
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cyclopropane-to-organoboron conversion via C–H and C–C bond activation
Shuyu
Kang†
,
Xueli
Lv†
and
Zhuangzhi
Shi
 *
*
State Key Laboratory of Coordination Chemistry, Chemistry and Biomedicine Innovation Center (ChemBIC), School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210093, China. E-mail: shiz@nju.edu.cn
First published on 13th January 2026
Abstract
Organoborons have emerged as a class of privileged building blocks in modern organic synthesis, enabling unparalleled molecular diversity and serving as versatile carboxylic acid bioisosteres with profound implications in drug discovery. Concurrently, cyclopropanes have garnered sustained attention as unique synthetic platforms, with their rigid and highly strained three-membered ring structures conferring distinctive steric and electronic properties that facilitate selective C–H and C–C activation processes. The strategic transformation of cyclopropanes into organoborons represents a particularly appealing synthetic approach, offering access to valuable molecular architectures. This review systematically examines the seminal advancements of cyclopropane-to-organoboron conversion over recent years, employing a structured classification based on two fundamental activation modes: C–H borylation and C–C borylation. The review provides in-depth mechanistic elucidation, with particular emphasis on catalytic cycles, key reactive intermediates, and stereodiscrimination processes, thereby offering fundamental insights into the governing principles of these transformations. Looking forward, continued innovation in catalyst design and the exploration of novel reaction pathways are anticipated to significantly expand the synthetic utility and scope of conversions, potentially opening new frontiers in organic synthesis and medicinal chemistry.
 Shuyu Kang | Shuyu Kang was born in Henan Province, People's Republic of China, in 1997. He received his BS degree from Zhengzhou University in 2020. He then moved to Nanjing University, where he is currently pursuing his PhD under the supervision of Prof. Zhuangzhi Shi. His current research focuses on boron chemistry. |
 Xueli Lv | Xueli Lv was born in Shandong province, People's Republic of China, in 1995. She received her BS and MS degrees in chemistry and organic chemistry from Qingdao University of Science & Technology and Nankai University in 2017 and 2020. Then, she moved to Nanjing University and completed her PhD in organic chemistry with Prof. Zhuangzhi Shi. Her current research efforts are focused on asymmetric C–H activation. |
 Zhuangzhi Shi | Zhuangzhi Shi was born in Jiangsu province, People's Republic of China, in 1983. He received his BS and MS degrees in chemistry and organic chemistry from Yangzhou University in 2005 and 2008. Then, he moved to Peking University and completed his PhD in medicinal chemistry with Prof. Ning Jiao. In 2011, he joined the group of Prof. Frank Glorius at Munster University (Germany), as an Alexander von Humboldt Research Fellow. In 2014, he joined the Department of Chemistry at Nanjing University as a full professor. His current research efforts are focused on synthetic methodology, including the activation of inert chemical bonds, main group chemistry and asymmetric catalysis. |
Key learning points(1) Highlight the strategic significance of cyclopropane-to-organoboron conversion in advancing organic synthesis and drug discovery frontiers.(2) Discern the applicable cyclopropane types for organoboron preparation and grasp their inherent characteristics and reactivities. (3) Reveal the intricate mechanisms, influencing elements, and practical applications of cyclopropane C–H activation in organoboron synthesis. (4) Probe into the specific procedures, reaction intermediates, and product selectivities during cyclopropane C–C activation for organoboron generation. (5) Evaluate the current limitations and challenges of cyclopropane-to-organoboron conversion and project its future developmental trajectories. |
1. Introduction
Cyclopropane, a deceptively simple molecule, holds significant importance in organic chemistry due to its highly strained three-membered ring structure.1–4 This unique architecture, akin to a tightly coiled spring, is the foundation of its exceptional chemical reactivity.5,6 Cyclopropane units frequently serve as key structural motifs in natural products and pharmaceuticals (for selected examples, see Fig. 1).7 For example, they can enhance antibiotic activity by improving target binding or modulate biological activity in drug molecules by influencing conformation and receptor interactions. Their controlled flexibility also allows for better fit within protein active sites. Such properties of cyclopropane, from its strained ring structure to its versatile role in modulating biological activity, make it an indispensable component in the realm of organic chemistry. | ||
| Fig. 1 Selective examples of cyclopropane-based drugs and natural products. | ||
Well-established methods exist for their preparation, often under mild conditions with good functional group tolerance (Fig. 2). Synthetic strategies for cyclopropane derivatives can generally be classified into three main categories: [2+1] cycloaddition of carbenes with alkenes, such as Simmons–Smith reaction;8 ring-contraction reactions;9 and ring-closing cyclopropanation of acyclic precursors, such as the Corey–Chaykovsky reaction.10 In addition to these principal approaches, other useful routes include functionalization of cyclopropenes,11 contraction of cyclobutenes,12 the use of cyclopropyl Grignard reagents or cyclopropyl halides,13,14 and the Kulinkovich reaction employing esters or amides with Grignard reagents.15 Given their diverse synthetic accessibility, ease of derivatization, and unique strain-driven reactivity, cyclopropanes are ideal substrates for further functionalization.
 | ||
| Fig. 2 Methods for the synthesis of cyclopropanes. | ||
Organoborons hold a position of paramount importance across numerous domains of chemical science.16 In the pharmaceutical industry, the integration of boron groups has spurred the advancement of a multitude of contemporary drugs (Fig. 3).17 Specifically, in the realm of anticancer agents, the boron group can be meticulously designed to engage with specific cancer-related proteins. This interaction disrupts the normal function of these proteins, thereby effectively inhibiting tumor growth. In the case of antibacterial drugs, boron-containing compounds can target unique bacterial enzymes. This provides novel strategies for combating drug-resistant bacteria. In synthetic chemistry, which serves as the cornerstone for constructing complex organic molecules, organoboronates assume a central role. They are capable of forming C–C and C–heteroatom bonds with high precision, making them reliable building blocks in the synthesis of a wide variety of organic compounds.
 | ||
| Fig. 3 Selective examples of organoboron-based biologically and pharmacologically active molecules. | ||
The synthesis of organoboron compounds continues to be a pivotal area in organic chemistry, driven by their extensive applications.18,19 Among the established methodologies, the Brown hydroboration of olefins is particularly notable for its classic anti-Markovnikov transformation (Fig. 4A).20 This reaction features the simultaneous addition of hydrogen and boron atoms to the same face of the double bond via a four-membered concerted transition state. In contrast, the conversion of cyclopropanes to organoboron compounds introduces greater complexity and diversity, presenting both challenges and opportunities. Historically, cyclopropane borylation relied on pre-functionalized substrates like cyclopropyl halides or Grignard reagents.21,22 They were often limited by a narrow substrate scope, restricting the variety of cyclopropane derivatives that could be effectively utilized. Recent advancements have significantly expanded the synthesis of organoboron compounds from cyclopropanes through the activation of C–H and C–C bonds (Fig. 4B). However, achieving precise selectivity in these transformations remains a critical challenge. This includes differentiating between C–H and C–C activation and controlling the selectivity of distal and proximal σ-bonds within the cyclopropane ring. Through innovative strategies such as catalyst design, the use of directing groups, and the development of novel reaction systems, it is now possible to selectively synthesize a diverse range of organoboron compounds from cyclopropanes.
 | ||
| Fig. 4 (A) Classical hydroboration of alkenes with borane reagents. (B) Modern strategies for synthesis of organoboron compounds from cyclopropanes by C–H and C–C activation. | ||
This review aims to comprehensively cover the two main strategies for the cyclopropane-to-organoboron conversion: C–H borylation and C–C borylation. Given the vast number of C–C activation cases, we have further subclassified these reactions based on the types of cyclopropanes involved: (1) alk(en/yn)ylcyclopropanes; (2) gem-difluorocyclopropanes; (3) aminocyclopropanes; (4) iminocyclopropanes; (5) simple cyclopropanes. For each category, we will not only examine the reaction inception and evolution but also delve into the scope and limitations. We will support our analysis with representative reaction pathways derived from experimental data and density functional theory (DFT) calculations. In the concluding section, we will highlight the remaining challenges and opportunities in this field. By identifying the areas that require further breakthroughs, we hope to provide a clear roadmap for future research endeavors, inspiring scientists to continue exploring the fascinating intersection of cyclopropane and boron chemistry.
2. C–H borylation of cyclopropanes
Compared with traditional C–H borylation substrates such as arenes, alkenes, and linear alkanes, cyclopropanes represent a particularly intriguing class of aliphatic substrates owing to their distinctive bonding framework and pronounced ring strain.23 The planar arrangement of the three carbon atoms imposes severe angular strain (60° bond angles), coupled with torsional strain from the eclipsed C–H bonds. As a result, the C–C bonds exhibit substantial π-character and behave more like C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds, whereas the C–H bonds display greater σ-character, shorter bond lengths, and higher bond dissociation energies (106 kcal mol−1) compared with typical alkanes (101 kcal mol−1 for the C–H bond in ethane).24 These structural and electronic features render C–H borylation of cyclopropanes especially challenging, yet the resulting borylated cyclopropanes can participate in diverse, high-value transformations, offering significant opportunities for molecular diversification and synthetic innovation.
C bonds, whereas the C–H bonds display greater σ-character, shorter bond lengths, and higher bond dissociation energies (106 kcal mol−1) compared with typical alkanes (101 kcal mol−1 for the C–H bond in ethane).24 These structural and electronic features render C–H borylation of cyclopropanes especially challenging, yet the resulting borylated cyclopropanes can participate in diverse, high-value transformations, offering significant opportunities for molecular diversification and synthetic innovation.
Early borylation strategies relied on pre-functionalized cyclopropanes, such as halides or organometallics, often requiring stoichiometric metal reagents.25 While effective, these approaches offer limited substrate scope and raise sustainability concerns. In contrast, transition metal-catalyzed C–H borylation of cyclopropanes has emerged as a more direct and versatile alternative. Leveraging the ring strain and rigidity of the three-membered scaffold, these reactions enable site-selective functionalization without prior activation, providing efficient access to structurally defined organoboron compounds.
Among the various transition metal systems explored for C–H borylation, iridium catalysis has proven particularly effective.26 In 2013, Hartwig and colleagues reported an Ir-catalyzed borylation of C–H bonds in cyclopropanes using B2pin2 (Scheme 1).27 Considering the broad utility of the 4,4′-dtbpy/Ir catalyst combination in aromatic C–H activation, the authors conducted a bidentate nitrogen ligand screen that identified L1 as the optimal ligand for this system and subsequently refined the reaction conditions through solvent evaluation. The reaction was also shown to be compatible with a more accessible catalyst, [Ir(cod)(OMe)]2, albeit at the cost of slightly reduced yields. Notably, substrates having bulkier groups such as bromo or carbonyl groups exhibited higher diastereoselectivity compared to those containing less hindered groups like nitrile. The resulting boryl cyclopropanes act as versatile synthetic intermediates, as they can be readily converted into a range of derivatives, including trifluoroborate salts, boronic acids, and hydroxylated products. Furthermore, the cyclopropylboronate ester bearing a carbonyl substituent was found to undergo Suzuki–Miyaura cross-coupling in a moderate yield.
 | ||
| Scheme 1 Iridium-catalyzed C–H borylation of cyclopropanes. | ||
One year later, Sawamura and his team developed silica-supported monophosphane–Ir catalyst systems (Scheme 2).28 Different from Hartwig's approach, these systems enabled C–H borylation of cyclopropane chelation assisted by N or O atom. With the assistance of various directing groups, including N-heteroarenes, oximes, imines, and amides, the borylation occurred with remarkable regio- and stereoselectivities. This conversion led to the formation of cis-substituted cyclopropyl- and cyclobutylboronates. The successful borylation of sterically congested C–H bonds in substituted cyclopropanes, even including a tertiary C–H bond, showcases the potential of this directed C–H activation strategy for the functionalization of small-ring systems. Furthermore, beyond arylation and trifluoroborate formation, cyclopropylboronate esters can be further converted into primary alcohols through a one-carbon homologation/oxidation sequence.
