
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optical probes of hot carriers in halide perovskites
Zengshan
Xing†
 ,
Rui
Cai†
and
Tze Chien
Sum
*
,
Rui
Cai†
and
Tze Chien
Sum
*
Division of Physics and Applied Physics, School of Physical and Mathematical Sciences, Nanyang Technological University, 21 Nanyang Link, Singapore 637371, Singapore. E-mail: Tzechien@ntu.edu.sg
First published on 12th March 2026
Abstract
The unusual slow cooling of hot carriers (HCs) in halide perovskites presents a promising route to harvest the excess carrier energy and exceed the Shockley–Queisser (SQ) efficiency limit of single-junction perovskite solar cells. Spectroscopic interrogation of HC dynamics is a crucial step toward an in-depth understanding of the HC properties and underlying cooling mechanisms in these materials. A range of spectroscopy techniques – most notably pump–probe (or transient absorption (TA)) spectroscopy, pump–push–probe spectroscopy, steady-state photoluminescence (PL) and time-resolved PL spectroscopy – have been employed to study HC behaviour. However, variations in experimental implementations and data analyses can lead to conflicting or even erroneous interpretations, impeding meaningful comparisons across studies and hindering progress in the field. This tutorial review provides an overview of the most commonly used optical spectroscopy techniques for probing HC dynamics in the representative halide perovskite systems, detailing the underlying physical principles and models used to extract HC temperatures and cooling rates. We also discuss best practices for experimental design and data interpretation, aiming to minimize methodological pitfalls. By consolidating current approaches and offering guidance for their application, this tutorial serves as a resource for researchers seeking to explore HC phenomena and advance HC-based optoelectronic technologies in emergent semiconductor systems.
 Zengshan Xing | Dr Zengshan Xing obtained his PhD degree in Physics from the Hong Kong University of Science and Technology, Hong Kong SAR, China, in 2023. He is currently a Postdoctoral Research Fellow at the School of Physical and Mathematical Sciences (SPMS), Nanyang Technological University (NTU), Singapore. His current research focuses on excited-state carrier dynamics and photophysical properties of semiconductors via ultrafast spectroscopy. |
 Rui Cai | Dr Rui Cai obtained his PhD in Applied Physics from Nanyang Technological University (NTU), Singapore, in 2025, and has continued his postdoctoral research there. His research focuses on ultrafast, coherent light–matter interactions in emerging semiconductors. |
 Tze Chien Sum | Tze Chien Sum is a Professor of Physics at the School of Physical and Mathematical Sciences (SPMS), Nanyang Technological University (NTU), where he leads the Femtosecond Dynamics Laboratory. He is also the Director of the Institute of Advanced Studies (IAS) at NTU and an Associate Dean (Research) at the College of Science. His current research focuses on investigating light–matter interactions; energy and charge transfer mechanisms; and probing carrier and quasi-particle dynamics in a broad range of emergent nanoscale, light harvesting, and light emitting systems. He received his BSc (1999), MSc (2000), and PhD (2005) degrees in Physics from the National University of Singapore. |
Key learning points(1) The hot carrier (HC) cooling processes and the underlying mechanisms responsible for slow HC relaxation in halide perovskites.(2) The principles and experimental implementations of transient absorption (TA)-based spectroscopy (pump–probe and pump–push–probe schemes) and photoluminescence (PL) spectroscopy for HC characterization. (3) Proper application of physical models for evaluating HC temperatures. (4) Various approaches and models for analysing HC dynamics under different conditions, providing physical insights into mechanisms that retard HC cooling. (5) The strengths and limitations of different methodologies, enabling comparative and complementary studies of HC properties in halide perovskites. |
1. Introduction
Halide perovskites have been extensively studied for various optoelectronic applications for more than a decade. They are still under intense scrutiny due to their superior optoelectronic properties and device performances.1–3 In particular, halide perovskites are excellent light absorbers with their high absorption coefficients and desirable bandgaps for photovoltaic applications.4,5 Photons with energy above optical bandgaps can be absorbed to generate electrons and holes, where any excess energy leads to energetic carriers. Following photoexcitation, these energetic carriers will redistribute their energy to reach a quasi-equilibrium state among themselves via carrier–carrier or carrier–ion elastic scattering, known as thermalization.6,7 The thermalized carriers follow the Fermi–Dirac (FD) distribution with a characteristic temperature (Tc), where carriers with a carrier temperature notably higher than the lattice temperature are termed “hot” carriers.Following the thermalization process, the hot carriers (HCs) will release their excess energy to equilibrate with the lattice temperature, mainly through inelastic carrier–phonon scattering and phonon relaxation.8,9 Due to the polar nature of halide perovskites, HCs dominantly interact with the longitudinal optical (LO) phonons through the Fröhlich mechanism, which arises from Coulombic interaction between the carriers and the macroscopic electric field of the lattices caused by the LO phonon modes. Such carrier-phonon scattering persists until the excess energy is less than the energy of the LO phonons. The emitted LO phonons near the zone-centre will decay into daughter acoustic phonons or transverse optical (TO) phonons. In polar semiconductors, LO phonon relaxation can occur via (i) the Klemens channel (i.e., one LO phonon to two longitudinal acoustic (LA) phonons), and/or (ii) the Ridley channel (i.e., one LO phonon to one TO phonon and one transverse/longitudinal acoustic phonon).10,11 The Klemens channel is believed to be the more efficient pathway for LO phonon relaxations, due to the quick thermalization of LA phonons with the lattice.8 Subsequently, acoustic phonons further equilibrate with the environment by dissipating the excess energy as heat. On the other hand, carrier multiplication (CM) or multiple exciton generation (MEG) can occur if the energetic carriers possess excess energy above the threshold (> 2Eg). This results in creation of multiple electron–hole pairs following absorption of a single high-energy photon. CM/MEG is a competing process to HC cooling; thus, slower HC cooling is favourable for CM/MEG.12,13
HC cooling is an ultrafast process, occurring over hundreds of femtoseconds (fs) to picoseconds (ps) time scales. In conventional semiconductors, HC cooling generally occurs in the sub-ps timescale,6 making the effective utilization of HC energy extremely challenging. In contrast, the slower HC cooling observed in halide perovskites provides an exciting opportunity for harvesting the HCs before they could lose their excess energy as heat.14,15 This excess energy can be harvested by properly designing the HC extraction layers in hot-carrier solar cells or exploiting the CM/MEG effects. Theoretical calculations have revealed that a single-junction solar cell can reach a power conversion efficiency (PCE) of 66% if the HCs are extracted before cooling down to the lattice temperature.16 Besides, solar cells exploiting the CM effect have a theoretical PCE as high as 44.4%.17,18 This tantalizing possibility of surpassing the Shockley–Queisser (SQ) efficiency limit16 of 33% has stimulated tremendous research interest into HC photovoltaics as well as HC related optoelectronic applications.19 Correspondingly, significant efforts have been devoted to elucidating the mechanisms governing HC dynamics to guide the advancements of HC devices.
Several mechanisms have been proposed for the slow HC cooling process in halide perovskites, such as the (i) hot-phonon effect (or hot-phonon bottleneck effect), (ii) intrinsic phonon bottleneck effect in quantum-confined nanocrystals, (iii) Auger heating effect, (iv) band filling effect, and (v) polaron screening of phonon scattering, etc.20
(i) The hot-phonon effect slows both the LO phonon decay and HC relaxation due to the enhanced reabsorption of non-equilibrium LO phonons at high HC densities. This arises from a rate imbalance between electron–phonon scattering and LO phonon decay, since HC relaxation proceeds through the emission of LO phonons (energy transfer from HCs to LO phonons via electron–phonon scattering) and the LO phonons then decay into acoustic phonons. At low carrier concentrations, the emitted LO phonons remain within the phonon modes that are in thermal equilibrium with the lattice (Tp = TL).8,21 Under these conditions, HC cooling is mainly governed by LO phonon emission via electron–phonon scattering.10,22,23 With high carrier densities, however, the rapid LO phonon emission drives their phonon occupation number out of equilibrium, leading to the accumulation of hot-phonons.24,25 This arises because LO phonon decay is typically slower than the characteristic LO phonon emission.22,25,26 The resulting build-up of a large non-equilibrium LO phonon (hot phonon) population promotes LO-phonon reabsorption by excited carriers, thereby slowing HC relaxation and increasing the net LO phonon decay time. In halide perovskites, the slow LO phonon relaxation could originate from the following factors. Fu et al. demonstrated the hot-phonon effect at carrier densities of ∼1018 cm−3 and showed that it arises from the suppression of the efficient Klemens relaxation pathway for LO-phonon decay, caused by the mismatch of the LO-phonon energy with twice the acoustic-phonon energy (ELO > 2ELA).8 Upconversion of acoustic phonons to optical phonons has also been proposed as a contributing origin of the hot-phonon effect, due to the presence of hybrid phonon modes and the intrinsically low thermal conductivity of organic–inorganic halide perovskites.10
Do note that while the hot-phonon effect has also been reported in other semiconductor systems (e.g., group-III nitrides,27 III–V semiconductors etc.28) and in various superconducting systems (e.g., MgB2, graphene etc.),29–32 the underlying origins could differ substantially from those in halide perovskites. Thus, we will not discuss them here.
(ii) In contrast to the hot-phonon effect, the intrinsic phonon bottleneck effect typically appears in quantum-confined systems (e.g., quantum dots) and at a low excitation fluence (e.g., 〈N〉 ∼0.1).33 When the energy spacing between discrete energy levels exceeds the LO-phonon energy, energy relaxation requiring multiple phonon emissions becomes less efficient. As a result, HC cooling slows down as the degree of quantum confinement increases. This intrinsic phonon bottleneck effect has been observed in perovskite nanocrystals, where smaller nanocrystals exhibit longer HC relaxation times.33,34
(iii) The Auger heating effect occurs at high carrier densities >1019 cm−3, where carriers are reheated by absorbing energy from electron–hole recombination. The influence of Auger heating generally manifests as a prolonged HC relaxation tail extending to tens of picoseconds, as demonstrated in various halide perovskite structures.8,33,35
(iv) The band filling effect refers to the partial filling of band-edge density of states, leading to an increased average energy of excited carriers and a blue-shift of the photoluminescence spectrum. This effect was proposed to account for the long-lived hot-carrier emission with ns-lifetimes in FASnI3 at high carrier densities (>1018 cm−3), where the detailed mechanism warrants further investigation.36
(v) Polaron screening of phonon scattering occurs at low carrier densities below the Mott density (<1018 cm−3). Zhu et al. proposed that phonon scattering can be screened by Coulombic potential of large polarons, slowing down the HC relaxations.37 This was derived from the large increase in dielectric constants upon large polaron formation, which mediates electron-LO phonon interactions.7 Nevertheless, this mechanism is still under debate.20
It is noteworthy that HC cooling dynamics is not only affected by the excitation carrier density, but also other factors, such as chemical compositions, dimensionalities, sample morphologies etc.6,38 Therefore, case studies of HC dynamics are fundamentally critical for understanding the HC behaviour in a material system.
HC properties in halide perovskites have been explored using various techniques, such as transient absorption (TA) or pump–probe spectroscopy,6,39–42 photoluminescence (PL) spectroscopy,36,43,44 two-photon photoemission spectroscopy,37,45 THz spectroscopy46,47etc. Among them, TA and PL spectroscopies are more prevalent, likely due to their wide availability and ease of implementation. Additionally, TA-based pump-push-probe (PPP) spectroscopy has also been utilized for HC characterization.48,49 Different techniques involve distinct photophysical processes, rendering them appropriate for different experimental scenarios. For instance, PL spectroscopy is applicable only when appreciable HC light emission is detectable, while TA spectroscopy is incapable of distinguishing the individual contributions of holes and electrons to the carrier temperatures. Moreover, discrepancies may arise from different analysis schemes, even when using the same technique. For instance, Lim et al. have shown that the values of HC temperature can vary depending on the fitting schemes applied to TA spectra.40 Therefore, having a detailed understanding of the methodologies and underlying physical principles is pivotal for proper usage of the toolkits and fair comparison of experimental results.
