
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical reduction of N-(arylthio)succinimides: the effect of aryl substituents
Michael A. Saley and
Abdelaziz Houmam *
*
Electrochemical Technology Centre, Department of Chemistry, University of Guelph, Guelph, Ontario, N1G 2W1, Canada. E-mail: ahoumam@uoguelph.ca
First published on 11th May 2026
Abstract
The electrochemical reduction of a series of N-(arylthio)succinimides (1a–e) was investigated using cyclic voltammetry and constant-potential electrolysis. These results in conjunction with a computational investigation and application of electron transfer theories allowed elucidation of the initial electron transfer mechanisms, global reaction mechanisms, rationalization of trends and differences, and determination of the effect of the substituent on the aromatic ring on the reduction reactions. The study also revealed the effect of the leaving group through comparison with investigations of similar structures with other leaving groups. The study shows that for N-(arylthio)succinimides with an electron donating or a weakly withdrawing group, the first electron transfer is concerted with dissociation of the S–N chemical bond, and produces the disulfide, through a one-electron process. A totally different behavior was observed for the nitro-substituted derivative (1e), where the initial transfer follows a stepwise mechanism, involving the intermediate formation of a radical anion, yielding the 4-nitrophenyl thiolate instead of the disulfide, through a two-electron process. For the latter compound (1e), the reduction mechanism involves an interesting autocatalysis process, where the parent molecule is competitively consumed at the electrode and in solution. The occurrence of the autocatalytic mechanism depends on the effect of the substituent on the reduction potentials of the parent molecule as well as on the corresponding disulfide and is affected by the concentration and the scan rate.
Introduction
Succinimide derivatives are utilized in many different chemical applications. Succinimide is an excellent leaving group, making N-substituted derivatives useful synthetic reagents for a variety of reactions.1–3 These structures are well known pharmacophores with many derivatives having anticancer,4 antibiotic,5 anticonvulsant,6 and antihypertensive activity,7 and have also been shown to be effective enzyme inhibitors.8,9 In addition to these applications succinimide derivatives have been utilized in surface modification,10,11 catalysis,12 polymer chemistry,13 organic semiconductors,14 silver electroplating,15 and high temperature lubricants.16 It has been demonstrated that the N-substituent can directly correlate to relevant chemical properties of N-substituted succinimides.9 Given their relevance a thorough understanding of their electron transfer (ET) chemistry and the associated substituent effects is highly desirable for new and improved applications. This is also relevant since electron transfer processes may be involved in their vast biological activity.Electrochemical methods are ideal for the investigation of all aspects of ET initiated reactions, and for the elucidation of the associated substituent effects. However, few electrochemical studies of N-substituted succinimides exist in literature. Early studies investigated the reduction of succinimide in aqueous environments such as the first investigation by Tafel and Stern,17 and the later polarographic study by Chasle-Pommeret and coworkers.18 However, more modern electrochemical investigations employing cyclic voltammetry in aprotic solvents regarding succinimide derivatives are limited to succinimide, N-chlorosuccinimide, and N-bromosuccinimide.19–21 A more recent investigation by Božić and coworkers studied the oxidation of a variety of aromatic and chloro substituted succinimide derivatives using cyclic and square wave voltammetry.22 Furthermore, apart from the study from Božić, these studies do not apply dissociative electron transfer theory nor modern computational methods to aid in the investigation of ET reactions. Electrochemical studies should be conducted on derivatives with more complex substituents to further our understanding of succinimide chemistry. Herein we report the electrochemical reduction of a series of N-(arylthio)succinimides with a range of electron-donating and electron-withdrawing substituents (Chart 1). The goal of this study is to conduct a detailed investigation of the thermodynamics and kinetics which govern the dissociative electron transfer mechanism of N-(arylthio)succinimides 1a–e, and to elucidate any substituent effects. This is accomplished using cyclic voltammetry and analysis of the electrochemical data supported by quantum chemical calculations and the application of Savéant's dissociative electron transfer (DET) theory.
 | ||
| Chart 1 General structure of the investigated N-(arylthio)succinmides 1a–e. | ||
Dissociative electron transfer can follow one of three mechanisms, specifically the stepwise, concerted, or radical/ion pair (“sticky”) mechanism. The stepwise mechanism is characterized by an initial ET resulting in the formation of an intermediate species, which subsequently dissociates; an accurate description is provided by the Marcus–Hush theory when the initial ET step is the rate-determining step.23,24 The concerted mechanism features simultaneous ET and bond dissociation and is described by Savéant's DET Theory. An extension of the DET theory can also be used to describe the dissociation of radical ions produced by a stepwise ET mechanism.25 The radical/ion pair mechanism also involves the simultaneous ET and bond dissociation; however, the radical and ion fragments associate for a period of time. This process is described by the “sticky” DET theory also developed by Savéant.26–28
The standard activation energy (intrinsic barrier) for the stepwise mechanism (ΔG‡0,s) depends on the inner sphere and solvent reorganization energies depicted in eqn (1), λi and λ0 respectively, whereas for a concerted mechanism the intrinsic barrier (ΔG‡0,c) depends on the solvent reorganization (λ0) and bond dissociation (DR) energies depicted in eqn (2).
 | (1) |
 | (2) |
The activation energy-free energy quadratic relationship for a concerted or stepwise mechanism is given by eqn (3). The activation energy-free energy quadratic relationship for the radical/ion pair mechanism considers the energy attributed to the radical/ion association, DP, known as the radical-ion pairing energy depicted in eqn (4).
 | (3) |
 | (4) |
The transfer coefficient (α), which is directly related to the intrinsic barrier (eqn (5)), is a sensitive probe of the mechanism of the first electron transfer in ET processes involving bond dissociation. A value of 0.5 or above has been proved to be associated with a stepwise ET, and a value much lower than 0.5 is associated with a concerted ET process. Experimentally, the transfer coefficient can readily be determined from the electrochemical peak characteristics (peak width, Ep–Ep/2),29 or the variation of the peak potential, Ep, with the scan rate, ν.30
 | (5) |
The difference in the reaction free energy of the stepwise and concerted mechanism is expressed in eqn (6). A weaker bond (DR), a more negative standard reduction potential  , and a more positive
, and a more positive  (less stable leaving group) favour a concerted mechanism.