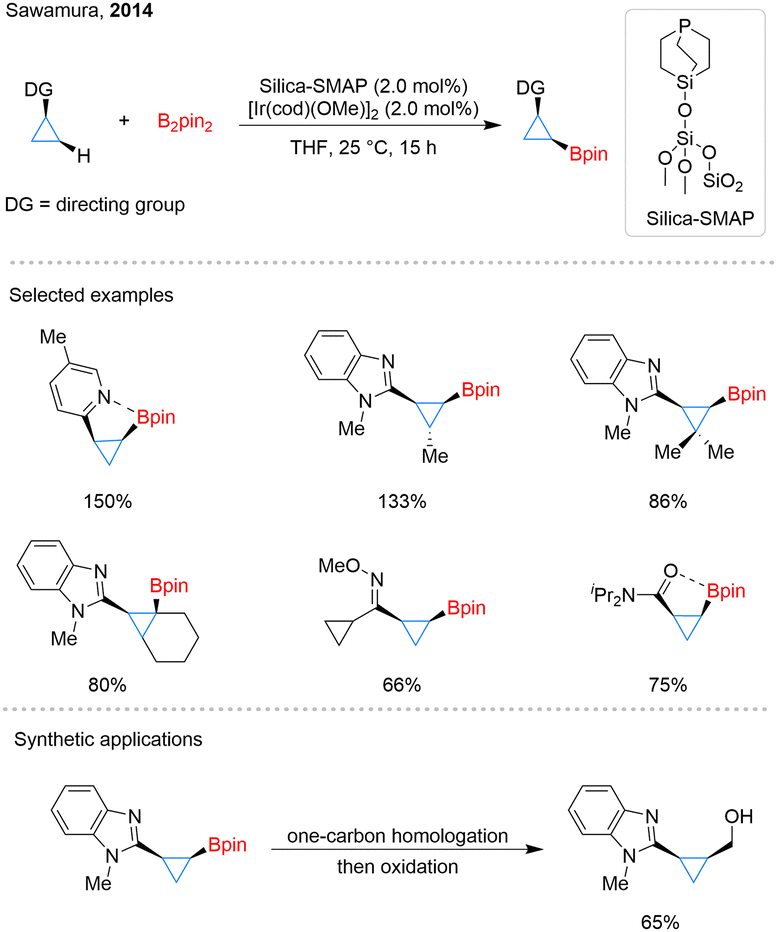 | ||
| Scheme 2 Silica-supported catalytic C–H borylation of cyclopropanes. | ||
Cyclopropylamines (CPAs) represent a particularly intriguing class of readily accessible cyclopropane derivatives, distinguished by their synthetic versatility.29 In 2015, Yamaguchi and Itami pioneered a significant advancement in this field through the development of an Ir-catalyzed, directed C–H borylation of CPAs 1, employing ligand L2 to efficiently access cyclopropyl boronate esters 2 (Scheme 3).30 In the course of reaction optimization, the authors found that the use of L1 or 4,4′-dtbpy led only to poor outcomes. Switching the ligand to L2 significantly improved the reactivity, and further adjustments of the boron source and the catalyst/ligand loadings, among other parameters, enabled the identification of the optimal conditions. This methodology enabled subsequent diversification via Suzuki–Miyaura coupling reactions, leading to the synthesis of valuable 2-arylcyclopropylamine derivatives. Notably, the stereochemical outcome of this transformation exhibited remarkable sensitivity to atmospheric conditions: under inert nitrogen atmosphere, partial epimerization occurred at the nitrogen center, while oxidative conditions (O2 atmosphere) effectively suppressed this process. This atmospheric control allowed for precise access to either cis- or trans-configured products (3 or 4, respectively), providing a unique stereochemical modulation strategy. These findings not only demonstrated the profound influence of reaction conditions on stereoselectivity in cyclopropane functionalization but also underscored the strategic importance of boron chemistry in controlling molecular architecture.
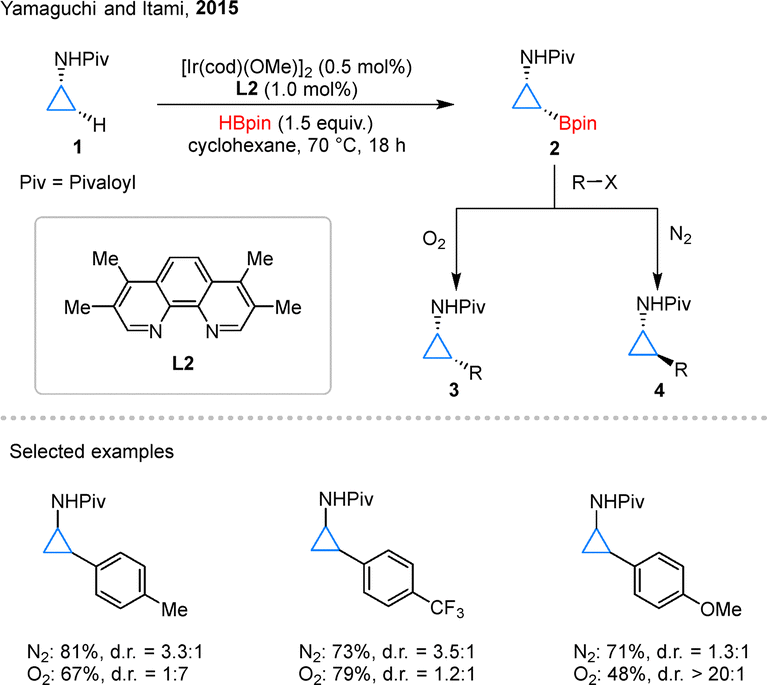 | ||
| Scheme 3 Iridium-catalyzed C–H borylation of CPAs and the follow-up transformation. | ||
Chiral cyclopropylboronates have garnered increasing attention due to the versatile diversification potential of the C–B bond.31 In 2017, Yu et al. reported a Pd-catalyzed enantioselective C–H borylation reaction using an acetyl-protected aminomethyl oxazoline ligand L3 (Scheme 4).32 Using B2pin2 as the boron source, the reaction achieved high enantioselectivity for the borylation of various cyclic amides, including cyclobutanes and cyclohexanes. Notably, subjecting cyclopropanecarboxylic amide 5 to these conditions afforded cyclopropylboronate 6 with excellent enantioselectivity, albeit in 32% yield. The reaction was also accompanied by the formation of the ring-opening byproduct, the geminal diboron compound 7. This work represented an early foray into the enantioselective C–H borylation of cyclopropanes.
 | ||
| Scheme 4 Asymmetric palladium-catalyzed C–H borylation of cyclopropane. | ||
In 2019, Xu and colleagues developed the enantioselective C–H borylation of cyclopropanecarboxamides using iridium catalyst with a chiral bidentate boryl ligand (CBL) L4 developed by themselves (Scheme 5).33 Unlike the above palladium catalysis, no ring-opening byproducts were observed in the systems. During reaction optimization, the authors primarily focused on the influence of CBLs on both the yield and enantioselectivity. They found that the steric bulk of the aryl and alkyl substituents on the CBLs was the key factor responsible for the high performance of the reaction. In substrate scope studies, cyclopropanes with imide, alkyl, or aryl substituents were generally compatible with the reaction. Mechanistic studies revealed that ligand L4 reacts with [Ir(cod)Cl]2 and B2pin2 to generate a 14-electron Ir(III) complex 10 bearing vacant coordination sites. Upon coordination with substrate 8, the complex forms intermediate 11, in which the β-C–H bond is preactivated through an agostic interaction. Subsequent oxidative addition affords Ir(V) species 12, which then undergoes reductive elimination to deliver the borylated product 9 and intermediate 13. Finally, 13 reacts with B2pin2 to regenerate 10. The authors further demonstrated a series of transformations of the boryl cyclopropanes, including Suzuki–Miyaura cross-coupling, alkenylation, and carbon homologation/oxidation. Notably, the primary alcohols obtained via oxidation could be elaborated through a three-step sequence to afford the antidepressant drug Levomilnacipran.
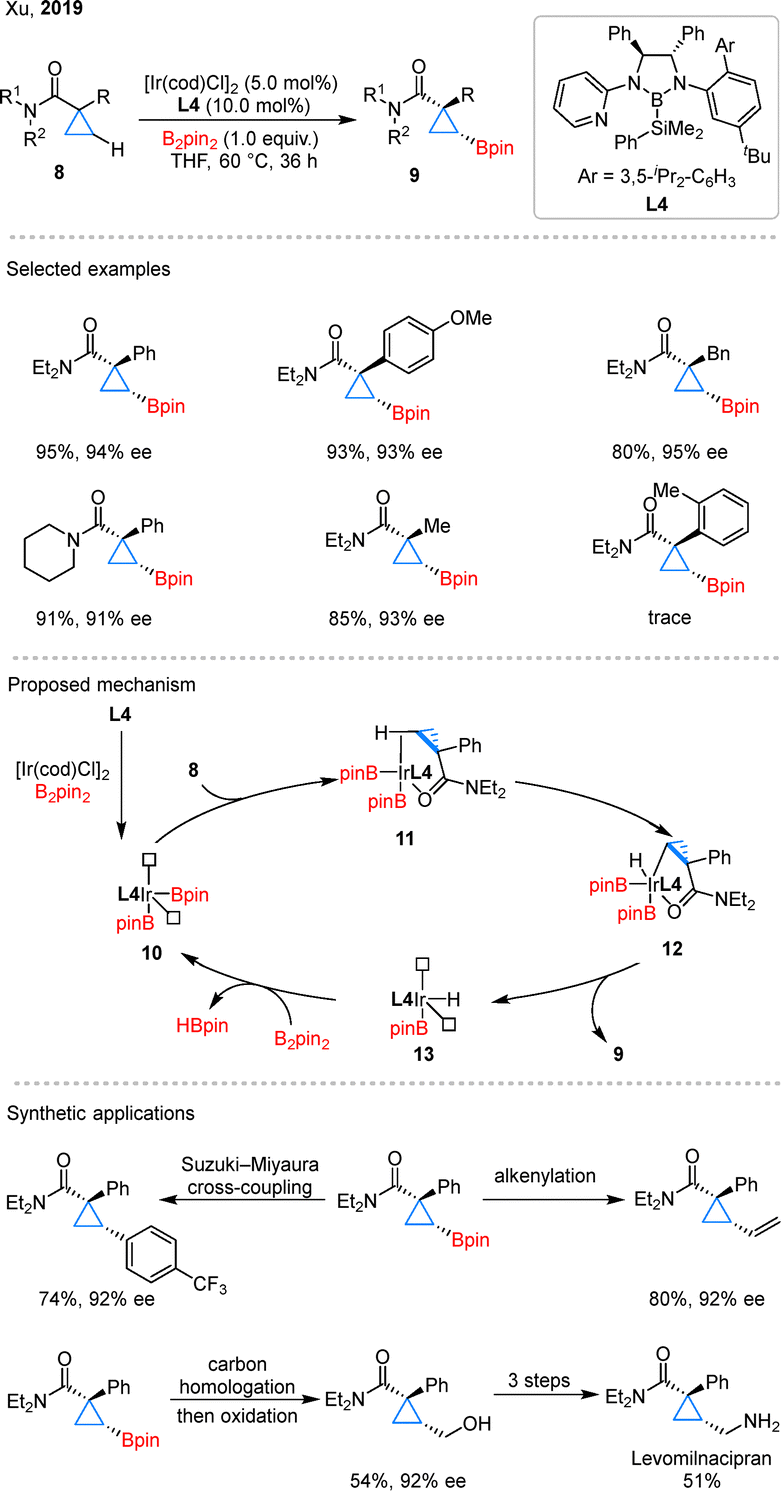 | ||
| Scheme 5 Asymmetric Ir-catalyzed C–H borylation of cyclopropanecarboxamides. | ||
The versatility of this catalytic system has been further demonstrated through its successful extension to CPAs, enabling the formation of optically active boronates.34 In 2022, Xu and colleagues achieved the asymmetric borylation of CPAs 14, marking a notable progression in stereocontrolled cyclopropane functionalization (Scheme 6).35 By strategically employing succinimide directing groups in conjunction with CBL L5, they successfully accessed chiral products 15 with high diaster- and enantio-selectivity. It is noteworthy that the use of acetyl group proved ineffective, highlighting the crucial role of this cyclic directing groups. Mechanistically, the succinimide group serves a dual purpose: it coordinates to the iridium center and modulates the electronic properties of the substrate, thereby facilitating selective β-C–H activation through the formation of a six-membered iridacycle intermediate.
