
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle crystal covalent organic frameworks
Yongzhi Chen†
,
Zhihao Zhang†
and
Donglin Jiang
 *
*
Department of Chemistry, Faculty of Science, National University of Singapore, 3 Science Drive 3, Singapore 117543, Singapore. E-mail: chmjd@nus.edu.sg
First published on 13th May 2026
Abstract
Covalent organic frameworks (COFs) are crystalline porous polymers that provide a versatile platform for designing dimension-specific organic architectures via topology diagrams and constructing diverse materials with structure-originated unique functions. Their synthesis relies on the organization of organic building units through coupled physicochemical processes, including covalent polymerization, supramolecular polymerization, nucleation and crystallization. As a result, the polymerization system develops a highly complex reaction landscape, characterized by the coexistence of oligomers and polymers of diverse sizes and shapes, concurrent covalent bond formations and noncovalent interactions, and competing thermodynamic and kinetic pathways that respond sensitively and distinctively to conditions. This intrinsic complexity limits precise structural control in polycrystalline COFs and poses great challenges for the synthesis of single crystal COFs, which remain scarce in terms of structural diversity, scalability and accessibility. In this review, we inspect current strategies and methodologies for the design and synthesis of single crystal COFs, scrutinize polymerization, nucleation and crystallization mechanisms to identify key control parameters, correlate structure–property relationships and compare their properties and functions with polycrystalline COFs to elucidate the impact of structural integrity on performance. We envision the key challenges and outline future major directions. By building a full picture, this review provides a conceptual framework and roadmap for advancing single crystal COFs toward fundamental innovation and practical implementation.
 Yongzhi Chen | Yongzhi Chen obtained his bachelor's degree at Wuhan Polytechnic University in 2016 and his master's degree at South China University of Technology in 2019. He began his PhD studies the same year in Professor Donglin Jiang's research group at the National University of Singapore. In 2023, he completed his PhD degree and continued with postdoctoral research in the same group, where his efforts are devoted to the design, synthesis and photocatalytic applications of covalent organic frameworks. |
 Zhihao Zhang | Zhihao Zhang obtained his double bachelor's degree at Northeastern University and Univeristé Toulouse III-Paul Sabatier in 2022 and his master's degree at National University of Singapore. He is currently a PhD student under the supervision of Professor Donglin Jiang at the National University of Singapore. His research interests include the design and synthesis of covalent organic frameworks. |
 Donglin Jiang | Donglin Jiang obtained his Bachelor's degree in Chemistry at Zhejiang University in 1989 and his PhD. degree at The University of Tokyo in 1998. He was supported by a JSPS fellowship (DC2) during the years 1997–1998 and was appointed as an assistant professor at The University of Tokyo in 1998. In 2000, he was appointed as a group leader in the JST ERATO Nanospace project, and in 2005 he moved to the Institute for Molecular Science, National Institutes of Natural Sciences as an associate professor to set up an independent laboratory at Okazaki and initiated a study on covalent organic frameworks, where he developed and deepened the field. In 2016 he joined Japan Advanced Institute of Science and Technology as a full professor. In 2018 he moved to the National University of Singapore as a professor. He was the world's most highly cited researcher in chemistry from 2018 to 2025, as determined by Clarivate Analytics. |
1. Introduction
Single crystals offer an irreplaceable platform for both basic research and advanced applications. They share a general yet unique structural feature, i.e. extended and well-defined lattices across the material. Their grain-boundary free nature paves an excellent approach to inherent characteristics, such as mechanical, optical and electrical properties. These distinct features make single crystals invaluable, playing indispensable roles in various industries, particularly in optics and electronics.1–4 Whilst great progress has been made in synthesizing single crystals of organic and inorganic compounds, there still lacks efficient methodology to grow single crystals of macromolecules, especially extended covalent polymers obtained through step-growth polymerization. The difficulty lies in the complexity of the polymerization system, which consists of rapidly generated oligomers of different sizes and shapes that undergo uncontrollable reactions among themselves, and concurrently involves different processes such as step-growth polymerization, supramolecular polymerization, aggregation, precipitation, nucleation and crystallization. Developing single crystals from such complexity remains a substantial challenge. Achieving breakthroughs in these methods and syntheses would not only uncover the hidden structure–function relationship but also shift the regime toward materials design and implementations.As novel crystalline porous polymers, covalent organic frameworks (COFs) are attracting ever increasing attention due to their predesignable structures and unique functions (Fig. 1).5–11 COFs are diverse in structure in terms of both their skeleton and pores, offering an irreplaceable molecular platform for designing specific properties and functions, ranging from gas adsorption12–21 to molecular separation,22–29 light emission,30–37 proton conduction,38–49 semiconductors,50–56 sensors,57–63 catalysis,64–96 drug delivery97–104 and energy conversion and storage.105–115 COFs are composed of light elements, such as C, H, B, N, O and S, synthesized via one-pot polymerization reactions and obtained as insoluble powders. These powders are usually in a polycrystalline form other than the expected single crystal.116–119 This precludes the possibility of using single-crystal X-ray diffraction (SCXRD) analysis to resolve their atomic coordinates, leading to large uncertainties regarding their true structures. An essential subject of the field is how to synthesize single crystals.
 | ||
| Fig. 1 Growth patterns of COFs. (a) Topology Design of 1D COFs. (b) Topology design of 2D COFs. (c) Topology design of 3D COFs. | ||
One distinct feature of COFs in terms of design and synthesis is that their building units are diverse in geometry (Fig. 2).3 Depending on the geometric combination of building blocks, COFs form covalently linked one-dimensional, two-dimensional or three-dimensional (1D, 2D or 3D) structures. This covalently-linked bonding feature is unique and distiguishes COFs from metal-organic frameworks (MOFs), in which coodination bonds between metal ions and organic ligands drive the structural formation. Noticeably, the structural growth patterns of 1D, 2D and 3D COFs are different.3–5 For 1D COFs, the four-substituted C2-or C4-symmetric knots combine with V-shaped linkers to form extended porous belts (Fig. 1a, x dimension). For 2D COFs, knots and linkers are connected via covalent bonds to generate polygonal backbones, which extend over 2D planes to form polymeric networks (Fig. 1b, x–y dimension).116 The resultant 2D networks stack at sub-nanometre intervals along the z direction through noncovalent interactions (Fig. 1b, z dimension), creating extended crystalline porous materials.116 In contrast, 3D COFs require at least one building unit with 3D or orthogonal geometry to ensure 3D covalent growth of polymeric backbones (Fig. 1c, x–y–z dimensions).119–125
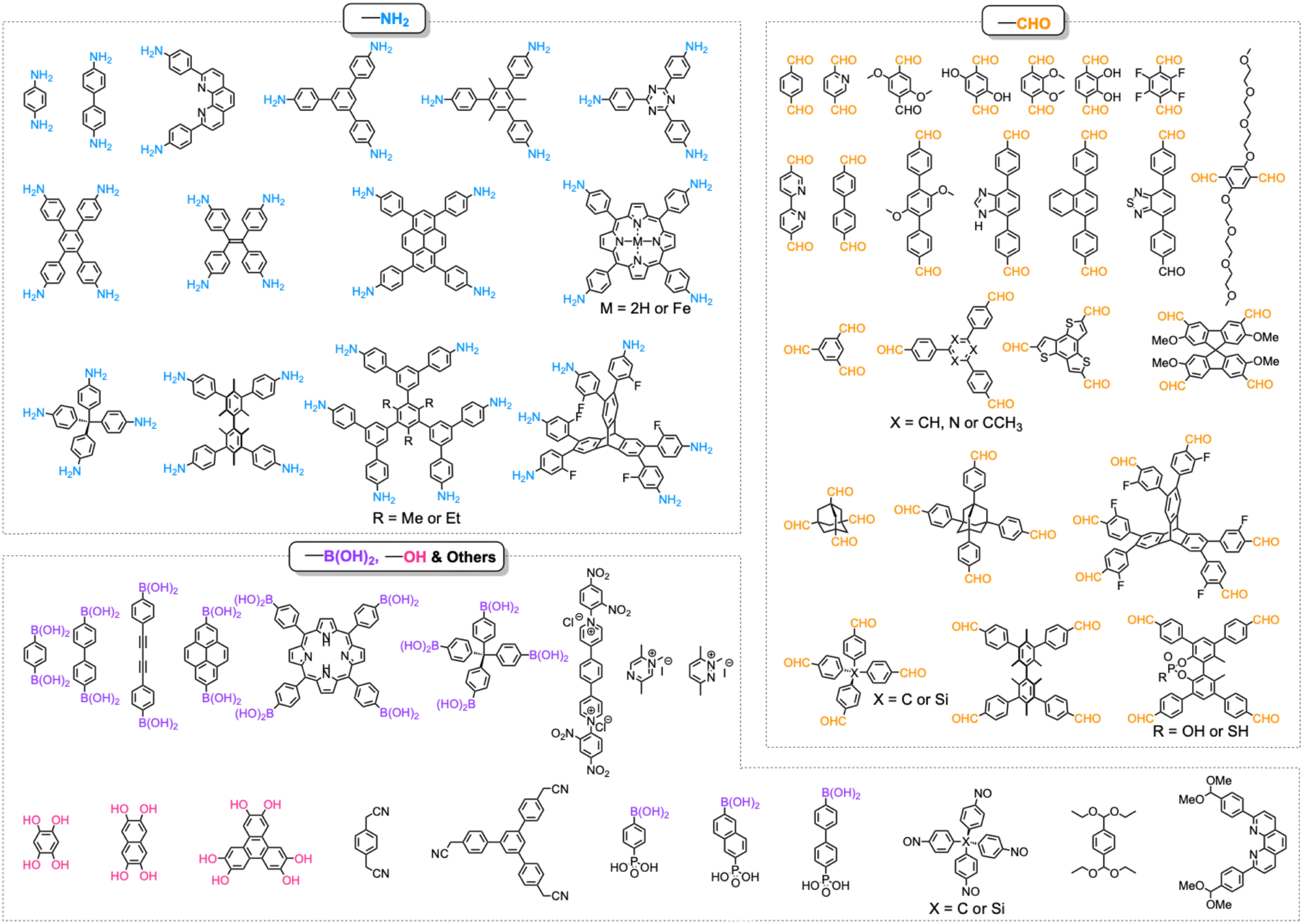 | ||
| Fig. 2 Monomer structures for the synthesis of single crystal COFs. | ||
Notably, the different growth patterns inherently require different design considerations. The synthesis of single crystal COFs must therefore be approached from different perspectives, with careful attention to the specific types of interactions involved and the strategies used to regulate them. For single crystal 2D COFs, their growth is anisotropic due to the involvement of both covalent bond formations and noncovalent interactions, which exhibit distinct yet related responses to synthetic conditions. It is worth noting that the noncovalent
interaction, i.e. π–π stacking, is not merely an auxiliary interaction in 2D COFs but constitutes a structural prerequisite for stabilizing their layered periodic framework. Indeed, 2D COFs reported universally to date rely on interlayer π–π stacking to maintain long-range order. In contrast, the growth of 3D COFs is predominantly governed by the covalent chemistry, though supramolecular interpenetration with less effective π–π stacking is commonly observed in the crystal structures of 3D COFs. Non-interpenetrated single crystal 3D COFs were successfully synthesized,126 and hence the absence of this noncovalent interaction has less effects on the formation of 3D COFs. Therefore, the supramolecular interpenetration is neither essential nor the structural driving force determining the synthesis of single crystal 3D COFs. Consequently, in view of complicated interactions in 2D systems, 3D COFs exhibit comparatively fewer barriers to single crystal growth, originating from the fact that their lattice growth is driven simply by covalent bonds with less effects from noncovalent interactions. Synthesizing single crystal 2D COFs requires control over both covalent bond formations and noncovalent interactions, which are distinctively and strongly associated with the kinetics and thermodynamics of the polymerization systems. Thus, in the case of 2D COFs, the key is to balance the growth of 2D covalent polymers over the x–y plane via polymerization and the extension of layers along the z axis through supramolecular polymerization, as well as nucleation and crystallization processes (Fig. 1b).127 These processes are affected by synthetic conditions with different sensitivities.127
To grow single crystal 2D COFs, synthetic conditions, including monomer concentration, temperature, catalyst, solvent, reaction time and reactor, need to be extensively optimized to find a critical zone, in which covalent polymerization, supramolecular polymerization, nucleation and crystallization can be well matched for the growth of single crystals other than polycrystals or amorphous polymers. Nonetheless, this critical zone is determined by multidimensional factors, while in most cases, a specific synthetic condition either favours the covalent growth or facilitates the supramolecular polymerization process. Thereby, systematic and concurrent control over the above two processes is a prerequisite for the formation of single crystal 2D COFs. In contrast, 3D COFs simplify the reaction system to form single crystals due to the reduced involvement of supramolecular polymerization (Fig. 1c). From the viewpoint of chemistry, the growth rate of single crystals in each dimension is equal due to the same chemical reactivity of functional groups, benefiting the formation of single crystal 3D COFs.
Progress over the past two decades in the design, synthesis and structural explorations has established the chemistry basis of the COF field, establishing a molecular platform for designed construction of diverse functional materials and systems.3–5,128–133 Looking toward the next decade, single crystal COFs are becoming important to advancements in design, functional exploration and practical applications, whilst synthetic methods towards single crystals need to be further explored. In this review, we summarize the advancements in single crystal COFs, by highlighting the unique structural features in these single crystals in comparison with their polycrystalline and/or amorphous counterparts. We scrutinize different synthetic strategies from the design of building units to the screening of reaction conditions, showing insights into diverse chemical approaches to single crystals. We compare limitations of different synthetic methods and predict future directions, with an aim to develop high-quality single crystal COFs and unlock their unique structures and intrinsic potentials.
2. Fundamentals, thermodynamics and kinetics
Single crystal COFs represent the highest degree of structural perfection in this class of materials, enabling precise determination of their atomic arrangements and correlation of structure–property relationships.134–138 The synthesis of single crystal COFs differs significantly from their polycrystalline counterparts in terms of reaction conditions, where regulating the thermodynamics and kinetics is critical throughout the whole process of polymerization, nucleation and crystallization (Fig. 3). Nonetheless, the inherent trade-offs and interdependencies between these processes make the growth of single crystal COFs challenging.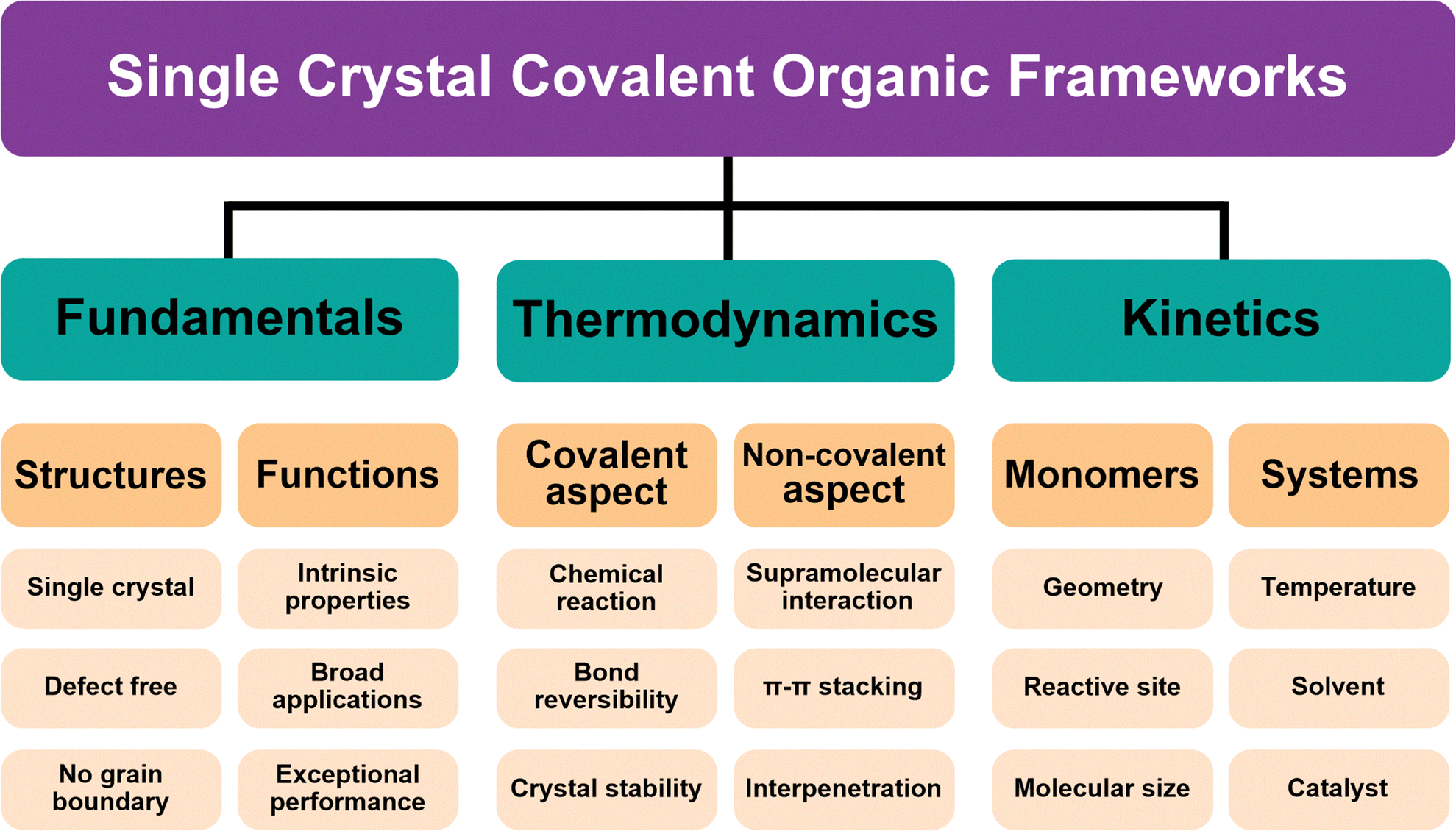 | ||
| Fig. 3 Single crystal COFs: fundamentals, thermodynamics and kinetics. | ||
2.1. Fundamental aspects
The development of single crystal COFs has revolutionized structural studies, shifting them from uncertainty to certainty. The pure and true structures from single crystals reveal intrinsic properties of COFs, which are inaccessible from polycrystalline and amorphous materials. In particular, polycrystalline COFs usually exhibit structural defects and disordered grain boundaries, which set additional barriers to accessing the ordered structures and pose great uncertainty to the structural accuracy. The properties and functions derived from the polycrystalline COFs represent an average level of both ordered and disordered structures. Unlike polycrystalline COFs, single crystal COFs exhibit rare or even no structural defects and exhibit excellent structural integrity throughout the whole crystal (Fig. 3). This marks a cornerstone in research, where single crystal COFs provide a unique platform for exploring their hidden structural features and original functions.127From the aspect of chemistry, developing methodologies for synthesizing single crystal COFs is challenging but highly desirable. It not only advances our capabilities in solving crystallization problems in crystallology but also provides new knowledge for better understanding of polymerization and supramolecular chemistry. Notably, innovation in such methodologies would inspire other fields, with transformative and long-lasting impacts.
From a materials science perspective, single-crystal COFs reveal their true nature to the field. Particularly for 2D COFs, their macroscopic morphology remained unknown until the synthesis of single crystal COFs. Remarkably, single crystal COFs with perfect structures do not exhibit the phase impurity-induced side effects of polycrystalline and amorphous materials and reveal the most relevant structure–function correlations.
From an application perspective, single crystal COFs have broad scopes ranging from optics to electronics, adsorption, separation, energy conversion and storage and many others. Notably, they usually show exceptional performances compared with their polycrystalline and amorphous counterparts.
2.2. Thermodynamics
Single crystal COFs have been synthesized through highly reversible chemical reactions to facilitate the error-correcting process, which is important throughout the nucleation and crystallization processes. Indeed, one-pot polymerization generates rather complicated reaction systems, in which crystallites, oligomers of different sizes, aggregates of different species and disordered segments inevitably co-exist. In this context, highly reversible reactions are necessary for the de-composition or de-assembly of structural defects, allowing monomers to be covalently linked and crystalize into single crystal COFs (Fig. 3).5,129Noteworthily, crystal formation must follow the thermodynamic criteria, which determine the equilibrium state of a crystallization system, generating thermodynamically stable crystals. For a specific polymerization reaction (ΔG = ΔH − TΔS), polymerizing small molecular units into large and continuous polymeric networks and macromolecules in an ordered manner is an entropy-decreasing process (ΔS < 0) but usually releases large amounts of heat (ΔH < 0). In this context, the reaction temperature determines whether this process can proceed spontaneously or not. Although lowering the temperature favours the supramolecular polymerization process, as monomers and oligomers need to have sufficient activation energy to overcome the energy barrier to promote the covalent polymerization, elevated temperatures are favourable for the kinetic process. Thermodynamics drives the formation of the most stable product and minimizes the surface energy to govern the morphological structures of single crystals (Fig. 3), and prolonging the reaction time is essential to reach a near-thermodynamic minimum and to reach the final equilibrium state.
2.3. Kinetics
The synthesis of single crystal COFs requires slowing down the kinetic processes, where nucleation, crystallization and crystal growth need to be carefully controlled to avoid and/or minimize structural defects, disorders and wrong connections. Nonetheless, there are many external and internal factors affecting kinetic processes (Fig. 3), rendering the synthesis of single crystal COFs a challenge.134Usually, a fully dissolved system is highly desired, as it provides a homogeneous solution to achieve suitable monomer concentrations. This is critical as nucleation occurs at the initial stage and is sensitive to the monomer concentration.127 On the one hand, a highly concentrated solution accelerates the polymerization and nucleation, resulting in rapid precipitation of products and unbalancing the concentration of different species in the reaction system. Consequently, polycrystalline COFs with disordered boundaries would generate concurrently. On the other hand, a too-diluted system slows down both polymerization and crystallization, hardly forming large enough single crystals due to the lack of sufficient building blocks. Therefore, a suitable concentration of monomers is necessary for the synthesis of single crystal COFs. For this reason, the solvent selection is key to the growth of single crystals, as concentration is related to the solubility of monomers and oligomers, where molecular backbone, size, rigidity, polarity and functional groups are factors that change the solubility.127
In addition, reaction kinetics is also affected by other external factors (Fig. 3), such as temperature, modulator, catalyst and reaction time. Among these factors, reaction temperature is one of the most important conditions to evaluate. For a typical 2D COF system, increasing the temperature usually accelerates covalent polymerization but disfavours supramolecular polymerization, resulting in low crystalline or even amorphous products. On the other hand, decreasing the temperature slows down polymerization but promotes noncovalent interactions, leading to -precipitates or aggregates of oligomers other than single crystals.
The above considerations unveil that the key is to find the reaction parameters that can achieve a good balance between polymerization, defect correction and supramolecular polymerization. Only systems that establish such a balance have the chance to generate high quality single crystals.
2.4. Phase diagram
The growth of single-crystal COFs is complex due to competitive thermodynamic and kinetic factors. Crystallization phase diagrams are useful tools in understanding the formation of single crystal COFs by providing specific zones of monomers, oligomers, polymers, temperature, catalyst and solvent. Clearly, the phase diagram is multi-component in nature, and should be divided into monomer–temperature, monomer–catalyst, monomer–polymer, polymer–temperature and polymer–catalyst diagrams.Crystallization phase diagrams have four typical zones – undersaturated zone, metastable zone, liable zone and precipitation zone.139–141 In the undersaturated zone, the monomer is fully dissolved, and there is no crystal growth. The metastable zone allows sluggish nucleation and slow growth of crystals. In contrast, rapid nucleation and fast growth of crystals occur in the labile zone, which usually results in polycrystalline or amorphous polymers due to supersaturation. In the precipitation zone only polycrystalline and disordered polymers are generated because of supersaturation.
Although crystallization phase diagrams guide the growth of single crystal COFs, not much is known due to the complicated polymerization systems and crystallization processes involved. In contrast, phase diagrams have been investigated for supramolecular assemblies, linear polymer systems and branched polymer networks, providing valuable conceptual frameworks for understanding complex self-assembly and crystallization processes.141–151 In supramolecular systems, phase diagrams are typically established via systematic variation of temperature, concentration, solvent composition or pH, combined with in situ spectroscopic and scattering techniques to resolve assembly pathways, nucleation thresholds and metastable intermediates.142–144 Accordingly, how cooperative and assembly mechanisms govern the emergence of ordered architectures can be revealed. In linear polymer systems, phase behaviour has been revealed by using thermodynamic models such as Flory–Huggins theory, which enables the mapping of miscibility windows and phase separation boundaries.141,145–147 Furthermore, block copolymer systems have demonstrated how competing interactions and compositional parameters dictate the formation of distinct ordered domains and morphologies, including lamellar, cylindrical and gyroid structures,148,149 which are commonly represented in composition–interaction parameter phase diagrams. In branched and network polymers, phase diagram studies reveal gelation transitions and connectivity percolation thresholds. Combining rheological analysis with theoretical gelation models identifies network formation pathways.150,151
These approaches highlight that phase diagram construction generally relies on multidimensional parameter mapping coupled with multiscale structural characterization and theoretical modelling. Such methodologies offer important guidance for investigating crystallization phase behaviour in COFs. Given the complex processes involving covalent bonding, supramolecular polymerization, nucleation and crystallization, adopting similar systematic mapping strategies and in situ analytical techniques may provide insights into nucleation regimes, crystallization windows and growth pathways in framework materials, revealing the phase diagram.
3. Design principles and structural resolution
3.1. Design principles
The development of single crystal COFs is more difficult than the synthesis of their polycrystalline and amorphous counterparts, and the monomer, topology and reaction conditions required are critical factors to be investigated.127,130,134,138 The requirements for building units (Fig. 2) are three-fold: (1) a rigid backbone enables a specific growth direction, for which polyaromatic skeletons are frequently deployed; (2) geometry-matched monomers generate well-defined yet extended polygonal networks, where the topology diagram guides the formation of frameworks; (3) the polymerization of monomers under suitable reaction conditions forms linkages, which are able to correct defects through a self-healing process.One important factor is the solubility of monomers and the resultant oligomers, which exert profound effects on polymerization and crystallization.117,152 Monomers with high solubility accelerate polymerization, quickly generating large amounts of oligomers, which however, tend to precipitate out from the reaction system; it is likely difficult to form single crystal COFs from these precipitates and may even result in amorphous polymers. On the other hand, poorly soluble monomers are difficult for polymerization, so that harsh conditions, such as polar solvents and/or high temperature, are needed to enhance the solubility. As a result, these systems hardly form single crystals. The monomers deployed for the synthesis of single crystal COFs to date are summarized in Fig. 2. Therefore, designing building units with structure and geometry to balance covalent and supramolecular polymerizations, nucleation and crystallization in a controllable fashion is important.
By introducing suitable functional groups, the crystallization process can be tuned to improve the crystallinity of COFs and even produce single crystal COFs. For example, as a milestone in the field of COFs, TPB-DMTP-COF (Fig. 4) has proven to be a great success in both synthesis and applications.78 TPB-DMTP-COF was synthesized through condensation of 1,3,5-tri(4-aminophenyl) benzene (TAPB) with 2,5-dimethoxyterephthalaldehyde (DMTP) and obtained as polycrystalline powders. TPB-DMTP-COF is unique as it is easily synthesized, tolerant of different conditions and usually exhibits high crystallinity, porosity and stability. This contrasts to TAPB-PDA-COF, an analogue synthesized by condensing TAPB and terephthalaldehyde (TA).153 Introducing two electron-donating methoxy groups to the pore walls induces a resonance effect of the two lone pairs from the oxygen atoms over the central phenyl ring, weakens the polarization of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds and softens the interlayer charge repulsion (Fig. 4), consequently facilitating the crystallization to improve crystallinity and porosity and stabilizing TPB-DMTP-COF against water, organic solvents, strong acids and strong bases.78 TPB-DMTP-COF has become a landmark for COF research, ranging from fundamental synthetic chemistry to various applications.154–157 Through tuning the reaction conditions, single crystals of TPB-DMTP-COF have been obtained by many research groups, highlighting its exceptional reliability and versatility.158–160
N bonds and softens the interlayer charge repulsion (Fig. 4), consequently facilitating the crystallization to improve crystallinity and porosity and stabilizing TPB-DMTP-COF against water, organic solvents, strong acids and strong bases.78 TPB-DMTP-COF has become a landmark for COF research, ranging from fundamental synthetic chemistry to various applications.154–157 Through tuning the reaction conditions, single crystals of TPB-DMTP-COF have been obtained by many research groups, highlighting its exceptional reliability and versatility.158–160
 | ||
| Fig. 4 A typical and representative design principle for the synthesis of stable TPB-DMTP-COFs. | ||
Stacking mode is important for 2D COFs because it affects their pore structures as well as physicochemical properties and functions. The stacking mode can be identified by the diffraction patterns, while controlled synthesis of stacking modes is still difficult and has not yet been fully explored. This is because the stacking mode is associated with the configuration of polygons116,161 and dominated by noncovalent interactions, such as π–π interactions, hydrogen bonds and van der Waals forces. In general, there are AA-, AB- and ABC-stacking modes reported to date for COFs.161–164 The AA-stacking manifold consists of eclipsed AA stacking, inclined AA stacking and antiparallel AA (= reversed AA) stacking modes.165 Notably, these modes are affected by both monomer structures and reaction conditions, and probably exist in a mixed form in polycrystalline COFs.116,117
Tuning reaction conditions, such as catalyst, temperature and solvent, influences the stacking mode through the thermodynamic process, forming the most stable frameworks. In comparison, designing monomers by introducing additional yet prominent interactions, such as steric hindrance, dipole–dipole forces and hydrophobic/hydrophilic interactions, affects the stacking mode via internal factors.161
Interestingly, the frequently used monomers with aromatic structures provide rigid backbones to ensure the correct propagation of the framework over the x–y plane and promote the stacking process along the z dimension via π–π interactions to favour an AA stacking mode, which is the dominant stacking mode in the single crystal 2D COFs reported so far. Notably, a strong π–π interaction from building blocks does not always aid in the formation of single crystals, as it accelerates the nucleation and crystallization process over the z direction, resulting in heterogeneous polycrystalline aggregates. Tuning the strength and rate of interlayer interactions via elaborate design of monomers has proven to be an effective way to synthesize single crystal 2D COFs. For instance, by introducing three methyl groups at the core of triphenylbenzene, the resultant 1,3,5-trimethyl-2,4,6-tris(4-aminophenyl)benzene (TTAB) and 1,3,5-trimethyl-2,4,6-tris(4-formylphenyl)benzene (TTFB) (Fig. 2) weaken the π–π interactions, balances covalent and supramolecular polymerization processes and slows down the nucleation and crystallization, thereby leading to the formation of single crystals of 2D COFs.166
The formation of 3D COFs is driven by polymerization over a 3D fashion and in principle does not involve a z-direction supramolecular polymerization. Indeed, the development of non-interpenetrated 3D COFs has proven that fully covalent connectivity along three spatial directions (Fig. 1c, x–y–z dimension) can independently sustain the framework without relying on supramolecular stacking as the primary structural driving force. Therefore, weak noncovalent interactions are not the structural foundation for 3D COFs, whereas for 2D COFs, interlayer supramolecular interactions are indispensable to the formation and stabilization of the extended frameworks. Thus, tuning reaction conditions to balance polymerization and crystallization becomes the main factor to grow single crystals of 3D COFs.126 Exploring reaction conditions that reduce and/or eliminate defects offer a direct route to single crystal 3D COFs. Moreover, the growth of single crystals is often reported to be more feasible for 3D COFs relative to 2D COFs, accompanying well-defined shapes of single crystals under the identical synthetic conditions.167,168
Topological design of 2D COFs affects the pore structures and density of π-units significantly.169 A valid topological combination of monomers generates 2D COFs with well-defined and extended lattices, including the representative trigonal,169 tetragonal,96 rhombic,170 pentagonal,171–173 hexagonal51 and Kagome topologies (Fig. 5).33 Notably, the pore size, shape and wall can be predesigned by the combination of monomers.116 In particular, pore surface engineering offers a general strategy to construct tailor-made pores.64,69,133,174 In general, the trigonal topology produces COFs with the smallest pore size and most confined pore environment as well as the highest density of π-units among different polygons,71 while hexagonal topology gives COFs the biggest pore size, largest pore volume and lowest density of π-units.
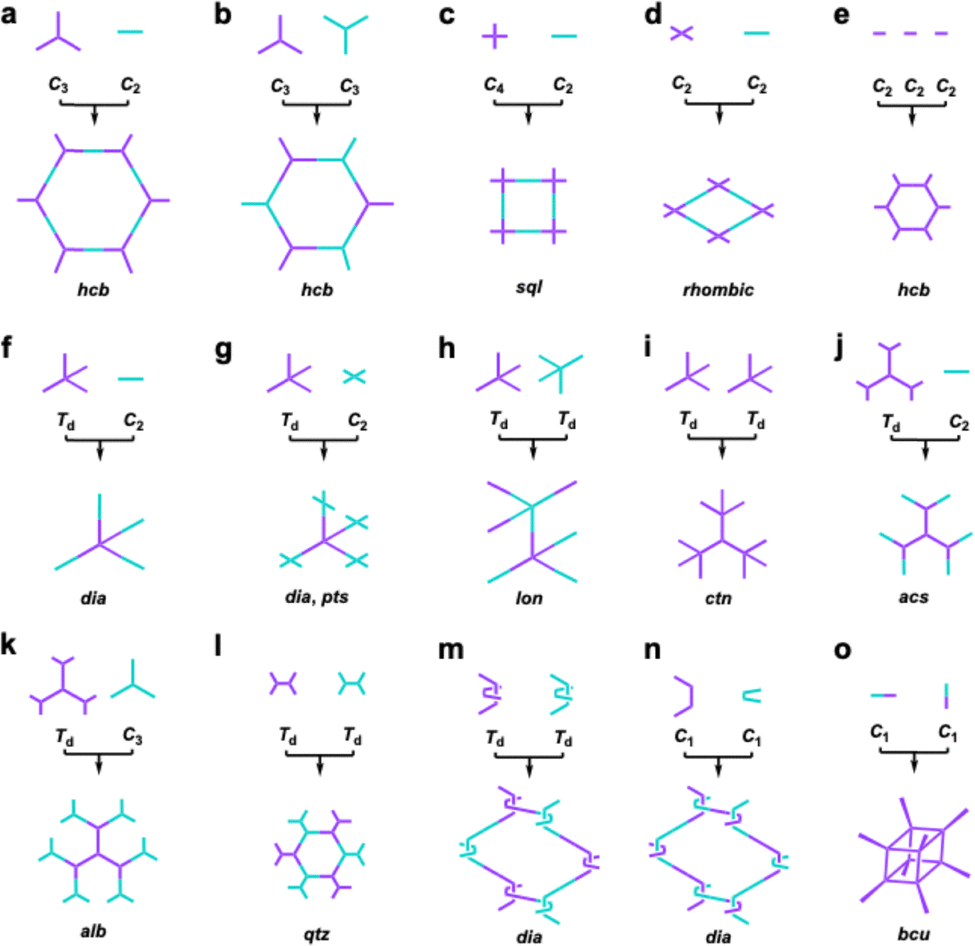 | ||
| Fig. 5 Topology diagrams for designing 2D and 3D COFs. (a)–(e) Typical topology diagrams for 2D COFs. (f)–(o) Typical topology diagrams for 3D COFs. | ||
Until now, there have still been limited topologies reported for single crystal COFs due to the synthetic difficulties. Typically, a [C3 + C2] strategy generates a hexagonal topology (Fig. 5a),127 which can also be realized by co-condensation of two C3-symmetric knots, i.e., the [C3 + C3] strategy (Fig. 5b).166 The combination of a C4-symmetric knot and C2-symmetric linker formulates a [C4 + C2] scheme,175 which yields a tetragonal diagram (Fig. 5c), while decreasing the knot geometry from C4-symmetry to C2-symmetry generates a [C2 + C2] diagram (Fig. 5d),176 leading to a rhombic topology. Self-condensation of C2-symmetric monomers generates a hexagonal lattice (Fig. 5e).177
Different from 2D COFs, the synthesis of 3D single crystal COFs needs 3D or orthogonal knots to enable the 3D propagation of lattices. A [Td + C2] strategy generates a dia network (Fig. 5f),178 while keeping the symmetry but increasing the number of reaction sites to 4 gives another [Td + C2] combination, generating an pts topology (Fig. 5g). Combination of two different Td monomers, i.e., [Td + Td] creates lon topology (Fig. 5h),179 while self-condensation of three Td monomers produces a ctn topology (Fig. 5i).177 Using another [Td + C2] strategy, in which the Td-symmetric monomer has a higher valency of 6 formed by structural rotation, due to the steric hindrance of methyl groups, produces an acs topology (Fig. 5j).180 Using this twisted Td to condense with a C3-symmetric monomer generates a [Td + C3] combination, which produces a rare self-catenated alb topology (Fig. 5k).181 Correspondingly, using two monomers twisted by methyl groups gives another [Td + Td] combination, resulting in qtz topology (Fig. 5l).182 Thus, the development of twisted high-valent monomers brings a new approach to synthesize 3D COFs.
Interestingly, weaving helical organic units to generate a new [Td + Td] formula produces woven COFs with dia topologies (Fig. 5m).183 In contrast, disassembling the helical organic units generates two monomers with crescent shapes to offer a new combination, i.e., [C1 + C1]. The crescent monomers with different reactive sites can be copolymerized under the guidance of dynamic covalent chemistry and coordination bonds to form 1D woven COFs (Fig. 5n).184 Self-condensation of bifunctional monomers [C1 + C1] which contain high-valency atoms, forms COFs with bcu topology (Fig. 5o).185
Connecting a knot and linker through different covalent bonds forms COFs with different physicochemical properties. To date, around thirty linkages have been developed,4 including boronate ester, boroxine, borosilicate, borazine, spiroborate, borophosphonate, imide, amide, ester, amine, dioxin, hydrazone, urea, triazine, imine, squaraine, azine, thiazole, oxazole, azodioxide, azobenzene, viologen, phenazine, vinylene, ketazine, nitrone, polyphenyl, ethynyl and diacetylene linkages. Nonetheless, only boroxine, boronate ester and imine linkages have been applied for direct synthesis of single crystal COFs due to their high reversibility (Fig. 6).178,186 Linkage conversion via post-synthetic modification results in more stable yet irreversible chemical bonds, such as amide, amine, triazole-functionalized amine, aminonitrile, olefin and formamide (Fig. 6).182,187–191 Developing single crystal COFs with other linkages is highly desired to broaden their applications but remains a great challenge due to the low reaction reversibility and strong covalent bonds.
 | ||
| Fig. 6 Reactions for the synthesis of single crystal COFs. | ||
3.2. Structural resolution
Single crystals offer a distinct form of materials that provide more precise structural information on COFs compared to polycrystalline analogues. The most straightforward techniques used to resolute structures of single crystals are single-crystal X-ray diffraction (SCXRD) and electron diffraction (ED).138The fundamental principle behind SCXRD is based on the interaction between X-rays and the electrons in a crystal. When a monochromatic beam of X-rays is directed at a single crystal, the X-rays are scattered by the atoms in the crystal lattice. The resulting diffraction pattern has information about the positions of the atoms within the crystal. By measuring the angles and intensities of the diffracted beams, it reconstructs the electron density distribution within the crystal, determining the positions of the atoms with atomic precision. The diffraction pattern produced by SCXRD has Bragg peaks, which represent constructive interference between X-rays scattered by different planes of atoms in the crystal. The angle at which these peaks appear provides crucial information about the spacing between the planes (the d-spacing), while the intensity of each peak is related to the number of atoms in the planes and the type of atoms present. Using this information, the phase problem and the positions of all atoms in the unit cell can be deduced, yielding a complete structural model.
For skeleton resolution, SCXRD is particularly important as COFs exhibit complex yet periodic skeletal structures that need to be characterized at the atomic level. SCXRD can accurately show the connectivity between building units, the arrangement of functional groups and the lattice topology.
For porous structural analysis, SCXRD enables the precise measurement of pore size, shape, distribution and connectivity, offering key insights into the performance in these applications. By analysing the diffraction patterns, the exact arrangement of atoms that form the pores can be determined. COFs often confine guest molecules, such as solvents or even gases, within their pores. SCXRD allows for the study of host–guest interactions at the atomic level, revealing how guest molecules are confined in the pores and how they interact with the framework. This information is crucial for optimizing COFs for molecular adsorption, catalysis and drug delivery.13,73,97,157
For dynamic transformation, SCXRD enables structural changes to be captured and offers insights into the adaptability of single crystal COFs. This is specially informative by showing dynamic behaviour, such as flexibility or reversible structural changes in response to external stimuli, such as temperature, pressure or humidity.61,182,192–194 By conducting SCXRD at different temperatures or under different environmental conditions, it is facile to uncover structural transformation and elucidate the properties and applications of materials.182,187
For quality control, SCXRD offers an ideal tool for assessing the quality of COF crystals. It allows for the detection of defects, imperfections and disorders within the crystal lattice. Understanding the nature and extent of defects is important because they can affect or even control properties, such as stability, porosity and reactivity. SCXRD reveals phase purity and ensures that the targeted structure has been successfully achieved in the COF samples.
For establishing structure–function correlations, SCXRD is unparalleled in providing atomic-level structural details to unveil the origin of properties. For COFs, this kind of precision is essential for understanding how their topology, pore structure and functionality are related to their properties. SCXRD reveals intricate details about the 3D structure of COFs, including the spatial arrangement of functional groups, the degree of interpenetration and the connectivity of building blocks.127,159 This comprehensive structural information is key to correlating structure–property relationships and provides a base on which to design COFs with specific properties.
SCXRD requires high-quality single crystals, which can be difficult to obtain for COFs. Most COFs tend to yield small-size polycrystalline products, making it challenging to conduct SCXRD analysis. Advances in synthetic methods have improved the situation; for example, seeded growth or modulator-assisted synthesis177,186 produces single crystals of large enough and high quality for SCXRD. COFs with high structural flexibility or multiple interpenetrating frameworks may pose challenges in obtaining an unambiguous structural solution. In such cases, complementary techniques like solid-state 13C nuclear magnetic resonance (ssNMR) or ED may be used alongside SCXRD to clarify the structure.127,178
Techniques such as RED and continuous-rotation MicroED allow the collection of high-quality diffraction data from sub-micrometre single crystals.201 These methods effectively sample reciprocal space, reducing artifacts from dynamical scattering and yielding reliable structural information.
ED can determine stacking arrangements, topologies and interlayer intervals. For example, stacking faults and interlayer slipped shifts in 2D COFs can significantly influence their electronic and optical properties. ED provides detailed insights into stacking modes, such as AA, AB or other alignments, which are challenging to discern through PXRD.127,159 Additionally, ED is highly sensitive to pore structures and framework distortions, enabling accurate resolution of pore size and symmetry.159
The use of ED has expanded the scope of functional COFs. For instance, chemically modified COFs and those with intricate interpenetrated structures have been successfully analysed using ED.182 These studies reveal how functional groups and framework connectivity affect the overall stability and porosity. In cases where solvent exchange, post-synthetic modifications or dynamic transformations are involved, ED captures stepwise structural changes, offering a spatial and time-resolved perspective on framework structural evolution.138
4. Polymerization method
Single crystal COFs provide high-resolution structural information, exhibiting distinct and intrinsic physicochemical properties, in comparison with polycrystalline analogues. Hence, exploring synthetic methods and properties of single crystal COFs has attracted tremendous attention over the past decade. In this section, we summarize representative methods for the synthesis of single crystal COFs and compare different strategies by disclosing reaction conditions and deploying unit cell parameters to unveil structural features.4.1. Modulator-assisted polymerization
Modulators are mono-functionalized monomer analogues that influence the reaction kinetics and thermodynamics, allowing for controlled polycondensation, nucleation and growth of large-size single crystals. The use of modulators in the synthesis of single crystal COFs has emerged as a useful tool to overcome challenges associated with polymerization and crystallization, such as defect formation and rapid nucleation.178,179 Acting as competitive inhibitors or reversibility enhancers, modulators facilitate the formation of crystalline structures with long-range order. As a result, single crystal COFs with high structural precision and complexity became accessible. The modulator-assisted synthesis controls the size of single crystal COFs at both micro and macro scales. Introducing suitable modulators allows control over key characteristics, such as crystallinity, porosity, morphology and surface functionality. These advancements address the longstanding challenges of COF synthesis, including rapid nucleation, structural defects and limited crystallinity.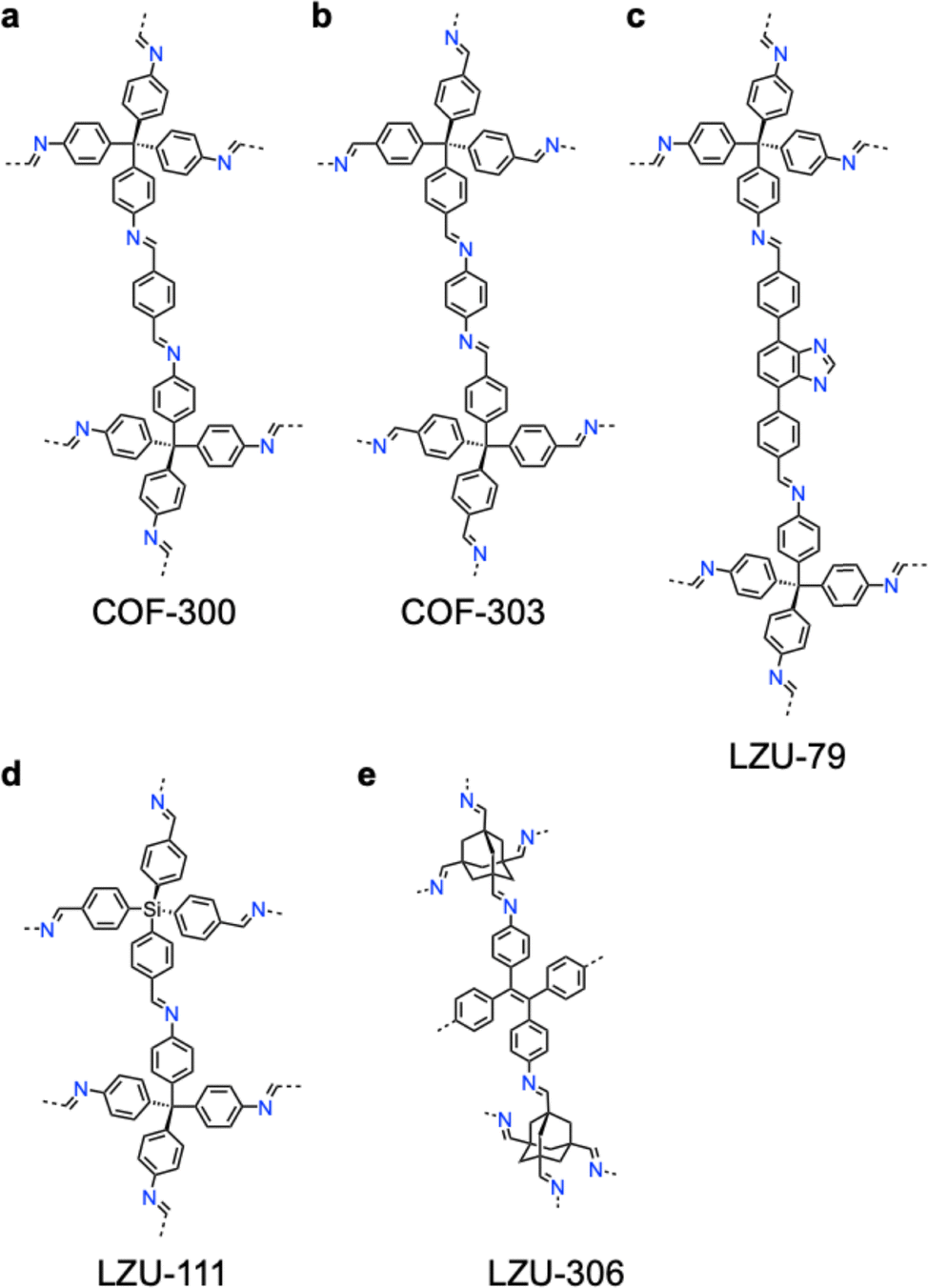 | ||
| Fig. 7 Modulator-assisted synthesis of single crystal 3D COFs. (a)–(e) Structures of (a) COF-300, (b) COF-303, (c) LZU-79, (d) LZU-111 and (e) LZU-306. | ||
A variety of imine-linked 3D COFs, including COF-300 (Fig. 7a), COF-303 (Fig. 7b), LZU-79 (Fig. 7c) and LZU-111 (Fig. 7d), with single crystals between 10 and 100 microns in size (Fig. 8), have been synthesized.178 Interestingly, the crystal size can be controlled by adjusting the added aniline amount. For example, in the case of COF-300, with tetra(4-aminophenyl)methane (TAM) and TA as monomers and in the absence of aniline, the reaction forms polycrystalline COF-300 with a size of 500 µm. Adding aniline of 0.6 to 15 equivalents forms high-quality single crystals of COF-300 to reach a maximum size of 100 microns after 80 days of reaction. The single crystal structure is resolved with a resolution of 0.85 Å through SCXRD technology. Table 1 summarizes the reaction conditions and structural resolutions of COF-300, COF-303, LZU-79 and LZU-111.
 | ||
| Fig. 8 SEM and optical microscopy images of (a) and (b) single crystal COF-300, (c) and (d) COF-303, (e) and (f) LZU-79 and (g) and (h) LZU-111. Reprinted with permission from ref. 178. Copyright 2018 American Association for the Advancement of Science. | ||
| COFs | Synthetic conditions | Crystal parameters | Resolution | Method | Ref. |
|---|---|---|---|---|---|
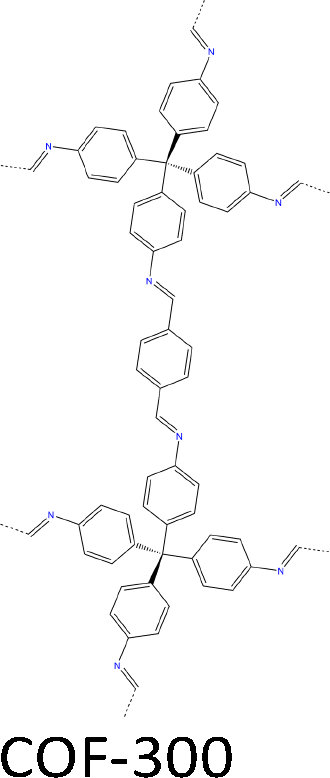 |
A mixture of TA (12.0 mg, 0.089 mmol) and aniline (0.12 mL, 15 equiv.) in 1,4-dioxane (0.5 mL) and AcOH (0.2 mL, 6 M) with TAM (20.0 mg, 0.052 mmol) dissolved in 1,4-dioxane (0.5 mL) formed 60 µm crystals within 30 to 40 days. | a = b = 26.23 Å | 0.85 Å | SCXRD | 178 |
| c = 7.57 Å | |||||
| A mixture of TA (12.0 mg, 0.089 mmol) and aniline (81 µL, 10 equiv.) in 1,4-dioxane (0.5 mL) with CF3CO2H (0.1 mL, 6 M) with TAM (20.0 mg, 0.052 mmol) dissolved in 1,4-dioxane (0.5 mL) formed 10 µm crystals within 2 hours at 40 °C. | a = b = 26.27 Å | 0.83 Å | SCXRD | 179 | |
| c = 7.56 Å | |||||
| A mixture of TA (12.0 mg, 0.089 mmol) and CF3CH2NH2 (70 µL, 10 equiv.) in 1,4-dioxane (0.5 mL) and CF3CO2H (0.1 mL, 6 M) with TAM (20.0 mg, 0.052 mmol) in 1,4-dioxane (0.5 mL) formed 60 µm crystals in 2 days at 40 °C. | 179 | ||||
| C16-L-AlaA (32.8 mg, 0.1 mmol) and 0.1 mmol sodium hydroxide were dissolved in 10 mL of water. Stirring at 50 °C for 10 minutes to obtain an emulsion, HCl (0.5 mL, 0.2 mol L−1) was added, then linker (6.7 mg, 110.05 mmol) was added and stirred in. A solution of TAM (9.5 mg, 0.025 mmol) in PTSA (0.5 mL, 0.2 mol L−1) was added and stirred for 5 minutes. Single crystals were collected by centrifugation and washed with THF and water three times before immersing in THF for 24 h. The sample was dried at ambient temperature for 12 h and 100 °C for 12 h to afford COF-300 powder. | Not available (NA) | 1.1 Å | SCXRD cRED | 167 | |
| At room temperature, terephthalaldehydebis(diethylacetal) (29 mg, 0.1 mmol), TAM (19 mg, 0.25 mmol), aniline (10, 20, 40, 60, 80, 100, 120, 140 or 160 µL) and 1,4-dioxane (1 mL) were added into a sample vial (10.0 mL). At this moment, a series of light-yellow transparent solutions were obtained. AcOH (1 mL) was added into this sample vial and sealed after being degassed in a liquid N2 bath for 20 min, flamed sealed, and warmed to room temperature. The sample vial was placed in an oven preheated to 80 °C. After 48 hours, the vials were opened. Next, a dropper was used to separate the liquid and solid phases. | a = b = 26.5565 Å | 0.79 Å | SCXRD | 203 | |
| c = 7.4431 Å | |||||
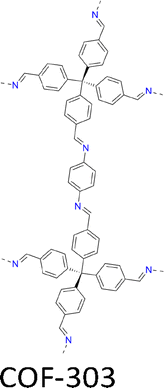 |
A mixture of tetrakis(4-formylphenyl)methane (TFM, 10.8 mg, 0.025 mmol), aniline (0.12 mL, 52 equiv.) in 1,4-dioxane (0.25 mL) and AcOH (0.1 mL, 6 M) with phenylenediamine (PDA, 5.4 mg, 0.05 mmol) in 1,4-dioxane (0.25 mL) formed 15 µm crystals within 15 days. | a = b = 26.47 Å | 1.10 Å | SCXRD | 178 |
| c = 7.45 Å | |||||
| A mixture of TFM (10.8 mg, 0.025 mmol), CF3CH2NH2 (50 µL, 25 equiv.), THF (0.25 mL), MeCN (0.25 mL) and CF3CO2H (0.1 mL, 6 M) with PDA (5.4 mg, 0.05 mmol) in THF (0.25 mL), MeCN (0.25 mL), formed 100 µm crystals in 2 days at 30 °C. | a = b = 25.87 Å | 0.81 Å | SCXRD | 179 | |
| c = 7.7 Å | |||||
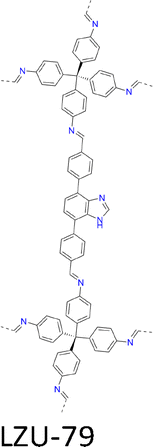 |
A mixture of 4,7-bis(4-formylbenzyl)-1H-benzimidazole (BFBZ, 17.0 mg, 0.052 mmol), aniline (0.12 mL, 25 equiv.), 1,4-dioxane (0.75 mL) and AcOH (0.2 mL, 6 M) with TAM (10.0 mg, 0.026 mmol) in 1,4-dioxane (0.75 mL), formed 50 µm crystals in 20 days and further reached 100 µm in 50 days. | a = b = 27.84 Å | 1.25 Å | SCXRD | 178 |
| c = 7.51 Å | |||||
| A mixture of BFBZ (8.5 mg), THF (0.25 mL), MeCN (0.25 mL) and CF3CO2H (0.1 mL, 6 M) with TAM (5 mg, 0.013 mmol) in THF (0.5 mL) and MeCN (0.5 mL) formed 100 µm crystals within 2 days at 30 °C. | NA | NA | 179 | ||
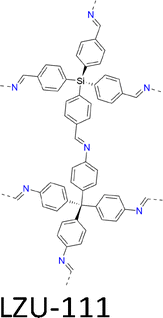 |
A mixture of tetrakis(4-formylphenyl)silane (TFS, 22.4 mg, 0.05 mmol), aniline (0.36 mL, 80 equiv.), 1,4 dioxane (0.5 mL) and AcOH (0.2 mL, 6 M) with TAM (19.0 mg, 0.05 mmol) in 1,4 dioxane (0.5 mL) formed 45 µm crystals in 47 days and further reached 55 µm in 80 days. | a = b = 20.40 Å | 1.20 Å | SCXRD | 178 |
| c = 33.77 Å | |||||
| A mixture of TFS (11.2 mg, 0.025 mmol), CF3CH2NH2 (102 µL, 52 equiv.), THF (0.25 mL), MeCN (0.25 mL) and CF3CO2H (0.1 mL, 6 M) with TAM (9.5mg, 0.025 mmol) in THF (0.25 mL) and MeCN (0.25 mL) formed 50 µm crystals within 1.5 days and further reached 50 µm in 1.5 days at 30 °C. | a = b = 20.56 Å | 1.20 Å | 179 | ||
| c = 33.72 Å |
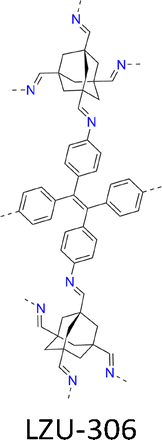
Synthesizing single crystal 3D COFs without interpenetrated structures remains a challenging task due to the use of Td and/or orthogonal knots. The single crystal LZU-306 COF (Fig. 7e and Table 2) was synthesized using an aniline modulator in the reaction of adamantane-1,3,5,7-tetracarbaldehyde (ADA-CHO) as the tetrahedral node and tetrakis(4-aminophenyl)ethylene (TPE-NH2) as the quadrilateral linker, generating a non-interpenetrated 3D COF with pts topology.126
| COFs | Synthetic conditions | Crystal parameters | Resolution | Method | Ref. |
|---|---|---|---|---|---|
 |
Adamantane-1,3,5,7-tetracarbaldehyde (ADA-CHO, 9.9 mg, 0.04 mmol), aniline (184 µL, 50 equiv.), 1,4-dioxane (0.5 mL) and AcOH (0.2 mL, 15 M) with tetrakis(4-aminophenyl)ethene (TPE-NH2, 15.7 mg, 0.020 mmol) in 1,4-dioxane (1 mL) formed 50 µm crystals in 25 days at 30 °C. | a = b = 15.32 Å c = 24.89 Å a = b = 15.72 Å c = 24.05 Å | 1.80 Å | SCXRD | 126 |
| A mixture of ADA-CHO (5 mg, 0.02 mmol), aniline (64 µL, 35 equiv.), 1,4-dioxane (0.25 mL), MeCN (0.25 mL) and CF3CO2H (0.1 mL, 9M) with TPE-NH2 (7.8 mg, 0.020 mmol) in 1,4-dioxane (0.25 mL) and MeCN (0.25 mL), formed 54 µm crystals in 12 hours at 30 °C. | NA | NA | NA | 179 | |
| A mixture of ADA-CHO (5 mg, 0.02 mmol), CF3CH2NH2 (71 µL, 45 equiv.), 1,4-dioxane (0.25 mL), MeCN (0.25 mL) and CF3CO2H (0.1 mL, 9 M) with TPE-NH2 (7.8 mg, 0.020 mmol) in 1,4-dioxane (0.25 mL) and MeCN (0.25 mL) formed 150 µm crystals in 1.5 day at 30 °C. In addition, comparable products can be obtained under conditions of CF3CO2H (0.1 mL, 6 M)/CF3CH2NH2 (55 µL, 35 equiv.). | a = 15.4155 Å | 1.15 Å | SCXRD | 179 | |
| b = 15.4155 Å | |||||
| c = 24.741 Å | |||||
| α = β = γ = 90° |
This non-interpenetrated COF features an open framework constructed entirely through covalent bonds and exhibits a high void volume of 80% and a low density of 0.33 g cm−3, outperforming typical interpenetrated 3D COFs. The shortest π–π distance in LZU-306 is 10.6 Å, which is much larger than those (4.6–5.1 Å) of interpenetrated 3D COFs.
