
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effects of external electric fields on proteins
Michal Cifra *a,
Saurabh Kumar Pandeya,
Tomás Zakara,
Michaela Poplováa,
Daniela G. Blanco Campoy†
a,
Jiří Průšaa,
Daniel Havelkaa,
Paolo Marracinob,
Micaela Libertic,
Francesca Apollonio*c,
Niall J. English*d,
Carl Calemanef and
Erik G. Marklund*g
*a,
Saurabh Kumar Pandeya,
Tomás Zakara,
Michaela Poplováa,
Daniela G. Blanco Campoy†
a,
Jiří Průšaa,
Daniel Havelkaa,
Paolo Marracinob,
Micaela Libertic,
Francesca Apollonio*c,
Niall J. English*d,
Carl Calemanef and
Erik G. Marklund*g
aInstitute of Photonics and Electronics of the Czech Academy of Sciences, Prague 18200, Czechia. E-mail: cifra@ufe.cz
bRise Technology srl, Lungomare Paolo Toscanelli, 170, 00121 Rome, Italy
cDepartment of Information Engineering, Electronics, and Telecommunications, Sapienza University of Rome, 00184 Rome, Italy. E-mail: francesca.apollonio@uniroma1.it
dUniversity College Dublin, Ireland. E-mail: niall.english@ucd.ie
eDepartment of Physics & Astronomy, Uppsala University, Box 516, SE-75120, Uppsala, Sweden
fCenter for Free-Electron Laser Science, DESY, Notkestrasse 85, DE-22607 Hamburg, Germany
gDepartment of Chemistry for Life Sciences, Uppsala University, Box 576, SE-75123 Uppsala, Sweden. E-mail: erik.marklund@kemi.uu.se
First published on 17th April 2026
Abstract
Proteins are highly abundant and, as a biomolecular class, have very versatile functions in living systems. Protein structures contain electrically charged residues and proteins’ electrostatics are crucial for their function. Although protein activity is commonly modulated through a variety of ligands and post-translational modifications, an external electric field (EF) represents an alternative, physical approach. By exerting forces on charged and dipolar regions, EFs can reshape the energetic landscape and dynamic behavior of proteins. This approach offers a mass-free, rapidly switchable, spatially precise, non-contact, and reagent-free way to control protein conformation and function – features increasingly appealing for applications in green bioprocessing, neuromodulation, ultrafast structural biology, and in studying proteins without clearly ligandable sites. Despite the growing evidence for diverse and reversible control of proteins by EF, the mechanisms are still underexplored and applications have not yet grown to their full potential. This review focuses on molecular mechanisms and integrates the findings of the effects of external EFs on proteins from both computational simulations and experimental studies. The literature shows that the EF acts on protein charged and dipolar groups, and when the EF parameters are well tailored, the EF consequently triggers effects on protein rigid body motion, secondary structure, tertiary structure, quaternary structure and molecular conformation, ultimately leading to changes in protein interactions and function (enzymatic, ion channelling, switching, …). These effects are being utilized not only on proteins as food components but also for bionanotechnological applications, e.g. in membrane proteins for controlling their transport properties, and in structural and force-generating proteins to steer self-assembly pathways and dynamic behavior. The compiled evidence clarifies key mechanisms by which EFs influence proteins and identifies promising directions for biomedical, food-processing, and biotechnological applications.
1 Introduction
Accounting for roughly half of the dry mass of most metabolically active cells,1,2 proteins form the single largest class of macromolecules, and play a key roles in biological systems, for example, in catalysis, regulation, and spatiotemporal organisation. At a cellular level, the vast majority of structural aspects and main functions of the human body and all kingdoms of life are carried out by proteins.3The function of a protein is determined by its primary sequence and the explicit conformation of the polypeptide chain.4 Accordingly, the structure, conformational dynamics, and function of proteins and their assemblies,5 as well as strategies to modulate them,6,7 have been the focus of considerable research attention. Anomalies in these protein systems result in serious disruptions of cellular function and give rise to a variety of diseases.8–10 For example, Alzheimer's and Parkinson's diseases are proteinopathies associated with protein misfolding and subsequent amyloid formation.11 Although protein malfunction underlies many pathological conditions, properly functioning proteins act as highly efficient molecular machines – both in vivo (for example, flagellar motors, myosin, DNA and RNA polymerases) and in bionanotechnological applications: serving as nanomotors,12 nanoscissors (lytic and cleavage enzymes),13 nanoassembly lines (synthase enzymes),14 and photo-electric transducers (light-harvesting protein complexes).15
Therefore, having the ability to control the protein structure and conformation, hence protein function, using an electric field (EF), as is possible in other molecules,16–19 is a formidable opportunity for the development of methods in biotechnology, structural biology, physical chemistry, nanotechnology, biosensing, analytics, the food industry and medicine, which have features unattainable by and complementary to chemical control only.7,20 External EFs offer a fundamentally mass-free, energy-only, parallel handle on proteins: the EF delivers force to every copy of the macromolecule without adding reagents, avoiding the stoichiometric constraints, toxicity, and downstream purification inherent to ligand or covalent chemistries: an advantage already exploited in pulsed EF (PEF) bioprocessing, where sub-millisecond bursts reshape proteins in aqueous media with no added solvent.21 Voltage control also translates into electronic speed: nanosecond EFs of ~100 MV m−1 have driven Å-scale conformational transitions that can be imaged in real time by electric-field-stimulated X-ray crystallography, a timescale orders of magnitude faster than ligand binding or covalent chemistry.22 Crucially, the stimulus can be delivered entirely without contact: picosecond PEFs radiated from dielectric-rod or wide-band antennas have been shown to trigger cellular responses without electrodes23 and microwave and THz EF bursts drive conformational changes in enzymes or disassemble non-covalent protein polymers24,25 and even modulate cellular activity.26 Because EFs couple to charge and permanent or induced dipoles rather than to a defined binding pocket, they can bias catalytic and allosteric energy landscapes even in “undruggable” proteins. This review surveys recent studies on how external EFs (pulsed, static, and oscillating up to the low-THz range) influence protein structure, conformation, and function, both in isolated molecules and in assemblies.
Why can EFs influence and potentially control protein function? Typically, proteins are charged dielectric nano-objects with a complex multipolar charge distribution.27–31 Consequently, the protein electrostatics (the distribution and interaction of electric charges within a protein) is a critical feature influencing its structure, stability, and function.32–34 Strong molecular EFs are known to play an essential role in protein folding,33 protein–ligand,35 protein–solvent,32,36 and protein–protein37 interactions, molecular recognition,38 as well as enzyme catalysis.39–42 Some proteins’ conformation is naturally regulated by EFs in vivo: voltage-gated ion channels switch between open and closed states depending on the voltage across the membranes in which they are incorporated.43–45 For these reasons, it is reasonable to assume that external EFs with appropriately chosen parameters of EF strength, waveform and spatial structure could modulate protein function or be used to probe the biophysical properties of proteins.
Our major focus is on works that provide a mechanistic (protein structure and dynamics-based) understanding of the observed effects so that the electromagnetic-field-based control of protein function and geometry can be rationally developed for the benefit of biomedicine and bionanotechnology, exemplified by our recent works.21,46,47 While there are reviews which cover some selected form of EF on proteins or application focus, they lack the comprehensive focus on mechanisms and often include complex samples (emphasizing food products or protein extraction from tissues,48–63 or focusing on protein structures in cells64,65) which prevents drawing mechanistic conclusions about the EF effects on proteins.
The structure of our paper is as follows: after an introduction, we move step-wise from fundamental mechanisms of EF action on proteins (Fig. 1), covering effects on protein motion, structure, energetics, electrostatics, interactions, and function. We provide detailed comprehensive tables of experimental (Table 2) and computational works (Table 3), with necessary methodical parameters, focusing on pure or well-defined protein samples. We include sections on indirect electrophysical and electrochemical influences, and conclude by addressing outstanding challenges, and outlining future research directions.
 | ||
| Fig. 1 An overview scheme of the physical effects of an EF on proteins as observed in molecular dynamics simulations (Table 3) and/or in experiments (Table 2). The blue shade denotes the protein status before and the orange color is the status of the protein during or shortly after EF action. The EF acts by force on the protein as an electrically charged particle by causing translational motion and via the protein electric dipole causing rotational motion of the protein. A sufficiently strong EF causes (in a field strength increasing manner) changes in protein structure ranging from slight conformational changes to alteration and disruptions in tertiary and even secondary structure leading to protein size and shape changes and ultimately can lead to unfolding. In another, complementary perspective the EF modulates the free energy landscape of the protein. The structural changes then lead to the modulation of the protein’s electrostatic, nanomechanical, dynamic and vibrational properties across all levels of protein organization. The EF can also act (often in parallel with effects on single protein chain structure) on the interactions between proteins either causing aggregation (via partial unfolding), or disassembly of homotypic or heterotypic protein poly/multi/oligomers, eventually perturbing or enhancing interaction of proteins with non-peptide interaction partners. Ultimately, the EF can disrupt, modulate or even enhance the function of proteins in their enzymatic, self-assembling roles or affect photonic (such as fluorescence) or channeling properties. | ||
2 Mechanisms of EF action and effects on proteins
At the length and energy scales of chemistry and biology, every force (direct or emergent from many body interactions) that holds atoms together inside a molecule (covalent, ionic) or brings separate molecules together (hydrogen bonds, electrostatic, van der Waals, …) in solution is ultimately a manifestation of electromagnetic interactions.66 Many non-covalent molecular interactions are a direct consequence of electrostatic and (quantum) electrodynamic interactions.67 Therefore, the EF naturally has a supreme position in the understanding of and action on the behavior of atoms and molecules. The interaction of an EF with matter fundamentally arises from its coupling to electric charges (ionic or electronic). In proteins, the direct interaction with an external field occurs due to the presence of electric charges on specific ionizable groups (fixed charges) of the constituent amino acids. Examples include glutamate and aspartate, which carry negative charges, and lysine, arginine, and histidine, which carry positive charges. The ionization state and the actual electric charge of these groups depend on the pH of the environment. A mechanistic molecular dynamics (MD) simulation study demonstrated on model peptides68 that the effect of the EF on a peptide (namely on root-mean-square deviation of the peptide) is proportional not to the net charge of a protein, but to the number of charged residues, directly confirming the role of charged residues presences in EF effects on proteins.Furthermore, an EF can interact with mobile electrons, such as the delocalized π-electrons found in amino acids such as tryptophan, tyrosine, and phenylalanine. The EF ![[E with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0045_20d1.gif) exerts a force
exerts a force ![[F with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0046_20d1.gif) = q· on electric charge q. As a result, the atomic groups with zero net charge or dipole are unaffected directly by the EF. It can be helpful to visualize the charged groups in a protein as the points where the EF exerts its influence. However, even non-charged groups may be affected indirectly. This occurs through interactions mediated by the surrounding solvent, nearby charged groups, or even distant charged groups within the protein structure.
= q· on electric charge q. As a result, the atomic groups with zero net charge or dipole are unaffected directly by the EF. It can be helpful to visualize the charged groups in a protein as the points where the EF exerts its influence. However, even non-charged groups may be affected indirectly. This occurs through interactions mediated by the surrounding solvent, nearby charged groups, or even distant charged groups within the protein structure.
The characteristics of an EF can vary based on its temporal and spatial parameters, which significantly influence its effects on proteins. Key parameters include the EF's vector (magnitude and direction), spectral (temporal) content and spatial distribution. The EF can be modulated in time in various ways, delivered as a complex waveform, or applied as a train of pulses, among other variations. For example, when the EF is delivered in pulses,69 the number of pulses and the intervals between them (determined by an inverse parameter known as the firing frequency) are crucial factors. Each of these parameters can influence how the EF interacts with individual protein molecules or protein assemblies. Beyond temporal structuring, the electromagnetic field can also be spatially shaped.70–72 However, creating complex EF spatial patterns, particularly at molecular scales and in three dimensions, presents significant technical challenges. Such efforts approach the physical limits of current nanotechnology73–75 and photonics, and have therefore not been extensively explored for protein control. Here a great portion of inspiration can be absorbed from nature, as biomolecular interactions are largely enabled by the fact that the biomolecules including proteins themselves are nanoscale devices generating spatially complex EFs34,76 with a dynamic range across 12 orders of magnitude in time77 and Ångström-level spatial precision.
2.1 Effects on protein rigid body motions
These works demonstrate that an EF can act on protein polymer fibers, leading to their linear and turning motions, hence enabling molecular manipulation at the single protein fiber level. Those results have a potential significance for bio-nanotechnological manipulation techniques and provide inspiration for what effects one could expect on the single protein level.
The tendency of a protein’s dipole moment to rotate and align with an external EF is the fundamental physical mechanism underlying EF-induced protein rotation. The same principle also forms the basis of many dielectric spectroscopy techniques, where the relaxation of the real part of the permittivity of protein solutions reflects the rotational diffusion dynamics of the proteins.103,104 The rotation of proteins by an EF has also been observed using optical methods, by analyzing the anisotropy of the optical signal, which is caused by EF-induced rotation of proteins, as illustrated in Fig. 2.81,105–117 However, the EF turning effects on proteins are poorly experimentally investigated at the single protein level. The two major reasons are (i) the difficulty of observing or imaging a single protein orientation on short time scales and (ii) the high EF strength that might be required to overcome the rotational diffusion due to thermal motion. So far, there have only been a few computational works that have demonstrated this effect, see Table 3, which may guide future experiments along these lines. An emerging area where rotational manipulation of proteins with EFs would have a large impact is X-ray imaging of single molecules.79 In X-ray single particle imaging (SPI), individual proteins are injected with random orientation into vacuum where they are exposed to the extremely intense (up to 1012 photons per pulse) femtosecond pulse delivered by the XFEL, each giving rise to a diffraction pattern reflecting its orientation at the time of exposure.118 From the diffraction data the three-dimensional (3D) structures of the proteins can in principle be determined, alleviating the need for crystallization or other limitations that restrict conventional techniques for structural biology. Powerful algorithms119 exist to assemble the data and enable structure determination, but they can be stumped by sparse or missing data, or noise from radiation damage,120 electronics or sample heterogeneity.121 One way to improve the performance of these algorithms is to control the rotation of the protein as it is exposed to the XFEL pulse. This could make SPI work with smaller sets of useful diffraction data, which would extend the application range of this technique.122 The protein rotation can be controlled by using static EFs that couple to the intrinsic dipole moment of the protein,79,123 or using lasers via a protein's anisotropic polarizability.124 However, these techniques have so far been described only theoretically. Efforts are currently being made to integrate EF-mediated orientation with native mass spectrometry to deliver well-defined and homogeneous mass-selected and oriented protein complexes from heterogeneous mixtures for SPI at the European XFEL.128,129 Marklund et al.79 simulated a gas-phase Trp-cage, a ribosome protein domain, ubiquitin and lysozyme under the influence of strong EFs and showed that there is an “orientation window” of field strengths where proteins become oriented but with intact structures. At field strengths extending into the GV m−1 range the proteins started to unfold on a 10-ns timescale, whereas for longer exposure times (50 ns) the window appeared to shift towards lower fields, Fig. 2. Subsequent simulations using ramped fields that gradually increase from zero field strength, which is more realistic than an instantaneous onset, indicated that ramping is better for preserving the structure, but also that protein orientation happens before unfolding – “orientation before destruction” – meaning that a well-timed pulse could be used to image native-like structures also with destructive field strengths.123 With the purpose of increasing the dipole of the gas phase samples, to make the orientation easier, simulation studies have explored the effect of adding a layer of water around the protein.130 This seems to have some positive effects, at least for the protein simulated. The water increases the total dipole slightly, but more importantly it tends to make the dipole more stable and decreases the deviation between the protein structures, which is crucial for reaching high resolution SPI. Another way to increase the dipole of proteins would be to add dipole enriching segments to the molecule itself, like chromophores,131 but then again, it's important to understand how these affect the structure.
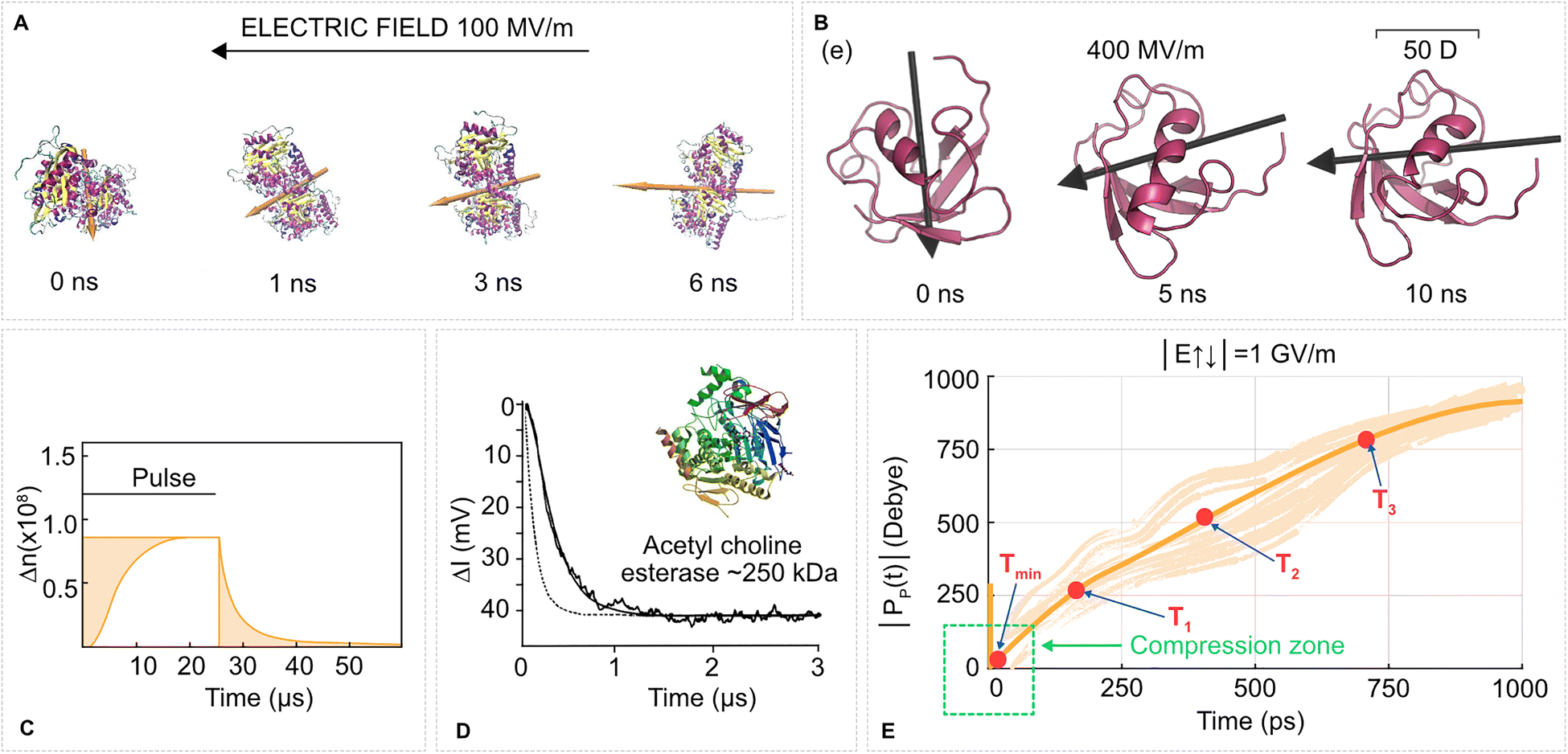 | ||
| Fig. 2 Protein rotation due to the torque of an EF on a protein permanent dipole is one of the most straightforward yet underutilized effects. (A) Tubulin protein in a molecular dynamics simulation (water and ions not shown) in a 100 MV m−1 EF aligns its dipole moment (orange arrow) with the EF vector (black arrow on the top) within a few nanoseconds. Based on our data from ref. 78. (B) Ubiquitin in a gas phase molecular dynamics simulation rotates to align its dipole moment with the EF vector. Adapted with permission from ref. 79. © (2017) American Chemical Society. (C) A pulsed EF (0.4 MV m−1, 25 µs) induces birefringence of a water solution of tubulins due to alignment of tubulin proteins with the EF. Adapted with permission from ref. 80. © (1985) Elsevier. (D) The same experimental method as in C (birefringence signal in mV), relaxation of esterase solutions after a 2.46 MV m−1 EF pulse. Adapted with permission from ref. 81. © (1996) Elsevier. (E) Time evolution of the dipole moment magnitude of ubiquitin from molecular dynamics simulations. The protein is initially positioned with its dipole moment vector antiparallel to the EF vector, and hence undergoes fast compression. Then the protein swiftly rotates to parallel with the increase of dipole moment magnitude. T1, T2, T3 corresponds to time points when the dipole magnitude was 1×, 2×, 3× the initial magnitude, respectively. The thick line is the mean of the traces from individual MD simulations. Adapted with permission from ref. 82. © (2022) Wiley. | ||
2.2 Effects on protein structure
Beyond translational and rotational rigid body-like motions, intense EFs can significantly influence protein structure at various levels, including overall conformation, tertiary structure, and secondary structure, as well as size, shape, surface area, and unfolding behavior, see Tables 2 and 3.MD simulations have shown extensively that intense EFs can affect protein conformation.68,78,82,123,126,133–158 In larger proteins, even moderate external fields (105–107 V m−1) influence the protein conformation. For example, fields two orders of magnitude weaker than typically required for protein damage caused the SARS-CoV-2 spike's receptor-binding β-sheet to unravel into an unstructured coil, abolishing its ability to bind ACE2 (Fig. 3).125 Similarly, a 30 MV m−1 static field applied to a GPCR (5-HT1A receptor) drove an atypical inward shift of its transmembrane helix 6 and disrupted the normal activation by its agonist.135 Simulations have also shown that oscillating fields can have distinct effects on the protein structure. A study of the effect on the structure of Alzheimer’s amyloid-β aggregates revealed that oscillating EFs (Os-EEF) in the low-frequency MHz–GHz range induce an “explosive” dissociation of peptide plaques, accompanied by the loss of their secondary structure, into monomers that do not reform (Fig. 4). In contrast static fields (St-EEF) tend to align peptides into dipole-stabilized parallel bundles that recombine once the field is removed.133 This superiority of oscillating fields for irreversible disaggregation was observed consistently for both short fragments and full-length Aβ, and higher frequencies (THz) were paradoxically less effective. Notably, some proteins can tolerate very strong fields – enzymatic active sites can generate internal fields on the order of 100 MV m−1 – suggesting that external fields in the 0.1–1 MV m−1 range can be harnessed without catastrophic damage.
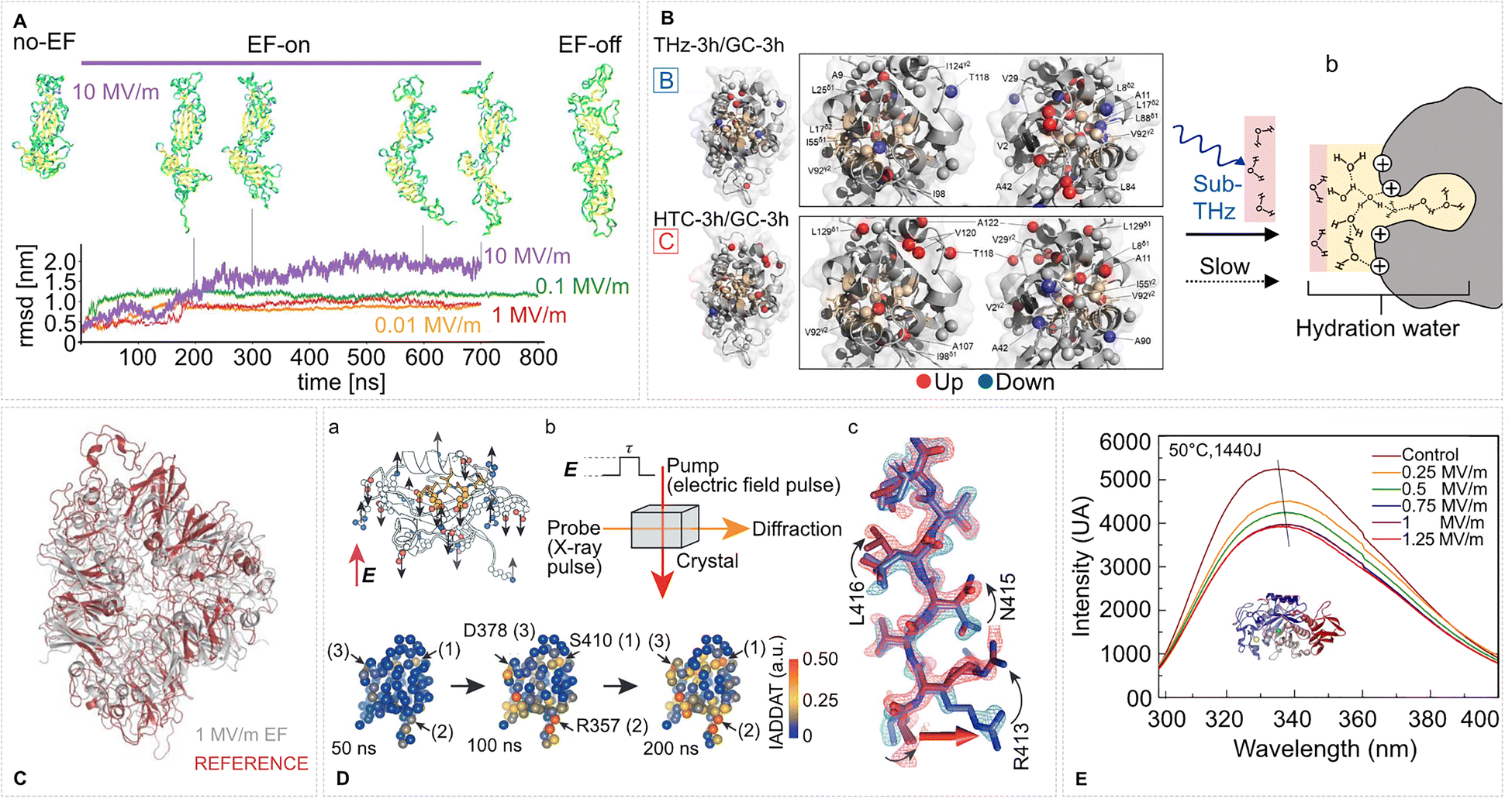 | ||
| Fig. 3 Conformation and tertiary structure changes in proteins induced by intense EFs. (A) EFs induce global conformational changes in the SARS-CoV-2 spike glycoprotein. Shown are representative RMSD trajectories under various EF strengths and structural snapshots under an EF of 10 MV m−1 at selected time points during and after field exposure. Adapted from ref. 125. Springer Nature (2024), CC BY 4.0. (B) Tertiary structure response of lysozyme to subTHz EF perturbation. Pathway B (THz-3 h vs. GC-3 h): 0.1 THz irradiation strengthens signals of core methyl groups (red spheres) while peripheral sites weaken (blue), indicating core tightening. Pathway C (HTC-3 h vs. GC-3 h): mild heating to 31 °C causes the inverse effect—core weakening and peripheral strengthening—suggesting cavity loosening. (b) Sub-THz radiation modifies lysozyme hydration. Schematic shows altered hydration 24 h post-irradiation. Fast, H-bond-broken water transitions to structured hydration (ochre) at the heterogeneous surface, promoting hydrophobic shielding and altered surface energetics. Adapted from ref. 24. Springer Nature (2024), CC BY 4.0. (C) EF-driven conformational changes in pea protein after 25 ns of 1 MV m−1 MD simulated EF exposure. Adapted with permission from ref. 126. © (2023) Elsevier. (D) EF effects on the LNX2PDZ2 protein (PDB ID: 2VWR) revealed by time-resolved X-ray crystallography. (a) Sampling of charged residues (highlighted in red) illustrates potential EF-responsive sites; the ligand is shown in yellow. (b) In EF-X, protein crystals are subjected to an EF pulse of duration τ (pump), followed by X-ray probing (probe) to detect induced structural changes. (c) Example of local rearrangements in hydrogen bonding visualized in the electron density map. Lower: Time evolution of EF-induced structural responses is mapped onto the protein's tertiary structure using Cα atom positions; color intensity reflects IADDAT (integration of absolute difference density above threshold) values. Adapted with permission from ref. 22. © (2016) Springer Nature. (E) Fluorescence spectra of α-amylase after PEF exposure show field-strength-dependent red shifts, indicating tertiary structural rearrangement. Adapted with permission from ref. 127. © (2021) Elsevier. | ||
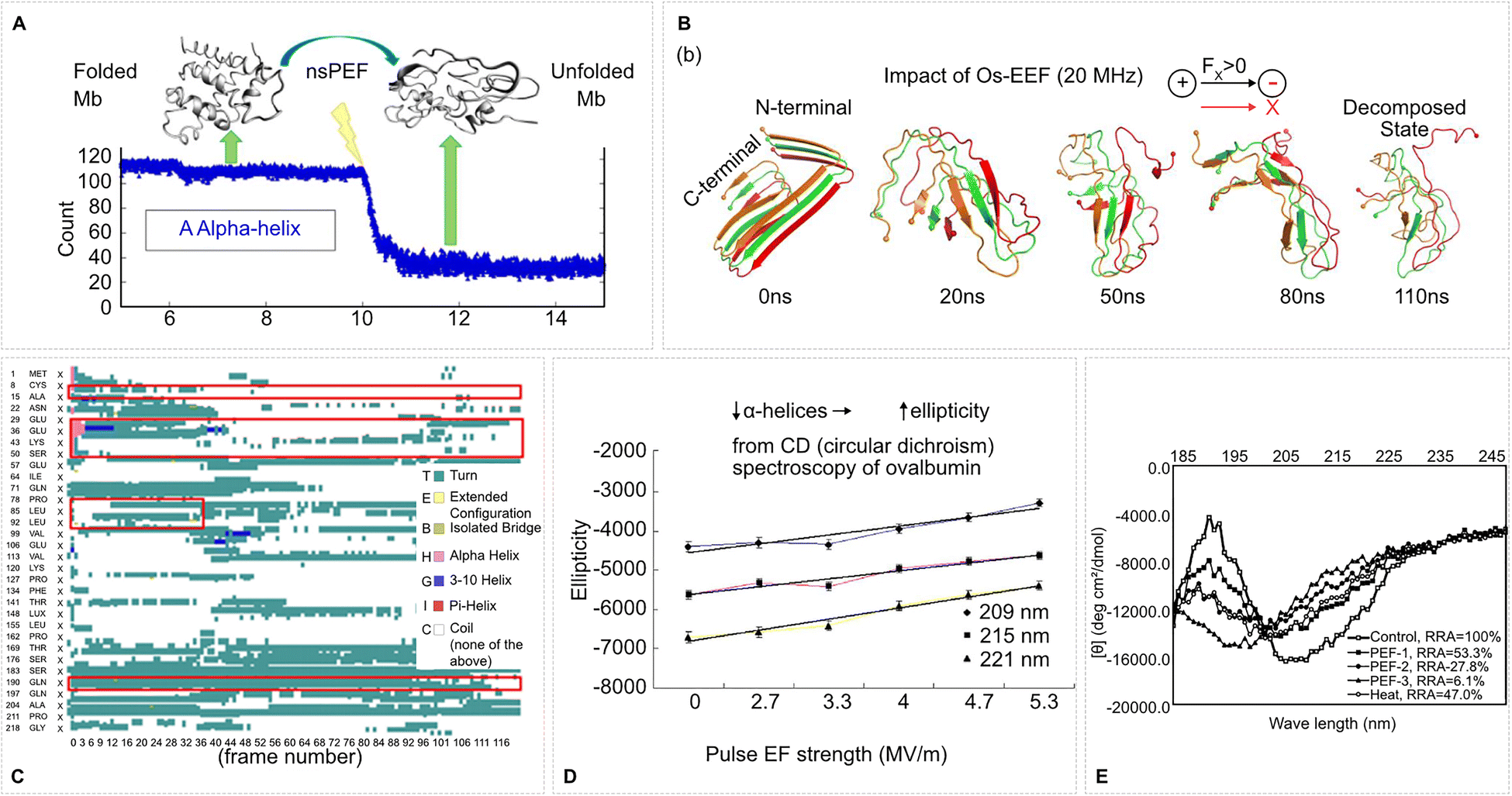 | ||
| Fig. 4 EF-induced secondary structure transitions in proteins. (A) Nanosecond PEFs (nsPEFs) unfold myoglobin (Mb), as seen by the sharp drop in α-helical content. The structural transition from folded to unfolded Mb correlates with the timing of the nsPEF exposure. Adapted with permission from ref. 159. © (2013) American Chemical Society. (B) Oscillating EFs (20 MHz) progressively disrupt the secondary structure of an Aβ-42 peptide fragment, leading to decomposition over 110 ns. Snapshots show the gradual destabilization from N- to C-terminal under directional field-induced force. Adapted from ref. 133. CC BY 4 (2025) American Chemical Society. (C) Time-resolved secondary structure evolution of β-casein under a pulsed EF from MD simulations. Frame-by-frame analysis reveals EF-induced transitions from α-helix (pink) to coil (cyan) and other motifs. Adapted with permission from ref. 160. © (2023) Elsevier. (D) Circular dichroism spectra of ovalbumin show increased ellipticity and loss of α-helicity with rising PEF strength at characteristic wavelengths (209, 215, and 221 nm), indicating unfolding or loss of ordered structure. Adapted with permission from ref. 161. © (2018) Elsevier. (E) CD spectra of pepsin exposed to varying PEF treatments show field-strength-dependent loss of secondary structure, reflected in reduced relative retention of activity (RRA) and spectral shifts, compared with thermal denaturation. Adapted with permission from ref. 162. © (2004) American Chemical Society. | ||
Experimental studies strongly support these predictions.21,22,24,127,134,163,164 Time-resolved crystallography using MV m−1 pulses (EF-X) has directly visualized Å-scale backbone and side-chain movements throughout a protein crystal, mirroring allosteric conformational changes.22 Terahertz (THz) absorption and NMR experiments have shown that intense sub-THz irradiation perturbs protein–water dynamics nonthermally: in ubiquitin, a 0.1 THz field accelerates hydrogen–deuterium exchange at buried amides while slowing exchange at surface loops, an effect opposite to uniform heating.163 Dielectric relaxation measurements combined with NMR spectroscopy on lysozyme solutions similarly found that sub-THz fields gradually lower the solvent permittivity by reorienting hydration water into a more “hydrophobic” structuring around the protein, without bulk heating,24 and affect protein conformation (Fig. 3).