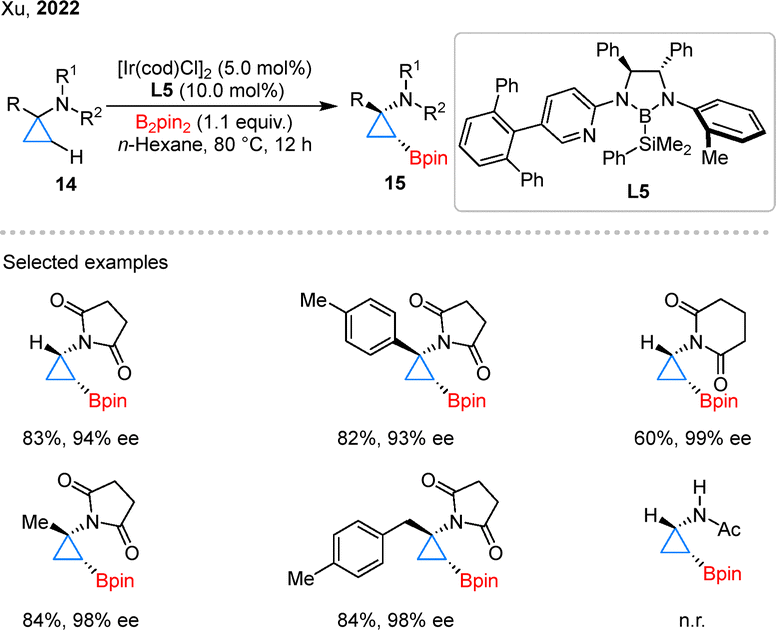 | ||
| Scheme 6 Asymmetric Iridium-catalyzed C–H borylation of CPAs. | ||
Ether motifs are prevalent in bioactive molecules and synthetic intermediates; however, their weak coordinating ability has traditionally limited their application in asymmetric C–H activation.36 In 2023, the Xu group reported an Ir-catalyzed asymmetric C–H borylation of cyclopropanes directed by ether group using CBL L6 as the ligand (Scheme 7).37 In substrate scope studies, the MeO group enabled efficient C–H borylation of both aryl- and alkyl-substituted cyclopropanes, whereas the EtO group proved significantly less effective. Notably, alkyl-substituted substrates required higher temperatures and longer reaction times for satisfactory conversion. This result highlights the versatility of CBL-type ligands in C–H borylation, underscoring their remarkable compatibility with diverse directing groups.
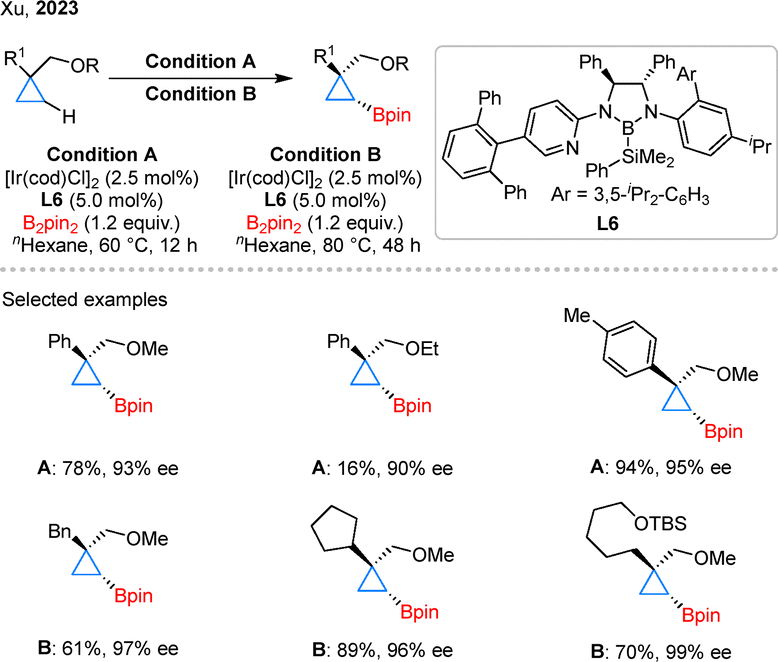 | ||
| Scheme 7 Asymmetric ether-directed C–H borylation of cyclopropanes. | ||
Analogous to cyclopropanes, cyclobutanes represent another structurally constrained motif of considerable importance in medicinal chemistry.38 Indeed, research on the C–H borylation of cyclopropanes and cyclobutanes has often advanced in parallel. As noted above, both Sawamura's and Yu's studies on cyclopropane C–H borylation were accompanied by related investigations on cyclobutanes.28,32 Furthermore, in 2020, Xu et al. employed their developed CBLs to achieve an asymmetric C–H borylation of cyclobutanes.39 Subsequently, Engle's group extended this concept to the methylenecyclobutanes.40 In contrast to cyclopropanes, the development of ring-opening borylation of cyclobutanes has been relatively limited, likely because the ring strain released upon cleavage of the four-membered ring is much smaller. Recently, inspired by Engle's findings, Lu and Ge achieved ring-opening borylation of methylenecyclobutanes.41
3. C–C borylation of cyclopropanes
The significant ring-strain inherent in cyclopropanes acts as a strong driving force for ring-opening reactions.42,43 This property has been extensively exploited as a strategic method in organic synthesis. As a result, cyclopropanes have emerged as highly versatile platforms for selective C–C borylation, enabling the synthesis of a diverse array of regio- and stereodefined organoboron compounds. Given the multitude of C–C activation instances, we have conducted a more meticulous classification of these reactions based on the types of cyclopropanes involved. These cyclopropane derivatives are systematically divided into five categories: (1) alk(en/yn)ylcyclopropanes; (2) gem-difluorocyclopropanes; (3) aminocyclopropanes; (4) iminocyclopropanes; and (5) simple cyclopropanes. The mechanisms of C–C activation typically encompass β-carbon elimination, oxidative addition, and nucleophilic borylation.3.1. Alk(en/yn)ylcyclopropanes
A wide spectrum of cyclopropanes incorporating unsaturated carbon–carbon bonds has been extensively utilized in C–C borylation reactions, demonstrating remarkable structural diversity. As illustrated in Fig. 5, this includes vinylcyclopropanes (VCPs), benzylidenecyclopropanes (BCPs), vinylidenecyclopropanes, propargylic cyclopropanes, alkylidenecyclopropanes (ACPs), and methylcyclopropanes (MCPs). To facilitate a more systematic and insightful analysis of these transformations, we have implemented a comprehensive classification framework based on reaction patterns. Specifically, these reactions have been categorized into three distinct subgroups: (1) monoborylation, involving the introduction of a single boryl group; (2) diborylation, characterized by the incorporation of two boryl moieties; and (3) borylative functionalization, which encompasses tandem borylation and subsequent functional group transformations.44 | ||
| Fig. 5 Structural diversity of cyclopropanes bearing unsaturated C–C bonds. | ||
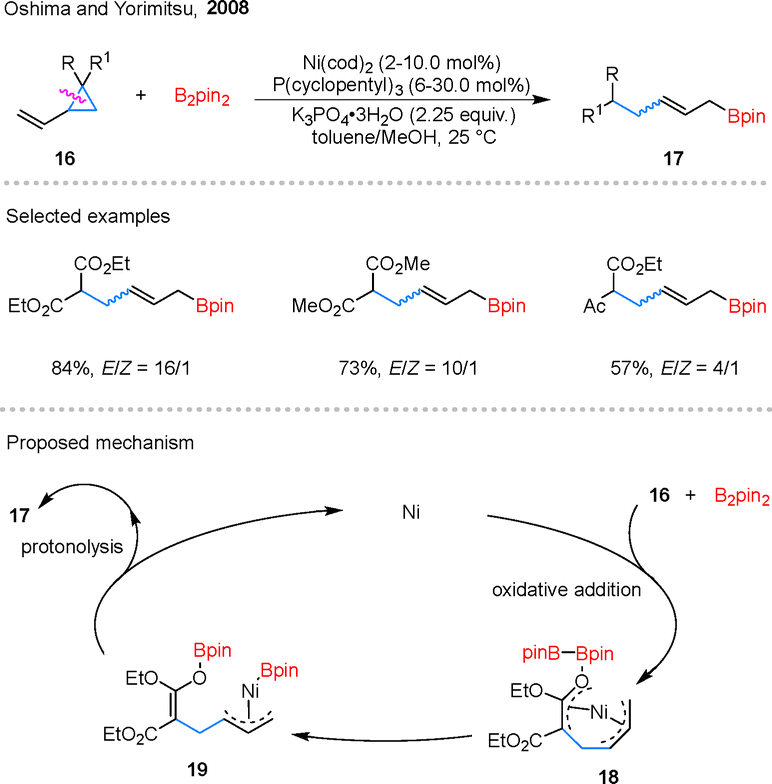 | ||
| Scheme 8 Nickel-catalyzed ring-opening borylation of VCPs. | ||
In 2019, Engle et al. also developed a copper-catalyzed hydroborylation of BCPs 20, giving two different products 21 and 22 (Scheme 9).47 The distinct outcomes observed with different ligands L7 and L8. DFT calculations suggest that the reaction pathway is primarily controlled by the energy difference between two competing steps: β-carbon elimination and protodecupration. Both originate from the same benzylic copper intermediate 23. The L7 ligand significantly increases the barrier for β-carbon elimination, while having little effect on protodecupration. As a result, the reaction shifts toward protodecupration, giving cyclopropylboronic ester 21. In contrast, with the L8 ligand, β-carbon elimination is more favorable, which aligns with the formation of alkenylboronate 22.
