Phase behaviour of liquid CO2 with an impurity of water: influence of CO2 hydrate
Received
24th March 2026
, Accepted 20th May 2026
First published on 22nd May 2026
Abstract
The solubility of water in liquid CO2 coexisting with CO2 hydrate or liquid water is evaluated in order to investigate the thermodynamic conditions to avoid the formation of CO2 hydrate in the transportation processes of liquid CO2. To this end, theoretical calculations have been carried out to obtain the chemical potentials of water and CO2 in all the phases involved in their coexistence. The solubility of water in liquid CO2 coexisting with liquid water decreases with decreasing temperature over a wide range of temperature and pressure, except for in the vicinity of the critical point of CO2. The decrease in the solubility is further enhanced by the formation of hydrate. We estimate the Gibbs energy of hydrate formation, which is an important property for sequestration of CO2, for cases where the temperature or pressure of water-saturated liquid CO2 decreases. We also estimate the amount of water precipitated as hydrate during these processes, which has a direct bearing on flow assurance in CO2 transportation. The present study will contribute to the development of a low-energy, safe CO2 transport network aiming at achieving large-scale carbon neutrality.
Introduction
Carbon dioxide (CO2) is often used in industries as a non-toxic solvent to extract precious substances, mostly under supercritical conditions.1 Advantage is taken of the fact that its critical temperature, 304.2 K, is well within the room temperature range and supercritical CO2 acts as a good solvent for a variety of substances. It is also utilised as a cooling agent owing to the large latent heat of the sublimation of dry ice, 25 kJ mol−1,2 which is associated with its triple point pressure (0.517 MPa) being higher than atmospheric pressure.3
One of the key concerns regarding CO2 is how emitted greenhouse gases can be reduced through CO2 capture and storage (CCS).4 Formation of clathrate hydrate (hereafter hydrate) at low temperatures and high pressures poses a potential risk of causing pipeline blockage during transportation of CO2 containing water as an impurity in the CCS process.5 It is possible to suppress hydrate formation by dosing with kinetic inhibitors such as polyvinyl caprolactam or thermodynamic inhibitors such as methanol. Extensive efforts have been devoted to finding efficient inhibitors and exploring possible mechanisms for the inhibitors to work.6–10 Another way is the removal of water prior to transportation. To implement this strategy to exploit safe and feasible CCS, it is an urgent task to gain a comprehensive understanding of the various features of water dissolved in liquid CO2.
A hydrate is a crystal composed of water molecules forming a host lattice structure in which small guest molecules are encapsulated.11 It is a nonstoichiometric compound with respect to guest composition, i.e., the number of guest molecules in the host lattice. Its thermodynamic stability and composition of the guest significantly depend on temperature and pressure. We have studied the thermodynamic stabilities of hydrates encaging various kinds of guest species based on atomistic models of intermolecular interactions.12–18 Our theory stems from the van der Waals and Platteeuw (vdWP) theory19–21 originally developed to predict three-phase coexistence (water, hydrate, and guest fluid phases) but is different in the ensemble to which the relevant partition function is linked. The free energy derived from our theory enables the evaluation of the two two-phase coexistences between the hydrate and aqueous phases and between the hydrate and guest fluid phases. We reveal that this approach, which requires only a few empirical parameters, plays a significant role in achieving this objective.
Hydrates have been utilised for storing various gases of small molecular size, desalination of seawater, and energy resources.11 Natural CH4 hydrate is expected to be an energy resource. Recovering CH4 from hydrate is essential to mitigating global warming; otherwise, the release of CH4 into the atmosphere from the hydrate accelerates it because CH4 has a stronger greenhouse effect than CO2 does. Replacing CH4 in hydrate with CO2 may provide an efficient way to contribute to CCS. Hence, the phase behaviour of CH4 hydrate would also be a key issue in CCS.
The solubility of water in liquid CO2 is an important property for CCS. Although a fairly large amount of measured solubility data has been accumulated,22,23 the solubility values depend on individual measurements, and the deviation among them is considerably large. The solubility of water in liquid CO2 is approximately on the order of 10−3 in mole fraction at 280 K around a pressure of 10 MPa, which is one order lower than that of CO2 in liquid water.24,25 A serious concern that has not been fully addressed is the influence of hydrate formation on the solubility. Liquid CO2 coexists with water in the high-temperature and low-pressure region, while it coexists with CO2 hydrate in the low-temperature and high-pressure region. We have developed a method to evaluate the solubility under the coexistence of hydrate taking into account the change in the chemical potential of water due to the formation of hydrate.17 We expect that an application of this method to an extended range of thermodynamic conditions could be useful for the practical transportation and storage of liquid CO2 containing water.
In the present study, we address the following three issues. First, the solubility of water in liquid CO2 is calculated in the presence or absence of hydrate. Our calculations are performed in a temperature range between 220 and 325 K and at pressures of 5, 10, 20, 30, 50 and 100 MPa. In contrast to our previous study,17 this range includes temperatures and pressures above the critical point of CO2. This extent of the coverage is essential since some operations in CO2 transportation would be made under such conditions to avoid bubbling. Next, we examine various properties of CO2 hydrate that affect the thermodynamic properties of the coexisting phases and compare them with those of CH4 hydrate obtained in our previous studies.14–16 Finally, we investigate the amount of CO2 hydrate formed from a supersaturated state prepared by rapid cooling or decompression along with the corresponding formation Gibbs energy of the hydrate.
Theory and methods
Since we have presented most of the methods to calculate the phase behaviours of CO2 hydrate and the solubility of water in our previous papers,14–18 they are described below only briefly. A CO2 hydrate coexists with either an aqueous solution or a CO2-rich liquid. The compositions in the aqueous solution and liquid CO2 are denoted by 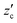 (mole fraction of CO2) and
(mole fraction of CO2) and 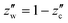 (mole fraction of water), respectively. Those in CO2 hydrate are represented by the mole fraction of CO2 as
(mole fraction of water), respectively. Those in CO2 hydrate are represented by the mole fraction of CO2 as 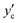 on the phase boundary between the aqueous solution and the hydrate (water/hydrate boundary) and as
on the phase boundary between the aqueous solution and the hydrate (water/hydrate boundary) and as 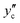 on the phase boundary between the hydrate and liquid CO2 (hydrate/CO2 boundary).
on the phase boundary between the hydrate and liquid CO2 (hydrate/CO2 boundary).
