
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCluster-based materials: design and applications
Hong
Fang
 abc,
Qiang
Sun
d and
Puru
Jena
*ef
abc,
Qiang
Sun
d and
Puru
Jena
*ef
aDepartment of Physics, Rutgers University-Camden, Camden, New Jersey 08102, USA. E-mail: hong.fang@rutgers.edu
bCenter for Computational and Integrative Biology, Rutgers University, Camden, New Jersey 08103, USA
cDepartment of Materials Science and Engineering, Rutgers University-New Brunswick, Piscataway, New Jersey 08854, USA
dSchool of Materials Science and Engineering, Peking University, Beijing 100871, China. E-mail: qsun@vcu.edu
eDepartment of Physics, Virginia Commonwealth University, Richmond, Virginia 23284, USA. E-mail: pjena@vcu.edu
fInstitute for Sustainable Energy and Environment, Virginia Commonwealth University, Richmond, Virginia 23284, USA
First published on 20th March 2026
Abstract
Nanoclusters constitute an interdisciplinary field bridging chemistry, physics, biology, medicine, and materials science. Its broad appeal is fueled not only by the expectation that one can gain a fundamental understanding of how the structure and properties of matter evolve one atom at a time, but also, how they could be designed, with specific size and composition, to mimic the chemistry of atoms in the periodic table. Referred to as superatoms, these clusters can function as modular building units for a new generation of cluster-assembled materials, provided they retain their structural identity upon integration into extended architectures. C60 fullerene is an example of such a cluster. Clusters, protected by ligands or supported on a substrate, can also form cluster-assembled materials with unique properties. In contrast, atoms in an existing crystal can be replaced by corresponding superatoms to form “cluster-based materials”. While several perspectives and review articles have been published to highlight the unique structure- and composition-specific properties of isolated clusters and cluster-assembled materials, it is during the past few years that attention has been focused on cluster-based materials. This review focuses on the recent developments in the design, synthesis, and applications of clusters with uncommon properties and highlights their potential as building blocks of cluster-based materials. The topics include isolated clusters enabling unusual reactions, their role in the design and synthesis of materials such as super-electrides, solid state electrolytes with fast ionic conductivity and stable interfaces, moisture resistant hybrid perovskite solar cells, thermoelectric materials with high figure of merit, and a new class of single-superatom catalysts. Also outlined are the challenges and opportunities going forward.
 Hong Fang | Hong Fang is an Assistant Professor of Physics in Rutgers University-Camden. He received his Doctorate in materials physics from the University of Cambridge and his Bachelor of Science in physics from East China Normal University and Bielefeld University. His research is in theories and modeling of solid-state materials and biopolymers, especially in discovering and understanding energy materials beyond Li/Na systems as well as behaviors of ion transport and kinetics at the interfaces. He has over 60 peer-reviewed publications in the related fields. |
 Qiang Sun | Dr Qiang Sun is a professor at Peking University. He received PhD in condensed matter physics from Nanjing University. His research focuses on cluster physics, low-dimensional magnetism, and phonon transport. He also works on machine-learning-assisted design of functional materials for hydrogen storage, battery electrodes and electrolytes, thermoelectric materials, and CO2 capture and conversion. Professor Sun and his collaborators made the first theoretical prediction of stable ferromagnetism in CrI3 monolayer, later confirmed experimentally, initiating the field of 2D magnetism. He was elected an IAAM Fellow in 2023 and was recognized among the top 0.1% Highly Cited Scientists in 2025. |
 Puru Jena | Dr Puru Jena is Distinguished Professor of Physics and Director of the Institute for Sustainable Energy and Environment at Virginia Commonwealth University. He received his PhD in Physics from the University of California at Riverside. He also served as a Program Director at the National Science Foundation and as a Jefferson Science Fellow and Senior Science Advisor at the US Department of State. Dr Jena's research covers a wide range of topics in nano-structured materials including atomic clusters, condensed matter Physics, chemistry, and materials Science. He is the author of more than 700 papers including 14 edited books. |
I. Introduction
The periodic table of elements is the foundation of all materials, however, its scope is constrained by the finite number of naturally occurring elements, many of which are rare or pose environmental and toxicity concerns. The Alchemists’ dream to replace gold with base metals never materialized because the chemistry of gold is pre-determined by its atomic orbitals that cannot be altered. Fortunately, advancement in new experimental and theoretical techniques over the past few decades have enabled researchers to manipulate and control properties of matter at the nanoscale by tailoring their size, shape, and composition and producing materials with unique properties. However, at this length scale, properties are not influenced by the addition or removal of a single atom.Atomic clusters, defined as an aggregate of atoms with finite size and composition, bridge not only this gap but also their properties can be changed by the addition or removal of a single atom or even a single electron.1–15 With developments in experimental techniques, one can now produce clusters of most elements with varying size and composition in the gas phase.16–22 Soft-landing of mass-isolated clusters on substrates23–25 as well as those assisted with ligands26–34 or guided by molecular templates35–38 have made it possible to synthesize cluster-assembled materials with unique properties. These experimental techniques combined with high-throughput calculations based on the density functional theory have enabled researchers to study the structure and properties of homo- and hetero-atomic clusters containing many atoms.39–42 The early motivation in studying clusters as a function of size was to understand how the properties of matter evolve, one atom at a time.43–45 While some understanding has been achieved in this regard, the answer to “when a cluster becomes a crystal” remains largely unanswered. This is because different properties evolve differently, making it difficult to find a unique answer. For example, a recent study showed that five H2O molecules can make a water drop,46,47 but five gold atoms do not make an inert gold. However, past studies have revealed many unusual size- and composition-specific properties of clusters. These include clusters mimicking the properties of atoms,5 otherwise nonmagnetic elements becoming magnetic,48 chemically inert elements becoming reactive,49 atoms achieving oxidation states higher than nature intended,50 clusters with like charges attracting,51 noble gas atoms reacting under ambient conditions,52etc. These findings have fueled much interest in cluster science.53–56
One of the unique properties of clusters this review focuses on is clusters as “superatoms” and their role in the design and synthesis of cluster-based materials. The concept of superatoms originated from the pioneering work of Khanna and Jena,57,58 who demonstrated that atomic clusters with specific size and composition can emulate the chemical properties of elements in the periodic table. This idea was motivated by two key experiments: (1) the observation of magic numbers in sodium clusters by Walter Knight and coworkers who explained their unusual stability using the jellium model to be due to electronic shell closure (e.g., 1S, 1P, 1D).59 (2) The observation by Will Castleman and co-workers60 that the Al13− cluster is highly stable and chemically inert towards oxygen due to atomic and electronic shell closure. Building on these findings, Jena and Khanna proposed that nearly spherical clusters with delocalized electrons filling superatomic orbitals according to an Aufbau principle could be designed to mimic atomic behavior. They further suggested that such clusters could serve as building blocks for cluster-assembled materials with uncommon properties, provided their structural and electronic integrity is retained upon assembly. Studies of supertoms have increased over the past 30 years as evidenced by the growing number of publications and their unusual properties (see Fig. 1).
 | ||
| Fig. 1 (A) Increasing number of papers containing the key word of superatoms either in the title or in the abstract published the past 30 years. (B) Works on isolated clusters and cluster-assembled materials bridging physics, chemistry, materials science, biology, and medicine, and enriching our understanding of the physical and chemical properties of matter at the nano-scale. | ||
The superatom concept has since expanded to include superalkalis and superhalogens, first proposed by Gutsev and Boldyrev in 1980's.61,62 Superalkalis (e.g., Li2F) have ionization potentials lower than that of alkali metals, while superhalogens (e.g., LiF2) have electron affinities exceeding those of halogens. Although these clusters are not spherical and do not exhibit superatomic orbitals, they do mimic the chemistry of alkali and halogen atoms, respectively. Some ions such as BH4−, CN−, CB11H12−, traditionally known in chemistry as polyatomic ions or complex ions, also exhibit superhalogen properties and have been used to substitute halogens, yielding materials with uncommon properties. In the past few years, numerous superhalogens and superalkalis have been designed using the octet rule,63,64 18-electron rule,65,66 32-electron rule,67,68 Hückel's rule,69,70 and Wade's rule.71–74 Some of these have been synthesized, forming novel materials. In addition, core–shell structures such as ligated metal chalcogenides75–78 and thiolated noble metal clusters26–34,79–83 have been classified as superatoms as they are nearly spherical and do exhibit superatomic orbitals, even though they may not mimic the chemistry of a particular atom in the periodic table. The bonding in these clusters has been characterized as concentric bonding in superatoms.84 This refers to a unique bonding pattern where atoms within a cluster arrange in nested spherical layers, resembling the electronic shell structures of noble gas atoms. In addition to the ligated clusters, some other examples of concentric bonding in metallic clusters include Au13, Au18, Pd6O4, and a cage-like boron cluster such as B12. Inspired by the quark model, a grand unified model for ligated Au clusters has also been proposed that accounts for the structure of 71 such clusters.85 Regardless of whether the above species are termed superatoms, polyatomic ions, or complex ions, their fundamental role as building blocks for new materials remains unchanged. To quote William Shakespeare, “a rose by any other name would smell as sweet”. The ligated clusters have rich chemistry and have been used to synthesize materials with unique properties.
In Table 1, we summarize the key reasons why superatomic clusters, instead of atoms whose chemistry they mimic, can serve as transformative building blocks for constructing novel materials. However, atomic clusters produced in the gas phase under inert condition are typically unstable once removed from that environment. Upon exposure to ambient conditions, they tend to react with surrounding gases and/or coalesce with neighboring clusters, thereby losing their individual identity, structure, and properties. To retain their identity, mass-isolated clusters could be soft-landed on substrates,23–25 protected by ligands,26–34 or guided by molecular templates.35–38 However, in these cases, interaction of clusters with the substrates or ligands must be considered which can also be used for further functionalization of the material properties. Another way to use clusters as a building block is to replace an atom in an existing material with a corresponding superatomic cluster. We refer to such materials as “cluster-based materials,” which forms the focus of this review. Given that several recent reviews have already comprehensively addressed core metal clusters protected by complex ligands (including metal chalcogenide clusters, Au/Ag nanoclusters, and mass-isolated clusters on substrates), we do not discuss these topics further here due to space constraints.26–34,75–83,86–104
| Atoms as building blocks | Superatoms as building blocks |
|---|---|
| Number of atoms is fixed. | Number of superatoms is unlimited. |
| Properties of atoms are governed by their atomic orbitals which cannot be changed. | Molecular orbitals of superatoms mimicking the chemistry of an atom can be changed by varying their size and composition. |
| Ionization potentials of alkali atoms and electron affinities of halogen atoms are fixed. | Superalkalis are characterized by ionization potentials lower than those of alkali metals, and superhalogens display electron affinities surpassing those of halogens. Such extreme electronic properties are not fixed; rather, they can be engineered by tailoring the cluster's size and chemical composition. |
| Most atoms cannot accommodate more than one extra electron. | Superatoms can accommodate multiple electrons. |
| In atom-assembled solids, the phonons are due to the vibration of atoms. | In cluster-based solids, phonons are due to intra-cluster as well as inter-cluster vibrations, thus fundamentally altering electron–phonon coupling. |
| Lattice constants and lattice symmetries are fixed. | Lattice constants and symmetries can be varied with size and composition. |
| Energy bands are formed by overlap of atomic orbitals. | Energy bands are formed by overlap of superatomic orbitals. |
Although several review articles and perspectives on superatoms have been published,53–56,105 this review focuses primarily on the developments over the past 5–10 years and illustrates how superatoms can promote unusual reactions enabling noble gas atoms to form chemical bonds at room temperature, and lead to unconventional electrides, solid state electrolytes with fast ionic conductivity and stable interfaces, moisture resistant hybrid perovskite solar cells, thermoelectric materials with high figure of merit, and superior catalysts. Also discussed are challenges and opportunities moving forward.
In Section II, we briefly outline the design rules and synthesis techniques of superatoms. Their unusual chemical and physical properties are described in Sections III and IV, respectively. Section V deals with the applications of cluster-based materials in energy production and storage, focusing on light absorbers in solar cells and solid-state electrolytes in metal-ion batteries. Superatoms with unusual ferroelectric, piezoelectric and thermoelectric properties are discussed in Section VI. A summary of our conclusions and the outlook focusing on challenges and opportunities are presented in Section VII.
II. Design rules and synthesis of superatoms and cluster-based materials
In this section, we discuss briefly how the superatomic clusters and cluster-based materials are theoretically designed and experimentally synthesized. In Table 2, we summarize the various electron counting rules used to design superatoms. The design rule that was initially used exclusively for simple metals is the jellium rule, originally used to describe the magic numbers in Na clusters.59 A good example of this design is the Al13 cluster.60 With 39 valence electrons, Al13 requires only one electron to complete its 1S 1P 1D 2S 1F 2P electronic shells. This is similar to halogens, which also require one electron to complete their outermost np shell. Hence, Al13 behaves like a halogen atom. Indeed, the electron affinity of Al13 cluster, namely, 3.6 eV, is very close to that of the chorine atom. Later, the octet,63,64 18-electron,65,66 and 32-electron rules67,68 have been used to design superatoms composed of low-atomic-weight atoms, transition metal atoms, and rare-earth atoms, respectively. Similarly, Wade's rule71–74 and Hückel's and Hirsch's rule69,70,106 have been used for boranes and aromatic molecules, respectively.107,108 Several superatoms mimicking the chemistry of halogens, chalcogens, pnictogens, and alkalis, as well as transition metals, have been designed and will be discussed in the following sections. Unlike in atoms, multiple electrons counting rules can be simultaneously used to create superatoms with uncommon properties. An example of such a system is B12(CN)12, where both the Wade's rule and the octet rule are simultaneously used to create an extraordinarily stable dianion. This will be discussed at length in the forthcoming sections.| Rule | Principle | Conditions | Best for | Properties | Examples | Notes |
|---|---|---|---|---|---|---|
| a No single rule universally applies; a hybrid approach sometimes is needed due to complexity from bonding and compositions (transition metals, main-group elements, and ligands). b Design rules are guidelines not absolutes, real superatoms can defy them due to factors such as geometry, ligand effects, relativistic stabilization, and electronic correlation. c Ref. 79. | ||||||
| Jellium rule | Stability arises from closed-shell electronic configurations in a spherical potential well: 2, 8, 18, 20, 34, 40, 58, 92… electrons. | – Delocalized valence electrons | Alkali, noble metal clusters (free or ligand-protected) | – Ionization energies peak at magic numbers | Na8, Na20, Mg40, Al13−, Au25(SR)18−, Ag44(SR)30 | The count of ‘free’ or delocalized electrons: n = NVm − YVX + SVL − z, where N is the number of metal atoms (M); S is the number of charge-donating ligands (L), Y is the number of charge-withdrawing ligands (X); Z is the overall charge; VM, VX and VL are the valence numbers of the metal and the ligands.c |
| – Nearly spherical geometry | – Delocalized metallic bonding | Au102(SR)44 | ||||
| – Uniform positive charge background | Au144(SR)60 | |||||
| SiAl14(C5Me5)6 | ||||||
| Wade's rule | Polyhedral skeletal structures require specific numbers of bonding electron pairs (NBEP) based on cluster geometry (closo, nido, arachno, hypho). | – Polyhedral or cage-like clusters | Boranes, carboranes, metallo-boranes ligand-protected clusters | – NBEP = n + 1: closo (complete polyhedron) | B6H62−, C2B10H12 | NBEP = (M × e− per metal) − xL × (ligand electrons) ± cluster charge |
| – Mostly applies to boranes, metallo boranes, and ligand-stabilized metal clusters | – NBEP = n + 2: nido (one missing vertex from closo) | Fe3(CO)12 | – Subtract non-skeletal electrons (e.g., ligands). | |||
| – NBEP = n + 3: arachno (two missing vertices) | Au25(SR)18−, Ag25(SR)18− Al7C−, [Pd6(CO)12]2+ | – divide remaining electrons by 2 to get skeletal electron pairs. | ||||