Real-time CD and autofluorescence studies were carried out for bovine serum albumin (BSA) and lysozyme in solution for low-strength EFs. It was conjectured that for the low-intensity fields, unfolding was caused by a frictional force generated by the electrophoretic motion of the proteins. Prolonged exposure to an EF led the proteins to dissipate a large amount of frictional energy – indicating that protein unfolding is a crucial first step for protein aggregation and also possibly for the formation of amyloid fibrils.165 Complimentarily, dynamic light scattering measurements resolved field-dependent dynamic modes of BSA and interpreted them in terms of field-induced intermolecular interactions in solution.166
PEFs in the 0.1 MV m−1 range have also been widely applied to proteins in solution, revealing diverse effects on protein conformation. Spectroscopic analyses (intrinsic fluorescence, circular dichroism (CD), Fourier transform infrared (FTIR)) show that PEF treatments often partially unfold proteins, reducing α-helix content and increasing disordered or β-sheet structures. This enhanced flexibility can increase the exposure of functional sites: for instance, 1 MV m−1 PEF on pea protein cut its helix fraction by 23% while doubling its binding capacity for the polyphenol EGCG (from 4 to 10 binding sites, confirmed by docking), resulting in higher antioxidant performance,126 as illustrated in Fig. 3b. In some cases, high-intensity PEFs can even promote refolding; prawn tropomyosin treated at 2 MV m−1 underwent “extensive spiralization” into a tightly packed conformation, effectively masking its IgE-binding epitopes and lowering allergenicity.134 By contrast, milder 0.5–1 MV m−1 pulses applied to the same protein increased its flexibility and surface exposure, initially raising antibody reactivity before eventual epitope masking at higher fields.134
Functional assays corroborate that conformational changes translate into altered activity. Nonthermal PEF inactivation of enzymes is one compelling example: a 0.25–1.25 MV m−1 pulse regime targeting α-amylase caused 70% loss of activity by specifically disrupting a tryptophan-rich active-site region, without the aggregation observed in thermal denaturation.127 Likewise, a brief 2 MV m−1 PEF was found to affect conformation of α-amylase eliminating 80% of its activity when the enzyme was bound in an electrostatic complex with pectin.167 Despite this extensive conformational damage, the PEF-treated enzyme–pectin complexes did not precipitate; instead, they assembled into larger but soluble branched structures. Classical studies used intense EFs on proteins or proteins-in-membrane fragments in solutions. They used changes in protein sample refractive index anisotropy as a real-time measure for protein conformational changes. For example, an electric pulse can induce bacteriorhodopsin to transiently release and rebind protons at distinct sites, indicating that fields perturb its proton-binding equilibrium.168 In dried purple membrane films, alternating high-voltage fields cause complex absorbance changes that imply a multi-step cyclic conformational transition in bacteriorhodopsin.169 Strong static fields drive a reversible spectral shift of purple bacteriorhodopsin to its deprotonated “blue” form, with only a minor retinal rotation observed.170 In solution, microsecond field pulses reveal two distinct conformational phases in bacteriorhodopsin: an ultrafast retinal reorientation followed by a slower protein charge rearrangement.171 Early pulsed-field experiments detected intramembrane structural changes with restricted chromophore motion in bacteriorhodopsin, and inferred an unusually high dipole polarizability as well as a five-state conformational cycle.172 Even relatively weak fields (15 kV m−1) can trigger a cooperative conformational change in bacteriorhodopsin (on a millisecond timescale).115
These studies demonstrate that strong EFs – whether static, pulsed, or oscillating – offer a precise means to modulate protein conformation and often also a function. Potential uses range from physically inactivating pathogens and enzymes (e.g. “electro-denaturation” to weaken viral spikes or halt spoilage enzymes),125 to enhancing food proteins’ digestibility and binding properties through conformational loosening,126 to novel therapeutic approaches like disrupting amyloid aggregates with oscillating fields.133 In parallel, the ability to trigger and probe conformational changes with fields (as in EF-X) is opening new avenues for mapping protein energy landscapes and allosteric pathways.22
 | ||
| Fig. 5 One of the most common effects seen in MD simulations with EFs is protein unfolding. Here we show an example of extensive analysis from ref. 123, where unfolding pathways of ubiquitin in an EF were obtained. Adapted from ref. 47. CC BY 3 (2021) Royal Society of Chemistry. (A) The native structure of ubiquitin with the α-helix in red and the five β-strands in blue and labelled with roman numerals. (B) Physical connections (labelled B–E) between strands I–V in the native structure and the residues involved. (C) An unfolding pathway at 500 MV m−1. The sate of connections are indicated as a five-bit binary number *BCDE, where 0 and 1 indicate a connection being intact and broken, respectively. D) Transition map between (un)folding states in a 300 MV m−1 EF, where node sizes indicate population sizes and edge widths indicate transition frequencies. Green represents transitions towards the unfolded state (downwards) and blue the opposite direction. The underlying data for this map is the aggregated data of 100 short simulations. Here, the unfolding takes place via a quite well-defined pathway, except that the C connection is sometimes ruptured before D and sometimes not. At higher field strengths the unfolding clearly prefers breaking the D connection first.123 | ||
On the experimental front, a range of spectroscopic and biophysical techniques have been employed to probe EF-induced secondary structure changes and unfolding. Far-UV CD and FTIR spectroscopy provide direct readouts of secondary structure content (helix, sheet, turn), while intrinsic fluorescence (tryptophan emission) and extrinsic dye fluorescence assays (e.g., ANS or thioflavin T binding) report on tertiary structure exposure and amyloid formation; complementary methods such as light scattering, electron microscopy, and X-ray scattering have also been used to characterize aggregation state and structural dimensions under EFs.173,176,177 Using such methods, numerous studies have confirmed that EFs can induce significant secondary structure alterations and unfolding in practice. In an in situ CD study, Rodrigues et al. showed that applying a low-frequency moderate EF during heating lowers the thermal denaturation midpoint of β-lactoglobulin and alters its unfolding pathway, as evidenced by changes in CD spectra and increased hydrophobic surface exposure.178 Peng et al. observed that high-frequency terahertz waves (∼35 THz) act as a non-thermal denaturing agent on an Aβ amyloid, breaking apart β-sheet-rich fibrils and slowing fibrillization by ∼80%, as indicated by reduced thioflavin T fluorescence and changes in the FTIR spectra.177 PEFs, which deliver transient high-voltage bursts, have emerged as an effective tool for modifying proteins in solution. PEF treatments generally induce partial unfolding: for instance, mild PEF pretreatment (1.5 MV m−1 pulses) of BSA caused the protein to become more disordered and partially unfolded, exposing hydrophobic regions that greatly enhanced its binding affinity and loading capacity for the hydrophobic nutraceutical curcumin.179 At higher PEF intensities (≥2.5 MV m−1) or longer exposures, however, BSA's structure was overly disrupted, leading to diminished binding—illustrating that there is an optimal degree of unfolding for functional enhancement.179 Likewise, pulsed fields applied to soybean protein isolates initially induced unfolding of quaternary and tertiary structures, depolymerizing large protein aggregates via increased electrostatic repulsion, but above a threshold field strength this unfolding became extensive enough to expose reactive groups that promoted irreversible aggregation (through hydrophobic interactions and disulfide bond formation).180 The ability to controllably perturb secondary structure with EFs carries broad implications and applications. In biotechnology and food science, electric-field-induced unfolding offers a novel means to tune protein functionality – improving solubility, emulsification, or ingredient binding by exposing buried segments, or guiding protein assembly and gelation via controlled denaturation.178–180 In biomedical contexts, strong EFs or electromagnetic fields provide a noninvasive strategy to modulate proteins: for example, to weaken viral infection by structurally damaging key viral proteins,125 or to dissolve pathogenic protein aggregates implicated in neurodegenerative diseases.175 Ongoing research combining external fields with advanced time-resolved structural probes (e.g. ultrafast X-ray scattering or single-molecule imaging) promises to further illuminate the mechanisms of EF-induced protein unfolding and to harness these effects for innovative therapeutic and industrial applications.47
2.3 Effects on energetic and thermodynamic properties
![[small mu, Greek, vector]](https://www.rsc.org/images/entities/i_char_e0e9.gif) · = −μE
· = −μE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ, that stabilises conformers with dipoles aligned to the field. Atomistic simulations consistently show that this energetic bias can split or funnel the landscape, modifying both basin depths and barrier heights. In amyloidogenic apoC-II60–70, a static field of 10–100 MV m−1 forced ∼94% dipole alignment, converting a single folded minimum into two distinct minima (compact vs. extended) on the Rg-end-to-end free energy landscape and lowering the extended basin by ∼5 kJ mol−1.153 Model β-sheets display a comparable field sensitivity: above ∼5 GV m−1 dielectric saturation attenuates water shielding, shifts the balance of hydrophobic and electrostatic terms, and changes the sheet–sheet binding free energy by tens of kJ mol−1.184 Field-induced thermodynamic shifts are not confined to backbone states. In thioredoxin, static EF as low as 20 MV m−1 were shown to lower the pKa of the buried Cys35 residue by 0.5–1.0 units, corresponding to a stabilization of the thiolate form by 3–6 kJ mol−1 in free energy. This field-induced shift reflects enhanced electrostatic polarization of the active site, promoting deprotonation and thus favoring the catalytically competent state,182 the PCA space was also significantly affected by a 120 MV m−1 EF, see Fig. 6D. Nonequilibrium MD of chignolin revealed a striking contrast between static and 2.45 GHz alternating EFs: whereas the static EF deepened the native basin, the oscillating EF flattened it and created a new metastable minimum, effectively redrawing the principal-component energy map (see Fig. 6) and accelerating state-hopping by an order of magnitude.181 Replica-exchange MD on diphenylalanine showed an even stronger effect: a modest static EF collapsed a three-state landscape into a single dominant basin, reducing conformational entropy and producing a “funneled” surface across 310–373 K.144 Such entropy–enthalpy trade-offs underline how EFs bias both minima and inter-state barriers. One strategy to quantify how an EF modifies protein stability is to compute field-dependent free-energy landscapes along a selected structural coordinate ξ – such as the root-mean-square deviation (RMSD), radius of gyration Rg, or a backbone principal component. Starting from a distribution P0(ξ) obtained under zero-field conditions, each protein conformation is assigned an interaction energy Ufield(ξ) = −(ξ)·, where (ξ) is the dipole moment at coordinate value ξ and is the applied field vector. This energetic bias is used to re-weight P0(ξ) using the Boltzmann factor e−βUfield(ξ) (with β = 1/kBT), yielding a field-biased probability PE(ξ) and corresponding free energy ΔG(ξ,E) = −kBTlnPE(ξ). This projection approach reveals how specific conformations are stabilized or destabilized by the EF. In a representative application,185 Baumketner employed umbrella sampling to map the folding free energy of polyalanine under a static field of 0.1 GV m−1, showing a ∼10 kJ mol−1 reduction in the folding barrier. At the same time, this bias destabilized pre-assembled β-sheets, providing a thermodynamic explanation for field-induced disaggregation of amyloid structures. Free-energy perturbation calculations likewise show that GHz–THz fields can shift the minima associated with hydration-layer dipole orientations by several kJ mol−1 without measurable bulk heating.186
cosθ, that stabilises conformers with dipoles aligned to the field. Atomistic simulations consistently show that this energetic bias can split or funnel the landscape, modifying both basin depths and barrier heights. In amyloidogenic apoC-II60–70, a static field of 10–100 MV m−1 forced ∼94% dipole alignment, converting a single folded minimum into two distinct minima (compact vs. extended) on the Rg-end-to-end free energy landscape and lowering the extended basin by ∼5 kJ mol−1.153 Model β-sheets display a comparable field sensitivity: above ∼5 GV m−1 dielectric saturation attenuates water shielding, shifts the balance of hydrophobic and electrostatic terms, and changes the sheet–sheet binding free energy by tens of kJ mol−1.184 Field-induced thermodynamic shifts are not confined to backbone states. In thioredoxin, static EF as low as 20 MV m−1 were shown to lower the pKa of the buried Cys35 residue by 0.5–1.0 units, corresponding to a stabilization of the thiolate form by 3–6 kJ mol−1 in free energy. This field-induced shift reflects enhanced electrostatic polarization of the active site, promoting deprotonation and thus favoring the catalytically competent state,182 the PCA space was also significantly affected by a 120 MV m−1 EF, see Fig. 6D. Nonequilibrium MD of chignolin revealed a striking contrast between static and 2.45 GHz alternating EFs: whereas the static EF deepened the native basin, the oscillating EF flattened it and created a new metastable minimum, effectively redrawing the principal-component energy map (see Fig. 6) and accelerating state-hopping by an order of magnitude.181 Replica-exchange MD on diphenylalanine showed an even stronger effect: a modest static EF collapsed a three-state landscape into a single dominant basin, reducing conformational entropy and producing a “funneled” surface across 310–373 K.144 Such entropy–enthalpy trade-offs underline how EFs bias both minima and inter-state barriers. One strategy to quantify how an EF modifies protein stability is to compute field-dependent free-energy landscapes along a selected structural coordinate ξ – such as the root-mean-square deviation (RMSD), radius of gyration Rg, or a backbone principal component. Starting from a distribution P0(ξ) obtained under zero-field conditions, each protein conformation is assigned an interaction energy Ufield(ξ) = −(ξ)·, where (ξ) is the dipole moment at coordinate value ξ and is the applied field vector. This energetic bias is used to re-weight P0(ξ) using the Boltzmann factor e−βUfield(ξ) (with β = 1/kBT), yielding a field-biased probability PE(ξ) and corresponding free energy ΔG(ξ,E) = −kBTlnPE(ξ). This projection approach reveals how specific conformations are stabilized or destabilized by the EF. In a representative application,185 Baumketner employed umbrella sampling to map the folding free energy of polyalanine under a static field of 0.1 GV m−1, showing a ∼10 kJ mol−1 reduction in the folding barrier. At the same time, this bias destabilized pre-assembled β-sheets, providing a thermodynamic explanation for field-induced disaggregation of amyloid structures. Free-energy perturbation calculations likewise show that GHz–THz fields can shift the minima associated with hydration-layer dipole orientations by several kJ mol−1 without measurable bulk heating.186
 | ||
| Fig. 6 EF effects on protein energetics and the free-energy landscape. (A) and (C) Snapshots of the opening detail (1st trajectory of –X 100 MV m−1 data): between alpha-tubulin 2 and 3 of an MT wall ring, see Fig. 11 and ref. 46. Time evolution of the binding energy contribution of six highly contributing selected amino acids. Mean values (thick line) and the standard deviation (shaded envelope) are from N = 3 trajectories. Adapted with permission from ref. 46 © (2021) Elsevier. (B) Sampled free-energy landscape of the peptide chignolin (see inset for the structure) for zero field, oscillating field, and static field. Adapted from ref. 181 under CC BY 4. (D) Principle component analysis comparison for the C-α of thioredoxin: no field (blue), E = 60 MV m−1 (yellow), E = 120 MV m−1 (violet). Adapted from ref. 182 under CC BY 4. (E) Potential of mean force (PMF) for the Cav2.1 channel with and without the presence of external THz fields (EF strength 0.6 GV m−1) at different frequencies. The PMF here characterizes the capability of Ca2+ ions to pass through the channel. The black dashed lines represent the position of each site. Site 1 (319Gly, 668Glu, 1460Gly, 1756Ala), site 2 (318Glu, 667Glu, 1459Glu, 1755Glu), site 3 (317Met, 666Gly, 1458Gly, 1754Gly), site 4 (316Thr, 665Thr, 1457Thr,1753Thr). Adapted with permission from ref. 183. © (2025) Elsevier. | ||
In the case of free-energy profiles of water-transport proteins, MD simulations of both static and alternating EF effects have been illuminating in the case of studying consequent dipolar orientation and the role this has on regulating water transport through human aquaporin 4.187,188 The potentials of mean force were evaluated, as well as by using the more efficient metadynamics sampling methods, to manipulate efficiently dipole-orientation in the transport channel, and studying water–flux effects, in a case of “field-enhanced” thermodynamic-state control for water–flux regulation. Direct sampling was required for assessing the shift in the potential of mean force by alternating EFs – ipso facto, owing to their time-dependent nature – whilst metadynamics approaches were found in both of these studies to be effective in enhancing statistical sampling and efficiency in the case of static (time-independent) fields. Indeed, the underlying statistical-mechanics framework for direct dipole-coupling to external EFs has been accomplished in non-equilibrium metadynamics, i.e., in using dipolar orientation as a direct collective variable per se, and this external-field control was applied to manipulate dipolar orientations of alanine.189
Not all field strengths produce sizeable effects: QM/MM simulations of CO binding to solvated myoglobin found that a 1 GHz field below 10 MV m−1 altered the binding free energy by <0.2 kJ mol−1, implying a threshold above which thermal noise is overcome.190 Nevertheless, the 42.8 THz EF has been shown computationally to lower the permeation barrier of Ca2+ in CaV2.1 channels,183 and weak microwave fields subtly redistribute orientation-dependent collision free energies in carnosine solutions.191 Collectively, these studies demonstrate that the EF, when suitably tuned, can deepen, flatten or bifurcate protein free-energy surfaces, thereby steering folding, binding, and aggregation pathways in ways that would be inaccessible through temperature or chemical perturbation alone.
 | ||
| Fig. 7 Electric-field effects on lysozyme phase behaviour, nucleation, and crystal quality. (A) Left: Phase diagram of lysozyme in protein–salt concentration space with or without an EF (1 kHz, 6 kV m−1). Blue circles: homogeneous protein solution; orange crosses: crystals; pink circled crosses: metastable liquid–liquid phase separation (LLPS). Red dash-dotted lines and arrows mark the field-induced downward shift of the crystallization and LLPS boundaries. Right: Growth of a single crystal, plotted as the normalized length versus time for field-off and field-on conditions. Adapted from ref. 192 under CC BY 4. (B) Results of 24 h nucleation assays under various AC fields. Red squares indicate conditions where crystals appeared; blue diamonds indicate no observable nuclei. Adapted with permission from ref. 193. © (2008) AIP Publishing. (C) Mechanistic model illustrating how lysozyme dimers (impurities) incorporate at step edges during growth, generating misoriented subgrains; an external field inhibits dimer incorporation, reducing misorientation and enlarging subgrain domains. Adapted with permission from ref. 194. © (2014) American Chemical Society. (D) Optical micrographs of hen-egg-white lysozyme crystals grown with NiCl2 under (a) no field, (b) a 500 kHz AC field, and (c) a 1 MHz AC field. Adapted with permission from ref. 195. © (2009) American Chemical Society. | ||
In typical DC-field crystallization, the protein (often carrying net charge in solution) electro-migrates toward an electrode, quickly establishing a high-concentration (supersaturated) region near that electrode.200,205 Consequently, nucleation is preferentially localized at the electrode and is largely suppressed in the bulk or at the anode, yielding a reduced number of crystals, usually only one or a few, growing at the cathodic interface.200,206 This method drastically reduces the overall nucleation rate while accelerating the first nucleation event (shortening induction time) and produces larger crystals than for zero-field controls.200,206 Indeed, crystals grown under a static EF (DC) are often significantly bigger and exhibit improved diffraction quality (e.g., lower “mosaicity” and higher resolution) due to the minimization of simultaneous nucleation events.200,207 In contrast, an alternating-current EF (especially at high frequency) can be applied without net electro-migration, influencing crystallization through pure electrostatic effects on the free-energy landscape.195,196 AC fields have been shown to either enhance or inhibit protein nucleation by tuning the field frequency to adjust the electrostatic contribution to the phase free energies.195,202 Notably, under a 1 MHz AC field, lysozyme crystals grew more slowly (with reduced growth kinetics), despite an increased supersaturation driving force, as the field effectively raised the step-edge free energy and produced flatter, more orderly crystal faces; this led to substantially improved crystal quality (fewer defects and lower mosaic spread) in the tetragonal lysozyme form.196,208 Many studies report that EFs can expedite the nucleation of proteins that are otherwise difficult to crystallize: for instance, the application of an extremely low-voltage (∼1 V, 20 Hz) internal field was sufficient to induce thaumatin crystallization in a regime where no crystals normally appeared.209 Likewise, microfluidic experiments with patterned ITO electrodes demonstrated that certain AC field configurations could both reduce nucleation induction time and localize crystal formation to specific sites, even enabling insulin crystals to form in cases where control samples showed none.197,210
The presence of an EF offers remarkable control over crystallization kinetics: it can reduce induction times, control the number and location of nuclei, increase crystal size, and enhance crystal quality (yielding larger and more orderly-packed crystals) under a broad range of conditions.193,199,211 This approach has been successfully leveraged to improve protein crystal homogeneity and diffraction properties for structural analysis,207,211 to manipulate crystal polymorphs by stabilizing a desired solid phase through field-induced shifts of the nucleation barrier,202 and even to achieve oriented crystallization for better crystal mounting or data collection.198,199 Continued advances in electric-field-assisted crystallization techniques – including scaling up to continuous crystallizers and combining fields with other stimuli – highlight the significant implications of this approach for both fundamental protein crystallography and practical protein manufacturing.199,212
2.4 Effects on electrostatic, mechanical and dynamic properties
 | ||
| Fig. 8 EF effects on local electrostatic properties of proteins and on the protein dipole moment. (A) 2D maps of the local electrostatic field distribution around the active site of SOD1. The maps show EF shifts on the representative planes π′ (top row) and π″ (bottom row) due to a monopolar nsPEF pulse (left) and bipolar one (right). Both signals alter the electrostatic map around the central copper ion, with the monopolar pulse producing localized changes and the bipolar pulse resulting in a more diffuse and uniform distribution, suggesting the possible rotation of specific protein residues and charge redistribution under the applied field. Adapted from ref. 216 under CC BY 4. (B) Average x-component of the dipole moment of hen egg-white lysozyme (HEWL) under different EF conditions, computed across multiple simulation replicates. Adapted from ref. 181 under CC BY 4. (C) Time evolution of the dipole moment magnitude of kinesin under five different EF conditions: no field (black), +100 MV m−1 along the X axis (red), −100 MV m−1 along the X axis (magenta), +100 MV m−1 along the Y axis (blue), and −100 MV m−1 along the Y axis (cyan). The EF is applied continuously for the entire duration of the molecular dynamics simulations. Solid lines represent the mean dipole magnitude, and the shaded regions indicate the standard deviation computed from N = 3 independent replicates. Insets show kinesin bound to a tubulin protofilament at the end of the simulation; black arrows denote the kinesin dipole vector, and colored arrows indicate the direction of the applied EF, using the same color scheme as the main plot. Adapted from ref. 217 under CC BY. (D) Representative structures of the chignolin peptide and surrounding hydration layers under applied EFs of (c) 0.7 GV m−1 and (d) 0.7 MV m−1. Dipole moment vectors of the peptide and water molecules are shown as yellow and green arrows, respectively. The directions of the applied electric E and magnetic B fields are indicated with arrows. Adapted with permission from ref. 153. © (2016) AIP Publishing. | ||
Tubulin proteins, carrying an unusually high charge and dipole moment,78 are the building unit of MTs, which makes them a target for the investigation of EF effects. Apart from the effects of the EF on the mechanical properties of the tubulin protein mentioned above,218,219 Marracino et al. studied the temporal evolution of the dipole moment of a tubulin dimer as well as of individual residues that form the binding sites for various drug molecules and cofactors such as GTP/GDP.78 Applying an EF of strength 10 MV m−1 to 300 MV m−1 resulted in the polarization of the dimer, which led to a change in the shape and orientation of the tubulin dimer. Data showed that the dipole component along the median axis (containing a highly charged C-terminal tail) is affected most, causing the C-terminal tail to extend beyond the dimer surface. The EF also affected the dipole moment of residues in the binding sites and this polarization effect varied with the strength of the applied EF. The analysis of the dipole moment in this study could be helpful in other cases such as protein–drug interactions, in which the binding of a drug can be controlled by applying an EF, or in ion channels, in which the conductance can be manipulated by the effect of an external EF on the orientation of charged residues in loops.
Marracino et al.220 used MD simulations to test whether classical Onsager–Kirkwood theory – valid under “weak field” linear-response conditions – still holds for nanosecond, high-intensity continuous-wave (CW) EFs. Unlike the static or ideal rectangular fields common in MD studies, real nsPEF waveforms possess finite rise/fall times that broaden their spectra into the 100 MHz–100 GHz range. The authors showed that CW amplitudes above ∼150 MV m−1 drive pronounced nonlinear behaviour throughout this band: higher-order polarization harmonics emerge, especially at the lower frequencies, but fade beyond the dipolar relaxation limit. The simulated polarization follows a Langevin-type curve; by adapting the Langevin function and introducing a frequency-dependent correlation P(ω), they built a predictive dipolar-interaction model (R2 ≈ 1) that naturally reproduces the Debye-like water relaxation time. Their results highlight the need to keep field strengths below ∼200 MV m−1 in protein MD studies to remain within the weak-field regime and avoid nonlinear or saturation artefacts.221
Protein electrostatics plays a fundamental role in shaping the structure, maintaining the stability, and governing the function of proteins through the spatial arrangement and interaction of internal charge distributions. As outlined in the Introduction, protein electrostatics is essential for processes such as molecular recognition and catalytic activity.40,41 Intense EFs interact with proteins and lead to altered charge distributions, often mediated by water polarization. MD simulations reveal that EFs in the range of 0.1–1 GV m−1 can modify electrostatic environments without structural damage at lower intensities, while higher intensities may induce unfolding. For example, in superoxide dismutase (SOD1),216 fields of 100 MV m−1 affect the electrostatic environment of the active site, enhancing substrate interaction. Similarly, in tubulin, fields above 20 MV m−1 induce significant changes of dipole components of several functionally important residues.78 Stark spectroscopy, particularly the vibrational Stark effect,222 is a key technique for studying protein electrostatics by measuring how local EFs influence vibrational probe frequencies, such as nitrile stretches. This spectroscopy offers insights into electrostatic forces within proteins, revealing their magnitude and direction, and was demonstrated to sense changes in internal protein electrostatics when applying a 100 MV m−1 EF (myoglobin in glycerol/water glass at 74 K).223,224
MTs form the cellular skeleton and guide mitosis, motility, and organelle transport (Section 18, ref. 132). Since the subunits of MT, αβ-tubulin dimers, are strongly charged, they can couple to an external EF. Saedi et al.218 pulled the tubulin dimer via a virtual spring while subject to an EF in a MD simulation: a static 30 MV m−1 field softened the dimer, whereas a 1 GHz field of the same amplitude stiffened it; at 10 GHz the response was negligible because the field direction flipped too rapidly for effective coupling. Using the same protocol, Setayandeh & Lohrasebi219 showed that 30 MV m−1 fields at 5 GHz increased rigidity of dimer pairs along the MT axis, whereas 1 and 6 GHz fields enhanced flexibility, mimicking taxol-stabilising effect. These studies demonstrate that EFs have the potential to modulate protein nanomechanics to achieve diverse functional outcomes, and can also be harnessed as a force handle in electromechanical force spectroscopy.