Thermodynamic conditions for hydrate formation
The phase behaviour of CO2 hydrate is represented in the space of temperature, T, pressure, p, and composition, y. The original vdWP theory was proposed to calculate the dissociation pressure of hydrate at the three-phase equilibrium, where the water, hydrate, and liquid CO2 phases coexist in a certain range of temperature.19 Actually, it is a locus of the intersections between the two two-phase boundaries (water/hydrate and hydrate/CO2) projected onto the T–p plane.12,13 The dissociation pressure for CO2 hydrate is depicted against temperature in Fig. 1. The present theoretical calculation successfully agrees with experimental measurements.26–28 The upper-left area delimited by the curve is the stable zone of the hydrate. The highest temperature of the three-phase coexistence for CO2 hydrate is around 297 K.
 |
| | Fig. 1 Dissociation pressure of CO2 hydrate, by which the CO2 hydrate formation and dissociation zones are separated, obtained from theoretical calculations (solid line). Experimental results are shown by filled circles. | |
The agreement of the dissociation pressures at high pressure is moderate compared with those at low pressures in the range of p < 20 MPa. Although the reentrant character in the experimental observations is reproduced by our theoretical calculations, the deviation from experimental measurement becomes large in the very high-pressure region above 200 MPa. As will be described below, the equilibrium condition is determined using the chemical potential of ice Ih. However, this ice is no longer stable under such high pressures. Thus, it is reasonable to consider that our calculations are sound and at least semi-qualitatively correct at pressures lower than ∼100 MPa.
Phase diagram for binary mixture of water and CO2
In Fig. 2, we display a global phase diagram of the binary mixture of water and CO2 at 10 MPa. This figure shows all the phases in T–y space involved in the present study. The stable phase at the low composition of CO2 is the aqueous solution occupying the left region separated by the blue line. It shrinks to zero below the freezing temperature of water for no incorporation of CO2 in ice. The stable phase in the middle is the hydrate phase, which is not given as a single line but is surrounded by the boundary of the aqueous solution with the hydrate (water/hydrate) and that of the hydrate with the CO2-rich solution (hydrate/CO2). The CO2-rich solution including the gaseous phase, called hereafter liquid CO2, exists at high composition of CO2, though it occupies only a narrow region beyond the red line. When the temperature is higher than the dissociation temperature denoted by the horizontal dotted line, no hydrate forms, and the aqueous phase coexists with liquid CO2 (water/CO2). The hydrate intervenes between the aqueous phase and the liquid CO2 phase below the dissociation temperature. Each phase boundary is determined by the equivalence of the chemical potential of either water or CO2 between the two phases. CO2 is regarded as the solute species in the aqueous solution, while it is treated as the solvent in liquid CO2. The hydrate, guest liquid, aqueous, and ice phases are designated by the superscripts (hy), (lq), (aq), and (ice) in the following description of the phase equilibria.
 |
| | Fig. 2 Phase diagram of the binary mixture of water and CO2 at 10 MPa. The boundaries of the stable phases are represented by a blue line for the aqueous solution, black lines for the hydrate, and red line for liquid CO2. The horizontal dotted line indicates the dissociation temperature of CO2 hydrate. | |
Liquid CO2 in equilibrium with aqueous solution without CO2 hydrate
First, we consider the thermodynamic conditions where T is high and/or p is low, and therefore CO2 hydrate is not formed. Liquid CO2 is in direct contact with the aqueous solution. It is assumed that the chemical potential of water in the aqueous solution, 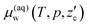 , can be approximated to that of pure water, μ0w(T,p). Likewise, that of CO2 in liquid CO2,
, can be approximated to that of pure water, μ0w(T,p). Likewise, that of CO2 in liquid CO2, 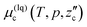 , is assumed to be equal to that of pure liquid CO2, μ0c(T,p). The rationale of these approximations is based on the low mutual solubilities.22–25 The methods to calculate the chemical potentials of the two pure substances are given elsewhere.17 The equilibrium condition for each two-phase coexistence is calculated from
, is assumed to be equal to that of pure liquid CO2, μ0c(T,p). The rationale of these approximations is based on the low mutual solubilities.22–25 The methods to calculate the chemical potentials of the two pure substances are given elsewhere.17 The equilibrium condition for each two-phase coexistence is calculated from| |
μ0j(T,p) = μ(k)j(T,p,z) = kBT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(ρjλj3) + μej(T,p) + f(r)j(T), ln(ρjλj3) + μej(T,p) + f(r)j(T),
| (1) |
where kB denotes the Boltzmann constant and μ(k)j(T,p,z), ρj, λj, f(r)j, and μej(T,p) stand for the chemical potential in solvent k, number density, thermal de Broglie wavelength, free energy of the rotational motion of the rigid model for the solute, and excess chemical potential arising from the interaction of the solute with surrounding solvent molecules for species j. The excess chemical potential, μej(T,p), is calculated assuming infinite dilution of the solute in the solvent of the counterpart species. In the practical calculation, the density of the solute is given by| |
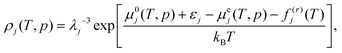 | (2) |
where εj is an energy correction associated with self-polarization. It is set to 7.8 kJ mol−1 for j = w (water) with the TIP4P/ice model and 0 kJ mol−1 for j = c (CO2) with the TraPPE model, accounting for the different environments.17,29,30 The solubility of solute species j in solvent k is| |
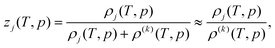 | (3) |
where ρ(k) is the density of the pure liquid k.
CO2 hydrate in equilibrium with either aqueous solution or liquid CO2
Cooling and compression promote the formation of CO2 hydrate. Such a hydrate has the two boundaries in the composition (mole fraction) space at a given T and p. It coexists with the aqueous phase on one boundary at y′ and with the liquid CO2 phase on the other boundary at y″.
The water/hydrate boundary is obtained from the equivalence of the chemical potentials of water in the aqueous solution and in the hydrate as
| |
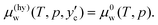 | (4) |
The chemical potential of water changes by Δ
μ(hy)w while traversing from the water/hydrate boundary fixed by
eqn (4) to the hydrate/CO
2 boundary. The chemical potential of water on the hydrate/CO
2 boundary is given by
| |
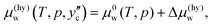 | (5) |
which should be equal to
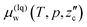
.
13,16
On the hydrate/CO2 boundary, the chemical potential of CO2 in the hydrate equals that in the liquid CO2 phase. The composition in the hydrate on this boundary is calculated from
| |
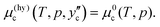 | (6) |
The chemical potential of CO
2 changes by Δ
μ(hy)c due to the decrease in composition from the hydrate/CO
2 boundary determined by
eqn (6) to the water/hydrate boundary. The chemical potential of CO
2 on the water/hydrate boundary is given as
| |
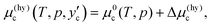 | (7) |
which should be equal to
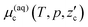
.
17
Eqn (5) and (7) have the same form. Therefore, the density of CO2 in the liquid water phase and the density of water in the liquid CO2 phase in the presence of the hydrate are calculated using the common form given by
| |
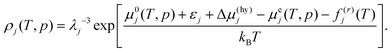 | (8) |
Excess chemical potential of water in liquid CO2
The excess chemical potential of solute species j is calculated according to the particle insertion31 as| |
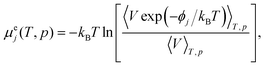 | (9) |
where ϕj is the interaction potential energy of a randomly inserted solute molecule with all the surrounding solvent molecules and V indicates the volume of the pure solvent. A molecular dynamics (MD) simulation is performed for 5 to 110 ns to generate molecular coordinates at each T and p for liquid water and CO2.32–34 The numbers of water and CO2 molecules are set to 1024 and 864, respectively. The number of solute insertions is maximally 8 × 1012 for each thermodynamic condition. All the intermolecular interactions are smoothly truncated at 1.2 nm by multiplying a switching function35 so that the lattice dynamics for the free energy calculation of the host hydrate and ice36 are performed without suffering from the discontinuities associated with the abrupt cutoff of the interaction.
