtained the opposite conclusion. The Fan group studied how DNA coverage on AuNPs affected the sensing performance of a stochastic DNA walker and found that loosely packed SNA gave the lowest LOD.133 In Fig. 8a, an exonuclease III (Exo III)-powered stochastic DNA walker autonomously move on a SNA-based 3D track. The driving force is derived from the unidirectional digestion of DNA tracks with Exo III. As DNA tracks on AuNPs were tagged with FAM, the digestion of DNA tracks led to FAM release and a corresponding increase in fluorescence intensity. The amount of DNA on AuNPs was regulated via ionic strengths of immobilization buffer.33,134 The low, medium, and high densities of DNA on 15 nm AuNPs were 25, 56, and 100 DNA per AuNP, respectively. LODs were 10 fM, 1 pM, and 200 pM for AuNPs–DNA conjugates with coverages of 25, 56, and 100 DNA per particle. Similarly, Liang et al. investigated the impact of DNA coverage on an enzyme-assisted target recycling assay to detect DNA via colorimetric assay.113 In the presence of target DNA, Exo III rapidly cleaved hybridized probes from AuNPs and induced the nanoparticle aggregation. The colour of AuNP solution changed from red into purple and even blue. AuNPs–DNA conjugates with low (30 strands per NP), medium (43 strands per NP), and high (54 strands per NP) coverage of DNA were prepared by adding different amounts of dithiothreitol. The LODs of low, medium, and high DNA covered AuNPs were 100 nM (red to purple), 250 nM (red to blue) and 50 nM (red to blue), respectively. Meanwhile, the decrease in DNA coverage also markedly shortened the response time. Specifically, AuNPs–DNA conjugates with 53 and 29 strands per NP changed into purple colour after 17 and 6 min, respectively. Overall, AuNPs with lower surface coverage imparted shorter response time and lower LOD in the above studies.
 | ||
| Fig. 8 (a) Schematics of Exo III-powered stochastic DNA walkers on SNA and response curves of AuNPs–DNA conjugates with different amounts of DNA. Reproduced with permission,133 Copyright 2017 Wiley-VCH. (b) Schematic illustration of AuNPs–DNA conjugate based colorimetric biosensors with controlled amounts of ssDNA. (c) Images (middle) and normalized redness values (right) of AuNP solutions with different densities of DNA. Reproduced with permission,135 Copyright 2023 The Royal Society of Chemistry. (d) Effect of probe DNA density on sensing performances for DNA with two different structures. (e) Representative images and redness values of AuNP solutions functionalized with different amounts of immobilized ssDNA, with linear DNA on top and stem-loop DNA on bottom. Reproduced with permission,137 Copyright 2025 Springer Nature. | ||
The Zako group performed a comprehensive investigation into the critical role of probe DNA density in the performance of AuNPs–DNA conjugate-based colorimetric biosensors. Consistent experimental condition was employed in their series of studies: 40 nm AuNPs–ssDNA conjugates were prepared via a freeze–thaw method, with various concentrations of ethylene glycol (EG) added to control the density of ssDNA. The colour of AuNP solution transited from red to blue/purple, quantified using redness value from digital image analysis and/or UV-vis spectroscopy (OD530/OD630). The effect of linear ssDNA probe density in salt-induced non-crosslinking aggregation was initially investigated (Fig. 8b).135 As AuNPs–ssDNA conjugates possessed superior dispersion stability against salts compared to AuNPs–dsDNA conjugates, increasing the amount of target ssDNA caused the colour of AuNP–DNA solution to change from red to blue/transparent. When the amount of ssDNA per AuNP increased from 654 to 1042, the detection sensitivity significantly decreased, confirmed by the normalized redness value of solutions (Fig. 8c). The LODs of AuNPs–ssDNA conjugates with 654 and 1042 ssDNA per AuNP were 17.5 nM and 10 nM, respectively. Subsequently, this sensing strategy was extended to the crosslinking aggregation, in which target DNA hybridized with two different AuNPs–ssDNA conjugates and induced the aggregation of AuNPs.136 Both the colour change and varying OD530/OD630 ratios indicated that sensitivity increased as the density of immobilized ssDNA decreased. This trend was ascribed to the alleviated electrostatic repulsion and steric hindrance at low probe density, leading to the improved hybridization efficiency. They further extended the work to explore the interplay between density and the secondary structure of probe DNA in non-crosslinking aggregation assay (Fig. 8d).137 Notably, the benefit of low probe density is structure-dependent: low probe density enhanced the sensitivity for linear probes, but decreased the sensitivity for stem-loop and G-quadruplex probes, as their stable secondary structures hindered the target hybridization (Fig. 8e). The above nanoparticle-based DNA biosensors highlight that both the sensing strategy and the configuration of probe DNA are key determinants when optimizing the coverage of DNA on nanoparticles.
 | ||
| Fig. 9 (a) DNA valency sorting by gel electrophoresis and affinity chromatography. Reproduced with permission,144,145 Copyright 2022 and 2001, American Chemical Society. (b) Structure of a plasmon ruler composed of two AuNPs linked by an MMP3 aptamer. The plasmonic resonance wavelength of an aptamer-Au plasmon ruler in the presence and absence of MMP3. Reproduced with permission,146 Copyright 2015, American Chemical Society. (c) One-pot synthesis of chimeric DNA-functionalized QDs. Chimeric DNA includes a ligand domain (ps, in orange) and a recognition domain (po, in green). Reproduced with permission,147 Copyright 2008, Springer Nature. (d) DNA scaffolds self-assembled from the different sets of chimeric DNAs for valence-engineered QDs. Reproduced with permission,150 Copyright 2017, Wiley-VCH. (e) Programmable atom-like nanoparticles prepared with ssDNA encoders containing polyA. Reproduced with permission,151 Copyright 2019, Springer Nature. | ||
Modified DNA with specific sequences can be utilized to obtain valence-controlled nanoparticles. For instance, a chimeric DNA containing two moieties, phosphorothioates (ps) and phosophodiester (po) backbone, has been utilized to prepare monovalent QDs (Fig. 9c).147 Since metal ions of QDs (e.g. Cd2+ and Zn2+) exhibit thousand-fold higher affinity for sulfur than that for oxygen, domains with the ps backbone direct the passivation of nanocrystals while domains with the po backbone mediate biorecognition.148 The number of ps oligomers can be regulated to obtain QDs with a range of valences. Compared with all po DNA, a significant increase in target binding efficiency was observed for the ps–po oligonucleotide. He and colleagues further assembled multi-color ps–po DNA-QDs to construct a QD-based computing system. Seven fundamental logic gates (AND, OR, NOR, NAND, INH, XNOR, and XOR) were achieved using binary and ternary QD complexes based on DNA strand displacement reactions.149 Beyond monovalent QDs prepared with ps–po DNA, multiple ps–po DNAs could be firstly assembled into programmable scaffolds and then employed to fabricate bivalent or multivalent QDs (Fig. 9d).150 More recently, the Fan group constructed AuNPs with a controlled valence bond using ssDNA containing polyA as an encoder (Fig. 9e).151,152 Atom-like nanoparticles with programmable n-valence were obtained by regulating the order, length and sequence of each encoder with alternating polyA/non-polyA domains. Subsequently, AuNPs with a precisely programmable bond length and bond energy were successfully fabricated using polyA-based encoders.153 In summary, nanoparticles with defined DNA valency can be assembled into complex nanostructures or used to implement Boolean logic operations, which have enormous potential in smart sensing applications.154
In summary, the coverage of bioreceptors on nanoparticles could influence sensitivity, LOD and response time of biosensors, with related examples in Table 2. The optimal coverage of bioreceptors depends largely on the configuration of bioreceptors and the adopted sensing strategy in the given biosensor. As for monovalent nanoparticles, delicate design of DNA sequences or modified nucleotide bases, such as DNA sequences containing ps–po oligonucleotides or polyA, enables precise control over DNA valency on QDs or AuNPs. The obtained programmable atom-like nanoparticles have shown great potential in nanoarchitecture assembly, distance-based energy transfer and intelligent biosensing.155 Compared with various preparing strategies of nanoparticles with well-defined DNA valency, controlling the valence of antibody still depends on stoichiometric conjugation followed by purification.142,156,157 Recently, Choi and colleagues developed a one-pot strategy to conjugate tandem repeat protein chains on AuNPs, enabling controlled valency without purification.158 The specific designed proteins contain multiple and regularly spaced hexa-histidine tags, which can bind multivalently onto nanoparticles. However, controlling the number of antibodies per nanoparticle directly without purification and protein engineering remains challenging.
| Surface-bioreceptor | Tailoring approach | Analyte | Signal strategy | Enhanced sensing performance | Ref. |
|---|---|---|---|---|---|
| MNPs–antibody | Ratio of antibody/NPs | cTnI | Scattering | Sensitivity: 5-fold increase for high antibody coverage | 130 |
| Abs. | Response time: 2-fold faster for high antibody coverage | ||||
| AuNPs–aptamer | Ratio of aptamer/diluent DNA | Interferon-gamma | FL | Sensitivity: 2 times increase for high aptamer coverage (256 vs. 9.6 aptamers/per AuNP) | 131 |
| Response time: 4 times decrease (36 min vs. 137 min) | |||||
| AuNPs–antibody | Ionic strength of the ligand | α-fetoprotein | OD | Lower LOD in the pg mL−1 range and good stability | 132 |
| 100-fold enhancement in sensitivity | |||||
| AuNPs–DNA | Ionic strength | DNA | FL | LOD: 200 pM, 1 pM, and 10 fM for AuNPs–DNA conjugates with 100, 56, and 25 strands per NP | 133 |
| AuNPs–DNA | Dithiothreitol amount | DNA | Abs. | LOD: 100 nM, 250 nM and 50 nM for AuNPs–DNA conjugates with 54, 43 and 30 strands per NP | 113 |
| Response time: 17 and 6 min for AuNPs–DNA conjugates with 53 and 29 strands per NP, respectively | |||||
| AuNPs–DNA | EG amount | DNA | Redness Value, OD | Increase in sensitivity for low DNA coverage | 135 |
| LOD: 17.5 nM and 10 nM for AuNPs–DNA conjugates with 654 and 1042 strands per NP, respectively | |||||
| AuNPs–aptamer | HPLC separation | MMP3 | Plasmonic λ | Detect single MMP3 based on a plasmonic ruler | 146 |
| QDs–DNA | po–ps DNA | DNA | FL | Intelligent molecular diagnostics | 149 |
| AuNPs–DNA | PolyA | DNA | FL | Intelligent sensing | 151 |
3.3. Composition of mixed ligands
 | ||
| Fig. 10 (a) Schematic diagram of a multifunctional isopeptide based electrochemical biosensor for annexin A1 detection. MISP represents the multifunctional isopeptide. Reproduced with permission,171 Copyright 2023, American Chemical Society. (b) Chemical structure of the designed multifunctional peptide for MDM2 detection. (c) Schematic representation of Pt–Se, Au–S, and Pt–S interfaces and their sensing performances. Reproduced with permission,172 Copyright 2024, American Chemical Society. (d) Utilization of mixed ligands for detecting multiple analytes based on nanoparticle aggregation. Reproduced with permission,159 Copyright 2020, American Chemical Society. (e) The principle of a SERS-based sandwich assay for one (i) and two (ii) target DNA detection. Reproduced with permission,173 Copyright 2011, The Royal Society of Chemistry. (f) SERS encoded Ag-pyramidal detection of multiple biomarkers. Reproduced with permission,174 Copyright 2015, Wiley-VCH. | ||
 | ||
| Fig. 11 (a) Schematic representation of antigen detection using the bio-barcode assay. Reproduced with permission,175 Copyright 2006, Springer Nature. (b) Preparation of dual aptamer-modified Fe3O4 MNPs and clinical applications. Reproduced with permission,178 Copyright 2022, American Chemical Society. (c) Schematic diagram of GOD/HRP-DNA@MMS. Reproduced with permission,180 Copyright 2023, Wiley-VCH. | ||
Nanoparticles can serve as versatile platforms for the co-immobilization or encapsulation of multiple enzymes, enabling their cooperative operation in cascade reactions. Yang's group developed polyphenol-encapsulated enzyme–DNA conjugates on SiO2@Fe3O4 magnetic microspheres (MMSs) with different enzymes confined within separate compartments.179 Two model enzymes, glucose oxidase (GOx) and HRP, were precisely immobilized on opposing hemispheres of MMS. This system exhibited a Michaelis constant (Km) of 1.19 mM, which was lower than those of randomly immobilized (1.47 mM) and free enzymes (3.45 mM), indicating the enhanced substrate affinity. Moreover, its catalytic efficiency (kcat/Km) was 5.32 × 10−8 s−1 mM−1, significantly higher than that of randomly immobilized enzymes (4.01 s−1 mM−1) and free enzymes (0.41 s−1 mM−1), highlighting the enhanced performance afforded by spatial organization. In a more refined design, GOx and HRP were co-immobilized on epoxy modified polyvinyl acetate (PVAC) MMSs utilizing Y-shaped DNA scaffolds assembled from two partially complementary ssDNA (Fig. 11c).180 The cascade activity was strongly dependent on the inter-enzyme distance, which could be precisely adjusted by varying the length of the DNA strand. Maximum activity (0.97 U µg−1) was achieved at an optimal spacing of 13.6 nm, 3.5-fold enhancement compared with that of free enzymes. The maximum reaction rate of GOD/HRP-DNA@MMS (0.97 U µg−1) exceeded that of free enzymes (0.28 U µg−1), while its Km (9.04 mg mL−1) was lower than that of the free system (14.71 mg mL−1), reflecting higher apparent cascade activity. The turnover number (kcat) of 385.6 min−1 further demonstrated the exceptional catalytic efficiency of this spatially organized enzyme system. Beyond post-synthetic immobilization, multi-enzyme systems have also been co-encapsulated within MOFs for intracellular lactate detection, achieving a 650-fold signal-to-noise ratio higher than that of conventional approaches.181 Moreover, multi-shelled MOFs provide a hierarchical scaffold for the spatial organization of enzymes at the nanoscale, leading to substantial enhancements in catalytic efficiency. For example, a GOx-HRP cascade encapsulated in multi-shelled ZIF-8 showed a 5.8- to 13.5-fold increase in catalytic efficiency compared to free enzymes in solution.182
Collectively, the composition of mixed ligands on nanoparticles affects the specificity, sensitivity, stability, and multiplexing capacity of biosensors (Table 3). The integration of antifouling agents into nano–bio interfaces can significantly enhance the specificity, sensitivity and operational stability of biosensors. More importantly, the antifouling molecules enable the nanoparticle-based biosensor to operate reliably in complex samples with matrix effects, such as human serum and whole blood, with satisfactory accuracy. Furthermore, nanoparticles co-functionalized with multiple bioreceptors and Raman reporters can serve as ideal platforms for multiplexed detection. By developing additional Raman reporters with distinct characteristic peaks, the number of detectable analytes can be expanded from a few to several tens. Lastly, mixed bioreceptor layers, such as combinations of antibodies and DNA and dual-aptamer or multi-enzyme systems, can act cooperatively to enhance the sensitivity and precision of detection.