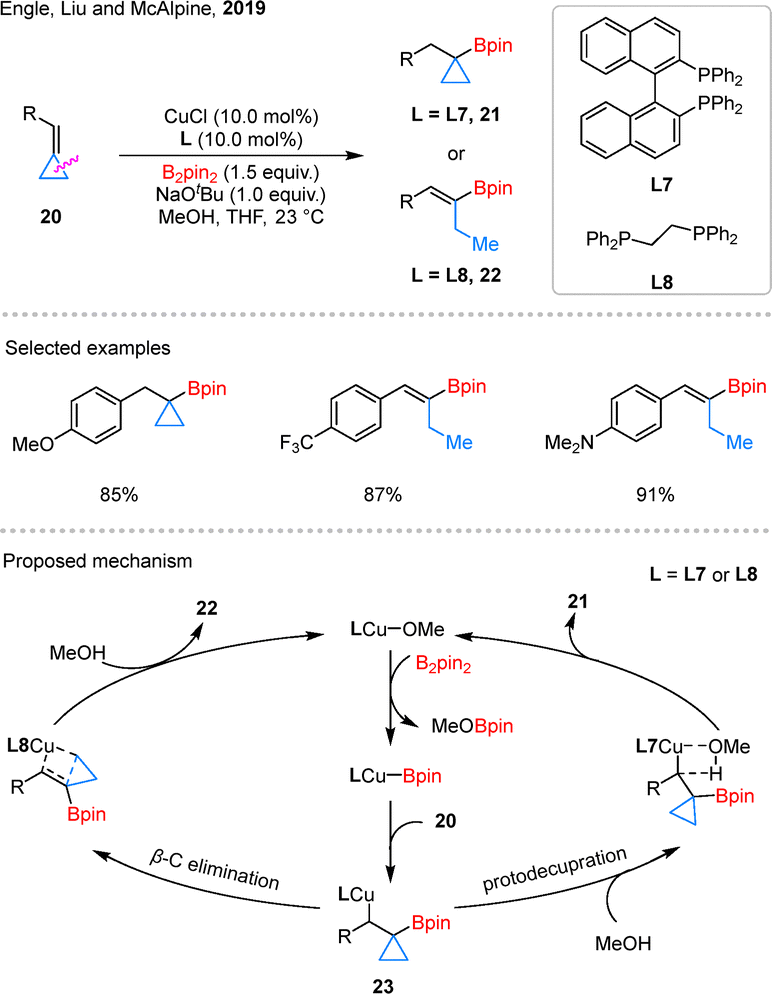 | ||
| Scheme 9 Copper-catalyzed selective hydroborylation of BCPs. | ||
The introduction of chiral catalysts has enabled the realization of asymmetric hydroborylation of VCPs, marking a stereoselective synthesis. In 2017, Lu and coworkers achieved an advancement in the field by developing an iron-catalyzed hydroborylation of VCPs 24 (Scheme 10).48 This innovative approach utilized pre-catalyst 25 to efficiently produce homoallylic boronates 26, achieving high enantiocontrol in the process. In the proposed mechanism, the active Fe–H species forms from 25 in the presence of NaHBEt3. Subsequent coordination of 24 to complex 25 generates intermediate 27, which undergoes alkene insertion into the Fe–H bond to afford intermediate 28. A regioselective β-carbon elimination from 28 produces alkyl iron species 29, which may undergo β-hydride elimination to give diene byproducts. Finally, ligand exchange between 29 and HBpin regenerates active Fe–H species and releases 26.
 | ||
| Scheme 10 Asymmetric iron-catalyzed borylation of VCPs. | ||
Beyond the well-established VCPs, vinylidene cyclopropanes have emerged as another versatile class of substrates in organic synthesis.49 In a notable contribution, Chen and coworkers demonstrated the copper-catalyzed synthesis of homopropargylic boronates 31 using vinylidene cyclopropanes 30 and 1,1-bisborylmethane as starting materials (Scheme 11).50 This methodology showcased broad substrate compatibility, accommodating vinylidene cyclopropanes with substituents at diverse positions under optimized reaction conditions. In the proposed mechanism, Cu-based carbon nucleophiles undergo migratory insertion with substrate 30 to generate vinyl copper intermediate 32, which subsequently undergoes β-carbon elimination to form open-chain intermediates (33 or 34). These intermediates then react with a proton source to afford functionalized products 31. Building upon this work, the same research group later reported the selective borylation of vinylidene cyclopropanes using unsymmetrical diboron compounds, further expanding the synthetic utility of this approach.51
 | ||
| Scheme 11 Copper-catalyzed borylative ring-opening of vinylidene cyclopropanes. | ||
 | ||
| Scheme 12 Copper-catalyzed borylation of propargylic cyclopropanes. | ||
ACPs and BCPs are known for their heightened ring strain and exhibit distinct reactivity patterns in C–C borylation.54 In 2018, Jin and colleagues demonstrated that nanoporous gold (AuNPore) could catalyze the diborylation of BCPs or ACPs in a heterogeneous manner, selectively cleaving the distal C–C bond (Scheme 13).55 Remarkably, this transformation proceeds in the absence of ligands or additives, highlighting the unique surface-mediated reactivity of AuNPore. Beyond its regioselectivity, the catalyst exhibits recyclability and operational stability, underscoring its potential for sustainable and green catalytic applications. However, the substrate scope was largely limited to simple BCPs, while alkyl-substituted analogues generally exhibited lower reactivity and required prolonged reaction times to achieve acceptable yields.
 | ||
| Scheme 13 Nanoporous gold-catalyzed selective diborylation of cyclopropanes. | ||
In 2023, Ge and coworkers developed a regiodivergent ring-opening dihydroborylation of BCPs 42 by cobalt catalysis, enabling access to both 1,3- and 1,4-diboronate products 43 and 44 (Scheme 14).56 Aryl groups with different electronic properties were well tolerated, though bulky substituents required higher temperatures to improve yields. The high regioselectivity is attributed to β-C elimination, which forms unique homoallylic cobalt intermediates. In the catalytic cycle, Co(acac)2 activated by HBpin and L generates LCo–H, which inserts into 42 to give benzylcobalt intermediate 45, followed by β-C elimination to yields homoallylic species 46. Ligand choice directs the subsequent steps: with rigid L11, 46 reacts with HBpin to form 47, which undergoes σ-bond metathesis to afford 1,4-diboronate 43, while β-H elimination is suppressed. In contrast, flexible L12 enables 46 to adopt a coplanar geometry, promoting β-H elimination to generate 48. Reinsertion of the diene into the Co–H bond generates allylcobalt species 49, which reacts with HBpin to form 50. Subsequent Co–H insertion yields 51, which undergoes σ-bond metathesis with HBpin to give 1,3-diboronate 44 and regenerate the active LCo–H catalyst.
 | ||
| Scheme 14 Cobalt-catalyzed diborylation of BCPs. | ||
In addition, Tu and Peng also developed an Ir-catalyzed 1,n-diborylation of cyclopropanes using their newly designed spirocyclic NHC Ir catalyst 53 or 56 (Scheme 15).57 A range of mono- and diaryl-substituted cyclopropanes 52 or 55 were well tolerated, affording previously rare 1,1-diborylated products 54 or 57, whereas alkyl-substituted ACPs failed to undergo the transformation. Furthermore, the scope of this diborylation strategy was extended to two additional substrate classes. Specifically, a series of MCPs 58 underwent smooth transformation to deliver the corresponding 1,4-diborylated products 59. In parallel, cyclopropyl-substituted ACPs 60 were successfully converted into 1,7-diborylated compounds 61 under milder reaction conditions. DFT calculations were performed to investigate the mechanism of the reaction using diaryl-substituted BCPs. The process begins with the form of hemilabile Ir(I) species 62. This intermediate reacts with B2pin2 to form the Ir(III) complex 63. The reductive elimination of 63 affords the Ir(I) species 64, which underwent coordinates and oxidative addition to 52 to yield the cyclic Ir(III) intermediate 65. Next, a concerted reductive elimination and oxidative addition step converts 65 into the vinyl-Ir(III) complex 66. A σ-bond metathesis between the Ir–H bond and the phenyl C–B bond then gives 67. The system undergoes σ-metathesis between the Ir–C bond and the α-C–H bond of the Bpin group, forming another four-membered Ir(III) complex 68. This intermediate undergoes intramolecular ligand exchange to form 69, which subsequently reacts with HBpin to yield 70. Finally, reductive elimination from 70, followed by dissociation of the catalyst, furnishes the product 54.
 | ||
| Scheme 15 Iridium-catalyzed regioselective 1,n-diborylation of cyclopropanes. | ||
Enantioselective diborylation has recently emerged as a significant advancement within this topic A notable contribution by Lu and Ge demonstrated a Cu-catalyzed asymmetric diborylation of BCPs, achieving selective formation of both 1,3- and 1,4-diboronates with chiral bisphosphine ligands L13 and L14 (Scheme 16).58 This methodology efficiently transformed BCPs with various substituents into either 1,4-diboration products 71 or 1,3-diboration products 72 with excellent enantioselectivity. Mechanistic studies suggest that the reaction begins with LCu–H insertion into BCPs to form benzyl copper species 73, which undergoes β-carbon elimination to give homoallylic cuprate 74. σ-Bond metathesis with HBpin generates a Z/E mixture of 75 and regenerates LCu–H. In the 1,4-pathway, (E)-75 inserts into Cu–H to form 76, leading to product 71. In contrast, (E)-75 inserts into LCu–Bpin yields 77, which affords 1,3-diboronate 72.
 | ||
| Scheme 16 Copper-catalyzed asymmetric diborylation of BCPs. | ||
C migratory insertion followed by β-carbon elimination to yield homoallylic intermediates 83. However, this pathway fails to explain the high Z-selectivity observed in Pt-catalyzed reactions with various substrates. An alternative mechanism involving oxidative addition of the proximal C–C bond may better account for this selectivity, as the electronic nature of substituents can influence the cleavage site. This pathway is likely favored when steric or electronic factors hinder migratory insertion. A similar oxidative addition may also apply to distal C–C bond cleavage, forming product 82.
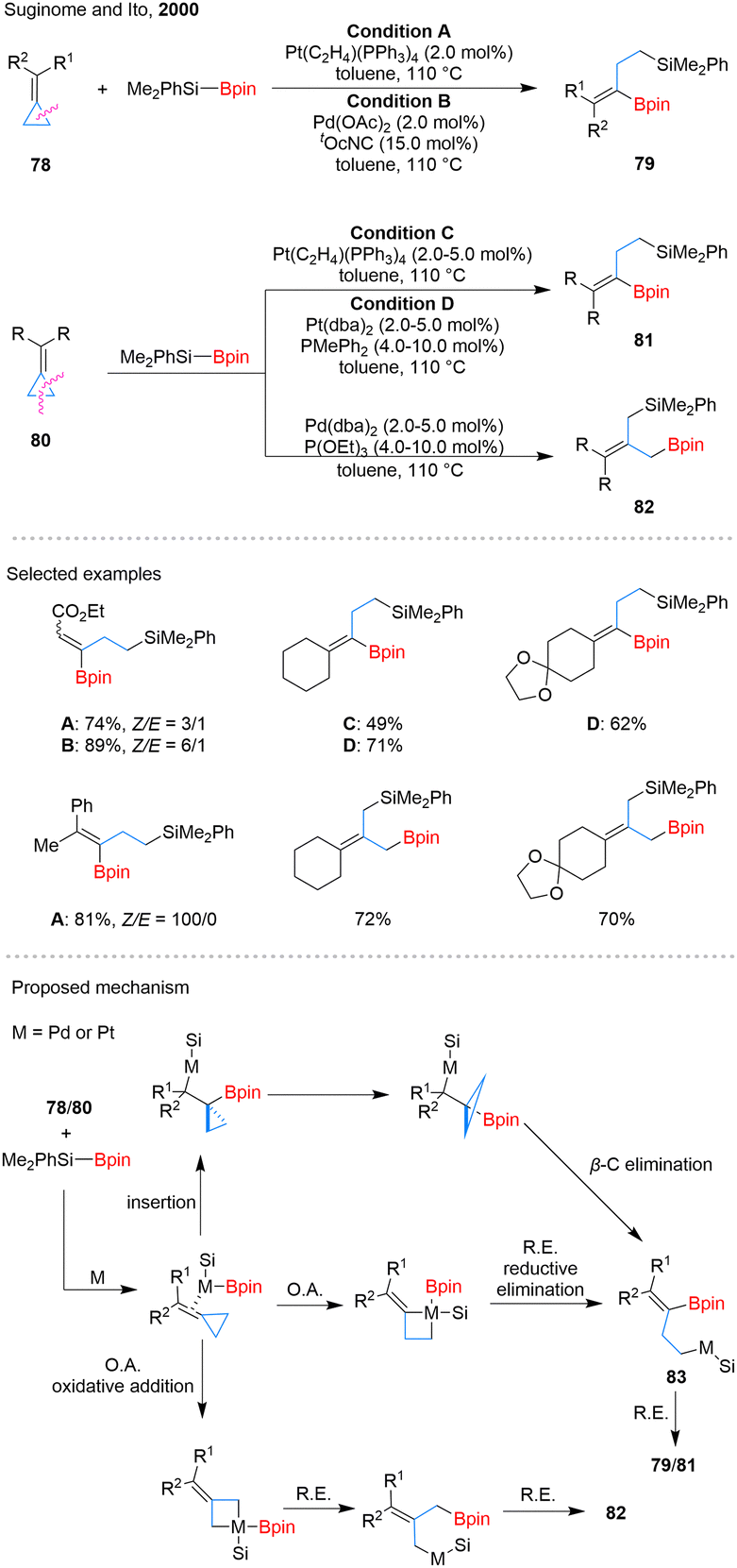 | ||
| Scheme 17 Catalytic silaborylation of ACPs. | ||
In 2007, the same research group successfully developed an asymmetric variant of the silaborylation of MCPs, employing palladium catalysis in conjunction with chiral ligand L15 to yield enantioenriched products (Scheme 18A).61 The reaction demonstrated excellent efficiency with bicyclic MCPs, consistently delivering the desired products. However, substrates lacking ring fusion or those containing cyclic acetal motifs exhibited reduced enantioselectivities and yields. The use of polymer-supported chiral ligands offers significant advantages, including ease of recovery and potential for reuse.62 Building on this concept, the group further explored Pd-catalyzed asymmetric silaborylation of MCPs utilizing a helically chiral polymeric ligand L16 (Scheme 18B).63 Remarkably, the application of L16 consistently provided higher enantioselectivity compared to previously reported systems. This enhancement is attributed to the polymeric ligand's ability to create a long-range chiral environment around the palladium center. In contrast to conventional small-molecule ligands, this unique feature not only improves catalytic reactivity but also enhances stereocontrol, offering a more efficient and sustainable approach to asymmetric synthesis.