Composition of CO2 hydrate
The vdWP theory19 establishes a relation between the occupancy of a cage of type j, xj, and the chemical potential of water in hydrate, μ(hy)w(T,p,y), with the aid of the chemical potential of water in empty hydrate, μ0h(T,p), as| |
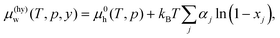 | (10) |
where αj is the ratio of the number of j type cages to the number of water molecules. This is applicable even to a thermodynamic condition where only the hydrate exists as a stable phase.12–18 The occupancy is calculated according to| |
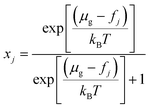 | (11) |
where μg and fj stand for the chemical potential of the guest in the hydrate and the free energy of occupation of the type j cage, which is either large 14- or small 12-hedron for CS-I CO2 hydrate. The free energy of cage occupation, fj, for the linear rigid CO2 molecule is given by| |
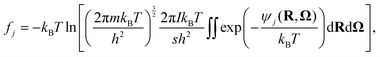 | (12) |
where m and I are the mass and the moment of inertia, h is the Planck constant, and s is the symmetry number, which is 2 for CO2. In this equation, ψj(R,Ω) represents the interaction energy of the guest CO2 molecule at position R and orientation Ω with all surrounding water molecules. The free energy of cage occupation is significantly affected by the host–guest interaction and the shape of the cage. A set of occupancies, xj, is connected with the composition, y, as| |
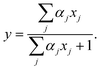 | (13) |
One of the significant differences from the original vdWP theory is that the chemical potential of guest species in the hydrate is calculated from eqn (13) combined with eqn (11) via the occupancies for each given composition, y. The chemical potential of the guest species in eqn (11) is substituted with that in its pure fluid state to determine the hydrate/CO2 boundary using eqn (10).12,13,29
Chemical potentials of ice, empty hydrate, and liquid water
The chemical potential of occupied hydrate is calculated according to eqn (10) once that of the empty hydrate and the occupancies are given. The free energy of empty hydrate, A(T,V,Nw), is assumed to be the sum of the cohesive energy, Uq(V,Nw), the harmonic vibrational free energy, FH(T,V,Nw), and the residual entropy term, TSr(Nw), given as18| | |
A(T,V,Nw) = Uq(V,Nw) + FH(T,V,Nw) − TSr(Nw).
| (14) |
The harmonic vibrational free energy is calculated from a set of the frequencies, νj, as| |
 | (15) |
where 〈〉 indicates the average over the generated hydrogen-disordered structures. The residual entropy does not affect the phase boundary since it is a function of only Nw. To obtain the equilibrium volume, 〈V〉, the following function is minimized with respect to V:| | |
Y(T,V,Nw,p) = A(T,V,Nw) + pV.
| (16) |
The chemical potential of empty hydrate, μ0h(T,p), is simply calculated as| |
 | (17) |
The same method is used to calculate the chemical potential of ice Ih. The anharmonic free energies of ice and hydrate are likely to be comparable. Since this component cancels out in eqn (4), we exclude it from eqn (14).
An extension to higher temperatures beyond the melting point of ice is required. The chemical potential of pure liquid water, μ0w(T,p), at temperature T = Tm + ΔT and pressure p can be estimated as
| |
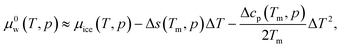 | (18) |
where Δ
s(
Tm,
p) and Δ
cp(
Tm,
p) are the differences in the entropy and heat capacity per molecule at constant pressure between ice and liquid water. Those properties are rather easily calculated from MD simulations.
18
The phase boundary between water and hydrate is found by equating the chemical potential of water in hydrate in eqn (10) with that of ice (or liquid water) according to eqn (4). The occupancy at this equilibrium is calculated from eqn (11) and (13). The other boundary between hydrate and guest species is given by the equivalence of the chemical potential of the guest species according to eqn (6).
Intermolecular interactions and structures of hydrate and ices
A hydrate is composed of host water and guest molecules. The water–water interaction is described by the TIP4P/ice model,29 which is superior in reproducing the melting temperature of ice Ih and several properties of hydrates.37,38 We adopt the TraPPE model for CO2,30 which is a rigid rotor with three interaction sites, and the united-atom OPLS model for CH4.39 The capability of this CO2 model to recreate the phase diagram around the triple point gives a rationale for its use under CO2-rich conditions instead of other models.40,41 Those parameters are listed in Table 1. A deviation from the Berthelot rule is introduced for the Lennard-Jones (LJ) interactions of the unlike pairs of molecules,13,16,42–44 while the Lorentz rule is always applied. A scaling factor, χ, is multiplied to the LJ energy parameter value from the Berthelot rule to recover the experimental dissociation pressure around 273 K.
Table 1 Potential models for CO2 and CH4. Partial charge (q), LJ size parameter (σ), and LJ energy parameter (ε) used in the models. A scaling factor, χ, is multiplied by the value obtained from the Berthelot rule for interactions with the oxygen atom of water. The C–O distance is fixed to 0.1160 nm
| Site |
q/e |
σ/nm |
ε/kJ mol−1 |
χ |
| C |
0.7000 |
0.2800 |
0.2245 |
1.13 |
| O |
−0.3500 |
0.3050 |
0.6568 |
1.13 |
| CH4 |
0.0 |
0.3730 |
1.231 |
0.980 |
One hundred hydrogen-disordered CS-I structures are generated using the GenIce tool45,46 for the calculation of the free energy and other properties of the hydrate. The CS-I structure consists of 368 water molecules. The generated configurations satisfy the ice rules having no net polarization.47,48 The chemical potentials of ice, empty hydrate, and liquid water were calculated previously.12–17
Results and discussion
Solubilities of CO2 in water and water in liquid CO2
In Fig. 3, we show the solubility of CO2 in the aqueous phase and the solubility of water in the liquid CO2 phase at 10 MPa. The aqueous phase is assumed to be in equilibrium with the hydrate or liquid CO2 in the whole range of temperature examined here, while the liquid CO2 is assumed to be in equilibrium with the hydrate or aqueous solution. Thus, each curve is extended to the range of the metastable state.17 As the temperature increases, the solubility curve shifts from the solid one to the dotted one at the crossover, the three-phase coexistence condition, indicated by the arrow. The agreement with the experimental measurements22 is excellent in the solubility of water in liquid CO2 once an appropriate value of the self-polarization energy of the TIP4P/ice29 model, εw = 7.8 kJ mol−1, is chosen.17 The experimental values at 0.1 MPa24,25 are compared to the theoretical ones in the inset of Fig. 3. The solubility of CO2 is reproduced within the range of accuracy required for our later discussion.
