
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Laser-induced polymer dynamics and applications
Daeyeon
Won†
ab,
Jiyong
Ahn†
a,
Jooyeun
Chong†
c,
Ahyoung
Cho
d,
Taehyun
Kim
a,
Seok Hwan
Choi
a,
Deog-Gyu
Seo
*d,
Jiheong
Kang
*c and
Seung Hwan
Ko
 *aef
*aef
aDepartment of Mechanical Engineering, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul 08826, Korea. E-mail: maxko@snu.ac.kr
bDepartment of Biomedical Engineering, Northwestern University, Evanston, Illinois 60208, USA
cDepartment of Chemistry, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul 08826, Korea. E-mail: jiheongkang@snu.ac.kr
dDepartment of Conservative Dentistry and Dental Research Institute, School of Dentistry, Seoul National University, 28 Yeongun-dong, Chongno-Gu, Seoul 03080, Korea. E-mail: dgseo@snu.ac.kr
eInterdisciplinary Program in Bioengineering, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul 08826, Korea
fInstitute of Engineering Research/Institute of Advanced Machinery and Design (SNU-IAMD), Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul 08826, Korea
First published on 30th April 2026
Abstract
Polymers play a crucial role in modern industry owing to their design flexibility and ease of processing. Recent advancements in synthesis have spurred the development of functional polymers, such as organic semiconductors and tough hydrogels, which excel in various applications compared to rigid materials. The performance of polymer-based devices depends on chain structures across multiple length scales, highlighting the need for processing technologies that can finely tune structures and properties. Laser-induced dynamics present an exciting avenue for achieving these objectives by enabling selective activation of pathways that modify functional polymers. By controlling laser parameters, properties such as electrical conductivity and surface morphology can be precisely engineered, minimizing the need for entirely new materials for different applications. This approach streamlines production, lowers costs, and improves material properties even under ambient conditions, setting it apart from conventional microfabrication techniques. Our review discusses the latest advancements in the interaction of lasers with various polymers that can be modified or enhanced by laser irradiation, focusing on energy delivery mechanisms and their influence on polymer properties. We explore how laser-driven structural changes can enhance electrical, mechanical, and optical characteristics. Finally, we discuss future applications of laser processes and design considerations necessary to meet specific application requirements that conventional methods often struggle to fulfill.
 Daeyeon Won | Daeyeon Won is currently a postdoctoral researcher in Biomedical Engineering at Northwestern University. He received his PhD in Mechanical Engineering from Seoul National University under the supervision of Prof. Seung Hwan Ko in 2024. He received his BS in Civil Engineering and Mechanical Engineering from Korea University in 2018. He also worked in the manufacturing industry as a Staff Engineer at the Semiconductor R&D Center of Samsung Electronics from 2024 to 2025. His research focuses on manufacturing processes and mechanical design of organic bioelectronics, including simple microfabrication of (semi)conducting hydrogels and organic electrochemical transistors. |
 Jiyong Ahn | Jiyong Ahn is currently a Staff Engineer at Samsung Electronics. He received his PhD in Mechanical Engineering from Seoul National University under the supervision of Prof. Seung Hwan Ko in 2026. He received his BS in Mechanical Engineering from Kyung Hee University in 2019. His research focuses on the manufacturing processes and mechanical design of functional devices based on polymer dynamics, including laser-induced polymers, molecular dynamics (MD) simulations, and organoid engineering for advanced biomedical applications. |
 Jooyeun Chong | Jooyeun Chong is a postdoctoral researcher in the Department of Chemistry at Seoul National University, working in the research group of Prof. Jiheong Kang. She received her PhD in Materials Science and Engineering from Korea Advanced Institute of Science and Technology in 2025 under the supervision of Prof. Jiheong Kang. She earned her BS in Materials Science and Engineering from Korea Advanced Institute of Science and Technology in 2021. Her research focuses on polymer mechanics based on structural modulation, particularly on tuning supramolecular interactions among polymer moieties to control and enhance mechanical properties. |
 Deog-Gyu Seo | Deog-Gyu Seo is currently a tenured Professor of the Department of Conservative Dentistry at Seoul National University. He received his BS and DDS (Doctor of Dental Surgery) degrees from the School of Dentistry at Yonsei University in 2001, followed by an MS degree in Conservative Dentistry from Yonsei University in 2004, along with specialist board certification from the Ministry of Health and Welfare. He then earned his PhD in Conservative Dentistry from Yonsei University in 2009. His research focuses on biomimetic materials such as bioremineralization with calcium silicate and catechol-based primers inspired by sea mussels as well as bonding mechanics analysis involving zirconia, ceramics, 3D printing resins, and composite adhesion. His clinical research includes cracked tooth analysis, dentin hypersensitivity, laser canal disinfection, and the development of machine learning devices for the diagnosis of root canal orifices and dental caries. |
 Jiheong Kang | Jiheong Kang is an Associate Professor in the Department of Chemistry at Seoul National University. He received his BS in Chemistry from Seoul National University in 2012 and his PhD in Supramolecular Chemistry from the University of Tokyo in 2017 under Prof. Takuzo Aida. He then worked as a postdoctoral associate with Prof. Zhenan Bao at Stanford University for 2 years. In 2020, he began his academic career at KAIST and moved to the Department of Chemistry at Seoul National University in 2024 as an Associate Professor. Jiheong's research focuses on dynamic polymer networks, soft materials for biomedical devices, and energy devices. His work has been published in top journals such as Science, Nature Nanotechnology, Nature Electronics, and Nature Communications. He has received the Reaxys PhD Award, MIT Technology Review TR35 Asia Pacific recognition, and the Young Scientist Award from the President of Korea. |
 Seung Hwan Ko | Seung Hwan Ko is a Professor at the Wearable Soft Electronics (WSE) Lab, Department of Mechanical Engineering, Seoul National University, South Korea and Director of the Global Leader Research Center for Soft Neural Interface. Before joining Seoul National University, he was a faculty member at KAIST, South Korea from 2009. He received his PhD in Mechanical Engineering from UC Berkeley in 2006 and worked as a postdoctoral researcher at UC Berkeley until 2009. The WSE Lab focuses on pioneering research in soft materials, innovative fabrication processes, developing flexible/stretchable electronic materials, and the development of wearable and bioelectronic devices to overcome existing challenges in current research fields. Currently, the group's primary research efforts are focused on the development of advanced soft wearable electronic devices, specifically designed for soft robotics, healthcare devices and neural interfaces. |
1. Introduction
Polymers have been widely utilized as essential materials in modern industry owing to their molecular design flexibility, processability, and low manufacturing cost.1 Beyond their traditional use, the advancement of synthetic chemistry and materials science has led to the rapid development of polymers with specific functionalities such as electrical conductivity or tissue-like mechanical properties. Organic (semi)conductors capable of transporting charges through conjugated backbones,2–4 hydrogels exhibiting outstanding toughness that can withstand substantial stress and strain,5–7 and photonic polymers displaying optical tunability6,8,9 have been attracting increasing attention, to name a few. Polymer-based devices such as wearable sensors or foldable displays offer clear advantages in dynamically operating environments where conventional rigid materials (e.g., metals and ceramics) struggle to perform. Their inherent softness and flexibility is opening up new application spaces in which polymers are not just beneficial but essential. These emerging research fields include bioelectronics that interface electronic devices with living systems10–12 and soft robotics that can delicately manipulate fragile objects.13–15The performances of these devices are determined by multi-length scales, ranging from the molecular structures of the polymer chains to their microstructures and morphologies.16–18 As a result, the importance of processing technologies that can coherently control polymer structures and properties across multiple length scales is becoming evident, and there is a clear need for strategies in which synthesis and processing are simultaneously considered to meet application-specific requirements. Traditionally, progress in polymer science has focused on the design and synthesis of new monomers and polymer architectures. However, the approach of synthesizing a new polymer for distinct set of applications has limitations in terms of time, cost, and synthetic complexity. While synthesis-centered strategies remain indispensable, there is now a pressing need for integrated approaches in which “well-established polymers” are prepared and then endowed with application-tailored properties through precise processing and functionalization. This requirement is particularly pronounced in applications such as bioelectronics, wearable devices, and energy systems, where often conflicting properties such as electrical conductivity and mechanical compliance must be simultaneously satisfied.19 In these cases, novel processing techniques that can finely tune structure–property relationships from the molecular scale to the device level are required.
Laser-induced polymer dynamics can provide a promising platform that aligns with these needs and can form a complementary process to conventional polymer chemistry. A laser is a monochromatic light source with a specific wavelength that delivers focused photon energy to desired spots selectively (Fig. 1A).20,21 By tuning parameters such as wavelength, pulse duration, fluence, irradiation atmosphere, and scanning strategy, laser processing can drive nonlinear reactions and induce unique structural modifications in functional polymers that are difficult to achieve with bulk thermal annealing and photochemical processes.22 Moreover, since a laser is an electromagnetic field,23 interference of its optical waves can be exploited to apply spatially patterned photon energies to polymers.24 In polymers with intrinsic polarization, the electric field of the laser can directly interact with the dipoles and thereby induce changes in the polarization state.
 | ||
| Fig. 1 Overview of laser–polymer processing and comparison with conventional fabrication approaches. (A) Benefits of laser–polymer interactions. The transient nature of laser-induced processes enables spatially confined modification of polymer surfaces with high temporal control. (B) Comparative summary of laser-based fabrication versus conventional fabrication techniques such as 3D printing and photolithography. (C) Representative applications are also highlighted, including micro/nanopatterning, selective annealing, and 3D hydrogel structuring, illustrating the versatility of laser-induced polymer processing. | ||
Laser–polymer interactions can be broadly categorized into photochemical, photothermal, pyrolysis, and ablation regimes, depending on how the deposited optical energy is converted into molecular and structural changes. Photochemical processes involve polymerization,25 crosslinking,26 and decomposition27 driven by photon absorption, typically under short-pulse irradiation. Photothermal processes arise from lattice heating and thermal diffusion, leading to melting, phase separation,28 and chain ordering.29 By further increasing the laser fluence and interaction, lasers can pyrolyze polymers into porous carbon structures,30 and under specific conditions, they bypass pyrolysis and instead ablate them. In practice, these regimes are not strictly isolated, and transitions between them can occur depending on the laser fluence, pulse duration, and material properties.
Properties of polymers including electrical conductivity, modulus, surface topography, and the density of chemical functional groups can be spatially engineered in a highly localized and programmable manner. The polymer backbone, side chains, and reactive functional groups can be predesigned in terms of their type and density at the synthesis stage, and then a laser can selectively reconfigure them only at the desired regions (Table 1). In this way, the burden of synthesizing entirely new materials for each application can be substantially alleviated. This approach leverages the monochromatic nature of lasers.
| Reactions | Types of polymers | Polymer examples | Laser processing | Ref. |
|---|---|---|---|---|
| Photothermal | Composite matrix/elastomers | PDMS, PCL, Ecoflex, silicone elastomers, SMPs | Local softening under laser heating for stretchable conductors, magnetic soft robotics, adhesion control and transfer process. | 203, 226 and 227 |
| Conductive/semiconducting polymers | PEDOT:PSS, PPy, P3HT | Conductivity enhancement and modulation for electronic and sensing devices. | 152, 170 and 187 | |
| Self-assembling/block copolymers | BCPs, PS-b-P2VP | Laser-induced phase separation, domain alignment, and nanopattern/nanotemplate formation | 153, 154 and 156 | |
| Liquid-crystalline/ferroelectric polymers | LCEs, PVDF | Molecular alignment, phase transition, dipole control, optical switching, and actuation in soft robotic or responsive optical systems. | 28 and 174 | |
| Photochemical | Photoresponsive polymer | Thiol–ene, norbornene-based, azobenzene-, hydrazone-, diarylethene-, spiropyran-, stilbene-, anthracene-, coumarin-, and cinnamate-based systems | Photochemical network formation, degradation, and reversible structural switching | 81–96, 101–106, 110–113, 122–128 and 132–137 |
| Acrylate-based hydrogels/elastomers | p(AM-co-PEGDA) hydrogel, polyacrylates | Photopolymerization and crosslinking | 5 and 144 | |
| Pyrolysis | Pyrolysis precursor polymers | PI, PDMS | Transformation by laser irradiation into new conductive or inorganic phases such as laser-induced graphene or SiC-based structures. | 132 and 179 |
Laser-derived dynamics can also be developed into promising alternatives that can complement the limitations of current microfabrication processes (Fig. 1B and C). When fabricating polymer-based electronic devices, multi-step processes such as photolithography and plasma etching are often essential,31 which require expensive vacuum equipment, leading to significantly high overall process costs. Besides, polymers can be easily damaged by heat and solvents, often imposing constraints on the applicable temperature range and solvent selection.32–34 In contrast, lasers can rapidly provide localized energy under ambient temperature and pressure conditions, allowing for direct control of polymer properties from the nanoscale to the microscale. This approach not only simplifies the processing stages but also has the advantage of achieving superior electrical and mechanical properties compared to traditional thermal processes by inducing nonlinear internal structural changes. These characteristics are particularly attractive in emerging fields of wearable and implantable devices, where rapid customized manufacturing is required, providing design freedom to spatially separate and implement conflicting properties such as high conductivity, high durability, biocompatibility, and environmental resistance by selectively tuning material properties on a single substrate.
In this review, we introduce the structural transformation in polymers resulting from their interaction with a laser. We have selected and classified representative polymers capable of significantly modulating their physical properties across interaction regimes (Fig. 2). For instance, under conditions of relatively low fluence and short pulse duration, molecular-scale interactions such as cross-linking and polymerization in tight regions become possible; these processes are critically utilized in the fabrication of nanoscale structures. At higher fluence and longer interaction times, sufficient thermal energy is imparted to polymers to induce changes in their nano-microstructure. This phenomenon enables the fabrication of electronic or optical polymers whose electrical and optical properties vary significantly depending on their phase.
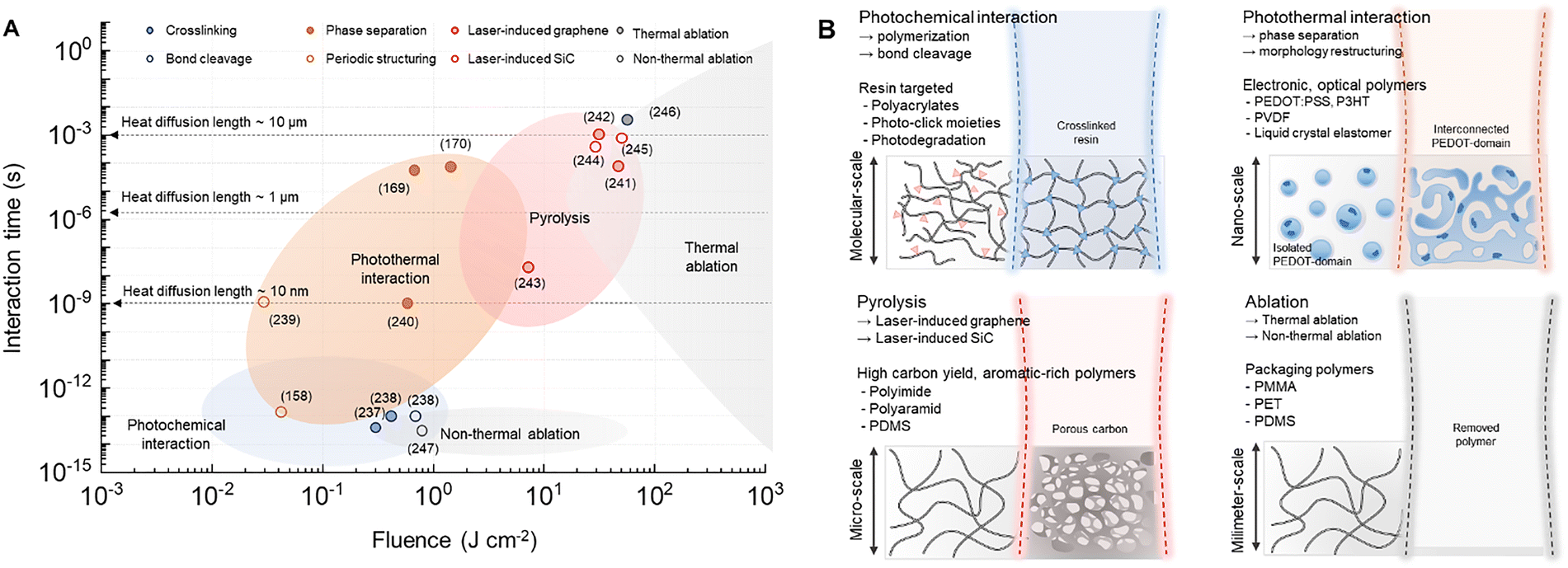 | ||
| Fig. 2 Laser-induced polymer dynamics. (A) Processing map of laser–polymer interactions as a function of fluence and interaction time, showing photochemical, photothermal, thermochemical, and ablation regimes (crosslinking, bond cleavage,237,238 periodic structuring,158,239 phase separation,169,170,240 laser-induced graphene,241–243 laser-induced SiC,244,245 thermal ablation,246 and non-thermal ablation247). Interaction time is defined as the pulse duration for pulsed systems and the local dwell time for CW scanning. Dashed lines indicate thermal diffusion lengths, and literature data points are overlaid for reference. (B) Schematic of each regime, explaining representative laser-polymer interactions and specific polymers. | ||
We discuss a comprehensive processing map that correlates laser parameters with the resulting interaction regimes. Specifically, the diagram plots laser fluence against interaction time, allowing the identification of dominant regimes such as photochemical, photothermal processes, pyrolysis, and ablation. To improve the physical interpretability of the map, we further clarify how the interaction time is defined for different laser platforms. For pulsed laser systems, the interaction time is taken as the pulse duration, whereas for CW scanning systems, it is recalculated by the local dwell time determined by the beam diameter and scan speed. For scanning pulsed systems, the pulse duration is used as the characteristic energy deposition time, while cumulative heating due to repeated pulse overlap is discussed separately in the text when relevant. In addition, we incorporated representative literature data points and included thermal diffusion boundaries to illustrate the transition from localized to diffusive energy transport (Table 2).
| # of Ref | Category | Fluence (J cm−2) | Interaction time (s) | Polymers/Pulse | Wavelength (nm) |
|---|---|---|---|---|---|
| 237 | Crosslinking | 0.3 | 7 × 10−14 | Gelatin-thiol-norbornene | 720 |
| 238 | Crosslinking | 0.4 | ∼10−13 | PEG-thiol–ene | 860 |
| Cleavage | 0.8 | ∼10−13 | PEG- o-nitrobenzyl ether | 740 | |
| 158 | Periodic structuring | 0.037 | 1.2 × 10−13 | PET/fs | 795 |
| 239 | Periodic structuring | 0.026 | ∼10−9 | P3HT/ns | 532 |
| 240 | Phase separation | 0.55 | ∼10−9 | PEDOT:PSS/ns | 355 |
| 170 | Phase separation | 1.5 | 6.67 × 10−5 | PEDOT:PSS/CW | 532 |
| 169 | Phase separation | 0.625 | 1.6 × 10−4 | PEDOT:PSS/CW | 532 |
| 241 | Laser-induced graphene | 48 | 1 × 10−4 | PI/µs | 405 |
| 242 | Laser-induced graphene | 33.7 | 1.35 × 10−3 | PI/µs | 1060 |
| 243 | Laser-induced graphene | 7.4 | 2.5 × 10−8 | PI/ns | 355 |
| 244 | Laser-induced SiC | 20–30 | 6.67 × 10−4 | PDMS/CW | 532 |
| 245 | Laser-induced SiC | 50 | 1–4 × 10−3 | PDMS/CW | 532 |
| 246 | Thermal ablation | 52 | 7.1 × 10−6 | PI/CW | 1060 |
| 247 | Non-thermal ablation | 0.8–2.0 | 6 × 10−14 | PET,PC,PVA,PMMA/fs | 795 |
We then discuss how these laser-driven structural changes translate into modifications of the electrical, mechanical, and optical properties of polymers, with the goal of providing insight into how processing parameters can be used to predict and tailor target properties. Finally, we highlight design considerations for meeting application-specific requirements that are difficult to achieve by conventional processing alone, and we illustrate how laser-enabled control of polymer properties can inspire new device architectures for researchers seeking to engineer high-precision polymer-based systems.
2. Laser as energy sources
Laser-based manufacturing technologies have greatly contributed to modern electronic devices and industrial production. For example, laser cutting and welding have become foundational tools in fields requiring high-precision machining,35 while in photolithography, laser light sources have long been used to induce photoacid generation in photoresists to define polymer patterns.36 This field has now advanced beyond the use of polymers merely as patterning tools toward an era in which laser-delivered energy is harnessed to actively enhance the properties of functional polymers and to use them as active device materials. In this context, understanding the characteristics of the energy supplied by lasers and how it is transferred into polymer systems becomes essential to understand laser–polymer interactions. Therefore, this chapter focuses on the role of lasers as energy-delivery tools in polymers and on the types of physical and chemical interactions that arise in this process.We introduce a comprehensive processing map that correlates laser parameters with the resulting polymer transformation regimes. Specifically, the diagram plots laser fluence against interaction time, allowing the identification of dominant regimes such as photochemical modification, photothermal processes, pyrolysis, and ablation (Fig. 2A). To improve the physical interpretability of the map, we further clarify how the interaction time is defined for different laser platforms. For pulsed laser systems, the interaction time is taken as the pulse duration, whereas for CW scanning systems it is recalculated by the local dwell time determined by the beam diameter and scan speed. For scanning pulsed systems, the pulse duration is used as the characteristic energy deposition time, while cumulative heating due to repeated pulse overlap is discussed separately in the text when relevant. In addition, we incorporated representative literature data points and included thermal diffusion boundaries to illustrate the transition from localized to diffusive energy transport (Table 2). We also introduce representative reactions and specific polymers corresponding to each regime (Fig. 2B).