(less stable leaving group) favour a concerted mechanism.
 | (6) |
If the ET follows a stepwise ET mechanism, then the dissociation of the intermediate species will follow either a homolytic or a heterolytic mechanism. If the electron is injected into the leaving group and after dissociation the electron is present on the leaving group, then the dissociation mechanism is considered homolytic. If the electron is injected into the main group and after dissociation the electron resides on the leaving group, the dissociation mechanism is considered heterolytic. These two dissociation mechanisms can be described by an extension of Savéant's DET theory, known as Intramolecular DET Theory.
This study aims to utilize electrochemical and computational methods to elucidate various electrochemical and thermodynamic parameters regarding the reduction of N-(arylthio)succinimides 1a–e. The mechanism of the initial ET will be deduced and the relevant factors affecting it will be discussed. The overall reduction mechanisms of all investigated compounds will be elucidated and the involved chemical and ET steps determined. The influence of the substituent effect on the ET mechanism and on the global reduction mechanism is of great interest and will be investigated in this study. Any observed changes as a function of the substituent will be rigorously investigated using electrochemical and computational methods and rationalized within the context of DET theory. Lastly, this study aims to compare the ET to the present N-(arylthio)succinimides (1a–e) to that of closely related species, such as the previously investigated N-(arylthio)phthalimides and aromatic sulfenyl chlorides.
Results and discussion
Voltammetric behaviour
Compounds 1a–e were synthesized according to literature procedures,31 and then recrystallized from acetonitrile. Detailed synthetic information and NMR characterization are available in the SI. Cyclic voltammetry was conducted in a dry electrochemical cell in anhydrous argon-saturated acetonitrile with tetrabutylammonium hexafluorophosphate (nBu4NPF6) as a supporting electrolyte. Compounds 1a–d exhibit two peaks in their cyclic voltammogram, whereas three peaks were observed for 1e. The reduction peak potential (Ep), the peak width (Ep–Ep/2), the number of electrons per molecule, and the corresponding transfer coefficient (α) are all determined and given in Table 1. All potentials are measured relative to the standard potential of the ferrocene/ferrocenium couple and reported relative to the SCE electrode.| Ep1 (V) | Ep2 (V) | Ep3 (V) | na | Slope | Ep1–Ep1/2 (mV) | αslope | αwidth | |
|---|---|---|---|---|---|---|---|---|
a Number of electrons consumed per molecule as determined by constant-potential electrolysis.b Upon increasing the scan rate the initial peak merges with the second making a Ep1 vs. log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ν plot unobtainable. ν plot unobtainable. |
||||||||
| 1a | −1.35 | −1.68 | — | 1 | −189 | 201 | 0.16 | 0.23 |
| 1b | −1.46 | −1.69 | — | 1 | −106 | 175 | 0.28 | 0.27 |
| 1c | −1.21 | −1.41 | — | 1 | —b | 152 | —b | 0.30 |
| 1d | −1.15 | −1.38 | — | 1 | —b | 160 | —b | 0.29 |
| 1e | −0.85 | −1.42 | −2.31 | 2 | −58 | 93 | 0.51 | 0.50 |
A typical cyclic voltammogram of N-(4-methylphenylthio)succinimide (1a) is shown in Fig. 1A. The initial peak is irreversible and is observed at a potential Ep1 = −1.35 V vs. SCE. It corresponds to the consumption of one electron per molecule by comparison to the ferrocene/ferrocenium couple as an internal standard. The variation of the first peak potential (Ep1) with the logarithm of the scan rate (logν) was determined (Fig. 1C) and a slope of −189 mV per unit logν was deduced. The deduced transfer coefficient value from this plot is 0.23, much lower than 0.5. The first reduction peak has a half-peak width (Ep–Ep/2) of 201 mV providing a transfer coefficient value of 0.16. These transfer coefficients suggest that the initial ET follows a concerted mechanism, where the electron transfer and a bond dissociation are simultaneous. A second reduction peak is observed at a potential Ep2 = −1.68 V vs. SCE and corresponds to the reduction of the bis(4-methylphenyl)disulfide as confirmed by comparison with an authentic sample shown in Fig. 1B. The formation of the disulfide indicates the dissociation of the S–N chemical bond in the initial dissociative ET to 1a. The formation of the disulfide upon reduction of aromatic sulfides with good leaving groups has been previously observed and has been found to result from the nucleophilic attack of an intermediate arylthiolate on the parent compound.32,33 This will be further discussed.
 | ||
| Fig. 1 Cyclic voltammograms, in acetonitrile in the presence of 0.1 M nBu4NPF6 at a scan rate of ν = 0.2 V s−1 using a glassy carbon electrode, of (A): N-(4-methylphenylthio)succinimide (1a, 2.48 mM) and (B): bis(4-methylphenyl)disulfide (2.08 mM), and (C): variation of the first reduction peak potential (Ep1) with logν for 1a. | ||
Similar cyclic voltametric investigations were performed for the rest of the N-(arylthio)succinimides series (1b–e) and the main cyclic voltammograms are reported in Fig. 2. Fig. 2A shows a typical cyclic voltammogram of N-(phenylthio)succinimide (1b). A first irreversible reduction peak is observed at a potential Ep1 = −1.46 V vs. SCE and corresponds to the consumption of one electron per molecule by comparison to the ferrocene/ferrocenium couple as an internal standard. The half peak width (Ep–Ep/2) value is 175 mV, providing a transfer coefficient value of 0.27. A very similar value (0.28) was also obtained from the slope of the Ep1 vs. logν plot, which was determined to be −106 mV per unit logv (see SI). These transfer coefficients suggest that the initial ET follows a concerted mechanism, similar to compound 1a as expected. A second irreversible reduction peak is observed at a potential Ep2 = −1.69 V vs. SCE and corresponds to the diphenyl disulfide by comparison with an authentic sample (see SI), also indicating the dissociation of the S–N chemical bond at the first reduction peak.