 |
| | Fig. 3 Solubility of CO2 in the aqueous solution (red) and that of water in the liquid CO2 (blue) in the presence (solid line) and absence (dotted line) of CO2 hydrate plotted against temperature at 10 MPa. The crossover of the coexistence curve is indicated by the arrow. Experimental values of the solubility of CO2 at 0.1 MPa (cross) are compared to the theoretical ones (open circle and dotted line) in the inset. The magenta open circles are experimental values of the solubility of water at 10.1 MPa.22 | |
The solubility of CO2 in liquid water coexisting with liquid CO2 decreases with rising temperature. This behaviour is commonly observed in the solubility of a solute substance having a low boiling point. However, the solubility increases with rising temperature when the aqueous solution coexists with CO2 hydrate. The crossover of the solubility curves against temperature has also been observed experimentally.49 The difference in the solubility between the dotted and solid curves arises from the decrease in the chemical potential of CO2 associated with traversing from the hydrate/CO2 boundary to the water/hydrate boundary in composition space, Δμ(hy)c in eqn (7).13,16 From a qualitative viewpoint, this is accounted for simply by the fact that the mole fraction of CO2 on the water/hydrate boundary is lower than that on the hydrate/CO2 boundary. As for the solubility of water in liquid CO2, the chemical potential of water on the water/hydrate boundary is higher, and therefore the solubility of water in the presence of the hydrate is lower than the value in the absence of the hydrate. A more quantitative argument on the difference in the solubility between CO2 and water will be made below in relation to the phase behaviours of the CO2 hydrate.
The solubility curves excluding the metastable regions are depicted in Fig. 4a. The agreement with experiments at 10.1 MPa is remarkably good, as shown previously.17,22 The solubility of CO2 in the absence of hydrate considerably depends on temperature. Formation of the hydrate not only changes the sign of the slope against temperature but also decreases the magnitude of the pressure dependence, as seen in Fig. 4a.16 This trend is also observed in experimental measurements.50 On the other hand, the formation of hydrate alters to some extent the slope of the solubility curve of water against temperature.17 Compression leads to a higher solubility of water even in the presence of hydrate.22,51
 |
| | Fig. 4 (a) Solubilities of CO2 in liquid water (upper) and water in liquid CO2 (lower) at 10 (blue), 30 (cyan), and 100 (red) MPa in the presence (solid line) and absence (dashed line) of CO2 hydrate plotted against temperature along with those obtained from experimental measurements at 10.1 MPa (magenta circle).22 (b) Solubilities of water in liquid CO2 with error bars at the same pressures along with those obtained from the fitting of experimental data (dashed line).23 | |
The standard deviations are calculated for the solubilities of water in CO2 by dividing a set of excess chemical potential values into 10 blocks. The error bars are shown in Fig. 4b. The standard deviations are fairly large at 100 MPa, which arises from the fact that particle insertion is difficult for very condensed liquids. On the other hand, those at low pressures such as 10 MPa are small. Fig. S1 demonstrates that our MD simulations are sufficiently long to obtain statistically reliable results even near the critical point where the molar volume fluctuates to a large extent.
It is appropriate to compare our results with solubility curves obtained from the fitting of accumulated experimental measurements.23 This comparison is made in Fig. 4b. The results are not in good agreement with ours unlike those by Seo et al.22 However, it is noted that the individual measurements used for the fitting differ from each other. In fact, the solubility values at 285 K and 10 MPa vary among sources: approximately 2.8 × 10−3 in the present study and ref. 22, 2.0 × 10−3 in ref. 23, and 2.6 × 10−3 in ref. 52 where an equation of state was used to calculate the solubility. This inevitably leads to a fairly large error. Therefore, we must be satisfied with the situation that the general features of the temperature and pressure dependence are at least semi-qualitatively reproduced. Another key issue in the fitting method is that the break in the solubility curve resulting from hydrate formation is effectively averaged out within a single smoothed curve.
Nonstoichiometric properties of CO2 hydrate
We examine some properties of hydrates which affect the solubility curve due to the non-stoichiometric nature characteristic to hydrates. Here, we compare our results for CO2 hydrate with those for CH4 hydrate. While CH4 hydrate was examined in our previous work14,15 using the OPLS model,39 the results for CO2 are newly obtained using the TraPPE model,30 which is capable of reproducing the phase behaviour of pure CO2 and therefore is advantageous to describing liquid CO2 containing a tiny amount of water.
One of the intriguing properties representing the non-stoichiometry of hydrates is the number of water molecules per guest species, called the hydration number. It has often been explored in studying the composition of hydrates.53,54 The pressure and temperature dependences of the hydration numbers along the three-phase coexistence curve are depicted for CO2 and CH4 hydrates in Fig. 5. The hydration number of CO2 hydrate is generally large. It decreases with compression for CO2 hydrate, while it has a peak around 3 MPa for CH4 hydrate. While the hydration number of CO2 hydrate relies heavily on the condition (temperature or pressure), that of CH4 hydrate is rather constant.53 The three-phase coexistence pressure of CO2 hydrate increases with temperature significantly around 280 K, as shown in Fig. 1. High pressure inevitably leads to encapsulation of the small cages. This results in the abrupt drop of hydration number in CO2 hydrate shown in Fig. 5.
 |
| | Fig. 5 Hydration numbers of (red) CO2 and (blue) CH4 hydrates along the three-phase coexistence curve as a function of (a) pressure and (b) temperature. | |
CO2 hydrate forms spontaneously in the upper-left area of the dissociation curve in Fig. 1. When the pressure is fixed at a certain value, the hydrate is in equilibrium with either the aqueous phase or the liquid CO2 phase depending on the composition. The aqueous solution coexists on the water/hydrate boundary while the liquid CO2 phase does on the hydrate/CO2 boundary. These two types of boundaries in temperature (T)–composition (y) space are depicted in Fig. 6a, and the corresponding results for CH4 hydrates are shown in Fig. 6b. The size of the stable region of CO2 hydrate in the T–y plane is comparable to that of CH4 at low pressures. The shape of the region, however, differs from that of CH4 and looks rather like that of C2H6 hydrate.13,18 The area increases with increasing pressure due to the gradual encapsulation of CO2 molecules in the small cages, and, therefore, the extent of increase by compression is more pronounced than that for CH4 hydrate. The composition of the enclosed region for CO2 hydrate calculated with the TraPPE model14,16,30 is lower than that estimated from a different potential model.55 The water/hydrate boundary curve has a break caused by the melting of ice except for at 1 MPa. The formation of hydrate rather than ice is a practical concern for flow assurance in the high-pressure region having the break.