SCXRD and RED (Fig. 9a and b) confirmed the non-interpenetrated pts structure with a tetragonal unit cell. N2 adsorption–desorption isotherms demonstrated a high Brunauer–Emmett–Teller (BET) surface area of 2059 m2 g−1 and a pore size of 10.9 Å (Fig. 9c and d).126 The ssNMR spectra indicated the completeness of covalent bonds with sharp and well-defined carbon signals, verifying the high degree of structural order. LZU-306 is thermally stable, retaining its structure up to 530 °C under N2 (Fig. 9e).
 | ||
| Fig. 9 (a) Reciprocal planes of SXRD data from a 50 µm-sized solvent-exchanged single crystal. (b) Reconstructed 3D RED data of a 500 nm-sized activated single crystal. (c) Pore size distributions of 50 µm-sized (black) and 200 nm-sized (red) LZU-306 calculated by NLDFT. (d) BET surface area plot for 50 µm-sized crystals calculated from the adsorption isotherm. (e) TGA curves of 50 µm-sized (black) and 200 nm-sized (red) LZU-306 under a N2 atmosphere. Reprinted with permission from ref. 126. Copyright 2020 Wiley-VCH. | ||
Employing a novel CF3CO2H/CF3CH2NH2 protocol with a CF3CO2H catalyst and CF3CH2NH2 as the modulator enables rapid synthesis of single crystal COFs (Fig. 10a and Tables 1–3).179 CF3CO2H is a strong catalyst for promoting backward reactions compared to acetic acid (AcOH), while CF3CH2NH2 is distinct from aniline and allows for rapid imine bond formation.
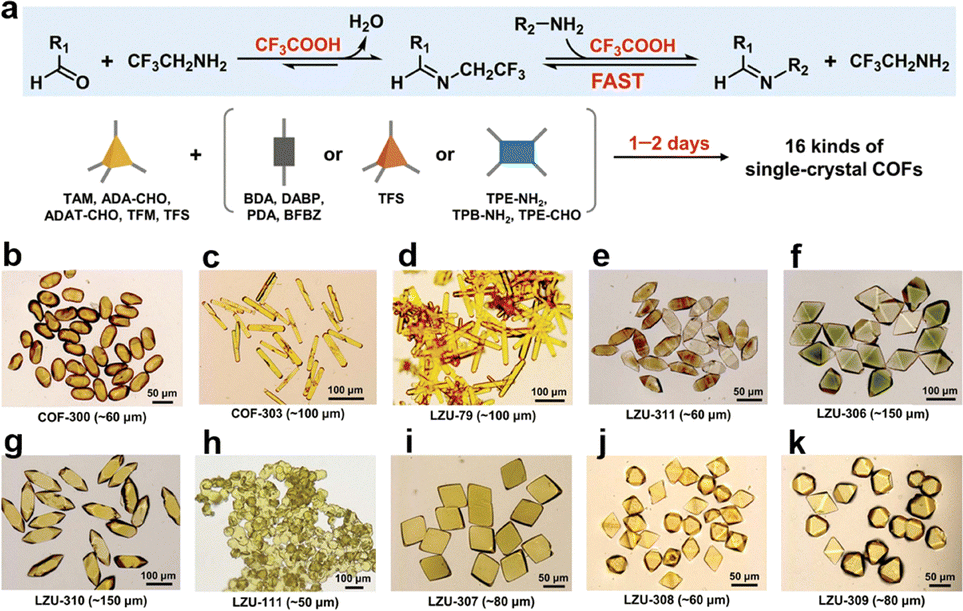 | ||
| Fig. 10 Rapid growth of large-sized single crystal COFs. (a) Protocol developed using CF3CO2H/CF3CH2NH2 for the rapid growth of single-crystal COFs in 1 to 2 days. (b)–(k) The optical microscopic images for single-crystal COFs obtained in 1 to 2 days with sizes of 50 to 150 mm: (b) COF-300, (c) COF-303, (d) LZU-79, (e) LZU-311, (f) LZU-306, (g) LZU-310, (h) LZU-111, (i) LZU-307, (j) LZU-308 and (k) LZU-309. Reprinted with permission from ref. 179. Copyright 2024 American Association for the Advancement of Science. | ||
| COFs | Synthetic conditions | Crystal parameters | Resolution | Method | Ref. |
|---|---|---|---|---|---|
 |
A sample vial (5.0 mL, body length 40 mm, neck length 18 mm) was sequentially charged with tetrakis(4-formylphenyl)silane (TFS, 22.4 mg, 0.050 mmol), BnOH (0.50 mL) and CF3CH2NH2 (106 µL, 27 equiv.). Then, 0.10 mL of CF3CO2H (9.0 M) was added. 4,4′-Diaminobiphenyl (DABP, 18.4 mg, 0.10 mmol) dissolved in BnOH (0.50 mL) was added to the solution. The mixture was filtered through a 0.20 µm Nylon-6 membrane filter, and the reaction was allowed to stand at a temperature of 30 °C. Yellow single crystals of LZU-310 were rapidly crystallized with uniform rod-like morphologies, and the crystal size reached ∼150 µm in 2 days. | a = 24.6955 Å | 0.81 Å | SCXRD | 179 |
| b = 24.6955 Å | |||||
| c = 8.6279 Å | |||||
| α = β = γ = 90° | |||||
 |
A sample vial (5.0 mL, body length 40 mm, neck length 18 mm) was sequentially charged with ADA-CHO (9.9 mg, 0.040 mmol), THF (0.50 mL) and CF3CH2NH2 (47 µL, 15 equiv.). Then, CF3CO2H (0.10 mL, 6.0 M) was added. DABP (14.7 mg, 0.080 mmol) dissolved in THF (0.50 mL) was added to the solution. The mixture was filtered through a 0.20 µm Nylon-6 membrane filter, and the reaction was allowed to stand at a temperature of 30 °C. Yellow single crystals of LZU-311 were rapidly crystallized with uniform rod-like morphologies and the crystal size reached ∼60 µm in 1.5 days. | a = 20.5618 Å | 1.20 Å | SCXRD | 179 |
| b = 20.5618 Å | |||||
| c = 33.719 Å | |||||
| α = β = 90° | |||||
| γ = 120° | |||||
 |
A sample vial (5.0 mL, body length 40 mm, neck length 18 mm) was sequentially charged with 1,3,5,7-tetrakis(4-formylphenyl)adamantane (ADAT-CHO, 11.2 mg, 0.020 mmol), THF (0.50 mL) and CF3CH2NH2 (16 µL, 10 equiv.). Then, CF3CO2H (0.11 mL, 6.0 M) was added. 1,2,4,5-Tetrakis(4-aminophenyl)benzene (TPE-NH2, 7.8 mg, 0.020 mmol) dissolved in THF (0.50 mL) was added to the solution. The mixture was filtered through a 0.20 µm Nylon-6 membrane filter, and the reaction was allowed to stand at a temperature of 30 °C. Yellow single crystals of LZU-307 were rapidly crystallized with uniform rhombohedral morphologies, and the crystal size reached ∼80 µm in 1 day. | a = 21.947 Å | 1.05 Å | SCXRD | 179 |
| b = 33.671 Å | |||||
| c = 23.432 Å | |||||
| α = β = γ = 90° | |||||
 |
A sample vial (5.0 mL, body length 40 mm, neck length 18 mm) was sequentially charged with ADA-CHO (5.0 mg, 0.020 mmol), THF (0.40 mL) and CF3CH2NH2 (16 µL, 10 equiv.). Then, CF3CO2H (0.090 mL, 6.0 M) was added. TPB-NH2 (8.9 mg, 0.020 mmol) dissolved in THF (0.40 mL) was added to the solution. The mixture was filtered through a 0.20 µm Nylon-6 membrane filter, and the reaction was allowed to stand at a temperature of 30 °C. Yellow single crystals of LZU-308 were rapidly crystallized with uniform octahedral morphologies, and the crystal size reached ∼60 µm in 1 day | a = 17.98 2Å | 1.65 Å | SCXRD | 179 |
| b = 17.982 Å | |||||
| c = 24.564 Å | |||||
| α = β = γ = 90° | |||||
 |
A sample vial (5.0 mL, body length 40 mm, neck length 18 mm) was sequentially charged with tetrakis(4-formylphenyl)ethylene (TPE-CHO, 8.8 mg, 0.020 mmol), THF (0.40 mL) and CF3CH2NH2 (16 µL, 10 equiv.). Then, CF3CO2H (0.10 mL, 6.0 M) was added. TAM (7.6 mg, 0.020 mmol) dissolved in THF (0.40 mL) was added to the solution. The mixture was filtered through a 0.20 µm Nylon-6 membrane filter, and the reaction was allowed to stand at a temperature of 30 °C. The yellow single crystals of LZU-309 were rapidly crystallized with uniform octahedral morphologies, and the crystal size reached ∼80 µm in 1 day. | a = 20.1697 Å | 1.40 Å | SCXRD | 179 |
| b = 20.1697 Å | |||||
| c = 30.506 Å | |||||
| α = β = γ = 90° |
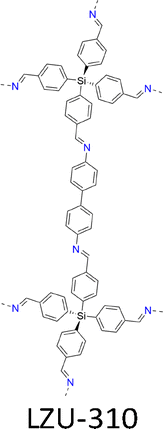
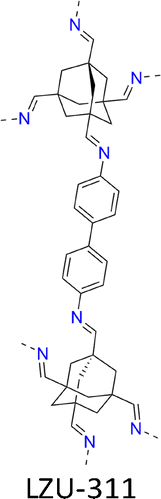
This method accelerates the crystallization process and produces single crystals in 1–2 days, which is 52 times faster compared to the previously reported 15–80 days under AcOH/aniline conditions. This system replaces weaker acids like AcOH and aniline to catalyse imine exchange more effectively. This method achieves rapid crystallization of 10 different COFs, including COF-300 (Fig. 7a and 10b), COF-303 (Fig. 7b and 10c), LZU-79 (Fig. 7c and 10d), LZU-111 (Fig. 7d and 10h), LZU-306 (Fig. 7e and 10f), LZU-310 (Fig. 11a and 10g), LZU-311 (Fig. 11b and 10e), LZU-307 (Fig. 11i and 10c), LZU-308 (Fig. 11d and 10j) and LZU-309 (Fig. 11e and 10k), producing single crystals up to 150 µm.8
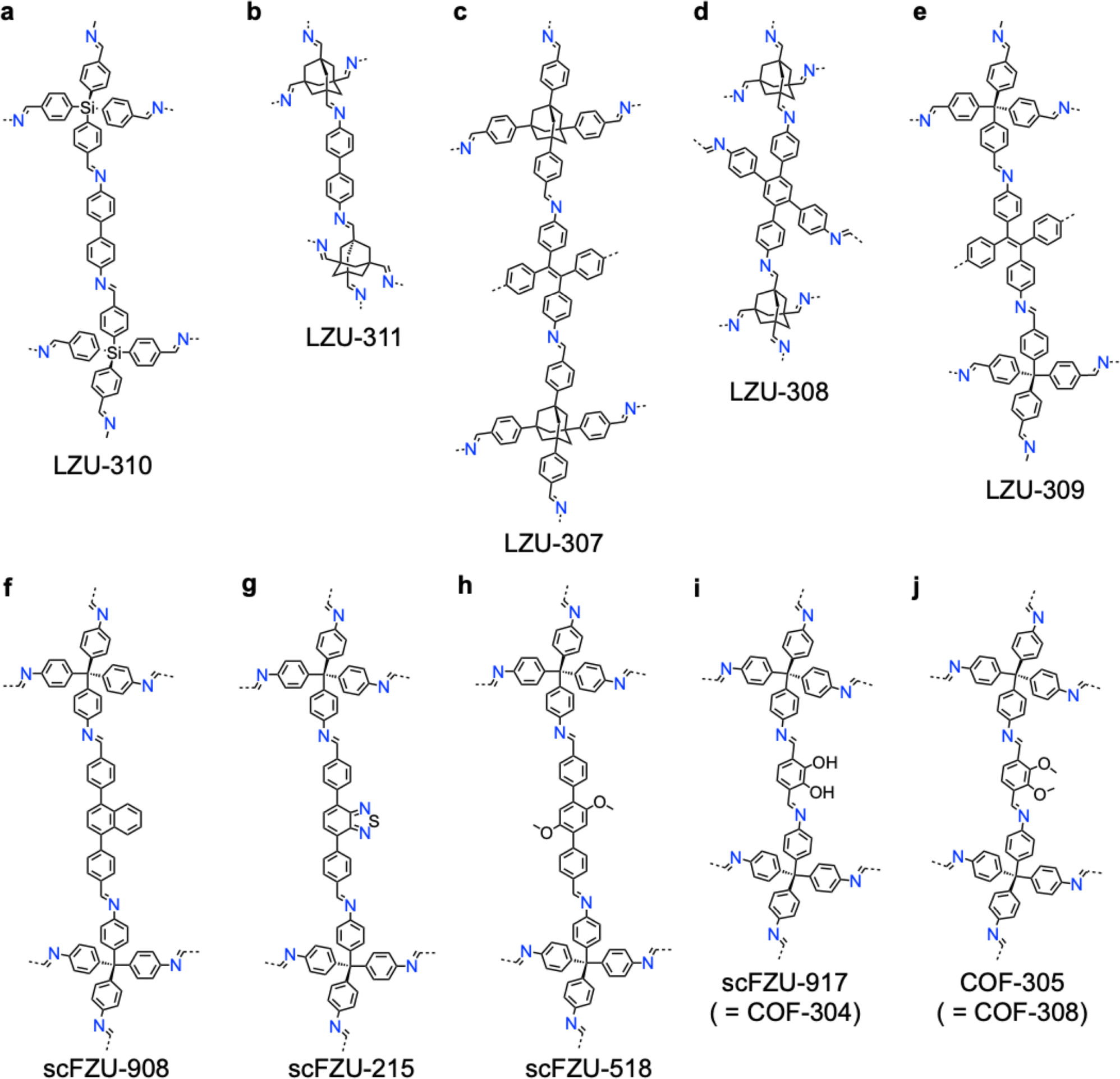 | ||
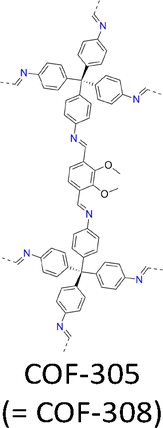
| Fig. 11 Structures of 3D COFs. (a)–(j) Structures of (a) LZU-310, (b) LZU-311, (c) LZU-307, (d) LZU-308, (e) LZU-309, (f) scFZU-908, (g) scFZU-215, (h) scFZU-518, (i) scFZU-917 (= COF-304) and (j) COF-305 (= COF-308). | ||
The high-resolution XRD data with a 0.79 Å resolution enables detailed structural analysis, including the identification of interpenetration patterns and host–guest interactions. The single-crystal structures revealed intricate topologies, such as the sevenfold-interpenetrated dia-c7 framework of COF-300 and a novel fourfold-interpenetrated pts framework in LZU-307 (Fig. 11c and 10i). Dynamic transformations are confirmed with reversible structural changes upon interaction with guest molecules, such as solvents or water.179
Rapid production of single crystal COFs is important to promote their applications. A binary solvent-supersaturation method has been developed for enabling fast growth of single crystal COFs within one week (Table 4).202 This method utilizes two kinds of solvents, i.e., 1,4-dioxane (D) as a good solvent and mesitylene (M) as a poor solvent, to tune their ratio, thereby accelerating a high supersaturation environment to facilitate rapid nucleation and crystal growth.
| COFs | Synthetic conditions | Crystal parameters | Resolution | Method | Ref. |
|---|---|---|---|---|---|
 |
A glass tube was charged with 1,4-di(4-aldehyde phenyl)naphthalene (Nap-CHO, 15.0 mg, 0.044 mmol), aniline (70 µL, 17.5 equiv.), mesitylene (0.7 mL) and 1,4-dioxane (0.2 mL). TAM (8.4 mg, 0.022 mmol) dissolved in 1,4-dioxane (0.1 mL) and AcOH (0.1 mL, 15 M) were added to the above solution. Then the mixture was allowed to further stand at ambient temperature (about 25 °C). The single crystals of scFZU-908 slowly crystallized out at ambient temperature. | a = b = 25.708 Å | NA | SCXRD | 202 |
| c = 7.632 Å | |||||
| α = β = γ = 90° | |||||
 |
A glass tube was charged with BTDZ-CHO (15.0 mg, 0.044 mmol), aniline (0.1 mL, 25 equiv.) and mesitylene (0.7 mL). Then TAM (8.4 mg, 0.022 mmol) dissolved in 1,4-dioxane (0.3 mL) and AcOH (0.1 mL, 15 M) were added to the above solution in succession. The mixed solution was allowed to further stand at ambient temperature (about 25 °C). Later, the scFZU-215 single crystal was obtained within 7 days and its size reached 200 µm-sized and some of them even reached ∼420 µm in 15 days. | NA | NA | NA | 202 |
 |
A glass tube was charged with 3PhOMe-CHO (11.4 mg, 0.033 mmol), aniline (50 µL, 16 equiv.) and mesitylene (0.7 mL). TAM (6.3 mg, 0.0165 mmol) dissolved in 1,4-dioxane (0.3 mL) and AcOH (0.1 mL, 15 M) were added to the above solution one after another. Then the mixture was allowed to further stand at ambient temperature (about 25 °C). The single crystal of scFZU-518 slowly grew from the mixed solution and its crystal size reached 200 µm within 15 days. | NA | NA | NA | 202 |
 |
A glass tube was charged with 2,3-dihydroxyterephthalaldehyde (23OH-CHO, 7.3 mg, 0.044 mmol), aniline (90 µL, 22.5 equiv.) and mesitylene (0.7 mL). TAM (8.4 mg, 0.022 mmol) dissolved in 1,4-dioxane (0.3 mL) and AcOH (0.1 mL, 15 M) was added to the above solution in succession. Then the mixture was allowed to further stand at ambient temperature (about 25 °C). The single crystals of scFZU-917 slowly grew at ambient temperature and the crystal size reached 100 µm within 15 days. | NA | NA | NA | 202 |
| DHPA (15 mg, 0.090 mmol) was dissolved in 1,4-dioxane (0.5 mL), and aniline (60 µL) was added. The mixture was transferred to an NMR tube, and then aqueous AcOH (0.2 mL, 15 M) was added. After being homogenously mixed, a red-crystalline precipitate was observed, and the solution of TAM (20 mg, 0.053 mmol) dissolved in 1,4-dioxane (0.75 mL) was added carefully. The reaction was placed at room temperature, standing still, for three weeks, and the dark red-coloured crystals slowly crystallized out. The as-synthesized single crystals were isolated and then exchanged by n-butanol three times for SXRD measurement. | a = b = 26.210Å | NA | SCXRD | 207 | |
| c = 7.582Å | |||||
 |
DMPA (17 mg, 0.088 mmol) was dissolved in 1,4-dioxane (0.5 mL), and then aniline (40 µL) and AcOH (200 µL, 15 M) were added to a vial (5 mL). After homogeneously mixing the solution, the solution of TAM (20 mg, 0.053 mmol) dissolved in 1,4-dioxane (0.75 mL) was added carefully. The reaction was placed and left to stand at room temperature. The yellow-coloured crystals of about 30 µm were crystallized out after 2 days, and the crystal sizes reached about 40 µm in 4 days. The as-synthesized single crystals were isolated and then exchanged by n-butanol three times for SXRD measurement. | a = 47.077 Å | 1 Å | SCXRD | 207 |
| b = 67.629 Å | |||||
| c = 42.547 Å | |||||
| α = β = γ = 90° | |||||
| TAM (5.6 mg, 0.01 mmol) was dissolved in 1,4-dioxane (200 µL). BDA-cis-OMe (3.88 mg, 0.02 mmol) was dissolved in 1,4-dioxane (200 µL) separately. Sc(OTf)3 (2.56 mg, 0.0052 mmol) was dissolved in acetonitrile (80 µL). In a 4 mL sample vial, 1,4-dioxane (400 µL), dichloromethane (300 µL), the BDA-cis-OMe solution, aniline (11 µL), the Sc(OTf)3 solution and the TAM solution were charged sequentially and then mixed under vibration. The reaction was allowed to stand at 25 °C for 4 days, giving octahedral shaped yellow crystals with sizes of 90–100 µm. | a = 41.05 Å | 0.95 Å | SCXRD | 208 | |
| b = 46.67 Å | |||||
| c = 40.94 Å | |||||
Accordingly, single crystal scFZU-908 (Fig. 11f) with different morphologies and sizes have been prepared through condensation of TAM with 4,4′-(naphthalene-1,4-diyl)dibenzaldehyde at D/M volume proportions of 10/0, 9/1, 8/2, 7/3, 6/4, 5/5, 4/6 and 3/7, respectively.
Increasing the volume ratio of mesitylene increases the crystal size of scFZU-908 from 12.5 µm to 140 µm (Fig. 12), while the morphologies of these crystals under different solvent ratios remained unchanged, showing elongated rhombic dodecahedrons.202 A mechanism study revealed that the growth kinetic of single crystal COFs gradually shifted from the reaction-controlled to the diffusion-controlled upon increasing the supersaturation of the reaction system. SCXRD revealed the tenfold interpenetrated dia structure of scFZU-908 with a tetragonal P42/n space group and unit parameters of a = b = 25.708 Å, c = 7.632 Å, α = β = γ = 90°, and a unit-cell volume of 5044.0 Å3. Notably, this approach showed excellent generality to produce isostructural single crystal scFZU-215 (Fig. 11g), scFZU-518 (Fig. 11h) and scFZU-917 (= COF-304, Fig. 11i) with the incorporation of benzothiadiazole, methoxy and phenolic hydroxyl groups into linker units, respectively.202 These single crystal COFs displayed prominent nonlinear optical properties due to the non-centrosymmetric nature of the crystal structures, which will be detailed in Section 5.2.
 | ||
| Fig. 12 Morphology and size of scFZU-908 after 5 days in various volume ratios (10/0, 9/1, 8/2, 7/3, 6/4, 5/5, 4/6 and 3/7) of 1,4-dioxane and mesitylene (scale bar = 50 µm). Reprinted with permission from ref. 202. Copyright 2025 Wiley-VCH. | ||
To accelerate the growth of single crystal COFs, an acetal/AcOH protocol has been developed.203 A notable difference from previous synthetic methods is the use of acetal-substituted aldehyde monomers as precursors. This strategic adjustment has several effects: (1) increasing the solubility of the monomer to provide a homogeneous reaction system, (2) preventing the direct polycondensation of amino and aldehyde monomers to slow down the reaction, and (3) avoiding concentration gradients and transient thermal fluctuations during the mixing of TA and TAM monomers in a hot-injection technique.
Remarkably, the acetal-mediated protocol allows the synthesis of single crystal COF-300 (Fig. 7a and Table 1) with increased growth rate, tuneable morphology and high crystal quality.203 Thus, this method decelerates the polymerization reaction, accelerates crystallization and enhances structural perfection. Single crystal COF-300 can be grown up to a large size of 60 µm within 1 h, 120 µm within 48 h and 300 µm after 30 days. This is much faster than the previous CF3CO2H/CF3CH2NH2 protocol, which affords single crystal COF-300, reaching sizes of 60 µm in 2 days (Fig. 13).203
 | ||
| Fig. 13 Morphological tuning of COF-300 with reaction conditions. Reprinted with permission from ref. 203. Copyright 2025 American Chemical Society. | ||
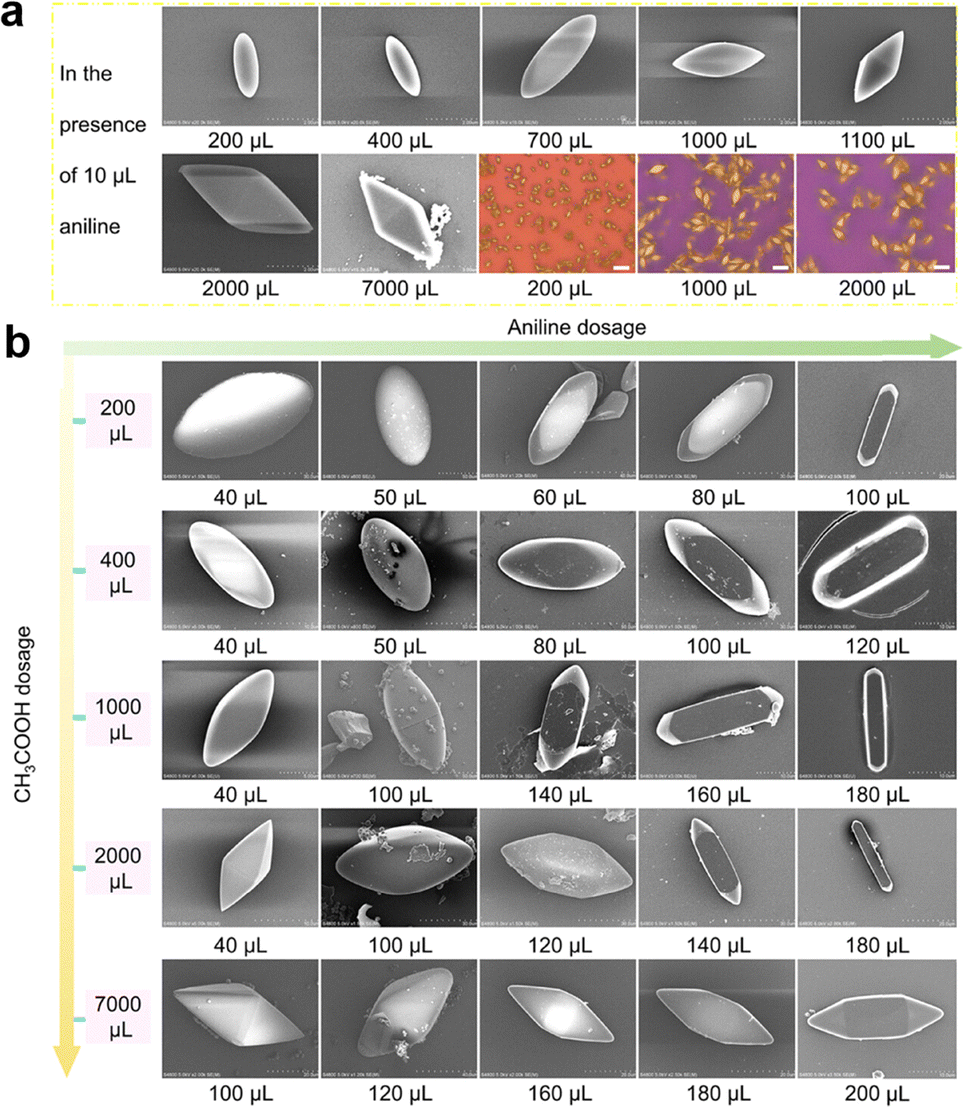
Interestingly, the shape of the single crystal COF-300 can be modulated by using different amines. The addition of a weak base 4-nitroaniline ((NO2)C6H4NH3+, pKa = 1.12, 55–276 mg, 2–10 equiv.) leads to the crystal shape of tetragonal dipyramid, which grows in crystallographic orientation of dia-topology due to the accelerated crystallization process. In comparison, using strong base p-aminophenol ((OH)C6H4NH3+, pKa = 5.65, 27–109 mg, 0.25–1 equiv.) yields spindle crystals of several micrometres in size. Moreover, under different AcOH/aniline dosages, the shape of single-crystal COF-300 is adjustable.
Keeping the aniline dosage at 10 µL and tuning the amount of AcOH (100–7000 µL) results in a shape transition from spindle (100–400 µL) to tetragonal dipyramid (500–7000 µL) (Fig. 13a). Furthermore, increasing the amount of aniline (40–200 µL) with a specific dosage of AcOH (100, 200, 300, 400, 500, 600, 1000, 1100, 1200, 2000 and 7000 µL) favours the shape transformation from spindle or tetragonal dipyramid to tetragonal quasi-prism (Fig. 13b).
This protocol generates high-quality crystals for laboratory analysis.203 In detail, COF-300 containing 1,4-dioxane as guest solvents exhibits unit cell parameters of a = b = 26.5565(16) Å, c = 7.4431(5) Å and a pore volume of 5249.2(7) Å3. Interestingly, replacing 1,4-dioxane with H2O results in the formation of intermolecular hydrogen bonds between H2O and N atoms in imine bonds, leading to significant framework shrinkage of COF-300 with unit cell parameters of a = b = 19.6219(10) Å, and c = 8.9048 (6) Å and a reduced pore volume of 3428.5(4) Å3. Noticeably, the hydrated COF-300 enables a better resolution of 0.80 Å, higher than that of the nascent COF-300 (0.83 Å).
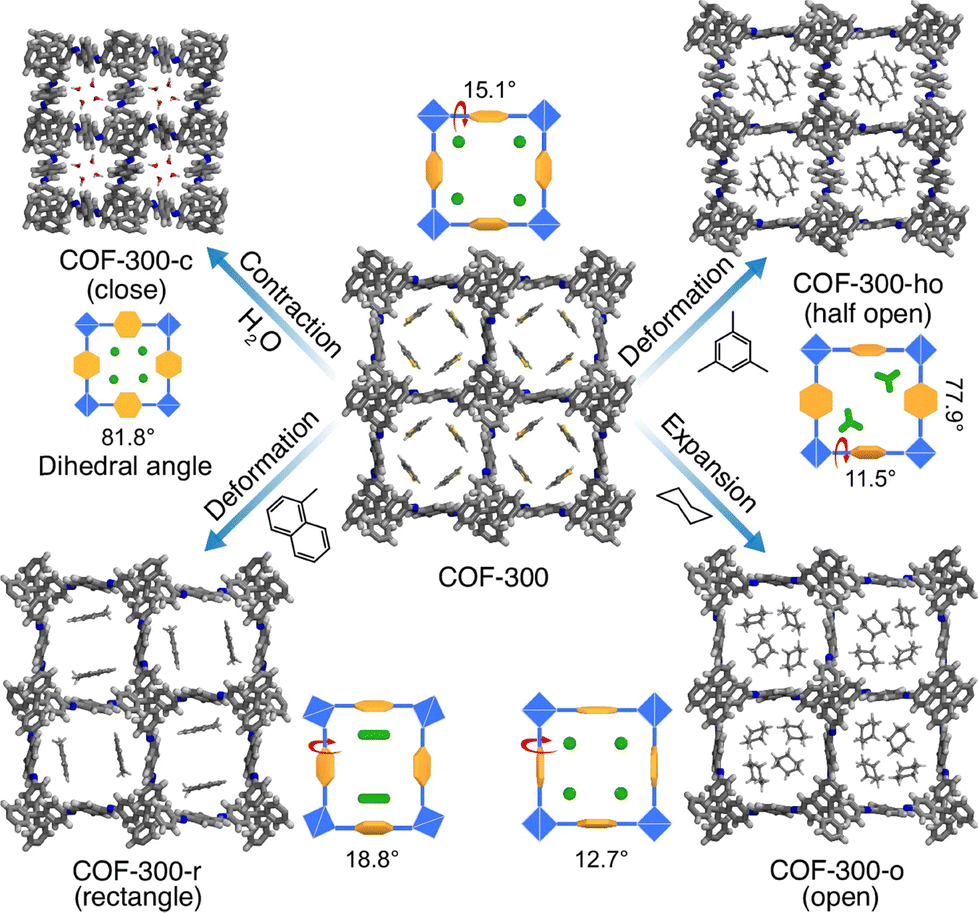
Noteworthily, COF-300 serves as a prototype for structural change investigation of single crystal COFs. Transformations induced by 21 different solvents generate five distinct structural conformations of single crystal COF-300 at room temperature.204 The original COF-300 exhibited a seven-fold interpenetrated dia topology with square channels measuring approximately 13.1 × 13.1 Å2. Interestingly, COF-300-c showed a closed-form structure that is induced by smaller molecules like water, leading to a considerable lattice contraction and shrinking the channels dramatically to 5.8 Å due to angular distortions of the tetrahedral nodes and rotation of aromatic rings. Using mesitylene as the molecular inducer generates the half-open form of COF-300-ho with partial opening channels (8.7 × 11.5 Å2), due to intermediate rotations of phenyl groups. Moreover, COF-300-r with rectangular deformed channels can be formed through the inclusion of 1,2,4-trimethylbenzene. Noticeably, COF-300-o with an open-form structure can be achieved through inclusion of large guest molecules like cyclohexane, resulting in maximal expansion of channels (14.1 × 14.1 Å2), the largest among these different forms (Fig. 14).204
 | ||
| Fig. 14 Schematics of the structural transformation of COF-300. Reprinted with permission from ref. 204. Copyright 2025 Springer Nature. | ||
Meanwhile, using polycyclic aromatic hydrocarbons (PAHs) such as naphthalene, anthracene, pyrene and perylene reveals the structural adaptability of COF-300 at elevated temperatures (Fig. 14). Remarkably, COF-300 maintains its crystallinity even at 280 °C, accommodating molten PAHs and demonstrating single-crystal-to-single-crystal transformations (Fig. 14). Each PAH orientates to unique spatial arrangements within the channels: naphthalene retains similar geometry to the original COF-300, while anthracene induces rectangular deformation due to its larger molecular size204 and pyrene and perylene develops further structural changes, allowing for only single-column arrangements (Fig. 14).
Noticeably, the dynamic conformational regulation of COF-300 (Fig. 15a) can be achieved by torsional adjustment of the imine linkages and biphenyl imines, which resembles a protein-folding-inspired design strategy for torsional motions in polypeptide backbones (Table 5).205 By resolving distinct anti-periplanar, syn-periplanar and anticlinal conformations within the same topological scaffold, COF-300 exhibits conformational polymorphism without altering the connectivity, revealing a structural basis for COF conformational dynamics.
 | ||
| Fig. 15 (a) Dynamic conformational regulation of COF-300. (b) Illustration of the formation of three TK-COF isomers. | ||
| COFs | Synthetic conditions | Crystal parameters | Resolution | Method | Ref. |
|---|---|---|---|---|---|
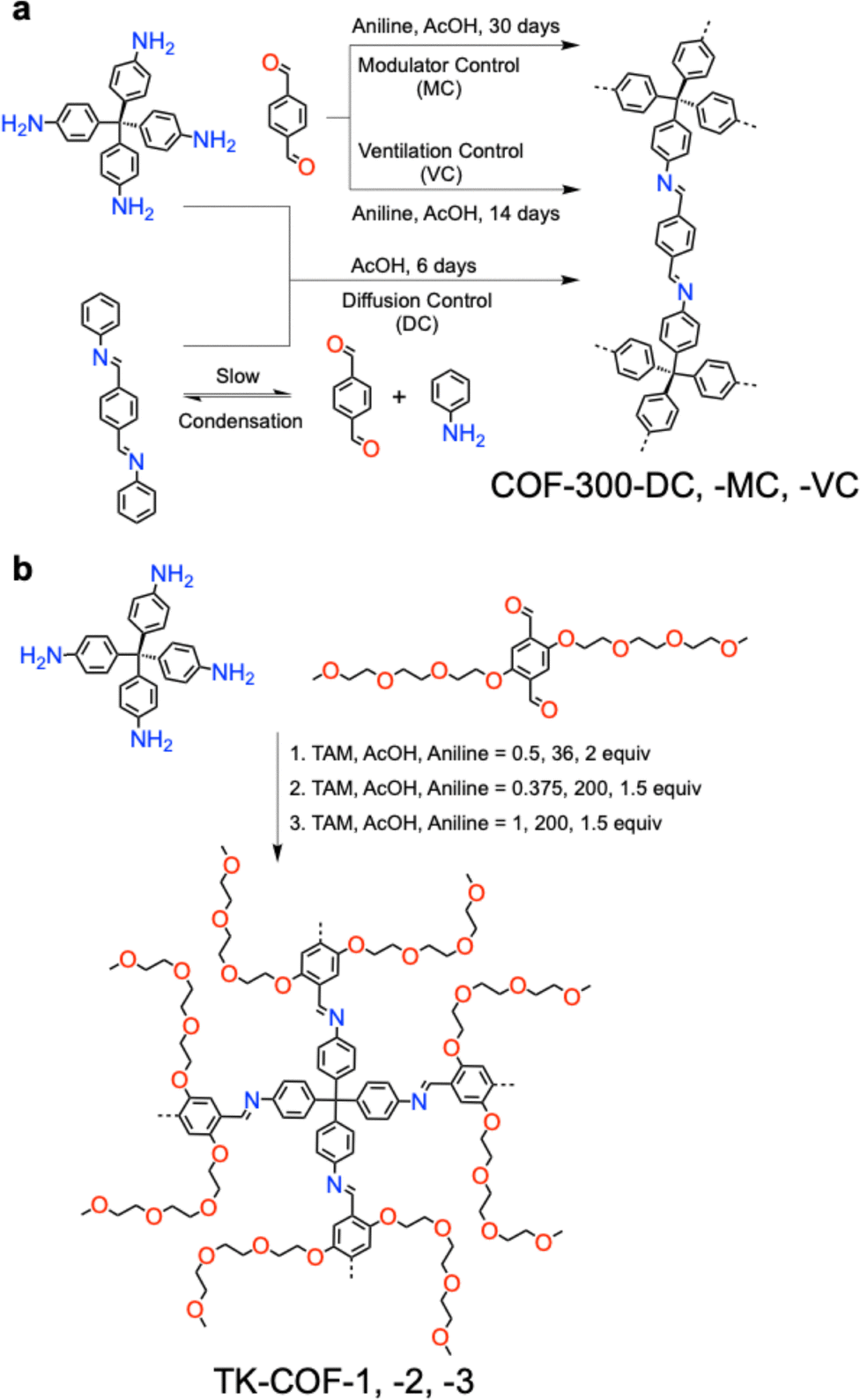
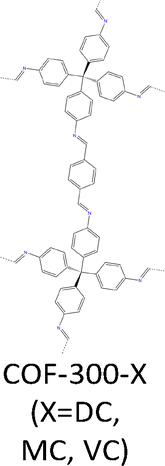 |
COF-300-DC: TA (12.0 mg, 0.089 mmol) is dissolved in 1,4-dioxane (0.5 mL), with aniline (0.12 mL, 15 equiv.) and aqueous AcOH (6 mol L−1, 0.2 mL) added, resulting in white coloured flake-like crystals precipitating out at the bottom of the glass diffusion tube (inner diameter ∼4.24 mm) standing vertically. Then, a solution of TAM (20.0 mg, 0.052 mmol) dissolved in 1,4-dioxane (0.5 mL, ultradry) was then added drop by drop onto the first layer to form stratification, yielding prismatic crystals of COF-300 with sizes of ∼50 µm within 6 days. To completely remove dioxane and keep the framework from collapsing, we used solvent exchange methods to completely remove the guest molecules in the framework and maintain the consistency of the sample crystal. THF, a low-boiling solvent, is used to exchange dioxane 3 times one day and then rocked on a shaker for 3 days. | a = b = 19.5107Å | 0.74 Å | SCXRD | 205 |
| c = 8.9850 Å | |||||
| α = β = γ = 90° | |||||
| COF-300-MC: TA (12.0 mg, 0.089 mmol), aniline (0.12 mL, 15 equiv.), and 0.5 mL of 1,4-dioxane were added to a vial, and 0.2 mL of AcOH (6 M) were added to the solution successively. Then TAM (20.0 mg, 0.052 mmol) dissolved in 1,4-dioxane (0.5 mL, ultradry) was added to the vial. The mixture was placed at 25 °C and the single crystals of COF-300 slowly crystallized out at room temperature and the crystal size reached ∼40 µm within 30 days, and 60 µm within 60 days. The single crystals were cleaned via the solvent exchange method. | NA | 0.85 Å | SCXRD | 205 | |
| COF-300-VC: the monomer addition order is the same as for COF-300-MC, while the vials were wrapped with perforated parafilm, allowing the original solution to volatilize gradually at the beginning of the reaction. Finally, single crystals grew to 50 µm within 14 days. The single crystals were cleaned via the solvent exchange method. | NA | 0.76 Å | SCXRD | 205 | |
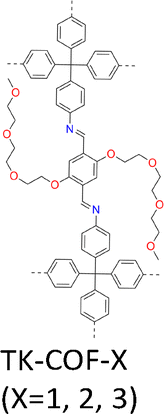 |
TK-COF-1: first, 4EBDA (1 equiv., 0.025 mmol, 11.46 mg) and aniline (2 equiv., 0.05 mmol, 4.5 µL) were mixed in 1,4-dioxane (0.25 mL), followed by ultrasonication for 5 min. Then, AcOH (36 equiv., 51 µL; H2O: 290 equiv., 130 µL) was added to the solution. Meanwhile, TAM (0.5 equiv., 0.0125 mmol, 4.76 mg) was dissolved in 1,4-dioxane (0.25 mL) using another glass vial, followed by ultrasonication for 5 min. Then, the mixture of 4EBDA, aniline, and catalyst was transferred into a 2 mL glass vial, followed by the addition of the TAM solution. A yellow solid was immediately precipitated from the solution. The obtained suspension was stored in a dark and noiseless Peltier incubator kept at 22 ± 0.5 °C for 2 days to yield TK-COF-1. | a = b = 30.470 Å | NA | HR PXRD using synchrotron radiation | 206 |
| c = 42.399 Å | |||||
| α = β = γ = 90° | |||||
| TK-COF-2: first, 4EBDA (1 equiv., 0.025 mmol, 11.46 mg) and aniline (1.5 equiv., 0.0375 mmol, 3.4 µL) were mixed in 1,4-dioxane (0.25 mL) in a 6 mL glass vial, followed by ultrasonication for 5 min. Then, AcOH (200 equiv., 286 µL; H2O: 290 equiv., 130 µL) was added to the solution. Meanwhile, TAM (0.375 equiv., 0.0125 mmol, 3.57 mg) was dissolved in 1,4-dioxane (0.25 mL) using another glass vial, followed by ultrasonication for 5 min. Then, the mixture of 4EBDA, aniline, and catalyst was transferred into a 2 mL glass vial, followed by the addition of the TAM solution. A yellow solid was immediately precipitated from the solution. The obtained suspension was stored in a dark and noiseless Peltier incubator kept at 8 ± 0.5 °C for 2 days to yield COF crystals. | a = 31.053 Å, | NA | HR PXRD using synchrotron radiation | 206 | |
| b = 26.517 Å, | |||||
| c = 28.200 Å | |||||
| α = 90° | |||||
| β = 17.719° | |||||
| γ = 90° | |||||
| TK-COF-3: first, 4EBDA (1 equiv., 0.025 mmol, 11.46 mg) and aniline (1.5 equiv., 0.0375 mmol, 3.4 µL) were mixed in 1,4-dioxane (0.25 mL) in a 6 mL glass vial, followed by ultrasonication for 5 min. Then, AcOH (200 equiv., 286 µL; H2O: 290 equiv., 130 µL) was added to the solution. Meanwhile, TAM (1 equiv., 0.025 mmol, 9.51 mg) was dissolved in 1,4-dioxane (0.25 mL) using another glass vial, followed by ultrasonication for 5 min. Then, the mixture of 4EBDA, aniline, and catalyst was transferred into a 2 mL glass vial, followed by the addition of the TAM solution. The obtained suspension was stored in a dark and noiseless Peltier incubator kept at 8 ± 0.5 °C for 2 days to obtain a mixture of TK-COF-2 and -3 crystals (the majority of the obtained crystals were TK-COF-2 crystals). Then, the samples were moved to another Peltier incubator kept at 22 ± 0.5 °C for 3–7 days for the dissolution of TK-COF-2 and precipitation of TK-COF3 crystals. Compared with the synthesis of pure TK-COF-2, the transition from TK-COF-2 to -3 can occur only when a relatively high concentration (≥2.5 × 10−2 M) of TAM is used. | a = b = 23.4519 Å | NA | SCXRD | 206 | |
| c = 57.759 Å | |||||
| α = β = γ = 90° |
Single crystal COF-300 was prepared using a diffusion gradient transimination method. First, a mixture of terephthalaldehyde (12.0 mg, 0.089 mmol), aniline (0.12 mL, 15 equiv.) and aqueous AcOH (0.2 mL, 6 M) in anhydrous 1,4-dioxane (0.5 mL) was placed at the bottom of a glass tube, whereupon white crystalline flakes form. A solution of TAM (20.0 mg, 0.052 mmol) in anhydrous 1,4-dioxane (0.5 mL) was then gently layered on top. The slow interdiffusion of reactants enables controlled nucleation and crystal growth via transimination (DC), yielding prismatic single crystals of COF-300-DC (Fig. 16a) with sizes of 50 µm in 6 days.205 In comparison, COF-300-MC (Fig. 16b) and COF-300-VC (Fig. 16c) were synthesized via imine-exchange (MC) and solvent-ventilation controlled (VC) crystal growth with a prolonged term of 30 and 14 days, respectively. SCXRD revealed their single crystal nature with a high resolution of 0.74 Å for COF-300-DC, 0.85 Å for COF-300-MC and 0.76 Å for COF-300-VC, respectively.
 | ||
| Fig. 16 (a)–(c) Optical images of single crystal COF-300 grown via (a) DC, (b) MC and (c) VC approaches, respectively. (d)–(f) Optical images of single crystals of (d) TK-COF-1, (e) TK-COF-2 and (f) TK-COF-3. (a)–(c) Reprinted with permission from ref. 205. Copyright 2024 American Chemical Society. (d)–(f) Reprinted with permission from ref. 206. Copyright 2024 American Chemical Society. | ||
This conformational adaptability is related to guest responses and structural breathing phenomena during gas adsorption. The ability to undergo reversible torsional adjustments without losing crystallinity suggests a mechanism for pore modulation, analogous to protein conformational transitions. Such dynamic flexibility adds insights to adsorption-induced phase behaviour, anisotropic pore response and stimuli-responsive properties in 3D COFs.205
Isomerism in single crystal COFs has scarcely been known due to the difficulties in controlling nucleation and crystallization manners. Distinct from the above method that uses transimination, imine-exchange and solvent-ventilation to grow COF-300 with different conformations, a strategy with flexible building blocks can induce multiple framework topologies from the same molecular components.206 A linear dialdehyde bearing two tetraethylene glycol (TEG) side chains (4EBDA) as the linker upon condensation with TAM generates TK-COFs (Fig. 15b). The flexibility of TAM combined with the conformational adaptability of the TEG side chains allows the framework to adopt different packing arrangements during crystal growth, thereby promoting structural isomerism.206
Indeed, this strategy generates three isomeric 3D COFs, denoted TK-COF-1 (Fig. 16d), TK-COF-2 (Fig. 16e) and TK-COF-3 (Fig. 16f) with distinct dia, qtz and dia-c3 nets, respectively, demonstrating that identical molecular components can afford multiple framework structures.
