
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formation of active sites in copper-exchanged zeolites for the direct methane-to-methanol conversion
Jörg W. A.
Fischer†‡
 a,
Andreas
Brenig‡
b,
Johannes
Wieser‡
b,
Jeroen A.
van Bokhoven
bc and
Vitaly L.
Sushkevich
*c
a,
Andreas
Brenig‡
b,
Johannes
Wieser‡
b,
Jeroen A.
van Bokhoven
bc and
Vitaly L.
Sushkevich
*c
aDepartment of Chemistry and Applied Biosciences, ETH Zurich, Vladimir-Prelog-Weg 1-5/10, 8093 Zurich, Switzerland
bInstitute for Chemical and Bioengineering, ETH Zurich, Vladimir-Prelog-Weg 1-5/10, 8093 Zurich, Switzerland
cCenter for Energy and Environmental Sciences, Paul Scherrer Institute, Forschungsstrasse 111, 5232 Villigen, Switzerland. E-mail: vitaly.sushkevich@psi.ch
First published on 27th February 2026
Abstract
Cu-exchanged zeolites are extensively studied for their promising properties in the direct and selective conversion of CH4 to CH3OH at low temperatures. Their performance has been attributed to the presence of multiple oxygenated Cu(II) active sites, which can co-exist in a given zeolite framework. Most emphasis has been placed on identifying these Cu-oxo centers, understanding their structural development during reaction with CH4, and establishing a correlation between the Cu speciation and the zeolite's Cu loading, Si/Al ratio, and topology. On the contrary, the processes underlying the oxidative formation of Cu(II) active sites have received comparatively little attention. This critical review presents an overview of the current understanding of the generation of Cu(II) active centers and highlights the impact of the oxidant type and the reaction conditions on the nature of the formed Cu(II) species. Key knowledge gaps are identified by addressing prevailing misconceptions and conflicting reports in the literature. To remedy these issues, strategies are proposed to resolve discrepancies and to gain new insight into the processes preceding the CH4-to-CH3OH conversion in Cu-exchanged zeolites, aiming to better understand and control the generation of these Cu(II) active sites in zeolites.
 Jörg W. A. Fischer | Jörg Fischer studied chemistry at the University of Konstanz, with academic stays at the University of Washington and at the University of Tübingen. He joined ETH in 2020 for his PhD with Prof. Jeschke. During that time, he focused on the development of operando EPR spectroscopy and its application to transition metal exchanged zeolites. In 2025, he obtained a postdoctoral fellowship from the Japan Society for the Promotion of Science (JSPS) and moved to Japan working at the Hokkaido University on CO2 hydrogenation over metal oxides. Recently he joined Utrecht University for a postdoctoral research stay, where he focuses on coke speciation in zeolites. |
 Andreas Brenig | Dr Andreas Brenig earned his Bachelor's and Master's degrees in Chemistry from the Technical University of Munich, where he specialized in Chemical Engineering and Catalysis. He then completed his doctoral studies at the Center for Energy and Environmental Sciences at Paul Scherrer Institute and is currently working as a postdoctoral researcher at the Institute for Chemical and Bioengineering at ETH Zurich. His research focuses on the physicochemical characterization of materials for energy applications and the implementation of chemometric methods. |
 Johannes Wieser | Johannes Wieser received his BSc and MSc degrees in Chemistry from the Technical University Munich (TUM). He subsequently joined the Department of Chemical and Applied Biosciences (D-CHAB) at ETH Zürich, where he is currently pursuing his PhD under the supervision of Professor Jeroen A. van Bokhoven. His research at ETH focuses on the development of processes for the selective oxidation of methane to value-added products, primarily via copper-exchanged zeolitic systems. |
 Jeroen A. van Bokhoven | Jeroen A. van Bokhoven completed a degree in chemistry at Utrecht University (Netherlands) in 1995 and went on to obtain a PhD in inorganic chemistry and catalysis from the same university in 2000 (with honours). From 1999 until 2002, he was head of the XAS (X-ray absorption spectroscopy) user-support group at Utrecht University. In 2002, he moved to the ETH, where he worked as a researcher in the group of Prof. Prins. In 2006, he obtained an SNF assistant professorship in the Department of Chemistry and Applied Biology. He was the 2008 recipient of the Swiss Chemical Society Werner Prize. Since 2010, Jeroen A. van Bokhoven has a Chair in Heterogeneous Catalysis at the Institute for Chemical and Bioengineering at ETH Zurich and is Head of Laboratory for Catalysis and Sustainable Chemistry at the Paul Scherrer Institute. |
 Vitaly L. Sushkevich | Vitaly Sushkevich earned his PhD from Lomonosov Moscow State University in 2014, where he led the development of novel catalysts and catalytic processes for producing monomers for synthetic rubbers from renewable feedstocks. In 2019, he joined the Paul Scherrer Institute as a Scientist, focusing on challenges in selective alkane oxidation and dehydrogenation. More recently, he has taken on a leading role in advancing research on the understanding of the mechanisms of synthesis of solid catalysts and porous materials. |
1. Introduction
Natural gas sources in remote or stranded locations are notoriously difficult to utilize, as the low volumetric energy density of CH4, the main constituent of natural gas, makes long-distance transportation economically unfeasible, frequently leading to gas flaring at extraction sites.1 Liquefaction of natural gas can be used for natural gas transportation from remote production facilities, but the relatively high cost of this process and the expensive infrastructure have limited its commercial application to large natural gas reserves. Alternatively, converting CH4 to condensable or value-added chemicals on-site is a promising route for CH4 valorization. The transformation of CH4 to CH3OH continues to attract attention as CH3OH is an important energy carrier (e.g., fuel, direct methanol fuel cells (DMFCs))2 and a chemical building block for producing olefins.3 The industrial production of CH3OH based on CH4 consists of two consecutive steps: (1) CH4 steam reforming into syngas (mixture of CO and H2) and (2) conversion of syngas to CH3OH at high pressures using a Cu/ZnO/Al2O3 (CZA) catalyst. This approach, especially the steam reforming of CH4 to syngas, is energy-intensive and only feasible for large-scale operations.4Scale-flexible and direct approaches for the CH4-to-CH3OH conversion are an attractive solution, but face one major challenge: the higher reactivity of CH3OH compared to CH4, leading to overoxidation of the former.5,6 Transition metal ion (TMI)-exchanged zeolites have emerged as promising materials for the selective oxidation of CH4 to CH3OH, attracting considerable attention over the past two decades.6,7 These materials can stabilize monomeric, dimeric, and multimeric metal-oxo sites that resemble the active centers of C–H bond activating enzymes, such as Cu containing particulate methane monooxygenase (pMMO) and Fe containing soluble methane monooxygenase (sMMO).8,9 The active species in TMI containing zeolites are usually generated by an oxidant such as O2, N2O, NO, or H2O2. Among the TMI-zeolites employed for the CH4-to-CH3OH conversion, Cu- and Fe-exchanged zeolites are the most studied ones. Cu-exchanged zeolites are considered to have the greatest economic potential, as the Cu-oxo sites can be generated using O2, whereas the activation of Fe-exchanged zeolites typically requires expensive oxidants such as N2O or H2O2, which are more costly than the produced CH3OH.10–12
The generation of these Cu(II) active centers is of fundamental importance, as they govern the reactivity of the material. While the existing literature predominantly focuses on the structural characterization of the Cu-oxo species and on optimizing the CH3OH productivity by varying the process parameters during CH4 conversion, it often overlooks how the activation conditions influence the formation and nature of the Cu(II) active sites. A comprehensive understanding of the mechanism of Cu–zeolite activation is essential for selectively generating specific Cu(II) active centers and maximizing their population within the zeolite framework. In general, the formation of Cu(II) active species in the (non-)isothermal chemical looping as well as in the continuous CH4-to-CH3OH processes (see Section 2.3) remains poorly understood. In particular, contradictory findings regarding the effect of the activation temperature and duration on the CH3OH productivity have been reported,13–22 and disagreements concerning the influence of the partial pressure of the oxidizing agent persist.16,17,21,23 Therefore, an in-depth understanding of the formation of Cu(II) active sites is essential to obtain a holistic picture of the overall redox cycle.
In this review, the formation of various Cu-oxo centers, which are active in the CH4-to-CH3OH conversion, is discussed in detail, and misconceptions and knowledge gaps are presented. The article is structured as follows: First, a brief introduction to zeolites and the Cu(II) sites stabilized within these materials is provided alongside their key spectroscopic fingerprints. This is followed by showcasing different approaches in which Cu–zeolites have been employed in the CH4-to-CH3OH transformation. Subsequently, the generation of distinct Cu(II) centers by various oxidizing agents, namely O2, N2O, NO, H2O, CO2, and H2O2, is reviewed, and their role in the stoichiometric stepwise chemical looping and catalytic processes is critically examined. To avoid ambiguities, the Lewis structures of each Cu(II) active site and selected intermediates are compiled in Scheme S1 in the Supporting Information, along with their corresponding names, potential resonance structures, and Cu oxidation states. Next, common misconceptions and conflicting literature reports are identified and discussed, and knowledge gaps are pointed out. Finally, recommendations are given to gain the needed in-depth understanding of the processes leading to the formation of Cu(II) active species. Whenever specific Cu–zeolites are addressed, their Cu/Al and Si/Al ratios are provided as reported in the original publications, using the notation CuXZEOY, where X, ZEO, and Y correspond to the Cu/Al ratio, the framework type code, and the Si/Al ratio. If either the Cu/Al or Si/Al ratio is not specified in the literature, it is omitted from the material description. In cases where the Cu/Al ratio is not provided, X is replaced by a hyphen, i.e., Cu-ZEOY, for improved readability.
2. Zeolites
Zeolites are microporous crystalline aluminosilicates built from primary building units (PBUs), consisting of a central Si or Al atom tetrahedrally coordinated by four O atoms (T-sites). These PBUs connect via corner-sharing O atoms to form unique secondary building units (SBUs), whose arrangement defines the framework-specific porous system. Depending on the particular arrangement of the SBUs, the porous network features regular cavities interconnected by channels ranging from 3 to 15 Å in diameter.24 These channels are either straight or puckered and can intersect each other. Generally, the diffusion through zeolites is governed by the molecule's size since a molecule can only pass through when the free diameter of the pore is equal to or greater than the molecule's dimensions (molecular-scale porosity).9 This size restriction imposes steric constraints on molecular diffusion through the microporous network to reach adsorption sites. Therefore, zeolites belong to a class of materials called “molecular sieves”. Depending on the pore interconnectivity/dimensionality, zeolites can be classified into 1, 2, or 3D materials. Scheme 1 shows three zeolite frameworks, which are employed most frequently in the CH4-to-CH3OH conversion, and which feature different dimensionalities. While the mordenite (MOR) framework is a 1D structure (due to its pore openings being larger than 3.4 Å), the chabazite (CHA) and MFI frameworks exhibit a 3D structure.25Scheme 1 also highlights the crystallographically distinct T-sites in each framework. | ||
| Scheme 1 Different zeolite frameworks employed most frequently in the CH4-to-CH3OH conversion: MOR (a), MFI (b), and CHA (c). The two largest ring openings, as well as characteristic lattice structures, are highlighted in transparent colors. Crystallographically distinct T-sites are indicated in different shades of blue. Si and framework O atoms are marked in grey and red, respectively. | ||
The distribution of Al over crystallographic T-sites obeys the Loewenstein rule, which states that whenever two T-sites are linked by one O atom of the framework (Ofw), only one of them can be an Al atom.26 Therefore, the resulting upper limit of isomorphous substitution of Si by Al is defined by a minimum Si/Al ratio of 1. Additionally, the Al substitution strives for an even distribution over the lattice in naturally occurring zeolites.27 However, experimental evidence has suggested that the Al distribution in synthetic zeolites exhibits non-random patterns and deviates from simple statistical distributions.28–30 While Loewenstein's rule does remain valid, Al substitutions are found to be in closer proximity than expected for a random distribution.9 The difference in bond length between Al–Ofw (∼1.7 Å) and Si–Ofw (∼1.6 Å) distorts the lattice locally and influences the shape and free diameter of the rings. Therefore, the fraction of Al can affect the stability of the zeolite, with MOR and MFI being stable only above a Si/A ratio of 5 and 10, respectively.31,32 For CHA, Si/Al ratios as low as 2 have been reported in gas absorption studies.33
The replacement of Si by Al introduces a net negative charge on the zeolite lattice, which requires an extra-framework cation for charge balance. Different extra-framework cations affect the properties of the zeolite and enable optimization of the material for a given application. When H+ serves as the counterion, a Brønsted acid site (BAS) is created, which imparts the characteristic acidity of the zeolite. Introduction of TMIs is a widely employed strategy to modify the zeolite's adsorption and redox properties.34 The most common approach for the incorporation of other cations, such as Cu(II), involves an ion exchange of the parent material in an aqueous medium, where the original cations, e.g., H+, NH4+, or Na+, are exchanged with a Cu(II)-aqua complex. Notably, exchange conditions critically influence the final speciation and material properties, and precise control is essential to prevent undesired side reactions. For example, at high pH, both the participation of deprotonated surface Si–OH groups in the exchange reaction and the precipitation of Cu(II)-aqua complexes, which form CuO clusters upon calcination, contribute to the formation of nonstoichiometric and over-exchanged zeolites. Stoichiometric exchange occurs when the positive charge of the incorporated TMIs exactly balances the framework's negative charge, corresponding to a TMI/Al ratio of 0.5 for a divalent TMI species such as a monomeric bare Cu2+ site or a dimeric Cu(II) center (see Sections 2.1.1 and 2.1.3). It is important to point out that the stoichiometric exchange with other Cu(II) species, such as monomeric [CuOH]+ or tri- and multimeric Cu(II) sites (see Sections 2.1.2 and 2.1.4), leads to a different TMI/Al ratio. Instead of aqueous ion exchange, Cu(II) can also be introduced into the zeolite pores via incipient wetness impregnation (IWI).35,36 Another possibility is the so-called “solid-state” ion exchange. Here, the parent zeolite is mechanically mixed with a TMI containing precursor, such as CuCl, and the mixture is subsequently heated up, leading to an ion exchange.37 While solid-state ion exchange is a relatively straightforward procedure, the method is limited by potential degradation of the zeolite framework at high temperatures and incomplete reaction of the precursor.38–40 Alternatively, TMIs can also be directly incorporated during the synthesis of the zeolite by employing TMI containing structure directing agents (SDAs). This approach has been demonstrated for the preparation of Cu-exchanged small-pore zeolites, such as Cu–CHA, where the narrow pore size might hinder diffusion of the Cu(II)-aqua complex during a conventional ion exchange, preventing a full ion exchange.41 All the above-mentioned methods for the introduction of Cu into zeolites typically give rise to a complex mixture of various co-existing Cu(II) active sites upon oxidative treatment. These species are discussed in more detail in the following section.
2.1 Definition of potential Cu(II) species in specific zeolite frameworks
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–12000 cm−1 in the ultraviolet-visible (UV-Vis) spectra of various Cu–zeolites with different frameworks has been attributed to these bare Cu2+ sites.50–54
000–12000 cm−1 in the ultraviolet-visible (UV-Vis) spectra of various Cu–zeolites with different frameworks has been attributed to these bare Cu2+ sites.50–54
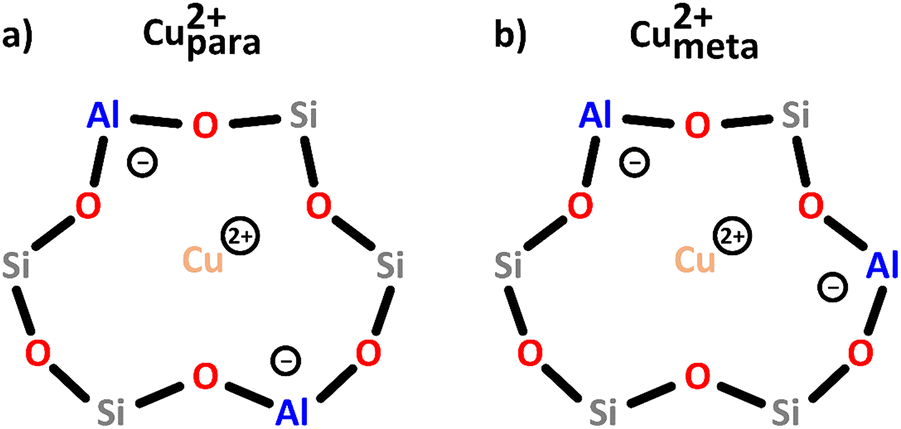 | ||
| Scheme 2 Bare monomeric Cu2+ charge-balanced by two Al T-sites positioned in either the para (a) or meta (b) arrangement. Color code: Al = blue, Si = grey, framework O = red, Cu = orange. | ||
In an operando EPR study, Godiksen et al. demonstrated that these two centers differ in their reactivity toward oxidation in gas mixtures relevant for the NH3-mediated selective catalytic reduction of NOx (NH3-SCR), indicating that the Al T-site arrangement has a major impact on their redox behavior.55 Furthermore, recent studies have suggested that even though these sites lack Oef ligands, they can still participate in the CH4-to-CH3OH conversion when present as a Cu2+/[CuOH]+ pair.46,50,56
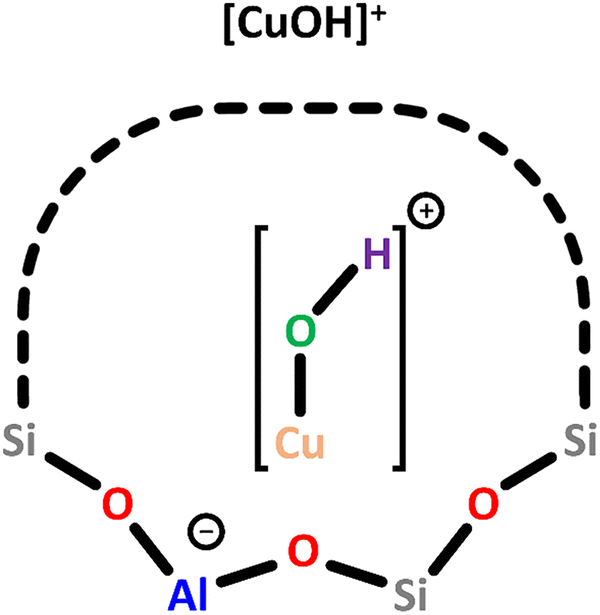 | ||
| Scheme 3 Monomeric [CuOH]+ charge-balanced by a single Al T-site. Color code: Al = blue, Si = grey, framework O = red, Cu = orange, extra-framework O = green, H = violet. | ||
In Cu–MOR, [CuOH]+ exhibits a gII value of 2.27, similar to that in Cu-MFI, and both species are believed to exhibit a bidentate coordination to the Ofw atoms of a single Al T-site. In contrast, DFT calculations have indicated that [CuOH]+ is coordinated by three Ofw atoms in Cu–CHA.58 Nevertheless, since the experimentally observed g value of [CuOH]+ in Cu–CHA (gII = 2.40) deviates significantly from that in Cu–MOR and Cu-MFI, a different coordination mode of [CuOH]+ in Cu–CHA compared to Cu–MOR and Cu-MFI seems plausible. Apart from the ligation, the zeolite framework also governs the location of [CuOH]+. In Cu–MOR, [CuOH]+ has originally been assumed to reside within a 6-MR, but a recent study by Heyer et al. has suggested that it is actually located in the 8-MR of the MOR side pocket (Scheme 1), facing the 8-MR channel.61,62 Other ion exchange positions were, however, not excluded by the authors. In Cu–CHA, [CuOH]+ is also positioned within the 8-MR, whereas it is situated in the 10-MR in Cu-MFI.46,50,58,59,64 In addition to EPR spectroscopy, [CuOH]+ has also been characterized by NO-FTIR and UV-Vis measurements, yielding absorption bands around 1900 cm−1 and between 16000 and 12000 cm−1, respectively.44,50,51,60 The participation of [CuOH]+ in CH4 partial oxidation has been demonstrated in the past years.46,50,56,62,63,65
700 cm−1, but subsequent Raman and DFT studies by the same research group revised this assignment to [Cu2(μ-O)]2+.66 Furthermore, Ipek et al. proposed the presence of [Cu2(trans-μ-1,2-O2)]2+ in Cu–CHA based on the identification of broad UV-Vis features in the 35000–22200 cm−1 range and distinct Raman signals (see Section 3).20 The different dimeric oxygenated Cu(II) centers are summarized in Scheme 4 and Scheme S1d–f. Depending on the zeolite's Si/Al ratio, these sites become dominant in materials characterized by a higher Cu loading than that needed for the generation of monomeric Cu(II) species.46,50,57 Notably, the spin–spin exchange interaction of the Cu(II) ions usually renders dimeric Cu-oxo motifs EPR invisible under standard measurement conditions, e.g., at X-band frequencies (9.5 GHz).46,47,67
 | ||
| Scheme 4 Dimeric [Cu2(µ-O)2]2+ (a), [Cu2(µ-O)]2+ (b) and [Cu2(trans-µ-1,2-O2)]2+ (c) charge-balanced by two Al T-sites. Color code: Al = blue, Si = grey, framework O = red, Cu = orange, extra-framework O = green. | ||
In Cu–MOR, three distinct [Cu2(μ-O)]2+ cores have been proposed based on Raman measurements by Vanelderen et al. and Plessers et al.51,68 One of these Cu(II) centers has been suggested to reside within the 8-MR of the MOR side pocket (Scheme 1), oriented toward the 12-MR channel, whereas the other two Cu-oxo sites have been shown to span across the 8-MR channel.68,69 Remarkably, [Cu2(μ-O)]2+ positioned in the more restricted 8-MR channel is more active in CH4 hydroxylation than its geometrically less confined counterpart.51,69 According to DFT calculations, the higher activity of the former Cu(II) species originates from the stronger van der Waals interaction between CH4 and the narrow zeolite lattice surrounding this Cu(II) active center. Notably, the nature of the dimeric Cu-oxo site has been shown to be influenced by the co-cation, which in turn affects the material's reactivity. In Cu–MOR and Cu-MFI, Brezicki et al. demonstrated that [Cu2(μ-O)]2+, characterized by a UV-Vis signal at ∼27500 cm−1, is the dominant dimeric Cu(II) species in the absence of Na+ co-cations, whereas [Cu2(trans-μ-1,2-O2)]2+, featuring a UV-Vis band at around 22000 cm−1, becomes the prevalent Cu(II) center in the presence of Na+.70 Compared to [Cu2(μ-O)]2+, [Cu2(trans-μ-1,2-O2)]2+ was found to promote CH4 overoxidation due to the greater number of reactive Oef atoms in this Cu(II) site. In contrast to Brezicki et al., Artsiusheuski et al. assigned the two features at approximately 27200 and 21900 cm−1 in the UV-Vis spectrum of Cu-MFI to two distinct [Cu2(μ-O)]2+ motifs within the 10-MR (Scheme 1), which differ in their reactivity toward CH4. Using Fourier-transform extended X-ray absorption fine structure (FT-EXAFS) spectroscopy, the Cu–Cu distance of the two [Cu2(μ-O)]2+ species was determined to amount to 2.9 and 3.2 Å.71
Two different Cu(II) dimers have been proposed to co-exist in Cu–CHA. Originally, one of them has been attributed to a [Cu2(trans-μ-1,2-O2)]2+ center, whereas the other one has been identified as a [Cu2(μ-O)]2+ species.20 However, a recent study has provided evidence that instead of [Cu2(trans-μ-1,2-O2)]2+ and [Cu2(μ-O)]2+, Cu–CHA might host two different [Cu2(μ-O)]2+ motifs, which differ in their activity, Cu–Cu distance, and location within the framework.69 Their exact structure and the origin of the variation in their activity remain elusive.20,50 Furthermore, Göltl et al. proposed the presence of two different [Cu2(μ-O)2]2+ centers in Cu–CHA to explain the difference in material reactivity after activation in either O2 or N2O.72 They concluded that activation with N2O leads to [Cu2(μ-O)2]2+, whereas activation with O2 results in the corresponding hydroxylated Cu(II) dimer ([Cu2(μ-OH)2]2+, Scheme S1g), which, however, requires the presence of H2O.
It is worth mentioning that a recent study by Heyer et al., focusing on dimeric Cu(II) species in Cu–CHA and Cu-MFI, contradicts the aforementioned observations by proposing the existence of only a single type of [Cu2(μ-O)]2+ located within the 8-MR of Cu–CHA and the 10-MR of Cu-MFI. This study attributes the differences in reactivity between frameworks to variations in the coordination of [Cu2(μ-O)]2+ to the zeolite lattice, which result in different ground spin states of S = 0 and S = 1 for [Cu2(μ-O)]2+ in Cu-MFI and Cu–CHA, respectively.67
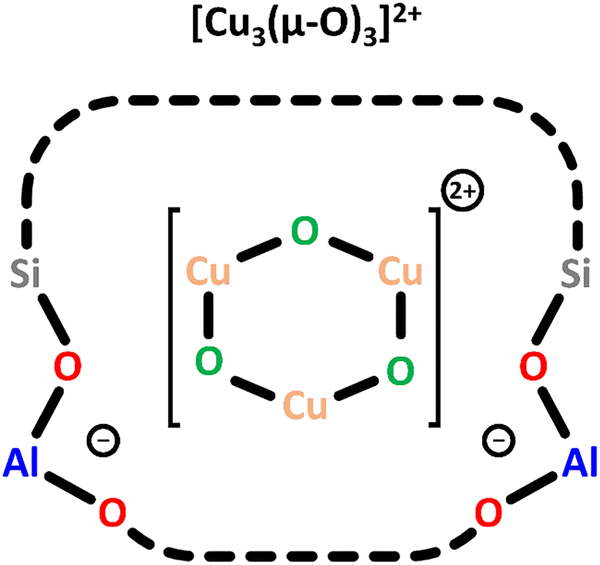 | ||
| Scheme 5 Trimeric [Cu3(µ-O)3]2+ charge-balanced by two Al T-sites. Color code: Al = blue, Si = grey, framework O = red, Cu = orange, extra-framework O = green. | ||
To enable the formation of these trimeric Cu(II) sites, the authors selected a MOR framework with a high concentration of Al T-sites in the 8-MR of the side pockets (Scheme 1) and employed an optimized synthesis procedure.73 In a subsequent theoretical study on Cu-MFI, the same research group proposed that trinuclear Cu-oxo clusters are the most stable Cu(II) species at elevated O2 partial pressures.74 A UV-Vis band around 31000 cm−1 has been assigned to the [Cu3(µ-O)3]2+ species, but no experimental EPR signature or NO-FTIR features have been reported yet. Notably, only Ikuno et al. and Kim et al. have observed similar UV-Vis absorption features in the range from 39000 to 30000 cm−1 and attributed them to [Cu3(µ-O)3]2+.19,21
Furthermore, theory-based studies have identified an increased stability of Cu-oxo clusters with even higher nuclearity.75 Using FT-EXAFS spectroscopy, Sushkevich et al. and Artsiusheuski et al. suggested the presence of CuXOY clusters in the large pores of Cu-FAU and Cu-exchanged beta (BEA), which also participate in the conversion of CH4 to CH3OH.76,77 In addition, Tomkins et al. suggested multimeric dispersed Cu-oxo clusters in Cu–MOR as active species based on transmission electron microscopy (TEM) and FT-EXAFS measurements, and the reduction of a large fraction of Cu(II) in the studied material after exposure to CH4 was reported.17
2.3 CH4-to-CH3OH conversion in Cu–zeolites
Scheme 6 illustrates a possible reaction mechanism for the selective CH4-to-CH3OH conversion via [Cu2(μ-O)]2+.78 The first steps include a dehydration of the material and the formation of the Cu(II) species with an oxidizing agent (1) and (2). Exposure to CH4 results in its adsorption on the Cu(II) active site (3), where it is subsequently converted to CH3OH, which induces a reduction of the Cu(II) center. The CH3OH formation can proceed either via homolytic (4.1) or heterolytic (4.2) C–H bond activation. Theoretical calculations by Phung et al., employing complete active space perturbation theory (CASPT2), favored the heterolytic pathway, thereby questioning the general validity of the radical rebound step in the homolytic reaction mechanism.79 After CH3OH formation (5), the material is exposed to H2O to facilitate the extraction of CH3OH from the framework (6). These steps complete the cycle, and the Cu(II) active site can be regenerated upon dehydration and oxidation.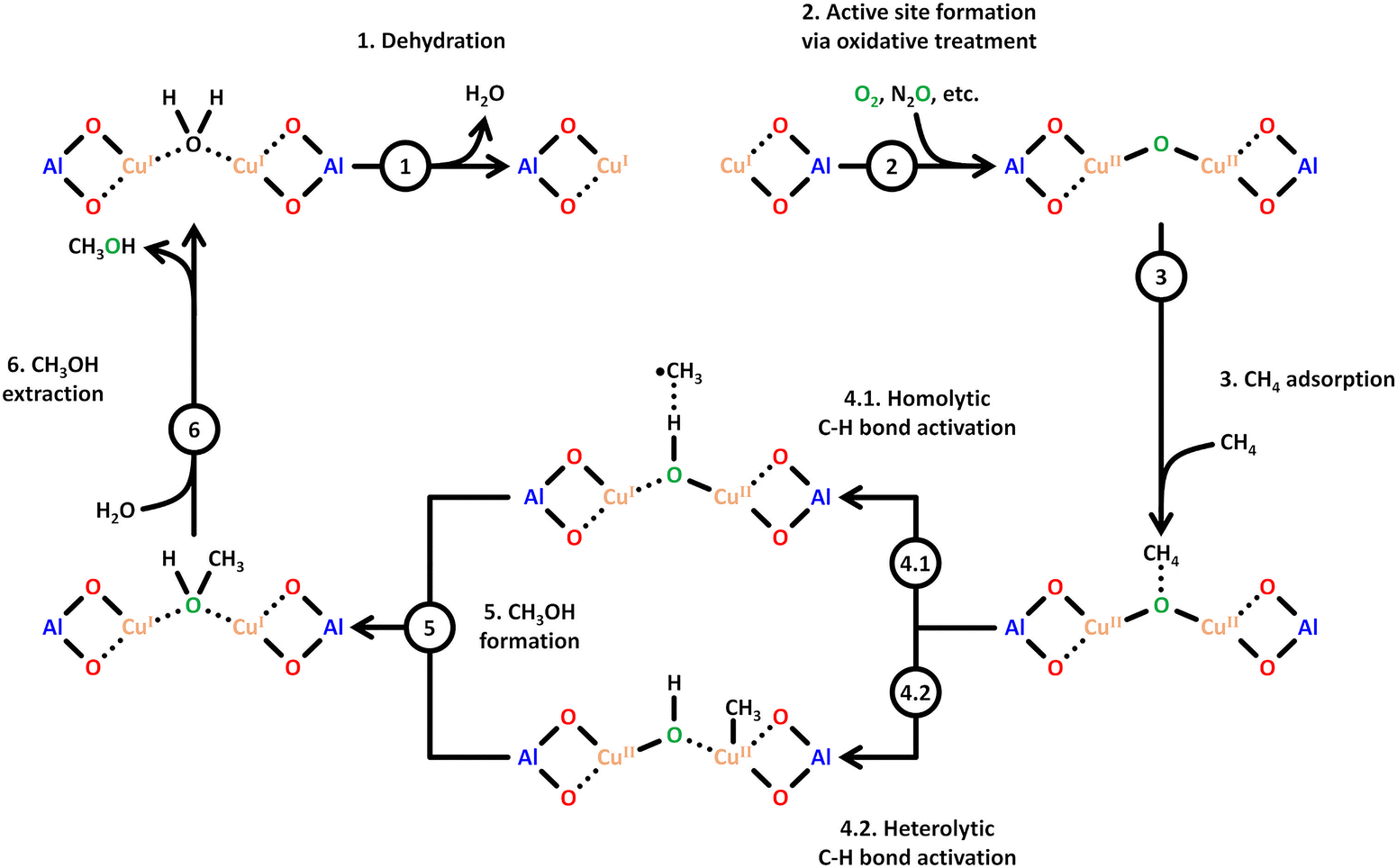 | ||
| Scheme 6 Proposed mechanism of CH4 hydroxylation by [Cu2(µ-O)]2+. Starting from a reduced state after H2O-assisted CH3OH extraction, the redox cycle consists of: dehydration (1), active site formation via exposure to an oxidizing agent such as O2 or N2O (2), CH4 adsorption at the Cu(II) active site (3), C–H bond activation via either homolytic or heterolytic (4.1 or 4.2) C–H bond activation, CH3OH formation (5), and CH3OH extraction with H2O (6). Dotted bonds correspond to dative interactions. Color code: Al = blue, framework O = red, Cu = orange, extra-framework O = green. Adapted from ref. 78 with permission from the American Chemical Society (Copyright 2018). | ||
Various strategies have been explored for the effective transformation of CH4 into CH3OH. In a stepwise process (chemical looping, vide infra), the outlined steps (activation (1) and (2), reaction (3)–(5), and product extraction (6)) are separated, while in the continuous (catalytic) operation mode, the oxidizing agent, e.g., O2, as well as CH4 and H2O, are introduced simultaneously. The primary issue of continuous CH4 hydroxylation lies in the significantly lower CH3OH yield and selectivity compared to the stepwise approach.13,80 Achieving high CH4 conversion at equally high CH3OH yield and selectivity is challenging due to both kinetic and thermodynamic constraints. High CH3OH yield is only attainable if the CH4 partial oxidation is faster than the subsequent oxidation of CH3OH to deeper oxidation products such as CO2. However, the C–H bond activation in CH4 is slower than in CH3OH, resulting in a substantial decrease in CH3OH selectivity at elevated CH4 conversion.8 This phenomenon is known as the “selectivity-conversion limit”.81–84
To overcome this limitation, product protection strategies can be employed. Of these, the chemical looping approach has been most frequently used for Cu–zeolites. This scheme involves a stoichiometric and stepwise process and is often combined with a temperature swing, but an isothermal operation mode is also possible.17,85,86 In detail, the stepwise process involves three consecutive steps: (1) generation of the active Cu(II) sites within the zeolite by an oxidant at temperatures between 423–753 K, (2) the reaction with CH4 at 423–623 K, and (3) the extraction of CH3OH using liquid H2O at ambient temperature or H2O vapor. After reaction with CH4, molecularly adsorbed CH3OH or an intermediate in the form of a CH3O methoxy species stabilized at a BAS or a reacted Cu(I) center may be present. The CH3O species are converted to CH3OH upon contact with H2O. The main drawback of this process is the non-continuous operation. Moreover, the desorption of CH3OH requires a solvent-based extraction technique, which leads to a dilute CH3OH solution and necessitates an energy-intensive concentration of CH3OH. Importantly, this issue is not limited to CH4 hydroxylation via chemical looping, but also arises in the continuous process. The main advantage of the stepwise process is the protection of the adsorbed CH3O against overoxidation, which generally leads to a higher CH3OH yield and selectivity compared to the catalytic process. For the sake of completeness, it should be mentioned that Panov et al. were the first to employ chemical looping for CH4 partial oxidation via Fe-exchanged zeolites.6
3. Formation of Cu(II) active sites during dehydration of as-prepared/hydrated Cu–zeolites under inert gas or vacuum
3.1 Generation of monomeric sites and spectroscopic characterization of Cu–zeolite dehydration
Considering that the oxidant represents an integral component in the CH4-to-CH3OH conversion for closing the Cu(II)/Cu(I) redox cycle and generating active Cu-oxo sites, it may seem counterintuitive to begin with a discussion of CH4 hydroxylation by Cu–zeolites that have been subjected to thermal treatment under inert gas or vacuum instead. Nevertheless, multiple studies have reported a non-negligible CH3OH productivity of various Cu–zeolites (Cu-ferrierite (FER), Cu–MOR, Cu–CHA) in CH4 partial oxidation following their activation in inert gas at elevated temperatures, occasionally reaching levels comparable to those achieved after conventional calcination.16,17,20,22,62,87–92 For example, Passini et al. observed that the CH3OH productivity of Cu0.23FER10.0 decreased only marginally from 7.3 to 6.5 µmolCH3OH gZEO−1 when treating the material in He instead of air at 823 K prior to interaction with CH4 at 483 K.87 Similarly, Tomkins et al. demonstrated that the performance of Cu–MOR6.0 during reaction with 6 bar CH4 at 473 K was comparable after an initial activation in either He or O2 at 473 K (20.0 and 21.2 µmolCH3OH gZEO−1, respectively).17 At first glance, these observations could suggest that the generation of oxygenated Cu(II) active species during thermal treatment of Cu–zeolites in inert gas is similarly efficient as in the presence of an oxidant. However, it is important to highlight that the Cu–zeolites in the aforementioned studies were synthesized via conventional aqueous ion exchange using Cu(NO3)2 or Cu(CH3COO)2 and remained in the as-prepared/hydrated state prior to treatment with inert gas at elevated temperatures.16,17,20,22,87–92 Under these conditions, Cu is already present in the oxidation state +2 and forms mobile [Cu(H2O)6]2+ and [Cu(OH)(H2O)5]+ adducts at low temperatures, which are detached from the zeolite lattice.57,93 The transformation of ligated H2O into OH− is facilitated by the electrostatic field imposed by the framework and yields H+ as a byproduct.94 As a result, the two hydrated complexes exist in a pH-dependent equilibrium as indicated by eqn (3.1) and Scheme 7.95–97| [Cu(H2O)6]2+ ↔ [Cu(OH)(H2O)5]+ + H+ | (3.1) |
46,50,106,107 and [CuOH]+/[CuOH]+62,65,108–111 have been demonstrated to participate in CH4 partial oxidation. The facile formation of Cu2+/[CuOH]+ as well as [CuOH]+/[CuOH]+via dehydration of Cu(II)-aqua complexes is illustrated in Scheme 7.
