
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNear-infrared spectroscopy of micro-solvated protonated methanol
Yaodi
Yang†
 a,
Qian-Rui
Huang†
b,
Jer-Lai
Kuo
*b and
Asuka
Fujii
*a
a,
Qian-Rui
Huang†
b,
Jer-Lai
Kuo
*b and
Asuka
Fujii
*a
aDepartment of Chemistry, Graduate School of Science, Tohoku University, Sendai, Japan. E-mail: asuka.fujii.c5@tohoku.ac.jp
bInstitute of Atomic and Molecular Sciences, Academia Sinica, Taipei, Taiwan. E-mail: jlkuo@pub.iams.sinica.edu.tw
First published on 20th April 2026
Abstract
Gas phase near-infrared (NIR) spectra of micro-solvated protonated methanol, MeOH2+⋯Xn (X = Ar, N2, and CO; n = 1 and 2), in the region of the first overtones of OH stretches were obtained via infrared photodissociation spectroscopy. Spectral details were analyzed with the support of high-precision ab initio anharmonic simulations. The free OH stretching overtones and combination bands of OH stretch and bend are the main features in this region. The hydrogen-bonded OH stretching overtone transitions exhibit weak and broad characteristics across all solvents (X). This trend increases with the proton affinity of X. Hot bands, intramolecular vibrational energy redistribution, and band congestion due to strong anharmonic couplings are proposed to have played crucial roles in this observation. Meanwhile, perturbations from low frequency modes, such as CH/OH rocking, CH bending, and solvent intramolecular vibrations, should not be overlooked in this cluster system. Our anharmonic algorithm has demonstrated its potential in reproducing precise NIR spectra, and the present system can serve as a benchmark for theoretical anharmonic computations in the NIR region.
Introduction
Near-infrared (NIR) spectroscopy, which directly probes higher vibrational levels, provides valuable and unique information on phenomena such as anharmonic couplings, the shape of the potential energy surface, and intramolecular vibrational energy redistribution (IVR).1–8 Gas phase molecular clusters, on the other hand, are well-defined systems that are highly useful for fundamental modeling of complex systems.9 Charged clusters can be readily size-selected by mass spectrometry and are often analyzed in combination with infrared (IR) spectroscopy to investigate their geometric and vibrational structures.9 However, applying NIR spectroscopy to size-selected gas phase clusters has been very limited. This is primarily due to the extremely weak transition intensities in this region.Furthermore, combination bands (CBs), overtones, and other vibrational couplings that arise from anharmonicities of clusters present additional challenges for spectral interpretation in the NIR region. Experimental vibrational spectra of gas phase clusters serve as direct benchmarks for high-precision computational chemistry, which in turn provides theoretical basis for elucidating observed spectral features. Although the theoretical modeling of NIR spectra can be traced back to the mid-twentieth century,10 reliable and in-depth analyses only become possible with the advent of high-precision anharmonic vibrational methods. Despite these advancements, reproducing NIR spectra still demands significant computational efforts, even for very simple molecular systems.
Recent advances in anharmonic vibrational approaches have enabled qualitative and even quantitative anharmonic analyses of small-sized protonated systems, such as protonated water, ammonia, alcohol, and amine clusters in the mid-infrared (MIR) region.11–31 These protonated hydrogen-bonded (H-bonded) systems have long been one of the major topics in gas phase vibrational spectroscopy, owing to the ubiquity and significance of proton solvation and the need for its microscopic understanding.
There have also been some efforts on NIR spectroscopy of protonated and/or neutral H-bonded species in the gas phase, including trials to verify the compatibility of anharmonic algorithms in the NIR region.32–39 McDonald et al. reported the NIR spectra of protonated water clusters, H+(H2O)n (n = 4–8), and Ar-tagged protonated water clusters, H+(H2O)n⋯Ar (n = 1–4), using infrared photodissociation (IRPD) spectroscopy in the frequency range of 3600–7300 cm−1.35 While the first overtones of the Ar-bound OH stretches were clearly visible in the Ar-tagged water clusters, overtones of the water-bound OH stretches were either very weak or entirely absent. In the case of protonated water dimer, the water-bound OH stretching overtone is expected to show an exceptionally low frequency transition owing to its symmetrically shared proton structure, making it understandably difficult to detect.40,41 However, such transitions of the clusters with n > 2 were still expected to be observed in the region probed by McDonald et al. From a micro-solvation perspective, the H2O molecules and the Ar messenger act as solvents filling the first solvation shell of the central H3O+. Since H2O is a much stronger proton acceptor compared to Ar, the effect of tuning the solvation environment on the OH stretching overtones of the solvated cation has emerged as a focal point for us.
Previously, we measured the NIR spectra of micro-solvated protonated water clusters, H3O+⋯Xn (X = Ar, N2 and CO; n = 1–3) by IRPD spectroscopy.37 Detailed spectral features were analyzed with the guidance of high-precision anharmonic vibrational calculations.37 The observed spectra of these clusters broadened significantly as proton affinity (PA) of the binding solvent increased (with PA following the order Ar < N2 < CO < H2O), regardless of the cluster size (n).
To test the generality of these findings, we plan to shift our focus to protonated methanol (MeOH2+). As an analog to the protonated water system, the spectra of protonated methanol are expected to show similar spectral patterns under the similar solvation environment. However, there are some key differences. The presence of only two OH bonds in a protonated methanol should, in principle, make its spectrum less complex compared to protonated water, which can act as a proton donor to form up to three OH⋯X H-bonds with surrounding solvent species. On the other hand, based on our previous work with MIR spectra of micro-solvated clusters of protonated methanol and protonated methylamine,27,31 the methyl group on MeOH2+ is expected to cause a non-negligible perturbation to the spectrum through the methyl-hydroxyl rocking motions.
In this work, we continue to use the same solvent species (X = Ar, N2 and CO) that were used for our previous study on the H3O+⋯Xn clusters. By varying the solvent species and the number of binding solvents (n = 1 and 2), we aim to evaluate the capability of our ab initio anharmonic algorithm for overtone analyses and to uncover the anharmonic coupling patterns in the NIR region of the MeOH2+⋯Xn clusters.