2.5 Effects on protein interactions
Protein interactions, encompassing both protein–protein interactions (PPIs) and protein–ligand (throughout our text, “ligand” refers to a non-protein molecule) interactions (PLIs), are fundamental to biological function and the assembly of quaternary structures. Such interactions drive the self-assembly of protein complexes, protein oligomers and polymers, and regulate processes ranging from enzymatic catalysis to signal transduction. The intermolecular forces stabilizing these complexes (hydrogen bonding, hydrophobic contacts, electrostatic interactions, etc.) can be sensitive to external perturbations like EFs, influencing their conformation and interaction propensity. From a thermodynamic standpoint, protein–protein interfaces are especially appealing targets for EF perturbation, because dissociating them entails overcoming a lower energy barrier than that required to trigger structural changes in individual proteins, such as unfolding.231 | ||
| Fig. 9 EFs can affect highly ordered protein structures, such as cytoskeleton protein polymers like MTs. (A) A schematic mechanism of EF action on a MT, where the opening of the MT wall occurs due to the action on dipoles of individual tubulin subunits (B) snapshots from a representative MD simulation. Adapted with permission from ref. 46. © (2021) Elsevier. | ||
Electrowetting-on-dielectric (EWOD) stacks platforms confine the applied voltage to the solid PDMS/Teflon dielectric and the nanometre-thin electrical double layer, leaving the interior of the micro- to nanolitre droplet virtually field-free. The resulting Maxwell stress at the three-phase line drives controllable droplet deformations that can be tracked in situ. Sen et al. used this oscillatory actuation to fragment pre-formed human serum albumin (HSA) fibrils in a sharply frequency-dependent manner, cutting ThT fluorescence by 60% at the 20 Hz optimum frequency.235 An analogous EWOD geometry biased with a static 8 MV m−1 EF227 produced a comparable loss of fibril-sheet content, confirmed by CD, AFM and TEM (Fig. 10E). Because the EF scarcely penetrates the liquid in EWOD platforms, both studies most likely induce bulk hydrodynamic shear (see Section 3 on indirect EF effects) and interfacial electro-compression rather than a direct bulk-field effect on the proteins.
 | ||
| Fig. 10 EFs affect protein–protein interactions. (A) A kinesin motor domain on a segment of an MT is detached within a few ns by a 100 MV m−1 EF in MD simulations. Adapted with permission from ref. 225. © (2023) Elsevier, (B) An EF disassembles protein isolate complexes in MD simulations which is corroborated by an increase of surface hydrophobicity in experiment (PEF firing frequency 1000 Hz, bipolar square wave: 40 µs pulse, 200 µs gap, 40 opposite polarity pulse, N = 25 pulses) as probed by ANS fluorescence – both effects are EF strength-dependent. Adapted with permission from ref. 180. © (2024) Elsevier. (C) Transmission electron microscopy evidences that an EF (2 + 2 µs bipolar pulses, 1 MV m−1, firing frequency 2 kHz) causes the disassembly of ferritin cage-like structures. Adapted with permission from ref. 141. © (2021) Elsevier. (D) A 120 MV m−1 (1 ns pulse width, 1.7 Hz firing rate, N = 1000 pulses) pulsed EF promotes degradation of transthyretin aggregates. Adapted from ref. 226 – CC BY 4. (E) Transmission electron microscopy images of human serum albumin fibrillar solutions: (a) without an applied EF, and (b) after exposure to an EF of approximately 8 MV m−1 for 10 min. Adapted with permission from ref. 227. © (2014) IOP. (F) Turbidity, reporting protein aggregation, of ovomucin-depleted egg white solutions (10% w/w, pH 4–9) after PEF treatment at varying EF strengths, while maintaining a constant specific energy input Wspec = 713 kJ kg−1. Adapted with permission from ref. 228. © (2017) Elsevier. | ||
Intense picosecond THz pulses provide a complementary route. Using 0.15–3 THz bursts with peak fields 40 MV m−1, Hough et al. showed that polymerised MTs disassembled within minutes (Fig. 11C); the rate scaled with both field amplitude and spectral content, and band-pass filtering revealed maximum efficacy near 0.5 THz.25 At the level of tubulin dimers, Chafai et al. exposed purified protein to a train of 2 MV m−1, 10 ns EF pulses and found a dose-dependent shift from normal nucleation–elongation kinetics to suppressed assembly, tracked by turbidimetry, intrinsic fluorescence and zeta-potential changes; below 200 pulses the effect was largely reversible, whereas ≥400 pulses produced persistent inhibition,21 with 800 pulses leading to a changed self-assembled tubulin structure as probed by AFM, see Fig. 11B. Analogous disassembly-promoting behaviour of an intense EF is observed with pathogenic protein aggregates. Jurgelevičiūté et al. treated isolated Sup35NM prion fibrils with PEF (10 pulses, either 50 or 1000 µs).238 The AFM imaging revealed progressive shortening and, at 2 MV m−1, complete breakup into oligomers, see Fig. 11A. Actin responded oppositely to continuous-wave 0.46 THz irradiation (power density ~0.6 W cm−2), which accelerated the elongation phase of in vitro actin polymerisation, generating 3.5-fold more filaments without detectable denaturation, as verified by SiR-actin microscopy and pyrene fluorescence.236
 | ||
| Fig. 11 Imaging of EF effects on protein fibers: (A) AFM imaging of the effects of pulse electric treatments of fiber-forming proteins. Sup35NM amyloid fibrils before and after PEF treatment (2 MV m−1, 10 pulses, 1 ms pulse duration, 1 Hz firing frequency) – adapted from ref. 238 – CC BY 4. (B) Control sample of self-assembled tubulin and the self-assembly from a tubulin treated by a PEF (2 MV m−1, 800 pulses, 10 ns pulse duration, 1 Hz firing frequency) – adapted with permission from ref. 21. © (2019) Wiley. (C) Intense picosecond (subTHz) electric pulses cause MT fragmentation (40.9 MV m−1, 1 ps pulse duration, 1 kHz firing frequency). Here we show a time dependence of this process captured using fluorescence microscopy – adapted from ref. 25 – Open Access license of The Optical Society/OPTICA. | ||
In the case of PPIs in amorphous-protein aggregation, aggregates of amorphous proteins were subjected to an oscillating EF. When their field response was observed using time-resolved polarized optical microscopy, they revealed the disintegration of large aggregate clusters as well as field-induced deformation, reorientation, and enhanced polarization. Further, amorphous aggregate suspensions' collective microscopic dynamics were investigated using small-angle dynamic light scattering. The intensity auto-correlation function exhibited field-enhanced local oscillations that were associated with two distinct elastic moduli – drawing attention to the potential of EFs to regulate protein-aggregation processes.237
PEFs (0.1–3 MV m−1, µs bursts) are widely used in food processing to affect PPIs: ovalbumin at neutral pH aggregated modestly, whereas at pH 4 or 9 it remained monomeric.228 Soybean proteins showed a biphasic trend, where 0.5–1.5 MV m−1 unfolded tetramers and released subunits, but ≥2 MV m−1 exposed hydrophobes and thiols that re-cross-linked the proteins180 (see also Fig. 10B). Time-resolved fluorescence and Ellman's assays pinpointed radical generation as a secondary pathway, indicating indirect electrochemical effects of EFs taking place as well (see Section 3.2). For the allergen tropomyosin, 0.5–1 MV m−1 bursts opened coiled-coil dimers and raised IgE binding by ∼30%, whereas prolonged or stronger treatment compacted the structure and lowered immunoreactivity.134
Together, these studies demonstrate that intense static, pulsed or THz-frequency EFs can decisively remodel protein–protein contacts (from weakening kinesin–tubulin binding and accelerating actin growth to fragmenting amyloids) highlighting a versatile, rapidly switchable handle for manipulating quaternary, multimeric and polymeric protein structure in vitro and in vivo: the selective dismantling of amyloid fibrils or weakening of motor-track contacts suggests non-invasive anti-aggregation therapies and neuromodulation strategies.133,237 Conversely, reinforcing or creating PPIs with tuned EFs could stabilise fragile enzyme assemblies in biosensors or power nanobiomachines. In agri-food technology, PEFs were demonstrated to deliver chemical-free control of gel texture, solubility and allergenicity.134,180,228
 | ||
| Fig. 12 Protein–ligand interactions modified by EFs. (A) In PEF-treated (1 MV m−1, 40 µs pulse, firing frequency 1 kHz, N = 20 pulses) pea protein isolate, the ligand (EGCG – epigallocatechin-3-gallate) occupies two high-affinity pockets (sites 1–2; ΔG = −9.1/−9.0 kcal mol−1), expanding the total docking sites (obtained from molecular modeling) from four in the control structure (no EF – not shown) to ten and increasing hydrophobic contacts from five to twelve through newly exposed residues such as Pro457 and Trp129. Adapted with permission from ref. 126. © (2023) Elsevier. (B) Schematic representation of the relationship between the curcumin-binding capacity and structural properties of BSA after PEF treatment. Photos: Visual inspection of free curcumin solution (left), BSA curcumin complex solution (middle) and PEF-treated BSA curcumin complex solution (right). Adapted with permission from ref. 179. © (2022) Elsevier. (C) Snapshots of the 5-HT1AR-serotonin complex at 600 ns in (from left) no EF, 10 MV m−1 EF, and 30 MV m−1 EF, serotonin is shown as sticks, and (right) overlay of 5-HT1A receptor structures from no EF (pink) and 30 MV m−1 EF system (yellow) MD simulations, spotlighting the conformational shift at the D3.32–Y7.53 lock. Adapted with permission from ref. 135. © (2025) Elsevier. (D) (Left) Relative intrinsic fluorescence quenching of α-amylase/pectin complexes after PEF treatment. The ordinate shows the fractional decrease in fluorescence, (F0 − F)/F0, while the abscissa lists the mass ratios of α-amylase to pectin (R32:1, R4:1, R1:1). PEF-treated samples (red circles) exhibit markedly greater quenching than non-treated controls (black squares), data represent mean ± SD (n = 3). (Right) Structure of α-amylase. Adapted with permission from ref. 167 © (2020) Elsevier with permission. | ||
These findings illustrate that strong EFs are potent modulators of protein–ligand/substrate binding, capable of enhancing interactions (by transiently altering conformations to improve ligand accessibility and binding affinity) or inhibiting them (via induced unfolding or misfolding). This knowledge opens up opportunities for innovative applications – from non-thermal control of enzymatic reactions and protein processing (e.g. tailored glycation and improved drug/nutrient binding) to the remote tuning of receptor activation and protein function in biomedical and biotechnological settings.
2.6 Effects on additional protein functions
 | ||
| Fig. 13 EF effects on proteins' enzymatic functions. (A) Inactivation of horseradish peroxidase (HRP) exposed to EF strengths varying from 0.5 to 2.5 MV m−1 with 207 pulses during storage at 4 °C, respectively (measured immediately, 24 h, or 48 h after PEF treatment). The treatment temperature did not exceed 40 °C. Adapted with permission from ref. 242. © (2005) Elsevier. (B) Inactivation of pepsin by PEFs. The width and total treatment time were 2 µs (bipolar pulses) and 126 µs, respectively. The media are 7.5 mM HCl with a pH of 2.0 and an electrical conductivity of 0.391 S m−1, and a water–phosphate–acetate mixture with a pH of 3.8 and an electrical conductivity of 0.345 S m−1. Adapted with permission from ref. 162. © (2004) American Chemical Society. (C) The relative activity of CPG2 after nsPEF exposure; relative activity (%) as a function of total exposure time t (s). *p < 0.05. Adapted with permission from ref. 243. © (2015) Elsevier. (D) Changes in the enzyme activity of alcalase after PEF treatment for different storage times. Data were expressed as the mean ± SD, n = 3. a, b, c indicated that the enzymatic activity of alcalase was significantly different (p < 0.05). The docking model between a substrate (casein) and the alcalase molecule was generated by MD simulations under EF strengths of 0 and 1 MV m−1. Adapted with permission from ref. 244. © (2022) Elsevier. (E) Left: Effect of PEF treatment (pulse width, 2.5 µs; frequency, 1128 Hz) at different EF intensities on the enzymatic activity of α-amylase (0.2 mM phosphate buffer, pH 6.0; enzyme concentration, 23 U mL−1). Error bars represent standard deviation. The different letters mean the data of two samples are significantly different (p < 0.05). Right: Temperature dependence of α-amylase enzymatic activity (enzyme concentration, 23 U mL−1) with and without PEF treatment. PEF treatment: EF strength, 1.5 MV m−1; flow rate, 100 mL min−1; pulse width, 2.5 µs; frequency, 1128 Hz. Error bars represent standard deviation. Enzyme activity at the optimum temperature is considered 100%. Adapted with permission from ref. 245. © (2016) Springer Nature. | ||
Similarly in ref. 266 it is shown that PEFs can induce pores in the voltage-sensor domains of different voltage gated transmembrane channels. Such fields can create conductive pores which evolve into complex pores stabilized by lipid molecules followed by unfolding of the voltage sensor domain from the channel. The mechanism is related to the degree of hydration of the sensor: the more hydrated the sensor is, the more electrostatically favorable for the entry of ions, and the more easily it porates (Fig. 14).
 | ||
| Fig. 14 EF effects on membrane protein functions. (A) Structure of the voltage-gated ion channels (VGICs); progression of the radius of complex pores generated by the increasing applied EF. Adapted with permission from ref. 266. © (2020) Elsevier. (B) The Michelis Mentin enzyme will behave like a Brownian motor. The E1 to E2 transition can be induced by an applied EF. Adapted with permission from ref. 267. © (2003) Association of Asia Pacific Physical Societies. (C) Central intraprotein electropore; evolution of the electropore's shape during the simulation with an applied EF. Adapted from ref. 268 – CC BY. (D) Conduction through an endogenous channel; approximate schematic of the electropore-channel radial shape. Adapted with permission from ref. 263. © (2019) Royal Society of Chemistry. (E) Depiction of layout of AQP4 and osmotic permeability static-field pulses applied at various EF strengths (V/A). Adapted from ref. 261 – CC BY. | ||
Experimental EF effects were demonstrated on membrane proteins as part of isolated membranes or in cells. One class of works exploited electro-optical approaches mentioned above. A common target protein was bacteriorhodopsin in purple membrane, in which a pulsed EF induced pH change,168,269,270 structural effects112,115,171,172 and DC- or AC-fields caused conformational changes.110,169,170 Another class of works focused on oscillating EF effects on ATPases: a pulsed EF drove the ATP synthesis271 and AC fields drove cation transport across the membrane in a resonant-like manner272 (Rb+,273 Na+.274 Follow-up works generalized the concept and proposed that enzymes can harness energy from various EF waveforms,275–279 including stochastic ones,280–282 using electroconformational coupling model.
3 Indirect effects of EF exposure
The EF applied to protein solutions can impart a variety of indirect stresses on the proteins via physical and chemical changes in the surrounding medium. These indirect effects are distinguished from the direct interactions of the EF with the protein's molecular structure described in the sections above, and instead involve secondary phenomena like heating, fluid motion, shockwave generation, electrolysis-driven pH shifts, reactive chemical species formation, and gas bubble formation. Such effects come into play for high-field exposures across the spectrum of DC, AC, pulsed, radio-frequency (RF), and microwave fields, especially at field strengths on the order of 0.1 MV m−1 and above. In this section, we review the major indirect mechanisms by which intense EFs can influence protein structure and stability. We exclude cases that involve plasma formation or dielectric breakdown (sparking), focusing instead on sub-breakdown conditions where the field-induced effects are non-ionizing. The indirect pathways are categorized into electrophysical effects – arising from the physical consequences of the EF (such as thermal and mechanical perturbations) – and electrochemical effects – arising from field-induced chemical reactions or species in the solution. All types of proteins (enzymes, structural proteins, antibodies, etc.) can be susceptible to these indirect stressors, although their specific responses may vary depending on factors like stability, size, and environment.3.1 Indirect electrophysical effects of EFs
In practice, electroosmotic contributions are strongest when charged interfaces (electrodes, channel walls, bubbles) are present and the electrical double layer can charge within the field period. The characteristic charging time τc sets a frequency crossover: for ωτc ≳ 1 (often ≳10–100 kHz in microscale geometries) AC electroosmosis diminishes markedly, whereas at 0.1–10 kHz it can generate steady vortical flows even at modest fields of order kV m−1.304–306 Consequently, flow-induced hydrodynamic interactions can be important in concentrated protein suspensions and near-wall geometries where long-ranged flows enhance collision rates and promote field-assisted aggregation or adsorption. In contrast, in dilute bulk solution away from boundaries (typical for many spectroscopic assays), direct electrophoretic/dielectrophoretic and dipolar interactions between proteins are usually the dominant field couplings, while electroosmotic flow mainly enters indirectly by redistributing heat and reactive species.299,308
These flows have dual consequences. On the one hand, they improve heat and species transport and mixing, preventing localized extremes of temperature or electrochemically produced species that would otherwise damage proteins. On the other hand, flow also introduces hydrodynamic shear and increases contact between proteins and interfaces, such as gas bubbles or electrodes, where adsorption and unfolding can occur.309 In summary, EF-induced flows can either protect or damage proteins depending on their magnitude and context. Controlling flow regimes is therefore essential in high-field experimental design.
3.2 Indirect electrochemical effects of EFs
| Anode: 2H2O → O2 + 4H+ + 4e− |
| Cathode: 2H2O + 2e− → H2 + 2OH− |
Within several ms pulses, when an intense EF is applied driving direct-current, the anodic boundary layer can acidify to low pH while the cathodic layer simultaneously alkalinises to high pH, provided the rate of electrolysis outstrips the local buffer capacity.317 Such pH changes rapidly protonate or de-protonate ionisable side chains, disrupting salt bridges, neutralising charge balance, and triggering unfolding or aggregation, see the review318 on general pH dependence of protein stability. For example, in electrospray ionisation capillaries acting as anodes, Konermann et al. detected >2-unit pH drops that unfolded cytochrome c, evidenced by higher charge-state spectra; raising flow or altering grounding decreased electrolysis and preserved the fold.319 This demonstrates that apparent EF effects may actually be hidden pH artefacts. Increasing buffer strength often increases electric conductivity and hence current, so H+/OH− production scales up and the width of the pH front changes only marginally.320 Thus “more buffer” rarely solves the problem. The pH shifts can be minimized (i) using short bipolar pulses, (ii) adding sacrificial or ion-exchange layers on electrodes,321 (iii) separating the sample via salt bridges,322 or accounted for by monitoring pH or applying matched chemical controls. Electrode-driven pH excursions, not the EF itself, often compromise protein integrity under intense EFs. Proper pH monitoring and mitigation are essential before attributing structural changes to direct EF–protein interactions.
Each indirect pathway: heating, electro-hydrodynamic flows, shock-cavitation, pH drifts, ROS/RNS reactions and bubble interfaces – can appear simultaneously in a high-EF protocol. Disentangling them through temperature probes, pH indicators, radical scavengers, acoustic sensors, in situ imaging and spectroscopy enables rational process design, whether the goal is to dissect the mechanism of EF action or for biotechnological or industrial applications.
4 Discussion
4.1 EF strength and timescale: their scaling and (mis)match between simulations and experiments
In most of the works aiming to probe the direct effects of EFs on proteins, the fields have to exceed certain threshold EF strength for the effect to manifest, see Fig. 15. In many cases, that limit would be EF strengths on the order of hundreds of MV m−1, although the exact threshold for such effects depends very much on the protein systems and experimental conditions and goes down to 10 kV m−1.125 It is insightful in this context to consider the strength of EFs that occur naturally in cells and proteins. Typical membrane potentials across the cell membranes of eukaryotic cells are approximately 100 mV, which if one assumes a membrane thickness of about 4 nm329 yields field strengths of up to tens of MV m−1. EFs inside the catalytic sites of enzymes reach EF strengths of GV m−1 and beyond.34,41,222 Therefore, we do not focus on weak and ultra-weak EF (< 0.1 kV m−1) effects, where direct effects of EF on molecules are expected to be negligible compared to thermal electric noise. For this reason, the weak electromagnetic field effects on biological systems present major controversies in the bioelectromagnetic research and have been a focus of other reviews.330–338 | ||
| Fig. 15 Primary EF effects on proteins depend on EF strength E and protein structure. The geometrical direction of effects is consistent with the fact that the EF vector is pointing from left to right. The protein always undergoes translation (electrophoretic force) as long as it has non-zero effective electric charge Q. Rotational force is also acting on the protein proportional to the dipole moment μ of the protein (blue arrow). Translation and rotation effects of the EF on whole proteins (rigid body motions) are taking place at any field strength values. When the torque magnitude T = E·μ significantly exceeds the energy due to rotational thermal motion (kT), the rotation becomes deterministic. In a multimeric protein complex, the translational force and rotation force due to the EF act also on the local charges QL and dipoles, respectively, of individual subunits. If the EF force is higher than some threshold force, (i.e. interaction energy between the EF and charges and dipoles and overcomes the binding energy (EB) between the monomer), it can cause disassembly of the complex, hence disrupting the quaternary structure. Within a protein, EF effects on the local charges and dipoles can cause deformation of the protein and affect tertiary structure. Similarly, the EF can affect the secondary structure and a very high EF strength can lead to protein partial unfolding and denaturation. One has to keep in mind that the real picture is statistical: the thresholds are not sharp since the EF changes the probability of events. | ||
Both computational modeling and experimental studies consistently demonstrate the influence of EFs on proteins, with clear evidence of translational and rotational motion, as well as strong indications of EF-induced changes in secondary, tertiary, and quaternary structures. However, a comparison of EF strengths and timescales used in these two approaches (Table 2 vs. 3) reveals substantial discrepancies. MD simulations typically employ EF strengths that are significantly (often several orders of magnitude) higher than those used in experiments, and they operate over much shorter timescales. These differences arise from fundamental limitations inherent to molecular simulation methods. MD simulations are restricted to nanosecond-to-microsecond timescales due to computational cost, which necessitates the use of elevated field intensities to induce observable structural changes within such brief periods. Furthermore, the systems modeled in MD are highly idealized: they are small in size and lack real-world complexities such as dislocations, grain boundaries, impurities, and environmental heterogeneity (e.g., variable solvent conditions). The use of empirical interatomic potentials (force fields), which approximate rather than fully capture quantum interactions, further limits accuracy. Additionally, temperature control mechanisms such as thermostats may artificially suppress slow conformational dynamics, further limiting the observable impact of low-intensity fields. As a result, simulations conducted at experimentally realistic field strengths, but at significantly smaller durations, frequently show only limited effects, such as electrophoretic motion, without substantial conformational changes. To overcome these limitations, MD simulations often rely on stronger EFs to accelerate rare events – such as folding, unfolding, or large-scale domain reorientation – that would otherwise occur over milliseconds or longer. This approach enables researchers to probe mechanisms of field-induced structural change within accessible simulation windows. Thus, the application of higher EF strengths in MD simulations, beyond those typically used in experimental settings, is a practical necessity driven by limitations in accessible timescales, system sizes, and the fidelity of current force fields. Although the quantitative thresholds for structural changes observed in simulations may not align directly with experimental values, the underlying mechanistic insights remain highly valuable. Bridging this gap requires the integration of multiscale modeling approaches, the refinement of interatomic potentials, and the development of scaling frameworks that enable a more accurate translation of computational findings into experimentally relevant predictions.
From a purely academic perspective, one approach is to bring the experimental time scales in line with the ones of simulation. That can be done using fast and ultra-fast spectroscopy techniques339,340 and advanced ultra-fast single molecule or super-resolution imaging and microscopy techniques,341,342 see Fig. 16 for a graphical overview of the variety of systems used to expose the proteins to EFs. For the advanced techniques to serve the research on EF effects on proteins, it is necessary to integrate them with EF exposure systems so that the effects can be analyzed in real time during the EF pulse and also shortly after to match the short simulation time scales. Still, there will be a physical limit to the strength of an EF that can be applied for a water-based protein sample for a given time before dielectric breakdown occurs. Although the dielectric strength for distilled water (at 25 °C and normal atmospheric pressure) is approximately 30 MV m−1 for a static (long duration) field, this threshold increases if the field acts only for a short time. For instance, small area and small gap systems can hold up to 100 MV m−1 up to 100 ns and up 400 MV m−1 up to 20 ns343 and this threshold might depend on conductivity.344
 | ||
| Fig. 16 Exposure systems delivering EFs to protein samples categorized according to the EF frequency range, bandwidth, observation capabilities under operando, and sample handling. Frequency range: DC-low frequency—electrode configurations that impose static or slowly varying fields on proteins, often in capacitor-like arrangement or as coplanar electrodes, such as in ref. 286, reproduced with permission, © (2005) AIP publishing; RF-microwave—cells that expose samples to radio-frequency or microwave range signals usually employing transmission lines or waveguides, example from ref. 22, reproduced with permission, © (2016) Springer Nature; Millimetre-wave/terahertz—quasi-optical platforms that illuminate proteins with high-frequency EF, as in, e.g., ref. 25, adapted with permission from © Optical Society of America. Bandwidth: broadband, pulsed – often transmission-line-based geometries for delivering nanosecond–microsecond field pulses with wide spectral content, such as in ref. 289, with permission; narrow-band, single frequency—resonator or waveguide, or for low-frequencies even a simple capacitor assemblies, providing monochromatic excitation at a selected frequency, e.g. ref. 290, reproduced under CC BY 4. Under-operando observation: none—closed reactors for bulk treatment without real-time monitoring, e.g. ref. 226, reproduced under CC BY 4. Spectroscopies—field-delivery setups compatible with simultaneous optical, vibrational, or electronic probes such as in ref. 291, adapted with permission from IEEE (©2024); imaging—transparent or open-access devices that enable concurrent microscopic visualisation, such as in ref. 292, adapted under CC BY 4. Sample handling: batch reactor—static chambers in which the sample remains stationary during exposure,163 adapted under CC BY 4; through-flow—flow-through reactors allowing continuous processing under field stimulation and in-line probing,293 adapted under CC BY 4. | ||
However, for practical applications, one might need to use longer duration electric pulses and lower EF strength. In that case, the molecular modeling strategies should comply and extend the time scale to bridge to the experiment. This is possible either by increasing computational power or applying further coarse-grained approaches in the simulations,345,346 which will enable the analysis of either longer time scales or larger protein systems. Longer simulations will also be able to address the question of the reversibility of the EF-induced conformational and structural changes in proteins.125,216,266,347,348 To increase the match between molecular modelling predictions and experiments, a promising avenue in a different direction to coarse-grained simulations is to include electronic effects, which are absent in classical MD simulations with standard (non-polarizable) force fields. Including electronic effects can be done by employing polarizable force fields349 or even more accurate approximations using ab initio calculations350,351 – such effects contribute to the molecular polarization due to external EFs and hence might influence the effects and their thresholds in simulations.
Theoretical considerations bringing together EF strength and time scales required for effects on proteins to take place have the ability to integrate the effects from molecular simulations and experiments. The classical work of EF effects on the chemical reactivity of molecules, including proteins, from a thermodynamic perspective, is that from E. Neumann.352 We recently proposed a related theoretical approach on a single molecule level and verified its utility in EF-mediated dissociation of a kinesin protein from an MT-model (lattice of tubulin heterodimers).225 There, we quantified the time needed to detach the kinesin from tubulin in each simulation. To rationalise the observed dependence of kinesin–MT dissociation on EF strength, we recast the detachment process and its time-scale within an Arrhenius framework assuming a single energy barrier. While demonstrated on this single protein complex system, we believe that this approach could be used generally for any process where the EF is acting to lower the energy barrier, hence modifying the rate of the process. In the absence of an applied EF, the rate (having units s−1) is (for the particular case in our work,225 it was the kinesin detachment rate from the MT)
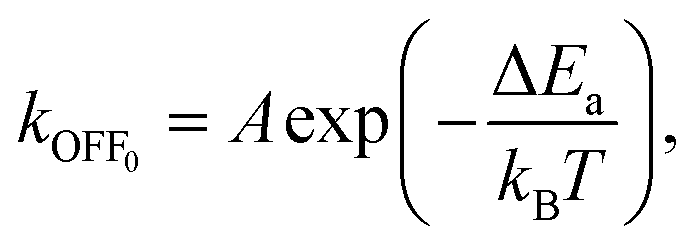 | (1) |
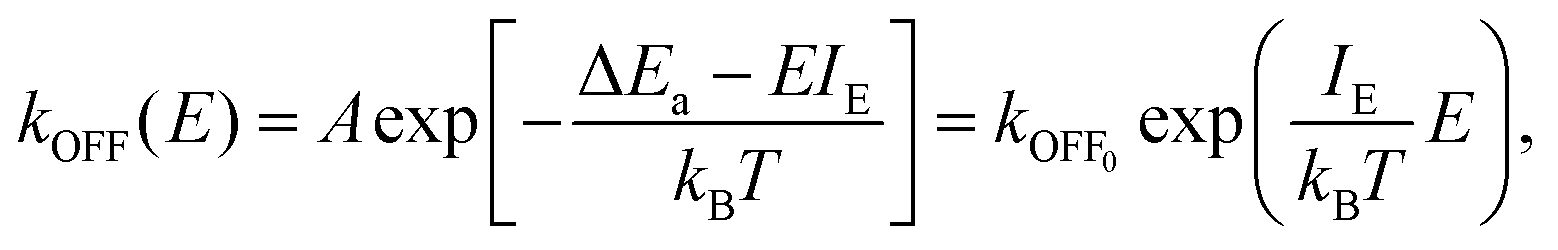 | (2) |
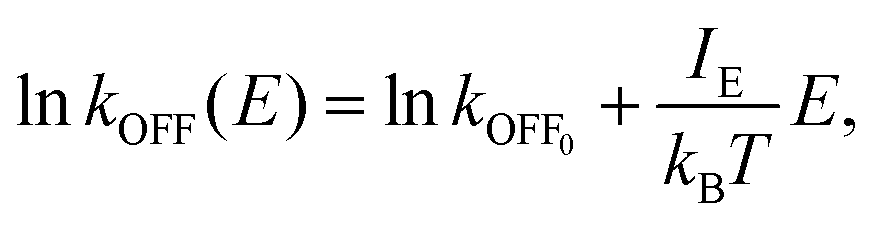 | (3) |
To provide an at-a-glance overview for non-specialists, Table 1 summarizes typical external-field strengths and effective forcing timescales across common modalities in protein–EF research. For pulsed modalities, we distinguish the single-pulse width τp from the cumulative “on-time” τon = Nτp (sum over N pulses); the elapsed experiment time can be much longer due to interpulse gaps.
| Modality | Typical E | Forcing protocol (τp, N, τon) | Notes/dominant couplings |
|---|---|---|---|
| All-atom MD (uniform static/AC) | 10 MV m−1–10 GV m−1 | Continuous (static/AC); τon ≈ 50 ps–5 µs | High E often used to access rare events on short trajectories; bulk periodic solvent, limited interfaces.79,138,181 |
| EF-X in crystals (nanosecond pulses) | 50–100 MV m−1 | τp ≈ 50–500 ns; N ≈ 1; τon ≈ τp | Direct, ultrafast perturbation; can drive coordinated motions along functional modes.22 |
| THz/mm-wave irradiation (pulsed or gated) | 0.1 kV m−1–100 MV m−1 | Ultrafast: τp ≈ 0.5–10 ps; gated trains: τp ∼ 0.1–1 µs; N ∼ 105–108; τon ∼ 1 µs–10 s | Couples to collective modes; can be nonthermal or thermal depending on absorption and duty cycle (instantaneous E vs. duty).24,25,163,177,293 |
| Electrode-based PEF (solutions/cells) | 0.1–10 MV m−1 | τp ≈ 1 ns–1 ms; N ∼ 10–104; τon ∼ 1 µs–100 ms | EF direct effects are often mixed with Joule heating and electrode reactions; elapsed treatment time can be ≫τon.134,243,343,355 |
| Low-frequency AC/DC (microfluidics, electrodes) | 1 V m−1–100 kV m−1 | Continuous/sinusoidal: τon ≈ 1 s–days (period ∼ 1 µs–1 s) | Electroosmosis/ICEO (induced charge electroosmosis) and electrohydrodynamic flows can dominate near interfaces at low f.165,173,304–306 |
4.2 Solvent- and ion-mediated contributions: bridging direct and indirect effects
External EFs always act on proteins within a solvent/ion matrix; thus even ostensibly “direct” EF–protein couplings are frequently mediated by the reorganization of hydration water and mobile ions. In bulk water, molecular simulations show that static EFs induce pronounced anisotropy in structure and dynamics (hydrogen-bond kinetics, reorientation and translation),356 while oscillatory fields can drive non-equilibrium responses that depend on frequency and field amplitude.357 At sufficiently strong fields, water can also exhibit enhanced proton-transfer/dissociation behavior and nontrivial thermodynamic (including entropic) contributions.358,359These solvent responses feed back on proteins by (i) changing effective dielectric screening and local electrostatic potentials, (ii) modifying hydration-shell friction and thus rotational diffusion and conformational kinetics, (iii) reshaping ion atmospheres and interfacial double-layer dynamics (thereby enabling electroosmotic/ICEO flows), and (iv) providing chemical pathways for electrochemistry-driven ROS/RNS formation in electrode-based geometries. A unified perspective is therefore to treat observed protein responses as a superposition of intramolecular EF coupling and solvent-/interface-mediated contributions, with the relative weight determined by waveform, frequency, conductivity, and boundary conditions. This framework also helps rationalize why similar nominal E can yield different outcomes in simulations (periodic bulk solvent, no electrodes) versus experiments (finite cells with interfaces and electrochemical constraints) and it is therefore important to consider such differences when comparing results obtained under seemingly identical conditions.