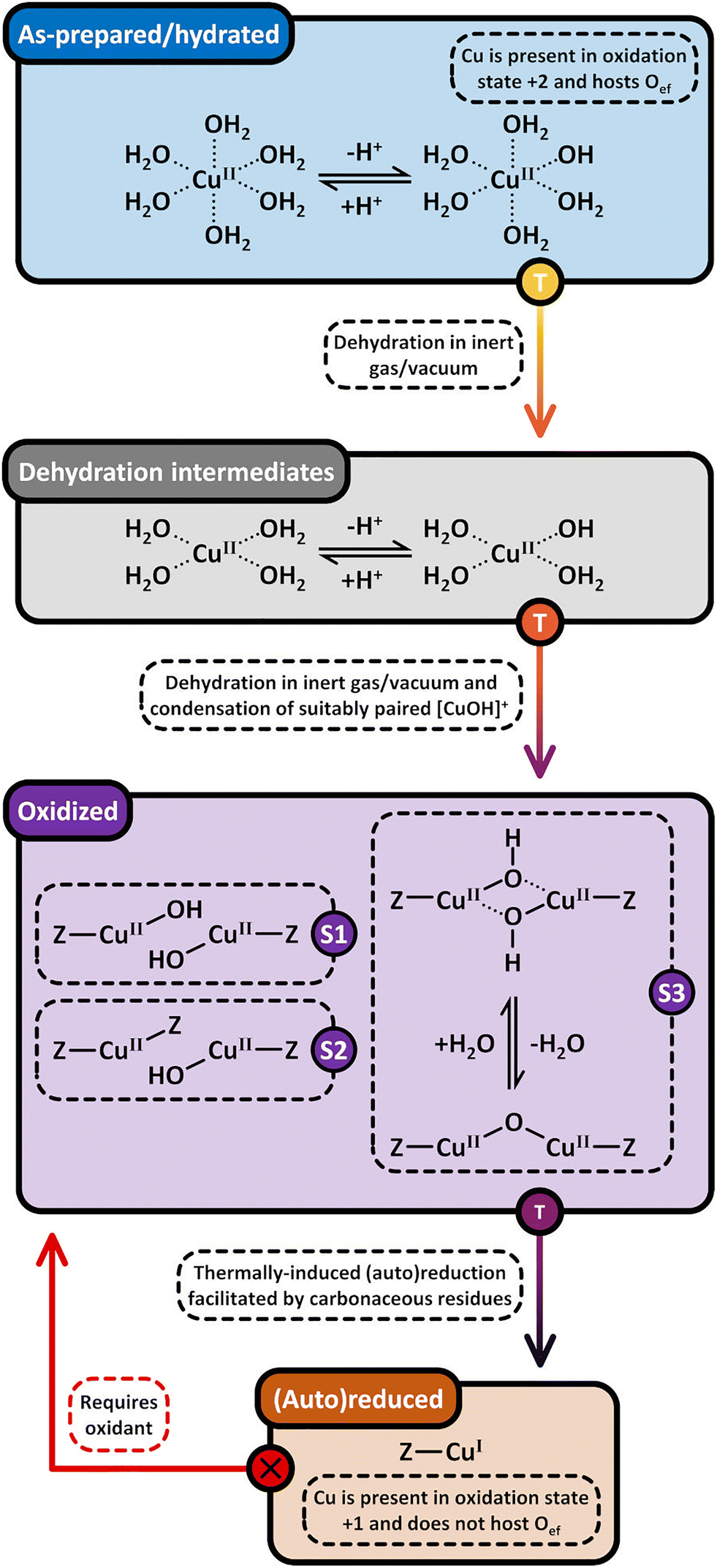 | ||
| Scheme 7 Formation pathways of [CuOH]+/[CuOH]+ (S1), Cu2+/[CuOH]+ (S2), and [Cu2(µ-O)]2+ (S3, violet background) via dehydration of [Cu(H2O)6]2+ and [Cu(OH)(H2O)5]+ (blue background) under inert gas or vacuum. [Cu2(µ-O)]2+ potentially evolves from the intramolecular condensation of [Cu2(µ-OH)2]2+. The dehydration proceeds through [Cu(H2O)4]2+ and [Cu(OH)(H2O)3]+ intermediates (grey background). Upon surpassing a material specific temperature threshold, the Cu-oxo sites are subjected to (auto)reduction in inert gas and vacuum, yielding Cu(I) (brown background). The latter can only be converted into oxygenated Cu(II) species in the presence of an oxidant (starting point marked by an “X”). Dotted bonds correspond to dative interactions. The term “Z” describes the negatively charged zeolite lattice. The color gradient from yellow to violet of the different starting points and arrows indicates the progressively increasing temperature. | ||
The transformation of [Cu(H2O)6]2+ and [Cu(OH)(H2O)5]+ into framework-bound Cu(II) has been extensively investigated in different Cu–zeolites using UV-Vis spectroscopy.15,53,98,112 In the as-prepared/hydrated state, the spectrum is dominated by a very intense signal at higher energies as well as a broad and asymmetric band at ∼12500 cm−1, which correspond to an Oef → Cu(II) ligand-to-metal charge transfer (LMCT) and unresolved d–d transitions of octahedral Cu(II)-aqua complexes, subjected to tetragonal elongation by Jahn–Teller distortion (Fig. 1a).15,53,98,112 Depending on the specific coordination environment of Cu(II) within the exchange positions, the substitution of H2O by Ofw throughout dehydration under inert gas or vacuum induces a topology-specific shift of the feature at ∼12500 cm−1.15,53,98,112 This is accompanied by an increase in the intensity of the d–d transitions due to symmetry reduction, i.e., rearrangement from a quasi-octahedral to a distorted four- or three-fold coordination mode. The ligand exchange is also evident from the bathochromic shift of the LMCT transition, arising from the lower optical electronegativity of Ofw relative to H2O.53,112–115
 | ||
| Fig. 1 UV-Vis spectra of Cu0.5CHA15.0 during interaction with N2 at temperatures in the range from 323 to 673 K (a). The thick blue spectrum corresponds to the as-prepared/hydrated material at 323 K, whereas thin grey spectra were recorded throughout the temperature increase. The dashed black spectrum indicates the state at 473 K at which the intensity of the d–d transitions is maximized. The thick black spectrum corresponds to the N2-treated material at 673 K. XANES spectra of Cu0.4CHA13.1 during dehydration in He from ambient temperature to 673 K (b). The thick blue spectrum corresponds to the as-prepared/hydrated material at ambient temperature, whereas thin blue and black spectra were recorded throughout the temperature increase between ambient temperature and 523 K, as well as 523 and 673 K, respectively. The thick black spectrum corresponds to the He-treated material at 673 K. The spectrum of the material after calcination at 673 K (thick dashed red) is provided for comparison. The inset in (b) depicts a magnified, background-subtracted region, where the Cu(II) pre-edge signal can be observed. CW X-band EPR spectra of Cu0.005CHA15.4 during evacuation at progressively increasing temperatures in the range from ambient temperature to 673 K (c). The spectral features of mobile [Cu(H2O)6]2+ and rigid [Cu(H2O)4(Ofw)2]2+ are indicated. The asterisk (*) corresponds to a carbon radical signal. Adapted from ref. 45, 53 and 104 with permission from Elsevier (Copyright 2019), the Royal Society of Chemistry (Copyright 2014), and the Nature Publishing Group (Copyright 2021). | ||
The insights provided by UV-Vis experiments have been further validated by X-ray absorption spectroscopy (XAS). The X-ray absorption near edge structure (XANES) spectrum of as-prepared/hydrated Cu–zeolites, collected at ambient temperature, is characterized by a smooth rising edge and an intense white line at ∼8997 eV, which are characteristic of Cu(II)-aqua complexes in a distorted octahedral environment (Fig. 1b).89,92,98–100,102,104 This is in accordance with the corresponding FT-EXAFS spectrum that displays a signal at ∼1.93 Å (phase uncorrected), corresponding to a first shell Cu–O scattering path with a Cu–O coordination number (CN) of 3.5–4.9.57,98–100,116,117 The absence of well-defined higher coordination shell peaks is indicative of the high degree of disorder and mobility of the solvated Cu(II).57,104,116,117 The decrease of the first shell CN due to the removal of H2O during dehydration in inert gas or vacuum is apparent from the gradual decline of the white line in the XANES spectrum and the simultaneous intensity loss of the first shell Cu–O signal in the FT-EXAFS spectrum.19,89,98–102,104,118 This occurs alongside a bathochromic shift of the rising edge, which emerges from the change in the Cu(II) coordination environment from a six- to a four-/three-fold binding mode.99,100,102 In addition, the rising edge becomes more structured and develops a distinct feature at ∼8986 eV, originating from the dipole-allowed 1s → 4p transition of Cu(II) combined with a shakedown charge transfer from a ligand p valence orbital into the metal 3d9 orbital.89,98,99,102,116,119,120 Moreover, a pre-edge signal at ∼8977 eV emerges, which stems from the dipole-forbidden but quadrupole-allowed 1s → 3d transition of non-centrosymmetric Cu(II).89,98–102,120,121 Based on multivariate curve resolution (MCR), Martini et al. proposed that the dehydration of [Cu(H2O)6]2+ and [Cu(OH)(H2O)5]+ into Cu2+ and [CuOH]+ proceeds via [Cu(H2O)4]2+ and [Cu(OH)(H2O)3]+ intermediates.53,102 The formation of these partially hydrated square planar complexes is in line with 1H hyperfine sublevel correlation (HYSCORE) experiments by Bruzzese et al., which indicated that the axially coordinated H2O ligands are lost first during vacuum treatment of Cu0.005CHA15.4 from ambient temperature to 323 K.45 The generation of these intermediates throughout the dehydration process is depicted in Scheme 7. Crucially, XANES spectroscopy demonstrates that, depending on the specific Cu–zeolite, dehydration under inert gas or vacuum at temperatures up to 473–673 K does not result in a pronounced Cu(II) (auto)reduction (vide infra), as evident from the absence of an intense rising edge feature at ∼8983 eV, corresponding to the 1s → 4px/y transition of quasi-linear Cu(I).16,19,51,89,90,92,98–104,106,122–125
EPR spectroscopy has provided complementary insight into the dehydration process. The continuous wave (CW) X-band EPR spectrum of as-prepared/hydrated Cu–zeolites, collected at ambient temperature, is characterized by a broad, motion-averaged isotropic signal in the range of g = 2.14–2.17 without a resolved parallel hyperfine structure, which originates from mobile, solvated Cu(II) (Fig. 1c).43,45,47,98,99 Interestingly, an additional anisotropic feature with a gII of 2.37 and a weak hyperfine structure has been identified in the CW X-band EPR spectrum of as-prepared/hydrated Cu0.005CHA15.4, arising from a [Cu(H2O)4(Ofw)2]2+ complex within the 8-MR (Scheme 1), which suggests that a certain fraction of Cu(II) already interacts with the zeolite lattice under these conditions.43,45,47 The transition from mobile [Cu(H2O)6]2+ and [Cu(OH)(H2O)5]+ into rigid fully framework-bound Cu(II) by dehydration under inert gas or vacuum is evident from the disappearance of the motion-averaged component and the simultaneous emergence of new signals featuring resolved hyperfine interactions, whose g and A parameters depend on the Cu(II) coordination geometry, the nature of the complexing ligands (Ofw, OH−), and the specific topology.43,45,47,98,99,126 This occurs alongside a decrease in overall spectral intensity. By combining EPR and XANES spectroscopy, Palomino et al. and Xamena et al. demonstrated that this effect does not arise from a conversion of paramagnetic Cu(II) (3d9, S = 1/2) into diamagnetic Cu(I) (3d10, S = 0) via (auto)reduction (vide infra), but from the formation of EPR-silent moieties instead.98,99 The latter have been associated with isolated [CuOH]+, adopting a distorted trigonal planar coordination.43,47,55,58,98,99 Owing to their near-degenerate ground state, these Cu(II) sites were proposed to be subjected to a pseudo Jahn–Teller effect, yielding a low-lying excited state. The EPR transition can couple to this vibration, which provides a pathway for fast energy dissipation that shortens the spin–lattice relaxation time. This results in a broadening of the spectral line shape, effectively rendering [CuOH]+ EPR-invisible. However, complementary CW X-band EPR and 1H HYSCORE experiments with Cu0.005CHA15.4 by Bruzzese et al. showed that [CuOH]+ is rather present in a distorted square-planar environment and does in fact exhibit a distinct EPR signature, which is in line with results from Sushkevich et al. and Fischer et al.46,50,58,109,127 The intensity drop can also be explained by the formation of [Cu2(µ-O)]2+, which is typically not observable by EPR spectroscopy regardless of its specific electronic configuration.43,58,98,99,128 In the S = 0 ground state, the unpaired electrons of the two Cu(II) ions are coupled antiferromagnetically, whereas in the S = 1 ground state, the large zero-field splitting of the ferromagnetically interacting electrons typically makes [Cu2(µ-O)]2+ undetectable by conventional CW X-band EPR spectroscopy.67 A noticeable exception to this are dimeric Cu(II) species in Y-type zeolites as reported by Chao et al.129 The exchange positions in Y-type zeolites allow for a unique geometric arrangement, resulting in ferromagnetically coupled Cu(II) pairs with a zero field splitting of D = 0.0476 cm−1, small enough to be detected at x-band frequencies.
3.2 Generation of dimeric sites via condensation or (auto)reduction
According to eqn (3.2), the generation of [Cu2(µ-O)]2+ throughout dehydration of as-prepared/hydrated Cu–zeolites under inert gas or vacuum can proceed via the condensation of adjacent [CuOH]+ sites, possibly via a [Cu2(µ-OH)2]2+ intermediate.16,20,54,92–94,98–101,106,130–134 The equilibrium between the latter two species is controlled by the H2O partial pressure, with [Cu2(µ-O)]2+ being favored at low H2O pressures.54,105,135| 2[Cu(OH)(H2O)5]+ → 2[CuOH]+ + 10H2O → [Cu2(µ-OH)2]2+ ↔ [Cu2(µ-O)]2+ + H2O | (3.2) |
 | ||
| Fig. 2 Raman spectrum (λex = 532 nm) of as-prepared/hydrated Cu0.45CHA12.0 (blue) as well as the corresponding spectra after interaction with O2 (black) and He (green) at 723 K (a). FT-EXAFS spectra (k2-weighted) of Cu0.31MOR11.0 during thermal treatment in He at temperatures between 323 and 723 K (b). Adapted from ref. 19 and 20 with permission from the American Chemical Society (Copyright 2017 and 2019). | ||
Further proof was presented by Ikuno et al., who monitored the evolution of the FT-EXAFS spectrum of Cu0.31MOR11.0 throughout dehydration in He in the temperature range from 323 to 723 K (Fig. 2b).19 From 373 K onward, the authors noted an increase in the intensity of a peak at ∼2.3 Å (phase uncorrected) that subsequently diminished as the temperature exceeded 573 K. This signal was correlated to the second shell Cu–Cu scattering path in [Cu2(µ-O)]2+, which develops below 573 K viaeqn (3.2) and undergoes (auto)reduction (vide infra) when surpassing this temperature threshold. The decline in the intensity of the Cu–Cu scattering peak during [Cu2(µ-O)]2+ (auto)reduction was linked to the dispersion of Cu(I) across the framework. Notably, an elongation of the Cu–Cu distance in [Cu2(µ-O)]2+ in Cu–MOR upon (auto)reduction has indeed been identified using wavelet-transform (WT) EXAFS spectroscopy.137–139
Above 473–673 K in inert gas or vacuum, Cu(II) is subjected to (auto)reduction as evident from the loss in absorbance of the LMCT and d–d transitions in the UV-Vis spectra (Fig. 1a),53,98,106 the appearance of the Cu(I) rising edge signal in the XANES spectra (Fig. 1b), which develops at the expanse of the features of Cu(II),16,19,89,98–102,104,106 and the intensity decrease of the first shell Cu–O scattering signal due to the loss of Oef ligands.16,19,90,92,98–104 As already indicated, this process is believed to involve the direct liberation of O2 from [Cu2(µ-O)]2+ (eqn (3.3)).93,130,134 At this point, it should already be mentioned that (auto)reduction is facilitated by potentially present carbonaceous impurities (see Section 4.2.1).101,128,140,141
| 2[Cu2(µ-O)]2+ → 4Cu+ + O2 | (3.3) |
| [CuOH]+ → Cu+ + ˙OH | (3.4) |
| [CuOH]+ + ˙OH → [CuO˙]+ + H2O | (3.5) |
| 2[CuO˙]+ → 2Cu+ + O2 | (3.6) |
| 2[CuO˙]+ → trans/cis [Cu2(µ-1,2-O2)]2+ | (3.7) |
| 2[CuO˙]+ → [Cu2(µ-O)2]2+ | (3.8) |
| 2[CuO˙]+ → [Cu2(µ-η2:η2-O2)]2+ → [Cu2(µ-O)]2+ + 1/2O2 | (3.9) |
Based on the preceding discussion, it can be concluded that the thermal treatment of as-prepared/hydrated Cu–zeolites under inert gas or vacuum is sufficient to induce the generation of Cu-oxo active sites as Cu is already present in the oxidation state +2 and bares reactive Oef in the form of OH− ligands.93,130 As illustrated in Scheme 7, the oxygenated Cu(II) active sites can manifest as paired Cu2+/[CuOH]+ and [CuOH]+/[CuOH]+, formed via simple dehydration of the corresponding Cu(II)-aqua complexes, or as [Cu2(µ-O)]2+, arising from the condensation of neighboring [CuOH]+. At elevated temperatures, [Cu2(µ-O)]2+ and [CuOH]+ are subjected to (auto)reduction, which decreases the CH3OH productivity of materials thermally treated in inert gas or vacuum compared to those activated in the presence of an oxidant. Crucially, the development of Cu(II) active sites in inert gas or vacuum is only feasible in as-prepared/hydrated samples, indicating that an oxidative activation is required if the Cu–zeolite is in an (auto)reduced state. As a final note of caution, the CH3OH productivity of materials, thermally treated under inert gas or vacuum, may be artificially inflated by the presence of O2 impurities.90 Nevertheless, since the extent of O2 contamination is usually undefined, it is challenging to assess the impact of residual O2 traces on the CH3OH output of these samples.
4. Utilization of O2 for the formation of active Cu(II) sites in chemical looping and continuous CH4 partial oxidation
4.1 Changing the multiplicity of O2 by reductive activation with Cu(I)
Conceptually, the transformation of CH4 and O2 into CH3OH is a spin-forbidden process.9,81,151–154 This arises from the fact that CH4 and CH3OH are both characterized by a closed-shell singlet (S = 0) electronic structure, whereas O2 is a diradical featuring a triplet (S = 1) ground state. As a result, the spin states of the reactants (|SCH4 ± SO2|) and product (|SCH3OH|) do not have a common term, rendering this reaction spin-forbidden according to Wigner's selection rule. Therefore, direct CH4 hydroxylation formally requires a change in the multiplicity of O2via its transformation into reactive ˙O2−, O22−, or O2− species upon interaction with a metal center. In the case of Cu-exchanged zeolites, the reductive activation of O2 involves the oxidation of Cu(I) into Cu(II), which represents a transition from a diamagnetic 3d10 into a paramagnetic 3d9 configuration.93 The possible ground states of the formed Cu-oxo sites are governed by their chemical formula and spin orientations. End-on/side-on [Cu(η-O2˙)]+ (Schemes S1l and m),155 [Cu2(µ-O)]2+,66,74,90,156–159trans/cis [Cu2(µ-1,2-O2)]2+70 as well as [Cu2(µ-η2:η2-O2)]2+70,156,160 can adopt either a triplet or open-shell singlet state, whereas [Cu3(µ-O)3]2+ may exist in either a doublet, quartet, or sextet state.73,74,158,159,161 Isolated Cu2+ and [CuOH]+ are exclusively present in the doublet state.56,62,107 However, since these species can participate in CH4 conversion only in the presence of a second proximal Cu(II) center (e.g., in [CuOH]+/[CuOH]+62,65,109–111,162 and Cu2+/[CuOH]+ pairs46,50,56,106,107), the total spin state of these active sites effectively corresponds to either a triplet or an open-shell singlet.56,62 In contrast to their high-spin counterparts, CH4 partial oxidation via dimeric Cu-oxo species in the singlet state and trimeric oxygenated Cu(II) centers in the doublet state is not accompanied by a spin inversion and hence does not violate Wigner's selection rule.74,150,157–159,163 For example, multiple theoretical studies have demonstrated that CH4 hydroxylation on triplet [Cu2(µ-O)]2+ proceeds via a spin flip at the radical rebound step in the homolytic C–H bond activation mechanism (Scheme 6), whereas no such transition is required in the case of singlet [Cu2(µ-O)]2+.157,158,164 Consequently, the reductive activation of O2 by Cu(I) and the transformation of the latter into a reactive Cu-oxo site with a suitable ground state are essential to facilitate the oxo-functionalization of CH4.9,81,151,153,163,165 Notably, Mahyuddin et al. determined that the different spin states of [Cu2(µ-O)]2+ and [Cu3(µ-O)3]2+ are essentially isoenergetic.154,157–159 Depending on the zeolite framework, the singlet–triplet splitting of [Cu2(µ-O)]2+ in Cu-exchanged AEI, AFX, CHA, MFI, MOR, and mazzite (MAZ) was found to amount to just 0.8–5.0 kJ mol−1.157,158 Likewise, the energetic difference between the quartet and doublet state of [Cu3(µ-O)3]2+ in Cu–MOR and Cu-MAZ is only 3.3–7.9 kJ mol−1.158 Theoretical studies by various other authors resulted in similarly small splittings between the high- and low-spin state of various Cu-oxo motifs in different Cu–zeolites.66,70,73,74,155 As a result, the formation of high-spin Cu-oxo species, whose participation in CH4 partial oxidation necessitates a spin crossover, is thermodynamically as feasible as the generation of their low-spin counterparts. In fact, the former have been reported to be even more active due to a higher spin density at the Oef ligand, which increases their reactivity in the H atom abstraction (HAA).67,73,74,95,107,157–159,161,166,167 Nevertheless, the magnitude of the energetic difference between distinct spin states is strongly affected by the chosen computational method and functional, as well as the location and geometry of the Cu(II) model structure.70,106,155,159,161,164,168 For the sake of completeness, Heyer et al. combined variable-temperature, variable-field magnetic circular dichroism (VTVHMCD) spectroscopy and DFT calculations to show that the ground state of [Cu2(µ-O)]2+ is governed by the relative orientation of the planes of the bidentate coordinating Al T-sites, which, in turn, depends on the specific zeolite framework (see Section 2.1.3).67 Thus, [Cu2(µ-O)]2+ hosted in Cu-MFI adopts a singlet state due to the parallel arrangement of the bidentate coordinating Al T-sites, which maximizes the superexchange interaction between the two half-filled Cu(II) 3d orbitals. On the contrary, this interaction is minimized in the case of [Cu2(µ-O)]2+ in Cu–CHA owing to the perpendicular orientation of the planes of the chelating framework ligands, which results in a triplet ground state.
4.2 Formation of Cu(II) sites using O2 during non-isothermal looping
As outlined in Section 2.3 and Scheme 6, the activation of Cu–zeolites for CH4 partial oxidation via non-isothermal looping is initiated by a heating ramp to a specific temperature, followed by a dwell period for a certain duration and a subsequent cool down (ideally in the presence of the oxidant) to a set temperature for CH4 hydroxylation.7,72,133,144 After CH3OH extraction, this procedure must be repeated to reactivate the material for the next reaction sequence. In the following sections, the available literature concerning each of the above mentioned periods will be reviewed, aiming to reconstruct the formation mechanism of different Cu(II) sites and to elucidate the impact of activation conditions on Cu speciation and material performance in CH4 hydroxylation. The behavior of Cu–zeolites in successive redox cycles is also addressed to assess the regenerability of the Cu(II) active centers.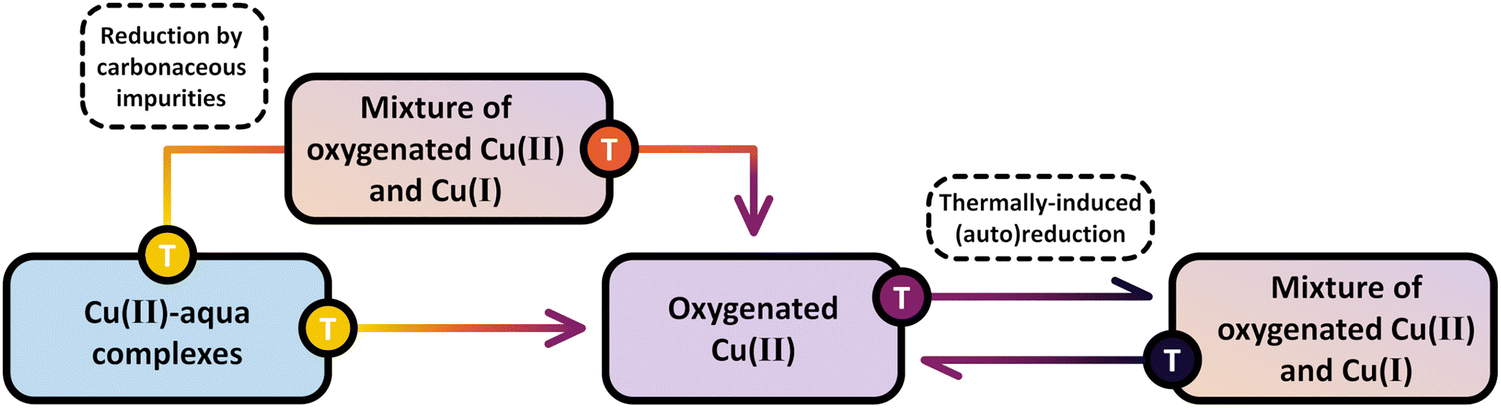 | ||
| Scheme 8 Evolution of Cu(II) species during treatment of as-prepared/hydrated Cu–zeolites (blue background) in O2 at progressively increasing temperatures. In the presence of carbonaceous impurities, calcination leads to the momentary reduction of a fraction of Cu(II) centers to Cu(I) (brown/violet background). Upon further increasing the temperature, Cu(I) is re-oxidized, and the material predominantly contains oxygenated Cu(II) species (violet background). This state is directly reached in the absence of carbonaceous impurities. At even higher temperatures, the Cu-oxo centers are subjected to a reversible temperature-controlled (auto)reduction in O2. The color gradient from yellow to violet of the different starting points and arrows indicates the progressively increasing temperature. | ||
Surprisingly, multiple studies have indicated that Cu(II) may still undergo (auto)reduction during thermal treatment in O2 despite the presence of an oxidant.21,51,103,127,132,174,176 Andersen et al., for example, analyzed the evolution of the XANES spectra of Cu0.48CHA15.1 during dehydration in O2 by linear combination fitting (LCF) and observed a decrease in the Cu(II) fraction by 37% upon raising the temperature from 683 to 773 K.103 Notably, the Cu(II) fraction did not recover when the material was kept in O2 at 773 K for ∼27 min, suggesting that the Cu(II) reduction did not originate from the oxidative decomposition of carbonaceous impurities (vide infra) but rather from a thermally driven change in the Cu(II)/Cu(I) equilibrium. Andersen et al. attributed this effect to the (auto)reduction of [CuOH]+via ˙OH loss as depicted in eqn (3.4). Nevertheless, this process does not necessarily need to be limited to [CuOH]+. Several theoretical studies have highlighted that the conversion of Cu(I) and O2 into various mono- and multimeric oxygenated Cu(II) centers is strongly exothermic.155,156,164,177–180 For instance, DFT calculations by Mahyuddin et al. revealed that the oxidation of two Cu(I) pairs, each situated in one of the opposing 8-MRs of the side pocket in Cu–MOR (Scheme 1), into two [Cu2(µ-O)]2+ moieties is characterized by a reaction enthalpy of about −463 kJ mol−1.179 Consequently, the reverse process, i.e., the transformation of Cu(II) into Cu(I) via Oef loss, is thermodynamically favorable at elevated temperatures, shifting the reaction equilibrium toward (auto)reduction.19,21 This correlation is supported by work from Brenig et al. focused on the generation of Cu(II) active sites in reduced Cu–MOR10.0, Cu-MFI11.5, and Cu–CHA11.0.127 After activating the materials in O2 at 753 K, these authors noted a progressive increase in the intensity of UV-Vis features between 27300–26400 and 2200–21800 cm−1, corresponding to the LMCT transition of two different classes of [Cu2(µ-O)]2+, during a cool down to 313 K in the presence of the oxidant. This clearly highlights that the formation of these dimeric Cu-oxo species is preferred at lower temperatures, whereas they are subjected to a reversible (auto)reduction at elevated temperatures. Additional evidence for this was provided by an LCF analysis of the XANES spectra recorded during temperature-programmed oxidation (TPO) of the three reduced samples with O2, which unveiled that the Cu(II) fraction first reaches a framework-dependent maximum at a certain temperature and then declines upon further raising the temperature. An analogy to this behavior is the redox equilibrium between Cu2O and CuO (eqn (4.1)), which shifts toward the former upon increasing the temperature.181–184
| 2Cu2O + O2 ↔ 4CuO | (4.1) |
A similar yet distinct phenomenon has been reported by Kvande et al. and Borfecchia et al., who tracked the evolution of the XANES spectra of Cu–MOR and Cu–CHA during dehydration from ambient temperature to 773 K in O2.139,175 Apart from the gradual loss of the white line intensity and the emergence of the rising and pre-edge feature of Cu(II), the authors observed a progressive development of the rising edge signal of Cu(I) upon increasing the temperature from ambient temperature to 623–653 K. Notably, the absorbance of the Cu(I) feature decreased again when further raising the temperature and completely vanished at 773 K. The same behavior, although in a different temperature regime, has been detected in the XANES spectra of Cu0.2FER11.0 collected during activation in O2 (Fig. 3a).169
 | ||
| Fig. 3 Cu K-edge XANES spectra of Cu0.2FER11.0 during activation in O2 at temperatures between 298 and 773 K, as well as after 120 min at 773 K (a). The inset in (a) shows a magnification of the spectral region where the transient appearance of the Cu(I) signal can be observed. MS response (left axis) during interaction with O2 as well as the temperature profile (right axis); the masses corresponding to H2O (m/z = 18), CO2 (m/z = 44), and CO (m/z = 28) were followed during the experiment (b). FTIR spectra of the ν(O–H) region of Cu0.53CHA14.8 during treatment in O2 in the 523–673 K range, as well as after 150 min at 673 K (c). The dotted line highlights the progressive decrease in intensity of the band at ∼3650 cm−1 corresponding to [CuOH]+. Adapted from ref. 16 and 169 with permission from Springer Nature Link (Copyright 2019) and the American Chemical Society (Copyright 2017). | ||
This transient Cu(II) reduction was interpreted based on the FTIR spectra of Cu0.53CHA14.8 recorded during dehydration from 303 to 673 K in flowing O2, which exhibited a continuous intensity loss of a feature at ∼3650 cm−1, assigned to the ν(O-H) stretching vibration of [CuOH]+ (Fig. 3b).15,16,53,57,104,112,130,174,190 Based on the apparent decomposition of [CuOH]+ at elevated temperatures in O2, the temporary appearance of the Cu(I) signal in the XANES spectra was associated with the (auto)reduction of [CuOH]+ into Cu(I) viaeqn (3.4).16,118,175,191 The subsequent diminution of this feature above 623–653 K was attributed to the re-oxidation of Cu(I) by O2, yielding [Cu(η1-O2˙)]+ (eqn (4.1)) or [Cu2(trans-µ-1,2-O2)]2+ (eqn (4.2)). Consequently, [CuOH]+ was envisioned as a precursor for the formation of mono- and dimeric Cu-oxo sites via its (auto)reduction into Cu(I). The tricoordinate [Cu(η1-O2˙)]+ and [Cu2(trans-µ-1,2-O2)]2+ were suggested to undergo a thermally-induced rearrangement into four-fold coordinated [Cu(η2-O2˙)]+ and [Cu2(µ-η2:η2-O2)]2+ during a cool down in O2 (see Section 4.2.5).16,172,175,192
| Cu+ + O2 → [Cu(η1-O2˙)]+ | (4.2) |
| 2Cu+ + O2 → [Cu2(trans-µ-1,2-O2)]2+ | (4.3) |
Scheme 8 highlights the differences between the reduction of Cu(II) by carbonaceous residues and the thermally-induced (auto)reduction of Cu(II) during calcination of Cu–zeolites at progressively increasing temperatures.
The origin of the beneficial impact of elevated activation temperatures on the CH3OH productivity was explored by Smeets et al. and Groothaert et al., who studied the development of the LMCT transition of [Cu2(µ-O)]2+ at ∼22700 cm−1 in as-prepared/hydrated Cu-MFI as a function of the calcination temperature by UV-Vis spectroscopy.133,136 Starting from ∼623 K, the intensity of this characteristic band progressively increased upon raising the temperature and reached a maximum at about 923 K (Fig. 4a). Notably, the absorbance of this feature started to decrease again upon further increasing the temperature to 1023 K. The loss in signal intensity could not be reversed during a subsequent O2 treatment at 723 K and was thus attributed to an irreversible decomposition of [Cu2(µ-O)]2+.
 | ||
| Fig. 4 UV-Vis spectra of as-prepared/hydrated Cu0.54MFI12.0 recorded at ambient temperature after calcination in O2 at different temperatures ranging from 523 to 1023 K. The spectrum marked by an asterisk (*) corresponds to the spectrum collected after activation in O2 at 1023 K, followed by interaction with O2 at 723 K (a). Total productivity of Cu0.31MOR11.0 in CH4 partial oxidation after activation in O2 at 773 K and after treatment in He at 773 or 623 K, followed by interaction with O2 at various temperatures ranging from 323 to 473 K (b). UV-Vis spectra of Cu–MOR10.0 after activation (dotted lines, recorded in O2) and reactivation (solid lines, recorded in O2) at temperatures in the range from 313 to 723 K. The characteristic LMCT transitions of the two different [Cu2(µ-O)]2+ motifs are highlighted in purple and orange (c). Adapted from ref. 19, 127 and 136 with permission from Elsevier (Copyright 2005), the American Chemical Society (Copyright 2019), and Wiley (Copyright 2025). | ||
Oord et al. observed a similar enhancement in the intensity of a band at 29000 cm−1 in the UV-Vis spectrum of Cu–CHA20.0, attributed to a not precisely defined dimeric Cu-oxo active site, when raising the O2 treatment temperature from 723 to 823 K.15 The necessity of high activation temperatures has also been highlighted by Wijerathne et al., who combined DFT calculations and Monte Carlo (MC) simulations to reveal that the transformation of the [Cu2(µ-OH)2]2+ intermediate into [Cu2(µ-O)]2+ in Cu–CHA and Cu–MOR, characterized by a random Al T-site arrangement, only takes place above ∼730 K at 1 × 10−5 mbar H2O and 200 mbar O2 (H2O/O2 ratio: 5 × 10−8).105 This is in qualitative agreement with theoretically predicted phase diagrams from Hutton et al., indicating that [Cu2(µ-O)]2+ becomes thermodynamically more stable than [Cu2(µ-OH)2]2+ above ∼680 K at the same H2O/O2 ratio in Cu–CHA, featuring a distinct Al T-site distribution.135 In contrast, a computationally derived phase diagram from Li et al. suggested that the conversion of [Cu2(µ-OH)2]2+ into [Cu2(µ-O)]2+ in Cu–CHA occurs already above 280 K, at the same H2O-to-O2 ratio.54 Using DFT, Suleiman et al. determined the minimal activation temperature required for the generation of [Cu2(µ-O)]2+, [Cu2(µ-O)2]2+, [Cu2(µ-η2:η2-O2)]2+, and [Cu3(µ-O)3]2+ in the 8-MR of as-prepared/hydrated Cu–MOR featuring H+ as co-cations.167 Depending on the specific Cu-oxo center, the temperature at which the Gibbs free energy of formation starts to become negative amounted to 618–733 K, demonstrating once again that elevated temperatures are necessary to remove H2O ligands from the Cu(II)-aqua complexes and to induce their aggregation into multimeric oxygenated Cu(II) species. Complete dehydration of the material is particularly important as the strongly hydrophilic Cu-oxo species readily adsorb H2O, which saturates the coordination sphere of the Cu(II) active sites, rendering them inaccessible for CH4 and effectively poisoning them.100,101,171–173 The gravity of H2O-induced passivation has been emphasized by Sushkevich et al., who combined FTIR spectroscopy and NO probe molecule adsorption to determine that the interaction of a single H2O molecule with [Cu2(µ-O)]2+ and [CuOH]+/[CuOH]+ is sufficient to completely deactivate these species at low CH4 pressures.101Scheme 9 highlights the necessity of high activation temperatures for the generation of various Cu-oxo sites throughout the treatment of as-prepared/hydrate Cu–zeolites with O2.