2.1. Overview of laser hardware and key process parameters
The laser–polymer interactions discussed throughout this review are realized through a variety of hardware platforms, each of which defines a distinct regime of energy delivery. Continuous-wave (CW) systems, including CO2, diode, and fiber lasers, provide a sustained thermal input over relatively long interaction times, making them well-suited for large-area photothermal transformations such as carbonization and annealing.37,38 Nanosecond-pulsed UV lasers offer sufficiently high photon energies to drive direct photochemical bond cleavage with heat-affected zones substantially smaller than those of CW systems. Ultrafast femtosecond systems reduce the pulse duration below the electron–phonon coupling timescale, enabling non-equilibrium energy deposition and nonlinear optical interactions including two-photon absorption, which confine polymerization to a sub-diffraction-limited focal volume for true three-dimensional nanofabrication.39,40 Interference-based platforms exploit the electromagnetic wave nature of lasers to generate spatially periodic intensity distributions across large areas without serial scanning.41 Line-beam configurations such as soft-shear laser zone annealing (SS-LZA) combine photothermal gradients with mechanical shear to drive the directed self-assembly of block copolymer microdomains at the macroscale.42 Because each platform accesses a fundamentally different interaction regime, the choice of hardware is not merely an experimental detail but a primary determinant of the resulting polymer transformation.Equally important is the laser process parameters, as the same nominal laser output can produce qualitatively different structural outcomes depending on how energy is delivered in space and time.43,44 Parameters such as wavelength, pulse duration, fluence, repetition rate, scan speed, beam spot size, and processing atmosphere collectively govern which of the photothermal, photochemical, or field-driven pathways will be activated in a given polymer system.45,46 In particular, the characteristic interaction time, defined by the pulse duration for ultrafast systems and by the beam dwell time for CW scanning systems, determines the boundary between thermally confined and thermally diffusive regimes.47,48 Therefore, understanding how these parameters map onto distinct physical mechanisms forms the necessary foundation for the mechanistic discussion of photothermal, photochemical, and field-driven laser–polymer interactions presented in the following sections.
2.2. Photothermal energy
The interaction between laser irradiation and polymers is fundamentally governed by the conversion of photon energy into thermal energy. Unlike conventional heating methods (e.g., hot plates or ovens), which rely on slow conductive or convective heat transfer from the surface, laser irradiation acts as a remote, coherent energy source capable of delivering energy into defined volumes with high spatiotemporal precision.49 This photothermal effect serves as the primary driving force for diverse polymer processing techniques, ranging from phase transformation and welding to ablation and carbonization.50To precisely control photothermal dynamics, one must first understand how laser energy is spatially distributed and absorbed within the material. Most laser systems used in polymer processing emit a fundamental transverse mode, characterized by a Gaussian intensity profile.51 The spatial intensity distribution I(r,z) is described as follows:
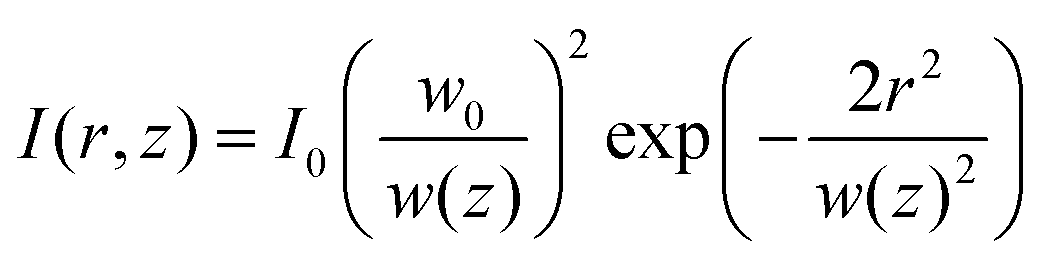
 intensity), and w(z) is the beam radius at distance z. The beam divergence and depth of focus are governed by the Rayleigh range (zR), defined as:
intensity), and w(z) is the beam radius at distance z. The beam divergence and depth of focus are governed by the Rayleigh range (zR), defined as:where λ is the wavelength. This Gaussian nature implies that the thermal energy input is spatially confined, creating a distinct thermal gradient from the center to the periphery, which is critical for high-resolution patterning.51,52
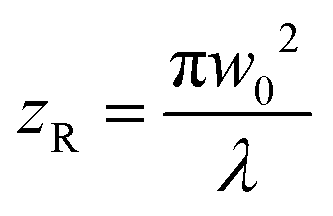
Once the beam interacts with the polymer, the volumetric heat generation rate Q(z) is governed by the Beer–Lambert law, which is dependent on the absorption coefficient (α) of the material, as follows:
| Q(z) = α(1 − R)I0exp(−αz) |
where D is the thermal diffusivity of the polymer.50,53 By tuning τp relative to the thermal relaxation time, one can switch between a thermal confinement and a thermal accumulation regime. The fundamental principle of photothermal conversion involves the excitation of electrons by incident photons (hv), followed by the relaxation of this energy into heat. However, the specific pathway of this conversion varies significantly depending on the electronic structure of the material class, including metals, semiconductors, and polymers (Fig. 3A).
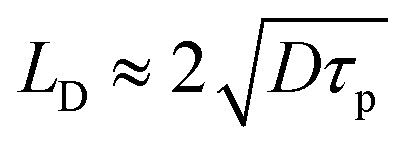
 | ||
| Fig. 3 Fundamentals of photothermal energy conversion. (A) Schematic of light-to-heat conversion mechanisms across different material classes. Localized surface plasmon resonance (LSPR)-induced heat generation in metals via Landau damping and electron–phonon scattering (left). Non-radiative recombination of excited electron–hole pairs across the bandgap in semiconductors (middle). Dissipation of absorbed photon energy into the lattice as heat through internal conversion and vibrational relaxation of excited molecular states in organic polymers (right). (B) Electronic transition types in polymers categorized by their dominant excitation pathways and relative transition energies. Unconjugated polymers (e.g., PDMS and PE) exhibit high-energy transitions requiring deep UV excitation, whereas conjugated polymers (e.g., PEDOT:PSS and PPy) possess delocalized electrons allowing for lower-energy transitions in the visible range. | ||
In metallic nanomaterials (e.g., Au and Ag), the abundance of free electrons allows for coherent oscillation with the incident electric field, known as localized surface plasmon resonance (LSPR). The energy absorbed by these oscillations decays via Landau damping, creating hot electron–hole pairs. These hot carriers subsequently scatter with the lattice via electron–phonon coupling on a picosecond timescale, effectively generating heat that diffuses into the surrounding medium.54–56 For inorganic semiconductors, photons with energy greater than the bandgap (Eg) excite electrons from the valence band to the conduction band. The photothermal effect arises primarily from non-radiative recombination processes, such as Auger recombination or defect-mediated recombination, where the excess energy of the excited carriers is released as phonons (lattice vibrations) rather than photons.54,57
In organic polymers, light absorption triggers transitions between molecular orbitals. The excited electrons in the singlet state (S1) rapidly undergo internal conversion and vibrational relaxation, dissipating energy into the polymer backbone as heat (Fig. 3B). Given that most polymers are dielectric insulators with wide bandgaps, their absorption characteristics are dictated by specific electronic transitions determined by their chemical structure.54,58 Understanding these transitions is essential for selecting the appropriate laser wavelength or designing the polymer matrix.59
The σ → σ* transition corresponds to the excitation of electrons in single bonds (C–C, C–H) and requires high energy, typically falling in the UV range.60,61 Saturated polymers such as polyethylene (PE), polydimethylsiloxane (PDMS), and polytetrafluoroethylene (PTFE) exhibit this transition.58,59,62 Consequently, these materials are highly transparent to standard visible and near-infrared (NIR) lasers, often necessitating high-fluence processing or the use of vacuum UV sources. Occurring in systems with conjugated double bonds, π → π* transitions require lower energy, covering the UV to Visible range.54 Conjugated polymers such as polyaniline (PANI), polypyrrole (PPy), polythiophene (PT), and poly(3,4-ethylenedioxythiophene):poly(styrene sulfonate) (PEDOT:PSS) strongly absorb light in this region.63,64 These materials exhibit inherent photothermal conversion capabilities due to their delocalized π-electrons, allowing them to be processed directly with visible lasers without additives. Polymers containing heteroatoms with lone pair electrons (e.g., O, N, and S) alongside π systems undergo n → π* transitions. Examples include polyimide (PI) and benzophenone.58,65 These transitions typically correspond to UV absorption but often possess Urbach tails extending into the visible range, facilitating laser ablation or modification at lower fluences compared to fully saturated polymers.59,66
A critical limitation in laser polymer processing is the inherent optical transparency of many functional polymers (e.g., elastomers and hydrogels) at the wavelengths of commercially available industrial lasers (typically vis/NIR). To induce effective photothermal dynamics such as melting (Tm), glass transition (Tg), or pyrolysis in these transparent materials, specific strategies are required to enhance local light absorption and focus heat generation (Fig. 4). Incorporating plasmonic nanoparticles (NPs) is a primary strategy to sensitize transparent polymers. As depicted in Fig. 4, metal NPs embedded in the polymer matrix act as efficient localized heat sources due to LSPR.55 When irradiated, the resonant oscillation of free electrons results in rapid, intense heating of the nanoparticle, which then diffuses into the surrounding polymer matrix. This allows for “cold” polymers to be processed using low-power lasers, enabling applications such as precision welding of transparent sheets or selective curing of hydrogels without damaging the bulk material.
 | ||
| Fig. 4 .Strategies for photothermal efficiency enhancement and spatiotemporal heat control. (left) Extrinsic absorption enhancement using plasmonic nanomaterials. Localized heat generation via surface plasmon resonance of metal nanoparticles embedded in optically transparent polymers. (middle) Intrinsic localization via two-photon absorption. Confinement of photothermal energy to the focal volume driven by nonlinear optical excitation, enabling 3D processing. (right) Thermal dose control via laser-scanning strategies. Manipulation of heat accumulation and dissipation by adjusting pulse repetition rates and scanning speeds to control the spatial extent of the heat-affected zone. | ||
For high-precision processing without physical additives, nonlinear optical effects such as two-photon absorption (TPA) are utilized (Fig. 4, middle). Unlike linear absorption, which involves a single photon bridging the energy gap (Egap = hv), TPA involves the simultaneous absorption of two photons with half the energy 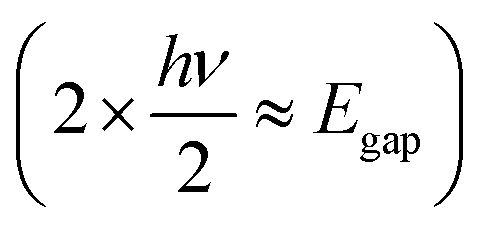 via a virtual state. This process depends on the square of the light intensity (I2), confining the absorption, and thus the heat generation, strictly to the focal volume of the laser (typically requiring femtosecond pulses).67,68 This strategy allows for sub-diffraction-limit resolution and true three-dimensional (3D) structuring within the bulk of transparent polymers, which is impossible with linear absorption.
via a virtual state. This process depends on the square of the light intensity (I2), confining the absorption, and thus the heat generation, strictly to the focal volume of the laser (typically requiring femtosecond pulses).67,68 This strategy allows for sub-diffraction-limit resolution and true three-dimensional (3D) structuring within the bulk of transparent polymers, which is impossible with linear absorption.
Once heat is generated, its spatial distribution determines the final material structure. The structural evolution is governed not just by the energy of a single pulse, but by the accumulation of heat over time and space (Fig. 4, right). By manipulating the laser scanning speed and pulse repetition rate relative to the thermal relaxation time of the material, one can precisely control the thermal history.69 When the time interval between pulses is longer than the thermal relaxation time, heat dissipates completely before the next pulse arrives. This regime minimizes the heat-affected zone, enabling high-resolution ablation or patterning.53 Conversely, when using high-repetition-rate lasers or CW lasers, or when the scanning speed is slow, residual heat from previous pulses accumulates. This thermal incubation effect raises the local background temperature, allowing the polymer to reach phase transition temperatures even at lower peak intensities.70 This strategy is crucial for inducing continuous phase changes, such as recrystallization or the formation of conductive carbon, over large areas by effectively managing the thermal dose.
2.3. Photochemical energy
The interaction between laser irradiation and polymeric materials is fundamentally governed by the conversion of photon energy into thermal energy. However, beyond this thermal dissipation, specific light–matter interactions can trigger direct molecular rearrangements through electronic excitation. Photo-responsive molecules undergo transformations when they absorb light of a specific wavelength. When the incident light provides energy that matches the molecular orbital gap, these molecules absorb the energy and transition to an excited state, initiating photochemical reactions. These reactions involve bond cleavage, bond formation, and reversible structural changes. Incorporating photo-responsive molecules into polymer networks enables precise modulation of the network structure and properties through controlled light exposure.71 In particular, the use of lasers as light sources offers distinct advantages, including exceptional spatiotemporal precision, high energy density, and non-contact processability.Photochemical reactions driven by photo-responsive molecules in polymer systems are generally categorized into three fundamental modes: network formation, network degradation, and reversible structural transformation (Fig. 5). We aim to discuss the underlying mechanisms of these three modes utilizing light sources such as lasers.
 | ||
| Fig. 5 Photochemical reactions in polymer. Schematic of photoresponsive molecules embedded in a polymer matrix (left) and the resulting structural changes in the polymer upon light irradiation (right). Upon light irradiation, a solution containing monomers and initiators undergoes polymerization, leading to curing. In addition, interchain crosslinking occurs through photoactive groups incorporated within the polymer chains, resulting in network formation. Network degradation can occur via photocleavable groups present within the chains, while reversible network transitions are enabled by photoresponsive moieties that undergo light-induced reversible transformations. | ||
Laser-induced photochemical reactions enable the formation of polymer networks (Fig. 6A and B). This process primarily occurs via two mechanisms: polymerization, in which monomers are propagated into polymer chains, and interchain crosslinking, in which existing polymer chains are covalently interconnected. Polymerization initiated by a laser enables highly controlled patterning and localized solidification, while the high photon flux allows extremely rapid initiation kinetics that accelerate fabrication processes (Fig. 6A).72,73 Photo-responsive molecules that are excited to the photo energy are photo-initiators, which generate radicals or photoacids to initiate the polymerization of monomers such as acrylates or epoxides.74 Upon light exposure, radical initiators such as Darocur 1173 are used to initiate the free-radical polymerization of vinyl monomers, such as acrylates and methacrylates. These photoinitiators absorb light and generate reactive radical species either via homolytic bond cleavage (Type I) or through interaction with a co-initiator (Type II), which then react with the carbon–carbon double bonds of monomers to produce new chain-end radicals. Continuous attack on the adjacent monomer drives chain propagation, leading to the growth of the polymer chain. Alternatively, photoacid generators (PAGs), such as iodonium PAG, are used to initiate the polymerization of electron-rich monomers, including epoxides, vinyl ethers, oxetanes, and cyclic siloxanes.75,76 PAGs release strong acids upon light exposure, enabling cationic polymerization. The absorption wavelengths of radical initiators and PAGs mostly lie in the ultraviolet region, although some can initiate under visible to near-infrared light. In photo-induced polymerization, key challenges include controlling light penetration, high-resolution patterning, and achieving rapid curing. The use of lasers has enabled advances in addressing these issues. In particular, complex three-dimensional microstructures can be fabricated through a two-photon femtosecond laser, where the simultaneous absorption of two non-resonant photons excites electrons to a higher electronic state equivalent to the combined energy of the two photons.67,77–79
 | ||
| Fig. 6 Polymer structural changes induced by photochemical reactions. (A) Polymerization of monomers via photo-initiator activation, including radical polymerization and photoacid generation mechanisms. (B) Formation of the polymer network through interchain crosslinking reaction. Chemical reactions of thiol–ene crosslinking and benzophenone crosslinking. (C) Deconstruction of polymer network through photocleavable groups and main chain degradation. Photo-induced degradation of ortho-nitrobenzyl and depolymerization of poly(olefin sulfone). (D) Reversible transformation of the polymer network through reversible photo-responsive groups. Mechanisms of photodimerization of anthracene and photoisomerization of azobenzene. | ||
Interchain crosslinking reactions transform an uncrosslinked polymer into a crosslinked polymer network when triggered by external energy sources such as light, heat, or mechanical force (Fig. 6B).80 Among them, photochemistry-driven interchain crosslinking is widely studied for microfabrication and spatial patterning (Table 3). The photo-responsive molecules in the polymer network absorb light and transition to an excited state, subsequently reacting with functional groups on nearby polymer chains to form new covalent linkages. These reactions can be broadly categorized into two types: those that form high-energy intermediates upon light absorption to perform insertion into bonds such as C–H, and click reactions such as the thiol–ene system. As an example of photo-induced insertion, perfluorophenyl azide (PFA) generates a highly reactive nitrene species upon exposure to UV light (254 nm), which subsequently inserts into the C–H bonds of alkyl groups. Thus, a di-PFA crosslinker can be incorporated into various polymer matrices, allowing selective crosslinking only in illuminated regions to produce a mechanically robust network.81–84 Similarly, other photo-reactive functionalities such as diazirines85–87 and benzophenone88–90 have been extensively explored for light-induced interchain crosslinking.
| Photoreactions | Photoreactive moieties | Mechanistic principle | Ref. |
|---|---|---|---|
| Insertion/H-abstraction | Perfluorophenyl azide (PFA), diazirine, benzophenone | Generation of high-energy intermediates (e.g., nitrenes, carbenes, triplet states) that insert into C–H bonds or undergo radical recombination. | 81–90 |
| Thiol-based click reactions | Thiol–ene | Photo-triggered thiol radical adds across a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C to form thioether. C to form thioether. |
91 |
| Thiol-Michael addition | Nucleophilic attack initiated by photobase generators. | 92 | |
| Thiol–yne | Photo-triggered two thiol molecules react with one alkyne triple bond to form a cross-linked 1,2-dithioether structure | 94 | |
| High-strain & specific click reactions | Norbornene-based system | Light activates a 'hidden' tetrazine to rapidly click with strained alkene like norbornene via a nitrogen-releasing Diels–Alder reaction. | 93 |
| Tetrazole-alkene cycloaddition | Conversion of a tetrazole into a highly reactive nitrile imine intermediate, then undergoes a rapid 1,3-dipolar cycloaddition with an alkene to form a pyrazoline. | 95 and 96 |
Among the photo-reactive interchain reactions, photo-induced click chemistries, such as thiol–ene,91 thiol-Michael addition,92 and norbornene-based systems,93 offer powerful tools for rapid and precise material synthesis. The radical thiol–ene reaction facilitates fast and oxygen-insensitive network formation through a sequential propagation-chain transfer mechanism, while the thiol-Michael addition utilizes photobase generators to initiate a nucleophilic attack on electron-deficient enes. Meanwhile, norbornene-based systems utilize the high ring strain energy of the norbornene moiety to achieve near-instantaneous reaction kinetics and high conversion while effectively suppressing homopolymerization, thereby promoting ideal crosslinking behavior. In addition, thiol-yne94 and photo-triggered tetrazole-alkene cycloadditions95,96 have been widely used, enabling the formation of photo-triggered cross-linked, uniform networks.
In contrast to constructing a polymer network, photo-induced destruction of the polymer network has also been investigated. Polymer degradation is crucial for sustainability, biomedical technologies, stimuli-responsive materials, and protective coatings. Among various degradation mechanisms, including enzyme,97 thermal,98 and mechanically99 driven pathways, light-driven degradation offers distinct advantages. It enables precise spatiotemporal control, operates under mild conditions, and can be programmed to degrade only upon light exposure.100 Photochemical degradation of polymers occurs through bond scission of photo-cleavable groups or photo-degradable backbones upon irradiation (Fig. 6C). As a result, the polymer network is degraded into lower-molecular-weight fragments such as oligomers or monomers. A representative photo-degradable is the ortho-nitrobenzyl group, which has been extensively studied due to its high photolysis efficiency and minimal side reactions.101–103 Upon irradiation, it forms an aci-nitro intermediate, which subsequently fragments to release the leaving group and generate a nitroso-aryl byproduct. Other examples of photocleavable groups include p-hydroxyphenacyl,104 coumarin,105 and boron-dipyrromethene,106 which are widely utilized in applications such as polymer degradation and controlled drug release.
Light-driven reactions can also cleave the polymer main chain, enabling depolymerization into monomers or self-immolative disassembly. Photo-depolymerization of polymers can be initiated at multiple sites along the polymer backbone upon irradiation.107–109 For example, poly(olefin sulfone) depolymerizes into olefin monomers and SO2 through backbone scission at numerous positions when exposed to light and heat.110,111 In contrast, self-immolative polymers are end-capped with photo-responsive degradation-triggering groups; upon photoactivation, cleavage at the chain end initiates domino-like depolymerization that proceeds from head to tail.112,113 These self-immolative polymers include polybenzylcarbamate, polybenzylether, and poly(benzyl carbonate).