 | ||
| Fig. 2 Cyclic voltammograms, in acetonitrile in the presence of 0.1 M nBu4NPF6 at a scan rate of ν = 0.2 V s−1 using a glassy carbon electrode, of (A): N-(phenylthio)succinimide (1b, 2.68 mM), (B): N-(4-chlorophenylthio)succinimide (1c, 2.66 mM), (C): N-(4-bromophenylthio)succinimide (1d, 2.20 mM), and (D): N-(4-nitrophenylthio)succinimide (1e, 2.24 mM). | ||
The N-(4-chlorophenylthio)succinimide (1c) shows a similar cyclic voltametric behavior. A typical cyclic voltammogram is shown in Fig. 2B. The first reduction peak is irreversible and is observed at a potential Ep1 = −1.21 V vs. SCE. It corresponds to the exchange of one electron per molecule by comparison to the ferrocene/ferrocenium couple as an internal standard. The half peak width (Ep − Ep/2) value is 152 mV and the deduced transfer coefficient value is 0.30, also indicating that the initial electron transfer to 1c is associated with the simultaneous dissociation of a chemical bond. The Ep1 vs. logν plot could not be obtained as the initial peak quickly merged with the second one upon elevating the scan rate. A second irreversible reduction peak, corresponding to the reduction of the bis(4-chlorophenyl)disulfide by comparison with an authentic sample, is observed at a potential Ep2 = −1.41 V vs. SCE, indicating the dissociation of the S–N chemical bond at the first reduction peak, similar to compounds 1a and 1b (see SI).
A typical cyclic voltammogram of N-(4-bromophenylthio)succinimide (1d) is shown in Fig. 2C. The cyclic voltametric behavior is also similar to the previous compounds (1a–c). A first irreversible reduction peak, corresponding to the exchange of one electron per molecule, is observed at a potential Ep1 = −1.15 V vs. SCE, followed by the irreversible reduction peak of bis(4-bromophenyl)disulfide at a potential Ep2 = −1.38 V vs. SCE. For the first peak, the half width is 160 mV, yielding a transfer coefficient of 0.29, indicating that the initial electron transfer follows a concerted mechanism. Like for compound 1c, it was not possible to obtain an Ep1 vs. logν plot as the first peak quickly merged with the second peak upon increasing the scan rate.
Fig. 2D shows the cyclic voltammograms of N-(4-nitrophenylthio)succinimide (1e). A first irreversible reduction peak is observed at Ep1 = −0.85 V vs. SCE and corresponds, unlike the other compounds (1a–d), to the consumption of two electrons per molecule by reference to the monoelectronic oxidation of ferrocene. Remember that all other compounds showed a monoelectronic first peak. The cyclic voltammetry data for compound 1e also shows other differences compared to the rest of the compounds (1a–d). The Ep1 vs. logν plot shows a slope of −58 mV per unit logv (see SI) and the peak width (Ep–Ep/2) value is 93 mV, providing transfer coefficients of 0.51 and 0.50, respectively. These transfer coefficient values suggest a stepwise ET mechanism involving the intermediate formation of the N-(4-nitrophenylthio)succinimide radical anion (1e˙−), before dissociation. Increasing the scan rate to 250 kV s−1 did not show reversibility of the first peak indicating that the dissociation of 1e˙− is very rapid. Another important peculiarity of the 4-nitrosubstituted compound 1e is the trace crossing observed when reversing the potential sweep after the initial peak (Red CV in Fig. 2D). This trace crossing is very reproducible and does not correspond to any adsorption at the electrode surface. It corresponds to an autocatalysis process, where the reduction of a small amount of the N-(4-nitrophenylthio)succinimide (1d), yields its own catalyst. This will be further investigated. The trace crossing affects the peak shape and therefore the calculation of the transfer coefficients, the peak width and the slope of the Ep1 vs. logν plot (see SI) were deduced from a voltammogram at a lower concentration of 0.5 mM where crossing was not present. In addition to the first peak, the cyclic voltammogram of 1e, shows a second peak at E0p2 = −1.42 V vs. SCE, which corresponds to the monoelectronic reversible reduction wave of the nitro group of the 4-nitrophenylthiolate anion. A third peak is also observed at Ep3 = −2.31 V vs. SCE and corresponds to the further irreversible reduction of the nitro group of the 4-nitrophenylthiolate anion.