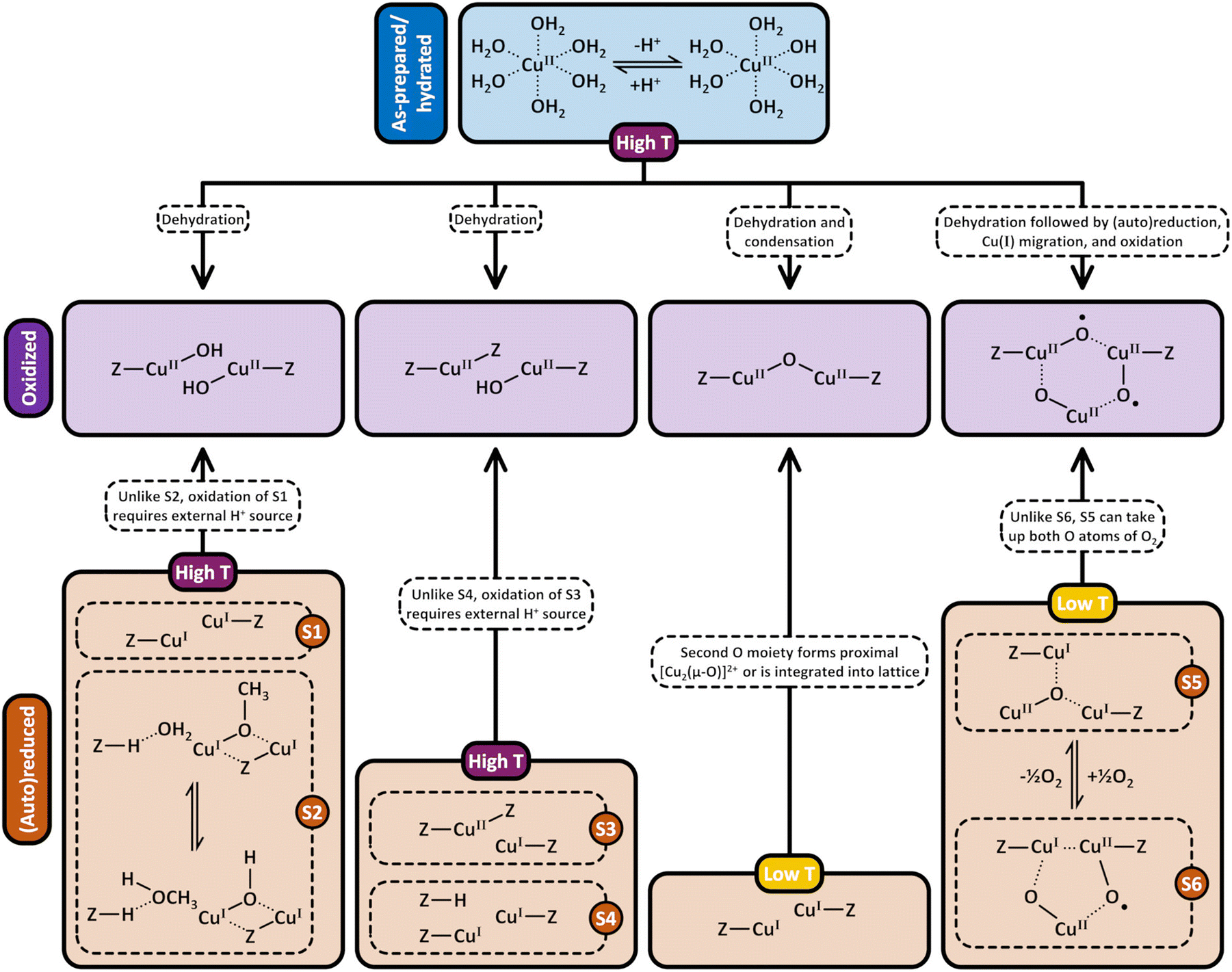 | ||
| Scheme 9 Formation pathways of [CuOH]+/[CuOH]+, Cu2+/[CuOH]+, [Cu2(µ-O)]2+, and [Cu3(µ-O)3]2+ (violet background) by treatment of as-prepared/hydrated (blue background) and (auto)reduced (brown background) Cu–zeolites with O2. After (auto)reduction of [CuOH]+/[CuOH]+, this site may exist either as two plain Cu(I) ions (S1) or as a BAS containing site (S2). This distinction is important as, unlike in the case of S2, the generation of [CuOH]+/[CuOH]+ from S1 necessitates an external H+ source. Similarly, the transformation of Cu2+/Cu+ (S3) into Cu2+/[CuOH]+ requires an additional H+ source, which is not necessary when starting from 2Cu+/BAS (S4). Unlike the reaction of [Cu3(µ-O)2]2+ (S6) with O2, the formation of [Cu3(µ-O)3]2+via interaction of [Cu3(µ-O)]2+ (S5) with O2 does not require a location to accommodate the second O moiety. Dotted bonds correspond to dative interactions. The term “Z” describes the negatively charged zeolite lattice. Depending on the necessary activation temperature, the different starting points are highlighted in either violet or yellow. | ||
Based on the results presented above, elevated calcination temperatures are essential for promoting the generation of di- and trimeric Cu(II) active sites in as-prepared/hydrated Cu–zeolites, which, in turn, results in an optimized performance in CH4 partial oxidation. However, this correlation may not necessarily apply to the formation of these species in (auto)reduced materials, as the activation of as-prepared/hydrated and (auto)reduced samples involves a fundamentally different series of elementary steps. In the former case, Cu is already present in the oxidation state +2 and the generation of mono-/multimeric oxygenated Cu(II) centers proceeds via dehydration of Cu(II)-aqua complexes and [CuOH]+ condensation (eqn (3.2)). In contrast, oxygenated Cu(II) centers in (auto)reduced Cu–zeolites develop via an actual oxidation of Cu(I) into Cu(II) in the presence of O2. Ikuno et al. emphasized this distinction by decoupling the oxidative formation of Cu-oxo species from other thermally driven processes, such as dehydration and (auto)reduction (Fig. 4b).19 They determined that the CH3OH output of Cu0.31MOR11.0, initially (auto)reduced in He at 773 K and subsequently exposed to O2 at temperatures ranging from 323 to 473 K, was comparable to that of the material directly calcined in O2 at 773 K. Conversely, a decrease in the temperature of the initial He treatment to 623 K followed by interaction with O2 at 473 K resulted in a substantial reduction of the CH3OH productivity. The authors rationalized this behavior by arguing that the initial thermal treatment in He promotes the formation of [CuOH]+ and [Cu2(µ-O)]2+ (eqn (3.2)), which are subsequently (auto)reduced viaeqn (3.3) and (3.4), yielding Cu(I) and ˙OH. Due to the weaker electrostatic interaction between Cu(I) and the zeolite lattice, the former is capable of dynamically reorganizing into a reduced precursor of a [Cu3(µ-O)3]2+ center. This precursor can be present either in the form of three Cu(I) ions or as a mixed [Cu2(µ-O)]2+/Cu(I) moiety and is rapidly oxidized by O2 and ˙OH into [Cu3(µ-O)3]2+viaeqn (4.4) and (4.5), regardless of the activation temperature. An alternative pathway for [Cu3(µ-O)3]2+ generation in Cu-MFI has been suggested by Li et al., which is based on the interaction of a 2Cu(I)/[CuOH]+ precursor with O2 (eqn (4.6)).54
| 3Cu+ + O2 + ˙OH → [Cu3(µ-O)3]2+ + H+ | (4.4) |
| [Cu2(µ-O)]2+ + Cu+ + 1/2O2 + ˙OH → [Cu3(µ-O)3]2+ + H+ | (4.5) |
| 2Cu+ + [CuOH]+ + O2 → [Cu3(µ-O)3]2+ + H+ | (4.6) |
Despite this conceptual inconsistency, the work by Ikuno et al. provides compelling evidence that, unlike in as-prepared/hydrated Cu–zeolites, the generation of Cu-oxo active sites in (auto)reduced materials does not require elevated temperatures. This is in contrast to analogous experiments from a study by Pappas et al.16 Compared to the CH3OH productivity of Cu0.49CHA12.1 activated in O2 at 773 K, these authors observed a decrease in the performance of the material in CH4 partial oxidation after He treatment at 773 K, followed by interaction with O2 at 473 K. This phenomenon was correlated to the temperature-controlled formation of less reactive four-fold coordinated Cu(II) species (see Section 4.2.5). Then again, Smeets et al. compared the minimal activation temperature necessary to detect the LMCT transition of [Cu2(µ-O)]2+ at ∼22700 cm−1 in as-prepared/hydrated and (auto)reduced Cu0.54MFI12.0.136 In the former case, the threshold temperature was 623 K, whereas it dropped to 553 K after treatment of the sample in He at 773 K, implying that the onset temperature of [Cu2(µ-O)]2+ formation is controlled by the initial state of the Cu–zeolite. Similarly, Woertink et al. observed the emergence of this characteristic feature already at 448 K when exposing (auto)reduced Cu0.54MFI12.0 to O2.66 The fact that the generation of oxygenated Cu(II) species in (auto)reduced Cu–zeolites takes place even at low temperatures has additionally been highlighted by an investigation of Brenig et al. focused on the activation of (auto)reduced Cu–MOR10.0, Cu-MFI11.5, and Cu–CHA11.0.127 By LCF analysis of the XANES spectra recorded throughout TPO, the authors registered a continuous increase in the Cu(II) fraction starting already at 240 K. Likewise, UV-Vis spectra collected after exposing the reduced materials to O2 at 313 K displayed a pronounced increase in the absorbance of two bands within the ranges of 27300–26400 and 22000–21800 cm−1, corresponding to the LMCT transition of two distinct [Cu2(µ-O)]2+ motifs (Fig. 4c). Compared to the spectra acquired after calcination at 753 K followed by a cool down to 313 K in the presence of the oxidant, the intensity in these regions was identical or even higher in the spectra of the reduced samples contacted with O2 at 313 K. This clearly illustrates that the generation of [Cu2(µ-O)]2+ in (auto)reduced Cu–zeolites is feasible at low temperatures and is equally or even more efficient than high-temperature activation. Notably, complementary CW X-band EPR experiments indicated that the oxidative formation of monomeric Cu(II) sites, including Cu2+para, Cu2+meta, and [CuOH]+, requires markedly higher temperatures than that of Cu(II) dimers. In fact, O2 treatment of the reduced materials at 723 K was necessary to attain the same signal intensity as in the spectra of as-prepared/hydrated samples calcined at 753 K. A similar phenomenon has been reported by Palomino et al., who observed that temperatures of up to 670 K were necessary to maximize the signal intensity of the CW X-band EPR spectrum of (auto)reduced Cu-MFI14.0 during interaction with O2.98 The origin of the nuclearity-dependent formation temperature of specific Cu(II) centers remains to be identified. The difference between the required temperature for Cu(II) di-/trimer and Cu(II) monomer formation during interaction of (auto)reduced Cu–zeolites with O2 is depicted in Scheme 9.
The mechanism of [Cu2(µ-O)]2+ generation in (auto)reduced Cu-MFI has been explored by Smeets et al.147 Unlike in the case of as-prepared/hydrated materials, where [Cu2(µ-O)]2+ evolves from [CuOH]+ condensation (eqn (3.2)), the formation of [Cu2(µ-O)]2+ in (auto)reduced samples is initiated by the reaction of O2 with neighboring Cu(I) ions, yielding a [Cu2(µ-η2:η2-O2)]2+ precursor (eqn (4.7)).
| Cu+ + O2 → [Cu2(µ-η2:η2-O2)]2+ | (4.7) |
| [Cu2(µ-η2:η2-O2)]2+ + 2Cu+ → 2Cu2+ + O2−fw + [Cu2(µ-O)]2+ | (4.8) |
| [Cu2(µ-η2:η2-O2)]2+ + [Si2(µ-O)]6+ → [Cu2(µ-O)]2+ + [Si2(µ-1,2-O2)]6+ | (4.9) |
| [Cu2(µ-η2:η2-O2)]2+ + 2Cu+ → 2[Cu2(µ-O)]2+ | (4.10) |
000 cm−1, Smeets et al. determined that this precursor develops within ∼2 min when contacting the (auto)reduced material with O2 at ambient temperature (Fig. 5a).
 | ||
| Fig. 5 UV-Vis spectra of Cu0.5MFI12.0, previously (auto)reduced in He at 723 K, during interaction with O2 at 298 K (a, top) as well as of Cu0.3MFI12.0 throughout heating in He from 298 to 648 K following treatment in He at 723 K and exposure to O2 at 298 K (a, bottom). Raman spectra (λex = 457.9 nm) of (auto)reduced Cu-MFI12.0 after interaction with O2 at ambient temperature (black) and after raising the temperature to 573 K (blue, b). MS signal of 16O2, 16,18O2, and 18O2 as a function of temperature during a TPD (2 K min−1) of activated Cu0.5MFI12.0 (c). The maximum in the 16,18O2 desorption profile occurs in the same temperature range where the LMCT band of [Cu2(µ-O)]2+ at ∼22700 cm−1 disappears. Adapted from ref. 147 with permission from the American Chemical Society (Copyright 2010). | ||
Upon increasing the temperature to 448 K in either O2 or He, the intensity of the band at ∼29000 cm−1 started to gradually decrease, which was accompanied by an absorbance gain at ∼22700 cm−1 (Fig. 5a). This transition was attributed to the progressive conversion of [Cu2(µ-η2:η2-O2)]2+ into [Cu2(µ-O)]2+. Importantly, complementary Raman experiments demonstrated that the characteristic signals of [Cu2(µ-O)]2+ at ∼870, 456, and 514 cm−1, corresponding to the νasym(Cu–O) and νsym(Cu–O) stretch as well as the vibration of a T-site ligand, can already be identified in the spectrum of the (auto)reduced sample after interaction with O2 at ambient temperature (Fig. 5b).66,147 This suggests that the transformation of the O2-bridged precursor into [Cu2(µ-O)]2+ can take place even below 448 K, which is in agreement with the results of the investigation by Brenig et al. of the Cu(II) dimer generation in Cu–MOR10.0, Cu-MFI11.5, and Cu–CHA11.0 by UV-Vis spectroscopy (vide supra).127
A key question in the formation of [Cu2(µ-O)]2+ concerns the fate of the second O moiety arising from the O–O bond rupture in [Cu2(µ-η2:η2-O2)]2+. Smeets et al. addressed this by analyzing the isotopic composition of O2 released during TPD of 18O2-activated Cu0.5MFI12.0 (Fig. 5c).147 The authors detected a prominent 16,18O2 signal in the same temperature range where the LMCT band of [Cu2(µ-O)]2+ at ∼22700 cm−1 began to disappear. Consequently, they argued that the 18Oef of [Cu2(µ-O)]2+ does not recombine with 18Oef from a second Cu(II) dimer but with 16Ofw instead. By invoking the concept of microscopic reversibility, Smeets et al. proposed that the second 18O moiety, originating from the O–O bond cleavage in [Cu2(µ-η2:η2-O2)]2+, is incorporated into the zeolite lattice as 18Ofw (eqn (4.8)). Due to the higher abundance of 16Ofw compared to 18Ofw, the 18Oef of [Cu2(µ-O)]2+ preferentially recombines with 16Ofw during the TPD, leading to the pronounced 16,18O2 signal. The two additional electrons required to convert the O22− group of [Cu2(µ-η2:η2-O2)]2+ into two O2− moieties were hypothesized to derive from two Cu(I) spectator ions, which are oxidized to Cu(II). At this stage, it remains unclear how the Cu(I) spectators are regenerated in the course of successive CH4-to-CH3OH cycles and whether the zeolite lattice can continuously serve as a reservoir for new Ofw groups. Moreover, the possibility of isotope scrambling between 18Oef of [Cu2(µ-O)]2+ and 16Ofw of the zeolite lattice, which could affect the 16,18O2 signal, was not considered.195,196
Two additional scenarios regarding the positioning of the second O moiety have been suggested by Mahyuddin et al., who employed DFT to investigate the generation of [Cu2(µ-O)]2+ in Cu–MOR.179 Following [Cu2(µ-η2:η2-O2)]2+ formation in the 8-MR of the MOR side pocket (Scheme 1) facing the 8-MR channel, the liberated O atom can be integrated into the same 8-MR of the zeolite lattice, yielding [Si2(trans-µ-1,2-O2)]6+ (Scheme S1n) and [Cu2(µ-O)]2+ (eqn (4.9)). Alternatively, the O moiety can be transferred to two Cu(I) ions situated in the opposite 8-MR, leading to the formation of a second proximal [Cu2(µ-O)]2+ center (eqn (4.10)). Compared to the mechanism proposed by Smeets et al., the incorporation of the released O atom into the framework via [Si2(trans-µ-1,2-O2)]6+ generation does not necessitate an electron transfer from Cu(I) spectators, as the new O22− group retains the same formal charge as the original O2− lattice species. Although the formation of [Cu2(µ-O)]2+via assimilation of the O moiety into the zeolite lattice is exothermic (−61.5 kJ mol−1), the true activation energy of the O–O bond cleavage is prohibitively high (250 kJ mol−1), rendering this reaction unfeasible even at 723 K. On the contrary, the true activation energy of O–O bond rupture leading to the generation of two [Cu2(µ-O)]2+ sites in opposing 8-MRs of the MOR side pocket (Scheme 1) is considerably smaller (43.9 kJ mol−1), allowing this reaction to proceed at lower temperatures. Notably, the mechanism of proximal [Cu2(µ-O)]2+ formation is characterized by two spin inversions. The first one takes place during the initial O2 adsorption and transforms the system from a triplet to an open-singlet state, whereas the second one occurs while passing the O atom from one 8-MR to the other and induces a change from the open-shell singlet to the quintet state. Accordingly, each individual Cu(II) dimer adopts the more reactive triplet state, which, however, necessitates a spin crossover throughout the subsequent CH4 partial oxidation (see Section 4.1).67,73,95,107,157–159,161,164,166,167 It is important to highlight that the transfer of the O moiety across the 8-MRs of the MOR side pocket is feasible due to the short distance separating the [Cu2(µ-η2:η2-O2)]2+ precursor from the neighboring Cu(I) pair.179 The migration of the second O atom might, however, not be possible anymore in Cu–zeolites featuring paired Cu(I) ions located at distant exchange positions. An intriguing hypothesis has been put forward by Alayon et al., who proposed that the interaction of the liberated O moiety with gas phase O2 may yield O3, which could diffuse to isolated Cu(I) pairs to oxidize them.100 Alternatively, Ikuno et al. mentioned that two O atoms could simply recombine to O2 again.19 Despite these considerations, it remains elusive at this stage how Cu-oxo generation by O2 is affected by the spacing of Cu(I) pairs.
In their study on the activation of (auto)reduced Cu–MOR by O2, Mahyuddin et al. also provided additional insight into the mechanism of [Cu3(µ-O)3]2+ generation (eqn (4.11)).179
| [Cu3(µ-O)]2+ + O2 → [Cu3(µ-O)(η1-O2)]2+ → [Cu3(µ-O)(µ-η2:η2-O2)]2+ → [Cu3(µ-O)3]2+ | (4.11) |
Finally, a conclusive reaction scheme for the oxidative generation of [CuOH]+/[CuOH]+ and Cu2+/[CuOH]+ in (auto)reduced Cu–zeolites has not been presented so far. Multiple theoretical studies have indicated that the interaction of O2 with isolated Cu(I) can result in end-on/side-on [Cu(η-O2˙)]+ (eqn (4.2) and (4.12)).155,156,178 Santra et al. even resolved the rovibrational branches of the ν(O–O) stretch of [Cu(η2-O2˙)]+ at about 1196, 1180, and 1170 cm−1 in the FTIR spectrum of Cu-FAU, prepared via solid-state ion exchange with CuCl, after adsorption of O2 at 80 K.155 Nevertheless, it is not clear if and how these monomeric Cu-oxo sites can further transform into [CuOH]+ or Cu2+. Envisioning a plausible pathway for the formation of [CuOH]+ is further complicated by the fact that a H+ source must be present either in the form of H2O or as a BAS.52,104,127 As depicted in Scheme 9, a potential H+ source is already present or must be supplied externally, depending on the state of the (auto)reduced [CuOH]+/[CuOH]+ and Cu2+/[CuOH]+ pairs.
| Cu+ + O2 → end-on/side-on [Cu(η-O2˙)]+ | (4.12) |
| End-on/side-on [Cu(η-O2˙)]+ + Cu+ → [Cu2(trans-µ-1,2-O2)]2+ or [Cu2(µ-η2:η2-O2)]2+ | (4.13) |
| [Cu2(trans-µ-1,2-O2)]2+ or [Cu2(µ-η2:η2-O2)]2+ → [Cu2(µ-O)]2+ + 1/2O2 | (4.14) |
 | ||
| Fig. 6 Time-resolved Raman spectra (λex = 532 nm) of Cu0.24CHA15.5, previously (auto)reduced in He at 723 K, during calcination in O2 at 723 K (a). Spectra have been scaled by the indicated constants. UV-Vis spectra of Cu0.54MFI12.0 throughout interaction with O2 at 773 K following treatment in He at the same temperature (b). Development of the normalized integrated signal intensity of monomeric (Cu2+ and [CuOH]+, solid lines) as well as dimeric (two types of [Cu2(µ-O)]2+, dotted lines) Cu(II) species in (auto)reduced Cu–MOR10.0 (blue), Cu-MFI11.5 (red), and Cu–CHA11.0 (green) during exposure to O2 at 313 K measured by EPR and UV-Vis spectroscopy (c). Adapted from ref. 22, 127 and 136 with permission from the American Chemical Society (Copyright 2021), Elsevier (Copyright 2005), and Wiley (Copyright 2025). | ||
Instead, these bands progressively emerge throughout the O2 treatment alongside the feature at 603 cm−1. Since the kinetic description of the [Cu2(µ-O)]2+ generation was based on MCR using a single spectral component, the extracted rate constant is convoluted by the temporal development of the signals arising from the framework vibrations. This raises the question whether the long calcination durations are actually related to the formation of [Cu2(µ-O)]2+. Moreover, if the O2-bridged precursor is present in a pseudo-steady state, one could expect to observe its corresponding features in the Raman spectra, which, however, is not the case.147 Additionally, since the materials studied by Bregante et al. were in an (auto)reduced state prior to their treatment with O2, it is crucial to keep in mind that these results cannot simply be transferred to the activation of as-prepared/hydrated Cu–zeolites. As highlighted in Section 3, the generation of [Cu2(µ-O)]2+ in as-prepared/hydrated materials proceeds via [CuOH]+ condensation (eqn (3.2)), which does not involve a potentially kinetically limiting O–O bond rupture.
Contrary to the findings of Bregante et al., Smeets et al. reported that the formation of [Cu2(µ-O)]2+ in (auto)reduced Cu0.54MFI12.0 is a rather quick process.136 Compared to the UV-Vis spectrum of Cu0.54MFI12.0 calcined at 773 K, these authors reported that the intensity of the LMCT transition of [Cu2(µ-O)]2+ at ∼22700 cm−1 can be fully restored within ∼1.5 min upon interaction of the (auto)reduced material with O2 at 773 K (Fig. 6b). The fact that the evolution of [Cu2(µ-O)]2+ in (auto)reduced Cu–zeolites proceeds at an exceptionally fast rate is further supported by a study of Brenig et al. focused on the activation of reduced Cu–MOR10.0, Cu-MFI11.5, and Cu–CHA11.0.127 Depending on the framework, these authors observed that the temporal development of the bands corresponding to the LMCT transition of two different [Cu2(µ-O)]2+ moieties reached a steady state after ∼1.6 h during treatment of the materials with O2 at just 313 K (Fig. 6c). Upon raising the activation temperature above 423 K, these features evolved almost instantaneously. Interestingly, the rate of [Cu2(µ-O)]2+ generation was found to be dictated by the zeolite topology. Throughout the interaction of the reduced samples with O2 at 313 K, the authors noted that the time required for the absorbance of the LMCT signals to reach 50% of their steady state intensity increased in the order of Cu–MOR10.0 (20 s) < Cu-MFI11.5 (410 s) < Cu–CHA11.0 (1310 s). A comparison of the temporal behavior of the simultaneously measured O2 consumption of the different Cu–zeolites revealed the same trend. Although the origin of this framework-dependent oxidizability remains uncertain, it can be speculated that it is related to the preferential stabilization of Cu in specific oxidation states within the distinct ion exchange positions or to variations in the proximity of Cu(I) centers. Nevertheless, a more in-depth analysis is necessary to fully reveal the influence of the zeolite's topology on the formation kinetics of specific Cu-oxo species as well as on the bulk oxidizability. Notably, complementary CW X-band EPR experiments revealed that the rate of Cu(II) monomer formation, including Cu2+para, Cu2+meta, and [CuOH]+, is equally affected by the framework but remains significantly slower than the rate of Cu(II) dimer generation.
 | (4.15) |
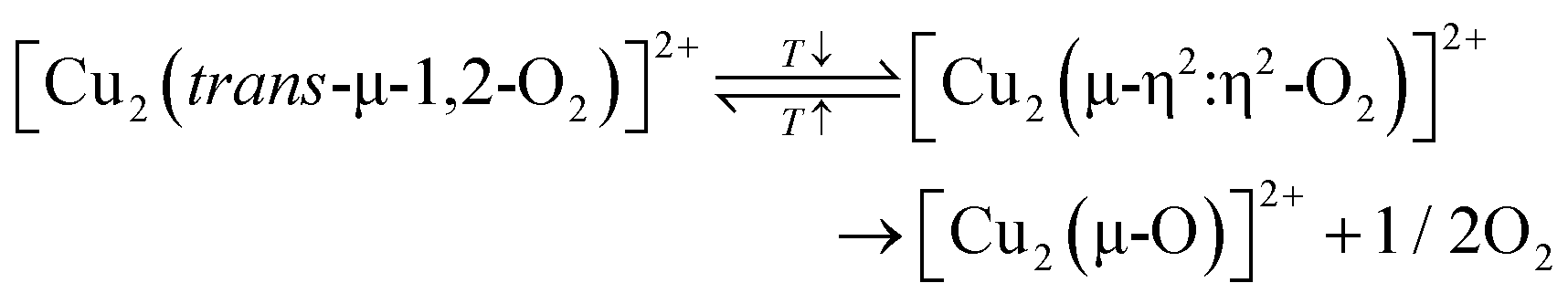 | (4.16) |
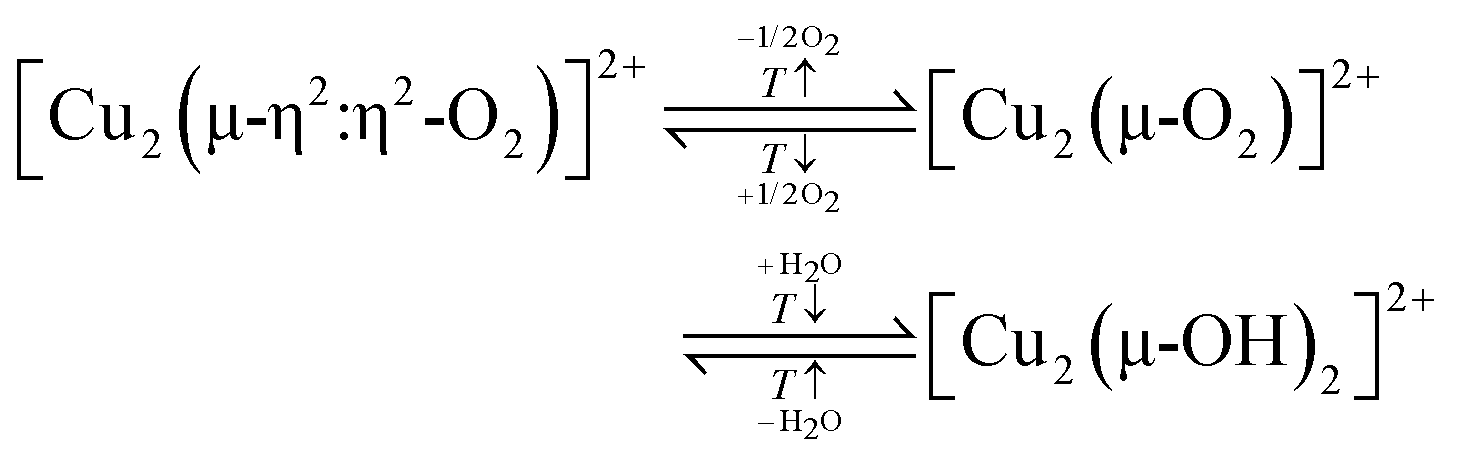 | (4.17) |
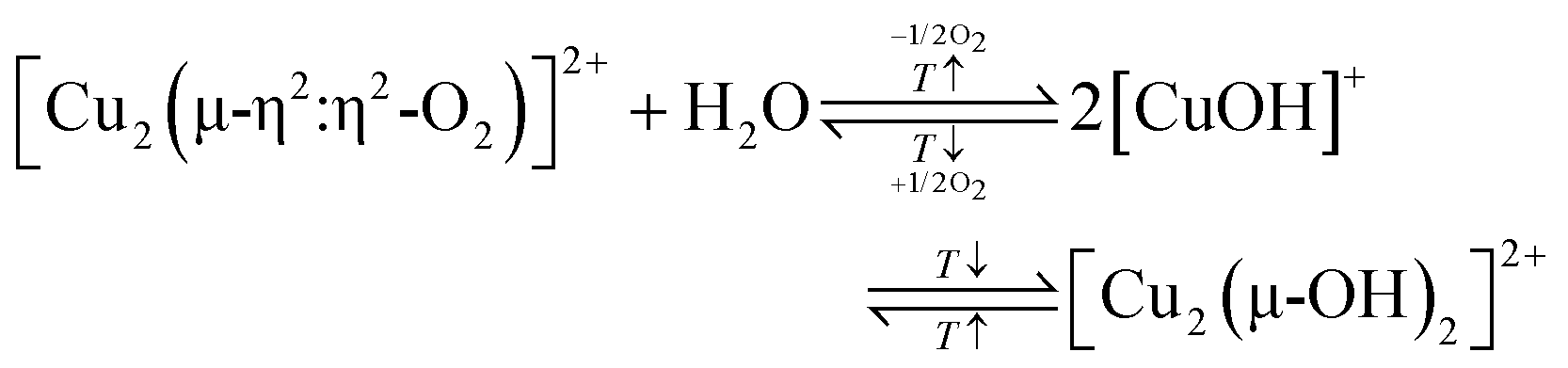 | (4.18) |
000 cm−1, associated with [Cu3(µ-O)3]2+, peaked at 723 K before declining again at 773 K. Based on this, Kim et al. argued that elevated temperatures favor the desorption of O2 from an intermediate of the [Cu3(µ-O)3]2+ site, resulting in the diminished CH3OH productivity after activation at temperatures higher than 723 K. Yet again, the CH4 partial oxidation experiments were conducted at 423 K after a cool down in O2, suggesting that this desorption effect should have been reverted.
Contrary to the work by Grundner et al., Bozbag et al. observed that the CH3OH productivity of Cu0.38MOR8.5 slightly increased by 5.7 µmolCH3OH gZEO−1 between the first (12.1 µmolCH3OH gZEO−1) and second (17.8 µmolCH3OH gZEO−1) redox cycle and remained approximately constant thereafter.199 They attributed this behavior to a facilitated Cu(II) active site formation in the second calcination period, which was related to the hydration of the material throughout the aqueous CH3OH extraction of the first cycle. However, it was not verified whether the sample's H2O content actually differed before the O2 treatment of the first and second cycle. Additionally, the reason for the beneficial role of H2O in the generation of the Cu(II) active sites was not elaborated in more detail. Notwithstanding these unresolved questions, an increase in the CH3OH output in the course of multiple redox cycles has also been reported by Pappas et al. for Cu-FER and Cu–CHA.16,89,169 This was related to a hydration-induced displacement of residual Cu(II) species from redox inert positions during the CH3OH extraction and their transformation into oxygenated Cu(II) active centers in the O2 treatment period of the subsequent reaction cycle. Nevertheless, inactive Cu(II) species usually correspond to isolated bare Cu2+ ions, which are charge-balanced by two Al T-sites positioned either in a 2NN or 3NN arrangement (see Section 2.1.1). Since the formation of these Cu(II) centers is characterized by a significantly lower Gibbs free energy compared to that of any other mono- or multimeric Cu-oxo species under the conditions of Cu–zeolite activation, the driving force for this redistribution phenomenon has yet to be determined.54,57,105,106
To further complicate matters, Wieser et al. and Knorpp et al. even noted a drop in the CH3OH output of Cu-MAZ4.3 by ∼40% during four consecutive cycles (from ∼159.8 to 96.4 µmolCH3OH gZEO−1), which they associated with a gradual unpairing of proximal [CuOH]+ centers and their migration from the 8-MR channel to the 6-MR of the gmelinite cage where they adopt the form of redox inert bare Cu2+ ions.111,200 The origin of this rearrangement phenomenon was related to the extensive dehydration of the material throughout activation at 723 K, which was proposed to enable the transfer of Cu between the different ion exchange positions.111
The discussion above highlights that more research is necessary to resolve the impact of the initial state, the zeolite framework, and the Cu speciation on the material regenerability.
4.3. Formation of Cu(II) sites using O2 during isothermal looping and continuous CH4 partial oxidation
Compared to non-isothermal looping, the processes occurring throughout the oxidative activation in isothermal CH4 partial oxidation and continuous CH4-to-CH3OH conversion have remained largely unexplored. As a result, a definitive correlation between the activation conditions, the nature of the generated Cu-oxo centers, and the Cu–zeolite's CH3OH productivity has yet to be established. Given the limited number of relevant studies, the following section will provide a combined overview of the formation and regeneration of oxygenated Cu(II) species in both isothermal CH4 hydroxylation and continuous CH4-to-CH3OH conversion. Where applicable, the effect of the activation conditions on the CH3OH productivity will be assessed.As depicted in Fig. 7a, increasing the temperature beyond 498 K during material treatment in O2 had little effect on the final amount of dehydrated Cu(II). However, rising temperatures significantly increased the amount of reduced Cu(I) throughout CH4 exposure (Fig. 7b). Increasing the temperature from 473 to 543 K resulted in an increase of the averaged CH3OH output from 75 to 277 µmolCH3OH gZEO−1 (Fig. 7c). Further raising the process temperature to 573 K led to a decline in the CH3OH productivity to 264 µmolCH3OH gZEO−1, highlighting the delicate balance between looping temperatures that are sufficiently high to enable dehydration and Cu-oxo active site generation, yet low enough to suppress CH4 overoxidation. Importantly, these constraints are less stringent in the case of Cu–zeolites, which are characterized by both a high activity in CH4 hydroxylation and CH3OH selectivity at elevated temperatures.204,206 An example for such a material is Cu-exchanged erionite (ERI) such as Cu0.3ERI5.6, whose CH3OH productivity after activation at either 573 or 723 K and subsequent reaction with 30 bar CH4 at 573 K differs by only ∼2 µmolCH3OH gZEO−1.206
 | ||
| Fig. 7 Development of the amount of dehydrated Cu(II) during exposure of Cu0.25MAZ4.3 to O2 at temperatures ranging from 473 to 573 K (a). Change in the amount of Cu(I) throughout reaction of activated Cu0.25MAZ4.3 with CH4 at the same temperatures (b). Corresponding CH3OH yield during aqueous product extraction (c). The CH3OH yield is averaged over four consecutively performed cycles at each temperature. Adapted from ref. 205 with permission from Wiley (Copyright 2023). | ||
The influence of the O2 pressure during calcination on the performance of Cu–MOR6.0 in isothermal CH4 partial oxidation has been studied by Tomkins et al.17 Consistent with their work on the impact of the O2 pressure on the CH3OH productivity of materials operating under non-isothermal conditions (see Section 4.2.3), these authors determined that the CH3OH output in isothermal CH4 hydroxylation is inversely proportional to the O2 pressure. This is illustrated by the fact that an increase in the O2 pressure from 1 to 6 bar during activation at 473 K, followed by reaction with 6 bar CH4 at the same temperature, induced a decrease in the CH3OH productivity from 21.2 to 16.8 µmolCH3OH gZEO−1. Nevertheless, elevated O2 pressures do not seem to impair the general oxidizability of Cu(I), as highlighted by Wieser et al., who investigated the effect of the O2 pressure on the development of the XANES spectra of reduced Cu0.25MAZ4.3 during activation at 523 K.205 By LCF analysis of the recorded spectra, these authors were able to show that the final Cu(I) content varies insignificantly upon increasing the O2 pressure from 1 to 4 bar. Moreover, their results suggest that the O2 pressure has a negligible effect on the Cu(I) oxidation rate.
Tomkins et al. also assessed the relation between the activation duration and the CH3OH output of Cu–MOR6.0.17 They demonstrated that the CH3OH productivity increases logarithmically from 10.4 to 25.5 µmolCH3OH/gZEO upon extending the activation time in O2 at 473 K from ∼3.3 (duration of heating ramp) to 42 h followed by reaction with 6 bar CH4 at the same temperature.
Similar to CH4 partial oxidation in a non-isothermal mode, the CH3OH productivity of most Cu–zeolites operating under isothermal conditions is relatively constant throughout multiple redox cycles.17,206 For example, Zhu et al. reported a stable CH3OH productivity of ∼113 µmolCH3OH gZEO−1 during four consecutive reaction cycles of Cu0.3ERI5.6 with 30 bar CH4 at 573 K, following activation in O2 at the same temperature.206 Likewise, Tomkins et al. observed a relatively steady CH3OH output of ∼20 µmolCH3OH gZEO−1 throughout four successive reactions of Cu–MOR6.0 with 6 bar CH4 at 473 K after O2 treatment.17 X-ray powder diffraction (XRPD) and N2 physisorption experiments highlighted that the Cu–zeolites are not subjected to hydrothermal degradation and retain their surface properties and crystallinity.17,206 These results imply that a constant fraction of Cu-oxo active centers can be regenerated after each redox cycle. A noteworthy exception to this is the work by Wieser et al., who studied the performance of Cu0.25MAZ4.3 in isothermal and isobaric CH4 partial oxidation at 523–573 K.111,205 Within this temperature regime, the authors observed a steady increase in the CH3OH productivity throughout four consecutive redox cycles. For example, the CH3OH output during CH4 hydroxylation at 563 K rose from 86 to 147 µmolCH3OH/gZEO−1 between the first and fourth reaction sequence.111 Wieser et al. attributed this trend to the gradual pairing of [CuOH]+ species via a hydration-induced migration through the MAZ framework.
The identification of paired [CuOH]+ centers in Cu-MAZ via anomalous XRPD (AXRPD) by Wieser et al. represents the first definitive structural characterization of an oxygenated Cu(II) active site involved in isothermal CH4 partial oxidation.111 The formation of [CuOH]+/[CuOH]+ pairs in as-prepared/hydrated and (auto)reduced Cu–zeolites under the conditions of isothermal CH4 hydroxylation, as well as their poisoning by residual H2O, is depicted in Scheme 10. For the sake of completeness, it should be mentioned that AXRPD has also enabled Knorpp et al. to assign [CuOH]+/[CuOH]+ pairs as the active centers in Cu-MAZ4.3 in non-isothermal CH4-to-CH3OH conversion.110 Synchrotron-based XRD has also been used by Andersen et al. and Ipek et al. to study the dehydration of Cu–CHA in O2 at temperatures up to 773 K.20,103 At present, it is not clear whether di- and trimeric Cu-oxo centers can also develop under the activation conditions employed in isothermal CH4 hydroxylation, and whether their structures resemble those of their counterparts generated during high-temperature calcination in non-isothermal CH4 hydroxylation. Tomkins et al. excluded the formation of dimeric Cu(II) species, as the UV-Vis spectra of Cu–MOR6.0 and Cu-MFI15.0, recorded after O2 treatment at 473 K for 13 h, did not exhibit the LMCT transition of [Cu2(µ-O)]2+ at 22200–22700 cm−1. Based on TEM, they proposed the participation of finely dispersed sub-nanometer CuXOY particles in the isothermal CH4-to-CH3OH conversion. Regarding the UV-Vis measurements, it should be noted that the adsorption of trace amounts of H2O on [Cu2(µ-O)]2+ can drastically reduce the intensity of its LMCT transition.50,80,133,144,207 For instance, Smeets et al. observed a complete disappearance of the characteristic feature of [Cu2(µ-O)]2+ at 22700 cm−1 in the UV-Vis spectrum of Cu0.56MFI12.0 upon contacting the activated material with a H2O-saturated stream of N2O at 637 and 723 K.207 This effect was fully reversible by switching to a pure N2O flow. Brenig et al. observed a similar effect in the UV-Vis spectrum of calcined Cu0.41CHA6.5 during interaction with H2O at 473 K, which resulted in a decrease in the absorbance of the LMCT bands of two different [Cu2(µ-O)]2+ motifs centered at about 26700 and 22400 cm−1.50 Currently, it is not known whether this intensity loss arises from the actual hydrolysis of [Cu2(µ-O)]2+ (inversion of eqn (3.2)) or whether it is merely a consequence of the H2O adsorption itself. If the latter is the case, virtually intact [Cu2(µ-O)]2+ centers could well be present after calcination at 473 K but remain undetectable by UV-Vis spectroscopy due to the high amount of residual H2O, emerging from the incomplete dehydration of the material. The potential generation of [Cu2(µ-O)]2+ throughout activation of as-prepared/hydrated and (auto)reduced Cu–zeolites in O2 under the conditions of isothermal CH4 partial oxidation is illustrated in Scheme 10. The poisoning of [Cu2(µ-O)]2+ by adsorption of H2O as well as the resulting adsorption complexes, which are characterized by a masked Oef → Cu(II) LMCT transition, are also shown.
 | ||
| Scheme 10 Formation of [Cu2(µ-O)(OH)2]2+, [(HO)Cu(µ-OH)Cu(O)]2+, [CuOH]+/[CuOH]+, and [Cu2(µ-O)]2+ (violet background) in O2 under the conditions of isothermal CH4 partial oxidation. [Cu2(µ-O)(OH)2]2+ is generated via multiple intramolecular H atom transfers in [Cu2(trans-µ-1,2-O2)(H2O)]2+, which, itself arises from the interaction of Cu+ (brown background) with O2 and H2O. A plausible mechanism for [Cu2(trans-µ-1,2-O2)(H2O)]2+ formation in as-prepared/hydrated Cu–zeolites has yet to be identified. In contrast, [CuOH]+/[CuOH]+ and [Cu2(µ-O)]2+ may evolve in both as-prepared/hydrated and (auto)reduced materials. Owing to the lower activation temperatures, the materials are not fully dehydrated, resulting in the adsorption of H2O on the Cu-oxo active sites. The ligation by H2O poisons the oxygenated Cu(II) centers and may obscure the characteristic LMCT feature of [Cu2(µ-O)]2+ in the UV-Vis spectra. [Cu2(μ-OH)2]2+ does not belong to the group of oxygenated Cu(II) species that are undetectable by UV-Vis spectroscopy as it features an (OH−)ef → Cu(II) LMCT transition at 32000–30000 cm−1.133,144 Notably, the development of this band at the expense of the characteristic LMCT signal of [Cu2(µ-O)]2+ upon hydration has not been observed so far.50,80,207 Dotted bonds correspond to dative interactions. The term “Z” describes the negatively charged zeolite lattice. | ||
An interesting proposal concerning the formation of dimeric Cu(II) active species in partially hydrated Cu–zeolites has been reported by Yumura et al., who studied the interaction of O2 and H2O with reduced Cu-MFI using DFT.208 According to these authors, the adsorption of O2 and H2O on Cu(I) ions, situated in the 10-MR and positioned in a 3NN arrangement, yields [Cu2(trans-µ-1,2-O2)(H2O)]2+ (eqn (4.19) and Scheme S1r). The latter is transformed into [Cu2(trans-µ-1,2-OOH)(OH)]2+ (Scheme S1s) via H atom transfer from the H2O ligand onto the bridging O2 moiety. Following another H atom transfer and a simultaneous O-OH bond cleavage, a [Cu2(µ-O)(OH)2]2+ (Scheme S1t) intermediate is generated, which further converts into [(HO)Cu(µ-OH)Cu(O)]2+ (Scheme S1u).
| 2Cu+ + O2 + H2O → [Cu2(trans-µ-1,2-O2)(H2O)]2+ → [Cu2(trans-µ-1,2-OOH)(OH)]2+ → [Cu2(µ-O)(OH)2]2+ → [(HO)Cu(µ-OH)Cu(O)]2+ | (4.19) |
010 mL gZEO−1 h−1 stream of 486 mbar CH4, 0.08 mbar H2O, 20 mbar O2, and 507 mbar N2 at 573 K.211 This resulted in a steady CH3OH output of 366 µmolCH3OH gZEO− h−1 for 24 h, albeit at a significantly reduced CH3OH selectivity of only 22%, thereby exemplifying the strict selectivity-conversion limit in catalytic CH4 partial oxidation (see Section 2.3).12,81–84 Notwithstanding the intrinsic restriction of the thermodynamically214 and kinetically215 favorable CH4 overoxidation on the CH3OH productivity, these results emphasize that a constant amount of oxygenated Cu(II) active sites can continuously be (re-)generated in an O2/CH4/H2O stream.