In this tutorial review, we aim to explicate the common experimental methods for investigating HC dynamics in halide perovskites, with a focus on TA- and PL-based techniques. We begin by introducing the principles and experimental details of TA, PPP and PL in probing the HC dynamics. Various approaches for characterizing the HC temperature and HC dynamics will be reviewed, accompanied by discussions on their respective strengths and limitations. Furthermore, we summarize the common strategies for analysing HC cooling rates, which not only provide critical insights into the HC properties, but also help clarify the underlying cooling mechanisms. Our comprehensive overview of the techniques and analysis methods offers invaluable guidance for exploring HC properties in a material system, which is essential for the development of HC-based applications.
2. Optical spectroscopy for HC characterization
2.1. Transient absorption (TA) spectroscopy
TA spectroscopy is one of the most widely used techniques for investigating HC dynamics. TA probes the absorption difference of a system under photoexcited state relative to the ground state on timescales down to sub-picosecond. The spectral features and dynamics reflect the photophysical processes undergone by the excited-state species. TA spectra are measured using the pump–probe technique, as illustrated in Fig. 1(a), where the pump beam is usually modulated at half the frequency of the probe beam. The pump and probe beams are typically femtosecond (fs) laser pulse trains, with a monochromatic pump and a broadband probe. When a pump pulse arrives before a probe pulse (t > 0), the detected ΔA (i.e., absorption difference between pump-on and pump-off states) exhibits the general spectral features as shown in Fig. 1(a). Negative signals correspond to photobleaching (PB), while positive signals signify photoinduced absorption (PA). Stimulated emission, if present, appears as a negative signal near the band-edge. The TA signal can also be expressed as the relative change in transmission, ΔT/T, which is related to ΔA as ΔA = −log(1 + ΔT/T). | ||
| Fig. 1 (a) Schematic of a TA setup and typical TA spectral features. (b)–(g) Hot carrier (HC) temperatures characterized by TA spectroscopy for MAPbBr3 nanocrystals (of size ∼14 nm, the Bohr diameter (BD) of MAPbBr3 is ∼9.8 nm).50 (b) representative pseudo-colour plot of the TA spectra; TA spectra fitted by (c) Scheme 1 (fitting starts from 2.41 eV to the energy of PA peak, spanning a range of ∼0.27 eV), (d) Scheme 2 (fitting starts at an intensity ratio of ∼0.3 and ends at the PA peak) and (f) Scheme 2 including PA slope (spanning a range of ∼0.3 eV); (e) and (g) the extracted carrier temperatures by different fitting schemes. | ||
| ΔA(E) = −A0(E)e(Ef − E)/kBTc | (1) |
Fig. 1(b) illustrates an example of the TA spectra of MAPbBr3 (MA = methylammonium) nanocrystals (of size ∼14 nm, equivalent to bulk) excited at 3.1 eV at early time scales. A PB peak appears upon photoexcitation, exhibiting a broad high-energy tail that narrows with increasing delay times because of HC cooling. The evolution of HC temperatures can be characterized by applying eqn (1) to the high-energy PB tails. Specifically, we highlight the various fitting schemes in the literature. Two general fitting schemes for the high energy-tail of PB signals are illustrated in Fig. 1(c) and (d). Scheme 1 fixes the lower-energy bounds of the fitting ranges while scheme 2 defines the starting points based on the specified ratio of the TA intensity to the PB peak intensity. Typically, a range of 0.1 to 0.3 eV will capture the exponential feature of the high-energy tails in the PB peaks. The use of different fitting schemes could incur discrepancies in the obtained carrier temperatures (Fig. 1(e)), which could complicate comparison between different studies. Lim et al. have shown that variations in the selected range or the starting intensity ratio can also affect the values of the fitted carrier temperatures.40 Additionally, the accuracy of the extracted carrier temperature is also influenced by the energy difference E − Ef, which determines how closely the FD distribution can be approximated to the MB distribution.
A further complication is the PA signal that appears above the PB peak during HC cooling, which arises primarily from the band gap renormalization (BGR) effect.25 This results in a rising slope in the normalized TA profile, which is not modelled in eqn (1). Therefore, eqn (1) fits with an upper bound defined by the minima of the PA as illustrated in Fig. 1(c) and (d). The line shape of PA from the BGR effect as a function of energy is defined as  .25,53 Therefore, to simulate the high-energy tail of PB with the BGR effect, eqn (1) is modified as
.25,53 Therefore, to simulate the high-energy tail of PB with the BGR effect, eqn (1) is modified as
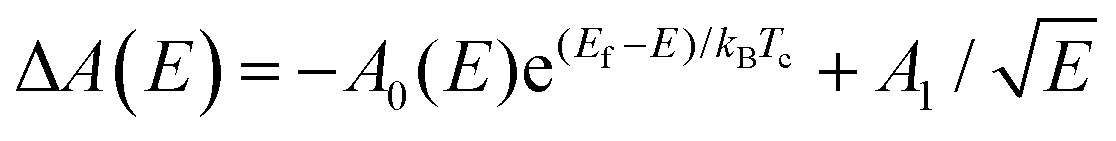 | (2) |
| ΔA(E) = (1 − fe(E, EF, Tc))2A(E, Eg + ΔEg) − A(E, Eg) | (3) |
 | (4) |
 | (5) |
 denotes the density of states for 3D systems. The excitonic absorption is modelled by n series of below-gap transitions with their intensities scaled by 1/n3. The continuum band absorption is simulated by the density of state modified by the Coulombic enhancement factor (i.e., the first term in eqn (5)). The transition linewidth broadening is generally modelled by a Gaussian function G(E, Γ), with Γ characterizing the extent of the broadening. The simulated absorption spectrum by convoluting G(E, Γ) with Aexc and Acon is presented in the inset of Fig. 2(a), illustrating the respective contributions from the exitonic and continuum bands.
denotes the density of states for 3D systems. The excitonic absorption is modelled by n series of below-gap transitions with their intensities scaled by 1/n3. The continuum band absorption is simulated by the density of state modified by the Coulombic enhancement factor (i.e., the first term in eqn (5)). The transition linewidth broadening is generally modelled by a Gaussian function G(E, Γ), with Γ characterizing the extent of the broadening. The simulated absorption spectrum by convoluting G(E, Γ) with Aexc and Acon is presented in the inset of Fig. 2(a), illustrating the respective contributions from the exitonic and continuum bands.
 | ||
| Fig. 2 (a) Globally-fitted TA spectra of MAPbBr3 nanocrystals (dashed lines). The inset shows the simulated absorption spectrum containing contributions from the excitonic and continuum bands. (b) Time-dependent carrier temperatures and quasi-Fermi levels from global fittings. Inset shows a comparison of the HC dynamics from different fitting methods. (c) Time-dependent variation of the bandgap energy and linewidth broadening, and (d) illustration of simulated TA at 300 K (solid lines in upper panel: JDOS; yellow dashed line: simulated TA at Tc = 500 K). | ||
The global-fitting of the full TA spectra (ΔA) with eqn (3)–(5) is illustrated in Fig. 2(a). The band gap shifts arising from the BGR effect (red-shift) and Burstein–Moss effect (BM, blue-shift), linewidth broadening and the PB shift are taken into consideration by the time-varying ΔEg, Γ, and EF. The bandgap energy (Eg), exciton binding energy (Eb), and linewidth at ground state (Γpump-off) are fixed as shared parameters, which can be obtained from fitting eqn (4) and (5) to the steady state absorption spectrum.40 In our example (Fig. 2(a) inset), the values are Eg = 2.385 ± 0.001 eV, Eb = 15 ± 1 meV and Γpump-off = 30 ± 1 meV. The corresponding time-dependent parameters are plotted in Fig. 2(b) and (c). The quasi-Fermi level increases along with the cooling of HC temperature due to its temperature dependent behaviour as observed in halide perovskites and other semiconductors.40,55 The band gap shrinks with increasing time delay as reflected in the increasing negative values of ΔEg. Fig. 2(d) presents the simulated changes in the absorption spectrum and the JDOS for the excited state at Tc = 300 K. The redshift in the excited-state absorption spectrum is due to the dominant BGR effect.25 Consequently, the JDOS is enhanced at higher energy, resulting in the above-gap PA signal at the higher-energy side of the PB peak. The simulated TA spectrum at Tc = 530 K is also presented in Fig. 2(d), showing the PB tail broadening toward the higher energy. The linewidth broadening (Γ) increases during HC cooling and is influenced by various factors, such as carrier-phonon interactions, energetic disorder, impurity scattering etc.40 The inset of Fig. 2(b) compares the HC dynamics obtained from different methods. The time-dependent carrier temperatures from this method exhibit slightly higher values than those obtained from fitting the high-energy PB tails at the early time scales before 0.6 ps. The discrepancies may arise from the effects of BGR and linewidth broadening. These effects are prominent before 0.6 ps, as shown in Fig. 2(c), and are not considered in the MB fitting of the high-energy tails. Despite slight differences in the values obtained from the different methods, their dynamics exhibits a similar trend with the HC cooling completing at ∼1 ps. Therefore, fitting the high-energy PB tails remains a common approach for analysing HC dynamics. Nevertheless, accurate estimation of the carrier temperature is crucial when HC energy must be considered for alignment in optoelectronic devices. Full-spectrum TA fitting provides high reproducibility, enabling reliable cross-comparisons across different studies.
 | (6) |
 | ||
| Fig. 3 Simulated TA spectrum with eqn (6) for 300 K (a) and 1000 K (b) with the same energy dispersion function; (c) an example of fitting eqn (6) to the TA spectrum. Reproduced with permission from ref. 56. Copyright 2024 Tiede et al. Advanced Optical Materials published by Wiley-VCH GmbH. | ||
Although the aforementioned equations for bulk material systems can be adopted in some low-dimensional systems,57,58 the reduced dimensionality can incur strong broadening effects and discrete energy levels, which deviate the TA profile from a Fermi-distribution and nullify the application of these models.59 For the widely studied mixed-phase quasi-2D/3D perovskites, in particular, the range of interest of TA signals for 3D domains can overlap with optical transitions of low-n phases (n refers to the number of metal-halide layers), which further complicates the shape analysis of HC dynamics.