Single crystals suitable for structure resolution were obtained through solvothermal imine condensation between TAM and 4EBDA under carefully optimized crystallization conditions (Table 5). The polymerization in 1,4-dioxane under acidic conditions promotes reversible imine formation and facilitates error correction during framework assembly.206 The slow crystallization process allows the growth of large single crystals observable with optical microscopy, enabling full structural determination by SCXRD.
SCXRD analysis revealed the single crystal structures of the three isomers. TK-COF-1 shows unit cell parameters of a = b = 30.470 Å, c = 42.399 Å, and α = β = γ = 90° and a unit-cell volume (V) of 39364 Å3, with the I41/a space group. TK-COF-3 exhibits a three-fold interpenetrated dia net and shares the same space group of I41/a, with unit cell parameters of a = b = 23.4519 Å, c = 57.759 Å, α = β = γ = 90° and V = 31767 Å3. In contrast, TK-COF-2 adopts the P2 space group, with unit cell parameters of a = 31.053 Å, b = 26.517 Å, c = 28.200 Å, α = 90°, β = 117.719°, γ = 90° and V = 20556 Å3.
All three COFs share the same chemical composition but exhibit distinct connectivity and topology due to differences in the spatial arrangement of the flexible building units. Morphological characterization by optical microscopy and SEM confirms that the COF crystals exhibit well-defined shapes and sizes in the tens-of-micrometres range. Interestingly, TK-COF-1 is a ditetragonal-dipyramid with four vertices truncated, while TKCOF-2 is a hexagonal prism and TK-COF-3 a slightly elongated octahedron. The presence of TEG side chains contributes to the structural adaptability during crystallization, inducing the framework to reorganize into multiple energetically accessible configurations.206 This strategy thus expands the structural diversity of single crystal 3D COFs and provides new insight into the relationship between molecular design, crystallization pathways and framework topology.
The structural complexity of COFs is important for function exploration. The configurational flexibility of tetrahedral TAM directs its polycondensation with linear dialdehyde linkers (2,3-dihydroxyterephthalaldehyde) (DHPA) for COF-304 (Fig. 11i) and DMPA for COF-305 (Fig. 11j), forming diamondoid structures with different degrees of structural complexity.207 The tetrahedral nodes capable of adopting multiple orientations propagate distinct configurations throughout the lattice, thereby encoding ordered structural diversity within 3D COFs.
Single crystal COF-304 and COF-305 were prepared through imine condensation reactions under solvothermal conditions (Table 4).207 In detail, for the synthesis of single crystal COF-305, DMPA (17 mg, 0.088 mmol) dissolved in 1,4-dioxane (0.5 mL) was mixed with aniline (40 µL) as the modulator and aqueous AcOH (15 M, 200 µL) as the catalyst. A solution of TAM (20 mg, 0.053 mmol) dissolved in 1,4-dioxane (0.75 mL) was carefully added into the reaction vial to minimize rapid nucleation. The reaction mixture was left standing at room temperature without disturbance. After 2 days, yellow crystals of 30 µm in size formed, which continued to grow to 40 µm after 4 days, a size suitable for SCXRD analysis. High-quality single crystal COF-304 was prepared by reacting DHPA and TAM under a similar condition.
SCXRD analysis at a resolution of 1 Å revealed that COF-305 with dia topology adopts the orthorhombic space group Fdd2 (no. 43) and exhibits an exceptionally large unit cell with parameters of a = 47.077 Å, b = 67.629 Å, c = 42.547 Å and α = β = γ = 90°, corresponding to a V value of 135460 Å3. This unit cell value is among the largest reported for 3D COFs.207 Notably, the TAM units within COF-305 contain multiple orientations to allow different rotational configurations and propagations in an ordered fashion throughout the structure, thereby enabling the co-existence of several independent network components and a more intricate interpenetration pattern. In contrast, although COF-304 adopts a dia topology it develops a periodic porous framework by connecting TAM with dialdehyde linkers. In this structure, the tetrahedral TAM maintains a uniform orientation throughout the lattice, forming a regular pore system and defined interpenetration pattern.207 Thus, single crystal COF-304 represents a conventional yet well-defined diamondoid COF, in which the building units adopt a uniform configuration.207
The above structural comparisons between single crystal COF-305 and COF-304 highlight how subtle differences in molecular configuration yield sharply different crystalline architectures.207 While COF-304 forms a uniform and symmetric porous network, COF-305 develops a hierarchically complex yet ordered framework. Both materials exhibit permanent porosity, while the structural complexity of single crystal COF-305 may generate diversified pore environments that may enhance host–guest interactions or molecular recognition.207
Interestingly, the same behaviour as for the single crystal COF-305 is observable for COF-308 (= COF-305, Fig. 11j).208 The synthesis is based on controlling the intermolecular arrangement of TAM. In contrast to a conventional COF-300 with TAM and TA units that exhibits flexible ‘edge-to-edge’ alignment for the TAM units, the introduction of methoxy-substituted dialdehyde (BDA-cis-OMe = DMPA) regulates intermolecular interactions and induces a ‘face-to-face’ packing mode (Fig. 17), thereby fixing the framework geometry. Furthermore, the introduction of a Lewis acid template stabilizes this packing configuration during crystallization, ultimately generating a different COF topology with enhanced rigidity.208
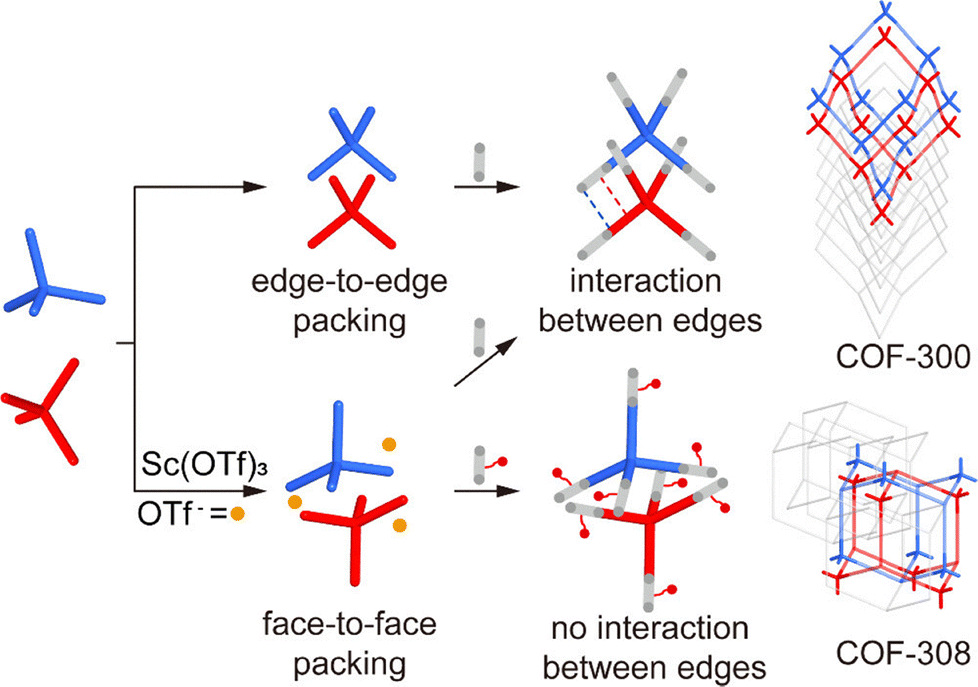 | ||
| Fig. 17 Different packing modes of COF-300 and COF-308. Reprinted with permission from ref. 208. Copyright 2025 American Chemical Society. | ||
Single crystal COF-308 was synthesized through condensation of TAM with BDA-cis-OMe.208 The reaction proceeds in a mixture of 1,4-dioxane, with aniline as a modulator and scandium trifluoromethanesulfonate (Sc(OTf)3) as the Lewis acid catalyst. The aniline modulator promotes reversible imine exchange and controls nucleation, while Sc(OTf)3 acts as a structure-directing agent to favour the ‘face-to-face’ arrangement of the TAM units.208 Large octahedral single crystals (Fig. 18a) with sizes up to 100 µm were obtained after slow crystallization.
 | ||
| Fig. 18 (a)–(f) Optical images of single crystals of (a) COF-308, (b) TK-COF-4, (c) TK-COF-5, (d) COF-300-AR, (e) COF-300-CHO and (f) NKCOF-155. (a) Reprinted with permission from ref. 208. Copyright 2025 American Chemical Society. (b) and (c) Reprinted with permission from ref. 168. Copyright 2025. Springer Nature. (d) and (e) Reprinted with permission from ref. 189. Copyright 2025 American Chemical Society. (f) Reprinted with permission from ref. 209. Copyright 2025 American Chemical Society. | ||
SCXRD analysis with a resolution of 0.95 Å reveals that COF-308 crystallizes in the orthorhombic space group Fddd with unit cell parameters of a = 41.05 Å, b = 46.67 Å, and c = 40.94 Å. COF-308 adopts a dia topology featuring an unusual [4+2] mixed interpenetration mode, composed of two independent sets of networks interlocked within the same crystal lattice.208 Detailed structural analysis indicates the presence of two crystallographically independent TAM nodes, which develop two interpenetrating networks, whose combined interpenetration pattern effectively locks the framework geometry. This structural arrangement restricts the relative sliding or twisting of adjacent frameworks, leading to a mechanically rigid architecture.208
The rigid structure of single crystal COF-308 is confirmed by gas adsorption. Nitrogen adsorption at 77 K displays a typical type-I isotherm with negligible hysteresis, in contrast to the flexible adsorption behaviour observed for single crystal COF-300. The BET surface area reaches 2060 m2 g−1, close to the theoretical value of 2260 m2 g−1, indicating the permanent porosity and structural integrity.208 The pore size distribution centered at 1.27 nm, is consistent with the crystallographically determined channels. These results demonstrate that controlling the packing of building blocks and framework interpenetration can effectively tune the dynamic behaviour of 3D single crystal COFs, offering a new strategy for designing rigid COFs suitable for gas storage, adsorption, crystallography and structure–property relationship investigations.208
A subtle modification in linker functional groups and the use of templating Lewis acids control the packing arrangement of building blocks. As a result, this transforms flexible frameworks into rigid architectures. This provides insights into the relationship between interpenetration, molecular packing and framework dynamics, which is critical for developing COFs with controllable structures.
2D COFs consist of layered architectures, while their crystal growth is typically limited to micro- or nanometre scales. In contrast, 3D COFs form large single crystals but lack functional advantages associated with layered frameworks. To reconcile these limitations, the 2.5D COFs (Fig. 19a and b) develop a stereoscopic bonding strategy by using tetrahedral tetratopic nodes and trigonal tritopic linkers to generate laterally extended skeletons, which are macroscopically 2D but microscopically 3D.168
 | ||
| Fig. 19 Structures of (a) TK-COF-4, (b) TK-COF-5, (c) COF-300-AR, (d) COF-300-CHO, (e) COF-300-1,2,3-triazole, (f) COF-300-1,2,4-triazole, (g) COF-300-pyrazole and (h) NKCOF-155. | ||
The tetra-functional TAM was reacted with triangular 2,4,6-tris(4-formylphenyl)-1,3,5-triazine (TFPT) or 1,3,5-tris(4-formylphenyl) benzene (TFPB) to form two COFs, TK-COF-4 (Fig. 19a) and TK-COF-5 (Fig. 19b), respectively. Unlike conventional 3D COFs constructed from similar building blocks, the reaction proceeds in a non-stoichiometric component mode, where only three of the four amine groups of TAM participate in imine formation, leaving one amine protruded from the layer plane.168 This configuration produces a layered architecture composed of laterally interpenetrated networks, while maintaining stereoscopic connectivity that promotes the growth of large single crystals.
The single crystal COFs were synthesized through a controlled solvothermal process in a mixed solvent system composed of 1,4-dioxane and o-DCB (Table 6), which provides an appropriate balance between solubility and crystallization kinetics.168 AcOH serves as the catalyst, while aniline or m-toluidine functions as the modulator to regulate nucleation and crystal growth. Optimizing conditions yields large prismatic single crystals (Fig. 18b and c) with sizes exceeding 0.1 mm, among one of the largest single crystal dimensions reported for layered COFs to date.168 The use of modulators plays a crucial role in slowing the polymerization kinetics and enabling defect correction through dynamic imine exchange, thereby facilitating the formation of highly ordered crystalline frameworks suitable for SCXRD analysis.168
| COFs | Synthetic conditions | Crystal parameters | Resolution | Method | Ref. |
|---|---|---|---|---|---|
 |
First, the powder of TFPT (9.44 mg, 0.024 mmol) was taken in a screw-cap glass vial (capacity: 5 or 10 mL). Then, o-DCB (2.16 mL) was added to the vial, which was sonicated for 10 min using an ultrasonic bath (3510-DTH, Branson). Next, AcOH (252 µL, 182 equiv.) and aniline (136 µL, 62 equiv.) were added to the vial, after which an additional 3 min sonication was applied. Subsequently, a 1,4-dioxane solution (360 µL) of TAM at 50 mM concentration, which was prepared in a separate vial and passed through a PTFE filter (pore size: 200 nm, SLLGX13NL, Merck-LG), was added to it. Finally, the mixture was homogenized quickly either by sonication for 1 min or by gently shaking the vial by hand. The capped vial was stored in a temperature-controlled incubator at 22 °C until crystals of sufficient size for the intended measurement were generated. | a = 9.8024 Å | NA | SCXRD | 168 |
| b = 36.6977 Å | |||||
| c = 18.2022 Å α = 90° | |||||
| β = 99.608° | |||||
| γ = 90° | |||||
 |
First, the powder of TFPB (9.36 mg, 0.024 mmol) was taken in a screw-cap glass vial (capacity: 5 or 10 mL). Then, o-DCB (2.16 mL) was added to the vial, which was sonicated for 5 min using an ultrasonic bath (3510-DTH, Branson). Next, AcOH (252 µL, 182 equiv.) and m-toluidine (110 µL, 42 equiv.) were added to the vial, after which an additional 3 min sonication was applied. Subsequently, a 1,4-dioxane solution (360 µL) of TAM at 50 mM concentration, which was prepared in a separate vial and passed through a PTFE filter (pore size: 200 nm, SLLGX13NL, Merck-LG), was added to it. Finally, the mixture was homogenized quickly either by sonication for 1 min or by gently shaking the vial by hand. The capped vial was stored in a temperature-controlled incubator at 22 °C until crystals of sufficient size for the intended measurement were generated. | a = 10.2481 Å | NA | SCXRD | 168 |
| b = 37.3978 Å | |||||
| c = 18.6451 Å | |||||
| α = 90° | |||||
| β = 103.078° | |||||
| γ = 90° | |||||
 |
The as-synthesized single-crystal COF-300 (50 mg) was dispersed in 25.0 mL of methanol. Terephthalic acid (BDC, 60 mg) was added to a single-crystal COF-300 suspension. The reaction mixture was cooled to 0 °C. NaBH4 (0.5 g) was added to small portions over a period of 20 min. The mixture was slowly stirred at 0 °C for 1 h and then allowed to warm to room temperature for another 10 h. The reduced COF-300-AR was washed with water and methanol. The resulting single-crystal COF-300-AR was immersed in DMF for 1 day and subjected to SXRD measurements. | a = b = 27.327 Å, c = 7.2710 Å | 0.91 Å | SCXRD | 189 |
| α = β = γ = 90° | |||||
 |
A mixture of formic acid (5.0 mL) and acetic anhydride (5.0 mL) was stirred at 70 °C for 1 h and then cooled to room temperature. The as-synthesized single-crystal COF-300-AR (50 mg) was dispersed in 5.0 mL of acetic anhydride. A single-crystal COF-300-AR suspension was added to a mixture of formic acid and acetic anhydride. The resulting mixture was stirred slowly at 50 °C for 10 h. After the reaction was complete, the resulting single-crystal COF-300-CHO was immersed in DMF for 1 day and subjected to SXRD measurement. | a = b = 25.8963 Å, c = 7.0109 Å | 0.98 Å | SCXRD | 189 |
| α = β = γ = 90° | |||||
SCXRD measurements unambiguously resolve the crystal structures of the 2.5D COFs. Single crystal TK-COF-4 adopts the monoclinic space group P21/n with unit cell parameters of a = 9.8024 Å, b = 36.6977 Å, c = 18.2022 Å, α = γ = 90°, β = 99.608° and V = 6455.95 Å3. Similarly, single crystal TK-COF-5 adopts the same P21/n space group but with slightly expanded lattice parameters (a = 10.2481 Å, b = 37.3978 Å, c = 18.6451 Å, α = γ = 90°, β = 103.078° and V = 6960.51 Å3).168 The crystal structures reveal a distinctive structural motif, in which two laterally displaced hcb-type networks interpenetrate within the same plane, forming a layered architecture. Importantly, the unreacted amines are periodically arranged and oriented perpendicular to the layers, producing an exceptionally high density of functional groups accessible to guest molecules.168 The frameworks develop interlayer N–H⋯N hydrogen bonds between primary amines and neighbouring imine nitrogen atoms, which contribute to the stabilization of layer growth.168
The development of new linkage chemistry in single crystal 3D COFs remains a challenging goal.189 Post synthetic modifications of single crystal COFs provide an available method but often cause partial loss of crystallinity. A multistep single crystal to single crystal (SC–SC) transformation enables the construction of the formamide-linked single crystal COF-300-CHO (Fig. 19d), from the imine-linked single crystal COF-300 (Fig. 7a).189
This approach sequentially transforms the dynamic imine linkage into more robust functional linkages while preserving the crystalline order.189 Thus, the imine bonds in single crystal COF-300 are transformed to amine bonds to yield COF-300-AR (Fig. 19c), followed by a formylation reaction that introduces formamide linkage to generate single crystal COF-300-CHO through a multistep SC–SC process.189
The parent COF-300 single crystals were synthesized through a reversible imine condensation between TAM and TA.189 The reaction is performed in 1,4-dioxane in the presence of glacial AcOH and aniline, together with ionic liquid 1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([BMIm][NTf2]) to regulate crystallization kinetics. The reaction mixture was maintained at room temperature for 5 days (Table 6), producing yellow prismatic single crystals suitable for SCXRD analysis. SCXRD analysis at a resolution of 0.90 Å revealed that COF-300 exhibits the tetragonal space group I41/a with a seven-fold interpenetration structure, showing unit cell parameters of a = b = 26.6062 Å, c = 7.4389 Å and α = β = γ = 90°.
The subsequent SC–SC transformation involves two sequential reaction steps. First, the imine linkages of COF-300 are reduced by NaBH4 to form the amine-linked COF-300-AR (Fig. 18d), while preserving the crystal morphology and long-range order.189 SCXRD analysis confirmed that COF-300-AR maintains the same I41/a space group as the parent single crystal COF-300 with slightly expanded lattice parameters of a = b = 27.327 Å and c = 7.2710 Å. In the second step, the amine linkages are converted into formamide groups by reaction in a solution of formic acid and acetic anhydride, generating the formamide-linked single crystal COF-300-CHO (Fig. 18e).189
Structural refinement revealed that the introduction of formamide groups induced significant lattice contraction due to steric effects and additional hydrogen-bond interactions. The lattice parameters decreased to a = b = 25.8963 Å and c = 7.0109 Å, resulting in a 13.4% reduction in the unit-cell volume. This could be attributed to the shortening of the organic linker and the formation of intermolecular C–H⋯O hydrogen bonds within the framework. Moreover, compared to the parent COF-300 and COF-300-AR, single crystal COF-300-CHO has improved chemical stability toward strong acids and bases, demonstrating that post-synthetic linkage transformation effectively enhances the robustness of COFs while preserving crystallinity.189
Noticeably, a solvent-free melt-phase PSM strategy using 1,2,3-triazole, 1,2,4-triazole and pyrazole enables the transformation of single crystal COF-300 (Fig. 7a) into amine-linked COF-300-1,2,3-triazole (Fig. 4e), COF-300-1,2,4-triazole (Fig. 4f) and COF-300-pyrazole (Fig. 4g), respectively (Table 7).190 This approach enables precise chemical modification of COF linkages while preserving single-crystal integrity, thereby allowing atomic-level characterization of chemical transformations in porous frameworks. Under molten conditions, these azoles diffuse uniformly throughout the framework without solvent interference, enabling efficient chemical modification of the imine linkages.
| COFs | Synthetic conditions | Crystal parameters | Resolution | Method | Ref. |
|---|---|---|---|---|---|
 |
The single crystals of COF-300 (1 mg) and azole reagents (100 mg), including 1,2,3-triazole (melting point: 25 °C), 1,2,4-triazole (121 °C), and pyrazole (70 °C), were combined in a sample vial (5.0 mL). The mixture was heated at 90 °C (1,2,3-triazole and pyrazole) and 125 °C (1,2,4-triazole) for 12 h. Subsequently, the mixture was washed with ethanol, and the resulting single crystals of COF-300-azole were isolated. The single crystal was sealed in a capillary tube for single-crystal X-ray crystallography analysis. | a = b = 26.01 Å, c = 54.30 Å | 0.86 Å | SCXRD | 190 |
 |
See above | NA | NA | NA | 190 |
 |
See above | NA | NA | NA | 190 |
 |
A sample vial (8.0 mL, body length 50 mm, neck length 18 mm) was sequentially charged with 2,2′,7,7′-tetramethoxy-9,9′-spirobi[fluorene]-3,3′,6,6′-tetracarbaldehyde (TMSFTA, 13.7 mg, 0.025 mmol), THF (0.25 mL), acetonitrile (0.25 mL) and CF3CH2NH2 (102 µL, 52 equiv.). Then, CF3COOH (0.10 mL, 6.0 M) was added. TAM (9.5 mg, 0.025 mmol) dissolved in THF (0.25 mL) and acetonitrile (0.25 mL) was added to the solution. The reaction was allowed to stand at 30 °C. The yellow single crystals of NKCOF-155 formed rapidly, exhibiting uniform morphology. The crystal size reached 80 µm in 3 days. The as-synthesized crystals were immersed in 1,4-dioxane to obtain fully solvent-occupied single crystals of NKCOF-155. | a = b =11.1981 Å, c = 18.3516 Å, α = β = γ = 90° | 0.79 Å | SCXRD | 209 |
The structural integrity of the resultant COFs is confirmed by SCXRD, which demonstrated that the framework topology remained intact after modification. The three COFs are isostructures and crystallize in a tetragonal system with the I41/a space group. Detailed crystallographic analysis on COF-300-1,2,3-triazole reveals the atomic-level structural changes associated with the modification. Its imine bond length (1.245 Å) is converted into an amine linkage with a bond length of 1.415 Å, while the attached azole groups form additional hydrogen-bond networks within the pores. Moreover, COF-300-1,2,3-triazole retains a similar structure to the parent COF-300, featuring a seven-fold interpenetrated dia-c7 topology.190
Developing biocomposites of single crystal 3D COFs with biomacromolecules is fundamentally important for application but remains a challenge. A new single crystal 3D COF designed for enzyme assembly and biomolecule integration highlights the potential of 3D COFs for biohybrid materials. The proposed strategy was based on the dynamic nature of imine bonds in COFs to enable covalent attachment of biomacromolecules through reversible bond exchange processes.209
Single crystal NKCOF-155 (Fig. 5h) was obtained through a condensation reaction between TAM (9.5 mg, 0.025 mmol) and spirobifluorene-based linker 2,2″,7,7″-tetramethoxy-9,9′-spirobi[fluorene]-3,3′,6,6′-tetracarbaldehyde (TMSFTA, 13.7 mg, 0.025 mmol) by using CF3COOH as the catalyst and CF3CH2NH2 as the modulator (Table 7). Noteworthily, the spirobifluorene core introduces a rigid and bulky geometry that stabilizes the resulting framework while generating well-defined pores suitable for biomolecule immobilization.209 After 3 days of reaction, the system yields well-defined melon-seed-shaped crystals of 80 µm in size (Fig. 18f), which can be directly observed by optical microscopy and SEM. The crystal structure of NKCOF-155 is obtained using SCXRD with a resolution of 0.79 Å. It adopts a tetragonal space group P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 2m, with unit cell parameters of a = b = 11.1981 Å, c = 18.3516 Å, α = β = γ = 90° and V = 2315.23 Å3.
2m, with unit cell parameters of a = b = 11.1981 Å, c = 18.3516 Å, α = β = γ = 90° and V = 2315.23 Å3.
The dynamic nature of imine linkages plays a key role in enabling enzyme immobilization.209 In aqueous environments, imine bonds undergo hydrolysis to generate aldehyde groups, which react with amino groups present on enzyme surfaces. This dynamic exchange mechanism allows enzymes to be covalently integrated into COFs without disrupting its crystallinity. The resulting enzyme–COF composites exhibit promising performance in biocatalytic reactions due to the synergistic combination of the ordered porous structure and the catalytic activity of the enzymes.209 Details of the reactions and catalytic performance are analyzed in Section 5.5.
These results show a strategy for assembling biomolecules within single crystal 3D COFs through dynamic covalent chemistry. The approach not only expands the functional scope of 3D COFs but also provides a new platform for designing biohybrid materials.
The valency of COFs has been long limited to 6. A series of single organic linkers, BPA-1 to 5, were designed to combine boronic and phosphonic acid functionalities in one molecule.185 Through an ingenious in situ strategy based on isoelectronic replacement, these linkers self-condensed to form borophosphonate cubes with a valency of 8, yielding a 3D polycubane COF, BP-COF-1 (Fig. 20a) and its isoreticular analogues BP-COF-2 to 5.185 The structure of microcrystalline BP-COF-1 was solved from PXRD data, with Pawley refinement confirming a body-cantered cubic (bcu) topology with a unit cell of a = 12.39(5) Å, b = 12.52(3) Å and c = 12.93(5) Å (Fig. 21a). Comprehensive characterization, including ssNMR (11B, 31P and 13C), revealed the tetrahedral coordination of boron within the cubes (Fig. 21b–d), while N2 sorption isotherm analysis showed a BET surface area of 519 m2 g−1, confirming permanent porosity.
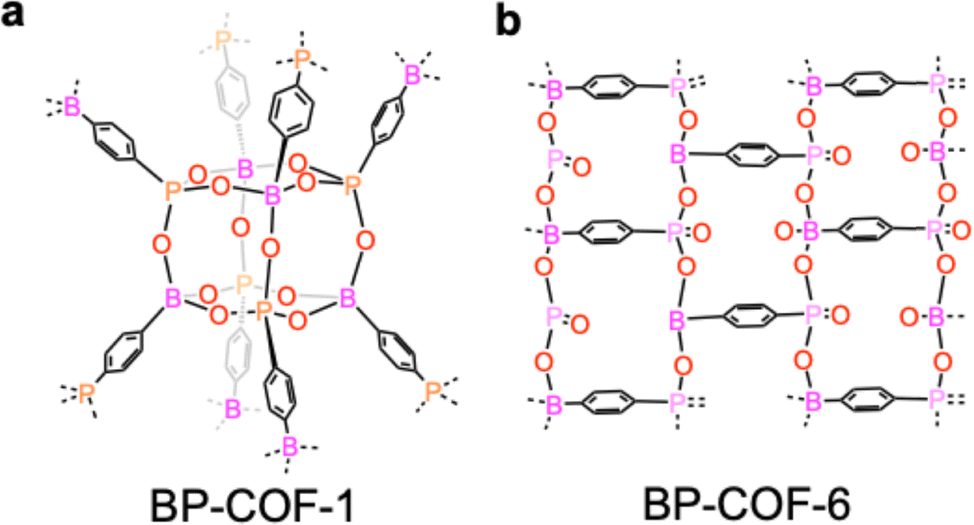 | ||
| Fig. 20 Structures of (a) BP-COF-1 and (b) BP-COF-6. | ||
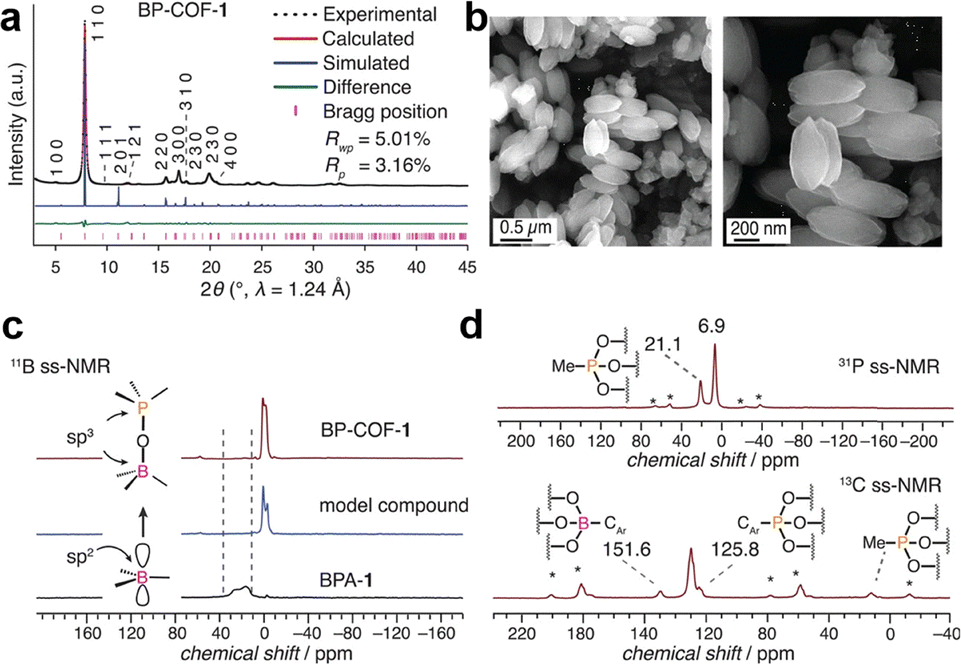 | ||
| Fig. 21 Characterization of BP-COF-1. (a) XRD refinement of BP-COF-1, showing the experimentally observed XRD pattern (black), Pawley fitting (red) and the simulated XRD pattern (blue). (b) SEM images of BP-COF-1. (c) Overlay of the 11B solid-state (ss) NMR spectra of BP-COF-1, a molecular borophosphonate model compound and BPA-1. (d) 31P and 13C NMR spectra of BP-COF-1. Reprinted with permission from ref. 185. Copyright 2020 American Association for the Advancement of Science. | ||
Interestingly, acid induces rearrangement of BP-COF-1 into BP-COF-6 (Fig. 20b), a constitutional isomer featuring infinite B–O–P rods (valency of ‘infinity’). Crucially, BP-COF-6 (Table 8) was obtained as large single crystals up to 50 µm (Fig. 22a), enabling its structure to be resolved via single-crystal X-ray diffraction.185
| COFs | Synthetic conditions | Crystal parameters | Resolution | Method | Ref. |
|---|---|---|---|---|---|
 |
(R)-BTAP-Cl (10.7 mg, 0.015 mmol) was added to a centrifuge tube (1 mL), followed by THF/acetonitrile (0.5 mL; 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by vol). The mixture was ultrasonicated until fully dissolved, then CF3CH2NH2 (47 µL, 40 equiv.) and CF3CO2H (100 µL, 1 M) were added sequentially. TAM (5.7 mg, 0.015 mmol) was added to the solution and shaken gently until dissolved. The mixture was filtered through a 0.45 µm PTFE filter membrane, and the clear filtrate was transferred to a 1-dram glass vial and sealed. The vial was left undisturbed at room temperature for 28 days, during which yellow single crystals of (R)-39 with uniform rice-like morphology were formed, with an average size of approximately 100 µm. The as-synthesized crystals were soaked in DMF for 24 h to exchange the guest molecules in the pores and then subjected to SCXRD measurements :1 by vol). The mixture was ultrasonicated until fully dissolved, then CF3CH2NH2 (47 µL, 40 equiv.) and CF3CO2H (100 µL, 1 M) were added sequentially. TAM (5.7 mg, 0.015 mmol) was added to the solution and shaken gently until dissolved. The mixture was filtered through a 0.45 µm PTFE filter membrane, and the clear filtrate was transferred to a 1-dram glass vial and sealed. The vial was left undisturbed at room temperature for 28 days, during which yellow single crystals of (R)-39 with uniform rice-like morphology were formed, with an average size of approximately 100 µm. The as-synthesized crystals were soaked in DMF for 24 h to exchange the guest molecules in the pores and then subjected to SCXRD measurements |
a = b = 27.31 Å, c = 23.57 Å, α = β = γ = 90° | 0.90 Å | SCXRD | 210 |
 |
(R)-BTAT-Cl (10.9 mg, 0.015 mmol) was added to a centrifuge tube (1 mL), followed by THF/acetonitrile (0.5 mL, 6:1 by vol). The mixture was ultrasonicated until fully dissolved, then CF3CH2NH2 (40 µL, 33 equiv.) and CF3CO2H (50 µL, 2 M) were added sequentially. TAM (5.7 mg, 0.015 mmol) was added to the solution and shaken gently until dissolved. The mixture was filtered through a 0.45 µm PTFE filter membrane, and the clear filtrate was transferred to a 1-dram glass vial and sealed. The vial was left undisturbed in a 35 °C oven for 28 days, during which colourless single crystals of (R)-39-SH with uniform polyhedron morphology were formed, with an average size of approximately 60 µm. The as-synthesized crystals were soaked in DMF for 24 h to exchange the guest molecules in the pores and then subjected to SCXRD measurements. |
a = b = 27.25 Å, c = 23.89 Å,α = β = γ = 90° | 1.1 Å | SCXRD | 210 |
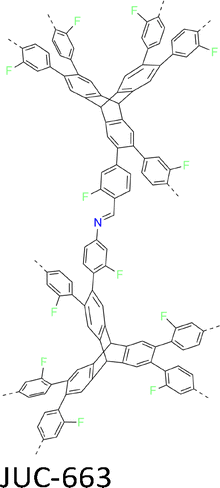 |
A vial (5 mL) was charged with HFFPTP (9.86 mg, 0.01 mmol), aniline (30–135 eq.) and anhydrous 1,4-dioxane (0.5 mL). The mixture was sonicated until the solid was just dissolved, and glacial AcOH (0.4 mL) was successively added into the vial. Immediately, HAFPTP (9.19 mg, 0.01 mmol) in anhydrous 1,4-dioxane (1 mL) was added to the solution. The mixture was allowed to stand at ambient temperature for crystallization. The light-yellow single crystals of JUC-663-X with sizes up to 5–30 µm appeared after 120 h | a = b = 32.4119 Å, c = 15.2924 Å, α = β = 90°, γ = 120° | NA | SAED/RED | 211 |
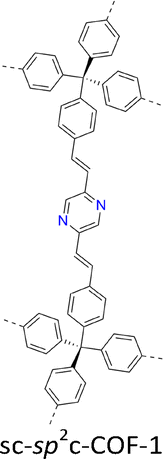 |
A pyrex tube (d = 10 mm) was charged with COF-303 (8 mg), 1,2,5-trimethylpyrazin1-ium iodide (p-TPI, 13.2 mg, 52.73 µmol), mesitylene (0.5 mL), 1,4-dioxane (0.5 mL) and acetonitrile (0.02 mL). Then CF3CO2H (0.18 mL) was added slowly along the interior surface of the pyrex tube. The pyrex tube was sealed under vacuum after pumping for 15 mins and set at 120 °C for three days. The as-synthesized sc-sp2c-COF-1 was washed with tetrahydrofuran and acetone six times with interval times of 2 h each and dried at 120 °C under vacuum conditions to yield a dark red powder with an average yield of 83%. | a = 20.4 Å, b = 20.4 Å, c = 8.8 Å, α = β =γ = 90° | 1.0 Å | SCXRD | 188 |
 | ||
| Fig. 22 Single crystal BP-COF-6. (a) Optical microscopy and SEM images of BP-COF-6. (b) Polycubane-to-1D rearrangement of BP-COF-1 to BP-COF-6. Reprinted with permission from ref. 185. Copyright 2020 American Association for the Advancement of Science. | ||
The structure in the space group Pc revealed unit cell parameters of a = 5.4728(16) Å, b = 4.3774(12) Å, c = 14.285(4) Å, β = 95.402(7)° and V = 340.71(16) Å3. Key structural details include B–O and P–O bond lengths of 1.47 Å and 1.54 Å within the rods and a layered architecture stabilized by interstitial water molecules via hydrogen bonds (O⋯O distances of 2.58–2.60 Å). The proposed ‘slice-and-slide’ mechanism for this topological transformation from a polycubane to a rod-based framework is illustrated in Fig. 22b.185 These results introduce a novel linkage chemistry for COFs and provides one of the rare atomic-level structural characterizations of a COF via single-crystal diffraction, thereby demonstrating a new aspect of higher valency and complexity in porous organic materials.185
Recently, two chiral 3D COFs (CCOFs) (Fig. 23 and Table 8) were successfully synthesized in the form of single crystals.210 In detail, (R)-39 and (R)-39-SH were obtained by condensing TAM with chiral tetraaldehyde building blocks derived from optically active 1,1′-biphenol-phosphoric/thiophosphoric acid ((R)-BTAP/(R)-(BTAT)), respectively, in the presence of CF3CH2NH2 as the modulator. Structurally, both frameworks crystallize in the chiral tetragonal space group I4. Single crystal COFs were prepared using either a solvothermal method or a modulation strategy, with sizes up to 100 µm (Fig. 24), which enabled structural resolution at 0.90 Å for (R)-39 (Fig. 23a). It crystallizes in the chiral space group I4 with unit cell parameters of a = b = 27.31 Å, c = 23.57 Å and α = β = γ = 90°. (R)-39-SH (Fig. 23b) has nearly identical building units to (R)-39, making them isostructural, with unit cell parameters of a = b = 27.25 Å, c = 23.89 Å and α = β = γ = 90°, achieving a resolution of 1.1 Å.210
 | ||
| Fig. 23 Single crystal chiral 3D COFs. (a) and (b) Structures of (a) (R)-39 and (b) (R)-39-SH. | ||
 | ||
| Fig. 24 Morphologies of single crystal (R)-39 and (R)-39-SH. (a)–(d) Optical microscopy and SEM images of (a) and (b) (R)-39 and (c) and (d) (R)-39-SH. Reprinted with permission from ref. 210. Copyright 2025 American Chemical Society. | ||
These two single crystal CCOFs demonstrated remarkable performance as heterogeneous Brønsted acid catalysts in asymmetric organic transformations, including asymmetric acetalization, transfer hydrogenation of ketimines and anti-selective Mannich reactions. The exceptional catalytic performance is largely attributed to the confined and precisely defined framework environments, which facilitate effective interaction and orientation of reactants at the catalytic sites, thereby ensuring high stereo-control and reactivity. Moreover, the catalysts exhibited remarkable durability and recyclability, retaining high catalytic efficiency and enantioselectivity after multiple cycles of use. The detailed catalytic performance will be presented in Section 5.
Morphological control in single crystal COFs has proven to be difficult due to the complicated nucleation and crystallization processes. By using fluorine-substituted triptycene derivatives with different amounts of aniline modulators, a series of high-quality single crystals of JUC-663-X (X = 30 to 135, the equivalent of aniline) have been synthesized (Fig. 25 and Table 8).211 The reason for using triptycene derivatives is that their aromatic backbones enable the precise directional propagation of networks throughout the crystallization process, thereby facilitating ordered structural formation. However, their low solubility, on the other hand, limits the development of large and high-quality single crystals.
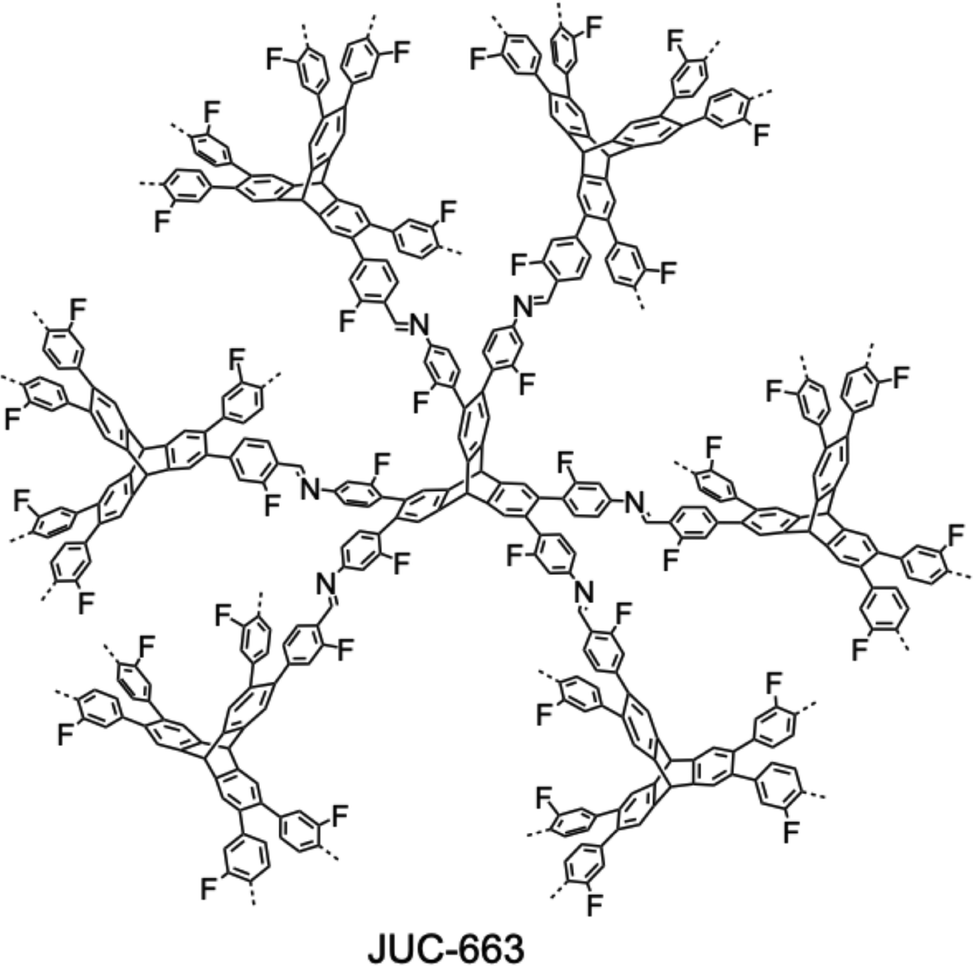 | ||
| Fig. 25 Schematic of JUC-663. | ||
To promote the solubility of triptycene derivatives, fluorine atoms were introduced into the triptycene structure, resulting in two monomers, i.e., 2,3,6,7,14,15-hexa (3′-fluoro-4′-formylphenyl) triptycene (HFFPTP) and 2,3,6,7,14,15-hexa (4′-amino-2′-fluorophenyl) triptycene (HAFPTP), with desirable solubility. Notably, through varying the aniline amount (30 to 130 equiv.), eight single crystals, i.e., JUC-663-30 (Fig. 26a), JUC-663-45 (Fig. 26b), JUC-663-60 (Fig. 26c), JUC-663-75 (Fig. 26d), JUC-663-90(Fig. 26e), JUC-663-105 (Fig. 26f), JUC-663-120 (Fig. 26g) and JUC-663-135 (Fig. 26h), were obtained by using 1,4-dioxane as the solvent and AcOH as the catalyst at room temperature for 5 days.211
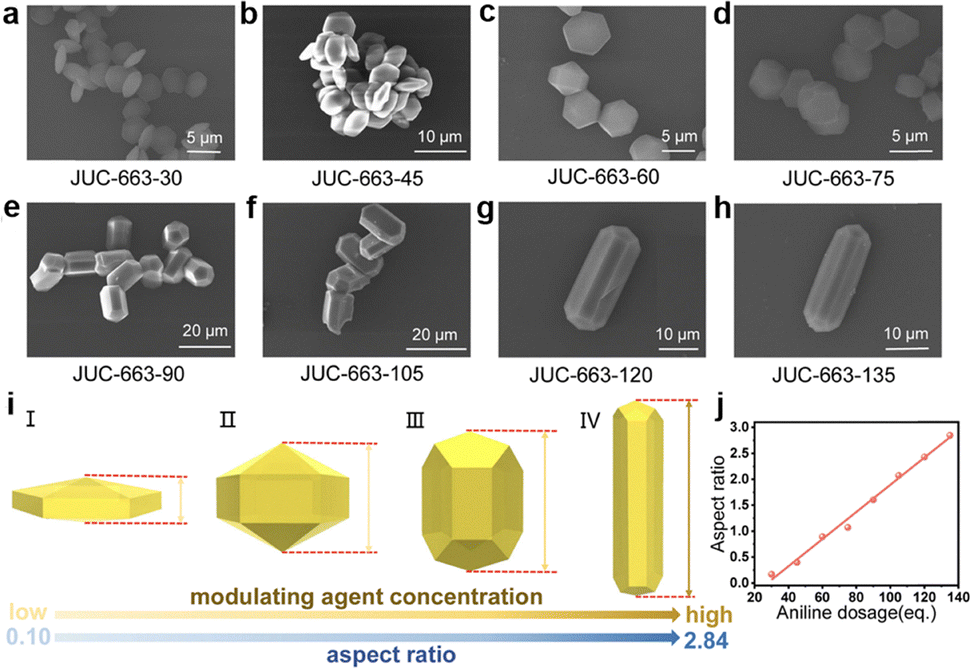 | ||
| Fig. 26 (a)–(h) Morphology tuning of JUC-663 by using different amounts of modulator. (i) Variation in aspect ratios of single crystals upon modulating agent concentration. (j) Correlation between aniline dosage and aspect ratio. Reprinted with permission from ref. 211. Copyright 2024 American Chemical Society. | ||
Notably, increasing the amounts of aniline resulted in notable variations in both crystal morphology and size (Fig. 26i). The aspect ratio of JUC-663-X increased almost linearly from 0.10 to 2.84 upon increasing the aniline dosage from 30 to 135 equiv. (Fig. 26j). SAED analysis with RED technology revealed unit cell parameters of a = b = 32.4119 Å, c = 15.2924 Å, α = β = 90° and γ = 120°. JUC-663-30 exhibited a two-fold interpenetrated acs topology with one mesopore of 2.3 nm and two micropores of 1.0 and 1.5 nm, respectively.
Using aniline below 30 equiv. yields amorphous polymers, while no solid is formed with aniline over 135 equiv. A mechanism study revealed that: (1) at low aniline concentration, the aldehyde groups of HFFPTP were not protected, resulting in high polymerization rates that typically generate amorphous products; (2) increasing the aniline concentration partially protects the aldehyde groups to slow down the reaction rate, thus favouring the growth of single crystals; (3) further increasing to excess amounts of aniline inhibits the polymerization, resulting in no products. This mechanism revealed the role of aniline in controlling the kinetics of single crystal growth.211
The single-crystal forms of 3D sp2c-COFs are fundamentally essential for many applications but have not yet been synthesized due to the irreversible and/or less reversible CC bond formation reactions.8,75,170,212 A post-synthetic modification method via imine-to-olefin transformation provided a feasible route for the synthesis of single-crystal sp2c-COFs (Table 8).188 In detail, single crystal COF-303 (Fig. 7b) was synthesized via condensation of tetrakis(4-formylphenyl) methane (TFM, 10.8 mg, 0.025 mmol) and p-phenylenediamine (PDA, 5.4 mg, 0.05 mmol) in the presence of aniline (0.12 mL) as the modulator and AcOH (6 M, 0.1 mL) as the catalyst at 40 °C for 3 days. The resultant single crystal COF-303 was isolated in yellow powder and was used as parent crystals for structural transformation. Structural analysis by using continuous cRED revealed its single crystal structure at a high resolution of 0.9 Å and the I2d space group, showing unit cell parameters of a = b = 23.87 Å and c = 7.83 Å.
Subsequently, structural transformation was conducted at ambient temperature via linker exchange, where the p-phenylenediamine linker was replaced by 1,2,6-trimethylpyrazin-1-ium iodide with trifluoroacetic acid (TFA) as the catalyst, resulting in single crystal sp2c-linked sc-sp2c-COF-1 (Fig. 27 and Table 8) with a red colour (Fig. 28a and b). This reaction is based on the imine-to-olefin transformation rather than the direct aldol polycondensation, thus slowing down the reaction rate of crystal growth. In this context, TFA is essential to control the hydrolysis rate of the imine bonds and the reconstruction rate of the olefin bonds. The successful linkage transformation was verified by using XRD, FTIR spectroscopy, Raman spectroscopy, ss NMR and XPS spectroscopy. Due to the isostructural crystals, sc-sp2c-COF-1 exhibited the same I2d space group with a structural resolution of 1.1 Å. Structural transformation resulted in crystal shrinkage with reduced unit cell parameters of a = b = 20.4 Å and c = 8.8 Å (Fig. 28c). This post synthetic method provided a robust and feasible protocol for the synthesis of single crystal sp2c-COFs that cannot be completed in a one-pot reaction.188 Notably, this strategy is applicable to the synthesis of another CC-linked 3D COF, i.e. sc-sp2c-COF-2 (Fig. 27), by using the 1,2,6-trimethylpyrazin-1-ium iodide (o-TPI) as the substitution linker. Interestingly, sc-sp2c-COF-2 adopts the same I2d space group with a five-fold interpenetration structure. sc-sp2c-COF-2 exhibits unit cell parameters of a = b = 19.9 Å, c = 8.7 Å and α = β = γ = 90°.
 | ||
| Fig. 27 Single-crystal-to-single-crystal transformation of COF-303 to sc-sp2c-COF-1 and sc-sp2c-COF-2. | ||
 | ||
| Fig. 28 (a) and (b) Colour change of COFs after structural transformation. (c) Crystal unit cell shrinkage after structural transformation. Reprinted with permission from ref. 188. Copyright 2025 Springer Nature. | ||
The fundamental difference between 2D and 3D COFs lies in their bonding and dimensionality. In 2D COFs, 2D layers are formed through covalent bonds within the x–y plane, but these layers undergo supramolecular polymerization along the z-axis driven by noncovalent interactions. The key problem is that this additional z-directional supramolecular polymerization of 2D layers is less correlated with the polycondensation reaction, requiring additional measures to control the z-directional growth into ordered structures. However, it remains difficult to form uniformly ordered structures via supramolecular polymerization, and it is especially difficult to form the same stacking mode across the entire crystals.
Dynamic covalent chemistry facilitates reversible bond formation, allowing for error correction. While this mechanism works efficiently within the x–y plane of the covalent 2D layers, it is less effective for affecting interlayer alignments along the z-axis due to the nature of noncovalent interactions. As a result, defects, such as misaligned layers, are more likely to persist and are hard to correct once formed in 2D COFs. Unlike 3D COFs formed by covalent bonds, 2D COFs involve interlayer interactions, making it difficult to achieve long-range order along the z direction and high-quality single crystals.
Indeed, in 2D COFs, the nucleation process within the x–y plane is rapid due to the high reactivity of building units and efficient covalent bond formation.116,119 However, the growth along the z-axis is much slower and less controlled. This imbalance between fast planar growth and slow vertical stack results in the formation of polycrystalline or amorphous products. The rapid nucleation leads to many small domains, making it challenging to merge them into large defect-free single crystals. In contrast, 3D COFs grow more uniformly because the covalent network extends isotropically, balancing the growth rates across all dimensions. The anisotropic growth nature of 2D COFs makes it difficult to control the crystallization process uniformly. Modulators, such as aniline, decrease polycondensation and nucleation to enhance crystallinity, but their influence is primarily limited to the x-y plane. Controlling the stacking mode along the z-axis remains challenging, as modulators are not directly involved in interlayer interactions.