No consensus concerning the influence of the O2 partial pressure on the performance of Cu–zeolites in catalytic CH4-to-CH3OH conversion has been reached yet. Narsimhan et al. varied the O2 partial pressure between 0.025 and 2.4 mbar during continuous CH4 hydroxylation by Cu0.37MFI13.6 at 483 K and determined that the reaction is zero order in O2.209 A similar result has been reported by Dinh et al., who employed Cu0.22CHA23.0 in catalytic CH4 partial oxidation at 0.08–0.87 mbar O2 and 543 K.212 On the contrary, Hirayama et al. observed a decrease in the CH3OH production rate from 376 to 322 µmolCH3OH gZEO−1 h−1 upon increasing the O2 partial pressure from 9 to 37 mbar during continuous CH4-to-CH3OH conversion by Cu–CHA10 at 573 K.211 Currently, it is not clear whether these variations arise from differences in the mechanism of the Cu(II) active site formation or the markedly contrasting reaction conditions. Nevertheless, the zero or even negative order dependency of O2 on the CH3OH production rate clearly highlights that the generation of oxygenated Cu(II) active species is not the rate-determining step in catalytic CH4 partial oxidation.212 As addressed in Section 4.2.4, this correlates well with the fact that the formation of [Cu2(µ-O)]2+ during the activation of (auto)reduced Cu–zeolites in non-isothermal CH4 hydroxylation proceeds at a high rate.127,136
The development of Cu-oxo active sites under the conditions of catalytic CH4 hydroxylation remains poorly understood. Narsimhan et al. exposed (auto)reduced Cu0.37MFI13.6 to an O2/CH4/H2O feed and observed that the onset of steady state CH3OH production was preceded by an initial induction period of ∼190 min.209 This behavior was attributed to the H2O-induced rearrangement of hydrated Cu(I) into the Cu-oxo active site.209,212 According to Narsimhan et al., this Cu(II) active species does not correspond to [Cu2(µ-O)]2+, as complementary UV-Vis spectra collected throughout the induction period did not display a pronounced LMCT band at ∼22700 cm−1. Nevertheless, considering the strong impact of H2O on the intensity of this band (see Sections 4.3.1 and 10.3), caution should be warranted when using the presence of this feature to assess the existence of [Cu2(µ-O)]2+ under the conditions of continuous CH4-to-CH3OH conversion. In a subsequent study by the same research group, [Cu2(µ-O)]2+ was identified as the Cu(II) active site for catalytic CH4 partial oxidation in Cu–CHA by analysis of the FT-EXAFS spectrum collected at 543 K under steady state CH3OH production.212 Similar to their previous work, the perpetual generation of the Cu(II) dimer was proposed to occur via the interaction of O2 with mobile hydrated Cu(I) ions, which are capable of dynamically migrating between the Al T-sites. The necessary charge balancing for the transfer of Cu(I) from one ion exchange position to the other was suggested to proceed via a simultaneous H2O-assisted transfer of H+ co-cations. At a sufficiently low Si/Al ratio and in the absence of co-cations that permanently block the Al T-sites (e.g. Na+ or NH4+), this mechanism enables the transfer of Cu(I) throughout the entire framework, thereby facilitating the evolution of [Cu2(µ-O)]2+ even in materials featuring a low Cu content. Whether the actual generation of [Cu2(µ-O)]2+ proceeds via one of the pathways depicted by eqn (4.7)–(4.10) or (4.12)–(4.14) remains a matter of debate. A different hypothesis regarding the nature and formation mechanism of the Cu-oxo active site in Cu–MOR10.0 for continuous CH4-to-CH3OH conversion has been presented by Ohyama et al.210 These authors collected the XANES spectrum of Cu–MOR10.0 after equilibration in an O2/H2O and O2/CH4/H2O stream at 573 K and computed the corresponding difference spectrum. This difference spectrum was compared to that obtained using simulated XANES spectra representing different Cu(II) and Cu(I) structural motifs. Based on this comparison, Ohyama et al. proposed that the oxygenated Cu(II) active site corresponds to a hydrated [Cu2(µ-1,2-O2)(OH)2(H2O)2] (Scheme S1v) complex, which develops via interaction of O2 with diffusion-paired mobile [Cu(OH)(H2O)] (eqn (4.20) and Scheme S1w).
 | (4.20) |
Finally, many mechanistic pathways dealing with the formation of oxygenated Cu(II) active sites during catalytic CH4 partial hydroxylation postulate the involvement of hydrated mobile Cu(I) ions.210,212 However, Cu(I)-aqua complexes are not stable and may disproportionate into metallic Cu and hydrated Cu(II) due to the larger hydration energy and higher CN of [Cu(H2O)6]2+ relative to [Cu(H2O)2]+.218–221 Instead, the mobile Cu(I) species may correspond to bare Cu(I) ions, which are capable of migrating through the zeolite lattice in the absence of any ligands.103,222 This has been demonstrated by Andersen et al., who monitored the dehydration of Cu0.48CHA15.1 during He treatment via simultaneous XRPD and XANES spectroscopy.103 Upon (auto)reduction of the material at temperatures above 636 K, Rietveld analysis of the corresponding XRPD patterns revealed a redistribution of Cu from the 8-MR to the 6-MR, indicating that Cu(I) could be dynamically migrating between ion exchange positions. Whether this process is relevant for the evolution of oxygenated Cu(II) species under conditions of continuous CH4-toCH3OH conversion, particularly concerning the migration timescale, remains an open question.
5. Usage of N2O in chemical looping and continuous CH4 hydroxylation for the formation of active Cu(II) species
5.1 Formation of Cu(II) sites using N2O during the stepwise CH4-to-CH3OH conversion via looping
Prior to the usage of N2O as an oxidizing agent for the formation of Cu-oxo active sites for CH4 partial oxidation, the interaction of N2O with Cu–zeolites was investigated in the context of the catalytic N2O decomposition reaction.133,145,207,223 Consequently, the oxidation of Cu(I) to Cu(II) by N2O has been studied far earlier than its direct application in the CH4-to-CH3OH conversion.700 cm−1 and a weaker LMCT band at ∼30000 cm−1, initially assigned to a [Cu2(µ-O)2]2+ center. The dimeric nature of this oxygenated Cu(II) species was additionally confirmed by in situ EPR spectroscopy, which revealed that up to 81% of the existing Cu(II) centers were EPR silent due to antiferromagnetic coupling (see Section 3.1). The presence of [Cu2(μ-O)2]2+ was further rationalized by its ability to release O2, thus serving as a potential Cu(II) active site for N2O decomposition. Reducing the temperature from 773 to 673 K caused a decrease in both the N2O conversion and the absorbance of the spectral feature at 22700 cm−1. Simultaneously, an increase of two signals centered at 30000 and 13000 cm−1, corresponding to an Ofw → Cu(II) LMCT and d–d transition of partially hydrated Cu(II), respectively, was observed. This behavior was attributed to the formation of a [Cu2(µ-O)]2+ core, which was postulated to be an intermediate of [Cu2(µ-O)2]2+. The authors suggested that below 773 K, the N2O decomposition reaction proceeds in two steps:| 2Cu+ + N2O → [Cu2(µ-O)]2+ + N2 | (5.1) |
| [Cu2(µ-O)]2+ + N2O → [Cu2(µ-O)2]2+ + N2 | (5.2) |
Following the initial identification of [Cu2(μ-O)2]2+ in Cu-MFI, in situ UV-Vis spectroscopy revealed that the intensity of the LMCT band at 22700 cm−1 gradually decreased during interaction with CH4, demonstrating the ability of this Cu(II) species to activate CH4.7 Woertink et al. employed Raman spectroscopy, using a laser with an appropriate wavelength to enhance the vibrations of this specific Cu(II) center, and identified both a pronounced νsym(Cu–O) at 456 cm−1 and a less intense νasym(Cu–O) at 870 cm−1, revealing that the Cu(II) active site previously assumed to be a [Cu2(µ-O)2]2+ species (vide supra) actually corresponds to a [Cu2(µ-O)]2+ center.66 Normal coordinate analysis (NCA) of these vibrations highlighted that the Cu–O–Cu angle in [Cu2(µ-O)]2+ is 140°. According to the authors, such a wide angle excludes the introduction of a second bridging Oef atom, as would be necessary to form the [Cu2(µ-O)2]2+ site. DFT calculations suggested that the [Cu2(µ-O)]2+ moiety is located within the 10-MR of MFI, with the two Al T-sites being separated by two intermediate Si T-sites. The assignment was based on a comparison of experimentally and theoretically determined geometric and electronic properties, revealing very similar features such as comparable Cu–O–Cu angles (138° theoretical vs. 140° experimental). Analyzing materials with various Cu/Al ratios revealed that the spectroscopic features of [Cu2(µ-O)]2+ became more prevalent in samples characterized by a higher Cu loading.136 This is quite intuitive, as the formation of a [Cu2(µ-O)]2+ active site requires two Cu(I) centers in close proximity to each other.
Further work has demonstrated that the [Cu2(µ-O)]2+ motif may be present in multiple other zeolite topologies besides Cu-MFI, such as Cu–MOR51,68 and Cu–CHA.194 However, dependent on the topology of the zeolitic host, distinct differences in the geometric and electronic properties of the [Cu2(µ-O)]2+ species have been shown to exist. Even within the same zeolite framework, slight variations in the geometric configuration of the [Cu2(µ-O)]2+ center have been observed. Vanelderen et al. have shown that upon exposure of (auto)reduced Cu0.43MOR5.0 to N2O, a LMCT band centered around 22200 cm−1 emerged.51 According to the authors, the band at 22200 cm−1 can be deconvoluted into two separate LMCT bands at 21900 and 23100 cm−1, suggesting that two [Cu2(µ-O)]2+ species distinguishable from each other may simultaneously be present in Cu–MOR. Raman spectroscopy revealed that although both Cu(II) species formally correspond to [Cu2(µ-O)]2+, they exhibit different νasym(Cu–O) and νsym(Cu–O) values. NCA highlighted that the two [Cu2(µ-O)]2+ centers exhibit slightly different Cu–O–Cu angles (137° vs. 141°) and Cu–O bond strengths. Notably, these two Cu(II) sites are characterized by a substantially different reactivity toward CH4; the Cu(II) species featuring a smaller Cu–O–Cu angle of 137° and a LMCT band at 21900 cm−1 reduced less rapidly compared to the Cu(II) center characterized by the LMCT band at 23100 cm−1 and the larger Cu–O–Cu angle of 141°. DFT models based on the experimental data suggested that two distinct [Cu2(µ-O)]2+ sites are located at different positions within the framework: the less reactive Cu(II) species at the intersection of the side pockets and the 12-MR channels, and the more reactive Cu(II) center in the intersections of the side pockets and the 8-MR channels (Scheme 1).51,69 The difference in reactivity of the two Cu(II) species toward HAA from CH4 has been suggested to originate from the zeolite framework itself, with a confinement of CH4 within the smaller side pockets of MOR suggested to lower the activation enthalpy required for HAA.51,69 Recently, Plessers et al. identified a third [Cu2(µ-O)]2+ site after interaction of (auto)reduced Cu–MOR9.0 with N2O.68 Alternation of both the Cu loading (Cu/Al ratio between 0.09 and 0.45) as well as the nature of the alkali co-cation employed to further charge balance the zeolite framework resulted in a variation of the ratio of different Cu(II) species, i.e., Cu2+vs. [CuOH]+vs. [Cu2(µ-O)]2+, in the studied Cu–MOR samples. The newly identified [Cu2(µ-O)]2+ species exhibits a LMCT band at ∼22300 cm−1 after N2O exposure. NCA of the νsym(Cu–O) and νasym(Cu–O) vibration of the new [Cu2(µ-O)]2+ moiety revealed that its Cu–O–Cu angle is 127°, and therefore significantly smaller than the Cu–O–Cu angles determined for the other two identified [Cu2(µ-O)]2+ centers in Cu–MOR (vide supra).51,68 In Cu–CHA, Rhoda et al. revealed a further [Cu2(µ-O)]2+ site by exposing (auto)reduced Cu0.29CHA15.7 to N2O at 823 K, leading to the emergence of an intense LMCT band at 22400 cm−1 as well as a weaker band at 6200 cm−1, as illustrated in Fig. 8.194 The reason for the high N2O treatment temperature was not discussed in detail, which is surprising given that N2O can already decompose into N2 and O2 at ∼623 K in the case of Cu0.4CHA13.0.13 As depicted in Fig. 8, exposure of activated Cu0.29CHA15.0 to CH4 at 473 K induced a significant decrease in intensity of the LMCT band at 22400 cm−1, which is indicative of the reduction of [Cu2(µ-O)]2+ to Cu(I). The combination of Raman spectroscopy of the N2O-activated sample (νsym(Cu–O) = 581 cm−1, νasym(Cu–O) = 837 cm−1) with NCA revealed that the Cu–O–Cu angle corresponds to 120°. DFT calculations highlighted that a [Cu2(µ-O)]2+ site with such a small angle can only be stabilized across the 8-MR of Cu–CHA, with three Si T-sites separating the Al T-sites. The bridging Oef atom was determined to point out of the 8-MR and into the CHA cages themselves (Scheme 1).
 | ||
| Fig. 8 UV-Vis spectra of Cu0.29CHA15.7 collected at ambient temperature after calcination in O2 at 723 K (blue), (auto)reduction in He at 973 K (black), N2O treatment at 823 K (red), and interaction with CH4 at 473 K (brown). Adapted from ref. 194 with permission from the American Chemical Society (Copyright 2021). | ||
A further factor affecting the performance of [Cu2(µ-O)]2+ in C–H bond activation was suggested to stem from the Cu–O–Cu angle itself, as elucidated by Mahyuddin et al.157 The authors suggested that a smaller Cu–O-Cu angle would result in greater overlap of the Cu d-orbitals, leading to a lowering of the energy of the acceptor orbital involved in C–H bond activation.
These discussed works show that a range of distinct [Cu2(µ-O)]2+ centers in various Cu–zeolites with different topologies can be formed via the oxidation of Cu(I) pairs with N2O.66,68,124,194 Tsai et al. investigated this process primarily using DFT calculations.124 Building on the previously determined geometric and spectroscopic features of [Cu2(µ-O)]2+ in Cu-MFI,66 the authors assessed which Cu(I) pair sites within the 10-MR of MFI would enable a low-barrier O2− transfer from N2O. A systematic variation of both the Ofw–Cu–Ofw bite angles, the CN (two and three) from the lattice to Cu(I), as well as the nature of the central atom (Si or Al) of the coordinating T-site, was performed. Two factors were determined to significantly increase the thermodynamic stability of a given Cu(I) pair: A linear Ofw–Cu–Ofw bite angle close to 180° and coordination to the Ofw atoms of Al T-sites instead of Si T-sites. The most stable Cu(I) pair configuration that is experimentally feasible exhibited two-coordinate Cu centers with an OSi–Cu–OAl bite angle of ∼149° and a Cu(I)–Cu(I) distance of 4.17 Å, thereby minimizing electrostatic repulsion between the two Cu(I) ions. The [Cu2(µ-O)]2+ species corresponding to a Cu(I) pair has a calculated Cu–O–Cu angle of 135°, and therefore is very similar to the value of 139° observed experimentally.224 Both the calculated Raman vibrations (451 cm−1 and 842 cm−1) and LMCT band (22926 cm−1) were very close to the experimentally obtained value by Woertink et al. (vide supra).224
Using the determined structure of the energetically favorable Cu(I) pair, three distinct N2O binding modes were evaluated via DFT: η1-N, μ-1,1-O, and μ-1,3-O,N, as depicted in Fig. 9a–c; all in the closed-shell singlet ground state. Notably, the η1-N and μ-1,3-O,N binding modes exhibited an apparent activation energy of 250 and 21 kJ mol−1, respectively, which is far from the experimentally determined value of 8 ± 2 kJ mol−1 for [Cu2(µ-O)]2+ formation. Remarkably, the calculated value in the case of the μ-1,1-O binding mode (8 kJ mol−1) matched the experiment well (Fig. 9a). The true activation energy for a μ-1,1-O binding mode was estimated at 46 kJ mol−1. It should be noted that Mahyuddin et al. also investigated the μ-1,1-O binding mode on a Cu(I) pair in MFI, though determining an even lower true activation energy of ∼26 kJ mol−1 for N–O bond cleavage.154 These results indicate that the exact configuration of N2O interaction with both Cu(I) centers is the determinative factor for the necessary activation energy, with non-bridging binding modes being characterized by a particularly high activation barrier. This, therefore, also directly influences the permittable Cu–Cu distance a Cu(I) pair may have for N2O activation. Tsai et al. determined that a Cu(I)–Cu(I) distance above 4.17 Å restricts μ-1,1-O binding mode, hence prohibiting N2O activation (Fig. 9d).124
 | ||
| Fig. 9 Potential binding configurations of N2O on Cu(I) pairs characterized by different Cu–Cu distances and coordination modes to the zeolite lattice. Structures (a)–(c) describe the interaction of N2O with a Cu(I) pair, which features a Cu–Cu distance of 4.17 Å prior to N2O adsorption. Each Cu(I) ion is coordinated by two Ofw atoms that are affiliated with an Al and Si T-site. In structure (d), the Cu–Cu distance before N2O adsorption is 5.85 Å. One of the involved Cu(I) ions is coordinated by three Ofw atoms, two belonging to a single Al T-site, whereas the third Ofw atom originates from a Si T-site. Adapted from ref. 124 with permission from the American Chemical Society (Copyright 2014). | ||
The reaction was suggested to be initiated via a one-electron transfer from one of the Cu(I)'s to a π* orbital of N2O, leading to a partial breaking of the N–O bond. This is followed by a further electron transfer from the second Cu(I), resulting in a cleavage of the N–O bond and formation of N2 and [Cu2(µ-O)]2+. The spin density distribution revealed that an electron is predominantly transferred from one of the two Cu(I)'s, thereby highlighting that it is a stepwise process, itself facilitated by the bent N–N–O geometry of 143° at the transition state (TS), stabilizing the π* and enhancing overlap with the d orbitals. Additionally, it is worth mentioning that during oxidation, a change in the coordination of Cu to the zeolite lattice occurs as well. Both Cu(I) centers are each coordinated to two Ofw separated by both an Al T-site and a Si T-site, while both of the resulting Cu(II)'s of [Cu2(µ-O)]2+ are coordinated to two Ofw solely separated by one Al T-site.
Mahyuddin et al. investigated the same three proposed N2O binding modes (Fig. 9a–c) via DFT calculations on a Cu(I) pair in Cu-AEI, with the site modelled to be located in the 8-MR of the zeolite.157 N2O would initially adsorb on one of the two Cu(I) centers with a binding energy of −25 to −47 kJ mol−1 (without and with dispersion correction). The μ-1,1-O binding mode (Fig. 9a) of N2O was determined to be the only feasible pathway for an oxo-transfer from N2O, which is highlighted in Scheme 11, with a determined true activation energy of ∼5 to 8 kJ mol−1, without and with dispersion correction, respectively, in the closed-shell singlet state.157 This value of 8 kJ mol−1 for Cu-AEI157 is significantly lower than that obtained for Cu-MFI by both Mahyuddin et al.154 and Tsai et al.124 (vide supra). Zhao et al. calculated an activation energy of 75 kJ mol−1 for the activation of a Cu(I) pair via the μ-1,1-O binding mode in Cu–MOR.164 This value was determined for a [Cu2(µ-O)]2+ species located in the 12-MR of MOR. The location of the Cu(I) pair within a given zeolite, as well as the topology, will significantly influence the characteristics of it, as well as the [Cu2(µ-O)]2+ resulting from said Cu(I) pair. Such is the case when comparing AEI and MFI; in the 8-MR of AEI, the Cu–Cu distance of the Cu(I) pair was determined to be only 2.7 Å, with the resulting [Cu2(µ-O)]2+ species exhibiting a Cu–O–Cu angle of ∼84° in comparison to ∼135° in the case of Cu-MFI.124,157 These effects also extend to the TS of N2O oxidation, with the N–O bond length at the TS evaluated to be slightly shorter at ∼1.2 Å in Cu-AEI than for Cu-MFI at ∼1.46 Å.124,157 This suggests that the necessary energy investments for an oxo-transfer to a Cu(I) pair, and thereby an oxidation and formation of [Cu2(µ-O)]2+, may significantly depend on framework topology, as well as location within a given topology. This is analogous to the influence that framework topology may have on a [Cu2(µ-O)]2+ sites ability for HAA from CH4.
 | ||
| Scheme 11 Formation pathways of [CuOH]+/[CuOH]+ (S1), Cu2+/[CuOH]+ (S2), [Cu2(µ-O)]2+ (S3), [Cu2(µ-O)2]2+ (S4), and [Cu3(µ-O)3]2+ (violet background) by treatment of as-prepared/hydrated (blue background) and (auto)reduced (brown background) Cu–zeolites with N2O in the chemical looping process. The transformation of Cu(II)-aqua complexes into the Cu-oxo species proceeds via the dehydration intermediates [Cu(H2O)4]2+ and [Cu(OH)(H2O)3]+ (grey background). Interaction of N2O with paired Cu(I) (S7) yields S3 and S4 via the adsorption intermediate (S5) (grey background). The reaction mechanism of [Cu3(µ-O)3]2+ generation starting from [Cu3(µ-O)]2+ (S6) has not been investigated yet, and potentially involved intermediates are not known (structure with transparent overlay and marked with a question mark). Similarly, it is not clear whether monomeric Cu(II) species can be re-oxidized by treatment of (auto)reduced Cu–zeolites with N2O, either from a state without a given H+ source (S7) or with (S8) (pathway marked with a question mark). Dotted bonds correspond to dative interactions. The term “Z” describes the negatively charged zeolite lattice. The color gradient from yellow to violet of the different starting points and arrows indicates the progressively increasing temperature. | ||
In the case of Cu-MFI, Tsai et al. elucidated that the N2O activation of paired Cu(I) proceeds along the singlet potential energy surface (PES).124 Although two singlet–triplet crossing points at 1.35 and 1.55 Å were identified during N–O bond elongation, the TS was located at the intermediate N–O distance of 1.46 Å. This would therefore result in a spin-crossover to a triplet state after reconstitution of the active site. Mahyuddin et al. suggested the same for Cu-AEI, with the re-oxidation occurring in the singlet state, followed by a spin crossover to a triplet state. This triplet state however, is almost isoenergetic to the singlet state (see Section 4.1).157 Heyer et al. further investigated the ground state of both [Cu2(µ-O)]2+ in Cu-MFI as well as Cu–CHA using VTVH MCD spectroscopy and DFT calculations.67 Cu-MFI was found to adopt a singlet ground state due to the parallel arrangement of the bidentate coordinating Al T-sites of the framework, thereby maximizing the superexchange interaction between the two half-filled Cu(II) 3d orbitals. This, however, stands in contrast to the theoretical works by Tsai et al., which determined [Cu2(µ-O)]2+ in Cu-MFI to be present in a triplet ground state.124 In Cu–CHA instead, Heyer et al.67 elucidated that the [Cu2(µ-O)]2+ site adopts a triplet ground state due to the perpendicular orientation of the coordinating Al T-sites of the framework, leading to a low orbital overlap of the O-bridged half-filled Cu(II) 3d orbitals. A triplet ground state for the [Cu2(µ-O)]2+ site has also been suggested for Cu–MOR by Zhao et al., due to the singlet state being 40 kJ mol−1 less stable.164 The reaction, however, proceeds on a singlet PES via a Cu(I) pair and N2O.164 The authors did not calculate the activation barrier for a Cu(I) pair and N2O via a triplet PES due to the instability of said state, resulting in a much higher activation barrier than in the case of a singlet PES. This implies that a spin inversion will occur after the TS if the [Cu2(µ-O)]2+ site has a triplet spin state.
Göltl et al. computationally investigated Cu–CHA for Si/Al > 8, with theoretically predicted phase diagrams being employed to examine a range of Al configurations inside CHA, with two Al T-sites either being in a 3NN or 4NN configuration, or one of the two Al T-sites located in adjacent 6-MR and 8-MR.72 The authors examined the effect of temperature and partial pressure of N2O on the stability of the species with different configurations of two Al T-sites. In the case of one or zero Al T-sites being present in the 6-MR, and therefore respectively the necessary other two or one Al T-sites located in the 8-MR, the [Cu2(µ-O)2]2+ species were present under all examined conditions, including at ambient temperature. If, however, two Al T-sites are allowed to be located in the same 6-MR, Cu(II) will be present in a hydrated state at ambient temperature. Higher temperatures and low N2O partial pressures could result in Cu migration and the formation of Cu2+, assumedly in the 6-MR. However, an increase in the partial pressure of N2O restricts such changes in Cu(II) speciation and will lead to the formation of [Cu2(µ-O)2]2+ instead. For example, at the typical activation temperature of 723 K, a partial pressure of ∼10 mbar N2O would suffice for the formation of [Cu2(µ-O)2]2+. When, however, the model was restricted to certain Al configurations, the phase diagrams showed that both [Cu2(cis-µ-1,2-O2)]2+ and [Cu2(µ-OH)2]2+, as well as bare and hydrated Cu(II), could all potentially be the thermodynamically preferred sites, dependent on both N2O partial pressure and temperature. As experimental verification, a UV-Vis spectrum of a Cu0.41CHA14.7 sample at ambient temperature, previously activated at 723 K in N2O, was collected, exhibiting both d–d transitions at 13400 cm−1 and a broad LMCT band between 29000–37000 cm−1. A comparison to theoretically predicted spectra suggested that these bands could be attributed to the suggested [Cu2(µ-O)2]2+ species. While the authors' theoretical findings showed that [Cu2(µ-OH)2]2+ can only form under an O2 and not N2O atmosphere, Raman spectroscopy revealed a band at 615 cm−1 attributed to said species. The authors, however, performed their activation procedure at 723 K, a temperature where Cu–CHA has been shown to be able to decompose N2O to O2 and N2,13 which could cause the resulting O2 to be responsible for the formation of [Cu2(µ-OH)2]2+. An explanation of why both species are able to exist simultaneously within CHA is, however, not present. Moreover, the source of H+ necessary to form [Cu2(µ-OH)2]2+ remains to be clarified. In addition, it needs to be noted that Zhao et al. calculated that for Cu–MOR that an O insertion into [Cu2(µ-O)]2+via a second N2O to form [Cu2(µ-1,2-O2)]2+, while being exergonic at a temperature of 523 K, presented an insurmountable activation barrier of 244 kJ mol−1.164
The vast majority of studies investigating the dimeric structures have been conducted on (auto)reduced materials that have been treated under He at elevated temperatures, followed by an exposure to N2O. An overview of the different starting conditions is provided in Scheme 11.51,66,68,194 Examining if the [Cu2(µ-O)]2+ active site can be formed from an as-prepared/hydrated state by N2O exposure has not received much attention, with solely one examination having been performed by Ipek et al.13 As-prepared/hydrated Cu0.4CHA12.0 was exposed to 30% N2O/He at 543 K, leading to absorption features at both 23100 and 21000 cm−1, attributed to different [Cu2(µ-O)]2+ species. The authors further examined the CH3OH productivity at 473 K of the same material in the chemical looping process, studying different activation temperatures by keeping the duration and the N2O concentration constant. An increase in activation temperature from 473 to 723 K led to an increase in CH3OH productivity from ∼13 to 35 µmolCH3OH gZEO−1. The authors also highlighted that at temperatures above 623 K N2O decomposition occurs, which would result in a lower amount of potential active sites for HAA. However, it should be noted that since the CH4 exposure was always conducted at 473 K, well below the temperature regime where N2O decomposition occurs, any resulting Cu(I) may be expected to undergo a re-oxidation by N2O during the cool down. The changes in productivity might therefore be more related to the preceding dehydration of the material at higher temperatures.
000, 34000, and 39000 cm−1, all attributed to [Cu3(µ-O)3]2+. The activation temperature during N2O exposure was shown to have a significant effect on the CH3OH productivity, with an increase from ∼30 to 97 µmolCH3OH gZEO−1 when increasing the activation temperature from 673 to 873 K. A consecutive cycle with a N2O activation at 873 K exhibited a similar productivity to the first cycle, as well as the presence of LMCT bands at 34000–39000 cm−1, thereby showing that the [Cu3(µ-O)3]2+ species can be reconstituted from a reduced state (Scheme 11). A slight bathochromic shift of the band at 39000 to 38500 cm−1 was observed, suggesting that the LMCT band at 34000 cm−1 was related to the [Cu3(µ-O)3]2+ site, with the other bands originating from inactive Cu-oxo clusters present in the material.
While the [Cu3(µ-O)3]2+ site may seemingly be generated from an as-prepared/hydrated state, Kim et al. suggested that during the activation procedure under N2O, an initial dehydration would lead to a reduction of Cu(II) to Cu(I), as well as the formation of square planar Cu(II). N2O activation would then lead to the formation of [Cu3(µ-O)3]2+ from these intermediates. Experimental verification was delivered by first exposing Cu0.4MOR20.0 to N2 at 723 K, leading to the appearance of a d–d transition at 16700 cm−1, attributed to square planar Cu(II). Exposure to N2O at 523 K led to an increase in absorbance at 34000 cm−1, and disappearance of the feature at 16700 cm−1, attributed to square planar Cu(II) being incorporated into a [Cu3(µ-O)3]2+ species (Scheme 11). Therefore, while [Cu3(µ-O)3]2+ can be formed from an as-prepared/hydrated material, an initial (auto)reduction of a fraction of the Cu is necessary. Therefore [Cu3(µ-O)3]2+ will not be able to be formed directly from the hydrated as-prepared state. Similar suggestions were made by Ikuno et al. for Cu0.31MOR11.0.19 While no spectroscopic characterization for the N2O-activated material is shown, the obtained CH3OH productivity of Cu0.31MOR11.0 is equivalent to the O2-activated material over a range of activation temperatures (473–773 K), for which a detailed characterization of the active site has been performed. The formation of [Cu3(µ-O)3]2+ under O2 exposure is discussed in greater detail in Section 4.2.2.
More definite characterizations of the [Cu3(µ-O)3]2+ active site formable by N2O have, to our knowledge, only been performed in the case of Cu–MOR.19,21 Whether this site is even able to be formed in other zeolitic frameworks is a matter of debate. Göltl et al., for one, suggested that in the case of Cu–CHA of Si/Al > 8, trimeric species cannot be stabilized under any conditions in the course of N2O treatment.72
500 cm−1, attributed to a defect-bound [CuOH]+. Plessers et al. delivered further evidence for both [CuOH]+ and bare Cu2+ being able to form from an as-prepared/hydrated state by N2O exposure. As-prepared/hydrated Cu–MOR9.0 with Cu/Al ratios varying from 0.09–0.45, was exposed to N2O at 523 K. EPR spectroscopy revealed spectroscopic fingerprints of both [CuOH]+ and Cu2+, with the fraction of [CuOH]+ increasing with the Cu/Al ratio.
Examining if [CuOH]+ can be re-constituted by oxidation with N2O from a reduced Cu(I) state has not yet been clearly shown. [CuOH]+ is typically suggested to be more resistant toward (auto)reduction in an inert atmosphere than [Cu2(µ-O)]2+, necessitating the use of reducing agents such as CH4.62 By simply performing one reaction cycle using the looping procedure followed by an exposure of the material to N2O, this question could easily be answered. However, such an examination is at this point lacking in the literature. Tsai et al., however, suggested that any mononuclear Cu(I) site would be unable to be activated by N2O, based on a very high activation energy due to the necessary η1-O binding mode (Fig. 9d).124 Such a binding mode would be necessary due to either a Cu(I) being completely isolated or sufficiently far (>5 Å) from another Cu(I), thereby excluding a bridged N2O binding mode. In any case, a bridged binding mode would only allow for the re-oxidation of one Cu(I) to form [CuOH]+, while HAA from CH4 would necessitate two proximal [CuOH]+. The determined activation energy for N2O activation in the case of a Cu–Cu distance of ∼5.13 Å, and therefore necessitating a terminal η1-O binding mode over a single Cu(I), was determined to be ∼67 kJ mol−1. While being higher than the activation energy of N2O activation via a Cu(I) pair (see Section 5.1.1), the value suggests that the reaction could be feasible at elevated temperatures.
Evidence for the involvement of monomeric Cu(I) species in N2O decomposition suggests that these species can undergo reversible redox processes in the presence of N2O. For example, Smeets et al. investigated N2O decomposition over a range of Cu–zeolites (MOR, MFI, FER, BEA). At Cu/Al ≤ 0.23, almost all Cu was EPR visible, indicating an exclusively monomeric configuration. At elevated temperatures of 723–773 K, N2O conversion was low (0–5%).225 However, adding 0.1% NO to the feed increased N2O conversion by ∼12–20% for all Cu–zeolites examined, suggesting that monomeric Cu species can be activated under these conditions. Notably, above ∼750 K, Cu0.22MFI12.0 began exhibiting intrinsic activity toward N2O decomposition, achieving ∼10% conversion at ∼830 K.207,225 It needs to be mentioned that the type of monomeric Cu(II) has not been revealed in this study (Scheme 11). Further, Fanning et al. performed extensive diffuse reflectance FTIR spectroscopy (DRIFTS) studies on Cu0.55–0.6MFI10.7–13.5 to examine the Cu(II) species formed by N2O oxidation of Cu(I).226 At the initial stage, the materials were exposed to Ar at 773 K. In the case of Cu0.6MFI13.5, a hypsochromic shift of a band at ∼930 to ∼970 cm−1 was associated with the reduction of Cu(II) to Cu(I). Further evidence for the reduction of Cu(II) was provided by complementary EPR measurements, which showed that 96% of the signal intensity decreased during the pretreatment. The observation of CO in the DRFITS suggests that the primary origin of the reduction is the presence of carbonaceous species from the Cu(CH3COO)2 precursors. A bathochromic shift of the band at 970 to 930–910 cm−1 was observed after the exposure to N2O at 583–643 K. This band was assigned to asymmetric framework vibrations perturbed by either [CuOH]+ or [CuO˙]+, a species for which any definite characterization is lacking in the literature (see Section 3.2). However, it should be noted that the assignment of the 930–910 cm−1 bands to monomeric species is contested, as dimeric sites have been assigned to similar bands.227 Further, this study suggests that the identified [CuOH]+ species, or their precursors, are reducible during an inert gas treatment at 773 K, which stands in contrast to other literature.42,62
The potential source of the necessary H atom for the reconstitution of [CuOH]+, as is the case for the formation of said species by O2 oxidation in Section 4.2.2, remains unresolved. Fanning et al. therefore suggested that Cu(I) could be re-oxidized by N2O, with a BAS or terminal Si–OH group donating a H atom for [CuOH]+ formation (eqn (5.3)). Another feasible source could be the simultaneous conversion of a Cu(I) pair via both N2O and H2O, as depicted in eqn (5.4), which would therefore require a close proximity of the two Cu(I)'s. However, below a certain Cu–Cu distance, the more likely result would be the formation of [Cu2(µ-O)]2+ species, as discussed in Section 5.1.1 and depicted in eqn (5.1).
| Cu+ + N2O + SiOH → [CuOH]+ + N2 + SiO˙ | (5.3) |
| 2Cu+ + N2O + H2O → 2[CuOH]+ + N2 | (5.4) |
5.2 Formation of Cu(II) sites using N2O during catalytic selective CH4 oxidation
While the continuous CH4-to-CH3OH conversion using N2O as an oxidant has received some attention and has been shown to be a plausible route, identification of the potential active sites responsible for HAA from CH4 is often lacking. This is primarily due to many spectroscopic techniques not being able to derive the same amount of information in continuous vs. cyclic CH4-to-CH3OH conversion. A prime example of this is UV-Vis spectroscopy. As mentioned in section 4.3.1, and will be discussed in more detail in Section 10.3, the presence of H2O traces may already significantly reduce the LMCT transitions of [Cu2(µ-O)]2+. As the feed-gas composition in catalytic operational mode often contains >3 vol% H2O, an identification of [Cu2(µ-O)]2+ using UV-Vis spectroscopy becomes impossible for the continuous CH4-to-CH3OH conversion. Significant emphasis has therefore been placed on drawing comparisons to active sites characterized in stepwise operational mode, as well as a reliance on DFT calculations.Ipek et al., as well as Memioglu et al., investigated various Cu–zeolites, including Cu-exchanged MOR, CHA, AEI, MFI, and MAZ, which all exhibited activity in the continuous CH4-to-CH3OH conversion using N2O as the oxidant.13,228 Most emphasis was placed on Cu0.4CHA12.0.13,228 Increasing the reaction temperature from 523 to 573 K entailed an increase in productivity from 12 to 55 µmolCH3OH gZEO−1 h−1. In situ UV-Vis spectroscopy under an N2O atmosphere at 543 K revealed absorption features at 21000 and 23100 cm−1, both attributed to [Cu2(µ-O)]2+ species near to the 8-MR of the CHA topology. This could be considered valid evidence that [Cu2(µ-O)]2+ species in Cu0.4CHA12.0 would be responsible for HAA from CH4 in continuous operational mode. The authors, however, also suggested that a different active species could form due to the presence of H2O in the feed, which is still able to perform an HAA from CH4, as previously suggested by Alayon et al.173 The authors also posited that, just as suggested by Yumura et al., a [Cu2(µ-O)(OH)2]2+ or [(HO)Cu(µ-OH)Cu(O)]2+ species could be the true active species under continuous conditions, and even cleave the C–H bond of CH4 more efficiently than the [Cu2(µ-O)]2+ species, as evidenced from theoretical calculations.208 However, experimental verification of such species remains elusive (see Section 12.3).