Experimental setup
The protonated methanol clusters were produced by the collision of an electron beam with a pulsed supersonic jet expansion of a gaseous mixture containing methanol, solvent X (X = Ar, N2 and CO), and carrier gas (Ar or He). An Even-Lavie pulsed valve,42 operating at a stagnation pressure of 80 atm, was used for the jet expansion. Such a high operating pressure facilitates more collisions, which ensures effective cooling of the weakly-bound clusters. Parent ions (MeOH2+⋯Xn) were mass-selected by the first quadrupole mass analyzer of a tandem type mass spectrometer. Subsequently, they were transferred into an octupole ion guide, in which vibrational predissociation occurred following exposure to a pulsed beam of tunable NIR light. The NIR light source used in this work was the output of an Nd:YAG laser (Spectra Physics GGR230) pumped IR-OPO/OPA (LaserVision). The resulting fragment ions (MeOH2+⋯Xn−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) orn−2), which had lost one or two solvent molecules, were selected at the second quadrupole mass analyzer before being detected by an ion detector at the end of the mass-spectrometer. The signal difference of the fragment ions, measured with and without irradiation of the NIR light was monitored, power-normalized, and plotted against scanned frequency for the dissociation spectrum. A more detailed description of the experimental setups can be found elsewhere.43
orn−2), which had lost one or two solvent molecules, were selected at the second quadrupole mass analyzer before being detected by an ion detector at the end of the mass-spectrometer. The signal difference of the fragment ions, measured with and without irradiation of the NIR light was monitored, power-normalized, and plotted against scanned frequency for the dissociation spectrum. A more detailed description of the experimental setups can be found elsewhere.43
Computational details
Geometric structures of the protonated methanol clusters, MeOH2+⋯Xn (X = Ar, N2 and CO), were optimized at the MP2/aug-cc-pVDZ level using Gaussian 16.44 Cartesian coordinates of the optimized structures are provided in Table S1 of the SI. The number of plausible isomers for small-sized clusters is limited. Only one low energy isomer was found for each individual cluster in this work, where each OH bond is solvated by a single solvent molecule. Other isomers, e.g., those in which a solvent molecule dimer is bound to one of the OH bonds, are much higher in energy, and their contribution to the NIR region is negligible.All vibrational calculations were carried out at the MP2/aug-cc-pVDZ equilibrium geometry. The partial Hessian vibrational analysis (PHVA) was used to obtain localized normal modes (LNM) for reduced-dimensional vibrational analysis.45 These LNMs provide a fragment-focused representation of the vibrational motions which allows us to isolate the most relevant motions for high-level anharmonic analysis. The applicability of this approach has been demonstrated in our previous studies, including the analysis of the MIR spectra of MeOH2+···Xn.31,46,47 Here, the same strategy is adopted. The LNMs used in this work are summarized in Table S2.
A subset of vibrational modes was selected to explicitly describe the NIR spectral region of interest. These include 2 OH stretching modes (s), 1 HOH bending mode (b), 1 or 2 intermolecular stretching modes (t), 4 CH/OH rocking motions (R), and, when applicable, the intramolecular stretching modes (sX) of the solvent molecule (N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N or CO). The selected localized normal modes of MeOH2+⋯N2 used in the calculations are visualized in Fig. S1 as examples. Each selected normal mode was represented using discrete variable representation (DVR) based on Gauss–Hermite quadrature.48,49 Seven grid points were employed for each mode coordinate. This choice was motivated by previous convergence tests on protonated water clusters.37 It was demonstrated that this grid size provides sufficient accuracy for describing anharmonic vibrational features in the NIR region.
N or CO). The selected localized normal modes of MeOH2+⋯N2 used in the calculations are visualized in Fig. S1 as examples. Each selected normal mode was represented using discrete variable representation (DVR) based on Gauss–Hermite quadrature.48,49 Seven grid points were employed for each mode coordinate. This choice was motivated by previous convergence tests on protonated water clusters.37 It was demonstrated that this grid size provides sufficient accuracy for describing anharmonic vibrational features in the NIR region.
A direct-product DVR grid for the full-dimensional system would formally contain up to 711 ≈ 2 × 109 grid points, which would make direct diagonalization infeasible. This led to the application of n-mode representation (nMR) to the PES, followed by a transformation into finite basis representation (FBR) and subsequent truncation into a smaller Hamiltonian. The PES was expanded using the nMR scheme proposed by Carter et al.50 as follows

To enable efficient diagonalization for up to 11 explicitly treated modes, we employed a finite-basis representation (FBR) built from eigenfunctions of the one-dimensional single-mode DVR Hamiltonians.48 For each selected normal coordinate qi, the corresponding 1D Hamiltonian, Ĥi = ![[T with combining circumflex]](https://www.rsc.org/images/entities/i_char_0054_0302.gif) i + Vi(1), was represented on a Gauss–Hermite DVR grid, and diagonalized to obtain a set of single-mode eigenfunctions, {ϕni(qi)}. The multi-dimensional vibrational basis functions were then expressed as direct products
i + Vi(1), was represented on a Gauss–Hermite DVR grid, and diagonalized to obtain a set of single-mode eigenfunctions, {ϕni(qi)}. The multi-dimensional vibrational basis functions were then expressed as direct products  . Rather than using the full direct-product basis, a configuration-selected basis was employed using two filters:
. Rather than using the full direct-product basis, a configuration-selected basis was employed using two filters:
(1) VCI-type total-quanta truncation: only configurations satisfying  were retained. In this work, Qmax = 7 or 8 was used to balance accuracy and computational cost. Production calculations employed Qmax = 7 to enable an efficient projection of eigenvectors onto another FBR basis built from products of selected n-dimensional eigenfunctions. Convergence was assessed by repeating representative calculations with Qmax = 8 with the primitive basis (see Fig. S2 in the SI).
were retained. In this work, Qmax = 7 or 8 was used to balance accuracy and computational cost. Production calculations employed Qmax = 7 to enable an efficient projection of eigenvectors onto another FBR basis built from products of selected n-dimensional eigenfunctions. Convergence was assessed by repeating representative calculations with Qmax = 8 with the primitive basis (see Fig. S2 in the SI).
(2) Energy-window truncation: each product basis function  was assigned an approximate energy,
was assigned an approximate energy,  , where εni is the eigenvalue of the 1D eigenfunction ϕni. Only configurations with Eestn ≤ 25000 cm−1 were included in the final FBR basis.
, where εni is the eigenvalue of the 1D eigenfunction ϕni. Only configurations with Eestn ≤ 25000 cm−1 were included in the final FBR basis.
Off-diagonal Hamiltonian couplings  were also neglected when both basis functions
were also neglected when both basis functions  and
and  have estimated energies in the interval 20000–25000 cm−1. This screening affects only couplings among highly excited configurations well above the targeted spectral range (0–8000 cm−1). Diagonal elements were retained for all selected basis functions.
have estimated energies in the interval 20000–25000 cm−1. This screening affects only couplings among highly excited configurations well above the targeted spectral range (0–8000 cm−1). Diagonal elements were retained for all selected basis functions.
The 4MR PES enables the Hamiltonian matrix elements in the FBR basis to be evaluated using low-dimensional integrals. Specifically, for each n-mode coupling term ΔV(n){i}, we evaluated the corresponding matrix elements in the product basis of the involved modes,  . These low-dimensional blocks were generated once and reused. The full-dimensional Hamiltonian elements,
. These low-dimensional blocks were generated once and reused. The full-dimensional Hamiltonian elements,  , were assembled by combining contributions from the relevant n-mode potential blocks, exploiting the fact that each n-mode term only couples a limited subset of quantum indices. This strategy avoids explicit construction of the full direct-product Hamiltonian on the DVR grid and substantially reduces the computational cost. Meanwhile, the matrix-element generation and assembly were embarrassingly parallelized over multiple CPU cores.