| Surface-bioreceptor | Tailoring approach | Analyte | Signal strategy | Enhanced sensing performance | Ref |
|---|---|---|---|---|---|
| AuNPs–aptamer | Antifouling peptide | Cancer antigen125 | Current | Antifouling performance can last 1–2 days; applicable to clinical serum samples | 169 |
| AuNPs–peptide | Multifunctional peptide | Annexin A1 | Current | LOD: 0.43 pg mL−1 in 50% human blood; applicable to clinical blood samples | 171 |
| PtNPs–peptide | Multifunctional peptide | MDM2 | Current | Linear range and LOD unchanged from PBS to undiluted serum | 172 |
| Satisfactory specificity and reproducibility | |||||
| AgNPs–aptamer | Multiple aptamer | PSA, Mucin-1, Thrombin | SERS | Multiplex detection and rapid response | 174 |
| AuNPs–antibody | Antibody and DNA | Protein | Optical | Sensitivity comparable with PCR | 175 |
| Fe3O4 NPs–aptamer | Dual aptamer | CTCs | FL | Capture heterogeneous CTCs, including EpCAM+ and PTK7+ subtypes | 178 |
| Clinical validation with 72 patient samples | |||||
| MNPs–enzyme | GOx and HRP | Cascade activity | Abs. | 3.5-fold enhanced activity compared with those of free enzymes | 180 |
| Reaction rate: 0.97 U µg−1 for GOD/HRP-DNA@MMS and 0.28 U µg−1 for free enzymes | |||||
| Km value: 9.04 mg mL−1 for GOD/HRP-DNA@MMS and 14.71 mg mL−1 for free enzymes |
3.4. Spatial distribution of bioreceptors
 | ||
| Fig. 12 (a) Schematic illustration of fabricating an AuNR-antibody based SERS label and its performance in food matrices. Reproduced with permission,187 Copyright 2021, American Chemical Society. (b) Schematic representation of regioselective modification of AuNRs and three types of AuNP–AuNR assemblies. (c) Raman spectra of HeLa cells with three types of assemblies. Non-assemblies represent the mixture of AuNPs and AuNRs modified without complementary DNA. Reproduced with permission,184 Copyright 2012, American Chemical Society. | ||
The anisotropy of Janus NPs can also be utilized to modify two distinct bioreceptors to achieve multiplex detection. The Yuan group fabricated Janus SiO2 NPs for the simultaneous and sensitive fluorescence detection of dual miRNAs in cancer cells (Fig. 13a).190 One hemisphere of Janus NPs is functionalized with amine groups while the other hemisphere is decorated with AuNPs. Carboxyl-labeled H2 DNA strands are immobilized on the amine-functionalized hemisphere through amide bonds, whereas sulfhydryl-labeled H1 DNA strands selectively anchor onto the AuNP-functionalized hemisphere via the Au–S bond. Target miRNAs, miRNA-21 and miRNA-155, act as catalysts to initiate catalytic hairpin assembly reactions. Even trace amounts of target miRNA can result in numerous dsDNA walker complexes. These DNA walkers subsequently traverse along the 3D DNA tracks on their respective hemispheres of Janus NPs, driven by strand displacement amplification. During this process, the fluorophores (FAM and Cy5) are spatially separated from their quenchers (AuNPs or black hole quencher), leading to a significant fluorescence recovery. The spatial segregation of the two DNA probes on Janus SiO2 NPs effectively prevents single molecule Förster resonance energy transfer (FRET) and minimizes signal crosstalk between distinct fluorophores, a common issue in homogeneous nanoparticle-based biosensors. The integration of catalytic hairpin assembly reaction and the 3D DNA walker provides dual amplification, enabling ultra-sensitive detection with LODs as low as 0.35 pM for miRNA-21 and 0.48 pM for miRNA-155. Compared with homogeneous SiO2 NPs, Janus SiO2 NPs exhibited higher walking efficiency and accelerated reaction kinetics due to the reduced steric hindrance and ordered probe arrangement. Notably, the fluorescence signal of homogeneous SiO2 NPs failed to reach equilibrium even after 5000 s while the Janus SiO2 NPs attained a stable plateau within only 2500 s (Fig. 13b). Additionally, the Janus architecture allows for the precise visualization of both miRNAs in the same subcellular location within living cells, providing more accurate and reliable co-localization information than the conventional fluorescence in situ hybridization and single-probe approaches (Fig. 13c).
 | ||
| Fig. 13 (a) Schematic illustration of a Janus DNA nanomachine for simultaneous detection of miRNA-21 and miRNA-155. (b) Reaction rate of homogeneous and Janus DNA nanostructure in the presence of target miRNA. (c) 3D and individual enlarged 3D images of dual-color fluorescent Janus NPs. Reproduced with permission,190 Copyright 2020, The Royal Society of Chemistry. (d) Schematic representation of enterobacterial contamination detection using a self-propelled Janus microsensor. (e) Time-lapse fluorescence images and (f) Fluorescence decay curves of Janus micromotors. Reproduced with permission,192 Copyright 2018, American Chemical Society. (g) Scheme of catalytic and magnetic driven GO/PtNPs/Fe2O3 Janus micromotors modified with Nisin for S. aureus. (h) SEM images of modified moving catalytic micromotors in S. aureus. and E. coli cultures, static modified micromotors, unmodified moving micromotors, modified magnetic moving micromotors (from left to right) in S. aureus and E. coli cultures. (i) Selectivity and capture efficiency of Nisin modified micromotors in bubble and magnetic modes. Reproduced with permission,194 Copyright 2021, Wiley-VCH. | ||
The spatial distribution of bioreceptors can also accelerate response time and reduce sample volume in combination with self-propelled nanoparticles. Alberto Escarpa's group has engineered a series of self-propelled Janus nanoparticles as mobile sensors with satisfactory sensing metrics in terms of extremely low sample volumes and ultra-fast response time.191 The micromotors are partially functionalized with catalytic layers to produce bubbles and propulsion forces, while the remaining surface is functionalized with bioreceptors to enable selective detection. For instance, the group pioneered a polycaprolactone micromotor encapsulated with PtNPs for bubble propulsion and 3-aminophenylboronic acid functionalized QDs for selectively binding to the lipopolysaccharide core of Salmonella enterica (Fig. 13d).192 The interaction between the lipopolysaccharide core and QDs induced rapid fluorescence quenching. The LOD of this micromotor-based assay is as low as 0.07 ng mL−1, far below the level considered toxic to humans (275 µg mL−1). Additionally, this micromotor can successfully detect Salmonella toxin in food within 15 min, compared with approximately 24 h required by the existing gold-standard Limulus amoebocyte lysate test (Fig. 13e and f). Subsequently, the same group further developed a smartphone-based portable device in combination with Janus micromotors for glutathione detection.193 The detection can be completed in less than 30 s, requiring only dropping human serum or plasma, along with the micromotors and fuel solution, onto a glass slide. The average cost per analysis is estimated to be 0.05€, considering the cost of fuel, micromotors and surfactants. Furthermore, GO/PtNPs/Fe2O3 wrapped Janus micromotors of dual-mode propulsion, including catalytic self-propulsion and fuel-free magnetic actuation, have been constructed (Fig. 13g).194 The surface of GO was modified with lanbiotic Nisin for specific binding to Lipid II in the cell walls of Gram-positive bacteria and biofilms. This Janus micromotor exhibited ∼85% capture efficiency for Staphylococcus aureus (S. aureus), compared with only 20–28% capture efficiency for Escherichia coli (E. coli) (Fig. 13h and 13i). Over 90% inactivation of S. aureus and approximately 80% removal and inhibition of S. aureus biofilms were achieved under dual propulsion modes. Additionally, the GO/PtNPs/Fe2O3 Janus micromotors remained effective in complex media, such as juice, serum, tap water, and even whole blood.
 | ||
| Fig. 14 (a) Construction of DNA framework probes with an equal amount of H2 and tunable numbers of H1. (b) Time-dependent fluorescence intensity, (c) LOD, and (d) dynamic range of DNA framework probes with controlled valency. Reproduced with permission,201 Copyright 2019, American Chemical Society. (e) Scheme of DNA frameworks with precise recognition and high-affinity binding for target cells. (f) Determination of Kd of different DNA frameworks. Reproduced with permission,202 Copyright 2023, American Chemical Society. (g) Preparation of SF-AuNCs with the precisely controlled number and position of DNA and SF-AuNC-AuNP nanoassemblies with different gap morphologies. (h) Comparison of ten sets of Raman intensities taken from three types of SF-AuNC–AuNP nanoassemblies. Reproduced with permission,206 Copyright 2021 Wiley-VCH. | ||
Mao and colleagues have developed DNA origami with multivalent aptamers to boost the binding affinity for tumor cells (Fig. 14e).202 The effect of aptamer valency and interspacing of multiple aptamers on cellular binding affinity was investigated, including the EpCAM aptamer, epidermal growth factor receptor (EGFR) aptamer and HER2 aptamer. By precisely adjusting the valency and spatial arrangement of multiple aptamers, a specific topology to match spatial distribution of target membrane protein clusters on cancer cells was constructed, termed as O(I)3O(II)3O(III)3. The binding sites O(I), O(II), and O(III) on DNA origami refer to inner loop regions with radii of 19 nm, 35 nm, and 62 nm, respectively (Fig. 14f). Specifically, the DNA nanostructure O(I)3O(II)3O(III)3 was functionalized with three EpCAM aptamers at a 19 nm radius region, two EGFR aptamers and one HER2 aptamer at a 35 nm radius and three EpCAM aptamers on a 62 nm radius region. For comparison, O(III)3, O(I)3, and O(I)3O(III)3 were all modified by EpCAM aptamers. The Kd values of designed DNA nanostructures against the Michigan Cancer Foundation-7 cell were 263.9 pM for O(I)3O(II)3O(III)3, 613.7 pM for O(III)3, 2089.5 pM for O(I)3, and 4889 pM for O(I)3O(III)3, respectively. O(I)3O(II)3O(III)3 possessed 3000-fold higher binding affinity towards the Michigan Cancer Foundation-7 cell than that of the DNA nanostructures with a monovalent aptamer. Hu et al. designed a series of adjustable multivalent aptamer-based DNA frameworks with tube or patch-like structure to precisely regulate their interactions with the receptor of tumor cells. These frameworks enabled multi-heteroreceptor-mediated recognition through precise control of aptamer type, valency, and geometric arrangement. This strategy not only favored the specific binding of nanostructures to tumor cells but also promoted clathrin-dependent endocytosis and intracellular uptake. Notably, tube-like structures with different diheteroaptamer pairs exhibited more than 5-fold higher uptake across various tumor cell types, demonstrating their strong potential for targeted drug delivery. In contrast, patch-like structures with triheteroaptamers were able to selectively direct specific macrophage towards tumor cells, leading to effective immune clearance.203
Advances in DNA nanotechnology enable precisely patterning DNA onto nanoparticles regarding coverage, components and spatial position. For instance, Sleiman et al. transferred DNA strand motifs from a parent 3D DNA template onto AuNPs, which provided arbitrary control over sequence, DNA number and their relative placement and directionality.204 Instead placing AuNPs onto DNA templates, Xie and coworkers encapsulated AuNPs into a DNA icosahedron cage and then transferred the molecular recognition elements from DNA nanocages to the inner AuNP surface.205 Various combinations of overhangs extended from the DNA icosahedron cage can be retained onto AuNPs via Au–S bonds. The DNA nanocage allows DNA to be transferred into a larger area on AuNPs compared with placing AuNPs onto DNA templates. Recently, the asymmetric pattern on DNA origami templates has been accurately transferred onto the surface of thiolated DNA functionalized gold nanocubes (TF-AuNCs) (Fig. 14g).206 The prepared specific functionalized gold nanocubes (SF-AuNCs) were named V1 to F9, with 9 DNA strands at their vertices, edges and faces. When small AuNPs with densely packed complementary DNA were hybridized with the DNA strands on the vertices, edges and face of SF-AuNCs, a series of high-order and discrete nanostructures formed. Single molecule Raman spectra of three representative types of SF–AuNCs–AuNP nanoassemblies with different gap morphologies were studied. Raman signals of on-vertex and on-edge SF–AuNC–AuNP nanoassemblies were significantly stronger than that of on-face SF–AuNC–AuNP nanoassemblies, while the former two exhibited comparable intensities (Fig. 14h). Due to the high SERS enhancement factor, these plasmonic AuNC–AuNP nanoassemblies provide an ideal platform for ultra-sensitive sensing and single-molecule research.
In summary, the approaches of preparing site-specific biofunctionalized nanoparticles primarily rely on controllable surface accessibility or DNA frameworks listed. DNA strands can be site-selectively modified on the surface of anisotropic nanoparticles and further assembled into well-defined architectures, mainly improving the sensitivity of SERS-based biosensing. Janus NPs with two different types of bioreceptors on opposite hemispheres can achieve multiplexed detection, thereby facilitating precision medicine. Beyond multiple detection, self-propelled Janus NPs integrating both bioreceptor and catalytic regions can significantly accelerate response time and minimize sample volume. Additionally, DNA frameworks allow precise control over the number, the type and the spatial distribution of DNA strands, either within the framework or by transferring predefined DNA patterns onto nanoparticle surfaces. This powerful capability provides substantial opportunities for biosensors with tunable analytical performances. Although regioselective modification of bioreceptors on nanoparticles has opened new avenues for improving various sensing merits, current research in this field has mainly focused on developing stable and easily implementable synthetic strategies, while their applications in quantitative biosensors remain at an early stage. In contrast to DNA, precise control over antibody functionalization on nanoparticles is more challenging due to the larger molecular size, structural diversity and unpredictable physicochemical properties of antibodies.158 Moreover, the antibody bioactivity is more susceptible to perturbation induced by nanoparticle surfaces. Therefore, there is a longer journey to realizing precise and reliable control of antibodies on nanoparticles (Table 4).