Xiao et al. investigated the potential of Cu0.03–0.22AEI7.7–11.6 for the catalytic CH4 oxidation using N2O as the oxidant.229,230 By increasing the calcination temperature from 823 to 1223 K, the authors decreased the amount of Al T-site pairs, from 57% to 8%, together with the fraction of total Al T-sites, from 98% to 51%. All examined Cu-AEI samples exhibited activity, with the highest activity achieved by the sample with the highest fraction of Al T-site pairs, that being the sample calcined at the lowest temperature of 823 K. Alternating these parameters, as well as varying Cu/Al ratio, culminated in Cu0.09AEI11.4 achieving impressive CH3OH productivities up to 2448 µmolCH3OH gZEO−1 h−1 at a selectivity of 45% at 623 K. In samples with Al T-site pairs, the authors suggested the [Cu2(µ-O)2]2+ species to be responsible for CH4 activation. Based on the results of FTIR spectroscopy conducted at 298 K following an evacuation procedure at 773 K, the authors concluded that N2O would bind to a [Cu2(µ-O)]2+ site in either a η1-N or η1-O binding mode (Fig. 9). It should, however, be noted that such high-temperature conditions have previously been shown to be able to cause (auto)reduction of [Cu2(µ-O)]2+ species.62,66 The authors may therefore have been probing N2O binding modes to (auto)reduced Cu(I) sites rather than the intended [Cu2(µ-O)]2+ species, which raises questions about their structural assignments. Nevertheless, characteristic bands at 2292 and 2248 cm−1, attributed to η1-N and η1-O binding modes, respectively, were observed (Fig. 9). The authors also did not exclude the possibility of a μ-1,3-O,N binding mode. According to their proposed mechanism, the resulting [Cu2(µ-O)2]2+ species would serve as the active site for HAA from CH4.230 Critically, however, no spectral evidence for the formation of this proposed active [Cu2(µ-O)2]2+ species was reported.229 The suggested reaction scheme is depicted in eqn (5.5).
 | (5.5) |
Chen et al., Xu et al., and Dai et al. examined the mechanism of the active site formation in the catalytic CH4-to-CH3OH conversion using N2O in multiple Cu-exchanged zeolite topologies, such as Cu-BEA, Cu–CHA, and Cu-MFI, in more detail.231–233 In a combinatorial approach of experiments and DFT calculations, the authors suggested that for Cu-BEA and Cu-MFI a re-oxidation of the [Cu2(µ-O)]2+ core via a μ-1,3-O,N binding mode to a Cu(I) pair would occur (Fig. 9). Furthermore, a mechanism of N2O activation over isolated Cu(I) sites was proposed as well.231,232 A Cu–CHA sample was prepared via solid-state ion exchange under a wet NH3 atmosphere with the aim of obtaining exclusively isolated Cu(II) in the 6-MR of the zeolite.232 Due to the lack of proximal Cu(I) the authors claim that the mechanism would proceed via a η1-O binding mode, as shown in Fig. 9d. The activation energy was calculated to be ∼130 kJ mol−1 for both single Cu(I) in CHA and MFI, and the reaction would proceed viaeqn (5.6).
| Cu+ + N2O → [CuO˙]+ + N2 | (5.6) |
The authors proposed that the active species is an oxo-monomeric [CuO˙]+ site rather than [CuOH]+. However, their suggested mechanism for Cu–CHA contains a critical thermodynamic inconsistency. According to their data, after CH3OH desorption occurs, there is an enormous energy barrier of approximately 800 kJ mol−1 between the resulting Cu(I) species and the subsequent N2O adsorption step (via η1-O binding mode in the pre-TS). This energy barrier is thermodynamically insurmountable under realistic reaction conditions. A second cycle would thus not be possible, thereby making the claims doubtful.
6. Cu(II) active site formation using either O2 or N2O and impact on CH3OH productivity
In light of the discussion in Sections 4 and 5, it is clear that the activation of (auto)reduced Cu–zeolites using either O2 or N2O may proceed in fundamentally different manners. Apart from the distinct formation mechanism of isostructural Cu-oxo sites (e.g., [Cu2(µ-O)]2+, see Sections 4.2.2 and 5.1.1), activation with O2 or N2O has been reported to result in the development of entirely different oxygenated Cu(II) centers within the same material. Owing to the oxidant-dependent variation in the Cu speciation, the CH3OH productivity of a given sample may significantly differ following treatment with O2 or N2O under otherwise identical conditions. In the following section, the impact of the oxidant type on the CH3OH output will be assessed and correlated to the differences in the nature and abundance of the generated Cu-oxo sites. This analysis will primarily be framed in the context of CH4 partial oxidation via non-isothermal looping, as the limited amount of available literature does not allow for a direct comparison of activation by O2 and N2O in other operation modes of CH4 hydroxylation.As outlined in Section 4.2.2, the CH3OH productivity of most Cu–zeolites increases upon raising the calcination temperature.13–16,18,19 A similar relationship between the material performance in CH4 partial oxidation and the temperature during N2O treatment has been established.13,19,21 However, conflicting observations regarding the relative CH3OH productivity after interaction with either O2 or N2O in specific temperature regimes have been reported. Ipek et al. investigated the behavior of Cu0.4CHA12.0 in CH4 partial oxidation following activation in O2 or N2O at temperatures ranging from 473 to 723 K.13 At 473 and 573 K, treatment with N2O resulted in a higher CH3OH output compared to calcination. The greater CH3OH productivity after N2O treatment in this temperature regime was attributed to a higher fraction of formed oxygenated Cu(II) active centers. This was supported by complementary UV-Vis experiments, which demonstrated that the absorbance of two spectral features at 23100 and 21000 cm−1, attributed to two different [Cu2(µ-O)]2+ motifs, was more intense after N2O exposure at 543 K than after O2 treatment. Notably, upon increasing the activation temperature to 723 K, the reverse trend was observed, with the CH3OH output being higher after interaction with O2 than after N2O treatment. This phenomenon was related to the (auto)reduction of [Cu2(µ-O)]2+, driven by the catalytic decomposition of N2O into N2 and O2, which effectively decreases the number of dimeric Cu(II) active sites. Nevertheless, Ipek et al. conducted the CH4 partial oxidation experiments at 473 K following a cool down from the activation temperature in the presence of the oxidant. Considering the discussion in Sections 4.2.5 and 5.1.1, the [Cu2(µ-O)]2+ (auto)reduction should have been reversed throughout the temperature decrease prior to the CH4 hydroxylation, assuming that its re-oxidation via N2O is not kinetically limited.
Rhoda et al. reported an increase in the CH3OH productivity from 64 to 82 µmolCH3OH gZEO−1 after activation of Cu0.29CHA15.7 at 823 K in N2O vs. O2.194 Similar to the UV-Vis study by Ipek et al., an amplification of the band of the LMCT transition of [Cu2(µ-O)]2+ at 22400 cm−1 upon exchanging O2 for N2O was observed. This coincides with observations from Brenig et al., who detected a significant gain in the absorbance of the LMCT transition of a [Cu2(µ-O)]2+ species at 22100 cm−1 after interaction of Cu0.35CHA11.0 with N2O instead of O2 at 723 K.50 Likewise, Plessers et al. identified that the intensity of the LMCT transition of three distinct types of [Cu2(µ-O)]2+ sites centered at 22300 cm−1 was more pronounced after activation of Cu–MOR in N2O at 523 K than after exposure to O2.51,68 A noteworthy exception to this general trend is the work by Sheppard et al., who determined that the band of [Cu2(µ-O)]2+ in Cu0.28MFI12.0 at 22200–22000 cm−1 was more intense after exposure of the (auto)reduced sample to O2 at 673 K than after interaction with N2O.85
Based on the spectroscopic studies by Ipek et al., Rhoda et al., and Brenig et al., the enhanced CH3OH output of N2O-activated Cu–zeolites originates from the more facile [Cu2(µ-O)]2+ generation compared to O2-treated samples. This is also evident from the variation in the threshold temperature required to observe the LMCT transition of the dimeric Cu-oxo centers during interaction of (auto)reduced Cu–zeolites with either N2O or O2. As mentioned in Section 4.2.2, Woertink et al. demonstrated that a minimal temperature of 448 K is necessary to trigger the appearance of the feature at 22700 cm−1 throughout interaction of (auto)reduced Cu0.54MFI12.0 with O2 (Fig. 10).224 On the contrary, this signal could already be identified at ambient temperature upon exposing the material to N2O.147
 | ||
| Fig. 10 Raman spectra (λex = 457.9 nm) of Cu0.54MFI12.0 activated in either O2 or N2O. The inset displays UV-Vis spectra recorded at ambient temperature after interaction of Cu0.54MFI12.0 with either O2 or N2O at temperatures ranging from 373 to 473 K and 323 to 398 K, respectively. The arrows in the inset represent the increasing temperature of the O2 and N2O treatment. Adapted from ref. 224 with permission from the National Academy of Sciences (Copyright 2009). | ||
The difference in the efficiency of [Cu2(µ-O)]2+ formation by N2O and O2 has also been highlighted by DFT calculations of Mahyuddin et al., which indicate that the nature of the oxidant governs the magnitude of the activation energy of the rate-determining step in the generation mechanism of the oxygenated Cu(II) species.157,179 For example, the transformation of paired Cu(I) into [Cu2(µ-O)]2+ in Cu-AEI and Cu-MFI by N2O exhibits a true activation energy of approximately 8 and 26 kJ mol−1, respectively.157,163 On the contrary, the activation energy of the rate-determining step in the formation of two [Cu2(µ-O)]2+ sites via oxidation of proximal Cu(I) pairs, situated in the opposing 8-MRs of the MOR side pocket (Scheme 1), with O2 amounts to ∼50 kJ mol−1.179 Although the energetic barrier of [Cu2(µ-O)]2+ formation by O2 is still low enough to proceed at moderate temperatures, the considerably smaller activation energy of [Cu2(µ-O)]2+ generation by N2O indicates that this reaction is more facile. The difference between the true activation energy of [Cu2(µ-O)]2+ formation by O2 and N2O is highlighted in Scheme 12, which also illustrates the corresponding oxidation intermediates. Notably, a higher true activation energy of 46 kJ mol−1 for the formation of [Cu2(µ-O)]2+ during activation of Cu-MFI with N2O has been reported by Tsai et al.124 At present, the origin of the considerable difference between the true activation energies for the development of [Cu2(µ-O)]2+ in Cu-MFI via interaction with N2O, as determined by Mahyuddin et al. and Tsai et al., remains unclear.
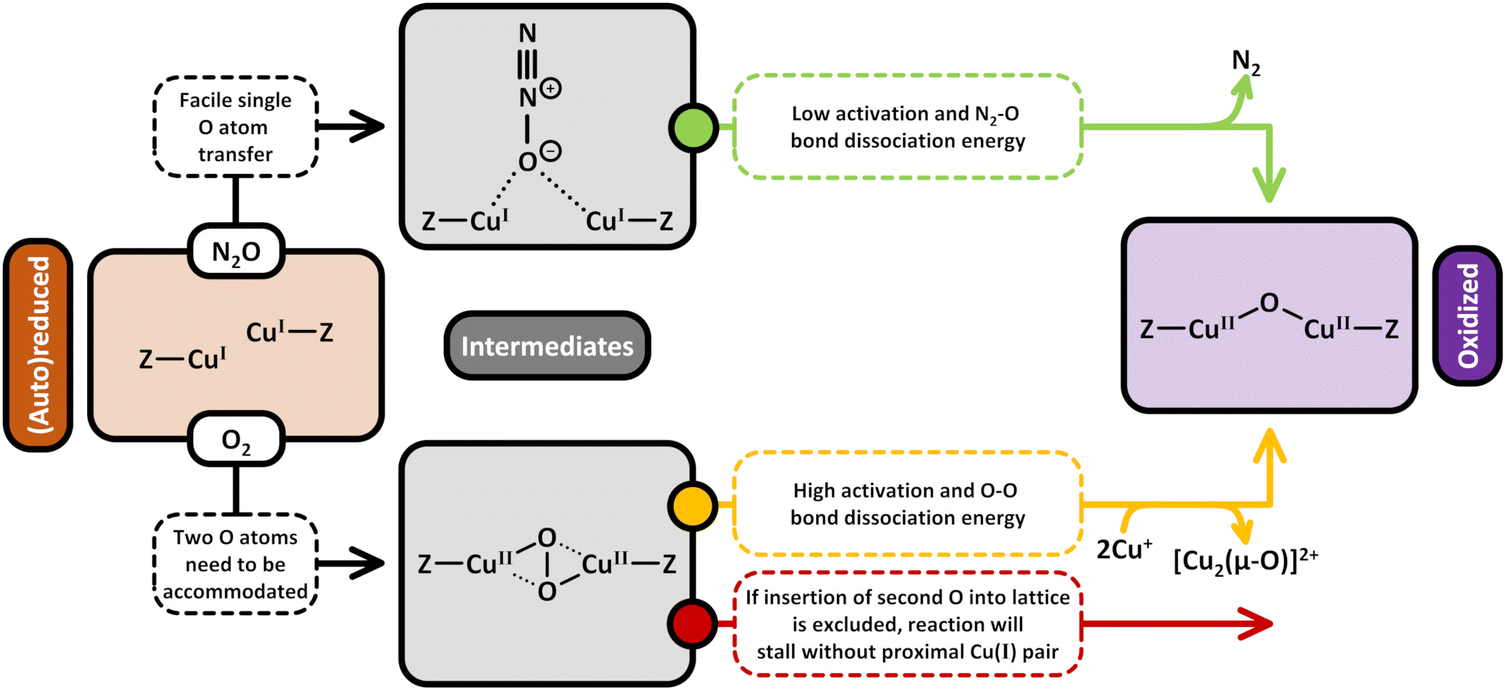 | ||
| Scheme 12 Transformation of Cu(I) (brown background) into [Cu2(µ-O)]2+ (violet background) using either O2 or N2O as the oxidant. The generation of [Cu2(µ-O)]2+via interaction of (auto)reduced Cu–zeolites with O2 proceeds via a [Cu2(µ-O)2]2+ intermediate (grey background) and cannot resume without a location to accommodate the second O moiety. In contrast, material activation by N2O, which proceeds via a particular adsorption complex (grey background), enables the formation of isolated [Cu2(µ-O)]2+ as only a single O atom is transferred. Compared to treatment in O2, [Cu2(µ-O)]2+ generation by N2O is thermodynamically more facile as the true activation energy and N2–O bond dissociation energy are lower. Dotted bonds correspond to dative interactions. The term “Z” describes the negatively charged zeolite lattice. | ||
The preferential formation of [Cu2(µ-O)]2+via treatment of Cu–zeolites in N2O instead of O2 also becomes apparent when comparing the generation mechanisms of the dimeric Cu-oxo site in the presence of the two oxidants (eqn (5.1) and (4.7)–(4.10)). In the case of N2O, the reaction proceeds via a simple two-electron redox process and involves the direct transfer of a single O moiety to two Cu(I) ions as well as the simultaneous release of N2.9,19,51,52,95,124,149,158 On the contrary, the O–O bond cleavage occurring throughout [Cu2(µ-O)]2+ evolution in O2 requires that the second O moiety must be accommodated either within the zeolite lattice or at a nearby Cu(I) pair.147,179 Keeping in mind that the former process is characterized by an excessively high true activation energy of 250 kJ mol−1 (see Section 4.2.2), it is reasonable to assume that the transformation of a proximal Cu(I) pair into another oxygenated Cu(II) center by the liberated O group is preferred. However, this implies that the development of [Cu2(µ-O)]2+ in O2 might be inhibited in the absence of neighboring Cu(I) ions, as there is no place to host the second O moiety. Activation in N2O thus enables the generation of isolated [Cu2(µ-O)]2+, which might not be possible in the case of O2 treatment. Scheme 12 illustrates the distinct formation mechanism of [Cu2(µ-O)]2+ in either O2 or N2O and emphasizes the necessity of a location to accommodate the liberated O group in the case of Cu–zeolite activation by O2.
The greater efficiency of [Cu2(µ-O)]2+ generation using N2O compared to O2 can also be rationalized by taking into account that the energy required for N2–O bond cleavage is significantly smaller than that of O–O bond rupture. Based on tabulated formation enthalpies at 0 K, the N2–O bond dissociation energy amounts to just ∼161 kJ mol−1, whereas that of O–O equals 494 kJ mol−1, which is also described in Scheme 12.234 Importantly, this consideration is not exclusive to the generation of [Cu2(µ-O)]2+ since N2–O and O–O bond cleavage is involved in the formation mechanism of other Cu-oxo species by N2O or O2, too. Similar to the results of Ipek et al., Kim et al. found that the CH3OH productivity of Cu0.4MOR20.0 was higher after interaction with N2O than after calcination in O2 at activation temperatures in the 573–623 K range. These authors also observed that this trend is reversed when further increasing the activation temperature to 673 K.21 However, upon raising the activation temperature to 773 K, the CH3OH output was again greater following exposure to N2O than after O2 treatment. Kim et al. ascribed the better performance in CH4 partial oxidation after activation in N2O at temperatures below 673 K and above 773 K to the thermodynamically favored transformation of a reduced Cu precursor into [Cu3(µ-O)3]2+. On the contrary, the formation of [Cu3(µ-O)3]2+ in the presence of O2 was proposed to occur via an O2-bridged intermediate (see Section 4.2.2), which was deemed to be responsible for the diminished CH3OH productivity. At low activation temperatures, the necessary O–O bond scission was suggested to be kinetically limited, whereas high activation temperatures were hypothesized to induce the desorption of O2 from the intermediate. The reason for the higher CH3OH productivity after interaction with O2 compared to N2O at intermediate activation temperatures from 673 to 723 K has yet to be explained.
Then again, Ikuno et al. did not observe any strong variation in the CH3OH output of Cu0.31MOR11.0 after activation of the material in either N2O or O2 at temperatures ranging from 473 to 773 K.19 Hence, these authors argued that the generation of the Cu-oxo active site, which was proposed to correspond to a [Cu3(µ-O)3]2+ species, is equally efficient in O2 and N2O, suggesting that O–O bond cleavage is not a limiting factor.
Finally, Göltl et al. noted a rise in the CH3OH productivity from 10 to 23 µmolCH3OH gZEO−1 when activating Cu0.41CHA14.7 in N2O at 723 K instead of O2.72 This was attributed to an oxidant-controlled variation in the nature of the Cu(II) active center. Theoretically predicated phase diagrams revealed that, depending on the specific Al T-site arrangement, [Cu2(µ-O)2]2+ and [Cu2(cis-µ-1,2-O2)]2+ are the thermodynamically preferred Cu-oxo species in Cu–CHA at 0.9 mbar N2O and 473 K, i.e., the conditions prior to CH4 hydroxylation. On the contrary, [Cu2(µ-OH)2]2+ as well as [Cu2(µ-OH)]2+ (Scheme S1x) were identified as the most favorable Cu(II) sites at 0.9 mbar O2 and 473 K.72,168 Complementary DFT calculations indicated that [Cu2(µ-O)2]2+ and [Cu2(cis-µ-1,2-O2)]2+ are capable of performing two consecutive CH4 partial oxidations. In contrast, [Cu2(µ-OH)2]2+ can convert only a single CH4 molecule into CH3OH, whereas [Cu2(µ-OH)]2+ was found to be inactive toward CH4 hydroxylation.72 Göltl et al. thus suggested that the higher CH3OH productivity of N2O-activated materials arises from the formation of oxygenated Cu(II) centers, which intrinsically yield more CH3OH compared to the Cu-oxo species generated in the presence of O2. Unfortunately, no mechanism for the formation of any of the proposed oxygenated Cu(II) active sites was provided. Consequently, the source of H+ necessary for the (re-)generation of [Cu2(µ-OH)2]2+ is not clear. It could be envisioned that the latter is formed via interaction of [Cu2(µ-O)]2+ with H2O, though this remains purely speculative. Moreover, it remains elusive how the O–O bond in [Cu2(cis-µ-1,2-O2)]2+ during interaction of the sample with N2O is formed. The latter question is particularly important since theoretical calculations by Zhao et al. have demonstrated that the formation of [Cu2(µ-1,2-O2)]2+via oxidation of [Cu2(µ-O)]2+ with N2O is characterized by an insurmountable free energy barrier of 244 kJ mol−1 at 523 K.164 The inability of N2O to generate Cu-oxo centers featuring O–O bonds has also been emphasized by Rhoda et al., who noted that, unlike activation in O2, treatment with N2O cannot result in the development of end-on/side-on [Cu(η-O2˙)]+.194 Likewise, the formation of [Cu2(µ-η2:η2-O)]2+ has only been observed through activation in the presence of O2 but not N2O.147
7. Formation of active Cu(II) species with H2O in stepwise and catalytic CH4-to-CH3OH conversion
7.1. Formation of Cu(II) sites using H2O during the stepwise CH4-to-CH3OH conversion via looping
A discussion on whether H2O is able to oxidize Cu(I) centers in zeolites has been present long before Cu(II) zeolites were shown to be active for CH4 activation. Anaerobic oxidation of Cu(I) to Cu(II) in zeolites has been suggested to be both impossible99 and possible,143 as well as both O2 and H2O being necessary simultaneously.98,99,235 This discussion has continued into the field of CH4-to-CH3OH conversion via Cu–zeolites.Alayon et al., using Cu0.38MOR, reported the first anaerobic oxidation of Cu(I) to Cu(II) by H2O, following a Cu(II) reduction in CH4.116 During the following H2O exposure, a marked decrease in the Cu(I) 1s → 4p transition at ∼8983 eV and an increase in the 1s → 3d transition of Cu(II) at ∼8978 eV via in situ high-energy resolution fluorescence detection (HERFD) XAS were observed. In addition, a hypsochromic shift of the edge was observed, a further indication of Cu oxidation occurring. Pappas et al., also using in situ XAS, showed that an anaerobic Cu(I) to Cu(II) oxidation was also observable for both Cu0.2FER11.0169 and Cu0.5CHA12.0.16 It also was the case for Cu0.38MOR as reported by Alayon et al.,116 H2O was not able to fully re-oxidize all Cu(I) reduced during the preceding CH4 exposure over the examined timeframes.
 | ||
| Fig. 11 In situ XANES spectra of Cu–MOR13.0 recorded during interaction with H2O at 473 K after reaction with CH4 at the same temperature (a). The arrow describes the temporal development of the Cu(I) pre-edge feature. FTIR spectra recorded at 100 K of CO adsorbed on Cu–MOR13.0 after treatment in vacuum at 673 K (bottom, b), reaction with CH4 at 473 K (middle, b) and re-oxidation with H2O at 473 K (top, b). The color gradient indicates the progressively increasing CO dosage. Arrows describe the evolution of the intensity of absorption bands, originating from the interaction of CO with Cu(I), after material exposure to CH4 and H2O. FTIR spectra recorded at 100 K of NO adsorbed on Cu–MOR13.0 after treatment in vacuum at 673 K (bottom, c), reaction with CH4 at 473 K (middle, c) and re-oxidation with H2O at 473 K (top, c). The color gradient indicates the progressively increasing NO dosage. Arrows describe the evolution of the intensity of absorption bands, originating from the interaction of NO with Cu(I) and Cu(II), after material exposure to CH4 and H2O. MS responses of unlabeled (m/z = 31) and 18O-labeled (m/z = 33) CH3OH after two consecutive reaction cycles of Cu–MOR13.0, using labeled H218O for CH3OH desorption and reactivation (d). MS responses of H2 (m/z = 2), H2O (m/z = 18), and CH3OH (m/z = 31) after interaction of Cu–MOR13.0 with 7 bar CH4 at 473 K, followed by a purge with H2O in He (2.6 vol%, 1 bar, total flow of 40 ml min−1, e). CH3OH yield and selectivity across multiple cycles, each involving a He activation at either 673 K (red line and blue bars) or 473 K (yellow line and green bars), followed by CH4 reaction and then catalyst reactivation by H2O at 473 K (f). Adapted from ref. 88 with permission from Science (Copyright 2017). | ||
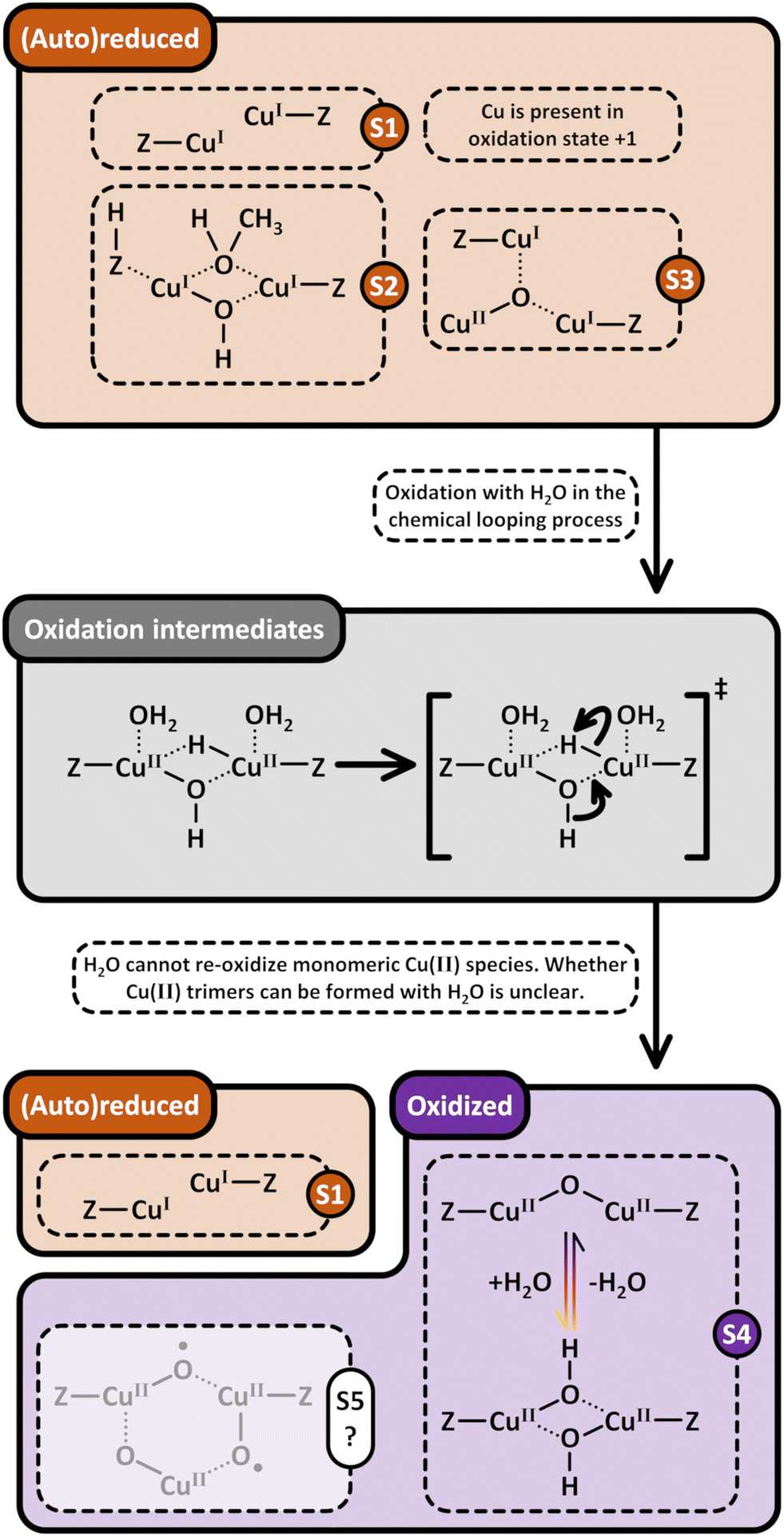 | ||
| Scheme 13 Formation pathways of Cu-oxo centers (violet background) via activation of (auto)reduced Cu–zeolites (brown background) with H2O in the chemical looping process. (Auto)reduced monomeric Cu(II) active sites (S1), such as [CuOH]+/[CuOH]+ or Cu2+/[CuOH]+, cannot be re-oxidized with H2O. Dimeric Cu(II) active sites (S1), (auto)reduced to a Cu(I) pair under high temperature inert conditions or vacuum, can also not be re-oxidized with H2O. The transformation of the Cu(I) pair (S2), which hosts CH3OH, into [Cu2(µ-O)]2+ and [Cu2(µ-OH)2]2+ (S4) proceeds via a hydride intermediate (grey background). Whether H2O is capable of re-oxidizing [Cu3(µ-O)]2+ into [Cu3(µ-O)3]2+ (S5) has not been investigated yet (structure is highlighted by a transparent overlay and marked with a question mark). Dotted bonds correspond to dative interactions. The term “Z” describes the negatively charged zeolite lattice. The color gradient from yellow to violet of the arrows in (S4) indicates the progressively increasing temperature. | ||
The proposed mechanism was verified by using H218O during steam exposure, with the O insertion by H2O into the Cu(I) pair being confirmed by the subsequent catalytic cycle yielding CH318OH, as shown in Fig. 11d.
| 2Cu+ + H2O → [Cu2(µ-O)]2+ + H2 | (7.1) |
The claim that H2O may act as an oxidant for Cu(I) centers in zeolites has, however, proven to be very contentious. One source of scrutiny stems from the re-oxidation of Cu(I) being thermodynamically unfavorable at 473 K under standard conditions (ΔG473K = 130 kJ mol−1), as detailed by Periana.236 While a valid criticism, these values are only applicable to bulk crystalline copper oxides and not the [Cu2(µ-O)]2+ species itself, and do not include any stabilizing effects that additional H2O molecules would exhibit. In response, a more thorough explanation of the anaerobic re-oxidation mechanism was presented by Sushkevich et al.189 Following the formation of [Cu2(µ-O)]2+ from one H2O molecule and a binuclear Cu(I) center, as shown in eqn (7.1), a second H2O molecule would lead to the formation of a [Cu2(µ-OH)2]2+ species, calculated to be significantly more stable than the [Cu2(µ-O)]2+ species (ΔΔG473K = −117 kJ mol−1). A ΔG473K = 58 kJ mol−1 was calculated as the upper limit for the re-oxidation to the [Cu2(µ-OH)2]2+ species. While endergonic, the reaction was suggested to still be able to proceed at 473 K primarily due to the further stabilization effects of additional H2O molecules on Cu(II), as well as the low adsorption of H2 relative to H2O. Both these factors would lead to a significant decrease in ΔG, making the reaction thermodynamically feasible. A high-temperature activation (673 K in He) would result in the [Cu2(µ-O)]2+ active site via a dehydration and unpoisoning of the [Cu2(µ-OH)2]2+ species, as depicted in eqn (7.2).88,202
| [Cu2(µ-OCH3)]+ + H+ + 2H2O → [Cu2(µ-OH)2]2+ + CH3OH + H2 → [Cu2(µ-O)]2+ + H2O | (7.2) |
 | ||
| Fig. 12 Suggested mechanism of [Cu2(µ-O)]2+ and [Cu2(µ-OH)2]2+ formation via oxidation of a Cu(I) pair, hosting CH3OH, with H2O. Adapted from ref. 237 with permission from the American Chemical Society (Copyright 2019). | ||
A first H2O molecule is responsible for the conversion of a CH3O group to molecular CH3OH adsorbed on a [Cu2(µ-OH)]+ species. The Cu oxidation would be initiated by the adsorption of a second H2O molecule, as depicted in Fig. 12. The second H2O molecule transfers a H+ from a proximal BAS, whose own formation was shown to be coupled to CH3O group formation (Fig. 13),88 to the [Cu2(µ-OH)]+ species. This transfer results in the oxidation of both Cu(I) centers of [Cu2(µ-OH)]+, resulting in the formation of a [Cu2(µ-OH)(µ-H)]2+ intermediate. The activation energy for Cu(I) oxidation was calculated at ∼80–105 kJ mol−1, dependent on the number of H2O molecules coordinated to the intermediate. These values are significantly lower than the activation energy of 290 kJ mol−1 as suggested by Mahyuddin et al. (vide supra).158,237 A third H2O molecule would be necessary in the release of H2, leading to a reformation of the [Cu2(µ-O)]2+ active site. However, the formed [Cu2(µ-O)]2+ will be poisoned by transformation into the energetically more favorable [Cu2(µ-OH)2]2+ species via the addition of a fourth H2O molecule. A regeneration of the active site able to activate CH4 would then proceed viaeqn (7.2) by employment of a high-temperature activation procedure. This transformation from Cu(I) pair to [Cu2(µ-O)]2+via the H2O poisoned intermediate [Cu2(µ-OH)2]2+ is further illustrated in Scheme 13. However, it needs to be noted that a definite spectroscopic identification of the [Cu2(µ-OH)2]2+ intermediate is lacking, and therefore the suggested transformation into the [Cu2(µ-O)]2+ active site upon dehydration remains to be proven under the examined conditions.
 | ||
| Fig. 13 Time-resolved in situ FTIR spectra of adsorbed surface species formed during the interaction of Cu–MOR13.0 (pretreated in a flow of He) with 7 bar CH4 at 473 K (a). Relative number of CH3O species vs. number of BASs formed during the interaction of CH4 with Cu–MOR13.0 at 473 K within 5 to 120 min (b). Adapted from ref. 88 with permission from Science (Copyright 2017). | ||
Heyer et al. experimentally investigated the anaerobic Cu(I) oxidation in Cu0.45MOR9.0.238 On the basis of the amount of EPR silent Cu(II) sites, the amount of [Cu2(µ-O)]2+ species in the system was estimated to be 36 ± 14% of the total Cu present in the material. Following an activation in He at 773 K, UV-Vis and Raman spectroscopies determined that solely mononuclear Cu(II) remains oxidized, with the [Cu2(µ-O)]2+ sites in the system having been (auto)reduced.62,238 No Cu(I) to Cu(II) oxidation was observed via in situ XAS when the (auto)reduced material was exposed to H2O at 473 K, the same temperature at which Sushkevich et al. first characterized the anaerobic oxidation of a Cu(I) pair to [Cu2(µ-O)]2+ (vide supra). At first glance, the results of Heyer et al. and Sushkevich et al. may therefore seem contradictory.88,238 A marked difference, however, lies in the fact that Sushkevich et al. performed a full cycle, consisting of the subsequent dosage of O2–CH4–H2O, before examining a re-oxidation of Cu–MOR13.0. Heyer et al. instead investigated the oxidation of an (auto)reduced Cu(I) pair to [Cu2(µ-O)]2+ by H2O in Cu0.45MOR9.0. While this difference may sound inconsequential, the proposed mechanism suggested by Palagin et al.237 requires the transfer of H+ from a proximal BAS to form the necessary [Cu2(µ-OH)(µ-H)]2+ intermediate, with the proximal BAS only forming following a C–H bond cleavage of CH4, as has been determined experimentally (Fig. 13).88 An (auto)reduction procedure will instead lead to no change in the charge balancing ability of Cu toward the framework, and therefore no BAS formation. The high Cu/Al exchange degree of 0.45 also suggests that a low amount of BAS would be present before the (auto)reduction procedure. However, attributing the conflicting observations reported by Sushkevich et al.88 and Heyer et al.238 to the lack of a proximal BAS to a Cu(I) pair entirely relies on the premise that the mechanism proposed by Palagin et al.237 is in fact valid. An alternative source of the discrepancy between these works may instead stem from Cu–MOR samples with different characteristics. Cu–MOR exhibits a variety of [Cu2(µ-O)]2+ active sites, which exhibit different abilities toward being oxidized and (auto)reduced.51,68 It may be entirely possible that one variation of the active site is able to be re-oxidized by H2O under the examined conditions due to favorable geometric effects by, for example, a variation in Al T-site distribution throughout the zeolite lattice. The significant impact that the Al T-site distribution in MOR may have is illustrated by the fact that only certain Al T-site arrangements, as well as the need for non-conventional ion-exchange procedures, allow for the [Cu3(µ-O)3]2+ species to be formed.96 In addition, the in situ XANES spectra of Cu–MOR13.0 recorded during H2O exposure has highlighted that not all Cu(II) reduced via CH4 is able to be re-oxidized by H2O (Fig. 13). While this may also stem from the inability of other active sites to be re-oxidized via H2O exposure (see Section 7.1.2), this may also indicate the inability of certain dimeric Cu(I) motifs to be re-oxidized via H2O exposure.
A further point of criticism regarding the feasibility of H2O as an oxidant for Cu(I) in zeolites has been the source of the molecular H2 formed during re-oxidation. An alternative source of H2 was proposed to stem from a decomposition of HCOOH to CO2 and H2 (ΔG473K = −63 kJ mol−1) over the material.90,238 HCOO− bands were detected by FTIR in the original work by Sushkevich et al.,88 which may lead to HCOOH formation during H2O exposure, which could subsequently decompose into CO2 and H2 over Cu(I).90,238 When Heyer included a CH4 exposure following the activation in He at 773 K (vide supra), the authors observed that CO2 and H2 are produced in a ratio of 1:1 during H2O exposure, a strong indication that the source is from an oxidation of HCOOH (ΔG473K = −63 kJ mol−1) instead of a re-oxidation of Cu(I) pairs.238 Any oxidation of Cu(I) witnessed in the work by Sushkevich et al. under H2O was instead suggested to stem from O2 impurities or potential leaks, or could also stem from a water-gas shift reaction occurring during H2O exposure (ΔG473K = −21 kJ mol−1).90,238 Any presence of O2 will definitely lead to re-oxidation. If these, however, were the sole sources of H2, it would be reasonable for Cu–zeolites solely exhibiting [CuOH]+ sites to also result in H2 formation during H2O exposure. This, however, is not the case, as shown by Sushkevich et al.65 [CuOH]+ sites are suggested not to be able to be re-oxidized via H2O from their Cu(I) precursor (see Section 7.1.2). Cu0.6MOR46.0, assumed to solely exhibit [CuOH]+ sites due to its high Si/Al ratio, was shown not to release H2 during H2O exposure, while FTIR was still able to identify both CO species (2157 cm−1) and HCOO− species (1619 cm−1) during CH4 exposure. If both a decomposition of HCOOH or a water-gas shift reaction are the only potential sources of H2, then it would be only logical that Cu0.6MOR46.0 would also lead to H2 formation. The conflicting results and interpretations of said results emphasize that to make definite statements on the ability, or inability, of H2O to be able to oxidize a Cu(I) motif to result in a [Cu2(µ-O)]2+ species, further investigations will be needed to identify the origin of this divergence in findings reported in this section.
Based on these observations, Sushkevich et al. investigated Cu–MOR at varying Si/Al and Cu/Al ratios, resulting in samples with differing ratios of monomeric vs. dimeric Cu(II) sites.65 A high Si/Al ratio would entail a low fraction of proximal Al T-sites throughout the zeolite lattice, thereby reducing the amount of dimeric Cu species present in MOR. Cu0.6MOR46.0 would therefore result in primarily monomeric Cu(II), with active Cu being present as [CuOH]+. Cu0.38MOR6.5 and Cu0.4MOR10.0 with lower Si/Al and higher Cu/Al would shift the equilibrium toward the formation of dimeric active sites. While the first cycle following a high-temperature activation procedure (673 K in He) resulted in CH3OH production in the case of Cu0.6MOR46.0, no quantifiable CH3OH productivity was observed in a second cycle activated under the same conditions (673 K in He) following H2O exposure to desorb CH3OH. Both Cu0.38MOR6.5 and Cu0.4MOR10.0, however, exhibited CH3OH productivity under the same conditions as Cu0.6MOR46.0. This is a strong indication that in the case of monomeric Cu(I), H2O is not able to re-oxidize Cu and reconstitute the [CuOH]+ species, as depicted in Scheme 13.
Heyer et al. arrived at conclusions similar to those of Sushkevich et al.88,238 As previously mentioned in Section 7.1.1, an initial activation procedure (773 K in He) resulted in solely mononuclear [CuOH]+ remaining in a +2 oxidation state. H2O exposure at 473 K following CH4 exposure at the same temperature led to an increase in absorption in the XANES spectrum at ∼8985 eV, associated with the 1s → 4p transition of Cu(I). An oxidation of Cu(I) to Cu(II) is typically associated with the opposite behavior, namely a decrease in the 1s → 4p transition at ∼8985 eV. This increase was instead suggested to be a spectroscopic fingerprint of a linear two-coordinate Cu(I)-aqua complex. In situ EPR spectroscopy distinguished a Cu species with a gII value of 2.4, attributed to hydrated Cu(II) in zeolites. More importantly, however, no increase in the amount of EPR active Cu was observed, thereby highlighting that no increase in EPR-visible Cu(II), such as [CuOH]+, occurred (Scheme 13).
Contrasting claims regarding the ability of [CuOH]+ to be reconstituted via an oxidation of a mononuclear Cu(I) by H2O have been made by Pereira et al. for Cu-MAZ2.7 samples with Cu/Al ratios ranging from 0.15 to 0.27.239 UV-Vis spectra of Cu0.23MAZ2.7 following O2 exposure at 723 K revealed broad LMCT bands at ∼30000–45000 cm−1, with an absorbance maximum at ∼ 41200 cm−1. A comparison to DFT calculated UV-Vis spectra suggested that these bands could be associated with [CuOH]+. Following a high-temperature activation under either O2 or CO2, the sample was exposed to CH4 at 523 K. A following H2O exposure at 523 K was suggested to re-oxidize Cu(I) to [CuOH]+. To unpoison the active sites, the material was exposed to CO2 at 723 K. The obtained yield of the following cycle was equivalent to the yield of the first cycle. No spectra are provided following H2O exposure, as well as following the consecutive reactivation under CO2 atmosphere at 723 K. As CO2 is considered a potential oxidant for Cu(I) to Cu(II) in zeolites (see Section 8), it is impossible to evaluate which of the two suggested oxidants is responsible for a Cu(I) re-oxidation.