, were assembled by combining contributions from the relevant n-mode potential blocks, exploiting the fact that each n-mode term only couples a limited subset of quantum indices. This strategy avoids explicit construction of the full direct-product Hamiltonian on the DVR grid and substantially reduces the computational cost. Meanwhile, the matrix-element generation and assembly were embarrassingly parallelized over multiple CPU cores.
The resulting sparse Hamiltonian was solved using Extended Eigensolver in Intel MKL, which is based on Krylov–Schur algorithm.52,53 Eigenpairs were computed for states up to 8000 cm−1 above the vibrational ground state. No empirical scaling factors were applied to the calculated vibrational frequencies. Simulated spectra were obtained by applying Lorentzian broadening with a full width at half maximum (FWHM) of 10 cm−1.
Results and discussion
The IRPD spectra for both the singly and doubly solvated protonated methanol clusters were measured in the first overtone region of the OH stretching modes (4700–7200 cm−1). Additionally, spectra in the frequency range of 3800–4700 cm−1 were also obtained primarily to assist with band assignments. In practice, an inevitable gap (<20 cm−1) exists between these two frequency ranges due to the photon splitting process of OPA. Consequently, the absolute intensity ratios of observed IR signals from the two distinct frequency ranges cannot be determined.Singly solvated protonated methanol MeOH2+⋯X
The observed NIR spectra of the singly Ar-solvated protonated methanol, MeOH2+⋯Ar, is shown by the black trace in Fig. 1a. The signal intensity of the photodissociation fragment ions was about two orders of magnitude weaker than that observed in the OH stretching fundamental region. Hence, extensive spectrum accumulation (over 10 scans) was required to improve the signal-to-noise (S/N) ratio of the observed spectrum. The sharp band at the high energy end of the spectrum (6948 cm−1) is assigned to the first overtone transition of the free OH stretching vibration, . Its fundamental transition was previously found at 3555 cm−1 in our study of the MIR spectra of MeOH2+⋯Xn.31 The most intense band in the observed spectrum appears at 5170 cm−1. This frequency region is consistent with the CB of one quantum HOH bend (transition at ∼1600 cm−1) and one quantum free OH stretch,
. Its fundamental transition was previously found at 3555 cm−1 in our study of the MIR spectra of MeOH2+⋯Xn.31 The most intense band in the observed spectrum appears at 5170 cm−1. This frequency region is consistent with the CB of one quantum HOH bend (transition at ∼1600 cm−1) and one quantum free OH stretch,  . While the strongest transition in the MIR region is the Ar-bound (H-bonded) OH stretch (∼3300 cm−1), its overtone transition,
. While the strongest transition in the MIR region is the Ar-bound (H-bonded) OH stretch (∼3300 cm−1), its overtone transition,  , peaking around 6430 cm−1, is significantly weaker than either
, peaking around 6430 cm−1, is significantly weaker than either  or the major CB of
or the major CB of  . Such a profound suppression of the
. Such a profound suppression of the  transition was not clearly observed in the micro-solvated hydronium ion clusters until solvation by CO,37 though similar suppressions have been generally reported in condensed phase studies of H-bonded species.32,54–59 Despite this, the gross feature of the observed NIR spectrum of MeOH2+⋯Ar basically resembles that of H3O+⋯Ar.35,37
transition was not clearly observed in the micro-solvated hydronium ion clusters until solvation by CO,37 though similar suppressions have been generally reported in condensed phase studies of H-bonded species.32,54–59 Despite this, the gross feature of the observed NIR spectrum of MeOH2+⋯Ar basically resembles that of H3O+⋯Ar.35,37
 | ||
| Fig. 1 Observed (black) and calculated (red) NIR spectra of MeOH2+⋯X, (a) X = Ar, (b) X = N2, and (c) X = CO. Each observed spectrum was obtained by monitoring its one-solvent-loss dissociation channel. All numbers labeled on the spectra are observed band frequencies. | ||
The simulated DVR-FBR spectrum of MeOH2+⋯Ar (red trace in Fig. 1a) is in overall good agreement with the observed spectrum. A few minor experimental features are not reproduced, most notably near 6000 cm−1, where overtones and combination bands involving CH3 bending and/or stretching vibrations are expected. These transitions are not captured in the present reduced-dimensionality vibrational analysis because the corresponding CH3 modes were not included explicitly in the DVR–FBR treatment. Their harmonic fundamentals are listed in Table S2 for reference. According to the assignment table for pronounced calculated transitions (Table S3), the assignments based on the simulation are consistent with the qualitative interpretation given above. The relatively weaker band at 6875 cm−1 next to  can be attributed to the CB of
can be attributed to the CB of  . The structured band patterns in the lower frequency region (3800–5000 cm−1) primarily result from combinations of the CH/OH rocking modes R with sfree or sHB. A typical example is the
. The structured band patterns in the lower frequency region (3800–5000 cm−1) primarily result from combinations of the CH/OH rocking modes R with sfree or sHB. A typical example is the  peak centered at 4812 cm−1, which also carries an intensity comparable to the
peak centered at 4812 cm−1, which also carries an intensity comparable to the  transition.
transition.
Fig. 1b displays the observed and calculated NIR spectra of MeOH2+⋯N2. The  transition appears at 6960 cm−1, and the dominant CB of
transition appears at 6960 cm−1, and the dominant CB of  is observed at 5196 cm−1. These features exhibit only slight blueshifts relative to MeOH2+⋯Ar. The overtone of N2-bound OH stretching band,
is observed at 5196 cm−1. These features exhibit only slight blueshifts relative to MeOH2+⋯Ar. The overtone of N2-bound OH stretching band,  , centered at 5810 cm−1 (the corresponding fundamental was found at ∼3100 cm−1)31 is red-shifted by ∼600 cm−1 relative to
, centered at 5810 cm−1 (the corresponding fundamental was found at ∼3100 cm−1)31 is red-shifted by ∼600 cm−1 relative to  in MeOH2+⋯Ar. This pronounced redshift reflects a weakening of the OH covalent bond due to the strengthened OH⋯N2 H-bond. Consequently, the CB
in MeOH2+⋯Ar. This pronounced redshift reflects a weakening of the OH covalent bond due to the strengthened OH⋯N2 H-bond. Consequently, the CB  also shifts to 6640 cm−1. In contrast, the
also shifts to 6640 cm−1. In contrast, the  band is hardly affected by the change of solvent species.
band is hardly affected by the change of solvent species.
One of the most intriguing observations for MeOH2+⋯N2 is the relatively broad and weak appearance of the  transitions. The DVR-FBR calculation suggests that
transitions. The DVR-FBR calculation suggests that  is strongly mixed with nearby vibrational states, leading to pronounced spectral congestion. In the observed spectrum of MeOH2+⋯N2, there are a few resolvable minor peaks surrounding
is strongly mixed with nearby vibrational states, leading to pronounced spectral congestion. In the observed spectrum of MeOH2+⋯N2, there are a few resolvable minor peaks surrounding  at 5810 cm−1. According to Table S3, the main intensity carriers of these bands originate from
at 5810 cm−1. According to Table S3, the main intensity carriers of these bands originate from  , which strongly mixes with other states such as
, which strongly mixes with other states such as  ,
,  , and so on. This mixing results in intensity borrowing from the bright state
, and so on. This mixing results in intensity borrowing from the bright state  to various dark states, primarily through mechanical anharmonicity. The deep involvement of the R modes again highlights the contribution of the methyl group in the NIR region of protonated methanol clusters. In contrast, the
to various dark states, primarily through mechanical anharmonicity. The deep involvement of the R modes again highlights the contribution of the methyl group in the NIR region of protonated methanol clusters. In contrast, the  transition of MeOH2+⋯N2 remains sharp, and its projection shows that it is almost free from couplings. Meanwhile, several weaker bands under the
transition of MeOH2+⋯N2 remains sharp, and its projection shows that it is almost free from couplings. Meanwhile, several weaker bands under the  envelope show nearly negligible contribution from
envelope show nearly negligible contribution from  . Their presence adds another layer of complexity to the assignment of the OH stretching overtone features.
. Their presence adds another layer of complexity to the assignment of the OH stretching overtone features.
An analogous situation occurs for the broad feature centered at ∼5000 cm−1 in the spectrum of MeOH2+⋯Ar in Fig. 1a, which lies between the  and
and  peaks. This feature is loosely attributed to the CB
peaks. This feature is loosely attributed to the CB  . However, a more careful review of its composition reveals that similar state mixing effects, including Fermi resonance (FR)-like interactions between the
. However, a more careful review of its composition reveals that similar state mixing effects, including Fermi resonance (FR)-like interactions between the  state and
state and  or
or  , have manifested themselves. These interactions appear in the form of intensity suppression of the main
, have manifested themselves. These interactions appear in the form of intensity suppression of the main  transition and a broadened lineshape. Upon replacing Ar with N2, the
transition and a broadened lineshape. Upon replacing Ar with N2, the  band shifts away from
band shifts away from  , giving rise to a wider gap between
, giving rise to a wider gap between  and
and  . The CB of
. The CB of  is expected to shift accordingly to ∼4700 cm−1, which unfortunately falls into the gap separating the two experimental scan ranges.
is expected to shift accordingly to ∼4700 cm−1, which unfortunately falls into the gap separating the two experimental scan ranges.
Fig. 1c shows the observed and calculated spectra of MeOH2+⋯CO. The  band slightly blueshifts to 6972 cm−1, and the intense feature at 5200 cm−1 can be assigned to the CB
band slightly blueshifts to 6972 cm−1, and the intense feature at 5200 cm−1 can be assigned to the CB  . The CB
. The CB  appears at 4835 cm−1. These bands remain essentially unshifted. However, assigning the CO-bound OH stretching overtone
appears at 4835 cm−1. These bands remain essentially unshifted. However, assigning the CO-bound OH stretching overtone  is not straightforward without the guidance from theoretical simulations. Given that CO has a larger PA than N2,60 the
is not straightforward without the guidance from theoretical simulations. Given that CO has a larger PA than N2,60 the 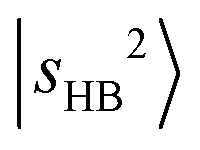 band is expected to exhibit an additional redshift and it is therefore likely buried under the broad feature in the 4800–5200 cm−1 region (its fundamental transition was found at ∼2600 cm−1).31 The simulation (Table S3) indicates that
band is expected to exhibit an additional redshift and it is therefore likely buried under the broad feature in the 4800–5200 cm−1 region (its fundamental transition was found at ∼2600 cm−1).31 The simulation (Table S3) indicates that  couples with many dark overtone/combination bands, but most of the projections of
couples with many dark overtone/combination bands, but most of the projections of  are very low (less than 5%) in each case. In the lower frequency part, two groups of weak peaks around ∼4200 cm−1 and ∼4400 cm−1 are assigned as the CB
are very low (less than 5%) in each case. In the lower frequency part, two groups of weak peaks around ∼4200 cm−1 and ∼4400 cm−1 are assigned as the CB  , showing reasonable correspondence with the broad feature in the experimental spectrum.
, showing reasonable correspondence with the broad feature in the experimental spectrum.
Doubly solvated protonated methanol MeOH2+⋯(X)2
Fig. 2 shows the observed and calculated NIR spectra of MeOH2+⋯(X)2. In the case of the singly solvated clusters, the free OH stretching overtones serve as intensity metrics due to their less perturbed character. However, these transitions are absent in the n = 2 spectra because both OH groups are solvated. Consequently, directly comparing the IR absorption intensities of the H-bonded OH stretching overtones across different solvent species is more challenging. | ||
| Fig. 2 Observed (black) and calculated (red) NIR spectra of MeOH2+⋯(X)2. (a) X = Ar. The observed spectrum was obtained by monitoring the 2Ar-loss dissociation channel. (b) X = N2. The main spectrum was obtained by monitoring the N2-loss dissociation channel. The inset shows the spectrum obtained by monitoring the 2N2-loss dissociation channel. (c) X = CO. The spectrum was obtained by monitoring the CO-loss dissociation channel. All numbers are observed band frequencies. | ||
For MeOH2+⋯(Ar)2 (Fig. 2a), the features observed at 6497 cm−1 and 6662 cm−1 are assigned to the first overtones of the Ar-bound OH stretches,  . Both bands exhibit slight blueshifts relative to the
. Both bands exhibit slight blueshifts relative to the  transition in MeOH2+⋯Ar. This observation demonstrates the anti-cooperative effect of H-bonds,61 in which the positive charge is delocalized to both proton acceptors during the formation of the second H-bond. This delocalization weakens the first H-bond and, in turn, strengthens the corresponding OH bond, which leads to an increase in its stretching frequency. Table S3 further indicates that the OH stretching overtones are strongly coupled with nearby vibrational states. The intense feature at 5002 cm−1 is likely a CB of one-quanta OH bending and one-quanta OH stretching mode,
transition in MeOH2+⋯Ar. This observation demonstrates the anti-cooperative effect of H-bonds,61 in which the positive charge is delocalized to both proton acceptors during the formation of the second H-bond. This delocalization weakens the first H-bond and, in turn, strengthens the corresponding OH bond, which leads to an increase in its stretching frequency. Table S3 further indicates that the OH stretching overtones are strongly coupled with nearby vibrational states. The intense feature at 5002 cm−1 is likely a CB of one-quanta OH bending and one-quanta OH stretching mode,  .
.
The black trace in Fig. 2b displays the experimental NIR spectrum of MeOH2+⋯(N2)2 measured by monitoring the single N2-loss channel, while the inset presents the spectrum obtained by monitoring the 2N2-loss channel. For photon energy above ∼6000 cm−1, absorption provides sufficient energy to trigger a switch in the major dissociation channel, which explains the absence of the OH stretching overtone bands in the single N2-loss channel spectrum. Accordingly, the bands centered at 6090 and 6320 cm−1 in the inset are assigned to the N2-bound  transitions. In the calculations, these bands are associated with many CBs. These overtone frequencies are redshifted by about 300–400 cm−1 compared to those of MeOH2+⋯(Ar)2. The CB of
transitions. In the calculations, these bands are associated with many CBs. These overtone frequencies are redshifted by about 300–400 cm−1 compared to those of MeOH2+⋯(Ar)2. The CB of  is found at 4860 cm−1, and is broader than the corresponding band in the spectrum of MeOH2+⋯(Ar)2. Our calculation suggests that it is engaged in couplings with the
is found at 4860 cm−1, and is broader than the corresponding band in the spectrum of MeOH2+⋯(Ar)2. Our calculation suggests that it is engaged in couplings with the  and
and  states. The origin of the doublet at 5550 cm−1 is assigned to the CBs involving one-quanta NN stretching modes (sX) and one-quanta H-bonded OH stretching modes,
states. The origin of the doublet at 5550 cm−1 is assigned to the CBs involving one-quanta NN stretching modes (sX) and one-quanta H-bonded OH stretching modes,  .
.
Fig. 2c presents the NIR spectrum of MeOH2+⋯(CO)2, in which pronounced bands are concentrated in the 5000–6000 cm−1 region with a maximum at ∼5200 cm−1, which is attributed to overtones or CB of H-bonded OH stretches. The observed bands are substantially broadened, with linewidths in the order of a few hundred wavenumbers. The DVR-FBR simulation noticeably overestimated the major band position  to ∼5423 cm−1 in this case. To evaluate whether this discrepancy can be attributed to hot bands, we carried out a hot band simulation by initiating transitions from an excited H-bond stretching mode t, which has the lowest excitation energy among the vibrational modes included in our reduced-dimensional analysis and is therefore most thermally populated. The resulting simulated hot band spectra is shown in Fig. S3. For the case of MeOH2+⋯(CO)2 (Fig. S3b), the simulated transitions from
to ∼5423 cm−1 in this case. To evaluate whether this discrepancy can be attributed to hot bands, we carried out a hot band simulation by initiating transitions from an excited H-bond stretching mode t, which has the lowest excitation energy among the vibrational modes included in our reduced-dimensional analysis and is therefore most thermally populated. The resulting simulated hot band spectra is shown in Fig. S3. For the case of MeOH2+⋯(CO)2 (Fig. S3b), the simulated transitions from  and
and  show intense bands at ∼5320 cm−1 and ∼5500 cm−1, improving the overall outline of the calculated spectrum to some extent, but mismatch in band maxima remains between the experimental and the calculated spectra, along with discrepancies in the intensity distribution.
show intense bands at ∼5320 cm−1 and ∼5500 cm−1, improving the overall outline of the calculated spectrum to some extent, but mismatch in band maxima remains between the experimental and the calculated spectra, along with discrepancies in the intensity distribution.
Three conspicuous neighboring peaks are observed in the lower frequency region of MeOH2+⋯(CO)2. For the peak at 4513 cm−1, a similar band is observed in the other two spectra of the n = 2 clusters with an almost unchanged frequency. It can be found at ∼4520 cm−1 also in the spectra of the n = 1 clusters. The same situation holds for the peak at 4233 cm−1. These bands likely originate from the same vibrational source, suggesting a link to the CH vibrational motions excluded in the present calculations, given their consistent frequency and absence in every calculated spectrum. On the other hand, a pronounced peak is observed at 4391 cm−1, to which corresponding bands are missing in the other n = 2 spectra. Though this band seems to be assigned to  because this CB is expected to be a major band in this region, this assignment cannot be reliable. Theoretically, the
because this CB is expected to be a major band in this region, this assignment cannot be reliable. Theoretically, the  transition should undergo coupling mechanisms resembling the situation in MeOH2+⋯CO with the dark states, which would lead to an appreciable broadening and redistribution of intensity rather than a narrow, well-isolated feature. Instead, this band may be attributed to the CB of the symmetric and antisymmetric CO stretches, which is reproduced at 4412 cm−1 by the DVR-FBR simulation. The
transition should undergo coupling mechanisms resembling the situation in MeOH2+⋯CO with the dark states, which would lead to an appreciable broadening and redistribution of intensity rather than a narrow, well-isolated feature. Instead, this band may be attributed to the CB of the symmetric and antisymmetric CO stretches, which is reproduced at 4412 cm−1 by the DVR-FBR simulation. The  transition likely contributes to the broad background enveloping the prominent peaks around 4400 cm−1.
transition likely contributes to the broad background enveloping the prominent peaks around 4400 cm−1.
Spectral broadening
Overall, both the NIR spectra reported here and the MIR spectra reported earlier31 of MeOH2+⋯Xn exhibit significant spectral broadening. The extent of broadening tends to become more remarkable as the PA of the binding solvents increases. To examine whether thermal effects contribute to this broadening, we focused on the MeOH2+⋯(CO)n clusters, for which the broadening effects are most pronounced.The internal energy of a cluster is capped by the weakest intermolecular interaction in the cluster (here, the hydrogen bond), because clusters with internal energy surpassing this limit would undergo spontaneous dissociation before the spectroscopic measurement. We therefore compare the spectra of “bare” and Ar-“tagged” CO-solvated clusters. The additional Ar atom introduces a much weaker intermolecular bond, thereby limiting their internal energy. However, as the weakly-tagged clusters are thermodynamically unstable, the signal intensity of the parent ions is greatly reduced. This makes a direct comparison in the NIR region practically difficult. Instead, we compare the spectra in the MIR region by assuming their broadening mechanisms are similar to that of the NIR region.
Fig. 3a and b present the MIR spectra of MeOH2+⋯CO and MeOH2+⋯(CO)2, whereas Fig. 3c and d display the spectra of their Ar-tagged counterparts. Ar-tagging (i.e., cooling) in Fig. 3c has substantially narrowed the linewidths in the H-bonded OH stretching region (2500–3000 cm−1) for the MeOH2+⋯CO spectrum, supporting an inhomogeneous contribution to the broadening, that is expected to also influence the NIR region. In contrast, the lack of such narrowing for the “free” OH stretching bands at ∼3400 cm−1 and ∼3600 cm−1 (noted that in the Ar-tagged MeOH2+⋯(CO) cluster, the Ar atom binds to the free OH site, causing a red-shift of the band from ∼3600 cm−1 to ∼3400 cm−1); these modes are less sensitive to the cooling in this situation. On the other hand, the cooling effect on MeOH2+⋯(CO)2 in Fig. 3d is less significant across the entire spectral range, suggesting a smaller contribution of inhomogeneous broadening in the NIR region for this cluster.
 | ||
| Fig. 3 Experimental IRPD spectra of (a) MeOH2+⋯CO, (b) MeOH2+⋯(CO)2, (c) MeOH2+⋯CO⋯Ar, and (d) MeOH2+⋯(CO)2⋯Ar in the MIR region. | ||
An alternative source for the spectral broadening is the highly coupled low frequency vibrational modes such as t, R, and sX modes. These modes can give rise to not only additional bands, but they also serve as pathways for IVR-based homogeneous broadenings. Here, the sX and R modes, which have relatively higher frequencies, are isoenergetic with the OH stretching mode at several quanta; thus, these modes are expected to act as promoting modes in the IVR theory.62 In contrast, the t mode, characterized by its extremely low frequency, functions as a typical bath mode.
The peak density distribution in the present NIR spectra of the protonated methanol clusters appears to be much higher in the 4000–5000 cm−1 region. As the solvent PA increases, the H-bonded OH stretching overtones gradually shift to this region where numerous minor states involving the low frequency modes are present. The further the  transition shifts toward this region, the more resonance/energy matching opportunities arise for couplings between
transition shifts toward this region, the more resonance/energy matching opportunities arise for couplings between  and other states. In extreme cases such as MeOH2+⋯CO and MeOH2+⋯(CO)2, the OH stretching overtone evolves into a strongly mixed manifold where mode-specific assignments are nearly inapplicable. Under such circumstances, intensities of the bright states are severely diluted to form numerous “daughter” bands, ultimately leading to accelerated IVR and congested spectral profiles. In the regions near and below 5000 cm−1,
and other states. In extreme cases such as MeOH2+⋯CO and MeOH2+⋯(CO)2, the OH stretching overtone evolves into a strongly mixed manifold where mode-specific assignments are nearly inapplicable. Under such circumstances, intensities of the bright states are severely diluted to form numerous “daughter” bands, ultimately leading to accelerated IVR and congested spectral profiles. In the regions near and below 5000 cm−1,  is another major contributor to spectral broadening resembling the mechanisms of
is another major contributor to spectral broadening resembling the mechanisms of  . State densities in the doubly solvated protonated methanol clusters are generally higher than those in the singly solvated ones due to the presence of an additional H-bonded solvent, which suggests even more of the above-mentioned coupling opportunities in the clusters with n = 2. However, there has been no strong evidence showing clear size dependence of spectral broadening at present. Such trends may be better viewed in solvated ammonium clusters in which the first solvation shell of the central cation may accommodate 4 solvent molecules.
. State densities in the doubly solvated protonated methanol clusters are generally higher than those in the singly solvated ones due to the presence of an additional H-bonded solvent, which suggests even more of the above-mentioned coupling opportunities in the clusters with n = 2. However, there has been no strong evidence showing clear size dependence of spectral broadening at present. Such trends may be better viewed in solvated ammonium clusters in which the first solvation shell of the central cation may accommodate 4 solvent molecules.
Summary
We reported the NIR spectra of micro-solvated protonated methanol clusters in the region of the first OH stretching overtone. The H-bonded OH stretching overtones are significantly weaker than both the free OH stretching overtones and several pronounced CBs, which is consistent with the previously reported H-bonded systems. As the solvent species changes from Ar to CO (in the order of ascending solvent PA) at a fixed solvation number, the H-bonded OH stretching overtone bands become less intense, and a clear trend of spectral broadening was observed. The broadening can be attributed to a combination of hot band, IVR, and increased band congestion induced by numerous anharmonic couplings. Our reduce-dimensional DVR-FBR anharmonic analysis reproduces the overall spectral patterns of the NIR spectra. For the present system, our results also show the importance of explicitly including the relevant low-frequency modes, such as intermolecular H-bond stretching, CH bending, and solvent intramolecular vibrations, since the overtone region is governed by extensive anharmonic mixings with these degrees of freedom.Author contributions
Yaodi Yang: investigation, formal analysis, data curation, writing – original draft. Qian-Rui Huang: methodology, software, formal analysis, data curation, writing – original draft. Jer-Lai Kuo: conceptualization, methodology, supervision, funding acquisition, writing – review & editing. Asuka Fujii: conceptualization, methodology, supervision, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article are included in the supplementary information (SI), which contains hot-band simulations, optimized geometry of all species, localized normal modes used in the anharmonic vibrational analysis and assignment table of pronounced calculated transitions. See DOI: https://doi.org/10.1039/d6cp00987e.Acknowledgements
This study is supported by a Grant-in-Aid for Scientific Research (Project No. 21H04671 and 25K03402) from JSPS. Y. Y was supported by the AGS RISE program of Tohoku University. Q.-R. H. and J.-L. K are supported by National Science and Technology Council, Taiwan (113-2113-M-001-032-MY3 and 114-2639-M-A49-002-ASP).References
- D. Boyall and K. L. Reid, Modern Studies of Intramolecular Vibrational Energy Redistribution, Chem. Soc. Rev., 1997, 26, 223–232 RSC.
- H. Siesler, Y. Ozaki, S. Kawata and H. Heise, Near-Infrared Spectroscopy: Principles, Instruments, Applications, John Wiley & Sons, Inc., 2002 Search PubMed.
- C. Pasquini, Near Infrared Spectroscopy: Fundamentals, Practical Aspects and Analytical Applications, J. Braz. Chem. Soc., 2003, 14, 198–219 CrossRef CAS.
- K. B. Beć and C. W. Huck, Breakthrough Potential in Near-Infrared Spectroscopy: Spectra Simulation. A Review of Recent Developments, Front. Chem., 2019, 7, 48 CrossRef PubMed.
- Y. Ozaki, Recent Advances in Molecular Spectroscopy of Electronic and Vibrational Transitions in Condensed Phase and Its Application to Chemistry, Bull. Chem. Soc. Jpn., 2019, 92, 629–654 CrossRef CAS.
- K. B. Beć, J. Grabska and Y. Ozaki, in Frontiers of Quantum Chemistry, ed. M. J. Wójcik, H. Nakatsuji, B. Kirtman and Y. Ozaki, Springer, Singapore, 2018, pp. 483–512 Search PubMed.
- P. Atkins, J. de Paula and J. Keeler, Atkins’ Physical Chemistry, Oxford University Press, New York, 11th edn, 2018 Search PubMed.
- ed. Y. Ozaki, C. Huck, S. Tsuchikawa and S. B. Engelsen, Near-Infrared Spectroscopy: Theory, Spectral Analysis, Instrumentation, and Applications, Springer Nature, Singapore, 2020 Search PubMed.
- ed. T. Ebata and M. Fujii, Physical Chemistry of Cold Gas-Phase Functional Molecules and Clusters, Springer, Singapore, 1st edn, 2019 Search PubMed.
- K. B. Beć, J. Grabska and C. W. Huck, Current and Future Research Directions in Computer-Aided Near-Infrared Spectroscopy: A Perspective, Spectrochim. Acta, Part A, 2021, 254, 119625–119635 CrossRef PubMed.
- M. Park, I. Shin, N. J. Singh and K. S. Kim, Eigen and Zundel Forms of Small Protonated Water Clusters: Structures and Infrared Spectra, J. Phys. Chem. A, 2007, 111, 10692–10702 CrossRef CAS PubMed.
- M. Torrent-Sucarrat and J. M. Anglada, Anharmonicity and the Eigen-Zundel Dilemma in the IR Spectrum of the Protonated 21 Water Cluster, J. Chem. Theor. Comput., 2011, 7, 467–472 CrossRef CAS PubMed.
- C. T. Wolke, J. A. Fournier, L. C. Dzugan, M. R. Fagiani, T. T. Odbadrakh, H. Knorke, K. D. Jordan, A. B. McCoy, K. R. Asmis and M. A. Johnson, Spectroscopic Snapshots of the Proton-Transfer Mechanism in Water, Science, 2016, 354, 1131–1135 CrossRef CAS PubMed.
- M. R. Fagiani, H. Knorke, T. K. Esser, N. Heine, C. T. Wolke, S. Gewinner, W. Schöllkopf, M.-P. Gaigeot, R. Spezia, M. A. Johnson and K. R. Asmis, Gas Phase Vibrational Spectroscopy of the Protonated Water Pentamer: The Role of Isomers and Nuclear Quantum Effects, Phys. Chem. Chem. Phys., 2016, 18, 26743–26754 RSC.
- K. Yagi and B. Thomsen, Infrared Spectra of Protonated Water Clusters, H+(H2O)4, in Eigen and Zundel Forms Studied by Vibrational Quasi-Degenerate Perturbation Theory, J. Phys. Chem. A, 2017, 121, 2386–2398 CrossRef CAS PubMed.
- H. Wang and N. Agmon, Reinvestigation of the Infrared Spectrum of the Gas-Phase Protonated Water Tetramer, J. Phys. Chem. A, 2017, 121, 3056–3070 CrossRef CAS PubMed.
- Q.-R. Huang, T. Nishigori, M. Katada, A. Fujii and J.-L. Kuo, Fermi Resonance in Solvated H3O+: A Counter-Intuitive Trend Confirmed via a Joint Experimental and Theoretical Investigation, Phys. Chem. Chem. Phys., 2018, 20, 13836–13844 RSC.
- Q.-R. Huang, Y.-C. Li, T. Nishigori, M. Katada, A. Fujii and J.-L. Kuo, Vibrational Coupling in Solvated H3O+: Interplay between Fermi Resonance and Combination Band, J. Phys. Chem. Lett., 2020, 11, 10067–10072 CrossRef CAS PubMed.
- Y. Zhang, Y. Wang, X. Xu, Z. Chen and Y. Yang, Vibrational Spectra of Highly Anharmonic Water Clusters: Molecular Dynamics and Harmonic Analysis Revisited with Constrained Nuclear-Electronic Orbital Methods, J. Chem. Theor. Comput., 2023, 19, 9358–9368 CrossRef CAS PubMed.
- K. R. Asmis, Y. Yang, G. Santambrogio, M. Brümmer, J. R. Roscioli, L. R. McCunn, M. A. Johnson and O. Kühn, Gas-Phase Infrared Spectroscopy and Multidimensional Quantum Calculations of the Protonated Ammonia Dimer N2H7, Angew. Chem., Int. Ed., 2007, 46, 8691–8694 CrossRef CAS PubMed.
- Y. Yang, O. Kühn, G. Santambrogio, D. J. Goebbert and K. R. Asmis, Vibrational Signatures of Hydrogen Bonding in the Protonated Ammonia Clusters NH4+(NH3)1−4, J. Chem. Phys., 2008, 129, 224302–224308 CrossRef CAS PubMed.
- H. Wang and N. Agmon, Complete Assignment of the Infrared Spectrum of the Gas-Phase Protonated Ammonia Dimer, J. Phys. Chem. A, 2016, 120, 3117–3135 CrossRef CAS PubMed.
- C.-K. Lin, Q.-R. Huang and J.-L. Kuo, Anharmonic Coupling behind Vibrational Spectra of Solvated Ammonium: Lighting up Overtone States by Fermi Resonance through Tuning Solvation Environments, Phys. Chem. Chem. Phys., 2020, 22, 24059–24069 RSC.
- C.-K. Lin and J.-L. Kuo, Anharmonic IR Spectra of Solvated Ammonium and Aminium Ions: Resemblance between Water and Bisulfate Solvations, Phys. Chem. Chem. Phys., 2022, 24, 20318–20325 RSC.
- R. Shishido, J.-L. Kuo and A. Fujii, Structures and Dissociation Channels of Protonated Mixed Clusters around a Small Magic Number: Infrared Spectroscopy of ((CH3)3N)n–H+–H2O (n = 1–3), J. Phys. Chem. A, 2012, 116, 6740–6749 CrossRef CAS PubMed.
- M. Schütz, A. Bouchet and O. Dopfer, Infrared Spectrum of the Cold Ortho-Fluorinated Protonated Neurotransmitter 2-Phenylethylamine: Competition between NH+⋯π and NH+⋯F interactions, Phys. Chem. Chem. Phys., 2016, 18, 26980–26989 RSC.
- C.-K. Lin, R. Shishido, Q.-R. Huang, A. Fujii and J.-L. Kuo, Vibrational Spectroscopy of Protonated Amine–Water Clusters: Tuning Fermi Resonance and Lighting up Dark States, Phys. Chem. Chem. Phys., 2020, 22, 22035–22046 RSC.
- C.-K. Lin, Q.-R. Huang, M. Hayashi and J.-L. Kuo, An Ab Initio Anharmonic Approach to IR, Raman and SFG Spectra of the Solvated Methylammonium Ion, Phys. Chem. Chem. Phys., 2021, 23, 25736–25747 RSC.
- J. R. Roscioli, L. R. McCunn and M. A. Johnson, Quantum Structure of the Intermolecular Proton Bond, Science, 2007, 316, 249–254 CrossRef CAS PubMed.
- J. A. Tan and J.-L. Kuo, Strong Quantum Coupling in the Vibrational Signatures of a Symmetric Ionic Hydrogen Bond: The Case of (CH3OH)2H+, J. Phys. Chem. A, 2015, 119, 11320–11328 CrossRef CAS PubMed.
- C.-K. Lin, Q.-R. Huang, Y.-C. Li, H.-Q. Nguyen, J.-L. Kuo and A. Fujii, Anharmonic Coupling Revealed by the Vibrational Spectra of Solvated Protonated Methanol: Fermi Resonance, Combination Bands, and Isotope Effect, J. Phys. Chem. A, 2021, 125, 1910–1918 CrossRef CAS PubMed.
- D. L. Howard and H. G. Kjaergaard, Influence of Intramolecular Hydrogen Bond Strength on OH-Stretching Overtones, J. Phys. Chem. A, 2006, 110, 10245–10250 CrossRef CAS PubMed.
- C. C. Wu, C. Chaudhuri, J. C. Jiang, Y. T. Lee and H. C. Chang, On the First Overtone Spectra of Protonated Water Clusters [H+(H2O)3–5] in the Free-OH Stretch Region, J. Chin. Chem. Soc., 2002, 49, 769–775 CrossRef CAS.
- J. P. Wagner, D. C. McDonald and M. A. Duncan, Near-Infrared Spectroscopy and Anharmonic Theory of the H2O+Ar1,2 Cation Complexes, J. Chem. Phys., 2017, 147, 104302–104307 CrossRef PubMed.
- D. C. McDonald, J. P. Wagner, A. B. McCoy and M. A. Duncan, Near-Infrared Spectroscopy and Anharmonic Theory of Protonated Water Clusters: Higher Elevations in the Hydrogen Bonding Landscape, J. Phys. Chem. Lett., 2018, 9, 5664–5671 CrossRef CAS PubMed.
- C. Schleif, H. A. Bunn, M. Jiménez-Redondo, P. Caselli and P. Jusko, Near-Infrared High Resolution Overtone Spectroscopy of the Hydronium Ion H3O+: The 2ν32+ and 2ν32− Bands, Phys. Chem. Chem. Phys., 2025, 27, 21307–21314 RSC.
- Q.-R. Huang, K. Yano, Y. Yang, A. Fujii and J.-L. Kuo, Near-Infrared Spectroscopy of H3O+⋯Xn (X = Ar, N2, and CO, n = 1–3), Phys. Chem. Chem. Phys., 2024, 26, 10757–10768 RSC.
- F. Kollipost, K. Papendorf, Y.-F. Lee, Y.-P. Lee and M. A. Suhm, Alcohol Dimers – How Much Diagonal OH Anharmonicity?, Phys. Chem. Chem. Phys., 2014, 16, 15948–15956 RSC.
- H. G. Kjaergaard, E. Vogt, A. S. Bogomolov and C. Lauzin, The Elusive Bound OH-Stretching First Overtone of Water Dimer, J. Phys. Chem. A, 2026, 130, 845–851 CrossRef CAS PubMed.
- P. Schuster, G. Zundel and C. Sandorfy, The Hydrogen Bond: Recent Developments in Theory and Experiments, North-Holland Pub. Co., Amsterdam, 1976 Search PubMed.
- J. M. Headrick, J. C. Bopp and M. A. Johnson, Predissociation Spectroscopy of the Argon-Solvated H5O2+ “Zundel” Cation in the 1000–1900 cm−1 Region, J. Chem. Phys., 2004, 121, 11523–11526 CrossRef CAS PubMed.
- U. Even, The Even-Lavie Valve as a Source for High Intensity Supersonic Beam, EPJ Tech. Instrum., 2015, 2, 17 CrossRef.
- K. Mizuse and A. Fujii, Infrared Photodissociation Spectroscopy of H+(H2O)6·Mm (M = Ne, Ar, Kr, Xe, H2, N2, and CH4): Messenger-Dependent Balance between H3O+ and H5O2+ Core Isomers, Phys. Chem. Chem. Phys., 2011, 13, 7129–7135 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- J. D. Head, Computation of Vibrational Frequencies for Adsorbates on Surfaces, Int. J. Quantum Chem., 1997, 65, 827–838 CrossRef CAS.
- Q.-R. Huang, Y.-C. Li, K.-L. Ho and J.-L. Kuo, Vibrational Spectra of Small Methylamine Clusters Accessed by an ab initio Anharmonic Approach, Phys. Chem. Chem. Phys., 2018, 20, 7653–7660 RSC.
- Q. Huang, R. Shishido, C. Lin, C. Tsai, J. A. Tan, A. Fujii and J. Kuo, Strong Fermi Resonance Associated with Proton Motions Revealed by Vibrational Spectra of Asymmetric Proton-Bound Dimers, Angew. Chem., Int. Ed., 2021, 60, 1936–1941 CrossRef CAS PubMed.
- J. C. Light and T. Carrington, in Advances in Chemical Physics, ed. I. Prigogine and S. A. Rice, John Wiley & Sons, Inc., 2000, vol. 114, pp. 263–310 Search PubMed.
- B. Shizgal, Spectral Methods in Chemistry and Physics, Springer, Netherlands, Dordrecht, 2015 Search PubMed.
- S. Carter, J. M. Bowman and N. C. Handy, Extensions and Tests of ‘Multimode’: A Code to Obtain Accurate Vibration/Rotation Energies of Many-Mode Molecules, Theor. Chem. Acc., 1998, 100, 191–198 Search PubMed.
- F. Neese, Software Update: The ORCA Program System—Version 6.0, WIREs Comput. Mol. Sci., 2025, 15, e70019 Search PubMed.
- Intel® oneAPI Math Kernel Library, ver. 2022.0.2, Intel Corporation, Santa Clara, CA, 2022.
- G. W. Stewart, A Krylov-Schur Algorithm for Large Eigenproblems, SIAM J. Matrix Anal. Appl., 2002, 23, 601–614 CrossRef.
- G. E. Hilbert, O. R. Wulf, S. B. Hendricks and U. Liddel, A Spectroscopic Method for Detecting some Forms of Chelation, Nature, 1935, 135, 147–148 CrossRef CAS.
- G. E. Hilbert, O. R. Wulf, S. B. Hendricks and U. Liddel, The Hydrogen Bond between Oxygen Atoms in Some Organic Compounds, J. Am. Chem. Soc., 1936, 58, 548–555 CrossRef CAS.
- A. Foldes and C. Sandorfy, Anharmonicity and Hydrogen Bonding: Part III. Examples of Strong Bonds. General Discussion, J. Mol. Spectrosc., 1966, 20, 262–275 CrossRef CAS.
- T. Di Paolo, C. Bourdéron and C. Sandorfy, Model Calculations on the Influence of Mechanical and Electrical Anharmonicity on Infrared Intensities: Relation to Hydrogen Bonding, Can. J. Chem., 1972, 50, 3161–3166 CrossRef CAS.
- T. Gonjo, Y. Futami, Y. Morisawa, M. J. Wojcik and Y. Ozaki, Hydrogen Bonding Effects on the Wavenumbers and Absorption Intensities of the OH Fundamental and the First, Second, and Third Overtones of Phenol and 2,6-Dihalogenated Phenols Studied by Visible/Near-Infrared/Infrared Spectroscopy, J. Phys. Chem. A, 2011, 115, 9845–9853 CrossRef CAS PubMed.
- Y.-L. Cheng, H.-Y. Chen and K. Takahashi, Theoretical Calculation of the OH Vibrational Overtone Spectra of 1-n Alkane Diols (n = 2–4): Origin of Disappearing Hydrogen-Bonded OH Peak, J. Phys. Chem. A, 2011, 115, 5641–5653 CrossRef CAS PubMed.
- E. P. L. Hunter and S. G. Lias, Evaluated Gas Phase Basicities and Proton Affinities of Molecules: An Update, J. Phys. Chem. Ref. Data, 1998, 27, 413–656 CrossRef CAS.
- D. Bing, T. Hamashima, A. Fujii and J.-L. Kuo, Anticooperative Effect Induced by Mixed Solvation in H+(CH3OH)m(H2O)n (m + n = 5 and 6): A Theoretical and Infrared Spectroscopic Study, J. Phys. Chem. A, 2010, 114, 8170–8177 CrossRef CAS PubMed.
- J. Jortner, S. A. Rice and R. M. Hochostrasser, in Advances in Photochemistry, ed. J. N. Pitts, G. S. Hammond and W. A. Noyes, John Wiley & Sons, Inc., 1969, vol. 7, pp. 149–309 Search PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © the Owner Societies 2026 |