4.3 Research at higher complexity scales: protein-containing biosystems and biosamples
There are classes of research works that observe the effects of EFs on proteins in complex systems. While the complexity of the systems (cells, tissues, complex food matrices) makes it difficult to assess whether direct or indirect EF effects are in place or the effects were taking place indirectly through other non-protein biomolecular components or indirect EF effects, these works seem to share common effects at the level of proteins so we briefly review them here. In one class of the works, typically from food research and biotechnology,51,54–57,59–63,241,360–364 often the complex samples containing proteins (mostly enzymes) were exposed to EFs. We excluded these studies from Table 2 in cases when the protein type and content of the protein-containing sample (intact or homogenized fruit, plant, or meat tissue) is undefined or the EF is used also for substantial thermal treatment, hence obscuring the mechanism of EF action. However, the results therein are relevant for research in the food industry and also provide an indication of and inspiration for research on the EF effects on proteins. The techniques employed to explore EF effects on proteins included native PAGE,365 SDS–PAGE,366,367 UV absorption spectroscopy,366,367 intrinsic (Trp or Tyr) fluorescence,368–371 CD spectroscopy,371,372 FTIR spectroscopy,366,373–375 Raman spectroscopy,376 accessible sulfhydryl group content determination,176,365–367,371,372,377,378 high-performance size-exclusion chromatography,372 high-performance liquid chromatography,365 protein surface hydrophobicity assay,367–369,377 dynamic light scattering,176,365,372,378,379 turbidity analysis,367 transmission electron microscopy,379 viscosity measurement,378,379 and enzyme activity assay.380–383 The results in these studies align with those in Section 2 and Table 2: the EF exerted effects on the conformational change,366–369,371,375,376,378 protein secondary structure,366,371–375 partial or complete protein unfolding,367,370,371,375–377 and affected the enzymatic activity,382,383 aggregation,176,365,367,375,378 changed immunoreactivity371 and rheological properties of the sample.176,365,377,379| Protein [reference] | EF strength | Pulse width/duration | Frequency | Exposure | Techniques used | Effects observed | Temp. |
|---|---|---|---|---|---|---|---|
| Alcohol dehydrogenase405 | 0, 1.2–2.8 MV m−1 | 40 µs (bipolar square wave, details NR) | 1 kHz | 75 (calculated from effective treatment time 0–4.5 ms) | Enzymatic activity assay, FTIR, CD, TRP/TYR fluorescence, UV-absorption, SDS–PAGE | Secondary structure, tertiary structure, enzymatic activity | 10–35 °C |
| α-Amylase245 | 0.3–2.1 MV m−1 | 2.5–20 µs | 1128 Hz | 80 pulses | Enzyme activity assay, CD, Trp fluorescence | Enzymatic activity (increased/decreased); structural: secondary structure (increase of α-helices and turns, decrease of β-sheets), tertiary structure (increase in Trp fluorescence) | 25 °C |
| α-Amylase246 | 100 V m−1 | Continuous sine wave | 1 Hz to 1 MHz | 250 s | Enzyme activity assay | Enzymatic activity (increased at lower frequencies) | 60 °C |
| α-Amylase127 | 0.25–1.25 MV m−1 | 10 µs | 1–30 Hz | 40 min | Enzyme activity assay, UV-absorption and fluorescence spectroscopy | Enzymatic activity (reduced); structural: conformation change near the tryptophan residues | 50 °C |
| α-Amylase/-pectin complexes167 | 0–2 MV m−1 | Bipolar 40 µs pulses | 1 kHz | 1.02 (2.03) ms | Enzyme activity assay, DLS and zeta-potential, intrinsic fluorescence, FTIR, DSC, confocal light scanning microscopy (CLSM), SEM | Enzymatic activity (decreased); structural: secondary structure (decrease of α-helices and increase of β-sheets), polarity of Trp microenvironment, charge redistribution, and strengthen of protein–polysaccharide association | 30 ± 0.5 °C |
| α-Amylase, lysozyme, lipase, peroxidase, alkaline phosphatase, polyphenol oxidase, glucose oxidase, pepsin249 | 1.3–8.7 MV m−1 | 2 µs | 0.5 Hz | 30 and 100 pulses at 2 s pulse period | Enzyme activity assays | Enzymatic activity was reduced either vastly (lipase, glucose oxidase, and heat-stable α-amylase), moderately (peroxidase and polyphenol oxidase), or slightly (alkaline phosphatase), lysozyme and pepsin showed diverse activity patterns under certain voltages | 20 °C |
| β-Lactoglobulin406 | 1.25 MV m−1 | 2 ms | 0.07 Hz | 1–10 pulses | DSC, SDS–PAGE, TEM, sol–gel transition assay | Thermal stability was greatly reduced; the gelation rate was enhanced; structural: denaturation, aggregation | 4–35 °C |
| β-Lactoglobulin407 | 800 W | Continuous irradiation | 2.45 GHz | 5 s | Polarimeter | Enhanced kinetics of the folding and denaturation processes | 4–44 °C |
| β-Lactoglobulin252 | Up to 4 × 105 kW | 10 µs | 1 Hz | 1 and 10 min (30 and 300 pulses) | OPA assay, determination of the kinetic parameters, CD | The tryptic and chymotryptic hydrolysis was significantly improved, the catalytic efficiency was improved; structural: electrical pretreatments induced changes in the 3D structure of the protein bringing it to a transient or a pseudo-unfolded state | 37 and 85 °C |
| β-Lactoglobulin178 | 0–1 kV m−1 | NR | 50 Hz–1 MHz | NR | CD, Trp fluorescence, ANS fluorescence, quenching analysis-retinol | Secondary structure (increase of random coil fraction, decrease of β-sheets); tertiary structure: (unfolding - Trp fluorescence was increased at 90 °C and decreased at 70 °C, higher affinity of hydrophobic compounds when exposed to higher temperatures); higher affinity for retinol (especially at lower frequencies) | 20–95 °C |
| Actin236 | 0.46 THz radiation with an energy dose of 5.7 mJ cm−2 | 10 ms | 1 Hz | 1200 pulses (20 min duration of experiment) | Polymerization assay, fluorescence microscopy | Activation of the elongation phase of actin polymerization | 25 °C |
| Alkaline phosphatase (AP), antidinitrophenyl (anti-DNP) with its antigen164 | 0.08 W m−2 | Continuous irradiation | 100 GHz | 1–2 h | Enzyme activity and kinetics assay | AP: irradiation may lead to other structural alterations of the enzyme that impose conformational changes affecting the turnover of the enzyme, thereby reducing the number of active enzyme molecules in the solution, anti-DNP: no effects | 25 °C |
| Amyloid-β (Aβ)177 | 0.73 mW mm−2 [E = 2340 V m−1] | 2 ms | Repetition rate 100 kHz (34.88 THz) | NR | ThT fluorescence binding assay, FTIR | The aggregation speed ratio decreased to approximately 80%; the THz wave at 34.88 THz significantly slowed down the fibrotic progression; secondary structure (the structure of the β-sheets was transforming to a looser structure with more coil and bend regions) | 37 °C |
| Amyloidogenic fragment (1–27) and non-amyloidogenic fragment408 | 177 V m−1 (calculated from spatial dose 150 mJ cm−2 and duty cycle) | 1 ms | 5 Hz | Up to 30 min | Scanning electron microscopy, thioflavin T fluorescence, polarized light microscopy (congo-red staining), synchrotron-radiation IR micro-spectroscopy | Fibril aggregation of the peptides, increased formation of β-sheets. The effect present in amyloidogenic sequence, not in non-amyloidogenic ones. Equivalent heating did not cause fibril formation | 27–30 °C |
| Citrate synthase-α-crystallin409 | 1.443 kV m−1 (calculated from average SAR 4.85 kW kg−1, assuming water as the material) | 10, 15, and 20 s | 2.45 GHz | Continuous irradiation (as a characterized artifact the MW EF peaked by 3–8-fold above the average amplitude each 50 Hz) | Surface plasmon resonance | Unfolding occurred at significantly lower temperatures | 25 °C, 29.5–44 °C |
| BSA179 | 0, 1, 1.5, 2, 2.5 and 3 MV m−1 | 40 µs | 1 kHz | 0.34–1.02 ms | Fluorescence spectroscopy (fluorescence quenching, intrinsic fluorescence), CD, zeta potential, UV-absorption, ANS fluorescence, Ellman's assay, BSA/PBSA-curcumin binding affinity assays | Moderate PEF treatment induces the partial unfolding of BSA, exposing more hydrophobic cavities, thereby facilitating the non-covalent binding of curcumin to the protein (surface SH content was increased as well); however rigorous PEF treatment conditions caused BSA aggregation and weakened the binding ability of BSA to curcumin (increase of intermolecular disulphide interactions, excessive exposure of hydrophobic groups – hydrophobic collapse); secondary structure (decrease of α-helix structures, increase of β-sheet, β-coil and random coil content) | ≤30 °C |
| BSA240 | 0, 0.5, 1 and 1.5 MV m−1 | 20 µs | 1000 Hz | 15, 20, 25, 30, 35, 40 and 45 min | SDS–PAGE, CD, attenuated total reflectance-FTIR, intrinsic fluorescence spectroscopy, UV-absorption, ANS fluorescence, Ellman's assay, DLS and zeta potential | The polarization effect of the PEF enhanced the emulsifying properties of BSA–glucose conjugates by accelerating the glycation reaction, while the ionization effect significantly suppressed the browning; secondary structure (decrease of α-helix content, increase of β-sheet, β-coil and random coil content); tertiary structure: unfolding | 90 °C |
| BSA241 | 0.35, 0.45, 0.57, 0.81 MV m−1 | 50 µs | NR | 10 pulses | DLS, CLSM, ANS fluorescence, DSC, intrinsic fluorescence spectroscopy | At lower electric field strengths (0.35–0.57 MV m−1), PEF treatment improved the browning intensity, the protein solubility and the grafting degree, and reduced the particle sizes, the fluorescence intensity and surface hydrophobicity of BSA/starch conjugates. At a higher EF strength (>0.57 MV m−1), BSA/starch conjugates exhibited decreased protein solubility, grafting degree, and particle size increase | 22.5–25.8 °C |
| BSA and bovine insulin410 | 15–20 mW kg−1, 0.5 W | — | 1.0 GHz | 3 to 48 h | Light scattering measurements, TEM, thioflavin T (ThT) assay | BSA: aggregate formation; bovine insulin: amyloid fibril formation | BSA: 25 to 45 °C, bovine insulin: 60 °C |
| BSA, MTs293 | 156 V m−1 (from 6.5 mW cm−2 beam power density) | CW | 500 GHz radiation | 0.4 s irradiation time | SAXS | No structural changes | NR |
| BSA, MTs293 | 1.8 MV m−1 | 600 fs | 100 MHz pulse repetition rate, effective irradiation spectrum 0.1 to 6.0 THz | 0.2–0.4 s | SAXS | No structural changes | NR, temperature rise due irradiation <0.12 °C |
| BSA lyophilized239 | 5–20 mW (only laser power value was provided) | NR | 3.6 THz | 60 min irradiation time | CD, UV-absorption, Trp fluorescence | Conformational changes – secondary structure (changes in the intensities of characteristic bands in UV absorption and CD spectra); modification of its functional characteristics – ligand binding parameters; as a ligand is added to the treated protein solution, fluorescence quenching is observed | NR |
| BSA, lysozyme165 | 78, 150, 300 and 500 V m−1 | NR | 10 and 500 Hz | 3 h. | CD, intrinsic (Trp) fluorescence | The tertiary structure of both lysozyme and BSA was affected, the secondary structure was perturbed in lysozyme (decrease of α-helices and increase of β-sheets); the electric force applied during EF exposure was significant to perturbing hydrogen bonds stabilizing the native structure of both proteins | 22.7 °C to 24.2 °C |
| Carboxypeptidase-G2 (CPG2)243 | 1 MV m−1 | 8, 16, 24 ns | 10 Hz | 20, 40, 60, 90, 120, 180 or 240 s | Enzyme activity assay, SEC, reverse phase-HPLC, SDS–PAGE, MS and CD, inductively coupled plasma-MS | Enzymatic activity (decreased) | NR |
| Cytochrome c, myoglobin, trypsin411 | 28–130 V m−1 | Static field | Static field | ∼90 min | Intrinsic fluorescence spectroscopy, attenuated total reflection infrared (ATR-IR), UV-VIS absorbtion, ThT fluorescence, CD, SEM | Secondary – (both the α-helix and β-sheet content decreased) and tertiary structure unfolding; amorphous aggregation | 25 °C |
| Apoazurin286,288 | 1–10 MV m−1 | Static field | — | — | Intrinsic (TRP) fluorescence spectroscopy | Change (decrease) of fluorescence intensity, however no conformational change in the protein | 20 °C |
| Ovomucin-depleted egg white228 | 70–170 kV m−1 | 20 µs | 300 Hz | NR | SDS–PAGE, turbidimetry | Aggregation | Below 50 °C |
| Elastin173 | 12 and 20 kV m−1 | Continuous exposure | Continuous exposure | More than 5 days | DLS and zeta-potential, field emission-SEM, FTIR and photoluminescence (PL) spectroscopy | Conformational changes in the secondary structure (increase of α-helices and decrease of β-sheets and turns); unfolding, denaturation and the size of the aggregates were reduced | RT |
| (Enhanced) green fluorescent protein (EGFP)284 | 250 mW | Continuous microwave irradiation (100 ps) | 9.5 GHz | ∼4.5 min | Fluorescence emission anisotropy | Microwave irradiation affects polarization of the EGFP fluorescence | 9.7–12.5 K |
| Ferritin141 | 1, 2, 3 or 4 MV m−1 | — | 2000 Hz | — | Fluorescence spectroscopy, CD, intrinsic fluorescence spectroscopy, Raman spectroscopy, cold field emission-SEM, TEM | Secondary structure: as the EF intensity increased, the α-helix content showed a trend of first decreasing and then increasing, and the β-sheet content trend was the opposite; tertiary structure: partial unfolding, decrease in the stability; reversible self-assembly; aggregation (the solubility of ferritin decreased with increasing exposure of hydrophobic groups – breakage of hydrogen bonds and the transformation of SS bridges) | RT |
| Green fluorescent protein (GFPuv5)283 | 50 and 70 MV m−1 | ∼60 ps | 40 Hz | 30 ms | Electric field modulation spectroscopy | Field-induced quenching of the fluorescence was observed, changes in the fluorescence decay profile – decrease in the fluorescence lifetime | RT |
| Horseradish peroxidase (HRP)242 | 0.5–2.5 MV m−1 | 1.5 µs | 10 Hz | 207–1242 pulses | Enzyme activity assay, CD, intrinsic fluorescence spectroscopy | Enzymatic activity (decreased); secondary structure (decrease of α-helix content); tertiary structure (intrinsic fluorescence intensity increased) | Below 40 °C |
| HSA fibrils227 | 0–23 MV m−1 | Static field | — | 10 min | ThT fluorescence, CD, FTIR spectroscopy, TEM, fluorescence microscopy | Secondary structure (decrease of β-sheet content), the initial propagation of the fibrils was followed by the disintegration of the fibrillar network into smaller aggregates | RT |
| HSA fibrils235 | ∼8 MV m−1 | Continuous exposure | 5 to 100 Hz | 1–6 min | ThT fluorescence, CD, turbidimetry, DLS, FTIR spectroscopy, fluorescence microscopy, high resolution-TEM | An initial fibrillar growth was followed by the destruction of amyloid fibrils into smaller aggregates; secondary structure (reduction of β-sheets, increase of α-helix, β-turns and random structure content) | 25 ± 0.5 °C |
| Luciferase412 | ∼0.75 MV m−1 | — | 100 kHz continuous sine wave | EF 5 × 100 s ON and OFF, for signal integration | Enzymatic assay, spectrometer | Decreased luciferase activity | 22 °C |
| WT/A4V SOD1413 | 25 kV m−1 | — | DC (via saltbridge, 14–18 µA) | 2–30 min | Electrophoresis, UV-VIS spectroscopy | A4V Zn4-SOD1 monomerized and partially unfolded | 15 °C cooling of the capillary |
| Lysozyme414 | 3.5 MV m−1 | Bipolar square: positive 2 µs, 20 µs gap, negative 2 µs | — | 300, 600, 900, 1200 µs | Activity assay, CD, micro-Raman spectroscopy | Activity of lysozyme decreased, changes in the secondary (loss of α-helices increase of β sheets and random coil) and tertiary structure (transition of S–S conformation) | Max 20 °C |
| Lysozyme415 | 3.5 MV m−1 | Bipolar: positive 2 µs, 15 µs gap, negative 2 µs | 1 kHz | Total exposure 300, 600, 900, 1200 µs | Lysozyme activity assay, aggregation analysis, sulfhydryl groups analysis, surface hydrophobicity analysis, fluorescence analysis, circular dichroism | Activity of lysozyme decreased-irreversible, aggregation effect only for 1200 µs exposure, increasing number of accessible sulfhydryl groups (cleavage of the disulfide bonds), hydrophobicity increased with subsequently hydrophobic collapse for longer EF exposure than 600 µs, increasing intensity in fluorescence spectra and red shifts after EF (changes in tertiary structure), changes in secondary structure (loss of α-helices) | Below 60 °C |
| Lysozyme416 | 3.5 MV m−1 | 2 µs | 1000 Hz | Total exposure 300, 600, 900, 1200 µs | Activity assay, ANS-binding fluorescence, intrinsic fluorescence spectra, CD | Decreasing activity of lysozyme, hydrophobicity increased with subsequently hydrophobic collapse for longer EF exposure 1200 µs, increases of fluorescent emission intensity and red shifts, changes in tertiary structures according to near-UV CD analysis, conformational changes in secondary structure (loss of α-helices) | ≤60 °C |
| Lysozyme (crystal)417 | 62 or 78 mW cm−2 (483 or 542 V m−1) | 25 ms | 0.4 THz | Effect studied expected within a single pulse | Structural analysis, X-ray crystallography | Electron density changes – consistent with a subtle longitudinal compression of the α-helix | Lower than 25 °C |
| Lysozyme24 | 0.15 kV m−1 | 0.8 µs pulse filled 0.1 THz pulses | 10 kHz | 10 min | Microwave dielectric relaxation spectroscopy, NMR | Decrease of the dielectric permittivity, irradiation probably enhanced protein hydration – water with reduced mobility and more H-bonds, | 25 °C |
| Hen egg-white lysozyme (HEWL)418 | 0, 1–2 MV m−1 (1 MV m−1 for H2O hydration; 2 MV m−1 for D2O hydration) | Static, N.A. | — | Continuous during QENS (quasi-elastic neutron scattering) acquisition (duration NR) | Incoherent QENS (BASIS; ΔE ≈ 3.5 µeV; Q = 0.3–1.1 Å−1) | No measurable change in protein dynamics (up to 2 MV m−1) or hydration-water relaxation times (1 MV m−1); authors suggest threshold Ec ∼ 2–3 MV m−1 (breakdown-limited) | 210–270 K |
| Lysozyme from egg white, albumin from bovine serum and urease from jack beans419 | 10, 15, 20, 25, 30 MV m−1 | 5 ns | 3 Hz | 500 pulses | SDS–PAGE, native–PAGE, enzyme activity assay | Lysozyme and albumin without effect, urease: trimer and dimer shifted downward from 250 kV cm−1 in SDS–PAGE (quaternary and tertiary structure disruption), decreased enzymatic activity from 250 kV cm−1 | NR, max. rise 3 °C |
| Multi-protein system (lysozyme, ovalbumin, ovotransferrin)420 | 2.5 MV m−1 | 2 µs | 100 Hz | 200, 400, 600, 800 µs | Chromatography, SDS–PAGE, turbidimetry | Turbidity measurement and SDS–PAGE shows that lysozyme could be the reason for aggregation in the multiprotein system during the PEF exposure due to electrostatic interactions and disulfide bonds, chromatography does not indicate aggregates inside samples | ≤18 °C |
| Multi-protein system (diluted egg white)421 | 2.5 MV m−1 | 2 µs | 100 Hz | 200, 400, 600, 800 µs | Soluble protein content determination, particle size distribution, and Z-average size measurement, zeta potential, protein carbonyl content measurement, free accessible sulfhydryl content determination, electrophoresis (native/SDS–PAGE) | Soluble protein content decrease, Z-average particle size increases after 400 µm, creating aggregates, no increase in protein carbonyl content, slight increase in free sulfhydryl content creating aggregates, no increase in protein carbonyl content, a slight increase in free sulfhydryl content, electrophoresis showed that lysozyme was prone to precipitation with other protein components (ovalbumin, ovotransferrin) | ≤18 °C |
| Lysozyme, pepsin422 | 3.5 MV m−1 | Bipolar: positive 2 µs, negative 2 µs, gap 20 µs | NR | 100–1200 µs (lys.)/600 µs (pep.) | CD, enzymatic activity assay, sorbitol effect | Disruption of secondary structure (far-UV CD): lysozyme loss of α-helical structure and pepsin loss of β-sheet structure, unfolded tertiary structure (near-UV CD), sorbitol has a structural protective effect on both proteins against the PEF, enzymatic activity decreasing with a longer exposure to the PEF | ≤45 °C |
| MTs25 | 40.9 MV m−1 | 1 ps | Broad 1 THz spectrum (due to 1 ps pulse width), 1~kHz pulse repetition rate | Up to 11 min exposure (i.e. N = 660000 pulses) |
Light and fluorescence microscopy | MTs disassembly | RT |
| MTs25 | 10 MV m−1 | 2.9 ps | 1.5 THz, pulse 1 kHz firing frequency | Up to 11 min exposure (i.e. N = 660000 pulses) |
Light and fluorescence microscopy | MTs disassembly | RT |
| MTs25 | 3.7 MV m−1 | 6.3 ps | 0.5 THz, pulse 1 kHz firing frequency | Up to 11 min exposure (i.e. N = 660000 pulses) |
Light and fluorescence microscopy | MTs disassembly | RT |
| MTs423 | 0.6–2 kV m−1 | Continuous harmonic wave | 3.5, 20, 29 GHz | 200 s exposure time | Turbidimetry assay of MT polymerization kinetics | — | 32–39 °C |
| Tubulin during polymerization424 | 2.5 V m−1 | 2 ms | 10 Hz | 10 min during polymerization | Negative stain electron microscopy | Formation of parallel oriented arrays of MTs in the direction of the field | 37 °C |
| MTs425 | <50–190 kV m−1 | NR | 10 kHz–MHz (sinus) | NR | Dark field microscopy | Parallel arrays of MTs are formed due to the dipole moment induced along the long axis of the MTs | 25–28 °C |
| Ovalbumin (OVA), BSA, OVA + BSA426 | 2, 2.5, 3 and 3.5 MV m−1 | 2 µs | 200 Hz | 400 µs | High-performance size exclusion chromatography, native/SDS–PAGE, sulfhydryl groups analysis | Protein aggregation of OVA caused by decreasing sulphydryl groups (≤2.5 MV m−1); no aggregation effect of BSA probably because of fewer sulfhydryl groups; aggregation effect of OVA + BSA caused by disulfide bond between proteins (≤2.5 MV m−1) | ≤30 °C |
| Ovalbumin + divalent metal ions (Ca2+, Cu2+, Ba2+)427 | 0 to 5 MV m−1 | 40 µs | 1 kHz | 565, 1130, 1695, 2260 µs | Raman spectroscopy, FTIR, fluorescence spectroscopy, surface tension, surface hydrophobicity, | Surface hydrophobicity and surface tension showed an enhanced trend at first and then reduced with pulse time. The spectra showed at the beginning of the pulse treatment, some hydrophobic amino acid side chains tended to be exposed. As PEF processing continued, ovalbumin's side chain groups were more inclined to be more intermolecular. Different metal ion types and pulse treatment times modified the protein structure of ovalbumin. | 4 °C |
| Ovalbumin ± Ca2+ (ref. 161) | 0 to 5 MV m−1 | 15 µs | 1 kHz | 720 to 4320 µs | CD, TEM | PEF’s linearly unfolding of secondary structure with increasing pulse intensity or treatment time (loss α-helix and decrease β-sheets) and perturbation on the tertiary structure was more significant than that on the secondary structure. the synergetic effect PEF + Ca2+ increases the effect of unfolding of the secondary structure + creating nanotubes followed by an aggregation effect with increasing conductivity. | 20 to 45 °C |
| Ovalbumin/egg white399 | 2.7–3.3 MV m−1 | 0.3 or 0.9 µs | 1.1 Hz | 50–400 pulses (exponential decay) | Free sulfhydryl group content analysis by DTNB, UV-spectroscopy, gelation effect of egg white | Probably reversible partial protein unfolding, not caused any visible aggregation, without changes in gelation properties of egg white | <29 °C |
| Ovomucoid (IgG1 and IgE antibodies)428 | 1, 2, 3 or 4 MV m−1 | NR (similar works from the related groups use ![[2 with combining tilde]](https://www.rsc.org/images/entities/char_0032_0303.gif) µs) µs) |
2000 Hz | Flow rate 3.2 mL min−1 (neither the chamber volume nor the residence time is provided) | SDS–PAGE, SEM, contact angle assay, CD, FTIR spectroscopy | PEF reduced the binding ability of IgG1 and IgE in OVM, no changes in primary structure, changes in secondary structure – α-helix and β-sheets decrease, PEF treatment induced the expansion of protein molecules, and increased intermolecular forces in solution, surface hydrophobicity increased, total accessible -SH content increased, SEM showed changes in microstructure of OVM (rod-shaped and spherical shape was transformed into irregular flake structure structures under the influence of the PEF) | 25 °C |
| Pea protein isolate126 | 0, 0.5–2.5 MV m−1 | 40 µs (monopolar square wave) | 1 kHz | 20 (calculated from effective treatment time 0.8 ms) | Surface hydrophobicity analysis, CD, intrinsic fluorescence spectroscopy, UV-absorption, antioxidant activity analysis | Secondary structure, tertiary structure, unfolding, increased antioxidant capacity | <30 °C |
| Papain247 | 2–5 MV m−1 | 4 µs | 1.5 kHz | 200 or 500 pulses, total time 0.8 or 2 ms | Far UV CD, determination of papain activity and free cysteine content, reducing agent prevents oxidation | Significant reduction of activity after 24 hours, decrease of free cysteine content after 24 hours, no change of pH after PEF exposure, major inactivation effect of papain was related to the conformational change of the α-helix, irreversibly inactivated | 10 °C |