To characterize the HC dynamics in the 3D domains of a quasi-2D/3D system, Fan, et al. performed global fitting to the full TA spectra of quasi2D/3D lead iodide NCs, separately including the contributions from the 3D and quasi-2D phases, given as ΔA = ΔA3D + ΔAquasi-2D.60 The ΔA3D is described by eqn (3) (see above) and the contributions from the quasi-2D phases are modelled by a set of Gaussian functions,
 | (7) |
 | ||
| Fig. 4 (a) TA spectrum of mixed 3D/quasi-2D CsPbI3 perovskite NCs at a pump–probe delay of 0.3 ps; (b) TA spectra of the quasi-2D PEAMAPI and 3D MAPI at 0.5 ps and 100 ps, and (c) the calculated excess energy of different systems; (d) extracted HC dynamics in the 3D domains in mixed quasi-2D/3D CsPbI3 NCs and the excess energy of the carriers; (e) schematic illustration of the carrier relaxation processes in a quasi-2D/3D system; (f) comparison of the hot carrier temperatures evaluated from the methods of MB fitting and excess energy calculations in MAPbBr3 bulk. (a), (d) and (e) are reprinted with permission from ref. 60. Copyright 2025 American Chemical Society; (b) and (c) are reproduced with permission from ref. 61. Copyright 2022 Wiley-VCH GmbH; and (f) is reprinted with permission from ref. 59. Copyright 2024 American Chemical Society. | ||
Notably, carriers generated in the low-n phases can funnel to the 3D domains due to the cascaded energy levels when the excitation energy is above the transition energy of the low-n phases. Therefore, the energy relaxation in quasi-2D/3D systems mainly consists of HC cooling in the 3D domains and the energy funnelling from the low-n phases to the 3D domains. To capture the overall energy relaxation process, assuming that the TA intensity is proportional to the carrier density n(ε), the excess energy of the system referring to the 3D band gap can be calculated as:60,61
 | (8) |
| 〈E〉excess(t) = 〈E〉(t) − 〈E〉(t = ∞) | (9) |
Using eqn (8) and (9), the excess energy dynamics for the quasi-2D (BAMAPbI3 and PEAMAPbI3) and 3D MAPbI3 films have been investigated (see Fig. 4(b) and (c)).61 Benefitting from the cascaded energy funnelling, the excess energy relaxation is slowed by two orders of magnitude in the quasi-2D systems as compared to their 3D counterparts. A slower relaxation is observed in the larger organic spacer (PEA) as a result of its weaker inter-layer interactions. In the combined model, ΔA = ΔA3D + ΔAquasi-2D, Fan et al. characterized the excess energy dynamics of the quasi-2D/3D NCs and the respective HC dynamics in 3D domains, as shown in Fig. 4(d).60 They uncovered a remarkably longer thermalization process (∼0.9 ps) for the HCs in the 3D domain, contributed by the energy funnelling from low-n phases during the energy relaxation (see Fig. 4(e)).
For a system where 〈E〉excess(t) mainly characterizes the excess energy of the HCs (i.e., bulk materials), the carrier temperature can be estimated via the relationship of  . This method has been verified by comparing the estimated carrier temperatures to those obtained from MB fitting in a MAPbBr3 bulk, as shown in Fig. 4(f).59 While the application of the excess energy calculation is less restricted by the TA line shape, due care must be taken for the effects of band gap renormalization and photoinduced broadening, which affect the authenticity of the assumption n(ε) ∝ ΔA (PB regions).
. This method has been verified by comparing the estimated carrier temperatures to those obtained from MB fitting in a MAPbBr3 bulk, as shown in Fig. 4(f).59 While the application of the excess energy calculation is less restricted by the TA line shape, due care must be taken for the effects of band gap renormalization and photoinduced broadening, which affect the authenticity of the assumption n(ε) ∝ ΔA (PB regions).
Fig. 5(a) shows an example of TA spectra characterizing the HC cooling. Accompanying the band-edge PB signal are two PA bands that appear above and below the band gap at early times, where the sub-bandgap PA signal later evolves into a PB signal. The red shift in the PB band at a later delay time signifies the dominance of the BGR effect (red shift) over the BM effect (blue shift). The higher-energy PA band was initially ascribed to the photoinduced change in refractive index,42 while a later study suggests its origin from the BGR effect.54,63 The sub-bandgap PA has also been attributed to various origins in the literature, such as the BGR effect, hot biexciton effect and hot-carrier Stark effect, etc. The BGR effect refers to the creation of new optical transitions below the intrinsic band gap in ultrafast time scales (within ∼100 fs), resulting in a sub-gap PA.42 The hot biexciton effect describes the bandgap shrinkage due to the Coulombic interaction between hot excitons by the pump and cold excitons near the band edge by the probe.54,64 The hot-carrier Stark effect refers to the field-induced shift of the absorption spectrum in the presence of hot carriers,65 giving rise to a derivative shape in the TA spectra. A latest study suggests that the Coulombic interplay from the hot biexciton effect has a key role, given that the fluence-independent decay of the sub-gap PA is consistent with timescale of band-edge exciton dissociation.54 Despite the debates regarding its origins and the seemingly correlated trend with HC cooling as shown in Fig. 5(b), their rates are found to be different (Fig. 5(c)).42,54 Therefore, the HC cooling process is typically characterized by the rise of the PB signal.62,66,67 The illustrated PB rise for the MAPbBr3 in Fig. 5(b) occurs in a time scale of ∼1 ps, which is consistent with the trends from the aforementioned analyses.
 | ||
| Fig. 5 (a) TA spectra of MAPbBr3 NCs probed at different time delays and (b) the dynamics probed at P1 and P2. (c) Dynamics of the HC temperature and TA kinetics of below-gap PA of a MAPbI3 thin film, adapted with permission from ref. 42. Copyright 2015, Michael B. Price et al., Springer Nature. (d) HC dynamics extracted by taking the difference between the band-edge PB kinetics for excitation energies above and near the band gap of a MAPbI3 thin film, reproduced from ref. 68. Zhi Guo et al., Science, 10.1126/science.aam7744 [2017], AAAS. | ||
Alternatively, the HC dynamics can be obtained by taking the difference between the band-edge PB kinetics for excitation energies above and near the band gap. For example, in the case of a MAPbI3 thin film in Fig. 5(d),68 the kinetics from the above-gap excitation of 3.14 eV comprises the contributions from the HC relaxation and the recombination of carriers near the band-edge. In contrast, the kinetics from the near band-edge excitation of 1.68 eV tracks mainly the carrier recombination process. The HC kinetics can therefore be resolved by taking their difference, as shown in the bottom panel of Fig. 5(d). Do note that the HC dynamics obtained by probing the PB kinetics can be affected by the recombination processes, especially from Auger recombination under high carrier densities.
2.2. Pump-push-probe (PPP) spectroscopy
PPP spectroscopy is a technique based on TA spectroscopy. While HC dynamics probed from TA measurements could be influenced by multiband excitation and multiparticle effects,49 PPP spectroscopy can provide a more straightforward visualization of HC cooling dynamics. For PPP measurements, a third pulse (i.e., push pulse) is introduced following the pump pulse to re-excite the cold carriers from the band edge, creating a net population of HCs at a specific time defined by time delay between the pump pulse and the push pulse (Fig. 6). The signals are recorded as a transient absorption (−ΔA) before the push pulse arrives (Fig. 6(b)). Both the PB signal in the visible region and the carrier absorption at infrared wavelengths reflect the population dynamics of these cold carriers because the transitions from G to E1 and that from E1 to E2 share the same excited state E1. The push pulse changes the population of cold carriers and therefore the carrier absorption intensity (Fig. 6(c)). After a certain time delay, the push-induced HCs relax back to the band edge along with the recombination to the ground state (Fig. 6(d)). Although PPP signals can be obtained by modulating either the pump or push beams, the push is usually not modulated to avoid push-induced effects other than HC generation. A direct probe of HCs has been realized in CdSe quantum dots69,70 and halide perovskites.48,49 Generally, the PPP approach provides a more accurate interpretation of HC behaviour due to the reduced influence from multi-band and multi-particle effects that are present in TA measurements. | ||
| Fig. 6 Schematic of the population dynamics in pump-push-probe (PPP) experiments. (a) Standard TA analysis of HC dynamics probed at the band edge (the as-generated HCs relax to the band edge (E1) with a characteristic HC lifetime τpump). (b) The thermodynamic equilibrium state before the push pulse arrives. (c) HCs generated by the push pulse (defined at time zero, and τpush describes the cooling time constant). (d) The bleaching recovers with a slightly reduced amplitude due to carrier recombination. (e) Comparison of TA and PPP dynamics on a sample of CsPbBr3 NCs, where the bleaching amplitude Apush is proportional to the corresponding hot exciton occupation numbers. (e) is adapted with permission from ref. 34. Copyright 2024, American Chemical Society. | ||
The push-induced HC dynamics can be characterized by monitoring the evolution of the carrier absorption at near infrared wavelengths (Fig. 6(b–d)).48 Alternatively, the push-induced changes in absorption near the band gap energy (i.e., the PB band) can also be used to track the cooling of the re-excited carriers (Fig. 6(e)).34 The carrier temperature can be estimated from the PPP spectra in a similar manner analogous to the TA approach.49 By subtracting the TA data (i.e., ΔA), the push-induced absorption change, termed ΔΔA, can then be determined. The spectral profile of ΔΔA is subjected to both the BGR effect induced by the HC population and the energy distribution of the HCs. A key challenge in PPP is the determination of the initial pushed HC density (N0hot) which depends on the absorption cross-section of the cold carriers. This can be overcome by taking the dependence of the fraction of the pushed HCs to cold carriers (N0hot/N0cold) on the PB amplitude ratio of the push and pump pulses (Apush/Apump).34,49
Both the HC cooling time and the hot phonon effect can be investigated using the PPP technique. In addition, examining HC cooling processes under different cold carrier densities can reveal the underlying carrier-phonon and/or carrier-carrier interaction mechanisms. By adjusting the pump and push fluences, the hot-to-cold carrier ratio can be tuned. Our recent work using the PPP technique demonstrates the influence of the cold carrier bath on the hot exciton cooling in perovskite quantum dots of different sizes.34 A larger fraction of cold carriers leads to faster HC cooling, likely due to the enhanced many-body interactions between the hot carriers/excitons and cold carriers/excitons as observed in perovskite quantum dots. On the other hand, the Auger effect may also contribute to HC cooling, particularly in more strongly confined systems, where the energy loss rate of HCs is reduced.
2.3. Photoluminescence spectroscopy
Photoluminescence (PL) spectroscopy is a powerful and widely accessible technique for examining light-emitting states of photoactive materials. It provides rapid insights into the energy landscape of the emitted photons. In an absorbing medium, photoexcitation promotes electrons to higher energy levels and these electrons subsequently relax to the ground state by emitting photons whose energies are determined by the electronic structure of the material. Although PL predominantly traces the recombination of photoexcited carriers near the band edge, it can reveal HC behaviour under certain circumstances. HC PL occurs when energetic electrons and holes recombine before fully relaxing to the band edge. Fig. 7(a) schematically illustrates the band-to-band recombination in the absence and presence of HC PL, respectively. Intuitively, HC recombination contributes high-energy, above-gap emissions to the PL spectra. Over the past few decades, HC PL has been observed in both direct and indirect bandgap semiconductors.37,71–78 | ||
| Fig. 7 (a) Schematic for carrier recombination in semiconductors with parabolic bands. (b) PL spectra for different carrier temperatures, simulated by eqn (11) with a bandgap energy of 1 eV (the inset shows a semi-log plot of the same PL spectra). | ||
 | (10) |
Alternatively, the generalized Planck's radiation law, also known as the Lasher–Stern–Wurfel (LSW) equation,82,83 can be employed to incorporate the HC effects. Band-to-band emissions in a semiconductor is described by:
 | (11) |
Occasionally, fitting the PL spectrum can be challenging due to the discrete energy levels and other effects that modify the absorption spectrum. For example, in fluence-dependent measurements, A(E) may vary with carrier density as a result of the band filling effect. Therefore, an accurate form of A(E) is required for systems exhibiting complex PL mechanisms. Katahara and Hillhouse84 proposed a unified model to fit the room temperature PL of GaAs, described by:
 | (12) |
 | ||
| Fig. 8 (a) Power-dependent PL spectrum of a FAPbI3 thin film (the excitation source is a 473 nm CW laser). (b) Power-dependent carrier temperature and quasi-Fermi-level splitting obtained from fitting. (c) Comparison of the band gap energy (Eg) or exciton binding energy (Eb) obtained from different methodologies. (a)–(c) are reprinted with permission from ref. 43. Copyright 2022 American Chemical Society. | ||
Gating techniques generally provide higher temporal resolutions as determined by the duration of the gating pulses. Typically, a nonlinear crystal is used to mix the emitted PL with a gating pulse (Fig. 9(a)). The signal pulse is generated via sum frequency generation (SFG), governed by the second-order nonlinear susceptibility χ(2) and phase-matching conditions. The signal pulse, whose temporal width is defined by the gating pulse, is subsequently detected and analysed to reconstruct the time-resolved PL. Alternatively, PL gating can be implemented using a Kerr medium, where the gating pulse induces a transient change in the refractive index through the third-order nonlinear susceptibility χ(3). With temporal resolution comparable to that of TA setups, PL gating techniques have been applied to study the HC cooling processes on the ps timescale.87–91Fig. 9(b)–(d) present TRPL analyses of CsPbBr3 nanocrystals obtained using our PL up-conversion setup. A pronounced high-energy tail appears in the PL spectra at early times, which evolves within the first few picoseconds and disappears at longer times. Fig. 9(c) shows the fits of representative spectra using eqn (12), while the extracted HC temperatures and corresponding quasi-Fermi-level splittings are shown in Fig. 9(d). The picosecond-scale HC cooling times are consistent with those obtained from TA measurements.34
 | ||
| Fig. 9 TRPL measurements on perovskites. (a) A sketch of the up-conversion setup. (b) Representative TRPL spectra of CsPbBr3 nanocrystals. (c) PL spectra at several delay times extracted from (b). Dot line: experiment. Solid lines: fitting curves. (d) Time-dependent carrier temperature and quasi-Fermi-level splitting obtained from the fits in (c). (e) PL dynamics at different emission energies in a FASnI3 thin film measured by a streak camera. (e) is adapted with permission from ref. 36. Copyright 2018 Springer Nature. | ||
In a TRPL measurement using a TCSPC setup, a histogram of photon arrival times is constructed by recording the delay between pulse excitation and single photon detection, thereby reconstructing the PL dynamics. Similarly, a streak camera converts photon arrival times into spatial information, enabling time-resolved PL measurements. Owing to their limited temporal resolution, these two techniques are commonly used to study PL dynamics on several picoseconds (ps) and longer timescales. An example of using a streak camera to probe HC dynamics is reported in the literature,36 where long-lived HCs were observed in the tin-based perovskite FASnI3. The HC signature appears in the PL dynamics at the high-energy tail, exhibiting a distinct fast decay component that is absent below a certain energy (Fig. 9(e)). These observations provide important insights into the slow HC cooling processes in perovskites.