Electrolyses
Electrolyses of all investigated compounds were performed at the corresponding first reduction potential (Ep1) for each compound, in acetonitrile, in the presence of tetramethylammonium hexafluorophosphate (0.1 M) and at a glassy carbon electrode. The electrolyses were monitored by cyclic voltammetry and HPLC. For all compounds except the N-(4-nitrophenylthio)succinimide, the electrolyses show the complete consumption of the investigated compounds (1a–d) after the passage of one electron per molecule and yield the corresponding bis(4-substituted phenyl) disulfide (XC6H4SSC6H4X) and the succinimide anion. These results are in agreement with cyclic voltammetry results as they confirm (i) the exchange of one electron per molecule, (ii) the dissociation of the S–N chemical bond upon electron transfer to theses compounds (1a–d), and (iii) the formation of the corresponding disulfide at the first reduction peak, through an associated chemical process. For the N-(4-nitrophenylthio)succinimide (1e), the electrolysis yields the 4-nitrophenyl thiolate instead of the bis(4-nitrophenyl) disulfide and its complete consumption requires the exchange of two electrons per molecule, in agreement with the cyclic voltametric data.The above electrochemical data can already allow the proposal of the reduction mechanisms of the investigated N-(arylthio)succinimides. For compounds 1a–d the first electron transfer follows a concerted mechanism as indicated by the lack of reversibility and the very low values of the transfer coefficient. The ET is therefore simultaneous with the dissociation of the S–N chemical bond as indicated by both the cyclic voltametric and the electrolysis data. The dissociation of the S–N chemical bond can in principle lead to either a 4-substituted phenyl thiyl radical and succinimide anion or to a 4-substituted phenyl thiolate anion and succinimidyl radical. The large difference between the oxidation potentials of the substituted phenyl thiolate and the succinimide, which are around 0 V32 and +1.6 V19 vs. SCE, respectively, indicates that the dissociation would lead to the thiyl radical and succinimide anion (Scheme 1, Reaction 1). This will be discussed further based on the theoretical calculations. The produced 4-substituted phenyl thiyl radical is immediately reduced as it is easier to reduce that the parent N-(arylthio)succinimide, to yield the corresponding thiolate anion (Scheme 1, Reaction 2). The 4-substituted phenyl thiolate is a good nucleophile and attacks the parent molecule to yield the corresponding bis(4-substituted phenyl) disulfide (Scheme 1, Reaction 3). The formation of the disulfides is demonstrated by their reduction at the second peak in the cyclic voltammograms for compounds 1a–d and is confirmed by the electrolyses showing the quantitative formation of these disulfides.
 | ||
| Scheme 1 Mechanism of the electrochemical reduction of N-(arylthio)succinimides (1a–d). | ||
For the N-(4-nitrophenylthio)succinimide (1e), the first electron transfer follows a stepwise mechanism (Scheme 2, Reaction 1) leading the intermediate formation the corresponding radical anion (1e˙–), which decomposes very quickly, yielding the 4-nitrophenyl thiyl radical and the succinimide anion (Scheme 2, Reaction 2). The 4-nitrophenyl thiyl radical is reduced immediately to the corresponding thiolate anion (Scheme 2, Reaction 3). While the homogeneous reduction of this radical by the parent radical anion (1e˙–) can not be excluded (Scheme 2, Reaction 4), the reduction takes place mainly at the electrode, given the very fast dissociation of the radical anion close to the electrode. The generated thiolate then attacks the parent substrate, 1e, in a SN2 nucleophilic substitution to yield the bis(4-nitrophenyl) disulfide, 2e (Scheme 2, Reaction 5). The reduction of 2e is much easier than that of the disulfides corresponding to the other compounds (2a–d) due to the strong electron withdrawing nature of the nitro substituent. The cyclic voltammogram of the bis(4-nitrophenyl) disulfide (2e) is shown in Fig. 3, along with that of the parent N-(4-nitrophenylthio)succinimide (1e) for comparison. It shows that the reduction of the bis(4-nitrophenyl)disulfide takes place at a potential Ep1 = −0.80 V vs. SCE, which is slightly less negative than the potential corresponding to the N-(4-nitrophenylthio)succinimide (1e), contrary to the other investigated compounds which show a peak for the disulfide at a more negative potential.
 | ||
| Scheme 2 Mechanism of the electrochemical reduction of N-(4-nitrophenylthio)succinimide (1e). | ||
 | ||
| Fig. 3 Cyclic voltammograms in acetonitrile in the presence of 0.1 M nBu4NPF6 at a scan rate of ν = 0.2 V s−1 using a glassy carbon electrode of (A) N-(4-nitrophenylthio)succinimide (1e, 2.44 mM) and (B): of bis(4-nitrophenyl)disulfide (2e, 2.70 mM). | ||
Fig. 3B also shows that the initial reduction of the bis(4-methylphenyl)disulfide (2e) leads to the dissociation of the S–S bond and the formation of the same 4-nitrophenyl thiolate as for N-(4-nitrophenylthio)succinimide (1e), as evidenced by the same second and third reduction peaks corresponding, respectively, to the first and second reductions of the nitro group of the 4-nitrophenyl thiolate. It is also important to note that while the cyclic voltammogram of 1e shows a first reduction peak corresponding to 2 electrons per molecule and a second corresponding to 1 electron per molecule (the dissociation leads to only one 4-nitrophenyl thiolate anion), that of compound 2e shows that both the first and second peaks are associated with the transfer of 2 electrons per molecule. It is worth noting that the disulfide (2e) is slightly easier to reduce than the parent N-(4-nitrophenylthio)succinimide (1e). This specificity is responsible for the autocatalytic process observed for compound 1e. Unlike for compounds 1a-d, the disulfide 2e, generated in the reduction of 1e, is immediately reduced yielding the 4-nitrophenyl thiolate anion (Scheme 2, Reaction 6) which reacts with the N-(4-nitrophenylthio)succinimide (1e) (Scheme 2, Reaction 5), causing the autocatalysis process, evidenced by the trace crossing in the cyclic voltammograms. This effect has been previously demonstrated by our research group and has been shown to be dependent on both the concentration of the substrate, 1e in this case, and the experimental scan rate. Fig. 4 confirms that this is indeed the case for 1e. This figure shows that even at a low concentration of 1e (0.52 mM), the trace crossing is still clearly observed, at a low scan rate (0.2 V s−1) indicating the occurrence of the autocatalytic mechanism (Fig. 4A). However, this trace crossing is almost eliminated when the scan rate is only elevated to 0.5 V s−1 (Fig. 4B) and totally disappears when the scan rate is further elevated to just 1 V s−1 (Fig. 4B), indicating that at this scan rate compound 1e is entirely consumed by the reduction at the electrode. At a higher concentration of 1e (2.44 mM), the trace crossing is more pronounced at the low scan rate of 0.2 V s−1 (Fig. 4D) indicating a more efficient autocatalysis process. This trace crossing now, persists even when the scan rate is increased to 2 V s−1 (Fig. 4E), and it only disappears when the scan rate is increased to 10 V s−1 (Fig. 4F).