High-quality crystals of 2D COFs have been synthesized by using acetonitrile as the solvent, aniline as the modulator and benzoic acid as the catalyst (Table 9).213 The structures of TAPPy-PDA-COF (Fig. 29a, Py-1P-COF) and TAPB-DMPDA-COF (Fig. 29b, = TPB-DMTP-COF) are resolved by fast Fourier transform (FFT) at resolutions of 24 Å and 31.5 Å, respectively. At this resolution it is difficult to gain atomic coordinates. However, they improved porosity to achieve BET surface areas of 2600–2640 m2 g−1 and narrow pore size distributions (Fig. 30). The size of crystals is tuneable, ranging from hundreds of nanometres to several micrometres, depending on reaction conditions. High-resolution transmission electron microscopy (HR TEM) and cRED confirmed the single crystal nature in the x–y plane, while the z dimension consists of stacking disorder.213
| COFs | Synthetic conditions | Crystal parameters | Resolution | Method | Ref. |
|---|---|---|---|---|---|
 |
Terephthalaldehyde (TA, 5.4 mg, 0.04 mmol), aniline (41 µL, 0.44 mmol), 1,4-dioxane (0.3 mL) and AcOH (90 µL, 6 M) reacted with 1,3,6,8-tetrakis(4-aminophenyl) pyrene (TAPPy, 10.0 mg, 0.018 mmol) and 1,4-dioxane (0.5 mL) at 65 °C, 2–5µm crystals crystallized out within 1 or 3 months. | a = 3.93 Å, b = 23.39 Å, c = 23.54 Å | 0.76 Å | RED | 176 |
| TA (12 mg, 0.089 mmol) in benzonitrile (0.5 mL), aniline (0.7 M, 3.2 equiv.) in benzonitrile (0.460 mL) and benzoic acid (0.938 g, 7.7 mmol) in benzonitrile (4.54 mL) reacted with TAPPy (28 mg, 0.050 mmol) in benzonitrile (0.5 mL) at 100 °C, 2–4 µm crystals crystallized out within 2 days. | a = 32.55 Å, b = 34.29 Å, c = 3.85 Å | 4.4 Å | RED | 213 | |
| A mixture of TA (5.4 mg, 0.04 mmol) and TAPPy (11.3 mg, 0.02 mmol) was dissolved in n-BuOH (100 µL, 0.25 vol%, as a cosolvent) and AcOH (100 µL), then transferred to a stainless-steel reactor (40 mL). The system was charged with 8 MPa CO2, heated to 80 °C, and reacted for 5 min. After the reaction, the reactor was slowly depressurized at a rate of 1–2 MPa min−1. The precipitate was collected by filtration, washed with acetone and tetrahydrofuran, and dried in a vacuum oven at 100 °C. | NA | NA | SAED | 201 | |
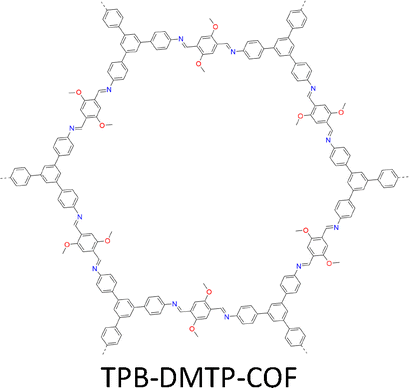 |
2,5-Dimethoxyterephthalaldehyde (DMTA, 11.6 mg, 0.06 mmol), anisole (0.5 mL), aniline (54 µL, 10 equiv.) and AcOH (200 µL) reacted with 1,3,5-tris(4-aminophenyl)benzene (TAPB, 14.0 mg, 0.04 mmol) and anisole (0.5 mL) at 150 °C. | a = b = 64.02 Å, c = 7.2 Å | 1.37 Å | RED | 127 |
| 2,5-Dimethoxyterephthalaldehyde (DMTA, 28 mg, 0.144 mmol) in benzonitrile (0.5 mL), aniline (0.4 mL, 0.7 M, 1.6 equiv.) in benzonitrile and benzoic acid (1.35 g, 11 mmol) in benzonitrile (4.72 mL) were reacted with 1,3,5-tris(4-aminophenyl)benzene (TAPB, 34 mg, 0.096 mmol) in benzonitrile (0.5 mL), and the solution was left to stand at 150 °C; 10–20 µm crystals crystallized out within 2 days. | a = b = 36.36 Å, c = 3.51 Å | 4 Å | RED | 159 |
 | ||
| Fig. 29 Modulator-assisted synthesis of 2D COFs. (a) and (b) Structures of (a) TAPPy-PDA-COF (= Py-1P-COF) and (b) TPB-DMTP-COF (= TAPB-DMPDA-COF). | ||
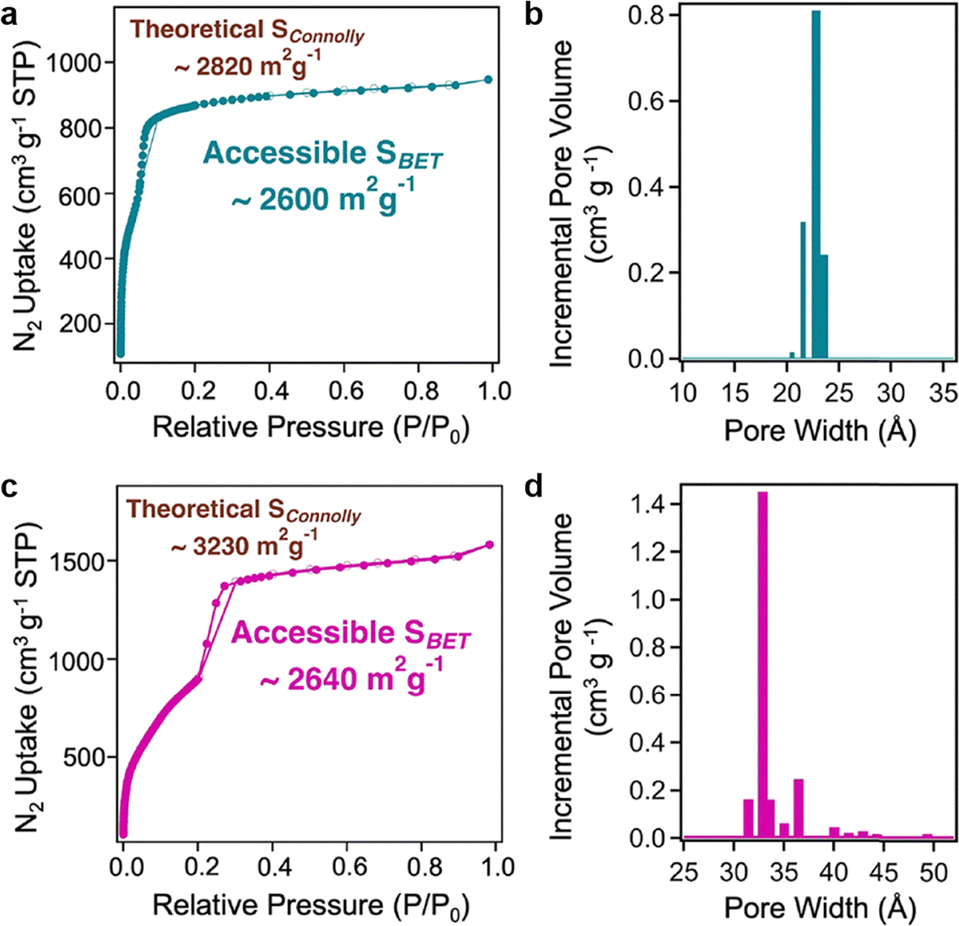 | ||
| Fig. 30 Porosity. (a) N2 sorption isotherm and (b) pore size distribution of single crystal TAPPy-PDA COF. (c) N2 sorption isotherm and (d) pore size distribution of single crystal TAPB-DMPDA COF. Reprinted with permission from ref. 213. Copyright 2022 American Chemical Society. | ||
Crystals of TAPPy-PDA-COF and TAPB-DMPDA-COF demonstrated efficient separation of benzene and cyclohexane – two industrially important compounds with close boiling points. Notably, TAPPy-PDA-COF exhibited reversed adsorption selectivity, preferentially retaining cyclohexane over benzene – a unique behaviour attributed to its high quality and well-defined pores.213 In contrast, polycrystalline samples showed poor or no separation, underscoring the importance of single crystal quality for achieving superior performance. The detailed separation performance will be discussed in Section 5.
By developing an intermediate tracing method, key factors driving the crystallization process, including nucleation and self-healing have been elucidated for the synthesis of single crystal Py-1P-COF (Fig. 29a).176 The combination of 1,3,5-tris(4-aminophenyl)benzene/terephthalaldehyde was used to synthesize a 2D COF (TAPB-TA) as an example for revealing the growth mechanism with mass spectrometry (Fig. 31a). Nucleation growth involving monomer condensation and self-assembly at the initial stage into small crystallite is heavily influenced by monomer concentration and solvent selection. During the self-healing growth stage, amorphous phases are converted into crystalline domains through partial decomposition and reassembly of intermediates, driven by reversible covalent bonding. By using mass spectrometry (MS), soluble growth intermediates in the reaction solution are traced to show the presence of TAPB monomers, intermediate oligomers and their aggregates, where the TAPB monomer exhibits distinct aggregation and self-assembly in different solvents.176 Aggregation is energetically favourable in 1,4-dioxane/mesitylene for crystallization, but unfavourable in methanol, leading to amorphous products (Fig. 31a). The self-assembly of TAPB monomers is energetically favourable in 1,4-dioxane/mesitylene, as revealed by DFT calculations and UV-vis spectroscopy.176 This environment promotes π–π stacking and ordered pre-arrangement of monomers, which reduces their degrees of freedom and directs polymerization toward crystalline COF structures. In contrast, methanol disrupts this self-assembly due to its strong polarity and hydrogen-bonding interactions with monomers, making self-assembly energetically unfavourable. As a result, random condensation occurs, leading to amorphous products.176
 | ||
| Fig. 31 (a) MS spectra of TAPB-TA growth solution using methanol versus 1,4-dioxane/mesitylene mixture as the solvent. (b)–(d) 3D reciprocal lattice of Py-1P reconstructed from the cRED data viewing along the (b) [100], (c) [010] and (d) [001] directions. Reprinted with permission from ref. 176. Copyright 2022 Springer Nature. | ||
These findings revealed that weak interlayer interactions and fast polymerization would result in poorly crystalline or amorphous domains. Density functional theory (DFT) calculations revealed that self-assembly of TPB reduces the degree of freedom of monomers, directing polymerization toward highly ordered structures. Guided by these insights, the researchers successfully grew single crystals of a Schiff-base COF, Py-1P (Fig. 29a), by controlling monomer structure and reaction speed. Using cRED, they resolved the framework's triclinic unit cell with parameters of a = 3.93 Å, b = 23.39 Å, c = 23.53 Å, α = 84.5°, β = 87.1° and γ = 87.1°, and revealed the AA stacking mode and a six-layer stacking arrangement, at a resolution of 0.76 Å (Fig. 31b–d).
The structural insights gained have profound implications for understanding its potential optoelectronic behaviour. The single-crystal structure of Py-1P, resolved for the first time by cRED, confirmed the existence of the long-predicted AA stacking model in a minority (∼10%) of crystals. More significantly, the discovery of a previously unknown, predominant multi-layer stacking structure (Py-1P-6, ∼90% of crystals) with 6 layers per unit cell reveals a much more complex structural landscape than previously assumed.176 This finding is critical because the interlayer stacking arrangement (e.g., AA vs. the complex offset in Py-1P-6) directly governs the through-space electronic coupling between adjacent layers, which is a fundamental factor dictating key optical properties such as exciton migration, energy transfer and ultimately electronic properties. Therefore, this structural disclosure offers more structural insights into the intrinsic optical properties of 2D COFs, which were previously inferred only from polycrystalline or predicted models.176
These results enable the molecular mechanisms that govern 2D COF growth to be revealed, highlighting the interplay of self-assembly, nucleation and self-healing. The findings not only enable the rational design of high-quality single crystals but also broaden the structural and functional versatility, paving the way to advanced applications.176
The scale-up synthesis of high-quality COFs is important for applications. A modulator method has been applied to continuous flow systems for scale-up synthesis of single crystal CF-TAPB-DMPDA and CF-TAPPy-PDA (Fig. 32).158 An optimized condition for continuous flow reaction involves heating the benzonitrile solutions to 90 °C.158 For the hexagonal CF-TAPB-DMPDA COF, a benzoic acid catalyst at 38.4 equivalents and aniline modulator at 1.6 equivalents per aldehyde group were used, with a residence time of 60 seconds in the flow reactor. For CF-TAPPy-PDA COF, the same catalyst loading was employed, while the aniline modulator is 2.0 equivalents to facilitate the formation of well-defined square-shaped particles. After exiting the reactor, both reaction mixtures were cooled at 25 °C for 60 minutes, during which smaller nanoparticles fused into uniform single crystal particles, achieving sizes up to 1.4 µm for CF-TAPB-DMPDA and 1.9 µm for CF-TAPPy-PDA.
 | ||
| Fig. 32 Continuous flow synthesis of single crystal COFs. (a) and (b) Schemes for the synthesis of (a) CF-TAPB-DMPDA COF and (b) CF-TAPPy-PDA COF. (c) and (d) Scanning electron microscopy images of (c) the hexagonal CF-TAPB-DMPDA COF and (d) the rhombic CF-TAPPy-PDA COF. Reprinted with permission from ref. 158. Copyright 2024 Royal Society of Chemistry. | ||
The single-crystal structures of these two COFs have not been resolved, while their high quality was confirmed through multiple complementary techniques, including PXRD data as the most direct structural evidence.158 For CF-TAPB-DMPDA, the PXRD results displayed a sharp primary diffraction peak at 2θ of 2.78°, corresponding to the (100) facet of a primitive hexagonal lattice, along with higher-order reflections that match well with the simulated pattern of an eclipsed stacking structure, indicating long-range order and high crystallinity. Similarly, CF-TAPPy-PDA exhibited a strong (100) diffraction peak at 2θ of 3.66°, consistent with a square planar lattice, also supported by additional higher-order peaks in agreement with simulated diffraction profiles. These sharp, well-defined peaks are absent in amorphous or polycrystalline materials, suggesting high quality crystals. Notably, this method achieves continuous production of single crystal COFs over 1 gram per hour, greatly improving the yield compared to batch synthesis.158
COFs synthesized under continuous flow achieve high BET surface areas exceeding 2000 m2 g−1 with narrow pore size distributions (3.2 nm for CF-TAPB-DMPDA). PXRD and 13C NMR spectroscopy confirmed the structural integrity and crystallinity. A demethylation reaction transforms CF-TAPB-DMPDA into hydroxyl-functionalized CF-TAPB-DHPDA.158 This modification preserves the hexagonal morphology, porosity and structural integrity of the original framework, while introducing solvent-dependent optical properties.
We have developed a methodology for synthesizing single crystal 2D COFs and identified key parameters that govern their crystallization.127 Using the imine-linked TPB-DMTP-COF (Fig. 29b) as a model system, we systematically varied synthetic conditions, including glassware setup, degassing method, solvent choice, temperature, modulator type and amount and reaction time, and found that each plays a critical role in polymerization and crystal formation (Fig. 33). Notably, a homogeneous reaction mixture with all components dissolved, combined with an aniline modulator and a reaction temperature of 50–70 °C, enables the growth of TPB-DMTP-COF single crystals as isolated rod-like structures with tunable diameters (200 nm–3 µm) and lengths (1–20 µm). An important finding is that structural characterization by 3DED revealed two linker conformations, i.e. trans and cis within the 2D polymer sheets, which stack antiparallel to form frameworks with double-sized unit cells. These findings reveal previously overlooked structural features and offer invaluable insights into the synthesis of high-quality single crystal 2D COFs.127
 | ||
| Fig. 33 Synthesis of single crystal TPB-DMTP-COF. Reprinted with permission from ref. 127. Copyright 2024 American Chemical Society. | ||
Moreover, to investigate the effects of solvothermal conditions, we systematically examined reaction temperatures, from room temperature to moderate (50–70 °C) and high (90–120 °C) ranges. We found that high temperatures accelerate polycondensation, giving rise to polycrystalline particles. In contrast, conducting the reaction at moderate temperatures (50–70 °C) allows for better control over polycondensation, supramolecular polymerization and crystallization, enabling the growth of high-quality single crystals.127
Over the past decade of experimenting with various heterogeneous solvothermal conditions for synthesizing TPB-DMTP-COF,78 we found that successful single-crystal growth requires starting from a homogeneous solution in which all components are fully dissolved at an appropriate concentration. To identify suitable solvents for dissolving TPB and DMTP, we tested a wide range of organic solvents, including o-dichlorobenzene (o-DCB), mesitylene, xylene, anisole, tetrahydrofuran (THF), dioxane, dichloromethane, chloroform, acetone, cyclohexane, dimethylformamide (DMF), dimethylacetamide (DMAc), dimethyl sulfoxide (DMSO) and N-methyl-2-pyrrolidone (NMP). Among these, anisole proved to be particularly effective, yielding clear solutions of both TPB and DMTP. Notably, anisole also demonstrated good solubility for intermediates and oligomers, offering a distinct advantage over the commonly used o-DCB/n-butanol solvent system.127
Through the systematic condition screening, we established a reliable protocol for synthesizing single crystals of TPB-DMTP-COF, as outlined below.127 First, DMTA and TAPB were each weighed into separate glass vials, followed by the addition of anisole. Aniline and AcOH were added to the vial containing DMTA. Both vials were sonicated and heated at 60 °C for at least 15 minutes to ensure complete dissolution and equilibration. Next, the two solutions were combined and transferred into a Schlenk tube pre-heated to 60 °C (Fig. 33). The tube was immediately degassed gently for 10 seconds, sealed with a Teflon cap and maintained at 60 °C. The initial reaction mixture appeared as a transparent red solution, with red precipitates beginning to form within 10–15 minutes. After the desired duration, the resulting red, rod-like precipitate was collected by centrifugation, washed via Soxhlet extraction with THF and activated with a supercritical carbon dioxide dryer.
The resulting TPB-DMTP-COF single crystals are readily dispersible in various solvents, such as THF, methanol, acetonitrile (MeCN), and dioxane.127 Optical microscopy revealed well-isolated rod-like crystals, exhibiting greenish-yellow rods up to 20 µm in length (Fig. 34a). Remarkably, these rods displayed high uniformity in both shape and size and were free of entanglement, standing in stark contrast to the irregular and heavily entangled nanoparticle aggregates typically observed in polycrystalline COFs (Fig. 34b and c).127
 | ||
| Fig. 34 Morphology of TPB-DMTP-COF single crystals. (a) OM image. (b) and (c) SEM images. (d)–(f) Cross-polarized OM images under a dark field with the stage rotated to 0, 45 and 90°, respectively. Reprinted with permission from ref. 127. Copyright 2024 American Chemical Society. | ||
The TPB-DMTP-COF microrods exhibit a characteristic type IV N2 adsorption isotherm, indicative of mesoporous materials.127 The BET surface area was measured to be 2730 m2 g−1, comparable to that of polycrystalline counterparts. Analysis using nonlocal density functional theory (NLDFT) revealed a narrow pore size distribution centred at 3.2 nm and a total pore volume of 1.18 cm3 g−1. Notably, the isotherm shows negligible gas uptake at relative pressures (P/P0) above 0.25, suggesting that N2 adsorption arises primarily from the uniform 3.2 nm mesopores.127
The single crystal nature of these microrods was verified using cross-polarized optical microscopy under dark-field conditions, where uniform extinction of light was observed across individual rods as the sample stage was rotated (Fig. 34d–f). FE SEM further revealed their morphology, showing well-defined hexagonal facets (Fig. 34c). The domain sizes exceeded 1 µm in diameter, corresponding to the underlying 2D polymer lattice structure of TPB-DMTP-COF.127
3DED is a state-of-the-art technique for structural analysis of nanosized crystals.138 The collected 3DED data achieved a resolution of 1.37 Å, the highest reported for TPB-DMTP-COF to date, with a data completeness of 91.9%. Despite this high resolution, ab initio structure determination was not feasible. Consequently, plausible structural models were manually constructed to interpret the framework.127
Notably, the reconstructed 3D reciprocal lattice revealed unit cell parameters of a = 65.319 Å, b = 64.959 Å, c = 7.232 Å, α = 90.84°, β = 90.28° and γ = 120.11°, indicating a unit cell that is approximately twice as large as those previously reported, both in the ab plane and along the c-axis stacking direction. Analysis of 2D slices from the 3D reciprocal lattice yielded the reflection conditions: h–h**l: l = 2n; hh**l: l = 2n; 00l: l = 2n. Based on these conditions, two candidate space groups were identified for TPB-DMTP-COF: P6cc (no. 184) and P6/mcc (no. 192).127
By carefully analysing the PXRD pattern of the TPB-DMTP-COF single crystals, we observed a weak diffraction peak at 2θ = 1.64°, which corresponds to the (100) reflection and is consistent with the large unit cell determined by 3DED.53 Furthermore, selected area electron diffraction (SAED) measurements yielded d spacings of d110 = 31.82 Å and d−120 = 31.39 Å, which closely match the theoretical values of d110 = 32.72 Å and d−120 = 32.72 Å calculated from the 3DED-derived unit cell model.127
These findings prompted the re-evaluation of the commonly assumed AA stacking model for 2D COFs (Fig. 35). To explore alternative structural possibilities, four candidate models were constructed based on the unit cell parameters, incorporating varying combinations of trans and cis connectivity between the TPB knot and DMTP linker units (Fig. 35).127 In these models, the in-plane framework exhibits alternating TPB–DMTP connectivity, consistent with the large a-axis parameter, while the layers are arranged in an antiparallel stacking fashion, accounting for the doubled c-axis parameter and accessible pore channels.
 | ||
| Fig. 35 Probable structural models of TPB-DMTP-COF. The trans and cis linker conformations and four different structures of TPB-DMTP-COF. Reprinted with permission from ref. 127. Copyright 2024 American Chemical Society. | ||
Although all four models share the same number of imine bonds, they differ in the spatial arrangement of the framework. Symmetrical analysis revealed that two of the models, AA′-3 and AA′-4, are incompatible with the space groups indicated by the 3DED data. Therefore, only the AA′-1 and AA′-2 models were subjected to further evaluation via least-squares refinement against the 3DED dataset.127 Refinement of the structural model AA′-1 was carried out using SHELXL, based on 3DED data with a resolution of 1.37 Å.53 The refinement successfully converged, yielding an R1 value of 0.331. In contrast, when the AA′-2 model was refined under identical conditions, convergence could not be achieved, and the refinement resulted in a higher R1 value of 0.401. Since refinement evaluates the agreement between calculated and observed structure factors from 3DED, these results indicate that AA′-1 provides a better fit to the experimental data and is therefore the more plausible model.127
The AA′-1 model features two distinct types of pores: one composed of six inward-facing trans linker conformations, and another formed by three pairs of alternating trans and cis linkers.126 The layers stack in an antiparallel fashion through π–π interactions, giving rise to a vertically extended framework. This arrangement accounts for the doubling of the c-axis parameter, a structural detail that was overlooked in previous reports.78,149,192 Additionally, the electrostatic potential map of the refined structure shows localized potential peaks surrounding the atomic positions, further supporting the accuracy and reliability of the AA′-1 structural model (Fig. 35).
To further validate the structural model of TPB-DMTP-COF, Pawley fitting was performed using the AA′-1 model to refine the unit cell parameters. The refinement yielded lattice parameters of a = b = 64.02(9) Å and c = 7.2(6) Å, with convergence to R values of Rp = 0.0667, Rwp = 0.0989 and Rexp = 0.0557.126 To assess the consistency of the structural model, the simulated PXRD pattern using the 3DED-refined atomic structure and the Pawley-refined unit cell were compared with the experimental PXRD data, showing excellent agreement and further supporting the validity of the AA′-1 model.
The observed doubled unit cell and antiparallel stacking structure are not unique to the synthetic method developed in this method.127 Similar PXRD features, including the characteristic peak at 2θ = 1.64°, were also observed in single crystals synthesized via alternative routes, as well as in polycrystalline samples. This suggests that the doubled unit cell and stacking mode may be a common structural motif among 2D COFs. For example, we obtained single crystals of TPB-BPTA-COF, a structural analogue of TPB-DMTP-COF under comparable conditions and identified a corresponding diffraction peak at 2θ = 1.53° in its PXRD pattern.127
This study revealed the key aspects in the synthesis of single crystal 2D COFs.127 (1) The single crystals exhibit well-defined hexagonal rod morphologies, with lengths reaching up to 20 µm and domain sizes extending to 3 µm – markedly distinct from the irregular, aggregated particles typically seen in polycrystalline samples. (2) Electron microscopy reveals that TPB-DMTP-COF exhibits an extended hexagonal lattice featuring uniform, well-ordered pores. (3) The framework adopts both trans and cis linker conformations, resulting in a significantly enlarged unit cell of 64.019 × 64.019 × 7.215 Å, which is approximately twice the dimensions reported for previously characterized single crystals and polycrystalline counterparts. (4) TPB-DMTP-COF favours an antiparallel stacking arrangement, representing a notable departure from the conventional AA stacking mode commonly observed in 2D COFs. (5) The structure of the single crystal was resolved at a resolution of 1.37 Å, the highest achieved for TPB-DMTP-COF to date. Further improvement in resolution remains an important goal for gaining deeper structural insights.127 The findings of trans and cis isomers of the linker sites and their ratio of 1/2 encourage further measures to control 2D covalent layers.
The same phenomenon was observed in the single crystal TPB-DMTP-COF synthesized by different methods and groups.159 The widely accepted imine-linked COF TPB-DMPDA (= TPB-DMTP-COF) featuring hexagonal pores aligned layer by layer is oversimplified. Using ultralow-dose HR TEM, SAXS, 3DED and ssNMR, a dual-pore structure with cis and trans diimine linkages coexisting in a 2/1 ratio forms two distinct types of 6-membered ring pores (Fig. 36). This dual-pore arrangement arises from an AA′-stacking mode where alternating layers are flipped relative to each other, doubling the c-axis of the unit cell. Supported by electron diffraction data and DFT calculations, the AA′ structure with enhanced π–π interactions is thermodynamically more stable than the conventional AA mode. This structural redefinition not only renewed the fundamental understanding of TAPB-DMPDA and related COFs but also has significant implications for their property and function.159
 | ||
| Fig. 36 Validation of the dual-pore AA′-stacking structure of TAPB-DMPDA. (a) Schematic representation of the dual-pore AA′-stacking structure. Each layer features two types of 6-MR pores, resulting from the coexistence of cis and trans conformations of the diimine linkage in a 2:1 ratio. Adjacent layers (i.e., A and A′) are oriented in a flipped arrangement relative to each other. (b) From left to right: Projected structural model, simulated HRTEM image, experimental HRTEM image and electrostatic potential map rendered from 3DED data. (c) Heteronuclear correlation 1H–13C NMR spectrum. The left panel shows the structural fragment of TAPB-DMPDA with two sets of chemically distinct carbon labelled. Reprinted with permission from ref. 159. Copyright 2024 American Chemical Society. | ||
The synthesis of imine-linked single crystal 2D COFs presents a significant step forward in the field of COFs, demonstrating superior structural and functional characteristics compared to polycrystalline counterparts. The innovative approach to achieving high-quality COFs sets a foundation for further exploration of their properties and applications.
Recently, single crystal COF-400 (Fig. 37a) with an unprecedented ABCD interlayer stacking mode was developed.214 The synthesis focuses on regulating both intralayer molecular conformation and interlayer interactions in COF-400, allowing the formation of moiré superstructures in stacked COF films. COF-400 was constructed from tetrakis(4-aminophenyl)ethene (ETTA) and DMTA, in which the two methoxy groups were strategically introduced to enhance interlayer interactions and promote ordered stacking. By carefully controlling the synthetic conditions, single crystals of COF-400 with sizes from 40 to 55 µm were obtained.214
 | ||
| Fig. 37 Structures of (a) COF-400 and (b) 2DPAV-DMP-TPB. | ||
The synthesis of single crystal COF-400 was achieved using a ‘cocktail method’ that combines dynamic covalent chemistry with catalytic acceleration.214 The condensation reaction between ETTA and DMTA was carried out in a solvothermal system containing organic solvents and additives (Table 10). Aniline was used as the modulator to enhance the reversibility of imine bond formation through transimination processes, while Sc(OTf)3 serves as a Lewis acid catalyst to accelerate the reaction kinetics. The combination of these additives significantly improves crystallization efficiency, reducing the crystal growth time from several months to one week, while producing high quality crystals (Fig. 38a).214
| COFs | Synthetic conditions | Crystal parameters | Resolution | Method | Ref. |
|---|---|---|---|---|---|
 |
Tetrakis(4-aminophenyl)ethene (ETTA) (7.2 mg, 0.01 mmol) was dissolved in 1,4-dioxane (200 µL). 2,5-Dimethoxyterephthalaldehyde (BDA-transOMe) (2.7 mg, 0.02 mmol) was dissolved in a mixture of dioxane and trichloromethane (120 µL, 1:2 in vol), respectively. Sc(OTf)3 (2 mg, 0.004 mmol) was dissolved in acetonitrile (26 µL). In a sample vial (4 mL), the BDA solution, aniline (15 µL), the Sc(OTf)3 solution and the ETTA solution were charged sequentially and then mixed by vibration. The reaction was allowed to stand at 30 °C for 5 days, giving spindle shaped orange crystals of COF-400, with a size range from 40 to 55 µm. |
a = 19.32 Å | 0.95 Å | SCXRD | 214 |
| b = 21.19 Å | HR-TEM | ||||
| c = 32.33 Å | |||||
 |
A glass ampoule was charged with 2DPI-DMP-TPB (5 mg), 2,2′-(5′-(4(cyanomethyl)phenyl)-[1,1′:3′,1″-terphenyl]-4,4″-diyl)diacetonitrile (10 mg, 1.2 equiv. per CN bond), Cs2CO3 (50 mg, 8 equiv. per CN bond), and a mixture of DMAc/H2O (0.5/0.1 mL). Afterwards, the mixture was sonicated for 10 minutes at room temperature and the ampoule was degassed by three freeze–pump–thaw cycles, sealed under vacuum, and heated at 130 °C for 10–12 days. After cooling to room temperature, the resulting powder was filtered and sequentially washed with dimethylformamide, H2O, ethanol and acetone. After Soxhlet extraction with acetone for 18 h, the sample was collected and dried under vacuum at 100 °C overnight to give 2DPAV-DMP-TPB (2.6 mg) as a dark-orange solid with a 48% yield. |
a = b = 36.505 Å | NA | HR-TEM | 191 |
| c = 7.094 Å | cRED | ||||
| α = β = 90° | |||||
| γ = 120° |
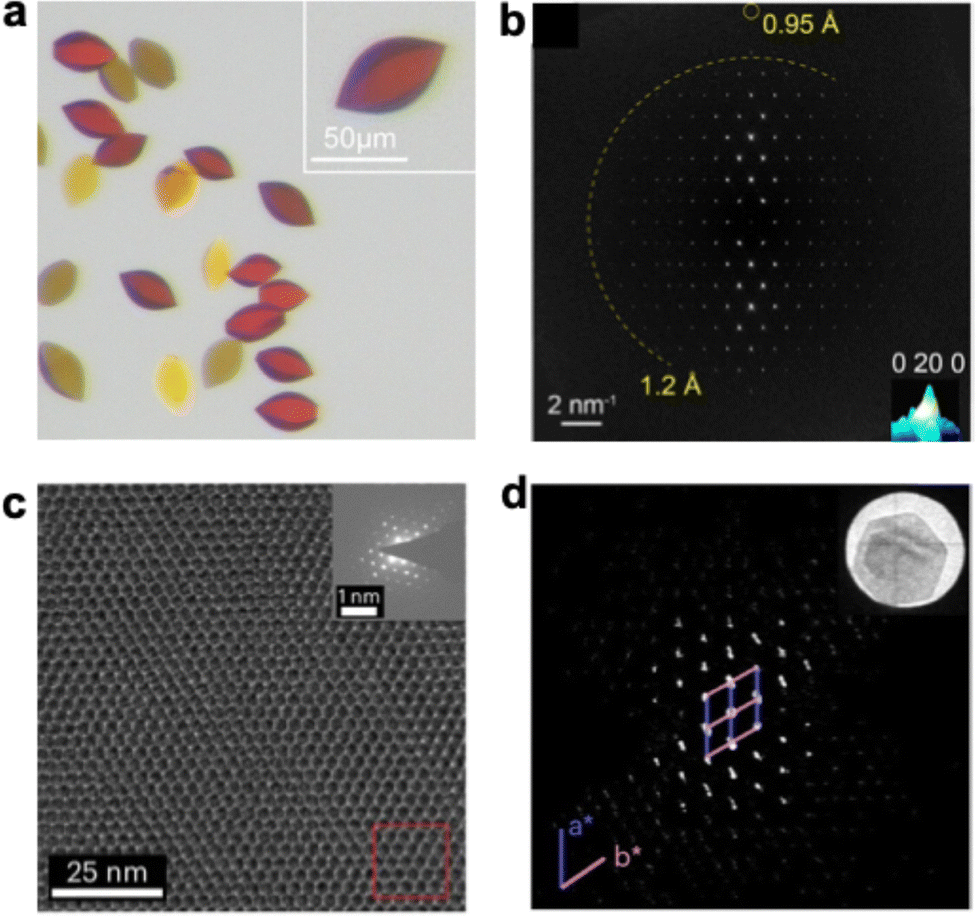 | ||
| Fig. 38 (a) Optical images of COF-400 single crystals and enlarged one single crystal image as the inset. (b) FFT of the raw HRTEM image of COF-400 with an interpretable resolution of 0.95 Å, marked by the 0 20 0 index. (c) Cs-corrected HRTEM image of 2DPAV-DMP-TPB along with the SAED pattern. (d) cRED of a hexagonal 2DPAV-DMP-TPB crystal. (a) and (b) Reprinted with permission from ref. 214. Copyright 2025 American Chemical Society. (c) and (d) Reprinted with permission from ref. 191. Copyright 2025 Springer Nature. | ||
SCXRD analysis revealed that COF-400 forms a layered two-dimensional network with a sql topology and an unusual ABCD stacking sequence along the stacking direction.214 The crystal structure was solved with a resolution as high as 0.95 Å (Fig. 38b), allowing precise determination of all non-hydrogen atoms. Detailed structural analysis revealed multiple possible conformations of the ETTA and DMTA units within the 2D networks. The experimentally determined structure shows ETTA with D2 symmetry, while DMTA adopts D2h symmetry. The imine linkages deviate slightly from the ideal plane of the 2D lattice, generating a waved sheet structure. Moreover, solvent molecules such as DMF occupy the interlayer space and interact with adjacent layers through non-covalent interactions, thereby affecting the interlayer distance and lattice parameters.214
Exfoliation of COF-400 resulted in 2D layers, which can be restacked with various twist angles and then generate moiré patterns analogous to those observed in layered inorganic materials such as twisted graphene. Noticeably, TEM revealed twisted superstructures with angles ranging from 6.4° to 69.7°, and distinct sets of COF lattices could be resolved with a resolution of 1.6 Å. These observations provide direct evidences of interlayer stacking relationships and demonstrate the ability to engineer superlattice structures through controlled stacking of 2D COF layers.214 These results deepen the understanding of interlayer interactions in 2D COFs and demonstrate that precise control of stacking sequences can generate emergent structural phenomena such as moiré superlattices. This not only elucidates the fundamental knowledge of COFs but also provides new opportunities for integrating 2D COF materials into electronic, optoelectronic and device-based applications, where controlled stacking and interlayer coupling are crucial.
In addition to the above bottom-up methods, post synthetic modification of 2S COFs has been explored for the single crystal to single crystal transformation of imine linkages into fully conjugated vinylene bonds.191 A Mannich–elimination strategy converts dynamic imine bonds into irreversible CC linkages, enabling the formation of poly(arylene vinylene) (PAV) 2D COFs while preserving the crystallinity of the parent materials. The design strategy relies on the inherent reversibility of imine-linked COFs, which can be grown as well-ordered crystals, followed by a controlled post-synthetic transformation that generates robust sp2-carbon conjugated frameworks.191
The synthetic procedure begins with the preparation of a single crystal imine-linked precursor COF, i.e. 2DPI-DMP-TPB (= TPB-DMTP-COF, Fig. 29b), obtained via solvothermal imine condensation. 2DPI-DMP-TPB is hexagonal flake-like single crystals with domain sizes exceeding 2 µm. Subsequently, the single crystal 2DPI-DMP-TPB undergoes a Mannich–elimination transformation to produce single crystal 2DPAV-DMP-TPB (Fig. 37b), in which the imine bonds react with activated methylene compounds to produce vinylene linkages by elimination of an amine species from the parent COFs.191 Mechanistic studies via model reactions revealed that the Mannich–elimination process proceeds efficiently in a DMAc/H2O (10:1 v/v) mixture at 120 °C for 8 h in the presence of a base (KOH or Cs2CO3) as the catalyst (Table 10).191
Remarkably, the resulting 2DPAV-DMP-TPB retains the morphology and crystallinity of the parent framework, demonstrating the feasibility of SC–SC transformation for constructing robust new linkage in 2D COFs. The crystal structure of the resulting 2DPAV-DMP-TPB was determined by cRED combined with Rietveld refinement (Fig. 38c). The material adopts a hexagonal lattice with refined unit cell parameters of a = b = 36.505 Å, c = 7.094 Å, α = β = 90° and γ = 120°, corresponding to a P6/mcc space group. The single crystal 2DPAV-DMP-TPB forms a honeycomb 2D polymer network with ordered π-conjugated vinylene linkages, stacked in an AA-serrated stacking arrangement along the c-axis. HRTEM measurements further revealed periodic lattice fringes with a spacing of approximately 3 nm (Fig. 38d), consistent with the simulated honeycomb framework structure.191
These results provide a method for synthesizing highly crystalline and fully π-conjugated 2D COFs, expanding the synthetic toolbox of COF chemistry and opens new opportunities for designing structurally well-defined and robust frameworks for various applications.
4.2. Steric hindrance modulation
Steric hindrance modulation is a method that introduces methyl groups to monomer units to enhance steric hindrance, which affects the solubility, planarity, rigidity, π-conjugation and rotation of monomers, offering a way to regulate the size, quality and crystallinity of COFs.166,180,182 This method has been developed for the synthesis of single crystal 2D and 3D COFs (Table 11).| COFs | Synthetic conditions | Crystal parameters | Resolution | Method | Ref. |
|---|---|---|---|---|---|
 |
TTAB (393 mg, 1 mmol) and aniline (3264 µL, 36 mmol) were dissolved in o-DCB/1,4-dioxane (100 mL, 3/1, by vol) under sonication. TTFB (432 mg, 1 mmol) and benzaldehyde (3642 µL, 36 mmol) were dissolved in another o-DCB/1,4-dioxane (100 mL, 3/1 by vol). The two solvents were mixed in a glass media bottle with a cap, followed by adding Sc(OTf)3 solution in dioxane (1 mL, 14.68 mg mL−1). The reaction was conducted at room temperature without any disturbance for 15 days. | NA | NA | HR TEM/SAED/FFT | 166 |
 |
A vial (5 mL) was charged with TFPB (17.0 mg, 0.026 mmol), aniline (60.0 µL, 25 equiv.) and anhydrous 1,4-dioxane (0.5 mL). The mixture was sonicated until the solid was just dissolved, and an additional portion of aniline (60.0 µL, 25 equiv.) and glacial AcOH (0.4 mL) were successively added into the vial. Immediately, TAPB (15.7 mg, 0.026 mmol) in anhydrous 1,4-dioxane (0.5 mL) was added to the solution. The mixture was left to stand at ambient temperature for crystallization. The light-yellow single crystals of USTB-5 with sizes up to ∼200 µm appeared after 24 h. | a = 17.9558 Å | 1.05 Å | SXRD | 182 |
| b = 17.9558 Å | |||||
| c = 19.8313 Å | |||||
| α = β = 90° | |||||
| γ = 120° | |||||
 |
USTB-5a (50.0 mg, containing 0.2 mmol imine groups) was dispersed in methanol (25.0 mL) and cracked into small pieces. To this mixture while slowly stirring at 0 °C after 5 min, NaBH4 (500.0 mg, 13.22 mmol, 78 equiv.) was added in small portions over a period of 2 h. After slowly stirring at 0 °C for 1 h, the mixture was slowly stirred at room temperature for another 10 h while monitoring with IR spectroscopy. Colourless crystalline solids of USTB-5r were collected by vacuum filtration and washed with water three times and then exchanged with fresh methanol three times in 24 h. | a = 18.2124 Å | 1.13 Å | SXRD | 182 |
| b = 18.2124 Å | |||||
| c = 17.975 Å | |||||
| α = β = 90° | |||||
| γ = 120° | |||||
 |
Before chemical amidation, USTB-5 was exchanged with 1,4-dioxane for 24 h. To a mixture of solvent-exchanged single crystals (ca. 10 mg) and 1,4-dioxane (1.0 mL), 2-methyl-2-butene (361.0 µL, 3.4 mmol), aqueous sodium chlorite solution (100.0 µL, 3.4 M, 0.34 mmol) and glacial AcOH (34.4 µL, 0.6 mmol) were added sequentially. The suspension was slowly stirred at room temperature in the dark for 24 h, then an additional portion of sodium chlorite solution (100.0 µL, 3.4 M, 0.34 mmol) and glacial AcOH (34.4 µL, 0.6 mmol) were slowly and successively added, and the mixture was slowly stirred for another 24 h. Sodium chlorite solution (100.0 µL, 3.4 M, 0.34 mmol) and glacial AcOH (34.4 µL, 0.6 mmol) were slowly and successively added again, and the mixture was slowly stirred for another 24 h. After amidation, the single crystals were directly selected from the reaction mixture for SXRD measurement. In the amidation period, the colour of the single crystals was changed from light yellow to orange red. | a = 31.0280 Å | 0.89 Å | SXRD | 182 |
| b = 31.0280 Å | |||||
| c = 39.8735 Å | |||||
| α = β = 90° | |||||
| γ = 120° | |||||
 |
A Pyrex tube was charged with TMTAPB (17.9 mg, 0.02 mmol), 1,4-phthalaldehyde (8.0 mg, 0.06 mmol), o-DCB (0.8 mL), chlorobenzene (0.2 mL) and AcOH (0.1 mL, 11 M). After being degassed via a freeze–pump–thaw technique three times, the tube was sealed under vacuum and placed in an oven at 120 °C for 3 d. The resulting precipitate was filtered, exhaustively washed by Soxhlet extractions with tetrahydrofuran and dichloromethane for 2 d, and dried at 120 °C under vacuum for 12 h to obtain yellow crystals (10.4 mg, 42% yield). | a = 16.15 Å | NA | cRED | 180 |
| b = 16.15 Å | |||||
| c = 36.93 Å | |||||
| α = β = 90° | |||||
| γ = 120° |
Classical nucleation theory predicts that both the surface energy and characteristic length of nuclei are related to the energetic barrier of nucleation.166 For 2D COFs, the surface energy includes the high-energy reactive group at the edge of the lattice and π–π stacking between layers. To minimize surface energy, introducing three methyl groups to the focal phenyl unit leads to the synthesis of C3-symmetric monomers, i.e. TTAB and TTFB. Unlike monomers without methyl groups, these monomers weaken their planarity, induce an inclined stacking mode to diminish the layer stacking and impede the nucleation, enabling a combinatorial strategy via thermodynamic control over dynamic control on the crystallization process.166 As revealed by monitoring the in-situ light absorption of precursor solutions, the nucleation time is 6 minutes for DiSTAP-1 (Fig. 39a), while monomers of TAPB and TFPB without methyl substituents form nuclei within 1 minute due to the planarity of TAPB and TFPB. In the case of TTAB-TFPB COF with only one nonplanar monomer, the nucleation time is 2 minutes. These results indicate that decreasing the monomer planarity reduces the nucleation rate.155 To prolong the crystallization time, two monofunctional modulators, i.e. aniline and benzaldehyde, have been applied to control polymerization, promoting the formation of more thermodynamically stable structure. Combining the two orthogonal strategies, the polymerization and crystallization processes are finely regulated to form polycrystals, mesocrystals and single crystals (Fig. 40a).166
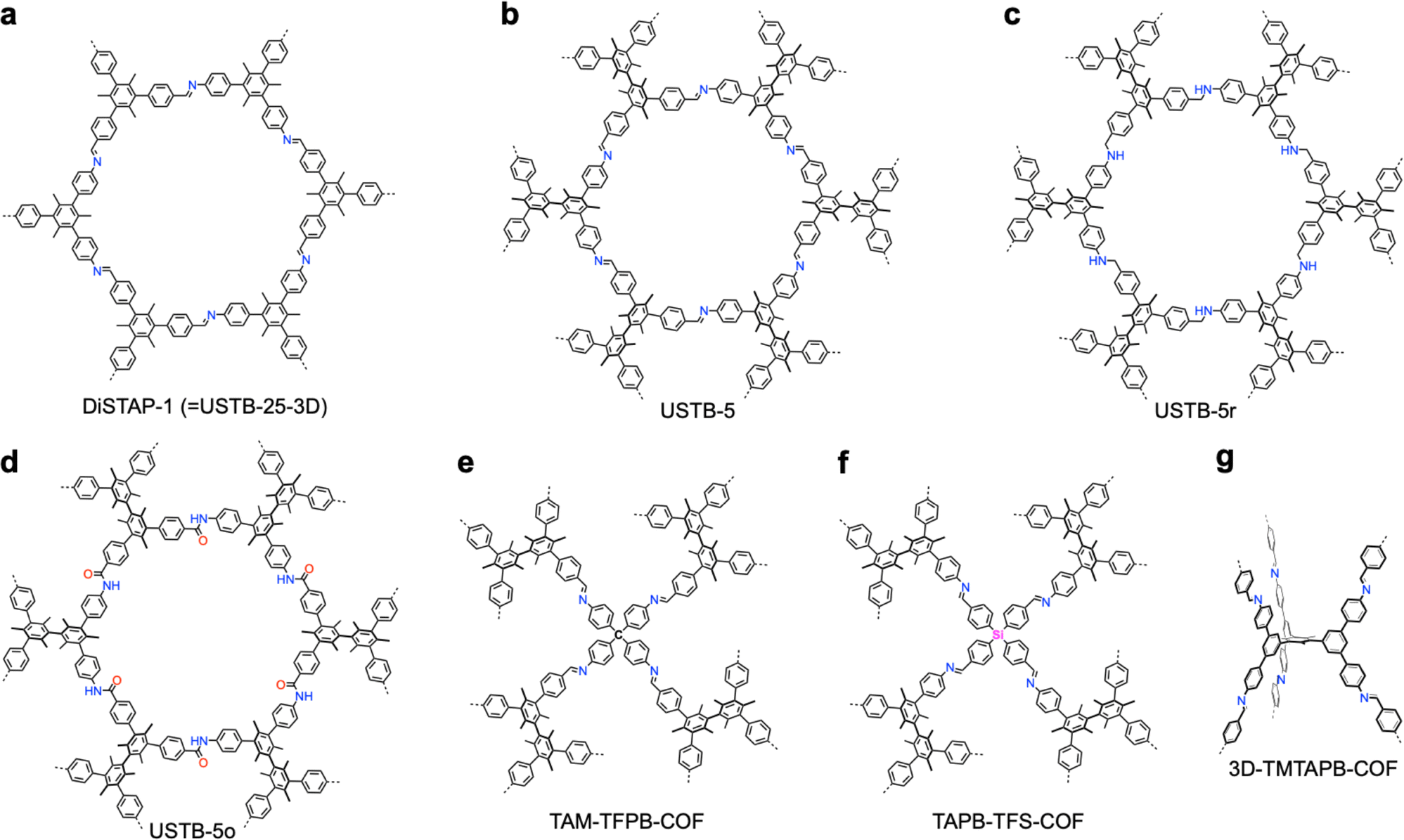 | ||
| Fig. 39 Structures of single-crystal COFs via modulation of monomer steric hindrance. (a)–(g) Structures of (a) DiSTAP-1, (b) USTB-5, (c) USTB-5r, (d) USTB-5o, (e) TAM-TFPB-COF, (f) TAPB-TFS-COF and (g) 3D-TMTAPB-COF. | ||
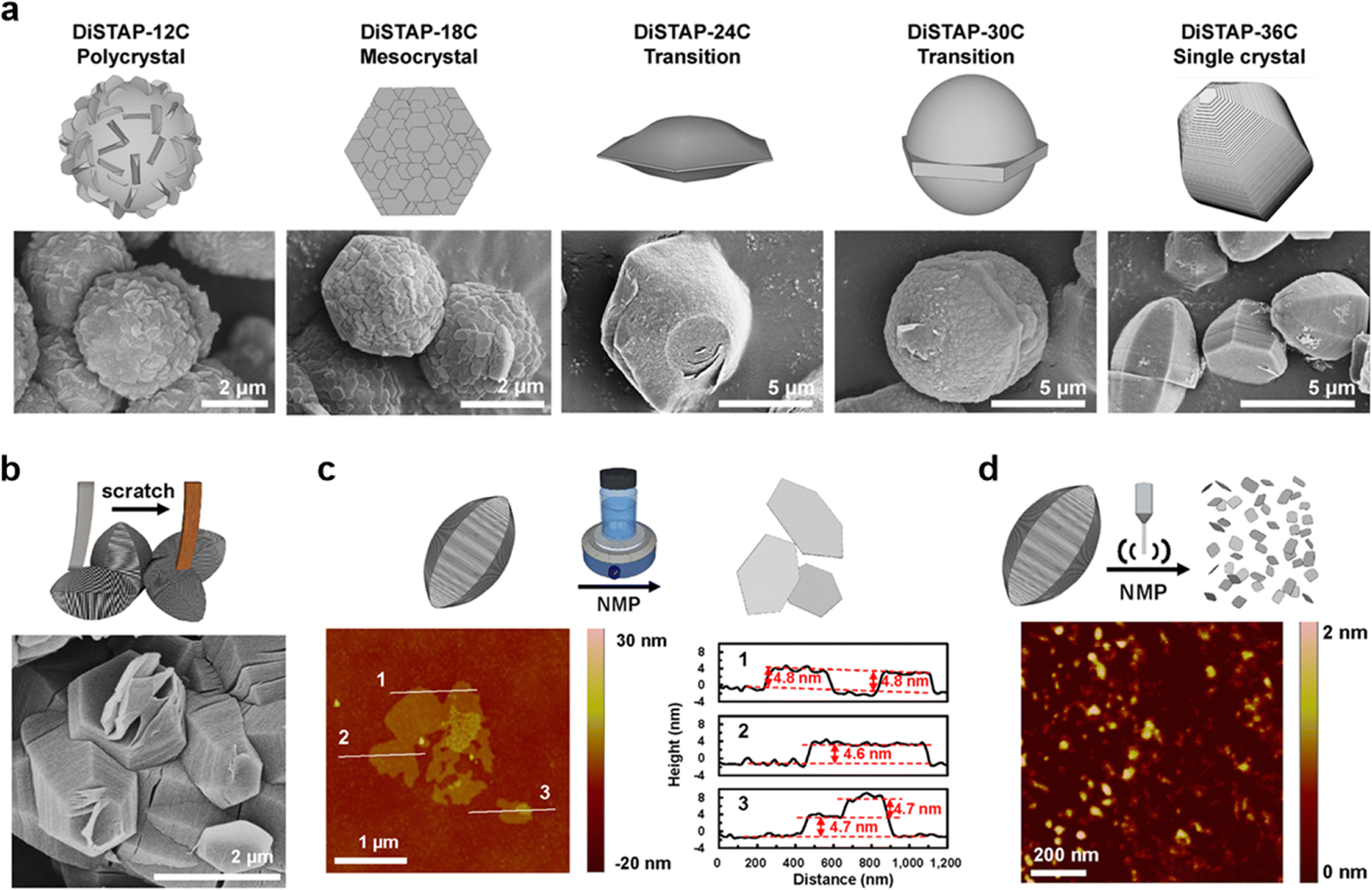 | ||
| Fig. 40 Nucleation and exfoliation of single-crystal DiSTAP-1. (a) Morphological illustrations (top panel) and SEM images (bottom panel) of DiSTAP-1 with different nucleation times. (b) Delamination of single crystal DiDTAP-1 by using a stick. (c) AFM image (left) and height profile (right) of exfoliated single-crystal DiSTAP-1 36C sheets by stirring in NMP for 1 week. (d) AFM image of DiSTAP-1 36C nanoflakes fabricated from deep exfoliation and broken up into single-crystal DiSTAP-1 36C by tip sonication in NMP. Reprinted with permission from ref. 166. Copyright 2023 American Chemical Society. | ||
The single crystal structure of DiSTAP-1 was investigated using HRTEM, optical microscopy with a polarized light source, and SEM, which reveal clear FFT patterns, transparent and regular particle shapes, different but uniform colour variation and regular morphology. SEAD analysis of DiSTAP-1 verifies a clear sixfold diffraction pattern, indicating uniform lattice orientation in the whole particle and the honeycomb-like crystal structures. Interestingly, the twisted monomer inclined stacking structure leads to a weak layer stacking in DiSTAP-1, so that delamination is easily achieved by stick scratching or stirring an N-methyl-2-pyrrolidone (NMP) dispersion of DiSTAP-1 at room temperature for one week (Fig. 40b–d).166 The resultant COF sheets are uniform with a thickness of 4.7 nm and sizes of micrometres (Fig. 40c). Moreover, 1 hour tip sonication (25 W) exfoliates DiSTAP-1 single crystals into single layer nanoflakes with a thickness of 0.6 or 1.3 nm and size of 32 (±15) nm (Fig. 40d). These nanoflakes are biocompatible and pass through the cell membrane of intact living plants, functioning as biocompatible nanocarriers for efficient delivery of essential biomolecular cargos such as genetic elements, agrochemicals and plant hormones (Fig. 40d).166
| COFs | Synthetic conditions | Crystal parameters | Resolution | Method | Ref. |
|---|---|---|---|---|---|
 |
A vial (5 mL) was charged with TTFB (11.3 mg, 0.026 mmol), aniline (0.10 mL, 42 equiv.) and TCM (0.5 mL). The mixture was sonicated until the solid was just dissolved, and AcOH (0.2 mL) was successively added into the vial. Immediately, TTAB (10.2 mg, 0.026 mmol) dissolved in TCM (0.5 mL) was added to this solution. Then the mixture was allowed to stand at ambient temperature for 3 days. A large number of USTB-25-3D single crystals with sizes up to 100 µm were obtained. The as-synthesized USTB-25-3D single crystals were exchanged in TCM, methanol, and acetone, respectively, for 24 h (fresh solvent was changed every eight hours during immersion). Then, the solvent-exchanged sample was filtered and activated for 12 h at 100 °C under vacuum oven, affording activated sample with a yield of 17.8 mg (88%). | a = b = 24.780(4) Å | 1.0 Å | HR 3D ED | 215 |
| c = 26.760(5) Å | |||||
| α = β = 90° | |||||
| γ = 120° | |||||
 |
A vial (20 mL) was charged with TFPB (170.0 mg, 0.26 mmol), aniline (0.6 mL), and anhydrous 1,4-dioxane (5.0 mL). The mixture was sonicated until the solid was just dissolved, and additional solutions of aniline (0.6 mL) and AcOH (2.0 mL) were successively added into the vial. Immediately, TAM (99.0 mg, 0.26 mmol) dissolved in anhydrous 1,4-dioxane (5.0 mL) was added to this solution. The mixture was then allowed to stand at ambient temperature for 15 days. A large number of TAM-TFPB-COF single crystals with sizes up to 50 µm were isolated. The as-synthesized TAM-TFPB-COF single crystals were exchanged in 1,4-dioxane, methanol, and acetone, respectively, for 24 h (fresh solvent was changed every eight hours during immersion). The solvent-exchanged sample was then filtered and activated for 12 h at 100 °C under vacuum, affording the activated sample with a yield of 110.7 mg (56%). | a = b = 23.101 Å | 1.5 Å | 3D ED | 216 |
| c = 36.875 Å | HR-TEM | ||||
| α = β = γ = 90° | Synchrotron PXRD | ||||
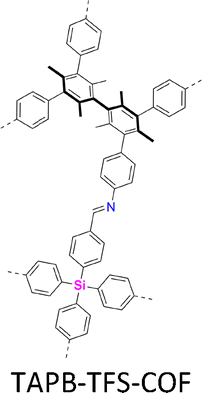 |
A 20 mL vial was charged with TFS (116.6 mg, 0.26 mmol), aniline (1.6 mL), and anhydrous 1,4-dioxane (5.0 mL). The mixture was sonicated until the solid was just dissolved and glacial AcOH (2.0 mL) was successively added into the vial. Immediately, TAPB (156.7 mg, 0.26 mmol) dissolved in anhydrous 1,4-dioxane (5.0 mL) was added to this solution. The mixture was then allowed to stand at ambient temperature for 30 days. A large number of TAPB-TFS-COF single crystals with sizes up to 100 µm were isolated. The as-synthesized TAPB-TFS-COF single crystals were exchanged in 1,4-dioxane, methanol, and acetone, respectively, for 24 h (fresh solvent was changed every eight hours during immersion). The solvent-exchanged sample was then filtered and activated for 12 h at 100 °C under dynamic vacuum, affording an activated sample with a yield of 87.9 mg (44%). | a = b = 25.412 Å | 1.7 Å | 3D ED | 216 |
| c = 32.766 Å | HR-TEM | ||||
| α = β = γ = 90° | Synchrotron PXRD | ||||
 |
A Pyrex tube was charged with TAPB-Me (17.9 mg, 0.02 mmol), 1,3,5-benzenetricarboxaldehyde (6.5 mg, 0.04 mmol), o-DCB (0.1 mL), chloroform (0.9 mL), aniline (10 µL) and 11 M aqueous AcOH (0.1 mL). After being degassed by a freeze–pump–thaw technique three times and sealed under vacuum, the tube was placed in an oven at 100 °C for 7 d. The resulting precipitate was filtered, exhaustively washed by Soxhlet extractions with tetrahydrofuran and dichloromethane for 2 days, and dried at 90 °C under vacuum for 12 h. The 3D-TFB-COF-Me was isolated as a light-yellow powder (13.6 mg, 61% yield). | a = 19.838 Å | 1.05 Å | cRED | 181 |
| b = 32.089 Å | |||||
| c = 27.530 Å | |||||
 |
A Pyrex tube was charged with TAPB-Et (18.7 mg, 0.02 mmol), 1,3,5-benzenetricarboxaldehyde (6.5 mg, 0.04 mmol), o-DCB (0.2 mL), chloroform (0.8 mL), aniline (10 µL) and aqueous AcOH (0.1 mL, 11 M). After being degassed by a freeze–pump–thaw technique three times and sealed under vacuum, the tube was placed in an oven at 110 °C for 3 days. The resulting precipitate was filtered, exhaustively washed by Soxhlet extractions with tetrahydrofuran and dichloromethane for 2 d, and dried at 90 °C under vacuum for 12 h. The 3D-TFB-COF-Et was isolated as a light-yellow powder (16.9 mg, 73% yield). | a = 19.857 Å | 1.40 Å | cRED | 181 |
| b = 32.164 Å | |||||
| c = 27.313 Å |
High-resolution 3D ED (Fig. 42d) collected at 1.0 Å revealed its single crystal structure with unit cell parameters of a = b = 25.197 Å, c = 26.455 Å, α = β = 90° and γ = 120° (Table 12). USTB-25-3D exhibits 1.13 nm micropores with a high surface area of 3039 m2 g−1 and pore volume of 1.16 cm3 g−1. The high surface area and narrow pore size enables USTB-25-3D with potential application in gas adsorption and separation at low pressure. By constructing the first single crystal 3D COF with three crossing sets of parallel layers, this work establishes inclined entanglement as a valuable approach to promote 3D covalent structural formation.215
Indeed, introducing steric hindrance to monomers induces largely twisted conformations, which leads to the formation of 3D COFs.182 For example, condensing 3,3′,5,5′-tetrakis(4-aminophenyl)bimesityl (TAPB) with 3,3′,5,5′-tetrakis(4-formylphenyl)bimesityl (TFPB) forms single crystal 3D COFs, due to the steric hindrance of the six methyl groups, which twisted the monomer from planar to tetrahedral configurations.182 Polycondensation of TAPB (15.7 mg, 0.026 mmol) and TFPB (17.0 mg, 0.026 mmol) with aniline as the modulator (50 equivalent) and glacial AcOH (0.4 mL) as the catalyst in 1,4-dioxane (1 mL) forms block-shaped single crystals of USTB-5 (Fig. 39b) with sizes up to 200 µm after 24 h at room temperature. More importantly, a gram-scale production of large-sized crystals up to 450 µm is achieved within 5 days (Fig. 41a), demonstrating the high efficiency and good reliability of the USTB-5 synthesis.