Nevertheless, several limitations constrain the application of PL-based approaches for HC analysis. Firstly, this method is limited to the materials that exhibit detectable HC light emission. Secondly, the presence of multiple luminescence mechanisms beyond the band-to-band carrier recombination, such as excitonic emission and defect-assisted emission, nonradiative channels etc., complicates HC analysis and poses a major challenge. Although excitonic effects can be neglected or explicitly included in eqn (12), other recombination channels can introduce significant uncertainties in the extracted carrier temperatures. Therefore, identifying the dominant PL mechanism is essential to mitigate these uncertainties.
In addition, PL line shapes can be influenced by a variety of factors that affect the accurate estimation of carrier temperatures. Interactions between charge carriers and their environment (e.g., phonons, excitons, traps, etc.) give rise to both inhomogeneous and homogeneous broadening of electronic states, ultimately modifying the PL profile. As discussed in ref. 80, neglecting linewidth broadening effects can lead to an overestimation of the carrier temperature when MB approximation is applied. Furthermore, applying eqn (10) to the high-energy tail of the PL spectrum requires careful selection of the fitting range to ensure reliable results. Improper use of the spectral window can result in either overestimation or underestimation of the carrier temperature.
3. Analysis of HC dynamics towards optoelectronic applications
A range of HC dynamics analysis toolkits is available for an in-depth investigation of the physical processes governing HC cooling. HC cooling time is a critical parameter that strongly influences the performance of HC solar cells. Longer cooling times allow HCs to diffuse more efficiently toward material interfaces, thereby facilitating more effective charge extraction. In contrast, slow HC cooling is generally detrimental to light-emitting applications where rapid cooling is instead desirable. Fast HC cooling can suppress HC trapping at defects and thus reduce non-radiative losses. For example, accelerated HC relaxation achieved by reducing excess excitation energy or through elemental doping has been shown to improve photoluminescence quantum yields.92–94 Moreover, fast HC cooling can promote the swift buildup of population inversion, thereby lowering the threshold for amplified spontaneous emission and lasing.87,95 On the other hand, the presence of hot carriers can also be detrimental to light-emitting devices. Hot carriers can degrade device performance by reducing carrier injection and enhancing nonradiative processes, such as Shockley–Read–Hall recombination and Auger recombination,92,96–98 leading to increased gain thresholds and efficiency roll-off.99 In addition, HCs can induce spectral broadening and shifts at high injection densities,36 and compromise operational stability through Joule heating.100 Therefore, analysis of HC dynamics provides essential insights into carrier behaviour and underlying cooling mechanisms, which are essential for the rational design of high performance hot-carrier devices. In the following sections, different approaches for HC dynamics analysis under various experimental conditions are reviewed.3.1. HC cooling rates in halide perovskites
HC cooling is often compared by examining the time required for the carrier temperature to decrease to 600 K across different systems.9,101 This benchmark is motivated by theoretical predictions indicating that HCs with temperatures above 600 K can still enable power conversion efficiencies exceeding 40%.33 However, HC cooling times are strongly influenced by various experimental parameters, such as excitation energy and carrier density, which complicates direct comparison between studies.6 To overcome this limitation, additional metrics have been introduced to characterize HC cooling rates, including cooling time constants, energy loss rates, and thermalization coefficient, etc. These metrics are discussed in the following sections. | ||
| Fig. 10 (a) Carrier temperatures from fitting the high-energy PB tail of the TA spectra for CsPb(Cl0.2Br0.8)3 at different carrier densities (solid lines: mono- or bi-exponential fits), adapted with permission from ref. 10 Copyright 2017, Yang et al. Springer Nature; (b) PB rise dynamics in TA signals for CsPbBr3 NCs of different sizes; (c) pseudo-colour map of τcool in relation to the hot (push fluences) and cool (pump fluences) carrier density, and (d) HC density dependent τcool under different initial cold carrier densities of the ZI-capped CsPbBr3 NCs, adapted with permission from ref. 105. Copyright 2020 American Chemical Society; (e) PPP bleaching dynamics for 12 nm CsPbBr3 NCs at different push fluences and (f) push-fluence-dependent cooling time constants (τcool) for different sizes of NCs (fluence is presented as a population ratio of hot to cold carriers). Figures (b) and (e) and (f) are reprinted with permission from ref. 34 Copyright 2024, American Chemical Society. | ||
Alternatively, band-edge PB dynamics probe state-filling near the CB minima and VB maxima, thereby reflecting the population buildup of cold carriers, as discussed earlier. Consequently, the rise time of the band-edge PB in TA measurements can be used to estimate the HC cooling rate. This approach is particularly valuable for quantum-confined systems, where discrete energy levels make accurate extraction of carrier temperatures from the high-energy tails of PB spectra challenging.33 In such cases, PB rise time provides a direct and robust measure of HC cooling. In halide perovskites, the PB rise times are typically on the sub-ps time scale (Fig. 10(b)), consistent with the cooling times extracted from time-dependent carrier temperature analysis. By examining PB rise dynamics, the dependence of HC cooling on factors such as material composition, NC size, excess excitation energy, etc. has been systematically investigated.51,62,66,106
Nonetheless, it should be noted that HC dynamics probed via band-edge PB in TA spectra are susceptible to multiple effects, including PB shifts induced by BGR and BM effects, spectral broadening due to phonon scattering and multi-exciton Auger processes etc.34,48,49 These effects can obscure the intrinsic HC cooling dynamics and complicate data interpretation. To mitigate such influences, HC dynamics can instead be directly investigated using PPP spectroscopy, as discussed earlier. In addition, PPP measurements facilitate comparative studies across materials with different bandgaps, as the excess energy can be precisely controlled via the push energy.48 The push-induced PB kinetics are typically described by an exponential decay with a time constant (τcool), which quantifies the HC cooling rate. Notably, experiments have demonstrated that τcool depends on both the pump and push fluences, corresponding to the initial cold-carrier and hot-carrier densities, respectively. This behaviour is illustrated in Fig. 10(c) and (d) for the ZI-capped CsPbBr3 NCs.105 Specifically, τcool increases linearly with the initial HC density (i.e., push fluence), while the slope of this dependence varies with the initial cold-carrier density set by the pump fluence, indicating a pronounced role of carrier–carrier interaction in HC cooling. To minimize such effects, it has been suggested to: (1) perform measurements at the lowest feasible pump fluence or (2) normalize the HC density to the cold-carrier density, Nhot0/Ncold0, and use this ratio as an independent parameter when correlating with τcool. This approach is exemplified in the study of HC dynamics in the CsPbBr3 NCs, as shown in Fig. 10(e) and (f).34 An increase in push fluence systematically slows the cooling process, consistent with the onset of the hot-phonon effect at higher carrier densities. The intercept τ0cool at zero normalized HC density reflects the intrinsic HC relaxation time, whereas a larger slope (β) signifies a more pronounced hot-phonon effect. Smaller NCs exhibit overall longer τcool values, consistent with trends observed from TA analysis (Fig. 10(b)), which can be attributed to an enhanced intrinsic phonon bottleneck effect arising from stronger quantum confinement.33,48
Table 1 summarizes the HC relaxation times of various perovskite systems characterized using different techniques. In the absence of a significant hot-phonon effect, similar sub-picosecond HC relaxation times are observed across these methods. Nevertheless, HC relaxation times can vary widely from sub-picoseconds to tens of picoseconds, depending on the excess energy and excitation fluence.65,66 Consequently, meaningful comparisons must be made under similar excitation conditions. For instance, under the same excitation excess energy in TA measurements (marked by (a) in Table 1), the HC cooling time is found to be faster in FAPbBr3, compared to the Cs- and MA-based structures. This trend is consistent with observations from PPP measurements (marked by (b) in Table 1). In addition, lead-iodide perovskites generally exhibit slower intrinsic HC relaxation time constants than lead-bromide counterparts, due to the weaker optical phonon scattering. Furthermore, the all-inorganic CsPbBr3 is more sensitive to the hot-phonon effect, as evidenced by the larger slope β.48 A more in-depth analysis of cooling dynamics and these intrinsic material properties can provide further insights into the HC relaxation mechanisms governing HC relaxation, as discussed in the following sections.