 | ||
| Fig. 4 Trace crossing behavior of the first peak in the cyclic voltammogram for N-(4-nitrophenylthio)succinimide (1e) at different concentrations and scan rates: (A) C = 0.52 mM, v = 0.2 V s−1; (B): C = 0.52 mM, v = 0.5 V s−1; (C) C = 0.52 mM, v = 1 V s−1; (D) C = 2.44 mM, v = 0.2 V s−1; (E) C = 2.44 mM, v = 2 V s−1; and (F) C = 2.44 mM, v = 10 V s−1. | ||
From the electrochemical results it is clear that the presence of the nitro substituent on the aromatic group induces a passage from a concerted to a stepwise mechanism, as well as the introduction of a very efficient autocatalytic process due to its substantial effect on the reduction of the nitro substituted disulfide formed from the homogenous SN2 reaction.
Theoretical calculations will allow better understanding and rationalizing some of the electrochemical results such as the mechanism of the first electron transfer and its dependence on the nature of the substituent on the aromatic ring of compounds 1a–e, and the products of the S–N bond dissociation.
Computational investigation
Computational investigations were conducted with Gaussian 1632 using DFT at the B3LYP level and a 6-311G++(2d,p) basis set in the solvent phase (acetonitrile) via the PCM. The neutral structures for all compounds 1a–e were optimized and their energies and molecular orbitals were determined (Fig. 5 and 6). All potential fragments including substituted phenyl thiyl radicals, their corresponding thiolate anions, the succinimidyl radical and its anion were optimized and their energies were calculated. The S–N bond dissociation energies for all investigated compounds were obtained (Table 2). | ||
| Fig. 5 Optimized molecular geometry and LUMO distributions for compounds 1a–e. | ||
 | ||
| Fig. 6 Structure of N-(4-nitrophenylthio)succinimide, 1e (A); its corresponding LUMO (B); the structure of its radical anion, 1e˙− (C) and the corresponding SOMO (D). | ||
| dS–Na | ΔEb | BDEc | |
|---|---|---|---|
| a S–N bond length from the optimized structures in Å.b ΔE = EproductspathwayA − EproductspathwayB in eV.c Bond dissociation energies (DXC6H4S-Succ), in kcal mol−1, determined from theoretical calculations. |
|||
| 1a | 1.73 | 1.8 | 64.0 |
| 1b | 1.73 | 1.7 | 65.0 |
| 1c | 1.73 | 1.6 | 64.7 |
| 1d | 1.73 | 1.6 | 64.8 |
| 1e | 1.71 | 1.1 | 64.7 |
Fig. 5 shows the optimized structures and the LUMOs of compounds 1a–d, which follow a concerted ET mechanism according to above-described electrochemical results. The optimized molecular geometries are very similar for these compounds. These LUMO distributions provide insight into the location where an incoming electron would be transferred. For compounds 1a–d, the LUMOs distributions are nearly identical with the orbital primarily distributed over the aromatic moiety and extending to the sulfur-nitrogen bond. This agrees with the suggested concerted electron transfer mechanism, as the extra electron is injected directly to the S–N bond causing its simultaneous dissociation. Calculations of the reduced forms (1a-d + e−) did not provide radical anions and only generated structures with the S–N bond fragmented. The absence of radical anions for compounds 1a-d is a clear indication that their electrochemical reduction can only follow a concerted electron transfer mechanism. This is exactly what the electrochemical results suggested as discussed in the previous section.
For the nitro-substituted compound (1e), which follows a stepwise electron transfer mechanism upon reduction according to the electrochemical data, some differences are worth noting. The molecular geometry is different, as the dihedral angle ϕ is rotated by 90° compared to the rest of the compounds (1a–d). The LUMO orbital is also strictly distributed on the aromatic moiety and does not overlap with the sulfur–nitrogen bond, unlike for compounds 1a–d. This suggests that the electron is injected into the nitrophenyl moiety to form an intermediate and only afterwards it is transferred to the sulfur–nitrogen σ* antibonding orbital where dissociation then occurs. Further confirmation of this comes from the calculations of the reduced form of the nitro-substituted compound (1e + e−), which in this case provides a real radical anion (Fig. 5). The structure of the radical anion (1e˙−) shows that the injection of the extra electron causes the rotation of the dihedral angle ϕ to generate a geometry similar to the neutral structures of 1a–d. It also causes the elongation of the S–N bond from 1.709 Å to 1.752 Å. The SOMO orbital of the radical anion (1e˙−) also confirms that the incoming electron is injected to the nitrophenyl moiety (Fig. 5). These results are in agreement with the electrochemical data and show indeed that while the N-(4-nitrophenylthio)succinimide (1e) is reduced following a stepwise ET mechanism, the rest of the compounds in this series follow a concerted ET mechanism.