| Surface-bioreceptor | Tailoring approach | Analyte | Signal strategy | Enhanced sensing performance | Ref. |
|---|---|---|---|---|---|
| AuNRs–antibody | Shape | Salmonella bacteria | SERS | Single molecule detection of Salmonella bacteria in complex food matrices | 187 |
| AuNRs–DNA | Shape | Lipid | SERS | Enhanced Raman signal and works as a Raman probe in live cells | 184 |
| SiO2 NPs–DNA | Janus NPs | miRNA-21, miRNA-155 | FL | Multiplex detection and fast response time | 190 |
| Precise visualization of two miRNAs and reliable co-localization | |||||
| QDs-saminophenylboronic acid | Janus NPs | Salmonella enterica | FL | Low LOD and fast response within 15 min | 192 |
| GO/PtNPs/Fe2O3-Nisin | Janus NPs | S. aureus | FL | Good specificity and effective in complex media | 194 |
| DNA framework-DNA | DNA framework | MicroRNA21 | FL | 8 times increase in sensitivity with increasing number of H1 | 201 |
| DNA framework-aptamer | DNA framework | Cancer cells | FL | 3000-fold higher binding affinity | 202 |
| SF-AuNCs | DNA framework | — | SERS | Enhanced Raman signal and ideal platform for ultra-sensitive sensing | 206 |
4. Characterization of the nano–bio interface
To further elucidate the effect of nano–bio interfacial factors on sensing performances, surface analysis techniques are required to precisely probe the orientation, coverage, spatial distribution of bioreceptors as well as the composition on nanoparticles. However, such characterization is still challenging and limited due to the nanoscale size, surface curvature, and inherent heterogeneity of nano–bio interfaces. This section summarizes available approaches for characterizing the nano–bio interfacial parameters discussed above, encompassing both widely accessible conventional techniques and advanced analytical approaches at the single particle level.4.1. Characterizing the orientation of bioreceptors
The orientation of the immobilized antibody is often indirectly estimated from antigen binding efficiency via immunoassay. However, conclusions inferred from binding efficiency are easily affected by other factors, such as stability of antibody–nanoparticle conjugates, antibody coverage and immunoassay conditions, which may obscure the true orientation of antibodies. To directly probe the orientation of antibodies, time-of-flight secondary ion mass spectrometry (TOF-SIMS) combined with principal component analysis has been employed. TOF-SIMS analyses the mass of secondary ions sputtered from a surface under primary-ion bombardment (Fig. 15a).207,208 TOF-SIMS serves a powerful surface characterization tool for both nanoparticles and biomolecules with a lateral resolution down to 30 nm. The principle of using TOF-SIMS to characterize antibody orientation relies on the fact that amino acid-related secondary-ion fragments reflect the outermost exposed region of antibodies. As the Fc and Fab domains possess different amino-acid compositions, multivariate principal component analysis of these fragments enables the identification of preferentially exposed domains and thereby the orientation of antibodies (Fig. 15b). Gajos et al. investigated how the antibody coverage governs the molecular orientation and immunological recognition on silicon substrates.209,210 Physical adsorption and two covalent coupling strategies—NHS-silane and amino-silane followed by glutaraldehyde activation–were compared. They demonstrated that the orientation of antibodies was affected by both surface coverage and immobilization approaches. NHS-silane promoted mixed orientations of head-on and tail-on due to intermolecular interactions, whereas glutaraldehyde activation favoured a dominant head-on orientation (Fig. 15c). More recently, they further observed that the orientations of both physical adsorbed and covalent bound antibodies were pH-dependent (Fig. 15d).211 The proportion of antibodies adopting tail-on and head-on orientations decreased with increasing pH. The actual fraction of the exposed Fc domain was approximately 0.1 for pH < 7 to about 0.35 for pH > 10. Overall, these studies provide a direct and quantitative framework to probe the antibody orientation that is affected by the immobilization protocol and antibody surface coverage. Such quantitative information on antibody orientation is valuable for the rational design of high-sensitivity biosensors. However, the above studies determined the antibody orientation at the silicon substrate rather than nanoparticles. Future investigations using TOF-SIMS to characterize the antibody orientation on nanoparticles are expected. Super-resolution fluorescence microscopy techniques can map the epitopes of antibodies on nanoparticles at a single particle level. DNA-point accumulation for imaging in nanoscale topography (PAINT), based on the short transient interaction of a labeled DNA probe (imager) to a complementary DNA target, has become a quantitative method to probe the orientation of antibodies on a single particle.212 Albertazzi and co-workers have quantified the orientation of the antibodies based on dual-color DNA-PAINT using a Fab-specific protein M coupled probe and Fc-specific protein G (Fig. 15e).213 The numbers of Fab and Fc binding sites per nanoparticle were quantified simultaneously, providing a powerful strategy for designing nanoparticles with controlled Fab and Fc exposure. | ||
| Fig. 15 (a) Schematic diagram of TOF-SIMS for probing nano–bio interfaces. Reproduced with permission,208 Copyright 2014, Wiley-VCH. (b) The principle of characterizing antibody orientation by TOF-SIMS. The exposed Fc and Fab domains in the outermost region exhibit distinct amino acid compositions. Reproduced with permission from ref. 210, under the CC BY 4.0 license. (c) TOF-SIMS analysis of the orientation of IgG immobilized by APTES/glugtaraldehyde and NHS-silane. Reproduced with permission,209 Copyright 2020, Elsevier. (d) Schematic representation of the vertical arrangement of the antibody immobilized by amino-silane at different pH values. Reproduced with permission,211 Copyright 2024, Elsevier. (e) Mapping the antibodies on the surface of nanoparticles using DNA-PAINT. Reproduced with permission,213 Copyright 2023, American Chemical Society. | ||
Approaches for monitoring the conformation of DNA generally rely on distance-dependent optical techniques, including single molecule FRET, SERS, and surface plasmon resonance. Single molecule FRET is a powerful technique for probing the conformation dynamics and interactions of biomolecules.214 In the context of nano–bio interfaces, Hildebrandt and coworkers employed time-resolved FRET to systematically investigate the conformation of DNA attached onto dihydrolipoic acid (DHLA)-PEG coated QDs (Fig. 16a).215 A terbium complex, serving as a donor, was conjugated to DNA hybridized at different positions along a 31-nucleotide DNA strand, which was linked to the QD acceptor via a peptide-His tag. Terbium-QD distances can be adjusted from 7 nm to 14 nm by varying the hybridization sites. It was found that ssDNA exhibited a flexible orientation near QDs with a compact structure of 0.15 nm per base, while dsDNA adopted a radial orientation with full extension of 0.31 nm per base pair. Subsequently, his group extended the terbium-QDs FRET platform to artificial bidomain proteins. The artificial bidomain proteins were labeled with a terbium complex on its C-terminus and a QD acceptor on its N-terminus.216 Specific binding of one or two protein targets to the artificial bidomain protein induced a conformational opening of the bidomain scaffold, accompanied by a corresponding fluorescence change. The Ju group has employed SERS to probe DNA conformation on multibranched gold nanostars to determine DNA orientation (Fig. 16b).217 Cy5 labelled DNA strands were assembled on gold nanostars via polyA, whose length controlled whether the DNA strands adopted a “lie-down” or “stand-up” conformation. As SERS enhancement is extremely sensitive to the distance between the Raman reporter and the gold surface, especially at electromagnetic “hot spots” located at the tips of nanostars, conformational changes in DNA translate directly into fluctuation in SERS intensity. Specifically, increasing the polyA length or inducing DNA hybridization extends the DNA away from the surface of gold nanostars, resulting in a reduced SERS signal. This study demonstrates that SERS enables in situ and quantitative monitoring of DNA conformation on nanoparticles. Similarly, surface plasmon resonance can also be utilized to probe the DNA conformation based on the distance-dependent plasmonic coupling between AuNPs and a neutral cysteine-tagged chimeric avidin (nChiAvd-Cys) modified planar gold surface (Fig. 16c).218 Dark-field scattering microscopy provides a direct and label-free optical readout of DNA conformational switching. The spectral shift of individual AuNPs correlates with the conformation of the hairpin DNA linker. By varying the applied electric field, the hairpin DNA can switch reversibly between folded and unfolded states, producing quantifiable shifts in the scattering responses. Owing to its single particle sensitivity and straightforward optical transduction, this surface plasmon resonance-based strategy is readily extended to monitor conformational changes of other biomolecules.
 | ||
| Fig. 16 (a) Schematic diagram of a terbium-QD based FRET nanoruler to investigate the conformation of ssDNA and dsDNA. Reproduced with permission,215 Copyright 2018, American Chemical Society. (b) Schematic illustration of SERS to monitor DNA conformation on gold nanostars. Reproduced with permission,217 Copyright 2017, Wiley-VCH. (c) Schematic representation of an AuNP–DNA nanoactuator on gold film and its application in assembly characterization based on surface plasmon resonance and dark-field scattering microscopy. Reproduced with permission,218 Copyright 2018, The Royal Society of Chemistry. (d) STM topographic image of CNT–DNA hybrids on a Si substrate. Reproduced with permission,220 Copyright 2009, American Chemical Society. (e) Schematic diagram of viral particles and virus-mimicking DNA origami epitopes for IgG binding. The bottom are high-speed AFM images of a three-stage binding process. Reproduced with permission,224 Copyright 2020, Springer Nature. | ||
Other techniques based on scanning probe microscopes, such as scanning tunneling microscopy (STM) and atomic force microscopy (AFM), have been used to explore the orientation or conformation of biomolecules at the sensing interface. STM enables the direct visualization of individual biomolecules with near-molecular resolution. For instance, the IgG antibody labeled with 5 nm AuNPs, as well as antibody–antigen complexes, has been imaged on atomically smooth pyrolytic graphite by STM.219 Yarotski and coworkers utilized STM to characterize the carbon nanotube (CNT)–DNA interface on a p-doped Si substrate and revealed both the spatial arrangement and interaction mechanism of DNA on CNTs (Fig. 16d).220 DNA-coated CNT segments appear as protruding islands, and the periodic height modulations in topography directly revealed the DNA wrapping geometry with a coiling period of 3.3 nm and a wrapping angle of 63° relative to the CNT axis. Additional width and height modulations along the DNA strand were also observed with characteristic lengths of 1.9 and 2.5 nm. This stable wrapping configuration was attributed to the optimal π-stacking, arising when DNA bases align with the helical orbital and charge-density pattern of CNTs. Compared with STM, AFM is not restricted to conductive samples and therefore more widely used for imaging biomolecules.221,222 The development of high-speed AFM has significantly improved the temporal resolution into the millisecond and even microsecond regime, making it suitable for observing dynamic processes.223 Fan's group investigated the transient binding conformations of IgG by high-speed AFM (Fig. 16e).224 DNA origami epitopes decorated with precisely spaced digoxin “spikes” were designed to capture IgG. Single-event transitions of IgG from wandering, monovalent binding, bivalent binding and wagging motions of Fc domains during the binding process were directly visualized by high-speed AFM. By varying the digoxin spacing from 3 to 16 nm, it was found that IgG molecules adopted conformations from highly compact to fully extended. Despite these advances, probing the orientation of bioreceptors on nanoparticles utilizing STM and AFM remains challenging. Both STM and AFM typically require atomically smooth surfaces as the tip convolution effect becomes significantly exacerbated on the highly curved surfaces of nanoparticles.225 In addition, the intrinsic heterogeneity of nano–bio interfaces complicates the discrimination between signal variations arising from particle heterogeneity and those induced by tip artifacts or environmental fluctuations. Therefore, most existing studies of sensing nano–bio interfaces by STM and AFM have focused on macroscopic and planar substrates rather than nanoparticles. Moreover, as STM relies on tunneling current, the presence of insulating bioreceptor layers on nanoparticles leads to fuzzy images and reduced image resolution. Finally, STM requires specialized operators in sample preparation, imaging parameter optimization, and data interpretation, which further limits its wide range of applications in nano–bio interfacial characterization.226
4.2. Characterizing the coverage of bioreceptors
Methods of quantifying the bioreceptor coverage on nanoparticles at the ensemble level include radio-labeled assay, UV-vis and fluorescence spectroscopy, mass spectrometry, surface plasmon resonance and quartz crystal microbalance.227–232 The nano–bio conjugates typically require preparatory treatments prior to the measurements. For instance, to quantify the amount of protein on nanoparticles, the nano–bio conjugates are subjected to acidic treatment that simultaneously hydrolyses the grafted protein and degrades nanoparticles. Subsequently, a spectrophotometric assay is performed to determine the concentration of primary amines in the resultant solution.233 Regarding bioconjugates of AuNPs, DNA strands or antibodies were commonly released from nanoparticles by ligand exchange or decomposing the nanoparticle core.229,234,235Besides quantification at the ensemble level, determining the number of bioreceptors at the single particle level is highly desired. TOF-SIMS can be operated in an event-by-event detection mode to quantify DNA loading on single gold nanostars in a label-free manner (Fig. 17a).236 Individual nanostars were analyzed using massive Au4004+ cluster projectiles, where each impact probed a nanometric emission volume with a lateral diameter of 10–15 nm and a depth of 5–10 nm. DNA loading was extracted from ion-coincidence analysis between gold cluster ions (e.g., Au7−) and DNA-specific phosphate and nucleobase-related ions. As co-emitted ions originate from the same nano-volume, their coincidence provides direct evidence of DNA present within the probed surface region. The coincidental yield, defined as the fraction of impacts on a nanoparticle that produces DNA-related ions, is utilized as a quantitative metric of DNA loading per particle. By normalizing the coincidental yields to reference particles of 50 nm gold nanospheres, DNA loading can be quantitatively compared across nanostars with different morphologies. This event-resolved TOF-SIMS enables direct determination of DNA loading on individual AuNPs, revealing how nanoscale curvature and geometry influence the loading density of bioreceptors. DNA-PAINT can also be adopted to quantify the coverage of bioreceptors at the single particle level. The Zijlstra group determined the number of DNA on individual AuNRs (a diameter of 22 nm and a length of 75 nm) in high-throughput via quantitative PAINT (Fig. 17b). AuNRs immobilized on a coverslip are functionalized with thiolated ssDNA docking strands. A fluorescent imager strand is introduced and imaged by total internal reflection fluorescent microscopy. Each transient binding event generates a burst, which is converted into a distribution of dark times between successive binding events. The dark-time histogram is fitted with a single-exponential to obtain the mean dark time, which scales inversely with the number of docking strands. Thus, with controlled imager concentration and known kinetic rates, the number of docking strands is obtained across large particle populations. A significant particle-to-particle difference in DNA coverage was observed among hundreds of AuNRs, ranging from 100 DNA per AuNR to nearly 600 DNA per AuNR.237 The Albertazzi group quantified both antibodies and streptavidin per NP via multiplexed DNA-PAINT.238 Similarly, significant heterogeneity of the available functional sites was found at both inter- and intraparticle levels.
 | ||
| Fig. 17 (a) Schematic diagram of TOF-SIMS to quantify the number of DNA on nanostars. Reproduced with permission,236 Copyright 2019, American Chemical Society. (b) Schematic illustration of functionalized nanoparticles for quantitative single-molecule counting based on DNA hybridization kinetics. The temporal analysis of the bursts reveals the number of functional groups on each individual particle. Reproduced with permission,237 Copyright 2020, The Royal Society of Chemistry. (c) Schematic representation of TOF-SIMS to investigate the composition distribution of copolymers and antibodies. Reproduced with permission,250 Copyright 2025, American Chemical Society. (d) DNA-PAINT based methodology for mapping active sites of anti-digoxigenin and anti-biotin antibodies on functional nanomaterials. Reproduced with permission,238 Copyright 2018, American Chemical Society. (e) 3D spatial characterization of biofunctionalized DNA on nanoparticles and calculated distribution score of DNA. Reproduced with permission,251 Copyright 2025, American Chemical Society. (f) NanoSIMS images of Au nanostructures with elemental distributions of 197Au− and 12C14N−. Reproduced with permission,252 Copyright 2016, Springer Nature. | ||
4.3. Characterizing the composition of mixed ligands
Spectroscopy-based approaches are widely adopted to identify the composition of functional layers on nanoparticles, including nuclear magnetic resonance (NMR), Fourier-transform infrared spectroscopy (FTIR), mass spectrometry, and fluorescence microscopy with fluorescence labeling.239–241 Although NMR is readily utilized to probe small molecules on nanoparticles, characterizing macromolecular bioreceptors such as antibodies or DNA by NMR remains challenging. The large molecular size of these bioreceptors and their slow rotational tumbling lead to severe line broadening and signal attenuation, which in turn reduce the signal-to-noise ratio and spectral resolution.242 Meanwhile, the peak overlap, complex data analysis, and critical requirements in sample purity and homogeneity further limit the applicability of NMR in characterizing nano–bio interfaces.243 In contrast, FTIR enables rapid assessment of the chemical composition of biofunctionalized layers on nanoparticles by probing the vibration and rotation mode of bioreceptors.244,245 Key spectral windows include the protein and peptide region (amide I/II, 1800–1500 cm−1), the carbohydrate region (C–O–C/C–O vibrations, 1200–900 cm−1) and the nucleic-acid region (phosphate groups, 600–1200 cm−1).246–248Mass spectrometry provides complementary and compositional information by measuring the mass-to-charge ratio of characteristic ions, thereby resolving the composition of bioreceptor layers. For instance, Chen and colleagues employed TOF-SIMS operated in event-by-event detection mode to determine the binding sites of nanoparticle–antibody conjugates and their surface binding density on cells.249 The binding sites between AuNPs and antibodies were inferred from coincidence mass spectra triggered by Au− and AuCN−. The simultaneous emission of cysteine ions indicated that AuNPs were bound by sulfur-terminal cysteine residues. Surface binding densities are then quantified using fractional coverage, defined as the ratio of effective projectile impacts on a given species to the total number of impacts. Coincidence of Au−/AuCN− ions was used to quantify the surface coverage of AuNP–antibody conjugates, whereas the coincidence of palmitate and oleate ions quantified the coverage of phospholipid layers of cell membranes. The lipid membrane and AuNP–antibody conjugates were found to cover 23% and 21% of the cell surface, respectively. More recently, TOF-SIMS has been applied as a surface and depth sensitive tool to characterize the interfacial composition of mixed antibody and antifouling polymers as well as antibody orientation on a silicon substrate (Fig. 17c).250 The characteristic secondary ions from 2-(2-methoxyethoxy) ethyl methacrylate (OEGMA) and methacrylic acid (MAA) confirm the composition of copolymers, and amino-acid fragments verify the existence of antibodies at the outermost interface. TOF-SIMS depth profiling further reveals a uniform copolymer composition through the polymer brush thickness. Furthermore, multivariate principal component analysis is applied to quantify the density and the orientation of interfacial antibodies.