 | ||
| Fig. 41 (a) Optical microscopy image of USTB-5 from a 5 day reaction. (b) Temperature-dependent unit cell parameters of USTB-5. (c) Conversion among COFs and their optical microscopy photos. (d) N2 adsorption curves of USTB-5, USTB-5r and USTB-5o. (e) Optical microscopy image of 3D-TMTAPB-COF. (f) 3D reciprocal lattice of 3D-TMTAPB-COF, the reflection conditions can be obtained as hhl, l = 2n, h–hl, l = 2n, 00l, and l = 2n. (a)–(d) were reprinted with permission from ref. 182. Copyright 2023 Springer Nature. (e) and (f) were reprinted with permission from ref. 180. Copyright 2023 American Chemical Society. | ||
SCXRD resolved the single crystal structure of USTB-5 with a resolution of 1.05 Å, showing a P6222 space group with unit cell parameters of a = 17.9558 Å, b = 17.9558 Å, c = 19.8313 Å, α = β = 90° and γ = 120°.182
Notably, USTB-5 exhibits two kinds of helices along the c axis, which are linked by imine bonds to generate complicated helical structures with unique chirality. As a result, an open helical tube with a pore size of 9.8 Å is formed through waving the double-strand helices, which exhibit a six-fold screw axis and a pitch of 39.66 Å. These structures provide intrinsic flexibility for USTB-5. Interestingly, SCXRD analysis at variable temperatures from 100 K to 300 K revealed the flexible structures of USTB-5, showing a thermal-induced unit cell expansion up to +263 Å3 (+5%) (Fig. 41b).182 In the temperature range of 100–200 K, the a/b axes exhibited a negative thermal expansion of −3%, while the c axis showed a positive thermal expansion of +10% (Fig. 41b). Accordingly, the linear thermal expansion coefficients along the a and c axes are αa = −82 × 10−6 K−1 and αc = +491 × 10−6 K−1, respectively. Noticeably, a larger anisotropic thermal expansion occurred along the c axis over the ab plane, due to the weak van der Waals forces among the three-fold interpenetrated nets in flexible porous USTB-5 along the c axis.182
Post-synthetic modification of single crystal USTB-5 by chemically reducing with NaBH4 or oxidating with NaClO2 transforms the imine linkage into amine and amide bonds, respectively (Fig. 41c).182 The resultant amine-linked USTB-5r (Fig. 39c) and amide-linked USTB-5o (Fig. 39d) present isoreticular frameworks, similar pore sizes, and the same topology as USTB-5. The imine-linked single crystals exhibit a stepwise N2 adsorption behaviour with a BET surface area of 1465 m2 g−1, pore size of 9.3 Å and a pore volume of 1.2 cm3 g−1 (Fig. 41d). In comparison, the amine-linked single crystal USTB-5r shows a type I adsorption curve with a BET surface area of 1149 m2 g−1, a pore size of 9.8 Å and a pore volume of 0.64 cm3 g−1, while the amide-linked single crystal USTB-5o exhibits a BET surface area of 1623 m2 g−1, a pore size of 8.6 Å and a pore volume of 1.26 cm3 g−1 (Fig. 41d).
Single crystal TAM-TFPB-COF and TAPB-TFS-COF (Fig. 39e) and TAPB-TFS-COF (Fig. 39f) were synthesized by condensing TAM (99.0 mg) with TFPB (170.0 mg), and TAPB (156.7 mg) with TFS (116.6 mg), respectively, in 1,4-dioxane containing aniline and AcOH at room temperature (Table 12).216 The single crystal structure of TAM-TFPB-COF was resolved by single crystal 3D ED at a high resolution of 1.5 Å (Fig. 42b and e), showing the unit cell parameters of a = b = 23.15 Å, c = 36.30 Å and V = 19457.3 Å3. In comparison, the single crystal structure of TAPB-TFS-COF was determined using synchrotron SCXRD at 100 K with a resolution of 1.7 Å. It shows the unit cell parameters of a = b = 25.53 Å, c = 32.69 Å and V = 21306.7 Å3.216 Notably, the two single crystals exhibit three-fold interpenetrated dia-b networks with crystal size ranging from 50 to 100 µm. Interestingly, replacing the central carbon atom in the TAM-TFPB-COF with a silicon atom, the resultant TAPB-TFS-COF presented 10.3% expansion in a/b axes and 9.9% contraction in the c axis. As a result, TAM-TFPB-COF and TAPB-TFS-COF showed a BET surface area of 3533 and 4107 m2 g−1, respectively, representing the highest so far recorded for imine-based COFs based on N2 sorption measurements at 77 K.216
 | ||
| Fig. 42 (a)–(c) SEM images of (a) USTB-25-3D, (b) TAM-TFPB-COF and (c) 3D-TFB-COF-Me. (d)–(f) Reconstructed 3D reciprocal lattice of (d) USTB-25-3D, (e) TAM-TFPB-COF and (f) 3D-TFB-COF-Me. (a) and (d) Reprinted with permission from ref. 215. Copyright 2024 Cell press. (b) and (e) Reprinted with permission from ref. 216. American Chemical Society. (c) and (f) Reprinted with permission from ref. 181. Copyright 2024 American Association for the Advancement of Science. Reprinted with permission from ref. 175. Copyright 2020 Wiley-VCH. | ||
Designing high-valency knots (>4 reaction sites) to expand the library of 3D building blocks is important for expanding the structural diversity of 3D COFs. A 6-connected 3D knot, i.e. 1,3,5-trimehyl-2,4,6-tri[3,5-di(4-aminophenyl-1-yl)phenyl-1-yl] benzene (TMTAPB), adopts a tetrahedral conformation as a result of steric hindrance-induced twisting.180 This is supported by the condensation of 2,4,6-tri[3,5-di(4-aminophenyl-1-yl)-phenyl-1-yl]benzene (TAPB) as a protype of the triangular prism node with 1,4-phthalaldehyde, which forms 2D-TAPB-COF with hcb topology and an eclipsed AA stacking mode, due to the planar conformation of the 6-connected TAPB knot.180 Subsequently, increasing the intramolecular steric hindrance by introducing a methyl group into TAPB develops TMTAPB with nonplanar a triangular prism conformation. As a result, condensing TMTAPB and 1,4-phthalaldehyde in a mixture of chlorobenzene and o-DCB at 120 °C for 3 days forms 3D-TMTAPB-COF (Fig. 39g). The single crystal structure of 3D-TMTAPB-COF has not been resolved, while high quality crystals are confirmed by optical microscopy and continuous rotational electron diffraction (cRED) (Fig. 41e and f). Noticeably, 3D-TMTAPB-COF adopts a rare 6-fold interpenetrated (Class IIIa interpenetration) acs topology. The BET surface area is 940 m2 g−1 and the pore size is 0.84 nm, as revealed by the N2 sorption isotherm. Notably, the microporous structures of 3D-TMTAPB-COF make it highly promising for separation applications, showing excellent performance over its 2D analogue, i.e. 2D-TAPB-COF.180
Notably, the twisted TMTAPB (= TAPB-R, R = Me) with spatial conformation allows a different [Td + C3] topological combination to generate two isostructural 3D-TFB-COF-Me (R = Me) (Fig. 43a) and 3D-TFB-COF-Et (R = Et) (Fig. 43b) with an ultrahigh BET surface area for methane storage.181
 | ||
| Fig. 43 Structures of single crystal (a) 3D-TFB-COF-Me and (b) 3D-TFB-COF-Et. | ||
Single crystal 3D-TFB-COF-Me was synthesized via the imine condensation between TAPB-Me (17.9 mg, 0.02 mmol) and 1,3,5-benzenetricarboxaldehyde (TFB, 6.5 mg, 0.04 mmol) in the mixed solvent of o-DCB/chloroform (0.1/0.9 mL), in the presence of an aniline modulator (10 µL) and aqueous AcOH catalyst (11 M, 0.1 mL) (Table 12).181 After a 7 day reaction at 100 °C, 3D-TFB-COF-Me was isolated as a light-yellow powder with uniform star-shaped morphology (Fig. 42c). Single crystal 3D-TFB-COF-Et was obtained by condensing TAPB-Et with TFB via a similar procedure. The single crystal structures of 3D-TFB-COF-Me and 3D-TFB-COF-Et were determined by cRED with high resolutions of 1.05 and 1.40 Å (Fig. 42f), respectively. 3D-TFB-COF-Me exhibited the space group of Ccc2, while Rietveld refinements generated lattice unit cell parameters of a = 19.838 Å, b = 32.089 Å and c = 27.530 Å. 3D-TFB-COF-Et also showed the same space group of Ccc2, with unit cell parameters of a = 19.857 Å, b = 32.164 Å and c = 27.313 Å.181
Noteworthily, 3D-TFB-COF-Me and 3D-TFB-COF-Et exhibit a rare self-catenated alb-3,6-Ccc2 topology, which enables both small pore sizes and ultrahigh surface areas.181 This is because self-pillaring avoids the close packing of different nets in an interpenetration topology. As a result, 3D-TFB-COF-Me and 3D-TFB-COF-Et present a recorded BET surface area of 4298 and 4502 m2 g−1, respectively. Additionally, both COFs show a narrow pore size of 1.1 nm, which is consistent with the values calculated from the crystal structures (1.1 nm).181 The pore volumes were calculated to be 1.83 and 1.86 cm3 g−1 for 3D-TFB-COF-Me and 3D-TFB-COF-Et, respectively. The high surface areas and microporous structures enable efficient gas storage, which will be detailed in Section 5.3. Using steric hindrance to design novel 3D nodes enables the creation of ultrahigh surface area and microporous COFs suitable for molecule storage.
4.2. Surfactant-assisted polymerization
Surfactant-assisted synthesis of single crystal COFs involves using surfactants to control the polymerization and crystallization processes. This method deploys surfactants as structure-directing agents by modulating self-assembly of building blocks and crystallization of COFs, stabilizing intermediate phases and reducing the nucleation rate, thereby enabling controlled growth of large-size single crystals.167,175,217–219 The surfactant-assisted synthesis presents a step forward in achieving single crystal COFs (Tables 13 and 14).| COFs | Synthetic conditions | Crystal parameters | Resolution | Method | Ref. |
|---|---|---|---|---|---|
 |
Sodium oleyl sulfate (SOS, 1 mg) was dissolved in chloroform (1 mL). 5,10,15,20-(Tetra-4-dihydroxyborylphenyl)porphyrin (monomer 1, 0.790 mg, 1 µmol) was dissolved in HCl (1 mL, 1 M). 1,2,4,5-Tetrahydroxybenzene (monomer 2, 0.567 mg, 4 µmol) was dissolved in HCl (1 mL, 1 M) and sonicated for 5 min. Polymerization reaction: SOS/chloroform solution (20 µL) was spread onto the surface of Milli-Q water (40 mL) in a beaker (60 mL). The chloroform solvent was allowed to evaporate from the water surface for 10 min, then a solution of monomer 1 (1 mL) was injected into the water phase. After 30 min, a solution of monomer 2 (1 mL) was injected into the water phase. The beaker was left into an oven at 50 °C for 7 days. | a = 24.90 Å | NA | HR TEM/SAED | 175 |
| b = 24.90 Å | |||||
| c = 7.6 Å | |||||
| α = β = γ = 90° | |||||
 |
Sodium oleyl sulfate (SOS, 1 mg) was dissolved in chloroform (1 mL). 5,10,15,20-(Tetra-4-dihydroxyborylphenyl)porphyrin (monomer 1, 0.790 mg, 1 µmol) was dissolved in HCl (1 mL, 1 M). 2,3,6,7-Tetrahydroxynaphthalene (monomer 3, 0.767 mg, 4 µmol) was dissolved in HCl (1 mL, 1 M) and sonicated for 5 min. Polymerization reaction: SOS/chloroform solution (20 µL) was spread onto the surface of Milli-Q water (40 mL) in a beaker (60 mL). The chloroform solvent was allowed to evaporate from the water surface for 10 min, then a solution of monomer 1 (1 mL) was injected into the water phase. After 30 min, a solution of monomer 3 (1 mL) was injected into the water phase. The beaker was left in an oven at 50 °C for 7 days. | a = 27.32 Å | NA | HR TEM/SAED | 175 |
| b = 27.32 Å | |||||
| c = 7.6 Å | |||||
| α = β = γ = 90° | |||||
 |
Milli-Q water (20 mL) was added to the Petri dish, then a solution of surfactants (20 µL, 1 mg mL−1) was spread onto the water surface. After waiting for 30 minutes, a diluted hydrochloric acid (400 µL, 0.001 mol mL−1) solution of 4,4′,4″,4‴-(porphyrin-5,10,15,20-tetrayl)tetraaniline (TAPP, 1 mg mL−1) was added to the water subphase. Ater waiting for another half an hour, an aqueous solution of 4,4′-(1,4-phenylene) bis (1-(2,4-dinitrophenyl) pyridilium) dichloride (600 µL, 1 mg mL−1, 1.5 equiv.) was added to the water subphase. The Petri dish was then sealed with a glass cover and was left undisturbed during the reaction. | a = 20.5 Å | 5 Å | HR TEM/SAED/FFT | 217 |
| b = 15.5 Å | |||||
| c = 3.95 Å | |||||
| α = β = γ = 90° |
| COFs | Synthetic conditions | Crystal parameters | Resolution | Method | Ref. |
|---|---|---|---|---|---|
 |
Water (26 mL) or ethanol/water mixtures (26 mL) with different volume ratios (1 vol%, 7.7 vol%, 15.4 vol%) were added into a Petri dish with a diameter of 56 mm and a height of 16 mm. An aqueous solution of PSS (60 µL, 1 mg mL−1) was spread at the air–water interface and left undisturbed for 30 minutes. A solution of TAPP protonated with PTSA (200 µL, 0.3 µmol) was added into the Petri dish and left undisturbed for 60 minutes. Then, an aqueous solution of aldehyde monomer (2,5-Ph, 3.69 mL, 1.8 µmol) was slowly injected into the container. 2D COF films were obtained after one week. | a = 25.0 Å | 3 Å | AC-HR TEM | 218 |
| c = 3.9 Å | SAED | ||||
| FFT | |||||
| Water (25 mL) was added to a Petri dish (diameter of 60 mm and height of 16 mm). An aqueous solution of polyacrylic acid (60 µL, 1 mg mL−1) was spread at the air–water interface and left undisturbed for 30 min. A solution of node (1 mg mL−1, 200 µL) protonated with PTSA (2 mol L−1) was added to the Petri dish and left undisturbed for 1 h. Next, we added diethylenetriamine aqueous solution (100 L, 1 mg mL−1) into the Petri dish and waited another hour. Then a terephthalaldehyde ethylene glycol solution (400 µL, 0.738 mg mL−1) was slowly injected into the vessel, and the Petri dish was sealed with a glass cover. 2D COF-1 films were obtained after 18 h of storage at room temperature. | a = 51.78 Å | NA | AC-HR TEM | 219 | |
| b = 51.73 Å | SAED | ||||
| c = 6.25 Å | FFT | ||||
| α = β = γ = 90° | |||||
 |
Water (26 mL) or ethanol/water mixtures (26 mL) with different volume ratios (1 vol%, 7.7 vol%, 15.4 vol%) were added into a Petri dish with a diameter of 56 mm and a height of 16 mm. An aqueous solution of PSS (60 µL,1 mg mL−1) was spread at the air–water interface and left undisturbed for 30 minutes. A solution of TAPP protonated with (200 µL, 0.3 µmol) was added to the Petri dish and left undisturbed for 60 minutes. Then, an aqueous solution of aldehyde monomer (2,3-Ph, 3.69 mL, 1.8 µmol) was slowly injected into the container. 2D COF films were obtained after one week. | a = 25.0 Å | 3 Å | AC-HR TEM | 218 |
| c = 3.9 Å | SAED | ||||
| FFT | |||||
 |
Water (26 mL) or ethanol/water mixtures (26 mL) with different volume ratios (1 vol%, 7.7 vol%, 15.4 vol%) were added to a Petri dish with a diameter of 56 mm and a height of 16 mm. An aqueous solution of PSS (60 µL, 1 mg mL−1) was spread at the air–water interface and left undisturbed for 30 minutes. A solution of TAPP protonated with (200 µL, 0.3 µmol) was added to the Petri dish and left undisturbed for 60 minutes. Then, an aqueous solution of aldehyde monomers (BPy, 3.72 mL, 1.8 µmol) was slowly injected into the container. 2D COF films were obtained after one week. | a = 29.0 Å | 3 Å | AC-HR TEM | 218 |
| c = 3.9 Å | SAED | ||||
| FFT | |||||
 |
Water (26 mL) or ethanol/water mixtures (26 mL) with different volume ratios (1 vol%, 7.7 vol%, 15.4 vol%) were added to a Petri dish with a diameter of 56 mm and a height of 16 mm. An aqueous solution of PSS (60 µL, 1 mg mL−1) was spread at the air–water interface and left undisturbed for 30 minutes. A solution of TAPP protonated with PTSA (200 µL, 0.3 µmol) was added to the Petri dish and left undisturbed for 60 minutes. Then, an aqueous solution of aldehyde monomers (BPh, 3.72 mL, 1.8 µmol) was slowly injected into the container. 2D COF films were obtained after one week. | a = 29.0 Å | 3 Å | AC-HR TEM | 218 |
| c = 3.8 Å | SAED | ||||
| FFT | |||||
 |
Water (25 mL) was added to a Petri dish (diameter of 60 mm and height of 16 mm). An aqueous solution of polyacrylic acid (60 µL, 1 mg mL−1) was spread at the air–water interface and left undisturbed for 30 min. A solution of node (1 mg mL−1, 200 µL) protonated with PTSA (2 mol L−1) was added to the Petri dish and left undisturbed for 1 h. Next, we added diethylenetriamine aqueous solution (100 µL, 1 mg mL−1) into the Petri dish and waited another hour. Then BPh ethylene glycol solution (400 µL, 1 mg mL−1) was slowly injected into the vessel, and the Petri dish was sealed with a glass cover. 2D COF-2 films were obtained after 18 h of storage at room temperature. | NA | NA | AC-HR TEM | 219 |
| SAED | |||||
| FFT |
 | ||
| Fig. 44 Synthetic protocol for single-crystalline 2D COF films using the SMAIS method. | ||
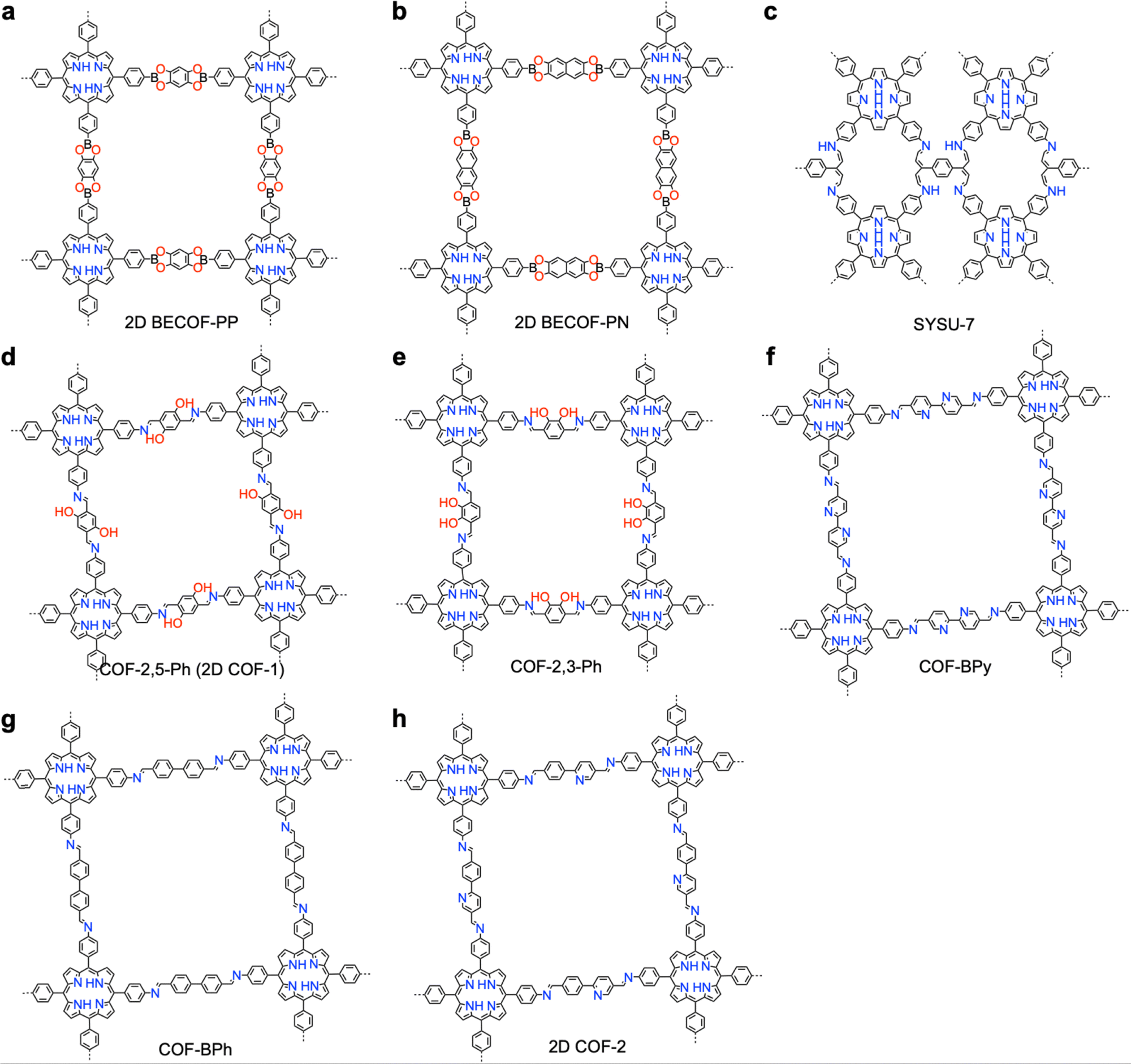 | ||
| Fig. 45 Surfactant-assisted synthesis of single crystal 2D COFs. (a)–(h) Structures of (a) 2D BECOF-PP, (b) 2D BECOF-PN, (c) SYSU-7, (d) COF-2,5-Ph (= 2D COF-1), (e) COF-2,3-Ph, (f) COF-BPy, (g) COF-BPh and (h) 2D COF-2. | ||
The resultant films of boronate ester-linked 2D BECOF-PP (Fig. 46a) have tuneable thicknesses from 6 to 16 nm and an average size of 10.3 ± 3.7 µm, as revealed by AFM (Fig. 46b).175 The single crystal nature of the COF films was characterized by HRTEM and SAED patterns. However, these small single crystal domains are not resolvable to gain atomic coordinates. In addition to SOS, sodium dodecylbenzene sulfonate produces crystalline films, while systems with other cationic, zwitterionic and nonionic surfactants or without surfactants result in only amorphous films.175
 | ||
| Fig. 46 Characterization of single crystals of COF films. (a) Atomic force microscopy image of crystalline domains on 2D BECOF-PP films. (b) Single crystal size distribution of 2D BECOF-PP. (c) Optical microscopy images of the SYSU-7 film suspended over a copper grid. (d) Scanning transmission electron microscopy high-angle annular dark-field image of the free-standing SYSU-7 film. (a) and (b) were reprinted with permission from ref. 175. Copyright 2020 Wiley-VCH. (c) and (d) were reprinted with permission from ref. 217. Copyright 2021 American Chemical Society. | ||
This method is applicable to the growth of free-standing films consisting of micrometre-sized single crystal ribbons at the air–water interface.217 Spreading sodium dodecyl sulfate (SDS, 1 mg mL−1) surfactant on the water interface and adding 4,4′,4″,4‴-(porphyrin-5,10,15,20-tetrayl) tetraaniline to the water subphase forms preassembled porphyrin ribbons consisting of a tetragonal lattice with an intermolecular distance of 15.0 Å. Injecting 4,4′-(1,4-phenylene)-bis(1-(2,4-dinitrophenyl) pyridilium) dichloride into the water subphase triggers an interfacial Zincke reaction, forming imine-linked single crystal ribbons of SYSU-7 COF (Fig. 45c), other than the viologen-linked COFs synthesized under solvothermal conditions.217,220 The single crystal ribbons (Fig. 46c and 35d) exhibit a typical length, width and thickness of 1.0 µm, 200 nm and 20 nm, respectively. The single crystal nature of the films is characterized by SAED and HRTEM.
Furthermore, a series of control experiments have been performed to investigate the growth of SYSU-7 films. (1) No film can be obtained without surfactant. (2) Using cationic surfactants, including hexadecyl trimethylammonium chloride, hexadecyl trimethylammonium bromide and hexadecyltrimethylammonium hydrogen sulfate, had the same issue that no product was generated. (3) Low-quality COF films with different morphologies, crystallinity and orientations were obtained when applying non-ionic surfactant (oleyl alcohol) and anionic surfactants with different chains. The above results manifested that synthesizing single-crystal SYSU-7 films at the surfactant–water interface requires control over both the length of the hydrophobic chains and the polar groups in the surfactants, which are significant to modulate the nucleation, polymerization and crystallization processes.217
The SMAIS method modulates the crystallization of COFs and relies on the preorganization of monomers and surfactants via noncovalent interactions. Thus, external and internal disturbances, such as temperature, concentration and pH, greatly affect the assembly, leaving the system with uncertainty in the crystal domain size and the single crystal integrity of the films. A macromolecule-mediated methodology by applying the charged polymers, i.e., poly(sodium 4-styrenesulfonate) (PSS) to direct the synthesis of thin films of COFs with large single crystal domains on the water surface has been developed.194 Compared to SMAIS, the charged polymers consist of repeating units through covalent bonds, which offer a chance to form long-range ordered structures along the polymer chains, while providing additional mechanical, thermal and chemical stability.
This strategy allows the assembly of protonated monomers as well as their polymerization and crystallization to form thin films.218 For example, PPS-mediated polymerization between 5,10,15,20-tetrakis(4-aminophenyl)-21H,23H-porphyrin (TAPP) and 2,5-dihydroxyterephthalaldehyde (2,5-Ph) generates a wafer-sized thin film of COF-2,5-Ph (Fig. 45d),218 which contains single crystal domains with an average size of 3.57(±2.57) µm2 (Fig. 47a). The single crystal nature of these domains is verified by aberration-corrected HRTEM (AC-HR TEM), SAED, and SEM (Fig. 47a and b). This strategy is applicable to the synthesis of thin films of another three 2D COFs, including COF-2,3-Ph (Fig. 45e), COF-BPy (Fig. 45f) and COF-BPh (Fig. 45g), via condensation of TAPP with dihydroxyterephthalaldehyde (2,3-Ph), 4,4′-biphenyl-dicarboxaldehyde (BPy), and 2,2’-bipyridine-5,5′-dicarbaldehde (BPh), respectively. These films consist of single crystal domains of 0.75(±0.5), 0.64(±0.33) and 0.077(±0.045) µm2, respectively, and their single crystal features are confirmed by AC-HR TEM, SAED and SEM, while their atomic coordinates are unavailable.
 | ||
| Fig. 47 Characterization of the single crystals of COF films. (a) Color-coded grain orientation map of COF-2,5-Ph. (b) AC-HRTEM image of COF-2,5-Ph with a resolution of 3 A. (c) AC-HRTEM image of the 2D COF-1 film. The inset shows the corresponding Fast Fourier transform pattern. (d) The AC-HRTEM image of the 2D COF-2 film. The inset shows the corresponding Fast Fourier transform pattern. (a) and (b) were reprinted with permission from ref. 218. Copyright 2022 American Chemical Society. (c) and (d) were reprinted with permission from ref. 219. Copyright 2024 Springer Nature. | ||
These 2D COFs films are fragile and brittle under folding, due to their discontinuous crystalline domain structure. Integrating elasticity and toughness into crystalline polymeric films brings new possibilities.219 Through a sacrificial go-between guided interfacial synthesis method, polycrystalline films of imine-linked 2D COFs with an interwoven structure for connecting single crystal domains were produced.219 Indeed, using polyacrylic acid as a polymer surfactant, the TAPP monomer pronated with PTSA, assembles underneath the surfactant via electrostatic and hydrogen-bonding interactions. Adding an aqueous solution of diethylenetriamine (1 mg mL−1) as a sacrificial go-between reagent to facilitate interweaving at the grain boundary in the reaction system, followed by injecting 2,5-Ph and 6-(4-formylphenyl) nicotinaldehyde linkers, results in the formation of thin films of 2D COF-1 (Fig. 45d) and 2D COF-2 (Fig. 45h), respectively.219
The single crystal nature of these domains is investigated by GIWAXS and AC-HRTEM, which show the reorientation of different domains, the gradual increase of the single-crystal domain size and the decrease in the number of grain boundaries. The resultant 2D COF films combine strength, toughness, and elasticity, originating from the interwoven structure at the grain boundaries (Fig. 47c and 36d).219
A micrometre-sized micelle method has been developed for the synthesis of single crystal 3D COFs (Fig. 48 and Table 15). This method assembles amphiphilic amino acid derivatives with long hydrophobic chains to form micelles in aqueous solution, which offer dynamic barriers to separate knots and linkers in aqueous solution and the hydrophobic cavity of micelles, respectively, depending on their hydrophilicity and hydrophobicity.167 For a typical procedure, the amino-acid derivative C16-GlyA (glycine with palmitoyl substituent group) forms 331 nm micelles upon dispersion in water under stirring.
 | ||
| Fig. 48 Synthetic strategy and morphology of single crystal COFs. (a) Schematic view of the micrometre-sized micelle method. Optical microscopy images of (b) COF-301, (c) COF-300, (d) SYSU-8, (e) SYSU-9, (f) COF-320 and (g) COF-TTA-BTA. Reprinted with permission from ref. 167. Copyright 2023 Springer Nature. | ||
| COFs | Synthetic conditions | Crystal parameters | Resolution | Method | Ref. |
|---|---|---|---|---|---|
 |
To grow the single-crystal COFs, amino-acid derivatives (0.1 mmol, C8-GlyA, C10-GlyA, C12-GlyA, C14-GlyA, C16-GlyA, C18-GlyA, C16-L-AlaA, C16-L-PhalaA, C16-L-ValA or C16-L-tLeuA) and sodium hydroxide (0.1 mmol) were dissolved in water (8.5 mL) at 50 °C under ambient conditions. The solution was stirred for 10 min to obtain an emulsion of uniform size, then linker (0.05 mmol) was added and stirred. Subsequently, a solution of TAM (9.5 mg, 0.025 mmol) in acid (PTSA/HCl/HOAc, 1.5 mL, 0.2 mol L−1) was added, then stirred for 5 min. After completion of the reaction, the product was collected by centrifugation and washed three times with water and THF. Further purification of the COFs was carried out by immersing THF for 24 h, followed by drying at ambient temperature for 12 h and 100 °C for 12 h to afford single-crystal COF powder. | a = 20.276 Å | 0.9 Å | SCXRD | 167 |
| b = 8.7098 Å | |||||
| c = 20.212 Å | |||||
| β = 99.308° | |||||
 |
To grow the single-crystal COFs, amino-acid derivatives (0.1 mmol, C8-GlyA, C10-GlyA, C12-GlyA, C14-GlyA, C16-GlyA, C18-GlyA, C16-L-AlaA, C16-L-PhalaA, C16-L-ValA or C16-L-tLeuA) and sodium hydroxide (0.1 mmol) were dissolved in water (8.5 mL) at 50 °C under ambient conditions. The solution was stirred for 10 min to obtain an emulsion of uniform size, then 0.05 mmol linker (linker A/B/C/D/E) was added and stirred. Subsequently, a solution of TAM (9.5 mg, 0.025 mmol) in acid (PTSA/HCl/HOAc, 1.5 mL, 0.2 mol L−1) was added, then stirred for 5 min. After completion of the reaction, the product was collected by centrifugation and washed three times with water and THF. Further purification of the COFs was carried out by immersing in THF for 24 h, followed by drying at ambient temperature for 12 h and 100 °C for 12 h to afford single-crystal COF powder. | NA | 0.9 Å | SCXRD | 167 |
 |
To grow the single-crystal COFs, amino-acid derivatives (0.1 mmol, C8-GlyA, C10-GlyA, C12-GlyA, C14-GlyA, C16-GlyA, C18-GlyA, C16-L-AlaA, C16-L-PhalaA, C16-L-ValA or C16-L-tLeuA) and sodium hydroxide (0.1 mmol) were dissolved in water (8.5 mL) at 50 °C under ambient conditions. The solution was stirred for 10 min to obtain an emulsion of uniform size, then 0.05 mmol linker (linker A/B/C/D/E) was added and stirred. Subsequently, a solution of TAM (9.5 mg, 0.025 mmol) in acid (PTSA/HCl/HOAc, 1.5 mL, 0.2 mol L−1) was added, then stirred for 5 min. After completion of the reaction, the product was collected by centrifugation and washed three times with water and THF. Further purification of the COFs was carried out by immersing in THF for 24 h, followed by drying at ambient temperature for 12 h and 100 °C for 12 h to afford single-crystal COF powder. | a = 26.461 Å | 1.1 Å | SCXRD | 167 |
| c = 7.4600 Å | |||||
 |
To grow the single-crystal COFs, amino-acid derivatives (0.1 mmol, C8-GlyA, C10-GlyA, C12-GlyA, C14-GlyA, C16-GlyA, C18-GlyA, C16-L-AlaA, C16-L-PhalaA, C16-L-ValA or C16-L-tLeuA) and sodium hydroxide (0.1 mmol) were dissolved in water (8.5 mL) at 50 °C under ambient conditions. The solution was stirred for 10 min to obtain an emulsion of uniform size, then 0.05 mmol linker (linker A/B/C/D/E) was added and stirred in. Subsequently, a solution of TAM (9.5 mg, 0.025 mmol) in 0.2 mol L−1 acid (PTSA/HCl/HOAc, 1.5 mL) was added, then stirred for 5 min. After completion of the reaction, the product was collected by centrifugation and washed three times with water and THF. Further purification of the COFs was carried out by immersing in THF for 24 h, followed by drying at ambient temperature for 12 h and 100 °C for 12 h to afford the single-crystal COF powder. | a = 23.360 Å | 0.9 Å | SCXRD | 167 |
| c = 8.4300 Å | |||||
 |
To grow the 2D COFs, C16-L-ValA (0.1 mmol) and sodium hydroxide (0.1 mmol) were dissolved in water (10 mL). This solution was stirred at 50 °C for 10 min to obtain an emulsion of uniform size, then benzene-1,3,5-tricarbaldehyde (BTA) (4.1 mg, 0.025 mmol) was added and stirred. A solution of 4,4′,4′-(1,3,5-triazine-2,4,6-triyl) trianiline (TTA, 8.85 mg, 0.025 mmol) in PTSA (1.5 mL, 0.2 mol L−1) was added and the solution was stirred for 5 min. After completion of the reaction, the product was collected by centrifugation and washed three times with water and THF. Further purification of the COFs was carried out by immersing in THF for 24 h, then drying at ambient temperature for 12 h and 100 °C for 12 h to afford 2DCOF-TTA-BTA powder. For gram-scale production, C16-L-ValA (3.556 g, 10 mmol), TTA (885 g, 2.5 mmol) and BTA (410 g, 2.5 mmol) were used to obtain a highly crystalline 2DCOF-TTA-BTA powder (yield: 92%, 1.07 g). Recovering rate of C16-L-ValA: 92%, 3.27 g. | a = 17.875 Å | NA | SCXRD | 167 |
| c = 3.4652 Å |
Adding 2,5-dihydroxy-1,4-benzenedicarboxaldehyde linker increases the micelle size to 503 nm, accompanied by a distinct colour change from white to yellow. Subsequent addition of the PTSA-protonated tetrakis(4-aminophenyl) methane (TAM) knot triggers polymerization and crystallization to form single crystal COF-301 (Fig. 48b and 49a), obtained as uniform cuboids with a typical length and width of 2 µm and 0.6 µm after a two-week reaction. The single crystal nature was investigated by SCXRD and SAED, which revealed that COF-301 exhibits a tetragonal space group of I41/a (a = 26.434(4) Å, c = 7.5876(15) Å), seven-fold interpenetration and diamond-type topology with a structural resolution up to 0.9 Å.167
Notably, this strategy enables the synthesis of a series of 3D COFs as well as 2D COFs (Fig. 48b–g). In detail, under identical synthetic conditions as for COF-301, polycondensation of TAM with TA, 2,3,5,6-tetrafluoroterephthalaldehyde, pyridine-2,5-dicarbaldehyde, and 4,4′-biphenyldicarboxaldehyde produces the single crystals of COF-300 (Fig. 7a and 48c), SYSU-8 (Fig. 48b and 49b), SYSU-9 (Fig. 48e and 49c) and COF-320 (Fig. 48f and 49d), respectively.167 Additionally, condensing 2,4,6-tris(4-aminophenyl)-1,3,5-triazine (TTA) with benzene-1,3,5-tricarbaldehyde (BTA) forms single crystals of 2D COF-TTA-TBA (Fig. 48g and 49e).167 These single crystals are confirmed by optical microscopy, SEM, HR TEM, SCXRD and SAED, revealing a regular shape, transparent crystal, precise atomic structure and single crystal nature.
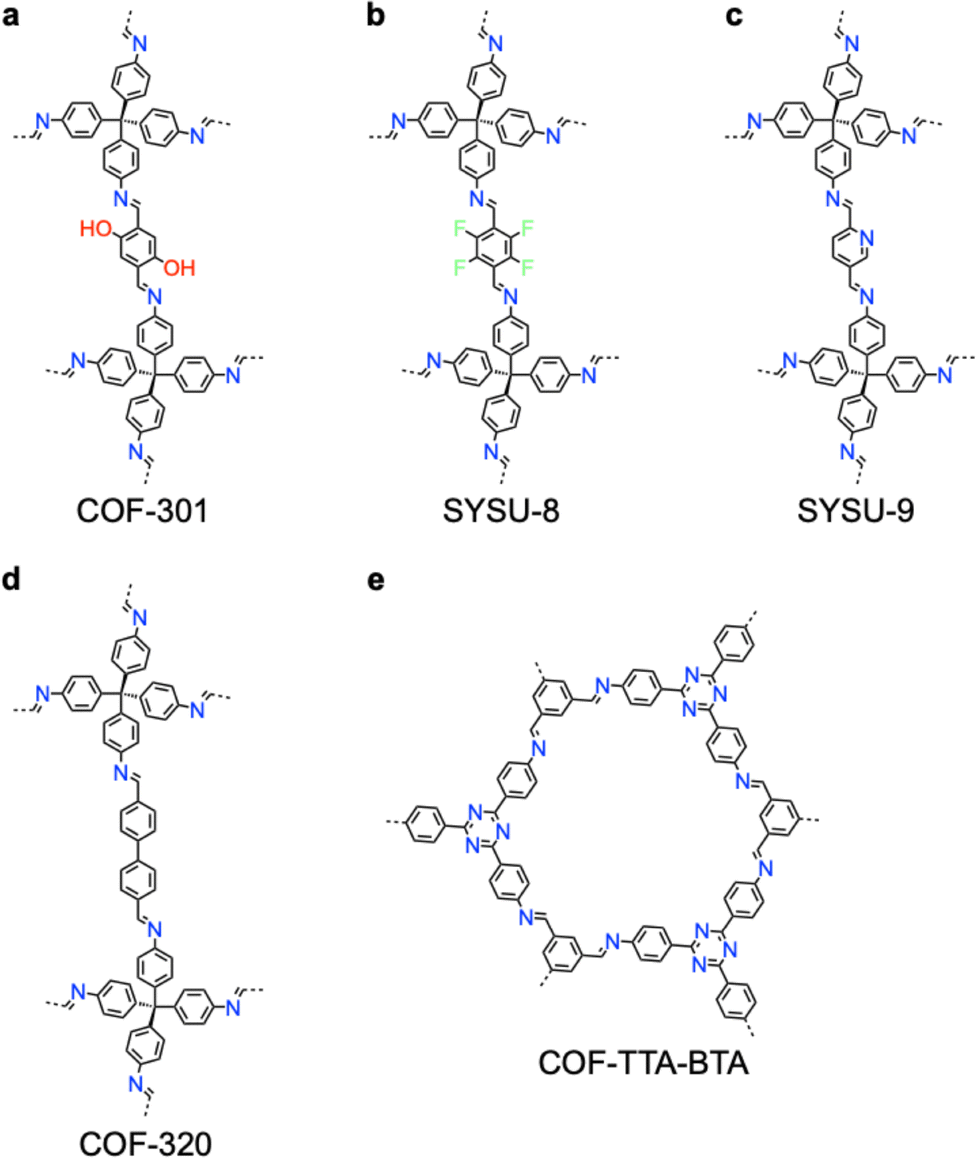 | ||
| Fig. 49 Structures of single crystal COFs obtained via a micrometre-sized micelle method. (a)–(f) Structures of (a) COF-301, (b) SYSU-8, (c) SYSU-9, (d) COF-320 and (e) COF-TTA-BTA. | ||
Noticeably, this strategy is dependent on the presence of amphiphilic amino acid derivatives with suitable length of hydrophobic tail. Without C16-GlyA, no solid product forms, even after prolonging the reaction time for over one month. Using amphiphilic amino acid derivatives with short chains such as octanoylglycine (C8-GlyA), decanoylglycine (C10-GlyA) and dodecanoylglycine (C12-GlyA) greatly decreases the yield with very little solid.195 The use of tetradecanoylglycine (C14-GlyA) increases the yield of the solid phase to 87%, which reaches a maximum of 92% yield for C16-GlyA but decreases to 79% using longer stearoylglycine (C18-GlyA).
These results indicate that the hydrophobicity and order of the assembled micellar structures affect the formation of single crystals by regulating the diffusion, polymerization and crystallization processes.167 The amino acid derivatives with short hydrophobic tails tend to form loose micelles that cannot regulate these processes, while the derivatives with too long hydrophobic chains result in larger micelles which hinder the efficiency of monomer dispersion in water, thereby decreasing the yield.167 On the other hand, using palmitoyl-L-alanine (C16-L-AlaA), palmitoyl-L-phenylalanine (C16-L-PhalaA), palmitoyl-L-valine (C16-L-ValA) and (S)-3,3-dimethy l-2-palmitamidobutanoic acid (C16-L-tLeuA) with different steric hindrances near the polar groups of the amino acid site has less effect on the morphology, size and yield of COFs.167 This result indicates that separation of amine- and aldehyde-monomers in different phases with surfactants is key to the formation of single crystal COFs.
4.3. Seed-assisted polymerization
Seed is important for nucleation and crystallite growth. Adding seeds to polymerization systems enables the synthesis of large-size single crystal COFs (Fig. 50 and Table 16).186 | ||
| Fig. 50 Seed-assisted synthetic method for single-crystal COFs. Reprinted with permission from ref. 186. Copyright 2018 American Association for the Advancement of Science. | ||
| COFs | Synthetic conditions | Crystal parameters | Resolution | Method | Ref. |
|---|---|---|---|---|---|
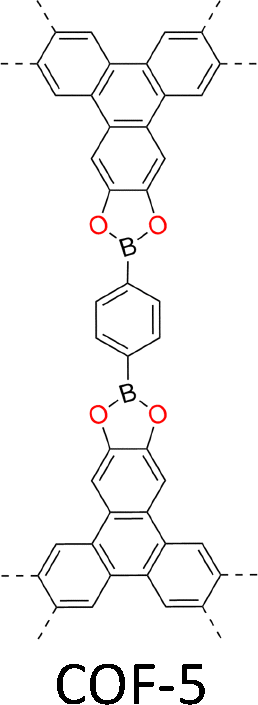 |
A mixture of HHTP (11 mg, 0.034 mmol) and benzene diboronic acid (BDBA) (8.28 mg, 0.05 mmol) was dissolved in n-BuOH (200 µL), and transferred into a stainless steel reactor (40 mL) and the system was heated to 90 °C and pressurized up to 9 MPa CO2, reacted for 90 min. After the reaction, the reactor was slowly depressurized at a rate of 1–2 MPa min−1. The precipitate was collected by filtration, washed with acetone, and dried in vacuum overnight. | NA | NA | SAED | 186 |
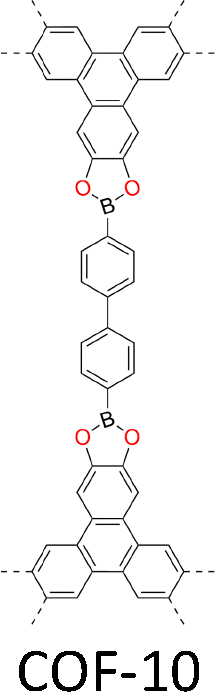 |
HHTP (0.02 mmol) and the appropriate corresponding boronic acid (BA, 0.03 mmol) were dissolved in a mixture of MeCN/1,4-dioxane/mesitylene (10 mL, 80/16/4 vol) and sonicated for 10 min. The solution was passed through a 0.45 micron PTFE syringe filter to remove insoluble particulates. This solution was put into a scintillation vial (20 mL) and sealed. This reaction mixture was heated without stirring to 90 °C for 18 h, which resulted in an indefinitely stable, translucent colloidal suspension. | a = b = 29.6 Å, c = 3.5 Å | NA | SAXS/WAXS FFT | 186 |
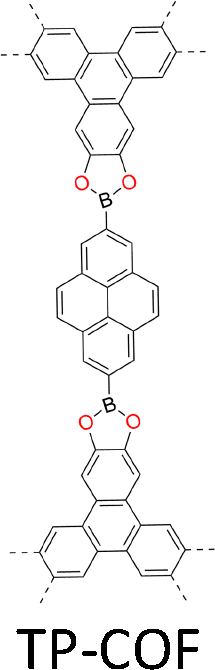 |
HHTP (4 mM) and diboronic acid (6 mM) were dissolved in separate solutions of MeCN/1,4-dioxane/mesitylene (80/16/4 vol) and sonicated for 10 min. These solutions were passed through a 0.45 micron PTFE syringe filter to remove insoluble particulates. These two solutions were then loaded into syringes (50 mL) on a calibrated syringe pump. A COF colloidal suspension was heated to 85 °C, then the diboronic acid and HHTP solutions were added at the noted rate. | a = b = 36.3 Å, c = 3.4 Å (COF-10) | NA | SAXS/WAXS FFT | 186 |
| a = b = 37.3 Å, c = 3.4 Å (TP-COF) | NA | SAXS/WAXS FFT | 186 | ||
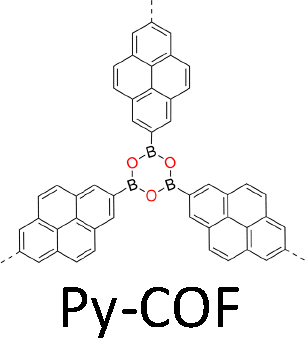 |
Phenylenebisboronic acid (PBBA, 33.2 mg, 10 mM) was dissolved in either a mixture of 4:1 1,4-dioxane:mesitylene (20 mL) when attempting powder synthesis of Ph-COF or a mixture of MeCN/1,4-dioxane/mesitylene (20 mL, 50/40/10 vol) when attempting colloidal syntheses. The PBBA solution was then heated to 70 °C for 72 hours. During this time, the clear solutions became white and opaque. |
NA | NA | SAXS/WAXS FFT | 177 |
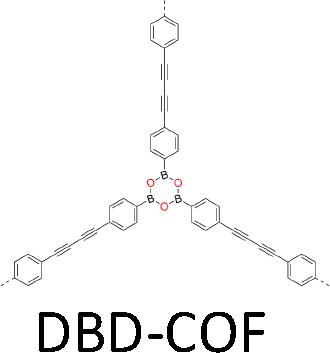 |
Biphenylenebisboronic acid (BBBA, 10 mM, 12 mg) was dissolved in a mixture of MeCN/1,4-dioxane/mesitylene (5 mL, 50/40/10 vol) when attempting colloidal syntheses. The BBBA solution was then heated to 70 °C for 72 hours. During this time, the clear solutions became white and opaque. | NA | NA | SAXS/WAXS FFT | 177 |
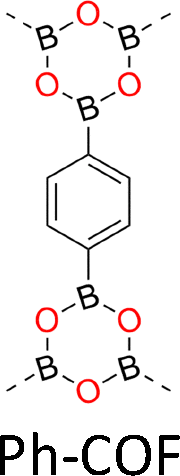 |
Diphenylbutadiynbisboronic acid (DBD-BA, 14.5 mg, 10 mM) was dissolved in a mixture of MeCN/1,4-dioxane/mesitylene (5 mL, 50/40/10 vol) when attempting colloidal syntheses. The DBDBA solution was then heated to 70 °C for 72 hours. During this time, the clear solutions became white and opaque. | NA | NA | SAXS/WAXS FFT | 177 |
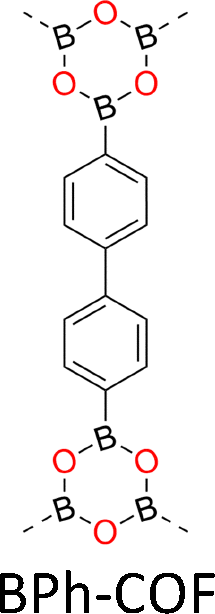 |
Pyrenebisboronic acid (PyBA, 14.5 mg, 10 mM) was dissolved in a mixture of MeCN/1,4-dioxane/mesitylene (5 mL, 50/40/10 vol) when attempting colloidal syntheses. The Py-BA solution was then heated to 70 °C for 72 hours. During this time, the clear solutions became white and opaque. | NA | NA | SAXS/WAXS FFT | 177 |
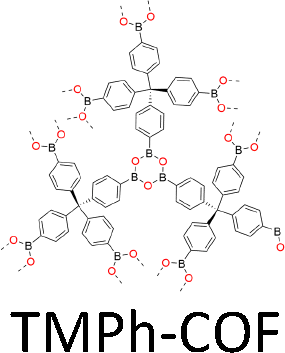 |
Tetrakis(phenlboronic acid)methane (TMPh-BA, 24.5 mg, 10 mM) was dissolved in a mixture of MeCN/1,4-dioxane/mesitylene (5 mL, 50/40/10 vol) when attempting colloidal syntheses. The TMPh-BA solution was then heated to 70 °C for 72 hours. During this time, the clear solutions became white and opaque. | NA | NA | SAXS/WAXS FFT | 177 |
Polymerization of 2,3,6,7,10,11-hexahydroxytriphenylene (HHTP) and 1,4-phenylenebis(boronic acid) (PBBA) using mesitylene, 1,4-dioxane and acetonitrile (4/16/80 in vol) as a solvent forms small nanoparticles with a size of 30 nm under solvothermal conditions.186 These nanoparticles are well-dispersed to form stable colloidal suspensions. Adding monomers slowly and in certain amounts to the colloidal suspension allows the nanoparticles to grow into large, faceted single crystals, ranging from 500 nm to 1.5 µm in size.