| Papain429 | 1 and 1.3 MV m−1 | NR | 0.2 Hz | 48–432 pulses, (flow rate 0.2, 0.4, 6.6 L min−1, chamber volume 0.13 mL estimated from electrodes dimensions) | Residual activity, relative protein concentration, sulfhydryl group content, fluorescence spectrophotometer | Enzymatic activity reduction, RPC and SH content decrease with the number of pulses for 0.2 and 0.4 L min−1 means secondary changes in structure, no significant changes in the ANS fluorescence – same surface hydrophobicity, higher peaks of fluorescence intensity for PEF means tertiary changes in structure, no emission shifts in wavelength | 20 °C (max 50 °C measured) |
| Pectinase430 | 0.3–2.1 MV m−1 (square monopolar pulses) | 5–20 µs | 1–10 kHz | 80 pulses, 50–160 mL min−1 (chamber volume NR) | Enzyme kinetics, far UV CD, fluorescence spectroscopy, SDS–PAGE | Pectinase activity increased with field strength (maximum at 12 kV cm−1) and flow rate (maximum 80 mL min−1), the PEF did not modify the polypeptide chain composition according to SDS–PAGE, in the secondary structure of the PEF sample α-helix and β-sheets increased and random coil content decreased, an increasing number of Trp residues on the surface of the pectinase molecules shows changes in the tertiary structure, the PEF increased the thermal stability of pectinase | RT |
| Horseradish peroxidase (POD), polyphenol oxidase (PPO)431 | 0.5–2.5 MV m−1 | POD: 1.4 and PPO: 0.6 µs | 10 Hz | POD: 290–1740 µs, PPO: 124–744 µs | Enzyme activity assay, CD | Enzymatic activity (decreased); secondary structure (decrease of α-helix content) | ≤40 °C |
| Pepsin162 | 1.78–3.56 MV m−1 | Bipolar square pulses: 2 µs positive, 18 µs gap, 2 µs negative | 800 Hz | Total treatment time 126–630 µs, i.e., N = 63–315 pulses | Enzymatic activity, UV absorption, CD, solubility analysis | Enzymatic activity deactivation, secondary structure loss, aggregation | <52 °C, pepsin claimed to be stable <60 °C, |
| Horseradish peroxidase, β-galactosidase, lactate dehydrogenase (LDH), glucoamylase, enolase, invertase432 | 0.4–2.3 MV m−1 | 2 µs | 50 Hz | 30 or 60 s | Enzyme activity assay, SDS–PAGE | Enzymatic activity (increased with lower and decreased with higher EF strength); the degree of the influence by PEF treatment seemed to be greatly different depending on the type of enzyme; refolding enhancement of the thermal denatured peroxidase | NR |
| The second PDZ domain of the human E3 ubiquitin ligase LNX2 (LNX2)PDZ2 (ref. 22) | ∼50–100 MV m−1 | 50–500 ns | – (effects observed within a single pulse) | — | X-ray crystallography | The symmetry of the protein crystal was reduced; perturbations of nearly every type of physical interaction throughout the protein structure—induction of side-chain rotamer flips, continuous displacement of backbone atoms, side chains and bound waters, propagated rotamer shifts suggesting collective motions through the structure, breaking and re-forming of hydrogen bonds, global motions of entire secondary structure elements, and complex coordinated changes in large regions | RT |
| Plasmin (fibrinolysis)248 | 0, 1.5–4.5 MV m−1 | 2 µs | 0.1 Hz | 10–50 pulses | Enzymatic activity assay | Enzymatic activity (decreased) | 10–15 °C |
| Polyphenol oxidase (PPO)433 | 1.8 MV m−1/0.9 MV m−1 | — | AC voltage, capacitively coupled: 10 Hz to 760 kHz | 0–6 min | Enzyme activity assay | Frequency-dependent inhibition or stimulation of PPO enzyme activity | NR, remained constant |
| Protamex (bacillolysin and subtilisin)251 | 1.4 and 1.82 MV m−1 | 20 µs | 10 Hz | 0–900 pulses | Enzymatic activity assay | Inactivation of the enzyme | 18 to 55 °C |
| Protease (from Pseudomonas fluorescens)434 | 0.62 MV m−1 | 700 µs | 0.1 Hz | 0, 10 and 20 pulses | Enzymatic activity assay | Inhibition of the enzymatic activity | 15 to 20 °C |
| S-Adenosylhomocysteine (AdoHcy), 5′-methylthiadenosine (MTA)250 | 325–583 V m−1 (calculated from 1.5–3.1 W g−1, assuming water as the material) | — | 10.4 GHz continuous irradiation | 0–90 min | Enzymatic activity assays, CD spectroscopy | Irreversible time-and temperature -dependent inactivation of both enzymes | 70 to 90 °C (below the denaturation temperature of the given enzymes) |
| Soy protein isolate180 | 0.5–3 MV m−1 | Pulse pair: 40 µs, 40 µs 200 gap, µs opposite polarity | 1 kHz | 51 (calculated from flow, volumes, total treatment time) | Turbidimetry, DLS, SDS–PAGE, size exclusion chromatography, carbonyl content (DNPH), sulfhydryl content (DTNB), surface hydrobhobicity (ANS), CD, Trp/Tyr fluorescence | Secondary structure, tertiary structure, protein–protein interaction, aggregation | 20 to 30 °C |
| Spike protein of SARS-CoV-2290 | <5 kV m−1 (estimated based on 700 W and geometry of the waveguide) | — | 2.45 GHz microwave | 2, 5 and 10 min | UV-Spectroscopy | Significant denaturation can be induced by microwave irradiation | 37 °C |
| Sup35NM (prion)238 | 1, 2 MV m−1 | 50, 1000 µs | NR | 10 pulses | AFM | Significant reductions in the length of prion fibrils, full disintegration of fibrils | RT (NR) + rise <4.6 °C |
| Transthyretin aggregates226 | 126 MV m−1 | 1 ns | 1.7 Hz | 0, 200–1000 pulses | Native/SDS–PAGE, BCA method, fluorescence microscopy | Protein aggregates disassembly | 18 °C |
| Tropomyosin134 | 0.5–2 MV m−1 | 10 µs | 50 Hz | Up to 4500 pulses (calculated from flow, volumes, total treatment time) | DLS, CD, PAGE, FTIR, Trp/Tyr fluorescence, carbonyl content (DNPH), surface hydrobhobicity (ANS) | Secondary structure, tertiary structure, interactions (depolymerization (from dimers), antibody binding) | NR/ice water bath |
| Tubulin21 | 2 MV m−1 | 11 ns | 1 Hz | 0–800 pulses | Intrinsic (Trp, Tyr) fluorescence, dynamic light scattering, zeta potential, atomic force microscopy, 2D-PAGE and immunoblotting | Protein conformation, hydrodynamic radius, effective charge, capability of self-assembly | 4 °C and slightly above |
| Tubulin, MTs80 | 100 to 400 kV m−1 | 25 to 300 µs | 1 | Effects are observed within N = 1 pulse | Electric birefringence method | Tubulin and MT orientation, affected by MT-associated proteins | 10 °C |
| Ubiquitin (Ub)163 | 18 and 90 mW cm−2 (E = 260, 582 V m−1) | 0.8 µs | 95 GHz radiation, 10 kHz pulse firing rate | 0–12 min total exposure, i.e. 5.76 s net exposure | NMR | Change of rate of hydrogen–deuterium exchange of the protein amides (accelerated in the protein interior and in hydrophobic surfaces while decelerated in the surface loop and short 310 helix regions) | 25 °C |
| Whey protein isolate355 | 1.5 and 3 MV m−1 | 25 µs | 1.04 kHz | 7.35 ms (N = 294 pulses) | SDS–PAGE, far-UV CD spectroscopy, measurement of free amino groups (o-pthalaldehyde (OPA) method) | EF promoted glycosylation of protein → decreased content of secondary structure and free amino groups, increase of glycosylation and emulsion stability | 30 ± 0.5 °C |
| System of study [reference] | Type, shape of field | Field strength | Force field and water type (where available) | Condition of simulation | Simulation length | Observable |
|---|---|---|---|---|---|---|
| Act d 2 kiwi fruit allergen435 | E, oscillating (2450 MHz) | 50 MV m−1 | CHARMM27, TIP3P | NPT (300, 325, 350, 375 K, Berendson, Parrinello–Rahman) | 2 ns | SS, RMSD, RMSF, SASA |
| Alanine polypeptide185 | E, static | 50–1000 MV m−1 | OPLS/AA, no water | NA | 400 ns | SS, entropy, enthalpy |
| Alcalase244 | E, static | 1 MV m−1 | CHARMM36 | NA | 200 ns | RMSD, H-bond, SASA, Rg |
| Amyloid-β peptide151 | E, static | 20 MV m−1 | Amber ff99SB-ILDN, TIP3P | NPT (310 K, V-rescale, Parrinello–Rahman) | 200 ns | SS |
| Amyloid-β peptide147 | E, static | 200–500 MV m−1 | Amber ff99SB-ILDN, TIP3P | NPT (300 K, V-rescale, Parrinello–Rahman) | 100 ns | Dipole moment |
| Amyloid-β peptide174 | E, oscillating (1 GHz) | 10, 100, 200 MV m−1 | NVT (300 K) | CHARMM36m | 300 ns | RMSD, RMSF, H-bond, SS, free energy |
| Amyloid-β peptide133 | E, static, oscillating (0.1–1 GHz) | 175, 200 MV m−1 | ff14SB, TIP3P | NVT(300 K, Monte Carlo) | 100 ns | Interpeptide distance, Rg, dispersion entropy, dipole moment, potential of mean force |
| Amyloid-β (1BA4) peptide214 | E, static | 100, 250, 500, 1000 MV m−1 | CHARMM, TIP3P | NPT | 10 ns | RMSD, dipole moment, SS, Rg |
| Amyloid-β peptide175 | E, static, oscillating (0.1, 1 GHZ) | 100, 200 MV m−1, | ff14SB, TIP3P | NVT (300 K, Langevin thermostat, Berendsen barostat) | 500 to 1200 ns | RMSD, H-bonds, dipole moment, SS |
| apoC-II peptide436 | E, EM, static, oscillating (1, 2.5 and 5 GHz) | 0.7 to 700 MV m−1 | GROMOS96-43a1, SPC | NPT (300 K, Berendson) | 200 ns | Rg, N–C termini distance, dipole moment, radial distribution functions (rdf), H-bond(s) |
| apoC-II peptide153 | E, EM, static | 7 kV m−1 to 700 MV m−1 | GROMOS96-43a1, SPC | NPT (300 K, Berendson) | 4.8 µs | RMSD, dipole moment, free energy map, Rg, amino acids conformations |
| Ara h 6 peanut protein155 | E, static, oscillating (2450 MHz) | 50 MV m−1 | CHARMM27, TIP3P | NPT (Berendsen, 300, 380, 425 K) | 1 ns | RMSD, Rg, dipole moment, SASA, SS |
| Aquaglyceroporin-7437 | E, static, oscillating (10 GHz) | 0.2 MV m−1 | CHARMM36, SPC/E | NPT (300 K, Nosé–Hoover, Parrinello–Rahman) | 71 ns | RMSD, RMSF, Rg, free energy, H-bonds, water permeation |
| Human aquaporin 4188 | E, static, oscillating (2.45, 20, 50, 100, 200, 500 GHz) | 65 MV m−1 to 25 GV m−1 | CHARMM27, TIP3P | NPT (298 K, Nosé–Hoover, Langevin dynamics) | 9 µs | Open and closed states of selectivity filter, dipole, potential-of-mean-force profile |
| Bovine insulin (2A3G)157 | E, static | 150–600 MV m−1 | Amber ff03, TIP3P | NPT (Berendsen, Parrinello–Rahman) | 1 µs | Dipole moment, SS, H-bonds |
| BSA, trypsin158 | E, static | 0.03–10000 MV m−1 |
Amber all-atom, TIP3P | NA | NA | RMSD |
| BSA240 | E, static | 1.2 MV m−1 | OPLS-AA, TIP3P | NPT | 30 ns | SASA, SS |
| Carnosine191 | EM, oscillating (2.45 GHz) | 10−6–102 V m−1 | OPLS-AA | NVT (velocity rescaling) | 20 ns | Electrostatic potential surface, dipole moment, molecular collision number, kinetic energy of collision |
| β-Casein (BCN)160 | E, sinusoidal | 4–4000 MV m−1 | CHARMM36, TIP3P | NPT (300 K, Berendsen, Parrinello–Rahman) | 10 ns | RMSD, RMSF, Rg, dipole moment, SS, distance matrix for residue pairs, SASA |
| Cav2.1 channel183 | E, oscillating (40.12, 42.5, 42.65, 42.83, 46.63, 48.5 THz) | 600 MV m−1 | CHARMM36, POPC, TIP3P | NPT (315 K, Nosé–Hoover, Parrinello–Rahman) | 3 ns | Free energy profile, H-bonds, vibrational spectra |
| Chignolin (1UAO)438 | E, static | 100, 250, 500 and 1000 MV m−1 | CHARMM27 and TIP3P | NPT (300 K, Berendsen thermostat) | 10 ns | RMSD, dipole moment, SS, Rg |
| Chignolin439 | E, static, oscillating (9–3600 ps period, i.e. 111 GHz–278 MHz) | 1000 MV m−1 | CHARMM27, TIP3P | NPT (300 K, Berendsen) | 10 ns | RMSD, SS, dipole moment |
| Chignolin181 | E, static, oscillating (2.45 GHz) | 200 MV m−1 | Amber ff99SB, TIP4P/2005 | NPT (300 K, Berendsen, Parrinello–Rahman) | 5 µs | RMSD, SASA, free energy, H-bond(s), Ramachandran plot analysis |
| Connexin 26 hemichannel (2ZW3)152 | E, static, oscillating (2.4 and 50 GHz) | 100, 175, 250, 325, 400 MV m−1 | CHARMM 36, TIP3 | NPT (310 K, Nosé Hoover, Parrinello–Rahman) | 50 ns | RMSD, dipole moment, H-bonds, tertiary structure |
| Cytochrome c154 | E, static | 500–2000 MV m−1 | CHARMM, TIP3P | NVT (310 K, Berendesen) | 40 ns | Protein–surface distance, protein orientation, radial distribution function, protein–surface interaction energy, RMSD |
| Dihydro-lipoamide dehydrogenase (1BBL)149 | E, static | 200–800 MV m−1 | CHARMM27, TIP3P | NPT (310 K) | 30 ns | RMSD, dipole moment, SS, Rg, H-bonds |
| Diphenyl-alanine peptide144 | E, static | 13 GV m−1, 19.5 GV m−1 | CHARMM 36-AA, TIP3P, REMD | NPT (Berendsen, Parrinello–Rahman) | 98 to 126 ns | Potential energy, RMSD, dipole moment |
| Dronpa348 | E, static | 0.4–1.3 GV m−1 | CHARMM27, water model not mentioned? | Langevin dynamics, 300 K | 85 ps | Dipole moment |
| Ferritin141 | E, static | 1–4 MV m−1 | CHARMM36 | NA | 350 ns | RMSD, SASA |
| Fibronectin-hydroxyapatite146 | E, static | 250–1000 MV m−1 | Amber ff99SB ILDN, TIP3P | NPT (300 K, Berendsen, Parrinello–Rahman) | 100 ns | Protein–ligand contacts, interaction energy, dipole moment, solvent radial distribution function |
| Hemoglobin440 | E, linear/circularly polarized (5.9 GHz) | 10, 7.1 V m−1 (looks like a typo, maybe authors meant GV m−1) | CHARMM36, TIP3P | NPT (310 K) | 51 ns | RMSD, SS |
| HEWL213 | E/M, oscillating (50–500 GHz) | 1–5 GV m−1 (RMS) | CHARMM27, TIP3P | NVT (300 K, Nosé Hoover) | 1 ns | RMSD, dipole moment |
| HEWL mutants441 | E/M, static, oscillating (2.45 GHz) | 100 to 1500 MV m−1 (RMS for osc, and amplitude for static) | GROMOS96 and SPC | NPT (Anderson–Hoover, 300 K) | 2.5–25 ns | RMSD, dipole moment |
| HEWL mutant (2LZT)401 | E/M, oscillating (2.45–100 GHz) | 500 MV m−1 (RMS) | GROMOS96, SPC | NPT (300 K) | 5 ns | H-bond |
| HEWL186 | E, static, oscillating (2.45, 10, and 100 GHz) | 50, 100, 200 MV m−1 | Amber ff99SB, TIP4P/2005 | NPT (V-rescale, Parrinello–Rahman) | 100 ns | Water density distribution, dipole moment |
| Human aquaporin 4187 | E, static, oscillating (2.45, 100 GHz) | 65 MV m−1 | CHARMM27, TIP3P | NPT (298 K, Nosé–Hoover, Langevin dynamics) | 50 ns | Dipole moment |
| Human aquaporin 4 (3GD8) in lipid bilayer442 | E, oscillating circularly polarized (100 GHz) | 500 MV m−1 | CHARMM27, TIP3P | NPT (298 K) | 60 ns | Pore radius, mean square displacement (MSD) of water |
| Human aquaporin 4263 | E, static | 200–500 MV m−1 | CHARMM27, TIP3P | NPT (298 K, Nosé–Hoover chains, Langevin dynamics piston) | 2–11 ns | Ion conductivity, pore radius profile |
| Insulin chain-B (1ZNI)443 | E, static, oscillating (2.45 GHz) | 10–1000 MV m−1 | CHARMM27, TIP3P | NPT (300 K, 400 K, Nosé–Hoover, Langevin dynamics) | 1.7–10 ns | RMSD, dipole moment, SS |
| Insulin chain-B (1ZNI)444 | E, oscillating (1.225, 4.9 GHz) | 10 to 500 MV m−1 (amplitude) | CHARMM27, TIP3P | NPT (300 K, Nosé–Hoover) | 10 ns | RMSD, dipole moment, SS, Rg |
| Integrins445 | E, static, oscillating (PEF) | 0.0015, 0.015, 0.15 MV m−1 for static; 0.3, 1, 3 MV m−1 for pulse (pulse width 10, 100, 1000 µs) | Monte Carlo simulations | NA | 20 s | Integrin clustering |
| Kinesin–tubulin225 | E, static | 30–100 MV m−1 | CHARMM36, TIP3P | NVT (300 K, Nosé–Hoover) | 30 ns | RMSD, intermolecular contacts, dipole moment, translational/rotational work, SS |
| Laccase446 | E, static | 0.01–1000 MV m−1 | OPLS/AA, SPC | NPT (300 K) | 50 ns | RMSD, H-bonds, dipole moment, SS, Rg, binding affinity |
| β-Lactoglobulin447 | E, oscillating (2.45 GHz) | 0.5 GV m−1 | Amber 99SB—ILDN | NPT (300, 333, 348, 363 K) | 2 ns | SS, RMSD, RMSF, Rg, dipole moment, SASA |
| β-Lactoglobulin148 | E, static | 3000 MV m−1 | Amberff14SB, TIP3P | NVT (300, 400, 450 K, Berendsen, Langevin dynamics) | 240 ns | RMSD, RMSF, SASA, dipole moment |
| L-type calcium channel448 | E, static, oscillating (1, 2.5 GHz) | 30 MV m−1 | GROMOS 96-43a1, SPC | NPT (310 K, Nosé–Hoover, Berendsen) | 10 ns | RMSD, RMSF, H-bonds |
| Lysozyme, myoglobin156 | E, static | 75, 180, 424 MV m−1 | Amber ff99SB, TIP3P | NVT (Langevin dynamics, 298 K) | 40 ns | Dipole moment, SASA |
| Model peptides400 | E, static | 1–5 GV m−1 | DFT/PCM, water, ethanol | Free energy, conformational stability, SS, H-bond(s) | ||
| Myofibrillar proteins145 | E, static | 0.8–2.8 MV m−1 | Gromacs 53a6, SPC | NPT (277 K, Berendsen thermostat and barostat) | 30 ns | H-bonds, Rg |
| Myoglobin190 | EM, oscillating (1 GHz) | 0.001 to 500 MV m−1 | Perturbed matrix method | NPT (300 K) | 10–20 ns | RMSD, free energy |
| Myoglobin159 | E, static, pulse | 100–1000 MV m−1 | GROMOS43a1, SPC | NVT (Berendsen) | 100 ns | Dipole moment, SS |
| Myoglobin449 | E, Gaussian pulse (time length = 8 ns), bipolar pulse (time length = 2 ns) | 100–1000 MV m−1 | GROMOS96-43a1, SPC | NVT (300 K, Berendsen) | 20 ns | Dipole moment, SS, H-bonds |
| Myoglobin221 | E, static | 100 V m−1 to 1000 MV m−1 | GROMOS96-43a1, SPC | NVT (300 K) | 100 ns | Dipole moment, SS |
| Myoglobin220 | E, oscillating (0.1–100 GHz) | 2–200 MV m−1 | ffG43a1, SPC | NVT (200 K, Berendsen thermostat) | 50 ns | Dipole moment |
| Voltage-gated ion channels: NavMs, NavPaS, HCN1266 | E, static, pulse | 97–108 MV m−1 | CHARMM36, TIP3P | NVT (300 K, Nosé–Hoover) | 200 ns | SS, pore size, salt-bridge, ion conduction |
| Ovalbumin140 | E, static, oscillating (2.5 GHz) | 500 MV m−1 | CHARMM33, TIP3P | NPT (300, 320, 340, 360 K, Nosé–Hoover) | 5 ns | SS, H-bond(s), saltbridge interactions, Rg, dipole moment, SASA |
| Peptides450 | E, static | 1.2–5 GV m−1 | DFT/PCM, vacuum | Free energy, geometry of the metal complex, dipole moment | ||
| α-Helix peptides68 | E, pulsed, oscillating (1–2 GHz) | 500 MV m−1 | CHARMM27, TIP3 | NPT (310 K) | 10 ns | RMSD, RMSF, dipole moment, SS |
| β-Sheet peptides184 | E, static | 4, 6, 10 GV m−1 | CHARMM27, TIP3P | NVE (300 K) | 4 ns | Potential of mean force (PMF) |
| Pancreatic trypsin inhibitor (BPTI)347 | E, static, square wave [pulse], oscillating | 100 MV m−1, 2 GV m−1 and 2.17 GV m−1 | CHARMM | NVE (100 K) | 150 ps | RMSD, dipole moment |
| Pea protein isolate126 | E, static | 1 MV m−1 | OPLS-AA, TIP3P | NPT (300 K, V-rescale, Parrinello–Rahman) | 25 ns | RMSD, RMSF, SASA, SS |
| Pectin methylesterase142 | E, static | 0.007–1000 MV m−1 | CHARMM27, TIP3P | NPT (323 K, Berendsen, Parrinello–Rahman) | 5 ns | RMSF, RMSD, Rg, SASA, B-factor, water dynamics, hydration of enzyme, intra/inter-molecular H-bonds, dipole moment, Ramachandran plots |
| Amyloid-β peptide (2BEG)234 | E, static | 250, 500 MV m−1 | CHARMM27, TIP3P | NPT (300 K, Berendsen, Langevin dynamics) | 50 ns | RMSD, SS, Rg, H-bonds |
| Polygalacturonase451 | E, static | 0.75–1.25 GV m−1 | CHARMM36, TIP3P | NPT | 10 ns | SS, RMSD, RMSF, H-bonds, hydrophobic SASA, dipole moment, free binding energy |
| Soybean hydrophobic protein452 | E, static | 2, 4, 3000 MV m−1 | CHARMM27, TIP3P | NPT (300 K, Berendsen) | 1 ns | RMSD, dipole moment, SS, Rg, SASA |
| SARS-CoV-2 spike protein125 | E, static | 0.1–10 MV m−1 | CHARMM36, TIP3P | NPT (300 K, Nosé Hoover, Parrinello–Rahman) | 700 ns | RMSD, RMSF, SS |
| SARS-CoV-2 spike protein139 | E, static | 0.01–10 MV m−1 | CHARMM36, TIP3P | NPT (300 K, Nosé Hoover, Parrinello–Rahman) | 700 ns | RMSD, RMSF, SS, free energy, electrostatic potential |
| Addlinespace serotonin 1A receptor (5-HT1AR)135 | E, static | 10–30 MV m−1 | Amber ff14SB, TIP3P | NPT (310 K, Nosé Hoover, Parrinello–Rahman) | 600 ns | RMSD, Rg, contact number between protein and ligand, dipole moment. |
| Serotonin 1A receptor (5-HT1AR)137 | E, static | 20 MV m−1 | Amber ff14SB, TIP3P | NPT (310 K, velocity-rescaling, Parrinello–Rahman) | 500 ns | RMSD, h-bond, residue contact count, dipole moment, SS |
| SOD1216 | E, monopolar, bipolar nsPEF (pulse duration 2 ns) | 100–700 MV m−1 | GROMOS96, SPC | NVT (300 K, V-rescale) | 200 ns | Dipole moment, SS, RMSD, SASA, Rg |
| Soybean beta-conglycinin, soybean glycinin180 | E, static | 100, 200, 300 MV m−1 | Gromos 53a6, TIP3P | NPT (300 K) | 30 ns | RMSD, H-bonds, SS |
| Amyloid-β fibrils136 | E, static, oscillating (0.2 GHz) | 200–400 MV m−1 | Amber ff99SB-ILDN, TIP3P | NPT (310 K, V-rescale, Parrinello–Rahman) | 500 ns | SS, dipole moment, dielectric constant, H-bonds, PCA |
| Thioredoxin182 | E, static | 20–120 MV m−1 | SPC | NPT (300–350 K) | 100 ns | pKa, free energy, PCA, SASA, H-bonds, dipole moment |
| Tropomyosin134 | E, static | 0.5, 1.0, 2.0 MV m−1 | CHARMM36, SPC/E | NPT (298.15 K) | 50 ns | SS, H-bonds, SASA |
| Tubulin218 | E, static, oscillating (1 GHz) | 30 MV m−1 | GROMOS96-43a1, SPC | NPT (300 K, V-rescale, Parrinello–Rahman) | 500 ps | Elastic constant, Young's modulus |
| Tubulin150 | E, static | 5–75 MV m−1 | CHARMM36, TIP3P | NPT (310 K, Langevin dynamics, Nosé–Hoover–Langevin) | 10 ns | RMSD, RMSF, dipole moment |
| Tubulin, kinesin233 | E, oscillating (1–10 GHz) | 30 MV m−1 | GROMOS96-43a1, SPC | NPT (310 K, Nosé–Hoover, Parrinello–Rahman) | 10 ns | Flexibility of MTs, kinesin’s affinity to tubulin |
| Tubulin–kinesin complex217 | E, static | 100 MV m−1 | CHARMM36, TIP3P | NVT (Nosé–Hoover thermostat) | 30 ns | Dipole moment, RMSF, contact surface |
| Tubulin78 | E, static | 10 to 300 MV m−1 | Amber03 force field, TIP3P | NVT (300 K) | 30 ns | Dipole moment, SS, shape and orientation (rotation) of protein |
| Tubulin ring46 | E, static | 50, 100 MV m−1 | Amberff14SB, TIP3P | NVT (Langevin thermostat, 0.5 ps coupling) | 50 ns | Dipole moment, binding energy |
| αβ-Tubulin and kinesin232 | E, oscillating (1 to 10 GHz) | 30 MV m−1 | GROMOS96-43a1, SPC | NPT (310 K, Parrinello–Rahman) | 500 ps | Kinesin’s affinity to tubulin dimer |
| αβ-Tubulin dimer pairs219 | E, oscillating (1–10 GHz) | 30 MV m−1 | GROMOS96-43a1, SPC | NPT (310 K, V-rescale, Parrinello–Rahman) | 300 ps | Elastic constant |
| αβ-Tubulin453 | E, oscillating (900 MHz, 2450 MHz) | 10 MV m−1 | GROMOS96-43a1, SPC | NPT (310 K, Nosé Hoover, Parrinello–Rahman) | 10 ns | RMSD, elastic constant, Young’s modulus |
| Trp-cage, Ctf, ubiquitin, lysozyme79 | E, static | 0 to 3 GV m−1 | OPLS-AA/L, vacuum | No-PBC (300 K, Berendsen) | 10 ns | RMSD, degree of orientation, total internal energy |
| Ubiquitin123 | E, time dependent | 100 to 3000 MV m−1 | OPLS-AA, vacuum | NVT (300 K, Berendsen thermostat) | 5 to 14 ns | Degree of orientation, RMSD |
| Ubiquitin47 | E, static | 3 GV m−1–11 GV m−1 | OPLS/AA, vacuum | NVT (300 K, Nose thermostat) | 50 ps | Unfolding, RMSD |
| Ubiquitin138 | E, static | 10–200 MV m−1 | Amber ff99sb-ILDN, SPC/E | NPT (310 K, Berendsen, Parrinello–Rahman) | 1 µs | RMSD, RMSF, H-bonds, dipole moment, Rg |
| Ubiquitin82 | E, static | 0.1–5 GV m−1 | CHARMM36, TIP3P | NPT (300 K, Langevin dynamics, Nosé–Hoover) | 5 ns | Dipole moment, SASA, RMSD, Rg, protein rotational diffusion, water diffusion |
| Ubiquitin130 | E, static | 0.05–3.0 GV m−1 | OPLS-AA, TIP4P | No-PBC (300 K, Berendsen) | 10 ns | Dipole orientation, dipole moment, RMSF, RMSD, diffraction pattern |
| Voltage sensing domain (VSD) of the NaV1.5 sodium cardiac channel454 | E, static | 100–200 MV m−1 | CHARMM36, POPC | NPT (310 K, Nosé–Hoover, Parrinello–Rahman) | 50 ns | RMSD, clustering structures, density profile of clusters based on RMSD and FNC (fraction of native contacts), free energy |
| Voltage-gated potassium (Kv) channel455 | E, static | 40–200 MV m−1 | CHARMM22, TIP3P | NPT (300 K, Langevin dynamics, Langevin piston) | 20 ns | SS, RMSD, water density, pore-radius profile |
| Voltage-gated potassium (Kv4) channel456 | E, static | 250 MV m−1 | CHARMM36, TIP3P | NPT (310 K, Nosé–Hoover, Parrinello–Rahman) | 4 ns | Pore radius, cross-sectional area, dipole moment |
| Voltage-gated potassium (Kv1.2) channel457 | E, oscillating (36.75 37.06, 37.68, and 38.2 THz) | 400 MV m−1 | CHARMM36m, CHARMM TIP3P | NPT (310 K, V-rescale, Parrinello–Rahman) | 400 ns | Dihedral angle, ion permeation, RMSD, RMSF, potassium ion occupancy |
| Voltage-gated Ca2+ channel143 | E, static | 100, 200 MV m−1 | CHARMM36, TIP3P | NPT (310 K, Nosé–Hoover, Parrinello–Rahman) | 50 ns | RMSD, fraction of native contacts (FNC), free energy |
Another class of works focused on the analysis of (mostly pulsed) EFs on proteins and their structures in cells. In such works, it was not possible to determine whether the effects of the EFs on the protein structures in the cells were direct or indirect (e.g., down-stream effects owing to plasma or internal membrane electroporation and change of intracellular ionic concentration or consequent cell swelling384). In these studies, most of the tools used to explore such effects were fluorescence-based microscopy techniques (epifluorescence, confocal), which enabled localization and visualization of the protein structures, such as cytoskeleton fibers. For instance, the effects of pulsed EFs on cytoskeleton structures were explored in several works.65 It was found that a MT network was disrupted289,385–388 or more densely packed389 either immediately or with some delay after electric pulses were applied. A recent study demonstrated the possibility to remodel the MT cytoskeleton in cells without a complete de-polymerization phase.390 Furthermore, the effects of a PEF were also observed on actin filaments391 in plant cells and in animal cell cultures.386,389,392–394 In some studies,395,396 the finding that the presence of Ca2+ in the buffer was required to obtain the EF effect strongly indicated that the EF acted only indirectly on actin filaments through modification of calcium signaling.