| – NBEP = n + 4: hypo (three missing vertices) | Al4H6, Al4H42−, Sn122−, Pb122−, AlPb12+, | – Match to NBEP formula to determine geometry type. | ||||
| Octet rule | Main-group atoms are most stable with 8 valence electrons (full s and p orbitals). | – Small main-group clusters | Main-group anions, superhalogens, molecular clusters | Superhalogens and superalkalis | ZrO, BO2−, AlO2−, BH4−, AlF4−, BF4−, MnO4− | Octet-rule superatoms often resemble covalent or ionic molecular systems with localized bonding. |
| – Atoms with accessible s and p orbitals | ||||||
| – No significant d-orbital participation | ||||||
| 18-Electron rule | Maximum stability occurs when a transition metal atom or cluster has 18 valence electrons (full s, p, d shells). | – Transition metal center | Organometallic complexes, transition metal-centered superatoms | Transition metal based 18-e superatom can be magnetic such as Mn@Si12 and Cr@Si12 | Fe(CO)5, Ni(CO)4, Mo(CO)6, V(CO)6 | Total valence electrons = metal valence electrons + ligand electrons ± charge. |
| – Ligand field strong enough to define orbital splitting | Ru3(CO)12 | |||||
| – No strong electron repulsion | TaAu12−, WSi12 | |||||
| IrCl(CO)(PPh3)2 | ||||||
| [Pd6(μ3-S)8(PPh3)6]2+ | ||||||
| 32-Electron rule | 32 valence electrons occupying cluster orbitals with spherical harmonics character, the cluster is stable with a closed-shell electronic configuration | – Spherical or near-spherical symmetry. | - 5f-Element encapsulated cages | Can be used to mimic xenon or a heavy element analog. | Pu@Pb12, Ge@Sn122−, Pu@C24 An@C28 (An = Th, Pa+, U2+, Pu4+), CBe8H12, U@B40 | – The 5f orbitals in actinides are chemically accessible, radially extended, and capable of covalent bonding, making them uniquely well-suited for stabilizing 32-electron clusters. |
| – Electrons are delocalized over the whole cluster | – Main-group-based cages with delocalized bonding, electron-rich ligand frameworks and high symmetry | – Due to the complex physics of f-orbitals, actinide-based clusters exhibit rich and complex magnetic properties | – Both the central atom's electrons and ligand/cage contributions are included in the counting. | |||
| Hückel's rule | Aromatic stability in planar, cyclic systems with (4n + 2) π-electrons | – Planar, cyclic geometry | π-Conjugated rings, metal rings, small planar clusters | Delocalized 2D aromaticity | Benzene, C4H42−, B62−, Al42−, LiAl4−, Na5+, Be3−, Cu42+, Ag42+, | Some B clusters are aromatic with 4n electrons due to multi-center bonding. |
| – Conjugated π-system | ||||||
| – Electron delocalization over ring | ||||||
| Hirsch's rule | A 3D superatom is aromatic containing 2(n + 1)2 delocalized π-electrons | Spherical symmetry, delocalized π-electrons | Spherical delocalization with correct number of π-electrons | Spherical electron cloud distributes electronic charge evenly across the structure to minimize repulsion | C202+, C6010+, [Sn12]2−, Au135+ | Often applies to charged species or zintl ions where π-electrons can be tuned. |
In Table 3 we summarize the different experimental techniques used for the synthesis of superatoms. In the gas phase, superatoms can be formed by using gas evaporation, laser vaporization, or sputtering techniques.16–25 Metal-encapsulated clusters such as M@Si16 (M = Sc, Ti, V, Zr, Ta, Hf, Lu, W) generated in the gas phase can be soft landed on substrates such as C60, preserving the cage structure of 16 Si atoms uniformly distributed around the central metal atom.24 Substrates can also be used to further tune the electronic and chemical properties of the deposited superatoms through cluster-surface interaction. For instance, a p-type organic substrate of hexa-tert-butyl-hexa-peri-hexabenzocoronene (HBHBC, C66H66) substrates is employed for the deposition of halogen-like superatom Lu@Si16, where the substrate can donate one electron to the superatom to close its electron shell.24 Similarly, a W@Si16 superatom, when deposited on an n-type substrate C60, is charge positive, while it is negatively charged with enhanced oxidation reactivity when deposited on a p-type HBHBC substrate.109 In addition, ligated superatomic M2@Au17 (M = Pd, Pt) clusters have been synthesized via a novel photoinduced fusion of M@Au12 superatoms triggered by photo-irradiation under visible light.110 The unprecedented M2@Au17 superatom has a structure of two M@Au12 cores sharing Au5 facets in the neutral form and exhibit an electronic configuration like that of the N2 molecule.110
| Methods | Features | Examples16–38,109–121 |
|---|---|---|
| Gas-phase synthesis of isolated clusters | Providing high precision in producing atomically precise superatomic clusters but being impractical for large-scale production. | Al13, Na8, Na20, Al13−, Au20, W@Au12, Nb11O15− |
| Supported clusters aided by gas phase synthesis | M@Si16 (M = Sc, Ti, V, Zr, Ta, Hf, Lu, W) | |
| Photo-induced fusion reaction in solution | Stabilizing atomically precise superatomic clusters by ligands and fusion, but this technique is valid for specific elements and sizes. | M@Au12 (M = Pd, Pt) |
| Solution-based synthesis of ligated clusters | Au25(SR)18, PdAu24(SR)18, PtAu24(SR)18, PtAg42, Co6Se8(PEt3)6, Co6S8(PEt3)6, PtAg18, MAg24(SR)18 (M = Ni, Pd, Pt), [IrHAg24(SPhMe2)18]2−, [OsH2Ag24(SPhMe2)18]2−, [RuH2Ag24(SPhMe2)18]2−, [Au6Al6](Cp*)6, [HAu7Al6](Cp*)6, | |
| Molecular template-guided synthesis | Employing molecular templates to direct the assembly of atoms into well-defined superatomic structures with precise atomic control, but limited heavily by the availability and design of suitable molecular templates | Al13−, PtBr4, FeSn12, Au25PET18 |
Ligand-protected nanoclusters with Au/Ag/Cu/Al metal cores constitute a large family of superatoms, where the metal core is electronically stabilized by ligands against aggregation in solution or in solid state. A well-known example is [Au25(SR)18]− (SR = thiolate), which exhibits an icosahedral Au13 core protected by six Au2(SR)3 ligands. There are many ligated metalloid gold clusters with different core sizes, such as Au13, Au20, Au23, Au24, Au25, Au26, Au30, Au36, Au38, Au55, Au60, Au99, Au102, Au103, Au133, and Au144.111 Among these, larger clusters can be converted to smaller ones by thermochemical etching with exogenous thiols, while smaller-to-larger conversion can be realized using fusion reactions in solutions.93 In general, three methods are used to synthesize ligated Au clusters, including reduction of gold precursors, conversion between different-sized clusters, and metallic Au evaporation.82 Other superatomic clusters can also be prepared by doping heterometals into the existing ligand-protected Au and Ag clusters. For example, a set of diphosphine-protected MAu12 (M = Ru, Rh, Ir) clusters have been synthesized by doping d-shell metals in the Au13 cluster,112 with new synthetic methods to make atomically precise open sites by controlled removal of ligands.30 Similarly, d-metal doped Ag clusters have been synthesized, including MAg24(SR)18 (M = Ni, Pd, Pt), [RhH@Ag24(SPhMe2)18]2−, [IrHAg24(SPhMe2)18]2−, [OsH2Ag24(SPhMe2)18]2−, and [RuH2Ag24(SPhMe2)18]2− (SPhMe2 = 2,4-dimethylbenzenethiolate).113–115 Most recently, new hydrocarbon ligated heteronuclear gold clusters with p-metals (e.g., Al), such as [Au6Al6](Cp*)6 and [HAu7Al6](Cp*)6, have also been synthesized by using various phosphines as additives on the reduction of the Au precursors.116
Another large family of superatoms is based on ligated metal chalcogenide clusters, M6E8 (M = metal, E = chalcogen), where the metal atoms are arranged on an octahedron and are contained in a cube of chalcogen atoms. Each metal atom in these clusters is coordinated by ligands L (e.g., phosphines), forming M6E8L6. These superatoms are typically synthesized by combining a precursor of a metal source and phosphine chalcogenide in solutions.77 By mixing such clusters of similar size, shape, and chemical properties, e.g., Co6Se8(PEt3)6 and Cr6Te8(PEt3)6 (PEt3 = triethylphosphine), solid solutions at the cluster level and the corresponding [cluster][C60]2 crystals can be synthesized with novel properties.28 Controlling the ligand coordination spheres of the metal chalcogenide clusters in the solution phase can lead to novel cluster fusion and materials, allowing rational control over the synthetic process beyond traditional photolytic or thermal methods.27 For example, carbene and multi-carbene adducts have been used to selectively modify the ligands of [Co6Se8L6] (L = PEt3, CO, C(H)SiMe3) clusters to tailor the cluster fusion reactivity and the shape of the cluster core, as well as create site-specific functionalized dimers and unprecedented cyanide/pyridine adducts that lead to novel solids.27
Atom-by-atom substitution in the metal core of the metal chalcogenide clusters has also proven to be a viable strategy for the design of new superatoms with tailored electronic, magnetic, and optical properties. For example, the stability, magnetic, and optical properties of cobalt sulfide superatom [Co6S8L6]+ ligated with triethylphosphine (L = PEt3) can be controlled by substituting Fe or Ni for Co in the core.78 Interestingly, it is found most recently that, by using NiCl2 as a precursor in the solution-phase synthesis, nickel sulfide clusters with a Ni3S3 core (with three Ni atoms arranged in a planar triangle and three bridging S atoms residing between Ni) are preferentially formed, instead of having the regular metal chalcogenide Ni6S8 core. The subsequently protonated Ni3S3H(PEt3)5 exhibits strong ferromagnetic ordering and coercivity.117
Solution-phase synthesis of superatomic cluster using dendrimer templates have become another attractive method, since the size of the generated clusters can be controlled in the state of the precursors.38,118–121 Compared to the conventional methods, such a template-guided method can support short-time synthesis, circumventing special conditions, such as low temperature or complex reactions. For example, the thiol-protected Au25, PdAu24, and PtAu24 superatoms can be facilely synthesized using AuCl3 and a phenylazomethine dendrimer template with a tetraphenyl methane core part.38 Most recently, the FeSn12 superatomic cluster that had only been synthesized in the gas phase before was synthesized in solution using FeCl3 and SnCl2 on the dendrimer templates.37
III. Unusual chemical properties of superatoms
The chemistry of atoms is governed primarily by the valence electrons which are fixed. On the other hand, the chemistry of the superatoms is governed by their molecular orbitals which can be tailored by their size and composition, hence enabling superatoms to exhibit rich chemistry. We demonstrate this by focusing on how a multiply charged superatom can be stable while the atom whose chemistry it mimics cannot hold more than one excess electron. Such superatoms can usher unusual reactions by making noble gas atoms to form chemical bonds at room temperature. Similarly, superalkalis can be used as building blocks of super electrides and a single superatom composed of earth-abundant materials can outperform single-atom catalysts such Pd.III.1 Stabilizing multiply charged anions
Although multiply charged anions, protected by charge compensating cations, can be stable in matrices or in solvents, isolated atoms cannot hold more than one electron. On the contrary, an isolated superatom can accommodate more than one electron if its binding energy is larger than the repulsive energy between the additional electrons. An intriguing question always has been: what is the smallest superatom that can be stable as a dianion? A classic example of a stable dianion is dodecaborane, B12H122−, whose second electron is bound by 0.9 eV, although C602−, which is larger in size, turns out to be unstable. Further, the second electron affinity of B12H122− can be systematically increased by replacing H with halogen atoms122 and can reach as high as 5.28 eV when H is replaced by CN.52,123 This highlights the importance of the electronic structure of the parent cluster as well as those of the ligands, suggesting that it may be possible to increase the stability of a multiply charged cluster by tailoring its size and composition. For example, it has been shown theoretically that by replacing a B atom by Be in B12(CN)122− cluster, the resulting BeB11(CN)123− trianion can be stabilized.124 This is consistent with the zintl-Klemm pseudo-element concept, given that Be− is isoelectronic with B.125Fang and Jena established a general model to design clusters that can be stable while accommodating as many as 5 extra electrons.126,127 They started with the underlying principle that in stabilizing a multiply charged anionic cluster in the gas phase, the excess electrons must occupy a series of bound states within a 3D potential well, whose depth can be approximated by the total energy required to reduce the highest negatively charged state to neutrality—that is, the cumulative sum of the electron affinities (EAn) associated with each added electron, while the width of the potential well can be measured by the radius of a cluster defined as half of its longest dimension, per Fig. 2A. The general trend is that a more negative state requires the cluster to be in a deeper and larger “well” to hold more bound electrons. In practice, the “well depth” as well as the “well width” of a cluster can be manipulated by tailoring the core and terminal groups attached to the periphery of the cluster. Thus, stable clusters carrying multiple charges (beyond “−2”) can be formed by assembling lesser-charged clusters and terminal groups each of which already satisfies a closed shell rule with high EA (at the corresponding charge state) and large size. For example, dodecaborane cluster B12H122−, stabilized at −2 charge state, can be readily functionalized and reach a variety of charge states and electron seizing powers by changing one of the core atoms as well as the ligands, e.g., B by Be and H by CN which can be accomplished by experiments.52
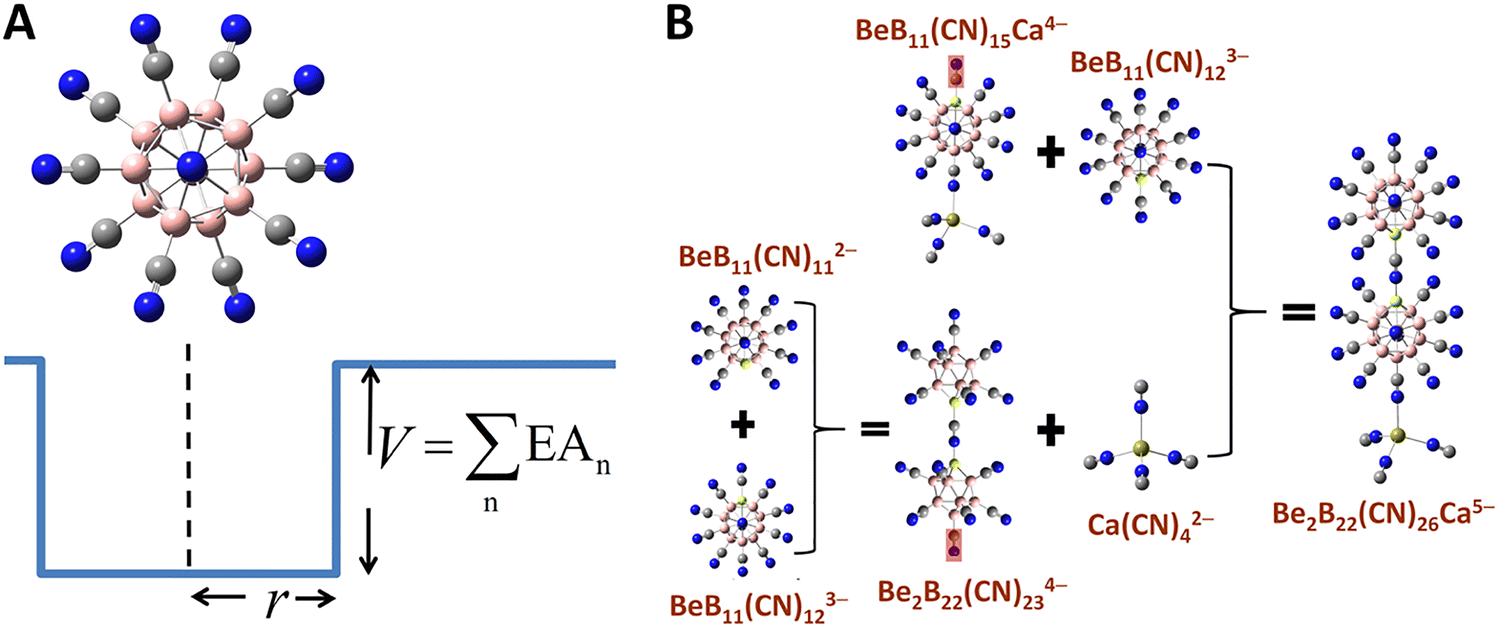 | ||
| Fig. 2 Systematic approach to design stable multiply charged clusters in the gas phase. (A) The general potential well model of a stable cluster with its dimension corresponding to the radius (R) and the sum of electron affinities (EAn) of the cluster.126 (B) Stable tetra- and penta-anions created by assembling lesser-charged clusters based on the general model, involving removal of CN− ligands in some cases (highlighted in red shades).126 | ||
In Table 4, we list the calculated electron affinities of clusters as successive electrons are added. Note that among all the atoms, halogens have the highest electron affinity, but superhalogens such as CN−, SCN−, BH4−, and BF4− (these are often referred to as polyatomic ions) have electron affinities even higher than that of the halogen atoms. However, none of these superhalogens are stable as di-anions such as M(CN)42− (M = Mg, Ca, Sr, Ba).128 Replacing H by CN in B12H122− increases the second electron affinity substantially, namely to 5.28 eV, in B12(CN)122− dianion. This prediction has been experimentally confirmed with measured second electron affinity at 5.5 eV.52 Replacing one B atom by Be in B12(CN)122− makes it possible for BeB11(CN)123− to be stable with the calculated third electron affinity of 2.65 eV. This trianion is a combination of monoanion (superhalogen) CN− and a stable dianion BeB11(CN)112−. According to the generalized theory given above,126 these stable dianions and trianions can be used to assemble tetra- and penta-anions that are stable at “−4” and “−5” charge states, respectively. Combining dianions BeB11(CN)112− and M(CN)42− (M = Ca, Sr, Ba) leads to a set of stable tetra-anions BeB11(CN)11M(CN)44− (M = Ca, Sr, Ba) with EA4 (the fourth electron affinity) being 0.79, 0.20 and 0.25 eV, respectively. Another stable tetra-anion is Be2B22(CN)234−, with EA4 as large as 1.48 eV by attaching BeB11(CN)112− to BeB11(CN)123−. A stable penta-anion Be2B22(CN)26Ca5− with EA5 of 30 meV and a vertical detachment energy of 180 meV is created by combining two BeB11(CN)112− with one Ca(CN)42−, as shown in Fig. 2B. These results are based on density functional theory and fully optimized clusters and have predictive capability as evidenced by the good agreement between theory and experiment for B12(CN)122−.
| Anion species | IR (Å) | EA1 | EA2 | EA3 | EA4 | EA5 |
|---|---|---|---|---|---|---|
| Cl− | 1.81 | 3.62 | — | — | ||
| Br− | 1.96 | 3.36 | — | — | ||
| I− | 2.20 | 3.06 | — | — | ||
| CN− | 1.40 | 4.10 | — | — | ||
| SCN− | 2.50 | 3.50 | — | — | ||
| SeCN− | 2.70 | 3.39 | — | — | ||
| BH4− | 2.24 | 3.42 | — | — | ||
| BF4− | 2.75 | 7.33 | — | — | ||
| BCl4− | 3.69 | 5.01 | — | — | ||
| B12(CN)11− | 5.86 | 8.49 | — | — | ||
| B12H122− | 3.91 | 4.57 | 0.86 | — | ||
| B12(CN)122− | 5.88 | 8.56 | 5.28 | — | ||
| B12(SCN)122− | 7.79 | 5.65 | 3.28 | — | ||
| BeB11(CN)112− | 5.89 | 8.10 | 4.62 | — | ||
| M(CN)42− (M = Ca, Sr, Ba) | 3.77–4.07 | 7.21–7.08 | 2.62–2.79 | — | ||
| BeB11(CN)123− | 6.11 | 8.44 | 4.76 | 2.65 | ||
| BeB23(CN)223− | 10.15 | 8.86 | 6.38 | 3.25 | ||
| BeB11(CN)11M(CN)44− (M = Ca, Sr, Ba) | 7.03–7.39 | 7.63–4.07 | 5.20–5.15 | 1.77–2.44 | 0.79–0.20 | — |
| Be2B22(CN)234− | 10.15 | 8.86 | 5.98 | 3.54 | 1.48 | — |
| Be2B22(CN)26Ca5− | 11.66 | 8.37 | 5.98 | 4.25 | 1.88 | 0.18 |
III.2 Super-electrophiles enabling Argon and N2 to form chemical bonds at room temperature
Here, we discuss how very stable multi-anionic clusters can be used to fundamentally change the chemistry of noble gas atoms, making them form chemical bonds at room temperature. Note that noble gas atoms are chemically inert due to the octet shell closure rule.63,64 They are characterized with high ionization potential and zero to negative electron affinity. Ways to make noble gas atoms to form chemical bonds has been a topic of interest for more than a century. Pauling first suggested that Kr and Xe, due to their large size, can be polarized and form chemical bonds.129,130 Thirty years later, this was experimentally confirmed by Bartlett.131 Since then, numerous unsuccessful efforts have been made to see if Ne and Ar can form chemical bonds at room temperature.132–139The prediction of highly stable B12(CN)122− (ref. 123) and its experimental discovery52 changed this scenario. Recent studies have shown that superatomic electrophiles can exert enough oxidative power to even bind the noble gases. When one of the ligands in B12(CN)122− is removed, the B atom without a ligand in B12(CN)11− remains positively charged and becomes super electrophilic. In addition, B12(CN)11− with the extra electron spread over the 11 CN ligands and the positive charge on the “naked” B atom, possesses a dipole moment. The superatomic electrophile becomes reactive, capable of binding an Ar atom chemically at room temperature, as shown in Fig. 3A. Consequently, argon loses its nobility.49,52,140 The binding energy of Ar to B12(CN)11− is 0.61 eV.