Slow addition of monomers is essential to maintain low concentrations, thereby suppressing nucleation and allowing the seeds to grow into larger crystals. Rapid monomer addition results in uncontrolled nucleation and smaller particle sizes.
The seeded growth method allows for precise control over the size and morphology of the COF crystals. For example, COF-5 crystals grew into well-defined, single crystal particles with hexagonal faceting and uniform sizes, exhibiting significantly improved crystallinity compared to polycrystalline samples. Characterization with TEM, wide-angle X-ray scattering (WAXS) and dynamic light scattering (DLS) confirmed the high quality and single crystal nature of the COF-5 (Fig. 51a), COF-10 (Fig. 51b) and TP-COF (Fig. 51c).186
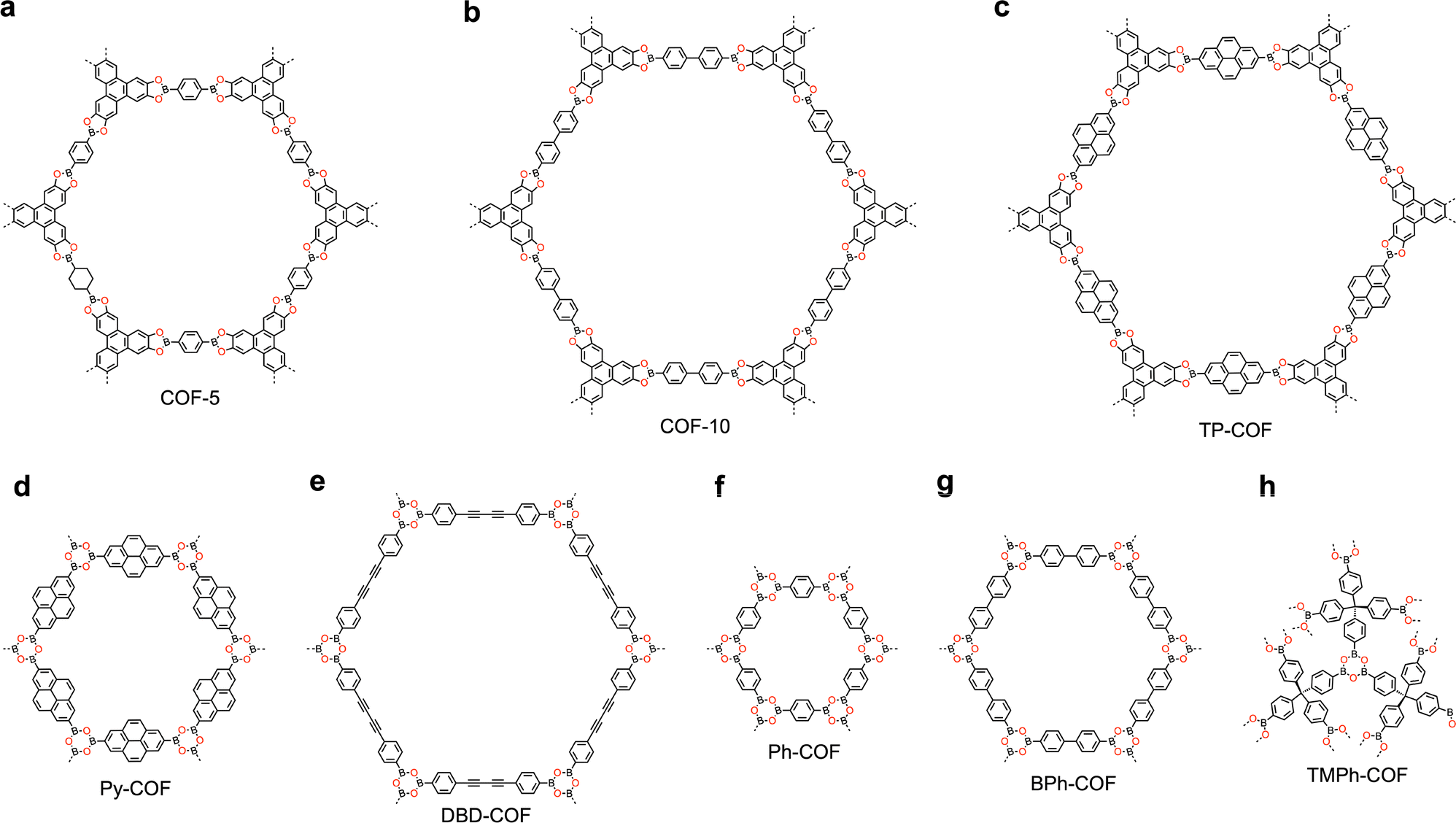 | ||
| Fig. 51 Structures of single-crystalline COFs obtained via seed-assisted synthesis. (a)–(g) Structures of (a) COF-5, (b) COF-10, (c) TP-COF, (d) Py-COF, (e) DBD-COF, (f) Ph-COF, (g) BPh-COF and (h) TMPh-COF. | ||
The TEM images (Fig. 52a–d) revealed well-defined lattice fringes extending throughout the crystals, confirming their single crystal structure. The WAXS data revealed sharp diffraction peaks, indicating long-range order and high crystallinity, while DLS measurements demonstrated uniform particle sizes, further validating the effectiveness of the seeded growth method (Fig. 52e–h). However, no atomic coordinates were given from the structural resolution. The single crystal COFs exhibited improved optical properties compared to polycrystalline samples, due to the high crystallinity and larger particle sizes of the single crystals allowing more efficient exciton diffusion (Fig. 52i).186
 | ||
| Fig. 52 (a) Low-magnification images of COF-5 particles. (b) Lattice resolution HRTEM image of a COF-5 particle. Inset: FFT of the image. (c) Low-magnification images of COF-10 particles. (d) Lattice resolution HRTEM image of a COF-10 particle. (e) The DLS number-average size of COF-5 particles as a function of added monomer equivalents. (f) DLS number-average size distributions obtained at three points shown in (e). (g) and (h) DLS number-average size distributions for the initial and final particle sizes of COF-10 (g) and TP-COF (h) during slow monomer addition. (i) Depiction of exciton distribution in COF-5 single crystals of different sizes. Reprinted with permission from ref. 186. Copyright 2018 American Association for the Advancement of Science. | ||
The seeded growth method is applicable to boronate ester-linked COF-5, COF-10 and TP-COF, as well as boroxine-linked 2D Py-COF (Fig. 51d), DBD-COF (Fig. 51e), Ph-COF (Fig. 51f), and BPh-COF (Fig. 51g) and 3D TMPh-COF (Fig. 51h) as single crystal particles (Fig. 52b).177 Low-dose HRTEM and synchrotron X-ray diffraction revealed the structures of these COFs. Ph-COF exhibits diffraction signals at 0.55 and 0.85 Å−1, corresponding to the (100) and (110) Bragg features from an AB offset structure, respectively. Py-COF and BPh-COF with an AA eclipsed structure have the same diffraction peak at 0.39 Å−1, which is due to their equivalent lattice dimensions and can be assigned to the (100) facet.
DBD-COF exhibits a hexagonal topology and the primary (200) diffraction signal at 0.21 Å−1, because of its longest linker among these 2D COFs. In contrast, the 3D boroxine-linked TMPh–COF exhibited diffractions at 0.59, 0.62, 0.91 and 0.96 Å−1, which were assigned to the (211), (220), (310) and (321) facets, respectively. Notably, Ph-COF has a preeminent lattice spacing of 10 Å (d110), as revealed by FFT images, and displays hexagonal morphology with clearly visible lattice fringes extending from one end of the particles to the other under TEM observation. Py-COF with a lattice fringe at 7.1 Å (d200) appears as agglomerated hexagonally faceted particles. The FFT pattern of Py-COF exhibited a d310 value at 4.1 Å.
Noticeably, BPh-COF displays a similar morphology as hexagonal particles but has a smaller interlayer distance than Py-COF, as indicated by a smaller d001 value. Interestingly, DBD-COF presents as predominantly trigonal particles and exhibits a lattice spacing of 11 Å, which is consistent with the results from structural reconstruction. The 3D TMPh-COF shows regularly faceted crystals with a lattice spacing of 10 Å. TEM imaging revealed the regular crystal morphologies and ordered lattice structures, indicating a single crystal feature.
4.4. Coordination-driven dynamic covalent polymerization
Single crystal 1D metallo-COFs (mCOF-Ag, Table 17) have been synthesized by combining labile metal coordination and dynamic covalent chemistry under solvothermal conditions.184 Specifically, to a 10 mL Schlenk tube, 4,4′-(1,10-phenanthroline-2,9-diyl)dianiline (17.4 mg, 0.048 mmol) and 2,9-bis(4-(dimethoxymethyl)phenyl)-1,10-phenanthroline (7.7 mg, 0.016 mmol) were weighted. Then, an o-DCB/n-BuOH (0.9/0.1 mL) mixture of AgBF4 (3.1 mg, 0.016 mmol) was added to the tube, which was subjected to sonification for 10 minutes before adding AcOH (0.1 mL, 6M). The polycondensation reaction was conducted at 120 °C for 3 days, yielding a light-yellow solid product, i.e., mCOF-Ag (Fig. 53).| COFs | Synthetic conditions | Crystal parameters | Resolution | Method | Ref. |
|---|---|---|---|---|---|
 |
To 4,4′-(1,10-phenanthroline-2,9-diyl)dianiline (17.4 mg, 0.048 mmol), 2,9-bis(4-(dimethoxymethyl)-phenyl)-1,10-phenanthroline (7.7 mg, 0.016 mmol) in a Schlenk tube (10 mL), was added AgBF4 (3.1 mg, 0.016 mmol) dissolved in n-BuOH (0.1 mL) and o-DCB (0.9 mL). After sonicating at room temperature for 10 min, AcOH (0.1 mL, 6 M) was added into the suspension. The Schlenk tube was frozen in a liquid N2 bath, evacuated to an internal pressure of 0 mbar and sealed. After warming to room temperature, the Schlenk tube was placed into an oven and heated at 120 °C for 3 days yielding a light-yellow solid at the bottom of the tube. | a = 15.66 Å | 0.95 Å | SAED | 184 |
| b = 15.66 Å | |||||
| c = 10.87 Å | |||||
 |
Cu(PDB-OMe)2(BF4) (17.8 mg, 0.016 mmol) and Cu(PDA)2(BF4) (14.0 mg, 0.016 mmol) in a Schlenk tube (10 mL) were added with n-BuOH (0.1 mL) and o-DCB (0.9 mL) and the resulting suspension was sonicated briefly. Then AcOH (0.1 mL, 6 M) was added. The Schlenk tube was frozen in a liquid N2 bath, evacuated to an internal pressure of 0 mbar and sealed. After warming to room temperature, the Schlenk tube was placed into an oven and heated at 120 °C for 3 days yielding a dark brown solid at the bottom of the tube. | a = 7.79 Å | 1.1 Å | 3DED | 183 |
| b = 31.44 Å | |||||
| c = 32.30 Å | |||||
 |
To Cu(PDA)2(BF4) (14.0 mg, 0.016 mmol) and PDB-OMe (15.4 mg, 0.032 mmol) in a Schlenk tube (10 mL), n-BuOH (0.1 mL) and o-DCB (0.9 mL) were added and the resulting suspension was sonicated briefly. Then AcOH (0.1 mL, 6 M) was added. The Schlenk tube was frozen in a liquid N2 bath, evacuated to an internal pressure of 0 mbar and sealed. After warming to room temperature, the Schlenk tube was placed into an oven and heated at 120 °C for 3 days yielding an orange-red solid at the bottom of the tube. | a = 15.42 Å | 1.4 Å | 3DED | 183 |
| b = 29.15 Å | |||||
| c = 10.84 Å |
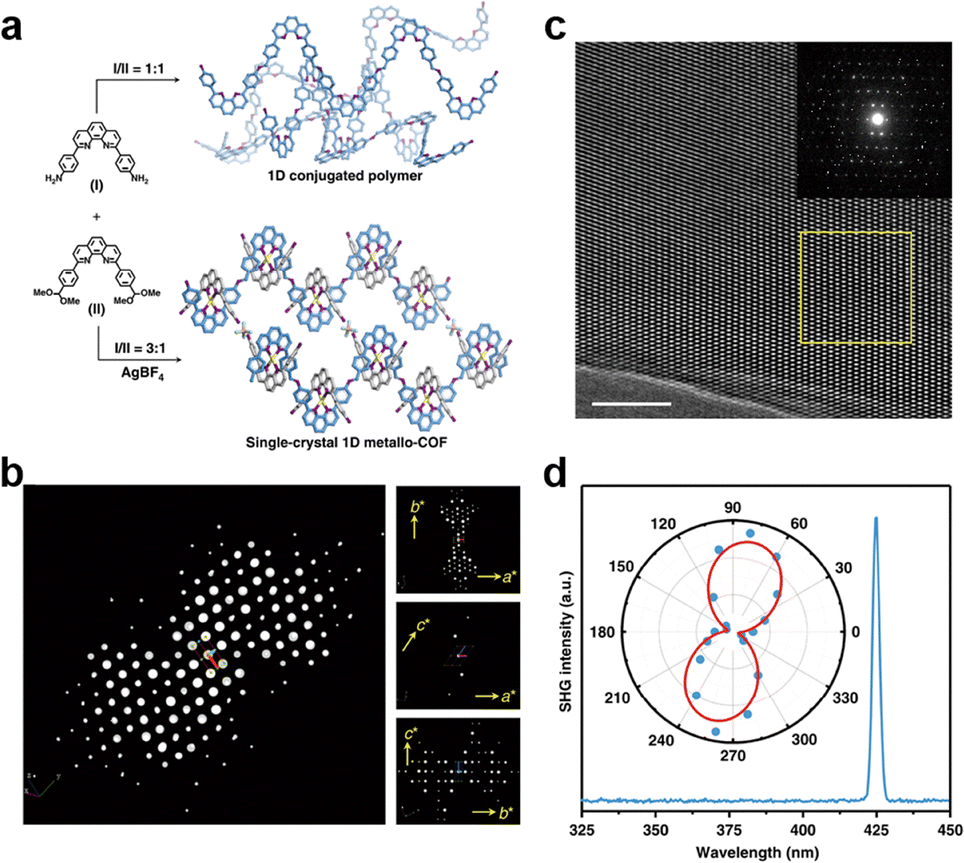 | ||
| Fig. 53 (a) Schematic representation for the preparation of single-crystalline mCOF. (b) 3D reciprocal lattice of mCOF-Ag reconstructed from the SCED data (left) and 2D slices cut from the reconstructed reciprocal lattice (right). (c) HRTEM image of mCOF-Ag and the SAED pattern (inset). Scale bar: 20 nm. (d) The second-harmonic generation (SHG) spectrum of an isolated crystal of mCOF-Ag. Reprinted with permission from ref. 184. Copyright 2020 Springer Nature. | ||
The single crystal structure of mCOF-Ag determined by SCED techniques revealed that mCOF-Ag has a space group of C2/c with unit cell parameters of a = 15.66 Å, b = 31.00 Å, c = 10.87 Å and β = 123.31°, respectively. The coordination of Ag+ ions to 1,10-phenanthroline units facilitates an additional reversible polycondensation process that promotes nucleation. The monomer 4,4′-(1,10-phenanthroline-2,9-diyl)diphenylamine in excess anchors to the polymer backbone via Ag+ coordination, forming periodic spacers that promote π–π stacking and hydrogen bonding between the one-dimensional chains.
Single crystal mCOF-Ag with micrometre size enables a structural resolution of 0.95 Å by SCED (Fig. 53b). HRTEM and SAED patterns confirm its single crystal nature (Fig. 53c). Due to its non-centrosymmetric structure, mCOF-Ag displays a distinct second harmonic generation signal at 425 nm upon excitation at 850 nm with a laser, showing its potential as a nonlinear optical material (Fig. 53d).
This method is applicable to 3D mCOFs (WCOF-Cu) (Fig. 54).183 Homoleptic tetrahedral complexes and polydentate linkers form a 3D woven COF (WCOF), where the metal ions in the complexes serve as templates to guide the assembly of the organic threads into a three-dimensional network. Imine condensation of copper(I) phenanthroline complexes forms a diamond-like COF under solvothermal conditions. The single crystal structure analysis via 3D ED (Fig. 54b) indicates that WCOF-Cu has a face-centred orthorhombic Bravais lattice and generates unit cell parameters of a = 7.79 Å, b = 31.44 Å, c = 32.30 Å and α = β = γ = 90°, respectively, with a resolution of 1.1 Å. WCOF-Cu adopts dia topology with the F222 space group and exhibits 3-fold interpenetrated structures. Noticeably, the HRTEM image observed from the [022] direction shows ordered lattice fringes with distances matched with the crystal structures (inset of Fig. 54c).183
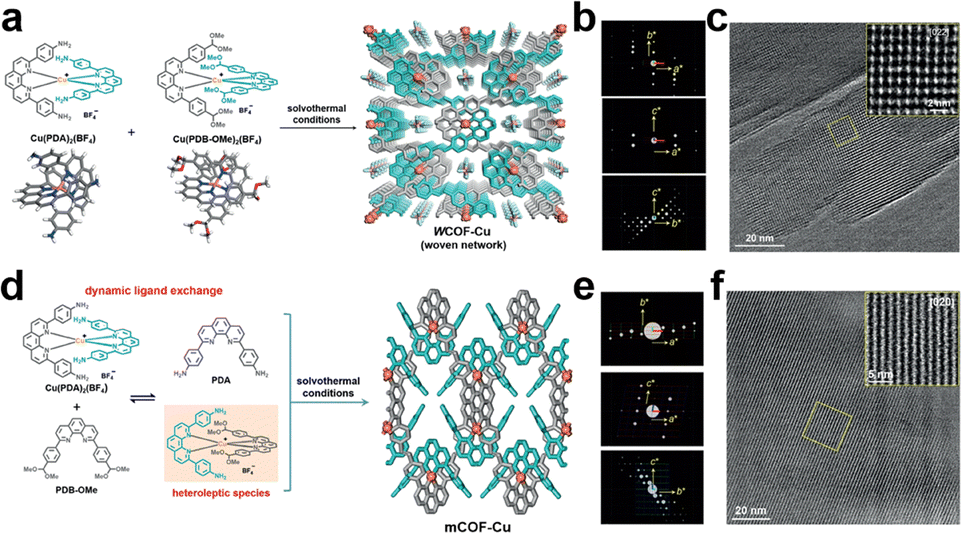 | ||
| Fig. 54 (a) Synthesis of WCOF-Cu. (b) 2D slices cut from the re-constructed reciprocal lattice of 3D ED data. (c) HRTEM image of WCOF-Cu, inset: magnified view of the highlighted area. (d) Synthesis of mCOF-Cu. (e) 2D slices cut from the re-constructed reciprocal lattice of 3D ED data. (f) HRTEM image of WCOF-Cu, inset: magnified view of the highlighted area. Reprinted with permission from ref. 183. Copyright 2020 Wiley-VCH. | ||
Interestingly, performing dynamic ligand exchange in the 3D WCOF-Cu generates the 1D mCOF-Cu (Fig. 54d and 44c) as an orange-red solid. The single crystal structure was investigated using 3D ED (Fig. 54e) and HRTEM (Fig. 54f). The 3D ED data reveals a monoclinic Bravais lattice with unit cell parameters of a = 15.42 Å, b = 29.15 Å, c = 10.84 Å and β = 124.38° (space groups: C2/c or Cc), which is in good agreement with that of mCOF-Ag (Fig. 53a). The HRTEM image of the ordered lattice fringes indicates its single crystalline nature with a structural resolution of 1.1 Å (Fig. 54f).
A straightforward and efficient approach enables the coordination-driven dynamic covalent polymerization (Table 18). By taking advantage of the easy crystallization of N-donor ligand-metal coordination bonds, a new class of highly crystalline porous materials is templated and guided to produce a new class of single crystals, which are hybrid MOF-COF (MOCOF).221 These MOCOFs exhibit dynamic covalent chemical properties, enhancing the reversibility of imine bond formation and dissociation, thereby promoting the growth of high-quality single crystals. This method reliably produces 14 distinct single-crystal MOCOFs (Fig. 55) within just 1–2 days, utilizing diverse combinations of imine-based COFs and nitrogen-donor ligands derived from MOFs. The resulting crystals range from 50 to 500 µm, enabling precise determination of their structures through single-crystal X-ray diffraction analysis with resolutions up to 0.79 Å. Meanwhile, the introduction of different chiral additives during the crystallization process allowed chirality transfer to the single crystal MOCOF framework (Fig. 55).
| COFs | Synthetic conditions | Crystal parameters | Resolution | Method | Ref. |
|---|---|---|---|---|---|
| MOCOF-100 | A Pyrex tube (10 × 8 mm, o.d. × i.d.) was charged with TAPP (17.0 mg, 0.025 mmol), BPDA (11.0 mg, 0.052 mmol), TPB (12.0 mg, 0.038 mmol), Fe(OAc)2 (5.0 mg, 0.029 mmol), 1,2-o-xylene (1.0 mL), n-BuOH (1.0 mL), DMF (0.1 mL) and aqueous para-toluenesulfonic acid (PTSA, 0.1 mL, 0.6 M). After sonication for 8 hours, the tube was flash-frozen at 77 K (liquid N2 bath). Following one freeze–pump–thaw cycle, the system was evacuated to an internal pressure of 50 mtorr and flame-sealed. Upon sealing, the length of the tube was reduced to approximately 10–14 cm. The reaction was heated at 120 °C for 48 hours, yielding brown rectangular-shaped single crystals, which were then isolated by filtration and washed with DMF (10 mL) and THF (10 mL). Yield: 28.6 mg (90% based on TAPP). | a = 24.7523 Å | 0.84 Å | SCXRD | 221 |
| b = 26.6928 Å | |||||
| c = 10.8382 Å | |||||
| α = 90° | |||||
| β = 97.672° | |||||
| γ = 90° | |||||
| MOCOF-101 | A Pyrex tube (10 × 8 mm, o.d. × i.d.) was charged with TAPP (17.0 mg, 0.025 mmol), BPyDA (11.0 mg, 0.052 mmol), TPB (12.0 mg, 0.038 mmol), Fe(OAc)2 (5.0 mg, 0.029 mmol), 1,2-o-xylene (1.0 mL), n-BuOH (1.0 mL), DMF (0.1 mL) and aqueous PTSA (0.1 mL, 0.6 M). After sonication for 8 hours, the tube was flash-frozen at 77 K (liquid N2 bath). Following one freeze–pump–thaw cycle, the system was evacuated to an internal pressure of 50 mtorr and flame-sealed. Upon sealing, the length of the tube was reduced to approximately 10–14 cm. The reaction was heated at 120 °C for 48 hours, yielding brown rectangular-shaped single crystals, which were then isolated by filtration and washed with DMF (10 mL) and THF (10 mL). Yield: 27.9 mg (89% based on TAPP). | a = 25.0868 Å | 0.89 Å | SCXRD | 221 |
| b = 26.7033 Å | |||||
| c = 10.8326 Å | |||||
| α = 90° | |||||
| β = 96.975° | |||||
| γ = 90° | |||||
| MOCOF-102 | A Pyrex tube (10 × 8 mm, o.d. × i.d.) was charged with TAPP (17.0 mg, 0.025 mmol), BDA (10.5 mg, 0.078 mmol), TPB (12.0 mg, 0.038 mmol), Fe(OAc)2 (5.0 mg, 0.029 mmol), 1,2-dichlorobenzene (1.0 mL), EtOH (1.0 mL), DMF (0.1 mL) and aqueous PTSA (0.1 mL, 0.6 M). After sonication for 8 hours, the tube was flash-frozen at 77 K (liquid N2 bath). Following one freeze–pump–thaw cycle, the system was evacuated to an internal pressure of 50 mtorr and flame-sealed. Upon sealing, the length of the tube was reduced to approximately 10–14 cm. The reaction was heated at 120 °C for 36 hours, yielding dark square-shaped single crystals, which were then isolated by filtration and washed with DMF (10 mL) and THF (10 mL). Yield: 25.7 mg (84% based on TAP). | a = 22.836 Å | 0.79 Å | SCXRD | 221 |
| b = 10.6811 Å | |||||
| c = 26.5704 Å | |||||
| α = 90° | |||||
| β = 105.462° | |||||
| γ = 90° | |||||
| MOCOF-103 | A Pyrex tube (10 × 8 mm, o.d. × i.d.) was charged with TAPP (17.0 mg, 0.025 mmol), BDA (7.0 mg, 0.052 mmol), Py (5.0 mg, 0.063 mmol), Fe(OAc)2 (5.0 mg, 0.029 mmol), mesitylene (1.0 mL), EtOH (1.0 mL), DMF (0.1 mL) and aqueous PTSA (0.15 mL, 0.6 M). After sonication for 8 hours, the tube was flash-frozen at 77 K (liquid N2 bath). Following one freeze–pump–thaw cycle, the system was evacuated to an internal pressure of 50 mtorr and flame-sealed. Upon sealing, the length of the tube was reduced to approximately 10–14 cm. The reaction was heated at 120 °C for 24 hours, yielding dark rod-like single crystals, which were then isolated by filtration and washed with DMF (10 mL) and THF (10 mL). Yield: 22.4 mg (80% based on TAPP). | a = 16.952 Å | 0.84 Å | SCXRD | 221 |
| b = 16.952 Å | |||||
| c = 13.3653 Å | |||||
| α = β = γ = 90° | |||||
| MOCOF-104 | A Pyrex tube (10 × 8 mm, o.d. × i.d.) was charged with TAP (17.0 mg, 0.025 mmol), BDA-2Me (8.5 mg, 0.052 mmol), Py (5.0 mg, 0.063 mmol), Fe(OAc)2 (5 mg, 0.029 mmol), mesitylene (1.0 mL), n-BuOH (1.0 mL), DMF (0.1 mL) and aqueous PTSA (0.15 mL, 0.6 M). After sonication for 8 hours, the tube was flash-frozen at 77 K (liquid N2 bath). Following one freeze–pump–thaw cycle, the system was evacuated to an internal pressure of 50 mtorr and flame-sealed. Upon sealing, the length of the tube was reduced to approximately 10–14 cm. The reaction was heated at 120 °C for 24 hours, yielding dark rod-like single crystals, which were then isolated by filtration and washed with DMF (10 mL) and THF (10 mL). Yield: 21.7 mg (78% based on TAPP). | a = 23.960 Å | 0.84 Å | SCXRD | 221 |
| b = 23.960 Å | |||||
| c = 13.410 Å | |||||
| α = β = γ = 90° | |||||
| MOCOF-105 | A Pyrex tube (10 × 8 mm, o.d. × i.d.) was charged with TAPP (17.0 mg, 0.025 mmol), BDA-4Me (9.9 mg, 0.052 mmol), Py (5.0 mg, 0.063 mmol), Fe(OAc)2 (5 mg, 0.029 mmol), mesitylene (1.0 mL), n-butanol (1.0 mL), DMF (0.1 mL) and aqueous PTSA (0.15 mL, 0.6 M). After sonication for 8 hours, the tube was flash-frozen at 77 K (liquid N2 bath). Following one freeze–pump–thaw cycle, the system was evacuated to an internal pressure of 50 mtorr and flame-sealed. Upon sealing, the length of the tube was reduced to approximately 10–14 cm. The reaction was heated at 120 °C for 24 hours, yielding dark rod-like single crystals, which were then isolated by filtration and washed with DMF (10 mL) and THF (10 mL). Yield: 21.1 mg (71% based on TAPP). | a = 17.409 Å | 0.84 Å | SCXRD | 221 |
| b = 17.409 Å | |||||
| c = 13.3992 Å | |||||
| α = β = γ = 90° | |||||
| MOCOF-106 | A Pyrex tube (10 × 8 mm, o.d. × i.d.) was charged with TAPP (17.0 mg, 0.025 mmol), BDA-4F (10.7 mg, 0.052 mmol), Py (5.0 mg, 0.063 mmol), Fe(OAc)2 (5 mg, 0.029 mmol), mesitylene (1.0 mL), n-butanol (1.0 mL), DMF (0.1 mL) and aqueous PTSA (0.15 mL, 0.6 M). After sonication for 8 hours, the tube was flash-frozen at 77 K (liquid N2 bath). Following one freeze–pump–thaw cycle, the system was evacuated to an internal pressure of 50 mtorr and flame-sealed. Upon sealing, the length of the tube was reduced to approximately 10–14 cm. The reaction was heated at 120 °C for 24 hours, yielding dark rod-like single crystals, which were then isolated by filtration and washed with DMF (10 mL) and THF (10 mL). Yield: 19.4 mg (68% based on TAPP). | a = 17.429 Å | 0.84 Å | SCXRD | 221 |
| b = 17.429 Å | |||||
| c = 13.3964 Å | |||||
| α = β = γ = 90° | |||||
| MOCOF-107 | A Pyrex tube (10 × 8 mm, o.d. × i.d.) was charged with TAPP (17.0 mg, 0.025 mmol), NBDA (9.6 mg, 0.052 mmol), Py (5.0 mg, 0.063 mmol), Fe(OAc)2 (5 mg, 0.029 mmol), 1,2-dichlorobenzene (1.0 mL), n-butanol (1.0 mL), DMF (0.1 mL) and aqueous PTSA (0.15 mL, 0.6 M). After sonication for 8 hours, the tube was flash-frozen at 77 K (liquid N2 bath). Following one freeze–pump–thaw cycle, the system was evacuated to an internal pressure of 50 mtorr and flame-sealed. Upon sealing, the length of the tube was reduced to approximately 10–14 cm. The reaction was heated at 120 °C for 48 hours, yielding dark rod-like single crystals, which were then isolated by filtration and washed with DMF (10 mL) and THF (10 mL). Yield: 20.5 mg (70% based on TAPP). | a = 35.3452 Å | 0.85 Å | SCXRD | 221 |
| b = 35.3452 Å | |||||
| c = 13.4264 Å | |||||
| α = β = γ = 90° | |||||
| MOCOF-108 | A Pyrex tube (10 × 8 mm, o.d. × i.d.) was charged with TAPP (17.0 mg, 0.025 mmol), ABDA (12.2 mg, 0.052 mmol), Py (5.0 mg, 0.063 mmol), Fe(OAc)2 (5 mg, 0.029 mmol), 1,2-dichlorobenzene (1.0 mL), n-butanol (1.0 mL), DMF (0.1 mL) and aqueous PTSA (0.15 mL, 0.6 M). After sonication for 8 hours, the tube was flash-frozen at 77 K (liquid N2 bath). Following one freeze–pump–thaw cycle, the system was evacuated to an internal pressure of 50 mtorr and flame-sealed. Upon sealing, the length of the tube was reduced to approximately 10–14 cm. The reaction was heated at 120 °C for 48 hours, yielding dark rod-like single crystals, which were then isolated by filtration and washed with DMF (10 mL) and THF (10 mL). Yield: 18.6 mg (55% based on TAP). | a = 16.952 Å | 0.84 Å | SCXRD | 221 |
| b = 16.952 Å | |||||
| c = 13.3653 Å | |||||
| α = β = γ = 90° | |||||
| MOCOF-109 R-MBA | A Pyrex tube (10 × 8 mm, o.d. × i.d.) was charged with TAPP (17.0 mg, 0.025 mmol), BDA (7 mg, 0.052 mmol), Py (5.0 mg, 0.063 mmol), Fe(OAc)2 (5 mg, 0.029 mmol), R-MBA (3.0 mg, 0.025 mmol), o-DCB (1.0 mL), n-butanol (1.0 mL), DMF (0.1 mL) and aqueous PTSA (0.2 mL, 0.6 M). After sonication for 8 hours, the tube was flash-frozen at 77 K (liquid N2 bath). Following one freeze–pump–thaw cycle, the system was evacuated to an internal pressure of 50 mtorr and flame-sealed. Upon sealing, the length of the tube was reduced to approximately 10–14 cm. The reaction was heated at 120 °C for 48 hours, yielding dark rod-like single crystals, which were then isolated by filtration and washed with DMF (10 mL) and THF (10 mL). Yield: 14.7 mg (53% based on TAPP). | a = 17.648 Å | 0.9 Å | SCXRD | 221 |
| b = 7.648 Å | |||||
| c = 6.7220 Å | |||||
| α = β = γ = 90° | |||||
| MOCOF-109 R-NEA | A Pyrex tube (10 × 8 mm, o.d. × i.d.) was charged with TAPP (17.0 mg, 0.025 mmol), BDA (7 mg, 0.052 mmol), Py (5.0 mg, 0.063 mmol), Fe(OAc)2 (5 mg, 0.029 mmol), R-NEA (4.3 mg, 0.025 mmol), o-DCB (1.0 mL), nBuOH (1.0 mL), DMF (0.1 mL) and aqueous PTSA (0.2 mL, 0.6 M). After sonication for 8 hours, the tube was flash-frozen at 77 K (liquid N2 bath). Following one freeze–pump–thaw cycle, the system was evacuated to an internal pressure of 50 mtorr and flame-sealed. Upon sealing, the length of the tube was reduced to approximately 10–14 cm. The reaction was heated at 120 °C for 48 hours, yielding dark rod-like single crystals, which were then isolated by filtration and washed with DMF (10 mL) and THF (10 mL). Yield: 15.1 mg (54% based on TAPP). | a = 17.6281 Å | 0.9 Å | SCXRD | 221 |
| b = 17.6281 Å | |||||
| c = 6.7289 Å | |||||
| α = β = γ = 90° | |||||
| MOCOF-109 D-PGly | A Pyrex tube (10 × 8 mm, o.d. × i.d.) was charged with TAPP (17.0 mg, 0.025 mmol), BDA (7 mg, 0.052 mmol), Py (5.0 mg, 0.063 mmol), Fe(OAc)2 (5 mg, 0.029 mmol), D-PGly (3.8 mg, 0.025 mmol), o-DCB (1.0 mL), n-BuOH (1.0 mL), DMF (0.1 mL) and aqueous PTSA (0.2 mL, 0.6 M). After sonication for 8 hours, the tube was flash-frozen at 77 K (liquid N2 bath). Following one freeze–pump–thaw cycle, the system was evacuated to an internal pressure of 50 mtorr and flame-sealed. Upon sealing, the length of the tube was reduced to approximately 10–14 cm. The reaction was heated at 120 °C for 48 hours, yielding dark rod-like single crystals, which were then isolated by filtration and washed with DMF (10 mL) and THF (10 mL). Yield: 13.4 mg (51% based on TAPP). | a = 17.6157 Å | 0.85 Å | SCXRD | 221 |
| b = 17.6157 Å | |||||
| c = 6.7078 Å | |||||
| α = β = γ = 90° | |||||
| MOCOF-109 L-Pro | A Pyrex tube (10 × 8 mm, o.d. × i.d.) was charged with TAPP (17.0 mg, 0.025 mmol), BDA (7 mg, 0.052 mmol), Py (5.0 mg, 0.063 mmol), Fe(OAc)2 (5 mg, 0.029 mmol), L-proline (2.9 mg, 0.025 mmol), o-DCB (1.0 mL), n-butanol (1.0 mL), DMF (0.1 mL) and aqueous PTSA (0.2 mL, 0.6 M). After sonication for 8 hours, the tube was flash-frozen at 77 K (liquid N2 bath). Following one freeze–pump–thaw cycle, the system was evacuated to an internal pressure of 50 mtorr and flame-sealed. Upon sealing, the length of the tube was reduced to approximately 10–14 cm. The reaction was heated at 120 °C for 48 hours, yielding dark rod-like single crystals, which were then isolated by filtration and washed with DMF (10 mL) and THF (10 mL). Yield: 16.7 mg (59% based on TAPP). | a = 17.5317 Å | 0.85 Å | SCXRD | 221 |
| b = 17.5317 Å | |||||
| c = 6.7248 Å | |||||
| α = β = γ = 90° | |||||
| MOCOF-110 | A Pyrex tube (10 × 8 mm, o.d. × i.d.) was charged with TAPP (17 mg, 0.025 mmol), BDA (7 mg, 0.052 mmol), Fe(OAc)2 (5.0 mg, 0.029 mmol), 4,4′-Bipyridine (BP) (11 mg, 0.070 mmol), o-DCB (0.9 mL), n-BuOH (0.1 mL), THF (0.1 mL) and aqueous AcOH (0.20 mL, 9 M). After 8 h of sonication, the tube was flash frozen at 77 K. Following one freeze–pump–thaw cycle, the system was evacuated to an internal pressure of 50 mtorr and flame sealed. The reaction was heated at 120 °C for 72 h, yielding a dark purple precipitate. The precipitate was isolated by filtration and washed with DMF (10 mL) and THF (10 mL). It was then transferred to a Soxhlet extractor and thoroughly washed with THF for 24 h. Finally, the product was dried under vacuum at 60 °C for 18 h at 10−2 mtorr to yield the activated sample. Yield: 28.62 mg, 90% based on Fe(TAPP). | a = 34.830 Å | 1.0 Å | SCXRD | 222 |
| b = 38.770 Å | |||||
| c = 11.270 Å | |||||
| α = β = γ = 90° | |||||
| MOCOF-111 | A Pyrex tube (10 × 8 mm, o.d. × i.d.) was charged with TAP (17 mg, 0.025 mmol), BDA (7 mg, 0.052 mmol), Fe(OAc)2 (5.0 mg, 0.029 mmol), 1,1,2,2-tetra(pyridin-4-yl)ethene (TPE) (14 mg, 0.041 mmol), o-DCB (0.7 mL), n-BuOH (0.3 mL), THF (0.1 mL) and aqueous AcOH (0.25 mL, 9 M). After 8 h of sonication, the tube was flash frozen at 77 K. Following one freeze–pump–thaw cycle, the system was evacuated to an internal pressure of 50 mtorr and flame sealed. The reaction was heated at 120 °C for 72 h, yielding a dark purple precipitate. The precipitate was isolated by filtration and washed with DMF (10 mL) and THF (10 mL). It was then transferred to a Soxhlet extractor and thoroughly washed with THF for 24 h. Finally, the product was dried under vacuum at 60 °C for 18 h at 10−2 mtorr to yield the activated sample. Yield: 28.47 mg, 88% based on Fe(TAPP). | a = 25.770 Å | 1.2 Å | SCXRD | 222 |
| b = 25.370 Å | |||||
| c = 13.500 Å | |||||
| α = 97.56° | |||||
| β = 79.15° | |||||
| γ = 91.13° | |||||
| MOCOF-112 | A Pyrex tube (10 × 8 mm, o.d. × i.d.) was charged with TAP (17 mg, 0.025 mmol), BDA (7 mg, 0.052 mmol), Fe(OAc)2 (5.0 mg, 0.029 mmol), 1,4-bis(pyrid-4-yl)benzene (BPB) (15 mg, 0.065 mmol), o-DCB (0.7 mL), n-butanol (0.3 mL), THF (0.1 mL) and aqueous AcOH (0.20 mL, 9 M). After sonication for 8 h the tube was flash frozen at 77 K. Following one freeze–pump thaw cycle, the system was evacuated to an internal pressure of 50 mtorr and flame sealed. The reaction was heated at 120 °C for 72 h, yielding a dark purple precipitate. The precipitate was isolated by filtration and washed with DMF (10 mL) and THF (10 mL). It was then transferred to a Soxhlet extractor and thoroughly washed with THF for 24 h. Finally, the product was dried under vacuum at 60 °C for 18 h at 10−2 mtorr to yield the activated sample. Yield: 32.75 mg, 92% based on Fe(TAPP). | a = 25.880 Å | 1.0 Å | SCXRD | 222 |
| b = 26.370 Å | |||||
| c = 15.400 Å | |||||
| α = 88.79° | |||||
| β = 96.75° | |||||
| γ = 88.51° | |||||
| MOCOF-113 | A Pyrex tube (10 × 8 mm, o.d. × i.d.) was charged with TAPP (17 mg, 0.025 mmol), BDA (7 mg, 0.052 mmol), Fe(OAc)2 (5.0 mg, 0.029 mmol), 5,10,15,20-tetra(4-pyridyl)-21H,23H-porphine (22 mg, 0.035 mmol), o-DCB (0.6 mL), n-BuOH (0.4 mL) and aqueous AcOH (0.20 mL, 12 M). After 8 h of sonication, the tube was flash frozen at 77 K. Following one freeze–pump–thaw cycle, the system was evacuated to an internal pressure of 50 mtorr and flame sealed. The reaction was heated at 120 °C for 72 h, yielding a dark purple precipitate. The precipitate was isolated by filtration and washed with DMF (10 mL) and THF (10 mL). It was then transferred to a Soxhlet extractor and thoroughly washed with THF for 24 h. Finally, the product was dried under vacuum at 60 °C for 18 h at 10−2 mtorr to yield the activated sample. Yield: 33.93 mg, 86% based on Fe(TAPP). | a = 30.080 Å | 1.0 Å | SCXRD | 222 |
| b = 25.530 Å | |||||
| c = 19.580 Å | |||||
| α = 89.86° | |||||
| β = 94.36° | |||||
| γ = 75.42° |
 | ||
| Fig. 55 (a)–(c) Chemical structures of (a) mCOF-Ag, (b) WCOF-Ag and (c) mCOF-Cu. (d) Construction of MOCOFs using a coordination-templated strategy involving metal coordination and imine condensation. (e) The assembly process involves Fe-porphyrin tetraamine and various aldehydes and N-donor ligands. | ||
These findings are indicative of chirality transfer from the guest to the MOCOF and can be attributed to (1) the uniformly dispersed imine linkages forming hydrogen bonds with the chiral additives and (2) the ability of the MOCOF to form single crystals, allowing the guest's chirality to be consistently expressed throughout the network.
Furthermore, by pre-organizing covalent and coordination bonding sites in the metal-porphyrin tetraamine building block, the researchers successfully synthesized other four micrometre-sized MOCOF single crystals, MOCOF-110, MOCOF-111, MOCOF-112 and MOCOF-113,222 from combinations of different building blocks (Fig. 55). The high quality of the crystals allowed their structures to be determined by single-crystal ED. The unique 2D ABC stacking arrangement in MOCOF-113 incorporates two distinct pairs of porphyrin macrocycles lacking coordination, separated by approximately 9.69 Å. Inspired by this configuration, post-synthetic metalation was conducted to introduce metal centres, creating heterogeneous metalloporphyrin catalysts capable of cooperative catalysis. In detail, MOCOF-113 was reacted with metal precursors (Co(OAc)2·4H2O, Cu(OAc)2·H2O or PtCl2) in DMF solvent at 80 °C for 12 hours, yielding metalated frameworks (113 M) with efficiencies exceeding 90%.222
4.5. Other polymerization methods
Single crystal COFs have been synthesized using other methods, such as dynamic reaction and supercritical CO2 methods, which are distinct from those described above.| COFs | Synthetic conditions | Crystal parameters | Resolution | Method | Ref. |
|---|---|---|---|---|---|
 |
To a solution of tetrakis[4-(hydroxylamino)phenyl]methane (111 mg, 0.250 mmol) in mesitylene (30 mL) and ethanol (20 mL) was added Celite-supported silver carbonate (1.67 mmol g−1, 1.20 g, 4.00 mmol). The suspension quickly turned black. The mixture was stirred for 5 min at room temperature and then filtered through a coarse fritted funnel. The green filtrate was then further filtered into an Erlenmeyer flask through a 0.45 µm PTFE filter. The flask was finally sealed with a rubber septum and left undisturbed for 48 h at room temperature to yield yellow crystals of NPN-1 in 48% yield. | NA | NA | SCXRD | 223 |
 |
To a solution of tetrakis[4-(hydroxylamino)phenyl]silane (115 mg, 0.250 mmol) in mesitylene (18 mL) and methanol (12 mL) was added Celite-supported silver carbonate (1.67 mmol g−1, 1.20 g, 4.00 mmol). The suspension quickly turned black. The mixture was stirred for 5 min at room temperature and then filtered through a coarse fritted funnel. The green filtrate was then further filtered into an Erlenmeyer flask through a 0.45 µm PTFE filter. The flask was finally sealed with a rubber septum and left undisturbed for 48 h at room temperature to yield yellow crystals of NPN-2 in 63% yield. | NA | NA | SCXRD | 223 |
 |
To a solution of 1,3,5,7-tetrakis(4-(hydroxylamino)phenyl) adamantane (141 mg, 0.250 mmol) in a mixture of mesitylene (20 mL), ethanol (20 mL) and tetrahydrofuran (10 mL) was added Celite-supported silver carbonate (1.67 mmol g−1, 1.20 g, 4.00 mmol). The suspension quickly turned black. The mixture was stirred for 5 min at room temperature and then filtered through a coarse fritted funnel. The green filtrate was then further filtered into an Erlenmeyer flask through a 0.45 µm PTFE filter and was diluted with mesitylene (20 mL) and ethanol (20 mL). The flask was finally sealed with a rubber septum and left undisturbed for one week at room temperature to yield yellow crystals of NPN-3 in 44% yield. | NA | NA | SCXRD | 223 |
 | ||
| Fig. 56 Nitroso polymer networks of single crystals. (a)–(c) Chemical structures of (a) NPN-1, (b) NPN-2 and (c) NPN-3. | ||
 | ||
| Fig. 57 Optical images of single crystals. (a) NPN-1 grown from a 3:2 (vol/vol) ratio of mesitylene/ethanol. (b) NPN-1 grown from a 3:2 (vol/vol) ratio of benzene/ethanol. (c) NPN-2 grown from a 3:2 (vol/vol) ratio of mesitylene/methanol. (d) NPN-3 grown from a 4:4:1 (vol/vol) ratio of mesitylene/ethanol/tetrahydrofuran. Reprinted with permission from ref. 223. Copyright 2013 Springer Nature. | ||
The SCXRD analysis revealed that NPN-1 (Fig. 57a), NPN-2 (Fig. 57b) and NPN-3 (Fig. 57c) exhibited diamondoid azodioxy networks, with significant four-fold interpenetration, while the unit cell parameters and structural resolutions were not provided. NPN-1 and NPN-2 crystallized in a tetragonal space group (Pb2), forming isostructural diamondoid networks held together by azodioxy linkages at short distances of 1.287 Å and 1.332 Å, respectively. The interpenetration of the networks allowed for the inclusion of guest molecules within channels parallel to the c-axis. This highlights the tuneable nature of the networks. The distances between the closest tetrahedral nodes in NPN-1 and NPN-2 were 12.26 Å and 12.76 Å, respectively, reflecting the influence of the central atom (carbon for NPN-1 and silicon for NPN-2). The networks retained significant porosity, with 36–39% of the crystal volume accessible for guest inclusion. The channel dimensions of NPN-1 and NPN-2 are 7.3 × 3.3 Å and 7.8 × 3.4 Å, respectively. In contrast, NPN-3 with an adamantane core, consists of six-fold interpenetration, larger node distances (15.39 Å) and smaller channel dimensions (5.2 × 5.2 Å), compared to NPN-1 and NPN-2.223
000-fold compared to conventional solvothermal methods (Fig. 58), achieving crystal growth rates of 40 µm min−1 to produce crystals up to 0.2 mm in size within 2–5 minutes.
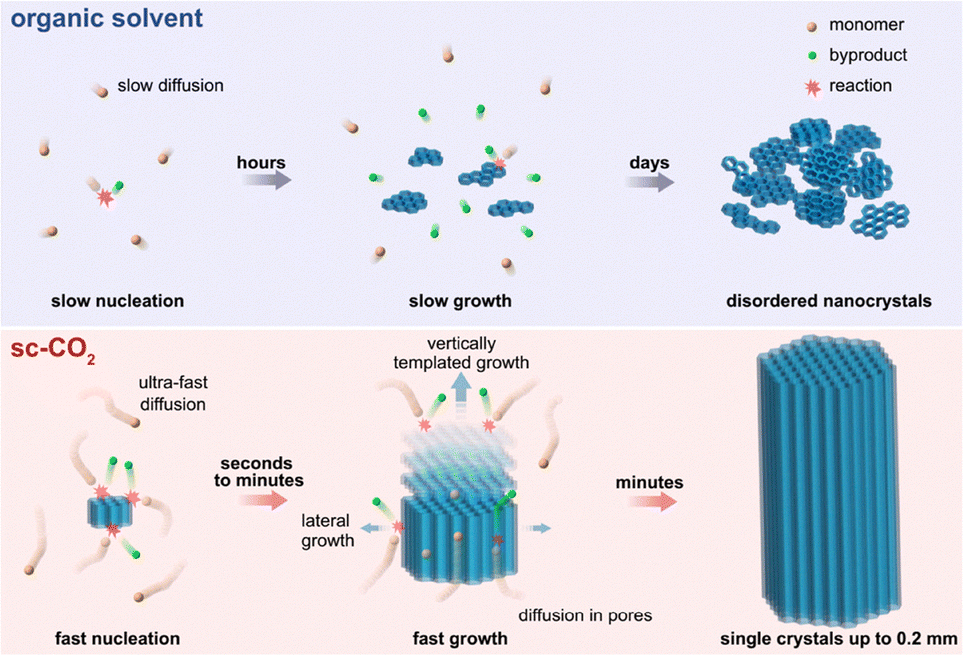 | ||
| Fig. 58 Polymerization mechanisms in different media of organic solvent and sc-CO2. Reprinted with permission from ref. 224. Copyright 2021 Springer Nature. | ||
The low viscosity and high diffusion coefficients of sc-CO2 facilitate the rapid transport of monomers to reaction sites and the efficient removal of by-products. This environment enables faster nucleation, polymerization and correction of defects, overcoming limitations posed by organic solvents. Specifically, a mixture of two monomers, i.e., TP (5.4 mg, 0.04 mmol) and 1,3,6,8-tretra(4-aminophenyle)pyrene (Py, 11.3 mg, 0.02 mmol), was dissolved in n-butyl alcohol (n-BuOH, 100 µL, 0.25 vol%) and AcOH (100 µL) and transferred to a stainless steel reactor (40 mL). The reactor was charged with 8 MPa CO2, heated to 80 °C and reacted for 5 min. After the reaction, the precipitate was collected by filtration, washed with acetone, tetrahydrofuran and dried in a vacuum oven at 100 °C. Control experiments, which were performed in a reaction tube with the same amount of monomers in n-BuOH/o-DCB/6M AcOH (0.5/0.5/0.1 mL) at 120 °C for 3 days, revealed the pivotal role of sc-CO2 in enhancing reaction kinetics and crystal quality. The single crystal structure is investigated by TEM, SAED and cross-polarized optical microscopy, but the structure was not resolved to identify atomic coordinates.224
The single crystals include a six-fold-symmetric FFT pattern, the same set of lattice fringe collected from different locations, and the uniform polarized light extinction. Moreover, these COFs demonstrated high photoluminescence polarization ratios and photoconductivity, indicative of their single-crystalline nature and superior optoelectronic properties.224
Supercritical CO2 accelerates the polymerization process by enhancing diffusion, increasing solubility and enabling faster error-checking (Fig. 58). This allows the synthesis of ultra-fast, large-sized and high-quality COF single crystals. This approach presents a new route toward single crystal COFs.