| Perovskite samples | TA (ps) | PPP (ps) | TRPL (ps) |
|---|---|---|---|
| a TA with the same excitation excess energy of 0.5 eV.117 b Intrinsic time constants evaluated from y-axis interceptions in the plots (time constants as function of push fluence) with the same push energy of 0.6 eV.48 c The HC lifetimes in nanocrystals are size-dependent. d Optical pump-IR push-THz probe. | |||
| MAPbI3 thin film | 0.6-60, ref. 8,25 | ∼0.32b–0.40, ref. 107 | 0.2–0.5, ref. 108 |
| 0.40a | |||
| FAPbI3 thin film | ∼0.2–304, ref. 10,25 | 0.22b–0.40, ref. 48 | — |
| CsPbI3 thin film | 1–30, ref. 15 | — | — |
| 0.44a | |||
| APbBr3 (A = MA, FA, Cs) thin film | ∼0.2–71, ref. 10 | 0.1–0.9, ref. 48,49 | ∼0.1 (ns), ref. 109 |
| (MA: 0.39)a | (MA:0.19)b | ||
| (FA: 0.15)a | (Cs:0.14)b | ||
| (FA:0.11)b | |||
| APbBrxI3−x (A = MA, FA, Cs) nanocrystalsc | ∼0.1–10, ref. 93 | 0.2–1.2, ref. 34,93 | 0.3–0.8, ref. 110 |
| APb1−xSnxI3 (A = MA, FA, Cs) thin film | ∼0.7–100, ref. 111,112 | 0.2–1, ref. 113![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) d d |
0.6–6 (ns), ref. 36,114 |
| APb1−xSnxI3 (A = MA, FA) nanocrystals | 0.2–14.3, ref. 106,115 | — | — |
| PEA2PbI4 thin film | ∼0.4, ref. 116 | — | — |
| (3AMP)PbI4, (4AMP)PbI4 thin film | 1.3–2.5, ref. 58 | — | — |
 | (13) |
 | (14) |
 | ||
| Fig. 11 HC temperature dynamics of the MAPbI3 thin film for (a) different excitation energies and (b) different carrier densities with pump energy at 3.1 eV; adapted with permission from ref. 25. Copyright 2015 Springer Nature. (c) Schematic of HC cooling governed by the hot-phonon effect and (d) push-fluence dependent dynamics of the HC temperature for the MAPbI3 thin film; adapted with permission from ref. 49. Copyright 2023 John Wiley and Sons. (e) Carrier-density-dependent carrier temperatures of the MAPbI3 thin film excited at 2.48 eV and (f) the effects of hot phonons and Auger heating to HC dynamics at high carrier density. Adapted with permission from ref. 8. Copyright 2017 Springer Nature. | ||
For the hot-phonon bottleneck effect occurring at high HC densities, non-equilibrium LO phonons rapidly accumulate because the intrinsic LO phonon decay time (τLO) is longer than the electron–phonon scattering time (τc), as illustrated in Fig. 11(c). Under steady-state conditions,91 the energy decay rate mediated by LO phonons decay into acoustic phonons is given by  . A large population of hot LO phonons can enhance LO phonon reabsorption, thereby increasing the effective LO phonon decay time. Under dynamic non-equilibrium conditions, the energy loss rate is modified by the ratio of Ceh/Cp relative to the static decay rate, with Ceh notably smaller than Cp.35,49,91 The energy loss rate can be expressed as:
. A large population of hot LO phonons can enhance LO phonon reabsorption, thereby increasing the effective LO phonon decay time. Under dynamic non-equilibrium conditions, the energy loss rate is modified by the ratio of Ceh/Cp relative to the static decay rate, with Ceh notably smaller than Cp.35,49,91 The energy loss rate can be expressed as:
 | (15) |
Alternatively, the energy loss rate can be evaluated by considering the generation and relaxation of LO phonons using Fermi's golden rule.8 This model accounts for phonon modes with a given wavevector q; accordingly, the corresponding energy loss rate is  . Assuming a spherical phonon distribution and dispersion-less LO phonon relationship in the Brillouin zone, the average energy loss rate per carrier for HCs in a parabolic band can be written as:8,123
. Assuming a spherical phonon distribution and dispersion-less LO phonon relationship in the Brillouin zone, the average energy loss rate per carrier for HCs in a parabolic band can be written as:8,123
 | (16) |
 is the transition matrix element with eE0 expressed as
is the transition matrix element with eE0 expressed as  , which characterizes polar optical phonon scattering via the Fröhlich mechanism, while neglecting polaron screen effects. Here,
, which characterizes polar optical phonon scattering via the Fröhlich mechanism, while neglecting polaron screen effects. Here,  is the Bose–Einstein distribution of LO phonon populations at Tc and
is the Bose–Einstein distribution of LO phonon populations at Tc and  is the quasi-Fermi level. The inverse quadratic dependence of the matrix element |Mq|2 on the wavevector q indicates that electrons primarily interact with zone-center LO phonons with small wavevectors.
is the quasi-Fermi level. The inverse quadratic dependence of the matrix element |Mq|2 on the wavevector q indicates that electrons primarily interact with zone-center LO phonons with small wavevectors.
In the presence of non-equilibrium phonons, Nq deviates from the equilibrium distribution Nq(TL). To estimate Nq, emitted LO phonons are assumed to decay with a characteristic time constant τph,
 | (17) |
In halide perovskites, LO phonons relax primarily via the more efficient Klemens channel, such that τph (equivalent to τLO in eqn (15)) is governed by the LO phonon energy and lattice temperature.8,124Eqn (17) shows the analytical solution:
 | (18) |
At higher carrier densities (>1019 cm−3), HC dynamics exhibit a distinct two-step decay process, further prolonging the cooling time to tens of picoseconds. The initial decay stage is dominated by the hot-phonon effect, whereas the subsequent slower decay at later times arises from Auger heating. The total energy loss rate, accounting for both hot-phonon and Auger heating effects can be expressed as:
 | (19) |
 | (20) |
Compared with bulk materials, the Auger effect is generally more pronounced in low dimensional structures due to confinement-enhanced Coulomb interactions and relaxed momentum conservation.98,126,127 As a result, the biexciton Auger lifetime decreases with decreasing NC volume (V), a trend observed across various material systems. This dependence can be described by τAug ∝ Vγ, where γ typically ranges from 0.5–1 depending on the degree of quantum confinement.33,128 Correspondingly, the Auger-related HC lifetime follows a similar scaling behaviour with NC volume. The temporal evolution of the HC population nhot(t) in the presence of Auger heating can be modelled as:
 | (21) |
| nhot(t) = nhot0(1 − D)e−At + De−3t/τAug | (22) |
 | (23) |
 | (24) |
 | ||
| Fig. 12 (a) HC dynamics of the MAPbBr3 thin film fitted with the two-temperature model (TTM), reproduced with permission from ref. 117. Copyright 2021 Chan et al. Advanced Energy Materials published by Wiley-VCH GmbH. (b) The calculated fast and slow decay time constants as a function of GPP/Cp at various Gep/Ce with a Cp = 20Ce. | ||
In the presence of the hot-phonon effect where energy relaxation is mediated by the decay of non-equilibrium phonons (Tp ≠ TL), the HC dynamics are modified as:112
 | (25) |
 | (26) |
 | (27) |
 .
.
As a result, Te(t) exhibits a biexponential decay comprising a fast (k+) and a slow (k−) component. In the absence of the hot-phonon effect, the fast decay rate k+ approaches k0, and Te(t) returns to a mono-exponential decay (A− → 0), as described by eqn (23) and (24). This model is consistent with experimental observations of Te(t) in mixed halide perovskite thin films, where a biexponential decay emerges at higher excitation fluences.112 The influence of the electron–phonon coupling constant Gep and phonon–phonon coupling constant Gpp on the HC relaxation time constants are illustrated in Fig. 12(b). Specifically, in the weak phonon–phonon coupling regime, the fast component τ+, which remains largely unaffected by Gpp/Cp, decreases with increasing Gep, whereas stronger phonon–phonon interaction (Gpp) leads to a shorter τ− that is insensitive to Gep/Ce. Moreover, when Gpp/Cp exceeds ∼1 THz, the τ+ begins to decrease while τ− is accelerated with larger Gep/Ce.
Beyond experimentally motivated analyses, the non-equilibrium dynamics of coupled HC and phonon populations can be investigated theoretically using ab initio approaches based on the time-dependent Boltzmann transport equation.134 Although such methods are well established for conventional semiconductors, their application to halide perovskite systems remains a frontier topic for future exploration.
 | (28) |
 | (29) |
Fig. 13(a) shows the extracted Q values obtained using this approach for perovskites with various chemical compositions, along with a comparison between halide perovskites and other semiconductor structures (Fig. 13(b)). Materials exhibiting low-Q values correspond to slower HC cooling rates and are therefore promising candidates for HC solar cell applications. The physical significance of Q is further demonstrated in a recent study on the effect from A-site cation engineering via guanidine (GA) incorporation. As shown in Fig. 13(c) and (d),11 a smaller Q value, indicating slower thermalization, is observed with increasing GA content. Importantly, the trend in Q extracted from steady-state PL measurements is consistent with the HC cooling time constants obtained independently from the TRPL analysis.
 | ||
| Fig. 13 Evaluation of the thermalization coefficient. (a) The plot of the excitation power versus ΔT for three different perovskite thin films. (b) Thermalization coefficients for various semiconductors. (a) and (b) are reprinted with permission from ref. 43. Copyright 2022 American Chemical Society. (c) Thermalization coefficients for halide perovskites with different ratios of GA cation modification and (d) corresponding HC dynamics extracted from TRPL measurements, reprinted from Ref. 11. Copyright 2025, with permission from Elsevier. | ||
3.2. Slow HC cooling for efficient carrier multiplication
Carrier multiplication (CM) or impact ionization, also referred to as multiple exciton generation (MEG) in confined systems, enables the generation of additional electron–hole pairs from a single high-energy photon, offering a promising route toward more efficient carrier-extraction devices. HCs possessing kinetic energies several times larger than the bandgap can re-excite additional carriers within a material by transferring the excess energy to other carriers (or excitons) rather than to the phonons. As a result, thermal loss is suppressed and an enhanced photocurrent can, in principle, be achieved. In an idealized framework, the CM rate constant kCM and cooling rate kcool are related by:135 | (30) |
 | ||
| Fig. 14 Simulated CM quantum yield (CMQY) (a) and the corresponding PCE (b). (c) Evidence of CM effects in FAPbI3 perovskites. Adapted with permission from ref. 137. Copyright 2018, Li et al. Springer Nature. Normalized band-edge bleaching dynamics for 7.5 nm FAPbI3 NCs under different pump photon energies, with average exciton occupation number 〈N〉 = 0.24. The inset shows similar measurements for the bulk-counterpart. (d) CM effects in a Cs0.05FA0.5MA0.45Pb0.5Sn0.5I3 thin film, showing initial bleaching amplitude |ΔA| as a function of the required photon flux IA for different pump energies. (a), (b) and (d) are adapted with permission from ref. 13. Copyright 2023, Wang et al. Springer Nature. | ||
Since carrier cooling and multiplication are intrinsically intertwined immediately after photoexcitation, TA spectroscopy is a powerful technique for capturing HC energy redistribution via carrier relaxation, reabsorption, or both. In TA measurements, the presence of CM manifests as an enhanced early-time band-edge bleaching amplitude when the pump photon flux is sufficiently low to avoid other nonlinear effects. Under CM conditions, the TA bleaching amplitude exhibits a stronger dependence on the pump fluence; fewer incident photons are required to achieve the same bleaching level compared to the case without CM.
Accordingly, two primary approaches are commonly employed to quantify CM. The first method compares the initial bleaching amplitude with the long-delay amplitude where carrier thermalization and cooling are complete, and extracts the CMQY from their ratio, as shown in Fig. 14(c). The second method fixes a reference bleaching amplitude and examines its dependence on the incident photon flux (Fig. 14(d)). This latter approach effectively mitigates unwanted nonlinear effects, such as photo-charging and spectral diffusion effect, although it does not directly resolve the carrier dynamics.
Halide perovskites have emerged as promising CM candidates for efficient electron–hole pair generation, supported by their characteristically slow HC cooling processes reported over the past decade.13,136–138 Nevertheless, CM is fundamentally constrained by the HC lifetime and competes with other scattering mechanisms during carrier cooling. A high initial carrier temperature combined with slow HC cooling is therefore favorable for efficient CM. Consequently, understanding and engineering HC behavior in CM-active materials remains a nontrivial challenge. For example, in Sn-based perovskites, cation alloying has been shown to effectively enhance both the initial carrier temperature and HC lifetime, thereby promoting CM.13 Using the Cs0.05FA0.5MA0.45Pb0.5Sn0.5I3 system and judicious device engineering, our group recently demonstrated perovskite solar cells with internal quantum efficiency exceeding 100%.13 CM/MEG studies in halide perovskites139 and other emergent semiconductors140–142 remain an active area of research.