 | ||
| Scheme 18 Palladium-catalyzed asymmetric silaborylation of MCPs. | ||
The borylative functionalization of cyclopropanes is not limited to silaborylation. In 2022, Peng and coworkers reported a Cu-catalyzed borylacylation of BCPs that proceeds through proximal C–C bond cleavage, delivering 1,3-borylacylated products (Scheme 19).64 Aryl- and naphthyl-substituted BCPs, along with a variety of chloroformate electrophiles, reacted efficiently under the standard conditions. Using cyclopropyl-substituted BCPs, the same group further developed a regioselective 1,5-borylacylation using a Cu/Pd dual-catalytic system.65
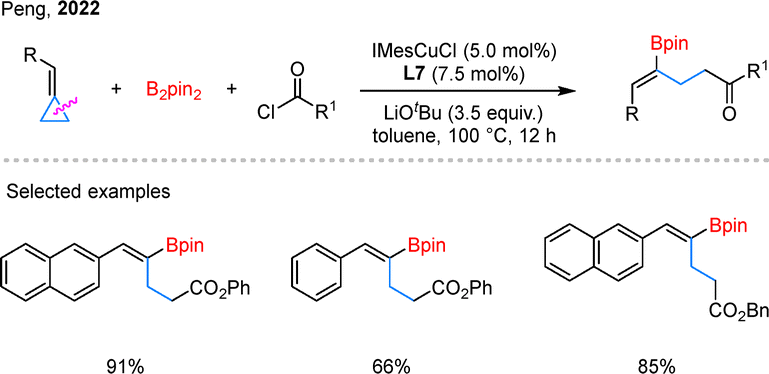 | ||
| Scheme 19 Copper-catalyzed borylacylation of BCPs. | ||
The substrate scope of alkene borocarbonylation has been largely limited to styrene derivatives,66 whereas BCPs, as a distinct alkene analogue, have shown potential as viable substrates for such transformations. In 2022, Wu and colleagues established a Cu/Pd dual-catalytic regiodivergent borylacylation of BCPs with aryl iodides and CO (Scheme 20).67 By carefully tuning the metal combination and ligand L11, the reaction of BCPs could be steered to furnish either γ-vinylboryl ketones 84 or β-cyclopropylboryl ketones 85. Under the standard conditions, the protocol exhibited broad substrate compatibility, tolerating BCPs bearing aryl or heteroaryl groups as well as various aryl iodides. The reaction showcases precise regioselectivity control enabled by tailored metal–ligand combinations, underscoring its unique mechanistic design.
 | ||
| Scheme 20 Catalytic regiodivergent borylacylation of BCPs. | ||
The development of asymmetric borylacylation has emerged as a powerful strategy for accessing enantiomerically enriched aminoboronates, marking a significant advancement in catalytic methodology.68 In a pioneering contribution, Su and colleagues reported a Cu-catalyzed 1,4-borylamination of BCPs through a cascade sequence involving hydroborylation followed by hydroamination (Scheme 21).69 This method demonstrated broad substrate tolerance, accommodating BCPs, as well as various aminating reagents. Mechanistic studies revealed that the transformation proceeds via two interconnected catalytic cycles. In the hydroborylation cycle, L17Cu–H inserts into BCPs to form benzyl copper intermediate 87, which undergoes β-carbon elimination and σ-bond metathesis to generate borylated intermediate 88. In the subsequent hydroamination cycle, 88 undergoes regio- and enantioselective hydrogenation to form intermediate 89, which then reacts with a hydroxylamine ester to yield the 1,4-borylaminated product 86. Later, Wu and coworkers further advanced the field by reporting a Cu-catalyzed borylation of BCPs, enabling the synthesis of both chiral 1,4-borylamination and 1,4-borylcarboxamidation products.70
 | ||
| Scheme 21 Copper-catalyzed asymmetric borylamination of BCPs. | ||
3.2. Gem-difluorocyclopropanes
Monofluoroalkenes are valuable fluorinated motifs often employed in medicinal chemistry as a metabolically stable bioisostere for amide functionalities (–NH–CO–).71 In 2021, Fu and Gong reported a Cu/Pd-cocatalyzed three-component coupling of gem-difluorocyclopropane 90 with alkynes and B2pin2, producing monofluorinated alkenes 92 (Scheme 22).72 Using an pre-catalyst 91, internal and terminal alkynes, as well as aryl-substituted gem-difluorocyclopropanes, were effectively converted into the desired products. Notably, natural products such as coumarin were also amenable to late-stage functionalization under the standard conditions. In the proposed mechanism, the reaction begins with borylcupration of the alkyne, generating a β-borylalkenyl copper species 93. Concurrently, the Pd(0) catalyst activates 90via C–C bond cleavage accompanied by C–F bond activation, forming an allyl–Pd(II) complex 94. Subsequent transmetalation between intermediates 93 and 94 produces a Pd(II) species 95, which undergoes reductive elimination to deliver the target product 92.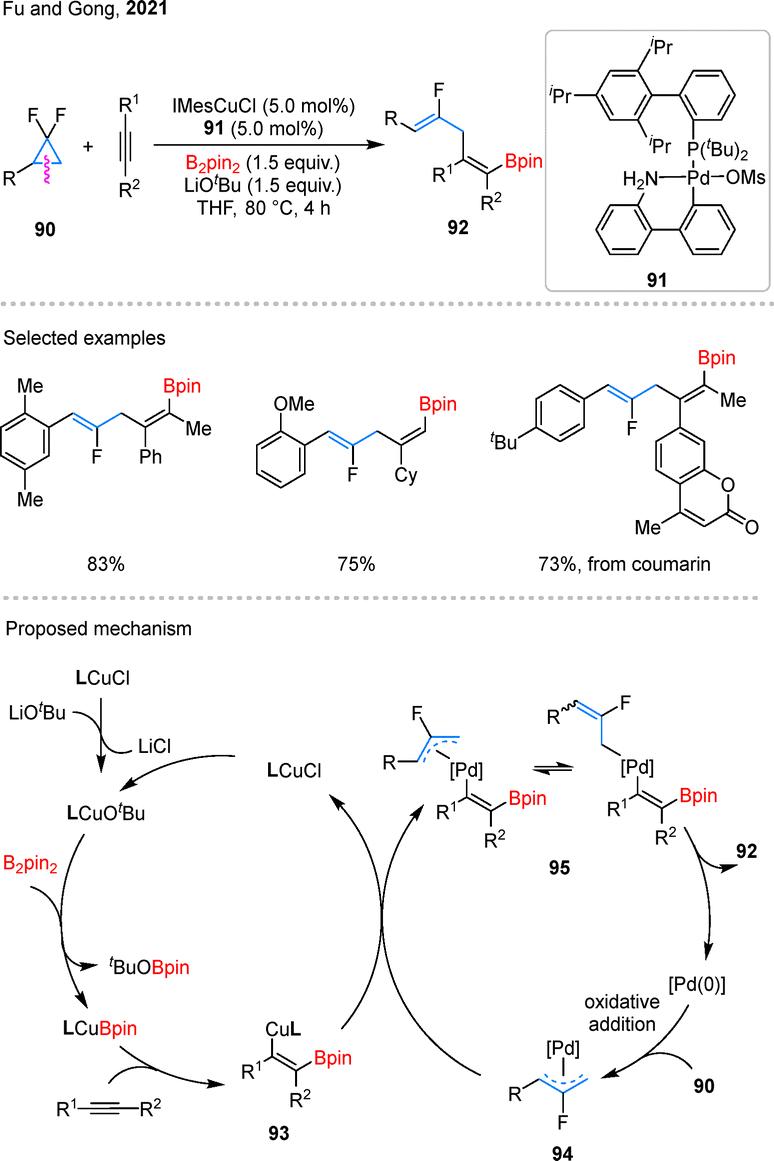 | ||
| Scheme 22 Catalytic cis-borylfluoroallylation of alkynes. | ||
Expanding their catalytic repertoire, the research team also uncovered t borylfluoroallylation of alkenes using dual Cu/Pd-catalysis. This transformation, which employs gem-difluorocyclopropanes and B2pin2 as key reagents, efficiently delivers monofluoroalkene scaffolds 96 with high precision (Scheme 23A).73 The reaction demonstrated broad compatibility with a range of substituted gem-difluorinated cyclopropanes and alkenes, showcasing its synthetic versatility. Notably, rather than isolating the boronate intermediates, the authors opted for their direct oxidation to the corresponding alcohols, streamlining the synthetic process. Building upon this success, they further developed the Pd-catalyzed coupling of gem-difluorocyclopropanes with gem-diborylalkanes 97, yielding boryl-substituted fluorinated alkenes 98 with excellent control over regioselectivity (Scheme 23B).74 The reaction exhibited remarkable tolerance to variations in the electronic properties of substituents. However, it was observed that more sterically demanding substrates, such as 1,2-disubstituted gem-difluorocyclopropanes, remained incompatible with the optimized reaction conditions.