The theoretical calculations allowed determination of important parameters such as the intrinsic barriers and the standard reduction potentials  of compounds 1a–d (Table 3).
of compounds 1a–d (Table 3).
| E0a |
TΔS0b |
λ0c |
ΔG‡0d |
|
|---|---|---|---|---|
a Standard reduction potential  in V vs. Ag/AgCl, determined from equation (10).b Entropy term in V vs. Ag/AgCl, determined from equation (10).b Entropy term  in eV determined from theoretical calculations.c Solvation energy in eV, calculated using equation (11).d Intrinsic barrier for the concerted ET in eV determined from theoretical calculations.c Solvation energy in eV, calculated using equation (11).d Intrinsic barrier for the concerted ET  in eV, calculated using eqn (2). in eV, calculated using eqn (2). |
||||
| 1a | −0.775 | 0.482 | 0.513 | 0.824 |
| 1b | −0.803 | 0.498 | 0.520 | 0.837 |
| 1c | −0.783 | 0.504 | 0.504 | 0.829 |
| 1d | −0.818 | 0.473 | 0.488 | 0.826 |
The standard reduction potentials are calculated for the N-(arylthio)succinimides 1a–d using eqn (7). The solvent reorganization energies were estimated using the ionic radius (as) in eqn (8) and used to calculate the intrinsic barriers using eqn (2).
 | (7) |
 | (8) |
The determined values (Table 3) are also in agreement with the previous results as the intrinsic barriers associated with the electron transfer to these compounds are large and in line with what is expected for concerted electron transfer mechanisms. The standard reduction potentials are much more positive than the observed peak potentials observed in the cyclic voltammograms of these compounds. These values further support the occurrence of a concerted electron transfer for these compounds.
The theoretical calculations also allowed further investigation of the dissociation of the S–N bond upon reduction of the investigated compounds 1a–e and the resulting products. The electrochemical results indicated the dissociation would lead to an arylthiyl radical and the succinimide anion (pathway A in Scheme 3) and not to an arylthiolate anion and the succinimidyl radical (pathway B in Scheme 3). This was based on the large difference between the oxidation potentials of the thiolate anions and the succinimide anion (around 1.6 V). All potential products (radicals and anions) of the two pathways were optimized their energies are reported in Table 2. The results clearly show that pathway A is thermodynamically favored over pathway B by at least 1.1 eV for all investigated compounds 1a–e (Fig. 7). This result along with the LUMOs determined earlier can allow elucidation of the dissociation mechanism of the intermediate radical anion formed in the reduction of the N-(4-nitrophenylthio)succinimide (1e). As discussed above the LUMO of 1e is strictly located on the aromatic moiety, indicating that the incoming electron is hosted by the π* orbital. The dissociation of the radical anion to the 4-nitrophenylthiyl radical and the succinimide anion hence involves a heterolytic dissociation mechanism where the electron is transferred from the aromatic moiety to the succinimidyl group causing the dissociation of the S–N chemical bond.
 | ||
| Scheme 3 Dissociation pathways for the N-(4-nitrophenylthio)succinimide radical anion. | ||
 | ||
| Fig. 7 Comparison of the energies of the potential products (arylthiyl radical + succinimide anion vs. arylthiolate + succinimidyl radical) of the reductive dissociation of compounds 1a–e. The reported energy difference (ΔE) is that corresponding to the two fragments minus that of the neutral molecule. | ||
The theoretical results can also help in rationalizing the passage from a concerted (for 1a–d) to a stepwise ET mechanism (for 1e) upon changing the substituent on the aromatic ring. The difference in the reaction free energy of the stepwise and concerted mechanism is expressed in eqn (6). The bond dissociation energy, DR, is similar for all investigated compounds and does not depend much on the nature of the substituent on the aromatic ring. The oxidation potential of the leaving group is identical since the same leaving group is ejected in the reduction of all compounds (1a–e). The differences in the entropy terms are negligible. This means that the main difference resides in the standard reduction potentials of the investigated compounds, directly related to the structure and LUMO of the N-(4-nitrophenylthio)succinimide (1e) compared to the rest of the investigated compounds (1a-d) as shown above by the theoretical calculations.
Previous investigations on structurally familiar compounds have also demonstrated that the nitro substituent has similar effects on ET processes.33–35 For the substituted-aryl thiocyanates,33 a striking change in the reductive mechanism as a function of the substituent on the aryl ring was observed. With electron-donating groups (methyl and methoxy) a transition between the concerted and stepwise mechanisms was encountered and a very efficient autocatalysis process was demonstrated. When nitro substituents are introduced, the initial electron transfer mechanism became stepwise and the autocatalytic mechanism was totally eliminated.33 In this case the introduction of 2 nitro groups greatly enhanced the lifetime of the initial radical anion intermediate and diminished the nucleophilicity of the intermediate thiolate towards the parent molecule, leading to the elimination of the autocatalysis process.
In the case of aromatic sulfenyl chlorides,34 the series followed a sticky ET mechanism except for the 2-nitro substituted species, which followed a stepwise ET mechanism. This, however, was due to the through space non-bonding interaction of the nitro substituent oxygen and the sulfur atom, which stabilized the intermediate species and made the stepwise mechanism more favourable.34
The N-(arylthio)succinimides 1a–e are structurally similar to the recently studied N-(arylthio)phthalimides which all follow a stepwise ET mechanism.35 The standard reduction potential  values of the N-(arylthio)phthalimides are significantly more positive than those of the presently investigated compounds (1a–d). The phthalimide anion also has a more positive oxidation potential than the succinimide anion. Both of these factors cause the N-(arylthio)phthalimides to follow a stepwise mechanism according to eqn (6). The bond dissociation energies are similar for both series and have a negligible effect. It is interesting to note that the radical anions of the N-(arylthio)phthalimides all follow a homolytic dissociation mechanism, whereas 1e, the only compound of the present series to follow a stepwise electron transfer mechanism, undergoes a heterolytic dissociation mechanism. This difference in dissociation mechanism can easily be attributed to the lack of a conjugated structure in the succinimidyl moiety capable of hosting an incoming electron, whereas in the case of phthalimide nearly the entire fused ring structure is a conjugated network capable of hosting incoming electrons.
values of the N-(arylthio)phthalimides are significantly more positive than those of the presently investigated compounds (1a–d). The phthalimide anion also has a more positive oxidation potential than the succinimide anion. Both of these factors cause the N-(arylthio)phthalimides to follow a stepwise mechanism according to eqn (6). The bond dissociation energies are similar for both series and have a negligible effect. It is interesting to note that the radical anions of the N-(arylthio)phthalimides all follow a homolytic dissociation mechanism, whereas 1e, the only compound of the present series to follow a stepwise electron transfer mechanism, undergoes a heterolytic dissociation mechanism. This difference in dissociation mechanism can easily be attributed to the lack of a conjugated structure in the succinimidyl moiety capable of hosting an incoming electron, whereas in the case of phthalimide nearly the entire fused ring structure is a conjugated network capable of hosting incoming electrons.
Conclusions
The electrochemical reduction of a series of N-(arylthio)succinimides (1a–e) was investigated using cyclic voltammetry and constant-potential electrolysis. With an electron donating or a weakly withdrawing group (Me, H, Cl and Br) the reduction follows a concerted electron transfer, where the ET and the bond dissociation are simultaneous. For all these compounds (1a–d), the products of the one ET process are the succinimide anion and an arylthiyl radical, whereas the overall product of the reduction is the corresponding disulfide obtained through an SN2 nucleophilic substitution of the intermediate substituted arylthiolate anion, obtained upon further reduction of the radical, and the parent molecule. The nitro-substituted derivative (1e) shows a totally different behavior. In this case, a stepwise initial transfer mechanism is followed, involving the intermediate formation of a radical anion, and the overall reduction yields the 4-nitrophenyl thiolate, not the disulfide, through a two-electron process. The bis(4-nitrophenyl)disulfide is generated during the reduction process, but is immediately reduced, since it is easier to reduce than the nitro-substituted parent compound (1e). This leads to an interesting autocatalysis process, where the parent molecule is competitively consumed at the electrode and in solution. The passage from concerted (for 1a–d) to stepwise (for 1e) ET mechanism is attributed to the effect of the nitro substituent which changes the structure of 1e compared to the rest of the compounds and lowers the associated LUMO energy, resulting in a less negative standard reduction potential. Computational investigations were utilized to determine the bond dissociation energy of the S–N cleaved bond for all compounds, the optimized molecular geometries, and the LUMO distributions. For compounds 1a–d, the LUMO is distributed on the aromatic moiety and along the sulfur-nitrogen bond, whereas for 1e the LUMO was not distributed along the sulfur–nitrogen bond. The latter compound is the only one to provide a radical intermediate. The determined intrinsic barriers and standard reduction potentials for compounds 1a–d further support the occurrence of the concerted ET mechanism for these compounds.Author contributions
The contributions of M. S. are conceptualization, data curation, formal analysis, investigation, methodology, visualization, validation, writing – original draft, and writing – review & editing. The contributions of A. H. are conceptualization, formal analysis, funding acquisition, project administration, supervision, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: it includes an Experimental Section, a description of the synthetic procedure of the investigated compounds, electrochemical data and theoretical data. See DOI: https://doi.org/10.1039/d6cp00898d.Notes and references
- H. Liu, X. He, Z. Chen, J. Zhang, X. Fang, Z. Sun and W. Chu, N-Trifluoromethyl succinimide as a new reagent for direct C−H trifluoromethylation of free anilines, Chem. – Asian J., 2023, 18(8), e202300039 CrossRef CAS PubMed.
- D. A. Rogers, J. M. Gallegos, M. D. Hopkins, A. A. Lignieres, A. K. Pitzel and A. A. Lamar, Visible-light photocatalytic activation of N-chlorosuccinimide by organic dyes for the chlorination of arenes and heteroarenes, Tetrahedron, 2019, 75(36), 130498 CrossRef.
- G. Zhang, R.-X. Bai, C.-H. Li, C.-G. Feng and G.-Q. Lin, Halogenation of 1,1-diarylethylenes by N-halosuccinimides, Tetrahedron, 2019, 75(12), 1658–1662 CrossRef CAS.
- I. H. Hall, O. T. Wong and J. P. Scovill, The cytotoxicity of N-pyridinyl and N-quinolinyl substituted derivatives of phthalimide and succinimide, Biomed. Pharmacother., 1995, 49, 251–258 CrossRef CAS PubMed.
- F. Zentz, A. Valla, R. Le Guillou, R. Labia, A. G. Mathot and D. Sirot, Synthesis and antimicrobial activities of N-substituted imides, Farmaco, 2002, 57(5), 421–426 CrossRef CAS PubMed.
- N. Banjac, N. Trisovic, N. Valentic, G. Uscumlic and S. Petrovic, Succinimides: Synthesis, properties and anticonvulsant activity, Hem. Ind., 2011, 65(4), 439–453 CrossRef CAS.
- M. I. Qayyum, S. Ullah, U. Rashid, A. Sadiq, M. H. Mahnashi, S. U. K. Khalil and M. M. Akhtar, N-phenyl and N-benzyl substituted succinimides: Preclinical evaluation for their antihypertensive effect and underlying mechanism, Eur. J. Pharmaco., 2024, 964, 176195 CrossRef CAS PubMed.
- B. Grodner, D. M. Pisklak and L. Szeleszczuk, Succinimide Derivatives as Acetylcholinesterase inhibitors - in silico and in vitro studies, Cur. Issues Mol. Biol., 2024, 46(6), 5117–5130 CrossRef CAS PubMed.
- J. A. Guevara-Salazar, M. Espinoza-Fonseca, H. I. Beltrán, J. Correa-Basurto, D. Q. Zavala and J. G. Trujillo-Ferrara, The electronic influence on the active site-directed inhibition of acetylcholinesterase by N-aryl-substituted succinimides, J. Mex. Chem. Soc., 2007, 51(4), 222–227 CAS.