5. Properties and functions
5.1. Semiconducting properties
Robust skeleton and rigid framework structures render sp2-c COFs an ideal platform for exploring semiconducting properties.158 Single crystal sc-sp2c-COF-1 (Fig. 27) derived from imine-linked single crystal COF-303 (Fig. 7b) via imine-to-olefin transformation exhibited prominent semiconducting properties.188 The optical band gaps of sc-sp2c-COF-1 and COF-303 were determined to be 1.72 and 2.61 eV, respectively. The narrowed band gap for sc-sp2c-COF-1 was attributed to the enhanced π-conjugation enabled by the CC linkages. Notably, sc-sp2c-COF-1 promotes the spin coupling to trigger magnetization. Electron spin resonance (ESR) spectroscopic measurements revealed a strong resonance peak with a signal linewidth of 110 G from unpaired free electrons in sc-sp2c-COF-1 (Fig. 59a). Moreover, the ESR signal exhibited a g value of 2.0036 and its intensity increased significantly as the temperature decreased from 300 K to 100 K (Fig. 59a). In comparison, COF-303 exhibited much weaker ESR signals with a linewidth of 77 G and g value of 2.0041 (Fig. 59b). Remarkably, the ESR signal of sc-sp2c-COF-1 is stable for over ten months in air. These results demonstrated the critical role of the fully π-conjugated linkage in determining semiconducting properties.188
 | ||
| Fig. 59 (a) and (b) Temperature-dependent ESR spectra of (a) sc-sp2c-COF-1 and (b) COF-303 from 300 K to 100 K. Insets: Corresponding enlarged spectra. g refers to the g factor and the numbers under the trace represent the magnetic field corresponding to the g factor. (c) and (d) Magnetic (M)-applied field (H) profiles of (c) sc-sp2c-COF-1 and (d) COF-303 at different temperatures. (e) Frequency-resolved complex terahertz photoconductivity for 2DPAV-BTT-BP. (f) Comparison of charge carrier mobilities and electrical conductivities of 2DPAV-BTT-BP, amorphous 2DPAV-BTT-BP (Kn) and 2DPI-BTT-BP. (a)–(d) Reprinted with permission from ref. 188. Copyright 2025 Springer Nature. (e) and (f) Reprinted with permission from ref. 191. Copyright 2026 Springer Nature. | ||
Subsequently, the magnetic properties of sc-sp2c-COF-1 and COF-303 were evaluated by using a vibrating sample magnetometer. The two COFs exhibited ferromagnetic-like hysteresis curves at 2 and 300 K. Noticeably, sc-sp2c-COF-1 exhibited enhanced magnetization at 2 K upon increasing the intensity of the magnetic field, attaining the highest value of 0.081 emu g−1 at 7 T (Fig. 59c), while COF-303 only showed a low value of 0.016 emu g−1 under identical conditions (Fig. 59d). Moreover, the M–H test revealed that sc-sp2c-COF-1 and COF-303 exhibited coercive force (Hc) values of 184 and 1294 Oe, respectively. A narrower Hc for sc-sp2c-COF-1 indicated a higher magnetic field conversion efficiency. Theoretical and experimental magnetic susceptibility studies revealed that sc-sp2c-COF-1 had a spin density of 0.39 per pyrazine unit. The Curie temperatures of sc-sp2c-COF-1 and COF-303 were estimated to be about 303 and 287 K, respectively, indicating that the magnetic properties were enhanced upon imine-to-olefin transformation.188
Indeed, the transformation from imine to olefin linkages significantly improves the electronic properties of the single crystal COFs.191 By employing an SC–SC transformation strategy, the resulting benzotrithiophene-based 2DPAV-BTT-BP (Fig. 59e) with highly ordered π-conjugated skeletons display greatly enhanced charge transport properties. Notably, single crystal 2DPAV-BTT-BP exhibited an intrinsic charge carrier mobility of approximately 10 cm2 V−1 s−1, as revealed by frequency-resolved photoconductivity measurements. The charge mobility is nearly one order of magnitude higher than that of imine-linked 2DPI-BTT-BP (Fig. 59f). Moreover, 2DPAV-BTT-BP displays a photoconductivity intensity at least ten times stronger than that of amorphous 2DPAV-BTT-BP(Kn).191
The increased π-conjugation and structural order enable efficient charge delocalization across the framework, making the single crystal olefin-linked COFs promising candidates for organic electronics, optoelectronic devices and photocatalytic applications.191
While intrinsic electronic and magnetic properties define the fundamental functionality of single crystal COFs, their processibility and high crystallinity also enable integration into electronic devices. Due to the excellent solution processibility via interfacial transfer, a single-crystal 2D BECOF-PP (Fig. 45a) film can be applied to semiconductor devices (Fig. 60a) such as field-effect transistors (FET).175 The 2D BECOF-PP film generated a transverse field with positive charge to provide a pseudo gate for emulating synaptic plasticity. Thus, incorporating an 11 nm-thick 2D BECOF-PP film into a silicon nanowire (SiNW) FET resulted in the hybrid device 2D BECOF-PP/SiNW (Fig. 60a), which mimics neuronal synapses in biological systems to show a learning-erasing-forgetting memory process. 2D BECOF-PP/SiNW exhibited charge trapping and storage capabilities and exhibited substantial hysteresis when applying and removing bias voltage (Fig. 60b). This hysteresis behaviour is essential for memory effects, which include three interlocked processes: potentiation (learning), depression (erasing) and relaxation (forgetting). Noticeably, a fast response time of around 20 s for the saturation of the potentiation was observed for 2D BECOF-PP/SiNW (Fig. 60c), while the depression time constant in the forgetting period (23 s, VG = 5 V → 0 V) is three times longer than the time constant in the erasing period (6 s, VG = 5 V → −5 V) (Fig. 60d).
 | ||
| Fig. 60 (a) Schematics of the 2D BECOF-PP/SiNW hybrid device. (b) Transfer characteristics of the 2D BECOF-PP/SiNW hybrid device. (c) Learn–erase/learn–forget cycles. (d) History of the input signal in the relaxation process. (e) Short-term potentiation with learning pulse. Reprinted with permission from ref. 175. Copyright 2020 Wiley-VCH. | ||
In comparison, using either the 2D BECOF-PP film alone or the bare SiNW device could not generate a hysteresis loop, implying no memory effects. Furthermore, the 2D BECOF-PP/SiNW hybrid device exhibited a feature of neuron to present a history-dependent memory (Fig. 60e), which is stored as an ionic state in the 2D BECOF-PP film.175 This work illustrates how structural order and charge storage capacity translate directly into neuromorphic device functionality.
In addition to lateral device architectures, the well-defined single-crystalline domains of 2D COF films provide a unique opportunity to investigate vertical charge transport across stacked layers.218 Unlike polycrystalline aggregates or pressed crystalline films, single crystal 2D COF films exhibited well-defined single crystalline domains with distinct grain boundaries and ordered molecular arrangements, enabling systematic evaluation of π–π stacking–mediated transport perpendicular to the layers. Such out-of-plane charge transport is critical for vertical electronic devices, including vertical transistors, organic LEDs, and photovoltaic cells.194
To probe vertical conductivity, conductive atomic force microscopy (C-AFM) was employed (Fig. 61a). COF-2,5-Ph (Fig. 45d) film with a thickness of 220 nm was spread onto ITO glass uniformly to eliminate the effect of morphology (Fig. 61b and c). The charge carrier mobility of the COF-2,5-Ph film was derived from the current versus ramping bias voltage curve at different crystalline domains (Fig. 61c). The COF-2,5-Ph film exhibited the out-of-plane carrier mobility of 3.31 × 10−6, 1.41 × 10−6, 5.98 × 10−7 and 2.02 × 10−7 cm2 V−1 s−1 at point 1, 2, 3 and 4 (Fig. 61d and e), respectively. These results suggested that large crystalline domains in 2D COF films contribute more to the carrier mobility. Interestingly, the charge carrier mobility showed an increasing tendency of COF-BPh < COF-BPy < COF-2,3-Ph < COF-2,5-Ph (Fig. 45). The reason for this tendency is that replacing the linker phenyl units with pyridine and increasing the length of the aldehyde knot decrease the efficient stacking, thereby decreasing the vertical conductivity.218
 | ||
| Fig. 61 (a) Diagram of the electronic conductivity measurement. (b) A typical current image acquired under a constant bias of 2.5 V and a scan rate of 0.5 Hz. (c) The corresponding topography image of (b). (d) Current vs. bias voltage curves acquired at marked areas with numbers in (a) under a constant bias from −2 to 2 V. (e) J0.5 vs. (Vapp–Vbi) plots at points 1–4 in (a). Reprinted with permission from ref. 218. Copyright 2022 American Chemical Society. | ||
Semiconducting COFs enable unique exciton dynamic, but the presence of grain boundaries limits the crystal domain size making the precise evaluation a challenging task.186 Transient absorption (TA) spectroscopy performed on the well-dispersed, single-crystalline COF-5 colloids revealed a dramatic improvement in data quality, with a signal-to-noise enhancement of approximately two to three orders of magnitude compared to polycrystalline powders, enabling the reliable characterization of their intrinsic photophysical properties (Fig. 62). The exciton dynamics showed a clear dependence on particle size and pump fluence: at high fluence, smaller COF crystallites exhibited faster exciton decay kinetics, which is attributed to exciton–exciton annihilation (Fig. 62b). This fluence- and size-dependent behaviour provides evidence that excitons can diffuse over significantly longer length scales within these large single-crystalline domains than was previously possible in materials with smaller crystallites.186
 | ||
| Fig. 62 (a) Transient absorption spectra (excitation wavelength 360 nm) as a function of indicated probe delay time for 110 nm COF-5 colloids. (b) Exciton decay kinetics (dots) and fits (lines) of COF-5 colloids of different sizes at high photon fluence (91 mJ pulse−1) observed at 410 nm. Inset: Exciton decay kinetics (dots) and fit (line) at low photon fluence (3 mJ pulse−1) observed at 410 nm. Reprinted with permission from ref. 186. Copyright 2018 American Association for the Advancement of Science. | ||
Beyond electronic and magnetic behaviour, single crystal COFs exhibit rich and tuneable optical properties arising from their ordered architectures and precisely arranged π units. In particular, nonlinear optical (NLO) responses are highly sensitive to symmetry and structural alignment, where single crystal COFs with highly ordered and symmetric structures exhibit potential applications in photonics, frequency conversion, optical switching and laser generation.225–227 To investigate the nonlinear optical properties of single crystal COFs, polarization-dependent second harmonic generation (SHG) images (Fig. 63) were recorded with a nonlinear optical microscopy system equipped with a 150 fs laser centred at 1030 nm. Notably, single crystal scFZU-908 (Fig. 11f) and scFZU-215 (Fig. 11g) displayed prominent SHG signals,202 due to the non-centrosymmetric local positions of naphthyl or benzothiadiazole functional groups. In comparison, no SHG signals are observable for single crystal scFZU-518 (Fig. 11h) and scFZU-917 (Fig. 11i).202
 | ||
| Fig. 63 Second harmonic generation (SHG) map. (a) SHG map of scFZU-908. (b) SHG map of scFZU-215. Reprinted with permission from ref. 202. Copyright 2025 Wiley-VCH. | ||
5.2. Optical properties
In addition to nonlinear responses, the layered and highly ordered structures of COFs significantly influence their photoluminescent behaviour. Single crystal boroxine-linked COF colloids exhibit enhanced and distinct optical properties compared to their molecular monomers, primarily characterized by intense fluorescence attributable to their layered structures.177 The 2D COF colloids are highly emissive under UV light, in contrast to their weakly fluorescent monomers. Excitation-emission matrix (EEM) spectroscopy (Fig. 64a and b) was used to probe the origin of this enhancement. For DBD-COF (Fig. 51e), the emission intensity was an order of magnitude higher than its monomer at the same excitation wavelength (Fig. 64c), a phenomenon attributed to aggregation-induced emission due to restricted molecular motion within the 2D framework. For Py-COF (Fig. 51d), a blue shift in emission and a red shift in absorption were observed (Fig. 64d), consistent with the formation of J-aggregates, along with an additional exciplex emission feature at 435 nm when excited at 350 nm (Fig. 64e). These observations demonstrate that the precise spatial organization of chromophores in the COF structure facilitates through-space electronic communication, leading to emergent and tuneable optoelectronic properties.177 | ||
| Fig. 64 (a) EEMS of DBD-COF. (Inset) Optical image of the DPBBA monomer (left) and DBD-COF (right) under UV irradiation. (b) EEMS of Py-COF. (Inset) Optical image of the PyBA monomer (left) and Py-COF (right) under UV irradiation. (c) Emission of DBD-COF compared to the DPBBA monomer at the same concentration. (d) Normalized absorption and emission intensity of Py-COF compared to the PyBA monomer showing a red shift of the absorption spectra and blue shift and broadening of the emission spectra. (e) Normalized emission of Py-COF under two excitation wavelengths showing the emergence of an excimer emission (430 nm). Reprinted with permission from ref. 177. Copyright 2019 American Chemical Society. | ||
The influence of structural order becomes even more evident when examining polarization-dependent emission. Single crystal COFs synthesized via supercritical solvothermal polymerization (sc-COFs) exhibit highly polarized photoluminescence (PL), a direct consequence of their high crystallinity and well-aligned molecular arrangement.224 As shown in Fig. 65a–c, angle-dependent PL measurements on a sc-COFTP-Py (Fig. 29a) crystal reveal strong polarization anisotropy: when the polarization angle (θ) is varied from 0° to 180°, the PL intensity changes periodically, with a maximum dichroic ratio (DR) (defined as IPL maximum/IPL minimum) of 43.5 and a polarization ratio (ρ) of 0.96. This is among the highest values reported for organic or polymeric crystalline materials and confirms the single-crystalline nature and high structural order of the sc-COFs. In contrast, the os-COFTP-Py samples do not show angle-dependent emission (ρ ≈ 0), highlighting the superior optical quality of the sc-COF single crystals. These results demonstrate the potential of sc-COF single crystals in polarization-sensitive optoelectronic applications.224
 | ||
| Fig. 65 (a) Polarized fluorescence microscopy images and schematic illustration of sc-COFTP-Py and os-COFTP-Py at θ = 0° and 90°. (b) Angle-dependent PL intensity at different θ. The inset is an optical microscopy image of a sc-COFTP-Py crystal. (c) PL spectra of the crystal when θ is 0°, 30°, 60° and 90°. The DR (defined as IPL(max)/IPL(min)) is calculated to be 43.5. The scale bars are 10 µm in (a) and 20 µm in (b). Reprinted with permission from ref. 224. Copyright 2021 Springer Nature. | ||
Noticeably, sc-COFTP-Py exhibits angle-dependent emission with a dichroic ratio (DR) (defined as IPL maximum/IPL minimum) of 43.5, while the solvothermal product os-COFTP-Py has no angle-dependent emission properties (Fig. 65b). Correspondingly, the ρ values of sc-COFTP-Py and os-COFTP-Py are 0.96 and 0, respectively. The sc-COFTP-Py attains a photoconductivity (ϕΣμ) of 6.8 × 10−6 cm2 V−1 s−1 after growth in sc-CO2 for 20 min. In comparison, the ϕΣμ of os-COFTP-Py is 6.0 × 10−6 cm2 V−1 s−1 even after 3 days of growth (Fig. 65c).224 This comparison further underscores that crystallinity and molecular alignment synergistically enhance both optical anisotropy and photoconductive performance.
5.3. Molecular adsorption and separation
The adsorption and separation performance of single crystal COFs is intrinsically governed by their well-defined morphology, pore architecture and structural order. In particular, crystal morphology directly influences mass transport pathways and diffusion efficiency.211 For example, JUC-663-45, JUC-663-90 and JUC-663-135 exhibit aspect ratios of 0.39, 1.58 and 2.74, respectively, and accommodates Rhodamine B (Rh B, molecular size: 9.24 Å × 14.06 Å × 17.57 Å) in their mesopore (2.3 nm) along the c-axis direction. Indeed, JLU-663-45 and JUC-663-90 (Fig. 25) adsorbed 75% and 50% Rh B from Rh B aqueous solution (20 mg L−1) within 10 min, respectively, and completed the 100% adsorption of Rh B within 60 min (Fig. 66). In contrast, JUC-663-135 exhibited sluggish removal rates to adsorb 60% Rh B within 30 min and 75% Rh B within 60 min, respectively. These results demonstrated that the length of the adsorption path affects the adsorption efficiency. Under identical conditions, single crystal COFs with shorter c-axis exhibited higher adsorbate flux due to the shortened length of the diffusion path.211 | ||
| Fig. 66 Rh B adsorption. (a) UV-vis spectra of the rapid adsorption of Rh B for JUC-663-45 in 60 min. (b) UV-vis spectra of the rapid adsorption of Rh B for JUC-663-90 in 60 min. (c) UV-vis spectra of the rapid adsorption of Rh B for JUC-663-135 in 60 min. (d) Removal efficiency of Rh B by JUC-663-45, 90, and 135 in 60 min. Reprinted with permission from ref. 211. Copyright 2024 American Chemical Society. | ||
Beyond morphology-controlled diffusion, the intrinsic uniformity of pore size, geometry and chemical environment in single crystal COFs enables precise investigation of adsorption thermodynamics and host–guest interactions.167 A series of single crystal COFs, including COF-300 (Fig. 7a), COF-301 (Fig. 49a), SYSU-8 (Fig. 49b), SYSU-9 (Fig. 49c) and COF-320 (Fig. 49d), exhibited distinct CO2 uptake at 195 and 273 K.167 Interestingly, at 195 K, all COFs showed a distinct ‘gate-opening’ behaviour for CO2 adsorption, demonstrated by a sharp rise in CO2 uptake at a certain pressure, while this feature disappeared when measured at 273 K. The threshold pressure for gate opening and the overall CO2 adsorption capacity could be modulated by altering the pore surface chemistry or the adsorption temperature, enabling systematic control of gas uptake. Indeed, COF-300 exhibited CO2 uptake of 480 cm3 g−1 (P/P0 = 0.95) at 195 K, which is significantly higher than the other single crystal analogues under the same temperature (Fig. 67a). However, increasing the temperature to 273 K remarkably decreased the CO2 adsorption capability for all COFs. COF-300 displayed the lowest CO2 uptake of 8.5 cm3 g−1 (P/P0 = 0.95) among these single crystals, while the CO2 adsorption capabilities are measured to be 25, 30, 53 and 62 cm3 g−1 for COF-320, SYSU-8, COF-301 and SYSU-9, respectively (Fig. 67b).167 These results illustrate how single crystal platforms allow systematic correlation between pore chemistry, temperature and cooperative adsorption behaviour.
 | ||
| Fig. 67 Gas adsorption. CO2 adsorption isotherms of (a) COF-300 and (b) COF-301. (c) N2 adsorption isotherms at 77 K and pore size distributions (inset) of (c) 3D-TFB-COFs. (d) Total gravimetric CH4 adsorption isotherms of 3D-TFB-COFs at 298 K. High-pressure total CH4 uptake of activated TAMTFPB-COF (e) and TAPB-TFS-COF (f) at 273, 283, 298 and 308 K, respectively. (a) and (b) Reprinted with permission from ref. 167. Copyright 2023 Springer Nature. (c) and (d) Reprinted with permission from ref. 181. Copyright 2024 American Association for the Advancement of Science. (e) and (f) Reprinted with permission from ref. 216. Copyright 2024 American Chemical Society. | ||
Developing single crystal COFs with ultrahigh surface areas and microporous structures for methane storage is fundamentally important but remains a challenging task. Single crystal 3D-TFB-COF-Me (Fig. 43a) and 3D-TFB-COF-Et (Fig. 43b) with a rare self-catenated topology show a recorded surface area of 4298 and 4502 m2 g−1, respectively, and the same micropore of 1.1 nm (Fig. 67c).181 The ultrahigh surface area originated from the self-catenated structures that can self-pillar to avoid the close packing of different nets in an interpenetration topology.181
Notably, 3D-TFB-COF-Me and 3D-TFB-COF-Et exhibited the gravimetric methane uptakes of 423 and 429 mg g−1 at 298 K and 100 bar (Fig. 67d), respectively. Under standard temperature and pressure (STP), the volumetric methane uptakes reached up to 249 cm3 (STP) cm−3 for 3D-TFB-COF-Me and 264 cm3 (STP) cm−3 for 3D-TFB-COF-Et.181 Noteworthily, 3D-TFB-COF-Et exhibits an exceptional volumetric adsorption capacity, surpassing the CH4 storage DOE target [263 cm3 (STP) cm−3] at 298 K and 100 bar. Furthermore, 3D-TFB-COF-Me and 3D-TFB-COF-Et maintained their high-pressure CH4 adsorption performance after three cycles of adsorption and desorption, without showing changes in PXRD patterns.181 Overall, this study demonstrates a protype of self-catenated COFs with an exceptional balance of gravimetric and volumetric surface areas for methane storage.
Single crystal TAM-TFPB-COF (Fig. 39e) and TAPB-TFS-COF (Fig. 39f) exhibited methane storage.216 The activated TAM-TFPB-COF exhibited a methane storage capacity of 284 cm3 g−1 (16.9 wt%) at 80 bar and 298 K, while under the identical conditions, the activated TAPB-TFS-COF showed a methane storage capacity of 271 cm3 g−1 (16.2 wt%).216 These capacities correspond to the total uptake of 403 cm3 g−1 (22.3 wt%) for TAM-TFPB-COF and 413 cm3 g−1 (22.8 wt%) for TAPB-TFS-COF, respectively. Remarkably, at 298 K and 200 bar, the total uptakes of activated TAM-TFPB-COF (Fig. 67e) and TAPB-TFS-COF (Fig. 67f) are boosted to 517 and 570 cm3 g−1 (369 and 407 mg g−1), respectively.
Moreover, under working pressures between 80 bar (adsorption) and 5.8 bar (desorption) at 298 K, the activated TAM-TFPB-COF and TAPB-TFS-COF exhibit working capacities of 332 and 354 cm3 g−1, respectively. Noticeably, when the pressure is under 60 bar, the activated TAM-TFPB-COF exhibited a higher total uptake than the activated TAPB-TFS-COF.216 This phenomenon indicated that the interaction between TAM-TFPB-COF and methane is stronger than the force between activated TAPB-TFS-COF and methane at a low relative pressure. Consequently, TAM-TFPB-COF showed higher isosteric heats of adsorption of 12.4 kJ mol−1, while TAPB-TFS-COF showed lower isosteric heats of adsorption of 11.1 kJ mol−1. Overall, this work showcases the preparation of two novel single crystal COFs with exceptional microporosity and represents an example of atom engineering to tune the permanent porosity for methane storage.216
Except for the tuneable adsorption capacity, the precisely defined microporous structures of single crystal COFs enable highly selective gas separation. A representative example is single crystal 3D-TMTAPB-COF (Fig. 39f), which exhibits a narrow pore size distribution suitable for SF6/N2 separation (Fig. 68a).180 The material shows strong affinity for SF6, with adsorption capacities of 81.2 and 60.9 cm3 g−1 at 273 and 298 K, respectively, while adsorbing only minimal N2 under identical conditions. Consequently, the selectivity of SF6 over N2 is 335 at 1 bar and 298 K (Fig. 68b), outperforming most crystalline porous materials.195 Moreover, the isosteric heats of adsorption (Qst) were evaluated to be 33.5 and 14.8 kJ mol−1 for SF6 and N2, respectively, implying a stronger affinity for SF6 in 3D-TMTAPB-COF. The above results manifested that 3D-TMTAPB-COF is promising for SF6/N2 separation, due to the matched pore size and interaction strength.180
 | ||
| Fig. 68 Gas uptake and selectivity. (a) SF6 (solid circle) and N2 (open triangle) sorption isotherms of 3D-TMTAPB-COF at 273 K (blue) and 298 K (red). (b) IAST selectivity of SF6/N2 at a ratio of 10:90 at 298 K for 3D-TMTAPB-COF. (c) C2H2 adsorption isotherms at 195 K for COF-300, COF-300-AR and COF-300-CHO. (d) C2H4 adsorption isotherms at 195 K for COF-300, COF-300-AR, and COF-300-CHO. (e) IAST CO2 selectivity of TK-COFs calculated for a mixed gas of CO2:N2 = 15:85 at 273 and 298 K. (f) Isosteric heat of adsorption of CO2 for TK-COFs. (a) and (b) Reprinted with permission from ref. 195. Copyright 2023 American Chemical Society. (c) and (d) Reprinted with permission from ref. 189. Copyright 2025 American Chemical Society. (e) and (f) Reprinted with permission from ref. 168. Copyright 2025 Springer Nature. | ||
A stepwise structural modification via imine-to-amine-to-formamide transformation tunes the pore environments of single crystal COFs for gas adsorption.189 The parent COF-300 (Fig. 7a) and the intermediate COF-300-AR (Fig. 19c) exhibited structural flexibility and breathing behaviour, leading to exceptionally high gas uptake capacities. For example, the volumetric CO2 capacities of single crystal COF-300 and COF-300-AR were evaluated to be 455 and 372 cm3 g−1 at 1 atm, respectively. In contrast, COF-300-CHO (Fig. 19d) exhibited a negligible adsorption capacity for CO2 due to its smaller pore size and more robust structure.189
The study further investigated the sorption properties of COF-300 and COF-300-AR towards acetylene (C2H2) and ethylene (C2H4), which are two important industrial raw materials.189 COF-300 displayed a record C2H2 adsorption capacity of 417.1 cm3 g−1 at 195 K and 1 atm, while COF-300-AR showed a low C2H2 adsorption capacity of 376.5 cm3 g−1 under the identical conditions. Noticeably, COF-300 and COF-300-AR underwent multiple phase variations induced by C2H2, as revealed by the three-stage C2H2 adsorption process (Fig. 68c).189 In contrast, a two-step gated behaviour was found for both COF-300 and COF-300-AR upon adsorption of C2H4 at 195 K (Fig. 68d). COF-300 and COF-300-AR displayed the C2H4 uptake amounts of 277.8 and 227.0 cm3 g−1 at 1 atm, respectively. No noticeable loss of C2H2 storage capacity was found after five cycles, indicating that COF-300 is a promising material for C2H2 storage.189 Overall, this PSM approaches open opportunities for tailoring crystal stability, pore environments and host–guest interactions, thereby advancing the rational design of single crystal COFs for applications in gas storage.189
The advantages of single crystals become even more pronounced when comparing intrinsic separation performance with that of polycrystalline counterparts. Single-crystalline imine-linked 2D COFs (TAPPy-PDA and TAPB-DMPDA, Fig. 29) exhibit markedly different molecular separation behaviour in gas chromatography experiments involving benzene and cyclohexane.213 As shown in Fig. 69a, the column coated with single-crystal TAPPy-PDA COF achieved complete baseline separation of the benzene/cyclohexane mixture with an unusual and reversed selectivity, preferentially retaining cyclohexane over benzene. In comparison, the polycrystalline TAPPy-PDA sample showed no separation under identical conditions. Similarly, Fig. 69b shows that single-crystal TAPB-DMPDA also preferentially adsorbed cyclohexane, achieving partial separation, whereas its polycrystalline form exhibited the opposite (normal) selectivity, retaining benzene more strongly. This reversal in the elution order and the superior separation performance of the single crystals is attributed to their highly accessible, ordered pore channels, which facilitate entropically driven selective interactions with cyclohexane, as opposed to the polycrystalline materials where non-selective surface interactions and grain boundaries dominate the adsorption behaviour. These findings highlight that high single-crystal quality is crucial for realizing the intrinsic, precision-based separation properties of COFs.213
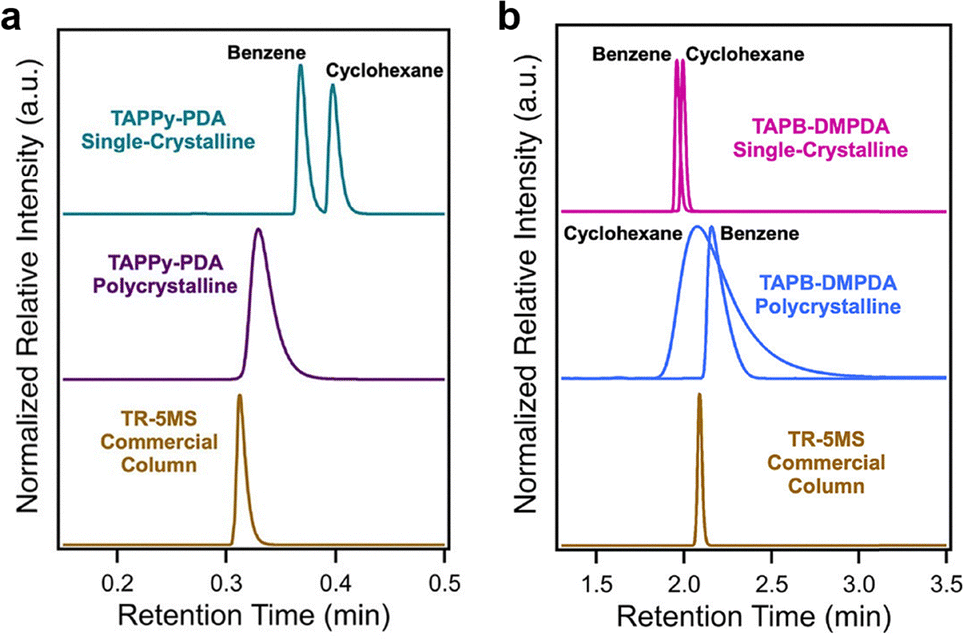 | ||
| Fig. 69 Gas chromatography separation performance of capillary columns (20 m length, 0.25 mm i.d.). (a) Column coated with the TAPPy-PDA COF (single and polycrystalline) and commercial standard TR-5MS column of the same dimensions. Data recorded at 423 K and 0.2 MPa. (b) Column coated with the TAPB-DMPDA COF (single and polycrystalline) and commercial standard TR-5MS column of the same dimensions. Data recorded at 393 K and 0.05 MPa. Reprinted with permission from ref. 213. Copyright 2022 American Chemical Society. | ||
Recently, single crystal 2.5D COFs, i.e. TK-COF-4 (Fig. 19a) and TK-COF-5 (Fig. 19b), were developed for selective adsorption of CO2.168 Gas sorption measurements revealed that TK-COF-4 and TK-COF-5 exhibit well-defined microporosity and show significant adsorption toward CO2 over N2. At 100 kPa, TK-COF-4 exhibited CO2 uptake capacities of 55 and 30 cm3 g−1 STP at 273 and 298 K, respectively, higher than TK-COF-5. The selectivity of CO2/N2 was calculated to be ca. 100 by using ideal adsorbed solution theory (IAST) at CO2/N2 = 15/85 (mol) (Fig. 68e). The average values of heat of adsorption (Qst) were evaluated to be 25.5 and 25.3 kJ mol−1 for TK-COF-4 and TK-COF-5 (Fig. 68f), respectively. The values of Qst are much lower than the aqueous amine method, suggesting a lower energy cost required by the adsorption-regeneration cycles.168 Notably, single crystal COFs display an advantageous balance between high CO2/N2 selectivity and relatively low Qst, which is typically difficult to achieve simultaneously in porous materials. This performance originates from the dense and periodically arranged primary amine sites, which enhance specific interactions with CO2 molecules while minimizing excessive binding strength. Furthermore, the highly crystalline nature of the materials enables faster CO2 adsorption kinetics compared to lower crystalline materials. The single crystal COFs also exhibit remarkable thermal and oxidative stability, maintaining both their crystal structure and CO2 adsorption capacity after thermal treatment at 100 °C in air, highlighting their potential for practical gas-capture applications.168
These studies establish a coherent structure–transport–selectivity relationship in single crystal COFs: morphology controls diffusion kinetics; uniform pore architecture enables cooperative and temperature-dependent adsorption; precise micropore matching governs gas selectivity; long-range crystallinity unlocks intrinsic, precision-based molecular separation that is inaccessible in polycrystalline materials. Such hierarchical structural control underscores the unique potential of single crystal COFs as model platforms for fundamental adsorption studies and high-performance separation technologies.
5.4. Proton conduction
Proton conducting materials are key components in proton-exchange membrane fuel cells, yet their practical application is often limited by insufficient thermal stability and structural degradation under strongly acidic conditions. In particular, when proton carriers such as phosphoric acid (H3PO4) are incorporated into porous frameworks, the host materials must exhibit robust acid/base resistance and strong covalent linkages to maintain structural integrity and ensure long-term conductivity. In this context, single crystal COFs provide an advantageous platform due to their well-defined pore environments and enhanced structural stability.Single crystal USTB-5r (Fig. 39c) and USTB-5o (Fig. 39d), derived from single crystal imine-linked USTB-5, exhibited stronger chemical linkage of amine and amide bonds, respectively, showing promise as effective supports for preparing proton-exchange membranes.182 Indeed, H3PO4 has been loaded into the pores of USTB-5r and USTB-5o under vacuum with high amounts of 194 wt% and 231 wt% based on their porosity, respectively, resulting in H3PO4@USTB-5r and H3PO4@USTB-5o.
Proton conductivity measurements were performed at different temperatures. H3PO4@USTB-5r exhibited anhydrous proton conductivities (Fig. 70a) of 5.3 × 10−2, 4.2 × 10−2, 3.8 × 10−2 and 3.5 × 10−2 S m−1 at 140, 130, 120, 110 and 100 °C, respectively. In contrast, H3PO4@USTB-5o exhibited the slightly higher proton conductivities (Fig. 70b) of 6.3 × 10−2, 5.6 × 10−2, 4.9 × 10−2, 4.2 × 10−2 and 3.8 × 10−2 S m−1 at 140, 130, 120, 110 and 100 °C, respectively. The anhydrous proton conductivities of H3PO4@USTB-5r and H3PO4@USTB-5o exceeded most COFs at 140 °C, excepting for H3PO4@TPB-DMeTP-COF (1.3 × 10−1 S m−1 at 140 °C)41 and H3PO4@TPB-DABI-COF (1.2 × 10−1 S m−1 at 140 °C). The activation energies of H3PO4@USTB-5r and H3PO4@USTB-5o were calculated to be 0.17 and 0.20 eV, respectively. Moreover, H3PO4@USTB-5r and H3PO4@USTB-5o exhibited high stability and reproducibility over both heating and cooling cycles within the temperature range of 0–140 °C (Fig. 70c and d).182
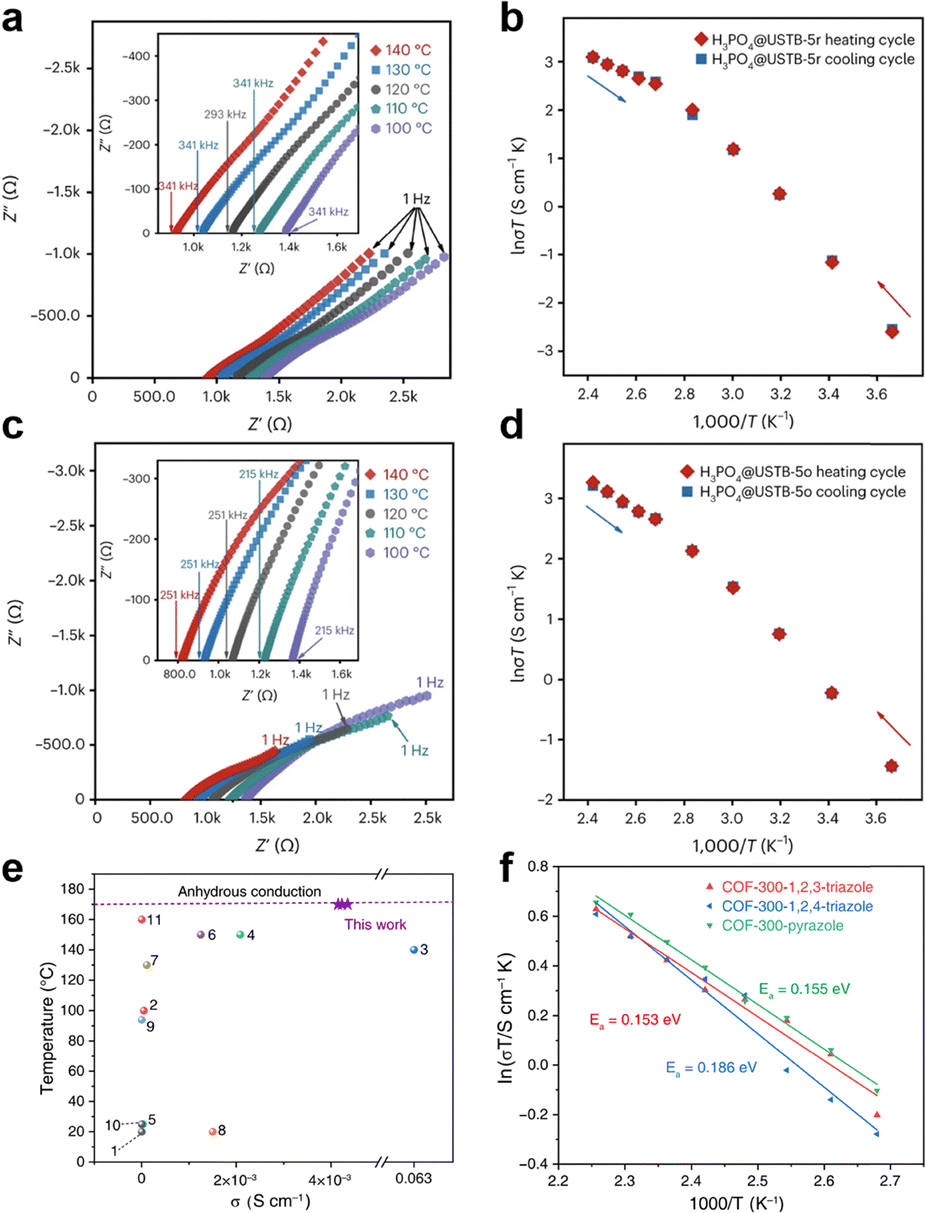 | ||
| Fig. 70 Proton conductivity. (a) Nyquist plots of H3PO4@USTB-5r at 140–100 °C. (b) Arrhenius plots of H3PO4@USTB-5r over both heating and cooling cycles within the temperature range of 140–0 °C. (c) Nyquist plots of H3PO4@USTB-5o at 140–100 °C. (d) Arrhenius plots of H3PO4@USTB-5o over both heating and cooling cycles within the temperature range of 140–0 °C. Insets: Enlarged Nyquist plots. (e) Proton conductivities of COF-300-azole in comparison to other single-crystal proton-conducting materials reported. (f) Activation energy (Ea) of COF-300-1,2,3-triazole, COF-300-1,2,4-triazole and COF-300-pyrazole. (a)–(d) Reprinted with permission from ref. 182. Copyright 2023 Springer Nature. (e) and (f) Reprinted with permission from ref. 190. Copyright 2026 Springer Nature. | ||
More recently, the well-known single crystal COF-300 was chemically modified to produce highly efficient proton-conducting materials while preserving single crystal order.190
A solvent-free melt-phase PSM strategy introduce azole molecules into the COF structure. The molten azole molecules diffuse through the pores and react with imine linkages in the framework, converting CN bonds into more stable amine linkages while simultaneously anchoring proton-conductive azole groups.190 The resulting COF-300-1,2,3-triazole (Fig. 19e), COF-300-1,2,4-triazole (Fig. 9f) and COF-300-pyrazole (Fig. 19g) contain extended hydrogen-bond networks formed between azole groups. These hydrogen-bond networks play a critical role in enabling efficient proton transport within the framework. Electrochemical measurements showed that COF-300-1,2,3-triazole, COF-300-1,2,4-triazole and COF-300-pyrazole exhibited anhydrous proton conductivities of 4.23 × 10−3, 4.15 × 10−3 and 4.32 × 10−3 S cm−1 at 170 °C (Fig. 70e), respectively. Mechanistic studies on proton conduction revealed a Grotthuss-type hopping mechanism with an activation energy of 0.153 eV for COF-300-1,2,3-triazole, 0.186 eV for COF-300-1,2,4-triazole and 0.155 eV for COF-300-pyrazole (Fig. 70f), respectively.190
These results highlight the advantage of single crystal COFs in studying structure–property relationships in proton conduction, as atomic-level functional units can be directly correlated with transport performance.190
5.5. Biological applications
Efficient delivery of functional molecules into plant cells remains a longstanding challenge in plant biotechnology. The cellulose microfibril network composing hemicellulose and pectin exhibited a cell wall penetration limit of 5–20 nm. This small size has limited many efforts on the development of delivery methods for biolistics and agrobacterium, where such attempts suffer from low efficiency and poor universality. Overcoming this physical barrier therefore requires nanocarriers that combine ultrasmall dimensions, structural robustness and biocompatibility.Exfoliation of single crystal 2D COFs into nanosheets extends their function in biological science for drug delivery due to the improved biocompatibility and cell membrane penetrability. However, due to high surface free energy and π–π interactions, the resultant 2D COF nanosheets are prone to aggregation, hence reducing their efficiency and functions.166
Introducing the steric hindrance to modulate the planar structure of single crystal 2D COFs weakens their interlayer stacking and prevents the aggregation of nanosheets, thereby leading to facile exfoliation and broad applications. Single crystal DiSTAP-1 (Fig. 39a) with poor planarity can be easily exfoliated into nanoflakes for plant biotechnology (Fig. 71a). Interestingly, a slit-shaped pore in the cell wall was generated, which can be ascribed to the penetration of 2D nanoflakes (Fig. 71b). Furthermore, infiltrating 3, 5 and 10 mg L−1 DiSTAP-1/quaternary chitosan (QC) into the leaves of N. benthamiana, kai-lan and spinach did not change the colour of leaves on both adaxial and abaxial sides, implying that DiSTAP-1/QC nanoflakes exhibited low phytotoxicity.166 In comparison, using chitosan-wrapped single-wall carbon nanotubes at 5 mg L−1 led to prominent leaf senescence, underscoring the superior biocompatibility of the COF-based nanocarrier.
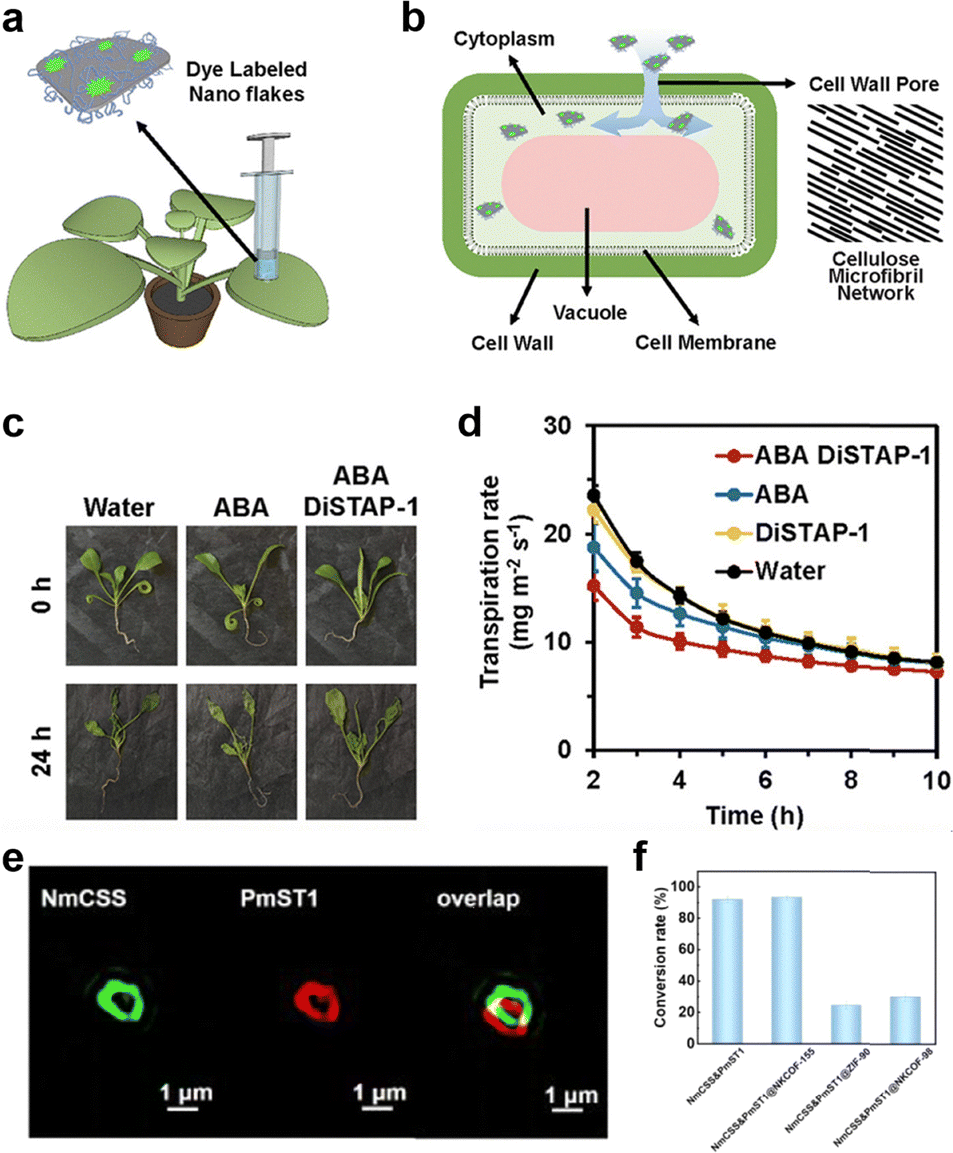 | ||
| Fig. 71 (a) Illustration of the infiltration of nanoflakes into intact plant leaves. (b) Illustration of the pathway of nanoflakes into the cytoplasm. (c) Photos of spinach before and after the drought experiment. (d) Transpiration rate of spinach leaves after infiltration of pure water, DiSTAP-1/QC nanocomplex solution, ABA solution, and ABA with the DiSTAP-1/QC nanocomplex solution. (e) SIM images of NmCSS&PmST1@NKCOF-155. (f) The conversion rate of the reactions. (a)–(d) Reprinted with permission from ref. 166. Copyright 2023 American Chemical Society. (e) and (f) Reprinted with permission from ref. 209. Copyright 2026 American Chemical Society. | ||
Furthermore, DiSTAP-1/QC has been used as a nanocarrier for loading and delivering plant hormone abscisic acid (ABA) in spinach plants. Control experiments via infiltration of pure water and DiSTAP-1/QC dispersion on spinach leaves showed a higher transpiration rate than that of spinach leaves infiltrated with free ABA in the first 8 h. Due to the fast degradation of ABA after 8 h, there was no difference among them (Fig. 71c). By comparison, using the ABA/DiSTAP-1/QC nanoflake composite to treat spinach leaves resulted in the lowest transpiration rate throughout the 10 h duration, highlighting the protecting function and enhanced cell wall penetration of the DiSTAP-1/QC nanoflakes.166 This sustained physiological response indicates that the nanoflakes not only enhance cell wall penetration but also protect ABA from rapid degradation, thereby prolonging its biological activity.
Single crystal COFs have recently emerged as promising platforms for constructing biohybrid materials, particularly for enzyme immobilization and biocatalysis.209 A representative example involves the construction of the single crystal NKCOF-155 (Fig. 19h), which hosts biomolecules within its ordered porous structure.209
The presence of dynamic imine linkages within the framework allows reversible bond exchange reactions, which can be exploited to covalently anchor biomolecules. Using this strategy, enzymes were successfully immobilized within the pores of NKCOF-155 through reaction between aldehyde groups and amino groups on the enzyme surface.209
The resulting enzyme–COF composite exhibits efficient catalytic activity while benefiting from the stability, porosity and structural order of the single crystal NKCOF-155. Indeed, two glycoenzymes (NmCSS and PmST1) were assembled with NKCOF-155 for the synthesis of human milk oligosaccharides (HMOs) (Fig. 71e). The catalytic conversion rate reaches the highest efficiency of 92% under the NmCSS/PmST1 molar ratio of 6:1 (Fig. 71f). Remarkably, NmCSS&PmST1@NKCOF-155 maintains a relative conversion rate of 60% after 5 cycles.209
These results demonstrate the potential of single crystal COFs as platforms for biohybrid catalysts and biomolecule immobilization, highlighting the growing intersection between COF chemistry and biological applications.209
5.6. Mechanical properties
2D polymers often exhibit either high strength or excellent toughness but seldom can have both simultaneously.219 When these materials are synthesized with high crystallinity, they typically become brittle and are prone to fragility. This is due to the presence of isolated and small crystalline domains as well as the grain boundaries. In comparison, amorphous polymers composed of linear polymers with entangled structures usually exhibit elastic yet tough mechanical properties.By using linear polymers as sacrificial go-betweens to interweave the isolated crystalline domains, strong, tough and elastic films of 2D COF-1 and 2D COF-2 (Fig. 45h) have been developed.112 Noticeably, aliphatic bi-amines were used for polymerization with an aldehyde linker to form linear chains, thereby facilitating the entanglement of adjacent domains of COFs at the grain boundary. As a result, 2D COF-1 and 2D COF-2 achieved Young's moduli of 56.7(±7.4) GPa and 73.4(±11.6) GPa, and breaking strengths of 82.2(±9.1) N m−1 and 29.5(±7.2) N m−1, respectively (Fig. 72).219 The simultaneous enhancement in stiffness and mechanical resilience highlights the effectiveness of the proposed strategy in mitigating brittleness without sacrificing crystallinity.
 | ||
| Fig. 72 (a) Schematic of AFM nanoindentation performed on a 2D COF-1 film suspended over a holey copper grid, with holes of about 47 µm in diameter, with an indentation depth of approximately 1.2 µm under a constant indentation rate. (b) Force–displacement curves of 20 cycles of loading (upper) and unloading of AFM tips with a radius of roughly 100 nm at the same position under a constant rate. (c) Force–displacement curve of the 2D COF-1 film until rupture at the same position as indented for b (purple curve) and another 10 cycles of loading (upper) and unloading of AFM tips adjacent to the edge of the rupture areas. Reprinted with permission from ref. 219. Copyright 2024 Springer Nature. | ||
Beyond grain-boundary engineering, post-synthetic modification of single-crystal COFs offers another powerful route to tailor mechanical properties at the molecular level. Single crystal 1D mCOF-Ag exhibits significantly enhanced mechanical properties after undergoing a post-synthetic crystalline-state polymerization process (Fig. 73a).184 The mechanical strength was quantitatively characterized by nano-indentation on isolated micro-sized crystals, which revealed that the Young's modulus of the pristine mCOF-Ag was 9.0 GPa (Fig. 73b). After cross-linking the pendant amine groups between adjacent chains using glyoxal to form the woven network WCOF-Ag, the Young's modulus more than doubled to 19.1 GPa (Fig. 73b). This dramatic increase in stiffness is attributed to the formation of a covalently connected 3D woven architecture (Fig. 73a), which imparts greater rigidity to the material compared to the original structure primarily held together by coordination bonds and non-covalent interactions. This demonstrates the unique potential of post-synthetic modification in single-crystal COFs to tailor their mechanical properties for advanced applications.184
 | ||
| Fig. 73 (a) Confined environments of mCOF-Ag (left) and the ideal structure of the cross-linked framework, WCOF-Ag (right). (b) The Young's moduli of mCOF-Ag and WCOF-Ag. Reprinted with permission from ref. 184. Copyright 2020 Springer Nature. | ||
5.7. Catalysis
The precise structural definition and long-range order of single crystal COFs provide unique opportunities for constructing heterogeneous catalysts with molecular-level control over active sites and reaction environments. In particular, the integration of well-defined chiral pores and tuneable acidic functionalities enables the development of solid catalysts capable of rivalling or even surpassing their homogeneous counterparts in asymmetric synthesis.In this context, two single crystal chiral COFs, (R)-39 and (R)-39-SH (Fig. 23), were developed as efficient and structured heterogeneous Brønsted acid catalysts.210 Notably, their enhanced acidity in conjugation with well-defined chiral pores, enable them to outperform homogeneous catalysts in three key asymmetric reactions: (1) asymmetric acetalization of 2-aminobenzamide and aldehydes, up to 99% yield and 99% ee, (2) enantioselective transfer hydrogenation of ketimines, up to 91% yield and 99.5% ee, and (3) anti-selective three-component Mannich reactions, up to 85% yield and 96% ee.