4.4. Characterizing the spatial distribution of bioreceptors
DNA-PAINT and SIMS-based approaches have been established to probe the spatial distribution of bioreceptors on nanoparticles. DNA-PAINT can resolve the nanoscale distribution of functionally active sites of anti-digoxigenin and anti-biotin antibodies on nanoparticles. Each antibody was selectively targeted by its cognate antigen conjugated to a distinct DNA docking strand. Sequential introduction of complementary fluorescent imager strands enables super-resolution localization of each antibody population in separate imaging rounds using the same fluorophore. Merging the localization datasets produces a dual-color nanoscale map that directly reveals intraparticle heterogeneity of the two receptor populations (Fig. 17d).238 More recently, the Prins group developed a quantitative workflow to determine the surface density and spatial organization of bioreceptors on planar substrates using single molecule DNA-PAINT.251 The spatial distribution of surface-tethered DNA is obtained by extracting single molecule localizations from transient binding of dye-labeled imager strands to complementary ssDNA. Within each small region of interest, the time-accumulated “localization clouds” are clustered to estimate the center coordinates of individual immobilized ssDNA, yielding a point pattern of molecular positions. These coordinates are evaluated using the nearest-neighbor test against the complete spatial randomness hypothesis. The standardized mean nearest-neighbor distance classifies the distribution as significantly clustered, random, or significantly dispersed. They further extended this framework to 3D DNA-PAINT for the full curved surface of particles, followed by clustering to assign localizations to individual particle and compensation for under-sampling to quantify both interparticle and intraparticle heterogeneity (Fig. 17e).252 In addition, the heterogeneity of DNA immobilized by two approaches, streptavidin–biotin coupling and PLL-g-PEG-N3/PLL-g-PEG coating followed by click coupling, is analysed. Stronger interparticle variability existed for PLL-g-PEG-based click chemistry, possibly due to the heterogeneous polymer coating and the dynamic nature of PLL-g-PEG adsorption. For intraparticle heterogeneity, both chemistries yielded clustered ssDNA distributions, with slightly stronger clustering for the PLL-g-PEG click system, consistent with clustered reactive sites for multivalent streptavidin binding sites and multiple azides per azide-PLL-g-PEG chain.In parallel, interfacial heterogeneity of different bioreceptors can be assessed by event-by-event nanoprojectile SIMS, generating independent mass spectrum per impact within nanoscale volume.253 By screening millions of events for coincidence emission of tag-specific ions from antibodies with different labels, whether species are truly co-localized within the same nanometric volume or random aggregation can be quantified. A co-localization factor derived from the yield ratio is used to estimate the nanoscale inhomogeneity degree of antibodies on silicon wafer. Tags with different hydrophobicities can affect their attachment on the surface, while chemically similar tags produced near-random attachment. Additionally, nanoscale SIMS (NanoSIMS) offers direct chemical imaging of interfaces with lateral resolution down to 50 nm, which can directly image characteristic native ions or labels as stable isotopes halogens, metal and rare-earth elements.254 Zhai et al. adopted NanoSIMS to probe the spatial distribution of polyvinylpyrrolidone on individual Au nanoprisms.255 12C14N− signals attributed to the adsorbed polyvinylpyrrolidone were localized at the perimeters of nanoprisms rather than on their top [111] facets (Fig. 17f).
5. AI-driven nano–bio interfacial design
AI, particularly machine learning and deep learning, has demonstrated considerable potential in both the realm of nanomaterial and biomolecule-related tasks, such as controllable synthesis and assembly, and the prediction of physicochemical and binding properties.256–260 However, within the biosensing field, AI remains concentrated on the downstream data analysis.261–264 The integration of AI into the upstream nano–bio interface design and sensing performance prediction is still in its infancy. In this section, we first provide an overview of the typical workflow and key challenges associated with AI-driven nano–bio interfacial design in biosensors. Essential computational concepts, such as descriptors, featurization, model selection and validation, are included to bridge the knowledge gap for researchers in the biosensing field. The second section focuses on CNT–DNA interfaces as a model system to illustrate how AI can improve the sensing performances of CNT–DNA biosensors.5.1. Workflow of AI-driven nano–bio interfacial design
The performance of nanoparticle-based biosensor depends not only on the nano–bio interaction but also on the specific bioreceptor–analyte interaction and the effective mitigation of nonspecific binding. Beyond understanding individual interactions, learning how they collectively interconnect and ultimately dictate key sensing metrics remains a considerably challenging task for AI models. In this context, the general workflow and associated challenges for AI-empowered nano–bio interface design in biosensing are summarized based on insights from protein corona studies and other nano–bio interaction systems (Fig. 18). The proposed framework begins with clarifying the analyte requirements and application scenarios, including the various analyte species (e.g., protein, peptide, RNA, DNA, or small molecule), the matrix (such as biological fluids, cell/tissue-related matrices, or artificial matrices) and the expected sensing performance criteria. This initial step establishes a well-defined objective that guides subsequent data acquisition and model development. | ||
| Fig. 18 General workflow and associated challenges for AI-empowered nano–bio interfacial design in biosensors. | ||
As a comprehensive dataset is indispensable for effective AI-assisted modeling, the next step is data acquisition. Experimental, empirical or simulation-derived data are acquired to describe the structural parameters and physiochemical properties of nanomaterials and biomolecules, binding attributes of nano–bio interfaces, and key indicators of sensing performances. Important parameters for nanomaterials include size, shape, composition, morphology, chirality, surface charge, surface modification, and assembly state. Molecular weight, isoelectric point, hydrophilic–hydrophobic property, and surface charge are involved in terms of bioreceptors.265 The nano–bio interfacial binding attributes encompass binding strength of bioreceptors (covalent or noncovalent bonds), bioreceptor–analyte affinity, orientation, coverage and spatial distribution of bioreceptors, and the driving forces underlying the undesired interactions. The output variables correspond to single, or multiple biosensing performance metrics as described above. A major challenge in this step is the scarcity of large-scale and high-quality, standardized datasets that jointly capture nano–bio interaction features and biosensing performances. Experimental values frequently vary across laboratories and protocols, compromising reliability and comparability in the absence of universally accepted references. The difficulty here also includes the lack of complex samples, in which matrix effects are more pronounced. Data obtained from complex biological samples are crucial for translating laboratory-based strategies into practical methods for detecting analytes in human fluids or cell/tissue-related matrices. Furthermore, while most biosensor studies emphasize the sensing performance of sensitivity and LOD, performance metrics related to stability and reproducibility remain relatively underexplored, further constraining the robust development of AI-driven models.
Subsequently, experimental or computational data are transformed into feature sets compatible with AI models, which requires deliberate encoding of nanomaterial properties. Compared with small organic molecules, generating meaningful descriptors for nanomaterials is challenging due to their structural complexity and morphological diversity. To address this issue, descriptors from experimental properties, quantum chemical calculations, periodic table properties and full nanostructure representations have been developed to capture the structural and physicochemical characteristics and to encode complex nanostructures.266 Common predictors encompass experimental parameters such as zeta potential, particle size, surface area, curvature, shape, and surface charge.267 Simulation-derived descriptors extracted from atomistic MD simulations, including ligand dynamics, solvation, hydrogen bonding, interaction energies, electrostatic potentials, are utilized for quantitative nanostructure–activity relationship models.268 Response-based descriptors, introduced by Zhong's group, exploit the fluorescence change of fluorescamine-labelled protein in the presence and absence of nanomaterials.269 When used as input to the machine learning models, these descriptors have demonstrated enhanced predictive performance compared with independent properties of nanomaterials or proteins. Apart from molecular or nano-level descriptors, the Zhu group pioneered the concept of virtual molecular projections to digitally represent complex nanostructures.270 Nanoparticles are simulated in three dimensions and converted into multidimensional arrays encoding atom types and spatial coordinates. When fed directly into convolutional neural network (CNN) models, these virtual projections enable accurate prediction of cellular uptake, lipophilicity, and zeta potential of nanoparticles. The same team further established PubVINAS, a publicly accessible online nanomaterial database integrating 705 annotated nanostructures across 11 material types.271 Despite these advances, the availability of robust, meaningful and universal nanodescriptors remains limited due to the inherent heterogeneity and incomplete characterization of nanomaterials. Moreover, the physicochemical characteristics of nanomaterials are highly sensitive to media—agglomerating, dissolving, and dynamically adsorbing or desorbing surrounding molecules—further complicating the development of efficient and generalizable descriptors.272 Therefore, incorporating both nanomaterial heterogeneity and environmental variables into descriptor design is essential for establishing reliable structure–function correlations.
Next, appropriate AI models are selected, trained, and iteratively refined according to specific biosensing tasks. Machine learning and deep learning are the two most prevalent AI models employed in materials science and biomedicine. Machine learning leverages statistical algorithms to learn patterns from data and generate predictions without being explicitly programmed, including decision trees (DTs), random forest (RF), support vector machine (SVM), k-nearest neighbour (kNN), logistic regression and Naive Bayes.272,273 In contrast, deep learning, as a subfield of machine learning, employs multi-layer artificial neural networks to automatically learn hierarchical data representations.272 Representative deep learning architectures include CNNs, recurrent neural networks (RNNs), graph neural networks (GNNs), as well as multi-task and transfer-learning frameworks. Deep learning typically requires larger datasets and often suffers from limited interpretability. Nevertheless, it is particularly effective in cases where the governing variables and underlying mechanisms are complex, highly interdependent, and not fully understood in advance. For deep and thorough insights into AI models, refer to previous works.274–276 As datasets describing nano–bio interfaces remain limited at the current stage, machine learning models are still adopted in most cases. However, deep learning offers distinct advantages for capturing the intricate and nonlinear relationships between nano–bio interfacial structures and biosensing performances, which is expected to play an increasingly important role in the future. The final step is experimental validation and closed-loop feedback.277 Experimental results are reintegrated into the training dataset, enabling retraining and performance refinement in subsequent cycles. The model performances are evaluated on unseen data, focusing on generalization and key metrics such as accuracy, precision, recall, and F1 score.
5.2. AI-driven interfacial design of CNT–DNA biosensors
CNT–DNA hybrids are powerful near-infrared nanosensors that exhibit robust optical signal modulation upon analyte binding.278 Several machine learning models have been developed to elucidate CNT–DNA interactions and their fluorescence modulation mechanism, offering a promising route to enhance sensing performance and facilitate rational biosensor design.279 From the perspective of DNA sequence design, Kelich et al. applied machine learning to discover CNT–DNA biosensors for serotonin detection (Fig. 19a).280 CNN classifiers and SVM regressors were trained to map sequence-derived features from 96 CNT–DNA conjugates with experimentally measured fluorescence responses at 1195 nm (Fig. 19b). The predictions were experimentally validated, and the validation results were used to retrain the models that identified DNA sequences with markedly enhanced sensitivity and selectivity (Fig. 19c). This study established a practical “experiment–machine learning–re-experiment” closed-loop framework, particularly suitable for the small-sample regime typical of nano–bio interface studies. Later, the same group further predicted the optical response of CNT–DNA for serotonin as a function of DNA sequence by exploiting the whole fluorescence spectra.281 Regression models trained with SVM successfully distinguished high and low response DNA sequences. Incorporating multiple spectral wavelengths into model increased the prediction robustness and facilitated the discovery of novel CNT–DNA sensors. Besides DNA sequence optimization, Kim et al. utilized the spectral fingerprinting of quantum-defect-modified CNTs for the detection of ovarian cancer.282 Organic colour centres (OCCs)—atomic-level quantum defects—are covalently functionalized onto nanotubes, creating well-defined and chemically tunable emissive states. Three OCC terminating groups and four ssDNA sequences were selected to construct a nanosensor array (Fig. 19d). Spectra from functionalized nanotubes were collected for patient and control sera, shown in Fig. 19e. A panel of supervised machine learning algorithms were evaluated, including logistic regression, artificial neural networks, DTs, RF and SVM. SVM approaches achieved the highest F-score among the tested algorithms. This sensing platform identified high-grade serous ovarian cancer with high positive and negative predictive values, achieving 87% sensitivity at 98% specificity. | ||
| Fig. 19 (a) Discovery of DNA-CNT optical sensors for serotonin detection. (b) Optical responses of 96 unique CNT-DNA conjugates to serotonin. (c) Main computational approach. Reproduced with permission,280 Copyright 2022, American Chemical Society. (d) OCC-DNA nanosensor array with different OCC terminating groups and four ssDNA sequences. (e) Representative fluorescence spectra of one OCC-DNA nanosensor. Reproduced with permission,282 Copyright 2022, Springer Nature. (f) Machine learning models to predict analyte-induced fluorescence changes in CNT-DNA nanosensors for dopamine. (g) Transformation of analytes into molecular fingerprints with RDKit for the predictive modeling of optical responses. (h) Support vector classifiers and regression models validated against the original dataset and an independent 21-molecule blind set. Reproduced with permission,284 Copyright 2026, American Chemical Society. | ||
In parallel, Beyene and co-workers systematically investigated the molecular determinant of catecholamine-like analytes for CNT–DNA based fluorescent sensors.283 The large optical responses strongly correlated with the electronic properties of bound ligands, such as electron density on the aryl motif, rather than the binding affinity alone. This insight highlights the importance of incorporating physicochemical descriptors of analytes and the local corona environment into machine learning models, moving beyond DNA sequence-based features toward physically informed representations. More recently, their group developed a machine learning model to predict the analyte-induced fluorescence response of a fixed CNT–(GT)6 dopamine nanosensor (Fig. 19f).284 Using a dataset of 63 small training molecules, each analyte was encoded using standard RDKit fingerprints with optional frontier molecular orbital energy descriptors. Principal component analysis of these molecular encodings was employed to map the chemical space and identify structural motifs associated with strong or weak fluorescence responses (Fig. 19g). Support vector regression and support vector classification were then utilized to predict continuous ΔF/F values and to discriminate strong or weak responses, respectively (Fig. 19h). Following cross-validation, the models were evaluated against an independent blind set of 21 molecules to assess their generalizability. While the classification model achieved robust discrimination (F1 ≈ 0.8), the regression performance was relatively modest (mean R2 ≈ 0.2–0.4). Collectively, this work demonstrates that analyte-centric molecular descriptors alone can capture meaningful structure-response relationships for a fixed CNT-DNA sensor. On the readout side, machine leaning can decode “hidden” information from full emission spectra of CNTs.285 By treating entire spectra as high-dimensional inputs, machine learning models resolve subtle peak shifts, intensity ratios, and line-shape changes and effectively turn the emission profile into a rich fingerprint of nano–bio interfacial state, enabling sensitive and multiplexed detection of analytes. In summary, the above studies outline a general paradigm for AI-driven CNT–DNA biosensor design, especially, how to choose small but high-quality datasets to train machine leaning models. Extending this framework to broader nano–bio interfaces promises to accelerate the discovery of sensitive, selective, fast response, and interpretable biosensors beyond CNT–DNA systems.