7.2 Formation of Cu(II) sites using H2O during catalytic selective CH4 oxidation
| [Cu2(µ-OH2)]2+ + H2O → [Cu2(µ-OH)2]2+ + H2 →[Cu2(µ-O)]2+ + H2O | (7.3) |
Koishybay et al. compared both anaerobic and aerobic CH4-to-CH3OH conditions for Cu0.33CHA10.0, and observed exclusive formation of CO2 under aerobic conditions.242 The authors assumed that solely non-selective sites, suggested to be [Cu3(µ-O)3]2+ and [Cu2(µ-O)2]2+, would be oxidized by O2 and not H2O. Therefore, selective sites, claimed to be represented by [CuOH]+ and [Cu2(µ-O)]2+ can be reconstituted from their reduced state under anaerobic CH4-to-CH3OH conversion conditions, leading to a selectivity of 100%. Jeong et al., using Cu–MOR without Na+ co-cations (vide supra), also solely observed the formation of CO2 when adding O2 in the feed, however, in tandem with a significant increase in the CH3OH formation rates.240 A low O2 amount (85 ppm) was shown to increase CH3OH productivity by a factor of ∼5.240 However, instead of suggesting that O2 is responsible for a more rapid Cu(I) re-oxidation in comparison to H2O, the authors suggested another entirely different role (see Section 7.2.3).
600 and 16500 cm−1, both attributed to bare Cu2+ by the authors. A decrease in absorbance was observed when the sample was exposed to a mixture of CH4 and H2O above 573 K, which originated from the reduction of Cu2+ sites. FTIR spectroscopy in the region of structural vibrations revealed two bands associated with different Cu species, namely at 900 cm−1, attributed to bare Cu2+, and at ∼950 cm−1, attributed to [CuOH]+. This, however, contrasts their findings using EPR spectroscopy, which, according to the authors, showed the presence of only bare Cu2+, while [CuOH]+ is an EPR visible species as well (see Section 2.1.2). When examining the system under an atmosphere of CH4 and H2O (573–673 K) using in situ FTIR spectroscopy, the band at ∼950 cm−1, previously assigned to a [CuOH]+ species, was instead attributed to a Cu-CH3 species in the 6-MR of CHA. On the basis of these findings, the authors postulated that bare Cu2+ in the 6-MR can be active for CH4 activation. To further support this claim, in situ FTIR spectroscopy of Cu–CHA10.0 in a quasi-looping manner was performed. Following an activation in He at 573–673 K, the sample was exposed to CH4 at the same temperature. A decrease in 900 cm−1 (bare Cu2+), and an increase in 950 cm−1 (Cu–CH3, no oxidation state of Cu suggested) and 2153 cm−1 ([Cu(CO)]+) bands were observed over the examined temperature range. Exposure of the sample to H2O resulted in a decrease in the band at 950 cm−1 and an increase in 900 cm−1. While no mechanistic scheme is presented, this would suggest that the reaction proceeds viaeqn (7.4).| [CuCH3]+ + H+ + H2O → Cu2+ + CH3OH + H2 | (7.4) |
000 cm−1. Based on these interpretations by the authors, the ability of bare Cu2+ to activate CH4, and therefore any re-oxidation of Cu(I) to bare Cu2+ being possible, remains to be proven. In a subsequent publication, Zhang et al. examined Cu0.05CHA10.0, which the authors suggested to solely exhibit bare Cu2+ sites in the 6-MR due to the low Cu/Al ratio.244 Reaction temperatures between 473–773 K were examined, with 673 K exhibiting the highest CH3OH productivity and selectivity (2678 mmolCH3OH molCu−1 h−1, 93% selectivity toward CH3OH). A comparison to Cu–CHA10.0 with higher Cu/Al showed that the site-specific yield at such a low Cu/Al greatly outperformed the other Cu–CHA10.0 catalysts at 673 K. Two species were detected using EPR spectroscopy, both attributed to bare Cu2+ in the 6-MR. Cu K-edge FT-EXAFS of the material revealed a Cu first shell CN of 4 for Cu0.05CHA10.0, which decreased with increasing Cu/Al. The conditions under which the XAS and EPR spectra were recorded were, however, not mentioned. Similar to their previous work,243 both the observed d–d transitions band at ∼13000 cm−1 in the UV-Vis spectra as well as a band at 900 cm−1 in the FTIR spectra were used to identify bare Cu2+ in the 6-MR of CHA as the potential active site. In contrast to previous work, which claimed bare Cu2+ to be the active site (vide supra),243 the authors suggested that in an initial stage, bare Cu(II) would be converted into a [CuOH]+ species through the combination of both H2O and CH4 exposure, as shown by eqn (7.5). | (7.5) |
 | (7.6) |
| CH4 + H2O ↔ CH3OH + H2 | (7.7) |
8. Less common oxidants (NO, CO2, H2O2) for the formation of active Cu-oxo species
8.1. NO
The use of NO as an oxidizing agent has not been explored thoroughly so far. One study by Sheppard et al.85 reported the isothermal looping at 423 K with NO as the oxidizing agent over a Cu0.28MFI12.0, followed by the reaction with CH4 and the extraction with H2O vapor. In a schematic description of the mechanism, the authors assume that NO binds to individual Cu(I) centers and forms N2O as well as a [Cu2(µ-O)]2+ species. Subsequently, the thus formed N2O is further capable of oxidizing another Cu(I) pair. The mechanism is based on the earlier work from Modén et al. on the NO decomposition over Cu-ZSM-5 (Fig. 14).245 | ||
| Fig. 14 Suggested mechanism of [Cu2(µ-O)]2+ formation via oxidation of a Cu(I) pair using NO or in situ generated N2O. Adapted from ref. 85 with permission from the Royal Society of Chemistry (Copyright 2014). | ||
Interestingly, in the same studies, the authors found that the amount of CH3OH extracted from Cu-MFI was 2–6 times greater when H2O vapor was used to perform the extraction compared to when liquid H2O was used. Using in situ UV-Vis spectroscopy, the authors monitored the re-oxidation by following the absorption band at 22000 cm−1. This band, attributed to the [Cu2(μ-O)]2+ active site, disappeared in the following exposure to CH4, demonstrating that the re-oxidized species is active in the CH4-to-CH3OH conversion. Further, the authors revealed that at these low temperatures, only a fraction of Cu species can be re-oxidized, since the intensity of the UV-Vis band was much more intense after high temperature activation in O2 or N2O. No details about the kinetics of monomeric species are provided, as UV-Vis is not able to give detailed insights into their behavior. However, given the fact that only a small fraction of the Cu can be oxidized, it can be speculated that the vast majority of them are dimeric species since their oxidation is generally believed to be faster. The generation of monomeric Cu(II) from Cu(I) with NO has been shown earlier by Lamberti et al.246 In this study, a series of MFI zeolites with low Al content (Si/Al = 180–100) was reacted with gaseous CuCl at 573 K, and an exchange level of 200% was assumed. After exposure to 8 mbar of NO for 10 min and again after evacuation for 60 min at ambient temperature, the EPR signal intensity showed an increase by a factor of 17 and 30, respectively, demonstrating that the oxidation of monomeric centers is possible. Complementary FTIR results indicate that this corresponds to a total amount of 28% of the Cu present in the sample. While the material has not been tested for the conversion of CH4 to CH3OH, it shows that, in principle, monomeric species can be formed. The authors pointed out that the coordination and subsequent oxidation of the Cu(I) sites with NO is complicated by the decomposition reaction of NO into N2O and NO2, which can take place even at ambient temperature.
8.2. CO2
The possibility of employing CO2 as an activation agent has been explored by Pereira et al.239 and Vieira et al.247 Starting from the initial as-prepared/hydrated state, Pereira et al. exposed Cu0.23MAZ2.7 to CO2 at 723 K, with UV-Vis spectra revealing a broad LMCT band around ∼41200 cm−1. A comparison to DFT calculated reference spectra suggested the species exhibiting the broad LMCT band to be [CuOH]+ pairs. A low intensity band at 10000–15000 cm−1 was attributed to d–d transitions of Cu2+. Using said activation procedure in CO2 at 723 K, followed by CH4 and H2O exposure at 523 K, resulted in increasing productivity values as a function of the Cu/Al ratio, with the highest value of 163 µmolCH3OH gZEO−1 achieved in the case of Cu0.27MAZ2.7. Notably, when an (auto)reduction procedure preceded CO2 exposure at 723 K, the productivity significantly decreased. In the case of Cu0.15MAZ2.7 the observed CH3OH productivity dropped from 94 to 43 µmolCH3OH gZEO−1, which would suggest that the species able to be (auto)reduced are not able to be re-oxidized via CO2 exposure. The authors further suggested that the species able to be re-oxidized via CO2 exposure from an (auto)reduced state would be Cu(x>3)Oy species, with [CuOH]+ suggested not to be able to be re-oxidized by CO2 directly. Instead, any LMCT bands attributed to [CuOH]+ observed after a high-temperature activation at 723 K in CO2 in a following cycle were instead attributed to stem from a re-oxidation by H2O during CH3OH extraction (see Section 7.1.2).
Vieira et al.247 also investigated the effect of CO2 on Cu0.1CHA4.7. The authors employed an isothermal looping protocol, using CO2 instead of O2, over a range of temperatures. The highest CH3OH productivity of 132 µmolCH3OH gZEO−1 was observed at 673 K, with however a second consecutively performed cycle leading to a drastic decline in activity to ∼ 40 µmolCH3OH gZEO−1. This stark decrease in productivity was attributed to both coke formation as well as incomplete Cu(I) oxidation. DFT calculations determined a true activation energy for the re-oxidation of a Cu(I) pair to [Cu2(µ-O)]2+ and CO at 259 kJ mol−1, therefore giving credence to the claim of incomplete Cu(I) oxidation. In addition, an (auto)reduction of [CuOH]+ at elevated temperatures under a CO2 atmosphere was suggested to occur. The authors, however, stated that the (auto)reduction of [CuOH]+ would not proceed in the first cycle due to an enhanced stability of [CuOH]+. The source of the enhanced stability in the first cycle was, however, not further elucidated. In comparison, using O2 as the oxidant led to an increase in CH3OH productivity over consecutive cycles at 673 K. When comparing the spectra obtained at 673 K in a CO2 atmosphere in Cu–CHA to the same conditions using O2, a broader band at 40000–45000 cm−1, attributed to both [CuOH]+ and [Cu2(µ-O)]2+ species, was observed. This broadening was assumed to mean a greater variety of the active Cu species following a CO2 activation. A performed peak fitting of the spectra suggested a higher fraction of [CuOH]+ pairs being present under a CO2 atmosphere than under a O2 atmosphere, which could indicate less condensation of [CuOH]+ pairs to [Cu2(µ-O)]2+ at 673 K. Since online MS or similar data are not provided, the role of CO2 remains elusive, especially since the H2O vapor from the product extraction might contribute to the oxidation, as suggested in the previous work of Pereira et al.239
8.3. H2O2
The use of H2O2 as an oxidizing agent for CH4-to-CH3OH conversion is more frequently reported over Fe-zeolites and over Cu-promoted Fe-zeolites.248–252 Recently, Cu–zeolites have been investigated.253–255 The use of H2O2 as an oxidizing agent has often been dismissed because of its higher economic value than CH3OH. However, the reaction still attracts scientific interest, due to its high selectivity and mild conditions. In an early study, Armstrong et al.,253 tested the low-temperature (323 K) conversion of CH4, C2H6, and C3H8 in a batch reactor over Cu-MFI with varying Cu/Al and Si/Al ratios under elevated pressure (30–20 bar). A Cu-MFI yielded 49.86 mmolCH3OH, which equals 96.1% selectivity towards CH3OH and a productivity of 3.7 molCH3OH kgZEO−1 h−1. They further found evidence by NO-FTIR measurements that isolated Cu(II) species characterized by a band at 1913 cm−1 are responsible for the selective conversion, however, the exact structure or sitting site has yet to be revealed. In over-exchanged zeolites, the authors reported an increase in overoxidation, while no further structural details have been provided on the active Cu site. Building on these results, Tang et al.256 reported a Cu-MFI material featuring mostly monomeric Cu(II) species yielding 12000 µmolCH3OH gZEO−1 within 30 min at 343 K. The authors utilized a combination of ex situ XAS, high-angle annular dark-filed scanning transmission electron microscopy (HAADF-STEM), and EPR to characterize the isolated Cu(II) atoms and concluded that it is a bare Cu2+ site coordinated by four Ofw moieties in the 6-MR in the MFI framework. It should, however, be pointed out that the material was hydrated during these spectroscopic experiments, and the formation of species with higher nuclearity during the reaction cannot be ruled out based on these data. They further showed that the low loading, which presumably leads to only bare Cu2+ centers, significantly reduced the overoxidation of reaction intermediates to CO2, which is observed with a reference material with higher Cu loading. This was further explained by DFT calculations that indicated that these proposed bare Cu2+ sites exhibit a uniquely reversed C–H bond activation trend, where the C–H bond of CH4 is more readily activated than that of CH3OH due to entropic and solvation contributions.256 However, experimental evidence for the proposed conversion of CH4-to-CH3OH with H2O2 at an isolated Cu2+ site still needs to be shown. Further, it should be noted that the turnover frequency for these materials is rather low, with up to 340 h-1 and an overall CH4 conversion of below 1%, accompanied by a H2O2 consumption of up to 80%. The reported CH3OH selectivity is about 80%. The ratio between C1 oxygenates and the consumed H2O2 was found to be between 0.2–0.4. Further support for the proposed monomeric site as the active centers comes from a theoretical study by Cheng et al.254 Their theoretical calculations revealed that the O–O bond of H2O2 can be broken to form surface-reactive hydroxyl groups through a H2O-mediated mechanism at mononuclear Cu–zeolites, forming monomeric [Cu(OH)2]+ species (Scheme 14 and Scheme S1z). The authors further performed kinetic analysis and found that the resulting homolytic C–H bond activation mechanism leads to a trade-off between selectivity and activity of these sites, which are also responsible for H2O2 self-decomposition.
 | ||
| Scheme 14 The reaction network of catalytic CH4-to-CH3OH conversion in Cu-MFI based on H2O2 as an oxidant. The Cu(II) active site in cycles (a) and (b) corresponds to [Cu(OH)2]+ or [CuOH]+, respectively. Adapted from ref. 254 with permission from Elsevier (Copyright 2023). | ||
9. Influence of the material's state on the possibility of forming active species
The formation pathways of the most commonly observed Cu active site motifs in Cu-exchanged zeolites, beginning from either the as-prepared/hydrated state (left side of the scheme) or the (auto-)reduced state (right side of the scheme) under various activation conditions employed in the CH4-to-CH3OH conversion, are illustrated in Scheme 15. As emphasized throughout this review, the initial state of the zeolite sample has a profound effect on the type of species formed during the activation procedure. | ||
| Scheme 15 The possibility to form Cu-oxo active sites (violet background) with a certain oxidant in Cu-exchanged zeolites, beginning from either the as-prepared/hydrated state (blue background) or the (auto)reduced state (brown background). Arrows and oxidants highlighted in green, red, and yellow indicate whether the generation of a certain oxygenated Cu(II) species by a specific oxidant is possible, not possible, or has not yet been verified. (Auto)reduced [CuOH]+/[CuOH]+ may exist either as two plain Cu(I) ions (S1), or as a BAS containing site (S2). This distinction is important as, unlike in the case of S2, the generation of [CuOH]+/[CuOH]+ from S1 necessitates an external H+ source. Unlike the reaction of [Cu3(µ-O)2]2+ (S3) with O2, the formation of [Cu3(µ-O)3]2+via interaction of [Cu3(µ-O)]2+ (S4) with O2 does not require a location to accommodate the second O moiety. Dotted bonds correspond to dative interactions. The term “Z” describes the negatively charged zeolite lattice. | ||
9.1 As-prepared/hydrated state
The as-prepared/hydrated state on the left side of Scheme 15 represents a common starting point for materials evaluation and spectroscopic investigations. In this state, the Cu(II) species exist primarily as [Cu(H2O)6]2+ and [Cu(OH)(H2O)5]+. Subjecting hydrated samples to heat and gas flow or vacuum will first lead to a partial dehydration of the Cu(II) ions and subsequently to their attachment to the zeolite framework, where the respective active sites are formed, as demonstrated by spectroscopic interrogation and quantitative mechanical modelling.45Samples used in their as-prepared/hydrated state without calcination exhibit markedly different activation behavior, particularly under inert conditions or vacuum, compared to properly calcined samples due to the presence of organic residues. The influence of sample pretreatment has been, for example, shown by Sushkevich et al.101 The authors employed in situ XANES spectroscopy to compare the temperature-induced Cu(I) generation in as-prepared/hydrated vs. pre-calcined Cu–MOR during treatment in He from 300 to 1100 K. A pronounced increase in the Cu(I) fraction at approximately 495 K was observed in the as-prepared/hydrated material, whereas temperatures above 650 K were necessary to induce noticeable decreases in Cu(II) in the pre-calcined samples. Exposing uncalcined samples to a heat treatment under an inert atmosphere or vacuum may therefore entail a substantial reduction of Cu(II) to Cu(I). This phenomenon has at times been misinterpreted to be a result of (auto)reduction.98,99 This reduction coincides with the formation of radical coke species, as evidenced in the EPR spectra, providing proof that carbonaceous residues are responsible for the reduction of Cu(II) during thermal treatment under inert conditions.
9.2 (Auto)reduced state
Starting from a fully (auto)reduced material (e.g., after CH4 treatment), oxidizing agents are necessary for the reformation of all Cu-oxo active sites, whereas treatment under inert conditions, e.g., He or vacuum, will not lead to the generation of active sites as indicated on the right-hand side of the Scheme 15.000–39000 cm−1, thereby showing that the trimeric species was able to be reconstituted from a reduced state.
10. The role of H2O in Cu–zeolite for CH4-to-CH3OH conversion
As outlined in previous sections, especially in Section 7, the formation of active Cu(II) sites in zeolites for CH4-to-CH3OH conversion is highly influenced by the presence of H2O, independent of the activation and starting conditions employed. However, the exact role, especially in continuous catalytic CH4-to-CH3OH conversion, remains a subject of debate. In the following, the different roles of H2O are outlined besides its discussed role as an oxidizing agent.10.1 H2O as an inhibitor of active sites
Complete dehydration of Cu–zeolite materials is particularly important to achieve high activity, as the strongly hydrophilic Cu-oxo species readily adsorb H2O. This adsorption saturates the coordination sphere of the Cu(II) active sites, rendering them inaccessible to CH4 and effectively poisoning them. Sushkevich et al. emphasized this phenomenon by combining FTIR spectroscopy with NO probe molecule adsorption, demonstrating that the interaction of even a single H2O molecule with [Cu2(μ-O)]2+ and [CuOH]+ species is sufficient to completely deactivate these active centers when a low CH4 partial pressure is employed (see Section 4.3).202 The impact of incomplete dehydration was further illustrated by Tomkins et al., who compared the CH3OH output of Cu–MOR during interaction with 1 bar CH4 at 473 K after activation in 1 bar O2 at either 723 K for 4 h or 473 K for 13 h.17 The authors observed a significant decrease in CH3OH productivity from approximately 45.3 to 2.1 µmolCH3OH gZEO−1 with the isothermal looping process, likely due to incomplete dehydration. The same was observed by Wieser et al. in the case of Cu-MAZ using the isothermal looping procedure.111,205 When increasing the cycling temperature from 473 to 548 K, a significant increase in yield from ∼75 to 275 µmolCH3OH gZEO−1 per cycle was observed. A LCF analysis of the spectra collected via in situ XANES spectroscopy further revealed a significant decrease in the fraction of hydrated Cu(II) during O2 exposure when increasing the cycling temperature. Although elevated CH4 pressures can partially mitigate the inhibitory effect of competitive H2O adsorption, the disparity in CH3OH output between the two procedures generally persists.17,201,203,20510.2 H2O-induced enhancement of CH3OH productivity
A distinction must be made between the documented poisoning effect of H2O on active sites and its role in enhancing CH3OH productivity across sequential redox cycles (see Section 4.2.6). Bozbag et al. reported that the CH3OH productivity of Cu–MOR increased by approximately 30% between the first and second cycle and remained relatively constant thereafter.199 This enhancement was attributed to facilitated Cu(II) active site formation during the second calcination period, resulting from material hydration throughout the aqueous CH3OH extraction of the first cycle. Similar productivity enhancements over multiple redox cycles have been reported by Pappas et al. for Cu-FER and Cu–CHA.89,169 These improvements were attributed to hydration-induced displacement of residual Cu(II) species from redox-inert positions during CH3OH extraction and their subsequent transformation into oxygenated Cu(II) active centers during O2 treatment in the following reaction cycle. Inactive Cu(II) species usually correspond to isolated bare Cu2+ ions, and the formation of these Cu(II) centers is characterized by a significantly lower Gibbs free energy compared to that of any active mono- or multimeric Cu-oxo species under the conditions of Cu–zeolite activation. Therefore, the driving force for this redistribution phenomenon is not clear and deserves further attention.103 Interestingly, a drop in the CH3OH output of Cu-MAZ by ∼40% during four consecutive cycles has been reported by Wieser et al. and Knorpp et al., which was associated with a gradual migration of active Cu(II) species from the 8-MR channel to an inert position in the 6-MR.111,200 The origin of this rearrangement phenomenon was related to the extensive dehydration of the material throughout activation at 723 K under an O2 atmosphere, which was proposed to enable the transfer of Cu between the different ion exchange positions. Notably, the migration of Cu(I) ions should not be influenced by H2O, since they exhibit diminishing affinity to H2O ligands, which makes migration during activation in the presence of an oxidation agent more likely. Nevertheless, it remains uncertain why this migration occurs over multiple redox cycles and is not complete after the very first activation from the hydrated or reduced state. As discussed above, for an accurate description of the effects related to Cu migration, a detailed analysis of the Cu speciation after each cycle is required to quantify the effect and evaluate the significance of these rearrangements, and whether they can explain the changes in activity. This is further complicated by the influence of H2O on the spectroscopic signatures of the active sites.10.3 Spectroscopic identification
Contact with H2O not only inhibits catalytic activity but also suppresses the characteristic spectroscopic UV-Vis fingerprints of active dimeric Cu(II) sites, complicating the identification of existing species. Smeets et al. observed the reversible disappearance of the UV-Vis band at 22700 cm−1, associated with the active [Cu2(μ-O)]2+ center in Cu-MFI, upon switching between H2O-saturated N2O streams at 637 and 723 K and pure N2O flow.207 Similarly, Brenig et al. observed decreased absorbance of the LMCT bands of two different [Cu2(μ-O)]2+ motifs at ∼26700 and ∼22400 cm−1 in the UV-Vis spectrum of calcined and O2-treated Cu–CHA during interaction with H2O at 473 K.127 This suggests that intact [Cu2(μ-O)]2+ centers could be present after calcination at 473 K but remain undetectable via UV-Vis spectroscopy due to high residual H2O content. Similarly, UV-Vis spectra from Sheppard et al. demonstrated the disappearance of the band at 22000 cm−1, attributed to [Cu2(μ-O)]2+ species, after switching from NO to H2O containing gas feed in Cu0.8MFI12.0.85 Remarkably, the introduction of NO after H2O exposure and purging with He led to the appearance of a weak UV-Vis signal despite incomplete dehydration in the same study. Similarly, the exposure to H2O is expected to alter the appearance of the EPR signal of the monomeric species. Even though no systematic study exists in which the EPR signal of activated zeolites was recorded upon dosing traces of H2O, the fact that the EPR signal is very different prior to dehydration indicates that H2O will have an effect on the EPR signal. These limitations pose significant challenges for identifying active species in the presence of H2O, particularly in catalytic CH4-to-CH3OH conversion and even more severe in liquid-phase processes employing H2O2 as an oxidizing agent. Under these conditions, the absence of UV-Vis fingerprints for active dimeric cores is not sufficient evidence to rule out their presence. For example, Narsimhan et al. exposed (auto)reduced Cu-MFI to an O2/CH4/H2O feed and observed the onset of steady-state CH3OH production. However, they ruled out the presence of [Cu2(μ-O)]2+ since complementary UV-Vis spectra collected throughout the induction period did not display a pronounced LMCT band at ∼22700 cm−1.209 Conversely, in a subsequent study by the same research group, [Cu2(μ-O)]2+ was identified as the Cu(II) active site for catalytic CH4 partial oxidation in Cu–CHA through analysis of EXAFS spectra collected at 543 K under steady-state CH3OH production.212 The extent to which species observed under dry conditions prevail under wet conditions remains debated, with theoretical investigations instead suggesting the emergence of distinct different species under wet conditions.
In that line, a [(HO)Cu(μ-OH)Cu(O)]2+ core has been proposed as the active species in partially hydrated Cu–zeolites by Yumura et al., who studied the interaction of O2 and H2O with reduced Cu-MFI using DFT.208 This species originates from a [Cu2(trans-μ-1,2-O2)(H2O)]2+ intermediate, which is transformed into [Cu2(trans-μ-1,2-OOH)(OH)]2+via H atom transfer from the H2O ligand onto the bridging O2 moiety. Further, another H atom transfer and simultaneous O-OH bond cleavage result in a [Cu2(μ-O)(OH)2]2+ species, which is the direct precursor for the active site. Similarly, Ipek et al. suggested this same species during continuous CH4-to-CH3OH conversion with N2O as an oxidizing agent, and theoretical calculations indicated that the site could cleave the C–H bond of CH4 more efficiently than the [Cu2(μ-O)]2+ species.13,228 However, experimental evidence for such a dimeric structure remains elusive. The first experimental identification of an active site during the cyclic, isothermal CH4-to-CH3OH conversion is a paired [CuOH]+ site in Cu-MAZ.111 Currently, it remains unclear whether dimeric and trimeric Cu-oxo centers can develop under the activation conditions employed in isothermal CH4 hydroxylation and whether their structures resemble those generated during high-temperature calcination in non-isothermal CH4 hydroxylation. Experimental evidence from EPR measurements of fully hydrated samples indicates that all or at least an overwhelming majority of the Cu(II) within the framework are present as monomeric species, raising questions about the formation of multimeric sites, at least in fully hydrated samples.43,46
10.4 H2O in liquid-phase H2O2 oxidation
The role of H2O becomes even less clear, and experimental evidence is scarcer, for active Cu(II) species existing in the liquid phase during H2O2 oxidation (see Section 8.3). Typically, materials are analyzed prior to catalytic testing in either the hydrated state or after dehydration, and spectroscopic signatures are assigned to distinct species by analogy to literature focusing on dehydrated species. However, these species might not be present in this composition under actual reaction conditions.A notable exception is a study by Yashnik et al., who employed mixed Fe-/Cu–zeolites and recorded ex situ UV-Vis spectra at specific points during the reaction, suggesting the presence of both monomeric and dimeric Cu(II) species.257 Their UV-Vis measurements indicated the existence of terminal peroxo complexes (Cu–OOH, LMCT at 36000 cm−1) originating from isolated Cu(II) ions in the presence of H2O2, as well as binuclear Cu(II) peroxo complexes connected via terminal OOH groups (LMCT at 20700 cm−1) or bridging OO groups (LMCT at 26700 cm−1). It should be noted, however, that the samples were measured ex situ, and contributions of Fe(III) species were not accounted for in the UV-Vis spectra, raising questions about the transferability of these results to pure Cu–zeolite materials.
Theoretical investigations by Cheng et al. demonstrated that the H2O2 O–O bond could be readily cleaved through an H2O-mediated mechanism at monomeric Cu(II) sites in zeolites to form reactive [Cu(OH)2]+.254 These sites were proposed to be responsible for C–H bond activation at low temperatures, overoxidation of CH3OH, and the self-decomposition of H2O2. Tang et al. also proposed monomeric Cu2+ sites attached solely to framework O atoms within the 6-MR as active sites even in the presence of H2O.256 This assignment, however, is in contrast to the EPR spectra of hydrated materials, which usually show no indication of a framework-bound species.43,45,46
11. The need for examination of Cu(I) speciation
As outlined in this review, substantial work has been conducted to elucidate the structural characteristics of the Cu(II) sites present in zeolitic hosts. The complexity of this task is highlighted by the ongoing debates regarding the exact location and configuration of various species. However, when compared to the knowledge gained on Cu(II) sites, the current insights into the nature of Cu(I) species are extremely limited. Very few studies have investigated Cu(I) speciation in Cu-exchanged zeolites. Notable exceptions include work by Dědeček et al. and Wichterlová et al., who utilized NO-FTIR and PL spectroscopy to provide indirect information about Cu(I) speciation and demonstrate correlations between different Cu(I) and Cu(II) sites.44,258 These investigations showed the existence of at least four distinct Cu(I) species, revealing that the speciation of Cu(I) sites exhibits a complexity and variety comparable to that of Cu(II) sites. Furthermore, complementary EPR measurements using NO as a probe molecule confirmed the presence of multiple Cu(I) sites in MFI and MCM-22 zeolites. This was evidenced by the formation of Cu(I)–NO adsorption complexes originating from two distinct Cu(I) sites within the zeolite framework, each coordinated by different numbers of framework oxygen ligands.259–261 Further work on the photocatalytic decomposition of NO over Cu(I) sites has revealed three distinct Cu(I) species by synchronous PL spectroscopy in MFI, including two monomers, previously identified with NO-EPR, and one Cu(I)–Cu(I) dimer structure.262 Further evidence for a distinct Cu(I) speciation and a resulting difference in reactivity comes from early work on photocatalytic decomposition of N2O. For example Anpo et al. investigated the reaction of N2O with Cu(I) species in Cu-FAU and found that dependent on the Si/Al ratio, two distinct isolated linear coordinated Cu(I) monomers as well as a dimer can exist and concluded that monomeric Cu(I) will be the more active of the two in the photocatalytic N2O decomposition at ambient temperature.263 Yamashita et al. investigated Cu–MOR14.9 with a weight loading of 1.9 wt% Cu, and by the utilization of CO probe molecules and XAS experiments revealed that isolated Cu(I) monomers with planar three-fold or linear two-fold coordinate geometry exist within the framework.264 Wieser et al. were able to pinpoint the exact locations of Cu during CH4 exposure with in situ AXRPD.111 While no verification of the oxidation state was delivered, Rietveld analysis was able to identify a CH3O species bound to a Cu moiety, thereby implying that a reduction to Cu(I) during CH4 exposure had occurred. In general, however, the exact location and structural arrangement of Cu(I) centers remain largely unresolved. Since Cu(I) species serve as precursors for both bare Cu(II) and active Cu(II)-oxo sites, their structural elucidation is a prerequisite to gaining mechanistic insights regarding Cu(II) formation. Therefore, methods suitable for direct structural characterization, such as advanced synchrotron techniques including EXAFS, AXRPD, or pair distribution function (PDF), should be applied. Additionally, complementary studies employing probe molecules and indirect spectroscopic methods, such as PL, NO-EPR, and NO-FTIR, reinforced by computational modeling, can contribute invaluable information and may allow for unambiguous identification of the respective location. Starting with materials with very low Cu loading and probing the reducibility of bare Cu2+ sites with CH4, the Cu(I) speciation can be varied by increasing Cu/Al and changing the Si/Al ratio. This approach can establish a path toward a comprehensive understanding of these critical precursors to active sites in Cu-exchanged zeolites for CH4-to-CH3OH conversion.12. Conclusion and outlook
Cu(II) species can form within zeolitic matrices, ranging from isolated bare Cu2+ and [CuOH]+ ions over a variety of different dimeric species, including [Cu2(μ-O)]2+ to trimeric [Cu3(µ-O)3]2+ species. The local environment of the zeolite topology, defined by the Al content and distribution throughout the framework, governs the type and the proximity of Cu(II) centers and therefore the redox activity and stability of these species. Their (re)generation is determined by complex (inter-)dependencies, which include the initial state of the material, the zeolite framework properties, as well as exposure temperature and duration to a given activation process.As demonstrated above, the starting condition of the material and the pretreatment play significant roles in the formation of the active Cu sites and, therefore, in the material's performance in CH4-to-CH3OH conversion. Unfortunately, in various studies, the starting conditions of the material are ill-defined, leading to ambiguities in interpretation and conflicting results. This also holds for the reproducibility test and the influence of the H2O steaming or wet extraction, which has been claimed to have various influences on the materials' performance. Authors need to be clearer about the starting states of their materials and catalysts at the beginning of each cycle to be able to unravel the source of the large number of conflicting reports in the literature. Systematic studies from well-defined states, such as carefully calcined hydrated samples or after an in situ calcination, will help clarify the influence of the material's state on its performance in the CH4-to-CH3OH conversion and lead to more consistent results. Importantly, the Cu speciation as well as the presence of carbonaceous residues need to be assessed prior to every cycle. Since no single spectroscopic technique is capable of providing complete insights, a combination of various methods, including but not limited to UV-Vis, EPR, XANES, FTIR, and Raman, is necessary to identify the relative presence of monomeric, dimeric, and multimeric species in a given sample. While in situ and operando measurements provide important information about the state of the samples, ex situ measurements of carefully prepared sealed samples can also contribute greatly. Furthermore, the influence of CHx and H2O on the active site formation needs to be assessed. One potential approach could be by using different reducing agents than CH4, such as CO and H2. Employing CO would preclude the formation of H2O, while H2 would preclude the formation of CHx species after careful pretreatment. A comparison of the different states could allow for further studies to unravel both the influence of CHx and H2O on active site formation.
Besides the importance of the material's state, as indicated in the Scheme 15, several open questions regarding the formation of specific sites under certain oxidation conditions persist. This includes a thorough investigation of the formation of [CuOH]+ upon exposure of reduced material to NO and N2O. This question is especially important to study in the present and absence of H2O as a potential proton source. Additional questions remain about the relative fraction of various Cu species after exposure to N2O and O2. Despite the belief that both oxidants form isostructural Cu-oxo sites, the activation of Cu–zeolites using either O2 or N2O proceeds through fundamentally different pathways, resulting in distinct Cu-oxo species with varying methanol productivity in the CH4-to-CH3OH conversion. Importantly, O2 activation requires accommodating the second oxygen atom from O–O bond cleavage at nearby Cu(I) sites, potentially limiting the formation of isolated [Cu2(μ-O)]2+ centers. Future research should focus on in situ characterization techniques that can directly observe the formation and transformation of active sites under reaction conditions, such as XANES or FTIR. For example, treatment at different temperatures in N2O and O2 could resolve the structural rearrangements that occur during the transformation of Cu(I) into active Cu(II) centers. Facilitating the identification and structural characterization of potential active site precursors.
Similarly, the active site identification and structural characterization of species existing in the presence of H2O in the isothermal (catalytic) schemes deserves some attention. While different sites such as the [(HO)Cu(μ-OH)Cu(O)]2+ core have been proposed, which could theoretically be active in the CH4-to-CH3OH, a detailed spectroscopic characterization is missing. A major factor for the lack of properly characterized active sites in the isothermal (catalytic) scheme is that a structural characterization remains challenging, especially because UV-Vis bands tend to diminish in the presence of H2O. Therefore, a set of spectroscopic tools needs to be developed that can trace the fate of Cu motifs under catalytic conditions, with high accuracy, to reveal the active fraction. It can be expected that only a small fraction of the present Cu(II) will be active, since the majority will exhibit a H2O coordination shell. This problem also extends to the liquid state and the use of H2O2, where the characterization of the potential Cu(II) active sites is even more difficult. The extraction of spectroscopic fingerprints of species that are obscured by spectator species can be done by applying spectroscopic methods under the paradigm of modulation-excitation with phase-resolved detection (MES-PSD).265–268 MES-PSD is a transient response method that relies on periodic perturbation of the system under investigation. This perturbation can be achieved by modulation of the gas feed of the reaction. After data treatment, the resulting spectra in the phase domain exclusively show those species undergoing reversible changes with the same frequency as the applied perturbation. The background, as well as static signals arising from spectator sites, are sufficiently suppressed. Variation in the gas feed between O2/N2O, H2O, and reducing agent, while monitoring the material with XANES, FTIR, or EPR, could lead to the identification of the spectroscopic fingerprints of the truly active species and subsequently to their full characterization. In situ AXRPD has also proven to be a very powerful technique, and is not restricted to any particular temperature range. A multimodal operational scheme of combining AXRPD with spectroscopic techniques, such as XANES and UV-Vis spectroscopy, may also prove to be a useful tool for unraveling the different Cu sites in a given zeolite framework.
In conclusion, this review summarizes the significant progress and also some persistent lack thereof made over the last decades in understanding the formation of the active Cu(II) site in CH4-to-CH3OH conversion and highlights common misconceptions and knowledge gaps. Common assumptions about Cu(II) site formation are challenged, and a more holistic view of the CH4-to-CH3OH, including the active site formation and Cu(I) speciation, is encouraged. Only by understanding and controlling all of these steps can a given material's performance be understood, and an efficient, selective, and scalable CH4 oxidation can be achieved.
Author contributions
J. W. A. F. conceptualization, writing the original draft, review, and editing. A. B. conceptualization, writing the original draft, review, and editing. J. W. conceptualization, writing the original draft, review, and editing. J. A. B. review and editing. V. L. S. conceptualization, supervision, review, and editing. All authors contributed to the editing of the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software, or code have been included, and no new data were generated or analyzed as part of this review.Acknowledgements
J. W. A. F. gratefully acknowledges support through a JSPS Postdoctoral Fellowship (PE24052). J. W. A. F., A. B., J. A. vB. and V. L. S. thank ETH Research Grant ETH-48 20–1.References
- S. Thomas and R. A. Dawe, Review of ways to transport natural gas energy from countries which do not need the gas for domestic use, Energy, 2003, 28, 1461–1477 CrossRef CAS.
- Z. Zakaria and S. K. Kamarudin, Direct conversion technologies of methane to methanol: An overview, Renewable Sustainable Energy Rev., 2016, 65, 250–261 CrossRef CAS.
- U. Olsbye, S. Svelle, M. Bjørgen, P. Beato, T. V. W. Janssens, F. Joensen, S. Bordiga and K. P. Lillerud, Conversion of Methanol to Hydrocarbons: How Zeolite Cavity and Pore Size Controls Product Selectivity, Angew. Chem., Int. Ed., 2012, 51, 5810–5831 CrossRef CAS PubMed.
- D. J. Wilhelm, D. R. Simbeck, A. D. Karp and R. L. Dickenson, Syngas production for gas-to-liquids applications: technologies, issues and outlook, Fuel Process. Technol., 2001, 71, 139–148 CrossRef CAS.
- R. J. Bunting, P. S. Rice, J. Thompson and P. Hu, Investigating the innate selectivity issues of methane to methanol: consideration of an aqueous environment, Chem. Sci., 2021, 12, 4443–4449 RSC.
- V. I. Sobolev, K. A. Dubkov, O. V. Panna and G. I. Panov, Selective oxidation of methane to methanol on a FeZSM-5 surface, Catal. Today, 1995, 24, 251–252 CrossRef CAS.
- M. H. Groothaert, P. J. Smeets, B. F. Sels, P. A. Jacobs and R. A. Schoonheydt, Selective Oxidation of Methane by the Bis(μ-oxo)dicopper Core Stabilized on ZSM-5 and Mordenite Zeolites, J. Am. Chem. Soc., 2005, 127, 1394–1395 CrossRef CAS PubMed.
- K. T. Dinh, M. M. Sullivan, P. Serna, R. J. Meyer, M. Dincă and Y. Román-Leshkov, Viewpoint on the Partial Oxidation of Methane to Methanol Using Cu- and Fe-Exchanged Zeolites, ACS Catal., 2018, 8, 8306–8313 CrossRef CAS.
