
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulticonfigurational non-adiabatic molecular dynamics towards photochemical-N2-extrusion reactions in borodiazenes
Christian
Salguero
 and
Steven A.
Lopez
*
and
Steven A.
Lopez
*
Department of Chemistry and Chemical Biology, Northeastern University, Boston, Massachusetts 02115, USA. E-mail: s.lopez@northeastern.edu
First published on 8th April 2026
Abstract
We used state-of-the-art quantum-chemical calculations to study the photodenitrogenation of 1-methylborodiazene to 1-methylborirane. Vertical excitation energy calculations indicate that the S0 → S1 (nN → πNN*) and S0 → S2 (nN → 2pB) transitions are accessible (318 nm and 287 nm, respectively), and the S0 → S1 transition is the bright state. The minimum-energy path indicates near-degeneracy between the S1 and S2 surfaces in the Franck–Condon region, leading to the steepest descent path on the S1 state to be towards fluorescent decay rather than denitrogenation. Our simulations indicate that denitrogenation primarily occurs shortly after an S1/S0 hopping event (79% of trajectories) and proceeds via three distinct pathways. The dominant pathway involves boron pyramidalization and partial πNN isomerization in the S1 state, with N2 elimination shortly after an S1/S0 hop. Minor pathways include either πNN isomerization or boron pyramidalization, with N2 elimination occurring on the S0 and S1 states, respectively. We also observed that some trajectories rearrange to form a diazoborete intermediate. Our results show that the labile N2 group can be used to access base-free boriranes.
Introduction
Photochemistry exemplifies principles of green chemistry by utilizing renewable solar energy to facilitate reactions under mild conditions. Light activation enables spatiotemporal control over the activation and deactivation of chemical processes, such as cycloadditions,1–3 isomerizations,4–6 and gas evolutions.7–9 These processes have excellent atom economy, minimal energy use, and high selectivity—traits that are highly valued in chemistry,10–12 biology,13–15 and materials science.1,16–18 Photochemical activation provides access to reactive intermediates that would otherwise be thermally inaccessible by breaking19–21 and forming bonds,22–24 causing isomerizations,25 and altering electronic properties (e.g., aromaticity/antiaromaticity26–28). These light-triggered modifications have led to the application of these reactions in energy storage,1,16,29 drug delivery,30–32 and total organic synthesis.33–35These photochemical reactions are used in organic synthesis to introduce ring strain23,36,37 into chemical systems that would otherwise be kinetically or thermally unfavorable. Researchers can then utilize these ring-strained compounds in additional chemical reactions to produce final products, such as grazoprevir,38 2-pyridylethylamine,39 and lasalocid.33,37,40,41 Three-membered rings, such as cyclopropanes,42 aziridines,43 and epoxides/oxiranes40,44,45 have garnered significant interest because of their distinctive reactivity, arising from inherent ring strain, which enables diverse synthetic applications.41,45,46 Despite the high ring strain, these three-member rings are commonly found in natural products,47–49 leading to increased interest in pharmaceutical chemistry43 and chemical biology44,50 (Fig. 1).
 | ||
| Fig. 1 Examples of cyclopropanes, aziridines, and oxiranes within pharmaceutical agents: (a) triglanol tiglate,47 (b) aziridines mitosol,49 and (c) triptolide,51 respectively. The three-membered rings are highlighted in red within the structures. | ||
The synthetic methods for these rings include cyclopropanations,52–54 aziridination,53,55–57 and epoxidations,53,58 each of which produces its respective three-membered ring and is well-established. The widespread application of aziridines and oxiranes has driven research into other hetero-analogues of cyclopropanes, such as thiiranes,59–61 phosphiranes,62–64 and siliranes.65–67 Like cyclopropanes, aziridines, and oxiranes, these compounds are highly reactive, often undergoing ring-opening reactions with nucleophiles62,65 or ring expansions with π-systems.67–69 As a result, these three-membered rings have found uses in pharmaceutical chemistry68,70 and polymer synthesis.62,65 One hetero-analogue of cyclopropane, whose chemistry is not fully understood, is borirane, the boron analogue.
Pioneering work on the synthesis of borirane was conducted by Berndt and co-workers in 1983, in which the first B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond was reported (Fig. 2a).71 Boriranes are highly reactive due to their ring strain and Lewis acidity.72,73 In 1991, Denmark and coworkers further expanded the field by introducing the first report of a Lewis base-stabilized borirane through the photoirradiation of diphenyl-(E)-2-phenylethenyl-borane (Fig. 2b).74
C double bond was reported (Fig. 2a).71 Boriranes are highly reactive due to their ring strain and Lewis acidity.72,73 In 1991, Denmark and coworkers further expanded the field by introducing the first report of a Lewis base-stabilized borirane through the photoirradiation of diphenyl-(E)-2-phenylethenyl-borane (Fig. 2b).74
 | ||
| Fig. 2 Comparison between the first (a) base-free borirane and (b) base-stabilized borirane. | ||
In the late 2000 s, Wang reported several N,C-chelated75,76 and N-heterocyclic carbene chelated boriranes77 (NHC-borirane) for potential use in optoelectronic and organic synthesis, stemming from their easy and reversible C–C/C–B bond rearrangements triggered by both light and heat. Braunschweig et al. reported the synthesis of NHC-boriranes through the salt elimination of NHC-dichloroboranes with the dianion of trans-stilbene towards trans-1,2,3-triphenyl NHC-boriranes.78 Curran and coworkers later reported a high-yield (80%) novel synthetic approach to NHC-boriranes via double hydroboration of acetylenedicarboxylates with NHC-boranes.73 The NHC-boriranes show exceptional inertness in gas-phase, aqueous, thermal, and photolytic conditions because of the Lewis base stabilization.73,78,79 In 2019, Curran et al. reported the first ring-opening reaction of base-stabilized borirane towards a novel class of NHC-boralactones.80 Recent works have reported access to borirane derivatives, such as carborane-fused boriranes,81,82 which have expanded the range of ligated boriranes. Nonetheless, base-free boriranes remain difficult to isolate.
Previous synthetic works have utilized denitrogenation reactions to synthesize three-member rings (i.e., cyclopropanes,11,83,84 aziridines,85 oxiranes,86 and thiiranes87,88). Similarly, Dowd described the photodenitrogenation of a five-membered ring, 4-methylene-Δ-pyrazoline, targeting the formation of trimethylenemethane; this highly reactive intermediate rapidly cyclized to produce cyclopropane at temperatures >77 K (Scheme 1a).83 Here, we present a proof of concept for using the labile azo group in a similar denitrogenation reaction toward a base-free borirane. We hypothesize that irradiation of a boron derivative of 4-methylene-Δ-pyrazoline, 1-borodiazene (bd), would promote N2 extrusion and produce 1-methylborirane, 5 (Scheme 1b).
 | ||
| Scheme 1 (a) Photodenitrogenation of 4-methylene-Δ-pyrazoline towards a 1,3-diyl and subsequent cycloaddition towards functionalized five-member ring (3) and/or ring-closure towards methyl-cyclopropane (4). (b) Proof-of-concept photodenitrogenation reaction of 1-methyl-borodiazene (bd) towards base-free 1-methylborirane (5). | ||
We conducted a computational investigation of the denitrogenation mechanism of bd using multiconfigurational (complete active space self-consistent field, CASSCF) and single-reference (time-dependent density functional theory, TD-DFT) quantum chemical methods. In this study, we comprehensively outlined the reaction pathways for the photochemical N2 extrusion of bd towards 5 using our open-source Python rapid artificial intelligence ab initio molecular dynamics (PyRAI2MD), which enabled quantum-mechanical nonadiabatic molecular dynamics (QM-NAMD).
Results and discussion
Active space and characterization of excitation on the optimized global minimum
The first step in the proposed photochemical denitrogenation is the absorption of a photon by bd, inducing electronic excitation. Therefore, we initially calculated the photophysical properties—vertical excitation energies and nature of transitions—using TD-DFT and EOM-CCSD. We carried out TD-DFT calculations using CAM-B3LYP-GD3(BJ) and ωB97X-D with the cc-pVDZ basis set. The predicted vertical excitation energy for the ground-state-optimized structure of bd, designated as bd-S0, was calculated to be 3.71 eV with CAM-B3LYP-GD3(BJ)/cc-pVDZ and 3.72 eV with ωB97X-D/cc-pVDZ. Both methods indicated that the S0 → S1 excitation is a nN → πNN* transition, with an oscillator strength of 0.003. To ensure the reliability of our TD-DFT results and due to the absence of experimental spectra, we compare them against EOM-CCSD calculations, which are considered a gold standard25,26,89 in excited-state single-reference computations.26,27 We calculated the S0 → S1 vertical excitation energy to be 4.07 eV using EOM-CCSD/cc-pVDZ. These results demonstrate that the TD-DFT values are 0.36 eV and 0.35 eV lower in energy than the EOM-CCSD values (CAM-B3LYP-GD3(BJ)/cc-pVDZ and ωB97X-D/cc-pVDZ, respectively).We used the complete active space self-consistent field (CASSCF) method and CASSCF with a second-order perturbation theory (CASPT2), both of which require an active space selection. We examined the orbital transitions predicted by TD-DFT and EOM-CCSD, and those involved in the photodenitrogenation of bd, to identify which orbitals to incorporate into the active space. We included twelve electrons distributed across 11 orbitals in the active space of bd; Fig. S1 shows the orbitals and their average orbital occupancies.
We compared the CASSCF and CASPT2 excitation energies and orbital transitions with those computed using single-reference methods (i.e., TD-DFT and EOM-CCSD) to ensure an accurate characterization of vertical excitation energies with the CASSCF(12,11) active space. When the vertical excitation energies and the nature of the transitions align between multiconfigurational and single-reference methods, this indicates that the selected active space sufficiently captures the photophysical properties of the molecule (i.e., bd-S0). Due to the lack of experimental spectra for bd, we used the gold-standard26,89 EOM-CCSD method as a reference. The vertical excitation energy for bd-S0 was calculated using state-averaged CAS(12,11) over the first five singlet states with the cc-pVDZ basis set (i.e., SA5-CASSCF(12,11)/cc-pVDZ) to be 4.50 eV. This indicates that the SA5-CASSCF(12,11)/cc-pVDZ method overestimates the S0 → S1 vertical excitation energy by 0.43 eV compared to EOM-CCSD/cc-pVDZ results. Additionally, it overestimates the S0 → S1 vertical excitation energy relative to CAM-B3LYP-GD3BJ/cc-pVDZ and ωB97X-D/cc-pVDZ by 0.79 and 0.78, respectively. The discrepancies observed in the SA5-CASSCF(12,11)/cc-pVDZ calculation and single-reference methods arise from the omission of dynamical correlation in the former, which the CASPT2 method corrects. For the CASPT2 correction, the calculation was performed as SA5-CASPT2(12,11)/cc-pVDZ//SA5-CASSCF(12,11)/cc-pVDZ; the S0 → S1 vertical excitation energy was 3.90 eV. This value is 0.17 eV lower than the EOM-CCSD/cc-pVDZ results and 0.19 eV and 0.18 eV higher than those predicted by CAM-B3LYP-D3(BJ)/cc-pVDZ and ωB97X-D/cc-pVDZ, respectively. The differences in vertical excitation energies among TD-DFT, EOM-CCSD, and CASPT2 arise because single-reference methods lack the static electronic correlation needed for multiconfigurational descriptions, which CASPT2 includes. This missing static correlation becomes more evident in states with multiconfigurational electronic character (i.e., bd-S0; see Fig. S1 for further discussion), underscoring the importance of using the CASSCF and CASPT2 methods for our photochemical study. The CASSCF and CASPT2 methods classify the S0 → S1 excitation as an nN → πNN* transition; this nature of the transition aligns with the TD-DFT and EOM-CCSD results. Table 1 summarizes these findings.
| Method | State | Energy (eV) | Wavelength (nm) | Oscillator strength | Nature |
|---|---|---|---|---|---|
| CAM-B3LYP-D3(BJ)/cc-pVDZ | S1 | 3.71 | 334 | 0.003 | nN → πNN* |
| S2 | 4.24 | 292 | 0.000 | πNN → 2pB | |
| S3 | 6.31 | 196 | 0.000 | nN → πNN* | |
| ωB97X-D/cc-pVDZ | S1 | 3.72 | 333 | 0.003 | nN → πNN* |
| S2 | 4.29 | 289 | 0.000 | πNN → 2pB | |
| S3 | 6.29 | 197 | 0.000 | nN → πNN* | |
| EOM-CCSD/cc-pVDZ | S1 | 4.07 | 305 | 0.004 | nN → πNN* |
| S2 | 4.47 | 277 | 0.000 | πNN → 2pB | |
| S3 | 6.58 | 188 | 0.000 | nN → πNN* | |
| SA5-CASSCF(12,11)/cc-pVDZ | S1 | 4.50 | 276 | 0.010 | nN → πNN* |
| S2 | 4.67 | 265 | 0.000 | πNN → 2pB | |
| S3 | 6.82 | 182 | 0.000 | nN → πNN* | |
| SA5-CASPT2(12,11)/cc-pVDZ//SA5-CASSCF(12,11)/cc-pVDZ | S1 | 3.90 | 318 | nN → πNN* | |
| S2 | 4.32 | 287 | πNN → 2pB | ||
| S3 | 5.57 | 223 | nN → πNN* |
After comparing the orbital transitions, vertical excitation energies, and oscillator strengths across CASSCF(12,11), CASPT2(12,11), TD-DFT, and EOM-CCSD methods, we determined that calculations using CAS(12,11), as illustrated in Fig. S1, effectively capture the vertical excitation energies of bd at equilibrium (i.e., bd-S0). Consequently, we employed SA5-CASSCF(12,11)/cc-pVDZ for all the following calculations.
Predicted absorption spectra and S1 minimum energy path
To confirm bd has an accessible S0 to S1 excitation, we calculate its absorption spectrum. We generated 500 Wigner-sampled non-equilibrium geometries based on the ground state frequencies of bd (Fig. 3a) and calculated the vertical excitation energies [S0 → Sn (n = 0–4)] for each structure using MS-CASPT2(12,11)/cc-pVDZ//CAS(12,11)/cc-pVDZ. We consolidate the results of the 500 vertical excitation calculations into the computed spectra shown in Fig. 3b. | ||
| Fig. 3 (a) The top view (top) and side view (bottom) overlay of 500 non-equilibrium structures from the Wigner ensemble of bd. (b) The predicted absorption spectrum was obtained by averaging the 500 vertical-excitation energy calculations. The absorption intensity is normalized to the peak with the highest oscillator strength, S1. All calculations utilized the MS-CASPT2(12,11)/cc-pVDZ//CASSCF(12,11)/cc-pVDZ method. | ||
The calculated spectrum exhibits three prominent peaks at 325 nm, 308 nm, and 200 nm, along with a minor peak at 188 nm. The red peak at 325 nm corresponds to the S0 → S1 (nN → πNN*) transition and is the most intense, despite being spectroscopically forbidden. The blue peak, centered at 308 nm, corresponds to an S0 → S2 (nN → 2pB) transition; this peak has the second-highest intensity and significant overlap with the S0 → S1 (nN → πNN*) peak. The purple peak at 200 nm corresponds to the S0 → S4 (nN → 2pB) transition; it has the lowest intensity of the three major peaks. The green peak at 188 nm corresponds to the S0 → S3 (nN → πNN*) transition. Its intensity is minimal compared to the other peaks [i.e., S0 → Sn (n = 1, 2, and 4)]; it significantly overlaps with the S0 → S4 peak. Like S0 → S1, the S0 → S3 transition is also spectroscopically forbidden (i.e., nN → πNN*). The variation in intensity between the S0 → S1 and S0 → S3 peaks arises from the different nN orbitals involved in the electronic transitions (i.e., out-of-phase and in-phase nN orbitals, respectively). The out-of-phase nN orbital in the S0 → S1 transition shows greater overlap with the πNN* orbital compared to the in-phase nN orbital in the S0 → S3 excitation (See SI Fig. S3). This leads to the S0 → S1 being the bright state, while the S0 → S3 peak has the lowest intensity of all the peaks. The S0 → S1 (nN → πNN*) and S0 → S2 (nN → 2pB) transitions are accessible via a 254 nm ultraviolet light source. In contrast, the S0 → S3 (nN → πNN*) and S0 → S4 (nN → 2pB) transitions are inaccessible because the required excitation energy exceeds the energy of the photon source. The overlap and accessibility of S0 → S1 (nN → πNN*) and S0 → S2 (nN → 2pB) peaks suggest a role in the photodenitrogenation of bd. We will focus on these excitations and their photochemistry.
We then focused on elucidating the photodenitrogenation mechanism. Since the S1 state is the accessible bright state, we calculated the minimum energy path (MEP) to find the steepest descent along the S1 surface, starting from the Franck–Condon point of bd-S0. We hypothesized that the diazo double bond (πNN) will elongate along the S1-MEP due to nN → πNN* excitation. Fig. 4 shows the S1-MEP, ground state geometry (bd-S0), final MEP geometry (bd-MEP-8), and the S1 minimum (bd-S1).
 | ||
| Fig. 4 (a) The calculated SA5-CASPT2(12,11)/cc-pVDZ minimum energy path of bd along the S1 surface. (b) The out-of-plane angle θB quantifies boron pyramidalization. The plane is defined by C8–B3–C11 (shown in red), with an orthogonal vector from this plane and the carbon of the methyl group defining the measured angle, θBP (highlighted in green). (c) The ground state structure, optimized with SA5-CASSCF(12,11)/cc-pVDZ, bd-S0. (d) The final step of the MEP, bd-MEP-8, and (e) the optimized S1 structure, bd-S1. The corresponding diazo double bond length (πNN) and boron pyramidalization (θBP) values are shown below each structure. | ||
Fig. 4 demonstrates the MEP along the S1 surface and the energies corresponding to the S0–S4 states; it includes 8 geometries [bd-MEP-X (X = 1–8)]. We observe that in the initial two steps of the S1-MEP (i.e., bd-MEP-1 and bd-MEP-2), the S1 surface is energetically close to the S2 surface; the S2–S1 energy gaps are 0.46 eV and 0.21 eV, respectively. The small S2–S1 energy gaps in bd-MEP-1 and bd-MEP-2 suggest a rapid crossing between the S1 and S2 surfaces immediately outside the Franck–Condon point. The S1-MEP converges to a structure, bd-MEP-8, 3.83 eV above bd-S0. At bd-MEP-8, the S1–S0 energy gap is 2.23 eV; this large gap indicates there is no significant coupling between the S1 and S0 surfaces (i.e., no S1/S0 crossing). Therefore, the steepest descent on the S1 surface leads toward a radiative decay channel rather than the proposed photodenitrogenation. The two geometries, bd-S0 and bd-MEP-8, shown in Fig. 4c and d have πNN bond lengths of 1.26 Å and 1.18 Å, respectively, reflecting a decrease of 0.08 Å from the Franck–Condon geometry to bd-MEP-8. Instead of the expected elongation of the πNN bond, we saw boron undergo a conformational change, shifting from a planar conformation in bd-S0 (Fig. 4d) to a pyramidalized conformation in bd-MEP-8 (Fig. 4e). These structures—bd-S0 and bd-MEP-8—demonstrate that boron pyramidalization outcompetes the elongation of the πNN bond along the S1-MEP. We attribute boron pyramidalization to an increase in electron occupancy in the 2pB orbital, along the S1-MEP, despite the initial nN → πNN* excitation.
To measure the extent of boron pyramidalization, we defined an out-of-plane angle, θBP, where a planar boron (unoccupied 2pB orbital) has a θBP of 90°, and an ideally pyramidalized boron (occupied 2pB orbital) has a θBP outside of 75°–105° (Fig. 4b). bd-S0 and bd-MEP-8 show θBP values of 89° and 69°, respectively. The 20° decrease in θBP from bd-S0 to bd-MEP-8 indicates a full pyramidalization of the boron along the S1-MEP. This boron pyramidalization occurs alongside the previously observed 0.08 Å reduction in the πNN bond length. The concurrent shortening of the πNN bond and boron pyramidalization along the S1 MEP suggests that the 2pB orbital is gaining electron occupancy along the steepest S1 path. This is supported by the notable overlap of the S0 → S1 (nN → πNN*) and S0 → S2 (nN → 2pB) peaks observed in the absorption spectra (Fig. 3b), suggesting that the S0 → S2 (nN → 2pB) transition is energetically accessible. These findings are in line with a charge transfer between the πNN* and 2pB orbitals.
Due to the large S1/S0 energy gap at bd-MEP-8, we optimized an S1 minimum, bd-S1, using the last step of the MEP as an initial guess. bd-S1 has a πNN of 1.18 Å and θBP of 69°; it is 3.61 eV above the bd-S0. These values show decreases of 0.08 Å and 20° for πNN and θBP, respectively, from bd-S0 to bd-S1. These measurements indicate that the boron in bd-S1 is pyramidalized relative to bd-S0. bd-S1 and bd-MEP-8 shared πNN and θBP values (1.18 Å and 69°, respectively) and had similar energies (3.83 eV and 3.61 eV, respectively), indicating that these structures occupy similar regions of the potential energy surface. At bd-S1, a 2.26 eV S1/S0 energy gap confirms a nonradiative decay channel via the S1 MEP, contradicting the hypothesis of nonradiative photodenitrogenation to 5. Although the S1-MEP offers detailed insight along the steepest S1-path, it neglects essential dynamical effects required to study the excited-state mechanism of bd.
We accounted for the missing dynamical effects by running quantum-mechanical nonadiabatic molecular dynamics (QM-NAMD) simulations using our open-source Python Rapid Artificial Intelligence Ab Initio Molecular Dynamics (PyRAI2MD) software.22,23 We generated 700 initial conditions for production trajectories using Wigner sampling based on bd-S0 frequencies. The fewest-switches surface-hopping (FSSH) algorithm captured nonadiabatic transitions over 1 ps with analytical nonadiabatic coupling vectors. A trajectory was considered complete if denitrogenation and an S1/S0 hop occurred, or if it reached 1 ps. The maximum energy drift allowed for these trajectories was 0.05 a.u.; 261 trajectories met this criterion. After 1 ps, 244 (94%) trajectories were on S0 surface, and 16 (6%) remained on the S1 state. Of those passing through an S1/S0 hop, 218 underwent denitrogenation to 5 (73) or a diradical, 6 (145). The remaining 26 either stayed as reactants (13) or formed diazoborete, 7 (13) (Scheme 2).
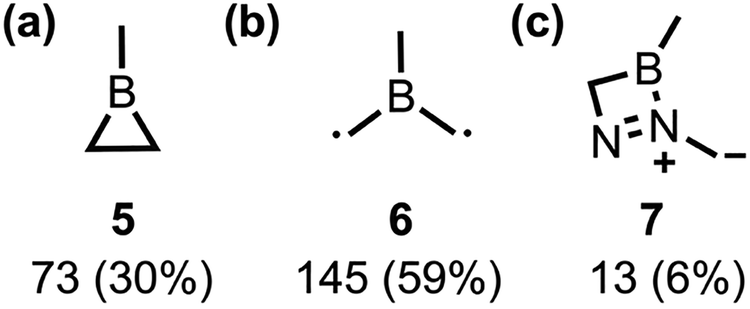 | ||
| Scheme 2 Product distribution of productive trajectories within our ensemble. The observed products include (a) borirane (5), (b) diradical (6), and (c) diazoborete (7). Below each structure are the number of trajectories leading to each product and their percentages (i.e., quantum yields). | ||
To investigate the photodenitrogenation mechanism, we monitored the two breaking CN bonds (σCN) throughout our ensemble of trajectories. We propose that denitrogenation may proceed through two possible mechanistic pathways: either both CN bonds break simultaneously (concerted), or one CN bond breaks first, followed shortly by the other (stepwise). We hypothesize that bd undergoes stepwise denitrogenation due to increased flexibility from nN → πNN* excitation, enabling πNN rotation (i.e., partial cis-to-trans isomerization) and σCN bond cleavage. We plotted the two σCN bond lengths from 244 trajectories ending in the ground state in Fig. 5. Additionally, two arbitrary profiles are shown as dashed lines, representing the ideal stepwise (red) and concerted (gray) denitrogenation pathways within this trajectory map.
 | ||
Fig. 5 Trajectory map of the photodenitrogenation reaction of bd. Each line represents a single trajectory, with black dots marking S1/S0 hopping points. We tracked the two CN bonds (σCN) that must break for photodenitrogenation during the NAMD simulations. To illustrate an ideal stepwise denitrogenation mechanism, we drew an arbitrary bilinear profile with red dashed lines indicating ±0.1 Å around the optimized C–N distance (1.54 Å) in the S0 state. A trajectory would follow this bilinear profile, with one CN bond elongating while the other stays within the red boundaries. Once the first CN bond breaks, the second CN bond then elongates and breaks as well. To depict an ideal concerted denitrogenation mechanism, we included an arbitrary 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 line, indicating that both CN bonds elongate simultaneously until denitrogenation occurs. :1 line, indicating that both CN bonds elongate simultaneously until denitrogenation occurs. | ||
In Fig. 5, both concerted and stepwise mechanisms are present, indicating that neither pathway dominates; instead, both are readily accessible for the denitrogenation reaction of bd. To clearly classify the denitrogenation mechanism in the ensemble of trajectories, we used the literature definition of dynamically stepwise or dynamically concerted mechanism, as reported by Yang et al.90 We determined that of the 244 trajectories that terminate in the S0 state, 218 undergo denitrogenation. 60 (28%) followed a dynamically stepwise mechanism, while 158 (72%) followed a dynamically concerted mechanism. Thus, denitrogenation of bd is mainly driven by the dynamically concerted mechanism.
Having established that the dynamically concerted mechanism (72%) outcompetes the dynamically stepwise mechanism (28%), we now examine the consistent boron pyramidalization in static (bd-S1 and bd-MEP-8) and dynamic (QM-NAMD) calculations. The pyramidilized boron in these geometries indicates that the 2pB orbital acquires additional electron occupancy along the S1 surface from the πNN* (i.e., a charge transfer). We hypothesize that trajectories will adopt one of two geometries (i.e., boron pyramidalization or partial cis-to-trans πNN isomerization) on the S1 surface, and those adopting boron pyramidalization will not denitrogenate.
To measure the extent of boron pyramidalization and partial cis-to-trans πNN isomerization in the denitrogenation mechanism within our trajectory ensemble, we used the geometrical parameters and thresholds presented in Fig. 6: θBP and θCNNC.
 | ||
| Fig. 6 (a) The out-of-plane angle θBP is defined as the angle between the methyl-boron and the normal vector (green) to the plane formed by the atoms highlighted in red. We defined a planar boron with θBP values of 75°–105°; otherwise, we classified the boron as pyramidilized. (b) The dihedral angle, θCNNC, is defined by the C–N–N–C (orange); we classify the cis-πNN as having θCNNC values from −25° to 25°, while other configurations are considered partial trans-πNN. | ||
Based on the geometrical descriptions shown in Fig. 6, we observed that 177 trajectories (82%) exhibit boron pyramidalization, while 182 (83%) undergo a partial cis-to-trans πNN isomerization. Out of 218 trajectories that underwent denitrogenation, 120 trajectories included both geometries, 37 only experienced boron pyramidalization, and 59 only experienced partial πNN isomerization. These statistics show that bd can undergo denitrogenation via either the boron pyramidalization or the partial πNN isomerization pathway; however, the major pathway includes both.
Out of 120 trajectories that experience both geometries, 12 initially undergo partial πNN isomerization, while 108 first experience boron pyramidalization. The πNN isomerization and boron pyramidalization conformations are linked to the electronic occupancy of the πNN* orbital and the 2pB orbital, respectively. The frequent occurrence of both geometries is associated with the near degeneracy between the S1 and S2 surfaces in the Franck–Condon region, which correspond to the nN → πNN* and nN → 2pB transitions, respectively. Furthermore, the favorable spatial overlap between the πNN* and 2pB orbitals facilitates charge transfer, thereby enabling the adoption of the associated conformations (i.e., partial πNN isomerization and boron pyramidalization). Statistically, the photodenitrogenation process begins with boron pyramidalization, followed by partial πNN isomerization, and ends with denitrogenation. We identified a trajectory that proceeds through partial πNN isomerization and boron pyramidalization during denitrogenation, characterized by the angles θCNNC and θBP, respectively. Fig. 7 provides a detailed analysis of this trajectory.
 | ||
| Fig. 7 Snapshots of a representative trajectory that undergoes the denitrogenation on the S1 surface via the mechanistic pathway that includes boron pyramidalization and partial cis-to-trans πNN isomerization. | ||
Fig. 7 shows snapshots of a trajectory undergoing denitrogenation via the dominant pathway that involves the partial πNN isomerization and boron pyramidalization. At 0 fs, θBP is 82°, and θCNNC is 10°. At 50 fs, θBP is 139°, and θCNNC is −5°. By 400 fs, θBP and θCNNC are 140° and 1°, respectively. At 500, θBP is 46°, and θCNNC is −16°. At 650 fs, θBP is 124°, and θCNNC is −21°. By 858.5 fs, θBP is 71°, and θCNNC is 57°. At 900 fs, θBP is 114°, and θCNNC is −8°. Between 0 fs and 50 fs, θBP and θCNNC increased by 57° and 15°, respectively. The rapid rise in θBP (57°) with a small decrease in θCNNC (15°) suggests the 2pB orbital gains electron occupancy, driving the mechanism, rather than the πNN* orbital. In the next 350 fs (50 fs to 400 fs), θBP and θCNNC increased by 1° and 6°, respectively, demonstrating that the geometry remains unchanged. We attribute this persistent geometry to the close resemblance between the 50 fs geometry and bd-S1. From 400 fs to 500 fs, θBP decreased by 94°, and θCNNC increased by 17°. The decrease in θBP from 140° to 46° indicates that the trajectory rehybridizes boron from sp2 to sp3, in the opposite direction to the initial pyramidalization. By 650 fs, θBP increased by 78°, and θCNNC decreased by 5°. Like the previous 100 fs window, the changes in θBP and θCNNC for the 500 fs to 650 fs period suggest that the sp3 boron has inverted, while the πNN bond remains in the cis-configuration. The fluctuation in the θBP indicates prolonged occupation of the 2pB orbital, whereas minimal changes in θCNNC suggest that the πNN* orbital remains empty during this period. Within the 650 fs to 858.5 fs time window, the θBP decreased by 53°, and θCNNC increased by 78°; at the 858.5 fs geometry, the trajectory underwent an S1/S0 hop. The lower θBP value (71°) is 4° below our planar boron threshold (75°), while the higher θCNNC (57°) indicates a partial cis-to-trans πNN isomerization. These geometric changes show that the 2pB orbital is essentially empty, while the πNN* orbital is occupied. 41.5 fs (i.e., at 900 fs) after the S1/S0 hop, the trajectory denitrogenates towards the 1,3-diyl. This trajectory shows the mechanistic pathway involving boron pyramidalization and partial πNN isomerization. Boron pyramidalization occurs first, followed by partial πNN isomerization, which leads to denitrogenation. Minor denitrogenation pathways are explored in Fig. S4 and S5.
Minor pathways
After analyzing the mechanisms for the major product, we investigate the pathway to the minor product, diazoborete (7). We identified a trajectory bypassing denitrogenation, leading to 7. To compare with earlier figures, we measured θBP and θCNNC along this trajectory. Thirteen trajectories (6%) showed this rearrangement. Fig. 8 examines a representative trajectory in detail. | ||
| Fig. 8 Snapshots of a representative trajectory that does not involve denitrogenation on the S1 surface; instead, it involves an insertion-like reaction that results in BN bond formation and BC bond cleavage, producing diazoborete (7). | ||
Fig. 8 provides snapshots of the representative trajectory. θBP and θCNNC are reported to facilitate direct comparison with previous figures (Fig. 7 and Fig. S4 and S5). At 0 fs, θBP measures 90° while θCNNC is −10°. By 50 fs, θBP is 83°, whereas θCNNC measures −54°. At 100 fs, θBP is 84°, with θCNNC equal to −35°. By 130 fs, θBP reaches 96°, and θCNNC is −70°. At 150 fs, θBP measures 93°, while θCNNC is at −92°. Finally, at 200 fs, θBP measures 53°, and θCNNC is −150°. Between 0 fs and 50 fs, θBP drops by 7°, and θCNNC decreases by 44°. This suggests that the 2pB orbital remains unoccupied during this period, while the πNN* orbital gains electron occupancy. In the following 50 fs (50 fs to 100 fs), θBP and θCNNC increased by 1° and decreased by 19°, respectively, indicating a prolonged occupancy of the πNN* orbital. From 100 fs to 130 fs, there is a 12° increase in θBP and a 35° decrease in θCNNC. Although the θBP increase is significant, boron remains in a planar conformation, not pyramidalizing per our geometric thresholds. At the 130 fs geometry, the trajectory undergoes an S1/S0 hop. Between 130 fs and 150 fs, the θBP decreases by 3°, while θCNNC decreases by 22°. Due to partial πNN isomerization, the nN orbital aligns with the 2pB orbital, leading to BN bond formation and BC bond cleavage; this rearrangement results in 7. Between 150 fs and 200 fs, the θBP drops by 40°, and θCNNC decreases by 58°. At this stage, the reference plane for θBP and the dihedral θCNNC have increased conformational freedom due to the rearrangement, rendering both measurements geometrically meaningless. This structure remains in the ground state and undergoes further rearrangements, as shown in Fig. S3.
Conclusion
This study provides an enumeration of the mechanistic pathways for the photochemical denitrogenation of 1-methylborodiazene (bd) towards base-free 1-methylborirane (5). We employed static multiconfigurational quantum-mechanical computations, combined with quantum–mechanical nonadiabatic molecular dynamics (QM-NAMD). The static MEP calculation showed a pyramidalizing (bd-MEP-8) on the S1-surface and optimized bd-MEP-8 to an S1-minimum. Static calculations indicate that the S1-MEP leads to radiative decay instead of photochemical denitrogenation. Static calculations, therefore, omit dynamical effects. As such, we used our open-source software, PyRAI2MD, to perform QM-NAMD simulations. Our QM-NAMD trajectories show that the denitrogenation reaction mainly follows a dynamically concerted mechanism (72%), involving two key geometries: partial πNN isomerization and boron pyramidalization. Of 218 trajectories, 120 (55%) involved both, 37 (17%) had only boron pyramidalization, and 59 (27%) only partial cis-to-trans πNN isomerization. As such, we concluded that denitrogenation can occur through either the boron pyramidalization pathway or the partial πNN isomerization path, but the dominant pathway involves both geometries. We attribute the high prevalence of these geometries to the near-degeneracy between S2- and S1-states near the Franck–Condon region. The S0 → S1(nN → πNN*) and S0 → S2(nN → 2pB) transitions cause bd to undergo conformational changes, such as partial πNN isomerization or boron pyramidalization, thereby facilitating denitrogenation. Most trajectories (71%) experience denitrogenation on the S0 surface, while a smaller portion (29%) fully denitrogenates on the S1 surface. We predicted quantum yields of 21% for 5 and 67% for 6.Computational methods
Single reference methods
All density functional electronic structure methods (i.e., DFT and TD-DFT) used Gaussian 16.B01.91 We used DFT to optimize the initial ground-state global minimum of bd using the PBE0/cc-pVDZ method. We used time-dependent density functional theory (TD-DFT) to calculate the vertical excitation energies of bd using the CAM-B3LYP-GD3(BJ) and ωB97X-D functionals with the cc-pVDZ basis set. We used Gaussian 16.B0191 to run the equation of motion coupled-cluster single doubles (EOM-CCSD) with the cc-pVDZ basis set.Multiconfigurational methods
We employed the state-average complete active space self-consistent field (SA-CASSCF) method, as implemented in OpenMolcas 19.11.92 This approach uses the format SA(N)-CASSCF(m,n), where N denotes the number of singlet states averaged in the calculation, while m and n denote the number of electrons and orbitals in the active space. Implementing the CASSCF method poses challenges, including appropriate active space selection and the inherent computational expense. The size of the active space is intrinsically connected to computational cost, scaling O(C(2n,m)) where n is the number of orbitals and m is the number of electrons.93 To the best of our knowledge, the current maximum active space size in contemporary computing hardware is 18 electrons distributed across 18 orbitals in OpenMolcas92 and NWChem.94,95Quantum mechanical nonadiabatic molecular dynamics
To incorporate the missing dynamical effects from previous static calculations—such as vertical excitation energies, computed absorption spectra, and the minimum energy path—we performed quantum mechanical nonadiabatic molecular dynamics (QM-NAMD) simulations. We generated 700 initial conditions for production trajectories using Wigner sampling based on bd-S0 frequencies. All computations were performed using OpenMolcas 19.1192 through our open-source software, PyRAI2MD.22,23 Trajectory propagation employed our SA5-CASSCF(12,11)/cc-pVDZ electronic-structure method with the Verlet algorithm, using a 0.5 fs time step. The fewest-switches surface-hopping (FSSH) algorithm tracked nonadiabatic transitions over 1 ps, utilizing analytical nonadiabatic coupling vectors. All trajectories were launched from the S1 surface; a trajectory was deemed complete if denitrogenation and an S1/S0 hop occurred, or if it reached 1 ps. Trajectories that had a maximal energy drift exceeding 0.05 a.u. were eliminated from the analysis. In total, 261 trajectories satisfied these criteria.Conflicts of interest
There are no conflicts of interest to declare.Data availability
The ML-photodynamics simulation code is open-sourced and released at: https://github.com/mlcclab/PyRAI2MD-hiam. The complete quantum mechanical data and initial conditions can be found at: https://doi.org/10.6084/m9.figshare.29294228.Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d6cp00519e.
Acknowledgements
The National Science Foundation Center under NSF-CHE-2144556 supported this work. We appreciate the assistance from the Northeastern Research Computing Team and the computing resources provided by the Massachusetts Life Science Center grant (G00006360). We dedicate this manuscript to Professor David Yarkony, who contributed 47 years to advancing theoretical chemistry and was very supportive of our research.References
- S. Cho, J. Usuba, S. Chakraborty, X. Li and G. G. D. Han, Solid-state photon energy storage via reversible [2 + 2] cycloaddition of donor-acceptor styrylpyrylium system, Chem, 2023, 9(11), 3159–3171, DOI:10.1016/j.chempr.2023.06.007.
- E. Skolia and C. G. Kokotos, Photochemical [2 + 2] Cycloaddition of Alkenes with Maleimides: Highlighting the Differences between N-Alkyl vs N-Aryl Maleimides, ACS Org. Inorg. Au, 2023, 3(2), 96–103, DOI:10.1021/acsorginorgau.2c00053.
- M. C. Paderes, M. Jeffrey Diaz, C. A. Pagtalunan, D. A. Bruzon and G. A. Tapang, Photo-Controlled [4 + 4] Cycloaddition of Anthryl-Polymer Systems: A Versatile Approach to Fabricate Functional Materials, Chem. – Asian J., 2022, 17(12), e202200193, DOI:10.1002/asia.202200193.
- F. Mukadum, Q. Nguyen, D. M. Adrion, G. Appleby, R. Chen, H. Dang, R. Chang, R. Garnett and S. A. Lopez, Efficient Discovery of Visible Light-Activated Azoarene Photoswitches with Long Half-Lives Using Active Search, J. Chem. Inf. Model., 2021, 61(11), 5524–5534, DOI:10.1021/acs.jcim.1c00954.
- X. Li, S. Cho, J. Wan and G. G. D. Han, Photoswitches and photochemical reactions for optically controlled phase transition and energy storage, Chem, 2023, 9(9), 2378–2389, DOI:10.1016/j.chempr.2023.05.029.
- H. Lee, J. Tessarolo, D. Langbehn, A. Baksi, R. Herges and G. H. Clever, Light-Powered Dissipative Assembly of Diazocine Coordination Cages, J. Am. Chem. Soc., 2022, 144(7), 3099–3105, DOI:10.1021/jacs.1c12011.
- D. M. Adrion, W. V. Karunaratne and S. A. Lopez, Multiconfigurational photodynamics simulations reveal the mechanism of photodecarbonylations of cyclopropenones in explicit aqueous environments, Chem. Sci., 2023, 14(45), 13205–13218, 10.1039/d3sc03805j.
- D. J. Shields, D. P. Karothu, K. Sambath, R. Ranaweera, S. Schramm, A. Duncan, B. Duncan, J. A. Krause, A. D. Gudmundsdottir and P. Naumov, Cracking under Internal Pressure: Photodynamic Behavior of Vinyl Azide Crystals through N(2) Release, J. Am. Chem. Soc., 2020, 142(43), 18565–18575, DOI:10.1021/jacs.0c07830.
- S. Datta and H. F. Davis, Photodissociation Dynamics of the Simplest Diazirine: Cyclo-CH(2)N(2) → CH(2) + N(2), J. Phys. Chem. A, 2025, 129(15), 3498–3507, DOI:10.1021/acs.jpca.5c00801.
- C. Hui, S. Wang and C. Xu, Dinitrogen extrusion from diazene in organic synthesis, Chin. Chem. Lett., 2022, 33(8), 3695–3700, DOI:10.1016/j.cclet.2022.03.073.
- T. Y. Chang, D. M. Adrion, A. R. Meyer, S. A. Lopez and M. A. Garcia-Garibay, A Green Chemistry Approach toward the Stereospecific Synthesis of Densely Functionalized Cyclopropanes via the Solid-State Photodenitrogenation of Crystalline 1-Pyrazolines, J. Org. Chem., 2022, 87(5), 2277–2288, DOI:10.1021/acs.joc.1c01808.
- S. Zhu, L. Jia, Q. Cheng, Q. Sun, X. Chen, H. Yu, Y. Han and H. Hou, Visible-Light-Induced Stereoselective Radical trans-Iodoalkylation of Terminal Alkyne with Iodoform, Org. Lett., 2024, 26(39), 8400–8404, DOI:10.1021/acs.orglett.4c03241.
- L. Pierau and D. L. Versace, Light and Hydrogels: A New Generation of Antimicrobial Materials, Materials, 2021, 14(4), 787, DOI:10.3390/ma14040787.
- M. López-Cano, M. Scortichini, D. K. Tosh, V. Salmaso, T. Ko, G. Salort, I. Filgaira, C. Soler, D. Trauner and J. Hernando, et al., Photoswitchable Diazocine Derivative for Adenosine A3 Receptor Activation in Psoriasis, J. Am. Chem. Soc., 2025, 147(1), 874–879, DOI:10.1021/jacs.4c13558.
- R. A. Homan, J. D. Lapek, C. M. Woo, S. Niessen, L. H. Jones and C. G. Parker, Photoaffinity labelling with small molecules, Nat. Rev. Methods Primers, 2024, 4(1), 30, DOI:10.1038/s43586-024-00308-4.
- F. J. Hernandez, J. M. Cox, J. Li, R. Crespo-Otero and S. A. Lopez, Multiconfigurational Calculations and Photodynamics Describe Norbornadiene Photochemistry, J. Org. Chem., 2023, 88(9), 5311–5320, DOI:10.1021/acs.joc.2c02758.
- P. J. Scott, C. R. Kasprzak, K. D. Feller, V. Meenakshisundaram, C. B. Williams and T. E. Long, Light and latex: advances in the photochemistry of polymer colloids, Polym. Chem., 2020, 11(21), 3498–3524, 10.1039/d0py00349b.
- T. Y. Xu, F. Tong, H. Xu, M. Q. Wang, H. Tian and D. H. Qu, Engineering Photomechanical Molecular Crystals to Achieve Extraordinary Expansion Based on Solid-State [2 + 2] Photocycloaddition, J. Am. Chem. Soc., 2022, 144(14), 6278–6290, DOI:10.1021/jacs.1c12485.
- D. M. Gatlin, W. L. Karney, M. Abe, B. S. Ault and A. D. Gudmundsdottir, Formation and Reactivity of Triplet Vinylnitrenes as a Function of Ring Size, J. Org. Chem., 2019, 84(14), 9215–9225, DOI:10.1021/acs.joc.9b01191.
- A. E. Ondrus and T. Zhang, Structure, Bonding, and Photoaffinity Labeling Applications of Dialkyldiazirines, Synlett, 2021,(11), 1053–1059, DOI:10.1055/a-1437-8202.
- A. A. Poloukhtine, N. E. Mbua, M. A. Wolfert, G.-J. Boons and V. V. Popik, Selective Labeling of Living Cells by a Photo-Triggered Click Reaction, J. Am. Chem. Soc., 2009, 131(43), 15769–15776, DOI:10.1021/ja9054096.
- S. Kobatake and D. Kitagawa, Photoresponsive Molecular Crystals for Light-Driven Photoactuators, in Photosynergetic Responses in Molecules and Molecular Aggregates, ed. H. Miyasaka, K. Matsuda, J. Abe and T. Kawai, Springer, Singapore, 2020, pp. 427–447 Search PubMed.
- A. Luque, J. Paternoga and T. Opatz, Strain Release Chemistry of Photogenerated Small-Ring Intermediates, Chemistry, 2021, 27(14), 4500–4516, DOI:10.1002/chem.202004178.
- F. Glarner, S. R. Thornton, D. Schärer, G. Bernardinelli and U. Burger, 6-Azabicyclo[3.1.0]hex-30-en-2-ol Derivative, photochemically generated building blocks for bicyclic β-lactams, Helv. Chim. Acta, 1997, 80(1), 121–127, DOI:10.1002/hlca.19970800112 (acccessed 2025/07/11).
- T. Jira, J. Janos and P. Slavicek, Sensitivity Analysis in Photodynamics: How Does the Electronic Structure Control cis-Stilbene Photodynamics?, J. Chem. Theory Comput., 2024, 20(24), 10972–10985, DOI:10.1021/acs.jctc.4c01008.
- J. M. Cox, M. Bain, M. Kellogg, S. E. Bradforth and S. A. Lopez, Role of the Perfluoro Effect in the Selective Photochemical Isomerization of Hexafluorobenzene, J. Am. Chem. Soc., 2021, 143(18), 7002–7012, DOI:10.1021/jacs.1c01506.
- M. Ueda, K. Jorner, Y. M. Sung, T. Mori, Q. Xiao, D. Kim, H. Ottosson, T. Aida and Y. Itoh, Energetics of Baird aromaticity supported by inversion of photoexcited chiral [4n]annulene derivatives, Nat. Commun., 2017, 8(1), 346, DOI:10.1038/s41467-017-00382-1.
- J. Yan, T. Slanina, J. Bergman and H. Ottosson, Photochemistry Driven by Excited-State Aromaticity Gain or Antiaromaticity Relief, Chemistry, 2023, 29(19), e202203748, DOI:10.1002/chem.202203748.
- F. Hemauer, H. P. Steinruck and C. Papp, The Norbornadiene/Quadricyclane Pair as Molecular Solar Thermal Energy Storage System: Surface Science Investigations, Chem. Phys. Chem., 2024, 25(9), e202300806, DOI:10.1002/cphc.202300806.
- N. M. Salkho, N. S. Awad, W. G. Pitt and G. A. Husseini, Photo-Induced Drug Release from Polymeric Micelles and Liposomes: Phototriggering Mechanisms in Drug Delivery Systems, Polymers, 2022, 14(7), 1286, DOI:10.3390/polym14071286.
- B. Sana, A. Finne-Wistrand and D. Pappalardo, Recent development in near infrared light-responsive polymeric materials for smart drug-delivery systems, Mater. Today Chem., 2022, 25, DOI:10.1016/j.mtchem.2022.100963.
- S. K. Choi, M. Verma, J. Silpe, R. E. Moody, K. Tang, J. J. Hanson and J. R. Baker Jr., A photochemical approach for controlled drug release in targeted drug delivery, Bioorg. Med. Chem., 2012, 20(3), 1281–1290, DOI:10.1016/j.bmc.2011.12.020.
- A. Petti, M. J. Karrasch, P. Chahar, F. H. Wessels, N. Holter, F. Boser, C. G. Daniliuc and F. Glorius, Cyclic Bifunctional Reagents Enabling a Strain-Release-Driven Formal [3 + 2] Cycloaddition of 2H-Azirines by Cascade Energy Transfer, J. Am. Chem. Soc., 2025, 147(16), 13276–13285, DOI:10.1021/jacs.4c18080.
- N. Rauscher, L. Naesborg, C. Jandl and T. Bach, Concise Total Synthesis of Agarozizanol B via a Strained Photocascade Intermediate, Angew. Chem., Int. Ed., 2021, 60(45), 24039–24042, DOI:10.1002/anie.202110009.
- J. Huang, S. Y. You, L. Z. Hu, Y. H. He and Z. Guan, One-Pot Photocascade Catalysis: Access to Pyrrole Derivatives from N-Arylglycines and Morita-Baylis-Hillman (MBH) Acetates, Org. Lett., 2024, 26(47), 10195–10200, DOI:10.1021/acs.orglett.4c04176.
- H. Maag, D. J. Lemcke and J. M. Wahl, Ring opening of photogenerated azetidinols as a strategy for the synthesis of aminodioxolanes, Beilstein J. Org. Chem., 2024, 20, 1671–1676, DOI:10.3762/bjoc.20.148.
- A. Luque, J. Gross, T. J. B. Zahringer, C. Kerzig and T. Opatz, Vinylcyclopropane [3 + 2] Cycloaddition with Acetylenic Sulfones Based on Visible Light Photocatalysis, Chemistry, 2022, 28(18), e202104329, DOI:10.1002/chem.202104329.
- M. Laktsevich-Iskryk, A. Hurski, M. Oseka and D. Kananovich, Recent advances in asymmetric synthesis via cyclopropanol intermediates, Org. Biomol. Chem., 2025, 23(5), 992–1015, 10.1039/d4ob01746c.
- D. R. Mishra and N. P. Mishra, Recent breakthroughs in ring-opening annulation reactions of aziridines, Org. Biomol. Chem., 2025, 23(13), 2967–2996, 10.1039/d4ob01577k.
- I. Vilotijevic and T. F. Jamison, Epoxide-opening cascades in the synthesis of polycyclic polyether natural products, Angew. Chem., Int. Ed., 2009, 48(29), 5250–5281, DOI:10.1002/anie.200900600.
- V. Palani and A. E. Wendlandt, Strain-Inducing Positional Alkene Isomerization, J. Am. Chem. Soc., 2023, 145(36), 20053–20061, DOI:10.1021/jacs.3c06935.
- S. Ma, D. Mandalapu, S. Wang and Q. Zhang, Biosynthesis of cyclopropane in natural products, Nat. Prod. Rep., 2022, 39(5), 926–945, 10.1039/d1np00065a.
- X. Ju, M. Lee, J. C. Leung and J. Lee, Synthesis and Functionalization of Aziridines: A Perspective View from Pharmaceutical Industries, Eur. J. Org. Chem., 2025, 28(15), e202401414, DOI:10.1002/ejoc.202401414.
- M. Hanif, A. F. Zahoor, M. J. Saif, U. Nazeer, K. G. Ali, B. Parveen, A. Mansha, A. R. Chaudhry and A. Irfan, Exploring the synthetic potential of epoxide ring opening reactions toward the synthesis of alkaloids and terpenoids: a review, RSC Adv., 2024, 14(19), 13100–13128, 10.1039/d4ra01834f.
- J. Herzberger, K. Niederer, H. Pohlit, J. Seiwert, M. Worm, F. R. Wurm and H. Frey, Polymerization of Ethylene Oxide, Propylene Oxide, and Other Alkylene Oxides: Synthesis, Novel Polymer Architectures, and Bioconjugation, Chem. Rev., 2016, 116(4), 2170–2243, DOI:10.1021/acs.chemrev.5b00441.
- S. S. Uthumange, A. J. H. Liew, X. W. Chee and K. Y. Yeong, Ringing medicinal chemistry: The importance of 3-membered rings in drug discovery, Bioorg. Med. Chem., 2024, 116, 117980, DOI:10.1016/j.bmc.2024.117980.
- N. Wang, J.-X. Zhao and J.-M. Yue, Total synthesis of cyclopropane-containing natural products: recent progress (2016–2024, Org. Chem. Front., 2025, 12(7), 2439–2456, 10.1039/d4qo02316a.
- J. Marco-Contelles, M. T. Molina and S. Anjum, Naturally Occurring Cyclohexane Epoxides: Sources, Biological Activities, and Synthesis, Chem. Rev., 2004, 104(6), 2857–2900, DOI:10.1021/cr980013j.
- S. J. Maurer, J. L. Petrarca de Albuquerque and M. E. McCallum, Recent Developments in the Biosynthesis of Aziridines, ChemBioChem, 2024, 25(16), e202400295, DOI:10.1002/cbic.202400295.
- C. J. Thibodeaux, W. C. Chang and H. W. Liu, Enzymatic chemistry of cyclopropane, epoxide, and aziridine biosynthesis, Chem. Rev., 2012, 112(3), 1681–1709, DOI:10.1021/cr200073d.
- H. Wang, S. Huangfu, D. Wei, Z. Sun, Y. Wu, X. Yu, B. Jiang and H. Chen, Triptolide-based cleavable antibody-drug conjugates for pancreatic cancer, Eur. J. Med. Chem., 2025, 295, 117798, DOI:10.1016/j.ejmech.2025.117798.
- A. G. Herraiz and M. G. Suero, New Alkene Cyclopropanation Reactions Enabled by Photoredox Catalysis via Radical Carbenoids, Synthesis, 2019,(14), 2821–2828, DOI:10.1055/s-0037-1611872.
- X. Han, N. Zhang, Q. Li, Y. Zhang and S. Das, The efficient synthesis of three-membered rings via photo- and electrochemical strategies, Chem. Sci., 2024,(34), 13576–13604, 10.1039/d4sc02512a.
- H. E. Simmons and R. D. Smith, A new synthesis of cyclopropanes from olefins, J. Am. Chem. Soc., 1958, 80(19), 5323–5324, DOI:10.1021/ja01552a080.
- J. Xu, X. Li and N. Chen, An Improved and Mild Wenker Synthesis of Aziridines, Synthesis, 2010,(20), 3423–3428, DOI:10.1055/s-0030-1257913.
- H. Wenker, The Preparation of Ethylene Imine from Monoethanolamine, J. Am. Chem. Soc., 1935, 57(11), 2328, DOI:10.1021/ja01314a504.
- V. V. Thakur and A. Sudalai, N-Bromoamides as versatile catalysts for aziridination of olefins using chloramine-T, Tetrahedron Lett., 2003, 44(5), 989–992, DOI:10.1016/S0040-4039(02)02729-6.
- Q. He, M. P. Pu, Z. Jiang, H. Wang, X. Feng and X. Liu, Asymmetric Epoxidation of Alkenes Catalyzed by a Cobalt Complex, J. Am. Chem. Soc., 2023, 145(28), 15611–15618, DOI:10.1021/jacs.3c05476.
- Y. Steudel, R. Steudel and M. W. Wong, The thermal decomposition of thiirane: a mechanistic study by ab initio MO theory, Chemistry, 2002, 8(1), 217–228, DOI:10.1002/1521-3765(20020104)8:1<217::aid-chem217>3.0.co;2-0.
- H. Kudo, K. Naritomi, S. Onishi, H. Maekawa, E. A. Q. Mondarte, K. Suthiwanich and T. Hayashi, Living Ring-Expansion Polymerization of Thiirane with Cyclic Monocarbamothioates, Macromolecules, 2020, 53(12), 4733–4740, DOI:10.1021/acs.macromol.0c00687.
- S. M. Song, J. Jin, J. H. Choi and W. J. Chung, Synthesis of cis-thiiranes as diastereoselective access to epoxide congeners via 4pi-electrocyclization of thiocarbonyl ylides, Nat. Commun., 2022, 13(1), 4818, DOI:10.1038/s41467-022-32499-3.
- A. Ghosh, T. H. Van Nguyen, C. Bellanger, S. Chelli, M. Ahmad, N. Saffon-Merceron, C. Taillier, V. Dalla, R. J. Mayer and I. M. Dixon, et al., Unraveling C-Selective Ring-Opening of Phosphiranes with Carboxylic Acids and Other Nucleophiles: A Mechanistically-Driven Approach, Angew. Chem., Int. Ed., 2025, 64(2), e202414172, DOI:10.1002/anie.202414172.
- T. Xin, M. B. Geeson, H. Zhu, Z. W. Qu, S. Grimme and C. C. Cummins, Synthesis of phosphiranes via organoiron-catalyzed phosphinidene transfer to electron-deficient olefins, Chem. Sci., 2022, 13(43), 12696–12702, 10.1039/d2sc05011k.
- X. Li, K. D. Robinson and P. P. Gaspar, A New Stereoselective Synthesis of Phosphiranes, J. Org. Chem., 1996, 61(22), 7702–7710, DOI:10.1021/jo9608533.
- F. A. D. Herz, M. Nobis, D. Wendel, P. Pahl, P. J. Altmann, J. Tillmann, R. Weidner, S. Inoue and B. Rieger, Application of multifunctional silylenes and siliranes as universal crosslinkers for metal-free curing of silicones, Green Chem., 2020, 22(14), 4489–4497, 10.1039/d0gc00272k.
- Y. Pan, S. Morisako, S. Aoyagi and T. Sasamori, Generation of Bis(ferrocenyl)silylenes from Siliranes, Molecules, 2020, 25(24), 5917, DOI:10.3390/molecules25245917.
- W. S. Palmer and K. A. Woerpel, Stereospecific Palladium-Catalyzed Reactions of Siliranes with Alkynes, Organometallics, 1997, 16(6), 1097–1099, DOI:10.1021/om960923w.
- J. Dong and J. Xu, Facile synthesis of thietanes via ring expansion of thiiranes, Org. Biomol. Chem., 2017, 15(4), 836–844, 10.1039/c6ob02387h.
- K. Banert, M. Chityala and M. Korb, Ring Enlargement of Three-Membered Heterocycles by Treatment with In Situ Formed Tricyanomethane, Chemistry, 2020, 26(28), 6158–6164, DOI:10.1002/chem.202000089.
- H. I. Kim, A. Veeramanoharan, B. Selvaraj, M. Olivier, E. Lee, J. W. Lee and C. M. Park, Thiiranes: Intelligent Molecules for S-Persulfidation, J. Am. Chem. Soc., 2024, 146(13), 8820–8825, DOI:10.1021/jacs.3c12908.
- H. Klusik and A. Berndt, A Boron-Carbon Double Bond, Angew. Chem., Int. Ed. Engl., 1983, 22(11), 877–878, DOI:10.1002/anie.198308771.
- U. M. Dzhemilev, L. I. Khusainova, K. S. Ryazanov and L. O. Khafizova, Boron-containing small rings: synthesis, properties, and application prospects, Russ. Chem. Bull., 2021, 70(10), 1851–1892, DOI:10.1007/s11172-021-3292-2.
- T. R. McFadden, C. Fang, S. J. Geib, E. Merling, P. Liu and D. P. Curran, Synthesis of Boriranes by Double Hydroboration Reactions of N-Heterocyclic Carbene Boranes and Dimethyl Acetylenedicarboxylate, J. Am. Chem. Soc., 2017, 139(5), 1726–1729, DOI:10.1021/jacs.6b09873.
- S. E. Denmark, K. Nishide and A. M. Faucher, On the generation and configurational stability of (2S,3S)-1,2,3-triphenylborirane, J. Am. Chem. Soc., 1991, 113(17), 6675–6676, DOI:10.1021/ja00017a050.
- C. Baik, Z. M. Hudson, H. Amarne and S. Wang, Enhancing the Photochemical Stability of N,C-Chelate Boryl Compounds: C−C Bond Formation versus CC Bond cis,trans-Isomerization, J. Am. Chem. Soc., 2009, 131(40), 14549–14559, DOI:10.1021/ja906430s.
- Y.-L. Rao, H. Amarne, S.-B. Zhao, T. M. McCormick, S. Martić, Y. Sun, R.-Y. Wang and S. Wang, Reversible Intramolecular C−C Bond Formation/Breaking and Color Switching Mediated by a N,C-Chelate in (2-ph-py)BMes2 and (5-BMes2-2-ph-py)BMes2, J. Am. Chem. Soc., 2008, 130(39), 12898–12900, DOI:10.1021/ja8052046.
- Y. L. Rao, L. D. Chen, N. J. Mosey and S. Wang, Stepwise intramolecular photoisomerization of NHC-chelate dimesitylboron compounds with C-C bond formation and C-H bond insertion, J. Am. Chem. Soc., 2012, 134(26), 11026–11034, DOI:10.1021/ja304211v.
- H. Braunschweig, C. Claes, A. Damme, A. Deissenberger, R. D. Dewhurst, C. Horl and T. Kramer, A facile and selective route to remarkably inert monocyclic NHC-stabilized boriranes, Chem. Commun., 2015, 51(9), 1627–1630, 10.1039/c4cc09036e.
- J. Wang and Q. Ye, Borirenes and Boriranes: Development and Perspectives, Chemistry, 2024, 30(11), e202303695, DOI:10.1002/chem.202303695.
- W. Dai, S. J. Geib and D. P. Curran, Ring-Opening Reactions of NHC-Boriranes with In Situ Generated HCl: Synthesis of a New Class of NHC-Boralactones, J. Am. Chem. Soc., 2019, 141(8), 3623–3629, DOI:10.1021/jacs.8b13010.
- H. Wang, J. Zhang and Z. Xie, Ring-opening and ring-expansion reactions of carborane-fused borirane, Chem. Sci., 2021, 12(39), 13187–13192, 10.1039/d1sc04453b.
- J. Wang, L. Xiang, X. Liu, A. Matler, Z. Lin and Q. Ye, Avenue to novel o-carboranyl boron compounds - reactivity study of o-carborane-fused aminoborirane towards organic azides, Chem. Sci., 2024, 15(13), 4839–4845, 10.1039/d4sc00489b.
- P. Dowd, Trimethylenemethane, Acc. Chem. Res., 1972, 5(7), 242–248, DOI:10.1021/ar50055a003.
- J. L. García Ruano, M. T. Peromingo, M. R. Martín and A. Tito, A New Entry to Enantiopure Polysubstituted Cyclopropanes: Stereoselective Denitrogenation of Sulfinylpyrazolines under Yb(OTf)3 Catalysis, Org. Lett., 2006, 8(15), 3295–3298, DOI:10.1021/ol061168d.
- D. de Loera and M. A. Garcia-Garibay, Efficient Aziridine Synthesis in Metastable Crystalline Phases by Photoinduced Denitrogenation of Crystalline Triazolines, Org. Lett., 2012, 14(15), 3874–3877, DOI:10.1021/ol301582n.
- Z. Wang, J. Wen, Q.-W. Bi, X.-Q. Xu, Z.-Q. Shen, X.-X. Li and Z. Chen, Oxirane synthesis from diazocarbonyl compounds via NHC-Ag+ catalysis, Tetrahedron Lett., 2014, 55(18), 2969–2972, DOI:10.1016/j.tetlet.2014.03.105.
- Z.-X. Yu and Y.-D. Wu, An SN2-like Transition State for Alkene Episulfidation by Dinitrogen Sulfide, J. Org. Chem., 2003, 68(15), 6049–6052, DOI:10.1021/jo034027n.
- W. Adam and R. M. Bargon, Episulfidation of Strained Cycloalkenes in the Thermolysis of 5-Aryloxy-1,2,3,4-thiatriazoles, Eur. J. Org. Chem., 2001,(10), 1959–1962, DOI:10.1002/1099-0690(200105)2001:10<1959::Aid-ejoc1959>3.0.Co;2-n.
- S. J. Bennie, B. F. E. Curchod, F. R. Manby and D. R. Glowacki, Pushing the Limits of EOM-CCSD with Projector-Based Embedding for Excitation Energies, J. Phys. Chem. Lett., 2017, 8(22), 5559–5565, DOI:10.1021/acs.jpclett.7b02500.
- Z. Yang, C. S. Jamieson, X.-S. Xue, M. Garcia-Borràs, T. Benton, X. Dong, F. Liu and K. N. Houk, Mechanisms and Dynamics of Reactions Involving Entropic Intermediates, Trends Chem., 2019, 1(1), 22–34, DOI:10.1016/j.trechm.2019.01.009.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, et al., Gaussian 16 Rev. B.01. 2016 Search PubMed.
- I. Fdez Galvan, M. Vacher, A. Alavi, C. Angeli, F. Aquilante, J. Autschbach, J. J. Bao, S. I. Bokarev, N. A. Bogdanov and R. K. Carlson, et al., OpenMolcas: From Source Code to Insight, J. Chem. Theory Comput., 2019, 15(11), 5925–5964, DOI:10.1021/acs.jctc.9b00532.
- F. Aquilante, J. Autschbach, R. K. Carlson, L. F. Chibotaru, M. G. Delcey, L. De Vico, I. Fdez Galvan, N. Ferre, L. M. Frutos and L. Gagliardi, et al., Molcas 8: New capabilities for multiconfigurational quantum chemical calculations across the periodic table, J. Comput. Chem., 2016, 37(5), 506–541, DOI:10.1002/jcc.24221.
- D. Mejia-Rodriguez, E. Apra, J. Autschbach, N. P. Bauman, E. J. Bylaska, N. Govind, J. R. Hammond, K. Kowalski, A. Kunitsa and A. Panyala, et al., NWChem: Recent and Ongoing Developments, J. Chem. Theory Comput., 2023, 19(20), 7077–7096, DOI:10.1021/acs.jctc.3c00421.
- J. Greiner, I. Gianni, T. Nottoli, F. Lipparini, J. J. Eriksen and J. Gauss, MBE-CASSCF Approach for the Accurate Treatment of Large Active Spaces, J. Chem. Theory Comput., 2024, 20(11), 4663–4675, DOI:10.1021/acs.jctc.4c00388.
| This journal is © the Owner Societies 2026 |