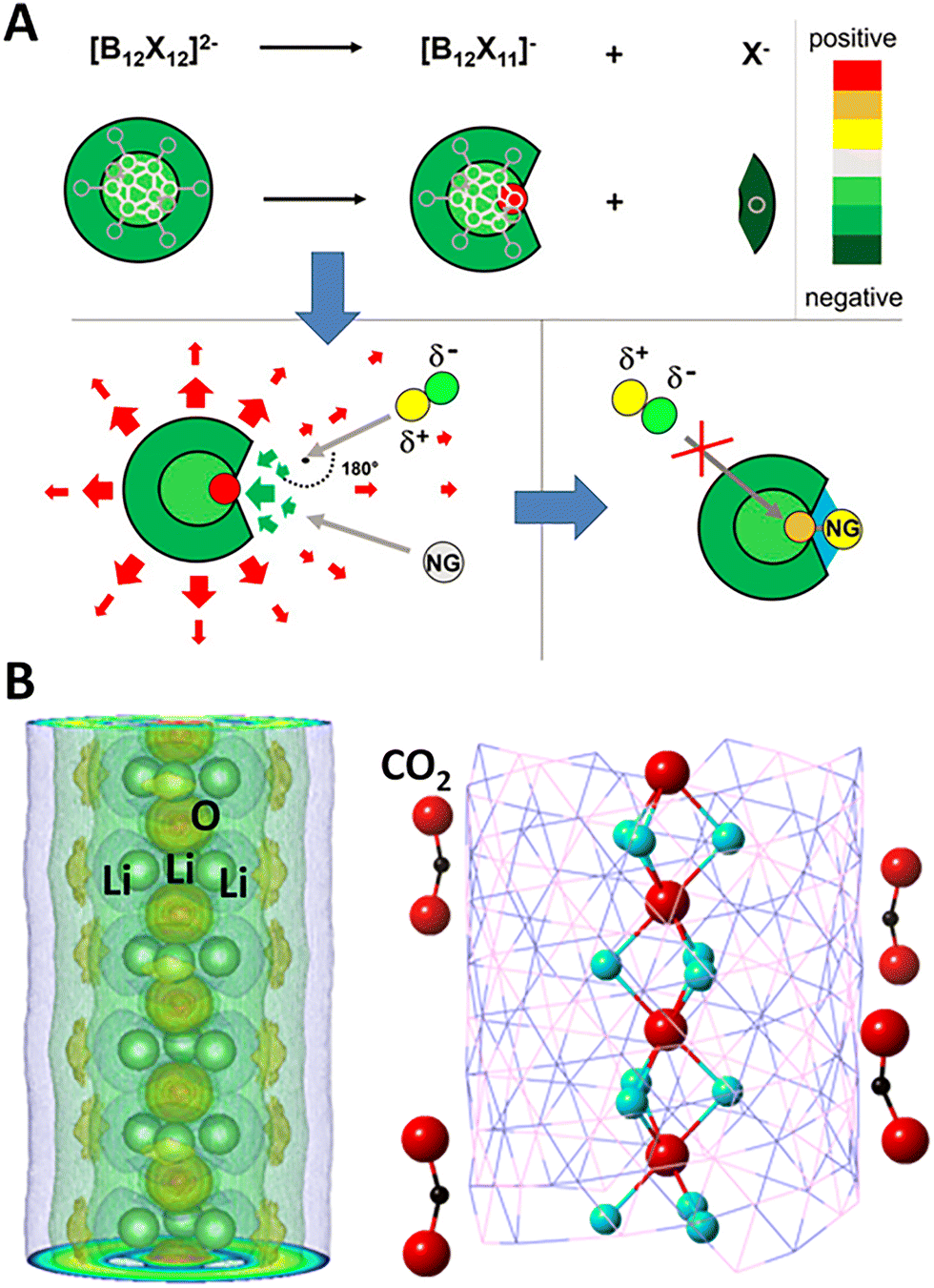 | ||
| Fig. 3 Superatoms as super-electrophiles and electrides. (A) Schematics showing the mechanism of binding noble gas atom by electrophilic clusters B12X11− (X = halogen, CN). Near the positively charged binding site (green arrows), the electric field is strong and attractive; at greater distances, it becomes repulsive (red arrows) owing to the overall negative charge. This leads to the Ng atom being attracted and bond to the positive binding site. Adapted with permission from ref. 52. Copyright 2019 Proc. Natl. Acad. Sci. U. S. A. (B) The 1D chain structure of Li3O+·e− electride, where the anionic electrons are shown by the yellow shells around the chain on the (Li3) planes. The formed nanostructure of Li3O+·e− electride grown in a boron–nitride nanotube can simultaneously activate (bend) multiple CO2 molecules. Adapted with permission from ref. 149. Copyright 2018 The Royal Society of Chemistry. | ||
In another experiment, it was shown that B12Br11−, with one Br removed from B12Br122−, can bind inert molecular nitrogen (N2) strongly to form B12Br11N2− at room temperature.141 More pronounced binding energies to the noble gas atoms can be achieved by incorporating highly electronegative terminal groups or/and replacing a B atom by Be in the boron cage of the clusters. According to the theoretical calculations, super-electrophilic BeB11(CN)112− dianion and BeB11(CN)10− monoanion can be formed by subsequently removing CN− terminal groups from a stable trianion BeB11(CN)123−. Both electrophiles can then interact strongly with noble gas atoms (Ng = He–Xe) at the exposed Be and B sites, forming [NgBeB11(CN)11]2− and [(Ng)2BeB11(CN)10]− compounds with the highest binding energies to Ne and Ar among the series being 0.15 and 0.38 eV, respectively.142 Even higher binding energies of 0.40 and 0.59 eV to Ne and Ar, respectively, can be achieved in trianions [BeB23(CN)22Ng]3−,143 where the superatomic electrophile BeB23(CN)223− is obtained by removing one CN− from the stable tetra-anion BeB23(CN)234−.126 Further computational studies show that more than one noble gas atoms can be bound simultaneously in [B12Y10–2Ng]0 (Y = CN− and BO−) with the highest binding energies of 0.25 and 0.84 eV per Ne and Ar, respectively.144
III.3 Superalkalis as super-electrides
Electrides are ionic compounds and are used as catalysts and reducing agents.145 Here, anion components are electrons (instead of atoms) localized in cavities in a periodic lattice.146 Given the extraordinarily low ionization potential, superalkalis can be used to form a new class of super-electrides. The non-nucleus-bound electrons exhibit high mobility and low work function, which can serve as powerful catalytic agents.147,148 Fang et al. have computationally shown that Li3O super-alkali can form a one-dimensional electride chain with every unit cell containing two Li3O+·e− units.149 The designed electride and its constructed nanostructures exhibit extremely low electronic work function of 2 eV and can, therefore, readily bind to and activate inert molecules such as CO2, as shown in Fig. 3B. Interestingly, the loosely bound anionic (p-type) electrons in this electride even carries a magnetic moment which is found to be 0.21µB/Li3O.149 Other superalkalis including Cs3O and Li4N can also form bulk electrides in 0-, 2-, and 3-dimensions.150,151 Most recently, 2[CaCl]+·2e− is theoretically designed to be a 0-D inorganic electride in which CaCl is a superalkali.152 The interstitial electrons of this electride form a tetragonal lattice between the atomic layers, leading to a highly anisotropic Dirac fermion at the X and M points, protected by symmetries. This electride also shows low work function of 3.4 eV which can effectively facilitate N2 cracking by electron donation.152 In another computational design, superalkli Ca4N2 can form a 1D electride [Ca4N2]2+·2e− with the anionic electrons forming 1D chains confined in the surface channels formed by Ca atoms.153 It is found that these anionic species are highly mobile, leading to exceptionally high conductance of the material at room temperature.III.4 Superatoms as super-catalysts
Catalysts play a critical role in the chemical industry as they are used in nearly 90% of all manufactured goods and are responsible for about 35% of the world GDP. Unfortunately, catalysts are usually composed of expensive metals like Pd, Pt, Rh, Ru, and Ir.154,155 Naturally, there is considerable interest in either reducing the amount of the metal used or finding earth-abundant metals to replace the expensive ones. As much of the chemical activity occurs on the surface, nanoparticles with large surface to volume ratio have been the catalysts of choice, smaller the particle greater is its reactivity (Fig. 4A).156 However, nanoparticles still have non-active atoms, and their size variation causes unwanted reactions. Consequently, in the past decade, attention has been focused on single-atom catalysts,157–162 where single metal atoms, the ultimate nanoparticle, are dispersed on a substrate. This too has limitations; single-atom catalysts only offer a single active site that may not be sufficient for reactions requiring the co-adsorption of multiple reactants.162–166 While dual- and triple-atom catalysts (DAC and TAC) have been suggested as a solution, there is no unique way to choose their composition.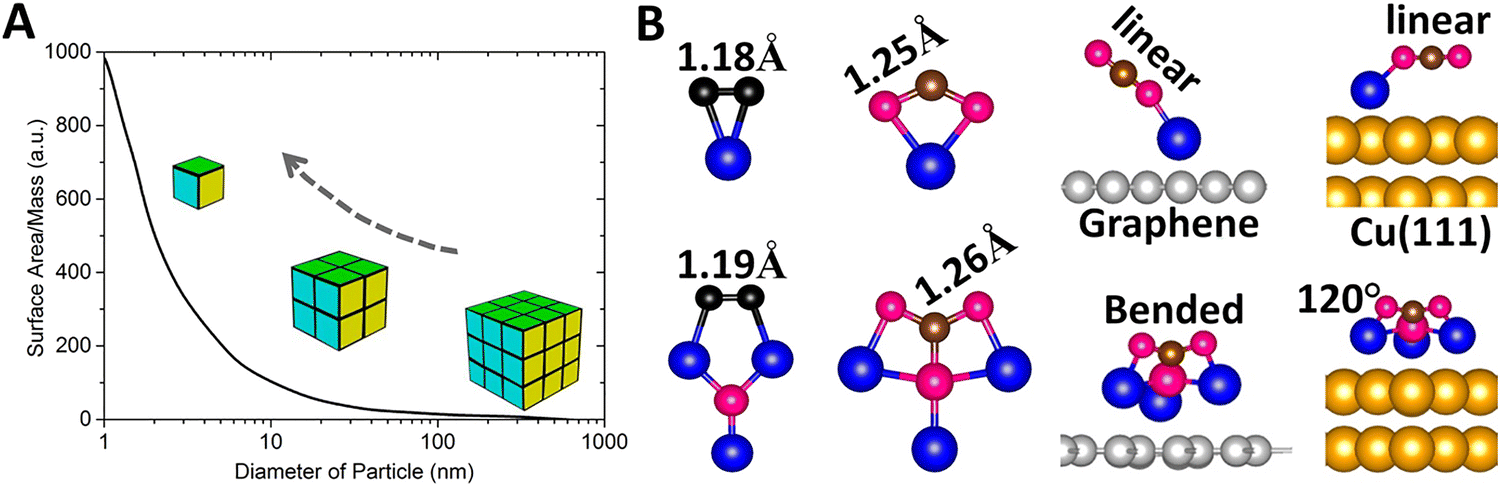 | ||
| Fig. 4 Superatoms as catalysts. (A) Schematics showing surface-mass ratio with the catalytic power increasing with the reduction of the particle size. Adapted with permission from ref. 156. Copyright 2007 AIP Publishing. (B) Interactions between chemically inert gas molecules (e.g., N2 and CO2) and superatom Li3O compared to that of atom Li, with N in black, O in red, C in brown, Li in blue, graphene in grey, and Cu in gold. Adapted with permission from ref. 182. Copyright 2024 Wiley-VCH. | ||
Superatoms offer an alternative. Their sizes can be very small with almost all the atoms lying on the surface and participating in chemical reactions. While several studies have demonstrated the benefits of using atomic clusters as catalysts, they deal with clusters in the gas phase.167–181 However, for practical applications, clusters need to be deposited on substrates which raises further concerns: (1) once deposited on a substrate, clusters would interact with the substrate atoms. This not only may change the structure and properties of the clusters but may also affect the substrate topology. Consequently, the reaction of molecules with clusters in the gas phase may be very different from those when supported on a substrate. (2) Secondly, the results may depend upon the adopted substrate itself.
Recently, Kilic and Jena182 addressed these issues by focusing on the reaction of small molecules such as H2, O2, N2, CO, and CO2 with Li and Li3O in the gas phase as well as when supported on graphene, Au(111) and Cu(111) substrates. Using calculations based on density functional theory, they showed that while the reactions of the above molecules with Li and Li3O in the gas phase are rather similar, Li3O binds and activates the above molecules more strongly than the Li atom (Fig. 4B). When deposited on the above substrates, the geometry of Li3O and that of the substrates are barely changed. As shown in Fig. 4B, the CO2 molecule maintains its linear geometry when interacting with the Li atom on the substrate, but undergoes significant bending when it interacts with the Li3O superatom, with the bond angle ∠OCO reducing to 120° on the Cu(111) substrate. This is due to the low ionization potential of Li3O, allowing it to readily donate an electron to the CO2 molecule and lead to increased binding energy and activation. Although neither Li nor Li3O are traditional catalysts, the above study demonstrates that a single-superatom catalyst can offer advantages over a single-atom catalyst, since they can provide multiple active sites with a facile electron transfer. As superatoms provide a new paradigm in catalyst design, identifying superatoms that can mimic the chemistry of commonly used catalysts, such as Pd, would be important.
In a photoelectron spectroscopy experiment on negatively charged ions, Castleman and coworkers compared the electronic structure of Pd atom with that of ZrO dimer.183 Considering the electronic configurations of Pd ([Kr]4d10), Zr ([Kr]4d25s2), and O ([He]2s22p4), one finds that the ZrO dimer is isoelectronic with the Pd atom. The good agreement in the electronic structures of these two species led the authors to suggest that ZrO could be a replacement catalyst for Pd. While early work of the interaction of gas molecules with isolated Pdn and ZrOn clusters casted doubt on this possibility,178 we should note that an understanding of the reactions of gas molecules with clusters in the gas phase may not be sufficient to ascertain their suitability as good catalysts, because in practical applications the catalysts need to be supported on a substrate. Recently, Kilic and Jena studied the hydrogen evolution reaction (HER), CO2 reduction reaction (CO2RR), and ammonia production by supporting ZrO dimer and Pd atom on graphene, Cu(111), Au(111) surfaces.182,184,185
First, it is important to ensure that the structure and properties of isolated clusters remain unaffected by their interaction with the substrate and that gas molecules bind and get activated by the supported clusters. Kilic and Jena confirmed that the geometry of ZrO remains unchanged when the dimer is supported on the above substrates and that the ZrO dimer is even more strongly bound to the substrate compared to the Pd atom, as shown in Fig. 5.184
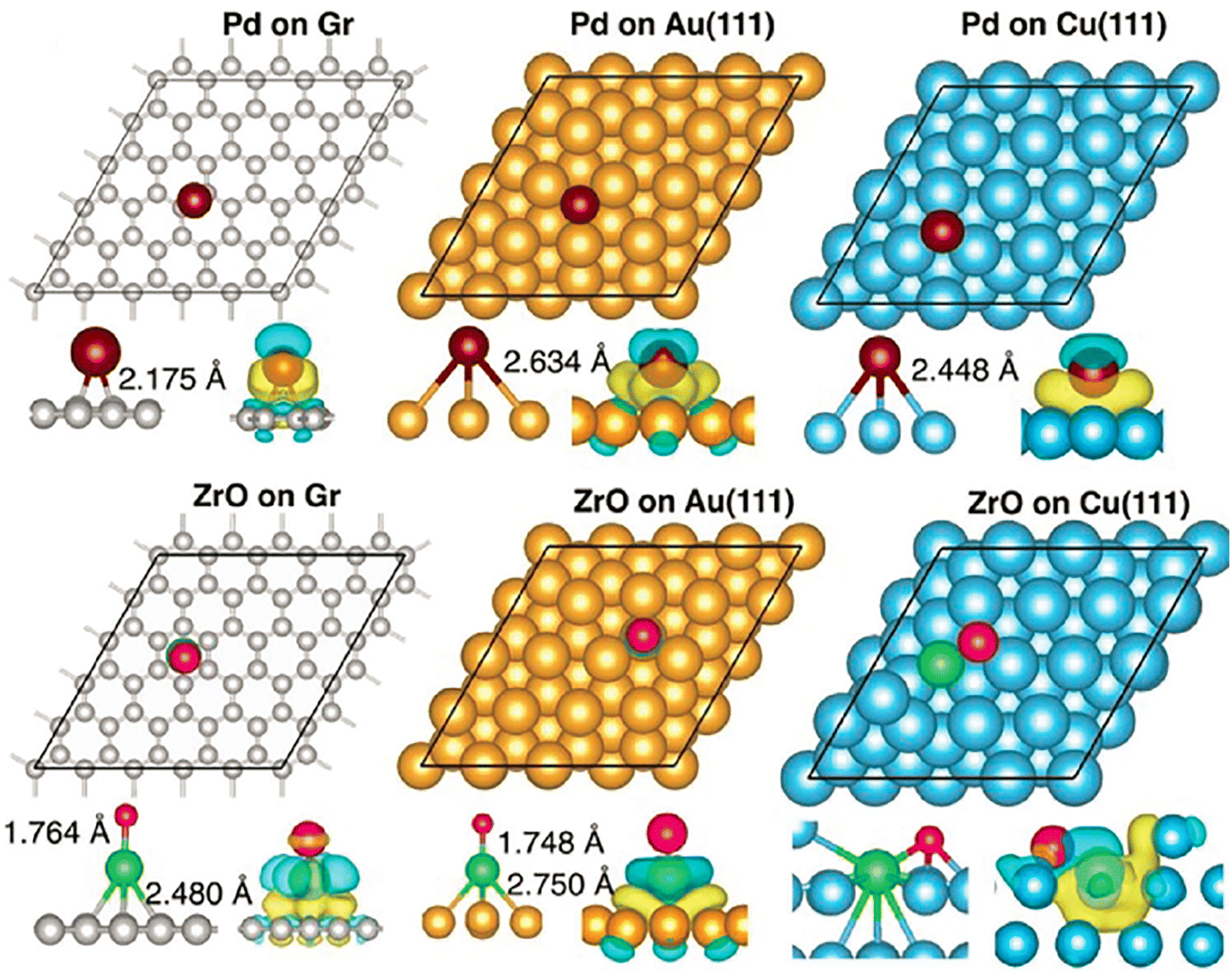 | ||
| Fig. 5 Optimized positions of Pd and ZrO supported on the graphene, Au(111), and Cu(111) surfaces, with C, Au, Cu, Pd, Zr, and O in gray, yellow, cyan, violet, green, and red, respectively. The corresponding charge density difference is shown in the lower panel, with yellow and cyan iso-surfaces representing charge accumulation and depletion, respectively. Adapted with permission from ref. 184. Copyright 2025 Wiley-VCH. | ||
In Fig. 6, we present the interaction CO2 with Pd atom and ZrO dimer supported on graphene, Cu(111) and Au(111) surfaces.184 The first step in a catalytic process is the binding and subsequent activation of the CO2 molecule. Note that CO2 is a linear molecule and very stable because of the covalent bond between C and O. Its activation requires charge transfer to the molecule so that not only the CO bond is elongated, but also, the O–C–O bond angle is decreased from 180°. For CO2 adsorbed on Pd (ZrO) doped graphene, Au, and Cu surfaces, the calculated C–O and C–O* bond lengths—where O* represents the oxygen atom bound to Pd or Zr—are 1.199/1.241 Å (1.215/1.329 Å), 1.174/1.183 Å (1.207/1.293 Å), and 1.229/1.236 Å (1.217/1.312 Å), respectively. The corresponding ∠OCO bond angles are 151° (132°), 179° (131°), 142° (130°). This clearly shows that ZrO can activate CO2 more strongly than the Pd atom, irrespective of the substrate used. The calculated CO2 reduction reaction and reaction pathways also shows that ZrO outperforms Pd as a catalyst, establishing the single-superatom catalysts are superior to single-atom catalysts. Investigations of NH3 synthesis further reveal that graphene-supported single-superatom catalysts (TiO, ZrO, and WC) outperform their single-atom counterparts (Ni, Pd, and Pt) in both stability and catalytic performance toward the electrochemical nitrogen reduction reaction.
 | ||
| Fig. 6 The top and lateral views of CO2 adsorbed on Pd single atom and ZrO speratom supported on the graphene, Au(111), and Cu(111) surfaces, with C (graphene), Au, Cu, Pd, Zr, and O in gray, yellow, cyan, violet, green, and red, respectively. For the CO2 molecule, C is in blue and O in red. Adapted with permission from ref. 184. Copyright 2025 Wiley-VCH. | ||
IV. Unusual physical properties of superatomic crystals – phonon transport and thermal conductivity
The transformative power of atomic clusters lies in their ability to serve as well-defined building blocks for the design of novel materials. By treating clusters as “superatoms” with tunable electronic structures, one can effectively extend and enrich the conventional periodic table, enabling access to chemical compositions and functionalities unattainable with individual elements alone. The principal classes of clusters employed as building units in experimentally synthesized assemblies include: (1) fullerenes and their analogues, such as C60, K3C60, and endohedral fullerenes (e.g., M@Si16 and M@Ge16);25,186–188 (2) boranes and carboranes, including B12H122− and C2B10H12;189,190 (3) metal chalcogenide clusters, such as M6X8 (M = Cr, Co, Re; X = S, Se, Te);28,77,191,192 (4) ligand-protected metal clusters,193–198 including Au25, AgAu24, Au1Ag22, Pt1Ag18, Ag14, and Pd3.As summarized in Table 1, materials assembled from cluster building blocks often exhibit properties markedly distinct from those of their atom-based counterparts, owing to their unique electronic shells, size-dependent effects, and inter-cluster coupling. In this section, we focus specifically on their phonon properties and the unique lattice dynamical behaviors that emerge from cluster-based architectures.