- B. E. R. Snyder, M. L. Bols, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Iron and Copper Active Sites in Zeolites and Their Correlation to Metalloenzymes, Chem. Rev., 2018, 118, 2718–2768 CrossRef CAS PubMed.
- J.-P. Lange and P. J. A. Tijm, Processes for converting methane to liquid fuels: Economic screening through energy management, Chem. Eng. Sci., 1996, 51, 2379–2387 CrossRef CAS.
- J.-P. Lange, Catalysis for biorefineries – performance criteria for industrial operation, Catal. Sci. Technol., 2016, 6, 4759–4767 RSC.
- M. Ravi, M. Ranocchiari and J. A. van Bokhoven, The Direct Catalytic Oxidation of Methane to Methanol—A Critical Assessment, Angew. Chem., Int. Ed., 2017, 56, 16464–16483 CrossRef CAS PubMed.
- B. Ipek and R. F. Lobo, Catalytic conversion of methane to methanol on Cu-SSZ-13 using N2O as oxidant, Chem. Commun., 2016, 52, 13401–13404 RSC.
- M. B. Park, S. H. Ahn, A. Mansouri, M. Ranocchiari and J. A. van Bokhoven, Comparative Study of Diverse Copper Zeolites for the Conversion of Methane into Methanol, ChemCatChem, 2017, 9, 3705–3713 CrossRef CAS.
- R. Oord, J. E. Schmidt and B. M. Weckhuysen, Methane-to-methanol conversion over zeolite Cu-SSZ-13, and its comparison with the selective catalytic reduction of NOx with NH3, Catal. Sci. Technol., 2018, 8, 1028–1038 RSC.
- D. K. Pappas, E. Borfecchia, M. Dyballa, I. A. Pankin, K. A. Lomachenko, A. Martini, M. Signorile, S. Teketel, B. Arstad, G. Berlier, C. Lamberti, S. Bordiga, U. Olsbye, K. P. Lillerud, S. Svelle and P. Beato, Methane to Methanol: Structure–Activity Relationships for Cu-CHA, J. Am. Chem. Soc., 2017, 139, 14961–14975 CrossRef CAS PubMed.
- P. Tomkins, A. Mansouri, S. E. Bozbag, F. Krumeich, M. B. Park, E. M. C. Alayon, M. Ranocchiari and J. A. van Bokhoven, Isothermal Cyclic Conversion of Methane into Methanol over Copper-Exchanged Zeolite at Low Temperature, Angew. Chem., Int. Ed., 2016, 55, 5467–5471 CrossRef CAS PubMed.
- J. Zheng, I. Lee, E. Khramenkova, M. Wang, B. Peng, O. Y. Gutiérrez, J. L. Fulton, D. M. Camaioni, R. Khare, A. Jentys, G. L. Haller, E. A. Pidko, M. Sanchez-Sanchez and J. A. Lercher, Importance of Methane Chemical Potential for Its Conversion to Methanol on Cu-Exchanged Mordenite, Chem. – Eur. J., 2020, 26, 7563–7567 CrossRef CAS PubMed.
- T. Ikuno, S. Grundner, A. Jentys, G. Li, E. Pidko, J. Fulton, M. Sanchez-Sanchez and J. A. Lercher, Formation of Active Cu-oxo Clusters for Methane Oxidation in Cu-Exchanged Mordenite, J. Phys. Chem. C, 2019, 123, 8759–8769 CrossRef CAS.
- B. Ipek, M. J. Wulfers, H. Kim, F. Göltl, I. Hermans, J. P. Smith, K. S. Booksh, C. M. Brown and R. F. Lobo, Formation of [Cu2O2]2+ and [Cu2O]2+ toward C–H Bond Activation in Cu-SSZ-13 and Cu-SSZ-39, ACS Catal., 2017, 7, 4291–4303 CrossRef CAS.
- Y. Kim, T. Y. Kim, H. Lee and J. Yi, Distinct activation of Cu-MOR for direct oxidation of methane to methanol, Chem. Commun., 2017, 53, 4116–4119 RSC.
- D. T. Bregante, L. N. Wilcox, C. Liu, C. Paolucci, R. Gounder and D. W. Flaherty, Dioxygen Activation Kinetics over Distinct Cu Site Types in Cu-Chabazite Zeolites, ACS Catal., 2021, 11, 11873–11884 CrossRef CAS.
- M. Álvarez, P. Marín and S. Ordóñez, Direct oxidation of methane to methanol over Cu-zeolites at mild conditions, Mol. Catal, 2020, 487, 110886 Search PubMed.
- C. Colella and W. S. Wise, The IZA Handbook of Natural Zeolites: A tool of knowledge on the most important family of porous minerals, Microporous Mesoporous Mater., 2014, 189, 4–10 CrossRef CAS.
- Christian Baerlocher, Lynne McCusker, Database of Zeolite Structures, can be found under https://www.iza-structure.org/databases/, (accessed 23 May 2025), n.d.
- W. Loewenstein, The distribution of aluminum in the tetrahedra of silicates and aluminates, Am. Mineral., 1954, 39, 92–96 CAS.
- M. Sato, K. Maeda and K. Hirasawa, in Studies in Surface Science and Catalysis, ed. J. Weitkamp, H. G. Karge, H. Pfeifer and W. Hölderich, Elsevier, 1994, pp. 589–596 Search PubMed.
- V. Gábová, J. Dědeček and J. Čejka, Control of Al distribution in ZSM-5 by conditions of zeolite synthesis, Chem. Commun., 2003, 1196–1197 RSC.
- D. E. Perea, I. Arslan, J. Liu, Z. Ristanović, L. Kovarik, B. W. Arey, J. A. Lercher, S. R. Bare and B. M. Weckhuysen, Determining the location and nearest neighbours of aluminium in zeolites with atom probe tomography, Nat. Commun., 2015, 6, 7589 CrossRef PubMed.
- O. H. Han, C.-S. Kim and S. B. Hong, Direct Evidence for the Nonrandom Nature of Al Substitution in Zeolite ZSM-5: An Investigation by 27Al MAS and MQ MAS NMR, Angew. Chem., Int. Ed., 2002, 41, 469–472 CrossRef CAS PubMed.
- R. Szostak, Molecular Sieves: Principles of Synthesis and Identification, Springer Netherlands, Dordrecht, 1989 Search PubMed.
- M. B. Z. Gili and M. T. Conato, Synthesis and characterization of mordenite-type zeolites with varying Si/Al ratio, Mater. Res. Express, 2018, 6, 015515 CrossRef.
- S. Ghojavand, E. B. Clatworthy, A. Vicente, E. Dib, V. Ruaux, M. Debost, J. El Fallah and S. Mintova, The role of mixed alkali metal cations on the formation of nanosized CHA zeolite from colloidal precursor suspension, J. Colloid Interface Sci., 2021, 604, 350–357 CrossRef CAS PubMed.
- Q.-F. Lin, Z. R. Gao, C. Lin, S. Zhang, J. Chen, Z. Li, X. Liu, W. Fan, J. Li, X. Chen, M. A. Camblor and F.-J. Chen, A stable aluminosilicate zeolite with intersecting three-dimensional extra-large pores, Science, 2021, 374, 1605–1608 CrossRef CAS PubMed.
- E. Jang, L. Choi, J. Kim, Y. Jeong, H. Baik, C. Y. Kang, C. H. Kim, K.-Y. Lee and J. Choi, A copper-impregnated BEA zeolite for adsorption and oxidation of aromatic species during vehicle cold starts, Appl. Catal., B, 2021, 287, 119951 CrossRef CAS.
- Z. Li, K. Xie and R. C. T. Slade, Studies of the interaction between CuCl and HY zeolite for preparing heterogeneous CuI catalyst, Appl. Catal., A, 2001, 209, 107–115 CrossRef CAS.
- R. G. Moore and J. M. Crawford, Isolated and Paired Metal Sites in Zeolites Using Solid-State Ion Exchange, Angew. Chem., Int. Ed., 2025, 64, e202505186 CrossRef CAS PubMed.
- Y. Xin, Q. Li and Z. Zhang, Zeolitic Materials for DeNO Selective Catalytic Reduction, ChemCatChem, 2018, 10, 29–41 CrossRef CAS.
- F. Gao, E. D. Walter, N. M. Washton, J. Szanyi and C. H. F. Peden, Synthesis and evaluation of Cu/SAPO-34 catalysts for NH3-SCR 2: Solid-state ion exchange and one-pot synthesis, Appl. Catal., B, 2015, 162, 501–514 CrossRef CAS.
- D. Wang, F. Gao, C. H. F. Peden, J. Li, K. Kamasamudram and W. S. Epling, Selective Catalytic Reduction of NO with NH3 over a Cu-SSZ-13 Catalyst Prepared by a Solid-State Ion-Exchange Method, ChemCatChem, 2014, 6, 1579–1583 CrossRef.
- L. Ren, Y. Zhang, S. Zeng, L. Zhu, Q. Sun, H. Zhang, C. Yang, X. Meng, X. Yang and F.-S. Xiao, Design and Synthesis of a Catalytically Active Cu-SSZ-13 Zeolite from a Copper-Amine Complex Template, Chin. J. Catal., 2012, 33, 92–105 CrossRef CAS.
- A. V. Kucherov, A. A. Slinkin, D. A. Kondrat’ev, T. N. Bondarenko, A. M. Rubinstein and Kh. M. Minachev, Cu2+-cation location and reactivity in mordenite and ZSM-5: e.s.r.-study, Zeolites, 1985, 5, 320–324 CrossRef CAS.
- A. Godiksen, F. N. Stappen, P. N. R. Vennestrøm, F. Giordanino, S. B. Rasmussen, L. F. Lundegaard and S. Mossin, Coordination Environment of Copper Sites in Cu-CHA Zeolite Investigated by Electron Paramagnetic Resonance, J. Phys. Chem. C, 2014, 118, 23126–23138 CrossRef CAS.
- J. Dedecek, Z. Sobalik, Z. Tvaruazkova, D. Kaucky and B. Wichterlova, Coordination of Cu Ions in High-Silica Zeolite Matrixes. Cu+ Photoluminescence, IR of NO Adsorbed on Cu2+, and Cu2+ ESR Study, J. Phys. Chem., 1995, 99, 16327–16337 CrossRef CAS.
- P. C. Bruzzese, E. Salvadori, S. Jäger, M. Hartmann, B. Civalleri, A. Pöppl and M. Chiesa, 17O-EPR determination of the structure and dynamics of copper single-metal sites in zeolites, Nat. Commun., 2021, 12, 4638 CrossRef CAS PubMed.
- J. W. A. Fischer, A. Brenig, D. Klose, J. A. van Bokhoven, V. L. Sushkevich and G. Jeschke, Methane Oxidation over Cu2+/[CuOH]+ Pairs and Site-Specific Kinetics in Copper Mordenite Revealed by Operando Electron Paramagnetic Resonance and UV/Visible Spectroscopy, Angew. Chem., Int. Ed., 2023, 135, e202303574 CrossRef.
- A. Godiksen, P. N. R. Vennestrøm, S. B. Rasmussen and S. Mossin, Identification and Quantification of Copper Sites in Zeolites by Electron Paramagnetic Resonance Spectroscopy, Top. Catal., 2017, 60, 13–29 CrossRef CAS.
- K. Pierloot, A. Delabie, M. H. Groothaert and R. A. Schoonheydt, A reinterpretation of the EPR spectra of Cu(II) in zeolites A, Y and ZK4, based on ab initio cluster model calculations, Phys. Chem. Chem. Phys., 2001, 3, 2174–2183 RSC.
- F. Göltl, P. Sautet and I. Hermans, Can Dynamics Be Responsible for the Complex Multipeak Infrared Spectra of NO Adsorbed to Copper(II) Sites in Zeolites?, Angew. Chem., Int. Ed., 2015, 54, 7799–7804 CrossRef PubMed.
- A. Brenig, J. W. A. Fischer, D. Klose, G. Jeschke, J. A. van Bokhoven and V. L. Sushkevich, Redox and Kinetic Properties of Composition-Dependent Active Sites in Copper-Exchanged Chabazite for Direct Methane-to-Methanol Oxidation, Angew. Chem., Int. Ed., 2024, 63, e202411662 CrossRef CAS PubMed.
- P. Vanelderen, B. E. R. Snyder, M.-L. Tsai, R. G. Hadt, J. Vancauwenbergh, O. Coussens, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Spectroscopic Definition of the Copper Active Sites in Mordenite: Selective Methane Oxidation, J. Am. Chem. Soc., 2015, 137, 6383–6392 CrossRef CAS PubMed.
- P. Vanelderen, J. Vancauwenbergh, B. F. Sels and R. A. Schoonheydt, Coordination chemistry and reactivity of copper in zeolites, Coord. Chem. Rev., 2013, 257, 483–494 CrossRef CAS.
- C. Negri, M. Signorile, N. G. Porcaro, E. Borfecchia, G. Berlier, T. V. W. Janssens and S. Bordiga, Dynamic CuII/CuI speciation in Cu-CHA catalysts by in situ Diffuse Reflectance UV-vis-NIR spectroscopy, Appl. Catal., A, 2019, 578, 1–9 CrossRef CAS.
- H. Li, C. Paolucci, I. Khurana, L. N. Wilcox, F. Göltl, J. D. Albarracin-Caballero, A. J. Shih, F. H. Ribeiro, R. Gounder and W. F. Schneider, Consequences of exchange-site heterogeneity and dynamics on the UV-visible spectrum of Cu-exchanged SSZ-13, Chem. Sci., 2019, 10, 2373–2384 RSC.
- A. Godiksen, O. L. Isaksen, S. B. Rasmussen, P. N. R. Vennestrøm and S. Mossin, Site-Specific Reactivity of Copper Chabazite Zeolites with Nitric Oxide, Ammonia, and Oxygen, ChemCatChem, 2018, 10, 366–370 CrossRef CAS.
- M. H. Mahyuddin, E. T. Lasiman, A. G. Saputro, S. V. Casuarina, N. Nugraha and H. K. Dipojono, Conditions to meet for the [CuOH]+ site to be favorable and reactive toward the conversion of methane to methanol over Cu-MOR zeolite, Catal. Sci. Technol., 2023, 13, 5767–5775 RSC.
- C. Paolucci, A. A. Parekh, I. Khurana, J. R. Di Iorio, H. Li, J. D. Albarracin Caballero, A. J. Shih, T. Anggara, W. N. Delgass, J. T. Miller, F. H. Ribeiro, R. Gounder and W. F. Schneider, Catalysis in a Cage: Condition-Dependent Speciation and Dynamics of Exchanged Cu Cations in SSZ-13 Zeolites, J. Am. Chem. Soc., 2016, 138, 6028–6048 CrossRef CAS PubMed.
- P. C. Bruzzese, E. Salvadori, B. Civalleri, S. Jäger, M. Hartmann, A. Pöppl and M. Chiesa, The Structure of Monomeric Hydroxo-CuII Species in Cu-CHA. A Quantitative Assessment, J. Am. Chem. Soc., 2022, 144, 13079–13083 Search PubMed.
- M. H. Groothaert, K. Pierloot, A. Delabie and R. A. Schoonheydt, Identification of Cu(II) coordination structures in Cu-ZSM-5, based on a DFT/ab initio assignment of the EPR spectra, Phys. Chem. Chem. Phys., 2003, 5, 2135–2144 Search PubMed.
- P. Vanelderen, J. Vancauwenbergh, M.-L. Tsai, R. G. Hadt, E. I. Solomon, R. A. Schoonheydt and B. F. Sels, Spectroscopy and Redox Chemistry of Copper in Mordenite, Chem. Phys. Chem., 2014, 15, 91–99 CrossRef CAS PubMed.
- A. Delabie, K. Pierloot, M. H. Groothaert, B. M. Weckhuysen and R. A. Schoonheydt, The siting of Cu(II) in mordenite: a theoretical spectroscopic study, Phys. Chem. Chem. Phys., 2002, 4, 134–145 RSC.
- A. J. Heyer, D. Plessers, A. Braun, H. M. Rhoda, M. L. Bols, B. Hedman, K. O. Hodgson, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Methane Activation by a Mononuclear Copper Active Site in the Zeolite Mordenite: Effect of Metal Nuclearity on Reactivity, J. Am. Chem. Soc., 2022, 144, 19305–19316 CrossRef CAS PubMed.
- V. L. Sushkevich, M. Artsiusheuski, D. Klose, G. Jeschke and J. A. Bokhoven, Identification of Kinetic and Spectroscopic Signatures of Copper Sites for Direct Oxidation of Methane to Methanol, Angew. Chem., Int. Ed., 2021, 60, 15944–15953 CrossRef CAS PubMed.
- D. Palagin, V. L. Sushkevich, A. J. Knorpp, M. Ranocchiari and J. A. van Bokhoven, Mapping Vibrational Spectra to the Structures of Copper Species in Zeolites Based on Calculated Stretching Frequencies of Adsorbed Nitrogen and Carbon Monoxides, J. Phys. Chem. C, 2021, 125, 12094–12106 CrossRef CAS.
- V. L. Sushkevich, D. Palagin and J. A. van Bokhoven, The Effect of the Active-Site Structure on the Activity of Copper Mordenite in the Aerobic and Anaerobic Conversion of Methane into Methanol, Angew. Chem., Int. Ed., 2018, 57, 8906–8910 CrossRef CAS PubMed.
- J. S. Woertink, P. J. Smeets, M. H. Groothaert, M. A. Vance, B. F. Sels, R. A. Schoonheydt and E. I. Solomon, A [Cu2O]2+ core in Cu-ZSM-5, the active site in the oxidation of methane to methanol, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18908–18913 CrossRef CAS PubMed.
- A. J. Heyer, D. Plessers, J. Ma, B. E. R. Snyder, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Magnetic Exchange Coupling in Zeolite Copper Dimers and Its Contribution to Methane Activation, J. Am. Chem. Soc., 2024, 146, 6061–6071 CrossRef CAS PubMed.
- D. Plessers, A. J. Heyer, H. M. Rhoda, M. L. Bols, E. I. Solomon, R. A. Schoonheydt and B. F. Sels, Tuning Copper Active Site Composition in Cu-MOR through Co-Cation Modification for Methane Activation, ACS Catal., 2023, 13, 1906–1915 CrossRef CAS PubMed.
- B. E. R. Snyder, P. Vanelderen, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Second-Sphere Effects on Methane Hydroxylation in Cu-Zeolites, J. Am. Chem. Soc., 2018, 140, 9236–9243 CrossRef CAS PubMed.
- G. Brezicki, J. Zheng, C. Paolucci, R. Schlögl and R. J. Davis, Effect of the Co-cation on Cu Speciation in Cu-Exchanged Mordenite and ZSM-5 Catalysts for the Oxidation of Methane to Methanol, ACS Catal., 2021, 11, 4973–4987 CrossRef CAS.
- M. A. Artsiusheuski, J. A. van Bokhoven and V. L. Sushkevich, Structure of Selective and Nonselective Dicopper(II) Sites in CuMFI for Methane Oxidation to Methanol, ACS Catal., 2022, 12, 15626–15637 CrossRef CAS.
- F. Göltl, S. Bhandari, E. A. Lebrón-Rodríguez, J. I. Gold, D. J. Hutton, S. I. Zones, I. Hermans, J. A. Dumesic and M. Mavrikakis, Exploring the Impact of Active Site Structure on the Conversion of Methane to Methanol in Cu-Exchanged Zeolites, Angew. Chem., Int. Ed., 2024, 63, e202403179 CrossRef PubMed.
- S. Grundner, M. A. C. Markovits, G. Li, M. Tromp, E. A. Pidko, E. J. M. Hensen, A. Jentys, M. Sanchez-Sanchez and J. A. Lercher, Single-site trinuclear copper oxygen clusters in mordenite for selective conversion of methane to methanol, Nat. Commun., 2015, 6, 7546–7555 CrossRef PubMed.
- G. Li, P. Vassilev, M. Sanchez-Sanchez, J. A. Lercher, E. J. M. Hensen and E. A. Pidko, Stability and reactivity of copper oxo-clusters in ZSM-5 zeolite for selective methane oxidation to methanol, J. Catal., 2016, 338, 305–312 CrossRef CAS.
- D. Palagin, A. J. Knorpp, A. B. Pinar, M. Ranocchiari and J. A. van Bokhoven, Assessing the relative stability of copper oxide clusters as active sites of a CuMOR zeolite for methane to methanol conversion: size matters?, Nanoscale, 2017, 9, 1144–1153 RSC.
- M. A. Artsiusheuski, O. Safonova, D. Palagin, J. A. van Bokhoven and V. L. Sushkevich, Structural Evolution of Copper-Oxo Sites in Zeolites upon the Reaction with Methane Investigated by Means of Cu K-edge X-ray Absorption Spectroscopy, J. Phys. Chem. C, 2023, 127, 9603–9615 CrossRef CAS.
- V. L. Sushkevich, O. V. Safonova, D. Palagin, M. A. Newton and J. A. van Bokhoven, Structure of copper sites in zeolites examined by Fourier and wavelet transform analysis of EXAFS, Chem. Sci., 2020, 11, 5299–5312 RSC.
- M. H. Mahyuddin, Y. Shiota, A. Staykov and K. Yoshizawa, Theoretical Overview of Methane Hydroxylation by Copper–Oxygen Species in Enzymatic and Zeolitic Catalysts, Acc. Chem. Res., 2018, 51, 2382–2390 CrossRef CAS PubMed.
- Q. M. Phung, T. Yanai, D. Plessers, B. F. Sels, R. A. Schoonheydt and K. Pierloot, Homolytic versus Heterolytic Methane Hydroxylation in Copper Zeolites, ACS Catal., 2025, 15, 1249–1264 CrossRef CAS.
- M. J. Wulfers, S. Teketel, B. Ipek and R. F. Lobo, Conversion of methane to methanol on copper-containing small-pore zeolites and zeotypes, Chem. Commun., 2015, 51, 4447–4450 RSC.
- M. Ravi, V. L. Sushkevich, A. J. Knorpp, M. A. Newton, D. Palagin, A. B. Pinar, M. Ranocchiari and J. A. van Bokhoven, Misconceptions and challenges in methane-to-methanol over transition-metal-exchanged zeolites, Nat. Catal, 2019, 2, 485–494 CrossRef CAS.
- A. A. Latimer, A. Kakekhani, A. R. Kulkarni and J. K. Nørskov, Direct Methane to Methanol: The Selectivity–Conversion Limit and Design Strategies, ACS Catal., 2018, 8, 6894–6907 CrossRef CAS.
- A. Blankenship, M. Artsiusheuski, V. Sushkevich and J. A. van Bokhoven, Recent trends, current challenges and future prospects for syngas-free methane partial oxidation, Nat. Catal., 2023, 6, 748–762 CrossRef CAS.
- M. Ahlquist, R. J. Nielsen, R. A. Periana and W. A. Goddard III, Product Protection, the Key to Developing High Performance Methane Selective Oxidation Catalysts, J. Am. Chem. Soc., 2009, 131, 17110–17115 CrossRef CAS PubMed.
- T. Sheppard, C. D. Hamill, A. Goguet, D. W. Rooney and J. M. Thompson, A low temperature, isothermal gas-phase system for conversion of methane to methanol over Cu–ZSM-5, Chem. Commun., 2014, 50, 11053–11055 RSC.
- P. Tomkins, A. Mansouri, V. L. Sushkevich, L. I. van der Wal, S. E. Bozbag, F. Krumeich, M. Ranocchiari and J. A. van Bokhoven, Increasing the activity of copper exchanged mordenite in the direct isothermal conversion of methane to methanol by Pt and Pd doping, Chem. Sci., 2018, 10, 167–171 RSC.
- R. J. Passini, M. Picinini, J. M. C. Bueno and E. A. Urquieta-Gonzalez, Direct methane to methanol stepwise conversion over Cu-oxo species in zeolites – Insights on the Cu-zeolite activation in air or helium from in situ UV-Vis analyses, Mol. Catal., 2022, 530, 112605 CAS.
- V. L. Sushkevich, D. Palagin, M. Ranocchiari and J. A. van Bokhoven, Selective Anaerobic Oxidation of Methane Enables Direct Synthesis of Methanol, Science, 2017, 356, 523–527 CrossRef CAS PubMed.
- D. K. Pappas, E. Borfecchia, M. Dyballa, K. A. Lomachenko, A. Martini, G. Berlier, B. Arstad, C. Lamberti, S. Bordiga, U. Olsbye, S. Svelle and P. Beato, Understanding and Optimizing the Performance of Cu-FER for The Direct CH4 to CH3OH Conversion, ChemCatChem, 2019, 11, 621–627 CrossRef CAS.
- G. Brezicki, J. D. Kammert, T. B. Gunnoe, C. Paolucci and R. J. Davis, Insights into the Speciation of Cu in the Cu-H-Mordenite Catalyst for the Oxidation of Methane to Methanol, ACS Catal., 2019, 9, 5308–5319 CrossRef CAS.
- M. C. Alvarez-Galvan, N. Mota, M. Ojeda, S. Rojas, R. M. Navarro and J. L. G. Fierro, Direct methane conversion routes to chemicals and fuels, Catal. Today, 2011, 171, 15–23 CrossRef CAS.
- D. K. Pappas, A. Martini, M. Dyballa, K. Kvande, S. Teketel, K. A. Lomachenko, R. Baran, P. Glatzel, B. Arstad, G. Berlier, C. Lamberti, S. Bordiga, U. Olsbye, S. Svelle, P. Beato and E. Borfecchia, The Nuclearity of the Active Site for Methane to Methanol Conversion in Cu-Mordenite: A Quantitative Assessment, J. Am. Chem. Soc., 2018, 140, 15270–15278 CrossRef CAS PubMed.
- J. O. Petunchi, G. Marcelin and W. K. Hall, Studies of the changes occurring on reduction and reoxidation of CuY zeolites, J. Phys. Chem., 1992, 96, 9967–9975 CrossRef CAS.
- C. M. Naccache and Y. B. Taarit, Oxidizing and acidic properties of copper-exchange Y zeolite, J. Catal., 1971, 22, 171–181 CrossRef CAS.
- L. Tao, I. Lee and M. Sanchez-Sanchez, Cu oxo nanoclusters for direct oxidation of methane to methanol: formation, structure and catalytic performance, Catal. Sci. Technol., 2020, 10, 7124–7141 RSC.
- S. Grundner, W. Luo, M. Sanchez-Sanchez and J. A. Lercher, Synthesis of single-site copper catalysts for methane partial oxidation, Chem. Commun., 2016, 52, 2553–2556 RSC.
- M. Lo Jacono, G. Fierro, R. Dragone, X. Feng, J. d’Itr and W. K. Hall, Zeolite Chemistry of CuZSM-5 Revisited, J. Phys. Chem. B, 1997, 101, 1979–1984 CrossRef CAS.
- G. Turnes Palomino, P. Fisicaro, S. Bordiga, A. Zecchina, E. Giamello and C. Lamberti, Oxidation States of Copper Ions in ZSM-5 Zeolites. A Multitechnique Investigation, J. Phys. Chem. B, 2000, 104, 4064–4073 CrossRef PubMed.
- F. X. Llabrés i Xamena, P. Fisicaro, G. Berlier, A. Zecchina, G. T. Palomino, C. Prestipino, S. Bordiga, E. Giamello and C. Lamberti, Thermal Reduction of Cu2+−Mordenite and Re-oxidation upon Interaction with H2O, O2, and NO, J. Phys. Chem. B, 2003, 107, 7036–7044 CrossRef.
- E. M. C. Alayon, M. Nachtegaal, A. Bodi, M. Ranocchiari and J. A. van Bokhoven, Bis(μ-oxo) versus mono(μ-oxo)dicopper cores in a zeolite for converting methane to methanol: an in situ XAS and DFT investigation, Phys. Chem. Chem. Phys., 2015, 17, 7681–7693 RSC.
- V. L. Sushkevich and J. A. van Bokhoven, Revisiting copper reduction in zeolites: the impact of autoreduction and sample synthesis procedure, Chem. Commun., 2018, 54, 7447–7450 RSC.
- A. Martini, E. Borfecchia, K. A. Lomachenko, I. A. Pankin, C. Negri, G. Berlier, P. Beato, H. Falsig, S. Bordiga and C. Lamberti, Composition-driven Cu-speciation and reducibility in Cu-CHA zeolite catalysts: a multivariate XAS/FTIR approach to complexity, Chem. Sci., 2017, 8, 6836–6851 RSC.
- C. W. Andersen, E. Borfecchia, M. Bremholm, M. R. V. Jørgensen, P. N. R. Vennestrøm, C. Lamberti, L. F. Lundegaard and B. B. Iversen, Redox-Driven Migration of Copper Ions in the Cu-CHA Zeolite as Shown by the In Situ PXRD/XANES Technique, Angew. Chem., Int. Ed., 2017, 56, 10367–10372 CrossRef CAS PubMed.
- E. Borfecchia, K. A. Lomachenko, F. Giordanino, H. Falsig, P. Beato, A. V. Soldatov, S. Bordiga and C. Lamberti, Revisiting the nature of Cu sites in the activated Cu-SSZ-13 catalyst for SCR reaction, Chem. Sci., 2014, 6, 548–563 RSC.
- A. Wijerathne, A. Sawyer, R. Daya and C. Paolucci, Competition between Mononuclear and Binuclear Copper Sites across Different Zeolite Topologies, JACS Au, 2024, 4, 197–215 CrossRef CAS PubMed.
- L. N. Wilcox, J. Rebolledo-Oyarce, A. D. Mikes, Y. Wang, W. F. Schneider and R. Gounder, Structure and Reactivity of Binuclear Cu Active Sites in Cu-CHA Zeolites for Stoichiometric Partial Methane Oxidation to Methanol, ACS Catal., 2024, 14, 3647–3663 CrossRef CAS.
- A. R. Kulkarni, Z.-J. Zhao, S. Siahrostami, J. K. Nørskov and F. Studt, Monocopper Active Site for Partial Methane Oxidation in Cu-Exchanged 8MR Zeolites, ACS Catal., 2016, 6, 6531–6536 CrossRef CAS.
- V. L. Sushkevich, R. Verel and J. A. Bokhoven, Pathways of Methane Transformation over Copper-Exchanged Mordenite as Revealed by In Situ NMR and IR Spectroscopy, Angew. Chem., 2019, 132, 920–928 CrossRef.
- V. L. Sushkevich, M. Artsiusheuski, D. Klose, G. Jeschke and J. A. van Bokhoven, Identification of Kinetic and Spectroscopic Signatures of Copper Sites for Direct Oxidation of Methane to Methanol, Angew. Chem., Int. Ed., 2021, 60, 15944–15953 CrossRef CAS PubMed.
- A. J. Knorpp, A. B. Pinar, C. Baerlocher, L. B. McCusker, N. Casati, M. A. Newton, S. Checchia, J. Meyet, D. Palagin and J. A. van Bokhoven, Paired Copper Monomers in Zeolite Omega: The Active Site for Methane-to-Methanol Conversion, Angew. Chem., Int. Ed., 2021, 60, 5854–5858 CrossRef CAS.
- J. Wieser, D. Wardecki, J. W. A. Fischer, M. A. Newton, C. Dejoie, A. J. Knorpp, G. Jeschke, P. Rzepka and J. A. van Bokhoven, Quantifying the Hydration-Dependent Dynamics of Copper Migration and Activity in Zeolite Omega for the Partial Oxidation of Methane, Angew. Chem., Int. Ed., 2024, e202407395 CAS.
- F. Giordanino, P. N. R. Vennestrøm, L. F. Lundegaard, F. N. Stappen, S. Mossin, P. Beato, S. Bordiga and C. Lamberti, Characterization of Cu-exchanged SSZ-13: a comparative FTIR, UV-Vis, and EPR study with Cu-ZSM-5 and Cu-β with similar Si/Al and Cu/Al ratios, Dalton Trans., 2013, 42, 12741–12761 RSC.
- J. A. Duffy, Novel Chemical Effects of Electronics Behaviour., Springer, Berlin, Heidelberg, 1977, pp. 147–166 Search PubMed.
- C. E. Housecroft and A. G. Sharpe, Inorganic Chemistry, Pearson Education Limited, Essex, 2008 Search PubMed.
- R. Boča, Electronic Energy Levels of Transition Metal Complexes, Elsevier, Amsterdam, 2025 Search PubMed.
- E. M. C. Alayon, M. Nachtegaal, E. Kleymenov and J. A. van Bokhoven, Determination of the electronic and geometric structure of Cu sites during methane conversion over Cu-MOR with X-ray absorption spectroscopy, Microporous Mesoporous Mater., 2013, 166, 131–136 CrossRef CAS.
- M. A. Newton, A. J. Knorpp, V. L. Sushkevich, D. Palagin and J. A. van Bokhoven, Active sites and mechanisms in the direct conversion of methane to methanol using Cu in zeolitic hosts: a critical examination, Chem. Soc. Rev., 2020, 49, 1449–1486 RSC.
- A. Martini, E. Alladio and E. Borfecchia, Determining Cu–Speciation in the Cu–CHA Zeolite Catalyst: The Potential of Multivariate Curve Resolution Analysis of In Situ XAS Data, Top. Catal., 2018, 61, 1396–1407 CrossRef CAS.
- J. L. DuBois, P. Mukherjee, T. D. P. Stack, B. Hedman, E. I. Solomon and K. O. Hodgson, A Systematic K-edge X-ray Absorption Spectroscopic Study of Cu(III) Sites, J. Am. Chem. Soc., 2000, 122, 5775–5787 CrossRef CAS.
- M. L. Baker, M. W. Mara, J. J. Yan, K. O. Hodgson, B. Hedman and E. I. Solomon, K- and L-edge X-ray absorption spectroscopy (XAS) and resonant inelastic X-ray scattering (RIXS) determination of differential orbital covalency (DOC) of transition metal sites, Coord. Chem. Rev., 2017, 345, 182–208 CrossRef CAS PubMed.
- H. Yamashita, M. Matsuoka, K. Tsuji, Y. Shioya, M. Anpo and M. Che, In Situ XAFS, Photoluminescence, and IR Investigations of Copper Ions Included within Various Kinds of Zeolites. Structure of Cu(I) Ions and Their Interaction with CO Molecules, J. Phys. Chem., 1996, 100, 397–402 CrossRef CAS.
- L. S. Kau, D. J. Spira-Solomon, J. E. Penner-Hahn, K. O. Hodgson and E. I. Solomon, X-ray absorption edge determination of the oxidation state and coordination number of copper. Application to the type 3 site in Rhus vernicifera laccase and its reaction with oxygen, J. Am. Chem. Soc., 1987, 109, 6433–6442 CrossRef CAS.
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Copper Active Sites in Biology, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS PubMed.
- M.-L. Tsai, R. G. Hadt, P. Vanelderen, B. F. Sels, R. A. Schoonheydt and E. I. Solomon, [Cu2O]2+ Active Site Formation in Cu–ZSM-5: Geometric and Electronic Structure Requirements for N2O Activation, J. Am. Chem. Soc., 2014, 136, 3522–3529 CrossRef CAS PubMed.
- C. Buono, A. Martini, I. A. Pankin, D. K. Pappas, C. Negri, K. Kvande, K. A. Lomachenko and E. Borfecchia, Local structure of Cu(I) ions in the MOR zeolite: A DFT-assisted XAS study, Radiat. Phys. Chem., 2020, 175, 108111 CrossRef CAS.
- F. Gao, E. D. Walter, M. Kollar, Y. Wang, J. Szanyi and C. H. F. Peden, Understanding ammonia selective catalytic reduction kinetics over Cu/SSZ-13 from motion of the Cu ions, J. Catal., 2014, 319, 1–14 CrossRef CAS.
- A. Brenig, J. W. A. Fischer, D. Klose, G. Jeschke, J. A. van Bokhoven and V. L. Sushkevich, Tracking Active Site Formation during Oxidative Activation of Copper-Exchanged Zeolites for Methane-to-Methanol Conversion, Adv. Sci., 2025, 12, 2413870–2413883 CrossRef CAS PubMed.
- M. Occhiuzzi, G. Fierro, G. Ferraris and G. Moretti, Unusual Complete Reduction of Cu2+ Species in Cu-ZSM-5 Zeolites under Vacuum Treatment at High Temperature, Chem. Mater., 2012, 24, 2022–2031 CrossRef CAS.
- C. Chao and J. H. Lunsford, EPR Study of Copper(II) Ion Pairs in Y-Type Zeolites, J. Chem. Phys., 1972, 57, 2890–2898 CrossRef CAS.
- J. Valyon and W. K. Hall, Effects of reduction and reoxidation on the infrared spectra from Cu-Y and Cu-ZSM-5 zeolites, J. Phys. Chem., 1993, 97, 7054–7060 CrossRef CAS.
- V. L. Sushkevich, A. V. Smirnov and J. A. van Bokhoven, Autoreduction of Copper in Zeolites: Role of Topology, Si/Al Ratio, and Copper Loading, J. Phys. Chem. C, 2019, 123, 9926–9934 CrossRef CAS.
- F. Gao, N. M. Washton, Y. Wang, M. Kollár, J. Szanyi and C. H. F. Peden, Effects of Si/Al ratio on Cu/SSZ-13 NH3-SCR catalysts: Implications for the active Cu species and the roles of Brønsted acidity, J. Catal., 2015, 331, 25–38 CrossRef CAS.
- M. H. Groothaert, K. Lievens, H. Leeman, B. M. Weckhuysen and R. A. Schoonheydt, An operando optical fiber UV-vis spectroscopic study of the catalytic decomposition of NO and N2O over Cu-ZSM-5, J. Catal., 2003, 220, 500–512 CrossRef CAS.
- M. Iwamoto, H. Yahiro, K. Tanda, N. Mizuno, Y. Mine and S. Kagawa, Removal of nitrogen monoxide through a novel catalytic process. 1. Decomposition on excessively copper-ion-exchanged ZSM-5 zeolites, J. Phys. Chem., 1991, 95, 3727–3730 CrossRef CAS.
- D. J. Hutton, D. H. Lopez and F. Göltl, Thermodynamic driving forces for autoreduction of Cu sites in the zeolite SSZ-13 React, Chem. Eng., 2024, 9, 1685–1695 CAS.
- P. J. Smeets, M. H. Groothaert and R. A. Schoonheydt, Cu based zeolites: A UV-vis study of the active site in the selective methane oxidation at low temperatures, Catal. Today, 2005, 110, 303–309 CrossRef CAS.
- G. Deplano, A. Martini, M. Signorile, E. Borfecchia, V. Crocellà, S. Svelle and S. Bordiga, Copper Pairing in the Mordenite Framework as a Function of the CuI/CuII Speciation, Angew. Chem., Int. Ed., 2021, 60, 25891–25896 CrossRef CAS PubMed.
- A. Martini, M. Signorile, C. Negri, K. Kvande, K. A. Lomachenko, S. Svelle, P. Beato, G. Berlier, E. Borfecchia and S. Bordiga, EXAFS wavelet transform analysis of Cu-MOR zeolites for the direct methane to methanol conversion, Phys. Chem. Chem. Phys., 2020, 22, 18950–18963 RSC.
- K. Kvande, B. Garetto, G. Deplano, M. Signorile, B. G. Solemsli, S. Prodinger, U. Olsbye, P. Beato, S. Bordiga, S. Svelle and E. Borfecchia, Understanding C–H activation in light alkanes over Cu-MOR zeolites by coupling advanced spectroscopy and temperature-programmed reduction experiments, Chem. Sci., 2023, 14, 9704–9723 RSC.
- C. Dossi, A. Fusi, G. Moretti, S. Recchia and R. Psaro, On the role of carbonaceous material in the reduction of Cu2+ to Cu+ in Cu-ZSM-5 catalysts, Appl. Catal., A, 1999, 188, 107–119 CrossRef CAS.