These in vivo (i.e., in cells) observations of effects of EFs on protein-based structures are biologically and biotechnologically significant, as they enable control of the cell fate with a physical EF handle. Although in most cases they can not provide a mechanistic understanding of the primary target of EF action, they provide inspiration for researchers to generate hypotheses for further in vitro research, such as in ref. 397.
4.4 Further outstanding questions for future research
In early PEF work, ovalbumin exposed to 2.7–3.3 MV m−1 exhibited transient activation of sulfhydryl groups and minimal spectroscopic changes, with recovery of the native-like state over minutes to hours,399 consistent with partial unfolding followed by refolding. In cytoskeletal systems, intense nanosecond pulses at 2 MV m−1 modulated tubulin conformation and in vitro MT assembly; recovery of repolymerization capacity after cold depolymerization was observed at modest pulse numbers, while high-dose exposures led to incomplete restoration, indicating a graded shift from reversible to irreversible.21 The recent study by Wang et al.180 on soy protein isolates mapped a critical intensity near 1–1.5 MV m−1, below which depolymerization and aggregation changes reversed within ∼30 min, while at ≥2 MV m−1 reaggregation was stabilized by hydrophobic and disulfide crosslinks.
Atomistic simulations extend these experimental findings, albeit in a different range of EF strengths and timescales. Density functional theory/PCM (polarizable continuum model) calculations on model α-helices revealed a threshold near ≈2.6 GV m−1 for complete helix disruption when the EF is aligned with the helix dipole; importantly, removing the field allowed the misfolded peptide to refold to near-native geometry, even in polar solvents.400 Constant-field MD of lysozyme in the 0.5 GV m−1 range under static and oscillating conditions showed accelerated hydrogen-bond breakage/reformation cycles, largely reversible except for some persistent losses in solvent-exposed charged regions at specific driving frequencies.401 For ubiquitin, moderate fields (≲0.5 GV m−1) induced reversible dipole reorientation and minor secondary-structure changes, while higher fields (1–2 GV m−1) could still be reversible if switched off prior to overstretching.140
Analogies can be drawn – cautiously – from protein refolding after mechanical unfolding,229 reassembly of supramolecular protein complexes, and reversible disruption of peptide–peptide interfaces. Across these contexts, reversibility hinges on perturbations that do not cross irreversible kinetic barriers such as extensive hydrophobic-core exposure or covalent bond scission. For EF perturbations, key determinants include the field vector relative to the intrinsic protein dipole, temporal waveform, and structural context (e.g., solvent-exposed flexible tails versus buried secondary-structure cores). Designing EF protocols that exploit such “safe windows” could enable reversible, on-demand modulation of protein structure and function.
Across field modalities, the characteristic recovery time and dominant sources of irreversibility span many orders of magnitude. In EF-X, perturbations delivered on nanosecond timescales can drive reversible, collective motions in crystals without apparent structural degradation.22 In sub-THz/THz irradiation, the primary solvent relaxation occurs on ps–ns timescales, suggesting that reversible modulation is plausible provided that the field does not trigger large-scale unfolding or chemical modification.24,163 By contrast, in electrode-based PEF experiments, irreversibility often originates from indirect pathways (Joule heating, pH gradients, ROS, and bubble interfaces), which act on ms–min timescales and can accumulate under repeated exposures. This is particularly relevant for emerging protein/peptide-based piezoelectric platforms,402 where longevity may be limited by cumulative electrochemical and hydration changes; mitigating strategies include minimizing duty cycle (an E2-weighted “dose”), using bipolar pulses, and isolating electrodes from the protein-containing volume.266
5 Conclusion
External EFs offer a powerful, reagent-free handle to perturb proteins and to modulate their interactions and function. Across the studies reviewed here, EFs couple directly to charged and dipolar groups, biasing rigid-body motion, reshaping intra- and inter-molecular electrostatics, and – when sufficiently strong or resonant with intrinsic motions – modifying secondary/tertiary/quaternary structure. In parallel, high-field protocols often introduce indirect electrophysical and electrochemical stresses – Joule heating, electrohydrodynamic flows, shock/cavitation, electrolysis-driven pH gradients, reactive oxygen/nitrogen species (ROS/RNS), and bubble interfaces – that can dominate the observed outcome if not controlled.A central message is that “EF effects on proteins” should be interpreted as a competition between direct EF–protein coupling and solvent-/interface-mediated pathways. The balance is set by the field strength, waveform and frequency, exposure time (or duty cycle), and geometry (electrode-based vs. contactless), together with protein stability and solution conditions (conductivity, ionic strength, buffering capacity, redox environment). When indirect pathways are mitigated, reversible control becomes feasible: ultrafast nanosecond perturbations in EF-X can drive coordinated motions along functional modes,22 while pulsed MV m−1 regimes and THz exposures can modulate binding, catalysis and supramolecular assembly on experimentally accessible timescales.24,25,225 When indirect stresses accumulate (heating, electrochemical byproducts, interface adsorption), irreversible unfolding and aggregation are more likely.
Preliminary design rules for EF-based protein control:
• Match the field vector to the target degree of freedom. Orientation and torque couple to the permanent dipole moment and charge distribution, while local polarization and bond-scale rearrangements couple to induced dipoles and heterogeneous internal electrostatics. Electro-optical experiments and modelling commonly place protein dipole moments in the ∼102–103 D range, so fields of order 1–10 MV m−1 already give μE ∼ kBT and can bias orientation, whereas remodeling tertiary interfaces, oligomer contacts, or buried secondary structure typically requires substantially larger effective fields and/or longer forcing, and is strongly direction-dependent.22,81,113,116,225
• Treat dose as a coupled strength–time problem. For many indirect pathways (Joule/dielectric heating, electrolysis, electrode corrosion, electrothermal/electrohydrodynamic flows), the accumulated “damage” or energy deposition scales approximately with an E2-type integral over the duty cycle (e.g. 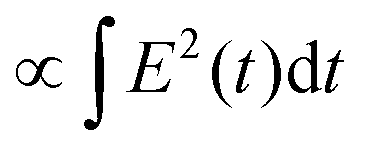 or ∝σE2 for Ohmic heating), so short pulses can access large peak fields while limiting the bulk temperature rise and electrochemical accumulation.294,299,403 For direct EF-driven conformational/functional changes, however, the field–time trade-off can be much steeper: if the EF lowers an effective activation barrier, the response rate can follow an Arrhenius-like dependence
or ∝σE2 for Ohmic heating), so short pulses can access large peak fields while limiting the bulk temperature rise and electrochemical accumulation.294,299,403 For direct EF-driven conformational/functional changes, however, the field–time trade-off can be much steeper: if the EF lowers an effective activation barrier, the response rate can follow an Arrhenius-like dependence 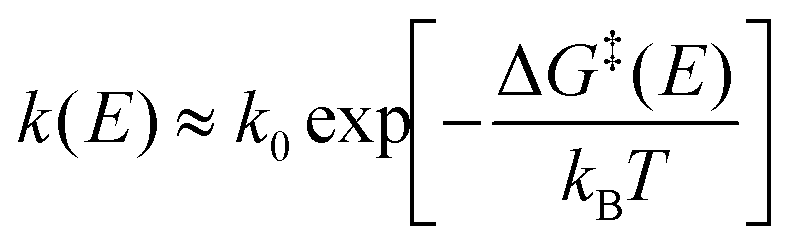 , with ΔG‡(E) ≃ ΔG‡0 − Δμ‡E (or related forms), implying an exponential sensitivity to E.22,225,352–354 For pulsed protocols, explicitly report both the single-pulse width τp and the cumulative on-time τon = Nτp (and also the total elapsed exposure time), because large-N pulse trains can have τon ≪ elapsed time yet still produce a substantial thermal/chemical dose.
, with ΔG‡(E) ≃ ΔG‡0 − Δμ‡E (or related forms), implying an exponential sensitivity to E.22,225,352–354 For pulsed protocols, explicitly report both the single-pulse width τp and the cumulative on-time τon = Nτp (and also the total elapsed exposure time), because large-N pulse trains can have τon ≪ elapsed time yet still produce a substantial thermal/chemical dose.
• Tune spectral content (frequency and waveform) to the target timescale, and to suppress unwanted interfacial physics. Protein dynamics spans picoseconds to days, so efficient forcing requires matching the stimulus bandwidth to the relaxation time of the targeted coordinate; narrowband drives can preferentially address a chosen dynamic window, whereas fast-rise pulses are inherently broadband and can excite multiple dispersive processes in water and biomolecules.77,220,404 Frequency also selects which macroscopic couplings dominate near interfaces: at low frequencies (typically ≲10 kHz) electrical-double-layer charging and induced-charge electroosmosis are strong, while at higher frequencies the double layer cannot follow and electroosmotic contributions diminish.304–306 In the GHz–THz range, coupling can shift toward fast polarization dynamics and collective modes; outcomes then depend critically on absorption and duty cycle (thermal vs. nonthermal regimes).24,177
• Control the matrix chemistry and interfaces. Buffer conductivity and thermal properties set Joule heating and electrothermal flows; buffer capacity sets pH excursions; electrode composition and dissolved oxygen/metal ions/peroxides set susceptibility to electrolysis, corrosion, and ROS/RNS chemistry. Use matched chemical controls (conductivity, buffering, scavengers), minimize direct electrode contact when possible (coatings, salt bridges), and manage polarity/pulse structure (e.g. bipolar or burst protocols) to separate direct EF effects from chemistry-driven artifacts.317,319–324
• Account for solvent-mediated thermodynamics and nonlinear dielectric response. Strong static and oscillating fields can reorganize water structure/dynamics (dielectric saturation, anisotropy, altered hydrogen-bond statistics), changing entropic contributions and effective local fields that feed back on protein stability and kinetics.356–359,404 These solvent responses may also limit durability in repeated-use bioelectronic and piezoelectric protein/peptide platforms.402
• Validate “direct” EF mechanisms with orthogonal controls and quantitative modelling. Because indirect pathways can mimic “nonthermal” effects, combine structural/functional readouts with monitoring of temperature, pH, and electrochemical/ROS markers, and report geometry, conductivity, waveform details, and local-field estimates (or EM/thermal simulations) to enable reproducibility and cross-modality comparison.47,297–299,403
Looking forward, closing the gap between simulations and experiments will require multiscale modelling (including explicit interfaces and realistic conductivity), improved force fields for polarization and charge transfer, and experimental platforms that deliver well-calibrated fields while simultaneously reporting structural and functional observables. Such advances should enable EF protocols that are not generic denaturants but rather tunable physical stimuli for selective, reversible and sustainable control of protein function.
Author contributions
M. C.: conceptualization; funding acquisition; project administration; resources; supervision; data curation; visualization; writing – original draft; writing – review & editing. S. K. P.: visualization; writing – review & editing. T. Z.: visualization; writing – original draft. M. P.: visualization; writing – original draft. D. G. B. C.: visualization; writing – original draft. J. P.: visualization; writing – original draft. D. H.: visualization. P. M.: writing – review & editing. M. L.: writing – review & editing. F. A.: visualization; writing – original draft; writing – review & editing. N. J. E.: writing – original draft. C. C.: writing – original draft. E. G. M.: writing – review & editing. Contributor roles follow the ANSI/NISO CRediT taxonomy.Conflicts of interest
There are no conflicts to declare.List of abbreviations
| Aβ | Amyloid-beta |
| AC | Alternating current |
| AQP | Aquaporin |
| AdoHcy | S-Adenosyl-L-homocysteine |
| AFM | Atomic force microscopy |
| ANS | 8-anilino-1-naphthalenesulfonic acid |
| anti-DNP | Antidinitrophenyl |
| AP | Alkaline phosphatase |
| ATR-FTIR | Attenuated total reflectance Fourier-transform IR |
| BCA | Bicinchoninic-acid protein assay |
| BSA | Bovine serum albumin |
| CD | Circular dichroism |
| CLSM | Confocal laser scanning microscopy |
| CW | Continuous wave |
| DC | Direct current |
| DFT | Density functional theory |
| DLS | Dynamic light scattering |
| DNPH | 2,4-Dinitrophenyl-hydrazine (carbonyl assay) |
| DSC | Differential scanning calorimetry |
| DTNB | 5,5′-Dithiobis-(2-nitrobenzoic acid) |
| EF | Electric field |
| EF-X | EF X-ray crystallography |
| EGFP | Enhanced green fluorescent protein |
| EM | Electromagnetic |
| EMC | Expand–maximize–compress |
| EWOD | Electrowetting-on-dielectric |
| FNC | Fraction of native contacts |
| FTIR | Fourier-transform infrared |
| GFP | Green fluorescent protein |
| GFPuv5 | Ultraviolet-excitable GFP variant (uv5) |
| GPCR | G-protein-coupled receptor |
| H-bond | Hydrogen bond |
| HEWL | Hen egg-white lysozyme |
| HPLC | High-performance liquid chromatography |
| HRTEM | High-resolution transmission EM |
| HRP | Horseradish peroxidase |
| HSA | Human serum albumin |
| HPG | High-power generator |
| HV | High voltage |
| ITO | Indium tin oxide |
| LDH | Lactate dehydrogenase |
| LLPS | Liquid–liquid phase separation |
| MD | Molecular dynamics |
| Mb | Myoglobin |
| MT | Microtubule |
| MTA | 5′-Methylthioadenosine |
| NEMD | Non-equilibrium molecular dynamics |
| NMR | Nuclear magnetic resonance |
| nsPEF | Nanosecond pulsed electric field |
| OPA | o-Phthalaldehyde |
| OVA | Ovalbumin |
| PCA | Principal component analysis |
| PDB | Protein data bank |
| PDMS | Poly(dimethylsiloxane) |
| PEF | Pulsed electric field |
| POD | Peroxidase |
| PPI | Protein–protein interaction |
| PPO | Polyphenol oxidase |
| PL | Photoluminescence |
| PMF | Potential of mean force |
| Rg | Radius of gyration |
| RMSD | Root-mean-square deviation |
| RMSF | Root-mean-square fluctuation |
| ROA | Raman optical activity |
| ROS | Reactive oxygen species |
| RNS | Reactive nitrogen species |
| RPC | Relative protein concentration |
| RXS | Reactive species (generic) |
| SASA | Solvent-accessible surface area |
| SAXS | Small-angle X-ray scattering |
| SEC | Size-exclusion chromatography |
| SEM | Scanning electron microscopy |
| SDS–PAGE | Sodium dodecyl-sulfate polyacrylamide-gel electrophoresis |
| SiR | Silicon–rhodamine (fluorophore) |
| SPI | Single-particle imaging |
| SPR | Surface plasmon resonance |
| TEM | Transmission electron microscopy |
| ThT | Thioflavin T |
| Trp | Tryptophan |
| Tyr | Tyrosine |
| Ub | Ubiquitin |
| UV-vis | Ultraviolet-visible spectroscopy |
| VCD | Vibrational circular dichroism |
| VG | Voltage generator |
| VGIC | Voltage-gated ion channel |
| VSD | Voltage-sensing domain |
| XFEL | X-Ray free-electron laser |
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Acknowledgements
This research was funded by Czech Science Foundation grant number 20-06873X (MC, SKP, TZ, MP, DGBC, JP), 25-16700S (DH), Czech Academy of Sciences PPLZ program, project n. L100672501 (MP) and (CC) by the Swedish Research Council via projects 2018-00740, 2023-04346 and (CC, EGM) via the Röntgen-Ångström Cluster (2019-03935). CC also acknowledges the Helmholtz Association through the Center for Free-Electron Laser Science at DESY. Tasnia Tarana and Neuron Collective are acknowledged for graphical design. Ondřej Kučera is acknowledged for valuable comments on the manuscript. ChatGPT tools were used as assistants for writing and rephrasing based on the inputs and prompts by the authors.Notes and references
- G. E. Neurohr and A. Amon, Trends Cell Biol., 2020, 30, 213–225 CrossRef CAS PubMed.
- R. Milo and R. Phillips, Cell biology by the numbers, Garland Science, 2015 Search PubMed.
- B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, Mol. Biol. Cell, Garland Science, 4th edn, 2002 Search PubMed.
- R. Nussinov, Y. Liu, W. Zhang and H. Jang, RSC Chem. Biol., 2023, 4, 850–864 RSC.
- L. Orellana, Front. Mol. Biosci., 2019, 6, 117 CrossRef CAS.
- H. Ai, M. Pan and L. Liu, ACS Cent. Sci., 2024, 10, 1442–1459 CrossRef CAS.
- H. Nada, Y. Choi, S. Kim, K. S. Jeong, N. A. Meanwell and K. Lee, Signal Transduction Targeted Ther., 2024, 9, 341 CrossRef.
- D. Vasudevan, Textbook of Biochemistry, Jaypee Brothers Medical Publishers (P) Ltd., 6th edn, 2011, pp. 27–39 Search PubMed.
- C. Soto and S. Pritzkow, Nat. Neurosci., 2018, 21, 1332–1340 CrossRef CAS PubMed.
- C. Scheckel and A. Aguzzi, Nat. Rev. Genet., 2018, 19, 405–418 CrossRef CAS PubMed.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2017, 86, 27–68 CrossRef CAS.
- S. Verbrugge, Z. Lansky and E. J. G. Peterman, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17741–17746 CrossRef CAS PubMed.
- W. A. M. Loenen, D. T. F. Dryden, E. A. Raleigh, G. G. Wilson and N. E. Murray, Nucleic Acids Res., 2014, 42, 3–19 CrossRef CAS.
- O. Brandman and R. S. Hegde, Nat. Struct. Mol. Biol., 2016, 23, 7–15 CrossRef CAS.
- R. Croce and H. van Amerongen, Photosynth. Res., 2013, 116, 153–166 CrossRef CAS.
- S. G. Boxer and T. Head-Gordon, Chem. Rev., 2025, 125, 6871–6873 CrossRef CAS.
- M. A. Rothermund, S. J. Koehler and V. Vaissier Welborn, Chem. Rev., 2024, 124, 13331–13369 CrossRef CAS.
- C. Yang, Y. Guo, H. Zhang and X. Guo, Chem. Rev., 2025, 125, 223–293 CrossRef CAS.
- Z. Long, J. Meng, L. R. Weddle, P. E. Videla, J. P. Menzel, D. G. A. Cabral, J. Liu, T. Qiu, J. M. Palasz, D. Bhattacharyya, C. P. Kubiak, V. S. Batista and T. Lian, Chem. Rev., 2025, 125, 1604–1628 CrossRef CAS PubMed.
- S. Ishizawa, K. Fujimura, K. Oisaki, S. Sato and J. Ohata, ChemCatChem, 2025, 17, e202402125 CrossRef CAS PubMed.
- D. E. Chafai, V. Sulimenko, D. Havelka, L. Kubínová, P. Dráber and M. Cifra, Adv. Mater., 2019, 31, 1903636 CrossRef.
- D. R. Hekstra, K. I. White, M. A. Socolich, R. W. Henning, V. Šrajer and R. Ranganathan, Nature, 2016, 540, 400–405 CrossRef CAS PubMed.
- R. A. Petrella, K. H. Schoenbach and S. Xiao, IEEE Trans. Plasma Sci., 2016, 44, 708–714 CAS.
- J.-i Sugiyama, Y. Tokunaga, M. Hishida, M. Tanaka, K. Takeuchi, D. Satoh and M. Imashimizu, Nat. Commun., 2023, 14, 2825 CrossRef CAS.
- C. M. Hough, D. N. Purschke, C. Bell, A. P. Kalra, P. J. Oliva, C. Huang, J. A. Tuszynski, B. J. Warkentin and F. A. Hegmann, Biomed. Opt. Express, 2021, 12, 5812–5828 CrossRef CAS.
- C. Marar, Y. Jiang, Y. Li, L. Lan, N. Zheng, G. Chen, C. Yang and J.-X. Cheng, Sci. Adv., 2024, 10, eado5560 CrossRef CAS.
- A. Wada and H. Nakamura, Nature, 1981, 293, 757–758 CrossRef CAS PubMed.
- M. N. Alves, S. E. Neto, A. S. Alves, B. M. Fonseca, A. Carrêlo, I. Pacheco, C. M. Paquete, C. M. Soares and R. O. Louro, Front. Microbiol., 2015, 6, 665 Search PubMed.
- L. Li, Z. Jia, Y. Peng, S. Godar, I. Getov, S. Teng, J. Alper and E. Alexov, Sci. Rep., 2017, 7, 8237 CrossRef PubMed.
- T. G. Castro, F.-D. Munteanu and A. Cavaco-Paulo, Biomolecules, 2019, 9, 116 CrossRef CAS PubMed.
- C. Kweyu, L. Feng, M. Stein and P. Benner, Comput. Visualization Sci., 2020, 23, 15 CrossRef.
- P. Ren, J. Chun, D. G. Thomas, M. J. Schnieders, M. Marucho, J. Zhang and N. A. Baker, Q. Rev. Biophys., 2012, 45, 427–491 CrossRef.
- M.-Y. Tsai, W. Zheng, D. Balamurugan, N. P. Schafer, B. L. Kim, M. S. Cheung and P. G. Wolynes, Protein Sci., 2016, 25, 255–269 CrossRef CAS PubMed.
- R. H. W. Funk and F. Scholkmann, Prog. Biophys. Mol. Biol., 2023, 177, 185–201 CrossRef CAS.
- M. T. Neves-Petersen and S. B. Petersen, Biotechnology Annual Review, Elsevier, 2003, vol. 9, pp. 315–395 Search PubMed.
- A. Chakravorty, Z. Jia, L. Li, S. Zhao and E. Alexov, J. Chem. Theory Comput., 2018, 14, 1020–1032 CrossRef CAS.
- Z. Zhang, S. Witham and E. Alexov, Phys. Biol., 2011, 8, 035001 CrossRef PubMed.
- A. Macchiarulo, G. Costantino, R. Sbaglia, S. Aiello, M. Meniconi and R. Pellicciari, Proteins: Struct., Funct., Bioinf., 2003, 50, 609–619 CrossRef CAS.
- A. Warshel, P. K. Sharma, M. Kato, Y. Xiang, H. Liu and M. H. M. Olsson, Chem. Rev., 2006, 106, 3210–3235 CrossRef CAS PubMed.
- S. D. Fried, S. Bagchi and S. G. Boxer, Science, 2014, 346, 1510–1514 CrossRef CAS PubMed.
- S. D. Fried and S. G. Boxer, Annu. Rev. Biochem., 2017, 86, 387–415 CrossRef CAS.
- C. Zheng, Z. Ji, I. I. Mathews and S. G. Boxer, Nat. Chem., 2023, 15, 1715–1721 CrossRef CAS.
- F. Bezanilla, Physiol. Rev., 2000, 80, 555–592 CrossRef CAS.
- A. L. Hodgkin and A. F. Huxley, J. Physiol., 1952, 116, 449–472 CrossRef CAS.
- H. C. Lai and L. Y. Jan, Nat. Rev. Neurosci., 2006, 7, 548–562 CrossRef CAS PubMed.
- J. Průša, A. T. Ayoub, D. E. Chafai, D. Havelka and M. Cifra, Comput. Struct. Biotechnol. J., 2021, 19, 1488–1496 CrossRef PubMed.
- A. Sinelnikova, T. Mandl, C. Östlin, O. Grånäs, M. N. Brodmerkel, E. G. Marklund and C. Caleman, Chem. Sci., 2021, 12, 2030–2038 RSC.
- M. A. Malik, M. A. Sheikh and N. A. Mir, Food Biosci., 2024, 61, 104636 CrossRef CAS.
- J. Marín-Sánchez, A. Berzosa, I. Álvarez, C. Sánchez-Gimeno and J. Raso, Bioelectricity, 2024, 6, 154–166 CrossRef.
- C. Zhang, X. Lyu, R. N. Arshad, R. M. Aadil, Y. Tong, W. Zhao and R. Yang, Food Chem., 2023, 403, 134367 CrossRef CAS PubMed.
- Y.-F. Liu, I. Oey, P. Bremer, A. Carne and P. Silcock, Compr. Rev. Food Sci. Food Saf., 2019, 18, 986–1002 CrossRef CAS.
- A. Taha, F. Casanova, P. Šimonis, V. Stankevič, M. A. E. Gomaa and A. Stirkė, Foods, 2022, 11, 1556 CrossRef CAS PubMed.
- X. Jiao, W. Chen and D. Fan, Curr. Opin. Food Sci., 2022, 48, 100936 CrossRef CAS.
- K. Takaki, K. Takahashi, A. Guionet and T. Ohshima, Molecules, 2021, 26, 6288 CrossRef CAS.
- S. Zhang, L. Sun, H. Ju, Z. Bao, X.-A. Zeng and S. Lin, Food Res. Int., 2021, 139, 109914 CrossRef CAS PubMed.
- S. K. Vanga, J. Wang, S. Jayaram and V. Raghavan, Processes, 2021, 9, 722 CrossRef CAS.
- R. M. Rodrigues, Z. Avelar, L. Machado, R. N. Pereira and A. A. Vicente, Food Res. Int., 2020, 137, 109709 CrossRef CAS.
- D. Niu, X.-A. Zeng, E.-F. Ren, F.-Y. Xu, J. Li, M.-S. Wang and R. Wang, Food Res. Int., 2020, 137, 109715 CrossRef CAS.
- L. H. Fasolin, R. N. Pereira, A. C. Pinheiro, J. T. Martins, C. C. P. Andrade, O. L. Ramos and A. A. Vicente, Food Res. Int., 2019, 125, 108586 CrossRef CAS.
- R. N. Pereira, R. M. Rodrigues, Ó. L. Ramos, A. C. Pinheiro, J. T. Martins, J. A. Teixeira and A. A. Vicente, J. Agric. Food Chem., 2018, 66, 11227–11233 CrossRef CAS.
- Z. Han, M.-J. Cai, J.-H. Cheng and D.-W. Sun, Trends Food Sci. Technol., 2018, 75, 1–9 CrossRef CAS.
- S. G. Giteru, I. Oey and M. A. Ali, Trends Food Sci. Technol., 2018, 72, 91–113 CrossRef CAS.
- L. Barsotti, E. Dumay, T. H. Mu, M. D. Fernandez Diaz and J. C. Cheftel, Trends Food Sci. Technol., 2001, 12, 136–144 CrossRef CAS.
- S. J. Beebe, Bioelectrochemistry, 2015, 103, 52–59 CrossRef CAS.
- P. M. Graybill and R. V. Davalos, Cancers, 2020, 12, 1132 CrossRef CAS.
- J. N. Israelachvili, Intermolecular and Surface Forces, Elsevier, 3rd edn, 2011 Search PubMed.
- R. S. Fisher, J. Liao, S. Y. Ahn, N. Modi, A. K. Kidane and A. C. Obermeyer, Annu. Rev. Chem. Biomol. Eng., 2025, 16, 119–145 CrossRef CAS PubMed.
- Q. Zhang, D. Shao, P. Xu and Z. Jiang, Polymers, 2022, 14, 123 CrossRef CAS PubMed.
- D. Miklavcic, Handbook of electroporation, Springer, Cham, 2019 Search PubMed.
- Y. Shen, H. Wang and S. Fan, Adv. Opt. Photonics, 2025, 17, 295 CrossRef.
- H. Rubinsztein-Dunlop, A. Forbes, M. V. Berry, M. R. Dennis, D. L. Andrews, M. Mansuripur, C. Denz, C. Alpmann, P. Banzer, T. Bauer, E. Karimi, L. Marrucci, M. Padgett, M. Ritsch-Marte, N. M. Litchinitser, N. P. Bigelow, C. Rosales-Guzmán, A. Belmonte, J. P. Torres, T. W. Neely, M. Baker, R. Gordon, A. B. Stilgoe, J. Romero, A. G. White, R. Fickler, A. E. Willner, G. Xie, B. McMorran and A. M. Weiner, J. Opt., 2016, 19, 013001 CrossRef.
- S. B. Glybovski, S. A. Tretyakov, P. A. Belov, Y. S. Kivshar and C. R. Simovski, Phys. Rep., 2016, 634, 1–72 CrossRef CAS.
- L. Vincenti, P. Pellegrino, M. Cascione, V. De Matteis, I. Farella, F. Quaranta and R. Rinaldi, Mater. Des., 2024, 243, 113036 CrossRef CAS.
- S. Toyama, T. Seki, Y. Kohno, Y. O. Murakami, Y. Ikuhara and N. Shibata, Nat. Rev. Electr. Eng., 2025, 2, 27–41 CrossRef.
- E. Oswald, K. Palanisamy and C. Kranz, Curr. Opin. Electrochem., 2022, 34, 100965 CrossRef CAS.
- H.-X. Zhou and X. Pang, Chem. Rev., 2018, 118, 1691–1741 CrossRef CAS PubMed.
- D. Nettels, N. Galvanetto, M. T. Ivanović, M. Nüesch, T. Yang and B. Schuler, Nat. Rev. Phys., 2024, 6, 587–605 CrossRef CAS.
- P. Marracino, D. Havelka, J. Průša, M. Liberti, J. Tuszynski, A. T. Ayoub, F. Apollonio and M. Cifra, Sci. Rep., 2019, 9, 10477 CrossRef PubMed.
- E. G. Marklund, T. Ekeberg, M. Moog, J. L. P. Benesch and C. Caleman, J. Phys. Chem. Lett., 2017, 8, 4540–4544 CrossRef CAS PubMed.
- G. Mithieux, F. Chauvin, B. Roux and B. Rousset, Biophys. Chem., 1985, 22, 307–316 CrossRef CAS.
- D. Porschke, C. Créminon, X. Cousin, C. Bon, J. Sussman and I. Silman, Biophys. J., 1996, 70, 1603–1608 CrossRef CAS.
- A. Chakraborty and R. Venkatramani, ChemPhysChem, 2023, 24, e202200646 CrossRef CAS PubMed.
- J. P. Landers, Handbook of capillary electrophoresis, CRC Press, 1996 Search PubMed.
- H. Rehm and H. Rehm, Protein biochemistry and proteomics, Elsevier, Academic Press, Amsterdam, Boston, 4th edn, 2006 Search PubMed.
- M. Uppalapati, Y.-M. Huang, V. Aravamuthan, T. N. Jackson and W. O. Hancock, Integr. Biol., 2011, 3, 57–64 CrossRef CAS.
- M. Uppalapati, Y.-M. Huang, T. N. Jackson and W. O. Hancock, Small, 2008, 4, 1371–1381 CrossRef CAS.
- K. J. Böhm, N. E. Mavromatos, A. Michette, R. Stracke and E. Unger, Electromagn. Biol. Med., 2005, 24, 319–330 CrossRef.