Phonons are collective excitations of vibrational modes in solids; they carry energy and momentum and can exhibit wave-particle duality. The ability to manipulate phonons holds significant implications for tailoring intrinsic properties of materials. However, phonons are bosons and do not follow the conventional polarity regulation used for fermions like electrons. Superatomic crystals (SACs)186,199 provide a unique platform for phonon manipulation. In conventional materials, phonons arise from atomic vibrations. In contrast, SACs, where superatomic clusters serve as the building blocks, exhibit two distinct vibrational modes: (1) intra-cluster vibrations of atoms within a superatomic unit, and (2) inter-cluster vibrations of the clusters themselves. These combined vibrational modes can alter the phonon spectra and influence phonon scattering and coherence, leading to properties distinct from those of conventional atomic crystals as described in Table 5. In addition, the interactions between phonons and electrons are also different as demonstrated in K3C60. Neither K nor C60 is superconducting, but K3C60 is where the C60 fullerenes composed of 60 carbon atoms play a fundamental role.200
| Feature | Superatomic crystals | Conventional atomic crystals |
|---|---|---|
| Building blocks | Large, complex superatoms | Individual atoms |
| Unit cell size | Large | Small (atomic scale) |
| Phonon dispersion | Flattened bands with many localized modes | Well-defined dispersive phonon bands |
| Phonon group velocity | Low (flat dispersion) | High (steep dispersion) |
| Phonon scattering | Strong 3- and 4-phonon scattering caused by structural complexity | Lower scattering |
| Phonon coherence | Weak in most cases | Strong in most cases |
| Dominant phonon modes | Localized intra-superatom modes and low-frequency inter-superatom modes | Acoustic and optical phonons |
| Anharmonicity | High | Low for most systems |
| Phonon mean free path (MFP) | Short | Long |
| Thermal conductivity (κ) | Ultralow | Moderate to high (depending on materials) |
| Temperature dependence of κ | Weak or plateau-like | Strong (κ ∝ 1/T at high T for most systems) |
Coupling between intra-superatom and inter-superatom vibrations can effectively suppress phonon mobility, while enhancing higher-order phonon scattering and phonon coherence. These factors contribute to intriguing properties such as ultralow lattice thermal conductivity and remarkable thermal rectification. Experimentally, Ong et al.201 conducted the first measurement of thermal transport in unary SACs of Co6X8(PEt3)6 [X = S, Se, Te; PEt3 = triethylphosphine, Et = (CH2CH3)3] and the binary SACs of [Co6X8(PEt3)6][C60]2. Their results indicate that heat conduction in SACs is mediated by collective inter-superatom phonons, with mean free paths determined by the inter-superatom separation and coupling strength. Upon transitioning from an orientationally disordered to an ordered structure, phonon coherence is enhanced, resulting in longer mean free paths and increased thermal conductivity at lower temperatures. Additionally, they found that the hierarchical bonding features expand the phonon space by combining localized 0D modes with the delocalized 2D and 3D modes. This phenomenon is observed in a SAC consisting of Re6Se8 as the superatomic building block, forming a layered van der Waals lattice.202 Theoretically, it was also shown that the rotational dynamics and orientational ordering of the superatoms significantly impact the lattice thermal conductivity.203
Moreover, the bonding hierarchy in SACs introduce significant four-phonon anharmonicity. For instance, in a superatom-based superionic conductor Na3OBH4, strong four-phonon scattering extends the phonon scattering phase space by 33% at 300 K, leading to a 24% reduction in lattice thermal conductivity.204 A similar phenomenon is observed in calculations of supersalt NaNO3 monolayer,205 which is composed of superhalogen NO3, significantly reducing the lattice thermal conductivity from 4.13 to 1.65 W m−1 K−1 at 300 K. Moving from atom-superatom hybrid systems to a solely superatom-based supersalt PH4AlBr4 composed of superalkali PH4 and superhalogen AlBr4, Du et al.206 showed theoretically that the structural and bonding hierarchies are significantly enhanced with a much more complex geometric configuration. Notably, the phonon mean-free paths of several modes are comparable to the interatomic distances. This, known as the Ioffe–Regel limit, results in strong phonon coherence. Consequently, the lattice thermal conductivity is controlled by a two-channel mechanism, incorporating both phonon scattering and phonon coherence.
These unusual phonon transport characteristics make SACs promising for applications in phononic devices. One example is the thermal rectification (TR), which is similar to the diode-like effect observed in electronics. TR occurs in structures that transfer heat in a nonreciprocal manner, driven by the interplay between structural gradient and lattice nonlinearity. Recent computational studies have shown that the superatom-based TR devices exhibit two notable features that distinguish them from atom-based systems with atomically flat surfaces:207,208 (1) the large size and non-spherical shape of superatoms not only make their assembled structures porous but also induce significantly high roughness on both internal and external surfaces. This enhances the tuning of phonon transport, resulting in unconventional thermal rectification behavior. (2) SACs-based fractals have high surface-to-volume ratio and surface roughness that changes exponentially with the fractal levels. This leads to highly nonlinear temperature variations with spatial positions, inducing nonlinear heat transfer; and causing the thermal conductivity to exhibit high asymmetry, which is sensitive to the direction of the heat current.
V. Polyatomic ions as building blocks of energy materials
Polyatomic ions such as BH4− hold the key to overcome some of the key challenges in developing materials for energy production and storage. As mentioned before, these ions not only mimic the chemistry of halogens, but also their electron affinities are larger than those of halogens. In this context, they can be regarded as superatoms. Each being a group of atoms (cluster) stabilized at a certain charge state; polyatomic ions can provide a much wider materials design palette than employing individual atoms. Instead of limited number of elements in each group of the periodic table, many more polyatomic species are available due to mix-and-match of elements in their compositions. Compared to the spherical shape and periodic bonding characteristics of atomic ions, ployatomic ions carry various chemical compositions, symmetries (shapes), sizes, charge distributions among the composite atoms, and electron affinities. Accordingly, polyatomic ions serving as dopants or substitutes in materials can bring additional degrees of freedom to tailor and optimize the bulk and interface properties. Significant breakthroughs in application and understanding of polyatomic ions as building blocks of energy materials have been achieved, especially in the development of solid-state electrolytes for batteries and hybrid halide perovskites for solar cells. These studies are highlighted by synergy between theory and experiment.V.1 Solid-state electrolytes
Solid-state electrolytes hold the promise to achieve safe, cost-effective, and high energy electrochemical systems that are essential for next-generation long-duration energy storage. Two major challenges remain in the development of solid electrolytes – to achieve adequate ionic conductivity at room temperature on par with those of liquid electrolytes and to have stable interface against metal anode for high energy density. However, solid electrolytes with high ionic conductivities are often characterized by weak anion–cation interactions and high formation energies, and, therefore, exhibit poor interfacial stability against the metal anode. Recent theoretical studies have shown that substituting halogen (a ubiquitous composite in solid electrolytes) by selected polyatomic ions (e.g., BH4−, BF4−, CN−etc.) can simultaneously enhance the ionic conductivity and stabilize the interfacial stability of solid electrolytes. Doping polyatomic ions to the halogen sites was first proposed for anitperovskite solid electrolytes due to their large structural tolerance.209,210 The strategy was later extended to the argyrodite family due to their well-known high ionic conductivity but poor interfacial stability against Li metal anode.211–213 Once incorporated into the structure of a solid electrolyte, the polyatomic ions can introduce motional dynamics into the host structure, with an extraordinarily large length scale that is 1–2 orders of magnitude higher than the regular thermal vibrational amplitude (0.01–0.1 Å) of individual atoms, as shown in Fig. 7A and B. The translational and rotational dynamics of the non-spherical polyatomic ions effectively alter the surrounding energy landscape, which can significantly lower the diffusion barrier and enhance the ionic diffusion in the electrolyte.209,210 Additional channel spaces for ionic diffusions can be formed around smaller halogen ions when they are co-exiting with relatively large polyatomic ions in a structure.210 Upon interfacial decomposition of the polyatom-based solid electrolyte, the polyatomic ions can form interphases that are ionically conductive yet electronically insulating, which can effectively stabilize the interface against metal anode,212 as shown in Fig. 7C–F.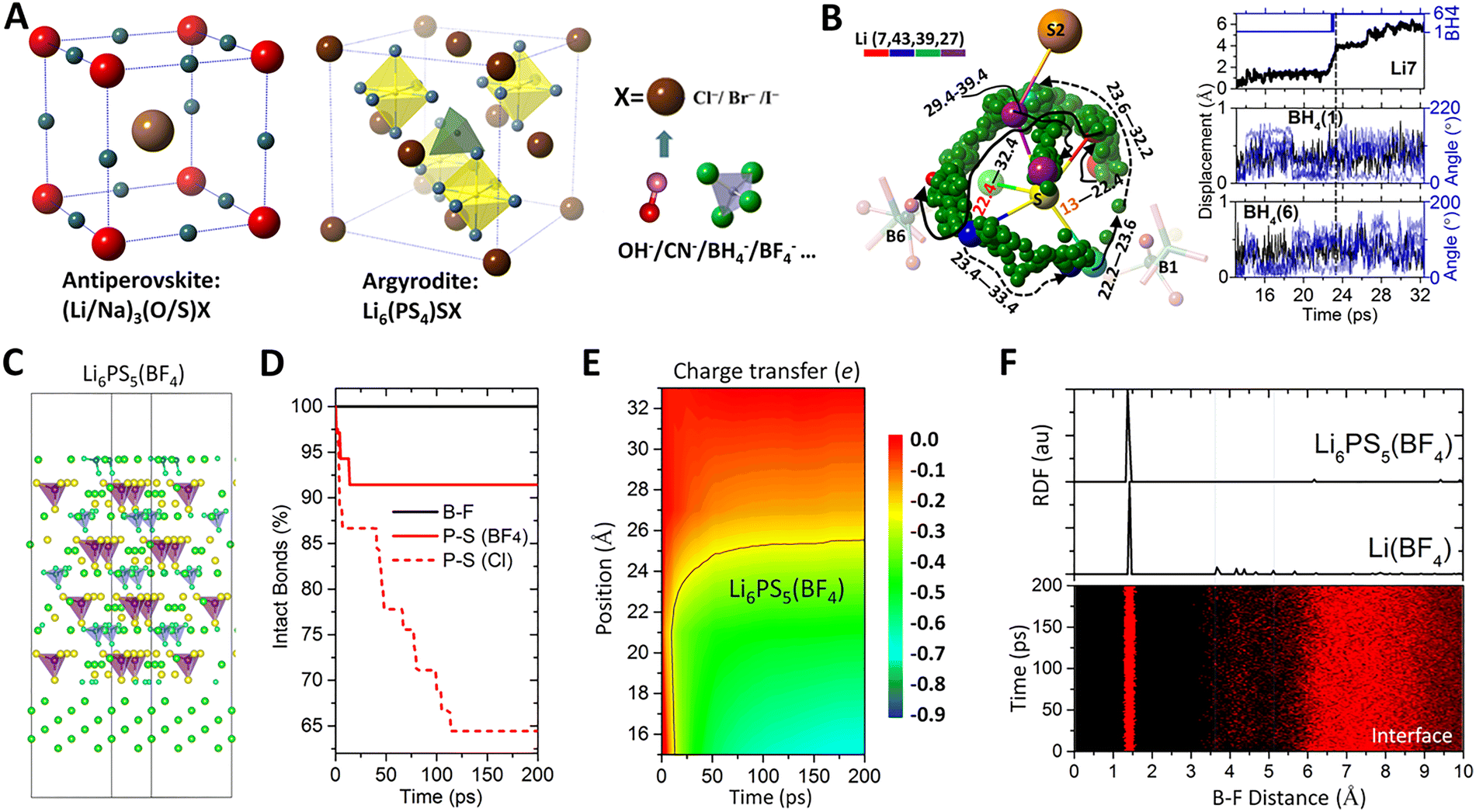 | ||
| Fig. 7 Solid state electrolytes based on superatomic ions. (A) Design of solid electrolytes by substituting halogen for selected polyatomic ions. (B) Diffusion trajectory (from 22.4 to 39.4 ps) of Li+ (indexed by 7, 43, 39 and 27 in the simulation cell) displacements (up to 6 Å) coupled with translational displacements (∼0.5 Å) and rotations (∼200°) of BH4− clusters (B1 and B6). Adapted with permission from ref. 211. Copyright 2022 Springer Nature. (C) An interface model between argyrodite solid electrolyte Li6PS5(BF4) based on polyatomic BF4− (violet tetrahedra) and Li metal (in green). (D) Explicit interface modelling showing that incorporating BF4− can stabilize the P–S bonds in Li6PS5(BF4), in contrast to the case of Li6PS5Cl based on Cl. (E) Presence of BF4− can inhibit electron transfer from Li metal into the solid electrolyte across the interface. (F) Radial distribution function analysis at the Li6PS5(BF4)–Li interface suggests the formation of Li(BF4) interphase which is electronically insulating and ionically conductive. Adapted with permission from ref. 212. Copyright 2025 American Physical Society. | ||
Inspired by these early theoretical studies, recent experiments214–219 have substituted the halogen site with polyatomic ions including BF4− and BH4− in the antiperovskite solid electrolytes M3YX and M3−xOHxX (M = alkali metal; Y = chalcogen; X = halogen). A mechanochemical synthesis is typically used, with ball milling powder mixing of metal borohydride/tetrafluoroborate, metal halides, and metal oxides.214,215 It is found that, among the doping series Li2OHCl1−x(BF4)x (x = 0.02, 0.05, 0.1), Li2OHCl0.98(BF4)0.02 exhibits the lowest activation energy and the highest ionic conductivity which is about 2 times higher than that of Li2OHCl. The polyatom-doped system also exhibits much enhanced stability against Li metal anode.214 The doping concentration of the polyatomic ion is crucial to achieve improved properties. High concentration of the polyatomic dopants can be detrimental to the ionic conductivity. This is evidenced by another recent experiment, where Na3O(BH4) with 100% substitution of Br in antiperovskite Na3OBr by BH4− exhibits even lower ionic conductivity than that of Na3OBr.215
The polyatomic doping strategy has also been applied to the argyrodite solid electrolytes Li6PS5X (X = halogen) in recent experiments. With the halogen replaced by polyatomic CN−, Li6PS5CN is synthesized using low-temperature solution-based processing.216 It is found that Li6PS5CN exhibits lower activation barrier than that of Li6PS5Br which is the lowest among the halide-argyrodite family.216 In another experimental work, Li6PS5Cl0.9(BH4)0.1 is mechanochemically synthesized with ball milling.217 It is found that the partial substitution of Cl− by BH4− greatly improves the ionic conductivity to 2.83 mS cm−1 which is twice as high as that of Li6PS5Cl. The experiment also shows that the Li/Li6PS5Cl0.9(BH4)0.1/Li symmetric cell can stably run for more than 400 h, with a critical current density of up to 3.5 mA cm−2.217 Doping high concentration of BH4− in argyrodite may lead to mixed phases, as shown by the recently synthesized thiophosphate solid electrolytes (1 − x)Li3PS4·2xLiBH4 using a simple milling method without heat treatment.218 The ionic conductivity increases, and the activation energy decreases with increasing BH4− up to x = 0.54 (with the highest room-temperature ionic conductivity of 11 mS cm−1), beyond which the trends are reversed. The resulting solid electrolyte also exhibits superior cycle and rate performances in a Li–In/NCM all-solid-state cell.218 Further experiments find that even higher ionic conductivities can be achieved in the argyrodite by simultaneously substituting halogen and BH4− for S2− using room-temperature ball milling.218 Among the resulting series, Li7−a−bPS6−a−b(BH4)aXb (X = Cl, Br, I; a + b ≤ 1.8), Li5.35PS4.35(BH4)1.15Cl0.5 exhibits the highest ionic conductivity of 26.1 mS cm−1 at room temperature. The corresponding symmetric cell maintains stable lithium plating/stripping over 2000 hours at 1 mA cm−2 and delivers a high critical current density of 2.1 mA cm−2,219 underscoring its excellent interfacial stability. A most recent experiment shows that LiBH4 doping into Li6PS5Cl creates to a tri-layer solid electrolyte interphase of Li3P/LiBH4/Li2S that blocks electrons while facilitating ion diffusion.220 This leads to greatly enhanced electrochemical stability, with the symmetric cells achieving a critical current density (CCD) of 7.3 mA cm−2 which is almost three times that of the baseline without doping.220
The working mechanism of lowering activation energy and improving ionic conductivity demonstrated in the above experiments relies on the interplay between the doped anion cluster and the diffusing metal ions. The dynamical reorientation of the polyatomic anions in the solid electrolytes at room temperature are verified by experimental characterizations using neutron diffraction or high-resolution synchrotron powder X-ray diffraction.216,221 Modeling studies and data analysis are necessary to uncover the possible correlation between the dynamics of the anion clusters and the diffusion metal ions. Most recent theoretical studies explain the coupling between the dynamics of the doped polyatomic ions and the metal ion diffusion.211,222,223 Using light mono-anion clusters (BH4−, CN−, SH−, etc.) working in the argyrodite as a model system, it is found that the dynamics of the anion cluster inside the structure can be categorized into two types. One is the so-called “responsive dynamics”, where, due to their exceptionally large motional degrees of freedom in the structure, the polyatom can effectively accommodate the motion of a passing metal ion by rotation and translation. This can lower the migration barrier and the activation energy, and, therefore, enhance the ionic diffusion. The other is called “active dynamics”, where the polyatomic anions are rotating and translating under thermal excitations. This type of dynamics may inhibit rather than facilitate the diffusion of a passing ion, depending on the specific migration path at that moment. Increasing the concentration of polyatomic dopants will increase both types of dynamics at the same time. Therefore, the polyatomic concentration that can enable the optimal properties of a system can only be achieved when the “responsive dynamics” is maximized against the detrimental effect of the “active dynamics”.211 Such cluster dynamics has been extended most recently to heavy polyanion clusters (e.g., PS43−) in the sulfide electrolytes.222 Thus, the coupling between the dynamics of the polyatomic ions and the diffusing ion is beyond the so-called “paddle-wheel mechanism” that has always been assumed, where a rotating cluster ‘drags’ or ‘pulls’ its nearby metal ions. Realizing the difference between the “responsive dynamics” and the “active dynamics” is crucial to accurately understand the effect of the doped anion cluster on the ionic diffusion. Had the clusters’ dynamics always facilitate the ionic diffusion as depicted in the early “paddle-wheel mechanism”, ionic conduction would increase with elevated doping level of the cluster. This contradicts the observation in the above-mentioned experiments.
V.2 Hybrid perovskite solar cells
In another field of interest in energy materials, halide hybrid perovskite (e.g., MAPbX3, MA = CH3NH3, X = halogen) solar cells have attracted considerable attention due to their low-cost solution-based synthesis and decent power conversion efficiency (PCE). One of the main challenges for the commercialization of these materials is their instability against moisture and heat. Recent studies have found that the polyatomic substitution for the halogen inside the hybrid perovskites has great potential to overcome this challenge. Early theoretical studies showed that partial replacement of halogen by selected polyatomic ions (e.g., BH4−, SCN−, etc.) can greatly enhance the stability of the hybrid perovskites.224,225 Recent experimental works in this field have been summarized in two comprehensive reviews.226,227 Here, we focus on the underlying mechanism of the stabilization of hybrid perovskites by incorporating polyatomic ions, which has not been adequately emphasized in the published reviews.Based on the concept of superatoms, one can view a hybrid perovskite as a “super” alkali halide. The organic cation MA+ behaves as a superalkali with an ionization potential lower than that of Li, while the inorganic framework anion [MX3]− (M = Pb, Sn, Ge; X = halogen) functions as a superhalogen with electron affinity greater than that of Cl. This pronounced ionic character enhances Coulombic attraction toward a polarized water molecule, which becomes confined between MA+ and [PbX3]−. Water then mediates degradation by promoting I− removal from [PbI3]−; the released I− combines with H+ from the organic cation to generate HI and PbI2,225 as shown in Fig. 8A.
 | ||
| Fig. 8 Doping polyatomic ions in hybrid perovskites to improve properties. (A) Mechanism of the hybrid perovskite catalyzed by water at the molecular level. (B) Partial replacement of halogen by SCN- or SeCN- with the linear configuration. The charge distribution (values in e) along these polyatomic ions entails reduced attraction with water molecule and enhanced bonding with Pb in the functional group. (C) Calculated band gaps (1.13 and 1.14 eV) and band edges (Rashba splitting highlighted in red) of the polyatom-doped hybrid perovskites compared to those of MAPbI3. Adapted with permission from ref. 224. Copyright 2017 American Chemical Society. | ||
Partial replacement of the halogen (X) in [PbX3]− by selected polyatomic ions (e.g., BH4−, SCN−, SeCN−etc.) can break the above degradation process. Given that the polyatomic cluster contains multiple atoms, the ‘−1’ charge of the whole cluster is unevenly distributed among the constitute atoms (e.g., S/Se, C, and N in SCN−/SeCN−, or B and H in BH4−). It has been proved that such multi-centered charge distribution inside a polyatomic ion can effectively reduce the Coulombic attraction to water, so that the water molecule is unlikely to be trapped between the superalkali MA+ and superhalogen [PbX3]− in the first place. Meanwhile, such charge distribution enhances the bonding between Pb and X within [PbX3]− so that X− is unlikely to be dislodged from the group to react with H+ of the organic cation,224 as shown in Fig. 8B.
