
Exploiting Z-scheme charge-carrier dynamics in MA2Z4-based van der Waals heterostructures for bias-assisted water-splitting applications
 *a
Wenkai
Zhao
a
and
*a
Wenkai
Zhao
a
and
Abstract
The MA2Z4 (M = transition metal; A = Si, Ge; Z = N, P, As) monolayers, characterized by exceptional stability and tailorable electronic structures, offer a versatile platform for constructing van der Waals heterostructures for water-splitting-related applications. Here, we systematically investigate six MA2Z4-based heterostructures—specifically MoSi2P4/MoSi2As4, MoSi2P4/WSi2P4, WSi2P4/WSi2As4, CrSi2P4/CrSi2As4, CrSi2P4/WSi2P4 and CrSi2P4/MoSi2P4—using first-principles calculations. All candidates exhibit intrinsic direct bandgaps and robust type-II band alignment. The resultant interfacial built-in electric fields facilitate a Z-scheme migration pathway, which ensures the spatial separation of photogenerated carriers while preserving potent redox potentials essential for overall water splitting. Model-based theoretical solar-to-hydrogen efficiency 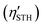 estimations give a maximum value of 38.65%. However, Gibbs free energy analyses for the hydrogen and oxygen evolution reactions reveal that, among the six systems, WSi2P4/WSi2As4, CrSi2P4/WSi2P4, and CrSi2P4/MoSi2P4 exhibit greater thermodynamic feasibility for photocatalytic water splitting, though the reactions are not spontaneous without bias. Furthermore, by integrating strain engineering with non-adiabatic molecular dynamics simulations, we delineate a competitive preservation of redox capacities: WSi2P4/WSi2As4 excels in reduction protection, whereas CrSi2P4/WSi2P4 prioritizes oxidation potency. Notably, the CrSi2P4/WSi2P4 heterostructure demonstrates ultrafast carrier dynamics with an interfacial electron–hole recombination time of 0.2 ps, significantly enhancing Z-scheme efficiency. Under 4% tensile strain, the
estimations give a maximum value of 38.65%. However, Gibbs free energy analyses for the hydrogen and oxygen evolution reactions reveal that, among the six systems, WSi2P4/WSi2As4, CrSi2P4/WSi2P4, and CrSi2P4/MoSi2P4 exhibit greater thermodynamic feasibility for photocatalytic water splitting, though the reactions are not spontaneous without bias. Furthermore, by integrating strain engineering with non-adiabatic molecular dynamics simulations, we delineate a competitive preservation of redox capacities: WSi2P4/WSi2As4 excels in reduction protection, whereas CrSi2P4/WSi2P4 prioritizes oxidation potency. Notably, the CrSi2P4/WSi2P4 heterostructure demonstrates ultrafast carrier dynamics with an interfacial electron–hole recombination time of 0.2 ps, significantly enhancing Z-scheme efficiency. Under 4% tensile strain, the 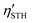 of CrSi2P4/WSi2P4 reaches 36.21% among the thermodynamically more favorable candidates, highlighting the tunability of MA2Z4-based heterostructures for bias-assisted water-splitting applications.
of CrSi2P4/WSi2P4 reaches 36.21% among the thermodynamically more favorable candidates, highlighting the tunability of MA2Z4-based heterostructures for bias-assisted water-splitting applications.

 Please wait while we load your content...
Please wait while we load your content...