6. Summary and outlook
Macroscopic sensing performances (sensitivity, LOD, specificity, selectivity, response time, multiplex, reproducibility and stability) of biosensors are ultimately connected to molecular scale interfacial parameters at nano–bio interfaces. Unraveling the relationship between macroscopic sensing performance and the molecular scale interfacial parameters is crucial for the rational design of biosensors. In this context, this review discussed why nano–bio interfaces are central to the bioreceptor–analyte interaction. The basic design rules for nano–bio interfaces facilitating the bioreceptor–analyte interaction are analyzed. Four key interfacial parameters, orientation, coverage, composition and spatial distribution of bioreceptor on nanoparticles, are elaborated to enhance the sensing performance of biosensors. Approaches to tailor these four interfacial parameters were summarized. In parallel, representative examples are presented to illustrate how rationally engineered nano–bio interfaces lead to the improved sensing performance.Although much progress has been made in tailoring nano–bio interfaces, challenges still exist in the interfacial design to boost the analytical performance in nanoparticle-based biosensors. Compared with interfacial properties of bioreceptors on macro-substrates like electrodes, glass coverslip and metal film, the approaches to control the interfacial parameters on nanoparticles require further exploration. For instance, the conformation of DNA or protein on a planar substrate can be regulated by an external electric field, while relevant studies on the nanoparticle surface were rare.218,286 Additionally, DNA nanostructures, nanoparticles and macromolecules all can serve as templates to control bioreceptor spacing on macro-substrates.287 In contrast, due to the small size and high curvature of nanoparticles, the approach to control the inter-bioreceptor distance on nanoparticles is currently limited to transferring predefined patterns from DNA nanostructures. Except for finite nano–bio interface tailoring approaches, approaches to characterize the interfacial parameters of bioreceptors on particles are also limited, particularly at the single particle level.288 Conventional characterization techniques, including UV-vis and fluorescence spectroscopy, FTIR, dynamic light scattering, mass spectrometry and SERS, are performed at the ensemble level, which conceals the heterogeneity of nano–bio interfaces.239,289–291 TOF-SIMS, a highly sensitive surface analytical technique, is a powerful tool for interfacial characterization, enabling the assessment of conformation, surface coverage, spatial distribution of bioreceptor as well as chemical composition. The emerging single molecule techniques, such as DNA-PAINT, single molecule FRET, SERS and plasmonic scattering imaging, enable direct display of bioreceptors at a single particle.237,238,292–294 Huge potential exists in investigating how the analytical performances of single particles are related to their interfacial parameters.295 For instance, how can the spatial distribution of bioreceptors on nanoparticles be quantitatively and precisely characterized? Given the same average bioreceptor density on a nanoparticle, a uniform or heterogenous spatial distribution of bioreceptor, which one can result in superior sensing outcome? Addressing these questions is crucial not only for the rational design of nano–bio interfaces but also for the stability and reproducibility of nanoparticle-based biosensors. Therefore, developing novel and facile approaches to tailor and characterize the nano–bio interfaces is required.
Another challenge is elucidating how various interfacial parameters correlate with multiple sensing metrics, a task requiring multivariate regression analysis. Additionally, experimental investigations of nano–bio interfaces, in certain cases, are difficult to implement as a multitude of interrelated variables must be simultaneously considered. For instance, variations in bioreceptor coverage on nanoparticles may be accompanied by concomitant changes in conformation and spatial distribution of bioreceptors. In this regard, AI-based modeling and simulation approaches are tremendously valuable, as they enable systematic exploration of numerous parameter permutations. Xu and coworkers developed a model of predicting DNA arrangement on AuNPs to achieve the optimal detecting activity of human telomerase.296 Key parameters, including the size of nanoparticles, spacing of probe DNA and distance between two catalytic pockets of telomerase, were incorporated in this model. The optimal DNA density obtained from the experiment matched well with the value from the model. The LOD of AuNPs–DNA conjugates with optimal DNA density was at least 1 order of magnitude lower than that of densely packed AuNPs, demonstrating the effectiveness of this model in guiding interfacial probe arrangement. Similarly, Sun and coworkers used atomistic MD simulation to optimize receptor structure and favourable binding-site on AuNPs.297 The binding between receptors and guests requires deep penetration of guests into the monolayer of receptors. Receptor design guided by the simulation exhibited 10-fold improvement in analyte affinity and a 100-fold decrease in LOD. With rapid advances in AI-based modeling and simulation, these approaches are expected to be extended to a broader range of nanoparticles and bioreceptors, playing an increasingly significant role in the design and optimization of nanoparticle-based biosensors.
In summary, this review dismantles the traditional “black box” of nanoparticle-based biosensors by systematically elucidating the intrinsic relationships between nano–bio interfaces and sensing performances (Fig. 20). The proposed closed-loop framework integrates three aspects: (1) nano–bio interface tailoring approaches, serving as the hand for fabrication; (2) nano–bio interface characterization at the single particle level, acting as the eye for direct observation; and (3) AI-driven modelling and simulation, working as the brain for rational analysis. We hope that this review can contribute to a deeper understanding of the pivotal role of the nano–bio interface in determining biosensing performances. Ongoing advances in customizing and characterizing the nano–bio interfaces, particularly at the single particle level, along with AI modelling and simulations, are poised to unlock profound insights into nano–bio interfaces and drive the development of next-generation nanoparticle-based biosensors with outstanding performance.
 | ||
| Fig. 20 Key aspects of nano–bio interfacial design for high-performance nanoparticle-based biosensors. | ||
Conflicts of interest
There are no conflicts to declare.Data availability
No primary data were used in the writing of this review paper and all data and information were sourced from the published literature.Acknowledgements
We acknowledge funding from the Australian Research Council Discovery Grant (DP250101047) and Industry Laureate Fellowship Programs (IL24010091) National Health and Medical Research Council Investigator Grant (GNT1196648), China Postdoctoral Science Foundation (2023M743164) and Youth Science Foundation of Henan Province (252300423738). We also thank Professor Chao Lu for his kind support.Notes and references
- Y. Wu, D. Bennett, R. D. Tilley and J. J. Gooding, Adv. Mater., 2019, 32, 1904339 CrossRef PubMed.
- H. Geng, S. Vilms Pedersen, Y. Ma, T. Haghighi, H. Dai, P. D. Howes and M. M. Stevens, Acc. Chem. Res., 2022, 55, 593–604 CrossRef CAS PubMed.
- Z. Gao, Y. Song, T. Y. Hsiao, J. He, C. Wang, J. Shen, A. MacLachlan, S. Dai, B. H. Singer, K. Kurabayashi and P. Chen, ACS Nano, 2021, 15, 18023–18036 CrossRef CAS PubMed.
- Z. Li, M. Guo and W. Zhong, Precis. Chem., 2025, 3, 297–318 CrossRef CAS PubMed.
- L. Gloag, M. Mehdipour, D. Chen, R. D. Tilley and J. J. Gooding, Adv. Mater., 2019, 31, 1904385 CrossRef CAS PubMed.
- D. Chen, Y. Wu, S. Hoque, R. D. Tilley and J. J. Gooding, Chem. Sci., 2021, 12, 5196–5201 RSC.
- Z. Farka, T. Juřík, D. Kovář, L. Trnková and P. Skládal, Chem. Rev., 2017, 117, 9973–10042 CrossRef CAS PubMed.
- J. Zhang, L. Mou and X. Jiang, Chem. Sci., 2020, 11, 923–936 RSC.
- J. J. Gooding and S. Ciampi, Chem. Soc. Rev., 2011, 40, 2704–2718 RSC.
- F. Yang, X. Zuo, C. Fan and X. Zhang, Natl. Sci. Rev., 2018, 5, 740–755 CrossRef CAS.
- R. Pardehkhorram and A. Andrieu-Brunsen, Chem. Commun., 2022, 58, 5188–5204 RSC.
- M. Singh, N. Kaur and E. Comini, J. Mater. Chem. C, 2020, 8, 3938–3955 RSC.
- K. Bhattacharjee and B. L. V. Prasad, Chem. Soc. Rev., 2023, 52, 2573–2595 RSC.
- L. Taiariol, C. Chaix, C. Farre and E. Moreau, Chem. Rev., 2022, 122, 340–384 CrossRef CAS PubMed.
- K. E. Sapsford, W. R. Algar, L. Berti, K. B. Gemmill, B. J. Casey, E. Oh, M. H. Stewart and I. L. Medintz, Chem. Rev., 2013, 113, 1904–2074 CrossRef CAS PubMed.
- P. Li, G.-H. Lee, S. Y. Kim, S. Y. Kwon, H.-R. Kim and S. Park, ACS Nano, 2021, 15, 1960–2004 CrossRef CAS PubMed.
- H. Zhou, J. Liu, J.-J. Xu, S.-S. Zhang and H.-Y. Chen, Chem. Soc. Rev., 2018, 47, 1996–2019 RSC.
- Y. Cao, L. Zhou, Z. Fang, Z. Zou, J. Zhao, X. Zuo and G. Li, Chem. Commun., 2023, 59, 3383–3398 RSC.
- C. J. Yang, S. Jockusch, M. Vicens, N. J. Turro and W. Tan, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17278 CrossRef CAS PubMed.
- S. O. Kelley, C. A. Mirkin, D. R. Walt, R. F. Ismagilov, M. Toner and E. H. Sargent, Nat. Nanotechnol., 2014, 9, 969–980 CrossRef CAS PubMed.
- K. B. Cederquist and C. D. Keating, Langmuir, 2010, 26, 18273–18280 CrossRef CAS PubMed.
- C. Chen, W. Wang, J. Ge and X. S. Zhao, Nucleic Acids Res., 2009, 37, 3756–3765 CrossRef CAS PubMed.
- A. K. R. Lytton-Jean and C. A. Mirkin, J. Am. Chem. Soc., 2005, 127, 12754–12755 CrossRef CAS PubMed.
- L. K. Fong, Z. Wang, G. C. Schatz, E. Luijten and C. A. Mirkin, J. Am. Chem. Soc., 2018, 140, 6226–6230 CrossRef CAS PubMed.
- E. Treasurer and R. Levicky, J. Phys. Chem. B, 2021, 125, 2976–2986 CrossRef CAS PubMed.
- A. Sedighi, P. C. H. Li, I. C. Pekcevik and B. D. Gates, ACS Nano, 2014, 8, 6765–6777 CrossRef CAS PubMed.
- Q. Gu, E. A. Josephs and T. Ye, Aggregate, 2022, e186 CrossRef CAS.
- Q. Gu, H. H. Cao, Y. Zhang, H. Wang, Z. J. Petrek, F. Shi, E. A. Josephs and T. Ye, ACS Sens., 2021, 6, 371–379 CrossRef CAS PubMed.
- S. Dey, R. Rivas-Barbosa, F. Sciortino, E. Zaccarelli and P. Zijlstra, Nanoscale, 2024, 16, 4872–4879 RSC.
- Z. Xia, E. Villarreal, H. Wang and B. L. T. Lau, Colloids Surf., B, 2020, 190, 110960 CrossRef CAS PubMed.
- S. R. Saptarshi, A. Duschl and A. L. Lopata, J. Nanobiotechnol., 2013, 11, 26 CrossRef CAS PubMed.
- K. B. Cederquist and C. D. Keating, ACS Nano, 2009, 3, 256–260 CrossRef CAS PubMed.
- H. D. Hill, J. E. Millstone, M. J. Banholzer and C. A. Mirkin, ACS Nano, 2009, 3, 418–424 CrossRef CAS PubMed.
- R. Münter, C. Stavnsbjerg, E. Christensen, M. E. Thomsen, A. Stensballe, A. E. Hansen, L. Parhamifar, K. Kristensen, J. B. Simonsen, J. B. Larsen and T. L. Andresen, Small, 2022, 18, 2106529 CrossRef PubMed.
- S. Tenzer, D. Docter, J. Kuharev, A. Musyanovych, V. Fetz, R. Hecht, F. Schlenk, D. Fischer, K. Kiouptsi, C. Reinhardt, K. Landfester, H. Schild, M. Maskos, S. K. Knauer and R. H. Stauber, Nat. Nanotechnol., 2013, 8, 772–781 CrossRef CAS PubMed.
- M. Mahmoudi, M. P. Landry, A. Moore and R. Coreas, Nat. Rev. Mater., 2023, 8, 422–438 CrossRef PubMed.
- J. H. Monserud and D. K. Schwartz, ACS Nano, 2014, 8, 4488–4499 CrossRef CAS PubMed.
- M. Oishi and K. Saito, ACS Nano, 2020, 14, 3477–3489 CrossRef CAS PubMed.
- J. Yang, X. Li, B. Jiang, R. Yuan and Y. Xiang, Anal. Chem., 2020, 92, 7893–7899 CrossRef CAS PubMed.
- B. C. Lagerholm and N. L. Thompson, Biophys. J., 1998, 74, 1215–1228 CrossRef CAS PubMed.
- J. C. Traeger, Z. Lamberty and D. K. Schwartz, ACS Nano, 2019, 13, 7850–7859 CrossRef CAS PubMed.
- S. Hwu, Y. Blickenstorfer, S. J. Ihle, M. Garzuel, C. Forró, L. Schmidheini, L. Demkó and J. Vörös, J. Phys. Chem. C, 2020, 124, 1566–1574 CrossRef CAS.
- C. Qiao, J. Wu, Z. Huang, X. Cao, J. Liu, B. Xiong, Y. He and E. S. Yeung, Anal. Chem., 2017, 89, 5592–5597 CrossRef CAS PubMed.
- Y. Chen, S. Sun, X. Liu, H. Li, S. Huan, B. Xiong and X.-B. Zhang, Anal. Chem., 2024, 96, 9551–9560 CrossRef CAS PubMed.
- S. J. Hurst, H. D. Hill and C. A. Mirkin, J. Am. Chem. Soc., 2008, 130, 12192–12200 CrossRef CAS PubMed.
- A. K. Pearce, T. R. Wilks, M. C. Arno and R. K. O’Reilly, Nat. Rev. Chem., 2021, 5, 21–45 CrossRef CAS PubMed.
- J. M. Rabanel, V. Adibnia, S. F. Tehrani, S. Sanche, P. Hildgen, X. Banquy and C. Ramassamy, Nanoscale, 2019, 11, 383–406 RSC.
- P. De Luna, S. S. Mahshid, J. Das, B. Luan, E. H. Sargent, S. O. Kelley and R. Zhou, Nano Lett., 2017, 17, 1289–1295 CrossRef CAS PubMed.
- Z. Shen, P. L. Xavier, R. Bean, J. Bielecki, M. Bergemann, B. J. Daurer, T. Ekeberg, A. D. Estillore, H. Fangohr, K. Giewekemeyer, M. Karnevskiy, R. A. Kirian, H. Kirkwood, Y. Kim, J. C. P. Koliyadu, H. Lange, R. Letrun, J. Lübke, A. Mall, T. Michelat, A. J. Morgan, N. Roth, A. K. Samanta, T. Sato, M. Sikorski, F. Schulz, P. Vagovic, T. Wollweber, L. Worbs, F. Maia, D. A. Horke, J. Küpper, A. P. Mancuso, H. N. Chapman, K. Ayyer and N. D. Loh, ACS Nano, 2024, 18, 15576–15589 CrossRef CAS PubMed.
- M. Horáček, R. E. Armstrong and P. Zijlstra, Langmuir, 2018, 34, 131–138 CrossRef PubMed.
- M. R. W. Scheepers, I. L. J. Van and M. W. J. Prins, Langmuir, 2019, 35, 14272–14281 CrossRef CAS PubMed.
- C. M. Platnich, F. J. Rizzuto, G. Cosa and H. F. Sleiman, Chem. Soc. Rev., 2020, 49, 4220–4233 RSC.
- C. C. Harper, J. S. Jordan, S. Papanu and E. R. Williams, ACS Nano, 2024, 18, 17806–17814 CrossRef CAS PubMed.
- J. E. Park, Y. Jung, M. Kim and J. M. Nam, ACS Cent. Sci., 2018, 4, 1303–1314 CrossRef CAS PubMed.
- R. M. Lubken, A. M. de Jong and M. W. J. Prins, ACS Nano, 2021, 15, 1331–1341 CrossRef CAS PubMed.
- A. Makaraviciute and A. Ramanaviciene, Biosens. Bioelectron., 2013, 50, 460–471 CrossRef CAS PubMed.
- F. M. Kelley, A. Ani, E. G. Pinlac, B. Linders, B. Favetta, M. Barai, Y. Ma, A. Singh, G. L. Dignon, Y. Gu and B. S. Schuster, Nat. Commun., 2025, 16, 3521 CrossRef CAS PubMed.
- M. Tonigold, J. Simon, D. Estupiñán, M. Kokkinopoulou, J. Reinholz, U. Kintzel, A. Kaltbeitzel, P. Renz, M. P. Domogalla, K. Steinbrink, I. Lieberwirth, D. Crespy, K. Landfester and V. Mailänder, Nat. Nanotechnol., 2018, 13, 862–869 CrossRef CAS PubMed.