4. Summary and outlook
Carrier temperature and HC cooling rate are fundamental physical parameters that directly determine the feasibility of HC-based optoelectronic and photovoltaic applications. Although a wide range of experimental techniques has been developed for characterizing HC dynamics, inconsistencies in data interpretation underscore the need for a deeper understanding of the assumptions, strengths and limitations associated with each method in order to establish reliable and reproducible practices.In this tutorial review, we have focused on halide perovskites as a representative material system to illustrate the characterization of HC properties using TA spectroscopy (including pump–probe and pump-push-probe spectroscopies) and PL spectroscopy. Upon photoexcitation, energetic carriers undergo rapid thermalization through carrier-carrier scattering, forming a FD distribution that can often be approximated by a MB distribution when carrier energies lie sufficiently above the quasi-Fermi level.33 This approximation is frequently adopted for halide perovskites, sometimes without rigorous validation.
These HCs broaden the high-energy tails of PB features in TA spectra and of emission profiles in PL spectra, as schematically illustrated in Fig. 15. Consequently, MB statistics are widely used to fit the high-energy tails of PB peaks in TA measurements or the high-energy tails of PL spectra to extract carrier temperatures. However, the extracted carrier temperatures are highly sensitive to the chosen fitting window, leading to considerable uncertainty and limited reproducibility.40 To address these issues, full-spectrum fitting approaches that explicitly incorporate the joint density of states, excitonic effects and phonon-induced linewidth broadening etc., have been developed and successfully applied to both TA and PL analyses.40,80
 | ||
| Fig. 15 Schematics of photophysical processes of HC cooling in TA, PPP and PL measurements. | ||
Overall, TA spectroscopy is generally regarded as the more robust and accurate technique for determining carrier temperatures, owing to its reduced susceptibility to recombination pathways and emission-related distortions. In contrast, while PL-based measurements are experimentally simpler and more widely accessible, they require sufficiently strong HC light emission and are more vulnerable to competing recombination channels, phonon broadening and transient bandgap renormalization, all of which can bias the extracted carrier temperatures.143 As an alternative and complementary approach, PPP spectroscopy enables direct interrogation of HC populations by selectively re-heating thermalized carriers. In PPP measurements, the push-induced HC population is reflected in the PB intensity, from which HC cooling dynamics can be inferred (Fig. 15 (middle panel)). Although PPP offers a conceptually straightforward route to tracking HC relaxation, careful consideration must be given to potential artefacts arising from multi-photon absorption induced by the push pulse.
Slower HC cooling is desirable for efficient HC extractions and carrier multiplication. The HC cooling rate can be inferred from the time-dependent carrier temperatures, the rise time of band-edge PB signals in TA spectroscopy, PPP bleaching dynamics, and thermalization coefficients, among other metrics. Analysing HC cooling rates across different carrier densities provides invaluable insights into the mechanisms that impede carrier cooling. In halide perovskites, HC cooling typically follows a monoexponentially decay at low carrier densities, with sub-ps time constants driven by intrinsic energy relaxation via LO phonon emission.53 At higher carrier densities, the hot-phonon bottleneck effect, involving non-equilibrium LO phonons, extends the cooling time to several ps. Furthermore, Auger heating at high carrier densities can further delay the cooling process, resulting in multi-exponential decay profiles extending to tens of ps. Beyond time-resolved measurements, fluence-dependent steady-state PL can also be used to extract the thermalization coefficients, where smaller values indicate slower HC cooling.
The mechanisms underlying slow HC cooling vary under different conditions. In bulk halide perovskites, at low carrier density (<1018 cm−3), HC relaxation is primarily governed by LO-phonon emission via electron–phonon scattering. Under these circumstances, slower HC relaxation indicates weaker electron–phonon coupling, which has been attributed to large polaron screening of phonon scattering. Studies on MAPbI3 thin films suggest that the hot-phonon effect begins to dominate at intermediate carrier densities (>1018 cm−3), while the Auger heating effect becomes significant at even higher carrier densities (∼1019 cm−3).8 For nanocrystals, quantum confinement leads to discrete energy levels that are more widely spaced than the LO-phonon energy, rendering intra-level energy relaxation via LO phonon emission less efficient and giving rise to an intrinsic phonon bottleneck effect. Therefore, HC cooling mechanisms must be carefully analysed on a case-by case basis.
Despite significant progress in understanding HC dynamics in lead halide perovskites, key challenges remain in fully harnessing HC energy while minimizing losses to cold carriers. Ongoing research efforts focus on tuning HC properties through material composition, dimensionality, nanostructuring, and phonon engineering, as well as in operando insights into hot carrier dynamics.144 Although different mechanisms govern HC cooling processes, this tutorial provides a framework for evaluating and screening of HC behaviour in emergent semiconductor systems, thereby supporting the development of next-generation HC devices. Looking forward, further advances in HC research will require the integration of multiple spectroscopic techniques with rigorous modelling frameworks that go beyond simplified MB approximations. Combining ultrafast spectroscopy with ab initio simulations, improved full spectrum analysis, and standardized fitting protocols will be essential for achieving quantitative consistency across different studies and material systems. Such efforts will not only deepen our understanding of HC physics in halide perovskites, but also provide critical design principles and modifications in conventional device architectures for next-generation HC devices, including carrier multiplication-based solar cells and hot-carrier-based optoelectronics.
Conflicts of interest
The authors declare no conflicts of interest.Data availability
Data for this article, including experimental and simulation data, are available at DR-NTU(Data) at https://doi.org/10.21979/N9/VWYDDT.Acknowledgements
This work is supported by the Ministry of Education under its AcRF Tier 1 grant (RG152/24) and AcRF Tier 2 grant (MOE-T2EP50123-0001) and the National Research Foundation (NRF) Singapore under its Competitive Research Programme (NRF-CRP25-2020-0004). Open access funding provided by Nanyang Technological University, Singapore (NTU Singapore).References
- K. Zhou, B. Qi, Z. Liu, X. Wang, Y. Sun and L. Zhang, Adv. Funct. Mater., 2024, 34, 2411671 CrossRef CAS.
- L. Chouhan, S. Ghimire, C. Subrahmanyam, T. Miyasaka and V. Biju, Chem. Soc. Rev., 2020, 49, 2869–2885 RSC.
- A. Rogalski, F. Wang, J. Wang, P. Martyniuk and W. Hu, Small Methods, 2025, 9, 2400709 CrossRef CAS.
- H. Sun, P. Dai, X. Li, J. Ning, S. Wang and Y. Qi, J. Energy Chem., 2021, 60, 300–333 CrossRef CAS.
- N. Torabi, A. Behjat, Y. Zhou, P. Docampo, R. J. Stoddard, H. W. Hillhouse and T. Ameri, Mater. Today Energy, 2019, 12, 70–94 CrossRef.
- M. Li, J. Fu, Q. Xu and T. C. Sum, Adv. Mater., 2019, 31, 1802486 CrossRef CAS PubMed.
- P. P. Joshi, S. F. Maehrlein and X. Zhu, Adv. Mater., 2019, 31, 1803054 CrossRef CAS PubMed.
- J. Fu, Q. Xu, G. Han, B. Wu, C. H. A. Huan, M. L. Leek and T. C. Sum, Nat. Commun., 2017, 8, 1300 CrossRef PubMed.
- W. Lin, S. E. Canton, K. Zheng and T. Pullerits, ACS Energy Lett., 2023, 9, 298–307 CrossRef.
- J. Yang, X. Wen, H. Xia, R. Sheng, Q. Ma, J. Kim, P. Tapping, T. Harada, T. W. Kee and F. Huang, Nat. Commun., 2017, 8, 14120 CrossRef CAS PubMed.
- Y. Zhang, H. Chen, J. Qu, R. Wang, S. Xu, J. Zhang and G. Conibeer, Sol. Energy Mater. Sol. Cells, 2025, 292, 113807 CrossRef CAS.
- S. Maiti, S. Ferro, D. Poonia, B. Ehrler, S. Kinge and L. D. Siebbeles, J. Phys. Chem. Lett., 2020, 11, 6146–6149 CrossRef CAS PubMed.
- Y. Wang, S. Ye, J. W. M. Lim, D. Giovanni, M. Feng, J. Fu, H. N. Krishnamoorthy, Q. Zhang, Q. Xu and R. Cai, Nat. Commun., 2023, 14, 6293 CrossRef CAS PubMed.
- K. Marjit, A. Das, D. Ghosh and A. Patra, ChemNanoMat, 2024, 10, e202300629 CrossRef CAS.
- Q. Shen, T. S. Ripolles, J. Even, Y. Ogomi, K. Nishinaka, T. Izuishi, N. Nakazawa, Y. Zhang, C. Ding, F. Liu, T. Toyoda, K. Yoshino, T. Minemoto, K. Katayama and S. Hayase, Appl. Phys. Lett., 2017, 111, 153903 CrossRef.
- R. T. Ross and A. J. Nozik, J. Appl. Phys., 1982, 53, 3813–3818 CrossRef CAS.
- S. Maiti, M. van der Laan, D. Poonia, P. Schall, S. Kinge and L. D. Siebbeles, Chem. Phys. Rev., 2020, 1, 011302 CrossRef.
- A. Kulkarni, W. H. Evers, S. Tomic, M. C. Beard, D. Vanmaekelbergh and L. D. Siebbeles, ACS Nano, 2018, 12, 378–384 CrossRef CAS PubMed.
- I. Ahmed, L. Shi, H. Pasanen, P. Vivo, P. Maity, M. Hatamvand and Y. Zhan, Light: Sci. Appl., 2021, 10, 174 CrossRef CAS PubMed.
- J. Fu, S. Ramesh, J. W. Melvin Lim and T. C. Sum, Chem. Rev., 2023, 123, 8154–8231 CrossRef CAS PubMed.
- A. Le Bris, L. Lombez, S. Laribi, G. Boissier, P. Christol and J.-F. Guillemoles, Energy Environ. Sci., 2012, 5, 6225–6232 RSC.
- T. Faber, L. Filipovic and L. Koster, J. Phys. Chem. Lett., 2024, 15, 12601–12607 CrossRef CAS PubMed.
- R. Joshi and D. Ferry, Phys. Rev. B:Condens. Matter Mater. Phys., 1989, 39, 1180 CrossRef PubMed.
- W. Pötz, Phys. Rev. B:Condens. Matter Mater. Phys., 1987, 36, 5016 CrossRef PubMed.
- Y. Yang, D. P. Ostrowski, R. M. France, K. Zhu, J. Van De Lagemaat, J. M. Luther and M. C. Beard, Nat. Photon., 2016, 10, 53–59 CrossRef CAS.
- P. Langot, N. Del Fatti, D. Christofilos, R. Tommasi and F. Vallée, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 14487 CrossRef PubMed.
- G. Srivastava, Phys. Rev. B:Condens. Matter Mater. Phys., 2008, 77, 155205 CrossRef.
- Y. Zhang, G. Conibeer, S. Liu, J. Zhang and J. F. Guillemoles, Prog. Photovoltaics Res. Appl., 2022, 30, 581–596 CrossRef CAS.
- S. Dal Conte, L. Vidmar, D. Golež, M. Mierzejewski, G. Soavi, S. Peli, F. Banfi, G. Ferrini, R. Comin and B. M. Ludbrook, Nat. Phys., 2015, 11, 421–426 Search PubMed.
- D. Novko, F. Caruso, C. Draxl and E. Cappelluti, Phys. Rev. Lett., 2020, 124, 077001 CrossRef CAS PubMed.
- E. Baldini, A. Mann, L. Benfatto, E. Cappelluti, A. Acocella, V. M. Silkin, S. V. Eremeev, A. B. Kuzmenko, S. Borroni and T. Tan, Phys. Rev. Lett., 2017, 119, 097002 CrossRef CAS PubMed.
- L. Perfetti, P. Loukakos, M. Lisowski, U. Bovensiepen, H. Eisaki and M. Wolf, Phys. Rev. Lett., 2007, 99, 197001 CrossRef CAS PubMed.