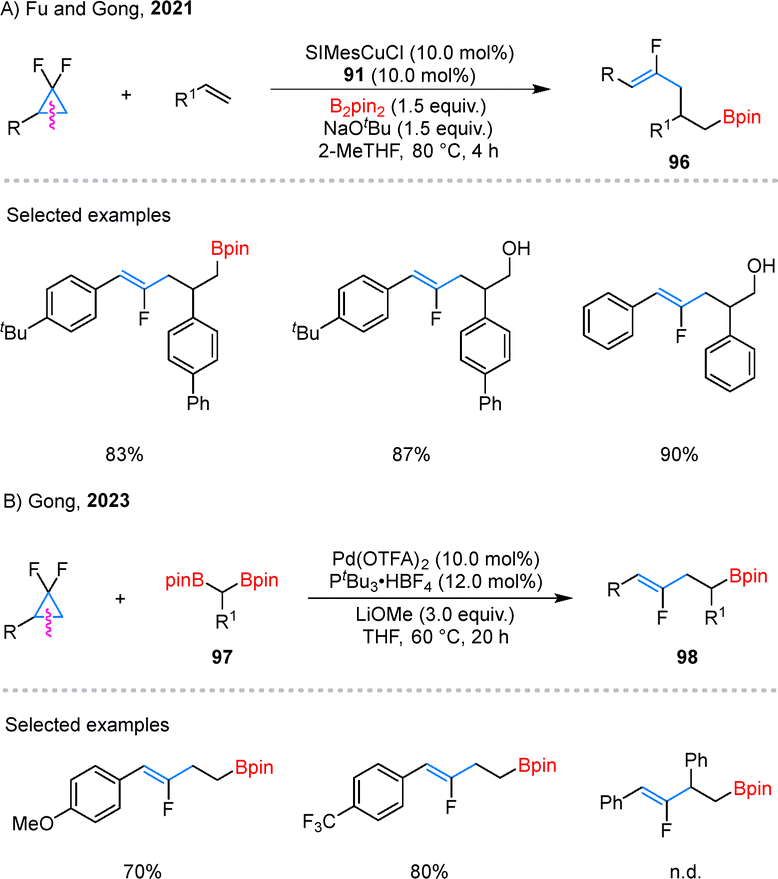 | ||
| Scheme 23 Catalytic borylation of gem-difluorinated cyclopropanes. | ||
3.3. Aminocyclopropanes
The nitrogen substituent in cyclopropanes can act as an internal driving force to facilitate C–C bond cleavage of the strained ring.5 In, 2021, Shi et al. disclosed a Rh-catalyzed proximal-selective C–C bond activation of NHPiv-substituted CPAs 99, affording γ-amino boronates 100 (Scheme 24).75 Substrate scope investigations showed that both alkyl and aryl groups at the R position were well tolerated. In contrast, substrates with substituents at the R1 position required higher catalyst and ligand loadings, as well as extended reaction times, to achieve satisfactory results. In the proposed mechanism, the reaction is initiate with ligand exchange between the cyclooctadiene (cod) ligand in complex 101 and substrate 99, affording intermediate 102. Chelation assisted by the amide oxygen, proximal C–C bond cleavage then occurs in 102 to generate intermediate 103. Intermediate 104 is subsequently formed through the reaction of 103 with HBpin. Computational analysis revealed that the hydroborylation step likely follows a σ-complex-assisted metathesis (σ-CAM) pathway rather than oxidative addition. From intermediate 104, re-coordination of the cod ligand affords intermediate 105, which undergoes reductive elimination of the C–H bond to deliver the product-coordinated 106. | ||
| Scheme 24 Rhodium-catalyzed proximal-selective C–C borylation of CPAs. | ||
Subsequently, Shi and colleagues further developed an enantioselective asymmetric hydroborylation of CPAs 107, affording chiral aminoboronates 108 (Scheme 25).76 The key to the high enantioselectivity is the combination of the chiral phosphite L18 and the anionic acac ligand with rhodium catalyst. The reaction system shows good tolerance towards aryl substituents with different electronic effects on the cyclopropane ring. However, when alkyl groups are present, the enantioselectivities are significantly lower. Mechanistic studies suggest that the active catalytic species 109, generated from cyclopropane 107, L18, and the Rh-bound acac ligand, undergoes proximal-selective C–C bond oxidative addition to form rhodacycle 110. Ligand exchange with HBpin produces intermediate 111, which undergoes σ-CAM to generate 112. Reductive elimination from 112via transition state 113 yields product 108 and regenerates catalyst 109 through ligand exchange with 107.
 | ||
| Scheme 25 Asymmetric rhodium-catalyzed hydroborylation of CPAs. | ||
In addition to rhodium catalysis, the same conversion can be achieved by inexpensive and earth-abundant group IV metals. In 2024, Wu and colleagues reported the example of zirconium-catalyzed hydroborylation of CPAs 114 (Scheme 26).77 Notably, hafnium also exhibited comparable catalytic efficiency for this transformation. Within the substrate scope, aryl-, alkyl-, and amide-substituted cyclopropanes were well tolerated. However, for 1,2-disubstituted cyclopropane substrates, the system exhibited limited diastereocontrol. Mechanistically, in situ generated Zr–H reacts with N–H bonds of 114 to form N–Zr species 116 with H2 release. β-Carbon elimination then cleaves the C–C bond, yielding zirconium species 117. Subsequently, 117 undergoes C–Zr and H–B bond metathesis with HBpin, producing intermediate 118 and regenerating the Zr–H species. In the second catalytic cycle, hydride transfer from Zr–H to intermediate 118 forms intermediate 119, which is further reduced by the previously released H2 to generate 115.
 | ||
| Scheme 26 Catalyzed hydroborylation of CPAs by earth-abundant group IV metals. | ||
Notably, Wu and colleagues recently discovered that the above hydroboration can proceed under a meta-free approach just using HBcat (Scheme 27).78 This method exhibits broad substrate compatibility, particularly in the late-stage modification of pharmaceutical compounds such as ibuprofen. However, substrates bearing carboxylic acids on the aryl ring showed poor reactivity. Mechanistic insights from DFT calculations indicate that the transformation begins with the formation of a van der Waals complex 120 between HBcat and the cyclopropane substrate. The approach of HBcat toward the less hindered C–C bond proceeds through a ring-opening transition state 121, which serves as the rate-determining step and leads to 122 stabilized by coordination with the oxygen atom of HBcat. A subsequent hydride transfer from the boron center to the adjacent carbocation via123 furnishes the borylated product 124.
 | ||
| Scheme 27 Metal-free hydroborylation of CPAs. | ||
Beyond the above hydroborylation strategies, a novel σ-C–C bond eliminative borylation of cyclopropanes has emerged as a powerful synthetic tool. In a significant contribution, Shi and coworkers introduced a stereoselective C–C bond activation of CPAs 125 mediated by BCl3, which enables the synthesis of γ-borylenamides 126 under metal-free conditions (Scheme 28).79 Notably, this stereoconvergent protocol is highly remarkable as it can tolerate cis/trans mixtures of substrates and produces products with high stereoselectivity. Under the optimized reaction conditions, a wide range of alkyl- and aryl-substituted substrates, including 1,2-disubstituted cyclopropanes, can successfully yield the desired products. However, substrates containing alkyne groups are not compatible with this reaction system. According to the proposed mechanism, substrate 125 first coordinates with BCl3 to form intermediate 127, where intramolecular O–B coordination enhances the acidity of the adjacent N–H bond, facilitating deprotonation by the CHMP via transition state 128. The resulting species 129 undergoes halide abstraction by BCl3 through transition state 130 to generate imine intermediate 131. Guided by the Piv group, 131 inserts regioselectively into the C–C bond to form intermediate 132. Subsequent deprotonation leads to 133, and a final rearrangement and proton dissociation affords product 134.
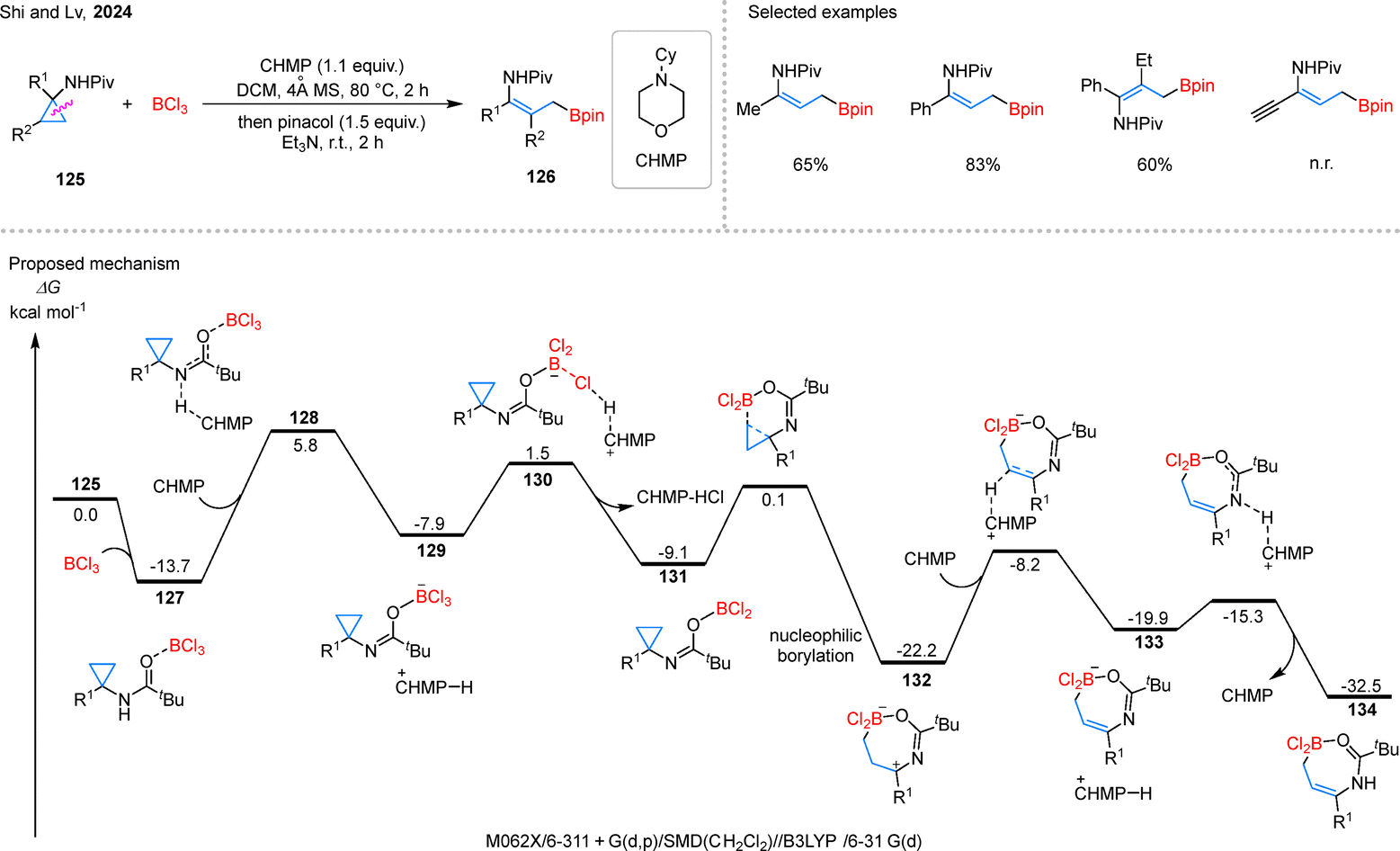 | ||
| Scheme 28 Stereoconvergent σ-C–C bond eliminative borylation of cyclopropanes. | ||
3.4. Iminocyclopropanes
1,2-Azaborines are aromatic BN heterocycles considered as benzene isosteres, and their increased polarity and altered electronic properties have been associated with enhanced solubility and biological performance–features that have inspired growing interest in their synthetic development.80 In 2023, Dong, Liu, and Houk have illuminated that cyclopropane adorned with electron-withdrawing moieties, such as imines, possess the capacity to undergo nucleophilic eliminative ring fission with the aid of a boron electrophile and a Lewis catalyst (Scheme 29).81 Aromatic and aliphatic amines were both well tolerated under the reaction conditions, and dibromoboranes bearing aryl or alkyl substituents proved to be suitable coupling partners. Moreover, substrates derived from drug molecules such as ibuprofen also participated smoothly in the reaction, although a few examples showed relatively low yields. In the proposed mechanism, 135 undergoes boron-mediated C–C bond cleavage to generate 137, which then undergoes reductive elimination to afford diene 138. DFT calculations suggest that diene s-trans-138 preferentially undergoes a 6π-electrocyclization via transition state 139 to form the cyclic N-borylimine 140. This intermediate then undergoes DBU-mediated elimination of HBr to furnish the pyridine borane product 136. | ||
| Scheme 29 Borylative ring-opening of cyclopropyl imines or ketones. | ||
3.5. Simple cyclopropanes
Although simple cyclopropanes are among the most prevalent three-membered ring motifs, methods for their selective ring-opening and borylation remain limited.82 In 2010, Stephan and colleagues disclosed the ring-opening of nonactivated cyclopropanes using frustrated Lewis pairs comprising phosphine and B(C6F5)3, exemplified by the direct electrophilic attack of B(C6F5)3 on cyclopropylbenzene to generate zwitterionic phosphonium borates (Scheme 30).83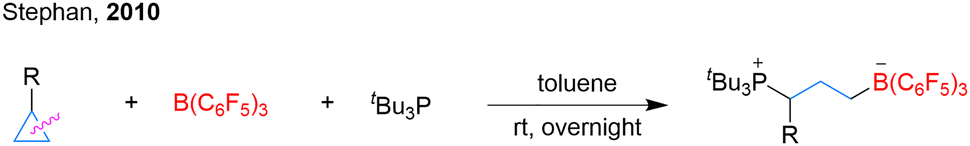 | ||
| Scheme 30 Borylation ring-opening of nonactivated cyclopropanes with B(C6F5)3. | ||
In 2018, Shi and Houk pioneered a metal-free σ-bond hydroboration of nonactivated cyclopropanes, employing BBr3 as a Lewis acid catalyst and PhSiH3 as the hydride donor (Scheme 31).84 This innovative methodology exhibits anti-Markovnikov selectivity, mirroring the classical Brown hydroboration. The optimized protocol demonstrates exceptional versatility, facilitating the efficient transformation of diverse cyclopropane derivatives, including spiro, aryl, and alkyl-substituted variants, into their corresponding borylated products. Notably, the system's robustness was further highlighted by the successful 1,3-hydroboration of cyclopropane gas under balloon pressure, yielding nPrBpin in an impressive 71% yield. Comprehensive mechanistic investigations, supported by DFT calculations, have unveiled a sophisticated reaction pathway. The transformation initiates with the electrophilic activation of cyclopropanes 141 by BBr3, generating reactive intermediate 142. This key species subsequently undergoes hydride abstraction from PhSiH3 through a well-defined transition state 143, ultimately delivering the borylated product 144.