The confinement effect of chiral pores is proven essential for high enantioselectivity, as reactions with bulky substrates that cannot access the active sites show a dramatic drop in yield. Furthermore, the CCOFs demonstrate excellent heterogeneity and recyclability, maintaining high crystallinity and catalytic performance over at least ten cycles without significant loss of activity or enantioselectivity (Fig. 74), underscoring their robustness and potential in asymmetric synthesis. This study represents an advancement in the design and synthesis of solid Brønsted acid catalysts, demonstrating the potential of structurally defined, porous and homochiral materials for applications in asymmetric catalysis.210
 | ||
| Fig. 74 (a) Recycling results of (R)-39 in the acetalization of 2-aminobenzamide with 4-fluorobenzaldehyde. (b) Recycling results of (R)-39 in the Mannich reaction of aniline, 4-fluorobenzaldehyde and tetrahydrothiopyran-4-one. Reprinted with permission from ref. 210. Copyright 2025 American Chemical Society. | ||
In addition to chiral organic catalysis, CO2 reduction has drawn tremendous research attention due to the increasing public concern in global warming.222 A series of single crystal MOCOFs for efficient photoelectrochemical (PEC) CO2 reduction to ethanol have been developed. The MOCOFs, including MOCOF-110, 111, 112 and 113 (Fig. 55), were constructed via a coordination-driven one-pot hetero-assembly strategy, forming well-defined 3D structures with pcu topology or layered stacking models. Post synthetic metalation of MOCOF-113 with Cu, Co or Pt yielded 113-Cu, 113-Co and 113-Pt. Among them, 113-Pt exhibited outstanding PEC CO2 reduction performance, achieving a high faradaic efficiency of 83.5% for ethanol at −1.0 V vs. RHE (Fig. 75) with a current density of −8.8 mA cm−2, significantly outperforming many reported catalysts. In situ Raman spectroscopy and DFT calculations revealed that the twin porphyrin-Pt sites facilitate C–C coupling via stabilized intermediates, enhancing ethanol selectivity. The catalysts showed stability over 100 hours of operation, while retaining 95% of its initial activity. These results highlighted the potential of single-crystalline MOCOFs as efficient catalysts for selective CO2-to-ethanol conversion.222
 | ||
| Fig. 75 (a) FE and the product distribution recorded at −1.0 V and 1 Sun illumination. (b) FE and the product distribution at different polarization potentials for 113-Pt. Reprinted with permission from ref. 222. Copyright 2025 American Chemical Society. | ||
5.8. Other properties and functions
Single-crystal COF films provide a uniform material platform to investigate their basic thermal properties.217 The SYSU-7 (Fig. 45c) film with a single crystal ribbon size of 3 × 0.2 µm2 achieves a thermal conductivity κ of 5.31(± 0.37) W m−1 K−1 at 305 K under vacuum, which is more than two orders of magnitude higher than that of other COFs (Fig. 76a). Noticeably, decreasing the ribbon size reduces the thermal conductivity. For example, when the ribbon size decreases to 0.1 × 0.02 µm2, the SYSU-7 film only attains a κ value below 0.4 W m−1 K−1 (Fig. 76b). The small-sized ribbon exhibits a high density of grain boundary, which increases phonon scattering by transmission and reflection, forming thermal boundary resistance (Kapitza resistance).217 The SYSU-7 film showed a Kapitza resistance value of 2.48 × 10−7 m2 K W−1, which is one magnitude larger than Al2O3 and three orders of magnitude higher than diamond.217 | ||
| Fig. 76 Thermal conductivity measurement. (a) Thermal conductivity of the suspended SYSU-7 film measured both in air and in vacuum as a function of temperature. (b) Thermal conductivity versus ribbon size at 305 and 317 K, respectively. Reprinted with permission from ref. 217. Copyright 2021 American Chemical Society. | ||
6. Conclusion and perspectives
In this review, we have surveyed the design, synthesis and functional exploration of single-crystal COFs. Single-crystal studies enable molecular design of building units to predetermine the topology, skeleton, linkage chemistry and pore architecture, unveiling intrinsic properties and establishing structure–property relationships. Despite substantial interdisciplinary and global efforts, the development of general and reliable synthetic methodologies for single-crystal COFs remains at an early stage, with challenges persisting for 1D and 2D systems. Over the past two decades, progress in the polymerization of molecular building blocks has shaped COFs into a distinct and rapidly evolving field. Nevertheless, several fundamental challenges remain unresolved, underscoring both the complexity of single-crystal COF formation and the significant opportunities for future breakthroughs in this area.(1) Single crystal 1D and 2D COFs: single crystal 3D COFs have been well established in terms of methodology and structure. In contrast, single crystal 1D COFs remained unprecedented,229 while 2D COFs are still limited in methodology, example, and structural resolution. This difference originates from their structures, where 3D COFs are formed by covalent bonds, while 2D COFs involve both covalent bonds and noncovalent interactions. The noncovalent interactions involved in the formation and crystallization of 2D COFs are essential yet complex and independent from covalent bonds, making the reaction system complex and requiring additional tools beyond covalent bonds to achieve effective control. These are frontiers in single crystal COFs that should be the focus of further efforts.
(2) Polymerization reactions: to date, the realization of single-crystal COFs has largely been confined to a narrow set of dynamic covalent linkages, including boronate esters, imines, amines and amides. Extending single-crystal COF synthesis to other types of linkages, particularly those formed via less reversible or irreversible reactions, and beyond conventional condensation chemistry to encompass coupling reactions,53,228 remains a fundamental and compelling challenge. Addressing this issue is not only of critical importance for expanding the structural and functional diversity of COFs but also represents an interesting subject for future investigation.
(3) Synthetic methodology: despite the availability of more than ten distinct synthetic routes for polycrystalline COFs, the synthesis of single-crystal COFs remains overwhelmingly reliant on solvothermal methods, underscoring a clear methodological bottleneck in the field. The development of fundamentally new synthetic strategies is therefore urgently needed not only to broaden the synthetic toolbox but also to enable more precise control over nucleation and crystal growth. This would improve scalability and reproducibility and ultimately accelerate the production of single crystals suitable for practical implementation and device integration. In addition, phase diagrams play a crucial role in understanding nucleation pathways, crystallization windows and the interplay between thermodynamic and kinetic factors in COFs. Establishing such diagrams for COF systems would provide valuable guidance for rational crystal growth and hence deserve more efforts.
(4) Artificial intelligence (AI): AI-driven approaches, including autonomous and robotic chemists, have emerged as powerful tools for accelerating chemical discovery and optimization. Their application to the synthesis of single-crystal COFs represents a particularly promising yet largely unexplored direction, given the current reliance on extensive empirical condition screening and labour-intensive experimentation. Integrating AI-guided design, optimization and closed-loop experimentation have the potential to transform single-crystal COF synthesis from a trial-and-error process into a more predictive, efficient and scalable paradigm.
In short, single-crystal COFs provide a robust foundation for definitive structural determination and for the systematic investigation of intrinsic structure–property relationships. Further exploration of single crystal 1D, 2D and 3D COFs is expected to deepen fundamental understanding of their molecular architectures and physicochemical behaviours, while gradually clarifying their applications in areas such as electronic, catalytic and quantum materials. In the coming 3–5 years, incremental but meaningful progress is likely to arise from improved control over covalent reactions and supramolecular polymerization processes, leading to a broader set of synthetic approaches, novel materials and implementations.
Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Acknowledgements
D.J. acknowledges the MOE Tier 1 grant (A-8003537-00-00) and Tier 2 grant (T2EP50224-0011).Notes and references
- C. Wang, H. Dong, L. Jiang and W. Hu, Chem. Soc. Rev., 2018, 47, 422–500 RSC.
- X. Zhang, H. Dong and W. Hu, Adv. Mater., 2018, 30, 1801048 CrossRef PubMed.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS.
- R. Liu, K. T. Tan, Y. Gong, Y. Chen, Z. Li, S. Xie, T. He, Z. Lu, H. Yang and D. Jiang, Chem. Soc. Rev., 2021, 50, 120–242 RSC.
- X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC.
- N. Huang, P. Wang and D. Jiang, Nat. Rev. Mater., 2016, 1, 1–19 Search PubMed.
- K. Geng, V. Arumugam, H. Xu, Y. Gao and D. Jiang, Prog. Polym. Sci., 2020, 108, 101288 CrossRef CAS.
- E. Jin, K. Geng, K. H. Lee, W. Jiang, J. Li, Q. Jiang, S. Irle and D. Jiang, Angew. Chem., Int. Ed., 2020, 59, 12162–12169 CrossRef CAS PubMed.
- X. Feng, Y. Dong and D. Jiang, CrystEngComm, 2013, 15, 1508–1511 RSC.
- T. He, K. Geng and D. Jiang, Trends Chem., 2021, 3, 431–444 CrossRef CAS.
- Y. Li, L. Guo, Y. Lv, Z. Zhao, Y. Ma, W. Chen, G. Xing, D. Jiang and L. Chen, Angew. Chem., Int. Ed., 2021, 60, 5363–5369 CrossRef CAS PubMed.
- N. Huang, X. Chen, R. Krishna and D. Jiang, Angew. Chem., Int. Ed., 2015, 54, 2986–2990 CrossRef CAS PubMed.
- L. Zhai, N. Huang, H. Xu, Q. Chen and D. Jiang, Chem. Commun., 2017, 53, 4242–4245 RSC.
- Y. Fu, Y. Wu, S. Chen, W. Zhang, Y. Zhang, T. Yan, B. Yang and H. Ma, ACS Nano, 2021, 15, 19743–19755 CrossRef CAS.
- Z. Zhou, T. Ma, H. Zhang, S. Chheda, H. Li, K. Wang, S. Ehrling, R. Giovine, C. Li and A. H. Alawadhi, Nature, 2024, 635, 96–101 CrossRef CAS PubMed.
- H. Lyu, H. Li, N. Hanikel, K. Wang and O. M. Yaghi, J. Am. Chem. Soc., 2022, 144, 12989–12995 CrossRef CAS PubMed.
- C. J. Doonan, D. J. Tranchemontagne, T. G. Glover, J. R. Hunt and O. M. Yaghi, Nat. Chem., 2010, 2, 235–238 CrossRef CAS PubMed.
- H. Li, A. Dilipkumar, S. Abubakar and D. Zhao, Chem. Soc. Rev., 2023, 52, 6294–6329 RSC.
- C. Sun, Y. Zhu, P. Shao, L. Chen, X. Huang, S. Zhao, D. Ma, X. Jing, B. Wang and X. Feng, Angew. Chem., Int. Ed., 2023, 62, e202217103 CrossRef CAS PubMed.
- C. Sun, D. Sheng, B. Wang and X. Feng, Angew. Chem., Int. Ed., 2023, 62, e202303378 CrossRef CAS PubMed.
- H. L. Nguyen, Adv. Mater., 2023, 35, 2300018 CrossRef CAS.
- P. Wang, X. Chen, Q. Jiang, M. Addicoat, N. Huang, S. Dalapati, T. Heine, F. Huo and D. Jiang, Angew. Chem., Int. Ed., 2019, 58, 15922–15927 CrossRef CAS PubMed.
- S. Xie, M. A. Addicoat and D. Jiang, J. Am. Chem. Soc., 2024, 146, 32640–32650 CrossRef CAS PubMed.
- D. Chen, W. Chen, Y. Wu, L. Wang, X. Wu, H. Xu and L. Chen, Angew. Chem., Int. Ed., 2023, 62, e202217479 CrossRef CAS PubMed.
- Z. Wang, S. Zhang, Y. Chen, Z. Zhang and S. Ma, Chem. Soc. Rev., 2020, 49, 708–735 RSC.
- J. Liu, G. Han, D. Zhao, K. Lu, J. Gao and T.-S. Chung, Sci. Adv., 2020, 6, eabb1110 CrossRef CAS PubMed.
- K. Dey, M. Pal, K. C. Rout, S. Kunjattu H, A. Das, R. Mukherjee, U. K. Kharul and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 13083–13091 CrossRef CAS.
- H. Yang, L. Yang, H. Wang, Z. Xu, Y. Zhao, Y. Luo, N. Nasir, Y. Song, H. Wu and F. Pan, Nat. Commun., 2019, 10, 2101 CrossRef PubMed.
- H. Wang, M. Wang, X. Liang, J. Yuan, H. Yang, S. Wang, Y. Ren, H. Wu, F. Pan and Z. Jiang, Chem. Soc. Rev., 2021, 50, 5468–5516 RSC.
- X. Mu, S. Xie, X. Ye, S. Tao, J. Li and D. Jiang, J. Am. Chem. Soc., 2024, 146, 25118–25124 CrossRef CAS PubMed.
- Z. Li, K. Geng, T. He, K. T. Tan, N. Huang, Q. Jiang, Y. Nagao and D. Jiang, Angew. Chem., Int. Ed., 2021, 60, 19419–19427 CrossRef CAS PubMed.
- Z. Li, N. Huang, K. H. Lee, Y. Feng, S. Tao, Q. Jiang, Y. Nagao, S. Irle and D. Jiang, J. Am. Chem. Soc., 2018, 140, 12374–12377 CrossRef CAS PubMed.
- S. Dalapati, E. Jin, M. Addicoat, T. Heine and D. Jiang, J. Am. Chem. Soc., 2016, 138, 5797–5800 CrossRef CAS PubMed.
- H. Ding, J. Li, G. Xie, G. Lin, R. Chen, Z. Peng, C. Yang, B. Wang, J. Sun and C. Wang, Nat. Commun., 2018, 9, 5234 CrossRef CAS PubMed.
- X. Li, Q. Gao, J. Wang, Y. Chen, Z.-H. Chen, H.-S. Xu, W. Tang, K. Leng, G.-H. Ning and J. Wu, Nat. Commun., 2018, 9, 2335 CrossRef PubMed.
- J. You, F. Yuan, S. Cheng, Q. Kong, Y. Jiang, X. Luo, Y. Xian and C. Zhang, Chem. Mater., 2022, 34, 7078–7089 CrossRef CAS.
- L. Zhang, W. Geng, X. Ou, S. He, X.-H. He, W. Li, Y. Jiao, Y. Zhou, C. Li and Y. Chen, J. Am. Chem. Soc., 2025, 147, 39999–40009 CrossRef CAS PubMed.
- H. Xu, S. Tao and D. Jiang, Nat. Mater., 2016, 15, 722–726 CrossRef CAS.
- K. C. Ranjeesh, R. Illathvalappil, S. D. Veer, J. Peter, V. C. Wakchaure, K. V. R. Goudappagouda, S. Kurungot and S. S. Babu, J. Am. Chem. Soc., 2019, 141, 14950–14954 CrossRef CAS PubMed.
- J. Li, J. Wang, Z. Wu, S. Tao and D. Jiang, Angew. Chem., Int. Ed., 2021, 60, 12918–12923 CrossRef CAS PubMed.
- S. Tao, L. Zhai, A. D. Dinga Wonanke, M. A. Addicoat, Q. Jiang and D. Jiang, Nat. Commun., 2020, 11, 1981 CrossRef CAS PubMed.
- S. Tao and D. Jiang, Angew. Chem., Int. Ed., 2024, 63, e202408296 CrossRef CAS PubMed.
- S. Tao and D. Jiang, J. Am. Chem. Soc., 2024, 146, 18151–18160 CrossRef CAS PubMed.
- Y. Yang, X. He, P. Zhang, Y. H. Andaloussi, H. Zhang, Z. Jiang, Y. Chen, S. Ma, P. Cheng and Z. Zhang, Angew. Chem., Int. Ed., 2020, 59, 3678 CrossRef CAS PubMed.
- L. Liu, L. Yin, D. Cheng, S. Zhao, H. Y. Zang, N. Zhang and G. Zhu, Angew. Chem., Int. Ed., 2021, 60, 14875 CrossRef CAS PubMed.
- Y. Peng, G. Xu, Z. Hu, Y. Cheng, C. Chi, D. Yuan, H. Cheng and D. Zhao, ACS Appl. Mater. Interface, 2016, 8, 18505–18512 CrossRef CAS PubMed.
- Z. Lu, C. Yang, L. He, J. Hong, C. Huang, T. Wu, X. Wang, Z. Wu, X. Liu, Z. Miao, B. Zeng, Y. Xu, C. Yuan and L. Dai, J. Am. Chem. Soc., 2022, 144, 9624–9633 CrossRef CAS PubMed.
- L. Hao, S. Jia, X. Qiao, E. Lin, Y. Yang, Y. Chen, P. Cheng and Z. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202217240 CrossRef CAS PubMed.
- H. Chen, Y. Liu, M. Zheng, C. Chen, H. Li, B. Niu, W. Wu and J. Wang, Adv. Funct. Mater., 2025, e20568 Search PubMed.
- E. Jin, K. Geng, S. Fu, M. A. Addicoat, W. Zheng, S. Xie, J.-S. Hu, X. Hou, X. Wu, Q. Jiang, Q.-H. Xu, H. I. Wang and D. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202115020 CrossRef CAS PubMed.
- S. Wan, J. Guo, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2008, 47, 8826–8830 CrossRef CAS PubMed.
- S. Wan, J. Guo, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2009, 48, 5439–5442 CrossRef CAS PubMed.
- Z. Li, S. Tao, M. Addicoat, T. Nakamura and D. Jiang, Nat. Synth., 2026, 5, 199–208 CrossRef CAS.
- D. Jiang, V. G. W. Tan, Y. Gong, H. Shao, X. Mu, Z. Luo and S. He, Chem. Rev., 2025, 125, 6203–6308 CrossRef CAS PubMed.
- Y. Li, S. Tao, Y. Chen, X. Ye, H. Shao, M. Lin, Y. Zhi and D. Jiang, Angew. Chem., Int. Ed., 2025, 64, e202425103 CrossRef CAS PubMed.
- Q. Yan, S. Tao, R. Liu, Y. Zhi and D. Jiang, Angew. Chem., Int. Ed., 2024, 63, e202316092 CrossRef CAS PubMed.
- J. X. Koh, K. Geng and D. Jiang, Chem. Commun., 2021, 57, 9418–9421 RSC.
- X. Liu, D. Huang, C. Lai, G. Zeng, L. Qin, H. Wang, H. Yi, B. Li, S. Liu, M. Zhang, R. Deng, Y. Fu, L. Li, W. Xue and S. Chen, Chem. Soc. Rev., 2019, 48, 5266–5302 RSC.
- D. Kaleeswaran, P. Vishnoi and R. Murugavel, J. Mater. Chem. C, 2015, 3, 7159–7171 RSC.
- H. Singh, V. K. Tomer, N. Jena, I. Bala, N. Sharma, D. Nepak, A. De Sarkar, K. Kailasam and S. K. Pal, J. Mater. Chem. A, 2017, 5, 21820–21827 RSC.
- S. Jhulki, A. M. Evans, X.-L. Hao, M. W. Cooper, C. H. Feriante, J. Leisen, H. Li, D. Lam, M. C. Hersam and S. Barlow, J. Am. Chem. Soc., 2020, 142, 783–791 CrossRef CAS PubMed.
- H.-L. Qian, C. Dai, C.-X. Yang and X.-P. Yan, ACS Appl. Mater. Interface, 2017, 9, 24999–25005 CrossRef CAS PubMed.
- T. Skorjanc, D. Shetty and M. Valant, ACS Sens., 2021, 6, 1461–1481 CrossRef CAS PubMed.
- T. He, C. Yang, Y. Chen, N. Huang, S. Duan, Z. Zhang, W. Hu and D. Jiang, Adv. Mater., 2022, 34, 2205186 CrossRef CAS PubMed.
- T. He, R. Liu, S. Wang, I. K. W. On, Y. Wu, Y. Xing, W. Yuan, J. Guo and Y. Zhao, J. Am. Chem. Soc., 2023, 145, 18015–18021 CrossRef CAS PubMed.
- T. He and Y. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202303086 CrossRef CAS PubMed.
- T. He, I. K. W. On, S. Bi, Z. Huang, J. Guo, Z. Wang and Y. Zhao, Angew. Chem., Int. Ed., 2024, 63, e202405769 CrossRef CAS PubMed.
- T. He, K. Geng and D. Jiang, ACS Mater. Lett., 2019, 1, 203–208 CrossRef CAS.
- T. He, W. Zhen, Y. Chen, Y. Guo, Z. Li, N. Huang, Z. Li, R. Liu, Y. Liu, X. Lian, C. Xue, T. C. Sum, W. Chen and D. Jiang, Nat. Commun., 2023, 14, 329 CrossRef CAS PubMed.
- R. Liu, Y. Chen, H. Yu, M. Položij, Y. Guo, T. C. Sum, T. Heine and D. Jiang, Nat. Catal., 2024, 7, 195–206 CrossRef CAS.
- Y. Chen, R. Liu, Y. Guo, G. Wu, T. C. Sum, S. W. Yang and D. Jiang, Nat. Synth., 2024, 3, 998–1010 CrossRef CAS.
- Y. Chen, Y. Guo, T. Wang, S. Ji, H. Shao, M. Lin, S. Seki, N. Yan and D. Jiang, Nat. Commun., 2025, 16, 6495 CrossRef CAS PubMed.
- Y. Chen and D. Jiang, Acc. Chem. Res., 2024, 57, 3182–3193 CrossRef CAS PubMed.
- Y. Chen and D. Jiang, Nat. Synth., 2024, 3, 939–940 CrossRef.
- E. Jin, Z. Lan, Q. Jiang, K. Geng, G. Li, X. Wang and D. Jiang, Chem, 2019, 5, 1632–1647 CAS.
- N. Huang, K. H. Lee, Y. Yue, X. Xu, S. Irle, Q. Jiang and D. Jiang, Angew. Chem., Int. Ed., 2020, 59, 16587–16593 CrossRef CAS PubMed.
- J. Guo and D. Jiang, ACS Cent. Sci., 2020, 6, 869–879 CrossRef CAS PubMed.
- H. Xu, J. Gao and D. Jiang, Nat. Chem., 2015, 7, 905–912 CrossRef CAS PubMed.
- Y. Zhi, P. Shao, X. Feng, H. Xia, Y. Zhang, Z. Shi, Y. Mu and X. Liu, J. Mater. Chem. A, 2018, 6, 374–382 RSC.
- S. Ma, Z. Li, J. Jia, Z. Zhang, H. Xia, H. Li, X. Chen, Y. Xu and X. Liu, Chin. J. Catal., 2021, 42, 2010–2019 CrossRef CAS.
- P.-F. Wei, M.-Z. Qi, Z.-P. Wang, S.-Y. Ding, W. Yu, Q. Liu, L.-K. Wang, H.-Z. Wang, W.-K. An and W. Wang, J. Am. Chem. Soc., 2018, 140, 4623–4631 CrossRef CAS PubMed.
- M. Li, J. Lech and P. Van Der Voort, Chem. Bio Eng., 2025, 2, 380–408 CrossRef CAS PubMed.
- G. Li, P. Fu, Q. Yue, F. Ma, X. Zhao, S. Dong, X. Han, Y. Zhou and J. Wang, Chem. Catal., 2022, 2, 1734–1747 CAS.
- X. Xu, R. Sa, W. Huang, Y. Sui, W. Chen, G. Zhou, X. Li, Y. Li and H. Zhong, ACS Catal., 2022, 12, 12954–12963 CrossRef CAS.
- R. Liu, D. Zhao, S. Ji, H. Shao, Y. Chen, M. Feng, T. Wang, J. Li, M. Lin, T. C. Sum, N. Yan, S. Seki and D. Jiang, Nat. Mater., 2025, 24, 1245–1257 CrossRef CAS PubMed.
- G. Pan, X. Hou, Z. Liu, C. Yang, J. Long, G. Huang, J. Bi, Y. Yu and L. Li, ACS Catal., 2022, 12, 14911–14917 CrossRef CAS.
- S. Feng, H. Cheng, F. Chen, X. Liu, Z. Wang, H. Xu and J. Hua, ACS Catal., 2024, 14, 7736–7745 CrossRef CAS.
- L. Wang, J. Sun, M. Deng, C. Liu, S. Ataberk Cayan, K. Molkens, P. Geiregat, R. Morent, N. De Geyter, J. Chakraborty and P. Van Der Voort, Catal. Sci. Technol., 2023, 13, 6463–6471 RSC.
- M. Deng, J. Chakraborty, G. Wang, K. S. Rawat, L. Bourda, J. Sun, I. Nath, Y. Ji, P. Geiregat, V. Van Speybroeck, X. Feng and P. Van Der Voort, J. Am. Chem. Soc., 2025, 147, 10219–10230 CrossRef CAS PubMed.
- M. Deng, J. Sun, A. Laemont, C. Liu, L. Wang, L. Bourda, J. Chakraborty, K. Van Hecke, R. Morent, N. De Geyter, K. Leus, H. Chen and P. Van Der Voort, Green Chem., 2023, 25, 3069–3076 RSC.
- J. Sun, H. Sekhar Jena, C. Krishnaraj, K. Singh Rawat, S. Abednatanzi, J. Chakraborty, A. Laemont, W. Liu, H. Chen, Y.-Y. Liu, K. Leus, H. Vrielinck, V. Van Speybroeck and P. Van Der Voort, Angew. Chem., Int. Ed., 2023, 62, e202216719 CrossRef CAS PubMed.
- M. P. Kou, Y. Y. Wang, Y. X. Xu, L. Q. Ye, Y. P. Huang, B. H. Jia, H. Li, J. Q. Ren, Y. Deng, J. H. Chen, Y. Zhou, K. Lei, L. Wang, W. Liu, H. W. Huang and T. Y. Ma, Angew. Chem., Int. Ed., 2022, 61, e202200413 CrossRef CAS PubMed.
- Z. Yong and T. Ma, Angew. Chem., Int. Ed., 2023, 62, e202308980 CrossRef CAS PubMed.
- L. Stegbauer, K. Schwinghammer and B. V. Lotsch, Chem. Sci., 2014, 5, 2789–2793 RSC.
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- A. Nagai, X. Chen, X. Feng, X. Ding, Z. Guo and D. Jiang, Angew. Chem., Int. Ed., 2013, 52, 3770–3774 CrossRef CAS PubMed.
- Q. Fang, J. Wang, S. Gu, R. B. Kaspar, Z. Zhuang, J. Zheng, H. Guo, S. Qiu and Y. Yan, J. Am. Chem. Soc., 2015, 137, 8352–8355 CrossRef CAS PubMed.
- L. Ge, C. Qiao, Y. Tang, X. Zhang and X. Jiang, Nano Lett, 2021, 21(7), 3218–3224 CrossRef CAS PubMed.
- G. Kaur and P. Kumar, Inorg. Chem. Commun., 2022, 145, 110043 CrossRef CAS.
- S. Das, T. Sekine, H. Mabuchi, T. Irie, J. Sakai, Y. Zhao, Q. Fang and Y. Negishi, ACS Appl. Mater. Interface, 2022, 14, 48045–48051 CrossRef CAS PubMed.
- R. Younas, F. Jubeen, N. Bano, S. Andreescu, H. Zhang and A. Hayat, Biotechnol. Bioeng., 2024, 121, 2017–2049 CrossRef CAS PubMed.
- G. Kaur and P. Kumar, Chem. Pap., 2024, 78, 3023–3032 CrossRef CAS.
- V. S. Vyas, M. Vishwakarma, I. Moudrakovski, F. Haase, G. Savasci, C. Ochsenfeld, J. P. Spatz and B. V. Lotsch, Adv. Mater., 2016, 28, 8749–8754 CrossRef CAS PubMed.
- G. Zhang, X. Li, Q. Liao, Y. Liu, K. Xi, W. Huang and X. Jia, Nat. Commun., 2018, 9, 2785 CrossRef PubMed.
- S. Feng, H. Xu, C. Zhang, Y. Chen, J. Zeng, D. Jiang and J.-X. Jiang, Chem. Commun., 2017, 53, 11334–11337 RSC.
- S. Tao and D. Jiang, CCS Chem., 2021, 3, 2003–2024 CrossRef CAS.
- F. Xu, S. Yang, X. Chen, Q. Liu, H. Li, H. Wang, B. Wei and D. Jiang, Chem. Sci., 2019, 10, 6001–6006 RSC.
- F. Xu, S. Jin, H. Zhong, D. Wu, X. Yang, X. Chen, H. Wei, R. Fu and D. Jiang, Sci. Rep., 2015, 5, 8225 CrossRef CAS PubMed.
- Q. Xu, Y. Tang, L. Zhai, Q. Chen and D. Jiang, Chem. Commun., 2017, 53, 11690–11693 RSC.
- C. Yin, X. Ye, S. Tao, D. Zhao, Y. Zhi and D. Jiang, Angew. Chem., Int. Ed., 2024, 63, e202411558 CrossRef CAS PubMed.
- J. Li, X. Jing, Q. Li, S. Li, X. Gao, X. Feng and B. Wang, Chem. Soc. Rev., 2020, 49, 3565–3604 RSC.
- C. R. DeBlase, K. E. Silberstein, T.-T. Truong, H. D. Abruña and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 16821–16824 CrossRef CAS PubMed.
- Z. Yang, J. Liu, Y. Li, G. Zhang, G. Xing and L. Chen, Angew. Chem., Int. Ed., 2021, 60, 20754–20759 CrossRef CAS PubMed.
- F. Xu, H. Xu, X. Chen, D. Wu, Y. Wu, H. Liu, C. Gu, R. Fu and D. Jiang, Angew. Chem., Int. Ed., 2015, 54, 6814–6818 CrossRef CAS PubMed.
- X. Zhao, P. Pachfule and A. Thomas, Chem. Soc. Rev., 2021, 50, 6871–6913 RSC.
- X. Chen, K. Geng, R. Liu, K. T. Tan, Y. Gong, Z. Li, S. Tao, Q. Jiang and D. Jiang, Angew. Chem., Int. Ed., 2020, 59, 5050–5091 CrossRef CAS PubMed.
- Y. Li, W. Chen, G. Xing, D. Jiang and L. Chen, Chem. Soc. Rev., 2020, 49, 2852–2868 RSC.
- C. He, S. Tao, R. Liu, Y. Zhi and D. Jiang, Angew. Chem., Int. Ed., 2024, 63, e202403472 CrossRef CAS PubMed.
- K. T. Tan, S. Ghosh, Z. Wang, F. Wen, D. Rodríguez-San-Miguel, J. Feng, N. Huang, W. Wang, F. Zamora and X. Feng, Nat. Rev. Methods Primers, 2023, 3, 1–19 CrossRef CAS.
- C. Wang, Y. Wang, R. Ge, X. Song, X. Xing, Q. Jiang, H. Lu, C. Hao, X. Guo and Y. Gao, Chem. – Eur. J., 2018, 24, 585–589 CrossRef CAS PubMed.
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortés, A. P. Côté, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Science, 2007, 316, 268–272 CrossRef CAS PubMed.
- B. Gui, G. Lin, H. Ding, C. Gao, A. Mal and C. Wang, Acc. Chem. Res., 2020, 53, 2225–2234 CrossRef CAS PubMed.
- X. Guan, F. Chen, Q. Fang and S. Qiu, Chem. Soc. Rev., 2020, 49, 1357–1384 RSC.
- Y. Zhang, J. Duan, D. Ma, P. Li, S. Li, H. Li, J. Zhou, X. Ma, X. Feng and B. Wang, Angew. Chem., Int. Ed., 2017, 56, 16313–16317 CrossRef CAS PubMed.
- X. Xu, P. Cai, H. Chen, H.-C. Zhou and N. Huang, J. Am. Chem. Soc., 2022, 144, 18511–18517 CrossRef CAS PubMed.
- L. Liang, Y. Qiu, W. D. Wang, J. Han, Y. Luo, W. Yu, G. L. Yin, Z. P. Wang, L. Zhang and J. Ni, Angew. Chem., Int. Ed., 2020, 59, 17991–17995 CrossRef CAS PubMed.
- L. Deng, W. Chen, G. Zhou, Y. Liu, L. Liu, Y. Han, Z. Huang and D. Jiang, J. Am. Chem. Soc., 2024, 146, 35427–35437 CrossRef PubMed.
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, eaal1585 CrossRef PubMed.
- P. J. Waller, F. Gándara and O. M. Yaghi, Acc. Chem. Res., 2015, 48, 3053–3063 CrossRef CAS PubMed.
- D. Jiang, Bull. Chem. Soc. Jpn., 2021, 94, 1215–1231 CrossRef CAS.
- L. Deng, Z. Ding, X. Ye and D. Jiang, Acc. Mater. Res., 2022, 3, 879–893 CrossRef CAS.
- D. Jiang, Chem, 2020, 6, 2461–2483 CAS.
- Z. Li, T. He, Y. Gong and D. Jiang, Acc. Chem. Res., 2020, 53, 1672–1685 CrossRef CAS PubMed.
- C. Qian, H. Wu, W. L. Teo, Y. Liao and Y. Zhao, Trends Chem., 2023, 5, 853–867 CrossRef CAS.
- M. Xue, J. Yang, F. Kang, X. Wang and Q. Zhang, J. Mater. Chem. C, 2022, 10, 17027–17047 RSC.
- M. Wang, Y. Jin, W. Zhang and Y. Zhao, Chem. Soc. Rev., 2023, 52, 8165–8193 RSC.
- T. Kim, J. Kim, N. Kim and H. Lim, Chem. Commun., 2025, 61, 17516–17529 RSC.
- Z. Huang, E. S. Grape, J. Li, A. K. Inge and X. Zou, Coord. Chem. Rev., 2021, 427, 213583 CrossRef CAS.
- J. S. Du, Y. Bae and J. J. De Yoreo, Nat. Rev. Mater., 2024, 9, 229–248 CrossRef CAS.
- Z.-H. Jiang and Q.-Y. Zhang, Prog. Mater. Sci., 2014, 61, 144–215 CrossRef CAS.
- G. Coquerel, Chem. Soc. Rev., 2014, 43, 2286–2300 RSC.
- N. A. Burger, G. Pembouong, L. Bouteiller, D. Vlassopoulos and B. Loppinet, Macromolecules, 2022, 55, 2609–2614 CrossRef CAS.
- T. F. De Greef, M. M. Smulders, M. Wolffs, A. P. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed.
- R. Contreras-Montoya, L. Á. de Cienfuegos, J. A. Gavira and J. W. Steed, Chem. Soc. Rev., 2024, 53, 10604–10619 RSC.
- G. Ungar and X.-B. Zeng, Chem. Rev., 2001, 101, 4157–4188 CrossRef CAS PubMed.
- A. Halperin, M. Kröger and F. M. Winnik, Angew. Chem., Int. Ed., 2015, 54, 15342–15367 CrossRef CAS PubMed.
- J. Ethier, E. R. Antoniuk and B. Brettmann, Soft Matter, 2024, 20, 5652–5669 RSC.
- H.-Y. Hsueh, C.-T. Yao and R.-M. Ho, Chem. Soc. Rev., 2015, 44, 1974–2018 RSC.
- D. Woo, H. Yoon and J. K. Kim, J. Polym. Sci., 2024, 62, 639–661 CrossRef CAS.
- S. Enders, K. Langenbach, P. Schrader and T. Zeiner, Polymers, 2012, 4, 72–115 CrossRef CAS.
- B. I. Voit and A. Lederer, Chem. Rev., 2009, 109, 5924–5973 CrossRef CAS PubMed.
- X. Ye, R. Liu, X. Mu, S. Tao, H. Yang, X. J. Gao, S.-W. Yang and D. Jiang, J. Am. Chem. Soc., 2025, 147, 6942–6957 CrossRef CAS PubMed.
- R. Roy and A. M. Evans, Commun. Mater., 2024, 5, 102 CrossRef CAS.
- L. Merí-Bofí, S. Royuela, F. Zamora, M. L. Ruiz-González, J. L. Segura, R. Muñoz-Olivas and M. J. Mancheño, J. Mater. Chem. A, 2017, 5, 17973–17981 RSC.
- X. Li, X. Ji, X. Zhang, X. Chen, H. Li, S. Zhang, F. Huo and W. Zhang, Nat. Commun., 2025, 16, 1223 CrossRef CAS PubMed.
- Q. Xu, Q. Li, Y. Guo, D. Luo, J. Qian, X. Li and Y. Wang, Small Methods, 2020, 4, 2000159 CrossRef CAS.
- K. T. Tan, S. Tao, N. Huang and D. Jiang, Nat. Commun., 2021, 12, 6747 CrossRef CAS PubMed.
- M. Traxler and W. R. Dichtel, Chem. Sci., 2024, 15, 7545–7551 RSC.
- C. Chen, L. Cao, Y. Liu, Z. Li, Z.-H. Li, G. Zhou, D. Zhang, X. Huang, Y. Wang, G. Li, L. Liu, Y.-Y. Yuan, Y. Zhang, Q. Wang, Y. Chen, Z. Shi, Q. Fang, Z. Huang, Z. Lai and Y. Han, J. Am. Chem. Soc., 2024, 146, 35504–35512 CrossRef CAS PubMed.
- A. Natraj, I. R. Landman, C. E. Pelkowski, D. W. Burke, S. Kewalramani and W. R. Dichtel, J. Am. Chem. Soc., 2024, 146, 16775–16786 CrossRef CAS PubMed.
- X. Wu, X. Han, Y. Liu, Y. Liu and Y. Cui, J. Am. Chem. Soc., 2018, 140, 16124–16133 CrossRef CAS PubMed.
- P.-Y. You, K.-M. Mo, Y.-M. Wang, Q. Gao, X.-C. Lin, J.-T. Lin, M. Xie, R.-J. Wei, G.-H. Ning and D. Li, Nat. Commun., 2024, 15, 194 CrossRef CAS PubMed.
- N. Huang, L. Zhai, D. E. Coupry, M. A. Addicoat, K. Okushita, K. Nishimura, T. Heine and D. Jiang, Nat. Commun., 2016, 7, 12325 CrossRef PubMed.
- Z. Wang, Y. Zhang, T. Wang, E. Lin, T. Wang, Y. Chen, P. Cheng and Z. Zhang, Small, 2023, 19, 2303684 CrossRef CAS PubMed.
- X. Li, J. Qiao, S. W. Chee, H.-S. Xu, X. Zhao, H. S. Choi, W. Yu, S. Y. Quek, U. Mirsaidov and K. P. Loh, J. Am. Chem. Soc., 2020, 142, 4932–4943 CrossRef CAS PubMed.
- S. Wang, V. A. Reddy, M. C.-Y. Ang, J. Cui, D. T. Khong, Y. Han, S. I. Loh, R. Cheerlavancha, G. P. Singh, S. Rajani and M. S. Strano, J. Am. Chem. Soc., 2023, 145, 12155–12163 CrossRef CAS PubMed.
- Z. Zhou, L. Zhang, Y. Yang, I. J. Vitorica-Yrezabal, H. Wang, F. Tan, L. Gong, Y. Li, P. Chen, X. Dong, Z. Liang, J. Yang, C. Wang, Y. Hong, Y. Qiu, A. Gölzhäuser, X. Chen, H. Qi, S. Yang, W. Liu, J. Sun and Z. Zheng, Nat. Chem., 2023, 15, 841–847 CrossRef CAS PubMed.
- T. Kitano, S. Goto, X. Wang, T. Kamihara, Y. Sei, Y. Kondo, T. Sannomiya, H. Uekusa and Y. Murakami, Nat. Commun., 2025, 16, 280 CrossRef CAS PubMed.
- S. Dalapati, M. Addicoat, S. Jin, T. Sakurai, J. Gao, H. Xu, S. Irle, S. Seki and D. Jiang, Nat. Commun., 2015, 6, 7786 CrossRef CAS PubMed.
- E. Jin, M. Asada, Q. Xu, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine and Q. Chen, Science, 2017, 357, 673–676 CrossRef CAS PubMed.
- F. Wen, K. Xu, Y. Feng and N. Huang, J. Am. Chem. Soc., 2024, 146, 19680–19685 CrossRef CAS PubMed.
- Y. Liu, L. Yuan, W. Chi, W.-K. Han, J. Zhang, H. Pang, Z. Wang and Z.-G. Gu, Nat. Commun., 2024, 15, 7150 CrossRef CAS PubMed.
- P.-J. Tian, X.-H. Han, Q.-Y. Qi and X. Zhao, Chem. Sci., 2024, 15, 9669–9675 RSC.
- A. Nagai, Z. Guo, X. Feng, S. Jin, X. Chen, X. Ding and D. Jiang, Nat. Commun., 2011, 2, 536 CrossRef PubMed.
- S. Park, Z. Liao, B. Ibarlucea, H. Qi, H. H. Lin, D. Becker, J. Melidonie, T. Zhang, H. Sahabudeen and L. Baraban, Angew. Chem., Int. Ed., 2020, 59, 8218 CrossRef CAS PubMed.
- C. Kang, K. Yang, Z. Zhang, A. K. Usadi, D. C. Calabro, L. S. Baugh, Y. Wang, J. Jiang, X. Zou, Z. Huang and D. Zhao, Nat. Commun., 2022, 13, 1370 CrossRef CAS PubMed.
- A. M. Evans, I. Castano, A. Brumberg, L. R. Parent, A. R. Corcos, R. L. Li, N. C. Flanders, D. J. Gosztola, N. C. Gianneschi and R. D. Schaller, J. Am. Chem. Soc., 2019, 141, 19728–19735 CrossRef CAS PubMed.
- T. Ma, E. A. Kapustin, S. X. Yin, L. Liang, Z. Zhou, J. Niu, L.-H. Li, Y. Wang, J. Su, J. Li, X. Wang, W. D. Wang, W. Wang, J. Sun and O. M. Yaghi, Science, 2018, 361, 48–52 CrossRef CAS PubMed.
- J. Han, J. Feng, J. Kang, J.-M. Chen, X.-Y. Du, S.-Y. Ding, L. Liang and W. Wang, Science, 2024, 383, 1014–1019 CrossRef CAS PubMed.
- Y. Yin, Y. Zhang, X. Zhou, B. Gui, G. Cai, J. Sun and C. Wang, J. Am. Chem. Soc., 2023, 145, 22329–22334 CrossRef CAS PubMed.
- Y. Yin, Y. Zhang, X. Zhou, B. Gui, W. Wang, W. Jiang, Y.-B. Zhang, J. Sun and C. Wang, Science, 2024, 386, 693–696 CrossRef CAS PubMed.
- B. Yu, R.-B. Lin, G. Xu, Z.-H. Fu, H. Wu, W. Zhou, S. Lu, Q.-W. Li, Y. Jin, J.-H. Li, Z. Zhang, H. Wang, Z. Yan, X. Liu, K. Wang, B. Chen and J. Jiang, Nat. Chem., 2024, 16, 114–121 CrossRef CAS PubMed.
- H. S. Xu, Y. Luo, P. Z. See, X. Li, Z. Chen, Y. Zhou, X. Zhao, K. Leng, I. H. Park and R. Li, Angew. Chem., Int. Ed., 2020, 59, 11527 CrossRef CAS PubMed.
- H.-S. Xu, Y. Luo, X. Li, P. Z. See, Z. Chen, T. Ma, L. Liang, K. Leng, I. Abdelwahab and L. Wang, Nat. Commun., 2020, 11, 1434 CrossRef CAS PubMed.
- C. Gropp, T. Ma, N. Hanikel and O. M. Yaghi, Science, 2020, 370, eabd6406 CrossRef CAS PubMed.
- A. M. Evans, L. R. Parent, N. C. Flanders, R. P. Bisbey, E. Vitaku, M. S. Kirschner, R. D. Schaller, L. X. Chen, N. C. Gianneschi and W. R. Dichtel, Science, 2018, 361, 52–57 CrossRef CAS PubMed.
- L. Yi, Y. Gao, S. Luo, T. Wang and H. Deng, J. Am. Chem. Soc., 2024, 146, 19643–19648 CrossRef CAS PubMed.
- S. Li, S. Xu, E. Lin, T. Wang, H. Yang, J. Han, Y. Zhao, Q. Xue, P. Samorì, Z. Zhang and T. Zhang, Nat. Chem., 2025, 17, 226–232 CrossRef CAS PubMed.
- Y. Li, Y. Wan, X. Deng, M. Zhao, B. Zhang and Z. Wang, J. Am. Chem. Soc., 2025, 147, 29526–29533 CrossRef CAS PubMed.
- A. Yao, C. Zhu, J. Liu, H. Xu, K. Shao, C. Sun, C. Qin, X. Wang, H. Zang and Z. Su, Nat. Commun., 2026, 17, 1115 CrossRef CAS PubMed.
- S. Ghouse, Z. Guo, S. Gámez-Valenzuela, D. Mücke, B. Zhang, L. Gao, S. Paasch, Y. Fu, C. Huang and C. Naisa, Nat. Chem., 2026, 1–10 Search PubMed.
- J. Fang, Z. Fu, X. Chen, Y. Liu, F. Chen, Y. Wang, H. Li, Y. Yusran, K. Wang and V. Valtchev, Angew. Chem., Int. Ed., 2023, 62, e202304234 CrossRef CAS PubMed.
- C. Ji, C. Kang, B. C. Patra and D. Zhao, CCS Chem., 2024, 6, 856–881 CrossRef CAS.
- M. Wang, T. Zeng, Y. Yu, X. Wang, Y. Zhao, H. Xi and Y.-B. Zhang, J. Am. Chem. Soc., 2023, 146, 1035–1041 CrossRef PubMed.
- Q. Chen, G. Zhou and Z. Huang, Acc. Chem. Res., 2024, 57, 2522–2531 CrossRef CAS PubMed.
- Z. Huang, T. Willhammar and X. Zou, Chem. Sci., 2021, 12, 1206–1219 RSC.
- T. Sun, W. Lei, Y. Ma and Y. B. Zhang, Chin. J. Chem., 2020, 38, 1153–1166 CrossRef CAS.
- A. Saha, S. S. Nia and J. A. Rodríguez, Chem. Rev., 2022, 122, 13883–13914 CrossRef CAS PubMed.
- L. Samperisi, X. Zou and Z. Huang, CrystEngComm, 2022, 24, 2719–2728 RSC.
- I. Andrusenko and M. Gemmi, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2022, 14, e1810 CAS.
- Y. Zhang, J. Su, H. Furukawa, Y. Yun, F. Gandara, A. Duong, X. Zou and O. M. Yaghi, J. Am. Chem. Soc., 2013, 135, 16336–16339 CrossRef CAS PubMed.
- R. Jia, R. Ye, Z. Chang, H. Yu, M. Wang, G. Xu, Z. Guo and H. Zhan, Chem. – Eur. J., 2025, 31, e202404423 CrossRef CAS PubMed.
- W. Gao, Z. Chen, J. Hong, Y. Zhang, Z. Yang, M. Liu, X. Wang, S. Shang, Z. You, Z. Shao, J. Dong, Y. Guo, J. Chen and Y. Liu, J. Am. Chem. Soc., 2025, 147, 15459–15468 CrossRef CAS PubMed.
- A. Yao, H. Xu, K. Shao, C. Sun, C. Qin, X. Wang and Z. Su, Nat. Commun., 2025, 16, 1385 CrossRef CAS PubMed.
- S. Liu, L. Wei, T. Zeng, W. Jiang, Y. Qiu, X. Yao, Q. Wang, Y. Zhao and Y.-B. Zhang, J. Am. Chem. Soc., 2024, 146, 34053–34063 CrossRef CAS PubMed.
- X. Wang, Y. Wada, T. Shimada, A. Kosaka, K. Adachi, D. Hashizume, K. Yazawa, H. Uekusa, Y. Shoji and T. Fukushima, J. Am. Chem. Soc., 2024, 146, 1832–1838 CrossRef CAS PubMed.
- L. Wei, X. Hai, T. Xu, Z. Wang, W. Jiang, S. Jiang, Q. Wang, Y.-B. Zhang and Y. Zhao, Nat. Commun., 2024, 15, 2411 CrossRef CAS PubMed.
- H. Wang, L. Yi, G. Hu, X. Zhou and H. Deng, J. Am. Chem. Soc., 2025, 147, 9067–9073 CrossRef CAS PubMed.
- S. Qiao, Y. Sun, K. Wang, S. Zhou, E. Lin, A. Zuo, X. Liu, J. Yu, Z. Zhang and Y. Chen, J. Am. Chem. Soc., 2026, 148, 9892–9900 CrossRef PubMed.
- B. Hou, X. Han, H. Xie, C. Yuan, Y. Guo, X. Chen, X. Tang, S. Su, H. Jiang, Z.-M. Ye, K. O. Kirlikovali, Y. Liu, O. K. Farha and Y. Cui, J. Am. Chem. Soc., 2025, 147, 12127–12137 CrossRef CAS PubMed.
- J. Zhang, Z. Wang, J. Suo, C. Tuo, F. Chen, J. Chang, H. Zheng, H. Li, D. Zhang and Q. Fang, J. Am. Chem. Soc., 2024, 146, 35090–35097 CrossRef CAS PubMed.
- E. Jin, J. Li, K. Geng, Q. Jiang, H. Xu, Q. Xu and D. Jiang, Nat. Commun., 2018, 9, 4143 CrossRef PubMed.
- A. Natraj, W. Ji, J. Xin, I. Castano, D. W. Burke, A. M. Evans, M. J. Strauss, M. Ateia, L. S. Hamachi, N. C. Gianneschi, Z. A. Alothman, J. Sun, K. Yusuf and W. R. Dichtel, J. Am. Chem. Soc., 2022, 144, 19813–19824 CrossRef CAS PubMed.
- L. Yi, Y. Gao, C. Yan, Y. Liu, S. Luo, T. Wang, L. Liu and H. Deng, J. Am. Chem. Soc., 2025, 147, 32145–32156 CrossRef CAS PubMed.
- B. Yu, S. Geng, X. Ding, X. Zhou, Y. Jin, H. Wang, X. Wang, T. Zheng, Z. Zhang and J. Jiang, Chem, 2024, 10, 2170–2179 CAS.
- B. Yu, Y. Tao, X. Yao, Y. Jin, S. Liu, T. Xu, H. Wang, H. Wu, W. Zhou and X. Zhou, J. Am. Chem. Soc., 2024, 146, 28932–28940 CrossRef CAS PubMed.
- F. Tan, S. Han, D. Peng, H. Wang, J. Yang, P. Zhao, X. Ye, X. Dong, Y. Zheng, N. Zheng, L. Gong, C. Liang, N. Frese, A. Gölzhäuser, H. Qi, S. Chen, W. Liu and Z. Zheng, J. Am. Chem. Soc., 2021, 143, 3927–3933 CrossRef CAS PubMed.
- Z. Ou, B. Liang, Z. Liang, F. Tan, X. Dong, L. Gong, P. Zhao, H. Wang, Y. Zou, Y. Xia, X. Chen, W. Liu, H. Qi, U. Kaiser and Z. Zheng, J. Am. Chem. Soc., 2022, 144, 3233–3241 CrossRef CAS PubMed.
- Y. Yang, B. Liang, J. Kreie, M. Hambsch, Z. Liang, C. Wang, S. Huang, X. Dong, L. Gong, C. Liang, D. Lou, Z. Zhou, J. Lu, Y. Yang, X. Zhuang, H. Qi, U. Kaiser, S. C. B. Mannsfeld, W. Liu, A. Gölzhäuser and Z. Zheng, Nature, 2024, 630, 878–883 CrossRef CAS PubMed.
- G. Das, T. Skorjanc, S. K. Sharma, F. Gándara, M. Lusi, D. S. Shankar Rao, S. Vimala, S. Krishna Prasad, J. Raya, D. S. Han, R. Jagannathan, J.-C. Olsen and A. Trabolsi, J. Am. Chem. Soc., 2017, 139, 9558–9565 CrossRef CAS PubMed.
- W. Zhang, Y. Zhang, W. Ma, X. Han, W. Gong, Y. Liu and Y. Cui, Chem, 2025, 11, 102398 CAS.
- W. Zhang, Z. Zhong, X. Wei, Y. Zhang, W. Ma, D. Liu, X. Han, J. Dong, W. Gong, F. Dai, Y. Liu, Y. Ma and Y. Cui, J. Am. Chem. Soc., 2025, 147, 17975–17984 CrossRef CAS PubMed.
- D. Beaudoin, T. Maris and J. D. Wuest, Nat. Chem., 2013, 5, 830–834 CrossRef CAS PubMed.
- L. Peng, Q. Guo, C. Song, S. Ghosh, H. Xu, L. Wang, D. Hu, L. Shi, L. Zhao and Q. Li, Nat. Commun., 2021, 12, 5077 CrossRef CAS PubMed.
- B. P. Biswal, S. Valligatla, M. Wang, T. Banerjee, N. A. Saad, B. M. K. Mariserla, N. Chandrasekhar, D. Becker, M. Addicoat and I. Senkovska, Angew. Chem., Int. Ed., 2019, 58, 6896–6900 CrossRef CAS PubMed.
- S. Nath, A. Puthukkudi, J. Mohapatra and B. P. Biswal, Angew. Chem., Int. Ed., 2023, 62, e202218974 CrossRef CAS PubMed.
- L. Mingabudinova, V. Vinogradov, V. Milichko, E. Hey-Hawkins and A. Vinogradov, Chem. Soc. Rev., 2016, 45, 5408–5431 RSC.
- H. Yang, S. Tao, S. He and D. Jiang, J. Am. Chem. Soc., 2025, 147, 19667–19674 CrossRef CAS PubMed.
- T. Wang, L. Hao, S. Wu, E. Lin, K. Wang, J. Wang, M. J. Zawarotko, P. Cheng, Y. Chen and Z. Zhang, Nat. Synth., 2026 DOI:10.1038/s44160-026-01021-8.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2026 |