- M. Li, S. Bhaumik, T. W. Goh, M. S. Kumar, N. Yantara, M. Grätzel, S. Mhaisalkar, N. Mathews and T. C. Sum, Nat. Commun., 2017, 8, 14350 CrossRef CAS.
- J. W. M. Lim, Y. Guo, M. Feng, R. Cai and T. C. Sum, J. Am. Chem. Soc., 2023, 146, 437–449 CrossRef PubMed.
- S. Kaniyankandy, Colloids Surf., A, 2022, 635, 128025 CrossRef CAS.
- H.-H. Fang, S. Adjokatse, S. Shao, J. Even and M. A. Loi, Nat. Commun., 2018, 9, 243 CrossRef PubMed.
- D. Niesner, H. Zhu, K. Miyata, P. P. Joshi, T. J. Evans, B. J. Kudisch, M. T. Trinh, M. Marks and X.-Y. Zhu, J. Am. Chem. Soc., 2016, 138, 15717–15726 CrossRef CAS.
- G. Kaur, A. Shukla, K. J. Babu and H. N. Ghosh, Chem. Rec., 2022, 22, e202200106 CrossRef CAS PubMed.
- G. Xing, N. Mathews, S. Sun, S. S. Lim, Y. M. Lam, M. Grätzel, S. Mhaisalkar and T. C. Sum, Science, 2013, 342, 344–347 CrossRef CAS PubMed.
- J. W. M. Lim, D. Giovanni, M. Righetto, M. Feng, S. G. Mhaisalkar, N. Mathews and T. C. Sum, J. Phys. Chem. Lett., 2020, 11, 2743–2750 CrossRef CAS PubMed.
- G. Yang, Y. Tu, J. Ye, R. Liu, Y. Zang, L. Zhang, Y. Wang, G. Li, Q. Zhou and L. Chu, J. Alloys Compd., 2023, 952, 170051 CrossRef CAS.
- M. B. Price, J. Butkus, T. C. Jellicoe, A. Sadhanala, A. Briane, J. E. Halpert, K. Broch, J. M. Hodgkiss, R. H. Friend and F. Deschler, Nat. Commun., 2015, 6, 8420 CrossRef CAS PubMed.
- J. W. M. Lim, Y. Wang, J. Fu, Q. Zhang and T. C. Sum, ACS Energy Lett., 2022, 7, 749–756 CrossRef CAS.
- S. Sourabh, H. Afshari, V. R. Whiteside, G. E. Eperon, R. A. Scheidt, T. D. Creason, M. Furis, A. R. Kirmani, B. Saparov and J. M. Luther, Prog. Photovoltaics Res. Appl., 2024, 32, 546–555 CrossRef CAS.
- T. J. Evans, K. Miyata, P. P. Joshi, S. Maehrlein, F. Liu and X.-Y. Zhu, J. Phys. Chem. C, 2018, 122, 13724–13730 CrossRef CAS.
- F. Sekiguchi, H. Hirori, G. Yumoto, A. Shimazaki, T. Nakamura, A. Wakamiya and Y. Kanemitsu, Phys. Rev. Lett., 2021, 126, 077401 CrossRef CAS PubMed.
- L. Gatto, I. Poli, D. Meggiolaro, F. Grandi, G. Folpini, A. Treglia, E. Cinquanta, A. Petrozza, F. De Angelis and C. Vozzi, ACS Energy Lett., 2025, 10, 1382–1388 CrossRef CAS PubMed.
- T. R. Hopper, A. Gorodetsky, J. M. Frost, C. Müller, R. Lovrincic and A. A. Bakulin, ACS Energy Lett., 2018, 3, 2199–2205 CrossRef CAS PubMed.
- M. Feng, S. Ye, J. W. M. Lim, Y. Guo, R. Cai, Q. Zhang, H. He and T. C. Sum, Small, 2023, 19, 2301831 CrossRef CAS PubMed.
- B. Wang, J. W. M. Lim, S. M. Loh, R. Mayengbam, S. Ye, M. Feng, H. He, X. Liang, R. Cai and Q. Zhang, ACS Nano, 2024, 18, 10807–10817 CrossRef CAS PubMed.
- B. T. Diroll and R. D. Schaller, Adv. Funct. Mater., 2019, 29, 1901725 CrossRef.
- K. Frohna, T. Deshpande, J. Harter, W. Peng, B. A. Barker, J. B. Neaton, S. G. Louie, O. M. Bakr, D. Hsieh and M. Bernardi, Nat. Commun., 2018, 9, 1829 CrossRef PubMed.
- A. Mondal, J. Aneesh, V. Kumar Ravi, R. Sharma, W. J. Mir, M. C. Beard, A. Nag and K. Adarsh, Phys. Rev. B, 2018, 98, 115418 CrossRef CAS.
- K. Fan, C. C. S. Chan, L. Yuan, K. Yan and K. S. Wong, ACS Photonics, 2022, 9, 2304–2314 CrossRef CAS.
- K. Lobato and L. Peter, J. Phys. Chem. B, 2006, 110, 21920–21923 CrossRef CAS PubMed.
- D. O. Tiede, K. A. Koch, C. Romero-Pérez, K. B. Ucer, M. E. Calvo, J. F. Galisteo-López, H. Míguez and A. R. Srimath Kandada, Adv. Opt. Mater., 2024, 12, 2401483 CrossRef CAS.
- V. A. Hintermayr, L. Polavarapu, A. S. Urban and J. Feldmann, ACS Nano, 2018, 12, 10151–10158 CrossRef CAS PubMed.
- J. Yin, R. Naphade, P. Maity, L. Gutiérrez-Arzaluz, D. Almalawi, I. S. Roqan, J.-L. Brédas, O. M. Bakr and O. F. Mohammed, Nat. Commun., 2021, 12, 3995 CrossRef CAS PubMed.
- K. Fan, K. A. Sergeeva, A. A. Sergeev, L. Zhang, C. C. Chan, Z. Li, X. Zhong, S. V. Kershaw, J. Liu and A. L. Rogach, ACS Nano, 2024, 18, 18011–18021 CrossRef CAS PubMed.
- K. Fan, H. Liu, T. Pan, Y. Wu, Y. Hai, K. Vighnesh, A. A. Sergeev, J. Wu, A. L. Rogach and K. S. Wong, ACS Nano, 2025, 19, 27552–27562 CrossRef CAS PubMed.
- S. Ramesh, D. Giovanni, M. Righetto, S. Ye, E. Fresch, Y. Wang, E. Collini, N. Mathews and T. C. Sum, Adv. Energy Mater., 2022, 12, 2103556 CrossRef CAS.
- H. Chung, S. I. Jung, H. J. Kim, W. Cha, E. Sim, D. Kim, W. K. Koh and J. Kim, Angew. Chem., Int. Ed., 2017, 129, 4224–4228 CrossRef.
- R. R. Tamming, J. Butkus, M. B. Price, P. Vashishtha, S. K. Prasad, J. E. Halpert, K. Chen and J. M. Hodgkiss, ACS Photonics, 2019, 6, 345–350 CrossRef CAS.
- G. Yumoto, H. Tahara, T. Kawawaki, M. Saruyama, R. Sato, T. Teranishi and Y. Kanemitsu, J. Phys. Chem. Lett., 2018, 9, 2222–2228 CrossRef CAS PubMed.
- M. T. Trinh, X. Wu, D. Niesner and X.-Y. Zhu, J. Mater. Chem. A, 2015, 3, 9285–9290 RSC.
- J. Chen, M. E. Messing, K. Zheng and T. Pullerits, J. Am. Chem. Soc., 2019, 141, 3532–3540 CrossRef CAS PubMed.
- K. Marjit, G. Ghosh, R. K. Biswas, S. Ghosh, S. K. Pati and A. Patra, J. Phys. Chem. Lett., 2022, 13, 5431–5440 CrossRef CAS PubMed.
- Z. Guo, Y. Wan, M. Yang, J. Snaider, K. Zhu and L. Huang, Science, 2017, 356, 59–62 CrossRef CAS PubMed.
- P. Guyot-Sionnest, M. Shim, C. Matranga and M. Hines, Phys. Rev. B:Condens. Matter Mater. Phys., 1999, 60, R2181 CrossRef CAS.
- F. T. Rabouw, R. Vaxenburg, A. A. Bakulin, R. J. van Dijk-Moes, H. J. Bakker, A. Rodina, E. Lifshitz, A. L. Efros, A. F. Koenderink and D. Vanmaekelbergh, ACS Nano, 2015, 9, 10366–10376 CrossRef CAS PubMed.
- J. Shah and R. Leite, Phys. Rev. Lett., 1969, 22, 1304 CrossRef CAS.
- K. Turvey and J. Allen, J. Phys. Chem. C, 1973, 6, 2887 CAS.
- E. Gross, S. Permogorov, Y. Morozenko and B. Kharlamov, Phys. Status Solidi B, 1973, 59, 551–560 CrossRef CAS.
- L. Selmi, M. Mastrapasqua, D. M. Boulin, J. D. Bude, M. Pavesi, E. Sangiorgi and M. R. Pinto, IEEE Trans. Electron Devices, 2002, 45, 802–808 CrossRef.
- M. Yang, Y. Chen, G. Shu, J. Shen, S. Hung, G. Chi, T. Lin, Y. Lee, C. Chen and C. Ko, Appl. Phys. A: Mater. Sci. Process., 2008, 90, 123–127 CrossRef CAS.
- J. Bude, N. Sano and A. Yoshii, Phys. Rev. B:Condens. Matter Mater. Phys., 1992, 45, 5848 CrossRef CAS PubMed.
- Y. Wang, X. Sun, Z. Chen, Y. Y. Sun, S. Zhang, T. M. Lu, E. Wertz and J. Shi, Adv. Mater., 2017, 29, 1702643 CrossRef PubMed.
- S. Kallatt, G. Umesh and K. Majumdar, J. Phys. Chem. Lett., 2016, 7, 2032–2038 CrossRef CAS PubMed.
- S. Lyon, J. Lumin., 1986, 35, 121–154 CrossRef CAS.
- K. J. Savill, M. T. Klug, R. L. Milot, H. J. Snaith and L. M. Herz, J. Phys. Chem. Lett., 2019, 10, 6038–6047 CrossRef CAS PubMed.
- S. Gogoi, S. Das, R. Gupta and S. D. Verma, J. Phys. Chem. Lett., 2025, 16, 3832–3839 CrossRef CAS.
- G. Lasher and F. Stern, Phys. Rev., 1964, 133, A553 CrossRef.
- P. Wurfel, J. Phys. Chem. C, 1982, 15, 3967 Search PubMed.
- J. K. Katahara and H. W. Hillhouse, J. Appl. Phys., 2014, 116, 173504 CrossRef.
- R. Elliott, Phys. Rev., 1957, 108, 1384 CrossRef CAS.
- H. Esmaielpour, L. Lombez, M. Giteau, J. F. Guillemoles and D. Suchet, Prog. Photovoltaics Res. Appl., 2022, 30, 1354–1362 CrossRef CAS.
- K. Chen, A. J. Barker, F. L. Morgan, J. E. Halpert and J. M. Hodgkiss, J. Phys. Chem. Lett., 2015, 6, 153–158 CrossRef CAS PubMed.
- T. Elsaesser, J. Shah, L. Rota and P. Lugli, Phys. Rev. Lett., 1991, 66, 1757 CrossRef CAS PubMed.
- A. Gregory, S. Usher, F. Majumder and R. Phillips, Solid State Commun., 1993, 87, 605–608 CrossRef CAS.
- S. Nüsse, P. H. Bolivar, H. Kurz, V. Klimov and F. Levy, Phys. Rev. B:Condens. Matter Mater. Phys., 1997, 56, 4578 CrossRef.
- V. Klimov, P. H. Bolivar and H. Kurz, Phys. Rev. B:Condens. Matter Mater. Phys., 1995, 52, 4728 CrossRef CAS PubMed.
- M. Righetto, S. S. Lim, D. Giovanni, J. W. M. Lim, Q. Zhang, S. Ramesh, Y. K. E. Tay and T. C. Sum, Nat. Commun., 2020, 11, 2712 CrossRef CAS PubMed.
- J. Ye, N. Mondal, B. P. Carwithen, Y. Zhang, L. Dai, X.-B. Fan, J. Mao, Z. Cui, P. Ghosh and C. Otero-Martínez, Nat. Commun., 2024, 15, 8120 CrossRef CAS PubMed.
- W. Niu, R. Chen, T. Pang, Y. Zheng, T. Wu, R. Zhang and D. Chen, Adv. Opt. Mater., 2024, 12, 2400307 CrossRef CAS.
- J. Qin, Y. Tang, J. Zhang, T. Shen, M. Karlsson, T. Zhang, W. Cai, L. Shi, W.-X. Ni and F. Gao, Mater. Horiz., 2023, 10, 1446–1453 RSC.
- E. M. Hutter, G. E. Eperon, S. D. Stranks and T. J. Savenije, J. Phys. Chem. Lett., 2015, 6, 3082–3090 CrossRef CAS PubMed.
- H. M. Jang, J.-S. Kim, J.-M. Heo and T.-W. Lee, APL Mater., 2020, 8, 020904 CrossRef CAS.
- Y. Jiang, M. Cui, S. Li, C. Sun, Y. Huang, J. Wei, L. Zhang, M. Lv, C. Qin and Y. Liu, Nat. Commun., 2021, 12, 336 CrossRef CAS PubMed.
- W. Zou, R. Li, S. Zhang, Y. Liu, N. Wang, Y. Cao, Y. Miao, M. Xu, Q. Guo and D. Di, Nat. Commun., 2018, 9, 608 CrossRef PubMed.
- H. Kim, L. Zhao, J. S. Price, A. J. Grede, K. Roh, A. N. Brigeman, M. Lopez, B. P. Rand and N. C. Giebink, Nat. Commun., 2018, 9, 4893 CrossRef.
- C. Wang, W. Chu, F. Ye, Z. Ou, Z. Li, Q. Guo, Z. Zheng, Z. Wang, X. Liu and G. Fang, Chem, 2022, 8, 3051–3063 CAS.
- X. Jia, J. Jiang, Y. Zhang, J. Qiu, S. Wang, Z. Chen, N. Yuan and J. Ding, Appl. Phys. Lett., 2018, 112, 143903 CrossRef.
- T. Wang, L. Jin, J. Hidalgo, W. Chu, J. M. Snaider, S. Deng, T. Zhu, B. Lai, O. Prezhdo and J.-P. Correa-Baena, Sci. Adv., 2020, 6, eabb1336 CrossRef CAS PubMed.
- P. Zeng, X. Ren, L. Wei, H. Zhao, X. Liu, X. Zhang, Y. Xu, L. Yan, K. Boldt and T. A. Smith, Angew. Chem., Int. Ed., 2022, 134, e202111443 CrossRef.
- T. R. Hopper, A. Gorodetsky, A. Jeong, F. Krieg, M. I. Bodnarchuk, M. Maimaris, M. Chaplain, T. J. Macdonald, X. Huang and R. Lovrincic, Nano Lett., 2020, 20, 2271–2278 CrossRef CAS PubMed.
- L. Dai, Z. Deng, F. Auras, H. Goodwin, Z. Zhang, J. C. Walmsley, P. D. Bristowe, F. Deschler and N. C. Greenham, Nat. Photon., 2021, 15, 696–702 CrossRef CAS.
- S. S. Lim, D. Giovanni, Q. Zhang, A. Solanki, N. F. Jamaludin, J. W. M. Lim, N. Mathews, S. Mhaisalkar, M. S. Pshenichnikov and T. C. Sum, Sci. Adv., 2019, 5, eaax3620 CrossRef CAS PubMed.
- H. Y. Hsu, C. Y. Wang, A. Fathi, J. W. Shiu, C. C. Chung, P. S. Shen, T. F. Guo, P. Chen, Y. P. Lee and E. W. G. Diau, Angew. Chem., Int. Ed., 2014, 126, 9493–9496 CrossRef.
- H. Zhu, K. Miyata, Y. Fu, J. Wang, P. P. Joshi, D. Niesner, K. W. Williams, S. Jin and X. Y. Zhu, Science, 2016, 353, 1409–1413 CrossRef CAS PubMed.
- S. Mishra, D. Acharjee, S. De, A. B. Mahato, M. K. Panda, D. Samanta, S. Ghosh and S. Ghosh, J. Phys. Chem. C, 2025, 129, 14458–14466 CrossRef CAS.
- W. Yan, C. Li, C. Peng, S. Tan, J. Zhang, H. Jiang, F. Xin, F. Yue and Z. Zhou, Adv. Mater., 2024, 36, 2312170 CrossRef CAS PubMed.
- M. Monti, K. I. Jayawardena, E. Butler-Caddle, R. M. Bandara, J. M. Woolley, M. Staniforth, S. R. P. Silva and J. Lloyd-Hughes, Phys. Rev. B, 2020, 102, 245204 CrossRef CAS.
- A. M. Ulatowski, M. D. Farrar, H. J. Snaith, M. B. Johnston and L. M. Herz, ACS Photonics, 2021, 8, 2509–2518 CrossRef CAS PubMed.
- L. J. van de Ven, E. K. Tekelenburg, M. Pitaro, J. Pinna and M. A. Loi, ACS Energy Lett., 2024, 9, 992–999 CrossRef CAS PubMed.
- L. Dai, J. Ye and N. C. Greenham, Light: Sci. Appl., 2023, 12, 208 CrossRef CAS PubMed.
- Q. Wei, H. Ren, J. Liu, Q. Liu, C. Wang, T. W. Lau, L. Zhou, T. Bian, Y. Zhou and P. Wang, ACS Energy Lett., 2023, 8, 4315–4322 CrossRef CAS.
- C. C. Chan, K. Fan, H. Wang, Z. Huang, D. Novko, K. Yan, J. Xu, W. C. Choy, I. Lončarić and K. S. Wong, Adv. Energy Mater., 2021, 11, 2003071 CrossRef CAS.
- J. Meng, Z. Lan, W. Lin, M. Liang, X. Zou, Q. Zhao, H. Geng, I. E. Castelli, S. E. Canton and T. Pullerits, Chem. Sci., 2022, 13, 1734–1745 RSC.
- C. Zhou, T. Zhang, C. Zhang, X. Liu, J. Wang, J. Lin and X. Chen, Adv. Sci., 2022, 9, 2103491 CrossRef CAS PubMed.
- H. Li, Q. Wang, Y. Oteki, C. Ding, D. Liu, Y. Guo, Y. Li, Y. Wei, D. Wang and Y. Yang, Adv. Mater., 2023, 35, 2301834 CrossRef CAS.
- D. Zanato, N. Balkan, B. Ridley, G. Hill and W. Schaff, Semicond. Sci. Technol., 2004, 19, 1024 CrossRef CAS.
- B. Ridley, Semicond. Sci. Technol., 1989, 4, 1142 CrossRef.
- J. T. Devreese and F. M. Peeters, The physics of the two-dimensional electron gas, Springer Science & Business Media, 2012 Search PubMed.
- P. Klemens, Phys. Rev., 1966, 148, 845 CrossRef CAS.
- M. Downer and C. Shank, Phys. Rev. Lett., 1986, 56, 761 CrossRef CAS PubMed.
- V. I. Klimov, A. A. Mikhailovsky, D. McBranch, C. A. Leatherdale and M. G. Bawendi, Science, 2000, 287, 1011–1013 CrossRef CAS PubMed.
- I. Robel, R. Gresback, U. Kortshagen, R. D. Schaller and V. I. Klimov, Phys. Rev. Lett., 2009, 102, 177404 CrossRef PubMed.
- Y. Li, X. Luo, T. Ding, X. Lu and K. Wu, Angew. Chem., Int. Ed., 2020, 132, 14398–14401 CrossRef.
- P. B. Allen, Phys. Rev. Lett., 1987, 59, 1460 CrossRef CAS PubMed.
- J. C. Johannsen, S. Ulstrup, F. Cilento, A. Crepaldi, M. Zacchigna, C. Cacho, I. E. Turcu, E. Springate, F. Fromm and C. Raidel, Phys. Rev. Lett., 2013, 111, 027403 CrossRef PubMed.
- L. Waldecker, R. Bertoni, R. Ernstorfer and J. Vorberger, Phys. Rev. X, 2016, 6, 021003 Search PubMed.
- S. Brorson, A. Kazeroonian, J. Moodera, D. Face, T. Cheng, E. Ippen, M. Dresselhaus and G. Dresselhaus, Phys. Rev. Lett., 1990, 64, 2172 CrossRef CAS PubMed.
- Z. Luo, T. Shu, Z. Chen, T. Jiang, H. Wu and T. Lai, Semicond. Sci. Technol., 2019, 34, 105011 CrossRef CAS.
- F. Caruso and D. Novko, Adv. Phys. X, 2022, 7, 2095925 Search PubMed.
- M. C. Beard, A. G. Midgett, M. C. Hanna, J. M. Luther, B. K. Hughes and A. J. Nozik, Nano Lett., 2010, 10, 3019–3027 CrossRef CAS PubMed.
- C. de Weerd, L. Gomez, A. Capretti, D. M. Lebrun, E. Matsubara, J. Lin, M. Ashida, F. C. Spoor, L. D. Siebbeles and A. J. Houtepen, Nat. Commun., 2018, 9, 4199 CrossRef PubMed.
- M. Li, R. Begum, J. Fu, Q. Xu, T. M. Koh, S. A. Veldhuis, M. Grätzel, N. Mathews, S. Mhaisalkar and T. C. Sum, Nat. Commun., 2018, 9, 4197 CrossRef PubMed.
- Y. Chen, J. Yin, Q. Wei, C. Wang, X. Wang, H. Ren, S. F. Yu, O. M. Bakr, O. F. Mohammed and M. Li, Nat. Photon., 2022, 16, 485–490 CrossRef CAS.
- Y. Chen, J. Yin, Q. Wei, C. Wang, X. Wang, H. Ren, S. F. Yu, M. Bakr, O. F. Mohammed and M. Li, Nat. Photonics, 2022, 16, 485–490 CrossRef CAS.
- R. Karmakar, P. Taank, D. Ghoshal, P. Yadav, D. Mandal, M. Shrivastava, A. Agarwal, M. C. Beard, E. M. Miller and K. V. Adarsh, Phys. Rev. Lett., 2025, 134, 026903 CrossRef CAS PubMed.
- J. Noh, C. Livache, D. Hahm, V. Pinchetti, H. Jin, C. Kim and V. I. Klimov, Nat. Commun., 2025, 16, 2952 CrossRef CAS PubMed.
- H. Jin, C. Livache, W. D. Kim, B. T. Diroll, R. D. Schaller and V. I. Klimov, Nat. Mater., 2023, 22, 1013–1021 CrossRef CAS PubMed.
- E. K. Tekelenburg, F. V. Camargo, A. Filippetti, A. Mattoni, L. J. van de Ven, M. Pitaro, G. Cerullo and M. A. Loi, Adv. Mater., 2025, 37, 2411892 CrossRef CAS PubMed.
- H. Afshari, V. Mapara, S. Sourabh, M. N. Khanal, V. R. Whitesides, R. A. Scheidt, M. C. Beard, G. E. Eperon, I. R. Sellers and M. Furis, EES Sol., 2025, 1, 828–838 RSC.
Footnote |
| † These authors contributed equally to this article. |
| This journal is © The Royal Society of Chemistry 2026 |