Optimal bandgaps and band edges are crucial to achieve high PCE in the hybrid perovskites. For the regular alkali halide ionic compounds AX (A = alkali, X = halogen), the bandgap decreases from X = Cl to I with increasing size of the halogen (X). Similarly, the bandgaps of hybrid perovskites viewed as “super” alkali halides also depend on the size of the doped superhalogen. Theoretical studies find that the polyatomic anions BH4−, SCN− and SeCN− all have ionic radii similar or smaller than that of I−.225 Therefore, the resulting MAPbI2.75Y0.25 (Y = BH4, SCN, SeCN) systems exhibit the optimal bandgaps for solar cell as that of MAPbI3. Besides the proper bandgap, the doped anion clusters can simultaneously achieve decent carrier mobility and the large Rashba splitting at the band edges, which are essential to reach high PCE in the hybrid perovskites,224 as shown in Fig. 8C.
Indeed, most recent experiments confirm the above theory. It has been demonstrated that the stability of the hybrid perovskite against humidity, thermal, and ultraviolet illumination are significantly enhanced by incorporating BH4−. According to the experiment, the polyatomic dopant not only effectively inhibits the evaporation and decomposition of the methylamine group (MA+ = CH3NH3+) in the resulting MAPbI3−x(BH4)x, but also increases the PCE from 18.43 to 21.10%, accompanied by increased short-circuit current density and open-circuit voltage with near-zero current–voltage hysteresis.228 In another telling experiment, it is demonstrated that adding appropriate alkali metal halide KSeCN into perovskite can result in MAPbI3−x(SeCN)x which exhibits significantly improved stability, PCE of 18.41%, hysteresis index of 1.5%, and efficiency of 85.3% after 500 h.229 More beneficiary effects of the polyatomic anion additives due to various mechanisms have been reported in recent experiments.230–232 High-quality perovskite CsPbBr3 nanocrystals featuring ultrahigh stability are obtained by incorporating polyatomic BF4− anions due to an in situ surface passivation.233 In another experiment, MABF4 serving as the BF4− additive can effectively improve the photovoltaic performance of the formamidine (FA) lead iodide (FAPbI3) perovskite solar cell by facilitating the perovskite crystal growth and enhancing the heterointerfaces between the FAPbI3 layer and carrier-transport materials, while retaining the favorable narrow bandgap of FAPbI3.234
VI. Superatom-based ferroelectric, piezoelectric and thermoelectric materials
A common characteristic of ferroelectric, piezoelectric and thermoelectric materials is that their properties are all related to electric polarization which can be enhanced in superatom-based structures due to the following factors: (1) the lack of spherical symmetry in superatoms can lead to anisotropic polarization; (2) the larger size of superatoms compared to atoms results in electron distribution over a broader phase space, making them more easily polarizable; (3) superatoms such as superhalogens and superalkalis have higher electron affinities and lower ionization potentials, respectively, facilitating charge transfer and thereby enhancing polarization.The early experimental study of ferroelectricity in the gas-phase Nb clusters was conducted by de Heer and colleagues in 2003.235 They observed ferroelectric-like state in cryogenically cooled neutral Nbn clusters (n = 2–150), with the transition temperature decreasing as cluster size increased. Additionally, pronounced even–odd alternations were noted for n > 38. Later, using molecular beam Stern–Gerlach deflection measurement, they found that the odd-n clusters deflected due to a single unpaired spin, and spin uncoupling coincided with the transition to ferroelectric state.236 Coexistence of ferroelectricity and ferromagnetism was also observed in rhodium clusters,237 where the transition temperatures exhibited size-dependent variations. Similar behavior was also predicted in Ta clusters.238 The ferroelectric state of free clusters can be further tuned through doping, as demonstrated in Nb clusters doped with Al, Au, Mn, Fe, and Co.239 These findings open avenues for designing new “multiferroic” materials composed of clusters. An intriguing question arises from these observations: as clusters exist in an intermediate phase between atoms and bulk matter, how many atoms are required to exhibit traditional ferroelectric effect? A recent experimental study determined this threshold to be approximately 5000 atoms in zero-dimensional systems.240 These advances have strongly motivated research on unconventional ferroelectricity in cluster-based materials.
Using an unbiased structure search combined with DFT calculations, Gao et al.241 theoretically proposed a family of stable three-dimensional ionic supersalts, PnH4MX4 (Pn = N, P; M = B, Al, Fe; X = Cl, Br), constructed from superalkali PnH4+ and superhalogen MX4− building blocks. The exothermic nature of the reactions MPn + 4HX → PnH4MX4 or PnH4X + MX3 → PnH4MX4 suggests the potential of efficient synthesis. These supersalts adopt distorted zinc blende (ZB) structures characterized by covalent-like directional bonding indued by the anisotropy induced by the aspherical geometry of the superatoms, as shown in Fig. 9A. Such a unique structural configuration enables ferroelectricity with significant ion displacement, as well as ferroelasticity with ultra-large reversible strain. Notably, the ionic bonding features reduce the switching barriers associated with large deformations, very different from conventional ferroelectric materials which are brittle in nature due to covalent bonding. PH4FeBr4 is particularly remarkable for its triferroic character, combining ferroelectricity, ferroelasticity, and antiferromagnetism within a single phase, where the spin configuration can be reversibly tuned by ferroelastic or 90° ferroelectric switching. Moreover, a supersalt crystal built exclusively from SbCl4 superhalogens is stabilized through valence disproportionation of Sb into Sb5+ and Sb3+ species.242 The coexistence of mixed valence and lone-pair activity generates an unprecedented form of ferroelectricity featuring multiple coupled polarization modes.
 | ||
| Fig. 9 Functional materials formed by superatomic ions. (A) Typical crystal structures of multiferroic supersalts featured by non-centrosymmetric superatomic ions NH4+ or PH4+ interacting with XY4− (X = B, Al, Fe; Y = Cl, Br). Adapted with permission from ref. 241. Copyright 2021 Springer Nature. (B) Monolayer structure of thermoelectric material Re6Se8I2 composed of superatomic cluster Re6Se82+ and two I− in unit cell. Adapted with permission from ref. 247. Copyright 2022 American Physical Society. | ||
This discovery of a purely superhalogen-based supersalt crystal and multimode ferroelectricity underscores the unique role of superatoms in designing novel functional materials.
Experimentally, Ai et al.243 successfully observed ferroelectricity in a fullerene adduct formed by C60 and S8 clusters. The combination of C60, the most symmetric molecule with Ih symmetry, and the highly symmetric S8 cluster with D4d symmetry resulted in the formation of a polar C60S8 adduct with C2v point group at room temperature. C60S8 undergoes polar-to-polar ferroelectric phase transition, the resulting ferroelectricity was confirmed through the observation of a ferroelectric hysteresis loop and ferroelectric domain switching. This experimental demonstration of ferroelectricity in 3D cluster assemblies is expected to inspire further research and exploration in this field.
For 2D assemblies, Huang et al.244 used ab initio molecular dynamics simulations and Monte Carlo methods to propose a 2D magnetic honeycomb lattice material TM2(C60)3 (TM = Mo, and W) consisted in exohedral metallofullerene clusters TM(C60)3, where the transition metal atoms deviate from the central plane, leading to a spontaneous electric dipole moment perpendicular to the plane. The energy barrier between the two dipolar states (up and down) was found to be 0.50 and 0.49 eV, respectively, low enough to allow switching between these states under an external electric field (gate voltage). Consequently, this system qualifies as a ferroelectric material with out-of-plane polarization.
For 1D assemblies, Zhao et al.245 computationally designed magnetic fullerene-based 1D chains of U2C@C80-M (M = Cr, Mn, Mo, and Ru), which exhibit enhanced spontaneous polarization (Ps) compared to isolated clusters. This enhancement ranges from two to four times and is attributed to the interaction between fullerene and metal atoms. The calculated Ps values (in pC m−1) for Cr, Mn, Mo, and Ru are 22.77, 34.60, 27.34, and 20.34, respectively, and the energy barrier EB's for polarization switching (in meV atom−1) are 7.98, 17.14, 3.45, and 2.74, respectively.
Compared to the advancements in cluster-based ferroelectricity, much less attention has been given to cluster-based piezoelectric materials. Recently, Wang et al.246 synthesized a superhalogen GaCl4 based piezoelectric material [(CH3)3NCH2Cl][GaCl4], which exhibits an exceptionally high piezoelectric coefficient (d33 = 226 pC N−1) and a remarkable piezoelectric voltage coefficient (g33 = 1318 × 10−3 V m N−1), along with outstanding mechanical durability and stability.
Unlike ferroelectricity and piezoelectricity, significant experimental works have been dedicated to synthesizing cluster-based thermoelectric materials as summarized in ref. 230. However, the maximum figure of merit (ZT) values reported so far remain below 1.0, leading to low conversion efficiency. To address this limitation, Li et al.247 studied the recently synthesized superatomic Re6Se8I2 monolayer,192 depicted in Fig. 9B. Their results reveal a clear phonon–electron decoupling mechanism: the heavy and anharmonic Re–I bonds strongly suppress lattice heat transport, reducing the thermal conductivity to 1.20 W m−1 K−1 at 300 K, while the stiff and harmonic Re–Se framework sustains high carrier mobility, producing a TE power factor of 4344 µW m−1 K−2 along the b axis under n-type doping—approximately an order of magnitude greater than that of comparable cluster-based systems. Consequently, superatomic Re6Se8I2 exhibits outstanding thermoelectric performance, with ZT values reaching 1.20 at 500 K and 1.43 at 900 K. As summarized in Table 6, the distinct merits of superatom-based ferroelectricity, piezoelectricity, and thermoelectricity underscore the immense potential of clusters in designing novel functional materials.
| Property | Ferroelectricity | Piezoelectricity | Thermoelectricity |
|---|---|---|---|
| Mechanism | Reversible spontaneous electric polarization | Polarization induced by mechanical stress | Electrical current generated from a temperature gradient |
| Required symmetry | Non-centrosymmetric with switchable dipoles | Non-centrosymmetric, polar axis | No strict symmetry requirement |
| Tunable features via superatom design | Electric dipole moment; switching field strength | Piezoelectric coefficient; mechanical compliance; anisotropy of response | Seebeck coefficient; electrical conductivity; phonon scattering for low thermal conductivity |
| Role of superatoms | (1) The internal dipole moments of superatoms enable strong and switchable polarization due to asymmetric charge distribution within superatoms. | (1) The internal dipole moments of superatoms provide built-in polarization that responds to mechanical strain. | (1) The internal dipoles of superatoms can affect carrier distribution. |
| (2) The high polarizability of superatoms enhances dielectric constant and strengthens electric field response. | (2) The high polarizability of superatoms improves electromechanical conversion efficiency. | (2) The high polarizability of superatoms modulates carrier behavior and contributes to thermopower. | |
| (3) The structural flexibility of superatoms allows low-energy switching between polar states. | (3) The structural flexibility enhances strain-to-polarization coupling via deformable bonds. | (3) The tunable electronic structure of superatoms boosts Seebeck coefficient and electrical conductivity through density-of-states tuning. | |
| (4) The anisotropic properties of superatoms enable directional polarization switching and control. | (4) The tunable electronic structure of superatoms allows engineered piezoelectric coefficients via electronic deformation. | (4) The anisotropic properties of superatoms support high power factor along optimal conduction directions. | |
| (5) The anisotropic properties of superatoms enhance the directional sensitivity to mechanical stimuli. | |||
| Performance metric | Polarization, coercive field | Piezoelectric coefficient, coupling factor | Figure of merit |
VII. Conclusion and outlook
In summary, we have reviewed the recent progress in elucidating the unique properties of superatomic clusters across disciplines and their potential as building blocks of novel materials. For example, boron-based superatomic clusters with unprecedented stability as multiply charged ions can enable noble gas atoms to react at room temperature. Distinctive properties of the superatoms, such as the non-spherical symmetry, variable chemical compositions, internal charge distribution, as well as extraordinary electronic structure and interaction, can render novel properties of the host materials and provide new understanding of the structure–property relationship. For example, a supersalt can be formed by only using a superhalogen anion such as SbCl4 as the building block. Superalkalis can be used to create a new class of electrides with enhanced catalytic properties while single-superatom catalysts can outperform single-atom catalysts. Superatoms can be used as building blocks of novel thermoelectric, piezoelectric, and multiferroic materials as well as materials for energy production and storage. Substitution of regular atomic ions by selected superatomic ions in energy materials can enhance both transport properties and stability of the bulk and interface. These achievements show that the future of this field is bright and, with new design and synthesis paradigm, superatoms with unique functionalities can unravel new science and applications.There are challenges as well as opportunities. While considerable work over the past two decades have showcased numerous superhalogens and superalkalis, similar progress has not been made in the design and synthesis of superatoms that mimic the chemistry of transition metal, noble metal, and rare-earth metal atoms. This is important because of the importance of these materials in technology such as catalysis and magnetism. For example, significant amount of work has been done on single atom-catalysts yet work on single-superatom catalysts is in its infancy. Work using ligands to manipulate the properties of core metal clusters is important for their applications in catalysis, magnetism, biomedical applications, and optical materials.
In the field of superatom-based materials, although many recent experiments have confirmed and supported the theoretical designs and mechanisms, many more systematic syntheses and characterizations are needed to further narrow the gap between the prototype modeling and experimental realization. In practice, doping superatomic ions into materials is often limited by the metastability and the creation of impurity phases. Besides optimizing the known synthetic routes, other non-equilibrium synthetic pathways are worth exploring to kinetically stabilize the metastable phases, such as laser deposition and ablation,248–251 high-temperature quenching,252 and high-energy ball milling.253 Beyond conventional techniques to characterize structural features, such as the X-ray diffraction and neutron scattering, optical spectroscopy, electron microscopy, and solid-state NMR, probing the dynamics of the superatomic ions inside a material would require development of characterization techniques with improved spatial and temporal resolutions. Meanwhile, new computational tools and machine-learning methods254 are needed to expedite property-oriented materials discovery based on different superatoms, as well as to enable accurate evaluation of phase stability. Large-scale molecular dynamics simulations are also needed to best capture the compositional and defect complexity possibly introduced by superatomic dopants in the bulk and interface.
Different from the conventional atom-based crystals, superatomic crystals have complex geometries with large unit cells and manifold orientations, resulting in strong phonon scattering (including higher order phonon scattering) and phonon coherence. To accurately calculate the phonon transport in superatomic crystals, machine-learning-based potentials are highly desirable. Such potentials are efficient for simulation but challenging for fitting, which is also true for simulating superatom-based thermoelectric materials involving both phonons and electrons. Accurate electronic band structure is also needed for calculating the figure of merit to measure the thermoelectric performance.
In any field of science, a common question to ask is about its future. For cluster science in general and superatoms in particular, the answer that comes to our mind is same as the one Walt Disney gave when asked about the future of Disney World. His answer, that Disney World will survive as long as there is human imagination, may very well apply to cluster science.
Author contributions
P. J., H. F., and Q. S. proposed the topic of the Review. All authors wrote and discussed the manuscript.Conflicts of interest
The authors declare no competing interests.Data availability
The data underlying this study are available in the published article.Acknowledgements
P. J. acknowledges partial support by the U.S. Department of Energy, Office of Basic Energy Sciences, Division of Materials Sciences and Engineering under Award DE-FG02-96ER45579. P. J. and H. F. acknowledge partial support by the U.S. Department of Energy award number DE-EE0008865. S. Q. acknowledges partial support by the National Natural Science Foundation of China [92372112 and 22373005].References
- E. E. B. Campbell, Proceedings of Nobel Symposium, World Scientific Publishing Co., Sweden, 2011 Search PubMed.
- P. Jena and A. Castleman Jr, Nanoclusters–A Bridge across Disciplines, Elsevier, 2010 Search PubMed.
- S. Sahu; R. Choudhury and P. Jena, Nano-scale materials: from science to technology, Nova Publishers, New York, 2006 Search PubMed.
- P. Jena; S. Khanna and B. Rao, Clusters and Nano-Assemblies: Physical and Biological Systems, World Scientific, Singapore, 2005 Search PubMed.
- Superatoms: Principles, Synthesis, and Applications, ed. P. Jena and Q. Sun, Wiley, 2022 Search PubMed.
- N. Gaston, Superatoms: An Introduction, CRC Press, 1st edn, 2023 Search PubMed.
- P. Milani and S. Iannotta, Cluster beam synthesis of nanostructured materials, Springer Science & Business Media, 2012 Search PubMed.
- Y. Kawazoe; T. Kondow and K. Ohno, Clusters and nanomaterials: theory and experiment, Springer Science & Business Media, 2013 Search PubMed.
- K. Sattler, Cluster Assembled Materials, CRC Press, New York, 1996 Search PubMed.
- J. Jellinek, Theory of atomic and molecular clusters: with a glimpse at experiments, Springer Science & Business Media, 2012 Search PubMed.
- C. V. Ciobanu; C.-Z. Wang and K.-M. Ho, Atomic Structure Prediction of Nanostructures, Clusters and Surfaces, John Wiley & Sons, New York, 2013 Search PubMed.
- J. A. Alonso, Structure and properties of atomic nanoclusters, World Scientific, 2nd edn, 2012 Search PubMed.
- P. K. Chattaraj, Aromaticity and Metal clusters, CRC Press, New York, 2010 Search PubMed.
- A. W. Castleman and S. N. Khanna, In Quantum Phenomena in Clusters and Nanostructures, Springer Berlin Heidelberg, Berlin, Heidelberg, 2003 Search PubMed.
- S. Sugano, Microcluster Physics, Springer Berlin Heidelberg, Berlin, Heidelberg, 1991 Search PubMed.
- W. A. de Heer, The physics of simple metal clusters: experimental aspects and simple models, Rev. Mod. Phys., 1993, 65, 611 CrossRef CAS.
- T. G. Dietz, M. A. Duncan, D. E. Powers and R. E. Smalley, Laser production of supersonic metal cluster beams, J. Chem. Phys., 1981, 74, 6511–6512 CrossRef CAS.
- S. C. Lee and R. H. Holm, The Clusters of Nitrogenase:
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Synthetic Methodology in the Construction of Weak-Field Clusters, Chem. Rev., 2004, 104, 1135 CrossRef CAS PubMed.
Synthetic Methodology in the Construction of Weak-Field Clusters, Chem. Rev., 2004, 104, 1135 CrossRef CAS PubMed. - B. T. Sterenberg, L. Scoles and A. J. Carty, Synthesis, structure, bonding and reactivity in clusters of the lower phosphorus oxides, Coord. Chem. Rev., 2002, 231, 183 CrossRef CAS.
- E. J. Bieske and O. Dopfer, High-Resolution Spectroscopy of Cluster Ions, Chem. Rev., 2000, 100, 3963 CrossRef CAS PubMed.
- G. Ganteför, H. R. Siekmann, H. O. Lutz and K. H. Meiwes-Broer, Pure metal and metal-doped rare-gas clusters grown in a pulsed ARC cluster ion source, Chem. Phys. Lett., 1990, 165, 293–296 CrossRef.
- X. Lei, H. Zhang, Y. Jia and Z. X. Luo, Gas-phase preparation and the stability of superatomic Nb11O15−, Phys. Chem. Chem. Phys., 2021, 23, 15766–15773 RSC.
- H. Tsunoyama, M. Shibuta, M. Nakaya, T. Eguchi and A. Nakajima, Synthesis and Characterization of Metal-Encapsulating Si16 Cage Superatoms, Acc. Chem. Res., 2018, 51, 1735–1745 CrossRef CAS PubMed.