 |
| | Fig. 6 Phase boundaries of (a) CO2 and (b) CH4 hydrates with the guest fluid (solid line) and water (dashed line) on the temperature–composition diagram at 1 (blue), 3 (cyan), 10 (green), 30 (orange) and 100 (red) MPa. The green bidirectional arrow denotes the composition range where the CO2 hydrate alone is stable at 273 K and 10 MPa. | |
The phase diagram shown in Fig. 6a is helpful to explore the origin of the change in the solubility upon formation of CO2 hydrate below the dissociation temperature, as represented in Fig. 3. The chemical potential of water decreases moving from the water/hydrate boundary to the hydrate/CO2 boundary along the green arrow in Fig. 6, i.e., Δμ(hy)w < 0 in eqn (5). The chemical potential of CO2 decreases (Δμ(hy)c < 0 in eqn (7)) moving in the opposite direction. The magnitudes of the changes in the chemical potentials are constrained by the Gibbs–Duhem equation as
| |
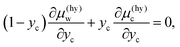 | (19) |
which implies that
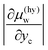
is smaller than
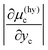
because
yc is smaller than 0.15 for CS-I hydrate, ruling out the multiple occupancy of cages. This relation, together with
eqn (8), explains why the change in the solubility of water upon formation of hydrate is smaller than that of CO
2. In fact, the difference in the chemical potentials between the two boundaries for CO
2, |Δ
μ(hy)c|, is several times larger than that for water, |Δ
μ(hy)w|, as tabulated in
Table 2. The chemical potential difference plays a decisive role in determining the magnitude of the effect of hydrate formation on the solubility.
Table 2 Chemical potential differences between the two boundaries for CO2, Δμ(hy)c, and for water, Δμ(hy)w, at 10 MPa
| T/K |
Δμ(hy)c/kJ mol−1 |
Δμ(hy)w/kJ mol−1 |
| 273 |
−2.03 |
−0.32 |
| 277 |
−1.25 |
−0.22 |
| 281 |
−0.44 |
−0.11 |
The composition of a hydrate is associated with partial filling of each cage. The occupancies for the individual cage types are calculated according to eqn (11). The occupancy substantially depends on the cage types, guest species, and thermodynamic conditions, as depicted in Fig. 7. Compression and/or cooling enhance the occupancies. The occupancies at two pressures for the large cage in CO2 hydrate nearly overlap each other. The large cages are more preferentially occupied in both CO2 and CH4 hydrates, although the magnitudes of the preference are different. While the large cages are almost fully occupied in CO2 hydrate so as to lower the chemical potential of water according to eqn (10), the small cages are of little use to stabilise the hydrate. This is in sharp contrast to the stabilization mechanism of CH4 hydrate, where both the large and small cages contribute to decrease the chemical potential of water in the hydrate.
 |
| | Fig. 7 Temperature dependence of cage occupancies for (a) CO2 and (b) CH4 hydrates at 10 (cyan: large, magenta: small) and 100 (blue: large, red: small) MPa. The solid and dashed lines represent the hydrate/guest and water/hydrate boundaries, respectively. | |
The preferential occupation of the large cages by CO2 molecules is substantiated by the orientationally averaged interaction energy of a guest with the surrounding water molecules, 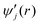 , against the radial distance, r, from the cage center defined as
, against the radial distance, r, from the cage center defined as
| |
 | (20) |
It is depicted in
Fig. 8a, along with the resultant temperature dependences of the free energies of cage occupation in
Fig. 8b. It is evident that a CO
2 molecule in the small cage has a higher interaction energy than that in the large cage due to its large molecular size. This leads to a much higher free energy of cage occupation, as shown in
Fig. 8b. A CH
4 molecule in a small cage is stabilized by the surrounding water to a similar extent as that in large cage. Consequently, the difference in the free energy of occupation between them is small.
 |
| | Fig. 8 (a) Orientationally averaged interaction energy of a guest with surrounding water molecules plotted against the radial distance from the cage center. (b) Free energies of cage occupation of (red) CO2 and (blue) CH4 for the (solid line) large and (dotted line) small cages. | |
Precipitation of CO2 hydrate under rapid cooling or decompression
Our main concern is the phase behaviour of liquid CO2 with an impurity of water, which corresponds to the region near zc = 1 in Fig. 2. The magnified temperature–composition diagram is depicted in Fig. 9. The diagram is divided into three regions, liquid (including gaseous) CO2 with a small amount of dissolved water, liquid CO2 coexisting with the hydrate (hydrate/CO2), and liquid CO2 coexisting with the aqueous solution (water/CO2). The regions are separated by the dotted line corresponding to the dissociation temperature of the hydrate and by the solid line corresponding to the solubility of water. The phase diagram is also shown in Fig. S2 for each pressure.
 |
| | Fig. 9 Magnified phase diagram of the binary mixture of water and CO2 for a region close to pure CO2 at 5 (blue), 10 (cyan), 20 (green), 30 (orange), and 100 MPa (red). The stable region of fluid CO2 is separated by the solid curve. Liquid CO2 coexists with either the aqueous solution or CO2 hydrate in the left region of each solid curve. Each dotted line indicates the dissociation temperature above which no hydrate is stable and the CO2 fluid is in equilibrium with the aqueous solution. The blue dot-dash line depicts the interpolated solubility curve taking into account the breaks due to the vaporization of liquid CO2 at 5 MPa. The magenta crosses denote the experimental solubilities at 10.1 MPa.22 | |
The liquid CO2 phase is in equilibrium with the aqueous solution at higher temperatures in the left region separated by the solubility curve in Fig. 9. The solubility of water in liquid CO2, 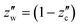 , decreases with decreasing temperature. Compression generally gives rise to a higher solubility of water. The curve at 10 MPa is nearly vertical around 310 K. This behaviour originates from the critical point of CO2 (304.2 K and 7.38 MPa) around which the density changes strongly. At 5 MPa, CO2 undergoes the phase transition from the liquid state to the gaseous one. This accompanies a sudden decline of the solubility, as illustrated in Fig. 9 where these two adjacent points are smoothly connected by the solid curve but the phase transition is represented by the dash-dot curve. The solubility of water in the gaseous CO2 phase is the vapor pressure of pure liquid water to a good approximation and therefore is expected to decrease with compression, according to the present definition of the solubility given by eqn (3), through an increase in ρ(k).
, decreases with decreasing temperature. Compression generally gives rise to a higher solubility of water. The curve at 10 MPa is nearly vertical around 310 K. This behaviour originates from the critical point of CO2 (304.2 K and 7.38 MPa) around which the density changes strongly. At 5 MPa, CO2 undergoes the phase transition from the liquid state to the gaseous one. This accompanies a sudden decline of the solubility, as illustrated in Fig. 9 where these two adjacent points are smoothly connected by the solid curve but the phase transition is represented by the dash-dot curve. The solubility of water in the gaseous CO2 phase is the vapor pressure of pure liquid water to a good approximation and therefore is expected to decrease with compression, according to the present definition of the solubility given by eqn (3), through an increase in ρ(k).