- Y. Tang, X. Zhao, Z. Nie, B. He and D. Deng, ACS Appl. Bio Mater., 2025, 8, 6252–6260 CrossRef CAS PubMed.
- B.-K. Jin, S. Odongo, M. Radwanska and S. Magez, Int. J. Mol. Sci., 2023, 24, 5994 CrossRef CAS PubMed.
- V. Mora-Sanz, L. Saa, N. Briz, M. Möller and V. Pavlov, ACS Appl. Mater. Interfaces, 2020, 12, 28993–28999 CAS.
- Q. Yu, Q. Wang, B. Li, Q. Lin and Y. Duan, Crit. Rev. Anal. Chem., 2015, 45, 62–75 CrossRef CAS.
- A. J. Sivaram, A. Wardiana, C. B. Howard, S. M. Mahler and K. J. Thurecht, Adv. Healthcare Mater., 2018, 7, 1700607 CrossRef PubMed.
- Z. Pei, H. Anderson, A. Myrskog, G. Dunér, B. Ingemarsson and T. Aastrup, Anal. Biochem., 2010, 398, 161–168 CrossRef CAS PubMed.
- D. Lou, L. Ji, L. Fan, Y. Ji, N. Gu and Y. Zhang, Langmuir, 2019, 35, 4860–4867 CrossRef CAS PubMed.
- C. Parolo, A. de la Escosura-Muñiz, E. Polo, V. Grazú, J. M. de la Fuente and A. Merkoçi, ACS Appl. Mater. Interfaces, 2013, 5, 10753–10759 CrossRef CAS PubMed.
- G. Ruiz, K. Tripathi, S. Okyem and J. D. Driskell, Bioconjugate Chem., 2019, 30, 1182–1191 CrossRef CAS PubMed.
- O. S. Wolfbeis, Chem. Soc. Rev., 2015, 44, 4743–4768 RSC.
- L. Zhang, Y. Mazouzi, M. Salmain, B. Liedberg and S. Boujday, Biosens. Bioelectron., 2020, 165, 112370 CrossRef CAS PubMed.
- M. Park, BioChip J., 2019, 13, 82–94 CrossRef CAS.
- A. A. Karyakin, G. V. Presnova, M. Y. Rubtsova and A. M. Egorov, Anal. Chem., 2000, 72, 3805–3811 CrossRef CAS PubMed.
- H. Sharma and R. Mutharasan, Anal. Chem., 2013, 85, 2472–2477 CrossRef CAS PubMed.
- S. Wu, H. Liu, X. M. Liang, X. Wu, B. Wang and Q. Zhang, Anal. Chem., 2014, 86, 4271–4277 CrossRef CAS PubMed.
- T. Nan, S. Wu, H. Zhao, W. Tan, Z. Li, Q. Zhang and B. Wang, Anal. Chem., 2012, 84, 4327–4333 CrossRef CAS PubMed.
- J. Sun, L. Li, R. Sun, H. Yin and J. Liu, Anal. Chem., 2025, 97, 16515–16524 CrossRef CAS PubMed.
- S. Gao, J. M. Guisán and J. Rocha-Martin, Anal. Chim. Acta, 2022, 1189, 338907 CrossRef CAS PubMed.
- S. K. Vashist, E. Lam, S. Hrapovic, K. B. Male and J. H. T. Luong, Chem. Rev., 2014, 114, 11083–11130 CrossRef CAS PubMed.
- L. Hu, Z. Chen, T. Li, X. Ye, Q. Luo and W. Lai, Food Chem., 2023, 429, 136816 CrossRef CAS PubMed.
- L. Cohen and D. R. Walt, Bioconjugate Chem., 2018, 29, 3452–3458 CrossRef CAS PubMed.
- I.-H. Cho, E.-H. Paek, H. Lee, J. Y. Kang, T. S. Kim and S.-H. Paek, Anal. Biochem., 2007, 365, 14–23 CrossRef CAS PubMed.
- E. von Witting, S. Hober and S. Kanje, Bioconjugate Chem., 2021, 32, 1515–1524 CrossRef CAS PubMed.
- J. M. Bolivar and B. Nidetzky, Biochim. Biophys. Acta, Proteins Proteomics, 2020, 1868, 140333 CrossRef CAS PubMed.
- W. Ma, A. Saccardo, D. Roccatano, D. Aboagye-Mensah, M. Alkaseem, M. Jewkes, F. Di Nezza, M. Baron, M. Soloviev and E. Ferrari, Nat. Commun., 2018, 9, 1489 CrossRef PubMed.
- A. H. Keeble, A. Banerjee, M. P. Ferla, S. C. Reddington, I. Anuar and M. Howarth, Angew. Chem., Int. Ed., 2017, 56, 16521–16525 CrossRef CAS PubMed.
- K. Guo, S. Wustoni, A. Koklu, E. Díaz-Galicia, M. Moser, A. Hama, A. A. Alqahtani, A. N. Ahmad, F. S. Alhamlan, M. Shuaib, A. Pain, I. McCulloch, S. T. Arold, R. Grünberg and S. Inal, Nat. Biomed. Eng., 2021, 5, 666–677 CrossRef CAS PubMed.
- G. P. Anderson, J. L. Liu, L. C. Shriver-Lake, D. Zabetakis, V. A. Sugiharto, H.-W. Chen, C.-R. Lee, G. N. Defang, S.-J. L. Wu, N. Venkateswaran and E. R. Goldman, Anal. Chem., 2019, 91, 9424–9429 CrossRef CAS PubMed.
- S. Kim, Y. Ahn, Y. Bae, S. Woo, J. Park, I. K. Han, H. Kim, S. Eom, S. Kang, W. Jung and J. Park, Biosens. Bioelectron., 2022, 213, 114441 CrossRef CAS PubMed.
- C. Albert, L. Bracaglia, A. Koide, J. DiRito, T. Lysyy, L. Harkins, C. Edwards, O. Richfield, J. Grundler and K. Zhou, Nat. Commun., 2022, 13, 5998 CrossRef CAS PubMed.
- N. G. Welch, J. A. Scoble, B. W. Muir and P. J. Pigram, Biointerphases, 2017, 12, 02D301 CrossRef PubMed.
- S. Y. Gan, G. J. Tye, A. L. Chew, W. K. Ng and N. S. Lai, Biosens. Bioelectron.: X, 2023, 14, 100379 CAS.
- S. Huang, W. Wang, J. Li, T. Zhang, Y. Liang, Q. Wang and Z. Jiang, Chem. Eng. J., 2021, 419, 129613 CrossRef CAS.
- X. Chen, Y. Huang, S. Yang, S. Wang, L. Chen, X. Yu, N. Gan and S. Huang, Biosens. Bioelectron., 2024, 266, 116738 CrossRef CAS PubMed.
- K. Alt, F. Carraro, E. Jap, M. Linares-Moreau, R. Riccò, M. Righetto, M. Bogar, H. Amenitsch, R. A. Hashad, C. Doonan, C. E. Hagemeyer and P. Falcaro, Adv. Mater., 2022, 34, 2106607 CrossRef CAS PubMed.
- Q. Zhang, J. Liang, A. Bongers, J. J. Richardson, K. Liang and Z. Gu, Adv. Sci., 2023, 10, 2206546 CrossRef CAS PubMed.
- G. Tian, Z. Zhou, M. Li, X. Li, T. Xu and X. Zhang, Anal. Chem., 2023, 95, 13250–13257 CrossRef CAS PubMed.
- Q.-Q. Zhuang, H.-H. Deng, S.-B. He, H.-P. Peng, Z. Lin, X.-H. Xia and W. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 31729–31734 CrossRef CAS PubMed.
- G. Hong, C. Su, Z. Huang, Q. Zhuang, C. Wei, H. Deng, W. Chen and H. Peng, Anal. Chem., 2021, 93, 13022–13028 CrossRef CAS PubMed.
- X. Chen, C. Su, Y. Yang, Z. Weng, Q. Zhuang, G. Hong, H. Peng and W. Chen, Anal. Chem., 2025, 97, 872–879 CrossRef CAS PubMed.
- H. Pei, X. Zuo, D. Pan, J. Shi, Q. Huang and C. Fan, NPG Asia Mater., 2013, 5, e51–e51 CrossRef CAS.
- B. Liu, Z. Huang and J. Liu, Angew. Chem., Int. Ed., 2018, 57, 9439–9442 CrossRef CAS PubMed.
- Q. He, Q. Wu, X. Feng, Z. Liao, W. Peng, Y. Liu, D. Peng, Z. Liu and M. Mo, Int. J. Biol. Macromol., 2020, 151, 757–780 CrossRef CAS PubMed.
- S. Stangherlin and J. Liu, J. Mater. Chem. B, 2023, 11, 6994–7003 RSC.
- X. Zhang, M. R. Servos and J. Liu, Langmuir, 2012, 28, 3896–3902 CrossRef CAS PubMed.
- B. Liu and J. Liu, Matter, 2019, 1, 825–847 CrossRef.
- J. I. Cutler, E. Auyeung and C. A. Mirkin, J. Am. Chem. Soc., 2012, 134, 1376–1391 CrossRef CAS PubMed.
- H. Kimura-Suda, D. Y. Petrovykh, M. J. Tarlov and L. J. Whitman, J. Am. Chem. Soc., 2003, 125, 9014–9015 CrossRef CAS PubMed.
- H. Pei, F. Li, Y. Wan, M. Wei, H. Liu, Y. Su, N. Chen, Q. Huang and C. Fan, J. Am. Chem. Soc., 2012, 134, 11876–11879 CrossRef CAS PubMed.
- D. Zhu, P. Song, J. Shen, S. Su, J. Chao, A. Aldalbahi, Z. Zhou, S. Song, C. Fan, X. Zuo, Y. Tian, L. Wang and H. Pei, Anal. Chem., 2016, 88, 4949–4954 CrossRef CAS PubMed.
- C.-Y. Lee, P. Gong, G. M. Harbers, D. W. Grainger, D. G. Castner and L. J. Gamble, Anal. Chem., 2006, 78, 3316–3325 CrossRef CAS PubMed.
- S. Choi and Y. S. Nam, Biosens. Bioelectron., 2022, 210, 114288 CrossRef CAS PubMed.
- A. Dillen, C. Scarpellini, W. Daenen, S. Driesen, P. Zijlstra and J. Lammertyn, ACS Sens., 2023, 8, 811–821 CrossRef CAS PubMed.
- R. Levicky, T. M. Herne, M. J. Tarlov and S. K. Satija, J. Am. Chem. Soc., 1998, 120, 9787–9792 CrossRef CAS.
- P. Liang, J. Canoura, H. Yu, O. Alkhamis and Y. Xiao, ACS Appl. Mater. Interfaces, 2018, 10, 4233–4242 CrossRef CAS PubMed.
- B. Liu, P. Wu, Z. Huang, L. Ma and J. Liu, J. Am. Chem. Soc., 2018, 140, 4499–4502 CrossRef CAS PubMed.
- T. L. Halo, K. M. McMahon, N. L. Angeloni, Y. Xu, W. Wang, A. B. Chinen, D. Malin, E. Strekalova, V. L. Cryns, C. Cheng, C. A. Mirkin and C. S. Thaxton, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 17104–17109 CrossRef CAS PubMed.
- B. Liu, T. Wu, Z. Huang, Y. Liu and J. Liu, Angew. Chem., Int. Ed., 2019, 58, 2109–2113 CrossRef CAS PubMed.
- B. Liu and J. Liu, Langmuir, 2019, 35, 6476–6482 CrossRef CAS PubMed.
- Z. Li, Y. Lv, X. Duan, B. Liu and Y. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202312975 CrossRef CAS PubMed.
- S. Shi, J. Chen, X. Wang, M. Xiao, A. R. Chandrasekaran, L. Li, C. Yi and H. Pei, Adv. Funct. Mater., 2022, 32, 2201069 CrossRef CAS.
- J. Su, W. Liu, S. Chen, W. Deng, Y. Dou, Z. Zhao, J. Li, Z. Li, H. Yin, X. Ding and S. Song, ACS Sens., 2020, 5, 3979–3987 CrossRef CAS PubMed.
- F. Li, X. Mao, F. Li, M. Li, J. Shen, Z. Ge, C. Fan and X. Zuo, J. Am. Chem. Soc., 2020, 142, 9975–9981 CrossRef CAS PubMed.
- M. Li, L. Song, M. Liu, R. Guo, M. Lin and X. Zuo, ACS Nano, 2025, 19, 23142–23150 CrossRef CAS PubMed.
- R. T. Busch, F. Karim, J. Weis, Y. Sun, C. Zhao and E. S. Vasquez, ACS Omega, 2019, 4, 15269–15279 CrossRef CAS PubMed.
- D. A. Cowan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2011, 49, 326–346 CrossRef CAS PubMed.
- P. Ghisellini, M. Caiazzo, A. Alessandrini, R. Eggenhöffner, M. Vassalli and P. Facci, Sci. Rep., 2016, 6, 37779 CrossRef CAS PubMed.
- H. J. Kim, D. Park, Y. Park, D.-H. Kim and J. Kim, Nano Lett., 2022, 22, 6537–6544 CrossRef CAS PubMed.
- E. A. Josephs and T. Ye, Nano Lett., 2012, 12, 5255–5261 CrossRef CAS PubMed.
- K. Abstiens, M. Gregoritza and A. M. Goepferich, ACS Appl. Mater. Interfaces, 2019, 11, 1311–1320 CrossRef CAS PubMed.
- Y. Cao, J. Wu, X. Zheng, Y. Lu, J. A. Piper, Y. Lu and N. H. Packer, Anal. Chim. Acta, 2022, 1209, 339863 CrossRef CAS PubMed.
- B. Saha, T. H. Evers and M. W. J. Prins, Anal. Chem., 2014, 86, 8158–8166 CrossRef CAS PubMed.
- X. Chen, F. Lisi, P. Bakthavathsalam, G. Longatte, S. Hoque, R. D. Tilley and J. J. Gooding, ACS Sens., 2021, 6, 538–545 CrossRef CAS PubMed.
- J. Zhang, F. Chai, J. A. Li, S. Wang, S. Zhang, F. Li, A. Liang, A. Luo, D. Wang and X. Jiang, Sci. Adv., 2024, 10, eadn5698 CrossRef CAS PubMed.
- X. Qu, D. Zhu, G. Yao, S. Su, J. Chao, H. Liu, X. Zuo, L. Wang, J. Shi, L. Wang, W. Huang, H. Pei and C. Fan, Angew. Chem., Int. Ed., 2017, 56, 1855–1858 CrossRef CAS PubMed.
- J. Li, B. Zhu, X. Yao, Y. Zhang, Z. Zhu, S. Tu, S. Jia, R. Liu, H. Kang and C. J. Yang, ACS Appl. Mater. Interfaces, 2014, 6, 16800–16807 CrossRef CAS PubMed.
- G. Hirao, N. Fukuzumi, A. Ogawa, T. Asahi, M. Mizuo and T. Zako, RSC Adv., 2023, 13, 30690–30695 RSC.
- Y. Tanaka, G. Hirao, N. Fukuzumi, T. Asahi, M. Maeda, A. Ogawa and T. Zako, Langmuir, 2025, 41, 4560–4568 CrossRef CAS PubMed.
- N. Fukuzumi, G. Hirao, A. Ogawa, T. Asahi, M. Maeda and T. Zako, Sci. Rep., 2025, 15, 8222 CrossRef CAS PubMed.
- G. Wang, Z. Li and N. Ma, ACS Chem. Bio., 2018, 13, 1705–1713 CrossRef CAS PubMed.
- M. P. Busson, B. Rolly, B. Stout, N. Bonod, E. Larquet, A. Polman and S. Bidault, Nano Lett., 2011, 11, 5060–5065 CrossRef CAS PubMed.