 | ||
| Scheme 31 Borylation ring-opening of nonactivated cyclopropanes with BBr3. | ||
In addition, transition-metal catalysis can also facilitate the hydroborylation of simple cyclopropanes. In 2020, Yamaguchi and coworkers reported an Ir-catalyzed distal-selective hydroborylation of cyclopropanes 145 (Scheme 32).85 Remarkably, this reaction does not require any directing groups and results in the formation of alkylboronates 146. The reaction accommodates amino, aryl, and silyl ether substituents on the cyclopropane ring, which broadens its applicability in organic synthesis. Mechanistically, a five-coordinate Ir complex 147 serves as the catalytically active species. In the presence of L19, oxidative addition of the Cβ–Cγ bond in 145 to complex 147 forms intermediate 148, which undergoes reductive elimination to give complex 149. From this point, three downstream pathways are proposed, interconverting through equilibria between intermediates 150, 151, and 152. Among these, the pathway proceeding via150 is the most favorable and ultimately furnishes the hydroborylated product 146. However, despite the use of the chiral ligand L19, enantioselectivity was not induced in this reaction.
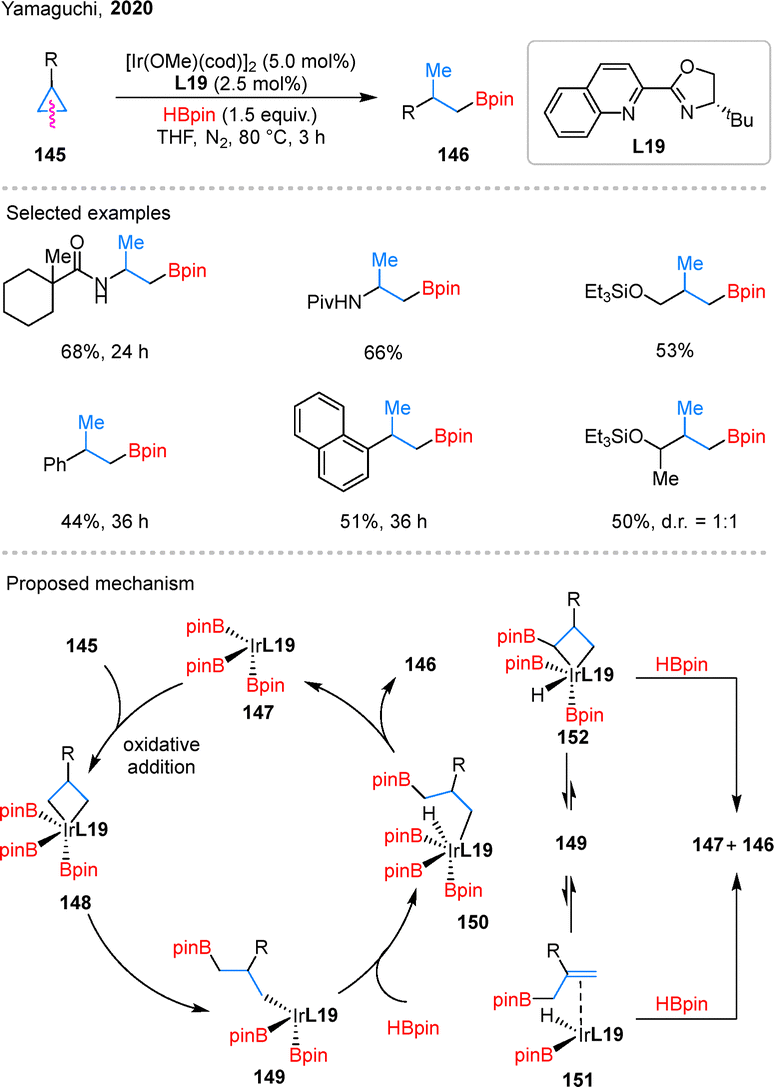 | ||
| Scheme 32 Iridium-catalyzed distal C–C borylation of cyclopropanes. | ||
4. Conclusions and perspectives
Over the past two decades, the cyclopropane-to-organoboron conversion has witnessed transformative progress, driven primarily by innovative strategies for C–H and C–C bond activation. This evolution has unlocked access to structurally complex and diverse boron-containing architectures from readily functionalized cyclopropanes, leveraging both catalyst-free and catalytic approaches. These advancements establish robust synthetic platforms with significant potential for rapid molecular construction.Despite these remarkable achievements, pivotal challenges remain that define the future trajectory of this dynamic field: (a) examples on C–H borylation of cyclopropanes remains significantly underdeveloped compared to C–C activation. Furthermore, current reliance on directing groups (e.g., amides, ethers) enables enantioselectivity but suffers from limited directing group diversity and a narrow scope of catalyst types. A fundamental frontier lies in achieving directing-group-independent asymmetric C–H borylation of simple, unactivated cyclopropanes. Overcoming this requires pioneering catalyst design (e.g., novel chiral ligands, earth-abundant metal complexes) and a deeper mechanistic understanding of stereoinduction in the absence of inherent directing effects. (b) Asymmetric C–C borylation strategies are currently restricted primarily to vinylcyclopropanes, proceeding via metal-boryl insertion followed by β-hydride elimination. While our group recently expanded this scope through the first directing-group-assisted asymmetric C–C borylation of cyclopropanes, such examples are exceedingly rare. The development of broadly applicable, catalytic methodologies for asymmetric C–C borylation across diverse cyclopropane scaffolds (e.g., alkyl-, aryl-, heteroatom-substituted) represents an imperative and fertile area for exploration. This necessitates exploring alternative activation modes beyond classical activation pathways. (c) The utilization of simple cyclopropanes lacking strong electronic biases or directing functionalities presents a formidable challenge. Achieving regio- and stereoselective borylation in these substrates demands innovative strategies to override inherent electronic and steric similarities. Research must focus on exploiting subtle conformational preferences, designing highly selective catalysts, or employing transient directing groups to impart the necessary control. (d) Current methodologies predominantly operate via conventional two-electron reaction manifolds. Integrating external fields-particularly photochemical and electrochemical activation-promises to revolutionize this chemistry. Such approaches could unlock radical-based mechanisms, single-electron transfer pathways, or redox-neutral processes, significantly broadening the reaction scope, enhancing functional group tolerance, and enabling novel disconnection strategies inaccessible via thermal routes. (e) Translating these sophisticated methodologies into practical industrial processes remains limited. Key hurdles include cost efficiency (catalyst loading, ligand cost, boron source), operational simplicity (reaction setup, workup), scalability, and environmental sustainability (solvent choice, waste generation). Although metal-free borylation offers advantages in yield and speed, the development of scalable, continuous-flow technologies and integration with downstream functionalization are critical priorities to accelerate the discovery and production of pharmaceuticals and bioactive molecules.
This review has chronicled the remarkable progress and persistent challenges in cyclopropane-to-organoboron conversion. We envision this field evolving into a versatile and transformative paradigm for synthetic organic chemistry. Addressing the outlined challenges will not only advance fundamental boron and cyclopropane chemistry but also foster the discovery of novel bioactive agents and functional materials. We anticipate that multidisciplinary collaborations-spanning catalysis, mechanistic studies, computational design, and process chemistry-will be essential to unlock the full potential of this powerful synthetic strategy. It is our sincere hope that this perspective serves as a catalyst, inspiring continued innovation and exploration in this exciting domain.
Conflicts of interest
The authors declare no competing financial interest.Data availability
No primary research results, software or code has been included and no new data were generated or analysed as part of this review.Acknowledgements
We would like to thank Financial support from the National Key R&D Program of China (2022YFA1503200), the National Natural Science Foundation of China (Grants 2024101703, 22025104, 22171134, and 21972064), the Jiangsu Funding Program for Excellent Postdoctoral Talent (Grant 2025ZB263), and the Fundamental Research Funds for the Central Universities (Grant 020514380326) for their financial support.References
- C. Ebner and E. M. Carreira, Chem. Rev., 2017, 117, 11651–11679 Search PubMed.
- A. L. Gabbey, K. Scotchburn and S. A. L. Rousseaux, Nat. Rev. Chem., 2023, 7, 548–560 CrossRef.
- A. U. Augustin and D. B. Werz, Acc. Chem. Res., 2021, 54, 1528–1541 Search PubMed.
- M.-M. Wang, T. V. T. Nguyen and J. Waser, Chem. Soc. Rev., 2022, 51, 7344–7357 RSC.
- O. O. Sokolova and J. F. Bower, Chem. Rev., 2021, 121, 80–109 CrossRef PubMed.
- Y. Cohen, A. Cohen and I. Marek, Chem. Rev., 2021, 121, 140–161 CrossRef.
- D. Nam, V. Steck, R. J. Potenzino and R. Fasan, J. Am. Chem. Soc., 2021, 143, 2221–2231 CrossRef PubMed.
- H. E. Simmons and R. D. Smith, J. Am. Chem. Soc., 1958, 80, 5323–5324 CrossRef.
- V. A. Petrov and W. Marshall, J. Fluorine Chem., 2012, 133, 61–66 CrossRef.
- E. J. Corey and M. Chaykovsky, J. Am. Chem. Soc., 1965, 87, 1353–1364 CrossRef.
- X. Bugaut, F. Liu and F. Glorius, J. Am. Chem. Soc., 2011, 133, 8130–8133 CrossRef.
- A. N. Baumann, F. Schüppel, M. Eisold, A. Kreppel, R. de Vivie-Riedle and D. Didier, J. Org. Chem., 2018, 83, 4905–4921 CrossRef PubMed.