- A. V. Kucherov, H. G. Karge and R. Schlögl, Quantitative ESR study of the CuH-ZSM-5 system: influence of preparation and pretreatment techniques on the valence state of copper, Microporous Mesoporous Mater., 1998, 25, 7–14 CrossRef CAS.
- P. D. Costa, B. Modén, G. D. Meitzner, D. K. Lee and E. Iglesia, Spectroscopic and chemical characterization of active and inactive Cu species in NO decomposition catalysts based on Cu-ZSM5, Phys. Chem. Chem. Phys., 2002, 4, 4590–4601 RSC.
- S. C. Larsen, A. Aylor, A. T. Bell and J. A. Reimer, Electron Paramagnetic Resonance Studies of Copper Ion-Exchanged ZSM-5, J. Phys. Chem., 1994, 98, 11533–11540 CrossRef CAS.
- M. H. Groothaert, J. A. van Bokhoven, A. A. Battiston, B. M. Weckhuysen and R. A. Schoonheydt, Bis(μ-oxo)dicopper in Cu-ZSM-5 and Its Role in the Decomposition of NO: A Combined in Situ XAFS, UV-Vis-Near-IR, and Kinetic Study, J. Am. Chem. Soc., 2003, 125, 7629–7640 CrossRef CAS PubMed.
- M. H. Groothaert, K. Lievens, J. A. van Bokhoven, A. A. Battiston, B. M. Weckhuysen, K. Pierloot and R. A. Schoonheydt, Bis(μ-oxo)dicopper as Key Intermediate in the Catalytic Decomposition of Nitric Oxide, Chem. Phys. Chem., 2003, 4, 626–630 CrossRef CAS PubMed.
- E. A. Pidko, E. J. M. Hensen and R. A. van Santen, Self-organization of extraframework cations in zeolites, Proc. R. Soc. Math. Phys. Eng. Sci., 2012, 468, 2070–2086 CAS.
- P. J. Smeets, R. G. Hadt, J. S. Woertink, P. Vanelderen, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Oxygen Precursor to the Reactive Intermediate in Methanol Synthesis by Cu-ZSM-5, J. Am. Chem. Soc., 2010, 132, 14736–14738 CrossRef CAS PubMed.
- A. Delabie, K. Pierloot, M. H. Groothaert, R. A. Schoonheydt and L. G. Vanquickenborne, The Coordination of Cu(II) in Zeolites − Structure and Spectroscopic Properties, Eur. J. Inorg. Chem., 2002, 515–530 CrossRef CAS.
- P. J. Smeets, J. S. Woertink, B. F. Sels, E. I. Solomon and R. A. Schoonheydt, Transition-Metal Ions in Zeolites: Coordination and Activation of Oxygen, Inorg. Chem., 2010, 49, 3573–3583 CrossRef CAS PubMed.
- U. Engedahl, A. Boje, H. Ström, H. Grönbeck and A. Hellman, Complete Reaction Cycle for Methane-to-Methanol Conversion over Cu-SSZ-13: First-Principles Calculations and Microkinetic Modeling, J. Phys. Chem. C, 2021, 125, 14681–14688 CrossRef CAS.
- M. Haris Mahyuddin, Y. Shiota and K. Yoshizawa, Methane selective oxidation to methanol by metal-exchanged zeolites: a review of active sites and their reactivity, Catal. Sci. Technol., 2019, 9, 1744–1768 RSC.
- H. Liu, J. Wang, T. Yu, S. Fan and M. Shen, The role of various iron species in Fe-β catalysts with low iron loadings for NH3-SCR, Catal. Sci. Technol., 2014, 4, 1350–1356 RSC.
- A. I. Olivos-Suarez, À. Szécsényi, E. J. M. Hensen, J. Ruiz-Martinez, E. A. Pidko and J. Gascon, Strategies for the Direct Catalytic Valorization of Methane Using Heterogeneous Catalysis: Challenges and Opportunities, ACS Catal., 2016, 6, 2965–2981 CrossRef CAS.
- M. H. Mahyuddin, H. K. Dipojono and K. Yoshizawa, in Direct Hydroxylation Methane Interplay Theory Experiment, ed. K. Yoshizawa, Springer, Singapore, 2020, pp. 75–86 Search PubMed.
- S. Santra, T. Archipov, A. B. Ene, H. Komnik, H. Stoll, E. Roduner and G. Rauhut, Adsorption of dioxygen to copper in CuHY zeolite, Phys. Chem. Chem. Phys., 2009, 11, 8855–8866 RSC.
- L. Vilella and F. Studt, The Stability of Copper Oxo Species in Zeolite Frameworks, Eur. J. Inorg. Chem., 2016, 1514–1520 CrossRef CAS.
- M. H. Mahyuddin, A. Staykov, Y. Shiota, M. Miyanishi and K. Yoshizawa, Roles of Zeolite Confinement and Cu–O–Cu Angle on the Direct Conversion of Methane to Methanol by [Cu2(μ-O)]2+-Exchanged AEI, CHA, AFX, and MFI Zeolites, ACS Catal., 2017, 7, 3741–3751 CrossRef CAS.
- M. H. Mahyuddin, T. Tanaka, Y. Shiota, A. Staykov and K. Yoshizawa, Methane Partial Oxidation over [Cu2(μ-O)]2+ and [Cu3(μ-O)3]2+ Active Species in Large-Pore Zeolites, ACS Catal., 2018, 8, 1500–1509 CrossRef CAS.
- M. H. Mahyuddin, A. Staykov, A. G. Saputro, M. K. Agusta, H. K. Dipojono and K. Yoshizawa, Novel Mechanistic Insights into Methane Activation over Fe and Cu Active Sites in Zeolites: A Comparative DFT Study Using Meta-GGA Functionals, J. Phys. Chem. C, 2020, 124, 18112–18125 CrossRef CAS.
- D. Panthi, O. Adeyiga and S. O. Odoh, DFT Analysis of Methane C−H Activation and Over-Oxidation by [Cu2O]2+ and [Cu2O2]2+ Sites in Zeolite Mordenite: Intra- versus Inter-site Over-Oxidation, ChemPhysChem, 2021, 22, 2517–2525 CrossRef CAS PubMed.
- K. D. Vogiatzis, G. Li, E. J. M. Hensen, L. Gagliardi and E. A. Pidko, Electronic Structure of the [Cu3(μ-O)3]2+ Cluster in Mordenite Zeolite and Its Effects on the Methane to Methanol Oxidation, J. Phys. Chem. C, 2017, 121, 22295–22302 CrossRef CAS PubMed.
- V. L. Sushkevich, R. Verel and J. A. van Bokhoven, Pathways of Methane Transformation over Copper-Exchanged Mordenite as Revealed by In Situ NMR and IR Spectroscopy, Angew. Chem., 2020, 132, 920–928 CrossRef.
- M. H. Mahyuddin, in Direct Hydroxylation Methane, ed. K. Yoshizawa, Springer Singapore, Singapore, 2020, pp. 87–100 Search PubMed.
- Z.-J. Zhao, A. Kulkarni, L. Vilella, J. K. Nørskov and F. Studt, Theoretical Insights into the Selective Oxidation of Methane to Methanol in Copper-Exchanged Mordenite, ACS Catal., 2016, 6, 3760–3766 CrossRef CAS.
- X. Liu, Y. Ryabenkova and M. Conte, Catalytic oxygen activation versus autoxidation for industrial applications: a physicochemical approach, Phys. Chem. Chem. Phys., 2014, 17, 715–731 RSC.
- N. Dietl, C. van der Linde, M. Schlangen, M. K. Beyer and H. Schwarz, Diatomic [CuO]+ and Its Role in the Spin-Selective Hydrogen- and Oxygen-Atom Transfers in the Thermal Activation of Methane, Angew. Chem., Int. Ed., 2011, 50, 4966–4969 CrossRef CAS PubMed.
- O. Suleiman, O. Adeyiga, D. Panthi and S. O. Odoh, Copper-Oxo Active Sites in the 8MR of Zeolite Mordenite: DFT Investigation of the Impact of Acid Sites on Methanol Yield and Selectivity, J. Phys. Chem. C, 2021, 125, 6684–6693 CrossRef CAS.
- F. Göltl, S. Bhandari and M. Mavrikakis, Thermodynamics Perspective on the Stepwise Conversion of Methane to Methanol over Cu-Exchanged SSZ-13, ACS Catal., 2021, 11, 7719–7734 CrossRef.
- D. K. Pappas, E. Borfecchia, K. A. Lomachenko, A. Lazzarini, E. S. Gutterød, M. Dyballa, A. Martini, G. Berlier, S. Bordiga, C. Lamberti, B. Arstad, U. Olsbye, P. Beato and S. Svelle, Cu-Exchanged Ferrierite Zeolite for the Direct CH4 to CH3OH Conversion: Insights on Cu Speciation from X-Ray Absorption Spectroscopy, Top. Catal., 2019, 62, 712–723 CrossRef CAS.
- A. J. Knorpp, A. B. Pinar, M. A. Newton, V. L. Sushkevich and J. A. van Bokhoven, Copper-Exchanged Omega (MAZ) Zeolite: Copper-concentration Dependent Active Sites and its Unprecedented Methane to Methanol Conversion, ChemCatChem, 2018, 10, 5593–5596 CrossRef CAS.
- A. J. Knorpp, M. A. Newton, A. B. Pinar and J. A. van Bokhoven, Conversion of Methane to Methanol on Copper Mordenite: Redox Mechanism of Isothermal and High-Temperature-Activation Procedures, Ind. Eng. Chem. Res., 2018, 57, 12036–12039 CrossRef CAS.
- E. Borfecchia, P. Beato, S. Svelle, U. Olsbye, C. Lamberti and S. Bordiga, Cu-CHA – a model system for applied selective redox catalysis, Chem. Soc. Rev., 2018, 47, 8097–8133 RSC.
- E. M. C. Alayon, M. Nachtegaal, A. Bodi and J. A. van Bokhoven, Reaction Conditions of Methane-to-Methanol Conversion Affect the Structure of Active Copper Sites, ACS Catal., 2014, 4, 16–22 CrossRef CAS.
- J. H. Kwak, T. Varga, C. H. F. Peden, F. Gao, J. C. Hanson and J. Szanyi, Following the movement of Cu ions in a SSZ-13 zeolite during dehydration, reduction and adsorption: A combined in situ TP-XRD, XANES/DRIFTS study, J. Catal., 2014, 314, 83–93 CrossRef CAS.
- E. Borfecchia, D. K. Pappas, M. Dyballa, K. A. Lomachenko, C. Negri, M. Signorile and G. Berlier, Evolution of active sites during selective oxidation of methane to methanol over Cu-CHA and Cu-MOR zeolites as monitored by operando XAS, Catal. Today, 2019, 333, 17–27 CrossRef CAS.
- R. Bulanek, K. Frolich, P. Cicmanec, D. Nachtigallova, A. Pulido and P. Nachtigall, Combined Experimental and Theoretical Investigations of Heterogeneous Dual Cation Sites in Cu,M-FER Zeolites, J. Phys. Chem. C, 2011, 115, 13312–13321 CrossRef CAS.
- U. Engedahl, H. Grönbeck and A. Hellman, First-Principles Study of Oxidation State and Coordination of Cu-Dimers in Cu-SSZ-13 during Methane-to-Methanol Reaction Conditions, J. Phys. Chem. C, 2019, 123, 26145–26150 CrossRef CAS.
- Y. Pérez-Badell, X. Solans-Monfort, M. Sodupe and L. A. Montero, A DFT periodic study on the interaction between O2 and cation exchanged chabazite MCHA (M = H+, Na+ or Cu+): effects in the triplet–singlet energy gap, Phys. Chem. Chem. Phys., 2009, 12, 442–452 RSC.
- M. H. Mahyuddin, T. Tanaka, A. Staykov, Y. Shiota and K. Yoshizawa, Dioxygen Activation on Cu-MOR Zeolite: Theoretical Insights into the Formation of Cu2O and Cu3O3 Active Species, Inorg. Chem., 2018, 57, 10146–10152 CrossRef CAS PubMed.
- T. Yumura, M. Takeuchi, H. Kobayashi and Y. Kuroda, Effects of ZSM-5 Zeolite Confinement on Reaction Intermediates during Dioxygen Activation by Enclosed Dicopper Cations, Inorg. Chem., 2009, 48, 508–517 CrossRef CAS PubMed.
- Y. Zhu, K. Mimura and M. Isshiki, Oxidation Mechanism of Cu2O to CuO at 600–1050 °C, Oxid. Met., 2004, 62, 207–222 CrossRef CAS.
- Z. Grzesik and M. Migdalska, Oxidation Mechanism of Cu2O and Defect Structure of CuO at High Temperatures, High Temp. Mater. Process, 2011, 30, 277–287 CrossRef CAS.
- C. K. Clayton and K. J. Whitty, Measurement and modeling of decomposition kinetics for copper oxide-based chemical looping with oxygen uncoupling, Appl. Energy, 2014, 116, 416–423 CrossRef CAS.
- C. K. Clayton, H. Y. Sohn and K. J. Whitty, Oxidation Kinetics of Cu2O in Oxygen Carriers for Chemical Looping with Oxygen Uncoupling, Ind. Eng. Chem. Res., 2014, 53, 2976–2986 CrossRef CAS.
- janaf.nist.gov/tables/O-029.html, can be found under https://janaf.nist.gov/tables/O-029.html, (accessed 14 January 2026), n.d.
- janaf.nist.gov/tables/Cu-019.html, can be found under https://janaf.nist.gov/tables/Cu-019.html, (accessed 14 January 2026), n.d.
- janaf.nist.gov/tables/Cu-015.html, can be found under https://janaf.nist.gov/tables/Cu-015.html, (accessed 14 January 2026), n.d.
- Y. Wang, H. Liu, Q. Duan, Z. Li, Y. Wang, H. Liu, Q. Duan and Z. Li, Understanding the Negative Apparent Activation Energy for Cu2O and CoO Oxidation Kinetics at High Temperature near Equilibrium, Catalysts, 2024, 14, 832 CrossRef CAS.
- V. L. Sushkevich, D. Palagin, M. Ranocchiari and J. A. van Bokhoven, Response to Comment on ‘Selective anaerobic oxidation of methane enables direct synthesis of methanol’, Science, 2017, 358, eaan6083 CrossRef PubMed.
- M. Dyballa, K. Thorshaug, D. K. Pappas, E. Borfecchia, K. Kvande, S. Bordiga, G. Berlier, A. Lazzarini, U. Olsbye, P. Beato, S. Svelle and B. Arstad, Zeolite Surface Methoxy Groups as Key Intermediates in the Stepwise Conversion of Methane to Methanol, ChemCatChem, 2019, 11, 5022–5026 CrossRef CAS.
- K. Kvande, D. K. Pappas, E. Borfecchia and K. A. Lomachenko, Advanced X-ray Absorption Spectroscopy Analysis to Determine Structure-Activity Relationships for Cu-Zeolites in the Direct Conversion of Methane to Methanol, ChemCatChem, 2020, 12, 2385–2405 CrossRef CAS.
- I. A. Pankin, E. Borfecchia, A. Martini, K. A. Lomachenko, C. Lamberti and A. V. Soldatov, DFT-assisted XANES simulations to discriminate different monomeric CuII species in CHA catalysts, Radiat. Phys. Chem., 2020, 175, 108510 CrossRef CAS.
- K. Kvande, D. K. Pappas, M. Dyballa, C. Buono, M. Signorile, E. Borfecchia, K. A. Lomachenko, B. Arstad, S. Bordiga, G. Berlier, U. Olsbye, P. Beato and S. Svelle, Comparing the Nature of Active Sites in Cu-loaded SAPO-34 and SSZ-13 for the Direct Conversion of Methane to Methanol, Catalysts, 2020, 10, 191 CrossRef CAS.
- H. M. Rhoda, D. Plessers, A. J. Heyer, M. L. Bols, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Spectroscopic Definition of a Highly Reactive Site in Cu-CHA for Selective Methane Oxidation: Tuning a Mono-μ-Oxo Dicopper(II) Active Site for Reactivity, J. Am. Chem. Soc., 2021, 143, 7531–7540 CrossRef CAS PubMed.
- M. A. Artsiusheuski, J. A. van Bokhoven and V. L. Sushkevich, Modeling and Experiment for Oxygen Isotope Exchange over Copper-Containing Mordenite, J. Phys. Chem. C, 2021, 125, 12366–12373 CrossRef CAS.
- M. A. Artsiusheuski, J. A. van Bokhoven and V. L. Sushkevich, Oxygen Isotope Exchange over Copper-Containing Mordenite: The Effect of Copper Loading and Si/Al Ratio, J. Phys. Chem. C, 2021, 125, 26512–26521 CrossRef CAS.
- K. A. Lomachenko, A. Martini, D. K. Pappas, C. Negri, M. Dyballa, G. Berlier, S. Bordiga, C. Lamberti, U. Olsbye, S. Svelle, P. Beato and E. Borfecchia, The impact of reaction conditions and material composition on the stepwise methane to methanol conversion over Cu-MOR: An operando XAS study, Catal. Today, 2019, 336, 99–108 CrossRef CAS.
- M. Dyballa, D. K. Pappas, K. Kvande, E. Borfecchia, B. Arstad, P. Beato, U. Olsbye and S. Svelle, On How Copper Mordenite Properties Govern the Framework Stability and Activity in the Methane-to-Methanol Conversion, ACS Catal., 2019, 9, 365–375 CrossRef CAS.
- S. E. Bozbag, E. M. C. Alayon, J. Pecháček, M. Nachtegaal, M. Ranocchiari and J. A. van Bokhoven, Methane to methanol over copper mordenite: yield improvement through multiple cycles and different synthesis techniques, Catal. Sci. Technol., 2016, 6, 5011–5022 RSC.
- A. J. Knorpp, Direct Conversion of Methane to Methanol over Copper-Exchanged Zeolite Omega (MAZ), ETH Zürich, 2019 Search PubMed.
- A. J. Knorpp, M. A. Newton, S. C. M. Mizuno, J. Zhu, H. Mebrate, A. B. Pinar and J. A. van Bokhoven, Comparative performance of Cu-zeolites in the isothermal conversion of methane to methanol, Chem. Commun., 2019, 55, 11794–11797 RSC.
- V. L. Sushkevich and J. A. van Bokhoven, Kinetic study and effect of water on methane oxidation to methanol over copper-exchanged mordenite, Catal. Sci. Technol., 2020, 10, 382–390 RSC.
- P. Tomkins, M. Ranocchiari and J. A. van Bokhoven, Direct Conversion of Methane to Methanol under Mild Conditions over Cu-Zeolites and beyond, Acc. Chem. Res., 2017, 50, 418–425 CrossRef CAS PubMed.
- V. L. Sushkevich and J. A. van Bokhoven, Methane-to-Methanol: Activity Descriptors in Copper-Exchanged Zeolites for the Rational Design of Materials, ACS Catal., 2019, 9, 6293–6304 CrossRef CAS.
- J. Wieser, A. J. Knorpp, D. C. Stoian, P. Rzepka, M. A. Newton and J. A. van Bokhoven, Assessing the Productivity of the Direct Conversion of Methane-to-Methanol over Copper-Exchanged Zeolite Omega (MAZ) via Oxygen Looping, Angew. Chem., Int. Ed., 2023, 62, e202305140 CrossRef CAS PubMed.
- J. Zhu, V. L. Sushkevich, A. J. Knorpp, M. A. Newton, S. C. M. Mizuno, T. Wakihara, T. Okubo, Z. Liu and J. A. van Bokhoven, Cu-Erionite Zeolite Achieves High Yield in Direct Oxidation of Methane to Methanol by Isothermal Chemical Looping, Chem. Mater., 2020, 32, 1448–1453 CrossRef CAS.
- P. J. Smeets, B. F. Sels, R. M. van Teeffelen, H. Leeman, E. J. M. Hensen and R. A. Schoonheydt, The catalytic performance of Cu-containing zeolites in N2O decomposition and the influence of O2, NO and H2O on recombination of oxygen, J. Catal., 2008, 256, 183–191 CrossRef CAS.
- T. Yumura, Y. Hirose, T. Wakasugi, Y. Kuroda and H. Kobayashi, Roles of Water Molecules in Modulating the Reactivity of Dioxygen-Bound Cu-ZSM-5 toward Methane: A Theoretical Prediction, ACS Catal., 2016, 6, 2487–2495 CrossRef CAS.
- K. Narsimhan, K. Iyoki, K. Dinh and Y. Román-Leshkov, Catalytic Oxidation of Methane into Methanol over Copper-Exchanged Zeolites with Oxygen at Low Temperature, ACS Cent. Sci., 2016, 2, 424–429 CrossRef CAS PubMed.
- J. Ohyama, A. Hirayama, Y. Tsuchimura, N. Kondou, H. Yoshida, M. Machida, S. Nishimura, K. Kato, I. Miyazato and K. Takahashi, Catalytic direct oxidation of methane to methanol by redox of copper mordenite, Catal. Sci. Technol., 2021, 11, 3437–3446 RSC.
- A. Hirayama, Y. Tsuchimura, H. Yoshida, M. Machida, S. Nishimura, K. Kato, K. Takahashi and J. Ohyama, Catalytic oxidation of methane to methanol over Cu-CHA with molecular oxygen, Catal. Sci. Technol., 2021, 11, 6217–6224 RSC.
- K. T. Dinh, M. M. Sullivan, K. Narsimhan, P. Serna, R. J. Meyer, M. Dincă and Y. Román-Leshkov, Continuous Partial Oxidation of Methane to Methanol Catalyzed by Diffusion-Paired Copper Dimers in Copper-Exchanged Zeolites, J. Am. Chem. Soc., 2019, 141, 11641–11650 CrossRef CAS PubMed.
- J. Pokhrel and D. F. Shantz, Continuous partial oxidation of methane to methanol over Cu-SSZ-39 catalysts, J. Catal., 2023, 421, 300–308 CrossRef CAS.
- Y. Fan, W. Zhou, X. Qiu, H. Li, Y. Jiang, Z. Sun, D. Han, L. Niu and Z. Tang, Selective photocatalytic oxidation of methane by quantum-sized bismuth vanadate, Nat. Sustainable, 2021, 4, 509–515 CrossRef.
- Z. R. Jovanovic, J.-P. Lange, M. Ravi, A. J. Knorpp, V. L. Sushkevich, M. A. Newton, D. Palagin and J. A. van Bokhoven, Oxidation of methane to methanol over Cu-exchanged zeolites: Scientia gratia scientiae or paradigm shift in natural gas valorization?, J. Catal., 2020, 385, 238–245 CrossRef CAS.
- C. Paolucci, I. Khurana, A. A. Parekh, S. Li, A. J. Shih, H. Li, J. R. Di Iorio, J. D. Albarracin-Caballero, A. Yezerets, J. T. Miller, W. N. Delgass, F. H. Ribeiro, W. F. Schneider and R. Gounder, Dynamic multinuclear sites formed by mobilized copper ions in NOx selective catalytic reduction, Science, 2017, 357, 898–903 CrossRef CAS PubMed.
- F. Gao, D. Mei, Y. Wang, J. Szanyi and C. H. F. Peden, Selective Catalytic Reduction over Cu/SSZ-13: Linking Homo- and Heterogeneous Catalysis, J. Am. Chem. Soc., 2017, 139, 4935–4942 CrossRef CAS PubMed.
- K. M. MacKay, R. A. MacKay and W. Henderson, Introduction to Modern Inorganic Chemistry, CRC Press, Cheltenham, 2017 Search PubMed.
- J. Blumberger, L. Bernasconi, I. Tavernelli, R. Vuilleumier and M. Sprik, Electronic Structure and Solvation of Copper and Silver Ions: A Theoretical Picture of a Model Aqueous Redox Reaction, J. Am. Chem. Soc., 2004, 126, 3928–3938 CrossRef CAS PubMed.
- Y. Kuroda, R. Kumashiro and M. Nagao, Prominent redox feature of copper ion exchanged in ZSM-5-type zeolite, Appl. Surf. Sci., 2002, 196, 408–422 CrossRef CAS.
- Y. Kuroda, S. Konno, Y. Yoshikawa, H. Maeda, Y. Kubozono, H. Hamano, R. Kumashiro and M. Nagao, Stabilization of copper metal clusters in mordenite micropores, J. Chem. Soc., Faraday Trans., 1997, 93, 2125–2130 RSC.
- M. Signorile, E. Borfecchia, S. Bordiga and G. Berlier, Influence of ion mobility on the redox and catalytic properties of Cu ions in zeolites, Chem. Sci., 2022, 13, 10238–10250 RSC.
- P. J. Smeets, M. H. Groothaert, R. M. van Teeffelen, H. Leeman, E. J. M. Hensen and R. A. Schoonheydt, in Studies in Surface Science and Catalysis, ed. R. Xu, Z. Gao, J. Chen and W. Yan, Elsevier, 2007, pp. 1080–1087 Search PubMed.
- J. S. Woertink, P. J. Smeets, M. H. Groothaert, M. A. Vance, B. F. Sels, R. A. Schoonheydt and E. I. Solomon, A [Cu2O]2+ core in Cu-ZSM-5, the active site in the oxidation of methane to methanol, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18908–18913 CrossRef CAS PubMed.
- P. J. Smeets, M. H. Groothaert, R. M. van Teeffelen, H. Leeman, E. J. M. Hensen and R. A. Schoonheydt, Direct NO and N2O decomposition and NO-assisted N2O decomposition over Cu-zeolites: Elucidating the influence of the CuCu distance on oxygen migration, J. Catal., 2007, 245, 358–368 CrossRef CAS.
- P. E. Fanning and M. A. Vannice, A DRIFTS Study of Cu–ZSM-5 Prior to and during Its Use for N2O Decomposition, J. Catal., 2002, 207, 166–182 CrossRef CAS.
- G. D. Lei, B. J. Adelman, J. Sarkany and W. M. H. Sachtler, Identification of copper(II) and copper(I) and their interconversion in Cu/ZSM-5 De-NOx catalysts, Appl. Catal., B, 1995, 5, 245–256 CrossRef CAS.
- O. Memioglu and B. Ipek, A potential catalyst for continuous methane partial oxidation to methanol using N2O: Cu-SSZ-39, Chem. Commun., 2021, 57, 1364–1367 RSC.
- P. Xiao, Y. Wang, Y. Lu, T. De Baerdemaeker, A.-N. Parvulescu, U. Müller, D. De Vos, X. Meng, F.-S. Xiao, W. Zhang, B. Marler, U. Kolb, H. Gies and T. Yokoi, Effects of Al distribution in the Cu-exchanged AEI zeolites on the reaction performance of continuous direct conversion of methane to methanol, Appl. Catal., B, 2023, 325, 122395 CrossRef CAS.
- P. Xiao, Y. Wang, K. Nakamura, Y. Lu, T. De Baerdemaeker, A.-N. Parvulescu, U. Müller, D. De Vos, X. Meng, F.-S. Xiao, W. Zhang, B. Marler, U. Kolb, R. Osuga, M. Nishibori, H. Gies and T. Yokoi, Highly Effective Cu/AEI Zeolite Catalysts Contribute to Continuous Conversion of Methane to Methanol, ACS Catal., 2023, 13, 11057–11068 CrossRef CAS.
- C. Dai, Y. Zhang, N. Liu, G. Yu, N. Wang, R. Xu and B. Chen, Mechanistic insight into the effect of active site motif structures on direct oxidation of methane to methanol over Cu-ZSM-5, Phys. Chem. Chem. Phys., 2023, 25, 24894–24903 RSC.
- Y. Chen, N. Liu, C. Dai, R. Xu, G. Yu, N. Wang and B. Chen, Continuous oxidation of methane into methanol by N2O over Cu-Zeolite: A combined experimental and theoretical study, Catal. Today, 2024, 442, 114934 CrossRef CAS.
- R. Xu, N. Liu, C. Dai, Y. Li, J. Zhang, B. Wu, G. Yu and B. Chen, H2O-Built Proton Transfer Bridge Enhances Continuous Methane Oxidation to Methanol over Cu-BEA Zeolite, Angew. Chem., 2021, 133, 16770–16776 CrossRef.
- D. D. Wagman, Selected Values of Chemical Thermodynamic Properties, Institute For Materials Research, National Bureau of Standards, 1965 Search PubMed.
- P. H. Kasai and R. J. Bishop Jr., Thermochemical decomposition of water catalyzed by zeolites, J. Phys. Chem., 1977, 81, 1527–1529 CrossRef CAS.
- R. A. Periana, Comment on ‘Selective anaerobic oxidation of methane enables direct synthesis of methanol’, Science, 2017, 358, eaan5970 CrossRef PubMed.
- D. Palagin, V. L. Sushkevich and J. A. van Bokhoven, Water Molecules Facilitate Hydrogen Release in Anaerobic Oxidation of Methane to Methanol over Cu/Mordenite, ACS Catal., 2019, 9, 10365–10374 CrossRef CAS.
- A. J. Heyer, J. Ma, D. Plessers, A. Braun, M. L. Bols, H. M. Rhoda, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Spectroscopic Investigation of the Role of Water in Copper Zeolite Methane Oxidation, J. Am. Chem. Soc., 2024, 146, 21208–21213 CrossRef PubMed.
- T. Caroline., P. Pereira, J. V. R. Vieira, C. H. F. Da Cunha, S. C. M. Mizuno, Y. O. Carvalho, T. Faheina, M. Picinini, A. L. Blanco, A. C. M. Tello, E. A. Urquieta-Gonzalez, A. Lopez-Castillo, A. M. De Lima, J. B. O. D. Santos and J. M. C. Bueno, Conversion of methane to methanol over Cu-MAZ (zeolite omega): An oxygen-free process using H2O and CO2 as oxidants, Appl. Catal., B, 2024, 342, 123370 CrossRef.
- Y. R. Jeong, H. Jung, J. Kang, J. W. Han and E. D. Park, Continuous Synthesis of Methanol from Methane and Steam over Copper-Mordenite, ACS Catal., 2021, 11, 1065–1070 CrossRef CAS.
- V. L. Sushkevich and J. A. van Bokhoven, Effect of Brønsted acid sites on the direct conversion of methane into methanol over copper-exchanged mordenite, Catal. Sci. Technol., 2018, 8, 4141–4150 RSC.
- A. Koishybay and D. F. Shantz, Water Is the Oxygen Source for Methanol Produced in Partial Oxidation of Methane in a Flow Reactor over Cu-SSZ-13, J. Am. Chem. Soc., 2020, 142, 11962–11966 CrossRef CAS PubMed.
- C. Zhou, S. Li, S. He, Z. Zhao, Y. Jiao and H. Zhang, Temperature-dependant active sites for methane continuous conversion to methanol over Cu-zeolite catalysts using water as the oxidant, Fuel, 2022, 329, 125483 CrossRef CAS.
- H. Zhang, P. Han, D. Wu, C. Du, J. Zhao, K. H. L. Zhang, J. Lin, S. Wan, J. Huang, S. Wang, H. Xiong and Y. Wang, Confined Cu–OH single sites in SSZ-13 zeolite for the direct oxidation of methane to methanol, Nat. Commun., 2023, 14, 7705 CrossRef CAS PubMed.
- B. Modén, P. Da Costa, B. Fonfé, D. K. Lee and E. Iglesia, Kinetics and Mechanism of Steady-State Catalytic NO Decomposition Reactions on Cu–ZSM5, J. Catal., 2002, 209, 75–86 CrossRef.
- C. Lamberti, S. Bordiga, M. Salvalaggio, G. Spoto, A. Zecchina, F. Geobaldo, G. Vlaic and M. Bellatreccia, XAFS, IR, and UV−Vis Study of the CuI Environment in CuI-ZSM-5, J. Phys. Chem. B, 1997, 101, 344–360 CrossRef CAS.
- J. V. R. Vieira, T. C. P. Pereira, C. H. F. da Cunha, D. D. Petrolini, A. C. M. Tello, A. M. Lima, Y. O. Carvalho, A. L. R. Garcia, E. A. Urquieta-Gonzalez, J. B. O. dos Santos, P. M. Lima and J. M. C. Bueno, Isothermal conversion of methane to methanol over Cu-CHA using different oxidants, Catal. Today, 2025, 446, 115121 CrossRef CAS.
- S. J. Freakley, N. Dimitratos, D. J. Willock, S. H. Taylor, C. J. Kiely and G. J. Hutchings, Methane Oxidation to Methanol in Water, Acc. Chem. Res., 2021, 54, 2614–2623 CrossRef CAS PubMed.
- C. Hammond, M. M. Forde, M. H. Ab Rahim, A. Thetford, Q. He, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, N. F. Dummer, D. M. Murphy, A. F. Carley, S. H. Taylor, D. J. Willock, E. E. Stangland, J. Kang, H. Hagen, C. J. Kiely and G. J. Hutchings, Direct Catalytic Conversion of Methane to Methanol in an Aqueous Medium by using Copper-Promoted Fe-ZSM-5, Angew. Chem., Int. Ed., 2012, 51, 5129–5133 CrossRef CAS PubMed.
- C. Hammond, N. Dimitratos, J. A. Lopez-Sanchez, R. L. Jenkins, G. Whiting, S. A. Kondrat, M. H. Ab Rahim, M. M. Forde, A. Thetford, H. Hagen, E. E. Stangland, J. M. Moulijn, S. H. Taylor, D. J. Willock and G. J. Hutchings, Aqueous-Phase Methane Oxidation over Fe-MFI Zeolites; Promotion through Isomorphous Framework Substitution, ACS Catal., 2013, 3, 1835–1844 CrossRef CAS.
- C. Hammond, N. Dimitratos, R. L. Jenkins, J. A. Lopez-Sanchez, S. A. Kondrat, M. Hasbi ab Rahim, M. M. Forde, A. Thetford, S. H. Taylor, H. Hagen, E. E. Stangland, J. H. Kang, J. M. Moulijn, D. J. Willock and G. J. Hutchings, Elucidation and Evolution of the Active Component within Cu/Fe/ZSM-5 for Catalytic Methane Oxidation: From Synthesis to Catalysis, ACS Catal., 2013, 3, 689–699 CrossRef CAS.
- T. Yu, Z. Li, L. Lin, S. Chu, Y. Su, W. Song, A. Wang, B. M. Weckhuysen and W. Luo, Highly Selective Oxidation of Methane into Methanol over Cu-Promoted Monomeric Fe/ZSM-5, ACS Catal., 2021, 11, 6684–6691 CrossRef CAS.
- R. D. Armstrong, V. Peneau, N. Ritterskamp, C. J. Kiely, S. H. Taylor and G. J. Hutchings, The Role of Copper Speciation in the Low Temperature Oxidative Upgrading of Short Chain Alkanes over Cu/ZSM-5 Catalysts, Chem. Phys. Chem., 2018, 19, 469–478 CrossRef CAS PubMed.
- L. Cheng, X. Chen, P. Hu and X.-M. Cao, Advantages and limitations of hydrogen peroxide for direct oxidation of methane to methanol at mono-copper active sites in Cu-exchanged zeolites, Chin. J. Catal., 2023, 51, 135–144 CrossRef CAS.
- J. Ohyama, A. Hirayama, N. Kondou, H. Yoshida, M. Machida, S. Nishimura, K. Hirai, I. Miyazato and K. Takahashi, Data science assisted investigation of catalytically active copper hydrate in zeolites for direct oxidation of methane to methanol using H2O2, Sci. Rep., 2021, 11, 2067 CrossRef CAS PubMed.
- X. Tang, L. Wang, B. Yang, C. Fei, T. Yao, W. Liu, Y. Lou, Q. Dai, Y. Cai, X.-M. Cao, W. Zhan, Y. Guo, X.-Q. Gong and Y. Guo, Direct oxidation of methane to oxygenates on supported single Cu atom catalyst, Appl. Catal., B, 2021, 285, 119827 CrossRef CAS.
- S. A. Yashnik, V. V. Boltenkov, D. E. Babushkin, O. P. Taran and V. N. Parmon, Methane Oxidation by H2O2 over Different Cu-Species of Cu-ZSM-5 Catalysts, Top. Catal., 2020, 63, 203–221 CrossRef CAS.
- B. Wichterlová, J. Dědeček, Z. Sobalík, A. Vondrová and K. Klier, On the Cu Site in ZSM-5 Active in Decomposition of NO: Luminescence, FTIR Study, and Redox Properties, J. Catal., 1997, 169, 194–202 CrossRef.
- V. Umamaheswari, M. Hartmann and A. Pöppl, EPR Spectroscopy of Cu(I)−NO Adsorption Complexes Formed over Cu−ZSM-5 and Cu−MCM-22 Zeolites, J. Phys. Chem. B, 2005, 109, 1537–1546 CrossRef CAS PubMed.
- E. Giamello, D. Murphy, G. Magnacca, C. Morterra, Y. Shioya, T. Nomura and M. Anpo, The interaction of NO with copper ions in ZSM5: An EPR and IR investigation, J. Catal., 1992, 136, 510–520 CrossRef CAS.
- Z. Sojka, M. Che and E. Giamello, EPR Investigation of the Electronic Structure of Mononuclear Copper(I) Nitric Oxide Adduct Formed upon Low-Pressure Adsorption of NO onto Cu/ZSM-5 Zeolite, J. Phys. Chem. B, 1997, 101, 4831–4838 CrossRef CAS.
- M. Anpo, Y. Shioya, H. Yamashita, E. Giamello, C. Morterra, M. Che, H. H. Patterson, S. Webber and S. Ouellette, Preparation and Characterization of the Cu+/ZSM-5 Catalyst and Its Reaction with NO under UV Irradiation at 275 K. In situ Photoluminescence, EPR, and FT-IR Investigations, J. Phys. Chem., 1994, 98, 5744–5750 CrossRef CAS.
- M. Anpo, M. Matsuoka, K. Hano, H. Mishima, T. Ono and H. Yamashita, Photocatalytic decomposition of N2O on Cu+/Y-zeolite catalysts prepared by ion-exchange, Korean J. Chem. Eng., 1997, 14, 498–501 CrossRef CAS.
- H. Yamashita, M. Matsuoka, K. Tsuji, Y. Shioya, M. Anpo and M. Che, In Situ XAFS, Photoluminescence, and IR Investigations of Copper Ions Included within Various Kinds of Zeolites. Structure of Cu(I) Ions and Their Interaction with CO Molecules, J. Phys. Chem., 1996, 100, 397–402 CrossRef CAS.
- D. Ferri, M. S. Kumar, R. Wirz, A. Eyssler, O. Korsak, P. Hug, A. Weidenkaff and M. A. Newton, First steps in combining modulation excitation spectroscopy with synchronous dispersive EXAFS/DRIFTS/mass spectrometry for in situ time resolved study of heterogeneous catalysts, Phys. Chem. Chem. Phys., 2010, 12, 5634–5646 RSC.
- J. W. A. Fischer, F. Buttignol, A. Garbujo, D. Ferri and G. Jeschke, Elucidation of site-specific red-ox kinetics in the CO-assisted N2O decomposition over Fe–ferrierite by combining modulation excitation with operando EPR spectroscopy, Chem. Sci., 2025, 16, 4884–4891 RSC.
- A. Urakawa, T. Bürgi and A. Baiker, Sensitivity enhancement and dynamic behavior analysis by modulation excitation spectroscopy: Principle and application in heterogeneous catalysis, Chem. Eng. Sci., 2008, 63, 4902–4909 CrossRef CAS.
- F. Buttignol, J. W. A. Fischer, A. H. Clark, M. Elsener, A. Garbujo, P. Biasi, I. Czekaj, M. Nachtegaal, G. Jeschke, O. Kröcher and D. Ferri, Iron-catalysed cooperative redox mechanism for the simultaneous conversion of nitrous oxide and nitric oxide, Nat. Catal., 2024, 1–11 Search PubMed.
Footnotes |
| † Current address: Institute for Catalysis, Hokkaido University, N-21, W-10, Sapporo 001-0021, Japan. |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2026 |