- M.-X. Li, C.-H. Xu, N. Zhang, G.-S. Qian, W. Zhao, J.-J. Xu and H.-Y. Chen, ACS Nano, 2018, 12, 3341–3350 CrossRef CAS PubMed.
- H. Xing, Y. Bai, Y. Bai, L. H. Tan, J. Tao, B. Pedretti, G. A. Vincil, Y. Lu and S. C. Zimmerman, J. Am. Chem. Soc., 2017, 139, 3623–3626 CrossRef CAS PubMed.
- M. Colombo, L. Fiandra, G. Alessio, S. Mazzucchelli, M. Nebuloni, C. De Palma, K. Kantner, B. Pelaz, R. Rotem and F. Corsi, Nat. Commun., 2016, 7, 1–14 Search PubMed.
- H. Wang, Y. Li, M. Liu, M. Gong and Z. Deng, Small, 2015, 11, 2247–2251 CrossRef CAS PubMed.
- N. T. Emerson and H. Yang, J. Am. Chem. Soc., 2022, 144, 12915–12923 CrossRef CAS PubMed.
- D. Zanchet, C. M. Micheel, W. J. Parak, D. Gerion and A. P. Alivisatos, Nano Lett., 2001, 1, 32–35 CrossRef CAS.
- S. E. Lee, Q. Chen, R. Bhat, S. Petkiewicz, J. M. Smith, V. E. Ferry, A. L. Correia, A. P. Alivisatos and M. J. Bissell, Nano Lett., 2015, 15, 4564–4570 CrossRef CAS PubMed.
- G. Tikhomirov, S. Hoogland, P. E. Lee, A. Fischer, E. H. Sargent and S. O. Kelley, Nat. Nanotechnol., 2011, 6, 485–490 CrossRef CAS PubMed.
- N. Ma, E. H. Sargent and S. O. Kelley, Nat. Nanotechnol., 2009, 4, 121–125 CrossRef CAS PubMed.
- X. He, Z. Li, M. Chen and N. Ma, Angew. Chem., Int. Ed., 2014, 53, 14447–14450 CrossRef CAS PubMed.
- J. Shen, Q. Tang, L. Li, J. Li, X. Zuo, X. Qu, H. Pei, L. Wang and C. Fan, Angew. Chem., Int. Ed., 2017, 56, 16077–16081 CrossRef CAS PubMed.
- G. Yao, J. Li, Q. Li, X. Chen, X. Liu, F. Wang, Z. Qu, Z. Ge, R. P. Narayanan, D. Williams, H. Pei, X. Zuo, L. Wang, H. Yan, B. L. Feringa and C. Fan, Nat. Mater., 2020, 19, 781–788 CrossRef CAS PubMed.
- G. Yao, H. Pei, J. Li, Y. Zhao, D. Zhu, Y. Zhang, Y. Lin, Q. Huang and C. Fan, NPG Asia Mater., 2015, 7, e159 CrossRef.
- X. Chen, X. Liu, G. Yao, Q. Li, R. Liu, H. Wu, Y. Lv, C. Fan, L. Wang and J. Li, NPG Asia Mater., 2020, 12, 49 CrossRef CAS.
- D. Fan, J. Wang, E. Wang and S. Dong, Adv. Sci., 2020, 7, 2001766 CrossRef CAS PubMed.
- L. Yu and H. Yan, Nat. Biomed. Eng., 2023, 7, 1535–1536 CrossRef CAS PubMed.
- K. B. Johnsen, M. Bak, F. Melander, M. S. Thomsen, A. Burkhart, P. J. Kempen, T. L. Andresen and T. Moos, J. Controlled Release, 2019, 295, 237–249 CrossRef CAS PubMed.
- D. J. Nieves, N. S. Azmi, R. Xu, R. Lévy, E. A. Yates and D. G. Fernig, Chem. Commun., 2014, 50, 13157–13160 RSC.
- H. Choi and Y. Jung, Chem. Sci., 2022, 13, 7552–7559 RSC.
- O. Zeiri, ACS Sens., 2020, 5, 3806–3820 CrossRef CAS PubMed.
- N. Dridi, Z. Jin, W. Perng and H. Mattoussi, ACS Nano, 2024, 18, 8649–8662 CrossRef CAS PubMed.
- M. Zheng and X. Huang, J. Am. Chem. Soc., 2004, 126, 12047–12054 CrossRef CAS PubMed.
- J. Baggerman, M. M. J. Smulders and H. Zuilhof, Langmuir, 2019, 35, 1072–1084 CrossRef CAS PubMed.
- S. Lowe, N. M. O'Brien-Simpson and L. A. Connal, Polym. Chem., 2015, 6, 198–212 RSC.
- C. Jiang, G. Wang, R. Hein, N. Liu, X. Luo and J. J. Davis, Chem. Rev., 2020, 120, 3852–3889 CrossRef CAS PubMed.
- A. K. Nowinski, F. Sun, A. D. White, A. J. Keefe and S. Jiang, J. Am. Chem. Soc., 2012, 134, 6000–6005 CrossRef CAS PubMed.
- C. Sanchez-Cano and M. Carril, Int. J. Mol. Sci., 2020, 21, 1007 CrossRef CAS PubMed.
- C. Guo, H. Ding, M. Xie, H. Zhang, X. Hong, L. Sun and F. Ding, Colloids Surf., A, 2021, 615, 126155 CrossRef CAS.
- Q. Dai, C. Walkey and W. C. Chan, Angew. Chem., Int. Ed., 2014, 53, 5093–5096 CrossRef CAS PubMed.
- M. Chen, R. Han, W. Wang, Y. Li and X. Luo, Anal. Chem., 2021, 93, 13555–13563 CrossRef CAS PubMed.
- S. Zhao, X. Qiao, M. Chen, Y. Li, X. Wang, Z. Xu, Y. Wu and X. Luo, ACS Sens., 2022, 7, 1740–1746 CrossRef CAS PubMed.
- R. Han, Y. Li, W. Hou, C. Ding and X. Luo, Anal. Chem., 2023, 95, 9025–9033 CrossRef CAS PubMed.
- R. Han, Y. Li, W. Wang, C. Ding, J. J. Davis and X. Luo, Anal. Chem., 2024, 96, 15511–15517 CAS.
- Z. Zhang, Y. Wen, Y. Ma, J. Luo, L. Jiang and Y. Song, Chem. Commun., 2011, 47, 7407–7409 RSC.
- L. Xu, W. Yan, W. Ma, H. Kuang, X. Wu, L. Liu, Y. Zhao, L. Wang and C. Xu, Adv. Mater., 2015, 27, 1706–1711 CrossRef CAS PubMed.
- H. D. Hill and C. A. Mirkin, Nat. Protoc., 2006, 1, 324–336 CrossRef CAS PubMed.
- Y. Wang, X. Liu, L. Wu, L. Ding, C. Y. Effah, Y. Wu, Y. Xiong and L. He, Biosens. Bioelectron., 2022, 195, 113661 CrossRef CAS PubMed.
- Q. Zhang, R. Ma, Y. Zhang, J. Zhao, Y. Wang and Z. Xu, ACS Sens., 2023, 8, 875–883 CrossRef CAS PubMed.
- C. Li, S. Yang, R. Li, S. Gong, M. Huang, Y. Sun, G. Xiong, D. Wu, M. Ji, Y. Chen, C. Gao and Y. Yu, ACS Appl. Mater. Interfaces, 2022, 14, 7646–7658 CrossRef CAS PubMed.
- H. Shen, X. Zheng, Z. Zhou, W. He, M. Li, P. Su, J. Song and Y. Yang, J. Mater. Chem. B, 2020, 8, 8467–8475 RSC.
- G.-B. Chu, W.-Y. Li, X.-X. Han, H.-H. Sun, Y. Han, G.-Y. Zhi and D.-H. Zhang, Small, 2023, 19, 2301413 CrossRef CAS PubMed.
- Y. Zhang, L. Xu and J. Ge, Nano Lett., 2022, 22, 5029–5036 CrossRef CAS PubMed.
- T. Man, C. Xu, X.-Y. Liu, D. Li, C.-K. Tsung, H. Pei, Y. Wan and L. Li, Nat. Commun., 2022, 13, 305 CrossRef CAS PubMed.
- L. Wang, Y. Zhu, L. Xu, W. Chen, H. Kuang, L. Liu, A. Agarwal, C. Xu and N. A. Kotov, Angew. Chem., Int. Ed., 2010, 49, 5472–5475 CrossRef CAS PubMed.
- L. Xu, H. Kuang, C. Xu, W. Ma, L. Wang and N. A. Kotov, J. Am. Chem. Soc., 2012, 134, 1699–1709 CrossRef CAS PubMed.
- S.-Y. Ding, J. Yi, J.-F. Li, B. Ren, D.-Y. Wu, R. Panneerselvam and Z.-Q. Tian, Nat. Rev. Mater., 2016, 1, 16021 CrossRef CAS.
- R. Pardehkhorram, S. Bonaccorsi, H. Zhu, V. R. Gonçales, Y. Wu, J. Liu, N. A. Lee, R. D. Tilley and J. J. Gooding, Chem. Commun., 2019, 55, 7707–7710 RSC.
- R. Pardehkhorram, F. A. Alshawawreh, V. R. Gonçales, N. A. Lee, R. D. Tilley and J. J. Gooding, Anal. Chem., 2021, 93, 12954–12965 CrossRef CAS PubMed.
- G. Wang, Y. Akiyama, N. Kanayama, T. Takarada and M. Maeda, Small, 2017, 13, 1702137 CrossRef PubMed.
- E. E. Coughlin, J. Hu, A. Lee and T. W. Odom, J. Am. Chem. Soc., 2021, 143, 3671–3676 CrossRef CAS PubMed.
- Z. Yang, X. Peng, P. Yang, Y. Zhuo, Y.-Q. Chai, W. Liang and R. Yuan, Chem. Sci., 2020, 11, 8482–8488 RSC.
- A. Escarpa and B. Jurado-Sánchez, Anal. Chem., 2025, 97, 12913–12924 CrossRef CAS PubMed.
- M. Pacheco, B. Jurado-Sánchez and A. Escarpa, Anal. Chem., 2018, 90, 2912–2917 CrossRef CAS PubMed.
- K. Yuan, C. Cuntín-Abal, B. Jurado-Sánchez and A. Escarpa, Anal. Chem., 2021, 93, 16385–16392 CrossRef CAS PubMed.
- K. Yuan, B. Jurado-Sánchez and A. Escarpa, Angew. Chem., Int. Ed., 2021, 60, 4915–4924 CrossRef CAS PubMed.
- X. Liu, F. Zhang, X. Jing, M. Pan, P. Liu, W. Li, B. Zhu, J. Li, H. Chen, L. Wang, J. Lin, Y. Liu, D. Zhao, H. Yan and C. Fan, Nature, 2018, 559, 593–598 CrossRef CAS PubMed.
- S. Wang, Z. Zhou, N. Ma, S. Yang, K. Li, C. Teng, Y. Ke and Y. Tian, Sensors, 2020, 20, 6899 CrossRef CAS PubMed.
- S. Fu, T. Zhang, H. Jiang, Y. Xu, J. Chen, L. Zhang and X. Su, TrAC, Trends Anal. Chem., 2021, 140, 116267 CrossRef CAS.
- Q. Liu, Z. Ge, X. Mao, G. Zhou, X. Zuo, J. Shen, J. Shi, J. Li, L. Wang, X. Chen and C. Fan, Angew. Chem., Int. Ed., 2018, 57, 7131–7135 CrossRef CAS PubMed.
- C. Chen, X. Wei, M. F. Parsons, J. Guo, J. L. Banal, Y. Zhao, M. N. Scott, G. S. Schlau-Cohen, R. Hernandez and M. Bathe, Nat. Commun., 2022, 13, 4935 CrossRef CAS PubMed.
- N. Chauhan, Y. Xiong, S. Ren, A. Dwivedy, N. Magazine, L. Zhou, X. Jin, T. Zhang, B. T. Cunningham, S. Yao, W. Huang and X. Wang, J. Am. Chem. Soc., 2023, 145, 20214–20228 CrossRef CAS PubMed.
- X. Hu, G. Ke, L. Liu, X. Fu, G. Kong, M. Xiong, M. Chen and X.-B. Zhang, Anal. Chem., 2019, 91, 11374–11379 CrossRef CAS PubMed.
- M. Mao, Z. Lin, L. Chen, Z. Zou, J. Zhang, Q. Dou, J. Wu, J. Chen, M. Wu, L. Niu, C. Fan and Y. Zhang, J. Am. Chem. Soc., 2023, 145, 5447–5455 CrossRef CAS PubMed.
- X. Hu, H. Chi, X. Fu, J. Chen, L. Dong, S. Jiang, Y. Li, J. Chen, M. Cheng, Q. Min, Y. Tian and P. Zhang, J. Am. Chem. Soc., 2024, 146, 2514–2523 CrossRef CAS PubMed.
- T. G. W. Edwardson, K. L. Lau, D. Bousmail, C. J. Serpell and H. F. Sleiman, Nat. Chem., 2016, 8, 162–170 CrossRef CAS PubMed.
- N. Xie, S. Liu, H. Fang, Y. Yang, K. Quan, J. Li, X. Yang, K. Wang and J. Huang, ACS Nano, 2019, 13, 4174–4182 CrossRef CAS PubMed.
- R. Niu, C. Song, F. Gao, W. Fang, X. Jiang, S. Ren, D. Zhu, S. Su, J. Chao, S. Chen, C. Fan and L. Wang, Angew. Chem., Int. Ed., 2021, 60, 11695–11701 CrossRef CAS PubMed.
- M. Senoner and W. E. S. Unger, J. Anal. At. Spectrom., 2012, 27, 1050–1068 RSC.
- Y.-P. Kim, H. K. Shon, S. K. Shin and T. G. Lee, Mass Spectrom. Rev., 2015, 34, 237–247 CrossRef CAS PubMed.
- K. Gajos, K. Szafraniec, P. Petrou and A. Budkowski, Appl. Surf. Sci., 2020, 518, 146269 CrossRef CAS.
- K. Gajos, P. Petrou and A. Budkowski, Molecules, 2022, 27, 3672 CrossRef CAS PubMed.
- K. Gajos, K. Sanocka, M. Wytrwał, P. Dąbczyński and A. Budkowski, Appl. Surf. Sci., 2024, 656, 159644 CrossRef CAS.
- R. Riera, E. Archontakis, G. Cremers, T. de Greef, P. Zijlstra and L. Albertazzi, ACS Sens., 2023, 8, 80–93 CrossRef CAS PubMed.
- M. M. E. Tholen, B. J. H. M. Rosier, R. T. Vermathen, C. A. N. Sewnath, C. Storm, L. Woythe, C. Izquierdo-Lozano, R. Riera, M. van Egmond, M. Merkx and L. Albertazzi, ACS Nano, 2023, 17, 11665–11678 CrossRef CAS PubMed.
- H. Son, J. Park, I. Hwang, Y. Jung, S. Bae and S. Lee, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2113747118 CrossRef CAS PubMed.
- J. Guo, X. Qiu, C. Mingoes, J. R. Deschamps, K. Susumu, I. L. Medintz and N. Hildebrandt, ACS Nano, 2019, 13, 505–514 CrossRef CAS PubMed.
- C. Léger, A. Yahia-Ammar, K. Susumu, I. L. Medintz, A. Urvoas, M. Valerio-Lepiniec, P. Minard and N. Hildebrandt, ACS Nano, 2020, 14, 5956–5967 CrossRef PubMed.
- J. Guo, Y. Chen, Y. Jiang and H. Ju, Chem. – Eur. J., 2017, 23, 9332–9337 CrossRef CAS PubMed.
- K. Tapio, D. Shao, S. Auer, J. Tuppurainen, M. Ahlskog, V. P. Hytönen and J. J. Toppari, Nanoscale, 2018, 10, 19297–19309 RSC.
- C. H. Olk, J. Heremans, P. S. Lee, D. Dziedzic and N. E. Sargent, J. Vac. Sci. Technol., B, 1991, 9, 1268–1271 CrossRef CAS.