- T. Kamoshida, M. Shibuta, T. Ohta, T. Eguchi and A. Nakajima, Molecularly Designed Cluster–Surface Interaction for Halogen-like and Alkali-like Metal-Encapsulating Silicon Cage Superatoms on n- and p-Type Organic Substrates, J. Phys. Chem. C, 2022, 126(26), 10889–10899 CrossRef CAS.
- T. Yokoyama and A. Nakajima, Bridging the Gas and Condensed Phases for Metal-Atom Encapsulating Silicon- and Germanium-Cage Superatoms: Electrical Properties of Assembled Superatoms, Phys. Chem. Chem. Phys., 2023, 25, 9738–9752 RSC.
- C. M. Aikens, R. Jin, X. Roy and T. Tsukuda, From atom-precise nanoclusters to superatom materials, J. Chem. Phys., 2022, 156, 170401 CrossRef CAS PubMed.
- D. A. Reed, T. J. Hochuli, N. A. Gadjieva, S. He, R. A. Wiscons, A. K. Bartholomew, A. M. Champsaur, M. L. Steigerwald, X. Roy and C. Nuckolls, Controlling Ligand Coordination Spheres and Cluster Fusion in Superatoms, J. Am. Chem. Soc., 2022, 144, 306–313 CrossRef CAS PubMed.
- J. Yang, J. C. Russell, S. Tao, M. Lessio, F. Wang, A. C. Hartnett, S. R. Peurifoy, E. A. Doud, E. S. O’Brien, N. Gadjieva, D. R. Reichman, X. Zhu, A. C. Crowther, S. J. L. Billinge, X. Roy, M. L. Steigerwald and C. Nuckolls, Superatomic solid solutions, Nat. Chem., 2021, 13, 607–613 CrossRef CAS PubMed.
- H. Hirai, S. Ito, S. Takano, K. Koyasu and T. Tsukuda, Ligand-protected gold/silver superatoms: current status and emerging trends, Chem. Sci., 2020, 11, 12233 RSC.
- Y. Fukumoto, T. Omoda, H. Hirai, S. Takano, K. Harano and T. Tsukuda, Diphosphine-Protected IrAu12 with Open Site(s): Synthesis and Programmed Stepwise Assembly, Angew. Chem., Int. Ed., 2024, 63, e20240202 CrossRef PubMed.
- T.-H. Chiu, J.-H. Liao, F. Gam, Y.-Y. Wu, X. Wang, S. Kahlal, J.-Y. Saillard and C. W. Liu, Hydride-Containing Eight-Electron Pt/Ag Superatoms: Structure, Bonding, and Multi-NMR Studies, J. Am. Chem. Soc., 2022, 144(23), 10599–10607 CrossRef CAS PubMed.
- H. Shen and T. Mizuta, An Atomically Precise Alkynyl-Protected PtAg42 Superatom Nanocluster and Its Structural Implications, Chem. – Asian J., 2017, 12, 2904–2907 CrossRef CAS PubMed.
- M. Wang, L. Wang, H. Wu, J. Sun, X. Xu, S. Guo, Y. Jia, S. Li, Z.-J. Guan and H. Shen, PtAg18 superatoms costabilized by phosphines and halides: synthesis, structure, and catalysis, Nanoscale, 2023, 15, 17818 RSC.
- I. Antsiburov, J. Stephan, R. J. J. Weininger, C. Gemel and R. A. Fischer, Copper Imidazolin-imine Coordination Compounds as Precursors for a Cu/Al Complex, Inorg. Chem., 2024, 63(38), 17331–17339 Search PubMed.
- K. Yamamoto and T. Imaoka, Precision Synthesis of Subnanoparticles Using Dendrimers as a Superatom Synthesizer, Acc. Chem. Res., 2014, 47, 1127–1136 CrossRef CAS PubMed.
- T. Kambe, N. Haruta, T. Imaoka and K. Yamamoto, Solution-phase synthesis of Al13− using a dendrimer template, Nat. Commun., 2017, 8, 2046 CrossRef PubMed.
- H. Muramatsu, T. Kambe, T. Tsukamoto, A. Kuzume, R. Hosono, T. Imaoka and K. Yamamoto, Synthesis and functionalities of FeSn12 superatom prepared by single atom introduction with a dendrimer template, Chem. – Eur. J., 2024, 30, e20240 CrossRef PubMed.
- H. Muramatsu, T. Kambe, T. Tsukamoto, T. Imaoka and K. Yamamoto, Controlled Synthesis of Au25 Superatom Using a Dendrimer Template, Molecules, 2022, 27, 3398 Search PubMed.
- R. Kubo, Electronic properties of metallic fine particles. I, J. Phys. Soc. Jpn., 1962, 17, 975–986 CrossRef CAS.
- X. Gu, M. Ji, S. H. Wei and X. G. Gong, AuN clusters (N = 32, 33, 34, 35): Cagelike structures of pure metal atoms, Phys. Rev. B:Condens. Matter Mater. Phys., 2004, 70, 205401 CrossRef.
- V. Bonačić-Koutecký and R. Mitrić, Theoretical Exploration of Ultrafast Dynamics in Atomic Clusters: Analysis and Control, Chem. Rev., 2005, 105, 11 Search PubMed.
- T. Maier, M. Jarrell, T. Pruschke and M. H. Hettler, Quantum cluster theories, Rev. Mod. Phys., 2005, 77, 1027 CrossRef CAS.
- M. Einax, W. Dieterich and P. Maass, Colloquium: Cluster growth on surfaces: Densities, size distributions, and morphologies, Rev. Mod. Phys., 2013, 85, 921 CrossRef CAS.
- L. Rozes and C. Sanchez, Titanium oxo-clusters: precursors for a Lego-like construction of nanostructured hybrid materials, Chem. Soc. Rev., 2011, 40, 1006 RSC.
- P. Jensen, Growth of nanostructures by cluster deposition: Experiments and simple models, Rev. Mod. Phys., 1999, 71, 1695 CrossRef CAS.
- X. Fan, D. S. Tikhonov and M. Schnell, Electric nuclear quadrupole coupling reveals dissociation of HCl with a few water molecules, Science, 2024, 384, 1435–1440 CrossRef PubMed.
- B. Zhang, Y. Yu, Y. Y. Zhang, S. Jiang, Q. Li, H. S. Hu, G. Li, Z. Zhao, C. Wang, H. Xie, W. Zhang, D. Dai, G. Wu, D. H. Zhang, L. Jiang, J. Li and X. Yang, Infrared spectroscopy of neutral water clusters at finite temperature: Evidence for a noncyclic pentamer, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 15423–15428 Search PubMed.
- F. Liu, S. N. Khanna and P. Jena, Magnetism of Small Vanadium Clusters, Phys. Rev. B:Condens. Matter Mater. Phys., 1991, 43, 8179 Search PubMed.
- M. Rohdenburg, V. A. Azov and J. Warneke, New Perspectives in the Noble Gas Chemistry Opened by Electrophilic Anions, Front. Chem., 2020, 8 Search PubMed.
- H. Fang, H. Banjade, Deepika and P. Jena, Realization of Zn3+ oxidation state, Nanoscale, 2021, 13, 14041 RSC.
- T. Zhao, J. Zhou, Q. Wang and P. Jena, Like Charges Attract?, J. Phys. Chem. Lett., 2016, 7(14), 2689–2695 CrossRef CAS PubMed.
- M. Mayer, V. van Lessen, M. Rohdenburg, G.-L. Hou, Z. Yang, R. M. Exner, E. Aprà, V. A. Azov, S. Grabowsky, S. S. Xantheas, K. R. Asmis, X. B. Wang, C. Jenne and J. Warneke, Rational Design of an Argon-Binding Superelectrophilic Anion, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(17), 8167–8172 CrossRef CAS PubMed.
- B. Xue, Y. Lai, L. Cai, Y. Liu, J.-F. Yin and P. Yin, Emergent Research Trends on the Structural Relaxation Dynamics of Molecular Clusters: From Structure–Property Relationship to New Function Prediction, Acc. Chem. Res., 2024, 57(20), 3057–3067 CrossRef CAS PubMed.
- A. V. Artem’ev and C. W. Liu, Recent progress in dichalcophosphate coinage metal clusters and superatoms, Chem. Commun., 2023, 59, 7182–7195 RSC.
- F. Yu, J. Li and Z. Liu, et al., From Atomic Physics to Superatomic Physics, J. Cluster Sci., 2023, 34, 1691–1708 CrossRef CAS.
- P. Jena and Q. Sun, Super Atomic Clusters: Design Rules and Potential for Building Blocks of Materials, Chem. Rev., 2018, 118(11), 5755–5870 CrossRef CAS PubMed.
- S. N. Khanna and P. Jena, Assembling crystals from clusters, Phys. Rev. Lett., 1992, 69, 1664–1667 CrossRef CAS PubMed.
- S. N. Khanna and P. Jena, Atomic Clusters: Building blocks for a class of solids, Phys. Rev. B:Condens. Matter Mater. Phys., 1995, 51, 13705–13716 CrossRef CAS PubMed.
- W. D. Knight, K. Clemenger, W. A. de Heer, W. A. Saunders, M. Y. Chou and M. L. Cohen, Phys. Rev. Lett., 1984, 52, 2141 CrossRef CAS.
- R. E. Leuchtner, A. C. Harms and A. W. Castleman, Thermal metal cluster anion reactions: Behavior of aluminum clusters with oxygen, J. Chem. Phys., 1989, 91(4), 2753–2754 CrossRef CAS.
- G. L. Gutsev and A. I. Boldyrev, DVM-Xα calculations on the ionization potentials of MXk+1–complex anions and the electron affinities of MXk+1 “superhalogens”, Chem. Phys., 1981, 56, 277–283 CrossRef CAS.
- G. L. Gutsev and A. I. Boldyrev, The electronic structure of superhalogens and superalkalies, Russ. Chem. Rev., 1987, 56, 519 CrossRef.
- G. N. Lewis, The atom and the molecule, J. Am. Chem. Soc., 1916, 38, 762–785 CrossRef CAS.
- I. Langmuir, The agreement of electrons in atoms and molecules, J. Am. Chem. Soc., 1919, 41, 868–934 CrossRef CAS.
- P. Pyykkö and N. Runeberg, Icosahedral WAu12: A predicted closed-shell species, stabilized by aurophilic attraction and relativity and in accord with the 18-electron rule, Angew. Chem., Int. Ed., 2002, 41, 2174–2176 CrossRef.
- X. Li, B. Kiran, J. Li, H.-J. Zhai and L.-S. Wang, Experimentalobservation and confirmation of icosahedral W@Au12 and Mo@Au12 molecules, Angew. Chem., Int. Ed., 2002, 41, 4786–4789 CrossRef CAS PubMed.
- J.-P. Dognon, C. Clavaguéra and P. Pyykkö, A Predicted Organometallic Series Following a 32-Electron Principle: An@C28 (An = Th, Pa+, U2+, Pu4+), J. Am. Chem. Soc., 2009, 131(1), 238–243 CrossRef CAS PubMed.
- D. Manna, A. Sirohiwal and T. K. Ghanty, Pu@C24: A New Example Satisfying the 32-Electron Principle, J. Phys. Chem. C, 2014, 118(13), 7211–7221 CrossRef CAS.
- E. Hückel, Quantentheoretische beiträge zum benzolproblem, Eur. Phys. J. A, 1931, 70, 204–286 Search PubMed.
- E. Hückel, Zur quantentheorie der doppelbindung, Eur. Phys. J. A, 1930, 60, 423–456 Search PubMed.
- K. Wade, The structural significance of the number of skeletal bonding electron-pairs in carboranes, the higher boranes and borane anions, and various transition-metal carbonyl cluster compounds, J. Chem. Soc. D, 1971, 792–793 RSC.
- K. Wade, in Advances in Inorganic Chemistry and Radiochemistry, ed. H. J. Emeléus and A. G. Sharpe, Academic Press, New York, 1976, vol. 18 Search PubMed.
- D. M. P. Mingos, A general theory for cluster and ring compounds of the main group and transition elements, Nat. Phys. Sci., 1972, 236, 99–102 CrossRef CAS.
- D. M. P. Mingos, Polyhedral skeletal electron pair approach, Acc. Chem. Res., 1984, 17, 311–319 CrossRef CAS.
- G. Liu, A. Pinkard, S. M. Ciborowski, V. Chauhan, Z. Zhu, A. P. Aydt, S. N. Khanna, X. Roy and K. H. Bowen, Tuning the electronic properties of hexanuclear cobalt sulfide superatoms via ligand substitution, Chem. Sci., 2019, 10, 1760 RSC.
- A. M. Champsaur, J. Yu, X. Roy, D. W. Paley, M. L. Steigerwald, C. Nuckolls and C. M. Bejger, Two-Dimensional Nanosheets from Redox-Active Superatoms, ACS Cent. Sci., 2017, 3, 1050–1055 CrossRef CAS PubMed.
- N. A. Gadjieva, A. M. Champsaur, M. L. Steigerwald, X. Roy and C. Nuckolls, Dimensional Control of Assembling Metal Chalcogenide Clusters, Eur. J. Inorg. Chem., 2020, 1245–1254 CrossRef CAS.
- H. Gholipour-Ranjbar, H. Fang, J. Guan, D. Peters, A. Seifert, P. Jena and J. Laskin, Designing New Metal Chalcogenide Nanoclusters through Atom-by-Atom Substitution, Small, 2021, 17, 2002927 CrossRef PubMed.
- M. Walter, J. Akola, O. Lopez-Acevedo, P. D. Jadzinsky, G. Calero, C. J. Ackerson, R. L. Whetten, H. Grönbeck and H. Häkkinen, A unified view of ligand-protected gold clusters as superatom complexes, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9157–9162 CrossRef CAS PubMed.
- Protected Metal Clusters: From Fundamentals to Applications, ed. T. Tsukuda and H. Häkkinen, Elsevier, ISBN: 9780081000861, eBook ISBN: 9780444635020, 1st edn, 2015, vol. 9 Search PubMed.
- M. W. Heaven, A. Dass, P. S. White, K. M. Holt and R. W. Murray, Crystal Structure of the Gold Nanoparticle [N(C8H17)4][Au25(SCH2CH2Ph)18], J. Am. Chem. Soc., 2008, 130(12), 3754–3755 CrossRef CAS PubMed.
- R. H. Adnan, J. M. L. Madridejos, A. S. Alotabi, G. F. Metha and G. G. Andersson, A Review of State of the Art in Phosphine Ligated Gold Clusters and Application in Catalysis, Adv. Sci., 2022, 9, 2105692 CrossRef CAS PubMed.
- I. Chakraborty and T. Pradeep, Atomically Precise Clusters of Noble Metals: Emerging Link between Atoms and Nanoparticles, Chem. Rev., 2017, 117(12), 8208–8271 CrossRef CAS PubMed.
- A. Muñoz-Castro, Bonding Interaction Within Concentric Structural Layers in Gold Superatoms. The Concentric Bond, ChemPhysChem, 2025, 26, e202400892 CrossRef PubMed.
- W. Xu, B. Zhu and X. Zeng, et al., A grand unified model for liganded gold clusters, Nat. Commun., 2016, 7, 13574 CrossRef CAS PubMed.
- Y. Xu, J. Chen, A. P. Aydt, L. Zhang, I. Sergeyev, E. G. Keeler, B. Choi, S. He, D. R. Reichman, R. A. Friesner, C. Nuckolls, M. L. Steigerwald, X. Roy and A. E. McDermott, Electron and Spin Delocalization in [Co6Se8(PEt3)6]0/+1 Superatoms, ChemPhysChem, 2024, 25, e202300064 CrossRef CAS PubMed.
- H. Gholipour-Ranjbar, L. Sertse, D. Forbes and J. Laskin, Effect of Ligands on the Reactivity of the Undercoordinated Fragment Ions of Co6S8(PEt3–xPhx)6+ (x = 0–3) Clusters on Surfaces, J. Phys. Chem. C, 2024, 128(20), 8232–8238 CrossRef CAS.
- H. Gholipour-Ranjbar, Deepika and P. Jena,
et al., Gas-phase fragmentation of single heteroatom-incorporated Co5MS8(PEt3)6+ (M = Mn, Fe, Co, Ni) nanoclusters, Commun. Chem., 2022, 5, 130 CrossRef CAS PubMed.
- Deepika, H. Gholipour-Ranjbar, H. Fang, L. Sertse, L. Laskin and P. Jena, Atomically Precise Core-Tailored Metal Chalcogenide Nanoclusters: Tuning the Electronic Structure and Magnetic Properties, J. Phys. Chem. C, 2022, 126(14), 6512–6522 CrossRef CAS.
- J. Cheng, Q. Feng, X. Li and J. Yang, Two-Dimensional Robust Ferromagnetic Semiconductors via Assembly of Magnetic Superatoms [Fe6S8(CN)6]5−, J. Phys. Chem. Lett., 2023, 14, 5048–5054 CrossRef CAS PubMed.
- H. Xiang, S.-H. Wei and X. Gong, Structures of [Ag7(SR)4]− and [Ag7(DMSA)4]−, J. Am. Chem. Soc., 2010, 132, 7355–7360 CrossRef CAS PubMed.
- Protected Metal Clusters: From Fundamentals to Applications, ed. T. Tsukuda and H. Häkkinen, 1st edn, 2015, vol. 9 Search PubMed.
- T. Dainese, S. Antonello, S. Bogialli, W. Fei, A. Venzo and F. Maran, Gold Fusion: From Au25(SR)18 to Au38(SR)24, the Most Unexpected Transformation of a Very Stable Nanocluster, ACS Nano, 2018, 12, 7057–7066 CrossRef CAS PubMed.
- A. Muñoz-Castro, Single, Double and Triple Intercluster Bonds: Analyses of M2Au36(SR)24 (M = Au, Pd, Pt) as 14-, 12- and 10-ve Superatomic Molecules, Chem. Commun., 2019, 55, 7307–7310 RSC.
- P. L. Rodríguez-Kessler and A. Muñoz-Castro, Electronic Communication in Au38(SR)24 Superatomic Molecules, ChemPhysChem, 2024, 25, e202400183 CrossRef PubMed.
- J. Chen, Q.-F. Zhang, T. A. Bonaccorso, P. G. Williard and L.-S. Wang, Controlling Gold Nanoclusters by Diphospine Ligands, J. Am. Chem. Soc., 2014, 136, 92–95 CrossRef CAS PubMed.
- A. Muñoz-Castro, Triple 1D ≡ 1D superatomic bonding. Au22(dppo)6 as a Π4- and Δ2-triply bonded cluster based on Au11 assembled units, Phys. Chem. Chem. Phys., 2020, 22, 1422–1426 RSC.
- X. Yang, W. Jiang, C. B. Knobler and M. F. Hawthorne, Rigid-Rod Molecules: Carborods. Synthesis of Tetrameric p-Carboranes and the Crystal Structure of Bis(tri-n-butylsilyl)tetra-p-carborane, J. Am. Chem. Soc., 1992, 114, 9719–9721 CrossRef CAS.
- J. Mueller, K. Base, T. F. Magnera and J. Michl, Rigid-rod oligo-p-carboranes for molecular tinkertoys. An inorganic Langmuir-Blodgett film with a functionalized outer surface, J. Am. Chem. Soc., 1992, 114, 9721–9722 CrossRef CAS.
- A. A. Buglak and M. T. Nguyen, Interactions of coinage metal nanoclusters with low-molecular-weight biocompounds, Biophys. Rev., 2024, 16, 441–477 CAS.
- S. Maity, S. Kolay, S. Chakraborty, A. Devi, Rashi and A. A. Patra, Comprehensive Review of Atomically Precise Metal Nanoclusters with Emergent Photophysical Properties Towards Diverse Applications, Chem. Soc. Rev., 2025, 54, 1785–1844 RSC.
- A.-M. Hada, M. Lamy de la Chapelle, M. Focsan and S. Astilean, Recent Advances in Metal Nanoclusters: From Novel Synthesis to Emerging Applications, Molecules, 2025, 30, 3848 CrossRef CAS PubMed.