- X. Gong, J. Zheng, Y. Zheng, S. Cao, H. Wen, B. Lin and Y. Sun, Succinimide-modified graphite as anode materials for lithium-ion batteries, Electrochim. Acta, 2020, 356, 136858 CrossRef CAS.
- M. Darder, K. Takada, F. Pariente, E. Lorenzo and H. D. Abruña, Dithiobissuccinimidyl propionate as an anchor for assembling peroxidases at electrodes surfaces and its application in a H2O2 biosensor, Anal. Chem., 1999, 71(24), 5530–5537 CrossRef CAS PubMed.
- C. Yang, L. A. Farmer, D. A. Pratt, S. Maldonado and C. R. J. Stephenson, Revisiting the reactivity of the dismissed hydrogen atom transfer catalyst succinimide-N-oxyl, J. Am. Chem. Soc., 2024, 146(18), 12511–12518 CrossRef CAS PubMed.
- W. Ryu, L. Xiang, K. S. Jin, H.-J. Kim, H.-C. Kim and M. Ree, Newly found digital memory characteristics of pyrrolidone-and succinimide-based polymers, Macromol. Rapid Commun., 2021, 42(14), e2100186 CrossRef PubMed.
- Y. Yuan, S. Flynn, X. Li, H. Liu, J. Wang and Y. Li, Wide bandgap polymer donors based on succinimide-substituted thiophene for nonfullerene organic solar cells, Macromol. Rapid Commun., 2024, 45(17), e2400275 CrossRef PubMed.
- A. Blair, Silver plating systems based on thiosulfate and succinimide, Met. Finish., 1995, 93(1), 290–297 CrossRef.
- S. Wang, S. Yu, J. Feng and S. Liu, Multifunctional lubricant additive derived from polyisobutylene succinimide dispersant, J. Dispers. Sci., 2021, 42(3), 396–406 CrossRef CAS.
- J. Tafel and M. Stern, Reduction von succinimiden zu pyrrolidonen, Ber. Dtsch. Chem. Ges., 1900, 33, 2224 CrossRef.
- M. F. Chasle-Pommeret, S. Jaouannet, A. Lebouc and A. Tallec, Reduction electrochimique de gem-dichloro-succinimides. Etude stereochimique, Electrochim. Acta, 1984, 29(9), 1287–1290 CrossRef CAS.
- M. W. Moore, M. Finklestein and S. D. Ross, The electrochemical reduction of succinimide: Reactivity of quaternary ammonium ions under electrolysis conditions, Tetrahedron, 1980, 36, 727–730 CrossRef.
- J. E. Barry, M. Finklestein, M. W. Moore and S. D. Ross, Rates and mechanism in the electron transfer mediated reduction of N-chlorosuccinimide by succinimide anion, J. Org. Chem., 1985, 50, 528–531 CrossRef CAS.
- J. E. Barry, M. Finklestein, M. W. Moore and S. D. Ross, The N-bromosuccinimide-succinimide anion complex, an intermediate in the electron transfer mediated reduction of N-bromosuccinimide by succinimide anion, Tetrahedron Lett., 1984, 25(27), 2847–2850 CrossRef CAS.
- B. Božić, J. Lović, N. Banjac, Ž. Vitnik, V. Vitnik, D. Mijin, G. Ušćumlić and M. A. Ivić, Voltammetric and quantum investigation of selected succinimide, Int. J. Electrochem. Sci., 2018, 13(5), 4285–4297 CrossRef.
- R. A. Marcus, Chemical and electrochemical electron-transfer theory, Annu. Rev. Phys. Chem., 1964, 15, 155 CrossRef CAS.
- R. A. Marcus and N. Sutin, Electron transfers in chemistry and biology, Biochim. Biophys. Acta, 1985, 811, 265 CrossRef CAS.
- J.-M. Savéant, Electron transfer, bond breaking, and bond formation, Acc. Chem. Res., 1993, 26, 455 CrossRef.
- L. Pause, M. Robert and J.-M. Savéant, Reductive cleavage of carbon tetrachloride in a polar solvent. An example of a dissociative electron transfer with significant attractive interaction between the caged product fragments, J. Am. Chem. Soc., 2000, 122, 9829 CrossRef CAS.
- J.-M. Savéant, Effect of ion pairing on the mechanism and rate of electron transfer. Electrochemical aspects, J. Phys. Chem. B, 2001, 105, 8995 CrossRef.
- C. P. Andrieux, J.-M. Savéant and C. Tardy, Cage and entropy effects in the dynamics of dissociative electron transfer, J. Am. Chem. Soc., 1998, 120, 4167 CrossRef CAS.
-
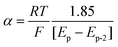 .
. -
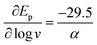 .
. - M. Furukawa, T. Suda and S. Hayashi, Behaviors of N-(substituted thio) phthalimides, N-(substituted thio)-succinimides, and N-(substituted thio) isatins toward some nucleophiles, Chem. Pharm. Bull., 1976, 24, 1708–1713 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 16, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. Houmam, E. M. Hamed and I. W. J. Still, A Unique autocatalytic process and evidence for a concerted-stepwise mechanism transition in the dissociative electron-transfer reduction of aryl thiocyanates, J. Am. Chem. Soc., 2003, 125, 7258–7265 CrossRef CAS PubMed.
- C. Ji, M. Ahmida, M. Chahma and A. Houmam, Radical/ion pair formation in the electrochemical reduction of arene sulfenyl chlorides, J. Am. Chem. Soc., 2006, 128, 15423–15431 CrossRef CAS PubMed.
- M. A. Saley, E. M. Hamed and A. Houmam, Dissociative electron transfer to N-(arylthio) phthalimides: Factors affecting the formation and dissociation of radical anions, ChemElectroChem, 2024, 11(8), 202300786 CrossRef.


| This journal is © the Owner Societies 2026 |