- D. A. Yarotski, S. V. Kilina, A. A. Talin, S. Tretiak, O. V. Prezhdo, A. V. Balatsky and A. J. Taylor, Nano Lett., 2009, 9, 12–17 CrossRef CAS PubMed.
- J. Hu, M. Gao, Z. Wang, Y. Chen, Z. Song and H. Xu, Appl. Nanosci., 2021, 11, 293–300 CrossRef CAS PubMed.
- O. Ouerghi, A. Touhami, A. Othmane, H. Ben Ouada, C. Martelet, C. Fretigny and N. Jaffrezic-Renault, Sens. Actuators, B, 2002, 84, 167–175 CrossRef CAS.
- G. R. Heath and S. Scheuring, Nat. Commun., 2018, 9, 4983 CrossRef PubMed.
- P. Zhang, X. Liu, P. Liu, F. Wang, H. Ariyama, T. Ando, J. Lin, L. Wang, J. Hu and B. Li, Nat. Commun., 2020, 11, 1–9 Search PubMed.
- H. S. N. Jayawardena, S. H. Liyanage, K. Rathnayake, U. Patel and M. Yan, Anal. Chem., 2021, 93, 1889–1911 CrossRef CAS PubMed.
- A. Rodríguez-Galván and F. F. Contreras-Torres, Nanomaterials, 2022, 12, 3013 CrossRef PubMed.
- H. Yu, X. Xu, P. Liang, K. Y. Loh, B. Guntupalli, D. Roncancio and Y. Xiao, Bioconjugate Chem., 2017, 28, 933–943 CrossRef CAS PubMed.
- B. Liu and J. Liu, Anal. Methods, 2017, 9, 2633–2643 RSC.
- S. J. Hurst, A. K. R. Lytton-Jean and C. A. Mirkin, Anal. Chem., 2006, 78, 8313–8318 CrossRef CAS PubMed.
- Y. Zu and Z. Gao, Anal. Chem., 2009, 81, 8523–8528 CrossRef CAS PubMed.
- D. Bouzas-Ramos, J. I. García-Alonso, J. M. Costa-Fernández and J. Ruiz Encinar, Anal. Chem., 2019, 91, 3567–3574 CrossRef CAS PubMed.
- Q. Wei, T. Becherer, S. Angioletti-Uberti, J. Dzubiella, C. Wischke, A. T. Neffe, A. Lendlein, M. Ballauff and R. Haag, Angew. Chem., Int. Ed., 2014, 53, 8004–8031 CrossRef CAS PubMed.
- R. Oliverio, B. Liberelle, F. Murschel, A. Garcia-Ac, X. Banquy and G. De Crescenzo, ACS Appl. Nano Mater., 2020, 3, 10497–10507 CrossRef CAS.
- B. L. Baldock and J. E. Hutchison, Anal. Chem., 2016, 88, 12072–12080 CrossRef CAS PubMed.
- R. Kozlowski, A. Ragupathi and R. B. Dyer, Bioconjugate Chem., 2018, 29, 2691–2700 CrossRef CAS PubMed.
- M. J. Eller, K. Chandra, E. E. Coughlin, T. W. Odom and E. A. Schweikert, Anal. Chem., 2019, 91, 5566–5572 CrossRef CAS PubMed.
- M. Horáček, D. J. Engels and P. Zijlstra, Nanoscale, 2020, 12, 4128–4136 RSC.
- P. Delcanale, B. Miret-Ontiveros, M. Arista-Romero, S. Pujals and L. Albertazzi, ACS Nano, 2018, 12, 7629–7637 CrossRef CAS PubMed.
- Q. Ong, Z. Luo and F. Stellacci, Acc. Chem. Res., 2017, 50, 1911–1919 CrossRef CAS PubMed.
- Y. Randika Perera, R. A. Hill and N. C. Fitzkee, Isr. J. Chem., 2019, 59, 962–979 CrossRef CAS PubMed.
- Y. Wang, P. E. D. Soto Rodriguez, L. Woythe, S. Sánchez, J. Samitier, P. Zijlstra and L. Albertazzi, ACS Appl. Mater. Interfaces, 2022, 14, 37345–37355 CrossRef CAS PubMed.
- L. Cerofolini, E. Ravera, C. Fischer, A. Trovato, F. Sacco, W. Palinsky, G. Angiuoni, M. Fragai and F. Baroni, Anal. Chem., 2023, 95, 9199–9206 CrossRef CAS PubMed.
- L. Poppe, J. B. Jordan, K. Lawson, M. Jerums, I. Apostol and P. D. Schnier, Anal. Chem., 2013, 85, 9623–9629 CrossRef CAS PubMed.
- M. Wang, C. Fu, X. Liu, Z. Lin, N. Yang and S. Yu, Nanoscale, 2015, 7, 15191–15196 RSC.
- M. Maddahfar, S. Wen, S. M. Hosseinpour Mashkani, L. Zhang, O. Shimoni, M. Stenzel, J. Zhou, B. Fazekas de St Groth and D. Jin, Bioconjugate Chem., 2021, 32, 1146–1155 CrossRef CAS PubMed.
- A. Derenne, K.-M. Derfoufi, B. Cowper, C. Delporte and E. Goormaghtigh, Anal. Chim. Acta, 2020, 1112, 62–71 CrossRef CAS PubMed.
- U. Böcker, S. G. Wubshet, D. Lindberg and N. K. Afseth, Analyst, 2017, 142, 2812–2818 RSC.
- F. Faghihzadeh, N. M. Anaya, L. A. Schifman and V. Oyanedel-Craver, Nanotechnol. Environ. Eng., 2016, 1, 1 CrossRef.
- L.-J. Chen, S. S. Shah, J. Silangcruz, M. J. Eller, S. V. Verkhoturov, A. Revzin and E. A. Schweikert, Int. J. Mass Spectrom., 2011, 303, 97–102 CrossRef CAS PubMed.
- K. Gajos, O. Lishchynskyi, P. D
![[a with combining cedilla]](https://www.rsc.org/images/entities/char_0061_0327.gif) bczyński, S. Tymetska, Ł. Bodek, Y. Shymborska, N. Janiszewska, Y. Stetsyshyn and A. Budkowski, ACS Appl. Mater. Interfaces, 2026, 18, 3122–3139 CAS.
bczyński, S. Tymetska, Ł. Bodek, Y. Shymborska, N. Janiszewska, Y. Stetsyshyn and A. Budkowski, ACS Appl. Mater. Interfaces, 2026, 18, 3122–3139 CAS. - W. S. Tan, A. M. de Jong and M. W. J. Prins, Langmuir, 2025, 41, 22181–22192 CrossRef CAS PubMed.
- Y. Zhai, J. S. DuChene, Y.-C. Wang, J. Qiu, A. C. Johnston-Peck, B. You, W. Guo, B. DiCiaccio, K. Qian, E. W. Zhao, F. Ooi, D. Hu, D. Su, E. A. Stach, Z. Zhu and W. D. Wei, Nat. Mater., 2016, 15, 889–895 CrossRef CAS PubMed.
- W. S. Tan, A. M. de Jong and M. W. J. Prins, ACS Appl. Mater. Interfaces, 2024, 16, 58191–58202 CrossRef CAS PubMed.
- D. S. Verkhoturov, M. J. Eller, Y. D. Han, B. Crulhas, S. V. Verkhoturov, A. Revzin and E. A. Schweikert, Langmuir, 2022, 38, 5626–5632 CrossRef CAS PubMed.
- B. L. Gorman and M. L. Kraft, Anal. Chem., 2020, 92, 1645–1652 CrossRef CAS PubMed.
- Y. Jiang, D. Salley, A. Sharma, G. Keenan, M. Mullin and L. Cronin, Sci. Adv., 2022, 8, eabo2626 CrossRef CAS PubMed.
- C. Li, L. Ma, Z. Xue, X. Li, S. Zhu and T. Wang, Adv. Sci., 2025, 12, 2501000 CrossRef CAS PubMed.
- N. Shirokii, Y. Din, I. Petrov, Y. Seregin, S. Sirotenko, J. Razlivina, N. Serov and V. Vinogradov, Small, 2023, 19, 2207106 CrossRef CAS PubMed.
- Z. Meng and K. Xia, Sci. Adv., 2021, 7, eabc5329 CrossRef CAS PubMed.
- N. Serov and V. Vinogradov, Adv. Drug Delivery Rev., 2022, 184, 114194 CrossRef CAS PubMed.
- F. Cui, Y. Yue, Y. Zhang, Z. Zhang and H. S. Zhou, ACS Sens., 2020, 5, 3346–3364 CrossRef CAS PubMed.
- M. Bhaiyya, D. Panigrahi, P. Rewatkar and H. Haick, ACS Sens., 2024, 9, 4495–4519 CrossRef CAS PubMed.
- S. Han, J. Yea, S. Song, S. Lee, J.-W. Choi, Y. K. Lee, J. Park and K.-I. Jang, Device, 2026, 4, 101017 CrossRef.
- T. Akkaş, M. Reshadsedghi, M. Şen, V. Kılıç and N. Horzum, Adv. Mater., 2025, 37, 2504796 CrossRef PubMed.
- J. Huzar, R. Coreas, M. P. Landry and G. Tikhomirov, ACS Nano, 2025, 19, 4333–4345 CrossRef CAS PubMed.
- K. T. Butler, D. W. Davies, H. Cartwright, O. Isayev and A. Walsh, Nature, 2018, 559, 547–555 CrossRef CAS PubMed.
- E. Wyrzykowska, A. Mikolajczyk, I. Lynch, N. Jeliazkova, N. Kochev, H. Sarimveis, P. Doganis, P. Karatzas, A. Afantitis, G. Melagraki, A. Serra, D. Greco, J. Subbotina, V. Lobaskin, M. A. Bañares, E. Valsami-Jones, K. Jagiello and T. Puzyn, Nat. Nanotechnol., 2022, 17, 924–932 CrossRef CAS PubMed.
- A. K. Chew, J. A. Pedersen and R. C. Van Lehn, ACS Nano, 2022, 16, 6282–6292 CrossRef CAS PubMed.
- Y. Duan, R. Coreas, Y. Liu, D. Bitounis, Z. Zhang, D. Parviz, M. Strano, P. Demokritou and W. Zhong, NanoImpact, 2020, 17, 100207 CrossRef PubMed.
- D. P. Russo, X. Yan, S. Shende, H. Huang, B. Yan and H. Zhu, Anal. Chem., 2020, 92, 13971–13979 CrossRef CAS PubMed.
- X. Yan, A. Sedykh, W. Wang, B. Yan and H. Zhu, Nat. Commun., 2020, 11, 2519 CrossRef CAS PubMed.
- X. Yan, T. Yue, D. A. Winkler, Y. Yin, H. Zhu, G. Jiang and B. Yan, Chem. Rev., 2023, 123, 8575–8637 CrossRef CAS PubMed.
- Y. Rong, A. V. Padron, K. J. Hagerty, N. Nelson, S. Chi, N. O. Keyhani, J. Katz, S. P. A. Datta, C. Gomes and E. S. McLamore, Analyst, 2018, 143, 2066–2075 RSC.
- H. Chen, H. Liu, L. Xing, D. Fan, N. Chen, P. Ma and X. Zhang, ACS Sens., 2025, 10, 2872–2882 CrossRef CAS PubMed.
- A. Bocan, R. Siavash Moakhar, C. del Real Mata, M. Petkun, T. De Iure-Grimmel, S. G. Yedire, H. Shieh, A. Khorrami Jahromi, S. S. Mahshid and S. Mahshid, Adv. Mater., 2025, 37, 2417520 CrossRef CAS PubMed.
- T. Wang, D. P. Russo, P. Demokritou, X. Jia, H. Huang, X. Yang and H. Zhu, Nano Lett., 2024, 24, 10228–10236 CrossRef CAS PubMed.
- F. Zivic, A. K. Malisic, N. Grujovic, B. Stojanovic and M. Ivanovic, Mater. Today Commun., 2025, 48, 113525 CrossRef CAS.
- J. Ackermann, J. T. Metternich, S. Herbertz and S. Kruss, Angew. Chem. Int. Ed., 2022, 61, e202112372 CrossRef CAS PubMed.
- Y. Yang, M. Zheng and A. Jagota, npj Comput. Mater., 2019, 5, 3 CrossRef.
- P. Kelich, S. Jeong, N. Navarro, J. Adams, X. Sun, H. Zhao, M. P. Landry and L. Vuković, ACS Nano, 2022, 16, 736–745 CrossRef CAS PubMed.
- P. Kelich, J. Adams, S. Jeong, N. Navarro, M. P. Landry and L. Vuković, J. Chem. Inf. Model., 2024, 64, 3992–4001 CrossRef CAS PubMed.
- M. Kim, C. Chen, P. Wang, J. J. Mulvey, Y. Yang, C. Wun, M. Antman-Passig, H.-B. Luo, S. Cho, K. Long-Roche, L. V. Ramanathan, A. Jagota, M. Zheng, Y. Wang and D. A. Heller, Nat. Biomed. Eng., 2022, 6, 267–275 CrossRef CAS PubMed.
- A. T. Krasley, S. Chakraborty, L. Vuković and A. G. Beyene, ACS Nano, 2025, 19, 7804–7820 CrossRef CAS PubMed.
- S. Chakraborty, A. T. Krasley, C. H. Smith, A. G. Beyene and L. Vuković, Nano Lett., 2026, 26, 787–794 CrossRef CAS PubMed.
- C. Tian, S. Lee, S. Park, Y. Cho, C. Baek and S.-Y. Cho, ACS Nano, 2025, 19, 23992–24004 CrossRef CAS PubMed.
- S. Ranallo, A. Amodio, A. Idili, A. Porchetta and F. Ricci, Chem. Sci., 2016, 7, 66–71 RSC.
- D. Roy and J. W. Park, J. Mater. Chem. B, 2015, 3, 5135–5149 RSC.
- J. J. Gooding, ACS Sens., 2023, 8, 1–2 CrossRef CAS PubMed.
- K. E. Sapsford, K. M. Tyner, B. J. Dair, J. R. Deschamps and I. L. Medintz, Anal. Chem., 2011, 83, 4453–4488 CrossRef CAS PubMed.
- S. Buckhout-White, M. Ancona, E. Oh, J. R. Deschamps, M. H. Stewart, J. B. Blanco-Canosa, P. E. Dawson, E. R. Goldman and I. L. Medintz, ACS Nano, 2012, 6, 1026–1043 CrossRef CAS PubMed.
- S. Mourdikoudis, R. M. Pallares and N. T. K. Thanh, Nanoscale, 2018, 10, 12871–12934 RSC.
- P. Zhang, G. Ma, W. Dong, Z. Wan, S. Wang and N. Tao, Nat. Methods, 2020, 17, 1010–1017 CrossRef CAS PubMed.
- S. Naudi-Fabra, M. Tengo, M. R. Jensen, M. Blackledge and S. Milles, J. Am. Chem. Soc., 2021, 143, 20109–20121 CrossRef CAS PubMed.
- G.-N. W. Gomes, M. Krzeminski, A. Namini, E. W. Martin, T. Mittag, T. Head-Gordon, J. D. Forman-Kay and C. C. Gradinaru, J. Am. Chem. Soc., 2020, 142, 15697–15710 CrossRef CAS PubMed.
- S. Dhiman, T. Andrian, B. S. Gonzalez, M. M. E. Tholen, Y. Wang and L. Albertazzi, Chem. Sci., 2022, 13, 2152–2166 RSC.
- X. Xu, L. Wang, Y. Huang, W. Gao, K. Li and W. Jiang, Anal. Chem., 2016, 88, 9885–9889 CrossRef CAS PubMed.
- X. Sun, L. Riccardi, F. De Biasi, F. Rastrelli, M. De Vivo and F. Mancin, Angew. Chem., Int. Ed., 2019, 58, 7702–7707 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |