
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-isocyanate polyurethane foams: where we stand and what comes next?
Bruno
Grignard
 *ab,
Maxime
Bourguignon
b,
Florent
Monie
bc,
Thomas
Vidil
c,
Henri
Cramail
c,
Gabriel
Perli
d,
Haritz
Sardon
d,
Jean-Marie
Raquez
e,
Sylvain
Caillol
f and
Christophe
Detrembleur
*bg
*ab,
Maxime
Bourguignon
b,
Florent
Monie
bc,
Thomas
Vidil
c,
Henri
Cramail
c,
Gabriel
Perli
d,
Haritz
Sardon
d,
Jean-Marie
Raquez
e,
Sylvain
Caillol
f and
Christophe
Detrembleur
*bg
aFederation of Researchers in Innovative Technologies for CO2 Transformation (FRITCO2T), Institutional CCU Technology Platform, 13 allée de la Chimie, 4000, Liège, Belgium. E-mail: bruno.grignard@uliege.be
bCenter for Education and Research on Macromolecules (CERM), Chemistry Department, 13 allée de la Chimie, 4000, Liège, Belgium. E-mail: Christophe.detrembleur@uliege.be
cUniv. Bordeaux, CNRS, Bordeaux INP, LCPO, UMR562, 16 avenue Pey-Berland,, 33600 Pessac, France
dPOLYMAT and Department of Polymers and Advanced Materials: Physics, Chemistry and Technology, Faculty of Chemistry, University of the Basque Country UPV/EHU, Paseo Manuel de Lardizabal 3, 20018 Donostia-San Sebastián, Spain
eLaboratory of Polymeric and Composite Materials (LPCM), Center of Innovation and Research in Materials and Polymers (CIRMAP), University of Mons (UMONS), Mons, Belgium
fInstitut Charles Gerhardt Montpellier (ICGM), CNRS, ENSCM, University of Montpellier, 34293 Montpellier, France
gWEL Research Institute, 1300 Wavre, Belgium
First published on 20th April 2026
Abstract
Polyurethane foam stakeholders are currently facing stringent regulatory constraints, such as the REACH regulation restricting the use of isocyanates and increasing environmental pressures calling for more sustainable materials. This dual challenge compels both academia and industry to innovate and redesign conventional PU chemistry and foaming toward non-isocyanate solutions while integrating eco-design principles to enhance recyclability and circularity. This tutorial review summarizes the current advances in synthetic strategies for producing non-isocyanate polyurethanes (NIPUs), with a particular focus on foaming concepts, which have witnessed a significant acceleration in development over the last five years. Emerging end-of-life management solutions via chemolysis or physical means – exploiting covalent adaptable network features – and applications of NIPU foams are also discussed. For each aspect, a comparative analysis between conventional PU and NIPU highlights the remaining technological, environmental and economic challenges that must be addressed to achieve competitive, scalable, and sustainable NIPU foams.
 Bruno Grignard | Bruno Grignard obtained his PhD from the University of Liège in 2007 in the field of CO2 utilization for macromolecular engineering through controlled polymerization processes. Then, he continued as an associate R&D scientist in charge of the development of the CO2 utilization technologies for polymer processing (foaming, solvent, drying, anti-solvents), polymer synthesis through novel conceptual routes for CO2 conversion and CO2-sourced materials design. He is now a principal research logistician within the “FRITCO2T” R&D platform dedicated to CO2 capture, utilization and recycling at ULiège. |
 Henri Cramail | Henri Cramail obtained his PhD from the University of Bordeaux (UB) in 1990 in ring-opening metathesis polymerization. After a post-doctoral stay at the University of Durham, U.K., he became an Assistant Professor of Polymer Chemistry at UB and was appointed as Professor in 1999. In 2004, he was awarded Junior Member of the ‘Institut Universitaire de France’. He was Director of the Laboratoire de Chimie des Polymères Organiques (LCPO), CNRS-University of Bordeaux (2007–2016). He is leading the team ‘Biopolymers and Bio-based Polymers’ focusing on the development of green pathways to bio-based polymers from renewable resources (vegetable oils, polysaccharides, lignin, CO2). |
 Haritz Sardon | Dr Haritz Sardon has been a professor at the UPV/EHU since 2018. He graduated from the UPV/EHU in 2011 before joining the group of Dr Hedrick at IBM-Almaden Research Center as a postdoc in 2012. In 2014, he returned to POLYMAT as a group leader. He has received several awards including Excellence of Young Researcher in Chemistry Award by the Spanish Royal Society (2021). His overall research aims to prepare new functional polymeric materials by combining sustainable polymerization based on organocatalysis with biobased monomers or monomers derived from plastic waste |
 Jean-Marie Raquez | Jean-Marie Raquez, PhD in Polymer Science, is the head of Laboratory Polymeric and Composite Materials at the University of Mons (Belgium) and Associated Professor at Polytechnique Montreal (canada). His research activities encompass all the general aspects about the realm of “bioplastics”, i.e., bio-based and/or biodegradable polymer materials, particularly ranging from their controlled and catalyzed polymerization reactions, production of high performance nanocomposites/nanohybrids via reactive processing (i.e., reactive extrusion and additive manufacturing), to their implementation in diverse applications (packaging, textile, automotive, etc.), including their end-life scenario. |
 Sylvain Caillol | Sylvain Caillol is Senior Research Director at CNRS, based at the Charles Gerhardt Institute of Montpellier, and a leading expert in biobased polymers and sustainable polymer design. He is the author of over 300 publications and recipient of multiple awards, and has made major contributions to the development of polyhydroxyurethanes. |
 Christophe Detrembleur | Christophe Detrembleur obtained his PhD in 2001 from CERM at the University of Liege (Belgium) in the field of controlled radical polymerization (CRP) techniques. He then moved to Bayer AG (Leverkusen, Germany) to develop polymers from CRP and low-viscous UV-curable polyurethane formulations. After 2.5 years in this company, he joined CERM as a permanent FNRS Researcher (currently working as FNRS Research Director) to create his own research group. His research focuses on the valorization of carbon dioxide as a C1-feedstock for producing low-carbon footprint, circular polymers (e.g., non-isocyanate polyurethanes and polycarbonates) and materials (coatings, adhesives, and foams). |
Key learning points(1) Fundamentals and core principles of synthetic methods for non-isocyanate polyurethanes (NIPUs)(2) Basics of non-isocyanate polyurethane (self-)foaming procedures (3) Practical approaches to end-of-life strategies for NIPU foams (4) Performance assessment of emerging applications of NIPU foams (5) Major differences between non-isocyanate and conventional PU foams; level of competitiveness and remaining challenges |
1. Introduction to PU foams
In 2023, the polyurethane (PU) foam market was valued at US$ ∼49.5 billion, with an expected growth projection up to US$ ∼67.8 billion by 2028.1 The PU foam market dynamic is mainly driven by the increasing demands of end-user industries from the building, automotive/aerospace, furniture, and other sectors.2–5 These key players are seeking low-cost, energy-efficient, lightweight and durable materials with chemical resistance and adaptable thermo-mechanical properties, particularly in thermal insulation, sealing, structural items of vehicles (seats, soundproofing, interior components, etc.), comfort (mattresses, footwear, etc.) or personal protective equipment (helmets, filtration, etc.) (Fig. 1). Industrially, the popularity of PU foams arises from their versatility and ease of fabrication from low-temperature reactive formulations (generally from room temperature to 40–60 °C) made of polyisocyanates and polyols.5,6 The PU foaming is achieved by the use of external chemical or physical blowing agents (i.e., exogenous chemical/physical blowing agent) or via a self-foaming approach,7 for which the blowing agent (CO2) originates from the isocyanate compound (i.e., endogenous chemical blowing agent).8 Upon simple addition of water, the hydrolysis of isocyanate releases CO2in situ, causing material expansion during PU network formation.5,6 Inherently, PU foaming benefits from the high exothermicity of the isocyanate/polyol reaction. The in situ heat generation rapidly raises the temperature of the formulation from r.T. to far above 100 °C (generally between 120 and 180 °C), promoting the rapid formation of a polymer network while simultaneously releasing the blowing agent. It also ensures that the polymer architecture is formed at a temperature above its final glass transition or crystallization temperature, as a key element for the preparation of stable rigid PU foams. Leveraging the intrinsic features of the PU chemistry, flexible and rigid foams are easily manufactured through continuous extrusion or slabstock processes, via discontinuous reactive injection molding (RIM) or via spray foaming.9 Continuous foam extrusion consists of mixing a pre-formed thermoplastic PU with a chemical or physical foaming agent in an extruder at a temperature high enough to melt the polymer and produce a gas, causing material expansion by a drop in pressure at the exit of the extruder. In the slabstock process, the PU expands and then solidifies within seconds after mixing the reactive ingredients (polyols, polyisocyanates and blowing agent) on a moving conveyor, yet delivering foams in the form of blocks or sandwich panels (e.g., for thermal insulation). In RIM, the reactive formulation is fed under pressure via a supply line into a mixing chamber before injection into a (pre-heated) closed mold, where PU expansion and curing occur within a short time. Unlike the continuous processes, RIM is adapted to the design of objects with complex 3D shapes. As an adaptation of RIM, the formed-in-place foam allows for soft foam sealing on location (gasket foaming) by directly applying the two-component formulation onto a substrate via a mobile mixing head. Spray foams, particularly suited for insulation applications, utilize a propellant to expel the reactive polyol/isocyanate formulation to surfaces through an application gun/pressurized can. These foaming technologies have been well-managed and exploited for decades. However, the PU foam sector is facing stringent legal regulatory compliance related to health and safety considerations associated with the exposure, handling and use of isocyanates.10 Further, obligations in creating an industrially viable PU end-of-life scenario to fulfill the sustainable objectives of our modern society, opportunities in exploiting chemicals of bio- and/or CO2 origin,10–14 and price volatility of raw isocyanates are additional drivers pushing these stakeholders to fundamentally reinvent the fabrication of PU foams without using isocyanates. Routes to isocyanate-free polyurethanes – also named non-isocyanate polyurethanes (NIPUs) – have been extensively described in the past15–20 and the last months have seen the publication of reviews dedicated to their foaming.21–24 While these documents provide a good insight into the current state of the technology, they generally lack a tutorial dimension. To bridge this gap, we propose a comprehensive and pedagogical review intended as a vade-mecum for newcomers to NIPU synthesis and foaming. This review delves into the underlying mechanisms of each foaming approach with a critical examination of their advantages and limitations compared to conventional PUs as well as their impact on the morpho-structural foam properties. In addition, we explore emerging end-of-life scenarios of NIPU foams and assess the foam utility for key real-world usages. | ||
| Fig. 1 Polyurethane foam applications and future deployment of NIPU foams. | ||
2. How to design non-isocyanate polyurethanes?
Herein, the most relevant NIPU synthetic approaches will be briefly illustrated, explaining only the key features of each concept and positioning them in light of their potential for NIPU foam fabrication. For a more complete and comprehensive overview of the state-of-the-art in NIPU synthesis, we invite the reader to consult previous accounts.15–20 Ring-opening (co)polymerization (RO(CO)P) methods and step-growth copolymerization are the most relevant tools to fabricate NIPUs.2.1. NIPUs by RO(CO)P
RO(CO)P refers to either the direct copolymerization of aziridines with CO2 or the ionic polymerization of 5- to 7-membered cyclic carbamates.25 Conceptually similar to the CO2/epoxy polymerization, the CO2/aziridine ROCOP is typically realized within the temperature range of 80 to 150 °C using compressed CO2 (pCO2 = 30–100 bar) with or without a catalyst (Scheme 1, path A).11,26–32 This synthetic approach produces PU chains containing a significant level of polyamine defects (even > 80%) but also branching. The polyamine defects are significantly minimized by using supercritical CO2 (100 < pCO2 < 220 bar) while providing both high molar mass chains (27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 < Mn < 210000 g mol−1) and urethane linkage content up to 60%.28,30 Enrichment of the polymer chains in urethane linkages up to 85% is also observed by using a complex Y(CCl3COO)3–ZnEt2–glycerine catalytic system26 and defect-free PUs are even accessible with (binary) organozinc catalysts including ZnEt2, EtZnOMe, ZnEt2/pyrogallol, ZnEt2/resorcinol, ZnEt2/o-aminophenol and ZnEt2/o-phenylenediamine, but at the expense of the NIPU molar mass being limited to oligomers of 400 to 4500 g mol−1, whatever the catalytic system.27
000 < Mn < 210000 g mol−1) and urethane linkage content up to 60%.28,30 Enrichment of the polymer chains in urethane linkages up to 85% is also observed by using a complex Y(CCl3COO)3–ZnEt2–glycerine catalytic system26 and defect-free PUs are even accessible with (binary) organozinc catalysts including ZnEt2, EtZnOMe, ZnEt2/pyrogallol, ZnEt2/resorcinol, ZnEt2/o-aminophenol and ZnEt2/o-phenylenediamine, but at the expense of the NIPU molar mass being limited to oligomers of 400 to 4500 g mol−1, whatever the catalytic system.27
 | ||
| Scheme 1 Non-isocyanate polyurethane synthesis via ring-opening (co)polymerization approaches. | ||
5-Membered cyclic carbamates display rather low reactivity, making their ROP challenging.33–35 Their polymerization is only possible by an anionic pathway with geometrically constrained oxazolidones fused with an alicyclic cyclohexene moiety. Unlike planar “conventional” oxazolidones, the bicyclic structure of the molecule creates a conformational deformation increasing its ring strain, then facilitating its ring-opening (Scheme 1, path B). By using BuLi as the initiator and N-acyloxazolidone as the co-initiator, linear NIPUs with Mn up to 8000 g mol−1 have been fabricated.33 Recently, Rieger designed five-membered cyclic carbamates fused in a trans configuration with a limonene unit, which increases the ring strain of the heterocycle and favors its ROP. With Sn(Oct)2 as a catalyst, low dispersity polyurethanes with Mn up to 16000 g mol−1 have been synthesized at 80–100 °C in 15–20 h.34 Besides, 6- and 7-membered cyclic carbamates undergo (cationic) ROP36–41 with various catalysts (Bu2SnO, Bu2Sn(OCH3)2)41 or electrophilic initiators (TfOH, TfOMe or BF3·OEt2)36,40 (Scheme 1, path C). When conducted in bulk at 70–120 °C, PUs with Mn of 14000–35000 g mol−1 or oligomers with Mn < 5000 g mol−1 have been designed from the 6- or 7-membered monomers, respectively.41 One difficulty in accessing higher molar mass PUs via cationic ROP arises from the competition between chain propagation and competitive transfer and termination reactions, creating dead chains.
Limited to rare examples, both RO(CO)P routes yield thermoplastic materials potentially valuable for physical or chemical foaming (e.g., via extrusion-foaming or foam molding). However, the challenges associated with CO2/aziridine or cyclic carbamate polymerization (presence of defects, polymerization sensitivity to moisture and air, poor catalyst efficiency) and the immaturity of both methodologies, limited to academic research, make their industrial adoption questionable. Furthermore, NIPUs synthesized using RO(CO)P techniques exhibit low molar masses, resulting in inadequate chain entanglement. This deficiency could potentially adversely affect their mechanical properties, which have not yet been documented. Given the current state of knowledge, large-scale industrial production of these NIPUs remains unlikely in the short term, and the major drawbacks associated with their synthesis severely limit their applicability in foam fabrication.
2.2. NIPUs by polycondensation
Polycondensation involves either two monomers (AA + BB monomers), each containing functional groups, or a single molecule bearing both functionalities (AB monomer) (Scheme 2). These monomers react with each other via a condensation reaction to form a covalent linkage, together with the concomitant release of a by-product.42 Repeated condensation reactions with simultaneous removal of the by-product drive chain formation, giving access to polymers of high molar mass. Such a step-growth polymerization generally proceeds in two stages, i.e., oligomerization at low temperature (below 100 °C) followed by chain extension at a higher temperature, typically under vacuum to eliminate the condensate. Melt transurethanization polycondensation has been exploited to fabricate high molar mass NIPUs utilizing either AB monomers (α-hydroxy-ω-O-hydroxyethyl or α-hydroxy-ω-O-hydroxyphenyl urethanes, dihydroxyethyl dicarbamates)43–47 (Scheme 2, path A) or AA–BB monomer mixtures (dimethyl dicarbamate/diol mixtures) (Scheme 2, path B).44,48–53 Upon proper elimination of ethylene glycol, the thermally driven self-polycondensation of α-hydroxy-ω-O-hydroxyethyl urethanes catalyzed by tin complexes (Bu2SnO or SnCl2) at T = 100–150 °C or dihydroxyethyl dicarbamates (T = 170–180 °C) furnishes various NIPUs with decent Mn up to 31000–64000 g mol−1. However, AB monomers or dihydroxyethyl dicarbamates favor the formation of ethylene carbonate, via a back-biting reaction, and amines that can further participate in condensation reactions to create up to 15% of urea defects within the chains. Switching to the polymerization of α-hydroxy-ω-O-hydroxyphenyl urethanes can suppress these back-biting reactions, together with the better leaving group ability of the phenol that allows for creating NIPUs at lower temperatures of 90–120 °C.46,54 Significant improvements in the NIPU synthesis via melt transurethanization polycondensation have been accomplished by combining dimethyl dicarbamate AA monomers with BB diols.48,51,52 Unlike previous AB monomers, alcoholysis of the AA monomer by a diol releases MeOH that is easier to eliminate than ethylene glycol or phenol. In addition, upon proper selection of the diol BB monomers, back-biting reactions are suppressed, limiting the formation of urea defects within the chains. Following this methodology, NIPUs with various microstructures and Mn > 50000 g mol−1 have been fabricated. For representative illustrations of these examples, we invite the reader to refer to existing reviews.42 Polycondensation via melt-transurethanization forms thermoplastic NIPUs with decent/high molar masses suitable for foaming processes. Advantageously, the AA or AB monomers are generally accessible from low-cost and commercially available ethylene carbonate or dimethyl carbonate. One has to mention that NIPUs are also accessible via a sequential terpolymerization approach involving diamines, CO2, and dihalides.55–57 The process involves the formation of a dicarbamate salt by the capture of CO2 by a diamine in the presence of a base. This salt further undergoes alkylation via an SN2 with alkyl dihalides to create urethane linkages. As this pathway generates equimolar amounts of waste and utilizes undesirable halide chemicals, we have disregarded this synthetic pathway from our survey/analysis.
 | ||
| Scheme 2 Non-isocyanate polyurethane synthesis via polycondensation methods. | ||
Polycondensation methods are already deployed at the industrial scale via reactive extrusion for the synthesis of other types of polymers (e.g., bisphenol A polycarbonate, polylactic acid, etc.). This technique should be adaptable to NIPUs provided that a sufficient supply of monomers is available in the future. Such features should then open perspectives in manufacturing NIPU foams in a single device via the reactive extrusion-continuous foaming process.
2.3. NIPUs by step-growth polyaddition
Step-growth polyaddition shares similarities with polycondensation except that no by-product is formed during the reaction. Over the last decade, this polymerization manifold has been extensively deployed to synthesize polyhydroxyurethanes (PHUs) (Scheme 3, path A).15–20 PHUs typically result from the moisture-tolerant polyaddition between 5-membered polycyclic carbonates and polyamines. As the aminolysis of the cyclic carbonate is rather slow even at T > 80 °C, various research teams have developed catalytic solutions to facilitate chain extension by using Lewis acids (Li salts, MgBr2, Yb(OTf)3, etc.),58 organobases (DBU, TBD, phosphazene)58 or hydrogen bond donors such as (thio)ureas.59 Recent discoveries have highlighted the multifaceted role of water as a highly efficient accelerator in thermoset PHU fabrication from r.T. reactive formulations.60 Besides acting as a catalyst for the aminolysis of cyclic carbonates, water also disrupts the multiple intra-/inter-molecular H-bonding interactions within the PHU clusters, enhancing the chains' mobility and delaying the vitrification via the hydroplasticization effect.60,61 Other strategies capitalize on microwave utilization62–64 or on the smart design of the monomers to facilitate the formation of urethane linkages. Introducing ether, ester or sulfur electron-withdrawing groups in the β-position of the carbonate moiety,16,65–68 expanding the ring size to 6-membered cyclic carbonates16,69–71 or inserting endo- or exo-cyclic vinylene groups within 5-membered cyclic carbonates allows for significant improvement of aminolysis,72–75 even enabling some r.T. transformations.16 Playing on the amine structure also shows benefits for the PHU synthesis, especially when using amine scaffolds with β-alcohols.76 All these reactivity considerations have been detailed and rationalized in a very recent tutorial review, which the reader can refer to.16 Except for NIPUs synthesized from exovinylene cyclic carbonates and primary amines (furnishing poly(hydroxyoxazolidone)s, a special class of PHUs embedding cyclic urethane linkages) or secondary amines (furnishing poly(oxo-urethane)) (Scheme 2, path B), access to high molar mass PHUs is challenging, with Mn being most often limited to <15000 g mol−1 under bulk or solvent conditions, but reaching up to 35000 g mol−1 by reactive extrusion.77,78 The cause for the low molar masses partly arises from the reversible nature of the hydroxy-urethane linkages, limiting chain growth to an equilibrium (see Section 4.2). Increasing extents of intermolecular hydrogen bonding over the reaction course also hinder chain mobility, ultimately contributing to slowing down the polymerization. While rather limiting for the fabrication of thermoplastic PHUs, this chemistry is, on the contrary, perfectly suited for the synthesis of thermoset PHUs from reactive polyfunctional precursors (with one of the comonomers displaying a functionality greater than 2).79
 | ||
| Scheme 3 Non-isocyanate polyurethane synthesis via step-growth polyaddition. | ||
The extensive structural diversity of accessible polyamines and polycyclic carbonate precursors, some of which originate from bio- and/or CO2 sources,80–84 enables the engineering of PHU thermosets with thermo-mechanical properties that are modular on demand, thereby complying with the specifications of targeted applications. With these assets in our hands, we possess a versatile chemical tool capable of not only supplanting conventional PUs with a non-isocyanate alternative but also adapting to the dominant industrial slabstock, RIM, formed-in-place, or spray foaming technologies of PUs, further allowing for the retrofitting of existing industrial foaming plants.
Take-home messageKey differences between conventional PUs and NIPUsDifference 1 – reaction kinetics. Synthetic routes to NIPUs display insufficient or no exothermicity and proceed more slowly than the fast and exothermic isocyanate/polyol reactions in conventional PUs. Difference 2 – microstructure. NIPUs lack biuret, allophanate, isocyanurate, and uretdione linkages providing rigidity in conventional PUs.6 The NIPU structures vary significantly from one to another, depending on the chosen synthetic route (e.g., chain-growth vs. step-growth polymerization). These structural variations translate into significantly different thermo-mechanical properties. Difference 3 – molar mass and architecture. Achieving high molar mass thermoplastic NIPUs remains difficult; the most mature and efficient methodologies yield thermosetting PHUs. Main challenges for NIPU (foam) development Challenge 1 – catalyst and monomer design. Optimize catalysts, design monomers or hybridize chemistry85–87 to accelerate urethane formation and offer more flexibility to synchronize polymerization with the release of the blowing agent for adequate foam expansion and curing. Challenge 2 – performance matching. Select monomer combinations that enable the fabrication of NIPUs with thermo-mechanical properties comparable to those of conventional PUs. Challenge 3 – synthetic protocols for industrial integration. Adapt NIPU synthetic methods to align with established PU technologies and processes, ensuring compatibility and smooth retrofitting of existing industrial plants. |
3. How to foam non-isocyanate polyurethanes?
3.1. Polymer foaming and underlying mechanism
Understanding and controlling the polymer foaming process is crucial to fabricating materials with adequate properties. Briefly, the foaming mechanism follows three steps, including cell nucleation, growth, and stabilization (Fig. 2).9,88 Cell nucleation consists of forming a polymer/gas solution by dissolving a blowing agent into a molten polymer/viscous reactive formulation. When reaching supersaturation, the gas dissolved within the polymer causes thermodynamic instability, creating nucleation nodes. This step is followed by cell growth. The increase in gas pressure within the cells and gas diffusion allows the “soft” viscoelastic matrix to expand, creating a porous structure.89,90 Ensuring proper viscosity control is crucial for uniform bubble growth. Unsuitable polymer viscosity/viscoelastic properties lead to cell drainage and coalescence of the gas bubbles, forming non-homogeneous foams with large cell sizes or even collapsed foams. Too high viscosities limit the polymer expansion. In the last step, cell stabilization prevents the foam from collapsing. This stage is usually controlled by adding stabilizers, by rapid material cooling (most often for thermoplastic foams) or curing (thermosetting foams). Dispersion of bio- (cellulose, lignin, etc.),91 inorganic (silica, fibers, CaCO3, clays, etc.)92–94 or carbon-based (nanotube, black carbon, graphite)95 fillers within the polymer offers an additional lever to adjust the foam's characteristics. These multi-role additives act as nucleation agents, (melt)viscosity modifiers and/or structural reinforcements. Finally, the proper selection of the blowing agent (physical or chemical, endogenous or exogenous)8,96 and the foaming process govern the formation of foams with closed- or open-cell structures and controlled cell geometry/size. For example, a physical blowing agent is generally preferred to design low-density foams with closed cells. The decomposition of a chemical blowing agent that releases small gaseous molecules (CO2, N2, or H2) is preferred for one-pot formulations and a simplified setup.7 The high diffusion rate of these gases within the polymer causes cell rupture, leading to foams with open cells.7 | ||
| Fig. 2 General polymer foaming mechanism. | ||
3.2. Physical vs. chemical blowing of polymers
Physical foaming leverages the physical change (generally endothermic) of a non-reactive chemical, commonly referred to as a physical blowing agent, blended in a polymer melt/reactive formulation to generate a gas that promotes material expansion.7 The most prevalent physical changes involve either the volatilization of low-boiling point liquids (endothermic blowing agents) or the release of compressed gas (CO2, N2) to atmospheric pressure. Industrially, the supercritical carbon dioxide-assisted blowing technology is the method of choice to fabricate elastomeric thermoplastic PU foams for high-performance shoe soles (e.g., BASF Infinergy®). Hydrocarbon blowing agents (e.g., pentane) are commonly used for the fabrication of insulation materials (panels, tapes, sealants, etc.) from reactive thermosetting formulations.Chemical foaming leverages the decomposition or chemical reaction of an inorganic salt (e.g. carbonate or borohydride salts), or an organic compound (hydrogen peroxide, azocarbodiimides, tetrazoles, semicarbazides, etc.), mixed with a polymer melt/reactive formulation, to generate in situ volatile compounds (e.g. CO2, N2, H2 or water) that promote material expansion.7 Most chemical blowing agents are classified as “endothermic” as their decomposition is generally triggered by heat and usually leads to foams with open-cell morphology found in items for comfort, protective equipment (shock absorption), acoustic insulators, etc.
Self-foaming refers to a particular endogenous chemical blowing process where the blowing agent is generated in situ by the (partial) decomposition of a monomer or another reactive foam component (e.g., reactive stabilizer) that is contributing to the polymer structure.7,8 This methodology is well-adapted to the foaming of conventional PUs from isocyanate/polyol formulations. The partial hydrolysis of the isocyanate releases CO2, causing material expansion simultaneously with PU network construction. Self-foaming methods are predominantly utilized at the industrial scale, occasionally in conjunction with the application of physical blowing agents (e.g., for spray foaming), to generate foams suitable for diverse applications.
Directly adapting a PU foaming process for NIPUs requires substantial adjustments. The limited accessibility of thermoplastic NIPUs confines foam fabrication almost exclusively to thermosetting formulations. Moreover, the inherently low reactivity of NIPU precursors in these formulations poses significant challenges in controlling the viscosity increase, the gelation rate of the NIPU matrix, and the release of the blowing agent (whether endogenous or exogenous). As discussed in the following sections, polyhydroxyurethanes are emerging as the most promising candidate for NIPU foam fabrication.
3.3. NIPU foams by physical blowing
 | ||
| Fig. 3 Supercritical CO2-assisted foaming of thermoplastic PHUs via a one-step or two-step procedure. SEM figures reproduced from ref. 100 with permission from the Royal Society of Chemistry. Copyright 2016, Royal Society of Chemistry Publisher. | ||
Efforts to design PHU foams in a single-step foaming process significantly alter the foam characteristics. This strategy involves a saturation step of the PHUs at 90 °C, a temperature close to their Tm, followed by a fast vessel depressurization. Due to the low CO2 density even at 300 bar, a long saturation time of 24 h is necessary to dissolve a sufficient amount of CO2 in the matrix. Upon release of the CO2 pressure, poorly defined foams with broad cell size distributions between 4 and 22 µm featuring irregular shapes or even collapsed cells are obtained.100 A similar trend was reported later by Rwei for the scCO2-assisted one-step foaming of branched PHUs synthesized from bis-phenol A diglycidyl carbonate, a blend of ethylene diamine with 7.5 or 10 mol% of a C36 alkylenediamine and ppm amounts of 1,2,4,5-benzenetetracarboxylic acid.101 Following the saturation of these amorphous polymers at a pCO2 of 150 bar for 6 h at 80 °C – a temperature far exceeding their Tg's estimated at ∼30 °C – ill-defined foams with densities of 215 to 432 kg m−3 and cell sizes in the 9 to 23 µm range were obtained upon CO2 release. The author correlated the lower foam density of the C36 alkylenediamine-rich PHU materials to the higher flexibility of the chain, facilitating the penetration of CO2 within the polymer matrix during the saturation step.
 | ||
| Fig. 4 Foaming of thermosetting PHU resins using Solkane® (A) or HFO-1233zd (B) as a physical blowing agent. SEM picture (A) reproduced from ref. 104 with permission from John Wiley and Sons. Copyright © 2016, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. SEM picture (B) reproduced from ref. 105 with permission from Elsevier. Copyright 2025, Elsevier Publisher Ltd. | ||
Following a procedure similar to Blattmann, Zhang exploited trans-1-chloro-3,3,3-trifluoropropylene (HFO-1233zd, 14 wt%) to foam a reactive NIPU formulation composed of 1,4-cyclohexanedimethanol bis(cyclic carbonate) (CD) and blends of m-XDA and PEI (Fig. 4B).105 Although the morpho-structural properties of the foams are governed by the content of highly reactive PEI, lower-density foams (d = 139 kg m−3) with a more uniform pore structure (cell size of 0.8 mm) are produced at a 40/60 m-XDA/PEI blend ratio (by mass). The originality of the work lies in the utilization of cellulose nanocrystals (CNCs) that function as a biosurfactant stabilizing cells and limiting Ostwald ripening. Adding 3.3–9.7 wt% of CNC contributes to the formation of foams with slightly reduced densities (∼130 kg m−3) and smaller cell diameters (0.6 mm). Furthermore, the presence of the filler enhanced the mechanical properties of the foams, particularly along the foaming direction, with a two- to three-fold increase in the compression modulus, from 0.15 MPa without the additive to 0.35 MPa with 9.7 wt% CNC.
Water, ethanol or binary water/ethanol mixtures have also been used for the fabrication of thermosetting foams.106 An aminotelechelic hydroxyurethane-co-urea oligomer (86% urea linkages) – made by stepwise transurethanization polycondensation of a dicarbamate monomer with a diol (pripol®2033) followed by chain extension with priamine®1074 – is blended with the blowing agent, a bis-aminopropyl-terminated PDMS stabilizer (2 wt%) and a polyepoxy resin as a crosslinker until a homogeneous solution is obtained before curing at 73–120 °C for 2 h. When using water as the blowing agent (content not specified in the original work), the optimal foaming temperature is determined to be 95 °C, yet resulting in a uniform, highly flexible foam with a density of 190 ± 40 kg m−3, an open-cell morphology and cells averaging 590 ± 270 µm. Substituting water with ethanol leads to foams with higher density (280 ± 25 kg m−3) and smaller cell diameters (280 ± 120 µm). A slight reduction in density (115 ± 40 kg m−3) is observed when a 75/25 water/ethanol mixture is used, although the corresponding foam retains a highly heterogeneous cell size distribution (430 ± 150 µm). Changing the stabilizer content (up to 10 wt%), reducing the foaming temperature (from 73 to 85 °C) or changing the water/ethanol composition doesn’t improve the morpho-structural characteristics, homogeneity or density of the foams.
3.4. NIPU foams by chemical blowing
 | ||
| Fig. 5 Chemical blowing of thermosetting PHU foams. (A) Size dependence of the decomposition temperature of NaHCO3 salt; (B) evolution of the foam density with the particle size and catalyst loading; (C) SEM characterization of various thermosetting PHU foams. SEM pictures reproduced from ref. 107 with permission from Elsevier. Copyright © 2023, Elsevier Ltd. | ||
Other studies, with limited or even no details on the foaming process or foam characteristics, have utilized bicarbonate salts in the production of bio-based PHU foams. Pizzi et al. developed glucose-based NIPU oligomers by condensing a dialkylcarbonate monomer derived from glucose with HMDA, followed by curing and foaming at 200 °C for 30 minutes using NaHCO3 (∼1 wt%) as the blowing agent and a reactive epoxysilane coupling agent/compatibilizer (<1 wt%).108 This formulation produced rigid foams with open cells and densities ranging from 75 to 127 kg m−3. However, it seems likely that the role of the silane additive in the foaming process is more significant than initially assumed. We believe that the methoxysilane groups can participate in (self-)condensation reactions, leading to the formation and release of methanol at 200 °C. Such an assumption is supported by results showing that the absence of NaHCO3 in the reactive NIPU formulation delivered foams with a density of 75 kg m−3, which is almost identical to the 88–90 kg m−3 density observed under the same conditions in the presence of the salt.
Similarly, some authors reported the dual use of NH4HCO3 with citric acid to create plant oil-based PHU foams from carbonated linseed oil/HMDA reactive formulations.109,110 The acid acts as a catalyst, accelerating the decomposition of the bicarbonate salt and releasing gas at 80 °C, while also leading to foam formation within 15 minutes. However, the foam properties evolved over several weeks, transitioning from a stiff material after 24 h to a more rigid structure with a Tg of 42 °C after weeks of equilibration. This gradual change in foam behavior is attributed to the incomplete reaction between the sterically congested internal carbonate moieties and amines within the initial 15 minutes, which slowly progresses over time. Unfortunately, no data on the density, cell size and morphology or mechanical performances of the so-produced expanded materials are provided.
3.5. How to produce NIPU foams via self-blowing methods?
 | ||
| Fig. 6 H2 self-foaming of NIPUs. (A) Foaming mechanism; (B) thermosetting PHU foams produced from 5-membered cyclic carbonates; (C) thermosetting PHU foams produced from 6-membered cyclic carbonates; (D) thermosetting foams produced from NIPU oligomers synthesized by polycondensation. SEM pictures reproduced (1) from ref. 114 with permission from Elsevier. Copyright © 2015, Elsevier Ltd; (2) from ref. 118 with permission from the Royal Society of Chemistry. Copyright © 2020, Royal Society of Chemistry Publisher; (3) from ref. 115 with permission from Elsevier. Copyright © 2016, Elsevier Ltd; (4) from ref. 116 with permission from Elsevier. Copyright © 2022, Elsevier Ltd; (5) from ref. 121 with permission from Elsevier. Copyright © 2022, Elsevier Ltd. | ||
To tackle the inherent low reactivity of 5CC, Coste et al. specifically designed a liquid tetra-functional six-membered cyclic carbonate precursor (T6CC) (Fig. 6C), which has been utilized to prepare PHU foams under catalyst-free conditions.116 The rapid gelation of the T6CC/cadaverine formulation at 50 °C in ∼5.5 min confirms the higher reactivity of six-membered rings vs. five-membered cyclic carbonates, as no gelation is observed in the comparative TMPTC/cadaverine system even after 3 hours. Then, still adopting a shorter sequential curing at 50 °C for 4 h followed by a post-treatment at 120 °C for 2 h, homogeneous high-density PHU foams with diverse material properties have been manufactured from T6CC and various diamines (cadaverine, m-XDA or IPDA) using PMH (5 mol% vs. amine). The rigid foam made from IPDA (Tg = 27 °C) has the largest pore size of 1.35 ± 0.32 mm and the lowest density of 170 kg m−3. The foam density increases with cadaverine (350 kg m−3) and m-XDA (530 kg m−3) while cell sizes reduce to 0.42 ± 0.19 mm and 0.21 ± 0.07 mm, respectively. Both foams display Tg values below r.T. (Tg = 7 and 15 °C), making them flexible. The authors correlated the morpho-structural properties of the foams to the reactivity of the diamines. Unlike the formulation made from IPDA (Tgel = 17 min), the fast gelation PHUs based on cadaverine (Tgel = 4 min) and m-XDA (Tgel = 7 min) limit material expansion upon gas release, leading to highly dense foams of smaller cell sizes. PMH has been further utilized for the fabrication of biobased PHU foams by mixing cyclocarbonated lignin with a commercial fatty-acid based dimer diamine (Priamine) as precursors.117,118 The TBD catalyst and DMSO were to speed up the curing and facilitate the compatibilization of the formulation components, respectively (Fig. 6B). The foaming is initiated by the addition of Momentive (1.5 to 3% in volume) and the curing of the reactive formulation at 150 °C for 12 h. Rigid foams with densities of 241–337 kg m−3 and a high Tg of ∼100 °C have been produced. The concept has been recently extended to the preparation of low-density (<100 kg m−3) flexible NIPU foams (Tg around −15 °C to −18 °C) containing 17.4 to 35.6% of lignin.119 A simple adaptation of these works enabled the fabrication of lignin-based nonisocyanate polyurethane/silver nanoparticle composite foams which exhibit antimicrobial properties (cf Section 5).120
Slightly different in concept is the utilization of PMH to produce thermosetting foams from (aminotelechelic) NIPU oligomers and polyepoxide crosslinkers (Fig. 6D). This strategy is exemplified by Valette et al., who adapted their previous formulations from physical blowing of NIPUs, by simply replacing water, ethanol or water/ethanol mixtures as a physical blowing agent (cf. Section 2.1) with reactive PMH.121 Foaming was realized at 100 °C for 30 minutes and the authors found that a 0.7% loading of PMH is optimal to fabricate foams with the lowest density (160 kg m−3), having open cells with a size of 430 ± 210 µm. Based on rheology experiments, the amine/epoxide reaction is fast and allows for gelation in just 1.5 min. Foams can also be produced from r.T. reactive formulations, but curing requires a longer time of 14 h. All foams display a negative Tg value ranging from −28 °C to −10 °C in line with the flexible nature of these materials. Ahmad et al. also reported a similar strategy by curing foam formulations made of PMH (5 wt% vs. amine), amine-terminated hydroxyurethane oligomers based on castor oil and ethylene diamine, HMDA or diethylene triamine and epoxy resin (structure not specified, EEW ∼190 g eq−1). Foams with densities of 155 to 235 kg m−3, open-cell morphology with cell sizes of 115 to 142 µm and Tg values of 28–35 °C have been produced at 90 °C in 1 h. As a trend, a rise in the epoxy content within the formulation densifies the NIPU foams, in line with a higher crosslinking density/rate of the network that limits its expansion.
3.5.2.1. From five-membered cyclic carbonates. North has conceptualized the first dual utilization of 5-membered cyclic carbonates (5CC) as both a monomer for the construction of PHUs and a source of CO2 to create pores within the matrix. In his system, the decarboxylation follows in cascade the aminolysis of a specific sorbitol-derived bis-carbonate with a fused ring structure.122 The ring-opening of the 5CC moieties delivers the two usual β-hydroxyurethane regioisomers. The one with primary alcohols then participates in an intramolecular secondary cyclization with the adjacent unreacted carbonate ring, resulting in the formation of an isosorbide-like structure and the release of CO2 (Scheme 4). When selecting cadaverine or HMDA as comonomers, porous semi-crystalline linear NIPUs (Tg of −31 and −6 °C, Tm of 190 and 180 °C, respectively) were formed in 20 h at 100 °C. Unfortunately, the authors did not evaluate or describe the foam's morphology nor characteristics.
 | ||
| Scheme 4 North's intramolecular decarboxylation concept. | ||
Thiol-induced foaming. North's findings highlight the ambident electrophilic character of 5CCs that can be rationalized by the hard and soft acid base (HSBA) theory of Pearson.123–127 Five-membered cyclic carbonates with a low substitution degree (like ethylene or propylene carbonates) feature two electrophilic sites, i.e., a hard carbonyl and a soft methylene carbon (Scheme 5). By playing with the hard or soft nature of the nucleophiles, it is possible to guide chemo- and regio-selective aminolysis and decarboxylation reactions. Hard amine nucleophiles undergo rapid acylation via carbonyl attack, while soft thiol nucleophiles engage in slower S-alkylation through methylene attack.128 This second reaction, forming thioethers, triggers in situ CO2 release via spontaneous decomposition of the unstable carbonic acid intermediate.
 | ||
| Scheme 5 CO2 self-foaming of thermosetting PHU resin induced by a thiol and influence of the amine and thiol structure on the reactivity. | ||
Following these considerations, Detrembleur et al. expanded the field of CO2 self-blowing poly(hydroxyurethane)s to diverse formulations (Fig. 7).128 By mixing stoichiometric amounts of complementary reactive groups found in TMPTC, diamine (jeffamine, m-XDA or HMDA) and an aliphatic dithiol ((2,2′(ethylenedioxy)diethanethiol) (EDT)), uniform open-cell foams with densities of 166–320 kg m−3 and cell sizes of 0.25–0.98 mm have been fabricated (Fig. 7A). The presence of thioether linkages within the network induces chain mobility, resulting in flexible and resilient foams of low Tg values between 2 and 24.6 °C. In this multi-step foaming protocol, the catalyst plays a primordial role in regulating aminolysis and decarboxylation. While aminolysis proceeds even without a catalyst, S-alkylation of thiols requires it. Besides, the reactivity of S-nucleophiles is influenced by a balance between acidity and sulfur electron delocalization.129 Aromatic thiols are more reactive than their aliphatic counterparts and a low basicity organobase, such as NEt3 or the diamine monomers used to fabricate NIPU foams, promotes S-alkylation.130 With much less reactive aliphatic thiols, only nitrogen superbases (DBU, DBN, or TBD) are effective, significantly enhancing the CO2 delivery while also allowing for moderate acceleration in hydroxyurethane formation.128,131,132 As a result, both reactions occur simultaneously but at different rates, with aminolysis being slightly faster than S-alkylation. This difference in thiol reactivity is demonstrated by comparative foaming experiments from Thiol/TMPTC/m-XDA formulations (T = 100 °C, t = 3 h, 5 mol% DBU vs. cyclic carbonate). The formulation made of the aliphatic thiol (EDT) exhibits a delayed foam expansion – initiated after 30 min – compared to those with aromatic thiols, such as bismuthiol (20 min) or 4,4′-thiobisbenzenethiol (10 min) (Fig. 7B).130 Unlike the foam made from EDT that is flexible (Tg = 8 °C) and high in density (355 kg m−3), those made from aromatic thiols are less dense (203–309 kg m−3) and more rigid (Tg of 30–41 °C). Enhancing the thiol functionality of the blowing agent has minimal impact on the morpho-structural characteristics of the foams but significantly improves their thermo-mechanical properties through additional crosslink formation.133 As demonstrated by Torkelson et al., processing fatty acid-derived biscyclic carbonate (Erisys® GS-120)/Jeffamine® T-403 formulations with di- (EDT), tri- (trimethylolpropane tris(3-mercaptopropionate) (TMPTSH) or tetra-functional pentaerythritol tetrakis(3-mercaptopropionate)thiols delivers foams of similar densities of 240–270 kg m−3, cell size averaging from 0.25 to 0.31 mm, cell density of 42–51 × 103 cells per cm3 and low content of open cells for high density materials (d = 530 kg m−3). All foams are flexible and highly resilient, with Tgs evolving from −30 to −23 °C with the increase of thiol functionality.133 This is consistent with a higher crosslinking density restricting chain mobility within the network. Additionally, tri- and tetra-functional thiols lead to foams four- to six-fold more resistant to deformation than those obtained from the EDT blowing agent.
 | ||
| Fig. 7 Thiol-induced foaming of thermosetting PHU resins. (A) Influence of the thiol content on the morpho-structural and mechanical properties of the foams; (B) influence of the thiol structure (aliphatic vs. aromatic) on the morphologies and thermal properties of the foams. SEM pictures reproduced (1) from ref. 131 with permission from Elsevier. Copyright © 2023, Elsevier Ltd; (2) from ref. 128 with permission from John Wiley and Sons. Copyright © 2020, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim; (3) from ref. 130 with permission from the Royal Society of Chemistry. Copyright © 2024, Royal Society of Chemistry Publisher. | ||
To mitigate the unpleasant smell of thiols, thiols masked under a “dormant” form, being either a thiolactone134 or a dithiocarbonate,135 have been proposed as blowing agents (Fig. 8). These heterocycles are more reactive than cyclic carbonates. They undergo rapid ring-opening by amines, creating in situ a thiol during the initial stage of the process (Fig. 8A). The foam formation results from the alignment of three divergent and regioselective domino reactions, i.e., the fast thiol generation by aminolysis of the masked precursor, followed by the slower PHU network construction and its blowing via S-alkylation. For stoichiometric TMPTC/Jeffamine EDR148 formulations, adding 25–30 mol% of thiolactone or dithiocarbonate (vs. cyclic carbonate) yields open cells and flexible foams of 167 kg m−3 (in 5 h at 80 °C) or 364 kg m−3 (in 24 h at 90 °C), respectively (Fig. 8B). Unlike formulations made of dithiocarbonates, DBU is optional with thiolactones. However, the absence of a catalyst leads to foams of higher density (442 kg m−3). This trend extends to other formulations replacing EDR148 by m-XDA, where foam density increases from 185 kg m−3 with DBU to 310 kg m−3 without a catalyst. Due to the inherent properties of dithiocarbonates, the resulting NIPU foams exhibit two Tg's, indicative of a two-phase material.135 Since dithiocarbonates copolymerize with diamines and are more reactive than analogous five-membered cyclic carbonates, it is postulated that low Tg (−45 to −31 °C) polythiourethane segments form early in the curing process. Subsequent chain extension and gelation through polyaddition with TMPTC create PHU sequences with a higher Tg of 11 to 19 °C.
 | ||
| Fig. 8 CO2 self-foaming of PHU thermosetting resins using masked thiols. (A) General domino concept; (B) key differences in PHU foaming using blowing agents masked under the form of a thiolactone or a dithiocarbonate (exemplified for a TMPTC/EDR148 formulation). SEM pictures reproduced from (1) ref. 134 with permission from the American Chemical Society. Copyright © 2022, American Chemical Society; (2) from ref. 135 with permission from John Wiley and Sons. Copyright © 2022, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Most previous protocols require a pre-curing step to adjust viscosity and/or prolonged foaming even at high temperatures to achieve an optimal balance between viscosity increase, gelation and foam formation. To shorten the foam fabrication, strategies such as decoupling gelation from foaming or preheating the foam components have been proposed. The first solution exploits oscillatory shear rheology to monitor at various temperatures the evolution of the shear loss (G″) and shear storage (G′) moduli of a non-stoichiometric reactive PHU foam formulation (without a thiol) and maintain tanδ values (tanδ = G″/G′) above 1, ideally from 2 to 4 (Fig. 7A).131,136 According to Torkelson, this value range supports uniform bubble growth in formulations made of TMPTC/Jeffamine EDR148131 or cashew nutshell-based cyclic carbonate/Jeffamine T-403.136 Then, since the matrix is not yet crosslinked, a thiol blowing agent (EDT) is added in the correct proportion to reach stoichiometry in the reactive group while providing sufficient viscosity for effective foaming. The formulation is then cured at elevated temperature (120 °C), forming foams within 20 to 120 min. Interestingly, materials produced using this accelerating foaming method exhibit morphologies and thermal properties comparable to those obtained with the original slower processes.
The second solution involves preheating all foam components separately at 100 °C,130 before mixing them at this temperature. For a stoichiometric TMPTC/m-XDA/EDT formulation, the foam expansion begins after 2 min in the presence of DBU. When aromatic thiols replace EDT, the expansion occurs significantly faster, within 15 sec with bismuthiol and instantaneously with 4,4′-thiobisbenzenethiol even in the absence of a catalyst. If gas generation is fast enough to induce a rapid rise of the foam in 2 to 10 min, a curing of 30 min is required to consume carbonate moieties and create sufficient crosslinked nodes. All foams are uniform with open-cells, with those made from aromatic thiols being less dense (166–185 kg m−3) than those derived from EDT (372 kg m−3). The foam formation is shortened by increasing the amine content. For example, in catalyst-free 1/1/0.25 TMPTC/m-XDA/aromatic thiol formulations, non-stoichiometric conditions – where amine and thiol reactive groups are in excess relative to cyclic carbonates – yield highly crosslinked foams (155–156 kg m−3) that can be demolded in just 5 minutes. The versatility of this approach is further exemplified by substituting m-XDA with Jeffamine EDR148, which results in a homogeneous, low-density foam (155 kg m−3).
Water-induced foaming. Five-membered cyclic carbonates are less sensitive to hydrolysis than isocyanates. Their decomposition into the corresponding vicinal diol and CO2 occurs slowly and typically requires elevated temperature, a strongly alkaline medium and an excess of water.137–139 A key distinction from conventional polyurethane (PU) foaming is that the vicinal diol formed during hydrolysis is unreactive toward both amines and cyclic carbonates. As a result, it does not participate in the crosslinking process of the PHU (Fig. 9A).
 | ||
| Fig. 9 Water-induced foaming of thermosetting PHU resins. (A) General concept; (B) examples of PHU foams produced using water or a hydrated protein as a blowing agent. SEM pictures reproduced from (1) ref. 140 with permission from John Wiley and Sons. Copyright © 2022, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim and (2) ref. 142 with permission from the Royal Society of Chemistry. Copyright © 2024, Royal Society of Chemistry Publisher. | ||
Thus, achieving self-foaming in PHU systems using water requires precise control over both aminolysis and decarboxylation by carefully adjusting the formulation composition and stoichiometry, water content, temperature (80–100 °C), and catalyst (e.g., DBU or KOH).140 Following these considerations, PHU foams have been produced by mixing a non-stoichiometric ratio of reactive amine and cyclic carbonate groups, with a fixed [NH2]/[5CC] ratio of 0.75. This ensures that (theoretically) 25% of the cyclic carbonates serve as sacrificial groups to generate CO2 through base-catalyzed hydrolysis. As illustrated for a TMPTC/m-XDA formulation using DBU as a catalyst (Fig. 9B), the morpho-structural and thermo-mechanical properties of the foams vary with the processing temperature and the water content. At 100 °C, a low-density foam (153 kg m−3) with large open cells averaging 2.3 mm is produced in 3 h using 1 eq. of water vs. sacrificial cyclic carbonates. The addition of hydrotalcite as a nucleation agent and viscosity modifier within the formulation improves the foam uniformity and decreases the cell size to 390 µm without impacting the density. Lowering the temperature to 80 °C slows down the CO2 release and necessitates prolonged curing times of 5 h to prevent foam collapse. This foaming procedure delivers twice denser foam (327–366 kg m−3). The slower hydrolysis can be compensated by a fourfold increase in the water amount (vs. sacrificial carbonates), yielding foams with lower densities (179 kg m−3) and more uniform open cells averaging 0.54 mm. In this foaming strategy, the extent of hydrolysis dictates the amount of vicinal diol produced, which in turn affects the PHU network crosslinking density. This is evidenced by the gel content values ranging from 86% to 95%. As a result of higher chain mobility, Tg values drop from 25 °C to 8 °C when the water content increases from 1 to 4 equivalents at foaming temperatures of 100 °C and 80 °C, respectively. Additionally, the presence of vicinal diol groups induces strong hydroplasticization under ambient humidity. Drying the PHU foams significantly raises their Tg's by 25–40 °C. Although foam density and cell size vary across foaming conditions, their mechanical performance also differs greatly due to varying degrees of crosslinking. For example, the compression modulus ranges from 6.66 MPa for a foam made at 100 °C with 1 eq. of water to just 0.149 MPa for a foam made at 80 °C with excess water. The versatility of the water-induced foaming approach, also applicable to PHU foaming in close molds, is proven by the fabrication of uniform low-density materials (226–480 kg m−3) from various formulations, either by replacing m-XDA with jeffamine EDR 148 or HMDA, or by valorizing other carbonates (neopentyl or PEG diglycidyl carbonate) and amine (TREN) precursors. Similar to the thiol system, pre-heating the components (e.g., TMPTC and Jeffamine EDR 148) to 100 °C before curing accelerates the foam fabrication in just 30 min with no impact on their morpho-structural and thermal properties compared to similar materials produced via the slow foaming procedure. Further improvements use TREN as a reactive curing accelerator. This polyamine has an “aminoethyl amine”-type structure with highly reactive amine groups. When mixed at 30% with another polyamine, it causes very rapid gelation of the network, preventing foam collapse. This results in a stable and uniform low-density material (193–209 kg m−3) within only 5 min when formulations are pre-heated to 100 °C.141
Interestingly, bio-based fillers derived from biomass waste streams such as proteins (gelatin, keratin, zein), lignin, cellulose or chitosan play a dual role in foam fabrication, acting simultaneously as a “reservoir” releasing water during foaming due to their inherent hydrophilicity and a biofiller improving cell nucleation and initial viscosity.142 Even a 1 wt% content of the biofiller is sufficient to obtain uniform open-cell foams with medium density between 240 and 320 kg m−3 (Fig. 9B). This open-cell porosity arises from the incorporation of bio-based fillers. These solid particles act as nucleation sites at the initial stage of the foaming. They typically increase the number of cells, resulting in thinner and mechanically weaker cell walls. In addition, these rigid particles generate local stress during bubble growth because they can’t accommodate the deformation of the expanding cell membranes, which ultimately promotes cell-wall rupture and the formation of open-cell foams. Well-crosslinked foams with a homogeneous porous structure were obtained with bio-filler contents of up to 30 wt%, demonstrating their potential to reduce production costs, as the bio-fillers originate from low-value biomass waste.
Room temperature-induced fast foaming. Conceptually, the foaming of PU benefits from the high exothermicity of the isocyanate/polyol reaction. The heat generated in situ creates a “snowball effect” that rapidly raises the temperature of the foam components from r.T. to far above 100 °C (generally >120–180 °C), further accelerating all reactions at play. This promotes the fast network formation within minutes while simultaneously delivering CO2 as the blowing agent. Unlike conventional PUs, the aminolysis of cyclic carbonates only generates a slight exotherm of ∼65 °C below the threshold of ∼100 °C. To kick-start the foaming from r.T. formulations, an extra “heat boost” is needed. Epoxide130,141,143 or exovinylene cyclic carbonate144 (αCC) additives proved to be effective by triggering additional heat generation (Fig. 10).
 | ||
| Fig. 10 Fast foaming of NIPUs from r.T. reactive formulations via cascade reactions with epoxides as heat release promoters. (A) Cascade reactions in water-induced self-blowing of PHUs; (B) cascade reactions in thiol-induced self-blowing of PHUs; (C) foaming parameters and properties of hybrid epoxy/PHU foams of various compositions produced by the water-induced fast foaming approach with an illustration of (i) the time evolution of the temperature with the epoxide content during foaming, (ii) the time for foam expansion and maximum temperature reached for various epoxide compositions, (iii) the evolution of the foam density with the epoxide content and (iv) the thermal characteristics of the hybrid PHU foams at various epoxide contents. | ||
Epoxide additives. The epoxy-amine reaction is recognized for its high exothermicity related to the strain release of the cyclic structure.145,146 In epoxy resins, it is common to witness an induction period during the curing process with little changes in viscoelastic properties, before a sudden exotherm and a fast increase in viscosity. It has been shown to result from an autocatalytic mechanism relying on the slow appearance of the first hydroxyl groups that further catalyze the network formation. This effect is further intensified by the rapid increase in viscosity, which inhibits efficient heat dissipation. To control the exothermicity during foaming, the hybridization of PHU chemistry through the addition of epoxides has proven effective. In a water-induced foaming process (Fig. 10A), partially substituting 25% of TMPTC with its epoxy precursor (TMPTE) in a TMPCT/m-XDA/TREN formulation is sufficient to elevate the temperature from r.T. to over 110 °C, producing a uniform, open-cell, medium-density foam (<300 kg m−3) within 3 min.141 Mechanistically, the tricomponent m-XDA/TMPTC/water reaction creates a first elevation of the temperature from r.T. to ∼87 °C. This heat rise triggers in cascade the aminolysis of the epoxy additive, leading to a second exothermic event elevating the temperature within the foaming zone to >100 °C. As a trend, increasing the TMPTE content influences both the foam expansion and the material thermo-mechanical properties. Epoxy-rich compositions exhibit significantly higher exothermicity, with temperatures even reaching 180 °C when TMPTC is replaced by 3 eq. of TMPTE. However, the exotherm is delayed, and foam formation slows down with increasing TMPTE content (e.g., 6 min with 3 eq. of TMPTE). Beyond 0.5 eq. of TMPTE, the foam density plateaus around 150 kg m−3, likely due to the higher degree of crosslinking (Fig. 10C). Lower epoxide content (≤0.5 eq. vs. TMPTC) produces foams with Tg's (∼20 °C) and compression moduli (0.5–0.6 MPa) similar to pure expanded PHUs. Conversely, increasing the epoxide concentration leads to materials with epoxy resin-like properties, exhibiting higher Tg's of 35–40 °C and enhanced stiffness (E = 3.7–4.7 MPa). Further research underscores the role of epoxide structure in the exothermic behavior of the foaming process. With aromatic epoxide (DER332), the presence of the electron-withdrawing group nearby the oxirane ring lowers the activation barrier required for its aminolysis, thereby increasing its reactivity compared to aliphatic epoxides (e.g., neopentyl glycol diglycidyl ether).143 As a result, formulations containing aromatic epoxides exhibit higher exothermic peaks than their aliphatic counterparts. The use of epoxide with aromatic moieties also introduces rigidity into the NIPU chains, further increasing the Tg of the resulting foams and improving their mechanical performances.
Adding epoxy additives to initiate the fast r.T. foaming of PHUs using thiol blowing agents is also applicable (Fig. 10B).130 However, this strategy necessitates the proper selection of the foam components and the careful adjustment of their composition. For successful foaming, it is essential to use epoxy/cyclic carbonate blends in a 50/50 molar ratio and acidic thiols such as bismuthiol and 4,4-thiobisbenzothiol. Deciphering the foaming mechanism highlights a complex interplay of multiple cascades and parallel reactions. In the initial stage of the process, a quick acid–base reaction takes place between the thiol and the amine, yet protecting the thiol in the form of a non-reactive salt while simultaneously releasing heat. The rise in temperature triggers the aminolysis of the cyclic carbonate and/or the epoxide. When reaching 80–100 °C in 1–2 min, the thiol-amine salt begins to decompose, releasing the reactive thiol. This species engages in competitive decarboxylation of 5CCs, driving foam expansion, and click reaction with the remaining epoxide, helping to maintain an elevated temperature (up to 170 °C) to ensure efficient material curing and prevent foam collapse. The fine-tuning of the formulation enables precise control over the morphology and thermo-mechanical properties of the foams by simply varying the chemical structure of the epoxide and amine components. Uniform rigid foams of 312 kg m−3, Tg's of 49 °C and compression modulus of 81 MPa have been produced by employing TMPTC with bismuthiol, aromatic DER332 and m-XDA. In contrast, switching to aliphatic epoxides (e.g., butanediol diglycidyl ether, BDGE) and (cyclo)aliphatic amines (such as Jeffamine EDR148 or 1,3-BAC) produces flexible to semi-rigid foams with densities of 227–400 kg m−3, low Tg values ranging from −17 to 24 °C, and compression moduli between 0.096 and 23.5 MPa.
Exovinylene cyclic carbonate additives. αCCs are five-membered heterocycles that can be efficiently synthesized from CO2 and propargylic alcohols (Fig. 11).144 Unlike conventional 5CCs, the presence of an exocyclic olefin moiety within the cyclic carbonate structure makes αCCs highly reactive toward N-, O-, or S-nucleophiles,72,75,147,148 unlocking diverse chemical transformations. In particular, their aminolysis with primary amines yields 5-membered cyclic urethanes (hydroxy-oxazolidones) rapidly.16,149–151 This reaction proceeds instantaneously at r.T., even without a catalyst, and is characterized by a significant exotherm. Leveraging these unique reactivity features in the water-induced CO2 self-foaming of cyclic carbonate/amine formulations allows raising the temperature to 110–130 °C within a few seconds by simply varying the αCC (4,4-dimethyl-5-methylene-1,3-dioxolan-2-one, DMACC) content vs. 5CC from 0.1 up to 0.3 eq. (Fig. 11A). However, both the aminolysis of αCC and the decarboxylation of 5CC can terminate NIPU chains, which reduces the crosslinking density of the networks and impacts the foam's morphology. To compensate for the adverse effects of these terminations and achieve an optimal balance between foam expansion and crosslinking, it is essential to use polyfunctional monomers with functionalities higher than two, such as TMPTC and TREN. For example, in a stoichiometric TMPTC/αCC/TREN formulation containing 0.15 eq. of water vs. TMPTC, decreasing the αCC content from 0.3 to 0.15 eq. increases the foam density from 173 to 323 kg m−3 while no foam is produced below 0.15 eq. Besides, at a fixed αCC content of 0.25 eq. relative to TMPTC, foams with densities of 188–233 kg m−3 are obtained and found uniform only when the water content is maintained between 10 and 15%. Substituting DMACC with sterically hindered αCC analogs (bearing cyclohexyl, phenyl, or isobutyl groups) does not significantly alter the exotherm or the foaming process. These analogs lead to the fabrication of homogeneous foams with slightly higher densities (∼270–390 kg m−3, Fig. 11B). Additionally, the choice of αCC influences the glass transition of the resulting materials, with values ranging between 28 and 38 °C. All foams display an open-cell morphology with a cell size averaging 85–180 µm.
 | ||
| Fig. 11 Fast foaming of NIPUs from r.T. reactive formulations via cascade reactions with exovinylene cyclic carbonates as heat release promoters. (A) Cascade reactions in water-induced self-blowing of PHUs; (B) foaming parameters and properties of NIPU foams of various compositions produced by the water-induced fast foaming approach with an illustration of (i) maximum temperature reached for various αCC compositions and related foam densities, (ii) Tg value evolution with the αCC content. | ||
3.5.2.2. Amine–CO2 adducts. Taking inspiration from reversible non-aqueous CO2–amine carbon capture technologies, Choong et al. have proposed carbamate salts as latent blowing agents for PHU self-foaming applications (Fig. 12A).152 Alkylamines have a strong affinity for various electrophiles and act as a CO2 acceptor, rapidly forming amine–CO2 adducts even at r.T. Further addition of a nitrogen superbase markedly enhances the reactivity of amine sorbent with CO2, allowing for the formation of amine–CO2-superbase “mixed” (bi)carbamates.153–155 Spontaneous CO2 desorption and amine/superbase regeneration are generally triggered at high temperature, often exceeding 100 °C. In the quest for liquid polyamine–CO2 adducts allowing for homogeneous mixing at r.T. with cyclic carbonate/amine formulations and tunable CO2 release, Choong et al. identified triethylenetetramine (TETA) as a promising candidate.152 In the absence of a nitrogen superbase, this polyamine absorbs 17.9% of CO2, providing a viscous liquid TETA–CO2 adduct (viscosity = 3500 mPa s−1). Adding DBU or DBN not only enhanced the absorption rate of CO2 but also allowed for higher CO2 uptake of 27.6% and 60.4%, respectively. Furthermore, these superbases facilitate CO2 desorption with desorption rate constants of 0.0021–0.0023 h−1 at a low temperature of 60 °C compared to 0.009 h−1 for the TETA–CO2 adduct at 100 °C. Mixing of TETA–CO2–DBN with two model cyclic carbonates substituted by an ester (M1) or an ether (M2) promotes hydroxyurethane formation (with high carbonate conversion of 93% for M1 and 68% for M2 after 2 h at 60 °C) and expedites CO2 desorption. This is reflected in the increased desorption rate constant values of 0.222 h−1 and 0.132 h−1 corresponding to CO2 releases of 87% and 77% respectively for M1 and M2 after 24 h at 60 °C. Learning from these model experiments provides guidelines for the fabrication of CO2 self-blowing PHUs using TETA–CO2–DBN as the latent amine. To synchronize CO2 desorption with PHU resin expansion, diamines (e.g., 1,2-bis(2-aminoethoxy)ethane or 1,5-diaminopentane) have been cured at 60 °C for 24 h with a blend of TMPTC and a furan-based biscyclic carbonate (FBC) (80/20 molar ratio). Notably, using TMPTC alone resulted in unsatisfactory foaming due to rapid gelation outpacing CO2 release. By analogy with M1, the ester groups within FBC enhance its reactivity against amines compared to the ether-substituted TMPTC, enabling good control over viscosity increase, gelation, and CO2 release. The use of 13 mol% of TETA–CO2–DBN vs. TMPTC leads to uniform open-cell foams with densities of 203–239 kg m−3 and a cell diameter of 232 ± 97 to 314 ± 116 µm (Fig. 12C). These foams exhibit flexibility, as reflected in their Tg values between 16 °C and 24 °C. Reducing the TETA–CO2–DBN content results in denser foamed materials (d = 401 kg m−3), while higher concentrations lead to rapid matrix expansion, collapse, and stabilization at 462 kg m−3. Increasing the temperature accelerates CO2 desorption, leading to non-homogeneous high-density foams (405 kg m−3) with large pores of ∼2 mm.
 | ||
| Fig. 12 Latent amine formation either via CO2 capture (A) or the oligomerization approach (B) and their thermal decomposition. (C) CO2 self-blown PHU foams produced from cyclic carbonates and latent amines or oligomeric PHU–carbamate adducts. SEM pictures reproduced from ref. 152 and 156 with permission from the American Chemical Society. Copyrights © 2023 and 2025, American Chemical Society. | ||
While elegant, Choong's concept is constrained by the limited availability of liquid polyamine–CO2 adducts suitable for PHU foaming and by the utilization of a significant amount of organobases, which are often costly and/or toxic. To address these limitations, Caillol revisited the foaming of thermosetting PHUs through a sequential methodology (Fig. 12B).156 In the first step, non-crosslinked PHU oligomers synthesized from polyfunctional monomers are infused with scCO2 under moderate conditions (45 °C, 100 bar, 1 h). Rheological analysis confirms that while the viscosity of the reactive formulation increases at 45 °C, no gelation occurs. During this stage, CO2 not only dissolves into the reactive mixture but also promotes the in situ formation of a carbamate salt as a latent blowing agent. After the saturation stage and cell depressurization (when cooled to 0 °C), the foaming is initiated by exposing the “infused” resin to elevated temperatures of 100–140 °C for 2–3 h. During this phase, the carbamate salt progressively decomposes and releases CO2 – promoting material expansion – and an amine – enabling further network curing and foam stabilization by reaction with the cyclic carbonate moieties. The success of this methodology is proven by the fabrication of rigid foams (227 kg m−3, Tg = 49 °C) with open cells averaging 0.58 ± 0.39 mm from TMPTC/m-XDA compositions added with 5 wt% of LAPONITE® fillers serving as a viscosity modifier and nucleation agent. The versatility of the approach is further illustrated by replacing up to 50% of TMPTC with a bis(5-membered cyclic carbonate), i.e., poly(propylene oxide) biscarbonate (PPOBC), which confers flexibility to the foams by the presence of the polyether backbone. This leads to the fabrication of foams with densities ranging from 285 to 491 kg m−3 and Tgs progressively decreasing to 28 °C for a 50/50 TMPTC/PPOBC molar ratio (Fig. 12C).
3.5.2.3. Carbonyldiimidazole. In 2023, Long introduced bis-carbonylimidazolides (BCI) as both latent CO2 blowing agents and monomers for the rapid construction of self-blowing NIPU foams devoid of pending hydroxyl groups (Fig. 13).157 These chemicals undergo β-hydrogen elimination by reaction with amines under basic catalysis, which liberates a carbamic acid at temperatures exceeding 160 °C, which subsequently undergoes entropically driven decarboxylation. The aminolysis of the carbonylimidazolide moiety, running in a parallel fashion, enables the formation of a new carbamate linkage with concomitant release of imidazole condensates (Fig. 13A). NIPU foams have been successfully fabricated within 30 seconds to 15 minutes by simply melting a (cyclo)aliphatic difunctional BCI monomer at 160–180 °C, followed by mixing with either an aliphatic (Jeffamine T403) or an aromatic (4-(4-aminophenoxy)benzene-1,3-diamine, TADE) triamine and their blend in various compositions. Manipulating the surfactant and catalyst content enables precise control over the morpho-structural properties of the foams. Without a surfactant, open-cell foams with cell sizes ranging from 500 µm to 1 mm were produced respectively when aliphatic or aromatic amines are used in combination with an aliphatic BCI monomer (Fig. 13B). Due to its stabilizing effect, the utilization of a silicone-ether type surfactant (DABCO DC193) at 0.8 to 5 wt% content significantly reduces the foam density, and for some formulations, allows for cell closure (Fig. 13C). Controlling the foam formulations by assorting the BCI and triamine monomers also allowed tuning the mechanical behavior of the foams. The combination of aliphatic precursors leads to flexible materials with low Tg (−3 °C) while mixing (blends of) monomers with cycloaliphatic and aromatic spacers, imparting restricted rotation, delivers rigid porous NIPUs with Tg up to 126 °C. Inherent to this concept is the formation of an equimolar imidazole condensate that requires removal by methanol extraction before foam characterization/utilization. Recent developments, reported in an abstract of papers from the ACS Spring 2024,158 utilize an orthogonal Michael addition to trap this by-product efficiently. This approach creates in situ small molecule additives acting as either flexible plasticizers or rigid fillers.
 | ||
| Fig. 13 CO2 self-blown NIPU foams by thermal decomposition of carbamate monomers (A) and flexible (B) or rigid (C) foams produced from biscarbonyldiimidazole monomers and various polyamines. SEM pictures reproduced from ref. 157 with permission from the Royal Society of Chemistry. Copyright © 2023, Royal Society of Chemistry Publisher. | ||
3.6. Porous NIPUs through “emulsion templated-like polymerization”
A series of bio-NIPU foams have been developed by Pizzi through a polycondensation process, which has been further reproduced by other researchers (Fig. 14). Decoding the foaming mechanism is challenging, as this aspect has not been properly addressed in the original publications, and the experimental protocols somehow lacked details and clarity. It is worth noting that the researchers used aqueous solutions of (bio-)monomers/oligomers in combination with glutaraldehyde and carboxylic acids as raw chemicals/crosslinkers. In this regard, we believe they exploited a kind of emulsion templated polymerization process – also referred to as “internal phase emulsion” polymerization – to create porous NIPUs.159 Conceptually, all foaming procedures exploit a similar methodology. A biomolecule in aqueous solution – selected from glucose,160,161 tannin,162–164 xylose165,166 – is transformed into a (poly)alkylcarbonate by condensation with dimethylcarbonate and then further reacted at 65–90 °C with an excess of diamine, e.g., hexamethylene diamine (70% in water), to create carbamates or NIPU oligomers. The addition of a carboxylic acid (e.g., maleic, citric, malic or aconitic acid)162/glutaraldehyde solution upon mixing then initiates the phase-separation qualified as “foaming”. We do believe that the foam formation responds to a complex interplay of various phenomena. The acid–base reaction between the carboxylic acid and amines – described as violent by the authors – generates a significant exotherm, causing partial evaporation of water, further expanding the liquid formulation. Concomitantly, the temperature rise promotes the crosslinking of the NIPU with an increase in the polymer viscosity. This results in the formation of a kind of stable emulsion, analogous to “mayonnaise”, composed of water droplets surrounded by the NIPU network under construction, whose curing is completed by a final high-temperature treatment (T = 70–103 °C) for 2–10 h (or overnight). During this thermal process, the water evaporates, yet delivering from flexible to rigid bio-NIPU foams. Besides the urethane linkages, amides and imines are identified, attesting to the condensation of amine moieties with carboxylic acids and aldehydes, respectively.164 The absence of decomposition products of citric acid excludes any participation of carboxylic acid as an endothermic blowing agent. This is further supported by the high decomposition temperature of citric acid, between 160 and 270 °C, which is not reached during the foaming process. | ||
| Fig. 14 Pizzi's foaming process of biobased NIPU precursors using glutaraldehyde/citric acid systems. SEM picture reproduced from ref. 164 from MDPI journal. | ||
The stability, density and cell morphology of the foams are governed by the contents of carboxylic acid and glutaraldehyde. Generally, increasing the carboxylic acid content (e.g., maleic acid) decreases the foam density. Pizzi has correlated this observation to the higher exotherm produced by the carboxylic acid/amine reaction at higher acid content. This likely enhances the water evaporation during the initial foaming stage, resulting in foams with lower density and larger cells. This is illustrated for glucose-160 or xylose-based166 NIPU materials, for which an increase of respectively the maleic (to 150%) or citric acid (to 200%) content in the monomer composition (vs. the corresponding benchmark samples) reduces the foam density from 130 kg m−3 to 80 kg m−3 and 467 kg m−3 to 303 kg m−3. Except for formulations containing citric acid, the use of glutaraldehyde is mandatory to get stable foams. Glutaraldehyde serves as an additional crosslinker, fastening the viscosity increase and gelation of the medium. When being absent, the foam collapses, while at too high a content, the density increases due to a weakening of the nucleation efficiency upon water evaporation and material drying. This is particularly observed for a tannin-based NIPU with a quasi-doubling of the density (from 150 to 260 kg m−3) by doubling the glutaraldehyde content.160,164 Considering only the publications specifying the NIPU material density, all foams display an open-cell morphology with large pores of heterogeneous size distribution between ∼500 µm and ∼2 mm.
Take-home messageKey differences between conventional PU and NIPU foamingDifference 1 – Foaming process control. (Self-)Foaming of PUs with physical or chemical blowing agents benefits from the high exothermicity of the isocyanate/polyol reaction, enabling rapid gelation of thermosetting resins and synchronization between PU crosslinking and expansion. Foams are produced within minutes with excellent control of morpho-structural and thermo-mechanical properties. PHU, made by step-growth copolymerization of polycyclic carbonates and polyamines, accounts for >90% of NIPU foams. PHU reactions are however slow and display low exothermicity, making it challenging to control viscosity increase, gelation rate, and blowing agent generation. Proper selection of catalysts, monomers, and the blowing system is essential to align PHU crosslinking with expansion. Physical blowing of PHU thermosetting resins has been achieved with hydrocarbons, (chloro)fluorocarbons, or water/ethanol mixtures, but prolonged curing (>14 h) at high temperature (>80 °C) is required to prevent material collapse. The addition of co-monomers, such as epoxy or acrylate, can be used to induce cascade exotherms and enable the crosslinking reaction under milder conditions. Concurrently, the scCO2-assisted foaming methodology is the only relevant approach for blowing thermoplastic PHUs. With chemical foaming agents (Na2CO3, NaHCO3/citric acid, azodicarbonamide, etc.), the foaming process remains slow, poorly managed and lacks versatility. Any variation in one processing parameter (e.g., size of the inorganic carbonate salt, foaming temperature) or in one monomer structure necessitates a complete readjustment of the foaming window. Self-blowing PHUs are prevalent and rely either on H2 release from hydrogenosiloxanes (e.g., Momentive® MH 15) or on CO2 generation from the partial decarboxylation of a foam precursor, such as a cyclic carbonate or carbonated amines. Hydrogenosiloxanes and (masked) thiol additives (used for decarboxylating cyclic carbonates) contribute to PHU crosslinking while reducing their Tg's, thereby enhancing foam flexibility. Similar to conventional PUs, water can induce the partial hydrolysis of cyclic carbonates with the release of CO2. This reaction is accompanied by the formation of unreactive vicinal diol moieties, which do not participate in foam crosslinking but increase its hydrophilicity. To prevent collapse and access uniform foams, the proportion of sacrificial cyclic carbonates should not exceed 25%. Water and also halide salt residues issued from the fabrication of cyclic carbonate monomers act as co-catalysts in PHU formation and accelerate the gelation. With carbonated amines, the control of the foaming process is strongly dependent on the decomposition rate of the amine–CO2 adducts and temperature. This foaming strategy is actually constrained by a limited range of suitable carbamate salts that decompose below 150 °C with kinetics compatible with the PHU network formation. Porous NIPU formation through “emulsion templated-like polymerization” is slow, restricting its utilization. Up to now, PHU chemistry hybridization is the only self-blowing process able to rapidly foam NIPU from r.T. formulations. Difference 2 – Foam density and morphology. In PU thermosetting foams, density (30 kg m−3 < d < 250 kg m−3), cell size (μm-scale), and porosity type (open or closed cells) are finely controlled through the careful adjustment of formulation parameters, including surfactant, catalyst and blowing agent. Morpho-structural control remains less efficient in PHU foams, which may exhibit non-uniformity and high densities exceeding 100–400 kg m−3. These foams typically have open porosity and large cells, with dimensions exceeding 100 µm to 1–2 mm. However, it is important to note that most reports do not utilize surfactants/foam stabilizers to assist foaming and control cell morphology. To date, closed-cell foams have been described for one carbonyldiimidazole/amine self-blowing formulation when used in combination with a surfactant and for the scCO2-assisted physical blowing of thermoplastic PHU. A nearly closed cell morphology was also described in the case of a thiol-assisted self-foamed PHU formulation, although the final density of the foam is relatively high (>500 kg m−3). As PHU foams exhibit higher hydrophilicity compared to conventional PU foams, surfactants or foam stabilizers used in PU foams do not display similar performance in PHU foaming. Therefore, identifying appropriate surfactants is paramount for the future optimal control of the morphology of PHU foams. Moreover, it is worth mentioning that progress must be made to precisely quantify the closed-to-open cell ratio, as conventional methodologies such as picnometry have not been properly used to this end in the field of NIPU foams to date. Difference 3 – Role of surfactants in controlling foaming. Silicone-based surfactants play a multifaceted role in PU foam formation by controlling the polyol/isocyanate emulsification, cell nucleation and stabilization, as well as foam density. Their low surface tension decreases the energy barrier to promote air bubble formation during mixing. This so-called “bubble dispersion stage” fixes the number of gas bubbles with no additional bubbles being formed during subsequent foam expansion.5,167 By adsorbing at the gas–liquid interface, the surfactants reduce the interfacial energy and limit the Ostwald ripening (diffusion of the smallest cells to the larger ones).168 They also prevent the cell wall drainage, i.e. the flowing of the monomers out of the cell wall to the exterior, forming the plateau borders (struts) separating bubbles in a foam.5,167,169 During the bubble expansion, a gradient in surfactant concentration is created along the cell surface. This triggers the movement of the surfactant from the higher to the lower concentration area (Marangoni effect). As the surfactants move, they drag liquids (monomers or low viscosity oligomers) with them, slowing film drainage. This phenomenon delays/limits cell wall rupture, coalescence and foam collapse, enhancing foam stability and uniformity during rise and curing. When evaluating NIPU foams, directly utilizing silicone-based surfactants that are effective in PU formulations presents a significant challenge in enhancing foaming properties. Indeed, the fundamental differences in the two distinct chemistries arise from monomer structure, miscibility, and polarity, initial viscosity, gelation rates, and also the hydrophilicity of the final polymer matrix. These factors are likely to alter surfactant interfacial behavior and therefore the bubble dispersion stage and cell stabilization. Surfactants specifically adapted for NIPU foaming are still to be discovered. Difference 4 – Thermo-mechanical properties. PHUs lack isocyanate-derived motifs (biuret, allophanate, isocyanurate, uretdione) and often have lower crosslinking density than conventional PU foams. This translates into low Tg and very low stiffness materials. Achieving rigid PHU foams with Tg > 60–80 °C remains a significant hurdle. Difference 5 – Hydrophilicity and humidity sensitivity. The OH-rich PHU backbone is intrinsically more hydrophilic than PU, making PHU foams susceptible to plasticization when exposed to humidity, affecting their mechanical properties and reducing their Tg value.170 Difference 6 – Industrial deployment. Most PU foam manufacturing processes are based on two-component diisocyanate/polyol formulations poured onto a conveyor belt, injected into molds or sprayed onto walls. These established technologies rely on the precise control of the initial viscosity of the reactive formulation and its homogeneous mixing, typically achieved using high-shear static mixers or high-pressure mixing heads. This mixing step is critical as it ensures the proper blending of the foam ingredients with the blowing agent and stabilizer, forming an emulsion that dictates the quality and morpho-structural characteristics of the foam. Foams are generally produced in 1 to 5 minutes at a temperature stabilized at 40–60 °C to ensure operational stability of the manufacturing process. To date, there is no report on testing NIPU foaming on pilot industrial lines using these technologies, and no NIPU foam can be found on the market yet. Within the frame of the fundamental developments reported here, only small-scale samples are usually investigated (few grams). Increasing the volume of the reactive formulation is known to impact the foaming parameter (e.g., viscosity, exothermicity) and thus the final properties of the foams (cell size, density, open/closed cell ratio). Scaling up the foaming processes is foreseen as a crucial step forward to better define the industrial applicability of the reported systems. Main challenges for NIPU foaming Challenge 1 – Low temperature, fast foaming. Hybridization of the NIPU technology has recently enabled the fast foaming (below 5 min) of NIPU from r.T. thermosetting formulations. To broaden the spectrum of NIPU foam structures, properties and thus applications, it is crucial to expand the scope of highly reactive co-monomers that function as heat-release promoters (e.g., acrylates). However, this expansion should be accompanied by careful consideration of their large-scale availability, origin, cost, and toxicity. Challenge 2 – Morpho-structural control of NIPU foams. Achieving NIPU foams with ultra-low densities (<50 kg m−3), closed porosity, small cell sizes, and high cell densities similar to conventional PUs (notably used for thermal insulation) will require several advancements. Improvements include the identification and use of surfactants to stabilize cell structures and/or suitable nucleating agents (fillers) to promote nucleation sites and generate a high number of cells. Optimizing processing conditions, such as emulsifying a physical blowing agent into the reactive formulation with adequate stabilizers or ensuring effective filler dispersion via high shear mixing, should enable the production of foams responding to these criteria. The wide range of surfactants and nucleating agents already developed for isocyanate-based foam chemistry can provide a valuable starting point for controlling the morpho-structural properties of NIPU foams. Further optimization of the morpho-structural properties of NIPU foams might be attained by adapting these well-established additives to the specific reactivity and physico-chemical characteristics of NIPU formulations. Challenge 3 – NIPU foams with tunable thermo-mechanical properties. Specific adjustments of both the NIPU microstructure and foam formulation composition are required here. Improvements involve exploiting the PHU hydroxyl groups for further chemical transformation to increase the crosslinking density of the foams, ideally during their formation by using simple and cost-effective methods. Mechanical performances can also be enhanced by utilizing monomers that introduce rigidity and/or improve thermal resistance through specific interactions. For example, aminotelechelic oligoamides or hydroxyoxazolidone precursors can promote hydrogen bonding, while (poly)aromatic- or lignin-derived monomers can facilitate π–π stacking. Similarly, the controlled dispersion of fillers such as chitin or cellulose nanofibers up to the percolation threshold may yield interpenetrated NIPU networks with superior mechanical strength. Accelerated aging and durability testing of NIPU foams have still to be considered. These tests simulate long-term use of the foams by exposing them to harsh conditions like elevated temperatures (thermo-oxidation), high humidity, UV and chemical agents to predict any variation in their thermo-mechanical properties (e.g., hardness, compression resistance, resilience, etc.). Challenge 4 – Suppressing or decreasing the hydrophilicity of PHU foam. This objective can be accomplished through various approaches. One potential method involves modifying the dangling hydroxyl (OH) groups with appropriate hydrophobic groups. Ideally, this modification should occur during the foaming process, as post-modification may not be relevant for industrial applications. This approach necessitates the identification of a chemoselective transformation that specifically targets hydroxyl groups and is compatible with the foaming conditions and the presence of amines. However, identifying a reactant able to react selectively with the OH moieties during foaming is extremely challenging and requires significant chemical engineering, particularly when simple and inexpensive transformations are desired. A relevant example for PHU hydrophobization by Shaplov et al. is the acetalization of their pending OH groups by aldehydes.171 However, this was only achieved on pre-formed PHU in solution, and the possibility to realize it in situ during PHU foaming remains challenging. Instead of transforming the existing OH groups, a promising strategy for hydrophobization involves incorporating reactive hydrophobic (nano)fillers into the foaming formulation. This has been recently demonstrated by Liu et al. by adding hydrophobic thiol-functionalized polyhedral oligomeric silsesquioxane (POSS-SH) to water-induced self-blown NIPU formulations.171 Besides imparting hydrophobicity to the NIPU matrix, this POSS-SH served as additional crosslinked nodes. This strategy is similar to the one reported by Gong et al., who highlighted that the dispersion of hydrophobic reduced graphene oxide significantly suppressed atmospheric moisture absorption by the PHU foams.172 The final proposed approach involves incorporating silicone-based surfactants into the NIPU formulations. If designed appropriately, these surfactants will stabilize the gas bubble/cell interface by covering the cell surface with a hydrophobic segment. Consequently, they will serve as protective coatings on the cell surface within the NIPU foams, thereby enhancing their humidity resistance. Challenge 5 – Retrofitting of industrial infrastructures. Implementing the foaming of NIPUs on existing industrial PU manufacturing lines requires significant adjustments. This includes the judicious selection of non-isocyanate PU chemistries to precisely control reaction kinetics to align with industrial processes; the efficient mixing/emulsification of all foam ingredients (monomers, blowing agent, stabilizers, catalyst and additives) into a homogeneous mixture via high-pressure mixing or high shear static mixers; the proper control of the initial formulation viscosity and mixing temperature, allowing the deposition, spraying or high-pressure injection and flowing of the formulation onto conveyors, onto walls or into closed molds, respectively. Although the fast NIPU foaming has recently been achieved via the hybrid formulations, the developments remain at the lab scale. Testing these NIPU foaming processes on pilot industrial lines is now necessary to identify the remaining technological barriers that need to be overcome to transfer the technology to industry. This will also necessitate the large-scale production of monomers (e.g., cyclic carbonates), still not commercially available at a large scale. |