Not only can polymer networks be constructed or degraded, but they can also be engineered to undergo reversible transformations through wavelength-dependent reactions of molecular photo-switches (Fig. 6D). A photo-switch is a molecule that reversibly switches between two or more distinct chemical states upon exposure to specific wavelengths of light, enabling controlled modulation of its physical or chemical properties. Among photo-switchable molecules, those capable of undergoing reversible shape transformations such as azobenzene,114 hydrazone,115 diarylethene,116 spiropyran,117 and stilbene118 can transition between two or more distinct states in response to light. Azobenzene, hydrazone, and stilbene undergo reversible trans–cis isomerization, whereas diarylethene and spiropyran exhibit photocyclization between open and closed forms. When incorporated into polymer networks, reversible photoswitching events can induce large-scale structural and mechanical changes in the material.119–122 These photoswitchable systems have enabled a wide range of advanced applications, including polymer assembly,123–125 photoprogramming,126 actuation,127 and light-controlled adhesion.122,128
Other photo-switchable molecules, photo-dimerizable molecules such as anthracene,129 coumarin,130 and cinnamate,131 are capable of forming dimers through light-induced bond formation and subsequently reverting to monomeric states via photocleavage. These photo-dimerizable molecules can be incorporated into polymer architectures to impart reversible polymer crosslinking behavior. For example, anthracene undergoes reversible photodimerization upon exposure to 364 nm light, which induces dimer formation, whereas irradiation at 254 nm or application of heat cleaves the dimer back into its monomeric state. When anthracene moieties are incorporated into polymer chains, the material remains in a monomeric, non-crosslinked state under one wavelength, but forms dimers, and thus interchain linkages, under another, enabling wavelength-dependent modulation of the network topology and dynamic mechanical behavior.132 These concepts have been demonstrated in various systems, such as phase-patterned materials,133 self-healable polymer networks,134,135 shape-memory polymers,136 and recyclable polymers.137
2.4. Localized electromagnetic field
Polymer processing through laser direct writing has a constraint in implementing sub-micron patterning due to the diffraction limit.138 While this limitation can be somewhat overcome by using high NA lenses139 or an immersion setup,140 the requirements for precise optics and alignment, as well as the narrow process window, make it less accessible. One approach to address this resolution limit is to use the electromagnetic wave nature of lasers to create a periodic light intensity distribution at the sub-micron scale. When two or more coherent beams intersect, an interference pattern is formed on the sample surface, with bright and dark regions repeating according to their phase relationship.141 This interference field acts like a virtual “optical mask”, allowing for the selective delivery of photon energy to the polymer on a nanoscale, which is called laser interference lithography (LIL) (Fig. 7A).24,142,143 | ||
| Fig. 7 Electromagnetic nature of the laser beam. (A) Schematic of laser interference lithography (LIL). By imposing two or more coherent beams, an interference pattern is formed on the sample surface, giving periodic light intensity distribution at the sub-micron scale. (B) Schematic of laser-induced periodic surface structure (LIPSS) process. By using a short-pulse laser, a single laser beam also can make an interference pattern with scattered surface plasmons, enabling nano-structuring on polymer surfaces. | ||
To implement LIL, a single laser beam is divided using a beam splitter, and each beam is then guided by reflectors onto the sample. In this 2-beam optics configuration, the optical setup is simple, and the pattern period can be easily adjusted by changing the angle of the rotating stage.24 When patterning photo-patternable polymers using LIL, the pattern period is determined by p = λ/2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ, where λ is the wavelength of the laser and θ is half of the angle at which the two beams intersect. LIL can even be applied to composite hydrogels with metallic nanoparticles to fabricate Bragg stack structures in approximately 10 min.144 This process employs a CW 532 nm laser in Denisyuk reflection mode to record a latent image within the hydrogel film. The Bragg stack consists of a photosensitive AgBr nanocrystal multilayer structure that is embedded within a p(AM-co-PEGDA) hydrogel film. The lattice spacing of the multilayer structure formed within the hydrogel matrix through laser interference is measured at approximately 200 nm. A lateral interference pattern with a periodicity of approximately 1.5 µm is also observed due to internal reflection.
sinθ, where λ is the wavelength of the laser and θ is half of the angle at which the two beams intersect. LIL can even be applied to composite hydrogels with metallic nanoparticles to fabricate Bragg stack structures in approximately 10 min.144 This process employs a CW 532 nm laser in Denisyuk reflection mode to record a latent image within the hydrogel film. The Bragg stack consists of a photosensitive AgBr nanocrystal multilayer structure that is embedded within a p(AM-co-PEGDA) hydrogel film. The lattice spacing of the multilayer structure formed within the hydrogel matrix through laser interference is measured at approximately 200 nm. A lateral interference pattern with a periodicity of approximately 1.5 µm is also observed due to internal reflection.
By using a four-beam setup where four ±1 order beams stemming from a diffraction grating on a phase mask are superimposed on the sample plane, much higher contrast dot or pillar structures can be fabricated.145 In this case, the spatial intensity profile drops close to zero between peaks, allowing for the formation of nanostructures in the form of disks or pillars arranged periodically in a photo-polymer. For example, when superimposed on gold nanoparticles (AuNP), hybrid plasmonic-hydrogel structures can be created.146 Utilizing poly(N-isopropylacrylamide)-based hydrogels that permit photo-crosslinking, selective crosslinking occurs only in the areas of maximum intensity within the interference field. Subsequently, non-exposed areas can be removed through development or cleaning processes, resulting in hydrogel patterns with feature sizes in the hundreds of nanometers.
Photothermal energy by LIL can also allow for the ablation of the polymers, thereby facilitating the formation of nanostructures in the polymer.147 Two UV pulse laser beams are interfered to deliver pulse shots onto the sample of the light-emitting conjugated polymer poly[(9,9-dioctylfluorenyl-2,7-diyl)-alt-co-(1,4-benzo-{2,1′,3}-thiadiazole)]. The bright fringes of the interference pattern lead to the ablation of the polymer, while the areas corresponding to the dark fringes retain the polymer, enabling the creation of a grating structure. The resulting patterns have a periodicity of approximately 350 nm, where laser ablation with line-by-line scanning is difficult to achieve.
Nano-structuring using laser interference can also be achieved with a single beam when employing ultrashort pulse lasers ranging from nanosecond to femtosecond lasers. When the incident laser beam interferes with surface plasmon polaritons scattered at the surface of a material, it can induce a periodic light intensity distribution, enabling nano-structuring in polymers, which is called laser-induced periodic surface structures (LIPSS) (Fig. 7B).148–150 The period of nanostructures is determined by laser wavelength and applied number of pulses. There are two main types of LIPSS. Low spatial frequency LIPSS (LSFL), which has a structural period similar to that of the laser wavelength, can be formed when the laser is incident vertically in materials with high light absorption. In the case of polymers, LSFL can be induced by repeatedly irradiating laser pulses at fluences significantly lower than the ablation threshold. This makes periodic nanostructures aligned parallel to the polarization direction of the laser.148 High spatial frequency LIPSS (HSFL) can fabricate smaller structures than LSFL, with some studies showing period dimensions as small as one-third of the laser wavelength.150 These are normally observed with femtosecond lasers, at fluences below the ablation threshold.
LIPSS can be applied to various polymers. For example, when a nanosecond UV laser is applied to PEDOT:PSS, a surface with a periodicity similar to 240 nm can be achieved.151 The irradiated PEDOT:PSS also undergoes molecular structural changes, and Raman spectroscopy reveals a shift in the peak at 1450 to 1441 cm−1, indicating that restructuring is possible. This nanostructured PEDOT:PSS could improve the signal quality when applied to sensors or unique cell differentiation platforms for biological applications. Similarly, the use of interference-based patterning techniques allows for the nanoscale structuring of polymers while maintaining the advantages of laser processing in terms of maskless fabrication, fast processing time and simple optical system.
Some polymers possess molecular structures that can directly respond to the oscillating electric field of a laser beam. In particular, polymers with permanent polarization in their main chain28 or side chain152 can be rearranged to the direction of an external electric field when a linearly polarized laser is applied. However, simply exposing the polymer to a laser is not sufficient for chain deformation. Enough photothermal energy is required for sufficient chain mobility to rotate and rearrange polarization followed by laser polarization. If the polymer does not absorb enough photothermal energy from the laser, it remains in a cold state below the Tg, which is not responsive to an electric field even with a high laser power. Therefore, selecting a suitable laser wavelength and absorber is important to induce a synergistic effect from the electric field and photothermal energy (Fig. 8A). Incorporating plasmonic nanoparticles, such as AuNPs, into the surface or interior of the polymer is a good example.
 | ||
| Fig. 8 Interaction of polarization in polymer chains with the laser-driven electric field. (A) Alignment and ordering of polymer chains depending on laser parameters. Sufficient photothermal energy is required for chain mobility to interact with external electric field provided by the laser beam. (B) Laser scan speed is a crucial parameter to induce chain alignment. With a slow writing speed, thermal fluctuation overwhelms the alignment effects of the electric field. Even if temporary alignment occurs, thermal rearrangement soon randomizes the polarization. | ||
The scan rate serves as a crucial process parameter that controls these dynamics. If the scan speed is too fast, sufficient cumulative heat cannot be generated, resulting in a slight increase in chain mobility. If the scan rate is too slow, thermal fluctuation overwhelms the alignment effects of the electric field. In such cases, even if temporary alignment occurs, thermal rearrangement soon randomizes the polarization direction, leading back to a disordered state.28 With the same electric field amplitude (i.e., same laser power), the scan rate can greatly change the polarization states by controlling the mixed condition of electric field and heat (Fig. 8B).
3. Polymeric structure change by laser-induced thermodynamics
Unlike bulk processing, which relies on incremental and slow heat transfer from the surface, a laser induces unconventional thermodynamic environments with extreme temperature gradients and fast cooling to room temperature. This induces previously unexplored structural changes in polymers in multiple length scales such as phase separation, chain ordering and pyrolysis. In this section, we discuss the multi-scale structural changes in various functional polymers by determining the key parameters that enable their active interaction with a laser.3.1. Nano-/micro-structural changes
During the formation of polymer networks, differences in miscibility between the polymer components often drive the aggregation of interactionally favorable species. This aggregation behavior of polymers leads to phase separation with distinct compositions. When the phase separation becomes large and spatially controlled, macroscopic phase patterning can be achieved on length scales ranging from tens of nanometers to several micrometers. These macroscopic polymer phase patterns can be directed through thermodynamically governed strategies. Therefore, laser-based photothermal heating can drive polymer phase separation with both rapid fabrication and precise spatial control. In laser-induced phase patterning, numerous parameters including the type of polymer, laser intensity and wavelength, beam geometry, and annealing temperature govern the resulting pattern formation. We aim to discuss the mechanism underlying laser-induced polymer patterning, explore how key parameters can be tuned to engineer diverse patterned polymer systems, and examine the resulting structural, mechanical, and functional properties.Block copolymers (BCPs) are polymers consisting of two or more chemically distinct polymer blocks covalently linked to a single chain. BCPs self-assemble into distinct phases because of their thermodynamically immiscible blocks, ultimately yielding ordered nanostructures. However, the structures generated solely by the spontaneous phase separation of block copolymers typically remain confined to short-range order. To induce larger-scale phase separation and achieve macroscopic ordering, additional processing such as thermal annealing above the Tg of the BCP is required. Laser zone annealing (LZA) uses a high-power laser that is tightly focused in one direction and broadened in the perpendicular direction to form a laser line, which is then scanned across the film.42 It enables rapid, large processing areas and produces significantly faster and more effective ordering than conventional bulk annealing.
In particular, soft-shear laser zone annealing (SS-LZA) combines LZA with a soft-shear mechanism to achieve rapid, large-area alignment (Fig. 9A).153,154 In this process, a focused laser beam is scanned across the substrate to generate a moving “hot zone,” enabling localized heating and subsequent rapid cooling. When a thick crosslinked PDMS pad is placed on top of the BCP thin film, the thermal expansion mismatch between the PDMS pad and the underlying silicon substrate induces planar shear within the BCP thin film. This soft shear aligns lamellar microdomains unidirectionally along the laser-scanning direction, producing uniform cm2-scale alignment within only seconds to minutes. SS-LZA is effective across various BCP systems as long as the timescale of photothermal shear surpasses the intrinsic morphological relaxation time of the polymer. Therefore, compared with other BCP alignment techniques such as directed self-assembly and solvent vapor annealing, SS-LZA offers a significantly simpler and faster processing route.155
 | ||
| Fig. 9 Laser-induced nano-/micro-structural changes in polymers. (A) Schematic of soft-shear laser zone annealing (SS-LZA) and the resulting platinum nanowires on Si substrates templated by the ordered structure of PS-b-P2VP. Optical microscopy images show the effects of annealing temperature and the number of SS-LZA cycles on PS-b-P2VP ordering. Insets display the corresponding FFT patterns with calculated orientational order parameters (η). Reproduced with permission.153 Copyright 2020, the American Chemical Society under CC BY 4.0 license (https://creativecommons.org/licenses/by/4.0/). (B) Scanning electron microscopy (SEM) image of platinum nano-meshes fabricated via SS-LZA. Platinum lattices with triangular, rectangular, oblique, and mixed composition. Reproduced with permission.156 Copyright 2015 Springer Nature under CC BY 4.0 license (https://creativecommons.org/licenses/by/4.0/). (C) AFM images of polymer film irradiated with linear polarized light and circular polarized light (top). AFM images of the PET film irradiated with linear, elliptical, and circular polarized light (bottom). Reproduced with permission.158 Copyright 2015, Elsevier. (D) Intensity distributions of optical vortex beans with different topological charges (left) and the resulting microstructures formed in azopolymer films, characterized by scanning probe microscopy (right). Reproduced with permission.168 Copyright 2023, Multidisciplinary Digital Publishing Institute under CC BY 4.0 license (https://creativecommons.org/licenses/by/4.0/). (E) Schematic of laser-induced phase separation and adhesion of PEDOT:PSS via localized photothermal heating. Reproduced with permission.169 (F) Schematic of multipoint frontal polymerization and three-dimensional scan of a polymerized sample showing two seam convergences, along with the corresponding height map. Reproduced with permission.171 | ||
When the phase of BCPs is patterned using SS-LZA, the resulting structures can serve as templates for fabricating metal nanowire patterns. For example, in SS-LZA-patterned polystyrene-b-poly(2-vinyl pyridine) (PS-b-P2VP), the metal salt selectively loads into the P2VP domains (Fig. 9B).156 Subsequent removal of the BCP and reduction of the metal ions yield nanowire arrays that replicate the original polymer morphology. By repeating this stack-by-stack process, a metal nanomesh can be constructed. Thus, SS-LZA serves as a powerful technique that enables the self-assembly of metals with well-defined periodicity and facilitates rapid large-area alignment of ordered nanostructures.
In the SS-LZA method, the photothermal effect of a laser is not applied directly to the polymer. Instead, the laser heats the underlying germanium or silicon substrate, and this metal/inorganic layer subsequently transfers heat to the BCP film positioned above it. Therefore, to effectively induce the photothermal response of the substrate, laser sources operating in the vis/NIR wavelength range are typically employed. In contrast, when the polymer directly absorbs laser photons, it generates periodic surface patterns that correspond to the laser wavelength, by LIPSS (Fig. 9C).157–159 Polymer LIPSS are formed when a short-pulse (nanosecond to femtosecond) UV laser is irradiated onto a polymer surface. The incident light undergoes scattering and absorption within the polymer, and the interference between these optical fields leads to a spatially periodic energy distribution. As a result, nanoscale periodic surface structures are formed on the polymer through localized photothermal modulation. LIPSS can be generated on a wide range of polymers, including poly(trimethylene terephthalate),160,161 polystyrene,162,163 poly(ethylene terephthalate) (PET),164 and poly(3-hexylthiophene).165,166 The formation of LIPSS is strongly influenced by factors such as the intrinsic light absorption characteristics of the polymer, the degree of UV irradiation, and the incident angle of the laser beam. In addition, the polarization state of the laser significantly affects the resulting morphology, where linearly polarized lasers produce linearly aligned periodic structures, whereas circularly polarized lasers generate circular nanodot patterns (Fig. 9C).158
Beyond circular polarization, tuning the orbital angular momentum of light, characterized by its topological charge, generates multi-spiral interference fringes associated with optical vortices (Fig. 9D).167,168 In this context, azo-containing polymers, which are highly sensitive to light polarization, can be patterned into spiral surface structures when irradiated with optical vortex beams. A recent study has demonstrated that combining a linearly polarized Gaussian beam with an optical vortex beam of adjustable topological charge enables the fabrication of multi-spiral patterns.168 By varying the topological charge of the vortex beam, the number of spirals formed in the azopolymer surface can be tuned, yielding a diverse array of chiral patterned polymer structures. Nanoscale polymer patterns fabricated by LIPSS have enabled broad technological applications, ranging from organic field-effect transistors and organic solar cells to various bio-related platforms.
Laser-induced phase separation can also be applied to conducting polymers such as PEDOT:PSS to form conductive pathways (Fig. 9E).169,170 PEDOT:PSS consists of conductive PEDOT-rich domains mixed with an excess of insulating PSS-rich domains, which act as a counter anion and water-stabilizing polymer, respectively. Therefore, to promote long-range PEDOT–PEDOT connectivity and achieve high conductivity, various strategies such as additive incorporation and thermal annealing are employed to remove excess PSS. Laser-induced phase separation offers a rapid and straightforward approach to facilitate PEDOT–PEDOT connectivity. When a PEDOT:PSS film is irradiated by a laser beam, only the irradiated region undergoes photothermal-driven annealing that activates the aggregation of PEDOT-rich domain. As a result, laser-induced patterning of conductive PEDOT networks becomes feasible, enabling broad applications in biomedical devices and other advanced soft electronic technologies. This will be discussed in more detail in Section 3, Electrical properties.
Macroscopic polymer patterning can be achieved not only in the polymer state but also directly in monomer solutions by controlling light-induced polymerization. Frontal polymerization refers to a polymerization process in which the reaction is initiated at a localized region and subsequently propagates outward through the surrounding medium. When a monomer solution containing photothermally active carbon black is irradiated with a laser, thermally activated polymerization is initiated locally and progresses outward (Fig. 9F).171 By applying multiple laser spots, multiple initiation sites are generated simultaneously, and the propagating reaction fronts converge to form seam-like boundaries between them. This strategy enables large-area polymer patterning on a macroscopic scale.
3.2. Molecular structure change
Laser irradiation also modifies polymers at the molecular level through photochemical reactions, thermally driven mobility changes, and field-assisted conformational transitions.49,52 Unlike bulk thermal processing, which creates thermodynamic equilibrium states, the ultrafast energy injection and rapid cooling rates inherent to laser processing facilitate access to non-equilibrium dynamics. This capability allows for the trapping of metastable molecular orientations and the precise modulation of reaction kinetics.67,172 Because these processes occur within confined volumes, they adjust bonding structure, chain orientation, and phase composition with spatiotemporal precision that conventional thermal or chemical treatments cannot achieve. By selectively overcoming activation barriers for specific covalent bond remodeling or non-covalent chain realignment, these molecular-scale mechanisms govern how laser processing imparts localized mechanical, chemical, and electronic functionality without compromising the structural integrity of the surrounding matrix.173Photochemical activation provides a direct means to alter network structure. In photo-responsive hydrogels, patterned irradiation drives functional group ligation, additional crosslink formation, or selective cleavage of photocaged linkages (Fig. 10A).137 These wavelength-specific pathways allow controlled modification of local crosslink density and functionality (Fig. 10B), while thiol–ene and norbornene-based chemistries demonstrate how monomer architecture and photoreaction kinetics dictate the resulting network configuration (Fig. 10C). These systems show that photochemically driven bond exchange and cleavage can establish molecular gradients without affecting adjacent regions.
 | ||
| Fig. 10 Mechanisms of laser-induced molecular structure modification. (A) Schematic of photochemical reaction pathways including ligation, crosslinking, and cleavage in photo-responsive hydrogels. (B) Spatiotemporal modulation of crosslinking density and functionality via wavelength-selective irradiation. (C) Dependence of network configuration and reaction kinetics on monomer architectures such as thiol–ene and norbornene systems. Reproduced with permission.137 Copyright 2025, the American Association for the Advancement of Science. (D) Activation of laser-induced dynamic crosslinking (LIDC), where photothermal heating above the nematic–isotropic transition initiates radical-mediated bond exchange in allyl sulfide networks. (E) Transformation from a kinetically trapped isotropic state (C-Iso) to a thermodynamically stable polydomain phase (R-Poly) via RAFT-mediated structural reconfiguration and stress relaxation. (F) Temporal evolution of the nematic order parameter, highlighting the role of dynamic bond exchange in recovering and amplifying mesogenic alignment. Reproduced with permission.174 Copyright 2024, Springer Nature. (G) Synergistic mechanism of electric-field alignment and photothermal mobility enhancing conformational transitions in ferroelectric polymers. (H) Molecular dynamics simulations illustrating the competition between field-driven polar alignment and thermally induced relaxation. (I) Phase diagram and spectroscopic evidence of reversible transformations. Reproduced with permission.28 Copyright 2022, Wiley-VCH. | ||
Laser-induced photothermal effects also contribute to molecular modification. Localized heating increases segmental mobility or promotes thermally activated bond formation, as shown in PDMS–Ag flake composites where photothermal input accelerates crosslinking and induces local network reconstruction.172 This demonstrates that molecular control can arise from thermal pathways alone, independent of photochemical activation. When photothermal and photochemical inputs coexist, they provide a broader design space for directing molecular transformations.
This combined mechanism is central to liquid crystal elastomers.174 Laser-induced heating promotes mesogen and backbone reorientation, with the extent of structural change governed by the depth of the thermal transition (Fig. 10D). When dynamic covalent bonds such as reversible addition–fragmentation chain transfer (RAFT)-mediated exchange are active, these reorganized states become chemically stabilized through bond exchange, yielding networks with significantly higher orientational order (Fig. 10E). Time-dependent measurements further show that systems containing dynamic bonds maintain and amplify alignment even after the temperature field dissipates (Fig. 10F). In this class of materials, photothermal mobility and photochemical exchange operate jointly to produce stable yet reprogrammable molecular architectures.