The Gibbs energy difference between the reactants (water and CO2) and the product (hydrate) plays an essential role in determining the rate of hydrate formation. It is also known as the thermodynamic driving force for the formation of hydrate and has been calculated with available thermodynamic properties from MD simulations.37,38 We have shown that it can also be calculated in a relatively simple way from the chemical potentials, which are available from the method we developed.16 The Gibbs energy of formation in our definition is measured on a per water molecule (mole) basis, reflecting the non-stoichiometric nature of hydrate. As stated above, we make a slightly rough approximation for the chemical potentials of water and CO2 such that water in hydrate on the water/hydrate boundary has the same chemical potential of pure water, although it contains a certain amount of CO2, and the guest species in the hydrate on the hydrate/CO2 boundary has the same chemical potential of pure CO2. The Gibbs energy of formation at a composition of yc is given as
| |
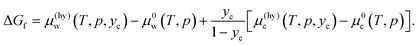 | (21) |
This quantity converges to zero on approaching the three-phase equilibrium. There are two extreme cases for the composition of the product hydrate,
yc. In the first one, we consider formation of the hydrate under a water-rich condition,
i.e., on the water/hydrate boundary where the composition of the guest in the hydrate is
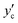
. The Gibbs energy of formation is given by
| |
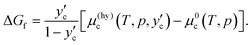 | (22) |
This is plotted against temperature in
Fig. 10. In the second, the hydrate is assumed to be formed under a guest-rich condition on the hydrate/guest boundary at a composition of
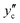
. The Gibbs energy of formation is given by
| |
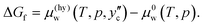 | (23) |
This is also shown in
Fig. 10. It differs only slightly from that found according to
eqn (22).
 |
| | Fig. 10 Gibbs energy of formation of CO2 hydrate versus temperature at a pressure of 10 MPa on the hydrate/CO2 (red) and water/hydrate (blue) boundaries. | |
Thus far, the initial state was chosen to be pure water and CO2 at a given temperature and pressure. We can consider a different formation process in which the initial state is liquid CO2 supersaturated with water prepared by a temperature drop. This process is schematically drawn in Fig. 11. The temperature is dropped vertically from a certain point on the phase boundary to a point below the dissociation temperature of CO2 hydrate. Consequently, an excessive amount of water precipitates as CO2 hydrate.
 |
| | Fig. 11 Isobaric cooling in the temperature–composition diagram. The red solid and dotted lines indicate the phase boundary of liquid CO2 and the dissociation temperatures of CO2 hydrate. The temperature is dropped along the vertical line from a point on the red solid curve to an arbitrary temperature, T in eqn (24). | |
The liquid CO2 at the initial state contains a certain amount of saturated water whose density is ρw or an equivalent composition of the corresponding solubility, zc. Since μ0w(T,p) in eqn (23) should be replaced with μ(lq)w(T,p,zc) in this process, the Gibbs energy of formation is calculated from 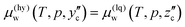 as
as
| |
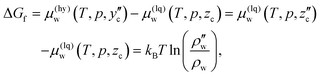 | (24) |
where
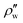
is the equilibrium density of water in liquid CO
2 coexisting with CO
2 hydrate at
T and
p.
This Gibbs energy of formation, the sign of which is limited to negative, is depicted as a function of the degree of cooling in Fig. 12 for (a) the initial temperature from which liquid CO2 undergoes rapid cooling and (b) the final one to which it will be settled. It decreases as the degree of cooling or equivalently the supersaturation is intensified. In other words, supersaturation significantly induces formation of CO2 hydrate to achieve equilibrium between hydrate and liquid CO2 at the final temperature. The Gibbs energy change is almost linear in both panels. A higher initial temperature requires a deeper cooling to have the same energy value as is seen in panel (a). CO2 hydrate does not appear at 300 or 320 K at a pressure of 10 MPa, and therefore the corresponding curves shift to the left side. The Gibbs energies for the various final temperatures seem to be laid on a common straight line passing through the origin in panel (b) under the fixed final temperature condition.
 |
| | Fig. 12 Gibbs energy of formation of CO2 hydrate plotted against the degree of cooling at 10 MPa for (a) four initial temperatures and (b) three final temperatures. | |
Another intriguing quantity from an industrial viewpoint is the amount of hydrate precipitated from liquid CO2 supersaturated with water. The formation of hydrate incurs the blockage of pipelines in liquid CO2 transportation. The amount of hydrate from one mole of liquid CO2 is plotted in Fig. 13 as a function of temperature drop. Again, we examine it for either (a) the initial temperature from which liquid CO2 is cooled down or (b) the final one to which it will be settled. As plotted in Fig. 13a, the amount of water increases simply with the extent of cooling, but the relation between the amount and the initial temperature at a given degree of cooling is complicated. However, the amount simply decreases with decreasing final temperature at a given degree of cooling, as seen in Fig. 13b.
 |
| | Fig. 13 Amount of wate precipitated as hydrate from one mole of liquid CO2 at 10 MPa, plotted against the degree of cooling for (a) four initial temperatures and (b) three final temperatures. | |
Next, we consider a pressure drop of liquid CO2. This may also happen in a pipeline going downstream. Here, we assume the pressure drop takes place isothermally, as drawn in Fig. 14. We calculate the amount of hydrate precipitated in this process in the same way as in the case of the temperature drop. The amount of precipitation due to decompression is shown in Fig. 15. The amount is small compared with that of the cooling process even when the initial pressure is an order magnitude larger than the final pressure. As is expected from Fig. 9, the precipitation amount becomes larger with increasing temperature.
 |
| | Fig. 14 Isothermal decompression in the temperature–composition diagram. The red and blue solid lines indicate the phase boundaries of liquid CO2. The pressure drops along the horizontal line from an initial point on the red curve to the final point on the blue curve. | |
 |
| | Fig. 15 Amount of water precipitated as hydrate from one mole of liquid CO2 by rapid decompression to a final pressure of 10 MPa, plotted against the initial pressure at 230 (blue), 240 (cyan), 250 (green), 260 (orange), and 270 K (red). | |
Conclusions
The solubility of water in liquid CO2 is calculated using atomistic intermolecular potential models. Liquid CO2 coexists with liquid water and CO2 hydrate above and below the dissociation temperature of the hydrate, respectively. The solubility of water in liquid CO2 increases with increasing temperature, and compression enhances the solubility regardless of the presence or absence of hydrate.
Several properties of CO2 hydrate are examined, focusing on the water/hydrate and hydrate/CO2 two-phase coexistences. Whereas CH4 hydrate is stabilised by moderate occupancy of large cages and the same degree of occupancy of small cages, the stability of CO2 hydrate is mostly due to the preferential occupation of large cages.
CO2 hydrate forms not only by a temperature drop but also by a pressure drop of liquid CO2 containing water as an impurity. The amount is seemingly small but can pose the risk of pipeline blockage. Another important factor for flow assurance is the nucleation kinetics of CO2 hydrate, but that is out of the scope of the present study.
The temperature and pressure ranges differ for each operational condition. The solubility of water and the amount of precipitated hydrate are heavily dependent on temperature and pressure. Hence, the allowable content of water in liquid CO2 may be set flexibly, avoiding overly conservative constraints. The Gibbs energy of formation and the associated properties in the present study are expected to provide a valuable insight into the conditions for safe CO2 transportation without hydrate formation.
Author contributions
Hideki Tanaka: conceptualization, methodology, investigation, interpretation, and writing – original draft. Masakazu Matsumoto: methodology, constructive discussions, writing – review and editing, and funding acquisition. Takuma Yagasaki: methodology, interpretation, constructive discussions, and writing – review and editing. Munetaka Takeuchi: constructive discussions, interpretation, and writing – review and editing. Yoshihito Mori: constructive discussions, interpretation, and writing – review and editing. Takumi Kono: constructive discussions, project administration, and funding acquisition.
Conflicts of interest
The authors have no conflicts to disclose.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request. All relevant figures are included in the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d6cp01072e.
Acknowledgements
The present work is supported by JSPS KAKENHI (Grant Number: 23K20830) and the Research Center for Computational Science for providing computational resources (Project: 23-IMS-C028).
References
- G. Brunner, Gas Extraction, Springer, New York, 1991 Search PubMed.
- S. Angus, B. Armstrong and K. M. de Reuck, International Thermodynamic Tables of the Fluid State – Carbon Dioxide, 1976 Search PubMed.
- Thermophysical properties of fluid systems, NIST Chemistry Webbook (National Institute of Standards and Technology), 2025, https://webbook.nist.gov/chemistry/, (accessed March 16, 2026).
- N. MacDowell, N. Florin, A. Buchard, J. Hallett, A. Galindo, G. Jackson, C. S. Adjiman, C. K. Williams, N. Shah and P. Fennell, Energy Environ. Sci., 2010, 3, 1645–1669 RSC.
- T. Kuznetsova, B. Jensen, B. Kvamme and S. Sjøblom, Phys. Chem. Chem. Phys., 2015, 17, 12683–12697 RSC.
- T. Yagasaki, M. Matsumoto and H. Tanaka, J. Am. Chem. Soc., 2015, 137, 12079–12085 CrossRef CAS PubMed.
- M. A. Kelland, Energy Fuels, 2018, 32, 12001–12012 CrossRef CAS.
- M. Aminnaji, M.-F. Qureshi, H. Dashti, A. Hase, A. Mosalanejad, A. Jahanbakhsh, M. Babaei, A. Amiri and M. Maroto-Valer, Energy, 2024, 300, 131579 CrossRef CAS.
- M. Aminnaji, M.-F. Qureshi, H. Dashti, A. Hase, A. Mosalanejad, A. Jahanbakhsh, M. Babaei, A. Amiri and M. Maroto-Valer, Energy, 2024, 300, 131580 CrossRef CAS.
- M. A. Kelland and J. Pomicpic, Energy Fuels, 2025, 39, 9802–9817 CAS.
- E. D. Sloan and C. A. Koh, Clathrate Hydrates of Natural Gases, CPC Press, Boca Raton, 2008 Search PubMed.
- H. Tanaka, T. Yagasaki and M. Matsumoto, J. Phys. Chem. B, 2018, 122, 297–305 Search PubMed.
- H. Tanaka, T. Yagasaki and M. Matsumoto, J. Chem. Phys., 2018, 149, 074502 CrossRef PubMed.
- H. Tanaka, M. Matsumoto and T. Yagasaki, J. Chem. Phys., 2023, 158, 224502 CrossRef CAS PubMed.
- H. Tanaka, M. Matsumoto and T. Yagasaki, J. Chem. Phys., 2023, 159, 194504 CrossRef CAS PubMed.
- H. Tanaka, M. Matsumoto and T. Yagasaki, J. Chem. Phys., 2024, 161, 214503 CrossRef CAS PubMed.
- H. Tanaka, M. Matsumoto, T. Yagasaki, M. Takeuchi, Y. Mori and T. Kono, J. Chem. Phys., 2025, 163, 124504 CrossRef CAS PubMed.
- T. Yagasaki, M. Matsumoto and H. Tanaka, J. Chem. Phys., 2026, 164, 020901 Search PubMed.
- J. H. van der Waals and J. C. Platteeuw, Adv. Chem. Phys., 1959, 2, 1–57 Search PubMed.
- J. I. Lunine and D. J. Stevenson, Astrophys. J., Suppl. Ser., 1985, 58, 493–531 CrossRef CAS PubMed.
- H. Tanaka and M. Matsumoto, Adv. Chem. Phys., 2013, 152, 421–462 CrossRef CAS.
- M. D. Seo, J. W. Kang and C. S. Lee, J. Chem. Eng. Data, 2011, 56, 2626–2629 CrossRef CAS.
- I. Aavatsmark and R. Kaufmann, Int. J. Greenhouse Gas Control, 2015, 32, 47–55 CrossRef CAS.
- E. Wilhelm, R. Battino and R. Wilcock, Chem. Rev., 1977, 77, 219–262 CrossRef CAS.
- R. Crovetto, J. Phys. Chem. Ref. Data, 1991, 20, 575–589 CrossRef CAS.
- C. H. Unruh and D. L. Katz, Trans. AIME, 1949, 186, 83–86 Search PubMed.
- S. Takenouchi and G. C. Kennedy, J. Geol., 1965, 73, 383–390 CrossRef CAS.
- S. Nakano, M. Moritoki and K. Ohgaki, J. Chem. Eng. Data, 1998, 43, 807–810 CrossRef CAS.
- J. L. F. Abascal, E. Sanz, R. García Fernández and C. Vega, J. Chem. Phys., 2005, 122, 234511 CrossRef CAS PubMed.
- J. J. Potoff and J. I. Siepmann, AIChE J., 2001, 47, 1676–1682 CrossRef CAS.
- B. Widom, J. Chem. Phys., 1963, 39, 2808 CrossRef CAS.
- S. Nosé, Mol. Phys., 1984, 52, 255–268 CrossRef.
- W. G. Hoover, Phys. Rev. A:At., Mol., Opt. Phys., 1985, 31, 1695–1697 CrossRef PubMed.
- H. C. Andersen, J. Chem. Phys., 1980, 72, 2384–2393 CrossRef CAS.
- I. Ohmine, H. Tanaka and P. G. Wolynes, J. Chem. Phys., 1988, 89, 5852–5860 CrossRef.
- A. Pohorille, L. R. Pratt, R. A. LaViolette, M. A. Wilson and R. D. MacElroy, J. Chem. Phys., 1987, 87, 6070–6077 Search PubMed.
- J. Grabowska, S. Blazquez, E. Sanz, I. M. Zerón, J. Algaba, J. M. Míguez, F. J. Blas and C. Vega, J. Phys. Chem. B, 2022, 126, 8553–8570 CrossRef CAS PubMed.
- J. Algaba, I. M. Zerón, J. M. Míguez, J. Grabowska, S. Blazquez, E. Sanz, C. Vega and F. J. Blas, J. Chem. Phys., 2023, 158, 184703 CrossRef CAS PubMed.
- W. L. Jorgensen, J. D. Madura and C. J. Swenson, J. Am. Chem. Soc., 1984, 106, 6638–6646 Search PubMed.
- G. Pérez-Sánchez, D. González-Salgado, M. M. Piñeiro and C. Vega, J. Chem. Phys., 2013, 138, 084506 CrossRef PubMed.
- H. Tanaka, M. Matsumoto, T. Yagasaki, M. Takeuchi, Y. Mori and T. Kono, J. Chem. Phys., 2024, 158, 224502 CrossRef PubMed.
- M. M. Conde and C. Vega, J. Chem. Phys., 2010, 133, 064507 CrossRef CAS PubMed.
- J. M. Míguez, M. M. Conde, J.-P. Torré, F. J. Blas, M. M. Piñeiro and C. Vega, J. Chem. Phys., 2015, 142, 124505 CrossRef PubMed.
- J. Grabowska, S. Blazquez, E. Sanz, I. M. Zerón, J. Algaba, J. M. Míguez, F. J. Blas and C. Vega, J. Phys. Chem. B, 2022, 126, 8553–8564 CrossRef CAS PubMed.
- M. Matsumoto, T. Yagasaki and H. Tanaka, J. Comput. Chem., 2018, 39, 61–64 CrossRef CAS PubMed.
- M. Matsumoto, T. Yagasaki and H. Tanaka, J. Chem. Inf. Model., 2021, 61, 2542–2546 CrossRef CAS PubMed.
- J. D. Bernal and R. H. Fowler, J. Chem. Phys., 1933, 1, 515–548 CrossRef CAS.
- L. Pauling, J. Am. Chem. Soc., 1935, 57, 2680–2684 CrossRef CAS.
- I. Aya, K. Yamane and H. Nariai, Energy, 1997, 22, 263–271 CrossRef CAS.
- P. Servio and P. Englezos, Fluid Phase Equilib., 2001, 90, 127–134 CrossRef.
- A. Chapoy, R. Burgass, B. Tohidi, J. M. Austell and C. Eickhoff, SPE J., 2011, 16, 921–930 CrossRef CAS.
- J. Gernert and R. Span, J. Chem. Thermodyn., 2016, 93, 274–293 CrossRef CAS.
- T. Uchida, Waste Manage., 1997, 17, 343–352 CrossRef CAS.
- A. Hachikubo, M. Kida, D. Yahagi and S. Takeya, Energy Fuels, 2024, 38, 9676–9682 CrossRef CAS.
- S. C. Velaga and B. J. Anderson, J. Phys. Chem. B, 2014, 118, 577–589 CrossRef CAS PubMed.
|
| This journal is © the Owner Societies 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab,
Masakazu Matsumoto
*ab,
Masakazu Matsumoto (mole fraction of CO2) and
(mole fraction of CO2) and  (mole fraction of water), respectively. Those in CO2 hydrate are represented by the mole fraction of CO2 as
(mole fraction of water), respectively. Those in CO2 hydrate are represented by the mole fraction of CO2 as  on the phase boundary between the aqueous solution and the hydrate (water/hydrate boundary) and as
on the phase boundary between the aqueous solution and the hydrate (water/hydrate boundary) and as  on the phase boundary between the hydrate and liquid CO2 (hydrate/CO2 boundary).
on the phase boundary between the hydrate and liquid CO2 (hydrate/CO2 boundary).


 , can be approximated to that of pure water, μ0w(T,p). Likewise, that of CO2 in liquid CO2,
, can be approximated to that of pure water, μ0w(T,p). Likewise, that of CO2 in liquid CO2,  , is assumed to be equal to that of pure liquid CO2, μ0c(T,p). The rationale of these approximations is based on the low mutual solubilities.22–25 The methods to calculate the chemical potentials of the two pure substances are given elsewhere.17 The equilibrium condition for each two-phase coexistence is calculated from
, is assumed to be equal to that of pure liquid CO2, μ0c(T,p). The rationale of these approximations is based on the low mutual solubilities.22–25 The methods to calculate the chemical potentials of the two pure substances are given elsewhere.17 The equilibrium condition for each two-phase coexistence is calculated from



 .13,16
.13,16


 .17
.17














 is smaller than
is smaller than  because yc is smaller than 0.15 for CS-I hydrate, ruling out the multiple occupancy of cages. This relation, together with eqn (8), explains why the change in the solubility of water upon formation of hydrate is smaller than that of CO2. In fact, the difference in the chemical potentials between the two boundaries for CO2, |Δμ(hy)c|, is several times larger than that for water, |Δμ(hy)w|, as tabulated in Table 2. The chemical potential difference plays a decisive role in determining the magnitude of the effect of hydrate formation on the solubility.
because yc is smaller than 0.15 for CS-I hydrate, ruling out the multiple occupancy of cages. This relation, together with eqn (8), explains why the change in the solubility of water upon formation of hydrate is smaller than that of CO2. In fact, the difference in the chemical potentials between the two boundaries for CO2, |Δμ(hy)c|, is several times larger than that for water, |Δμ(hy)w|, as tabulated in Table 2. The chemical potential difference plays a decisive role in determining the magnitude of the effect of hydrate formation on the solubility.

 , against the radial distance, r, from the cage center defined as
, against the radial distance, r, from the cage center defined as


 , decreases with decreasing temperature. Compression generally gives rise to a higher solubility of water. The curve at 10 MPa is nearly vertical around 310 K. This behaviour originates from the critical point of CO2 (304.2 K and 7.38 MPa) around which the density changes strongly. At 5 MPa, CO2 undergoes the phase transition from the liquid state to the gaseous one. This accompanies a sudden decline of the solubility, as illustrated in Fig. 9 where these two adjacent points are smoothly connected by the solid curve but the phase transition is represented by the dash-dot curve. The solubility of water in the gaseous CO2 phase is the vapor pressure of pure liquid water to a good approximation and therefore is expected to decrease with compression, according to the present definition of the solubility given by eqn (3), through an increase in ρ(k).
, decreases with decreasing temperature. Compression generally gives rise to a higher solubility of water. The curve at 10 MPa is nearly vertical around 310 K. This behaviour originates from the critical point of CO2 (304.2 K and 7.38 MPa) around which the density changes strongly. At 5 MPa, CO2 undergoes the phase transition from the liquid state to the gaseous one. This accompanies a sudden decline of the solubility, as illustrated in Fig. 9 where these two adjacent points are smoothly connected by the solid curve but the phase transition is represented by the dash-dot curve. The solubility of water in the gaseous CO2 phase is the vapor pressure of pure liquid water to a good approximation and therefore is expected to decrease with compression, according to the present definition of the solubility given by eqn (3), through an increase in ρ(k).
 . The Gibbs energy of formation is given by
. The Gibbs energy of formation is given by
 . The Gibbs energy of formation is given by
. The Gibbs energy of formation is given by


 as
as
 is the equilibrium density of water in liquid CO2 coexisting with CO2 hydrate at T and p.
is the equilibrium density of water in liquid CO2 coexisting with CO2 hydrate at T and p.