- M. F. Matus and H. Häkkinen, Understanding ligand-protected noble metal nanoclusters at work, Nat. Rev. Mater., 2023, 8, 372–389 CrossRef CAS.
- S. Li, N.-N. Li, X.-Y. Dong, S.-Q. Zang and T. C. W. Mak, Chemical Flexibility of Atomically Precise Metal Clusters, Chem. Rev., 2024, 124, 7262–7378 CrossRef CAS PubMed.
- J. Zhao, Q. Du, S. Zhou and V. Kumar, Endohedrally Doped Cage Clusters, Chem. Rev., 2020, 120, 9021–9163 CrossRef CAS PubMed.
- A. Hirsch, Z. Chen and H. Jiao, Spherical Aromaticity in Ih Symmetrical Fullerenes: The 2(N + 1)2 Rule, Angew. Chem., Int. Ed., 2000, 39, 3915–3917 CrossRef CAS PubMed.
- B. Z. Child, S. Giri, S. Gronert and P. Jena, Aromatic Superhalogens, Chem. – Eur. J., 2014, 20, 4736–4745 CrossRef CAS PubMed.
- B. Pathak, D. Samanta, R. Ahuja and P. Jena, Borane Derivatives: A New Class of Super- and Hyperhalogens, ChemPhysChem, 2011, 12, 2423–2428 CrossRef CAS PubMed.
- K. Terasaka, T. Kamoshida, T. Ichikawa, T. Yokoyama, M. Shibuta, M. Hatanaka and A. Nakajima, Alkaline Earth Metal Superatom of W@Si16: Characterization of Group 6 Metal Encapsulating Si16 Cage on Organic Substrates, J. Am. Chem. Soc., 2024, 146, 9605–9613 CrossRef CAS PubMed.
- R. Saito, K. Isozaki, Y. Mizuhata and M. Nakamura, Synthesis of N2-Type Superatomic Molecules, J. Am. Chem. Soc., 2024, 146, 20930–20936 CrossRef CAS PubMed.
- S. Kenzler and A. Schnepf, Metalloid gold clusters – past, current and future aspects, Chem. Sci., 2021, 12, 3116–3129 RSC.
- S. Takano, H. Hirai, T. Nakashima, T. Iwasa, T. Taketsugu and T. Tsukuda, Photoluminescence of Doped Superatoms M@Au12 (M = Ru, Rh, Ir) Homoleptically Capped by (Ph2)PCH2P(Ph2): Efficient Room-Temperature Phosphorescence from Ru@Au12, J. Am. Chem. Soc., 2021, 143(28), 10560–10564 CrossRef CAS PubMed.
- H. Yi, S. Song, S. M. Han, J. Lee, W. Kim, E. Sim and D. Lee, Superatom-in-Superatom Nanoclusters: Synthesis, Structure, and Photoluminescence, Angew. Chem., Int. Ed., 2023, 62, e202302591 CrossRef CAS PubMed.
- M. Han, J. Paik, Y. Kim, M.-G. Choi, C. M. Aikens and D. Lee, Insights into the Metal-Exchange Synthesis of MAg24(SR)18 (M = Ni, Pd, Pt) Nanoclusters, Chem. Mater., 2020, 32, 10216–10226 CrossRef.
- H. Yi, S. M. Han, S. Song, M. Kim, E. Sim and D. Lee, Superatom-in-Superatom [RhH@Ag24(SPhMe2)18]2− Nanocluster, Angew. Chem., Int. Ed., 2021, 60, 22293–22300 CrossRef CAS PubMed.
- I. Antsiburov, M. Schütz, R. Bühler, M. Muhr, J. Stephan, C. Gemel, W. Klein, S. Kahlal, J.-Y. Saillard and R. A. Fischer, All-Hydrocarbon-Ligated Superatomic Gold/Aluminum Clusters, Inorg. Chem., 2024, 63, 3749–3756 CrossRef CAS PubMed.
- X. Li, X. Wu, N. Dilley, H. Gholipour-Ranjbar, S. Lee, D. Zemlyanov, H. Fang, P. Jena and J. Laskin, Discovery of a Ferromagnetic Nickel Chalcogenide Nanocluster Ni3S3H(PEt3)5, Small, 2025, 21, 2500070 CrossRef CAS PubMed.
- K. Esumi, K. Miyamoto and T. Yoshimura, Comparison of PAMAM–Au and PPI–Au nanocomposites and their catalytic activity for reduction of 4-nitrophenol, J. Colloid Interface Sci., 2002, 254, 402–405 CrossRef CAS PubMed.
- H. Ye and R. M. Crooks, Electrocatalytic O2 reduction at glassy carbon electrodes modified with dendrimer-encapsulated Pt nanoparticles, J. Am. Chem. Soc., 2005, 127, 4930–4934 CrossRef CAS PubMed.
- C. Ornelas, J. R. Aranzaes, L. Salmon and D. Astruc, “Click” dendrimers: Synthesis, redox sensing of Pd(OAc)2, and remarkable catalytic hydrogenation activity of precise Pd nanoparticles stabilized by 1,2,3-triazole-containing dendrimers, Chem. – Eur. J., 2008, 14, 50–64 CrossRef CAS PubMed.
- J. C. Garcia-Martinez and R. M. Crooks, Extraction of Au nanoparticles having narrow size distributions from within dendrimer templates, J. Am. Chem. Soc., 2004, 126, 16170–16178 CrossRef CAS PubMed.
- J. Warneke, G.-L. Hou, E. Aprà, C. Jenne, Z. Yang, Z. Qin, K. Kowalski, X.-B. Wang and S. S. Xantheas, Electronic Structure and Stability of [B12X12]2− (X = F–At): A Combined Photoelectron Spectroscopic and Theoretical Study, J. Am. Chem. Soc., 2017, 139, 14749–14756 CrossRef CAS PubMed.
- H. Zhao, J. Zhou and P. Jena, Stability of B12(CN)122−: Implications for Lithium and Magnesium Ion Batteries, Angew. Chem., Int. Ed., 2016, 55, 3704 CrossRef CAS PubMed.
- T. Zhao, J. Zhou, Q. Wang and P. Jena, Colossal Stability of Gas-phase Tri-anions: Super-pnictogens, Angew. Chem., Int. Ed., 2017, 56, 13421 CrossRef CAS PubMed.
- R. Nesper, The Zintl-Klemm Concept – A Historical Survey, Z. Anorg. Allg. Chem., 2014, 640, 2639–2648 CrossRef CAS.
- H. Fang and P. Jena, Stable Tetra- and Penta-Anions in the Gas Phase, Angew. Chem., Int. Ed., 2019, 58, 11248 CrossRef CAS PubMed.
- M. Zhong, H. Fang and P. Jena, Record-high stability and compactness of multiply-charged clusters aided by selected terminal groups, Phys. Chem. Chem. Phys., 2020, 22, 4880 RSC.
- M. K. Scheller, R. N. Compton and L. S. Cederbaum, Gas-Phase Multiply Charged Anions, Science, 1995, 270, 1160–1166 Search PubMed.
- L. Pauling, The theoretical prediction of the physical properties of many electron atoms and ions. Mole refraction, diamagnetic susceptibility, and extension in space, Proc. R. Soc. London, Ser. A, 1927, 114181–114211 Search PubMed.
- L. Pauling, The formulas of antimonic acid and the antimonates, J. Am. Chem. Soc., 1933, 55, 1895–1900 CrossRef CAS.
- N. Bartlett, Xenon hexafluoroplatinate (V) Xe+[PtF6]−, Proc. Chem. Soc., 1962, 197–236 Search PubMed.
- L. Khriachtchev, M. Pettersson, N. Runeberg, J. Lundell and M. Rasanen, A stable argon compound, Nature, 2000, 406, 874–876 CrossRef CAS PubMed.
- M. Pettersson, J. Lundell and M. Rasanen, Neutral rare-gas containing charge transfer molecules in solid matrices. I. HXeCl, HXeBr, HXeI, and HKrCl in Kr and Xe, J. Chem. Phys., 1995, 102, 6423–6431 CrossRef CAS.
- J. Lundell, L. Khriachtchev, M. Pettersson and M. Rasanen, Formation and characterization of neutral krypton and xenon hydrides in low-temperature matrices, Low Temp. Phys., 2000, 26, 680–690 CrossRef CAS.
- L. Khriachtchev, K. Isokoski, A. Cohen, M. Rasanen and R. B. Gerber, A small neutral molecule with two noble-gas atoms: HXeOXeH, J. Am. Chem. Soc., 2008, 130, 6114–6118 CrossRef CAS PubMed.
- A. Lignell, L. Khriachtchev, J. Lundell, H. Tanskanen and M. Rasanen, On theoretical predictions of noble-gas hydrides, J. Chem. Phys., 2006, 125, 184514 CrossRef PubMed.
- M. Pettersson, L. Khriachtchev, A. Lignell, M. Rasanen, Z. Bihary and R. B. Gerber, HKrF in solid krypton, J. Chem. Phys., 2002, 116, 2508–2515 CrossRef CAS.
- M. Pettersson, J. Lundell, L. Khriachtchev and M. Rasanen, Neutral rare-gas containing charge-transfer molecules in solid matrices. III. HXeCN, HXeNC, and HKrCN in Kr and Xe, J. Chem. Phys., 1998, 109, 618–625 CrossRef CAS.
- D. Samanta, Prediction of superhalogen-stabilized noble gas compounds, J. Phys. Chem. Lett., 2014, 5, 3151–3156 CrossRef CAS PubMed.
- M. Mayer, M. Rohdenburg, V. van Lessen, M. C. Nierstenhöfer, E. Aprà, S. Grabowsky, K. R. Asmis, C. Jenne and J. Warneke, First steps towards a stable neon compound: observation and bonding analysis of [B12(CN)11Ne]−, Chem. Commun., 2020, 56, 4591 RSC.
- M. Rohdenburg, Z. Warneke, H. Knorke, M. Icker and J. Warneke, Chemical Synthesis with Gaseous Molecular Ions: Harvesting [B12Br11N2]− from a Mass Spectrometer, Angew. Chem., Int. Ed., 2023, 62, e202308600 CrossRef CAS PubMed.
- M. Joshi and T. K. Ghanty, Quantum chemical prediction of a superelectrophilic dianion and its binding with noble gas atoms, Chem. Commun., 2019, 55, 14379 Search PubMed.
- M. M. Zhong, H. Fang, Deepika and P. Jena, Super-electrophiles of tri- and tetra-anions stabilized by selected terminal groups and their role in binding noble gas atoms, Phys. Chem. Chem. Phys., 2021, 23, 21496 Search PubMed.
- H. Fang, Deepika and P. Jena, Binding of noble gas atoms by superhalogens, J. Chem. Phys., 2021, 155, 014304 Search PubMed.
- C. Liu, S. A. Nikolaev, W. Ren and L. A. Burton, Electrides: a review, J. Mater. Chem. C, 2020, 8, 10551–10567 Search PubMed.
- J. L. Dye, M. R. Yemen, M. G. DaGue and J. Lehn, Optical spectra of alkali metal anion and electride films, J. Chem. Phys., 1978, 68, 1665 CrossRef CAS.
- M. Kitano, et al., Ammonia Synthesis Using a Stable Electride as an Electron Donor and Reversible Hydrogen Store, Nat. Chem., 2012, 4, 934 CrossRef CAS PubMed.
- Y. Toda, H. Hirayama, N. Kuganathan, A. Torrisi, P. V. Sushko and H. Hosono, Activation and splitting of carbon dioxide on the surface of an inorganic electride material, Nat. Commun., 2013, 4, 2378 CrossRef PubMed.
- H. Fang, J. Zhou and P. Jena, Super-alkalis as building blocks of one-dimensional hierarchical electrides, Nanoscale, 2018, 10, 22963 Search PubMed.
- Y. Tsuji, P. L. V. K. Dasari, S. F. Elatresh, R. Hoffmann and N. W. Ashcroft, Structural diversity and electron confinement in Li4N: potential for 0-D, 2-D, and 3-D electrides, J. Am. Chem. Soc., 2016, 138, 14108–14120 Search PubMed.
- C. Park, S. W. Kim and M. Yoon, First-principle prediction of new electrides with nontrivial and topology based on one-dimensional building blocks, Phys. Rev. Lett., 2018, 120, 026401 CrossRef CAS PubMed.
- W. Meng, J. Jiang, Y. Jiao, F. Ma, Y. Yang, Z. Cheng and X. Wang, Zero-Dimensional Interstitial Electron-Induced Spin–Orbit Coupling Dirac States in Sandwich Electride, Small Sci., 2024, 2400131 CrossRef CAS PubMed.
- X. Liu, Z. Ding, J. Liu, W. Hu and J. Yang, Two-dimensional Ca4N2 as a one-dimensional electride [Ca4N2]2+·2e− with ultrahigh conductance, Nanoscale, 2020, 12, 5578 RSC.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Activenonmetallic Au and Pt species on ceria-based water-gas shift catalysts, Science, 2003, 301, 935–938 CrossRef CAS PubMed.
- J. Shan, M. Li, L. F. Allard, S. Lee and M. Flytzani-Stephanopoulos, Mild oxidation of methane to methanol or acetic acid on supported isolated rhodium catalysts, Nature, 2017, 551, 605–608 Search PubMed.
- C. Buzea, I. I. Pacheco and K. Robbie, Nanomaterials and nanoparticles: sources and toxicity, Biointerphases, 2007, 2(4), MR17–MR71 CrossRef PubMed.
- X.-F. Yang, et al., Single-Atom Catalysts: A New Frontier in Heterogeneous Catalysis, Acc. Chem. Res., 2013, 46, 1740–1748 Search PubMed.
- A. Wang, J. Li and T. Zhang, Heterogeneous single-atom catalysis, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- B. Qiao, et al., Single-atom catalysis of CO oxidation using Pt1/FeOx, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- R. T. Hannagan, G. Giannakakis, R. Réocreux, J. Schumann, J. Finzel, Y. Wang, A. Michaelides, P. Deshlahra, P. Christopher and M. Flytzani-Stephanopoulos, et al., First-principles design of a single-atom-alloy propane dehydrogenation catalyst, Science, 2021, 372, 1444–1447 CrossRef CAS.
- S. Ding, M. J. Hülsey, J. Pérez-Ramírez and N. Yan, Transforming energy with single-atom catalysts, Joule, 2019, 3, 2897–2929 Search PubMed.
- T. He, A. R. P. Santiago, Y. Kong, M. A. Ahsan, R. Luque, A. Du and H. Pan, Atomically Dispersed Heteronuclear Dual-Atom Catalysts: A New Rising Star in Atomic Catalysis, Small, 2022, 18, 2106091 CrossRef CAS PubMed.
- L. Lei, X. Guo, X. Han, L. Fei, X. Guo and D. Wang, From Synthesis to Mechanisms: In-Depth Exploration of the Dual-Atom Catalytic Mechanisms Toward Oxygen Electrocatalysis, Adv. Mater., 2024, 36, 2311434 CrossRef CAS PubMed.
- X. Wang, L. Xu, C. Li, C. Zhang, H. Yao, R. Xu, P. Cui, X. Zheng, M. Gu, J. Lee, H. Jiang and M. Huang, Developing a class of dual atom materials for multifunctional catalytic reactions, Nat. Commun., 2023, 14, 7210 CrossRef CAS PubMed.
- S. Lu, M. Mazur, K. Guo, D. C. Stoian, M. Gu, W. M. Tucho and Z. Yu, Breaking Scaling Relations for Highly Efficient Electroreduction of CO2 to CO on Atomically Dispersed Heteronuclear Dual-Atom Catalyst, Small, 2024, 20, 2309251 Search PubMed.
- Y. Shao, Q. Yuan and J. Zhou, Single-Atom Catalysts and Dual-Atom Catalysts for CO2 Electroreduction: Competition or Cooperation?, Small, 2023, 19, 2303446 Search PubMed.
- X. L. Zhang, L. Zhang, J. H. Chen, C. Y. Li and W. M. Sun, On the Interaction between Superatom Al12Be and DNA Nucleobases/Base Pairs: Bonding Nature and Potential Applications in O2 Activation and CO Oxidation, ACS Omega, 2020, 5, 15325–15334 Search PubMed.
- X. L. Zhang, L. Zhang, Y. L. Ye, X. H. Li, B. L. Ni, Y. Li and W. M. Sun, On the Role of Alkali-Metal-Like Superatom Al12P in Reduction and Conversion of Carbon Dioxide, Chem. – Eur. J., 2021, 27, 1039–1045 CrossRef CAS PubMed.
- X. L. Zhang, Y. L. Ye, L. Zhang, X. H. Li, D. Yu, J. H. Chen and W. M. Sun, Designing an alkali-metal-like superatom Ca3B for ambient nitrogen reduction to ammonia, Phys. Chem. Chem. Phys., 2021, 23, 18908–18915 RSC.
- Y. L. Ye, K. Y. Pan, W. L. Wang, B. L. Ni and W. M. Sun, On the Catalytic Performance of (ZrO)n (n = 1–4) Clusters for CO Oxidation: A DFT Study, ChemPhysChem, 2023, 24, e202200776 CrossRef CAS PubMed.
- T. Sengupta and S. N. Khanna, Converting CO2 to formic acid by tuning quantum states in metal chalcogenide clusters, Commun. Chem., 2023, 6, 53 CrossRef CAS PubMed.
- G. Liu, P. Poths, X. Zhang, Z. Zhu, M. Marshall, M. Blankenhorn, A. N. Alexandrova and K. H. Bowen, CO2 Hydrogenation to Formate and Formic Acid by Bimetallic Palladium–Copper Hydride Clusters, J. Am. Chem. Soc., 2020, 142, 7930–7936 CrossRef CAS PubMed.
- X. Zhang, G. Liu, K. Meiwes-Broer, G. Ganteför and K. Bowen, CO2 Activation and Hydrogenation by PtHn− Cluster Anions, Angew. Chem., Int. Ed., 2016, 55, 9644–9647 CrossRef CAS PubMed.
- C. Kerpal, D. J. Harding, J. T. Lyon, G. Meijer and A. Fielicke, N2 Activation by Neutral Ruthenium Clusters, J. Phys. Chem. C, 2013, 117, 12153–12158 CrossRef CAS.
- L.-H. Mou, Y. Li, Z.-Y. Li, Q.-Y. Liu, Y. Ren, H. Chen and S.-G. He, Dinitrogen Activation and Functionalization by Heteronuclear Metal Cluster Anions FeV2C2− at Room Temperature, J. Phys. Chem. Lett., 2020, 11, 9990–9994 CrossRef CAS PubMed.
- H. Himmel and M. Reiher, Intrinsic Dinitrogen Activation at Bare Metal Atoms, Angew. Chem., Int. Ed., 2006, 45, 6264–6288 CrossRef CAS PubMed.
- Y. Y. Wang, X. Ding, Z. Ji, X. Huang and W. Li, Heteronuclear Trimetallic MFe2 and M2Fe (M = V, Nb, and Ta) Clusters for Dinitrogen Activation, ChemPhysChem, 2023, 24, e202200952 CrossRef CAS PubMed.
- S. Behera, N. King, D. Samanta and P. Jena, Potential for ZrO clusters as replacement Pd catalyst, J. Chem. Phys., 2014, 141, 034301 CrossRef PubMed.
- Y. W. Wang, Y. D. Zhu, X. W. Zhu, J. L. Shi, X. Y. Ren, L. L. Zhang and S. F. Li, Machine Selective Hydrogenation of CO2 to CH3OH on a Dynamically Magic Single-Cluster Catalyst: Cu3/MoS2/Ag(111), ACS Catal., 2023, 13, 714–724 CrossRef CAS.
- B. Chen, Y.-F. Jiang, H. Xiao and J. Li, Selective CO2-to-HCOOH Electroreduction on Graphdiyne Supported Bimetallic Single-Cluster Catalysts, ACS Catal., 2024, 14, 10510–10518 CrossRef CAS.
- X. Li, S. Mitchell, Y. Fang, J. Li, J. Perez-Ramirez and J. Lu, Advances in heterogeneous single-cluster catalysis, Nat. Rev. Chem., 2023, 7, 754–767 CrossRef CAS PubMed.