Laser irradiation can also modify polymers through electric-field–assisted pathways coupled with photothermal effects. In ferroelectric polymers such as poly(vinylidene fluoride) (PVDF), strong local fields guide chains toward electrostatically favored trans conformations, while photothermal input increases their mobility to facilitate these conformational transitions (Fig. 10G).28 Molecular dynamic (MD) simulations indicate the rapid formation of polar conformers under field-dominated conditions, in contrast with the thermally weighted relaxation into gauche states during slower scans (Fig. 10H). Spectroscopic analysis confirm reversible shifts among α-, β-, and γ-phases depending on the balance of electric-field strength and thermal input, enabling spatial control over the polarization and piezoelectric response (Fig. 10I). This hybrid field- and heat-driven mechanism enables precise tuning of the dipole orientation and crystalline phase distribution.
Collectively, photochemical modification, photothermal activation, and field-assisted conformational control define how laser irradiation governs molecular architecture in polymers. By adjusting covalent structures, stabilizing aligned domains, or directing dipole orientations, laser processing provides a versatile platform for engineering spatially programmed functionality in soft materials.
3.3. Pyrolysis
Under certain laser parameters, the polymer structure can no longer be maintained, and pyrolysis begins to occur. This pyrolysis has generally been regarded as undesirable, but recent research has applied it to give electrical conductivity to insulating polymers. By using the polymer as a matrix, glassy carbon or graphene can be selectively fabricated, enabling monolithic electrode patterning within substrates. Conventional patterning of conductive carbon materials such as graphene is performed via chemical vapor deposition, which requires high temperatures and vacuum chambers for the production of electronics.175 However, by simply scanning lasers, the synthesis and patterning of graphene can be easily achieved at room temperature and atmospheric pressure, which is called laser-induced graphene (LIG).38,176,177LIG was discovered by irradiating a laser on PI films (Fig. 11A).178 The polymer acts as a precursor containing carbon, and under photothermal energy from the laser, the local temperature rises above 2500 °C. This breaks the chemical bonds within the polymer (e.g., C–O, CO, and N–C bonds), and the carbon atoms rearrange to form a graphene structure. Due to the rapid cooling rate after laser irradiation, there is insufficient time for the carbon atoms to settle into the preferred hexagonal honeycomb structure. Consequently, LIG remains as polycrystalline structures containing many defects, such as pentagon and heptagon ring pairs (Fig. 11B).178 Therefore, LIG consists of a heterogeneous mixture of graphitic domains, amorphous carbon, and residual heteroatoms originating from the precursor polymer. These heteroatoms and disordered phases play a critical role in defining the resulting material properties. For instance, residual oxygen and nitrogen can influence the carbonization process by modifying local bonding configurations and lowering the effective graphitization threshold. At the same time, the presence of amorphous carbon contributes to an increase in defect density, which impacts both electrical conductivity and thermal transport. As a result, LIG formation should be understood as a nonequilibrium carbonization process, where the final structure reflects a balance among graphitic ordering, defect generation, and incomplete decomposition of the precursor polymer.
 | ||
| Fig. 11 Laser-induced pyrolysis of polymers. (A) Schematic of the laser-induced graphene (LIG) process. (B) TEM image of LIG, which contains defects (e.g., heptagon and pentagon). (C) SEM image of porous LIG on the PI film. (D) Raman spectrum of LIG film showing clear 2D peaks. Reproduced with permission.178 Copyright 2014, Springer Nature. (E) Schematic of laser-induced successive pyrolysis of PDMS. (F) Photographs of the fabricated SiC from PDMS. (G) Schematic of the mechanism of successive pyrolysis of PDMS transforming it into SiC even at temperatures lower than those in a furnace. Reproduced with permission.183 Copyright 2020, Springer Nature. (H) Schematic of the laser-induced pyrolysis process depending on laser parameters. A high laser intensity transforms PDMS into an SiO2-rich surface, whereas a low laser intensity transforms it into carbon-rich surfaces. (I) SEM images of processed regions under each laser condition. Both images show highly porous microstructures due to the released gas during the process. (J) Photograph of the Janus film using a single PDMS slab and a single process by irradiating each side of the film with low-intensity and high-intensity lasers, respectively. Reproduced with permission.184 Copyright 2025, Elsevier. | ||
LIG acquires 3D porous structures. The high temperatures under laser exposure cause the non-carbon atoms (e.g., O and N) in polymers to recombine and be released as gases such as carbon monoxide (CO), carbon dioxide (CO2), and nitric oxide (NO). During release of these gases, LIG forms highly porous 3D structures, enabling high surface to volume ratio and permeability (Fig. 11C). The formation of graphene can also be verified by the D band (1350 cm−1), G band (1580 cm−1) and 2D (2700 cm−1) band in Raman spectroscopy (Fig. 11D). As the laser fluence increases, graphene begins to form, and the emergence of the 2D band with the G band indicates the creation of graphene. The structural quality of LIG can be evaluated through the relative intensity of these bands. LIG tends to have a highly defective, amorphous structure, resulting in a higher intensity ratio of the D band compared to typical high-quality monolayer graphene. A lower ID/IG ratio indicates a lower density of defects and a larger crystallite size. The I2D/IG ratio is associated with the structural quality and the number of layers in graphene. Generally, a higher I2D/IG ratio suggests that the graphene structure is the fewest-layered, indicating the least number of stacked layers.
Laser parameters play a crucial role in the conversion of polymers into LIG. To form LIG, photothermal energy exceeding a certain threshold must be supplied to induce the pyrolysis of the polymer structure and the recombination of carbon. For example, a fluence of over 5.5 J cm−2 is required to form LIG on a PI film.179 A variety of lasers have been used to generate LIG. A CO2 laser with a wavelength of 10.6 µm and a 405 nm CW laser can effectively induce photothermal energy to fabricate LIG. For polymers that are hard to break their chemical bonds, UV lasers, with shorter wavelengths and higher energies, have the advantage of being able to induce both photochemical and photothermal reactions, providing energies greater than the bond dissociation energy needed to break the chemical bonds within these materials.
For materials that lack aromatic structures, making it difficult to obtain LIG with a single irradiation (e.g., wood and coconut), a mechanism for generating LIG through multiple laser irradiations has been reported.180,181 The initial few laser pulses primarily serve to ablate the precursor material into amorphous carbon. This amorphous carbon strongly absorbs the infrared wavelength of CO2 lasers. Subsequent laser irradiations act through heat, converting the adequately heated amorphous carbon back into LIG. This mechanism can be implemented by inducing multiple exposures of laser light to the same area, either through multiple laser pulses or by defocusing the laser beam.
Polymers containing Si can yield SiC as a byproduct during pyrolysis. For example, in transparent PDMS, non-linear light absorption can be induced using a femtosecond laser to cut and recrystallize the chemical bonds of PDMS, resulting in the formation of SiC. The high repetition rate of the femtosecond laser such as 63 MHz induces heat accumulation on the PDMS surface, which can increase the temperature to that necessary for the transformation into SiC.182 Following laser pulse irradiation, Fourier transform infrared spectroscopy analysis (FTIR) revealed the disappearance of the sharp peaks corresponding to the characteristic bonds of PDMS, specifically C–H (2950 cm−1 and 2900 cm−1) and CH2 bonds (1400 cm−1). The loss of these peaks indicates that the laser pulse induced the cleavage of these chemical bonds, suggesting that carbon and hydrogen atoms were released as gases, such as hydrocarbon gases and CO2.
Importantly, the typical peaks of carbon materials (D band and G band) were not observed during this process, suggesting that the formation of the carbon material itself can be considered negligible. In contrast, the peak corresponding to the Si–O bond (1080 cm−1) remained or was observed to be broad after irradiation, indicating the possibility of silicon oxide materials, such as SiO2 or SiO, being formed. X-ray diffraction (XRD) analysis confirmed that the laser-fabricated black region is composed of β−SiC. Specifically, diffraction peaks observed at around 2θ values of 36°, 60°, and 72° correspond to the (111), (220), and (311) planes of crystalline β−SiC, respectively, proving that pure PDMS was directly transformed into SiC through femtosecond laser irradiation. The formed SiC exhibited electrical conductivity, which was confirmed through its linear current–voltage (I–V) curve that increased with applied voltage. Under optimal conditions, the average resistivity was measured to be about 5.3 Ω cm, which is approximately 40-times higher than that of bulk SiC (0.13 Ω cm). This low electrical conductivity is attributed to the weak interconnection of SiC, as well as the potential formation of secondary products.
The formation of SiC from PDMS is also influenced by the scanning rate of the femtosecond laser. A slow scanning speed, such as 0.5 mm s−1, can lead to excessive overlap of the pulses, which may result in the formation of cracks. These cracks can then transform the already formed β−SiC into amorphous SiC or other materials, ultimately decreasing the conductivity. Although β−SiC formation is possible at faster scanning speeds, such as 5 mm s−1, insufficient melting occurs between the formed structures, weakening the electrical connectivity. Therefore, it is crucial to find an appropriate scanning rate that allows for the synergy of electrical and mechanical properties.
Recent studies have reported the fabrication of SiC from PDMS through pyrolysis using CW 532 nm lasers (Fig. 11E). By marking a starting point on the transparent PDMS with black ink to enable laser absorption, when the CW laser beam passes through this starting point, the laser-induced photothermal energy initiates the pyrolysis of PDMS, leading to the formation of opaque β−SiC.183 The newly formed opaque SiC absorbs laser energy significantly more than transparent PDMS, enabling successive photothermal reactions. The laser beam irradiating SiC generates thermal energy, which is then conducted to the surrounding area, creating a localized heated zone (LHZ). This enhanced thermal energy induces pyrolysis in the PDMS area that is in close proximity, within a radius of a few micrometers, to the laser-irradiated spot, creating a conduction pyrolysis area (CPA). As the laser moves forward into the CPA, the photothermal center (the new irradiation point) created by the laser absorption transforms the CPA into a new pyrolytic center, simultaneously forming new LHZ and CPA around it, thus completing a successive cyclic sequence (Fig. 11F). The continuous progression of this repetitive photothermal pyrolysis not only facilitates the formation of SiC but also realizes a non-ablative fabrication method for transparent PDMS.
Looking more closely at the mechanism for SiC formation via laser-induced pyrolysis, traditional furnace heating methods require high temperatures above 1400 °C in an inert atmosphere to produce SiC. However, in the laser process, SiC formation has been observed at much lower temperatures, typically below 700 °C but with an extremely fast temperature rise of about 5000 °C s−1. At slow heating rates, a molecular mechanism predominates, leading to the cleavage of Si–O bonds and the formation of thermally stable cyclic oligomers. In contrast, rapid heating activates a radical mechanism before the molecular mechanism can be fully completed. Once the radical mechanism dominates and the radical pathway for SiOx formation begins, the generation of radicals induces additional cross-linking, which reduces chain flexibility and suppresses the molecular mechanism. As a result, homolytic scission of the Si–CH3 bond becomes predominant over cleavage of the Si–O bonds. As a result, rapid heating induces the condensation of the backbone chains, leading to the formation of SiOx at lower temperature ranges. In the final stage of SiC formation, this SiOx is reduced by the pyrocarbon generated during the pyrolysis process, producing SiC solid material and carbon monoxide (Fig. 11G). The formation of a thin layer of SiO2 on the surface of the laser scanning area is considered evidence supporting this SiC induction mechanism. Once the SiC created in the PDMS is removed, it can yield complex structures within the PDMS, which can then be applied to microfluidic channels and other applications.
Using this successive laser pyrolysis technique, research has shown that two distinct materials with different optical properties can be fabricated as byproducts from PDMS, depending on the conditions of laser irradiation (Fig. 11H).184 In the case of low energy density, the laser induces pyrolysis of PDMS to relatively shallow depths. After the bonds are cleaved in the initial stages of siloxane pyrolysis, the remaining carbon components become concentrated on the surface, resulting in the formation of a carbon-rich material. During the laser process, the release of gas molecules leads to the formation of a porous structure, with low energy allowing the reaction to proceed only to shallow depths. The surface, which is rich in carbon and possesses a hierarchical porous structure, effectively captures and absorbs incident solar light, enhancing the photothermal effect and exhibiting low reflectivity (Fig. 11I). Additionally, the porous layer induces an anti-reflective effect through the gradient refractive index effect, further increasing the absorption rate.
A high energy density facilitates pyrolysis to greater depths and accelerates the reaction of the residual Si with oxygen supplied from the surrounding air following the cleavage of Si–CH3 bonds, predominantly leading to the formation of SiO2 on the surface. FTIR and XRD analyses also show that the Si–O peaks and amorphous SiO2 peaks are more prominent under high energy density conditions compared to low energy density conditions. The pyrolysis byproduct produced in this manner can exhibit a hierarchical porous structure in the form of nanobeads, depending on the depth (Fig. 11I). This functions as a reflection-dominant material, strongly reflecting sunlight and being used for radiative cooling performance. The authors facilitated the easy fabrication of a Janus structure using a single material and a single process by irradiating each side of the film with low-intensity and high-intensity lasers, respectively (Fig. 11J). This allows the device to provide both cooling and heating functions through a simple operation mechanism of flipping it over.
4. Property modulation
Polymers undergo changes in their physical properties based on their structural modification occurring at various length scales, which are determined by the stages of polymer synthesis and processing. In this section, we aim to focus on how the structural changes in polymers induced by laser affect their three main physical properties (electrical, mechanical, and optical properties). Through the precise adjustment of laser parameters, different properties can be realized within the same polymer. We will explore the interactions between the polymer system and the laser that enable this implementation.4.1. Electrical properties
Most polymers are insulators and have stable chain structures made of covalent bonds, which strongly bind electrons. Their large bandgap makes it very difficult to excite these electrons. Nevertheless, synthesis methods and materials engineering have been developed to achieve electrical conductivity while maintaining the mechanical advantages of polymers. In this section, we introduce methodologies to enable charge transfer in polymers and discuss how to control or enhance it using lasers (Fig. 12A–C). | ||
| Fig. 12 Strategies to control electrical properties of polymers. (A) Designing bi-phasic composites containing a soft polymer as the mechanical phase and electrically conductive fillers as the electrical phase. (B) Directional charge transfer of intrinsically conductive conjugated polymers by controlling their molecular orientation. (C) Healing defects and increasing the crystal size in an amorphous or polycrystalline LIG film. | ||
The common approach to achieve electrical conductivity is to design composites by filling conductive fillers (e.g., metallic nanomaterials and carbon nanotubes) in insulating polymer chains.185 Consequently, the polymer backbones serve as a mechanical phase that alleviates mechanical stress, while the electrical fillers provide a stable pathway for charge flow as the electrical phase. The electrical properties of these bi-phase composite materials are determined by the percolation networks of the electrical phases, and an efficient network design can yield high electrical conductivity (Fig. 12A).186
Research has reported scanning laser on a composite of PDMS and Ag flakes to achieve high electrical conductivity while simultaneously patterning them on substrates.172 When infrared nanosecond laser pulses are irradiated onto the coated Ag flake-PDMS composite, the laser can penetrate through the PDMS, while the Ag flakes absorb the laser energy, generating photothermal energy that allows for the selective curing of the composite. As the laser beam scans the composite, rapid heating and cooling are repeated within the heat affected zone, resulting in the formation of smaller Ag flake-PDMS clusters (5–15 µm) compared to bulk annealing. This relatively intricate connectivity between small clusters not only leads to high electrical conductivity that is three-times higher than that achieved through bulk annealing, but also mitigates the destruction of the percolation networks during stretching.
Conjugated polymers consist of alternating single and double bonds, forming a π-conjugated system. The overlap of these conjugated π orbitals creates a one-dimensional energy band structure.3 As a result of this process, π electrons are delocalized across the entire chain rather than being confined to individual bonds, allowing charges to move along the chain. The representative conjugated polymer PEDOT:PSS has a core–shell distribution, where PEDOT-rich domains form the core, surrounded by PSS-rich domains that are constitutively designed for solvent dispersion and p-type doping. When a laser is irradiated onto PEDOT:PSS, the phase separation induced by the laser significantly enhances the connectivity between the PEDOT-rich domains, increasing the electrical conductivity to as high as 670 S cm−1 (Fig. 13A and B).170 For thin layers of PEDOT:PSS, their transparency to 532 nm is quite high, and thus AuNPs are mixed in to enhance laser absorption and maximize the photothermal effect.
 | ||
| Fig. 13 Electrical property modulation of polymers by laser-induced structural changes. (A) Schematic of laser-induced phase separation of PEDOT:PSS. (B) Increase in electrical conductivity of PEDOT:PSS by laser treatment. To enhance light absorption of transparent PEDOT:PSS film, it was mixed with AuNPs. Reproduced with permission.170 Copyright 2022, the American Association for the Advancement of Science. (C) MD simulation of the laser-induced phase separation process. Reproduced with permission.187 Copyright 2024, the American Chemical Society. (D) Laser-induced directional mobility enhancement of P3HT. AFM results showed that 0.020 J cm−2 of pulse laser made P3HT more conductive in the vertical direction without damaging the film. (E) Grazing incidence wide-angle X-ray scattering results depending on laser fluence. Reproduced with permission.152 Copyright 2018, the Royal Society of Chemistry. (F) Schematic of LIG and flash joule heating (FJH) process for healing defects inside film. (G) Increase in conductivity of LIG film after FJH treatment. (H) Raman spectroscopy results after FJH treatment. (I) TEM images showing an increase in the crystallinity of graphene after FJH treatment. Reproduced with permission.188 Copyright 2024, Springer Nature under CC BY 4.0 license (https://creativecommons.org/licenses/by/4.0/). | ||
Research has also explored the mechanism of phase separation in PEDOT:PSS using MD simulation. Due to the difference in light absorption between PEDOT and PSS, when a laser is irradiated onto PEDOT:PSS, the π–π interactions between the PEDOT-rich domains are disrupted, causing the PEDO-rich domains to vibrate unstably in place. At this time, adjacent vibrating PEDOT-rich domains come into contact, leading to the electrostatic attraction between the PEDOT and PSS components being drawn in, resulting in the two particles meeting (Fig. 13C).187 This process leverages the fast heat supply and dissipation characteristics unique to lasers, distinguishing it from bulk annealing, where both PEDOT and PSS are heated uniformly.
Conjugated polymers are generally used as channel layers for various types of organic semiconductor devices such as organic thin film transistors (OTFTs)4 and organic electrochemical transistors (OECTs).3,31 Organic semiconductor devices not only benefit from the flexible mechanical properties of polymers but can also achieve high transconductance when used as OECT channels due to their volumetric interactions with the electrolyte.3 However, due to their inherently low charge mobility, organic transistors have the limitation of slow operation speeds compared to inorganic transistors. Thus, the development of processing methods to enhance their mobility is necessary. Also, the advantage of charge transport directionality varies based on the device configuration. For in-plane devices, charge transport is favorable in the edge-on direction, while for vertical device configurations, it is more advantageous in the out-of-plane direction (Fig. 12B).
Research has reported the use of lasers to enhance the charge mobility of the semiconducting polymer poly(3-hexylthiophene) (P3HT) in a certain direction (Fig. 13D).152 The electrical conductivity of P3HT is determined by the degree of molecular stacking and orientation. When femtosecond laser irradiation is applied to P3HT below the ablation threshold, it has been confirmed that its molecular orientation effectively transforms from an edge-on to a face-on structure. The transformed face-on orientation allows for precise and selective transitions since the femtosecond laser minimizes thermal propagation, ensuring stability during the process.
This molecular orientation transition is attributed to the interaction between the electric field of the laser and the dipole moment of the molecules. The authors confirmed through density functional theory simulations that the dipole moment of the P3HT monomer is 1.06 D and it can react to an electric field in laser non-contact mode without an additional electrode pair. The directional transition was clearly confirmed through grazing incidence wide-angle X-ray scattering (Fig. 13E). By adjusting the angle of X-ray incidence to examine depth information, it was found that the effects of laser irradiation alter not only the film surface but also penetrate deeply into the interior of the film. Laser irradiation also increased the crystallinity, which is critical for the face-on arrangement, suggesting the potential for improved charge mobility in conductive polymer films. The P3HT films irradiated with the optimized fluence exhibited approximately three-times higher vertical conductivity compared to the films that were not irradiated, owing to their vertical stacking in the face-on orientation. These transformed P3HT films are expected to be effective in improving the performance of vertical charge transport devices such as organic photovoltaics, organic light-emitting diodes, and the active research area of vertical OECTs.
In the case of LIG, polymers can be easily pyrolyzed to pattern conductors. However, due to their rapid cooling after the laser beam is removed, there is insufficient time for the carbon atoms to form a hexagonal structure. As a result, the carbon exhibits an amorphous or polycrystalline structure with numerous defects, including five-membered and seven-membered rings. Research has reported improvements in the defects and crystallinity of LIG at the atomic level through flash Joule heating (FJH) treatment (Fig. 13F).188 These enhancements were confirmed by a reduction in defect density, an increase in the crystallite size, and changes in the atomic bonding structure.
FJH is a process that induces rapid and intense resistive heating by applying a high-voltage direct current (DC) pulse through conductive materials. After the LIG pattern is formed, millisecond-scale FJH is applied within a vacuum chamber to heal the topological defects of LIG. During this process, the DC pulse generates heat instantaneously, resulting in a flash of black-body radiation. As the applied voltage increases, the temperature increases from about 1300 °C at 150 V to 2500 °C at 190 V. At temperatures below 1700 °C, the rearrangement of carbon atoms is insufficient. Therefore, by applying a sufficiently high voltage of 190 V, rearrangement of the carbon atoms is promoted, allowing for the healing of structural defects within the LIG. This process occurs while preserving the overall structure and porosity of the LIG.
The resistance of the LIG pattern significantly decreased after FJH treatment, with the resistance of F-LIG processed at 190 V reducing markedly from 590 Ω to 120 Ω, resulting in a fivefold increase in conductivity (Fig. 13G). The ID/IG ratio, which indicates the defect density of the original LIG, was 0.84. In the case of F-LIG-190 V after FJH treatment, this ratio significantly decreased to 0.33, indicating a substantial reduction in the density of structural defects (Fig. 13H). The crystallite size of the original LIG was 22.9 nm, whereas for F-LIG-190 V, it reached approximately 60 nm, representing an increase of about 2.6-times compared to LIG. This indicates that the FJH treatment significantly promotes the healing of structural defects within graphene, leading to the growth and enlargement of crystalline regions. Analysis of the pair distribution function also revealed that the nearest C–C bond distance in the original LIG was concentrated at 1.458 Å, demonstrating a broad discrete range. In contrast, for F-LIG-190 V, this bond distance shifted to a shorter value of 1.425 Å. The reduction in bond distance and the concentrated distribution suggest an enhancement in the hexagonal graphitic structure, which was previously dominated by five-membered and seven-membered rings (Fig. 13I).
4.2. Mechanical properties
By tailoring the optical characteristics of the laser and the composition of light-responsive components, the mechanical behavior of the resulting polymer can be precisely engineered. In this section, we focus on how light-regulated polymerization kinetics dictate network formation and material properties, and we further examine the adhesion behavior arising from polymer interdiffusion driven by laser-induced melting.The mechanical properties of polymers are strongly governed by their molecular architecture, including molecular length and the formation of network structures. Polymers exhibit a critical entanglement molecular weight, above which sufficient chain entanglement can occur to impart robust mechanical strength (Fig. 14A).189,190 When the molecular weight is below this threshold, the polymer chains do not effectively entangle, resulting in a poor mechanical performance. Consequently, the properties of polymers vary significantly depending on their molecular weight. Recent studies have reported that laser-induced polymerization can influence the growth of the polymer molecular weight, thereby altering the resulting mechanical characteristics.
 | ||
| Fig. 14 Strategies to control mechanical properties through laser light-induced processes. (A) Schematic showing the effect of molecular weight on chain entanglement density and the corresponding stress–strain behavior. (B) Comparison between laser and LED light sources and their intensity-dependent conversion behaviour. (C) Schematic of laser-induced polymer interdiffusion via localized polymer melting. | ||
In photo-polymerization, the extent of the light-initiated reaction ultimately governs the degree of polymerization. The conversion efficiency is primarily determined by the irradiation intensity and curing time. At very low light intensities, monomer-to-polymer conversion is inefficient, requiring prolonged exposure (Fig. 14B).191 In contrast, high-intensity irradiation induces rapid polymerization, producing long polymer networks with much short exposure times. Consequently, polymers formed under low-intensity light generally exhibit lower mechanical strength, lower Tg, and more pronounced viscoelastic behavior as a result of their incomplete conversion (Fig. 15A).192,193 However, because lasers provide exceptionally high energy density and rapid reaction kinetics, they can achieve both fast curing and high molecular weights.194,195 Owing to these advantages, laser-based photopolymerization is widely employed in applications such as 3D printing and surface coating.196
 | ||
| Fig. 15 Mechanical property modulation of polymers. (A) Light intensity–dependent modulation of material properties under laser irradiation. Reproduced with permission.192 Copyright 2019, the American Chemical Society under CC BY NC ND 4.0 license (https://creativecommons.org/licenses/by/4.0/). (B) Comparison of the degree of conversion of dental resin composites (Filtek One, PowerFill, and PowerFlow) under LED and laser irradiation. Reproduced with permission.195 Copyright 2020, Multidisciplinary Digital Publishing Institute under CC BY 4.0 license (https://creativecommons.org/licenses/by/4.0/). (C) Effect of pulse frequency on the propagation rate in pulsed-laser polymerization. Reproduced with permission.199 Copyright 2022, the Royal Society of Chemistry. (D) Images of the highly entangled hydrogel and regular hydrogel. (E) Stress–strain curves of the highly entangled hydrogel, hard regular hydrogel, and soft regular hydrogel. Reproduced with permission.5 Copyright 2024, the American Association for the Advancement of Science. (F) Comparison of mechanical properties of the highly entangled hydrogel (blue) and low entangled hydrogel (red). (G) Images of adhered PEDOT:PSS on an SBS substrate after laser annealing, along with the resulting peel strength demonstrating strong adhesion. Reproduced with permission.169 Copyright 2024, Springer Nature. (H) Schematic of laser-induced adhesive and its enabled transfer printing process and the cyclic test of the adhesion. Reproduced with permission.203 Copyright 2024, the American Association for the Advancement of Science. | ||
Particularly, in composite materials, polymerization behaves differently under laser and LED irradiation due to differences in wavelength distribution, power density, and penetration depth (Fig. 14B).195,197 LEDs provide relatively broad wavelength distributions, lower intensities, and wide illumination areas, whereas lasers deliver highly coherent, narrowly distributed wavelengths with significantly higher power density focused on a localized region. Under identical irradiation intensities and exposure times, 449 nm laser curing yields higher conversion than broad-band 450 nm LED curing (Fig. 15B). Lasers can efficiently deliver energy even in scattering media such as particle-containing resins due to their higher power density and improved penetration depth.
In pulsed-laser polymerization, the pulse parameters such as pulse frequency can critically influence polymer growth.198,199 Each pulse generates radicals, after which chains grow during the interval between pulses. Newly formed radicals in subsequent pulses may terminate existing chains, meaning the chain length decreases as the pulse frequency increases. The theoretical polymer chain length is determined as follows:
| L0,i = ikp[M]tp |
Pulse frequency also influences side-reaction pathways.199–201 In radical polymerization of acrylates, although ideal growth occurs through end-chain propagation, intramolecular hydrogen abstraction leads to backbiting, producing mid-chain radicals that propagate far more slowly than end-chain radicals. Therefore, real polymerization involves initiation, end-chain propagation, backbiting, mid-chain propagation, and termination simultaneously. At very high pulse frequencies, the growth interval between pulses becomes too short for significant backbiting to occur, reducing the contribution of side reactions and making end-chain propagation dominant. At high pulse frequency, the effective propagation rate increases (Fig. 15C).199
In photo-induced polymerization, not only control of the light source but also adjustment of the concentration of photo-responsive initiators can significantly influence the final material properties.5,202 In conventional hydrogels, stiffness is typically increased by increasing the crosslinking density, which often leads to embrittlement and poor toughness. In contrast, entanglements act as slip-links that reinforce the polymer network while allowing chains to slide and distribute stress efficiently along their contour, thereby enhancing toughness. Therefore, achieving a highly entangled network requires careful optimization of the monomer, crosslinker, and initiator concentrations. Specifically, the initiator concentration must be much lower than the monomer concentration to allow long-chain growth and increased entanglement, and the crosslinker concentration must also be lower than the monomer concentration to ensure that entanglements outnumber crosslinks. Additionally, the initiator concentration should be markedly lower than the crosslinker concentration so that longer chains can effectively transmit mechanical loads throughout the network. Polymers synthesized under these conditions exhibit an exceptional mechanical performance, including high toughness, large fatigue thresholds, and superior strength (Fig. 15D–F).
In laser-based mechanical tuning, studies have demonstrated not only control of the polymer chain length but also modulation of polymer adhesion (Fig. 14C). Laser-induced adhesion is a technique that enables bonding between materials by utilizing laser irradiation. When two polymers are brought into contact, interchain diffusion leads to chain entanglement across their interface, effectively eliminating the boundary and producing strong adhesion. Efficient diffusion requires high chain mobility, which is typically achieved by elevating the temperature. Owing to their strong photothermal effect, lasers can rapidly and locally raise temperatures, enabling precise adhesion at targeted regions.
Leveraging this advantage, irradiation of 532 nm laser light onto PEDOT:PSS films coated on polymer substrates such as PET, PI, and thermoplastic elastomers induced strong localized heating due to light absorption by PEDOT:PSS (Fig. 15G).169 This photothermal energy promotes interdiffusion and chain entanglement between the coating and the substrate, resulting in exceptionally high adhesion strength. The bonded interface remains stable even in wet environments, allowing PEDOT:PSS patterns on insulating substrates to maintain integrity during in vivo operation, enabling reliable long-term neural signal recording for more than three weeks.
Laser-induced adhesion can also be applied to microchip transfer during device fabrication by changing the viscoelasticity of the polymer through the photothermal effect (Fig. 15H).203 Shape memory polymers (SMPs) are elastic at room temperature, but under compressive force and laser exposure, these polymer soften and become highly viscous due to the elevated temperature. They then become sticky and adhere strongly to the microchip. Releasing the preload and re-irradiating the laser allows the SMP to rapidly return to its original shape, detaching the microchip. This process occurs within ∼3 ms, provides high adhesion strength (∼1 MPa), and remains reliable over multiple transfer cycles. Laser-assisted transfer enables precise placement even on rough or curved surfaces, demonstrating strong potential for scalable mass-transfer manufacturing.
4.3. Optical properties
Optical responses in polymer networks are dictated by how molecular configurations and nanoscale architectures guide the interaction of light with matter.174,204–206 Because these optical behaviors originate from structural variables that can be redefined through localized thermal or photochemical activation, laser irradiation provides a deterministic means to program optical states within compliant polymer substrates. Unlike global stimuli such as electric fields or solvent swelling, which affect the entire material uniformly, the focused laser beam allows for the maskless inscription of arbitrary optical patterns.207,208 This pixel-by-pixel writing capability enables the monolithic integration of transparent, scattering, and colored regions within a single continuous film, offering a level of design freedom unattainable with conventional lithographic or chemical methods.174,204,205To achieve precise optical modulation, two primary structural strategies are employed: control of molecular orientation to tune the refractive index uniformity and regulation of nanoscale periodicity to manipulate structural color.
Firstly, transparency is governed by the refractive index contrast (Δn) across micro-domains.209 In an isotropic random coil state, refractive-index fluctuations are minimal, allowing light to pass without scattering. However, inducing anisotropic alignment of molecular units (e.g., mesogens) creates birefringence.210–212 The resulting mismatch in refractive index between domains acts as scattering centers, turning the material opaque. Laser irradiation serves as a tool to locally switch the material between these isotropic (transparent) and anisotropic (scattering) states by overcoming the rotational energy barriers of the polymer chains (Fig. 16A).174
 | ||
| Fig. 16 Strategies for laser-induced optical property modulation. (A) Transparency control via molecular orientational alignment. Modulation of light scattering intensity based on the refractive-index mismatch (Δn) between isotropic (transparent) and anisotropic (opaque) domains. (B) Structural color tuning via regulation of nanoscale periodicity. Wavelength-selective reflection governed by Bragg's law (mλ = 2ndsinθ), where the reflected color is determined by the controllable interlayer distance (d) of the periodic structures. | ||
Secondly, structural color arises from the constructive interference of light scattered from periodic nanostructures, governed by Bragg's law (mλ = 2ndsinθ) (Fig. 16B).213,214 The reflected wavelength (λ) is directly proportional to the lattice spacing (d). Laser processing modifies this periodicity by inducing localized volume changes, densification, or polymerization gradients.204,205 By precisely controlling the thermal or chemical dose, the inter-domain spacing in block copolymers or the inter-particle distance in colloidal assemblies can be expanded or contracted, thereby shifting the reflection band across the visible spectrum.
Recent studies have successfully translated these theoretical strategies into functional optical devices, demonstrating high-fidelity control over both transparency and coloration. The modulation of transparency through orientational control has been realized in liquid crystal networks.174 By utilizing the photothermal effect to locally heat a polymer above its transition temperature, its molecular arrangement can be reconfigured. Rapid cooling preserves specific orientational states, effectively writing transparent or opaque patterns based on the induced birefringence. As shown in Fig. 17A, laser irradiation selectively lowers the rotational order in targeted regions, reducing refractive-index mismatch and establishing spatial control over optical transmission. Fig. 17B presents the dose-dependent transmittance spectra, confirming that the transparent states can be reversibly written and erased by controlling the alignment of anisotropic domains through laser fluence. Furthermore, Fig. 17C demonstrates the spatial patterning of isotropic and anisotropic regions, highlighting the ability to locally switch between high-transmission and high-scattering optical states for display applications.
 | ||
| Fig. 17 Spatiotemporal modulation of optical properties via laser-induced structural reorganization. (A) Reduction of rotational barriers by laser irradiation for localized switching between isotropic and anisotropic states to modulate refractive-index mismatch. (B) Reversible writing and erasing of transparent windows confirmed by dose-dependent transmittance spectra via control of the mesogen alignment. Inset photograph shows spatially modified optical properties with varying dose. (C) Spatial patterning of high-transmission (isotropic) and high-scattering (anisotropic) regions for optical encoding. Reproduced with permission.174 Copyright 2024, Springer Nature. (D) Contraction or relaxation of periodic lamellar spacing driven by laser-induced thermal perturbation of microphase separation equilibrium. (E) Temperature-dependent shift of peak reflection wavelength (λmax) dictated by the laser-modulated microdomain spacing. (F) Stabilization of the reorganized photonic lattice into a uniform structural color pattern following thermal annealing. Reproduced with permission.204 Copyright 2019, Wiley-VCH. (G) Mechanical constraint of particle assembly during hydration by laser-defined crosslinking gradients for lattice constant setting. (H) Correlation between laser-programmed scaffold geometry and reflected color (λmax). (I) Multicolor patterning with high spatial fidelity and angle-dependent optical response in hydrogel-embedded particle systems. Inset is corresponding SEM image. Reproduced with permission.205 Copyright 2025, Wiley-VCH under CC BY 4.0 license (https://creativecommons.org/licenses/by/4.0/). | ||
In the domain of structural color, laser-driven reorganization has been demonstrated in BCP systems.204 The photonic spacing originates from the thermodynamic microphase separation of chemically distinct blocks. Local laser heating perturbs the equilibrium balance between enthalpic segregation and entropic mixing, driving the contraction or relaxation of the lamellar spacing (Fig. 17D). Notably, the steep spatiotemporal thermal gradients induced by laser irradiation accelerate the polymer chain mobility, achieving long-range ordering of microdomains on timescales significantly shorter than conventional oven annealing. This rapid ordering kinetics enables the high-throughput production of defect-free photonic crystals. Fig. 17E shows the temperature-dependent shift of the peak reflection wavelength (λmax), confirming that the structural color is directly dictated by the laser-modulated microdomain spacing. After thermal annealing, the reorganized photonic lattice stabilizes into a uniform structural color pattern, indicating the formation of a thermodynamically equilibrated photonic lattice (Fig. 17F).
A distinct photonic mechanism is observed in particle-based systems, where structural color is generated by the spatial arrangement of discrete colloidal particles embedded in a hydrogel matrix.205 In this approach, laser irradiation establishes spatial gradients in crosslinking density. These gradients mechanically constrain the equilibrium position of the particles during swelling or drying, thereby setting the final lattice constant (Fig. 17G). The structural parameters, such as the scaffold geometry and pore size defined by the laser, correspond directly to the resulting color (Fig. 17H). This allows for the creation of complex, multicolor patterns with high spatial fidelity and angle-dependent optical responses, independent of the intrinsic material chemistry (Fig. 17I).
Collectively, these demonstrations verify that the optical characteristics of polymers can be deterministically redefined by laser-induced transformations. By sequentially applying the principles of orientational control and periodic modulation, laser processing enables optical functions that are programmable, reversible, and compatible with polymer substrates. This establishes laser-induced optical modulation as a foundational strategy for next-generation polymer-based optical systems, paving the way for advancements in flexible displays, optical encryption, and biomimetic camouflage skins.
5. Applications
The structural and property changes induced by lasers enable application-targeted functionalities for polymers that are difficult to achieve with conventional processing methods. Nano-microfabrication of soft and wet hydrogels, which are challenging to process using lithography, can lead to the development of miniaturized implantable devices. By inducing various phases of polymers on a single plane, it is possible to create programmed motion that responds differently to external stimuli. Highly porous graphene conductors generated through laser pyrolysis can facilitate the rapid and efficient creation of battery electrodes with a high surface area. In this section, we will introduce polymers functionalized through laser techniques and discuss how these functionalities provide enhanced efficiency in applications compared to traditional processing methods.5.1. Bio-applications
Recent advances in laser processing of polymeric and conductive polymer systems have enabled bio-interfaces whose performance is determined mainly by their geometry and locally confined photothermal effects rather than additional surface chemistries.178,215,216 Ultrafast femtosecond and CO2 laser patterning of polymers such as PI, elastomer-supported films, and ultrathin hydrogels can generate micro- and nanoscale topographies or can convert these polymers into a porous conductive carbon network known as LIG.178,217 Because these processes are direct writing processes that are mask-free and solvent-free, they are suitable for the fabrication of implantable and wearable bioelectronics, where surface purity, adhesion, and mechanical compliance must be preserved.178,215A persistent difficulty in implantable electronics is the chronic foreign body response that leads to fibrotic encapsulation and signal degradation. Conventional hydrophilic or zwitterionic coatings can delay this response but often show limited stability or cause an increase in impedance.218 Laser-generated surface topography on PI provides a purely physical route to obtain immune-stealth surfaces. Multiphoton ablation can pattern ordered micro-grooves and nanoscale islands that disrupt the focal adhesion of macrophages and fibroblasts, thereby reducing cell attachment by more than one order of magnitude while the base polymer remains intact (Fig. 18A).219 Because the laser energy is confined within the focal volume, the surrounding film retains its flexibility and electrical integrity, enabling in vivo recordings for several weeks (Fig. 18B). This illustrates an important design principle. Spatially programmed surface roughness can provide long-term biocompatibility without additional surface chemistry, and the same approach can be extended to neural, cardiac and other implantable sensors.216
 | ||
| Fig. 18 Laser-induced polymer interfaces for advanced biomedical applications. (A) Immune-stealth capability via laser-generated multiscale topography disrupting focal adhesion of macrophages and fibroblasts. (B) Reduction of fibrous encapsulation and inflammation at patterned interfaces confirmed by histological evaluation. Reproduced with permission.219 Copyright 2025, Wiley-VCH under CC BY 4.0 license (https://creativecommons.org/licenses/by/4.0/). (C) Fabrication of ultrathin, adhesive hydrogel-based bioelectronics via cryogenic laser transfer. (D) Stable monitoring without delamination enabled by conformal attachment of graphene-hydrogel patches on beating hearts. (E) High-fidelity, biocompatible real-time ECG recording by soft patches. Reproduced with permission.221 Copyright 2023, Springer Nature. (F) Soft, stretchable NeuroString sensors fabricated via laser carbonization of metal-complexed polyimide. (G) Simultaneous detection of dopamine and serotonin facilitated by in situ-generated catalytic metal nanoparticles. Reproduced with permission.222 Copyright 2022, Springer Nature. (H) Intracellular delivery via laser-induced photoporation using transient vapor nanobubbles (VNBs) for membrane perforation. (I) Enhanced colloidal stability and photothermal reproducibility of GQD and rGO via PEG or PEI functionalization. (J) High-efficiency macromolecule delivery with preserved cell viability for safe non-viral cell engineering. Reproduced with permission.223 Copyright 2020, Multidisciplinary Digital Publishing Institute under CC BY 4.0 license (https://creativecommons.org/licenses/by/4.0/). | ||
At the device level, laser-induced graphene that is produced directly from PI by CO2 laser pyrolysis has advanced flexible bioelectronics toward tissue-like systems. The resulting porous graphene electrodes exhibit low impedance, high charge injection capacity, and robust resistance to repeated mechanical loading.178,220 Arrays prepared in this manner can record cortical activity and deliver electrical stimulation to the brain surface without tissue penetration. Delamination is minimized because the conductive phase is generated inside the original polymer.178,220 More recent cryogenic laser transfer processes have combined such porous graphene with very thin hydrogel layers that act simultaneously as an adhesive and as an energy-dissipation medium (Fig. 18C).221 These graphene–hydrogel composites can undergo large tensile deformation, guide crack propagation paths, and adhere conformally to moving organs such as a beating heart (Fig. 18D). This enables stable electrocardiogram monitoring while maintaining antibacterial and biocompatible properties (Fig. 18E). This development shows a shift to electronic materials whose mechanics and hydration state approach those of living tissue through the union of laser-textured carbon frameworks and hydrated polymer matrices.
Laser fabrication has also enabled multifunctional biosensors that connect mechanical compliance to electrochemical sensitivity. In the system that was reported as NeuroString, laser carbonization of a PI sheet complexed with a metal-porphyrin forms a conductive network of iron-oxide-decorated graphene fibers, which is embedded in a elastomer matrix (Fig. 18F).222 This sensor maintains low resistance under tensile strains exceeding 1500% and can detect neurotransmitters such as dopamine and serotonin at nanomolar levels in the brain and gut of freely moving animals. The graphene network provides high electrocatalytic activity for the oxidation of monoamines, and the elastomer provides extreme stretchability (Fig. 18G). This shows that laser patterning can integrate electrochemical performance, biocompatibility, and mechanical adaptability in a single architecture and can make biochemical monitoring possible in organs previously inaccessible to rigid probes.
Laser energy can also act directly on cells. In laser-induced photoporation, short optical pulses create localized heating and vapor nanobubbles around light-absorbing nanoparticles.223,224 These events open transient, nanometer-scale pores in the membrane and allow intracellular delivery (Fig. 18H).223 Graphene quantum dots and reduced graphene oxide that are functionalized with polyethylene glycol or polyethyleneimine are especially effective, as surface functionalization improves colloidal stability and makes photothermal conversion reproducible (Fig. 18I). Under nanosecond irradiation in the visible or near-infrared range, these materials allow the delivery of macromolecules such as dextran and nucleic acids with efficiencies higher than 80% while preserving high cell viability (Fig. 18J). This demonstrates that safe and nonviral intracellular transport can be achieved by combining the spatial precision of light with the photothermal responsiveness of graphene-based polymer systems, thereby linking laser science directly to cell and gene engineering.
Taken together, these results support a unified view of laser technology as a bridge among photons, polymers, and biology. Multiphoton ablation, direct laser carbonization, and photothermal transfection all rely on rapid, localized energy deposition that sculpts polymer matter at the length scales that control cell adhesion, ion transport, and protein adsorption. The materials that are obtained in this way exhibit hierarchical morphology, hydrated elasticity, and optical reactivity, thereby enabling functions that range from immune modulation to neural and cardiac sensing and to intracellular delivery. As laser sources become more programmable, and data-driven optimization of pulse width, repetition rate, and scan paths becomes common, the design space for such bio-interfaces will increase further. Prospective future directions include biodegradable or self-healing laser-patterned polymers for transient implants; adaptive graphene-hydrogel composites that respond to biological feedback; and closed-loop devices that integrate sensing and actuation in the same laser written circuit.
5.2. Soft robotics
Laser-based techniques such as photopolymerization, photothermal heating, phase patterning, and photochemical reactions are increasingly exploited in robotics. Because lasers enable highly localized heating and photoactivation, they serve as a powerful tool for precise material processing and on-demand actuation. In this context, we aim to examine how these laser-based strategies have been integrated into recent advances in micro-robotics.Magnetic microrobots are emerging as promising in vivo biomedical devices. However, embedding hard magnetic components such as neodymium–iron–boron (NdFeB) microparticles into microrobots has been technically challenging due to their high opacity, which severely hampers light penetration and makes photo-polymerization fabrication difficult. Recently, hard magnetic microrobots have been realized by introducing a microrobot cavity via an fs laser (Fig. 19A and B).225 Specifically, a helical microrobot loaded with NdFeB microparticles is fabricated by first writing a microrobot-shaped cavity into a positive photoresist using an fs laser. The resulting cavity is then infused under vacuum with a slurry of NdFeB powder, epoxy resin, and curing agent, and subsequently cured. The fabrication of various microrobot shapes is possible, including double helices and bacteria-inspired shapes. These microrobots show strong magnetic responsiveness and swimming performance even in highly viscous media and under upstream flow, demonstrating the utility of femtosecond laser processing as a powerful fabrication route for composite microrobots incorporating otherwise incompatible materials such as metal microparticles.
 | ||
| Fig. 19 Laser-enabled fabrication, actuation, and deconstruction in robotic systems. (A) Schematic of the fabrication of a magnetic helical microrobot using a femtosecond laser and a positive photoresist. (B) Schematic of microrobot fabrication with diverse geometries and their actuation in blood. Reproduced with permission.225 Copyright 2025, the American Association for the Advancement of Science. (C) Laser-induced α/β phase patterning in PVDF and their electromechanical responses to an electric field. (D) Actuation of PVDF patterned robot. Reproduced with permission.28 Copyright 2022, Wiley-VCH. (E) Schematic of a DPI-HFP–containing silicone robot and light-triggered depolymerization mechanism. (F) Images of silicone robots before light triggering and after light-induced decomposition. Reproduced with permission.227 Copyright 2023, the American Association for the Advancement of Science. | ||
When fabricating magnetic robots, magnetization is usually introduced by applying an external magnetic field while the polymer matrix is in a softened or mobile state. A recent approach leverages the photothermal effect of lasers to locally induce polymer mobility, thereby enabling spatially selective magnetization.226 In this method, NdFeB microparticles are encapsulated within polycaprolactone (PCL) and subsequently dispersed in a silicone matrix (Ecoflex). The NdFeB particles not only serve as magnetic fillers but also act as photothermal absorbers, where upon laser irradiation, they generate localized heat that softens the PCL, creating a mobile region within the composite. When an external magnetic field is applied, only the laser-heated regions become magnetized. By repeating this process in different areas, complex and multidirectional magnetization patterns can be encoded within a single film. This spatial programming of magnetization enables distinct actuation modes under uniform magnetic fields, allowing the realization of diverse three-dimensional deformations and locomotion behaviors.
These laser-enabled patterning strategies can be extended not only to magnetization programming but also to phase patterning within a single film. PVDF, a representative piezoelectric polymer with strong intrinsic dipoles, exhibits multiple crystalline phases, α, β, and γ, whose polarity increases in the order α < γ < β. Laser-induced phase transitions provide a means to spatially encode these phases within one continuous film (Fig. 19C and D).28 The scanning rate critically determines the resulting phase. Under ultrafast scanning, thermal effects are minimal, and the electromagnetic field of the laser promotes dipole alignment, yielding the highly polar β phase. In contrast, slow scanning induces substantial photothermal heating via the Au nanoparticles, increasing the conformational freedom to produce random alignment, which results in the low-polarity α phase. By patterning domains with distinct polarization states within the same film by using different scan rates, α and β phase patterning can be achieved. The electromechanical resonance frequencies of the α and β phases are different, where β-phase regions respond strongly near 40 Hz, whereas α-phase regions resonate near 50 Hz. Consequently, by switching the driving frequency, a single PVDF sheet can generate different actuation modes, enabling soft robotic systems capable of multimodal, directionally programmable motion.
Photochemical reactions can also be exploited to induce the degradation of soft robots. Conventional silicone elastomers used in soft robotics are chemically robust and essentially nondegradable, which complicates their degradation. By incorporating diphenyliodonium hexafluorophosphate (DPI-HFP) into the silicone matrix and subsequently applying 365 nm UV light together with heating to 120 °C, fluoride ions are produced that attack the Si–O–Si backbone, leading to breakdown of the elastomer network (Fig. 19E and F).227 In this way, a soft robot that behaves like a standard silicone actuator during operation can be engineered to undergo on-demand structural disintegration via a photochemically triggered degradation process.
5.3. Energy devices
Laser-induced polymers provide a direct and adaptable route for constructing energy devices by enabling localized modulation of electrical, thermal, and mechanical properties within polymer substrates. Because this process does not require masks, solvents, or elevated temperatures, it supports patterning on compliant materials and allows device architectures that depend on spatially discrete functionality. Recent studies have demonstrated conformal on-skin harvesters,228,229 emissivity-tuned thermoelectric modules,230 stretchable micro-supercapacitors,231,232 and polymer-derived carbon structures that enhance triboelectric or piezoelectric responses.230,233,234 These results indicate that laser processing can bypass the thermal and geometric constraints inherent in conventional fabrication methods and establish a unified framework for integrating heterogeneous energy-conversion mechanisms in flexible systems.Laser irradiation can generate conductive carbon networks in elastomeric stacks, enabling compact tactile–energy hybrid platforms.230 The porous laser-induced graphene formed within the elastomer matrix achieves stable charge transport and maintains strong interfacial bonding under deformation, allowing pressure inputs to be converted directly into voltage signals (Fig. 20A). The resulting electrical output remains consistent during repeated loading cycles, demonstrating reliable electromechanical coupling within the multilayer configuration (Fig. 20B). This example shows how laser-induced carbon structures can combine sensing and power-generation roles in a single, monolithic construct suited for distributed tactile mapping.
 | ||
| Fig. 20 Laser-induced polymer architectures for energy harvesting and storage. (A) Monolithic integration of laser-induced graphene LIG into elastomeric stacks for tactile sensing and energy harvesting. (B) Stable electromechanical coupling under cyclic loading demonstrating reliable pressure-to-voltage conversion. Reproduced with permission.230 Copyright 2023, Elsevier. (C) Freestanding, porous laser-induced carbon networks acting as mechanically compliant electrodes for flexible energy storage. (D) Adaptive circuit reconfiguration between high-voltage operation and long-duration discharge modes triggered by electrolyte introduction. Reproduced with permission.235 Copyright 2026, Elsevier. (E) Solar-driven thermoelectric generation enabled by spatially programmed high-absorption laser-induced pyrolyzed PDMS (LIPP-H) regions. (F) Continuous passive cooling performance of low-absorption LIPP-C regions, maintaining thermal gradients for energy harvesting even under low-irradiance conditions. Reproduced with permission.184 Copyright 2025, Elsevier. | ||
Laser-based carbonization also enables the formation of freestanding conductive films that function as lightweight and mechanically compliant electrodes.235 Controlled irradiation defines a continuous porous carbon network that can be detached from the polymer precursor while maintaining electrical continuity and structural flexibility (Fig. 20C). When electrolyte droplets are introduced, the freestanding patterned electrode reconfigures the circuit and switches between a high-voltage operating state and a long-duration discharge state. Each mode shows distinct electrical and optical signatures arising from voltage and capacity differences governed by the laser-defined geometry (Fig. 20D). This behavior demonstrates that laser-induced polymer architectures can support adaptive circuit functions through simple environmental inputs.
Laser pyrolysis further supports hybrids that couple photothermal and thermoelectric energy conversion. In these systems, laser-induced pyrolyzed PDMS (LIPP) arrays with spatially programmed absorption and radiative cooling are formed directly on the polymer surface, while conductive PEDOT:PSS pathways are patterned without additional processing.184 The high-absorption LIPP-H region establishes a sustained temperature gradient under solar illumination, enabling thermoelectric generation (Fig. 20E). In contrast, the low-absorption LIPP-C region maintains efficient radiative cooling, allowing the thermal gradient to persist during night-time or low-irradiance conditions, as confirmed by stable environmental measurements (Fig. 20F). These results illustrate how laser-defined optical and thermal responses can be combined to achieve continuous, environment-adaptive energy harvesting. Collectively, these demonstrations show that laser-induced polymer transformations enable energy devices with high mechanical compliance, functional heterogeneity, and strong material integration. Because a laser directly structures conductive pathways, thermal interfaces, and interfacial mechanics within the polymer matrix, the resulting devices can accommodate complex deformations while maintaining stable function. As laser systems become more programmable and polymer chemistries become increasingly responsive to photothermal, photochemical, and field-driven processes, the design space for flexible and adaptive energy technologies will continue to expand, positioning laser-induced polymers as a central platform for next-generation multifunctional power systems.
6. Conclusion and prospects
In this review, we have comprehensively discussed the emerging field of laser-induced polymer dynamics, a discipline that stands at the intersection of synthetic chemistry, materials science and electronics. For decades, polymer engineering has primarily relied on the synthesis of new monomers or the chemical modification of polymer chains to achieve desired functionalities. However, the advent of precision laser processing has introduced a transformative paradigm, the ability to “program” the physical and chemical properties of existing polymers with extreme spatial and temporal resolution.Our discussion has highlighted that laser-induced transformations are not merely thermal ablation processes. Instead, they involve a complex interplay among photothermal, photochemical, and electromagnetic field-driven mechanisms. By tuning the laser parameters, researchers can now induce local crystallization, phase separation, pyrolysis, and molecular alignment. These localized modifications enable the fabrication of multifunctional systems such as flexible sensors, soft actuators, and energy-harvesting devices on a single, monolithic substrate without the need for complex multi-step assembly or expensive cleanroom environments.
Despite the remarkable progress in the laboratory, several critical challenges must be addressed to transition laser processing technologies from academic demonstrations to large-scale industrial applications. We identify three primary technical bottlenecks: resolution, 3D processing capabilities, and throughput.
6.1. Enhancing spatial resolution
Overcoming the diffraction limit is the most immediate challenge in the laser processing of polymers. Most current laser direct writing systems are governed by the Rayleigh criterion, which limits the minimum feature size to approximately half the wavelength of the incident light. While this is sufficient for many flexible electronic applications, it falls short for the next generation of nanophotonic devices or high-density molecular sensors, which require submicron features. To push the boundaries of resolution, future research must focus on non-linear optical phenomena. Stimulated emission depletion laser lithography involves a dual-laser-beam system consisting of a central writing beam to initiate polymerization and a peripheral, doughnut-shaped inhibition beam to suppress the reaction at the focal edges, thereby effectively overcoming the optical diffraction limit to achieve a sub-diffraction-limit resolution as fine as 9 nm. This technique enables the maskless, precise fabrication of complex three-dimensional nanostructures under ambient conditions, offering a highly versatile and portable alternative to traditional electron-beam lithography for advanced nanofabrication. However, its application in modifying commercial polymers remains in its infancy.6.2. Advanced 3D processing from surface modification to volumetric architectures
Current laser-polymer interactions are predominantly surface-centric. Whether it is the formation of LIG or surface texturing (e.g., LIPSS), the functional changes are often confined to the top few micrometers of the material. For applications such as soft robotics, tissue engineering scaffolds, and 3D structural electronics, there is an urgent need to move into 3D. By precisely controlling the focal plane in the Z-axis, researchers can induce volumetric phase transitions or create internal conductive networks within a solid elastomer. This “volumetric additive manufacturing” approach would allow for the creation of truly biomimetic systems where sensors and actuators are embedded deep within a protective polymer skin, mimicking the complex internal structures of biological organisms. Recent studies have reported promising results combining additive manufacturing and laser direct writing, demonstrating a good example of this innovative approach. This approach combines the direct ink writing method with a laser system. As a micro-scale polymer jet exits the nozzle, the integrated laser precisely irradiates it. Through strong photothermal effects, the polymer is crosslinked and solidified within a very short time of less than 0.25 s. Since the ink solidifies immediately upon leaving the nozzle, complex three-dimensional curves or cantilever structures can be printed in mid-air without the need for separate supports.196 This technique achieves a fine resolution of around 50 µm, enabling the precise fabrication of devices. Future prospects also involve the development of “transparent-to-laser” polymer composites that contain specific localized absorbers or up-conversion nanoparticles that have synergies with synthetic approaches.6.3. Increasing throughput
To transition to large-scale manufacturing, perhaps the most significant barrier to industrial adoption is throughput. Laser direct writing is fundamentally a serial process, where the laser beam must hit every point of the pattern, which makes the fabrication of large-area devices (e.g., wafer-scale chip fabrication) prohibitively slow and expensive. To achieve industrial-scale throughput, we envision a transition from single-beam scanning to parallelized processing and hybrid manufacturing. The use of spatial light modulators or digital micromirror devices can transform a single laser beam into thousands of micro-beams, allowing for the simultaneous fabrication of repetitive patterns over large areas. Furthermore, combining laser processing with high-speed roll-to-roll (R2R) manufacturing could revolutionize the production of flexible electronics. In this vision, a polymer web moves at high speeds while an array of ultrafast lasers performs real-time functionalization, patterning, and cutting.A key challenge in translating laboratory-scale laser processing to industrial R2R manufacturing lies in the shift of governing laser–polymer interaction dynamics. In contrast to static, batch processing, R2R systems involve continuous substrate motion, which modifies both the temporal and thermal history experienced by the material. One of the most significant changes is the emergence of heat accumulation effects due to repeated or overlapping laser exposure, particularly at high repetition rates or under quasi-continuous irradiation. As a result, the effective thermal response may deviate substantially from that predicted based on single-pass interaction times. In addition, the effective interaction time becomes dependent not only on beam size and scan speed, but also on web speed and process synchronization, complicating the interpretation of processing windows. Pulse overlap and repetition rate further contribute to cumulative energy deposition, often leading to transitions toward more thermally dominated regimes. Finally, heat dissipation pathways are fundamentally altered in R2R systems, where moving substrates, mechanical tension, and thermal contact with supporting rollers can significantly influence the cooling dynamics. These factors highlight that scaling laser-induced polymer processing to R2R manufacturing requires not only engineering optimization, but also a re-evaluation of the underlying interaction regimes.
Taking this a step further, an aperture that allows for individual control of the laser beams can be implemented to enhance scalability by turning each beam on and off. In industry, a system has already been commercialized that incorporates micro-electro-mechanical system (MEMS)-made apertures with electron beam lithography to enable the on-and-off control of each beam.236 By introducing a similar electronic aperture, if individual laser beams can be adjusted, it will satisfy both the scalability of production and the high design flexibility of the laser devices simultaneously.
The field of laser-induced polymer dynamics is moving toward a future where materials are no longer static. Instead, they are dynamic systems that can be digitally defined and redefined. By addressing the challenges of resolution, 3D depth, and production speed, we can transition from “processing materials” to “programming matter”. The synergy between advanced laser optics, computational design, and responsive polymer chemistry will likely lead to a new era of manufacturing. In this era, the distinction between the “material” and the “device” will blur, as the laser provides the means to weave complex functions directly into the molecular fabric of our world. The journey from fundamental light–matter interactions to ubiquitous smart technology is well underway, and the next decade promises to be a period of unprecedented growth and innovation in this multidisciplinary domain.
Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or codes have been included, and no new data were generated or analysed as part of this review.Acknowledgements
This study was supported by the National Research Foundation of Korea (RS-2025-11092968, RS-2024-00406534, RS-2025-00552995, and RS-2025-02372998).Notes and references
- H. Tran, R. Gurnani, C. Kim, G. Pilania, H.-K. Kwon, R. P. Lively and R. Ramprasad, Nat. Rev. Mater., 2024, 9, 866–886 CrossRef CAS.
- H. Bronstein, C. B. Nielsen, B. C. Schroeder and I. McCulloch, Nat. Rev. Chem., 2020, 4, 66–77 CrossRef CAS PubMed.
- B. D. Paulsen, K. Tybrandt, E. Stavrinidou and J. Rivnay, Nat. Mater., 2020, 19, 13–26 CrossRef CAS.
- Y. Zhang, W. Zhang, Z. Chen, L. Wang and G. Yu, Chem. Soc. Rev., 2025, 54, 2483–2519 RSC.
- J. Kim, G. Zhang, M. Shi and Z. Suo, Science, 2021, 374, 212–216 CrossRef CAS PubMed.
- X. Li and J. P. Gong, Nat. Rev. Mater., 2024, 9, 380–398 CrossRef CAS.
- C. Liu, N. Morimoto, L. Jiang, S. Kawahara, T. Noritomi, H. Yokoyama, K. Mayumi and K. Ito, Science, 2021, 372, 1078–1081 CrossRef CAS PubMed.
- B. H. Miller, H. Liu and M. Kolle, Nat. Mater., 2022, 21, 1014–1018 CrossRef CAS PubMed.
- D. Won and S. H. Ko, Nat. Mater., 2022, 21, 997–998 CrossRef CAS PubMed.
- J. Deng, H. Yuk, J. Wu, C. E. Varela, X. Chen, E. T. Roche, C. F. Guo and X. Zhao, Nat. Mater., 2021, 20, 229–236 CrossRef CAS PubMed.
- M. Khatib, E. T. Zhao, S. Wei, J. Park, A. Abramson, E. S. Bishop, A.-L. Thomas, C.-H. Chen, P. Emengo and C. Xu, Nature, 2025, 645, 656–664 CrossRef CAS PubMed.
- J. Kim, D. Won, T. H. Kim, C.-Y. Kim and S. H. Ko, Biosens. Bioelectron., 2024, 258, 116327 CrossRef CAS PubMed.
- H. Kim, S.-k Ahn, D. M. Mackie, J. Kwon, S. H. Kim, C. Choi, Y. H. Moon, H. B. Lee and S. H. Ko, Mater. Today, 2020, 41, 243–269 CrossRef CAS.
- S. Terryn, J. Langenbach, E. Roels, J. Brancart, C. Bakkali-Hassani, Q.-A. Poutrel, A. Georgopoulou, T. G. Thuruthel, A. Safaei and P. Ferrentino, Mater. Today, 2021, 47, 187–205 CrossRef CAS.
- P. Won, K. K. Kim, H. Kim, J. J. Park, I. Ha, J. Shin, J. Jung, H. Cho, J. Kwon and H. Lee, Adv. Mater., 2021, 33, 2002397 CrossRef CAS PubMed.
- R. De, M. K. Mahata and K. T. Kim, Adv. Sci., 2022, 9, 2105373 CrossRef CAS PubMed.
- D. C. Kong, M. H. Yang, X. S. Zhang, Z. C. Du, Q. Fu, X. Q. Gao and J. W. Gong, Macromol. Mater. Eng., 2021, 306, 2100536 CrossRef CAS.
- D. Won, H. Kim, T.-S. Kim and S. H. Ko, Appl. Phys. Rev., 2025, 12 Search PubMed.
- D. Won, J. Bang, S. H. Choi, K. R. Pyun, S. Jeong, Y. Lee and S. H. Ko, Chem. Rev., 2023, 123, 9982–10078 CrossRef CAS PubMed.
- S. Hong, H. Lee, J. Yeo and S. H. Ko, Nano Today, 2016, 11, 547–564 CrossRef CAS.
- J. Zuo and X. Lin, Laser Photonics Rev., 2022, 16, 2100741 CrossRef.
- T. Pinheiro, M. Morais, S. Silvestre, E. Carlos, J. Coelho, H. V. Almeida, P. Barquinha, E. Fortunato and R. Martins, Adv. Mater., 2024, 36, 2402014 CrossRef CAS PubMed.
- F. Consoli, V. T. Tikhonchuk, M. Bardon, P. Bradford, D. C. Carroll, J. Cikhardt, M. Cipriani, R. J. Clarke, T. E. Cowan and C. N. Danson, High Power Laser Sci. Eng., 2020, 8, e22 CrossRef CAS.
- Q. Xie, M. Hong, H. Tan, G. Chen, L. Shi and T. Chong, J. Alloys Compd., 2008, 449, 261–264 CrossRef CAS.
- Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2010, 43, 6245–6260 CrossRef CAS.
- C. Mazzotta, C. Traversi, A. L. Paradiso, M. E. Latronico and M. Rechichi, J. Ophthalmol., 2014, 2014, 604731 Search PubMed.
- N. Bityurin, B. Luk'Yanchuk, M. Hong and T. Chong, Chem. Rev., 2003, 103, 519–552 CrossRef CAS PubMed.
- D. Won, H. Cho, H. Kim, G. Lee, J. Kwon, J. Kim, S. Hong, J. Choi, S. W. Kim and S. H. Ko, Adv. Opt. Mater., 2022, 10, 2201206 CrossRef CAS.
- S. Chae, K. H. Jo, S. W. Lee, H. S. Keum, H. J. Kim, J. Choi and H. H. Lee, Macromol. Chem. Phys., 2016, 217, 537–542 CrossRef CAS.
- J. Lim, S. Park, H. Cho, Y. Lee, I. Ha, Y. Kim, E. Hwang, H. Lee, J. Shin and J. Kwon, Chem. Eng. J., 2022, 428, 131050 CrossRef CAS.
- D. Ohayon, V. Druet and S. Inal, Chem. Soc. Rev., 2023, 52, 1001–1023 RSC.
- J. Kern, S. Venkatram, M. Banerjee, B. Brettmann and R. Ramprasad, Phys. Chem. Chem. Phys., 2022, 24, 26547–26555 RSC.
- H. Liu, Z. Zhang, D. Yuan, M. Chen, H. Jiang, J. Liang, X. Chen, D. Sun, L. Zhang and L. Liu, J. Mater. Chem. A, 2023, 11, 19860–19869 RSC.
- W. Yang, Z. Luo, R. Sun, J. Guo, T. Wang, Y. Wu, W. Wang, J. Guo, Q. Wu and M. Shi, Nat. Commun., 2020, 11, 1218 CrossRef CAS PubMed.
- J. Deepak, R. Anirudh and S. S. Sundar, Mater. Today: Proc., 2023 DOI:10.1016/j.matpr.2023.02.102.
- T. Ito and S. Okazaki, Nature, 2000, 406, 1027–1031 CrossRef CAS PubMed.
- M. G. Stanford, K. Yang, Y. Chyan, C. Kittrell and J. M. Tour, ACS Nano, 2019, 13, 3474–3482 CrossRef CAS PubMed.
- R. Ye, D. K. James and J. M. Tour, Acc. Chem. Res., 2018, 51, 1609–1620 CrossRef CAS PubMed.
- S. Jun, A. Herbst, K. Scheffter, N. John, J. Kolb, D. Wehner and H. Fattahi, Commun. Phys., 2024, 7, 161 CrossRef.
- K. S. Lee, D. Y. Yang, S. H. Park and R. H. Kim, Polym. Adv. Technol., 2006, 17, 72–82 CrossRef CAS.
- A. Klenke, M. Müller, H. Stark, M. Kienel, C. Jauregui, A. Tünnermann and J. Limpert, IEEE J. Sel. Top. Quantum Electron., 2018, 24, 1–9 Search PubMed.
- P. W. Majewski and K. G. Yager, ACS Nano, 2015, 9, 3896–3906 CrossRef CAS PubMed.
- H. Hiraoka, S. Lazare and A. Cros, J. Photochem. Photobiol., A, 1992, 65, 293–302 CrossRef CAS.
- S. S. Harilal, J. R. Freeman, P. K. Diwakar and A. Hassanein, in Laser-Induced Breakdown Spectroscopy: Theory and Applications, Springer, 2014, pp. 143–166 Search PubMed.
- M. S. Brown and C. B. Arnold, Laser precision microfabrication, Springer, 2010, pp. 91–120 Search PubMed.
- E. Akman, Y. Erdoğan, M. Ö. Bora, O. Coban, B. G. Oztoprak and A. Demir, Int. J. Adhes. Adhes., 2020, 98, 102548 CrossRef CAS.
- F. Brygo, C. Dutouquet, F. Le Guern, R. Oltra, A. Semerok and J. Weulersse, Appl. Surf. Sci., 2006, 252, 2131–2138 CrossRef CAS.
- R. S. Kappes, F. Schönfeld, C. Li, A. A. Golriz, M. Nagel, T. Lippert, H.-J. Butt and J. S. Gutmann, SpringerPlus, 2014, 3, 489 CrossRef PubMed.
- K. Sugioka and Y. Cheng, Light: Sci. Appl., 2014, 3, e149–e149 CrossRef CAS.
- D. Bäuerle, Laser processing and chemistry, Springer Science & Business Media, 2013 Search PubMed.
- A. E. Siegman, Lasers, University science books, 1986 Search PubMed.
- R. R. Gattass and E. Mazur, Nat. Photonics, 2008, 2, 219–225 CrossRef CAS.
- A. Vogel and V. Venugopalan, Chem. Rev., 2003, 103, 577–644 CrossRef CAS PubMed.
- X. Cui, Q. Ruan, X. Zhuo, X. Xia, J. Hu, R. Fu, Y. Li, J. Wang and H. Xu, Chem. Rev., 2023, 123, 6891–6952 CrossRef CAS PubMed.
- A. O. Govorov and H. H. Richardson, Nano Today, 2007, 2, 30–38 CrossRef.
- S. Link and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 8410–8426 CrossRef CAS.
- G. Chen, Nanoscale energy transport and conversion: a parallel treatment of electrons, molecules, phonons, and photons, Oxford university press, 2005 Search PubMed.
- T. Lippert, Polymers and Light, Springer, 2004, pp. 51–246 Search PubMed.
- J. F. Rabek, Photochemistry and Photophysics, CRC Press, 1991 Search PubMed.
- C. L. Anderson, T. Zhang, M. Qi, Z. Chen, C. Yang, S. J. Teat, N. S. Settineri, E. A. Dailing, A. Garzón-Ruiz and A. Navarro, J. Am. Chem. Soc., 2023, 145, 5474–5485 CrossRef CAS PubMed.
- Y. Kim and J. Choi, J. Mater. Chem. C, 2023, 11, 2196–2205 RSC.
- T. Bloomstein, M. Horn, M. Rothschild, R. Kunz, S. Palmacci and R. Goodman, J. Vac. Sci. Technol., B: Nanotechnol. Microelectron.:Mater., Process., Meas., Phenom., 1997, 15, 2112–2116 CAS.
- Y. He, Y. Cao and Y. Wang, Asian J. Org. Chem., 2018, 7, 2201–2212 CrossRef CAS.
- Y. Wang, H.-M. Meng, G. Song, Z. Li and X.-B. Zhang, ACS Appl. Polym. Mater., 2020, 2, 4258–4272 CrossRef CAS.
- J.-P. Fouassier and J. Lalevée, Photoinitiators for polymer synthesis: scope, reactivity, and efficiency, John Wiley & Sons, 2012 Search PubMed.
- T. Lippert and J. T. Dickinson, Chem. Rev., 2003, 103, 453–486 CrossRef CAS PubMed.
- S. Kawata, H.-B. Sun, T. Tanaka and K. Takada, Nature, 2001, 412, 697–698 CrossRef CAS PubMed.
- S. Maruo, O. Nakamura and S. Kawata, Opt. Lett., 1997, 22, 132–134 CrossRef CAS PubMed.
- S. M. Eaton, H. Zhang, P. R. Herman, F. Yoshino, L. Shah, J. Bovatsek and A. Y. Arai, Opt. Express, 2005, 13, 4708–4716 CrossRef PubMed.
- S. Richter, S. Döring, A. Tünnermann and S. Nolte, Appl. Phys. A: Mater. Sci. Process., 2011, 103, 257–261 CrossRef CAS.
- Z. Huang and H. Wang, J. Polym. Sci., 2024, 62, 969–997 CrossRef CAS.
- C. Decker, J. Polym. Sci., Polym. Chem. Ed., 1983, 21, 2451–2461 CrossRef CAS.
- C. Decker, Nucl. Instrum. Methods Phys. Res., Sect. B, 1999, 151, 22–28 CrossRef CAS.
- J.-P. Fouassier and J. Lalevée, Photoinitiators: Structures, reactivity and applications in polymerization, John Wiley & Sons, 2021 Search PubMed.
- N. Klikovits, P. Knaack, D. Bomze, I. Krossing and R. Liska, Polym. Chem., 2017, 8, 4414–4421 RSC.
- N. Zivic, P. K. Kuroishi, F. Dumur, D. Gigmes, A. P. Dove and H. Sardon, Angew. Chem., Int. Ed., 2019, 58, 10410–10422 CrossRef CAS PubMed.
- M. Hippler, E. D. Lemma, S. Bertels, E. Blasco, C. Barner-Kowollik, M. Wegener and M. Bastmeyer, Adv. Mater., 2019, 31, 1808110 CrossRef PubMed.
- E. Kim, S. Jeon, H.-K. An, M. Kianpour, S.-W. Yu, J.-Y. Kim, J.-C. Rah and H. Choi, Sci. Adv., 2020, 6, eabb5696 CrossRef CAS PubMed.
- Z.-C. Ma, Y.-L. Zhang, B. Han, X.-Y. Hu, C.-H. Li, Q.-D. Chen and H.-B. Sun, Nat. Commun., 2020, 11, 4536 CrossRef CAS PubMed.
- G. Tillet, B. Boutevin and B. Ameduri, Prog. Polym. Sci., 2011, 36, 191–217 CrossRef CAS.
- L.-H. Liu and M. Yan, Acc. Chem. Res., 2010, 43, 1434–1443 CrossRef CAS PubMed.
- Z.-S. Tan, Z. Jamal, D. W. Teo, H.-C. Ko, Z.-L. Seah, H.-Y. Phua, P. K. Ho, R.-Q. Png and L.-L. Chua, Nat. Commun., 2024, 15, 6354 CrossRef CAS PubMed.
- D. W. Teo, Z. Jamal, H.-Y. Phua, C. G. Tang, R.-Q. Png and L.-L. Chua, ACS Appl. Mater. Interfaces, 2019, 11, 48103–48112 CrossRef CAS PubMed.
- G.-J. N. Wang, Y. Zheng, S. Zhang, J. Kang, H.-C. Wu, A. Gasperini, H. Zhang, X. Gu and Z. Bao, Chem. Mater., 2018, 31, 6465–6475 CrossRef.
- K. Dey, S. R. Chowdhury, E. Dykstra, A. Koronatov, H. P. Lu, R. Shinar, J. Shinar and P. Anzenbacher, J. Mater. Chem. C, 2020, 8, 11988–11996 RSC.
- K. Dey, S. Roy Chowdhury, E. Dykstra, H. P. Lu, R. Shinar, J. Shinar and P. Anzenbacher Jr, ACS Appl. Electron. Mater., 2021, 3, 3365–3371 CrossRef CAS.
- Y. Jiang, X. Zhang, H. Nie, J. Fan, S. Di, H. Fu, X. Zhang, L. Wang and C. Tang, Nat. Commun., 2024, 15, 6060 CrossRef CAS PubMed.
- F. Deeg, J. Pinsl and C. Bräuchle, J. Phys. Chem., 1986, 90, 5710–5715 CrossRef CAS.
- Q. Liu and J. L. Locklin, ACS Omega, 2020, 5, 9204–9211 CrossRef CAS PubMed.
- S. Mukherjee, R. Xie, V. G. Reynolds, T. Uchiyama, A. E. Levi, E. Valois, H. Wang, M. L. Chabinyc and C. M. Bates, Macromolecules, 2020, 53, 1090–1097 CrossRef CAS.
- H. Shih, A. K. Fraser and C. C. Lin, ACS Appl. Mater. Interfaces, 2013, 5, 1673–1680 CrossRef CAS PubMed.
- X. Zhang, W. Xi, C. Wang, M. Podgorski and C. N. Bowman, ACS Macro Lett., 2016, 5, 229–233 CrossRef CAS PubMed.
- V. X. Truong, K. M. Tsang, F. Ercole and J. S. Forsythe, Chem. Mater., 2017, 29, 3678–3685 CrossRef CAS.
- A. B. Lowe, C. E. Hoyle and C. N. Bowman, J. Mater. Chem., 2010, 20, 4745–4750 RSC.
- Y. Fan, C. Deng, R. Cheng, F. Meng and Z. Zhong, Biomacromolecules, 2013, 14, 2814–2821 CrossRef CAS PubMed.
- A. P. Dhand, B. E. Kirkpatrick, M. Garay-Sarmiento, B. R. Nelson, C. E. Miksch, B. Meurer-Zeman, H. M. Zlotnick, A. Mandal, J. S. Lee, J. Cione, C. N. Bowman, K. S. Anseth and J. A. Burdick, Sci. Adv., 2025, 11, eadw9262 CrossRef CAS PubMed.
- C. DelRe, Y. Jiang, P. Kang, J. Kwon, A. Hall, I. Jayapurna, Z. Ruan, L. Ma, K. Zolkin and T. Li, Nature, 2021, 592, 558–563 CrossRef CAS PubMed.
- G. Gałko and M. Sajdak, Appl. Sci., 2022, 12, 9138 CrossRef.
- P. Liu, S. Jimaja, S. Immel, C. Thomas, M. Mayer, C. Weder and N. Bruns, Nat. Chem., 2024, 16, 1184–1192 CrossRef CAS PubMed.
- H. Masai, T. Nakagawa and J. Terao, Polym. J., 2024, 56, 297–307 CrossRef CAS.
- N. Kretschy, A. K. Holik, V. Somoza, K. P. Stengele and M. M. Somoza, Angew. Chem., Int. Ed., 2015, 54, 8555–8559 CrossRef CAS PubMed.
- T. Liu, B. Bao, Y. Li, Q. Lin and L. Zhu, Prog. Polym. Sci., 2023, 146, 101741 CrossRef CAS.
- M. P. O’Hagan, Z. Duan, F. Huang, S. Laps, J. Dong, F. Xia and I. Willner, Chem. Rev., 2023, 123, 6839–6887 CrossRef PubMed.
- R. S. Givens, M. Rubina and J. Wirz, Photochem. Photobiol. Sci., 2012, 11, 472–488 CrossRef CAS PubMed.
- W. Qiu, C. Gehre, J. P. Nepomuceno, Y. Bao, Z. Li, R. Müller and X. H. Qin, Angew. Chem., Int. Ed., 2024, 63, e202404599 CrossRef CAS PubMed.
- E. Korhonen, T. Kumpulainen, M. Pamula, A. Valkonen, E. See, M. Pettersson and M. Nissinen, Chem. – Eur. J., 2025, 31, e202500530 CrossRef CAS PubMed.
- T. Chen, H. Wang, Y. Chu, C. Boyer, J. Liu and J. Xu, ChemPhotoChem, 2019, 3, 1059–1076 CrossRef CAS.
- H. S. Wang, M. Agrachev, H. Kim, N. P. Truong, T.-L. Choi, G. Jeschke and A. Anastasaki, Science, 2025, 387, 874–880 CrossRef CAS PubMed.
- L. Wimberger, G. Ng and C. Boyer, Nat. Commun., 2024, 15, 2510 CrossRef CAS PubMed.
- S. Takemura, A. Seki, K. V. Le, Y. Naka and T. Sasaki, ACS Omega, 2025, 10(25), 27238–27249 CrossRef CAS PubMed.
- H. Yaguchi and T. Sasaki, Macromolecules, 2007, 40, 9332–9338 CrossRef CAS.
- A. G. Gavriel, M. R. Sambrook, A. T. Russell and W. Hayes, Polym. Chem., 2022, 13, 3188–3269 RSC.
- O. Shelef, S. Gnaim and D. Shabat, J. Am. Chem. Soc., 2021, 143, 21177–21188 CrossRef CAS PubMed.
- F. A. Jerca, V. V. Jerca and R. Hoogenboom, Nat. Rev. Chem., 2022, 6, 51–69 CrossRef PubMed.
- D. Sosnin, M. Izadyar, S. A. A. Abedi, X. Liu and I. Aprahamian, J. Am. Chem. Soc., 2025, 147, 14930–14935 CrossRef CAS PubMed.
- I. Hnid, D. Frath, F. Lafolet, X. Sun and J.-C. Lacroix, J. Am. Chem. Soc., 2020, 142, 7732–7736 CrossRef CAS PubMed.
- J. K. Rad, Z. Balzade and A. R. Mahdavian, J. Photochem. Photobiol., C, 2022, 51, 100487 CrossRef.
- F. Xu, J. Sheng, C. N. Stindt, S. Crespi, W. Danowski, M. F. Hilbers, W. J. Buma and B. L. Feringa, Chem. Sci., 2024, 15, 6763–6769 RSC.
- Y. Gu, E. A. Alt, H. Wang, X. Li, A. P. Willard and J. A. Johnson, Nature, 2018, 560, 65–69 CrossRef CAS PubMed.
- J. J. van der Tol, T. A. Engels, R. Cardinaels, G. Vantomme, E. Meijer and F. Eisenreich, Adv. Funct. Mater., 2023, 33, 2301246 CrossRef CAS.
- S. L. Walden, P. H. Nguyen, H.-K. Li, X. Liu, M. T. Le, L. Xian Jun, C. Barner-Kowollik and V. X. Truong, Nat. Commun., 2023, 14, 8298 CrossRef PubMed.
- H. Zhou, C. Xue, P. Weis, Y. Suzuki, S. Huang, K. Koynov, G. K. Auernhammer, R. Berger, H.-J. Butt and S. Wu, Nat. Chem., 2017, 9, 145–151 CrossRef CAS PubMed.
- R. Dong, Y. Liu, Y. Zhou, D. Yan and X. Zhu, Polym. Chem., 2011, 2, 2771–2774 RSC.
- F. Xu, M. Ovalle, Y. Fu, M. A. Stuart and B. L. Feringa, Nat. Commun., 2025, 16, 10017 CrossRef CAS PubMed.
- K. Yano, Y. Itoh, F. Araoka, G. Watanabe, T. Hikima and T. Aida, Science, 2019, 363, 161–165 CrossRef CAS PubMed.
- Z. Zheng, H. Hu, Z. Zhang, B. Liu, M. Li, D.-H. Qu, H. Tian, W.-H. Zhu and B. L. Feringa, Nat. Photonics, 2022, 16, 226–234 CrossRef CAS.
- R. R. Weber, R. Hein, A. Ryabchun, Y. Gisbert, D. Garcia Romero, M. A. Loi and B. L. Feringa, Nat. Commun., 2025, 16, 9106 CrossRef CAS PubMed.
- Y. Zhou, M. Chen, Q. Ban, Z. Zhang, S. Shuang, K. Koynov, H.-J. R. Butt, J. Kong and S. Wu, ACS Macro Lett., 2019, 8, 968–972 CrossRef CAS PubMed.
- J. Lee, D. Lee, C. H. Ahn and T. A. Kim, Adv. Funct. Mater., 2025, 35, 2414842 CrossRef CAS.
- X. He, Y. Tian, R. T. O’Neill, Y. Xu, Y. Lin, W. Weng and R. Boulatov, J. Am. Chem. Soc., 2023, 145, 23214–23226 CrossRef CAS PubMed.
- S. Karthikeyan and V. Ramamurthy, J. Org. Chem., 2007, 72, 452–458 CrossRef CAS PubMed.
- P. Sun, Z. Shi, W. Sima, X. Tang, T. Yuan, M. Yang, H. Xu and Z. Li, J. Mater. Chem. C, 2023, 11, 14217–14225 RSC.
- Q. Chen, Q. Yang, P. Gao, B. Chi, J. Nie and Y. He, Ind. Eng. Chem. Res., 2019, 58, 2970–2975 CrossRef CAS.
- Z. Jiang, M. L. Tan, M. Taheri, Q. Yan, T. Tsuzuki, M. G. Gardiner, B. Diggle and L. A. Connal, Angew. Chem., 2020, 132, 7115–7122 CrossRef.
- T. D. Nguyen, M. T. N. Nguyen and J. S. Lee, ACS Appl. Electron. Mater., 2025, 7, 8625–8635 CrossRef CAS.
- B. Jin, H. Song, R. Jiang, J. Song, Q. Zhao and T. Xie, Sci. Adv., 2018, 4, eaao3865 CrossRef PubMed.
- A. P. Dhand, B. E. Kirkpatrick, M. Garay-Sarmiento, B. R. Nelson, C. E. Miksch, B. Meurer-Zeman, H. M. Zlotnick, A. Mandal, J. S. Lee and J. Cione, Sci. Adv., 2025, 11, eadw9262 CrossRef CAS PubMed.
- J. Fischer and M. Wegener, Laser Photonics Rev., 2013, 7, 22–44 CrossRef CAS.
- S. Y. Liang, Y. F. Liu, S. Y. Wang, Z. K. Ji, H. Xia, B. F. Bai and H. B. Sun, Adv. Funct. Mater., 2022, 32, 0224957 CrossRef CAS.
- D. P. Sanders, Chem. Rev., 2010, 110, 321–360 CrossRef CAS PubMed.
- A. F. Lasagni, C. Gachot, K. E. Trinh, M. Hans, A. Rosenkranz, T. Roch, S. Eckhardt, T. Kunze, M. Bieda, D. Günther, V. Lang and F. Mücklich, Direct laser interference patterning, 20 years of development: from the basics to industrial applications, Laser-based micro- and nanoprocessing XI, SPIE, 2017, pp. 186–196.
- H. Shen, Y. Wang, L. Cao, Y. Xie, Y. Wang, Q. Zhang, W. Zhang, S. Wang, Z. Han and X. Zhu, Appl. Surf. Sci., 2021, 541, 148466 CrossRef CAS.
- D. Sola, C. Lavieja, A. Orera and M. J. Clemente, Opt. Lasers Eng., 2018, 106, 139–146 CrossRef.
- N. Jiang, H. Butt, Y. Montelongo, F. Liu, S. Afewerki, G. L. Ying, Q. Dai, S. H. Yun and A. K. Yetisen, Adv. Funct. Mater., 2018, 28, 1702715 CrossRef.
- E. Stankevičius, E. Daugnoraitė and G. Račiukaitis, Opt. Lasers Eng., 2019, 116, 41–46 CrossRef.
- N. G. Quilis, S. Hageneder, S. Fossati, S. K. Auer, P. Venugopalan, A. Bozdogan, C. Petri, A. Moreno-Cencerrado, J. L. Toca-Herrera and U. Jonas, J. Phys. Chem. C, 2020, 124, 3297–3305 CrossRef CAS PubMed.
- T. Zhai, X. Zhang, Z. Pang and F. Dou, Adv. Mater., 2011, 23, 1860–1864 CrossRef CAS PubMed.
- C. Florian, S. V. Kirner, J. Krüger and J. Bonse, J. Laser Appl., 2020, 32 Search PubMed.
- S. Gräf, C. Kunz, A. Undisz, R. Wonneberger, M. Rettenmayr and F. A. Müller, Appl. Surf. Sci., 2019, 471, 645–651 CrossRef.
- J. Heitz, B. Reisinger, M. Fahrner, C. Romanin, J. Siegel and V. Svorcik, Laser-induced periodic surface structures (LIPSS) on polymer surfaces, 2012 14th International Conference on Transparent Optical Networks (ICTON), IEEE, 2012, pp. 1–4 Search PubMed.
- E. Gutiérrez-Fernández, I. A. Gabaldón-Saucedo, Á. Rodríguez-Rodríguez, E. Solano, M. C. García-Gutiérrez, A. Nogales, A. Cirera, T. A. Ezquerra and E. Rebollar, Appl. Surf. Sci., 2020, 509, 145350 CrossRef.
- S. Chae, A. Yi, H. H. Lee, J. Choi and H. J. Kim, J. Mater. Chem. C, 2018, 6, 9374–9382 RSC.
- A. A. Leniart, P. Pula, A. Sitkiewicz and P. W. Majewski, ACS Nano, 2020, 14, 4805–4815 CrossRef CAS PubMed.
- P. W. Majewski and K. G. Yager, Macromolecules, 2015, 48, 4591–4598 CrossRef CAS.
- S. R. Nowak and K. G. Yager, Adv. Mater. Interfaces, 2020, 7, 1901679 CrossRef CAS.
- P. W. Majewski, A. Rahman, C. T. Black and K. G. Yager, Nat. Commun., 2015, 6, 7448 CrossRef PubMed.
- R. de Nalda, M. E. Corrales, P. Recio, I. M. Casasús, L. Banares, T. A. Ezquerra and E. Rebollar, Phys. Status Solidi A, 2024, 221, 2300721 CrossRef CAS.
- E. Rebollar, M. Castillejo and T. A. Ezquerra, Eur. Polym. J., 2015, 73, 162–174 CrossRef CAS.
- E. Rebollar, J. R. V. de Aldana, I. Martín-Fabiani, M. Hernández, D. R. Rueda, T. A. Ezquerra, C. Domingo, P. Moreno and M. Castillejo, Phys. Chem. Chem. Phys., 2013, 15, 11287–11298 RSC.
- I. Martin-Fabiani, E. Rebollar, S. Pérez, D. R. Rueda, M. C. Garcia-Gutierrez, A. Szymczyk, Z. Roslaniec, M. Castillejo and T. A. Ezquerra, Langmuir, 2012, 28, 7938–7945 CrossRef CAS PubMed.
- R. I. Rodríguez-Beltrán, J. Prada-Rodrigo, A. Crespo, T. A. Ezquerra, P. Moreno and E. Rebollar, Polymers, 2022, 14, 5243 CrossRef PubMed.
- D. Fajstavr, K. Neznalová, V. Švorčík and P. Slepička, Materials, 2019, 12, 3460 CrossRef CAS PubMed.
- X. Wang, C. A. Ohlin, Q. Lu and J. Hu, Biomaterials, 2008, 29, 2049–2059 CrossRef CAS PubMed.
- R. I. Rodríguez-Beltrán, M. Hernandez, S. Paszkiewicz, A. Szymczyk, Z. Rosłaniec, T. A. Ezquerra, M. Castillejo, P. Moreno and E. Rebollar, Appl. Surf. Sci., 2018, 436, 1193–1199 CrossRef.
- J. Cui, Á. Rodríguez-Rodríguez, M. Hernández, M.-C. García-Gutiérrez, A. Nogales, M. Castillejo, D. Mosegui Gonzalez, P. Müller-Buschbaum, T. A. Ezquerra and E. Rebollar, ACS Appl. Mater. Interfaces, 2016, 8, 31894–31901 CrossRef CAS PubMed.
- Á. Rodríguez-Rodríguez, E. Rebollar, M. Soccio, T. A. Ezquerra, D. R. Rueda, J. V. Garcia-Ramos, M. Castillejo and M.-C. Garcia-Gutierrez, Macromolecules, 2015, 48, 4024–4031 CrossRef.
- A. Porfirev, S. Khonina, N. Ivliev, A. Meshalkin, E. Achimova and A. Forbes, Sci. Rep., 2022, 12, 3477 CrossRef CAS PubMed.
- A. P. Porfirev, N. A. Ivliev, S. A. Fomchenkov and S. N. Khonina, Nanomaterials, 2023, 13, 612 CrossRef CAS PubMed.
- D. Won, H. Kim, J. Kim, H. Kim, M. W. Kim, J. Ahn, K. Min, Y. Lee, S. Hong and J. Choi, Nat. Electron., 2024, 7, 475–486 CrossRef CAS.
- D. Won, J. Kim, J. Choi, H. Kim, S. Han, I. Ha, J. Bang, K. K. Kim, Y. Lee and T.-S. Kim, Sci. Adv., 2022, 8, eabo3209 CrossRef CAS PubMed.
- A. L. Cook, M. A. Dearborn, T. M. Anderberg, K. Vaidya, J. E. Jureller, A. P. Esser-Kahn and A. H. Squires, ACS Appl. Mater. Interfaces, 2024, 16, 17973–17980 CrossRef CAS PubMed.
- S. Song, H. Hong, K. Y. Kim, K. K. Kim, J. Kim, D. Won, S. Yun, J. Choi, Y.-I. Ryu and K. Lee, ACS Nano, 2023, 17, 21443–21454 CrossRef PubMed.
- A. Selimis, V. Mironov and M. Farsari, Microelectron. Eng., 2015, 132, 83–89 CrossRef CAS.
- S. H. Choi, J. H. Kim, J. Ahn, T. Kim, Y. Jung, D. Won, J. Bang, K. R. Pyun, S. Jeong and H. Kim, Nat. Mater., 2024, 23, 834–843 CrossRef CAS PubMed.
- F. Liu, P. Li, H. An, P. Peng, B. McLean and F. Ding, Adv. Funct. Mater., 2022, 32, 2203191 CrossRef CAS.
- R. Ye, D. K. James and J. M. Tour, Adv. Mater., 2019, 31, 1803621 CrossRef PubMed.
- P. Zaccagnini, C. Ballin, M. Fontana, M. Parmeggiani, S. Bianco, S. Stassi, A. Pedico, S. Ferrero and A. Lamberti, Adv. Mater. Interfaces, 2021, 8, 2101046 CrossRef CAS.
- J. Lin, Z. Peng, Y. Liu, F. Ruiz-Zepeda, R. Ye, E. L. Samuel, M. J. Yacaman, B. I. Yakobson and J. M. Tour, Nat. Commun., 2014, 5, 5714 CrossRef CAS PubMed.
- Z. Zhang, H. Zhu, W. Zhang, Z. Zhang, J. Lu, K. Xu, Y. Liu and V. Saetang, Carbon, 2023, 214, 118356 CrossRef CAS.
- Y. Chyan, R. Ye, Y. Li, S. P. Singh, C. J. Arnusch and J. M. Tour, ACS Nano, 2018, 12, 2176–2183 CrossRef CAS PubMed.
- R. Ye, Y. Chyan, J. Zhang, Y. Li, X. Han, C. Kittrell and J. M. Tour, Adv. Mater., 2017, 29, 1702211 CrossRef PubMed.
- Y. Nakajima, S. Hayashi, A. Katayama, N. Nedyalkov and M. Terakawa, Nanomaterials, 2018, 8, 558 CrossRef PubMed.
- J. Shin, J. Ko, S. Jeong, P. Won, Y. Lee, J. Kim, S. Hong, N. L. Jeon and S. H. Ko, Nat. Mater., 2021, 20, 100–107 CrossRef CAS PubMed.
- Y. Jung, S. Jeong, G. Heo, K. R. Pyun, S. H. Choi, J. Bang, J. G. Lee, H. Kim, J. Shin and S. Hong, Joule, 2025, 9, 102007 CrossRef CAS.
- A. Roy, R. Afshari, S. Jain, Y. Zheng, M.-H. Lin, S. Zenkar, J. Yin, J. Chen, N. A. Peppas and N. Annabi, Chem. Soc. Rev., 2025, 54, 2595 RSC.
- J. Chong, C. Sung, K. S. Nam, T. Kang, H. Kim, H. Lee, H. Park, S. Park and J. Kang, Nat. Commun., 2023, 14, 2206 CrossRef CAS PubMed.
- H. Kim, D. Won, S. H. Ko and J. Choi, Macromolecules, 2024, 57, 2048–2056 CrossRef CAS.
- L. Cheng, C. S. Yeung, L. Huang, G. Ye, J. Yan, W. Li, C. Yiu, F.-R. Chen, H. Shen and B. Z. Tang, Nat. Commun., 2024, 15, 2925 CrossRef CAS PubMed.
- T. Kang, J. Dai, Y. Huang, H. Kim, S. Keten and J. Kim, Chem. Rev., 2025, 125(22), 11032–11057 CrossRef CAS PubMed.
- R. P. Wool, Macromolecules, 1993, 26, 1564–1569 CrossRef CAS.
- T. Luu, Z. Jia, A. Kanaev and L. Museur, J. Phys. Chem. B, 2020, 124, 6857–6866 CrossRef CAS PubMed.
- R. Anastasio, W. Peerbooms, R. Cardinaels and L. Van Breemen, Macromolecules, 2019, 52, 9220–9231 CrossRef CAS PubMed.
- J. Wu, Z. Zhao, C. M. Hamel, X. Mu, X. Kuang, Z. Guo and H. J. Qi, J. Mech. Phys. Solids, 2018, 112, 25–49 CrossRef CAS.
- L. Abhinandan, R. Chari, A. Nath and M. Trivedi, J. Laser Appl., 1999, 11, 248–257 CrossRef CAS.
- V. Negovetic Mandic, M. Par, D. Marovic, M. Rakić, Z. Tarle and E. Klarić Sever, Nanomaterials, 2023, 13, 303 CrossRef CAS PubMed.
- Q. Zhuang, Y. Zhang, X. Liu, W. Xiao, Z. Chen, L. Lu, Z. Ding, S. Chen, Q. Chen and S. Patel, Nat. Electron., 2025, 1–13 Search PubMed.
- A. Pahlevan, M. H. Tabatabaei, S. Arami and S. Valizadeh, Open Dent. J., 2016, 10, 538 CrossRef CAS PubMed.
- S. Pierik, A. Van Herk, C. Plessis, J. Van Steenis, T. Loonen and A. Bombeeck, Eur. Polym. J., 2005, 41, 1212–1218 CrossRef CAS.
- G. Quintens and T. Junkers, Polym. Chem., 2022, 13, 2019–2025 RSC.
- F. Lugo, L. Trossaert, Y. Marien, M. Sabbe, M. Edeleva, D. D'hooge and P. Van Steenberge, Polym. Chem., 2025, 16, 3496–3510 RSC.
- B. Wenn and T. Junkers, Macromol. Rapid Commun., 2016, 37, 781–787 CrossRef CAS PubMed.
- Y. Kamiyama, R. Tamate, T. Hiroi, S. Samitsu, K. Fujii and T. Ueki, Sci. Adv., 2022, 8, eadd0226 CrossRef CAS PubMed.
- C. Li, S. Zhang, J. Jiang, S. Wang, S. He and J. Song, Sci. Adv., 2024, 10, eads9226 CrossRef CAS PubMed.
- H. Eoh, H. S. Kang, M. J. Kim, M. Koo, T. H. Park, Y. Kim, H. Lim, D. Y. Ryu, E. Kim and J. Huh, Adv. Funct. Mater., 2019, 29, 1904055 CrossRef CAS.
- T. Faraone, J. Qian, S. Kolagatla, A. L. Bradley, L. Florea and C. Delaney, Adv. Mater., 2025, 2504116 CrossRef CAS PubMed.
- H. S. Kang, J. Lee, S. M. Cho, T. H. Park, M. J. Kim, C. Park, S. W. Lee, K. L. Kim, D. Y. Ryu and J. Huh, Adv. Mater., 2017, 29, 1700084 CrossRef PubMed.
- M. S. Khan, R. Lachmayer and B. Roth, OSA Continuum, 2020, 3, 2808–2816 CrossRef CAS.
- Y. Yuan, X. Li, L. Jiang, M. Liang, X. Zhang, S. Wu, J. Wu, M. Tian, Y. Zhao and L. Qu, Nat. Commun., 2023, 14, 3967 CrossRef CAS PubMed.
- A. M. Goodman, Appl. Opt., 1978, 17, 2779–2787 CrossRef CAS PubMed.
- J. Li, C.-H. Wen, S. Gauza, R. Lu and S.-T. Wu, J. Disp. Technol., 2005, 1, 51 CrossRef CAS.
- M. Mitov, Adv. Mater., 2012, 24, 6260–6276 CrossRef CAS PubMed.
- S.-T. Wu, U. Efron and L. D. Hess, Appl. Opt., 1984, 23, 3911–3915 CrossRef CAS PubMed.
- T.-D. Nguyen, E. Sierra, H. Eguiraun and E. Lizundia, Eur. J. Phys., 2018, 39, 045803 CrossRef.
- T. Xu, H. Shi, Y. K. Wu, A. F. Kaplan, J. G. Ok and L. J. Guo, Small, 2011, 7, 3128–3136 CrossRef CAS PubMed.
- M. B. Applegate, J. Coburn, B. P. Partlow, J. E. Moreau, J. P. Mondia, B. Marelli, D. L. Kaplan and F. G. Omenetto, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 12052–12057 CrossRef CAS PubMed.
- S. G. Rayner, C. C. Howard, C. J. Mandrycky, S. Stamenkovic, J. Himmelfarb, A. Y. Shih and Y. Zheng, Adv. Healthcare Mater., 2021, 10, 2100031 CrossRef CAS PubMed.
- J. Ma, J. Wu, Z. Lin, J. Wang, W. Yao, Y. Zhang, X. Zhang, L. Zhu, Y. Hayasaki and H. Zhang, Small Sci., 2025, 5, 2400400 CrossRef CAS PubMed.
- P. Yin, Y. Liu, L. Xiao and C. Zhang, Polymers, 2021, 13, 2834 CrossRef CAS PubMed.
- H. Seo, G. J. Ko, S. Song, J. H. Lee, Y. Seo, S. Han, C. H. Eom, H. Kim, S. Kim and K. S. Lee, Adv. Sci., 2025, e06482 CrossRef CAS PubMed.
- Y. Lu, H. Lyu, A. G. Richardson, T. H. Lucas and D. Kuzum, Sci. Rep., 2016, 6, 33526 CrossRef CAS PubMed.
- Y. Lu, G. Yang, S. Wang, Y. Zhang, Y. Jian, L. He, T. Yu, H. Luo, D. Kong and Y. Xianyu, Nat. Electron., 2024, 7, 51–65 CrossRef CAS.
- J. Li, Y. Liu, L. Yuan, B. Zhang, E. S. Bishop, K. Wang, J. Tang, Y.-Q. Zheng, W. Xu and S. Niu, Nature, 2022, 606, 94–101 CrossRef CAS PubMed.
- J. Liu, C. Li, T. Brans, A. Harizaj, S. Van de Steene, T. De Beer, S. De Smedt, S. Szunerits, R. Boukherroub and R. Xiong, Int. J. Mol. Sci., 2020, 21, 1540 CrossRef CAS PubMed.
- R. Xiong, K. Raemdonck, K. Peynshaert, I. Lentacker, I. De Cock, J. Demeester, S. C. De Smedt, A. G. Skirtach and K. Braeckmans, ACS Nano, 2014, 8, 6288–6296 CrossRef CAS PubMed.
- D. Wu, X. Wang, R. Li, C. Wang, Z. Ren, D. Pan, P. Ren, Y. Hu, C. Xin and L. Zhang, Sci. Adv., 2025, 11, eadw1272 CrossRef CAS PubMed.
- H. Deng, K. Sattari, Y. Xie, P. Liao, Z. Yan and J. Lin, Nat. Commun., 2020, 11, 6325 CrossRef CAS PubMed.
- M.-H. Oh, Y.-H. Kim, S.-M. Lee, G.-S. Hwang, K.-S. Kim, Y.-N. Kim, J.-Y. Bae, J.-Y. Kim, J.-Y. Lee and Y.-C. Kim, Sci. Adv., 2023, 9, eadh9962 CrossRef CAS PubMed.
- R. Hinchet and S.-W. Kim, ACS Nano, 2015, 9, 7742–7745 CrossRef CAS PubMed.
- R. Zhang, M. Wang, Z. Wan, Z. Wu and X. Xiao, Results Eng., 2023, 19, 101314 CrossRef CAS.
- W. Guo, Y. Xia, Y. Zhu, S. Han, Q. Li and X. Wang, Nano Energy, 2023, 108, 108229 CrossRef CAS.
- K.-W. Kim, S. J. Park, S.-J. Park, I. Kim, B. Park, S. H. Kim, U. Jeong, J. K. Kim and C. Yang, npj Flexible Electron., 2024, 8, 18 CrossRef CAS.
- C. Zhang, Z. Peng, C. Huang, B. Zhang, C. Xing, H. Chen, H. Cheng, J. Wang and S. Tang, Nano Energy, 2021, 81, 105609 CrossRef CAS.
- Y. Chen, B. Xie, J. Long, Y. Kuang, X. Chen, M. Hou, J. Gao, S. Zhou, B. Fan and Y. He, Adv. Mater., 2021, 33, 2104290 CrossRef CAS PubMed.
- S. Lee, E. V. Bordatchev and M. J. Zeman, J. Micromech. Microeng., 2008, 18, 045011 CrossRef.
- G. Choi, S. Yoon, Y. Jung, H. Park, D. Kim, S. Yu, M. Kim and S. H. Ko, Carbon, 2025, 120852 Search PubMed.
- H. Matsumoto, H. Inoue, H. Yamashita, H. Morita, S. Hirose, M. Ogasawara, H. Yamada and K. Hattori, Multi-beam mask writer MBM-1000 and its application field, Photomask Japan 2016: XXIII Symposium on Photomask and Next-Generation Lithography Mask Technology, SPIE, 2016, pp. 26–31.
- A. Dobos, J. Van Hoorick, W. Steiger, P. Gruber, M. Markovic, O. G. Andriotis, A. Rohatschek, P. Dubruel, P. J. Thurner, S. Van Vlierberghe, S. Baudis and A. Ovsianikov, Adv. Healthcare Mater., 2020, 9, e1900752 CrossRef PubMed.
- C. A. DeForest and K. S. Anseth, Nat. Chem., 2011, 3, 925–931 CrossRef CAS PubMed.
- J. Cui, A. Rodriguez-Rodriguez, M. Hernandez, M. C. Garcia-Gutierrez, A. Nogales, M. Castillejo, D. Mosegui Gonzalez, P. Muller-Buschbaum, T. A. Ezquerra and E. Rebollar, ACS Appl. Mater. Interfaces, 2016, 8, 31894–31901 CrossRef CAS PubMed.
- J. Kim, D. Won, T. H. Kim, C. Y. Kim and S. H. Ko, Biosens. Bioelectron., 2024, 258, 116327 CrossRef CAS PubMed.
- M. G. Stanford, C. Zhang, J. D. Fowlkes, A. Hoffman, I. N. Ivanov, P. D. Rack and J. M. Tour, ACS Appl. Mater. Interfaces, 2020, 12, 10902–10907 CrossRef CAS PubMed.
- J. Lin, Z. Peng, Y. Liu, F. Ruiz-Zepeda, R. Ye, E. L. Samuel, M. J. Yacaman, B. I. Yakobson and J. M. Tour, Nat. Commun., 2014, 5, 5714 CrossRef CAS PubMed.
- S. Y. Jeong, Y. W. Ma, J. U. Lee, G. J. Je and B. S. Shin, Sensors, 2019, 19(22), 4867 CrossRef CAS PubMed.
- J. Shin, J. Ko, S. Jeong, P. Won, Y. Lee, J. Kim, S. Hong, N. L. Jeon and S. H. Ko, Nat. Mater., 2021, 20, 100–107 CrossRef CAS PubMed.
- Y. Jung, S. Jeong, G. Heo, K. R. Pyun, S. H. Choi, J. Bang, J. G. Lee, H. Kim, J. Shin, S. Hong, J. Lee, D. Won, J. Song and S. H. Ko, Joule, 2025, 9, 102007 CrossRef CAS.
- M. Abdulhafez, G. N. Tomaraei and M. Bedewy, Carbon, 2026, 246, 120941 CrossRef CAS.
- T. Kumada, H. Akagi, R. Itakura, T. Otobe, M. Nishikino and A. Yokoyama, Appl. Phys. Lett., 2015, 106, 221605 CrossRef.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2026 |