- C. Andersen, V. Ferey, M. Daumas, P. Bernardelli, A. Guérinot and J. Cossy, Org. Lett., 2019, 21, 2285–2289 Search PubMed.
- B. M. Coleridge, C. S. Bello and A. Leitner, Tetrahedron Lett., 2009, 50, 4475–4477 Search PubMed.
- E. J. Corey, S. A. Rao and M. C. Noe, J. Am. Chem. Soc., 1994, 116, 9345–9346 Search PubMed.
- A. J. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412–443 Search PubMed.
- R. J. Grams, W. L. Santos, I. R. Scorei, A. Abad-García, C. A. Rosenblum, A. Bita, H. Cerecetto, C. Viñas and M. A. Soriano-Ursúa, Chem. Rev., 2024, 124, 2441–2511 CrossRef PubMed.
- T. Ishiyama, N. Matsuda, N. Miyaura and A. Suzuki, J. Am. Chem. Soc., 1993, 115, 11018–11019 CrossRef.
- I. F. Yu, J. W. Wilson and J. F. Hartwig, Chem. Rev., 2023, 123, 11619–11663 CrossRef PubMed.
- S. J. Geier, C. M. Vogels, J. A. Melanson and S. A. Westcott, Chem. Soc. Rev., 2022, 51, 8877–8922 RSC.
- B. Zhao, Z. Li, Y. Wu, Y. Wang, J. Qian, Y. Yuan and Z. Shi, Angew. Chem., Int. Ed., 2019, 58, 9448–9452 CrossRef.
- M. Shimizu, M. Schelper, I. Nagao, K. Shimono, T. Kurahashi and T. Hiyama, Chem. Lett., 2006, 35, 1222–1223 CrossRef.
- A. De Meijere, Angew. Chem., Int. Ed. Engl., 1979, 18, 809–826 Search PubMed.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef.
- S. Löhr and A. de Meijere, Synlett, 2001, 0489–0492 CrossRef.
- R. Bisht, C. Haldar, M. M. M. Hassan, M. E. Hoque, J. Chaturvedi and B. Chattopadhyay, Chem. Soc. Rev., 2022, 51, 5042–5100 RSC.
- C. W. Liskey and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 3375–3378 CrossRef.
- R. Murakami, K. Tsunoda, T. Iwai and M. Sawamura, Chem. – Eur. J., 2014, 20, 13127–13131 CrossRef.
- P. Bertus and J. Caillé, Chem. Rev., 2025, 125, 3242–3377 CrossRef PubMed.
- S. Miyamura, M. Araki, T. Suzuki, K. Itami and J. Yamaguchi, Angew. Chem., Int. Ed., 2015, 54, 846–851 CrossRef PubMed.
- D. Y.-K. Chen, R. H. Pouwer and J.-A. Richard, Chem. Soc. Rev., 2012, 41, 4631 RSC.
- J. He, Q. Shao, Q. Wu and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 3344–3347 CrossRef PubMed.
- Y. Shi, Q. Gao and S. Xu, J. Am. Chem. Soc., 2019, 141, 10599–10604 CrossRef CAS.
- T. T. Talele, J. Med. Chem., 2016, 59, 8712–8756 CrossRef CAS PubMed.
- Y. Shi, Y. Yang and S. Xu, Angew. Chem., Int. Ed., 2022, 61, e202201463 CrossRef CAS PubMed.
- K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788–802 CrossRef CAS PubMed.
- T. Xie, L. Chen, Z. Shen and S. Xu, Angew. Chem., Int. Ed., 2023, 62, e202300199 CrossRef CAS PubMed.
- E. Lee-Ruff and G. Mladenova, Chem. Rev., 2003, 103, 1449–1484 CrossRef CAS.
- X. Chen, L. Chen, H. Zhao, Q. Gao, Z. Shen and S. Xu, Chin. J. Chem., 2020, 38, 1533–1537 CrossRef CAS.
- T. Kang, T. G. Erbay, K. L. Xu, G. M. Gallego, A. Burtea, S. K. Nair, R. L. Patman, R. Zhou, S. C. Sutton, I. J. McAlpine, P. Liu and K. M. Engle, ACS Catal., 2020, 10, 13075–13083 CrossRef CAS.
- W. Zheng, Y. Cao, B. B. Tan, Y. Wang, S. Ge and Y. Lu, J. Am. Chem. Soc., 2025, 147, 12273–12284 CrossRef CAS PubMed.
- A. de Meijere, Chem. Rev., 2003, 103, 931–932 CrossRef CAS.
- M. Wang and Z. Shi, Chem. Rev., 2020, 120, 7348–7398 CrossRef CAS.
- Y. Zhu, J. Li, H. Zhao and B. Su, Chin. J. Chem., 2024, 42, 3588–3604 CrossRef CAS.
- V. Pirenne, B. Muriel and J. Waser, Chem. Rev., 2021, 121, 227–263 CrossRef CAS.
- K. Oshima, Y. Sumida and H. Yorimitsu, Org. Lett., 2008, 10, 4677–4679 Search PubMed.
- J. M. Medina, T. Kang, T. G. Erbay, H. Shao, G. M. Gallego, S. Yang, M. Tran-Dubé, P. F. Richardson, J. Derosa, R. T. Helsel, R. L. Patman, F. Wang, C. P. Ashcroft, J. F. Braganza, I. McAlpine, P. Liu and K. M. Engle, ACS Catal., 2019, 9, 11130–11136 CrossRef CAS PubMed.
- C. Chen, X. Shen, J. Chen, X. Hong and Z. Lu, Org. Lett., 2017, 19, 5422–5425 CrossRef CAS.
- M. Shi, L.-X. Shao, J.-M. Lu, Y. Wei, K. Mizuno and H. Maeda, Chem. Rev., 2010, 110, 5883–5913 CrossRef CAS.
- J. Chen, S. Gao and M. Chen, Chem. Sci., 2019, 10, 10601–10606 Search PubMed.
- J. Chen, S. Gao and M. Chen, Org. Lett., 2019, 21, 8800–8804 CrossRef CAS.
- M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117–3179 Search PubMed.
- J. Zhao and K. J. Szabó, Angew. Chem., Int. Ed., 2016, 55, 1502–1506 Search PubMed.
- A. Brandi, S. Cicchi, F. M. Cordero and A. Goti, Chem. Rev., 2014, 114, 7317–7420 Search PubMed.
- Q. Chen, X. Zhang, S. Su, Z. Xu, N. Li, Y. Li, H. Zhou, M. Bao, Y. Yamamoto and T. Jin, ACS Catal., 2018, 8, 5901–5906 Search PubMed.
- B. B. Tan, M. Hu and S. Ge, Angew. Chem., Int. Ed., 2023, 62, e202307176 Search PubMed.
- W.-F. Wang, K. Lu, P.-R. Liu, H.-H. Zeng, L.-M. Yang, A.-J. Ma, Y.-Q. Tu and J.-B. Peng, ACS Catal., 2024, 14, 5156–5166 CrossRef CAS.
- W. Zheng, B. B. Tan, S. Ge and Y. Lu, J. Am. Chem. Soc., 2024, 146, 5366–5374 Search PubMed.
- M. Oestreich, E. Hartmann and M. Mewald, Chem. Rev., 2013, 113, 402–441 Search PubMed.
- M. Suginome, T. Matsuda and Y. Ito, J. Am. Chem. Soc., 2000, 122, 11015–11016 Search PubMed.
- T. Ohmura, H. Taniguchi, Y. Kondo and M. Suginome, J. Am. Chem. Soc., 2007, 129, 3518–3519 CrossRef CAS.
- J. Lu and P. H. Toy, Chem. Rev., 2009, 109, 815–838 CrossRef CAS.
- Y. Akai, T. Yamamoto, Y. Nagata, T. Ohmura and M. Suginome, J. Am. Chem. Soc., 2012, 134, 11092–11095 CrossRef CAS PubMed.
- L.-M. Yang, H.-H. Zeng, X.-L. Liu, A.-J. Ma and J.-B. Peng, Chem. Sci., 2022, 13, 7304–7309 RSC.
- H.-H. Zeng, Y.-Q. Wang, Y.-Y. He, X.-L. Zhong, H. Li, A.-J. Ma and J.-B. Peng, J. Org. Chem., 2024, 89, 2637–2648 CrossRef CAS PubMed.
- Y. Yuan, F. Wu, J. Xu and X. Wu, Angew. Chem., Int. Ed., 2020, 59, 17055–17061 CrossRef CAS PubMed.
- F.-P. Wu and X.-F. Wu, Chem. Sci., 2022, 13, 4321–4326 RSC.
- W. Ming, H. S. Soor, X. Liu, A. Trofimova, A. K. Yudin and T. B. Marder, Chem. Soc. Rev., 2021, 50, 12151–12188 RSC.
- Y.-S. Zhu, Y.-L. Guo, Y.-Y. Zhu and B. Su, J. Am. Chem. Soc., 2024, 146, 32283–32291 CrossRef CAS.
- Y. Yuan, F. Ge, T. Yang and X.-F. Wu, J. Catal., 2025, 450, 116280 CrossRef CAS.
- S. Oishi, H. Kamitani, Y. Kodera, K. Watanabe, K. Kobayashi, T. Narumi, K. Tomita, H. Ohno, T. Naito, E. Kodama, M. Matsuoka and N. Fujii, Org. Biomol. Chem., 2009, 7, 2872 RSC.
- A. M. Y. Suliman, E.-A. M. A. Ahmed, T.-J. Gong and Y. Fu, Org. Lett., 2021, 23, 3259–3263 CrossRef CAS PubMed.
- A. M. Y. Suliman, E.-A. M. A. Ahmed, T.-J. Gong and Y. Fu, Chem. Commun., 2021, 57, 6400–6403 RSC.
- E.-A. M. A. Ahmed, H. Zhang, W.-G. Cao and T.-J. Gong, Org. Lett., 2023, 25, 9020–9024 Search PubMed.
- Y. Wang, J. Bai, Y. Yang, W. Zhao, Y. Liang, D. Wang, Y. Zhao and Z. Shi, Chem. Sci., 2021, 12, 3599–3607 RSC.
- T. Wang, M. Wang, Y. Wang, M. Li, Y. Zheng, Q. Chen, Y. Zhao and Z. Shi, Chem, 2023, 9, 130–142 Search PubMed.
- S. Li, H. Jiao, X.-Z. Shu and L. Wu, Nat. Commun., 2024, 15, 1846 CrossRef PubMed.
- S. Li, C. Hu, L. Leo Liu and L. Wu, Angew. Chem., Int. Ed., 2024, 63, e202412368 CrossRef PubMed.
- S. Kang, J. Lv, T. Wang, B. Wu, M. Wang and Z. Shi, Nat. Commun., 2024, 15, 7380 CrossRef PubMed.
- Z. X. Giustra and S.-Y. Liu, J. Am. Chem. Soc., 2018, 140, 1184–1194 CrossRef.
- H. Lyu, T. H. Tugwell, Z. Chen, G. A. Kukier, A. Turlik, Y. Wu, K. N. Houk, P. Liu and G. Dong, Nat. Chem., 2024, 16, 269–276 CrossRef.
- A. D. J. Calow, D. Dailler and J. F. Bower, J. Am. Chem. Soc., 2022, 144, 11069–11074 CrossRef PubMed.
- J. G. M. Morton, M. A. Dureen and D. W. Stephan, Chem. Commun., 2010, 46, 8947–8949 Search PubMed.
- D. Wang, X. Xue, K. N. Houk and Z. Shi, Angew. Chem., Int. Ed., 2018, 57, 16861–16865 Search PubMed.
- H. Kondo, S. Miyamura, K. Matsushita, H. Kato, C. Kobayashi, Arifin, K. Itami, D. Yokogawa and J. Yamaguchi, J. Am. Chem. Soc., 2020, 142, 11306–11313 Search PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2026 |