- K. Kim, A. Sikora, K. S. Nakayama, M. Umetsu, W. Hwang and W. Teizer, J. Appl. Phys., 2015, 117, 144701 CrossRef.
- M. G. L. Van den Heuvel, M. P. De Graaff, S. G. Lemay and C. Dekker, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7770–7775 CrossRef CAS.
- M. G. L. van den Heuvel, C. T. Butcher, S. G. Lemay, S. Diez and C. Dekker, Nano Lett., 2005, 5, 235–241 CrossRef CAS PubMed.
- N. Isozaki, H. Shintaku, H. Kotera, T. L. Hawkins, J. L. Ross and R. Yokokawa, Sci. Rob., 2017, 2, eaan4882 CrossRef.
- R. Stracke, K. Böhm, L. Wollweber, J. Tuszynski and E. Unger, Biochem. Biophys. Res. Commun., 2002, 293, 602–609 CrossRef CAS PubMed.
- S. Kobayashi, Biochim. Biophys. Acta, Spec. Sect. Biophys. Subj., 1964, 88, 541–552 CAS.
- D. Riveline, A. Ott, F. Jülicher, D. A. Winkelmann, O. Cardoso, J.-J. Lacapère, S. Magnúsdóttir, J.-L. Viovy, L. Gorre-Talini and J. Prost, Eur. Biophys. J., 1998, 27, 403–408 CrossRef CAS.
- M. E. Arsenault, H. Zhao, P. K. Purohit, Y. E. Goldman and H. H. Bau, Biophys. J., 2007, 93, L42–L44 CrossRef CAS PubMed.
- C. Wigge, H. Hinssen, G. Reiss and S. Herth, Appl. Phys. Lett., 2010, 96, 243703 CrossRef.
- A. M. Roseman, S. Chen, H. White, K. Braig and H. R. Saibil, Cell, 1996, 87, 241–251 CrossRef CAS.
- H. Noji, R. Yasuda, M. Yoshida and K. Kinosita, Nature, 1997, 386, 299–302 CrossRef CAS PubMed.
- W. Wriggers and K. Schulten, Proteins: Struct., Funct., Bioinf., 1997, 29, 1–14 CrossRef CAS.
- H. Omote, N. Sambonmatsu, K. Saito, Y. Sambongi, A. Iwamoto-Kihara, T. Yanagida, Y. Wada and M. Futai, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 7780–7784 CrossRef CAS PubMed.
- M. D. Daily, G. N. Phillips and Q. Cui, J. Mol. Biol., 2010, 400, 618–631 CrossRef CAS PubMed.
- J. R. Braxton, H. Shao, E. Tse, J. E. Gestwicki and D. R. Southworth, Nat. Struct. Mol. Biol., 2024, 31, 1848–1858 CrossRef CAS PubMed.
- A. Oleinikova, P. Sasisanker and H. Weingärtner, J. Phys. Chem. B, 2004, 108, 8467–8474 CrossRef CAS.
- V. Raicu and Y. Feldman, Dielectric relaxation in biological systems: Physical principles, methods, and applications, Oxford University Press, USA, 2015 Search PubMed.
- Molecular Electro-Optics, ed. S. Krause, Springer US, Boston, MA, 1981 Search PubMed.
- D. Porschke, Annu. Rev. Phys. Chem., 1985, 36, 159–178 CrossRef CAS.
- S. P. Stoylov and M. V. Stoimenova, Molecular and colloidal electro-optics, CRC Press, 2016, vol. 134 Search PubMed.
- L. Keszthelyi, Biochim. Biophys. Acta, Biomembr., 1980, 598, 429–436 CrossRef CAS PubMed.
- Y. Kimura, A. Ikegami, K. Ohno, S. Saigo and Y. Takeuchi, Photochem. Photobiol., 1981, 33, 435–439 CrossRef CAS.
- K. Barabás, A. Dér, Z. Dancsházy, P. Ormos, L. Keszthelyi and M. Marden, Biophys. J., 1983, 43, 5–11 CrossRef PubMed.
- Y. Kimura, M. Fujiwara and A. Ikegami, Biophys. J., 1984, 45, 615–625 Search PubMed.
- S. P. Stoylov, G. Todorov and A. Zhivkov, Bioelectrochem. Bioenerg., 1984, 12, 49–55 CrossRef.
- J. Antosiewicz and D. Porschke, Biochemistry, 1989, 28, 10072–10078 CrossRef CAS.
- A. Dér, R. Tóth-Boconádi, L. Keszthelyi, H. Kramer and W. Stoeckenius, FEBS Lett., 1995, 377, 419–420 CrossRef.
- D. Porschke, Biophys. J., 1996, 71, 3381–3391 CrossRef CAS PubMed.
- T. Schönknecht and D. Pörschke, Biophys. Chem., 1996, 58, 21–28 CrossRef.
- Y. Sugiyama, T. Inoue, M. Ikematsu, M. Iseki and T. S. T. Sekiguchi, Jpn. J. Appl. Phys., 1997, 36, 5674 CrossRef CAS.
- R. Neutze, R. Wouts, D. van der Spoel, E. Weckert and J. Hajdu, Nature, 2000, 406, 752–757 CrossRef CAS.
- N.-T. D. Loh and V. Elser, Phys. Rev. E:Stat., Nonlinear, Soft Matter Phys., 2009, 80, 026705 CrossRef.
- C. Östlin, N. Timneanu, C. Caleman and A. V. Martin, Struct. Dyn., 2019, 6, 044103 CrossRef.
- T. Mandl, C. Östlin, I. E. Dawod, M. N. Brodmerkel, E. G. Marklund, A. V. Martin, N. Timneanu and C. Caleman, J. Phys. Chem. Lett., 2020, 11, 6077–6083 CrossRef CAS.
- A. Wollter, E. De Santis, T. Ekeberg, E. G. Marklund and C. Caleman, J. Chem. Phys., 2024, 160, 114108 CrossRef CAS.
- A. Sinelnikova, T. Mandl, H. Agelii, O. Grånäs, E. G. Marklund, C. Caleman and E. De Santis, Biophys. J., 2021, 120, 3709–3717 CrossRef CAS.
- M. Amin, J.-M. Hartmann, A. K. Samanta and J. Küpper, J. Am. Chem. Soc., 2025, 147, 7445–7451 CrossRef CAS.
- C. R. Arbeitman, P. Rojas, P. Ojeda-May and M. E. Garcia, Nat. Commun., 2021, 12, 5407 CrossRef CAS.
- Z.-L. Chen, Y. Li, J.-H. Wang, R. Wang, Y.-X. Teng, J.-W. Lin, X.-A. Zeng, M.-W. Woo, L. Wang and Z. Han, Int. J. Biol. Macromol., 2023, 244, 125082 CrossRef CAS PubMed.
- A. Guionet, T. Fujiwara, H. Sato, K. Takahashi, K. Takaki, M. Matsui, T. Tanino and T. Ohshima, J. Electrost., 2021, 112, 103597 CrossRef CAS.
- A. Kadek, K. Lorenzen and C. Uetrecht, Drug Discovery Today: Technol., 2021, 39, 89–99 CrossRef CAS.
- T. Kierspel, A. Kadek, P. Barran, B. Bellina, A. Bijedic, M. N. Brodmerkel, J. Commandeur, C. Caleman, T. Damjanović and I. Dawod, et al., Anal. Bioanal. Chem., 2023, 415, 4209–4220 CrossRef CAS PubMed.
- H. Agelii, E. L. S. Jakobsson, E. D. Santis, G. Elfrink, T. Mandl, E. G. Marklund and C. Caleman, Phys. Chem. Chem. Phys., 2025, 27, 10939–10948 RSC.
- E. D. Santis, T. Mandl, J. C. K. Kung, K. Huynh, S. Daly, L. A. D'Alessandro, L. MacAleese, C. Uetrecht, E. G. Marklund and C. Caleman, Phys. Chem. Chem. Phys., 2025, 27, 14767–14776 RSC.
- B. Alberts, Molecular biology of the cell, Garland Science, 2017 Search PubMed.
- S. Kalita, D. Danovich and S. Shaik, J. Am. Chem. Soc., 2025, 147, 2626–2641 CrossRef CAS.
- X. Hu, H. Wang, Y. Hu, P. Wen, X. Wu and Z. Tu, Food Chem., 2025, 463, 141376 CrossRef CAS PubMed.
- L. Guan, B. Qi, J. Tan, Y. Chen, Y. Sun, Q. Zhang and Y. Zou, J. Chem. Inf. Model., 2025, 65, 2066–2079 CrossRef CAS.
- M. Makhkamov, A. Baev, E. Kurganov and J. Razzokov, Sci. Rep., 2024, 14, 22246 CrossRef CAS.
- L. Guan, J. Tan, B. Qi, Y. Chen, M. Cao, Q. Zhang and Y. Zou, Biophys. Chem., 2024, 312, 107283 CrossRef CAS PubMed.
- Y. Shuto, E. Walinda, D. Morimoto and K. Sugase, J. Phys. Chem. B, 2023, 127, 7417–7430 CrossRef CAS PubMed.
- A. Lipskij, C. Arbeitman, P. Rojas, P. Ojeda-May and M. E. Garcia, Viruses, 2023, 15, 2405 CrossRef CAS.
- W. A. Müller, J. R. Sarkis, L. D. F. Marczak and A. R. Muniz, Innovative Food Sci. Emerging Technol., 2022, 75, 102911 CrossRef.
- S. Zhang, Y. Li, Z. Bao, N. Sun and S. Lin, Int. J. Biol. Macromol., 2021, 182, 849–857 CrossRef CAS PubMed.
- C. P. Samaranayake and S. K. Sastry, Phys. Chem. Chem. Phys., 2021, 23, 14422–14432 RSC.
- A. R. Ruiz-Fernández, L. Campos, F. Villanelo, S. E. Gutiérrez-Maldonado and T. Perez-Acle, Membranes, 2021, 11, 473 CrossRef.
- B. Narayan, C. Herbert, B. J. Rodriguez, B. R. Brooks and N.-V. Buchete, J. Phys. Chem. B, 2021, 125, 5233–5242 CrossRef CAS PubMed.
- M. Dong, H. Tian, Y. Xu, M. Han and X. Xu, Food Chem., 2021, 342, 128306 CrossRef CAS PubMed.
- S. Basu, B. Gorai, B. Basu and P. K. Maiti, J. Phys. Chem. B, 2021, 125, 3–16 CrossRef CAS.
- S. Muscat, F. Stojceski and A. Danani, J. Mol. Graphics Modell., 2020, 96, 107535 CrossRef CAS PubMed.
- I. Baruah and G. Borgohain, Biophys. Chem., 2020, 106479 CrossRef CAS.
- Z. Jiang, L. You, W. Dou, T. Sun and P. Xu, Polymers, 2019, 11, 282 CrossRef.
- J. J. Timmons, J. Preto, J. A. Tuszynski and E. T. Wong, PLoS One, 2018, 13, e0202141 CrossRef PubMed.
- Y. Lu, X.-F. Shi, F. R. Salsbury and P. Derreumaux, J. Chem. Phys., 2017, 146, 145101 CrossRef PubMed.
- H. Alizadeh, J. Davoodi and H. Rafii-Tabar, J. Mol. Graphics Modell., 2017, 73, 108–114 CrossRef CAS.
- N. Todorova, A. Bentvelzen, N. J. English and I. Yarovsky, J. Chem. Phys., 2016, 144, 085101 CrossRef PubMed.
- Y. Xie, Y. Pan, R. Zhang, Y. Liang and Z. Li, Appl. Surf. Sci., 2015, 326, 55–65 CrossRef CAS.
- S. K. Vanga, A. Singh and V. Raghavan, Innovative Food Sci. Emerging Technol., 2015, 30, 79–88 CrossRef CAS.
- T. E. Benavidez, D. Torrente, M. Marucho and C. D. Garcia, Langmuir, 2015, 31, 2455–2462 CrossRef PubMed.
- X. Wang, Y. Li, X. He, S. Chen and J. Z. H. Zhang, J. Phys. Chem. A, 2014, 118, 8942–8952 CrossRef CAS.
- M. Damm, C. Nusshold, D. Cantillo, G. N. Rechberger, K. Gruber, W. Sattler and C. O. Kappe, J. Proteomics, 2012, 75, 5533–5543 CrossRef CAS PubMed.
- P. Marracino, F. Apollonio, M. Liberti, G. d'Inzeo and A. Amadei, J. Phys. Chem. B, 2013, 117, 2273–2279 CrossRef CAS.
- Y. Wu, S. Qin, Y. Zang, M. Zhou, S. Chen and S. Huang, Innovative Food Sci. Emerging Technol., 2023, 89, 103484 CrossRef CAS.
- J.-N. Wei, X.-A. Zeng, T. Tang, Z. Jiang and Y.-Y. Liu, Innovative Food Sci. Emerging Technol., 2018, 45, 249–254 CrossRef CAS.
- R. Yang, S.-Q. Li and Q. H. Zhang, J. Agric. Food Chem., 2004, 52, 7400–7406 CrossRef CAS.
- Y. Tokunaga, M. Tanaka, H. Iida, M. Kinoshita, Y. Tojima, K. Takeuchi and M. Imashimizu, Biophys. J., 2021, 120, 2386–2393 CrossRef CAS.
- A. Homenko, B. Kapilevich, R. Kornstein and M. Firer, Bioelectromagnetics, 2009, 30, 167–175 CrossRef CAS PubMed.
- I. Bekard and D. E. Dunstan, Soft Matter, 2014, 10, 431–437 RSC.
- K. Kang, Front. Phys., 2023, 11, 1282099 CrossRef.
- W. Jin, Z. Wang, D. Peng, W. Shen, Z. Zhu, S. Cheng, B. Li and Q. Huang, Food Hydrocolloids, 2020, 101, 105547 CrossRef CAS.
- K. Tsuji and B. Hess, Ferroelectrics, 1988, 86, 147–157 CrossRef CAS.
- K. Tsuji, S. C. Miiller and B. Hess, Eur. Biophys. J., 1988, 15, 329–337 CrossRef CAS.
- K. Tsuji and B. Hess, Eur. Biophys. J., 1986, 13, 273–280 CrossRef.
- K. Tsuji and E. Neumann, Biophys. Chem., 1983, 17, 153–163 CrossRef CAS PubMed.
- K. Tsuji and E. Neumann, Int. J. Biol. Macromol., 1981, 3, 231–242 CrossRef CAS.
- D. De, N. Pawar and A. N. Gupta, Phys. Chem. Chem. Phys., 2021, 23, 4195–4204 RSC.
- P. A. Vargas-Rosales, A. D'Addio, Y. Zhang and A. Caflisch, ACS Phys. Chem. Au, 2023, 3, 456–466 CrossRef CAS.
- S. Kalita, H. Bergman, K. D. Dubey and S. Shaik, J. Am. Chem. Soc., 2023, 145, 3543–3553 CrossRef CAS.
- R. M. Rodrigues, L. H. Fasolin, Z. Avelar, S. B. Petersen, A. A. Vicente and R. N. Pereira, Food Hydrocolloids, 2020, 101, 105505 CrossRef CAS.
- W. Peng, Z. Zhu, J. Lou, K. Chen, Y. Wu and C. Chang, eLight, 2023, 3, 18 CrossRef.
- R. M. Rodrigues, Z. Avelar, A. A. Vicente, S. B. Petersen and R. N. Pereira, Food Chem., 2020, 304, 125442 CrossRef CAS.
- R. Wang, Q.-H. Wen, X.-A. Zeng, J.-W. Lin, J. Li and F.-Y. Xu, Food Chem., 2022, 377, 131945 CrossRef CAS.
- R. Wang, P.-F. Guo, J.-P. Yang, Y.-Y. Huang, L.-H. Wang, J. Li, S.-Y. Lin, Q.-L. Sheng, X.-A. Zeng and Y.-X. Teng, Food Hydrocolloids, 2025, 160, 110761 CrossRef CAS.
- H. Wu, M. R. Ghaani, Z. Futera and N. J. English, J. Phys. Chem. B, 2022, 126, 376–386 CrossRef CAS PubMed.
- V. D'Annibale, D. Fracassi, P. Marracino, G. D'Inzeo and M. D'Abramo, Molecules, 2022, 27, 6454 CrossRef PubMed.
- Y. Sun, Y. Li, Y. Yang, S. Wang and Y. Gong, Spectrochim. Acta, Part A, 2025, 126039 CrossRef CAS PubMed.
- P.-K. Yang, Biopolymers, 2014, 101, 861–870 CrossRef CAS.
- A. Baumketner, J. Phys. Chem. B, 2014, 118, 14578–14589 CrossRef CAS.
- H. Wu, M. R. Ghaani, P. K. Nandi and N. J. English, J. Phys. Chem. B, 2022, 126, 858–868 CrossRef CAS PubMed.
- R. Reale, N. J. English, J.-A. Garate, P. Marracino, M. Liberti and F. Apollonio, J. Chem. Phys., 2013, 139, 205101 CrossRef.
- N. J. English and J.-A. Garate, J. Chem. Phys., 2016, 145, 085102 CrossRef PubMed.
- J. A. Garate, A. Bernardin, Y. Escalona, C. Yanez, N. J. English and T. Perez-Acle, J. Phys. Chem. B, 2019, 123, 2599–2608 CrossRef CAS PubMed.
- F. Apollonio, M. Liberti, A. Amadei, M. Aschi, M. Pellegrino, M. D'Alessandro, M. D'Abramo, A. Di Nola and G. d'Inzeo, IEEE Trans. Microwave Theory Tech., 2008, 56, 2511–2519 CAS.
- D. Gou, K. Huang, Y. Liu, H. Shi and Z. Wu, J. Phys. Chem. B, 2022, 126, 7686–7700 CrossRef CAS.
- D. Ray, M. Madani, J. K. G. Dhont, F. Platten and K. Kang, J. Phys. Chem. Lett., 2024, 15, 8108–8113 CrossRef CAS PubMed.
- D. Hou and H.-C. Chang, Appl. Phys. Lett., 2008, 92, 223902 CrossRef.
- H. Koizumi, S. Uda, K. Fujiwara, M. Tachibana, K. Kojima and J. Nozawa, Cryst. Growth Des., 2014, 14, 5662–5667 CrossRef CAS.
- H. Koizumi, K. Fujiwara and S. Uda, Cryst. Growth Des., 2009, 9, 2420–2424 CrossRef CAS.
- H. Koizumi, S. Uda, K. Fujiwara, J. Okada and J. Nozawa, Crystals, 2017, 7, 170 CrossRef.
- F. Li and R. Lakerveld, Cryst. Growth Des., 2017, 17, 3062–3070 CrossRef CAS.
- C. N. Nanev and A. Penkova, Colloids Surf., A, 2002, 209, 139–145 CrossRef CAS.
- L. Fiona Alexander and N. Radacsi, CrystEngComm, 2019, 21, 5014–5031 RSC.
- M. Taleb, C. Didierjean, C. Jelsch, J. P. Mangeot, B. Capelle and A. Aubry, J. Cryst. Grow., 1999, 200, 575–582 CrossRef CAS.
- M. Taleb, C. Didierjean, C. Jelsch, J. Mangeot and A. Aubry, J. Cryst. Grow., 2001, 232, 250–255 CrossRef CAS.
- Y. Tomita, H. Koizumi, S. Uda, K. Fujiwara and J. Nozawa, J. Appl. Crystallogr., 2012, 45, 207–212 CrossRef CAS.
- M. I. Al-haq, E. Lebrasseur, H. Tsuchiya and T. Torii, Crystallogr. Rev., 2007, 13, 29–64 CrossRef CAS.
- G. Rummel, A. Hardmeyer, C. Widmer, M. L. Chiu, P. Nollert, K. P. Locher, I. Pedruzzi, E. M. Landau and J. P. Rosenbusch, J. Struct. Biol., 1998, 121, 82–91 CrossRef CAS PubMed.
- E. Nieto-Mendoza, B. A. Frontana-Uribe, G. Sazaki and A. Moreno, J. Cryst. Grow., 2005, 275, e1437–e1446 CrossRef CAS.
- A. Moreno and G. Sazaki, J. Cryst. Grow., 2004, 264, 438–444 CrossRef CAS.
- E. Rubin, C. Owen and V. Stojanoff, Crystals, 2017, 7, 206 CrossRef.
- H. Koizumi, S. Uda, K. Fujiwara, M. Tachibana, K. Kojima and J. Nozawa, J. Appl. Crystallogr., 2013, 46, 25–29 CrossRef CAS.
- T. Wakamatsu, Trans. Mater. Res. Soc. Jpn., 2016, 41, 13–15 CrossRef CAS.
- H. Koizumi, Y. Tomita, S. Uda, K. Fujiwara and J. Nozawa, J. Cryst. Grow., 2012, 352, 155–157 CrossRef CAS.
- C. Pareja-Rivera, M. Cuéllar-Cruz, N. Esturau-Escofet, N. Demitri, M. Polentarutti, V. Stojanoff and A. Moreno, Cryst. Growth Des., 2017, 17, 135–145 CrossRef CAS.
- M. I. Al-Haq, E. Lebrasseur, W.-K. Choi, H. Tsuchiya, T. Torii, H. Yamazaki and E. Shinohara, J. Appl. Crystallogr., 2007, 40, 199–201 CrossRef CAS.
- N. J. English and D. A. Mooney, J. Chem. Phys., 2007, 126, 091105 CrossRef PubMed.
- F. Toschi, F. Lugli, F. Biscarini and F. Zerbetto, J. Phys. Chem. B, 2009, 113, 369–376 CrossRef CAS PubMed.
- N. J. English and C. J. Waldron, Phys. Chem. Chem. Phys., 2015, 17, 12407–12440 RSC.
- E. della Valle, P. Marracino, O. Pakhomova, M. Liberti and F. Apollonio, PLoS One, 2019, 14, e0221685 CrossRef CAS.
- J. Průša and M. Cifra, Sci. Rep., 2019, 9, 19721 CrossRef PubMed.
- H. R. Saeidi, A. Lohrasebi and K. Mahnam, J. Mol. Model., 2014, 20, 2395 CrossRef CAS PubMed.
- S. S. Setayandeh and A. Lohrasebi, J. Mol. Model., 2015, 21, 85 CrossRef CAS.
- P. Marracino, A. Paffi and G. d'Inzeo, Phys. Chem. Chem. Phys., 2022, 24, 11654–11661 RSC.
- A. Amadei and P. Marracino, RSC Adv., 2015, 5, 96551–96561 RSC.
- J. D. Slocum and L. J. Webb, Annu. Rev. Phys. Chem., 2018, 69, 253–271 CrossRef CAS.
- E. S. Park, S. S. Andrews, R. B. Hu and S. G. Boxer, J. Phys. Chem. B, 1999, 103, 9813–9817 CrossRef CAS.
- E. S. Park, M. R. Thomas and S. G. Boxer, J. Am. Chem. Soc., 2000, 122, 12297–12303 CrossRef CAS.
- J. Průša and M. Cifra, Comput. Struct. Biotechnol. J., 2023, 21, 1349–1361 CrossRef.
- G. Urabe, T. Sato, G. Nakamura, Y. Kobashigawa, H. Morioka and S. Katsuki, Sci. Rep., 2020, 10, 12003 CrossRef CAS.
- N. K. Pandey, S. Mitra, M. Chakraborty, S. Ghosh, S. Sen, S. Dasgupta and S. DasGupta, J. Phys. D: Appl. Phys., 2014, 47, 305401 CrossRef.
- Y.-F. Liu, I. Oey, P. Bremer, A. Carne and P. Silcock, Food Res. Int., 2017, 91, 161–170 CrossRef CAS.
- M. Mora, A. Stannard and S. Garcia-Manyes, Chem. Soc. Rev., 2020, 49, 6816–6832 RSC.
- J. Alegre-Cebollada, Biophys. Rev., 2021, 13, 435–454 CrossRef CAS PubMed.
- N. A. Chebotareva, S. G. Roman and B. I. Kurganov, Biophys. Rev., 2016, 8, 397–407 CrossRef CAS PubMed.
- H. R. Saeidi, S. S. Setayandeh and A. Lohrasebi, Chin. Phys. B, 2015, 24, 080701 CrossRef.
- S. S. Setayandeh and A. Lohrasebi, J. Nanosci. Nanotechnol., 2018, 18, 7902–7906 CrossRef CAS.
- F. Lugli, F. Toschi, F. Biscarini and F. Zerbetto, J. Chem. Theory Comput., 2010, 6, 3516–3526 CrossRef CAS.
- S. Sen, M. Chakraborty, S. Goley, S. Dasgupta and S. DasGupta, Biophys. Chem., 2017, 226, 23–33 CrossRef CAS PubMed.
- S. Yamazaki, M. Harata, T. Idehara, K. Konagaya, G. Yokoyama, H. Hoshina and Y. Ogawa, Sci. Rep., 2018, 8, 9990 CrossRef PubMed.
- K. Kang and F. Platten, Sci. Rep., 2022, 12, 3061 CrossRef CAS.
- J. Jurgelevičiūté, N. Bičkovas, A. Sakalauskas, V. Novickij, V. Smirnovas and E. Lastauskiené, Appl. Sci., 2021, 11, 2684 CrossRef.
- O. P. Cherkasova, V. I. Fedorov, E. F. Nemova and A. S. Pogodin, Opt. Spectrosc., 2009, 107, 534–537 CrossRef CAS.
- X. Xu, S. Xiao, L. Wang, D. Niu, W. Gao, X.-A. Zeng, M. Woo, Z. Han and R. Wang, Int. J. Biol. Macromol., 2024, 257, 128509 CrossRef CAS.
- A. Taha, F. Casanova, P. Šimonis, J. Jonikaité-Švégždiené, M. Jurkūnas, M. A. E. Gomaa and A. Stirké, Innovative Food Sci. Emerging Technol., 2022, 82, 103190 CrossRef CAS.
- K. Zhong, X. Hu, G. Zhao, F. Chen and X. Liao, Food Chem., 2005, 92, 473–479 CrossRef CAS.
- T. Yu and X. Fu, Bioelectrochemistry, 2015, 101, 42–45 CrossRef CAS PubMed.
- Y. Li, S. Zhang, Z. Bao, N. Sun and S. Lin, Innovative Food Sci. Emerging Technol., 2022, 76, 102918 CrossRef CAS.
- M.-L. Tian, T. Fang, M.-Y. Du and F.-S. Zhang, Protein J., 2016, 35, 154–162 CrossRef CAS.
- C. P. Samaranayake and S. K. Sastry, LWT–Food Sci. Technol., 2018, 90, 448–454 CrossRef CAS.
- H. W. Yeom, Q. H. Zhang and C. P. Dunne, Food Chem., 1999, 67, 53–59 CrossRef CAS.
- H. Vega-Mercado, J. R. Powers, G. V. Barbosa-Cánovas and B. G. Swanson, J. Food Sci., 1995, 60, 1143–1146 CrossRef CAS.
- S. Y. Ho, G. S. Mittal and J. D. Cross, J. Food Eng., 1997, 31, 69–84 CrossRef.
- M. Porcelli, G. Cacciapuoti, S. Fusco, R. Massa, G. d'Ambrosio, C. Bertoldo, M. De Rosa and V. Zappia, FEBS Lett., 1997, 402, 102–106 CrossRef CAS PubMed.
- C. Arroyo, T. M. Kennedy, J. G. Lyng and M. O'Sullivan, Food Bioprod. Process., 2017, 105, 95–103 CrossRef CAS.
- R.-S. Agoua, L. Bazinet, E. Vorobiev, N. Grimi and S. Mikhaylin, ACS Sustainable Chem. Eng., 2020, 8, 14775–14785 CrossRef CAS.
- Clinical Aspects of Electroporation, ed. S. T. Kee, J. Gehl and E. W. Lee, Springer New York, New York, NY, 2011 Search PubMed.
- N. J. English and D. G. Carroll, J. Chem. Inf. Comput. Sci., 2001, 41, 1150–1161 CrossRef CAS PubMed.
- N. J. English, Mol. Phys., 2005, 103, 1945–1960 CrossRef CAS.
- N. J. English, Mol. Phys., 2008, 106, 1887–1898 CrossRef CAS.
- H. Cao, N. J. English and J. M. D. MacElroy, J. Chem. Phys., 2013, 138, 094507 CrossRef.
- K. Ishibashi, S. Hara and S. Kondo, Clin. Exp. Nephrol., 2009, 13, 107–117 CrossRef CAS.
- A. S. Verkman, P.-W. Phuan, N. Asavapanumas and L. Tradtrantip, Brain Pathol., 2013, 23, 684–695 CrossRef CAS PubMed.
- A. Verkman, Annu. Rev. Med., 2012, 63, 303–316 CrossRef CAS PubMed.
- P. Marracino, M. Bernardi, M. Liberti, F. Del Signore, E. Trapani, J.-A. Gárate, C. J. Burnham, F. Apollonio and N. J. English, ACS Omega, 2018, 3, 15361–15369 CrossRef CAS.
- M. L. Fernández, M. Risk, R. Reigada and P. T. Vernier, Biochem. Biophys. Res. Commun., 2012, 423, 325–330 CrossRef.
- M. Bernardi, P. Marracino, M. Liberti, J. Garate, C. Burnham, F. Apollonio and N. English, Phys. Chem. Chem. Phys., 2019, 1–8 Search PubMed.
- M. Bernardi, P. Marracino, M. R. Ghaani, M. Liberti, F. Del Signore, C. J. Burnham, J.-A. Gárate, F. Apollonio and N. J. English, J. Chem. Phys., 2018, 149, 245102 CrossRef PubMed.
- M. Bernardi, M. R. Ghaani and N. J. English, Mol. Phys., 2019, 117, 3783–3790 CrossRef CAS.
- L. Rems, M. A. Kasimova, I. Testa and L. Delemotte, Biophys. J., 2020, 119, 190–205 CrossRef CAS.
- T. Y. Tsong and C.-H. Chang, Chin. J. Phys., 2005, 43, 273–284 CAS.
- P. Marracino, M. Liberti, E. Trapani, C. Burnham, M. Avena, J.-A. Garate, F. Apollonio and N. English, Int. J. Mol. Sci., 2016, 17, 1133 CrossRef PubMed.
- K. Tsuji and E. Neumann, FEBS Lett., 1981, 128, 265–268 CrossRef CAS.
- K. Tsuji and B. Hess, Eur. Biophys. J., 1990, 18, 63–69 CrossRef CAS PubMed.
- B. E. Knox and T. Tsong, J. Biol. Chem., 1984, 259, 4757–4763 CrossRef CAS PubMed.
- T. Y. Tsong, Biochim. Biophys. Acta, Rev. Biomembr., 1992, 1113, 53–70 CrossRef CAS.
- E. H. Serpersu and T. Y. Tsong, J. Biol. Chem., 1984, 259, 7155–7162 CrossRef CAS.
- D. S. Liu, R. D. Astumian and T. Y. Tsong, J. Biol. Chem., 1990, 265, 7260–7267 CrossRef CAS.
- H. V. Westerhoff, T. Y. Tsong, P. B. Chock, Y.-D. Chen and R. D. Astumian, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 4734–4738 CrossRef CAS PubMed.
- T. Y. Tsong, D.-S. Liu, F. Chauvin and R. D. Astumian, Biosci. Rep., 1989, 9, 13–26 CrossRef CAS PubMed.
- T. Y. Tsong, Annu. Rev. Biophys. Biophys. Chem., 1990, 19, 83–106 CrossRef CAS.
- V. S. Markin and T. Y. Tsong, Biophys. J., 1991, 59, 1308–1316 CrossRef CAS.
- V. Markin, D. Liu, M. Rosenberg and T. Tsong, Biophys. J., 1992, 61, 1045–1049 CrossRef CAS.
- T. Xie, P. Marszalek, Y. Chen and T. Y. Tsong, Biophys. J., 1994, 67, 1247–1251 CrossRef CAS PubMed.
- T. Xie, Y.-D. Chen, P. Marszalek and T. Y. Tsong, Biophys. J., 1997, 72, 2496–2502 CrossRef CAS.
- T. Y. Tsong and C.-H. Chang, AAPPS Bull., 2003, 13, 12–18 Search PubMed.
- T. Nakabayashi, M. Kinjo and N. Ohta, Chem. Phys. Lett., 2008, 457, 408–412 CrossRef CAS.
- I. Barak, M. Golosovsky and D. Davidov, Piers Online, 2009, 5, 561–566 Search PubMed.
- J. W. Park and Y. M. Rhee, J. Am. Chem. Soc., 2016, 138, 13619–13629 CrossRef CAS.
- P. P. Pompa, A. Bramanti, G. Maruccio, R. Cingolani, F. De Rienzo, S. Corni, R. Di Felice and R. Rinaldi, J. Chem. Phys., 2005, 122, 181102 CrossRef CAS PubMed.
- T. Nakabayashi, M. Wahadoszamen and N. Ohta, J. Am. Chem. Soc., 2005, 127, 7041–7052 CrossRef CAS.
- P. P. Pompa, A. Bramanti, G. Maruccio, L. L. del Mercato, R. Chiuri, R. Cingolani and R. Rinaldi, SPIE Proceedings, 2005, p. 171.
- D. Havelka, D. E. Chafai, O. Krivosudský, A. Klebanovych, F. Vostárek, L. Kubínová, P. Dráber and M. Cifra, Adv. Mater. Technol., 2020, 5, 1900669 CrossRef CAS.
- P. Afaghi, M. A. Lapolla and K. Ghandi, Sci. Rep., 2021, 11, 23373 CrossRef CAS.
- A. Intze, M. E. Temperini, G. Gregori, F. Verde, M. Ortolani and V. Giliberti, IEEE Trans. Terahertz Sci. Technol., 2024, 14, 652–660 CAS.
- D. Havelka, I. Zhernov, M. Teplan, Z. Lánský, D. E. Chafai and M. Cifra, Sci. Rep., 2022, 12, 2462 CrossRef CAS.
- M. A. Schroer, S. Schewa, A. Y. Gruzinov, C. Rönnau, J. M. Lahey-Rudolph, C. E. Blanchet, T. Zickmantel, Y.-H. Song, D. I. Svergun and M. Roessle, Sci. Rep., 2021, 11, 22311 CrossRef CAS PubMed.
- J. C. Bischof and X. He, Ann. N. Y. Acad. Sci., 2006, 1066, 12–33 CrossRef.
- F. Zhao, S. M. Matt, J. Bu, O. G. Rehrauer, D. Ben-Amotz and S. A. McLuckey, J. Am. Soc. Mass Spectrom., 2017, 28, 2001–2010 CrossRef CAS.
- J. Wu, Z. Zhou, Y. Huang, X. Deng, S. Zheng, S. He, G. Huang, B. Hu, M. Shi, W. Liao and N. Huang, MedComm, 2024, 5, e746 CrossRef CAS.
- Z. Haider, J. Modolo, M. Liberti, F. Apollonio and M. Zhadobov, IEEE J. Microwaves, 2023, 3, 170–180 Search PubMed.
- Y. Y. Chen and A. W. Wood, Bioelectromagnetics, 2009, 30, 583–590 CrossRef CAS.
- A. Salari, M. Navi, T. Lijnse and C. Dalton, Micromachines, 2019, 10, 762 CrossRef.
- A. González, A. Ramos, H. Morgan, N. G. Green and A. Castellanos, J. Fluid Mech., 2006, 564, 415–433 CrossRef.
- A. Khayari, M. Medrano, E. Verlage, M. C. Velázquez-Ahumada, M. J. Freire and A. Ramos, J. Appl. Phys., 2011, 110, 064912 CrossRef.
- R. Orlacchio, M. Zhadobov, S. I. Alekseev, D. Nikolayev, R. Sauleau, Y. Le Page and Y. Le Dréan, Bioelectromagnetics, 2019, 40, 553–568 CrossRef.
- R. Piazza, Soft Matter, 2008, 4, 1740 RSC.
- A. Ramos, H. Morgan, N. G. Green and A. Castellanos, J. Phys. D: Appl. Phys., 1998, 31, 2338 CrossRef CAS.
- N. G. Green, A. Ramos, A. González, H. Morgan and A. Castellanos, Phys. Rev. E:Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2000, 61, 4011–4018 CrossRef CAS.
- T. M. Squires and M. Z. Bazant, J. Fluid Mech., 2004, 509, 217–252 CrossRef.
- B. J. Kirby and E. F. Hasselbrink, Electrophoresis, 2004, 25, 187–202 CrossRef CAS.
- C.-C. Chang and R.-J. Yang, Microfluid. Nanofluid., 2007, 3, 501–525 CrossRef CAS.
- C. Moino, F. Artusio and R. Pisano, Int. J. Pharm., 2024, 650, 123679 CrossRef CAS.
- C. C. Roth, R. A. Barnes Jr., B. L. Ibey, H. T. Beier, L. Christopher Mimun, S. M. Maswadi, M. Shadaram and R. D. Glickman, Sci. Rep., 2015, 5, 15063 CrossRef CAS PubMed.
- B. Harish, R. E. Gillilan, J. Zou, J. Wang, D. P. Raleigh and C. A. Royer, Biophys. J., 2021, 120, 2592–2598 CrossRef CAS PubMed.
- A. Y. Starikovskiy and M. N. Shneider, Phys. Fluids, 2024, 36, 042011 CrossRef CAS.
- K. S. Suslick, Science, 1990, 247, 1439–1445 CrossRef CAS PubMed.
- P. Riesz, K. Takashi and C. M. Krishna, Free Radical Res. Commun., 1990, 10, 27–35 CrossRef CAS PubMed.
- T. W. Randolph, E. Schiltz, D. Sederstrom, D. Steinmann, O. Mozziconacci, C. Schöneich, E. Freund, M. S. Ricci, J. F. Carpenter and C. S. Lengsfeld, J. Pharm. Sci., 2015, 104, 602–611 CrossRef CAS.
- E. Y. Lau, M. L. Berkowitz and E. Schwegler, Biophys. J., 2016, 110, 147–156 CrossRef CAS.
- R. Šmerc, D. Miklavčič and S. Mahnič-Kalamiza, Electrochim. Acta, 2024, 145363 Search PubMed.
- K. Talley and E. Alexov, Proteins: Struct., Funct., Bioinf., 2010, 78, 2699–2706 CrossRef CAS.
- L. Konermann, E. A. Silva and O. F. Sogbein, Anal. Chem., 2001, 73, 4836–4844 CrossRef CAS.
- P. Turjanski, N. Olaiz, P. Abou-Adal, C. Suárez, M. Risk and G. Marshall, Electrochim. Acta, 2009, 54, 6199–6206 CrossRef CAS.
- P. G. Erlandsson and N. D. Robinson, Electrophoresis, 2011, 32, 784–790 CrossRef CAS PubMed.
- T. Kakiuchi, J. Solid State Electrochem., 2011, 15, 1661–1671 CrossRef CAS.
- K. Wang, W. Mao, X. Song, M. Chen, W. Feng, B. Peng and Y. Chen, Chem. Soc. Rev., 2023, 52, 6957–7035 RSC.
- K. Červinková, P. Vahalová, M. Poplová, T. Zakar, D. Havelka, M. Paidar, V. Kolivoška and M. Cifra, Sci. Rep., 2024, 14, 22649 CrossRef PubMed.
- E. R. Stadtman, Annu. Rev. Biochem., 1993, 62, 797–821 CrossRef CAS PubMed.
- M. J. Davies, Biochem. J., 2016, 473, 805–825 CrossRef CAS.
- P. Vahalová, D. Havelka, E. Vaněčková, T. Zakar, V. Kolivoška and M. Cifra, Sens. Actuators, B, 2023, 385, 133676 CrossRef.
- E. D'Imprima, D. Floris, M. Joppe, R. Sánchez, M. Grininger and W. Kühlbrandt, eLife, 2019, 8, e42747 CrossRef PubMed.
- K. Mitra, I. Ubarretxena-Belandia, T. Taguchi, G. Warren and D. M. Engelman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4083–4088 CrossRef CAS PubMed.
- D. Stuerga and P. Gaillard, J. Microwave Power Electromagn. Energy, 1996, 31, 101–113 Search PubMed.
- D. Stuerga and P. Gaillard, J. Microwave Power Electromagn. Energy, 1996, 31, 87–100 Search PubMed.
- I. Y. Belyaev, V. S. Shcheglov, E. D. Alipov and V. D. Ushakov, IEEE Trans. Microwave Theory Tech., 2000, 48, 2172–2179 CrossRef CAS.
- K. R. Foster, IEEE Trans. Plasma Sci., 2000, 28, 15–23 CrossRef.
- R. K. Adair, Bioelectromagnetics, 2003, 24, 39–48 CrossRef PubMed.
- M. Cifra, J. Z. Fields and A. Farhadi, Prog. Biophys. Mol. Biol., 2011, 105, 223–246 CrossRef CAS.
- O. Kučera and M. Cifra, J. Biol. Phys., 2016, 42, 1–8 CrossRef.
- M. Cifra, F. Apollonio, M. Liberti, T. García-Sánchez and L. M. Mir, Int. J. Biometeorol., 2021, 65, 59–67 CrossRef PubMed.
- F. Apollonio, M. Liberti, A. Paffi, C. Merla, P. Marracino, A. Denzi, C. Marino and G. d'Inzeo, IEEE Trans. Microwave Theory Tech., 2013, 61, 2031–2045 Search PubMed.
- N. T. Hunt, Chem. Soc. Rev., 2009, 38, 1837–1848 RSC.
- M. Maiuri, M. Garavelli and G. Cerullo, J. Am. Chem. Soc., 2020, 142, 3–15 CrossRef CAS.
- E. D. B. Foley, M. S. Kushwah, G. Young and P. Kukura, Nat. Methods, 2021, 18, 1–6 CrossRef.
- G. Young and P. Kukura, Annu. Rev. Phys. Chem., 2019, 70, 301–322 CrossRef CAS.
- K. Schoenbach, J. Kolb, S. Xiao, S. Katsuki, Y. Minamitani and R. Joshi, Plasma Sources Sci. Technol., 2008, 17, 024010 CrossRef.
- H. M. Jones and E. E. Kunhardt, J. Appl. Phys., 1995, 77, 795–805 CrossRef CAS.
- S. Y. Joshi and S. A. Deshmukh, Mol. Simul., 2021, 47, 786–803 CrossRef CAS.
- S. Kmiecik, D. Gront, M. Kolinski, L. Wieteska, A. E. Dawid and A. Kolinski, Chem. Rev., 2016, 116, 7898–7936 CrossRef CAS.
- D. Xu, J. C. Phillips and K. Schulten, J. Phys. Chem., 1996, 100, 12108–12121 CrossRef CAS.
- B. Rakos, Int. J. Circ. Theory Appl., 2015, 43, 60–72 CrossRef.
- Z. Jing, C. Liu, S. Y. Cheng, R. Qi, B. D. Walker, J.-P. Piquemal and P. Ren, Annu. Rev. Biophys., 2019, 48, 371–394 CrossRef CAS.
- J. Liu, T. Zhu, X. Wang, X. He and J. Z. H. Zhang, J. Chem. Theory Comput., 2015, 11, 5897–5905 CrossRef CAS.
- D. J. Cole and N. D. M. Hine, J. Phys.: Condens. Matter, 2016, 28, 393001 CrossRef PubMed.
- E. Neumann, Top. Bioelectrochem. Bioenerg., 1981, 4, 113–160 CAS.
- M. J. Schnitzer, K. Visscher and S. M. Block, Nat. Cell Biol., 2000, 2, 718–723 CrossRef CAS PubMed.
- T.-L. Kuo, S. Garcia-Manyes, J. Li, I. Barel, H. Lu, B. J. Berne, M. Urbakh, J. Klafter and J. M. Fernández, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11336–11340 CrossRef CAS.
- W.-W. Sun, S.-J. Yu, X.-A. Zeng, X.-Q. Yang and X. Jia, Food Res. Int., 2011, 44, 1052–1058 CrossRef CAS.
- M. Shafiei, M. von Domaros, D. Bratko and A. Luzar, J. Chem. Phys., 2019, 150, 074505 CrossRef PubMed.
- Z. Futera and N. J. English, J. Chem. Phys., 2017, 147, 031102 CrossRef PubMed.
- G. Cassone, J. Phys. Chem. Lett., 2020, 11, 8983–8988 CrossRef CAS.
- V. Conti Nibali, S. Maiti, F. Saija, M. Heyden and G. Cassone, J. Chem. Phys., 2023, 158, 184501 CrossRef CAS.
- H. Vega-Mercado, O. Martín-Belloso, B.-L. Qin, F. J. Chang, M. Marcela Góngora-Nieto, G. V. Barbosa-Cánovas and B. G. Swanson, Trends Food Sci. Technol., 1997, 8, 151–157 CrossRef CAS.
- G. V. Barbosa-Cánovas and Q. H. Zhang, Pulsed electric fields in food processing: fundamental aspects and applications, CRC Press, 2001 Search PubMed.
- A. Van Loey, B. Verachtert and M. Hendrickx, Trends Food Sci. Technol., 2001, 12, 94–102 CrossRef CAS.
- R. Soliva-Fortuny, A. Balasa, D. Knorr and O. Martín-Belloso, Trends Food Sci. Technol., 2009, 20, 544–556 CrossRef CAS.
- W. Zhao and R. Yang, in Handbook of Electroporation, ed. D. Miklavčič, Springer International Publishing, Cham, 2017, pp. 2239–2251 Search PubMed.
- R. M. Rodrigues, A. J. Martins, O. L. Ramos, F. X. Malcata, J. A. Teixeira, A. A. Vicente and R. N. Pereira, Food Hydrocolloids, 2015, 43, 329–339 CrossRef CAS.
- J.-Y. Qian, L.-J. Ma, L.-J. Wang and W. Jiang, LWT–Food Sci. Technol., 2016, 74, 331–337 CrossRef CAS.
- L. Wu, W. Zhao, R. Yang, W. Yan and Q. Sun, J. Sci. Food Agric., 2016, 96, 3334–3341 CrossRef CAS.
- B. Y. Xiang, M. O. Ngadi, L. A. Ochoa-Martinez and M. V. Simpson, Food Bioprocess Technol., 2011, 4, 1341–1348 CrossRef CAS.
- B. Y. Xiang, M. O. Ngadi, B. K. Simpson and M. V. Simpson, J. Food Process. Preserv., 2011, 35, 563–570 CrossRef CAS.
- R. N. Pereira, J. A. Teixeira and A. A. Vicente, J. Agric. Food Chem., 2011, 59, 11589–11597 CrossRef CAS.
- R. M. Rodrigues, A. A. Vicente, S. B. Petersen and R. N. Pereira, Innovative Food Sci. Emerging Technol., 2019, 52, 1–7 CrossRef CAS.
- Y. Li, Z. Chen and H. Mo, LWT–Food Sci. Technol., 2007, 40, 1167–1175 CrossRef CAS.
- A. Singh, R. Lahlali, S. K. Vanga, C. Karunakaran, V. Orsat and V. Raghavan, Int. J. Food Prop., 2016, 19, 1217–1226 CrossRef CAS.
- S. K. Vanga, A. Singh, F. Kalkan, Y. Gariepy, V. Orsat and V. Raghavan, Int. J. Food Prop., 2016, 19, 1259–1271 CrossRef CAS.
- R. N. Pereira, B. W. S. Souza, M. A. Cerqueira, J. A. Teixeira and A. A. Vicente, Biomacromolecules, 2010, 11, 2912–2918 CrossRef CAS.
- Y.-Q. Li, Phys. Procedia, 2012, 33, 132–137 CrossRef CAS.
- W. Zhao, R. Yang, Y. Tang and R. Lu, Int. J. Food Eng., 2007, 3, 12 Search PubMed.
- R. N. Pereira, R. M. Rodrigues, Ó. L. Ramos, F. Xavier Malcata, J. A. Teixeira and A. A. Vicente, Food Bioprocess Technol., 2016, 9, 576–587 CrossRef CAS.
- R. N. Pereira, R. M. Rodrigues, E. Altinok, Ó. L. Ramos, F. Xavier Malcata, P. Maresca, G. Ferrari, J. A. Teixeira and A. A. Vicente, Food Res. Int., 2017, 99, 435–443 CrossRef CAS.
- T. Grahl and H. Märkl, Appl. Microbiol. Biotechnol., 1996, 45, 148–157 CrossRef CAS.
- A. J. Castro, B. G. Swanson, G. V. Barbosa-Cánovas and Q. H. Zhang, Pulsed Electric Fields in Food Processing, CRC Press, 2001, pp. 65–82 Search PubMed.
- C. P. Samaranayake and S. K. Sastry, J. Food Eng., 2016, 186, 17–26 CrossRef CAS.
- C. P. Samaranayake and S. K. Sastry, Food Chem., 2016, 199, 265–272 CrossRef CAS PubMed.
- A. G. Pakhomov, S. Xiao, O. N. Pakhomova, I. Semenov, M. A. Kuipers and B. L. Ibey, Bioelectrochemistry, 2014, 100, 88–95 CrossRef CAS PubMed.
- C. Blangero, M. P. Rols and J. Teissié, Biochim. Biophys. Acta, Biomembr., 1989, 981, 295–302 CrossRef CAS.
- C. Kanthou, S. Kranjc, G. Sersa, G. Tozer, A. Zupanic and M. Cemazar, Mol. Cancer Ther., 2006, 5, 3145–3152 CrossRef CAS.
- L. Carr, PhD thesis, Université de Limoges, 2016.
- L. Carr, S. M. Bardet, R. C. Burke, D. Arnaud-Cormos, P. Leveque and R. P. O'Connor, Sci. Rep., 2017, 7, 41267 CrossRef CAS.
- C. J. W. Meulenberg, V. Todorovic and M. Cemazar, PLoS One, 2012, 7, e52713 CrossRef CAS PubMed.
- D. E. Chafai, F. Vostárek, E. Dráberová, D. Havelka, D. Arnaud-Cormos, P. Leveque, J. Janáček, L. Kubínová, M. Cifra and P. Dráber, Adv. Biosyst., 2020, 4, 2000070 CrossRef CAS.
- T. Berghöfer, C. Eing, B. Flickinger, P. Hohenberger, L. H. Wegner, W. Frey and P. Nick, Biochem. Biophys. Res. Commun., 2009, 387, 590–595 CrossRef.
- D. G. Harkin and E. D. Hay, Cell Motil., 1996, 35, 345–357 CrossRef CAS.
- G. P. Tolstykh, G. L. Thompson, H. T. Beier, Z. A. Steelman and B. L. Ibey, Biochem. Biophys. Rep., 2017, 9, 36–41 Search PubMed.
- M. Stacey, P. Fox, S. Buescher and J. Kolb, Bioelectrochemistry, 2011, 82, 131–134 CrossRef CAS PubMed.
- G. L. Thompson, C. C. Roth, D. R. Dalzell, M. Kuipers and B. L. Ibey, J. Biomed. Opt., 2014, 19, 055005 Search PubMed.
- G. L. Thompson, C. Roth, G. Tolstykh, M. Kuipers and B. L. Ibey, Terahertz and Ultrashort Electromagnetic Pulses for Biomedical Applications, 2013, p. 85850T Search PubMed.
- D. L. Perrier, A. Vahid, V. Kathavi, L. Stam, L. Rems, Y. Mulla, A. Muralidharan, G. H. Koenderink, M. T. Kreutzer and P. E. Boukany, Sci. Rep., 2019, 9, 8151 CrossRef.
- M. Rief, M. Gautel, F. Oesterhelt, J. M. Fernandez and H. E. Gaub, Science, 1997, 276, 1109–1112 CrossRef CAS PubMed.
- M. D. Fernandez-Diaz, L. Barsotti, E. Dumay and J. C. Cheftel, J. Agric. Food Chem., 2000, 48, 2332–2339 CrossRef CAS PubMed.
- S. Ilieva, D. Cheshmedzhieva and T. Dudev, Phys. Chem. Chem. Phys., 2019, 21, 16198–16206 RSC.
- G. Y. Solomentsev, N. J. English and D. A. Mooney, J. Chem. Phys., 2010, 133, 235102 CrossRef PubMed.
- C. A. Petroff, G. Cassone, J. Šponer and G. R. Hutchison, Adv. Mater., 2021, 33, 2007486 CrossRef CAS.
- D. E. Chafai, A. Mehle, A. Tilmatine, B. Maouche and D. Miklavčič, Bioelectrochemistry, 2015, 106, 249–257 CrossRef CAS.
- W. J. Ellison, J. Phys. Chem. Ref. Data, 2007, 36, 1–18 CrossRef CAS.
- D. Niu, Y. Feng, X.-A. Zeng and J.-H. Cheng, Innovative Food Sci. Emerging Technol., 2024, 97, 103826 CrossRef CAS.
- O. E. Perez and A. M. R. Pilosof, Food Res. Int., 2004, 37, 102–110 CrossRef CAS.
- H. Bohr and J. Bohr, Phys. Rev. E:Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2000, 61, 4310–4314 CrossRef CAS.
- T. Kawasaki, Y. Yamaguchi, T. Ueda, Y. Ishikawa, T. Yaji, T. Ohta, K. Tsukiyama, T. Idehara, M. Saiki and M. Tani, Biomed. Opt. Express, 2020, 11, 5341–5351 CrossRef CAS.
- D. F. George, M. M. Bilek and D. R. McKenzie, Bioelectromagnetics, 2008, 29, 324–330 CrossRef CAS.
- D. I. de Pomerai, B. Smith, A. Dawe, K. North, T. Smith, D. B. Archer, I. R. Duce, D. Jones and E. M. Candido, FEBS Lett., 2003, 543, 93–97 CrossRef CAS.
- N. Huda and A. K. Bhuyan, J. Phys. Chem. B, 2023, 127, 4386–4395 CrossRef CAS.
- D. M. Landry, PhD thesis, Brigham Young University – Provo, 2013.
- Y. Shi, M. J. Acerson, K. L. Shuford and B. F. Shaw, ACS Chem. Neurosci., 2015, 6, 1696–1707 CrossRef CAS.
- W. Zhao and R. Yang, J. Phys. Chem. B, 2010, 114, 503–510 CrossRef CAS.
- W. Zhao, R. Yang, R. Lu, Y. Tang and W. Zhang, J. Agric. Food Chem., 2007, 55, 9850–9858 CrossRef CAS.
- W. Zhao and R. Yang, Eur. Food Res. Technol., 2008, 228, 47–54 CrossRef CAS.
- I. V. Lundholm, H. Rodilla, W. Y. Wahlgren, A. Duelli, G. Bourenkov, J. Vukusic, R. Friedman, J. Stake, T. Schneider and G. Katona, Struct. Dyn., 2015, 2, 054702 CrossRef.
- P. M. Favi, Q. Zhang, H. O'Neill, E. Mamontov and S. O. Diallo, J. Biol. Phys., 2014, 40, 167–178 CrossRef CAS PubMed.
- G. Urabe, T. Katagiri and S. Katsuki, Bioelectricity, 2020, 2, 33–39 CrossRef PubMed.
- L. Wu, W. Zhao, R. Yang and W. Yan, Food Chem., 2015, 175, 115–120 CrossRef CAS PubMed.
- L. Wu, W. Zhao, R. Yang and X. Chen, J. Food Eng., 2014, 139, 13–18 CrossRef CAS.
- W. Zhao and R. Yang, J. Phys. Chem. B, 2008, 112, 14018–14025 CrossRef CAS.
- G. Hammarin, P. Norder, R. Harimoorthy, G. Chen, P. Berntsen, P. O. Widlund, C. Stoij, H. Rodilla, J. Swenson, G. Brändén and R. Neutze, Sci. Rep., 2024, 14, 18286 CrossRef CAS.
- P. M. Vassilev, R. T. Dronzine, M. P. Vassileva and G. A. Georgiev, Biosci. Rep., 1982, 2, 1025–1029 CrossRef CAS.
- I. Minoura and E. Muto, Biophys. J., 2006, 90, 3739–3748 CrossRef CAS PubMed.
- W. Zhao and R. Yang, Food Bioprocess Technol., 2012, 5, 1706–1714 CrossRef CAS.
- J. Zhang, T. Tang, Z. Jiang, Y. Liu and A. Jiang, Food Chem., 2019, 293, 455–462 CrossRef CAS.
- Y. Li, S. Zhang, P. Jiang, L. Zhong, S. Lin and N. Sun, LWT–Food Sci. Technol., 2021, 141, 110891 CrossRef CAS.
- M. D. L. Meza-Jiménez, P.-R. Pokhrel, R. R. Robles De La Torre, G. V. Barbosa-Canovas and H. Hernández-Sánchez, LWT–Food Sci. Technol., 2019, 109, 336–341 CrossRef.
- F. Zhang, M. Tian, M. Du and T. Fang, J. Food Eng., 2017, 205, 56–63 CrossRef CAS.
- K. Zhong, J. Wu, Z. Wang, F. Chen, X. Liao, X. Hu and Z. Zhang, Food Chem., 2007, 100, 115–123 CrossRef CAS.
- T. Ohshima, T. Tamura and M. Sato, J. Electrost., 2007, 65, 156–161 CrossRef CAS.
- J. Castorena-García, F. Martínez-Montes, M. Robles-López, J. Welti-Chanes, H. Hernandez-Sanchez and R. Robles-de-la Torre, Rev. Mex. Ing. Quim., 2013, 12, 391–400 Search PubMed.
- H. Vega-Mercado, J. R. Powers, G. V. Barbosa-Cánovas and B. G. Swanson, Pulsed Electric Fields in Food Processing, CRC Press, 2001, pp. 121–134 Search PubMed.
- J. Wang, S. K. Vanga and V. Raghavan, Food Funct., 2020, 11, 1373–1384 RSC.
- N. Todorova, A. Bentvelzen and I. Yarovsky, J. Chem. Phys., 2020, 152, 035104 CrossRef CAS PubMed.
- Z. Rahimi and A. Lohrasebi, Eur. Phys. J. E:Soft Matter Biol. Phys., 2023, 46, 3 CrossRef CAS.
- L. Astrakas, C. Gousias and M. Tzaphlidou, J. Appl. Phys., 2011, 109, 094702 CrossRef.
- L. G. Astrakas, C. Gousias and M. Tzaphlidou, J. Appl. Phys., 2012, 111, 074702 CrossRef.
- N. Hamedi, M. Saviz, A. Banaei, S. Shabani, M. Qafary, A. A. Moosavi-Movahedi and R. Faraji-Dana, J. Mol. Liq., 2020, 312, 113283 CrossRef CAS.
- N. J. English, G. Y. Solomentsev and P. O'Brien, J. Chem. Phys., 2009, 131, 035106 CrossRef PubMed.
- C. J. Burnham and N. J. English, J. Phys. Chem. Lett., 2017, 8, 4646–4651 CrossRef CAS.
- A. Budi, F. S. Legge, H. Treutlein and I. Yarovsky, J. Phys. Chem. B, 2005, 109, 22641–22648 CrossRef CAS PubMed.
- A. Budi, F. S. Legge, H. Treutlein and I. Yarovsky, J. Phys. Chem. B, 2007, 111, 5748–5756 CrossRef CAS PubMed.
- E. K. Massaro, I. Goswami, S. S. Verbridge and M. R. von Spakovsky, Bioelectrochemistry, 2021, 137, 107638 CrossRef CAS PubMed.
- T. Yoon, W. Park, Y. Kim and S. Na, Appl. Surf. Sci., 2023, 608, 155124 CrossRef CAS.
- R. Saxena, S. K. Vanga and V. Raghavan, J. Food Biochem., 2019, 43, e12898 CrossRef PubMed.
- Z. Rahimi and A. Lohrasebi, Adv. Sci., Eng. Med., 2018, 10, 1077–1084 CrossRef CAS.
- P. Marracino, J. Phys. Chem. Biophys., 2013, 03, 1000117 Search PubMed.
- T. Dudev, S. Ilieva and L. Doudeva, Phys. Chem. Chem. Phys., 2018, 20, 24633–24640 RSC.
- L. Wang, S. Qin, O. Wei, X. Liang, D. Fu, J. Yu, S. Huang and L. Xie, Int. J. Biol. Macromol., 2025, 307, 142398 CrossRef CAS PubMed.
- A. Singh, V. Orsat and V. Raghavan, Biomolecules, 2013, 3, 168–179 CrossRef CAS PubMed.
- S. S. Setayandeh and A. Lohrasebi, J. Theor. Comput. Chem., 2016, 15, 1650010 CrossRef CAS.
- A. R. Ruiz-Fernández, L. Campos, F. Villanelo, J. A. Garate and T. Perez-Acle, Int. J. Mol. Sci., 2023, 24, 11397 CrossRef PubMed.
- W. Treptow, B. Maigret, C. Chipot and M. Tarek, Biophys. J., 2004, 87, 2365–2379 CrossRef CAS PubMed.
- F. Guo, J. Xiang, Y. Zhuo and K. Pei, Biomacromolecules, 2025, 26, 1002–1011 CrossRef CAS PubMed.
- X. Zhao, W. Ding, H. Wang, Y. Wang, Y. Liu, Y. Li and C. Liu, ACS Omega, 2024, 9, 9702–9713 CrossRef CAS.
Footnote |
| † Present address: J. Heyrovský Institute of Physical Chemistry of the Czech Academy of Sciences, Prague, 18200, Czechia. |
| This journal is © The Royal Society of Chemistry 2026 |