- M. E. Kilic and P. Jena, Catalytic Potential of Supported Superatoms, Small, 2024, 2403888 CrossRef CAS PubMed.
- S. J. Peppernick, K. D. D. Gunaratne and A. W. Castleman, Superatom spectroscopy and the electronic state correlation between elements and isoelectronic molecular counterparts, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 975 CrossRef CAS PubMed.
- M. E. Kilic and P. Jena, Superatoms as Superior Catalysts: ZrO vs. Pd, Small, 2025, 2409289 CrossRef CAS PubMed.
- M. E. Kilic and P. Jena, Single-atom vs. Single-superatom as Catalysts for Ammonia Production, Chem. Commun., 2025, 61, 3127–3130 RSC.
- E. Meirzadeh, A. M. Evans and M. Rezaee, et al., A few-layer covalent network of fullerenes, Nature, 2023, 613, 71–76 CrossRef CAS PubMed.
- X. Chang, Y. Xu and M. von Delius, Recent advances in supramolecular fullerene chemistry, Chem. Soc. Rev., 2024, 53, 47–83 RSC.
- M. Nakaya, T. Iwasa, H. Tsunoyama, T. Eguchi and A. Nakajima, Formation of a superatom monolayer using gas-phase-synthesized Ta@Si16 nanocluster ions, Nanoscale, 2014, 6, 14702–14707 RSC.
- Y. Meng, X. Lin, J. Huang and L. Zhang, Recent Advances in Carborane-Based Crystalline Porous Materials, Molecules, 2024, 29, 3916 CrossRef CAS PubMed.
- R. Núñez, M. Tarrés, A. Ferrer-Ugalde, F. F. de Biani and F. Teixidor, Electrochemistry and Photoluminescence of Icosahedral Carboranes, Boranes, Metallacarboranes, and Their Derivatives, Chem. Rev., 2016, 116, 14307–14378 CrossRef PubMed.
- S. He, S. Okuno, F. W. Ng, X. Pang, D. V. Esposito, N. M. Orchanian, M. L. Steigerwald, X. Roy and C. Nuckolls, Functional Monolayers on a Superatomic Pegboard, J. Am. Chem. Soc., 2023, 145, 8314–8318 CrossRef CAS PubMed.
- S. He, A. M. Evans, E. Meirzadeh, S. Y. Han, J. C. Russell, R. A. Wiscons, A. K. Bartholomew, D. A. Reed, A. Zangiabadi, M. L. Steigerwald, C. Nuckolls and X. Roy, Site-Selective Surface Modification of 2D Superatomic Re6Se8, J. Am. Chem. Soc., 2022, 144, 74 CrossRef CAS PubMed.
- H. Wang, X. Liu, W. Yang, G. Mao, Z. Meng, Z. Wu and H.-L. Jiang, Surface-Clean Au25 Nanoclusters in Modulated Microenvironment Enabled by Metal–Organic Frameworks for Enhanced Catalysis, J. Am. Chem. Soc., 2022, 144, 22008–22017 CrossRef CAS PubMed.
- J. Lyu, J. Qian, Z. Yang and J. Xie, Synthesis planning for atomically precise metal nanoclusters, Nanoscale Horiz., 2025, 10, 2304–2339 RSC.
- H. Li, X. Kang and M. Zhu, Superlattice Assembly for Empowering Metal Nanoclusters, Acc. Chem. Res., 2024, 57, 3194–3205 CrossRef CAS PubMed.
- G. Deng, B. K. Teo and N. Zheng, Assembly of Chiral Cluster-Based Metal–Organic Frameworks and the Chirality Memory Effect during their Disassembly, J. Am. Chem. Soc., 2021, 143, 10214–10220 CrossRef CAS PubMed.
- Z.-E. Zhang, Y.-F. Zhang, Y.-Z. Zhang, H.-L. Li, L.-Y. Sun, L.-J. Wang and Y.-F. Han, Construction and Hierarchical Self-Assembly of Multifunctional Coordination Cages with Triangular Metal–Metal-Bonded Units, J. Am. Chem. Soc., 2023, 145, 7446–7453 CrossRef CAS PubMed.
- L.-J. Wang, Z.-E. Zhang, Y.-Z. Zhang and Y.-F. Han, Cavity-Partitioned Self-Assembled Cage for Sequential Separation in Aqueous Solutions, Angew. Chem., Int. Ed., 2024, 63, e202407278 CrossRef CAS PubMed.
- E. A. Doud, A. Voevodin and T. J. Hochuli, et al., Superatoms in materials science, Nat. Rev. Mater., 2020, 5, 371–387 CrossRef.
- A. Hebard, M. Rosseinsky, R. Haddon, D. W. Murphy, S. H. Glarum, T. T. M. Palstra, A. P. Ramirez and A. R. Kortan, Superconductivity at 18 K in potassium-doped C60, Nature, 1991, 350, 600–601 CrossRef CAS.
- W. L. Ong, E. S. O'Brien, P. S. Dougherty, D. W. Paley, C. Fred Higgs Iii, A. J. McGaughey, J. A. Malen and X. Roy, Orientational order controls crystalline and amorphous thermal transport in superatomic crystals, Nat. Mater., 2017, 16, 83–88 CrossRef CAS PubMed.
- K. Lee, S. F. Maehrlein, X. Zhong, D. Meggiolaro, J. C. Russell, D. A. Reed, B. Choi, F. De Angelis, X. Roy and X.-Y. Zhu, Hierarchical Coherent Phonons in a Superatomic Semiconductor, Adv. Mater., 2019, 31, 1903209 CrossRef PubMed.
- Q. Liang, M. Bartnof, Y.-L. He, J. A. Malen and A. J. H. McGaughey, Fullerene rotational dynamics generate disordered configurations that suppress thermal conductivity in superatomic crystals, Nanoscale Horiz., 2020, 5, 1524 RSC.
- P.-H. Du, C. Zhang, J. Sun, T. Li and Q. Sun, Phase Stability, Strong Four-Phonon Scattering, and Low Lattice Thermal Conductivity in Superatom-Based Superionic Conductor Na3OBH4, ACS Appl. Mater. Interfaces, 2022, 14(42), 47882–47891 CrossRef CAS PubMed.
- T. Li, P.-H. Du, L. Bai, Q. Sun and P. Jena, NaNO3 monolayer: A stable graphenelike supersalt with strong four-phonon scattering and low lattice thermal conductivity insensitive to temperature, Phys. Rev. Mater., 2022, 6, 064009 CrossRef CAS.
- P.-H. Du, C. Zhang, T. Li and Q. Sun, Low lattice thermal conductivity with two-channel thermal transport in the superatomic crystal PH4AlBr4, Phys. Rev. B, 2023, 107, 155204 CrossRef CAS.
- P.-H. Du, Q. Wang, Q. Sun and P. Jena, Thermal Rectification of Sierpiński Tetrahedron Fractals Assembled from Supertetrahedral T2-type Tin Selenide Clusters, Phys. Rev. B, 2025, 111, L121406 CrossRef CAS.
- P. H. Du, Y. Kawazoe and Q. Sun, Unconventional Thermal Rectification in Assemblies of Supertetrahedral T3-Type Sn4In6Se20 Clusters, Nano Lett., 2024, 24(49), 15501–15508 CrossRef CAS PubMed.
- H. Fang and P. Jena, Li-rich antiperovskite superionic conductors based on cluster ions, Proc. Natl. Acad. Sci. U. S. A., 2017, 14, 1047 Search PubMed.
- H. Fang and P. Jena, Sodium superionic conductors based on clusters, ACS Appl. Mater. Interfaces, 2019, 11, 963–972 CrossRef CAS PubMed.
- H. Fang and P. Jena, Argyrodite-type advanced lithium conductors and transport mechanisms beyond paddle-wheel effect, Nat. Commun., 2022, 13, 2078 CrossRef CAS PubMed.
- H. Fang and P. Jena, Kinetic effects of anion clusters on the interfacial stability between solid-state electrolyte and metal anode, PRX Energy, 2023, 4, 043013 CrossRef.
- B. He, F. Zhang and Y. Xin, et al., Halogen chemistry of solid electrolytes in all-solid-state batteries, Nat. Rev. Chem., 2023, 7, 826–842 Search PubMed.
- J. Bian, S. Ling, B. Deng, H. Lin, R. Zhao, L. Kong, H. Yuan, J. Zhu, S. Han, L. Wang, R.-Q. Zhang, Y. Zhao and Z. Lu, Ternary Rotational Polyanion Coupling Enables Fast Li Ion Dynamics in Tetrafluoroborate Ion Doped Antiperovskite Li2OHCl Solid Electrolyte, Angew. Chem., Int. Ed., 2024, 63, e202400144 CrossRef CAS PubMed.
- E. Ahiavi, J. A. Dawson, U. Kudu, M. Courty, M. Saiful Islam, O. Clemens, C. Masquelier and T. Famprikis, Mechanochemical synthesis and ion transport properties of Na3OX (X = Cl, Br, I and BH4) antiperovskite solid electrolytes, J. Power Sources, 2020, 471, 228489 CrossRef CAS.
- A. E. Maughan, Y. Ha, R. T. Pekarek and M. C. Schulze, Lowering the Activation Barriers for Lithium-Ion Conductivity through Orientational Disorder in the Cyanide Argyrodite Li6PS5CN, Chem. Mater., 2021, 33, 5127–5136 CrossRef CAS.
- L. Liu, H. Wang, D. Ye, H. Zhao, J. Zhang and Y. Tang, Fluorine-Like BH4-Doped Li6PS5Cl with Improved Ionic Conductivity and Electrochemical Stability, ACS Appl. Mater. Interfaces, 2024, 16, 31341–31347 CrossRef CAS PubMed.
- Y.-J. Jang, H. Seo, Y.-S. Lee, S. Kang, W. Cho, Y. W. Cho and J.-H. Kim, Lithium Superionic Conduction in BH4-Substituted Thiophosphate Solid Electrolytes, Adv. Sci., 2023, 10, 2204942 CrossRef CAS PubMed.
- J.-H. Han, et al., Borohydride and halide dual-substituted lithium argyrodites, Mater. Horiz., 2024, 11, 251–261 RSC.
- Y. Wang, V. Raj, Q. Yan, C. D. Fincher, Y. Li, R. Raj, H. Celio, A. Dolocan, G. Yang, F. A. Perras, Y.-M. Chiang, J. Watt, H. Fang, P. Jena and D. Mitlin, Understanding the Role of Borohydride Doping in Electrochemical Stability of Argyrodite Li6PS5Cl Solid-State Electrolyte, Adv. Mater., 2025, 2506095 CrossRef CAS PubMed.
- Y. Sun, Y. Wang, X. Liang, Y. Xia, L. Peng, H. Jia, H. Li, L. Bai, J. Feng, H. Jiang and J. Xie, Rotational Cluster Anion Enabling Superionic Conductivity in Sodium-Rich Antiperovskite Na3OBH4, J. Am. Chem. Soc., 2019, 141, 5640–5644 CrossRef CAS PubMed.
- K. Jun, B. Lee, R. L. Kam and G. Ceder, The nonexistence of a paddle-wheel effect in superionic conductors, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2316493121 CrossRef CAS PubMed.
- K. Jun, Y. Chen, G. Wei, X. Yang and G. Ceder, Diffusion mechanisms of fast lithium-ion conductors, Nat. Rev. Mater., 2024, 9, 887–905 CrossRef CAS.
- H. Fang and P. Jena, Atomic-Level Design of Water-Resistant Hybrid Perovskites for Solar Cells by Using Cluster Ions, J. Phys. Chem. Lett., 2017, 8(16), 3726–3733 CrossRef CAS PubMed.
- H. Fang and P. Jena, Super-ion inspired colorful hybrid perovskite solar cells, J. Mater. Chem. A, 2016, 4, 4728–4737 RSC.
- S. Wang, C. Wu, H. Yao, L. Ding and F. Hao, The nonhalides in perovskite solar cells, Mater. Chem. Front., 2023, 7, 789 RSC.
- P.-Y. Lin, A. Loganathan, I. Raifuku, M.-H. Li, Y.-Y. Chiu, S.-T. Chang, A. Fakharuddin, C.-F. Lin, T.-F. Guo, L. Schmidt-Mende and P. Chen, Pseudo-Halide Perovskite Solar Cells, Adv. Energy Mater., 2021, 11, 2100818 CrossRef CAS.
- S. Xu, G. Liu, H. Zheng, X. Xu, L. Zhang, H. Xu, L. Zhu, F. Kong, Y. Li and X. Pan, Boosting Photovoltaic Performance and Stability of Super-Halogen-Substituted Perovskite Solar Cells by Simultaneous Methylammonium Immobilization and Vacancy Compensation, ACS Appl. Mater. Interfaces, 2020, 12(7), 8249–8259 CrossRef CAS PubMed.
- Z. Ma, Z. Xiao, W. Zhou, L. Jin, D. Huang, H. Jiang, T. Yang, Y. Liu and Y. Huang, Efficient CH3NH3PbI3−x(SeCN)x perovskite solar cells with improved crystallization and defect passivation, J. Alloys Compd., 2020, 822, 153539 CrossRef CAS.
- S. Bai, P. Da, C. Li, Z. Wang, Z. Yuan, F. Fu, M. Kawecki, X. Liu, N. Sakai and J. T.-W. Wang, Planar Perovskite Solar Cells with Long-Term Stability using Ionic Liquid Additives, Nature, 2019, 571, 245–250 CrossRef CAS PubMed.
- J. Sanchez-Diaz, R. S. Sánchez, S. Masi, M. Kreĉmarová, A. O. Alvarez, E. M. Barea, J. Rodriguez-Romero, V. S. Chirvony, J. F. Sánchez-Royo, J. P. Martinez-Pastor and I. Mora-Seró, Tin Perovskite Solar Cells with 41, 300 h of Operational Stability in N2 through A Synergistic Chemical Engineering Approach, Joule, 2022, 6, 861–883 CrossRef CAS PubMed.
- H. Kim, J. W. Lee, G. R. Han, S. K. Kim and J. H. Oh, Synergistic Effects of Cation and Anion in an Ionic Imidazolium Tetrafluoroborate Additive for Improving the Efficiency and Stability of Half-Mixed Pb–Sn Perovskite Solar Cells, Adv. Funct. Mater., 2021, 31, 2008801 CrossRef CAS.
- T. Zhang, Y. Zu, B. Zeng, R. Gan, P. Liu, X. Li, F. Han, Y. Qian, L. Zhao, A. Feng and Z. Wu, Anion regulation for surface passivation enables ultrahigh-stability perovskite nanocrystals, J. Chem. Phys., 2024, 21(161), 164710 CrossRef PubMed.
- D. Kubota, R. Katoh, H. Kanda, H. Yaguchi, T. N. Murakami and N. Nishimura, Spontaneous Heterointerface Modulation by a Methylammonium Tetrafluoroborate Additive for a Narrow-Bandgap FAPbI3 Photoabsorber in Perovskite Solar Cells, ACS Appl. Mater. Interfaces, 2024, 16, 53918–53929 CrossRef PubMed.
- R. Moro, X. Xu, S. Yin and W. A. de Heer, Ferroelectricity in Free Niobium Clusters, Science, 2003, 300, 1265 CrossRef CAS PubMed.
- R. Moro, S. Yin, X. Xu and W. A. de Heer, Spin Uncoupling in Free Nb Clusters: Support for Nascent Superconductivity, Phys. Rev. Lett., 2004, 93, 086803 CrossRef PubMed.
- L. Ma, R. Moro, J. Bowlan, A. Kirilyuk and W. A. de Heer, Multiferroic Rhodium Clusters, Phys. Rev. Lett., 2014, 113, 157203 CrossRef PubMed.
- W. Fa, C. Luo and J. Dong, Coexistence of ferroelectricity and ferromagnetism in tantalum clusters, J. Chem. Phys., 2006, 125, 114305 CrossRef PubMed.
- S. Yin, X. Xu, A. Liang, J. Bowlan, R. Moro and W. A. de Heer, Electron Pairing in Ferroelectric Niobium and Niobium Alloy Clusters, J. Supercond. Novel Magn., 2008, 21, 265 CrossRef CAS.
- Y. Niu, L. Li, Z. Qi, H. Aung, X. Han, R. Tenne, Y. Yao, A. Zak and Y. Guo, 0D van der Waals interfacial ferroelectricity, Nat. Commun., 2023, 14, 5578 CrossRef CAS PubMed.
- Y. Gao, M. Wu and P. Jena, A family of ionic supersalts with covalent-like directionality and unconventional multiferroicity, Nat. Commun., 2021, 12, 1331 CrossRef CAS PubMed.
- Y. Gao, H. Banjade, M. Wu and P. Jena, SbCl4: An Exceptional Superhalogen as the Building Block of a Mixed Valence Supercrystal with Unconventional Ferroelectricity, J. Phys. Chem. Lett., 2022, 13, 1049 CrossRef CAS PubMed.
- Y. Ai, W. Liao, Y. Weng, H. Lv, X. Chen, X. Song, P. Li and R. Xiong, Discovery of Ferroelectricity in the Fullerene Adduct C60S8, J. Am. Chem. Soc., 2023, 145, 23292 CrossRef CAS PubMed.
- F. Huang, M. Xiong, J. Zhou and T. Yang, Assembly-inspired multiferroicity with nontrivial Chern insulating phase from exohedral metallofullerenes, J. Chem. Phys., 2024, 160, 184302 CrossRef CAS PubMed.
- Y. Zhao, Y. Guo, Y. Qi, X. Jiang, Y. Su and J. J. Zhao, Coexistence of Ferroelectricity and Ferromagnetism in Fullerene-Based One-Dimensional Chains, Adv. Sci., 2023, 10, 2301265 CrossRef CAS PubMed.
- B. Wang, J. Hong, Y. Yang, H. Zhao, L. Long and L. Zheng, Achievement of a giant piezoelectric coefficient and piezoelectric voltage coefficient through plastic molecular-based ferroelectric materials, Matter, 2022, 5, 1296–1304 CrossRef CAS.
- T. Li, P. Du, Q. Sun and P. Jena, Thermoelectric Figure of Merit of a Superatomic Crystal Re6Se8I2 Monolayer, Phys. Rev. Appl., 2022, 18, 064067 CrossRef CAS.
- R. Pfenninger, M. Struzik and I. Garbayo, et al., A low ride on processing temperature for fast lithium conduction in garnet solid-state battery films, Nat. Energy, 2019, 4, 475–483 CrossRef CAS.
- J. Johannes Kriegler, E. Jaimez-Farnham, L. Hille, E. Dashjav and M. F. Zaeh, Pulsed laser ablation of a ceramic electrolyte for all-solid-state batteries, Proc. CIRP, 2022, 111, 800–805 CrossRef.
- Y. Eatmon, J. W. Stiles, S. Hayashi, M. Rupp and C. Arnold, Air Stabilization of Li7P3S11 Solid-State Electrolytes through Laser-Based Processing, Nanomaterials, 2023, 13, 2210 CrossRef CAS PubMed.
- K. Takada, R. Ma, M. Osada, N. Ohta, L. Zhang and T. Sasaki, Formation of nano-sized particles of a solid electrolyte by laser ablation, J. Power Sources, 2005, 146, 703–706 CrossRef CAS.
- A. Gautam, M. Sadowski, N. Prinz, H. Eickhoff, N. Minafra, M. Ghidiu, S. P. Culver, K. Albe, T. F. Fässler, M. Zobel and W. G. Zeier, Rapid crystallization and kinetic freezing of site-disorder in the lithium superionic argyrodite Li6PS5Br, Chem. Mater., 2019, 31, 10178–10185 CrossRef CAS.
- M. Hofer, M. Grube, C. F. Burmeister, P. Michalowski, S. Zellmer and A. Kwade, Effective mechanochemical synthesis of sulfide solid electrolyte Li3PS4 in a high energy ball mill by process investigation, Adv. Powder Technol., 2023, 34, 104004 CrossRef CAS.
- L. Fu, Q. Du, L. Sai and J. Zhao, Accelerating Global Search of Large-Sized Silver Clusters Using Cluster Graph Attention Network, J. Phys. Chem. Lett., 2024, 15, 9160–9166 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |