
Superconductivity in topological Ψ-graphene
Xiaolong
Yu
a,
Zhongyan
Lu
a,
Zhaopeng
Guo
b and
Kang
Xia
 *a
*a
aCollege of Science, Nanjing Forestry University, Longpan Rd 159, Nanjing, 210037, China. E-mail: kangxia@njfu.edu.cn
bSchool of Physics, Hangzhou Normal University, Yuhangtang Rd 2318, Hangzhou, 311121, China
First published on 22nd December 2025
Abstract
Typical Dirac cones in graphene induce the absence of electronic density around the Fermi energy level (Ef), prohibiting intrinsic superconductivity. Here, we tested the theoretical superconducting properties of graphene after introducing pentagonal and heptagonal carbon rings into the structure. Generally, 5–7 polygons of a metastable Ψ-graphene monolayer break the hexagonal symmetry to form type-II Dirac cones by band crossings. The polyhedral structure maintains integrity under high temperatures. The large specific surface area of the Ψ-graphene monolayer facilitates the physical adsorption of NO molecules. Weak interactions of atomic bonding and antibonding features coexist with the Bader charge transfer in the carbon monolayer in close proximities to one another. The collective vibrations of carbon, nitrogen, and oxygen atoms provide good dynamic stability of the Ψ-graphene–NO adsorption system. In the Ψ-graphene monolayer, the shift of Dirac cones leads to the formation of visible Fermi surfaces, which motivates further investigation into their influences on the superconducting properties. Out-of-plane and in-plane carbon vibrations are attributed to phonon modes in mediation with electron couplings. After computing the Eliashberg function, we evaluated strong electron–phonon coupling, with the superconducting transition temperature reaching 22 K. These theoretical predictions can stimulate interests in exploring topological graphene allotropes of intrinsic superconductivity.
Introduction
Superconductivity (SC) in graphene significantly broadens its application in electronic and quantum devices,1–4 with high carrier mobility and quantum Hall effect.5,6 However, the hexagonal honeycomb structure of mirror symmetry protects the Dirac-type band structure, prohibiting the emergence of SC.7 Twisted graphene bilayer superlattices with a “magic-angle” (≈1.1°) produce robust SC at extremely low temperatures (<3 K).8–10 Flat bands appear around zero Fermi energy, which results in half-filling correlated insulating states. Experimental observations of quantum oscillations confirm small Fermi surfaces under a perpendicular magnetic field of 0.4 T. Superconducting proximity effects enhance spin–orbit interactions in the heterostructure of single-layer WSe2 and bernal bilayer graphene.11–13 Interestingly, the superconducting state of bilayer graphene can be observed even at zero magnetic field.Beyond bilayer Moiré superlattices and bernal graphene, SC is found in ABC-stacked trilayer graphene.14 By tuning electrostatic gates under laser excitation, infrared-active phonon vibrations have demonstrated strong coupling strength with electrons. Another route to induce superconductivity in graphitic systems involves electron doping via alkali or alkaline-earth intercalation (e.g., Li, Na, K, Ca, and Sr), where reduced interlayer spacing enhances electron–phonon coupling (EPC).15–22 Larger deformation potentials with closer distances between doped metal atoms and the graphite plane (h) lead to stronger electron–phonon coupling (EPC). For example, the superconducting transition temperature (TC) in bulk graphite intercalation compounds (GICs) increases from 1.65 K for SrC6 (h = 2.47 Å) to 11.5 K for CaC6 (h = 2.26 Å).16–20 A comparable h value of 2.32 Å for the GIC NaC4 produces π–electron coupling with low-frequency vibrations, with an estimated TC value of 41.2 K under a hydrostatic pressure of 5 GPa.22 However, quantum confinement between the carbon monolayer and adatom layers prevents π–electron occupation of the interlayer band and suppresses intrinsic SC in the GIC LiC6 within the closest h distance of 1.85 Å.
Nevertheless, Li deposition on graphene removes the quantum confinement from bulk GICs. Li-covered graphene in single and double side constructs of LiC6 and Li2C6 structures have TC values of 8.1 and 18 K, respectively.20 A downshift of the intercalant state to the Fermi energy level (Ef) is conducive to electron occupation for strong coupling between intercalant π electrons and carbon out-of-plane vibrations. Superconducting-gap measurements using angle-resolved photoemission spectroscopy provide further evidence regarding the superconducting enhancement of Li-decorated graphene LiC6, with a TC value of 5.9 K,21 which is close to the predicted value of 8.1 K. A tensile-strain level of 16.5% loaded for graphene is theoretically combined with a doping concentration of 4.65 × 1014 cm−2 for electrons and holes.23 EPC is enhanced with enlarged Fermi surfaces to achieve a high TC value approaching 30 K. Using diverse carbon rings as building blocks, various structural prediction methods such as AIRSS,24 USPEX,25–27 CALYPSO,28–30 MAGUS,31–33 and RG234 have been applied to predict abundant graphene allotropes with sp2 and sp3 hybridizations.35–41 The characteristics of Dirac dispersions, such as tilt, Fermi-surface morphology, and nesting are reported to strongly affect the mode-resolved electron–phonon coupling and pairing interaction.42–44 These anisotropic electronic responses are of particular interest in carbon allotropes with distorted Dirac cones. Especially, anisotropic Dirac topology is exhibited in PHA,38 SW,39 POP,40 and penta-graphene.41 Structure patterns specifically containing five-, seven-, and eight-fold rings break the hexagonal symmetry of a typical honeycomb structure. Robust metallicity emerges from delocalized π bands crossing over Ef.40,45,46 However, potential intrinsic SC combined with Dirac topology has been rarely investigated among the metallic graphene allotropes.
In this work, we intensively studied the electronic topology and intrinsic SC of the previously predicted psi (Ψ)-graphene structure.45 We evaluated the topological Dirac cone features around Ef. Phonon dispersions demonstrated dynamic stability of Ψ-graphene under the physical adsorption of nitric oxide (NO) gas molecules. The electrons strongly coupled with carbon out-of-plane and in-plane vibrations, and Ψ-graphene demonstrated strong intrinsic SC with a TC value of approximately 22 K.
Computational methods
We performed first-principles calculations using the Vienna ab initio simulation package (VASP)47 based on density functional theory (DFT). We used the Perdew–Burke–Ernzerhof exchange–correlation functional (PBE)48 under the generalized gradient approximation combined in VASP code. We set the total-energy cutoff to 600 eV, reaching a convergence level of 1 × 10−6 eV on each atom and a force of 5 × 10−3 eV Å−1 per atom. We sampled a k mesh of 2π × 0.03 Å−1 in the Brillouin zone (BZ). We accurately optimized the atom positions inner carbon plane, with the constant vacuum thickness reaching 25 Å along the normal direction. By adding the D4 package49,50 to the PBE functional (PBE-D4), we calculated the adsorption energy regarding van der Waals (vdW) interactions between the carbon sheet and gas molecules. We utilized Lobster software51 to calculate the crystal orbital Hamilton population (COHP).52 Weak interactions between adjacent atomic orbitals were quantitatively analyzed for bonding and antibonding contributions. We used a Bayesian-learning algorithm employed in VASP to generate on-the-fly machine learning force fields (MLFF).53–56 We updated the MLFF from ab initio calculations when the Bayesian-error estimation exceeded a precise threshold for force error of 2 × 10−3 eV Å−1 per atom. High-quality MLFF was employed in molecular dynamics simulations to discuss the stability of the large-scale structure under finite temperatures. We executed the tight-binding model from Wannier90 package57 and WannierTools58 to plot and analyze the topological band gap in the full BZ, respectively. By employing Quantum Espresso (QE) program,59 electron–phonon coupling was computed by interpolation within the Monkhorst–Pack q grid of (4 × 1 × 3) sampling over the BZ. We chose the norm-conserving pseudopotential for the carbon atom with a kinetic energy cutoff of 100 and 400 Rydberg (Ry) for wavefunctions and charge density, respectively. We calculated electronic Fermi surfaces within the eight-dense k grid of (16 × 4 × 12). We set the iterative threshold of 1 × 10−14 for the self-consistent calculation. We further solved the Eliashberg function and obtained the electron phonon coupling coefficient (λ) and superconducting transition temperature TC using the McMillan formula.60Results and discussion
Ψ-graphene sheets constructed with 5–6–7 carbon rings exhibited the space-group symmetry of Pmma (Number 51) with vacuum thickness of 25 Å. Four Wyckoff-position (WP) sites were occupied by carbon atoms labeled as C1/C11 (equivalent atomic positions), C2/C3/C9/C10, C4/C6/C7/C12, and C5/C8 in Fig. S1 and Table S1 of the SI. The bond lengths between neighboring carbon atoms ranged from 1.409 to 1.503 Å, with lattice constants of a = 4.833 Å and c = 6.705 Å. The Ψ-graphene structure possessed a total energy of −9.174 eV atom−1, which was between the energies of Dirac cone bearing allotropes SW (−9.191 eV atom−1) and PHA graphene (−9.133 eV atom−1) (Table S2). The intermediate energy state for Ψ-graphene indicates possible thermodynamic meta-stability.We investigated the structural stability of polygonal Ψ-graphene at high temperatures of 1500 K (Fig. 1) and 300 K (Fig. S2) through machine learning molecular dynamics simulations (MLMD). We extracted the averaged supercell of (3 × 1 × 3) as the initial structure from the last 1 picosecond (ps) during ab initio molecular dynamics (AIMD) simulations for the NVT ensemble61 used for MLFF training. We further updated MLFF from AIMD using the NPT ensemble62,63 with a simulation time of 5 ps.64 A large (4 × 1 × 4) scale of the initial supercell system containing over 1700 carbon atoms was extended to employ efficient MLFF into MLMD with a total simulation time of 30 ps. Bayesian linear regression applied in MLMD samples a smoothed potential energy surface by learning the variances of the predictions from ab initio data. High-frequency thermal noise can be effectively suppressed with smaller temperature fluctuations. As shown in the left panels of Fig. 1(a–c), MLMD simulations achieved equilibrium states after 2 ps. Small fluctuations for temperature (T), pressure (P), and total energy (E) parameters reached a range of ±100 K, ±1 GPa, and ±0.01 eV atom−1, respectively, at around 1500 K and 0 GPa. We suggest that MLMD of the large-scale system exhibits greater accuracy for estimations of T, P, and E than AIMD [right panels in Fig. 1(a–c)]. The first two peaks of the pair distribution function g(r) were located at carbon–carbon distances of 1.43 and 2.47 Å, which indicated the closest and next-closest carbon atoms, respectively [red curve in Fig. 1(d)]. The neighboring carbon in the polyhedral structure was consistent with the pure AIMD NPT simulation (black curve). Furthermore, the averaged large-scale supercell structure confirms that Ψ-graphene maintains integrity at 1500 K. After cooling to room temperature (300 K), the structure remained stable, and the 5–6–7 ring configuration was well preserved (Fig. S2). Efficient MLMD calculations illustrate excellent structure stability for the Ψ-graphene monolayer under high temperature.
 | ||
| Fig. 1 The evolution of (a) temperature, (b) pressure, and (c) total energy during MLMD (30 ps, left panel) and AIMD (12 ps, right) simulations. (d) Pair distribution function [g(r)] versus atomic distance at 1500 K. The inset graph is the averaged structure sketch obtained from the last 2 ps of MLMD. | ||
The Ψ-graphene sheet composed of 5–6–7 rings was predicted to have a larger specific surface area approaching 2.71 m2 mg−1 than pristine graphene (2.66 m2 mg−1). The enlarged surface area potentially favors physical gas adsorption performance. To investigate the adsorption capability, we optimized the adsorption configurations by constructing a (3 × 1 × 3) Ψ-graphene supercell with seven types of gas molecules. We took into consideration parallel (‖) and perpendicular (⊥) configurations for each molecule of H2, O2, N2, CO2, NO, NH3, and CO. Specifically, we used “H” to represent the center above the hollow site of carbon polygons and “C” to indicate the top site of WP carbon atoms. For instance, the adsorption site (AS) “H5” occupies the pentagonal center [Fig. S1(b)], while the AS “C1” represents the top site of WP C1 [Table S1 and Fig. S1(c)]. The initial distance (d) between the carbon monolayer and the molecule was set to 3.5 Å to calculate physical adsorption [Fig. S3(a–c)] and further to 1.1, 1.3, and 1.5 Å [Fig. S3(d–f)] to compute “chemical adsorption”. We defined the adsorption energy (Eads) as shown below: Eads = Etot − Emon − Emol. Here, Etot, Emon, and Emol represent the total energy of the adsorption system, Ψ-graphene monolayer, and isolated molecule, respectively. We obtained negative Eads for optimized adsorption states of Ψ-graphene-molecule systems by performing PBE-D4 calculations [Fig. 2(a)]. Among all molecules, we predicted that NO was the easiest to be adsorbed on Ψ-graphene sheet with the lowest Eads reaching −0.687 eV. O2 adsorption exhibited the next lowest Eads of −0.549 eV. Detailed calculations at shorter distances (d = 1.1, 1.3, and 1.5 Å) illustrated consistent Eads results for physical NO adsorption on Ψ-graphene, although there were slightly different ASs after in-plane optimization [Fig. S3(d–f)].
 | ||
| Fig. 2 (a) Adsorption energy by structure optimization for an initial distance of 3.5 Å. (b) Calculated phonon dispersion (left panel) and phonon density of states (PDOS, right) for NO adsorption of Ψ-graphene. (c) Bader charge transfer and adsorption distance between Ψ-graphene and adsorbed gas molecules. −pCOHP and −ICOHP results for Ψ-graphene–NO adsorption with a d of around (d) 2.2 Å between WP C4 and N, and (e) stretching d to 4.4 Å. | ||
The adsorption of NO molecules especially affects the vibrations of neighboring carbon in Ψ-graphene. We computed the phonon dispersions [left panel in Fig. 2(b)] and phonon projected density of states (PDOS, right panel) for the Ψ-graphene–NO system in the AS “C4-⊥” state. Compared with phonon dispersions of pristine Ψ-graphene (Fig. S4), carbon vibrations in the normal direction maintained the main contributions to phonon dispersions within a frequency range of 400–830 cm−1. However, the oxygen (O) atom mainly contributed to flat acoustic branches at low-frequency around 35 cm−1 [red curve in Fig. 2(b)]. Vibrations of nitrogen (N) atoms in the normal direction participated in phonon optical branches around 250 and 350 cm−1, as shown in the blue curve in Fig. 2(b). The opposing vibration along the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond resulted in the highest frequency of phonon mode, which approached 1840 cm−1 (red). The in-plane carbon vibrations from WP C1 and C2 atoms were suppressed below a frequency of 1600 cm−1 (Fig. S4). These calculations indicate that monolayer Ψ-graphene exhibits superior adsorption performance for the NO molecule, which is accompanied with significant phonon softening and hardening effects.
O bond resulted in the highest frequency of phonon mode, which approached 1840 cm−1 (red). The in-plane carbon vibrations from WP C1 and C2 atoms were suppressed below a frequency of 1600 cm−1 (Fig. S4). These calculations indicate that monolayer Ψ-graphene exhibits superior adsorption performance for the NO molecule, which is accompanied with significant phonon softening and hardening effects.
As shown in Fig. 2(c), Ψ-graphene adsorption of O2 or NO molecules at low total energy takes advantage of the close distance d (black triangles) for Bader charge transfer (red solid dots). Specifically, almost 1.85 and 0.03e of Bader charge are transferred from O2 and NO molecules, respectively, to C2, with d reaching 2.24 Å for NO. In particular, there are weak atomic interactions between neighboring C and N in the adsorption process. Bonding and antibonding from carbon 2p orbitals were quantitatively calculated by the projected COHP (pCOHP) and integral COHP (ICOHP) [Fig. 2(d)]. The vdW interactions mainly originate from the C bonding state of the pristine Ψ-graphene monolayer in an energy range of −2.8 eV below Ef, with the −ICOHP peak reaching 0.79 eV [black area in Fig. 2(d)]. The N p orbital was attributed to the antibonding state in an energy range of 3.0 eV above the Ef. By doubly stretching the distance to 4.4 Å between WP C2 and the N atom [Fig. 2(e)], the positive −pCOHP values decreased (black) and the negative values disappeared (red) in an energy range between −7.0 to 4.5 eV. Decrement of the −ICOHP value indicates weaker atomic interactions for bonding and antibonding states. We suggest that this graphene allotrope serves as an excellent sensor for NO gas, with weak interactions in close proximity to Bader charge transfer.
The adsorption event with non-covalent interactions in the charge transfer distance perturbs the electronic band structures.65 To further explore the superconducting properties after NO adsorption, we calculated the band structure along the high-symmetry line (HSL) of Γ–X–Z–Γ–Y–Z–Γ and partial density of states (pDOS) for NO adsorption (Fig. 3) onto pristine Ψ-graphene (Fig. S5). Upon NO physisorption in close proximity to the WP C4 site for carbon atom, pDOS revealed that the nitrogen and oxygen atoms contributed to the band dispersions in an energy-level range of ±1 eV. New Fermi surfaces open around Γ–Y and X–Z directions with elevated Ef, as shown in the inset graph in Fig. 3. Consequently, band dispersions attributed to C4 p electrons are repelled to be flattened around 1 eV above Ef along Z–Γ–Y–Z–Γ for the NO–Ψ-graphene system. Nevertheless, we suggest that the adsorption system still characterizes potential superconductivity with new Fermi surfaces under band regulations by NO physical adsorption.
 | ||
| Fig. 3 (a) Band structure and (b) partial density of states (pDOS) for NO adsorption of Ψ-graphene. The inset graph in (a) is the Fermi surface. | ||
We observed an oblique band crossing along the HSL Y–Z at energy levels of about 0.2 eV below Ef [Fig. S5(a)]. The isoenergy surface at the band-crossing point (down) showed high similarity with the Fermi surface (up), as shown in the insets of Fig. S5(a). We suggest that Dirac electrons participate in the formation of electron (red) and hole-pocket (green) types of intrinsic Fermi surfaces. The tight-binding models were constructed by s and p orbitals of C atoms using the Wannier90 package. The band crossing along HSL Y–Z particularly suggests potential topological semimetal features in the Ψ-graphene monolayer. We verified six type-II Dirac cones with the π Berry phase around the Y point marked as red and blue dots in Fig. 4(a). Two Dirac cones (marked by red dots) located at HSL Y–Z were connected with each other by mirror symmetry My of the same energy. Four Dirac cones of blue dots were connected by My and glide mirror symmetry Gx. After plotting band gaps in the BZ using WannierTools [Fig. 4(b)], there were no other band crossings in the full BZ. However, mediation with the adsorbed NO molecule breaks the original mirror symmetry in Ψ-graphene, which leads to the disappearance of the band crossing along the Y–Z path [Fig. 3(a)].
 | ||
| Fig. 4 (a) Energy dispersion around the Y point. The Dirac cones are marked by red and blue dots. (b) Band gap distribution in the full BZ, with the energy band dispersions around Y shown in the inset. | ||
Unique Fermi surfaces inspire us to explore EPC SC in the metallic Ψ-graphene monolayer. After solving the Eliashberg function of α2F(ω), we estimated the EPC parameter λ of 1.32 and logarithmic average frequency ωlog of 218.3 K [Fig. 5(a)] using the following formulas:
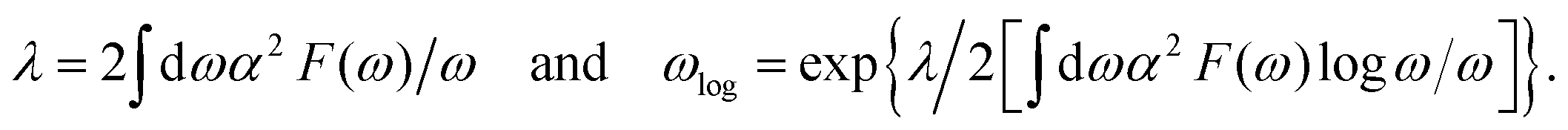
 | ||
| Fig. 5 (a) Calculations for EPC superconductivity, including the Eliashberg function α2F(ω), EPC (λ), logarithmic average frequency (ωlog), and superconducting transition temperature (TC). (b) TC value as a function of the Coulomb pseudopotential µ* and the parameter σ. The black dots denote the calculated data points, the red solid curve highlights the µ* = 0.10 across different σ, and the black solid line connects the data points at fixed σ = 0.60 across different µ*. | ||
The collective vibrations of WP C2 and C5 carbon atoms in the normal direction were attributed to the peaks of the α2F(ω) function (red curve) at phonon frequency ω of 220 and 415 cm−1, respectively (inset graphs). The ωlog value was quickly enhanced to reach about 11.5% and 26.1% of the total value with increasing phonon frequency (blue curve). A high ω > 1470 cm−1 contributed to 16.6% of the total ωlog, which originated from WP C1 in-plane vibrations for the α2F(ω) peak. Phonon modes of low frequency (ω < 220 cm−1) contributed to 68.2% of the total λ (green curve). We conducted TC estimation using the conventional McMillan formula:
| TC = (ωlog/1.2)exp[−1.04(1 + λ)/(λ − μ* − 0.62λμ*)]. |
We systematically tested different σ values for gaussian broadening from 0.02 to 0.60 Ry with the Coulomb pseudopotential µ* from 0.08 to 0.16 to reach the convergence of TC. Fig. 5(b) clearly shows a convergent TC value at a σ value of 0.60 Ry under considerable µ* values. Therefore, we used µ* = 0.10 and σ = 0.60 Ry with the Methfessel–Paxton smearing scheme. The TC value (black) increases with ω, which is consistent with the ωlog curve (blue). Out-of-plane phonon vibrations of low frequency and in-plane modes of high frequency in the Ψ-graphene monolayer were suggested to mediate electron couplings. We propose that Ψ-graphene has a strong intrinsic SC EPC with a TC value approaching 22 K, which is comparable to that of the predicted metastable PHH graphene (20 K).39 However, the broken symmetry of the system for NO-adsorbed Ψ-graphene makes the evaluation of quantities [λ and α2F(ω)] extremely time-consuming by performing QE code. We plan to further investigate this aspect in subsequent work.
Conclusions
Pentagonal and heptagonal carbon rings in Ψ-graphene break the hexagonal symmetry to bring about the emergence of electronic density of states at Ef. The 5–6–7 polyhedral structure remains intact under high temperature up to 1500 K during machine learning molecular dynamics simulations. The large specific surface area potentially facilitates the adsorption of gas molecules. In particular, single-layer Ψ-graphene exhibits the lowest adsorption energy of −0.687 eV for NO. Bader charge transfer occurs at the shortest distance around 2.2 Å. The Ψ-graphene–NO adsorption system possesses dynamic stability that involves weak chemical interactions based on COHP analyses. Band crossings below Ef originate in type-II Dirac cones, illustrating the topological semi-metallicity in Ψ-graphene. Shifted Dirac cones induce visible Fermi surfaces to encourage electron couplings with phonon mediation. We suggest that out-of-plane and in-plane carbon vibrations contribute to the low-frequency (220 and 415 cm−1) and high-frequency (1470 cm−1) phonon modes. We estimate that the electron–phonon coupling constant increases up to 1.32 with a logarithmic average frequency of 218.3 K. The conventional superconducting transition temperature approaches a peak value of 22 K. Therefore, Ψ-graphene possesses intrinsic SC with topological semi-metallicity.Author contributions
Xiaolong Yu: writing – original draft, methodology, investigation, formal analysis, data curation, conceptualization. Zhongyan Lu: methodology, data curation, Zhaopeng Guo: writing – original draft, methodology, visualization, formal analysis, conceptualization. Kang Xia: writing – review & editing, methodology, visualization, validation, supervision, resources, conceptualization.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article describes lattice parameter (Table S1), total energy (Table S2), crystal graphic (Fig. S1), MD simulation at 300 K (Fig. S2), adsorption-energy calculation details (Fig. S3), phonon dispersions and PDOS (Fig. S4), and electronic band structure and pDOS (Fig. S5). The supplementary data have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5cp04499e.Acknowledgements
The authors acknowledge the financial support from the National Natural Science Foundation of China under Grant No. 12004185 and 12304080, the Natural Science Foundation of the Jiangsu Higher Education Institutions of China under Grant No. 20KJB140016, and the Project funded by China Postdoctoral Science Foundation under Grant No. 2019M651767 and 2021M703461. The numerical calculations in this paper were performed on the computing facilities in the High Performance Computing Facility of Nanjing Forestry University.Notes and references
- J. Delahaye, J. Hassel, R. Lindell, M. Sillanpää, M. Paalanen, H. Seppä and P. Hakonen, Low-noise current amplifier based on mesoscopic Josephson junction, Science, 2023, 299(5909), 1045–1048 Search PubMed.
- O. P. Saira, M. Meschke, F. Giazotto, A. M. Savin, M. Möttönen and J. P. Pekola, Heat transistor: Demonstration of gate-controlled electronic refrigeration, Phys. Rev. Lett., 2007, 99(2), 027203 Search PubMed.
- M. Huefner, C. May, S. Kicin, K. Ensslin, T. Ihn, M. Hilke, K. Suter, N. F. de Rooij and U. Staufer, Scanning gate microscopy measurements on a superconducting single-electron transistor, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79(13), 134530 CrossRef.
- S. De Franceschi, L. Kouwenhoven, C. Schönenberger and W. Wernsdorfer, Hybrid superconductor-quantum dot devices, Nat. Nanotechnol., 2010, 5(10), 703–711 CrossRef.
- Y. B. Zhang, Y. W. Tan, H. L. Stormer and P. Kim, Experimental observation of the quantum hall effect and Berry's phase in graphene, Nature, 2005, 438(7065), 201–204 CrossRef.
- K. I. Bolotin, K. J. Sikes, Z. Jiang, M. Klima, G. Fudenberg, J. Hone, P. Kim and H. L. Stormer, Ultrahigh electron mobility in suspended graphene, Solid State Commun., 2008, 146(9–10), 351–355 CrossRef.
- J. M. Park, Y. Cao, K. Watanabe, T. Taniguchi and P. Jarillo-Herrero, Tunable strongly coupled superconductivity in magic-angle twisted trilayer graphene, Nature, 2021, 590(7845), 249–255 CrossRef.
- Y. Cao, V. Fatemi, A. Demir, S. Fang, S. L. Tomarken, J. Y. Luo, J. D. Sanchez-Yamagishi, K. Watanabe, T. Taniguchi, E. Kaxiras, R. C. Ashoori and P. Jarillo-Herrero, Correlated insulator behaviour at half-filling in magic-angle graphene superlattices, Nature, 2018, 556(7699), 80–84 CrossRef.
- Y. Cao, V. Fatemi, S. Fang, K. Watanabe, T. Taniguchi, E. Kaxiras and P. Jarillo-Herrero, Unconventional superconductivity in magic-angle graphene superlattices, Nature, 2018, 556(7699), 43–50 CrossRef PubMed.
- D. M. Kennes, J. Lischner and C. Karrasch, Strong correlations and d plus id superconductivity in twisted bilayer graphene, Phys. Rev. B, 2018, 98(24), 241407 CrossRef.
- C. S. Li, F. Xu, B. H. Li, J. Y. Li, G. A. Li, K. Watanabe, T. Taniguchi, B. B. Tong, J. Shen, L. Lu, J. F. Jia, F. C. Wu, X. X. Liu and T. X. Li, Tunable superconductivity in electron-and hole-doped bernal bilayer graphene, Nature, 2024, 631(8020), 300–306 CrossRef PubMed.
- Y. R. Zhang, R. Polski, A. Thomson, E. Lantagne-Hurtubise, C. Lewandowski, H. X. Zhou, K. Watanabe, T. Taniguchi, J. Alicea and S. Nadj-Perge, Enhanced superconductivity in spin-orbit proximitized bilayer graphene, Nature, 2023, 613(7943), 268–273 CrossRef PubMed.
- L. Holleis, C. L. Patterson, Y. R. Zhang, Y. Vituri, H. M. Yoo, H. X. Zhou, T. Taniguchi, K. Watanabe, E. Berg, S. Nadj-Perge and A. F. Young, Nematicity and orbital depairing in superconducting bernal bilayer graphene, Nat. Phys., 2025, 21(3), 444–450 Search PubMed.
- X. Z. Zan, X. D. Guo, A. L. Deng, Z. H. Huang, L. Liu, F. F. Wu, Y. L. Yuan, J. J. Zhao, Y. L. Peng, L. Li, Y. K. Zhang, X. Z. Li, J. D. Zhu, J. W. Dong, D. X. Shi, W. Yang, X. X. Yang, Z. W. Shi, L. J. Du, Q. Dai and G. Y. Zhang, Electron/infrared-phonon coupling in ABC trilayer graphene, Nat. Commun., 2024, 15(1), 1888 Search PubMed.
- N. B. Hannay, T. H. Geballe, B. T. Matthias, K. Andres, P. Schmidt and D. Macnair, Superconductivity in graphitic compounds, Phys. Rev. Lett., 1965, 14(7), 225–226 CrossRef.
- N. Emery, C. Hérold, M. d'Astuto, V. Garcia, C. Bellin, J. F. Marêché, P. Lagrange and G. Loupias, Superconductivity of bulk CaC6, Phys. Rev. Lett., 2005, 95(8), 087003 CrossRef PubMed.
- I. I. Mazin, Intercalant-driven superconductivity in YbC6 and CaC6, Phys. Rev. Lett., 2005, 95(22), 227001 CrossRef PubMed.
- T. E. Weller, M. Ellerby, S. S. Saxena, R. P. Smith and N. T. Skipper, Superconductivity in the intercalated graphite compounds C6Yb and C6Ca, Nat. Phys., 2005, 1(1), 39–41 Search PubMed.
- G. Csányi, P. B. Littlewood, A. H. Nevidomskyy, C. J. Pickard and B. D. Simons, The role of the interlayer state in the electronic structure of superconducting graphite intercalated compounds, Nat. Phys., 2005, 1(1), 42–45 Search PubMed.
- G. Profeta, M. Calandra and F. Mauri, Phonon-mediated superconductivity in graphene by lithium deposition, Nat. Phys., 2012, 8(2), 131–134 Search PubMed.
- B. M. Ludbrook, G. Levy, P. Nigge, M. Zonno, M. Schneider, D. J. Dvorak, C. N. Veenstra, S. Zhdanovich, D. Wong, P. Dosanjh, C. Strasser, A. Stöhr, S. Forti, C. R. Ast, U. Starke and A. Damascelli, Evidence for superconductivity in Li-decorated monolayer graphene, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(38), 11795–11799 CrossRef.
- C. M. Hao, X. Li, A. R. Oganov, J. Y. Hou, S. C. Ding, Y. F. Ge, L. Wang, X. Dong, H.-T. Wang, G. C. Yang, X.-F. Zhou and Y. J. Tian, Superconductivity in compounds of sodium-intercalated graphite, Phys. Rev. B, 2023, 108(21), 214507 CrossRef.
- C. Si, Z. Liu, W. H. Duan and F. Liu, First-principles calculations on the effect of doping and biaxial tensile strain on electron-phonon coupling in graphene, Phys. Rev. Lett., 2013, 111(19), 196802 CrossRef PubMed.
- C. J. Pickard and R. J. Needs, Ab initio random structure searching, J. Phys.: Condens. Matter, 2011, 23(5), 053201 CrossRef PubMed.
- A. R. Oganov and C. W. Glass, Crystal structure prediction using ab initio evolutionary techniques: Principles and applications, J. Chem. Phys., 2006, 124(24), 244704 Search PubMed.
- C. W. Glass, A. R. Oganov and N. Hansen, USPEX-evolutionary crystal structure prediction, Comput. Phys. Commun., 2006, 175(11–12), 713–720 Search PubMed.
- Q. Zhu, L. Li, A. R. Oganov and P. B. Allen, Evolutionary method for predicting surface reconstructions with variable stoichiometry, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87(19), 195317 Search PubMed.
- Y. C. Wang, J. Lv, L. Zhu and Y. M. Ma, Crystal structure prediction via particle-swarm optimization, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82(9), 094116 CrossRef.
- X. Y. Luo, J. H. Yang, H. Y. Liu, X. J. Wu, Y. C. Wang, Y. M. Ma, S.-H. Wei, X. G. Gong and H. J. Xiang, Predicting two-dimensional boron-carbon compounds by the global optimization method, J. Am. Chem. Soc., 2011, 133(40), 16285–16290 CrossRef.
- Y. C. Wang, J. Lv, L. Zhu and Y. M. Ma, CALYPSO: A method for crystal structure prediction, Comput. Phys. Commun., 2012, 183(10), 2062–2070 CrossRef.
- K. Xia, H. Gao, C. Liu, J. N. Yuan, J. Sun, H.-T. Wang and D. Y. Xing, A novel superhard tungsten nitride predicted by machine-learning accelerated crystal structure search, Sci. Bull., 2018, 63(13), 817–824 CrossRef.
- J. J. Wang, H. Gao, Y. Han, C. Ding, S. N. Pan, Y. Wang, Q. H. Jia, H.-T. Wang, D. Y. Xing and J. Sun, MAGUS: Machine learning and graph theory assisted universal structure searcher, Natl. Sci. Rev., 2013, 10(7), nwad128 CrossRef.
- Y. Han, C. Ding, J. J. Wang, H. Gao, J. Y. Shi, S. B. Yu, Q. H. Jia, S. N. Pan and J. Sun, Efficient crystal structure prediction based on the symmetry principle, Nat. Comput. Sci., 2025, 5(3), 255–267 CrossRef.
- X. Z. Shi, C. Y. He, C. J. Pickard, C. Tang and J. X. Zhong, Stochastic generation of complex crystal structures combining group and graph theory with application to carbon, Phys. Rev. B, 2018, 97(1), 014104 CrossRef.
- P. Borlido, C. Steigemann, N. N. Lathiotakis, M. A. L. Marques and S. Botti, Structural prediction of two-dimensional materials under strain, 2D Mater., 2017, 4(4), 045009 CrossRef.
- J. Lin, S. F. Li, T. Ouyang, C. X. Zhang, C. Tang, C. Y. He and J. X. Zhong, Two-dimensional carbon allotropes and nanoribbons based on 2,6-polyazulene chains: Stacking stabilities and electronic properties, J. Phys. Chem. Lett., 2021, 12(2), 732–738 CrossRef.
- K. Xia, X. L. Yu, C. Ding, Q. Lu, Z. W. Zhang and J. Sun, Superconductivity in monolayer carbon allotropes with high thermal stability, Nano Lett., 2024, 24(38), 11865–11872 CrossRef CAS.
- Z. H. Wang, X.-F. Zhou, X. M. Zhang, Q. Zhu, H. F. Dong, M. W. Zhao and A. R. Oganov, Phagraphene: A low-energy graphene allotrope composed of 5-6-7 carbon rings with distorted Dirac cones, Nano Lett., 2015, 15(9), 6182–6186 CrossRef CAS PubMed.
- H. C. Yin, X. Z. Shi, C. Y. He, M. Martinez-Canales, J. Li, C. J. Pickard, C. Tang, T. Ouyang, C. X. Zhang and J. X. Zhong, Stone-Wales graphene: A two-dimensional carbon semimetal with magic stability, Phys. Rev. B, 2019, 99(4), 041405 CrossRef CAS.
- C. Su, H. Jiang and J. Feng, Two-dimensional carbon allotrope with strong electronic anisotropy, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87(7), 075453 CrossRef.
- S. H. Zhang, J. Zhou, Q. Wang, X. S. Chen, Y. Kawazoe and P. Jena, Penta-graphene: A new carbon allotrope, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(8), 2372–2377 CrossRef CAS.
- P.-F. Liu, J. Y. Li, C. Zhang, X.-H. Tu, J. R. Zhang, P. Zhang, B.-T. Wang and J. S. David, Type-II Dirac cones and electron-phonon interaction in monolayer biphenylene from first-principles calculations, Phys. Rev. B, 2021, 104(23), 235422 CrossRef CAS.
- M. M. Möller, G. A. Sawatzky, M. Franz and M. Berciu, Type-II Dirac semimetal stabilized by electron-phonon coupling, Nat. Commun., 2017, 8(1), 2267 CrossRef.
- C. H. Li, N. K. Ravichandran, L. Lindsay and D. Broido, Fermi surface nesting and phonon frequency gap drive anomalous thermal transport, Phys. Rev. Lett., 2018, 121(17), 175901 CrossRef CAS.
- X. Y. Li, Q. Wang and P. Jena, Ψ-Graphene: A new metallic allotrope of planar carbon with potential applications as anode materials for lithium-ion batteries, J. Phys. Chem. Lett., 2017, 8(14), 3234–3241 CrossRef CAS.
- S. W. Wang, B. C. Yang, H. Y. Chen and E. Ruckenstein, Popgraphene: A new 2D planar carbon allotrope composed of 5-8-5 carbon rings for high-performance lithium-ion battery anodes from bottom-up programming, J. Mater. Chem. A, 2018, 6(16), 6815–6821 RSC.
- G. Kresse and J. Furthmüller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6(1), 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77(18), 3865–3868 CrossRef CAS.
- E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth and S. Grimme, A generally applicable atomic-charge dependent London dispersion correction, J. Chem. Phys., 2019, 150(15), 154122 CrossRef.
- E. Caldeweyher, J.-M. Mewes, S. Ehlert and S. Grimme, Extension and evaluation of the D4 London-dispersion model for periodic systems, Phys. Chem. Chem. Phys., 2020, 22(16), 8499–8512 RSC.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, Lobster: A tool to extract chemical bonding from plane-wave based DFT, J. Comput. Chem., 2016, 37(11), 1030–1035 CrossRef CAS.
- R. Dronskowski and P. E. Blöchl, Crystal orbital Hamilton populations (COHP): Energy-resolved visualization of chemical bonding in solids based on density-functional calculations, J. Phys. Chem., 1993, 97(33), 8617–8624 CrossRef CAS.
- R. Jinnouchi, F. Karsai and G. Kresse, On-the-fly machine learning force field generation: Application to melting points, Phys. Rev. B, 2019, 100(1), 014105 CrossRef CAS.
- R. Jinnouchi, J. Lahnsteiner, F. Karsai, G. Kresse and M. Bokdam, Phase transitions of hybrid perovskites simulated by machine-learning force fields trained on-the-fly with Bayesian inference, Phys. Rev. Lett., 2019, 122(22), 225701 CrossRef CAS.
- R. Jinnouchi, F. Karsai, C. Verdi, R. Asahi and G. Kresse, Descriptors representing two-and three-body atomic distributions and their effects on the accuracy of machine-learned inter-atomic potentials, J. Chem. Phys., 2020, 152(23), 234102 CrossRef CAS PubMed.
- P. T. Liu, J. T. Wang, N. Avargues, C. Verdi, A. Singraber, F. Karsai, X.-Q. Chen and G. Kresse, Combining machine learning and many-body calculations: Coverage-dependent adsorption of CO on Rh (111), Phys. Rev. Lett., 2023, 130(7), 078001 CrossRef CAS.
- G. Pizzi, V. Vitale, R. Arita, S. Blügel, F. Freimuth, G. Géranton, M. Gibertini, D. Gresch, C. Johnson, T. Koretsune, J. Ibañez-Azpiroz, H. Lee, J.-M. Lihm, D. Marchand, A. Marrazzo, Y. Nomura, L. Paulatto, S. Poncé, T. Ponweiser, J. F. Qiao, F. Thöle, S. S. Tsikin, M. Wierzbowska, N. Marzari, D. Vanderbilt, I. Souza, A. A. Mostofi and J. R. Yates, Wannier90 as a community code: New features and applications, J. Phys.: Condens. Matter, 2019, 32(16), 165902 CrossRef.
- Q. S. Wu, S. N. Zhang, H. F. Song, M. Troyer and A. A. Soluyanov, WannierTools: An open-source software package for novel topological materials, Comput. Phys. Commun., 2018, 224(5806), 405–416 CrossRef CAS.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, Quantum Espresso: A modular and open-source software project for quantum simulations of materials, J. Phys.: Condens. Matter, 2009, 21(39), 395502 CrossRef.
- W. L. Mcmillan, Transition temperature of strong-coupled superconductors, Phys. Rev., 1968, 167(2), 331–334 CrossRef CAS.
- V. V. Nauchitel’, Energy distribution function for the NVT canonical ensemble, Mol. Phys., 1981, 42(5), 1259–1265 CrossRef.
- M. Parrinello and A. Rahman, Crystal structure and pair potentials: A molecular-dynamics study, Phys. Rev. Lett., 1980, 45(14), 1196–1199 CrossRef CAS.
- M. Parrinello and A. Rahman, Polymorphic transitions in single crystals: A new molecular dynamics method, J. Appl. Phys., 1981, 52(12), 7182–7190 CrossRef CAS.
- O. T. Unke, S. Chmiela, H. E. Sauceda, M. Gastegger, I. Poltavsky, K. T. Schütt, K.-R. Müller and A. Tkatchenko, Machine learning force fields, Chem. Rev., 2021, 121(16), 10142–10186 CrossRef CAS.
- F. Schedin, A. K. Geim, S. V. Morozov, E. W. Hill, P. Blake, M. I. Katsnelson and K. S. Novoselov, Detection of individual gas molecules adsorbed on graphene, Nat. Mater., 2007, 6(9), 652–655 CrossRef PubMed.
| This journal is © the Owner Societies 2026 |