4. How to address the end-of-life of NIPU foams?
In the transition towards a circular economy preserving resources and lowering the environmental footprint of the products, waste polyurethane foams can be given a second life through recycling.173 There exist multiple options for treating and transforming end-of-life (EoL) PU foams. The most common is chemical recycling, with the end products (recyclates) being reintroduced into the production cycle of second-generation PUs (foams) as reusable chemicals (mainly polyols). These methods, reviewed recently by Malucelli et al. for conventional PU made from isocyanates,174 could virtually apply to all varieties of NIPUs, as illustrated for the recovery of small molecules from PHU thermosets via base-catalyzed methanolysis.175 As PU foams are generally thermosetting materials, they are also revalorized via a downcycling scenario – also named physical recycling – into new products. Generally, waste PU foams are mechanically converted into powders, flakes, or small particles and reused as additives/fillers in new (PU) matrices.174 Recently, Dichtel introduced carbamate exchange catalysts176,177 or exploited residual foam catalysts,178 allowing for the conversion of thermoset PU foams into covalent adaptable networks (CANs). Leveraging the melt-processability of the so-produced PU CAN and the use of a suitable blowing agent, he opened a new foam-to-foam recycling perspective via extrusion foaming.176 This CAN behavior is present and even facilitated in non-isocyanate polyhydroxyurethanes,179–183 and is exploited now to reprocess waste NIPU foams into films, adhesives, or structural composites.4.1. Chemolysis of NIPU foams
To date, the chemical recycling of NIPU foams has been reported only through high-pressure glycolysis, alkaline hydrolysis or hydroglycolysis and is exemplified with carbonated lignin/priamine-derived materials blown with Momentive (Fig. 15).117 These chemolytic methods break down the urethane linkages, releasing polyols, amine and CO2. When using cyclocarbonated lignins, the presence of additional labile ether and acyclic carbonate linkages within its chemical structure further aids in the foam decomposition. Glycolysis using ethylene glycol shows limited decomposition efficiency, even at 220 °C, with ∼30–40% of the foams being degraded after 3–6 h. Alkaline hydrolysis using 0.1 M KOH under the same conditions achieves significantly higher degradation, around 65–70%. The most effective approach is a hybrid hydroglycolysis system combining 10 wt% ethylene glycol with a 0.2 M KOH aqueous solution, which increases foam decomposition up to 90%. In this study, the authors focused only on characterizing, recovering and reusing lignin for the production of second-generation foams. Through in-depth characterization, the hydroglycolysis procedure yields an ethylene glycol-modified lignin structure with a high hydroxyl content, favoring the reformation of cyclic carbonate units by condensation with DMC, and molar mass comparable to native lignin, ensuring its total solubility within the foam formulation. These features are better suited for the revalorization of recycled lignin into second-generation foams. Lignin recovered through alkaline hydrolysis undergoes structural modification and can therefore only be reincorporated at a content of about 10–20% into new foam formulations. In contrast, lignin obtained via hydroglycolysis can be reintroduced at up to 100% in fresh formulations, enabling the production of foams with densities (251 kg m−3), compressive modulus (1.51 MPa) and compressive strength (0.126 MPa at 10% deformation) that are essentially identical to those of the original foams. | ||
| Fig. 15 Closed-loop chemical recycling of lignin-based NIPU foams. | ||
4.2. Covalent adaptable network NIPU foams
Covalent adaptable networks (CANs) provide an emerging solution to overcoming the end-of-life of thermosets. CANs are dynamic polymer systems capable of rearranging their network structures through reversible bonds and/or exchange reactions triggered by external stimuli, e.g., heat and pressure.184–188 In dissociative CANs, temporary scission of the crosslinked nodes upon heating reduces the network density and enables structural rearrangement. Conversely, associative CANs, also named vitrimers, maintain a constant network connectivity by forming new bonds simultaneously as existing ones break. In the absence of degradative side reactions, these dynamic thermosets can thus be repaired, reprocessed, and recycled without drastically compromising their material performance. For more details on CANs, we invite the reader to refer to the recently published user guide of Ladmiral, which serves as a vade mecum for neophytes.184 | ||
| Fig. 16 Concept of covalent adaptable networks (CANs) (A) and (B) reprocessing of CO2 self-blown PHU foams produced with thiol blowing agents into films. SEM pictures and foam micrographs reproduced from ref. 136 with permission from the American Chemical Society. Copyright © 2023, American Chemical Society. | ||
 | ||
| Fig. 17 Recycling of waste PHU foams containing additional dynamic disulfide linkages into films and adhesives. SEM pictures and other micrographs reproduced from ref. 190 with permission from Elsevier. Copyright © 2025, Elsevier Ltd. | ||
Take-home messageKey differences between the end-of-life options of conventional PUs and NIPUsDifference 1 – Chemical recycling. Chemolysis is the preferred method for decomposing thermosetting PU foams. Hydrolysis, glycolysis, aminolysis, ammonolysis, acidolysis, and phosphorolysis yield high-value, high-purity chemicals with diverse functionalities and physico-chemical properties. Among these methods, glycolysis of flexible PU foams has advanced to pilot-plant and industrial-scale operations. The chemical recycling of NIPU foams, apart from the hydroglycolysis of lignin-derived foams reported by Stemberg, remains nonexistent. Besides, data on the long-term hydrolytic stability of PHU foams are still lacking and could provide useful insights for future developments. Difference 2 – Physical recycling. High-temperature, high-pressure injection/compression molding techniques are effective for reprocessing PU foams of low crosslinking degree into structural parts, e.g., covers for motors and pumps, dashboard or door panels, etc. Thermosetting PU foams of high crosslinking degrees are generally ground into powders, particles or flakes that further serve as fillers for other resins or are rebonded with PU adhesives into the form of covers for tires, athletic mats, car linings, and carpet underlays, among other uses. Physical recycling of NIPU foams is limited to PHUs and exploits their covalent adaptable network properties to easily upcycle foams into films, structural composites and high-performance adhesives for metal, wood, glass and fabrics. Main challenges for NIPU recycling Challenge 1 – New chemical recycling strategies. Recycling waste NIPU foams faces challenges due to variations in NIPU chemical structures, crosslinking degree, and crystallinity. Specific recycling strategies should maximize yield and purity of recovered chemicals while minimizing by-products, environmental impact, and energy consumption. Conventional hydrolysis, glycolysis, aminolysis, or ammonolysis should be prioritized, but other chemolysis approaches could be engineered, especially for PHUs with pending OH moieties. Biodegradation using urethanases or designing NIPUs with labile linkages for facilitated recycling should also be considered for longer-term deployment. Challenge 2 – New physical recycling strategies. Going beyond the simple grinding of waste PU foams would capitalize on mechanochemistry. Mechanochemical recycling involves applying high-shear mechanical forces, e.g., via ball milling, to break covalent bonds in the NIPU network, promoting fragmentation, depolymerization or modification (e.g., hydroxyl group formation) reactions. This methodology might be applied to NIPU foams and its utility in producing (functional) powders or oligomers that might then be reused as fillers, chemical feedstocks, or precursors for new materials evaluated. Challenge 3 – Foam-to-foam recycling. Existing industrial “foam-to-foam” recycling methods for conventional PUs shred foams and rebind particles into new materials like carpet underlays. Implementing foam-to-foam recycling for NIPUs, especially thermosetting PHU foams, will exploit their CAN properties. Using suitable catalysts for transurethanization or urethane bond dissociation, or designing NIPU foams for easy recycling with labile linkages, could lead to reprocessable materials. Second-generation foams with new morpho-structural characteristics might be produced through extrusion-foaming with physical blowing agents. Challenge 4 – Integration within the current recycling streams. Short-term recycling of NIPUs should be standardized with existing PU methodologies to ensure their integration into industrial recycling streams. In this context, glycolysis, aminolysis and eventually ammonolysis, which are largely deployed to decompose conventional PUs, should be adapted to NIPUs. Mechanical grinding of NIPU foams into powder and their reuse as fillers or other foam components should be evaluated. |
5. What are the emerging applications of NIPU foams?
Over the last 5 years, NIPU foaming has witnessed a significant acceleration, especially with the adoption of physical or self-blowing methods, which have driven the emergence of products for cutting-edge applications. Despite being in their infancy, the elaboration of these practical applications is essential not only to validate the potential of these foams and benchmark their value with existing solutions but also to uncover performance limitations that can guide future material optimization.5.1. Fire retardant foams
Without flame retardants, conventional PU foams are highly flammable materials, producing a significant level of smoke during burning that contains CO2, toxic gases (HCN, NOx, CO), hydrocarbons (benzene, xylene, toluene, carbazole, etc.)192 and other hazardous chemicals (phenyl- or p-toluene-isocyanates, isoquinoline, nitriles, etc.).193 Their high flammability arises from their inherently porous structure, facilitating oxygen diffusion within the foam, and their intrinsically low limiting oxygen index (LOI) of 18% (LOI indicates the minimum oxygen concentration required to sustain combustion).194 Foam density also plays a significant role in the fire resistance properties.195,196 Low-density materials limit heat transfer from the surface to the core, leading to shorter ignition times. The open-cell structure of PU foams, the absence of aromaticity in the PU skeleton and the presence of hydrocarbon or H2 blowing agents within closed cells are additional factors worsening the resistance of the foam to fire.195,196 NIPU foams display flammability similar to conventional PU counterparts, as illustrated by LOI values as low as 17.5% or 22.3% for materials made from glucose161 or xylose-derived166 monomers, respectively, far away from 27%, the value at which the foam is considered fire-resistant. This ignitability is reduced by the insertion of aromatic monomers within the NIPU network.162 These aromatic species, like tannic acid or tannin, undergo carbonization in the condensed phase, creating a char that shields the underlying material from oxygen diffusion/exposition. The LOI values then increase to 24–25.5%,161,163 still below the 27% threshold deemed fire resistant but sufficient to make the materials self-extinguishing. Flame-retardant properties of tannin-based NIPU foams are achieved by adding an inorganic salt, such as Al(OH)3 (Fig. 18).163 Upon exposure to heat, this salt undergoes endothermic decomposition with concomitant water vapor release. The combined effects of endothermic decomposition, absorption of heat, and water vapor release create a protective interface of Al2O3 between the burning material and the surrounding environment, restricting oxygen contact and retarding the flame propagation rate. This translates into an LOI value of >27%, providing NIPU (bio)foams with fire retardancy (Fig. 18). | ||
| Fig. 18 Fire-retardant tannin-based NIPU foams formed by the use of Al(OH)3. SEM picture and burning tests reproduced from ref. 163 with permission from Elsevier. Copyright © 2023, Elsevier Ltd. | ||
Phosphorus-based flame retardants have also been exploited to fabricate fire-resistant PHU foams. Among them, phytic acid stands out as a multi-action flame retardant.197 When exposed to flame or heat, it decomposes in the gas phase into phosphorus-containing radicals (HPO˙, PO˙ and HPO2˙), which scavenge reactive OH and H radicals. This mechanism suppresses the thermo-oxidative degradation of polymers and reduces the formation of combustible volatiles. Additionally, the decomposition of phytic acid releases water vapor and other non-reactive gases, further diluting oxygen in the combustion zone. In the condensed phase, its degradation produces acids such as polyphosphoric acid, which catalyze the formation of intumescent char layers. The high efficiency of phytic acid in improving fire resistance is illustrated by an LOI of 29.1% for low-density closed-cell PHU foams (131 kg m−3) synthesized from cycloaliphatic biscyclic carbonates, m-XDA, and PEI using HFO-1233zd as a physical blowing agent.105 Novel trends explore now the utilization of phosphorus-containing biscyclic carbonates as analogs to 9,10-dihydro-9-oxa-10-phosphaphenanthrene-10-oxide to serve as reactive flame retardants. The insertion of these monomers at only 2 wt% into a PHU self-foaming formulation is sufficient to reduce the total smoke release during combustion by 31%.198
5.2. Heat storage and thermal insulation
5.3. Electromagnetic interference shielding
Electromagnetic interference (EMI) is a common problem that affects electronic components and communication or navigation systems. It is especially critical in sectors such as the military, aerospace, medical, and automotive industries. EMI results from the interaction of an electrical field with a magnetic field, forming electromagnetic waves (EM). These waves radiate in the surrounding environment and create electric and electromagnetic noises disturbing or even compromising the well-functioning of nearby systems such as smartphones, GPS devices, wireless tools, radars, or medical imaging equipment like X-rays and MRIs.208 Long-term exposure to EMI may also carry potential health risks, e.g., miscarriage.209 In this context, electromagnetic interference shielding across a wide range of frequencies is essential to protect electronic devices from parasitic signals. Traditional shielding films or coatings embedding magnetic and conductive particles primarily block EMI through reflection at the material interface, potentially causing secondary EM pollution.210,211 In contrast, modified polymer foams demonstrate superior EMI shielding performances.212–215 Their porous structure, combined with the presence of conductive fillers, promotes multiple internal reflections of EM waves, yet dissipates the energy in the form of heat through an attenuation process and limits the emissions of secondary EM waves (Fig. 19). Uniform open cell CO2 self-blown PHU foams made from TMPTC, bisphenol A diglycidyl carbonate, and m-XDA using water as a blowing agent have served as a scaffold to fabricate EMI shielding materials via a sequential immersion strategy (Fig. 19).172 First, insulation PHU foams have been transformed into a conductive material via deep coating in an EtOH solution containing silver nanowires (AgNWs) until the percolation threshold is surpassed. After 12 cycles, a maximum of 39.5 wt% of AgNWs has been incorporated within the porous materials, the filler being homogeneously dispersed all along the cell walls. In the second step, the conductive foams are impregnated with reduced graphene oxide@Fe3O4 composites. This filler, distributed at a 5–20 wt% loading across the cell walls, provides magnetic dipoles for EM radiation absorption. While the virgin PHU foam shows poor shielding efficiency due to its inherent electrical insulation properties, the incorporation of AgNWs yields a highly conductive foam (electrical conductivity of 4.54 S m−1) with an electromagnetic interference shielding effectiveness (SE) of 38 dB, surpassing the 20 dB of commercially available solutions. The addition of reduced graphene oxide@Fe3O4 filler further improves the EMI shielding by improving electromagnetic wave penetration and absorption loss. The best performance is observed with 5 wt% of reduced graphene oxide@Fe3O4, leading to a remarkable SE of 66 dB. This work represents the sole example of PHU-based foams where synergistic effects between dielectric loss from AgNWs and magnetic loss from Fe3O4 significantly boost EMI shielding performances. | ||
| Fig. 19 Conductive PHU/AgNW/RGO@Fe3O4 composite foams for EMI shielding applications. Content partly reproduced from ref. 172 with permission from the American Chemical Society. Copyright © 2025, American Chemical Society. | ||
5.4. Antibacterial foam dressing for wound healing
Hydrophilic PU foams have emerged as advanced care materials for the healing of chronic wounds caused by injuries, diabetes or burns. When used as dressings, they can absorb a large amount of exudate, reducing the risk of maceration, while maintaining an ideal moist/dry environment, promoting faster tissue regeneration.216–220 Their easy loading ability with bioactive ingredients such as Ag nanoparticles also imparts them with antimicrobial properties, minimizing bacterial infections and preventing healing complications. The intrinsic hydrophilic features of PHUs inspired Chen to design self-blowing open-cell foams (cell size of ∼300 µm) for antibacterial and wound healing applications from cyclocarbonated lignin, a mixture of polyetheramines, PMH blowing agent and silver nanoparticles (Fig. 20).120 The biocompatibility of the resulting foams is demonstrated through hemolysis tests on blood cells. The observed hemolysis below 3% indicates minimal disruption of red blood cells in the bloodstream and a low release of free hemoglobin, well within acceptable biocompatibility criteria. Besides, the foams showed no cytotoxicity, with fibroblast viability exceeding 100% after 24 h, further meaning the dressing is conducive for cell growth. The presence of Ag nanoparticles (10%) imparts to the foams antimicrobial properties with a bactericidal efficiency of 95% and 99% after 8 h exposure for S. aureus and E. coli (Fig. 20B–D), respectively. Finally, the wound healing potential of Ag-loaded PHU foams is attested in vivo in mice. Compared to commercial Tegaderm® films, the PHU dressings exhibited superior wound healing performance, as evidenced by enhanced collagen deposition and blood vessel proliferation, which accelerated the regeneration of fibrous connective tissue, resulting in faster wound closure. | ||
| Fig. 20 H2 self-blown PHU foams loaded with 6.5 or 10 wt% (vs. carbonated lignin) of Ag nanoparticles for antibacterial and wound healing applications. (A) Foam formulation; (B) antibacterial effect of AgNP-loaded PHU foams on E. coli; (C) bactericidal efficiency with time and (D) morphology of E. coli before and after 4 h of coculture on (AgNP-loaded) NIPU foams. Content partly reproduced from ref. 120 with permission from the American Chemical Society. Copyright © 2024, American Chemical Society. | ||
5.5. Sensing and pollutant capture
Exposure to pollutants such as formaldehyde, heavy metals, drugs, pesticide residues, etc. has become a major scourge of our modern society. Consequently, the removal of these substances from our environment represents a significant challenge. Porous materials221,222 and open-cell PU foams offer a promising solution by functioning as sponges that trap pollutants.223–225 Incorporating fluorophores (e.g., polyaromatic groups,226–228 lanthanide complexes,229,230 organic dyes231) within the foam leads to modified porous materials serving as fluorescent sensors for both the detection and quantification of pollutants. PHU foams, especially those self-blown using water or a physical blowing agent, exhibit a non-conventional fluorescence behavior with a multicolor emission even without the use of fluorescent probes.105,232 This non-conventional fluorescence behavior arises from aggregation-induced emission (AIE) luminogens that are naturally present within the foams. The AIE phenomenon is promoted by the abundance of hydroxyl groups along the PHU skeleton, prone to hydrogen bonding, yet favoring clusteroluminescence. Analytes or molecules interacting with these clusters may affect the fluorescence of the PHU foam. Consequently, the modification of the multicolor emission of PHU foams is advantageously exploited for multipurpose sensing applications.232 Functional open-cell foams (290 kg m−3) were specifically designed from TMPTC, EDR and the PEI polyamine using water as a blowing agent. The presence of multiple (reactive) amine moieties within the polymer backbone imparts formaldehyde-capturing and metal-complexing capabilities to the foam, leading to notable changes in both its thermo-mechanical and clusteroluminescence properties. The irreversible reaction between residual primary amines and formaldehyde139 (7 days of exposure at r.T.) reduces the pollutant concentration by 2/3 and forms stable adducts. These structural changes increase the foam Tg from −3 °C to 8 °C and form higher-order aggregates. These changes induce a red shift in fluorescence emission, with a maximum signal shifting from 450 to 495 nm upon excitation at a wavelength (λex) of 380 nm. Formaldehyde capture is further evidenced by a fluorescence color shift in the foam from cyan to green at λex = 454 nm. These PHU foams have also been used for metal ion sensing applications in water solutions (Fig. 21). Among a broad list of cations, foams could complex Cu2+, Fe2+, Fe3+ and Hg2+ ions as highlighted by quenching efficiencies of the fluorescence emission spectra of 73.2% to 90.5%. Monitoring the quenching of the fluorescence intensity (upon excitation at 380 nm) at various ion concentrations allowed for the precise quantification of the metal cation, with maximum foam/Cu2+, Fe2+ or Fe3+ binding efficiencies of respectively 81, 196.6 and 60.2 mg g−1. The utility of these nonconventional fluorescent foams is further illustrated by the ratiometric sensing and quantification of tetracycline, a bacteriostatic antibiotic active against various pathogens such as Streptococcus, Staphylococcus, etc. This drug forms a complex with the foam by H-bonding, which translates into an increase of the Tg from −3° to 11 °C and a red shift from 452 nm to 518 nm of the fluorescence emission spectrum upon excitation at 380 nm (for a drug concentration of 1.25 × 10−4 mol L−1 in water). Like metal ions, the quantification of drug residues has been realized by monitoring the decrease in intensity of the emission spectra, which revealed a limit of detection of the drug at a concentration as low as 3.17 × 10−8 mol L−1. Exposing the PHU foam to an increasing concentration of drug results in a multicolor fluorescence evolution from blue to green to yellow upon UV exposure (λex = 454 nm). This inspired the development of a smartphone-based detection device equipped with an RGB color scanning app to monitor fluorescence color changes, enabling rapid, on-site visual detection of tetracycline in aqueous solutions. When reshaped into PHU films, these hydrophilic, non-conventional fluorescent PHU foams can be utilized for monitoring relative humidity while simultaneously determining the Tg's of the PHU matrix. The fluorescence response of the PHU film is indeed influenced by the disruption of intramolecular hydrogen bonds caused by water molecules, directly impacting the clusteroluminescence of the material. Linear correlations between relative fluorescence intensity and RH and with Tg's of NIPU are indeed observed, facilitating the efficient and rapid monitoring of RH and hydroplasticization.233
Take-home messageKey differences between conventional PU and NIPU applicationsDifference 1 – Soft and rigid foams. PU foaming, a well-established and versatile technology, offers tailored foam solutions for comfort and insulation applications. Adjusting formulation and foaming processes allows for the production of soft elastomeric to rigid foams with controllable properties like crosslink density, cell porosity, thermal stability, resilience, flexibility, and thermal or acoustic insulation. In contrast, NIPU foaming chemistries and processes are still under development, challenging precise control over key foam characteristics like cell morphology, size, and density. Designing high Tg materials and mitigating hydroplasticization of PHU foams also pose difficulties. Open-cell flexible NIPU foams are accessible and exhibit excellent resilience and flexibility, promising seating and cushioning applications. Thermal insulating NIPU foams have been fabricated, but their performance is not yet comparable to commercial PU materials. Difference 2 – (Multi)functional foams. Advanced multi-functional PU foams find applications beyond comfort and insulation, including structural panels, electronic shielding, environmental filtration, biomedical dressings, antimicrobial foams, tissue engineering scaffolds, and smart materials. Emerging NIPU foam applications, limited to PHU materials, utilize their unique hydrophilic nature and clusteroluminescence for diverse functionalities. These foams can sense and quantify pollutants from wastewater, and when infused with phase change materials, they serve as efficient heat storage systems for r.T use. Furthermore, their exceptional biocompatibility and hemocompatibility render them highly appealing for the development of novel biomaterials. Difference 3 – Fire resistance. NIPU foams lack thermally stable isocyanurate structures found in conventional PU foams, promoting char formation and limiting oxygen diffusion during combustion. Achieving a limiting oxygen index value above 27% requires the addition of specific flame retardants. Main challenges for NIPU applications Challenge 1 – Performance improvement. Enhancing fatigue resistance and elasticity of flexible NIPU foams for cushioning, furniture, seats, and mattresses requires producing low-density materials (<80 kg m−3) with reduced cell size and controlled network crosslinking. Rigid insulating NIPU foams with closed cells, high cell density, and a density below 40–50 kg m−3 are crucial to reduce thermal conductivity below 25 mW m−1 K−1, provided a low-specific heat capacity blowing agent is used. Given the high Tg properties required for this application, a meticulous and challenging selection of monomers, as well as additives (e.g. surfactants, nucleating agents), is essential, accompanied by the intricate task of designing solvent-free formulations. Challenge 2 – Aging. The long-term stability and durability of optimized NIPU foams should be evaluated through standardized accelerated aging tests. These tests assess dimensional stability, surface cracking, morpho-structural changes and mechanical performance loss under various stresses. Resistance to UV, photo-oxidation, moisture, chemical/humidity exposure and heat must be systematically examined to ensure reliable long-term performance. Challenge 3 – Fire resistance. Combining nanofillers like nanoclays and expandable graphite is expected to enhance foam fire resistance by synergistically stabilizing the char layer and reducing dripping. Chemical modifications like PHU phosphorylation or targeted surface treatments might further improve fire resistance. Choosing the most efficient flame retardant should minimize loading, preserving the thermo-mechanical and morpho-structural properties of the foam. Reactive additives should be preferred to limit migration outside the foam. Challenge 4 – Compliance with industrial and regulatory obligations. Any new NIPU material reaching similar performances or outperforming conventional PU foams will have to comply with a series of standardized tests and specifications (ASTM, ISO) tailored to the targeted application, ensuring technical, safety, and regulatory compliance. |
 | ||
| Fig. 21 Non-conventional fluorescent CO2 self-blown PHU foams for metal capture and quantification (A). Potential interactions responsible for clusteroluminescence and SEM image of the foams at various excitation wavelengths (B). Capture of Fe2+, Cu2+ and Fe3+ ions by foams and quantification of their concentration via fluorescence (C). Content partly reproduced from ref. 232 with permission from John Wiley and Sons. Copyright © 2024, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
6. Conclusions and perspectives
The last decade has seen significant progress in non-isocyanate polyurethane chemistry and advanced NIPU materials design. While accessing high molar mass thermoplastic NIPUs remains difficult, most synthetic methods are especially suitable for producing thermosetting resins, like those used in traditional PU foams. Virtually, NIPUs, more particularly poly(hydroxyurethane)s made from the step-growth copolymerization of polycyclic carbonates with polyamines, have the potential to serve as alternatives to conventional PU foams. However, the polymerization kinetics of these PHU systems are generally slower than those of the traditional isocyanate/polyol (foaming) process. The microstructure of PHUs also differs from that of conventional PUs by the presence of dangling OH groups and the absence of biuret, allophanate, isocyanurate and uretdione linkages. All these structural motifs derived from the isocyanate chemistry play a crucial role in imparting the excellent thermo-mechanical properties of PU foams. Therefore, transferring a PU foaming process to PHUs is not straightforward and requires adjustments to account for these differences.Precise control of the viscosity of the formulation, the gelation and curing rates of the PHU matrix, and the release of the blowing agent (whether endogenous or exogenous) is critical for timely gas generation and matrix curing. This coordination ensures an efficient expansion of the matrix, minimizes cell drainage and foam collapse, and ultimately guarantees the dimensional stability of the final materials. Although the aminolysis of cyclic carbonates proceeds relatively slowly, it readily occurs within the temperature range of 80–140 °C. To ensure proper foaming under these conditions, physical blowing agents with high boiling points (e.g., (fluoro)hydrocarbons) and moderate vapor pressure, or chemical blowing agents with a controllable decomposition rate (e.g., Na2CO3), are preferred.
In self-foaming processes, the blowing agent, usually H2 or CO2, is generated in situ by the decomposition of one of the foam components (e.g., Momentive or the cyclic carbonate) contributing to the formation of the polymer matrix. As a trend, this decomposition reaction should be slightly delayed relative to the formation of urethane linkages to ensure the gas delivery when the matrix is sufficiently viscous and close to gelation. In CO2 self-blowing PHUs, the decarboxylation of the cyclic carbonates is initiated and adjusted either by adapting the concentration of the additive (water or thiol), the nucleophilic character of the thiol and the nature/content of the base catalyst. As Momentive and thiol participate in polymer network formation, their content in the formulation influences the thermo-mechanical properties of the final material. Usually, both components introduce flexibility within the network, favoring the fabrication of flexible foams. When water is used as an additive, part of the cyclic carbonate moieties are sacrificed to generate CO2in situ. To ensure sufficient network crosslinking, preventing the foam collapse, and decent mechanical properties of the foam, the level of sacrificial cyclic carbonates should not exceed 25%.
Catalysts, generally organobases, influence the foaming process of NIPUs by regulating the kinetics of all reactions at play. The control of these kinetics may be affected by “external” factors, especially for PHU foaming. Water, used to promote the in situ release of CO2, and/or halide salts, used to catalyze the production of cyclic carbonates by CO2/epoxide coupling, accelerate the gelation of the matrix. Water plays a dual role. It disrupts hydrogen bonds between PHU chains, increasing chain mobility, and enhances the electrophilicity of the carbonyl site by H-bonding, thereby facilitating amine attack and hydroxyurethane linkage formation. Cyclic carbonates are usually not purified before PHU synthesis and are thus contaminated by the catalyst. The halide salt residues may interact with the oxygen atom of the carbonyl, further influencing the aminolysis rate of the carbonate ring. This underlines the necessity to consider the co-catalytic role of these halide salt residues when optimizing foaming processes.
The foaming reactions of PHUs or other NIPUs display generally insufficient exothermicity, resulting in a prolonged curing procedure at elevated temperature to obtain stable foams. To address this limitation, hybridizing NIPU chemistry with additives such as epoxides or exovinylene cyclic carbonates, serving as heat-release promoters, offers a promising solution. These additives undergo highly exothermic reactions with amines, enabling the formulation to rapidly reach the foaming threshold within minutes, even under r.T. conditions. Compared to aliphatic epoxides, aromatic epoxides promote higher exotherms upon aminolysis and their use tends to improve the rigidity of the PHU foams. Exovinylene cyclic carbonates allow for rapid foam expansion (∼1 min). This hybridization approach not only enhances process efficiency but also paves the way to retrofitting existing industrial PU manufacturing lines where foaming should be very fast (i.e., within minutes). Importantly, self-blown NIPU foams of high potential bio-based contents (>90 wt%) are also easily accessible by valorizing carbon dioxide and bio-based products (e.g., vegetable oil derivatives, sugars) as raw materials, as well as abundant biowastes as biofillers (e.g., lignin derivatives, cellulose, keratin, chitosan, etc.).
Most foaming methodologies produce materials with densities typically >100–150 kg m−3 and open-cell structures. Access to very low-density materials (<50 kg m−3) with high cell density, small cell diameters and close porosity is necessary to match the performances and specifications of conventional foams, such as those used in thermal insulation panels. In this context, controlling the morpho-structural properties of NIPU foams will result from a complex interplay between surfactant properties and synchronization between the kinetics of gas generation/escape and NIPU curing. To date, most of the NIPU foaming approaches don’t use surfactants or exploit surfactants that have not been specifically designed for NIPU foaming. Without surfactants or with the use of poorly efficient surfactants, the gas–liquid interface cannot support any mechanical stress. Upon blowing agent release, the rising internal pressure drives rapid cell expansion, thinning the interface and leading to cell wall rupture. This further creates open porosity and even foam collapse if NIPU formation/solidification is not synchronized with bubble growth.
Silicone surfactants specifically tailored for NIPU are expected to control the initial bubble dispersion stage by stabilizing the air bubble/liquid interface created upon monomer mixing. Then, during the cell expansion step by release of the blowing agent, a surface tension gradient will be generated at the gas–liquid interface, slowing down the drainage flow rate of the thinning cell wall by the Marangoni effect. In low viscosity NIPU matrices lacking effective stabilization and mechanical resistance of the cell walls, the fast drainage leads to rapid cell rupture and foam collapse. Conversely, controlling the release of the blowing agent and the solidification of the NIPU, for instance by manipulating catalysis, should enable a finite cell expansion rate that provides sufficient time for surfactant diffusion and accommodation to alterations in the cell/polymer interface. The appropriate regulation of these phenomena should enable the formation of highly uniform low-density foams with closed-cell porosity, contingent upon the identification of suitable stabilizers.
Inherent to PHUs, the presence of OH moieties along the polymer skeleton makes the final material intrinsically hydrophilic. PHUs tend to easily absorb humidity, leading to plasticization with a significant drop in the foam's Tg values (up to 40 °C). Therefore, achieving rigid PHU foams with Tg > 80 °C remains challenging, even when using aromatic monomers, and foam properties can change upon exposure to different humidity conditions. The plasticization of PHU foams remains a substantial bottleneck, as it hinders the development of PHU foams that can effectively compete with PU foams in similar applications, resulting in less favorable performance. When similar applications are targeted, solutions addressing this issue, e.g., by suppressing or transforming the pending OH moieties, or conceptualizing new NIPU foaming strategies, should be considered in future works. PHU foams can also be advantageous in applications where hydrophilicity is required, as more hydrophobic PU foams may not be the ideal products. This has been demonstrated in designing wound dressings capable of absorbing large volumes of exudate and in developing shape-memory materials.156 Further applications necessitating this hydrophilicity characteristic should be identified and investigated to facilitate the penetration of PHU foams into this specific foam market.
Today, addressing the end-of-life of NIPU foams is key to creating sustainable materials. The challenges and priorities identified in conventional PU recycling by Malucelli provide a relevant framework that should also be considered for NIPUs, particularly when aiming to integrate into the same waste streams (including collection and sorting).174 Well-established, industrially viable recycling strategies based on physical and chemical recycling should then be adapted to NIPUs. The intrinsic CAN behavior of PHUs allows physical reprocessing of foams via compression molding into various second-life materials (films, adhesives). By taking inspiration from recent discoveries by Dichtel, the CAN properties of PHUs pave the way to the future direct reuse of EoL polyurethane foams as a raw material for new foam fabrication through extrusion-foaming (foam-to-foam recycling).176 Chemical recycling of NIPU foams, via hydroglycolysis, should focus on the design of efficient catalysts to transform waste foams into ready-to-reuse high-quality recycled raw materials, which in turn can be reincorporated in a loop into new foam formulations. Future recycling concepts should integrate the “designed-for-recycling” approach, leading to new added-value chemicals, far beyond conventional polyol or polyamine “liquors” typically formed via hydrolysis, glycolysis or aminolysis of PUs. Finally, life cycle assessment (LCA) of NIPU foams over the entire value chain, i.e., from monomer synthesis to material recycling, is essential to confirm their potential for reducing environmental impacts. As it stands, the only existing LCA of PHU bulk materials reveals a clear negative impact on three key indicators, i.e., fossil fuel consumption, greenhouse gas (GHG) emissions and water consumption.234 Energy consumption is 14.3% higher for PHUs than for conventional PUs. However, the emergence of r.T. foaming solutions for NIPU should lead to significant improvements in this key indicator. GHG emission penalty is expected to decrease by using renewable chemicals. This is particularly evident for PHUs for which a large diversity of cyclic carbonate precursors are now accessible from biogenic CO2 and bio-renewable raw epoxides. By considering biogenic chemicals in the simulation, a reduction in GHG emissions of 11.5% to 16.1% has been predicted for PHU production compared to PUs. Finally, the negative contribution of PHU towards water consumption stems from its inherently demanding use as a solvent during the depolymerization process. This underlines the strong need to improve/optimize the recycling scenario of NIPUs and adapt the protocols to the decomposition of foams. The emergence of predictive machine learning and artificial intelligence tools could play an important role in the future, not only in the development of environmentally friendlier solutions but also in facilitating the selection of monomers to guide the design of materials with specifications tailored on demand to the requirements of the targeted application. Pending the deployment of these tools, a “monomer selection guide” (Table 1) and two tables summarizing the morpho-structural and mechanical properties of NIPU foams, distinguishing PHUs (Table 2) from other types of non-isocyanate PUs (Table 3), are provided below.
| Water-induced CO2 self-foaming PHUs: formation of non-reactive vicinal diols upon hydrolysis of cyclic carbonates, the content of sacrificial cyclic carbonates should not exceed 25% to maintain network integrity. H2-induced self-foaming PHUs: use 1–5 wt% of Momentive MH15, Momentive brings flexibility to PHU foams. Latent amine blowing agents (i.e. amine/CO2 adducts): suffer from high decomposition temperature.a Other blowing systems: chemical and physical blowing agents do not affect the PHU microstructure, closed-cell porosity is accessible using scCO2 as a blowing agent, but this method is limited to the foaming of thermoplastics. | ||
|---|---|---|
| Monomers for PHU foams | Polycyclic carbonates |
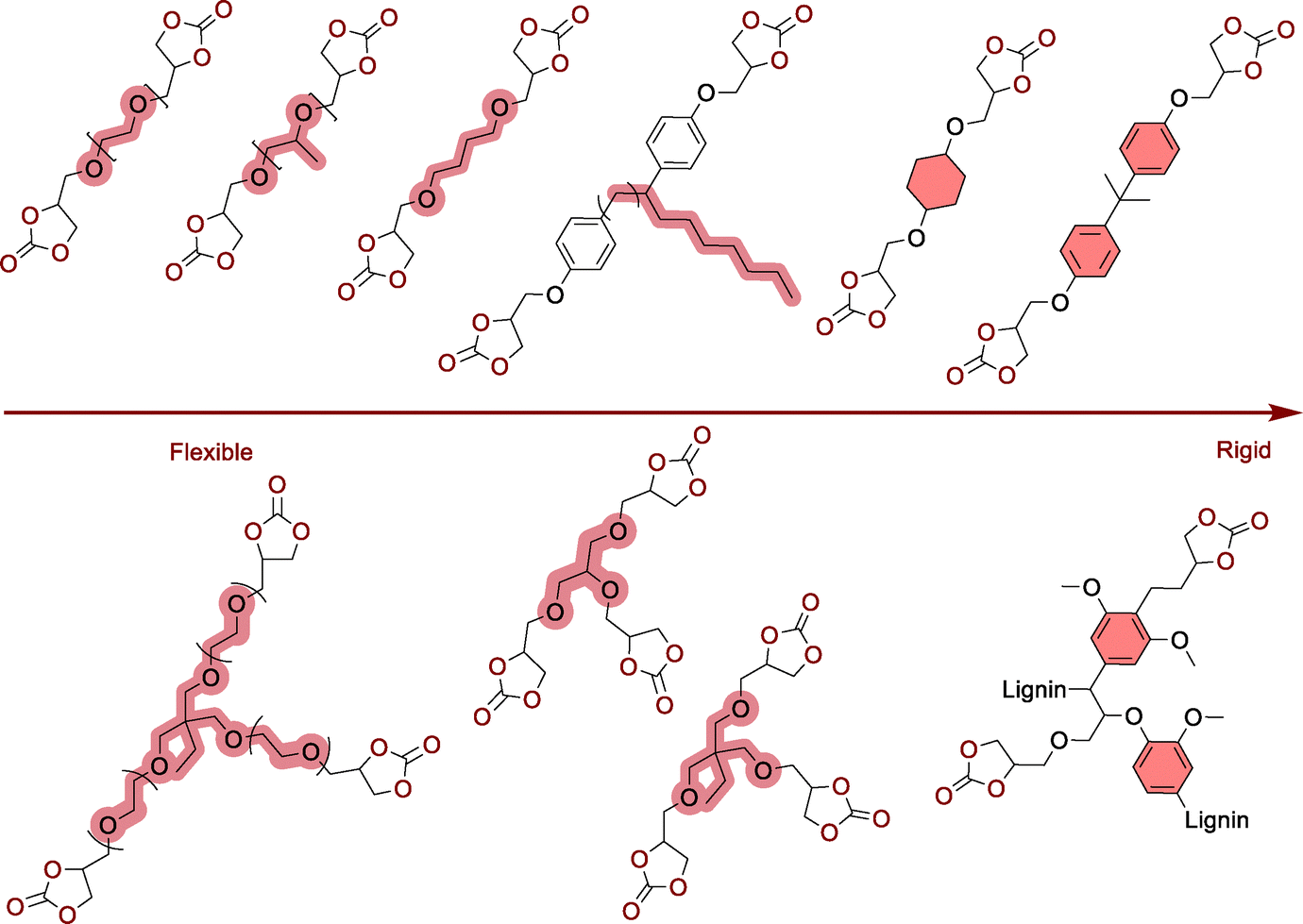
|
| Polyamines |
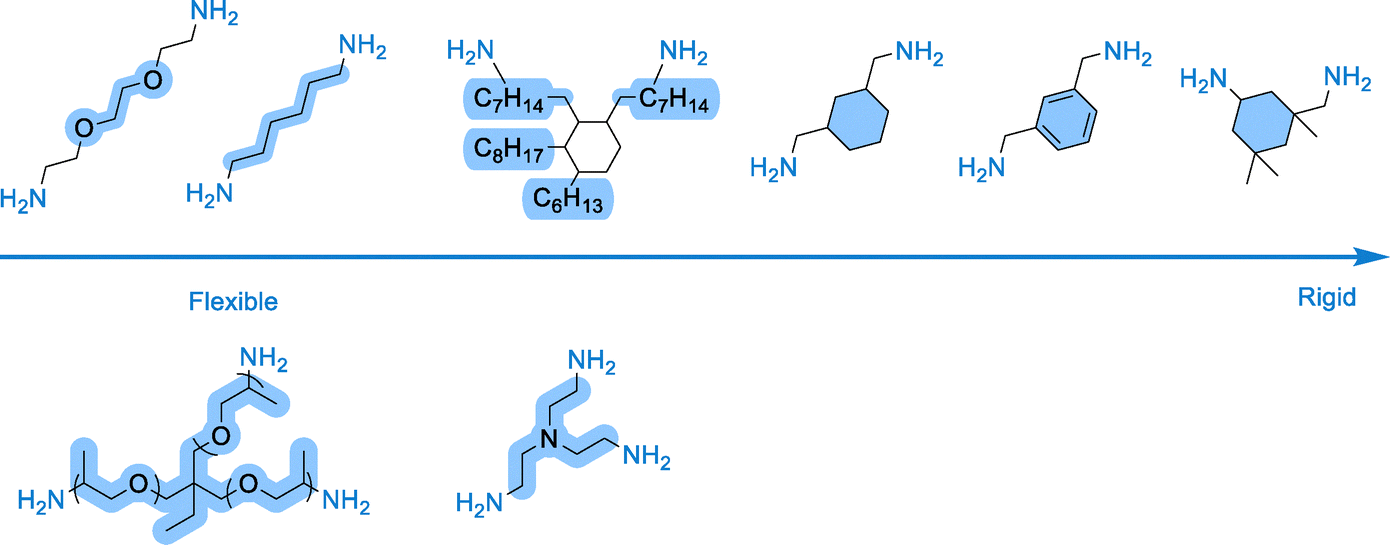
|
|
| Heat release promoters (r.T. foaming) | Polyepoxides |

|
| Blowing agenta | Polythiols (CO2 self-foaming) |
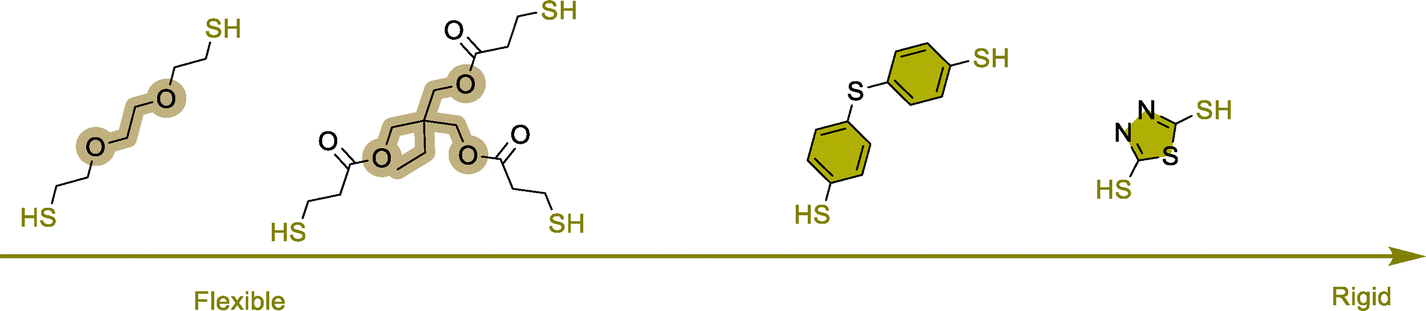
|
| NIPU type | Blowing technique | Blowing agent | Foaming procedure | Density (kg m−3) | Cell size and morphology (µm) | T g (°C) | Mechanical properties | |
|---|---|---|---|---|---|---|---|---|
| Thermoplastics | Physical foaming | scCO2 | Impregnation: 40 °C, 300 bar | 110–180 | Narrow distribution | −2 to −6 (Tm ∼ 88 °C) | Rigid | |
| Foaming: 80 °C100 | Closed cell 3–10 | Compression modulus 1.8–4.75 MPa (strain 80%) | ||||||
| Impregnation: 80 °C, 300 bar | 200–400 | Broad distribution | 30 | Rigid | ||||
| Foaming: 80 °C101 | Closed cell 9–23 | Compression modulus 0.123–0.149 MPa (strain 25%) | ||||||
| Acetone | (Tg – 5) °C | 35 | Open cell 125–150 | 80–110 | Highly rigid | |||
| Vacuum77 | Compression modulus 0.23–0.30 MPa (strain 15%) | |||||||
| Thermosets | Physical foaming | HFC (Solkane®365) | 80 °C, 14 h104 | 140–220 | Open cell 135–170 | 1 | Flexible | |
| Compression modulus 0.0175 MPa (strain 40%) | ||||||||
| HFO (1233zd) | 80 °C, 14 h105 | 140 | Closed cell 800 | 39 | Rigid | |||
| Compression modulus 0.15–0.35 MPa | ||||||||
| Chemical foaming | NaHCO3 | CO2 | 150 °C, 6 min107 | 140–480 | Open cell 15–330 | NA | NA | |
| H2O | ||||||||
| NH4HCO3 citric acid | CO2 | 80 °C, 15 min109 | NA | NA | 42 | Rigid | ||
| H2O | Compression modulus NA | |||||||
| Azo-compound ZnO | N2 | 120 °C, 30 min, then 60 °C, 30 min113 | 230–440 | Open cell 150–300 | 5–7 | Flexible | ||
| CO | Compression modulus 0.19–0.20 MPa (strain 50%) | |||||||
| NH3 | ||||||||
| Self-foaming | Poly(methylhydrogenosiloxane) | H2 | 80–150 °C | 190–330 | Closed cell and open cell | −18 to 100 | Flexible to rigid | |
| TBD catalysis114,117,118 | Compression modulus 0.0005–0.003 MPa (strain 20%) | |||||||
| 0.80–0.130 (strain 10%) | ||||||||
| r.T. (foaming: 3–7 days) | 270–300 | Open cell 140–1300 | 0–11 | Flexible | ||||
| Thiourea catalysis115 | Compression modulus 0.02–0.075 MPa (strain 30%) | |||||||
| 50 °C, overnight | 170–350 | Open cell 210–1350 | 7–27 | Flexible to rigid | ||||
| 120 °C, 2 h | Compression modulus 0.14–0.215 MPa (strain 50%) | |||||||
| 6-membered cyclic carbonates116 | ||||||||
| Thiol induced | CO2 | 100 °C | 140–175 | Open cell 250–980 | −30 to 25 | Flexible | ||
| Alkyl thiol, DBU catalysis128,133,134,136 | Compression modulus 0.008–0.065 MPa (strain 50%) | |||||||
| 100 °C | 200–300 | Open cell 900–1300 | 8–45 | Flexible to rigid | ||||
| Aromatic thiol (foaming: 5–10 min)130 | Compression modulus 0.35–0.4 MPa (strain 50%) | |||||||
| 80–100 °C, 4 h | 170–440 | Open cell 500–2000 | −25 to 15 | Flexible | ||||
| “Masked” thiol DBU or no catalysis134,135 | Compression modulus 0.001–0.21 MPa (strain 10%) | |||||||
| r.T. (foaming: 3–10 min) | 220–400 | Open cell 310–540 | −17 to 49 | Flexible to rigid | ||||
| Exotherm inducer: epoxy130 | Compression modulus 0.096–81 MPa (strain 50%) | |||||||
| Water induced | CO2 | 80–100 °C | 150–370 | Open cell 230–5400 | −7.1 to 25 | Flexible to semi-rigid | ||
| DBU or KOH catalysis140,232,235 | Compression modulus 0.03–0.149 MPa (strain 70%) | |||||||
| r.T. (foaming: 3–10 min) | 150–300 | Open cell 420–920 | −7 to 47 | Flexible to rigid | ||||
| Exotherm inducer: epoxy141,143 | Compression modulus 0.0067–1.9 MPa (strain 6%) | |||||||
| r.T. (foaming: 1–10 min) | 170–390 | Open cell 85–180 | −4 to 36 | Flexible to rigid | ||||
| Exotherm inducer: αCC carbonate144 | Compression modulus 0.02–8.9 MPa (strain 12%, 40% humidity) | |||||||
| Amine–CO2 adduct | CO2 | 60 °C | 200–460 | Open cell 180–2000 | 14–24 | Flexible | ||
| 1 step process: liquid amine/CO2 adduct + 5CC152 | Compression modulus NA | |||||||
| 100–140 °C | 270–450 | Open cell 330–910 | −3 to 0 (wet) | Flexible | ||||
| 2 step process: – Prepolymerization, scCO2 impregnation156 | 28–49 (dried) | Compression modulus 0.000025 (wet) – 9 MPa dried) (strain 50%) | ||||||
| NIPU type | Blowing technique | Blowing agent | Foaming procedure | Density (kg m−3) | Cell size and morphology (µm) | T g (°C) | Mechanical properties | |
|---|---|---|---|---|---|---|---|---|
| Thermosets | Physical | Water/ethanol | 73–120 °C, 2 h106 | 55–950 | Open cell 140–3000 | −20 to −18 | Highly flexible | |
| Compression modulus 0.0007–0.003 MPa (strain 25%) | ||||||||
| Amino-telechelic oligo-NIPU | Self-foaming | Poly(methylhydrogenosiloxane) | H2 | r.T. −100 °C121 | 130–400 | Open cell 210–430 | Self-foaming | Flexible |
| Epoxide crosslinker | Compression modulus 0.001–0.005 MPa (strain 50%) | |||||||
| Thermosets | “Emulsion-like” templated | Citric acid, glutaraldehyde, water | — | r.T. −160 °C166 | 80–470 | Open cell 200–2000 | NA–112 | Flexible to rigid |
| Amonio-telechelic oligo-NIPU | 2 step process: oligomerization, curing 80–160 °C for 2–10 h160,164 | Compression modulus 0.0015–1.2 MPa (strain 50%) | ||||||
| Glutaraldehyde crosslinker | ||||||||
Predicting the fabrication costs of NIPU foams presents a significant challenge due to the ongoing absence of mass production for certain key reactants, such as cyclic carbonates in PHU materials. Although there is no techno-economic study of NIPU foam production yet, a preliminary Aspen techno-economic simulation suggests that the production cost of PHU thermoplastic could approach a level of competitiveness comparable to conventional fossil-based flexible polyurethane (PU).234 In the proposed scenario, the thermoplastic was obtained by copolymerizing in dimethylformamide (DMF) a bis(cyclic carbonate), synthesized from 1,3-butadiene and carbon dioxide (CO2), with a mixture of diamines, i.e., amino-telechelic poly(butadiene-co-acrylonitrile) (PBN) and p-xylylenediamine (p-XDA). The minimum selling price (MSP) was estimated at 5.37 USD per kg, assuming a 20% internal rate of return. This MSP was higher than that of conventional PUs (3.53–4.49 USD per kg). Total production costs were primarily driven by raw materials, contributing to 59% of the overall cost. Operating expenses accounted for an additional 16%, while the remaining costs were attributed to taxes, utilities, and capital expenditures. With respect to the reactant cost, amines, particularly PBN, accounted for 62% of the total. A sensitivity analysis revealed that a reduction in amine prices or the replacement of PBN with less expensive alternatives, lower capital expenditures, and increased plant capacity could significantly decrease MSPs. Notably, scaling production to industrially relevant capacities up to 9000 t per year would facilitate economies of scale, resulting in an MSP reduction of 0.88 USD kg−1. Cumulatively, these changes could reduce the MSP of PHU to a production cost of 4.19 USD per kg. These preliminary techno-economic analyses suggest that, under optimized conditions, PHU-based materials could approach cost competitiveness with conventional fossil-based polyurethanes. More broadly, they underscore the strong long-term potential of NIPU chemistry, positioning it as a promising and forward-looking platform for the development of next-generation polyurethane materials.
Conflicts of interest
There are no conflicts to declare.Data availability
As this paper is a tutorial review, no primary research results, software or code have been included and no new data were generated or analysed.Acknowledgements
The authors thank the European Commission for its support in the frame of the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement no. 955700. B. G. thanks the FNRS for funding the CO2smos project (PDR – ID application 40028664). C. D. thanks the Region Wallonne for funding the WEL-T Advanced Grant “CHEMISTRY” project (application WEL-T-CR-2023 A – 02) and the Win2Wal project “ECOFOAM” (convention 2010130). C. D. and J. M. R. are FNRS Research Directors and thank FNRS for financial support and the European Regional Development Fund (ERDF-FEDER) for general support in the frame of UP_PLASTICS portfolio. H. S. thanks the European Union's Horizon 2020 in the frame of the Research and Innovation Program under the Grant TED2021-129852B-C22 funded by MCIU/AEI/10.13039/501100011033 and by the European Union NextGenerationEU/PRTR and the Grant PID2022-138199NB-I00 funded by MCIU/AEI/10.13039/501100011033. T. V. and H. C. would like to acknowledge the French government funding managed by the National Research Agency under the France 2030 program, reference ANR-23-PEXD-0008, project GreenFOAM.References
- MarketsandMarkets, Polyurethane Foams Market – Global Forecast to 2030, 2024.
- A. Mohammadi, A. Doctorsafaei, M. Ghodsieh and S. Beigi-Boroujeni, Polymeric Foams: Fundamentals and Types of Foams (Volume 1), American Chemical Society, 2023, vol. 1439, pp. 143–159, SE–7 Search PubMed.
- N. V. Gama, A. Ferreira and A. Barros-Timmons, Materials, 2018, 11, 1841 CrossRef PubMed.
- M. Ates, S. Karadag, A. A. Eker and B. Eker, Polym. Int., 2022, 71, 1157–1163 CrossRef CAS.
- J. Peyrton and L. Avérous, Mater. Sci. Eng., R, 2021, 145, 100608 CrossRef.
- E. Delebecq, J.-P. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2013, 113, 80–118 CrossRef CAS PubMed.
- G. Coste, C. Negrell and S. Caillol, Eur. Polym. J., 2020, 140, 110029 CrossRef CAS.
- F. Monie, T. Vidil, B. Grignard, H. Cramail and C. Detrembleur, Mater. Sci. Eng., R, 2021, 145, 100628 CrossRef.
- A. Ghosh, J. T. Orasugh, S. S. Ray and D. Chattopadhayay, Polymeric Foams: Fundamentals and Types of Foams (Volume 1), American Chemical Society, 2023, vol. 1439, pp. 4–63 Search PubMed.
- S. Merenyi, With an Introduction and Future Prospects Regarding the Area of Chemicals Legislation, GRIN Verlag ( 2012), Article 20123656293538, REACH: regulation (EC) No 1907/2006: consolidated version.
- B. Grignard, S. Gennen, C. Jérôme, A. W. Kleij and C. Detrembleur, Chem. Soc. Rev., 2019, 48, 4466–4514 RSC.
- A. Delavarde, G. Savin, P. Derkenne, M. Boursier, R. Morales-Cerrada, B. Nottelet, J. Pinaud and S. Caillol, Prog. Polym. Sci., 2024, 151, 101805 CrossRef CAS.
- X. Ou, Y. Niu, Q. Liu, L. Li, F. Wei, Y. Cui, Y. Zhou and F. Yan, Prog. Polym. Sci., 2024, 149, 101780 CrossRef CAS.
- A. Mouren and L. Avérous, Chem. Soc. Rev., 2023, 52, 277–317 RSC.
- F. D. Bobbink, A. P. van Muyden and P. J. Dyson, Chem. Commun., 2019, 55, 1360–1373 RSC.
- T. Habets, B. Grignard and C. Detrembleur, Prog. Polym. Sci., 2025, 165, 101968 CrossRef CAS.
- T. Theerathanagorn, T. Kessaratikoon, H. U. Rehman, V. D’Elia and D. Crespy, Chin. J. Chem., 2024, 42, 652–685 CrossRef CAS.
- A. Gomez-Lopez, F. Elizalde, I. Calvo and H. Sardon, Chem. Commun., 2021, 57, 12254–12265 RSC.
- L. Maisonneuve, O. Lamarzelle, E. Rix, E. Grau and H. Cramail, Chem. Rev., 2015, 115, 12407–12439 CrossRef CAS.
- C. Carré, Y. Ecochard, S. Caillol and L. Avérous, ChemSusChem, 2019, 12, 3410–3430 CrossRef PubMed.
- S. El Khezraji, H. Ben Youcef, L. Belachemi, M. A. Lopez Manchado, R. Verdejo and M. Lahcini, Polymers, 2023, 15 Search PubMed.
- P. Singh, M. Kour, G. Varshney and R. Kaur, Polymer, 2025, 333, 128658 CrossRef CAS.
- F. Orabona, F. Recupido, G. C. Lama, K. Polaczek, F. Taddeo, T. Salmi, M. Di Serio, L. Verdolotti and V. Russo, Green Chem., 2025, 27, 7403–7444 RSC.
- C. C. N. D. Lim, M. J. H. Ong, M. Wu, C.-L. K. Lee and P. Sen Choong, ACS Eng. Au, 2024, 4, 493–518 CrossRef CAS.
- M. L. Chaudhary and R. K. Gupta, Non-Isocyanate Polyurethanes: Chemistry, Progress, and Challenges, American Chemical Society, 2025, vol. 1507, pp. 5–91 Search PubMed.
- L. Gu, X. Wang, X. Chen, X. Zhao and F. Wang, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 5162–5168 CrossRef CAS.
- W. Kuran, A. Rokicki and D. Romanowska, J. Polym. Sci., Polym. Chem. Ed., 1979, 17, 2003–2011 CrossRef CAS.
- O. Ihata, Y. Kayaki and T. Ikariya, Angew. Chem., Int. Ed., 2004, 43, 717–719 CrossRef CAS PubMed.
- K. Soga, S. Hosoda and S. Ikeda, J. Polym. Sci., Polym. Chem. Ed., 1974, 12, 729–736 CrossRef CAS.
- O. Ihata, Y. Kayaki and T. Ikariya, Macromolecules, 2005, 38, 6429–6434 CrossRef CAS.
- K. Soga, W.-Y. Chiang and S. Ikeda, J. Polym. Sci., Polym. Chem. Ed., 1974, 12, 121–131 CrossRef CAS.
- K. Soga, S. Hosoda and S. Ikeda, Die Makromol. Chem., 1974, 175, 3309–3313 CrossRef CAS.
- D. Zhang, Y. Zhang, Y. Fan, M.-N. Rager, V. Guérineau, L. Bouteiller, M.-H. Li and C. M. Thomas, Macromolecules, 2019, 52, 2719–2724 CrossRef CAS.
- J. Futter, L. F. Richter, S. Hörl, M. Kränzlein and B. Rieger, Angew. Chem., Int. Ed., 2025, 64, e202502727 CrossRef CAS PubMed.
- L. Rubino, V. Patamia, A. Rescifina, M. Galimberti and V. Barbera, Sci. Rep., 2024, 14, 23521 CrossRef CAS PubMed.
- J. Kušan, H. Keul and H. Höcker, Macromolecules, 2001, 34, 389–395 CrossRef.
- B. Lebedev, V. Veridusova, H. Höcker and H. Keul, Macromol. Chem. Phys., 2002, 203, 1114–1125 CrossRef CAS.
- B. V. Lebedev, T. A. Bykova, E. G. Kiparisova, A. M. Kochetkov, H. Höcker, J. Kusan and H. Keul, Macromol. Chem. Phys., 1999, 200, 1863–1869 CrossRef CAS.
- B. V. Lebedev, N. N. Smirnova and E. G. Kiparisova, Macromol. Chem. Phys., 1997, 198, 41–58 CrossRef CAS.
- S. Neffgen, H. Keul and H. Höcker, Macromolecules, 1997, 30, 1289–1297 CrossRef CAS.
- G. Rokicki and A. Piotrowska, Polymer, 2002, 43, 2927–2935 CrossRef CAS.
- M. L. Chaudhary and R. K. Gupta, Non-Isocyanate Polyurethanes: Chemistry, Progress, and Challenges, American Chemical Society, 2025, vol. 1507, pp. 4–59 Search PubMed.
- S. Li, J. Zhao, Z. Zhang, J. Zhang and W. Yang, RSC Adv., 2015, 5, 6843–6852 RSC.
- N. Kébir, M. Benoit and F. Burel, Eur. Polym. J., 2018, 107, 155–163 CrossRef.
- B. Ochiai and T. Utsuno, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 525–533 CrossRef CAS.
- B. Sharma, L. Ubaghs, H. Keul, H. Höcker, T. Loontjens and R. van Benthem, Polymer, 2004, 45, 5427–5440 CrossRef CAS.
- S. Li, J. Zhao, Z. Zhang, J. Zhang and W. Yang, Polymer, 2015, 57, 164–172 CrossRef CAS.
- F. Meng and D. Tang, ACS Appl. Polym. Mater., 2023, 5, 5600–5608 CrossRef CAS.
- P. Dongdong and T. Hengshui, J. Appl. Polym. Sci., 2014, 132, 41377 CrossRef.
- D. Wołosz, P. G. Parzuchowski and A. Świderska, Eur. Polym. J., 2021, 155, 110574 CrossRef.
- N. Wybo, E. Cherasse, A. Duval and L. Avérous, J. Mater. Chem. A, 2025, 13, 11557–11572 RSC.
- N. G. Jaques, E. Grau, A. Llevot, T. Vidil, M. A. R. Meier and H. Cramail, Macromol. Chem. Phys., 2025, 226, e00207 CrossRef CAS.
- C. Duval, N. Kébir, A. Charvet, A. Martin and F. Burel, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1351–1359 CrossRef CAS.
- B. Sharma, H. Keul, H. Höcker, T. Loontjens and R. van Benthem, Polymer, 2005, 46, 1775–1783 CrossRef CAS.
- Z. Chen, N. Hadjichristidis, X. Feng and Y. Gnanou, Macromolecules, 2017, 50, 2320–2328 CrossRef CAS.
- S. Xie, Y. Li, Y. Chai, Q. Chen, M. North and H. Xie, ACS Macro Lett., 2024, 13, 14–20 CrossRef CAS PubMed.
- D. Wu, R. T. Martin, J. Piña, J. Kwon, M. P. Crockett, A. A. Thomas, O. Gutierrez, N. H. Park, J. L. Hedrick and L. M. Campos, Angew. Chem., Int. Ed., 2024, 63, e202401281 CrossRef CAS PubMed.
- M. Blain, L. Jean-Gérard, R. Auvergne, D. Benazet, S. Caillol and B. Andrioletti, Green Chem., 2014, 16, 4286–4291 RSC.
- M. Blain, H. Yau, L. Jean-Gérard, R. Auvergne, D. Benazet, P. R. Schreiner, S. Caillol and B. Andrioletti, ChemSusChem, 2016, 9, 2269–2272 CrossRef CAS PubMed.
- F. Monie, T. Vidil, E. Grau, B. Grignard, C. Detrembleur and H. Cramail, Macromolecules, 2024, 57, 8877–8888 CAS.
- M. Blain, A. Cornille, B. Boutevin, R. Auvergne, D. Benazet, B. Andrioletti and S. Caillol, J. Appl. Polym. Sci., 2017, 134, 44958 CrossRef.
- P.-L. Yang, S.-H. Tsai, Y.-Y. Han, H.-W. Chao, K.-N. Chen and D. S.-H. Wong, Ind. Eng. Chem. Res., 2024, 63, 17430–17440 CAS.
- J.-Z. Hwang, C.-L. Chen, C.-Y. Huang, J.-T. Yeh and K.-N. Chen, J. Polym. Res., 2013, 20, 195 CrossRef.
- P.-L. Yang, S.-H. Tsai, K.-N. Chen and D. S. Wong, Polymers, 2023, 15, 2499 CrossRef CAS PubMed.
- A. Cornille, M. Blain, R. Auvergne, B. Andrioletti, B. Boutevin and S. Caillol, Polym. Chem., 2017, 8, 592–604 RSC.
- O. Lamarzelle, P.-L. Durand, A.-L. Wirotius, G. Chollet, E. Grau and H. Cramail, Polym. Chem., 2016, 7, 1439–1451 RSC.
- O. Lamarzelle, G. Hibert, S. Lecommandoux, E. Grau and H. Cramail, Polym. Chem., 2017, 8, 3438–3447 RSC.
- H. Tomita, F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3678–3685 CrossRef CAS.
- R. H. Lambeth, S. M. Mathew, M. H. Baranoski, K. J. Housman, B. Tran and J. M. Oyler, J. Appl. Polym. Sci., 2017, 134, 44941 CrossRef.
- J. Liu, P. Miao, X. Leng, J. Che, Z. Wei and Y. Li, Macromol. Rapid Commun., 2023, 44, 2300263 CrossRef CAS PubMed.
- D. J. Fortman, J. P. Brutman, C. J. Cramer, M. A. Hillmyer and W. R. Dichtel, J. Am. Chem. Soc., 2015, 137, 14019–14022 CrossRef CAS PubMed.
- C. Ngassam Tounzoua, B. Grignard and C. Detrembleur, Angew. Chem., Int. Ed., 2022, 61, e202116066 CrossRef CAS PubMed.
- S. Gennen, B. Grignard, T. Tassaing, J. Ø. Christine and C. Detrembleur, Angew. Chem., Int. Ed., 2017, 56, 10394–10398 CrossRef CAS PubMed.
- J. Wu, Q. Guo, H. Hong, R. Xie and N. Zhu, J. CO2 Util., 2022, 65, 102192 CrossRef CAS.
- S. Dabral, U. Licht, P. Rudolf, G. Bollmann, A. S. K. Hashmi and T. Schaub, Green Chem., 2020, 22, 1553–1558 RSC.
- B. Quienne, R. Poli, J. Pinaud and S. Caillol, Green Chem., 2021, 23, 1678–1690 RSC.
- A. Datta Sarma, S. V. Zubkevich, F. Addiego, D. F. Schmidt, A. S. Shaplov and V. Berthé, Macromolecules, 2024, 57, 3423–3437 CrossRef CAS.
- F. Magliozzi, G. Chollet, E. Grau and H. Cramail, ACS Sustainable Chem. Eng., 2019, 7, 17282–17292 CrossRef CAS.
- P. Helbling, F. Hermant, M. Petit, T. Vidil and H. Cramail, Macromol. Chem. Phys., 2023, 224, 2300300 CrossRef CAS.
- V. Aomchad, À. Cristòfol, F. Della Monica, B. Limburg, V. D’Elia and A. W. Kleij, Green Chem., 2021, 23, 1077–1113 RSC.
- L. Guo, K. J. Lamb and M. North, Green Chem., 2021, 23, 77–118 RSC.
- C. Muzyka, D. V. Silva-Brenes, B. Grignard, C. Detrembleur and J.-C. M. Monbaliu, ACS Catal., 2024, 14, 12454–12493 CrossRef CAS.
- M. Alves, B. Grignard, R. Méreau, C. Jérôme, T. Tassaing and C. Detrembleur, Catal. Sci. Technol., 2017, 7, 2651–2684 RSC.
- M. Usman, A. Rehman, F. Saleem, A. Abbas, V. C. Eze and A. Harvey, RSC Adv., 2023, 13, 22717–22743 RSC.
- M. Decostanzi, Y. Ecochard and S. Caillol, Eur. Polym. J., 2018, 109, 1–7 CrossRef CAS.
- A. Cornille, Y. Ecochard, M. Blain, B. Boutevin and S. Caillol, Eur. Polym. J., 2017, 96, 370–382 CrossRef CAS.
- B. Bizet, E. Grau, J. M. Asua and H. Cramail, Macromol. Chem. Phys., 2022, 223, 2100437 CrossRef CAS.
- F. M. de Souza, Y. Desai and R. K. Gupta, Polymeric Foams: Fundamentals and Types of Foams (Volume 1), American Chemical Society, 2023, vol. 1439, p. 1 Search PubMed.
- A. Wong, R. K. M. Chu, S. N. Leung, C. B. Park and J. H. Zong, Chem. Eng. Sci., 2011, 66, 55–63 CrossRef CAS.
- C. B. Park, D. F. Baldwin and N. P. Suh, Polym. Eng. Sci., 1995, 35, 432–440 CrossRef CAS.
- G. C. DSouza, H. Ng, P. Charpentier and C. C. Xu, ChemBioEng Rev., 2024, 11, 7–38 CrossRef CAS.
- G. Harikrishnan, T. U. Patro and D. V. Khakhar, Ind. Eng. Chem. Res., 2006, 45, 7126–7134 CrossRef CAS.
- X. Cao, L. James Lee, T. Widya and C. Macosko, Polymer, 2005, 46, 775–783 CrossRef CAS.
- L. J. Lee, C. Zeng, X. Cao, X. Han, J. Shen and G. Xu, Compos. Sci. Technol., 2005, 65, 2344–2363 CrossRef CAS.
- A. Kausar, Polym. Plast. Technol. Eng., 2018, 57, 346–369 CrossRef CAS.
- M. Nofar, J. Utz, N. Geis, V. Altstädt and H. Ruckdäschel, Adv. Sci., 2022, 9, 2105701 CrossRef CAS PubMed.
- S. P. Nalawade, F. Picchioni and L. P. B. M. Janssen, Prog. Polym. Sci., 2006, 31, 19–43 CrossRef CAS.
- M. Dong, G. Wang, X. Zhang, D. Tan, D. Jaya Prasanna Kumar, J. Ren, H. Colorado, H. Hou, Z. Toktarbay and Z. Guo, Adv. Compos. Hybrid Mater., 2023, 6, 207 CrossRef CAS.
- M. Sauceau, J. Fages, A. Common, C. Nikitine and E. Rodier, Prog. Polym. Sci., 2011, 36, 749–766 CrossRef CAS.
- B. Grignard, J.-M. Thomassin, S. Gennen, L. Poussard, L. Bonnaud, J.-M. Raquez, P. Dubois, M.-P. Tran, C. B. Park, C. Jerome and C. Detrembleur, Green Chem., 2016, 18, 2206–2215 RSC.
- H.-I. Mao, C.-W. Chen, H.-C. Yan and S.-P. Rwei, J. Appl. Polym. Sci., 2022, 139, e52841 CrossRef CAS.
- F. N. Olang, US pat., US 2012/0183694 A1, 2012 Search PubMed.
- O. Figovsky, R. Potashnikov, A. Leykin, L. Shapovalovs and S. Sivokon, US pat., US 2015/0024138 Al, 2013 Search PubMed.
- H. Blattmann, M. Lauth and R. Mülhaupt, Macromol. Mater. Eng., 2016, 301, 944–952 CrossRef CAS.
- H. Ren, Q. Liao, Z. Zhou, S. Gao, Y. Wang, X. Du, B. Yuan and H. Zhang, Polymer, 2025, 334, 128740 CrossRef CAS.
- V. Valette, N. Kébir, F. Burel and L. Lecamp, eXPRESS Polym. Lett., 2023, 17, 974–990 CrossRef CAS.
- C. Amezúa-Arranz, M. Santiago-Calvo and M.-Á. Rodríguez-Pérez, Eur. Polym. J., 2023, 197, 112366 CrossRef.
- X. Xi, A. Pizzi, C. Gerardin and G. Du, J. Renew. Mater, 2019, 7, 301–312 CAS.
- T. Dong, E. Dheressa, M. Wiatrowski, A. P. Pereira, A. Zeller, L. M. L. Laurens and P. T. Pienkos, ACS Sustain. Chem. Eng., 2021, 9, 12858–12869 CrossRef CAS.
- C. H. Tran, J.-H. Park, S.-Y. Lee, B. Han, W. S. Jae, H. S. Lee, H. Paik and I. Kim, Polymer, 2025, 323, 128217 CrossRef CAS.
- J. A. Reyes-Labarta and A. Marcilla, J. Appl. Polym. Sci., 2008, 107, 339–346 CrossRef CAS.
- J. A. Reyes-Labarta and A. Marcilla, Ind. Eng. Chem. Res., 2012, 51, 9515–9530 CrossRef CAS.
- D. Kiełkiewicz, A. Siewniak, R. Gaida, M. Greif and A. Chrobok, Molecules, 2024, 29, 3908 CrossRef PubMed.
- A. Cornille, S. Dworakowska, D. Bogdal, B. Boutevin and S. Caillol, Eur. Polym. J., 2015, 66, 129–138 CrossRef CAS.
- A. Cornille, C. Guillet, S. Benyahya, C. Negrell, B. Boutevin and S. Caillol, Eur. Polym. J., 2016, 84, 873–888 CrossRef CAS.
- G. Coste, D. Berne, V. Ladmiral, C. Negrell and S. Caillol, Eur. Polym. J., 2022, 176, 111392 CrossRef CAS.
- J. Sternberg and S. Pilla, Nat. Sustain., 2023, 6, 316–324 CrossRef.
- J. Sternberg and S. Pilla, Green Chem., 2020, 22, 6922–6935 RSC.
- K. Evancho, R. S. Reiner, B. M. Bujanovic, S. Pilla and J. Sternberg, RSC Adv., 2025, 15, 38056–38068 RSC.
- J. Li, X. Xu, X. Ma, M. Cui, X. Wang, J. Chen, J. Zhu and J. Chen, ACS Appl. Bio Mater., 2024, 7, 1301–1310 CrossRef CAS PubMed.
- V. Valette, N. Kébir, F. B. Tiavarison, F. Burel and L. Lecamp, React. Funct. Polym., 2022, 181, 105416 CrossRef CAS.
- J. H. Clark, T. J. Farmer, I. D. V. Ingram, Y. Lie and M. North, Eur. J. Org. Chem., 2018, 4265–4271 CrossRef CAS.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- P. Tundo, F. Trotta, G. Moraglio and F. Ligorati, Ind. Eng. Chem. Res., 1988, 27, 1565–1571 CrossRef CAS.
- M. Selva and P. Tundo, J. Org. Chem., 2006, 71, 1464–1470 CrossRef CAS PubMed.
- M. Selva, A. Perosa, D. Rodríguez-Padrón and R. Luque, ACS Sustainable Chem. Eng., 2019, 7, 6471–6479 CrossRef CAS.
- P. Tundo, M. Musolino and F. Aricò, Green Chem., 2018, 20, 28–85 RSC.
- F. Monie, B. Grignard, J. M. Thomassin, R. Mereau, T. Tassaing, C. Jerome and C. Detrembleur, Angew. Chem., Int. Ed., 2020, 59, 17033–17041 CrossRef CAS PubMed.
- B. Grignard, P. Mampuys, J. Escudero, D. Masullo, F. Lemière, B. U. W. Maes and C. Detrembleur, Polym. Chem., 2022, 13, 6599–6605 RSC.
- M. Bourguignon, B. Grignard and C. Detrembleur, Polym. Chem., 2024, 16, 192–203 RSC.
- N. S. Purwanto, Y. Chen, T. Wang and J. M. Torkelson, Polymer, 2023, 272, 125858 CrossRef CAS.
- B. Lecart, C. Baumsteiger, F. Monie, A. Di Maria, C. Detrembleur, A. Richel and H. Vanderschuren, Green Chem., 2023, 25, 9282–9291 RSC.
- N. S. Purwanto, Y. Chen and J. M. Torkelson, Eur. Polym. J., 2024, 206, 112775 CrossRef CAS.
- F. Monie, B. Grignard and C. Detrembleur, ACS Macro Lett., 2022, 11, 236–242 CrossRef CAS PubMed.
- G. Coste, C. Negrell and S. Caillol, Macromol. Rapid Commun., 2022, 43, 1–10 CrossRef PubMed.
- N. S. Purwanto, Y. Chen and J. M. Torkelson, ACS Appl. Polym. Mater., 2023, 5, 6651–6661 CrossRef CAS.
- L.-F. Xiao, Q.-F. Yue, C.-G. Xia and L.-W. Xu, J. Mol. Catal. A: Chem., 2008, 279, 230–234 CrossRef CAS.
- M. Metzger, B. Strehle, S. Solchenbach and H. A. Gasteiger, J. Electrochem. Soc., 2016, 163, A1219 CrossRef CAS.
- M. Bourguignon, B. Grignard and C. Detrembleur, ACS Appl. Mater. Interfaces, 2021, 13, 54396–54408 CrossRef CAS PubMed.
- M. Bourguignon, B. Grignard and C. Detrembleur, Angew. Chem., Int. Ed., 2022, 61, e202213422 CrossRef CAS PubMed.
- M. Bourguignon, B. Grignard and C. Detrembleur, J. Am. Chem. Soc., 2024, 146, 988–1000 CrossRef CAS PubMed.
- D. Trojanowska, F. Monie, G. Perotto, A. Athanassiou, B. Grignard, E. Grau, T. Vidil, H. Cramail and C. Detrembleur, Green Chem., 2024, 26, 8383–8394 RSC.
- M. Chaib, S. Thakur, H. Ben Youcef, M. Lahcini and R. Verdejo, Eur. Polym. J., 2025, 229, 113843 CrossRef CAS.
- M. Makarov, M. Bourguignon, B. Grignard and C. Detrembleur, Macromolecules, 2025, 58, 1673–1685 CrossRef CAS.
- J. A. Marsella and W. E. Starner, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 921–930 CrossRef CAS.
- Z. Ran, X. Liu, X. Jiang, Y. Wu and H. Liao, Thermochim. Acta, 2020, 692, 178735 CrossRef CAS.
- F. Siragusa, E. Van Den Broeck, C. Ocando, A. J. Müller, G. De Smet, B. U. W. Maes, J. De Winter, V. Van Speybroeck, B. Grignard and C. Detrembleur, ACS Sustainable Chem. Eng., 2021, 9, 1714–1728 CrossRef CAS.
- F. Ouhib, B. Grignard, E. Van Den Broeck, A. Luxen, K. Robeyns, V. Van Speybroeck, C. Jerome and C. Detrembleur, Angew. Chem., Int. Ed., 2019, 58, 11768–11773 CrossRef CAS PubMed.
- L. Crane, T. Habets, B. Grignard, J.-C. M. Monbaliu, P. Stiernet and C. Detrembleur, ACS Sustain. Chem. Eng., 2025, 13, 16107–16116 CrossRef CAS.
- L. Narducci, T. Habets, Q. Grossman, B. Grignard, M. A. R. Meier and C. Detrembleur, Macromolecules, 2025, 58, 6452–6465 CrossRef CAS.
- T. Habets, F. Siragusa, B. Grignard and C. Detrembleur, Macromolecules, 2020, 53, 6396–6408 CrossRef CAS.
- P. Sen Choong, Y. L. E. Hui and C. C. Lim, ACS Macro Lett., 2023, 12, 1094–1099 CrossRef PubMed.
- L. Biancalana, G. Bresciani, C. Chiappe, F. Marchetti and G. Pampaloni, New J. Chem., 2017, 41, 1798–1805 RSC.
- P. V. Kortunov, L. S. Baugh, M. Siskin and D. C. Calabro, Energy Fuels, 2015, 29, 5967–5989 CrossRef CAS.
- J. K. Mannisto, L. Pavlovic, T. Tiainen, M. Nieger, A. Sahari, K. H. Hopmann and T. Repo, Catal. Sci. Technol., 2021, 11, 6877–6886 RSC.
- K. Gouveia, J. Vauloup, M. Colpaert, C. Ocando, P. Lacroix-Desmazes, V. Ladmiral, S. Caillol and J. M. Raquez, ACS Appl. Polym. Mater., 2025, 7, 6113–6124 CrossRef CAS.
- J. I. Sintas, J. D. Wolfgang and T. E. Long, Polym. Chem., 2023, 14, 1497–1506 RSC.
- T. Sintas, J. Wolfgang and T. E. Long, in ACS Spring 2024 – Sessions, 2024, p. 3990633.
- M. S. Silverstein, Prog. Polym. Sci., 2014, 39, 199–234 CrossRef CAS.
- X. Xi, A. Pizzi, C. Gerardin, H. Lei, X. Chen and S. Amirou, Polymers, 2019, 11, 1802 CrossRef CAS PubMed.
- X. Chen, J. Li, X. Xi, A. Pizzi, X. Zhou, E. Fredon, G. Du and C. Gerardin, Polym. Degrad. Stab., 2020, 175, 109121 CrossRef CAS.
- D. L. Smith, D. Rodriguez-melendez, S. M. Cotton, Y. Quan, Q. Wang and J. C. Grunlan, Polymers, 2022, 14, 5019 CrossRef CAS PubMed.
- Y. Zhao, Q. Zhang, H. Lei, X. Zhou, G. Du, A. Pizzi and X. Xi, Int. J. Biol. Macromol., 2024, 258, 128994 CrossRef CAS PubMed.
- X. Chen, X. Xi, A. Pizzi, E. Fredon, X. Zhou, J. Li, C. Gerardin and G. Du, Polymers, 2020, 12, 750 CrossRef CAS PubMed.
- P. Singh and R. Kaur, Mater. Lett., 2023, 331, 133433 CrossRef CAS.
- P. Singh and R. Kaur, J. Polym. Environ., 2023, 31, 743–753 CrossRef CAS.
- A. K. Malhotra and D. T. Wasan, Chem. Eng. Commun., 1987, 55, 95–128 CrossRef CAS.
- P. W. Voorhees, J. Stat. Phys., 1985, 38, 231–252 CrossRef.
- B. E. Obi, in Polymeric Foams Structure-Property-Performance A Design Guide, ed. B. E. Obi, William Andrew Publishing, Oxford, 2018, pp. 131–188 Search PubMed.
- C. F. Frias, A. C. Fonseca, J. F. J. Coelho and A. C. Serra, Prog. Org. Coatings, 2024, 187, 108100 CrossRef CAS.
- S. V. Zubkevich, M. Makarov, R. Dieden, L. Puchot, V. Berthé, S. Westermann, A. S. Shaplov and D. F. Schmidt, Macromolecules, 2024, 57, 2385–2393 CrossRef CAS PubMed.
- C. Pu, J. Yang, S. Jin, Y. Zhou and W. Gong, ACS Appl. Polym. Mater., 2025, 7, 7600–7611 CrossRef CAS.
- P. M. Stathatou, C. E. Athanasiou and M. J. Realff, J. Am. Chem. Soc., 2025, 147, 32299–32308 CrossRef CAS PubMed.
- G. Rossignolo, G. Malucelli and A. Lorenzetti, Green Chem., 2024, 26, 1132–1152 RSC.
- Y. Chen, M. Laporte and J. M. Torkelson, J. Polym. Res., 2024, 31, 264 CrossRef CAS.
- S. Kim, K. Li, A. Alsbaiee, J. P. Brutman and W. R. Dichtel, Adv. Mater., 2023, 35, 2305387 CrossRef CAS PubMed.
- D. T. Sheppard, K. Jin, L. S. Hamachi, W. Dean, D. J. Fortman, C. J. Ellison and W. R. Dichtel, ACS Cent. Sci., 2020, 6, 921–927 CrossRef CAS PubMed.
- S. Kim, L. M. Felsenthal, O. Sala, O. Welz, M. Bergeler, A. Alsbaiee and W. R. Dichtel, J. Am. Chem. Soc., 2025, 147, 35655–35663 CrossRef CAS PubMed.
- X. Chen, L. Li, K. Jin and J. M. Torkelson, Polym. Chem., 2017, 8, 6349–6355 RSC.
- C. Bakkali-Hassani, D. Berne, P. Bron, L. Irusta, H. Sardon, V. Ladmiral and S. Caillol, Polym. Chem., 2023, 14, 3610–3620 RSC.
- X. Chen, L. Li, T. Wei, D. C. Venerus and J. M. Torkelson, ACS Appl. Mater. Interfaces, 2019, 11, 2398–2407 CrossRef CAS PubMed.
- Y.-W. Huang and J. M. Torkelson, Macromolecules, 2025, 58, 5356–5367 CrossRef CAS.
- L. M. Nuñez Tapia, P. Y. Batista Peguero, E. Poisson, L. Bischoff, A. Ledoux and F. Burel, Chem. Eng. J., 2025, 520, 166382 CrossRef.
- D. Berne, S. Laviéville, E. Leclerc, S. Caillol, V. Ladmiral and C. Bakkali-Hassani, ACS Polym. Au, 2025, 5, 214–240 CrossRef CAS PubMed.
- R. G. Ricarte and S. Shanbhag, Polym. Chem., 2024, 15, 815–846 RSC.
- V. Zhang, B. Kang, J. V. Accardo and J. A. Kalow, J. Am. Chem. Soc., 2022, 144, 22358–22377 CrossRef CAS PubMed.
- G. M. Scheutz, J. J. Lessard, M. B. Sims and B. S. Sumerlin, J. Am. Chem. Soc., 2019, 141, 16181–16196 CrossRef CAS PubMed.
- B. R. Elling and W. R. Dichtel, ACS Cent. Sci., 2020, 6, 1488–1496 CrossRef CAS PubMed.
- N. S. Purwanto, Y. Chen, T. Wang and J. M. Torkelson, Polymer, 2023, 272, 125858 CrossRef CAS.
- V. D. Lechuga-Islas, E. Gillissen, M. Bourguignon, B. Grignard and C. Detrembleur, Chem. Eng. J., 2025, 516, 163998 CrossRef CAS.
- L. Li, X. Chen, K. Jin and J. M. Torkelson, Macromolecules, 2018, 51, 5537–5546 CrossRef CAS.
- S. T. McKenna and T. R. Hull, Fire Sci. Rev., 2016, 5, 3 CrossRef.
- K. Sałasińska, M. Leszczyńska, M. Celiński, P. Kozikowski, K. Kowiorski and L. Lipińska, Materials, 2021, 14, 1184 CrossRef PubMed.
- H. Singh and A. K. Jain, J. Appl. Polym. Sci., 2009, 111, 1115–1143 CrossRef CAS.
- J. Zhang, G. H. Yeoh and I. I. Kabir, Fire, 2025, 8, 64 CrossRef.
- A. Yadav, F. M. de Souza, T. Dawsey and R. K. Gupta, Ind. Eng. Chem. Res., 2022, 61, 15046–15065 CrossRef CAS.
- Y. Liu, A. Zhang, Y. Cheng, M. Li, Y. Cui and Z. Li, Polym. Test., 2023, 124, 108100 CrossRef CAS.
- G. Coste, M. Denis, R. Sonnier, S. Caillol and C. Negrell, Polym. Degrad. Stab., 2022, 202, 110031 CrossRef CAS.
- A. Sharma, V. V. Tyagi, C. R. Chen and D. Buddhi, Renew. Sustain. Energy Rev., 2009, 13, 318–345 CrossRef CAS.
- S. Ben Romdhane, A. Amamou, R. Ben Khalifa, N. M. Saïd, Z. Younsi and A. Jemni, J. Build. Eng., 2020, 32, 101563 CrossRef.
- K. Pielichowska and K. Pielichowski, Prog. Mater. Sci., 2014, 65, 67–123 CrossRef CAS.
- A. H. Alami, H. M. Maghrabie, M. A. Abdelkareem, E. T. Sayed, Z. Yasser, T. Salameh, S. M. A. Rahman, H. Rezk and A. G. Olabi, J. Energy Storage, 2022, 54, 105204 CrossRef.
- Y. Jing, Z. Zhao, X. Cao, Q. Sun, Y. Yuan and T. Li, Nat. Commun., 2023, 14, 8060 CrossRef CAS PubMed.
- M. Lee, S. D. Dahlhauser, C. Lucci, B. S. Donohoe, R. D. Allen and N. A. Rorrer, Adv. Funct. Mater., 2025, 35, 1–13 Search PubMed.
- L. J. Gibson and M. F. Ashby, Cellular Solids: Structure and Properties, Cambridge University Press, 2nd edn, 1997 Search PubMed.
- B. Hadała, B. Zygmunt-Kowalska, M. Kuźnia, A. Szajding and T. Telejko, Thermochim. Acta, 2024, 731, 179659 CrossRef.
- J. Zhu, Y. Gao, M. Zhang, K. Shen, J. Chen, C. Jin, G. Wu and G. Liu, Chem. Eng. J., 2025, 525, 170558 CrossRef CAS.
- S. B. Kondawar and P. R. Modak, in Materials for Potential EMI Shielding Applications Processing, Properties and Current Trends, ed. K. Joseph, R. Wilson, Elsevier, 2020, pp. 9–25 Search PubMed.
- D.-K. Li, H. Chen, J. R. Ferber, R. Odouli and C. Quesenberry, Sci. Rep., 2017, 7, 17541 CrossRef PubMed.
- A. A. Isari, A. Ghaffarkhah, S. A. Hashemi, S. Wuttke and M. Arjmand, Adv. Mater., 2024, 36, 2310683 CrossRef CAS PubMed.
- J. Kruželák, A. Kvasničáková, K. Hložeková and I. Hudec, Nanoscale Adv., 2021, 3, 123–172 RSC.
- M. Panahi-Sarmad, M. Noroozi, M. Abrisham, S. Eghbalinia, F. Teimoury, A. R. Bahramian, P. Dehghan, M. Sadri and V. Goodarzi, ACS Appl. Electron. Mater., 2020, 2, 2318–2350 CrossRef CAS.
- B. Zhao, M. Hamidinejad, S. Wang, P. Bai, R. Che, R. Zhang and C. B. Park, J. Mater. Chem. A, 2021, 9, 8896–8949 RSC.
- M. Panahi-Sarmad, M. Noroozi, X. Xiao and C. B. Park, Ind. Eng. Chem. Res., 2022, 61, 1545–1568 CrossRef CAS.
- H. M. S, P. Joseph and S. George, Nanoscale Adv., 2025, 7, 4510–4534 RSC.
- N. M. Petryk, N. L. B. Thai, L. V. Saldanha, S. T. Sutherland and M. B. B. Monroe, ACS Appl. Mater. Interfaces, 2025, 17, 26402–26415 CrossRef CAS PubMed.
- W. Liang, N. Ni, Y. Huang and C. Lin, Polymers, 2023, 15, 4301 CrossRef CAS PubMed.
- N. Sienkiewicz, Thermal Insulation and Radiation Control Technologies for Buildings, ed. J. Kośny and D. W. Yarbrough, Springer International Publishing, Cham, 2022, pp. 217–240, ISBN 978-3-030-98692-6 Search PubMed.
- S. Li, Y. Zhang, X. Ma, S. Qiu, J. Chen, G. Lu, Z. Jia, J. Zhu, Q. Yang, J. Chen and Y. Wei, Biomacromolecules, 2022, 23, 1622–1632 CrossRef CAS PubMed.
- Y. Ding, Z. Sun, R. Shi, H. Cui, Y. Liu, H. Mao, B. Wang, D. Zhu and F. Yan, ACS Appl. Mater. Interfaces, 2019, 11, 2860–2869 CrossRef CAS PubMed.
- S. Wang, H. Li, H. Huang, X. Cao, X. Chen and D. Cao, Chem. Soc. Rev., 2022, 51, 2031–2080 RSC.
- Q. Liu, Q. Sun, J. Shen, H. Li, Y. Zhang, W. Chen, S. Yu, X. Li and Y. Chen, Coord. Chem. Rev., 2023, 482, 215078 CrossRef CAS.
- C. Teodosiu, R. Wenkert, L. Tofan and C. Paduraru, Rev. Chem. Eng., 2014, 30, 403–420 CrossRef CAS.
- R. Selvasembian, W. Gwenzi, N. Chaukura and S. Mthembu, J. Hazard. Mater., 2021, 417, 125960 CrossRef CAS PubMed.
- C. Freitas, M. I. Severino, A. Al Mohtar, O. Kolmykov, V. Pimenta, F. Nouar, C. Serre and M. Pinto, ACS Mater. Lett., 2024, 6, 174–181 CrossRef CAS.
- Y. Yu, J. Wang, J. Lian and X. Cheng, RSC Adv., 2014, 4, 18222–18228 RSC.
- Z.-X. Fan, Q.-H. Zhao, S. Wang, Y. Bai, P.-P. Wang, J.-J. Li, Z.-W. Chu and G.-H. Chen, RSC Adv., 2016, 6, 26950–26953 RSC.
- Z. Chu, Z. Fan, X. Zhang, X. Tan, D. Li, G. Chen and Q. Zhao, Sensors, 2018, 18, 1565 CrossRef PubMed.
- Z. Zhou, Q. Wang, Z. Zeng, L. Yang, X. Ding, N. Lin and Z. Cheng, Anal. Methods, 2013, 5, 6045–6050 RSC.
- Y. Su, D. Zhang, P. Jia, W. Gao, Y. Li, Z. Bai, X. Liu, Q. Deng, J. Xu and C. Yang, Spectrochim. Acta, Part A, 2019, 217, 86–92 CrossRef CAS PubMed.
- Y. Chen, Y. Wu, Y. Zhu and S. Tian, Polym. Int., 2022, 71, 169–174 CrossRef CAS.
- M. Mahapatra, M. Bourguignon, B. Grignard, M. Vandevenne, M. Galleni and C. Detrembleur, Angew. Chem., Int. Ed., 2024, 64, e202413605 CrossRef PubMed.
- M. Mahapatra, M. Bourguignon, P. Stiernet, S. Melo, B. Grignard, M. Vandevenne, M. Galleni and C. Detrembleur, Adv. Funct. Mater., 2025, e23095 Search PubMed.
- C. Liang, Y. Jadidi, Y. Chen, U. Gracida-Alvarez, J. M. Torkelson, T. R. Hawkins and J. B. Dunn, ACS Sustainable Chem. Eng., 2024, 12, 12161–12170 CrossRef CAS PubMed.
- C. Pu, J. Yang, S. Jin, Y. Zhou and W. Gong, ACS Appl. Polym. Mater., 2025, 7, 7600–7611 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |