
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigating the kinetics of spin-crossover transitions using Raman spectroscopy
Gérald
Kämmerer
 a,
Lea
Kämmerer
a,
Stephan
Sleziona
a,
Senthil Kumar
Kuppusamy
b,
Mario
Ruben
bcd,
Marika
Schleberger
a,
Heiko
Wende
a and
Peter
Kratzer
*a
a,
Lea
Kämmerer
a,
Stephan
Sleziona
a,
Senthil Kumar
Kuppusamy
b,
Mario
Ruben
bcd,
Marika
Schleberger
a,
Heiko
Wende
a and
Peter
Kratzer
*a
aFaculty of Physics and Center for Nanointegration Duisburg-Essen (CENIDE), University Duisburg-Essen, Lotharstraße 1, 47057 Duisburg, Germany. E-mail: gerald.kaemmerer@uni-due.de; peter.kratzer@uni-due.de
bInstitute of Quantum Materials and Technologies (IQMT), Karlsruhe Institute of Technology (KIT), Kaiserstraße 12, 76131 Karlsruhe, Germany
cInstitute of Nanotechnology (IMT), Karlsruhe Institute of Technology (KIT), Kaiserstraße 12, 76131 Karlsruhe, Germany
dCentre Européen de Sciences Quantiques (CESQ), Institut de Science et d'Ingénierie, Supramoléculaires (ISIS), 8 allée Gaspard Monge, BP 70028, 67083 Strasbourg Cedex, France
First published on 21st February 2026
Abstract
The room-temperature spin-crossover iron(II) complex Fe(1-bpp-COOC2H5)2(BF4)2CH3CN exhibits reversible switching between the low-spin and high-spin states. The observation of coexisting spin domains within polycrystalline samples of Fe(1-bpp-COOC2H5)2(BF4)2CH3CN allows for real-time video recording of domain boundary propagation under an optical microscope. Quantitative analysis of this motion provides valuable information regarding the phenomenological transition involved in the spin-state switching process. To elucidate the intricate dynamics of this spin transition, a synergistic approach combining temperature-dependent Raman spectroscopy with ab initio computational methods is employed. Within this investigation, a comprehensive classification of Raman vibrational modes has been achieved, categorizing them based on their spin-state dependence and vibrational characteristics, symmetries, including stretching, bending, and tilting motions. Notably, low-frequency Raman modes associated with the iron center and its nitrogen ligand environment provide crucial insights into the local coordination and its role in the spin-crossover mechanism. This work reveals distinct spin-state-induced structural changes, including bond stretching and softening, which manifest as unique spectroscopic fingerprints for each spin state.
1 Introduction
Spin-crossover (SCO) molecules are a class of molecular materials exhibiting a bi-stable spin transition between low-spin (LS, S = 0) and high-spin (HS, S = 2) states. This transition, induced by external stimuli such as temperature, pressure, X-rays, static electric or magnetic fields, or inclusion of guest molecules, is accompanied by significant alterations in their electronic, optical and geometric properties.1–11 The SCO phenomenon for a single molecule centers on the Fe(II) ion, a transition metal with partially filled 3d orbitals.12–14 Ligand coordination induces splitting of the Fe 3d orbitals into eg and t2g sets. Temperature-induced transitions from the diamagnetic low-spin state (LS, S = 0) to the paramagnetic high-spin state (HS, S = 2) are accompanied by a decrease in ligand field strength (10 Dq) and an approximate 10% elongation of Fe–N bond lengths15–17 (see Fig. 1). Notably, N–N or C–C bond lengths are minimally affected by the spin transition. For practical applications, SCO complexes should exhibit abrupt spin-state switching with broad thermal hysteresis around room temperature and potential color changes.1–8,10,11,18–21 Recent synthesis of crystalline samples with the complex Fe(1-bpp-COOC2H5)2,18 containing two BF4− counter-ions and one CH3CN solvent molecule per unit cell, demonstrates spin-state switching near room temperature with pronounced hysteresis. This switching, occurring near T↑ = 321 K and accompanied by a color change from gray (LS) to orange (HS) (see Fig. 3, right part), is consistent with observations in other SCO materials.22–25 It is accompanied by a broad hysteresis making our SCO a promising candidate for applications in molecular switching and nanoelectronics devices, including light-emitting diodes.3,26–28 | ||
| Fig. 1 Top Panel: The crystallographic structure of Fe(1-bpp-COOC2H5)2(BF4)2CH3CN is presented, highlighting the average Fe–N bond distances calculated using density functional theory (DFT): the panel illustrates the spin-state-dependent conformational changes of the ethyl (C2H5) substituents, which adopt an out-of-plane configuration in the low-spin (LS) state and an in-plane configuration in the high-spin (HS) state. Additionally, the spatial position of the BF4− counter-anions and the CH3CN solvent molecule is shown, reflecting their respective position in the LS and HS states. Bottom Panel: A schematic energy level diagram illustrates the splitting of the Fe(II) 3d orbitals in a ligand field. The diagram demonstrates the formation of the t2g and antibonding eg* in the Oh representation, providing a simplified model for understanding the electronic transitions responsible for the thermally induced spin-state crossover in this complex. | ||
To lay the ground for the work presented in this paper, we summarize observations reported previously on similar samples. It is well known that the spin-state transition is reflected in the average Fe–N bond length, as corroborated by experimental X-ray diffraction (XRD) data9,18 and ab initio calculations, see Fig. 1 for detailed values. Moreover, the XRD data also reveals that the monoclinic cells of the LS and HS phases differ in both shape and volume, with the HS phase exhibiting a 2% volume increase. This hysteretic behavior is associated with a phase coexistence observable in optical microscopy due to the color change between the two phases. While the switching of individual molecules occur on the femtosecond time scale (e.g.,10,29–32), the much slower process of the growth of phase domains could provide further insight into the switching kinetics. Monitoring the speed of the domain boundary between LS and HS phases during hysteresis is an obvious way to access these kinetics.
From the experimental perspective, we employ a combination of techniques: optical microscopy provides insight into macro- and mesoscopic strain within the sample and its effect on crystal integrity, while Raman spectroscopy probes the motion of individual molecules and their interactions at the sub-molecular level. Raman spectroscopy, a non-destructive technique, offers valuable insight into the molecular structural evolution during spin-state switching.19 It has been demonstrated already that this method allows one to distinguish between LS and HS states, as exemplified by the pyridine ring breathing mode near 1020 cm−1, which exhibits significant shifts due to Fe–N bond elongation upon LS-to-HS transition.33,34
In the present work, we study polycrystalline material. In these samples, the LS and HS states show up as separate phase domains, growing, and shrinking as temperature is swept through the thermal hysteresis loop. The transition behavior depends on the material history, and the so-called “self-grinding” phenomenon35 is observed. We carried out a temperature-dependent analysis of the phase boundary motion. Simultaneously, the microscopic changes were monitored via Raman spectroscopy. This poses the question of how exactly the material's properties in the microstructure of this polycrystalline material and at the molecular level interact when the spin state is switched. Combining Raman spectroscopy with ab initio calculations enables the development of a unique structural fingerprint for Fe(1-bpp-COOC2H5)2(BF4)2CH3CN, similar to the approach used by Devid36 for other SCO complexes.36–41 By comparing Raman data sets with ab initio calculations, the typical spectral features can be classified, and the specific differences between LS/HS modes can be investigated, allowing a clear identification of distinct vibrational modes according to the atomic species in the bonding.38,40
2 Experimental and theoretical methods
2.1 Sample preparation and characterization
The preparation and characterization of our samples has been described previously,18 and here we just summarize the information relevant to us. Our polycrystalline sample (several hundred µm3, ρ ≈ 1.537 g cm−3) crystallizes in the LS state within the monoclinic space group P21/c (CCDC 1560719). Upon transition to the HS state, a crystallographic phase transformation to space group P21/n (CCDC 2330939) occurs. This phase transition involves a shearing of the cell (estimated from XRD18), resulting in a restructuring of the crystal lattice that is closely related to the spin-state transition and adjustments in molecular packing. The shearing and increasing of the unit cell causes a translation and rotation of the SCO molecules, counter-anions, and solvents. The compound, Fe(1-bpp-COOC2H5)2(BF4)2CH3CN, features a central iron(II) ion (Fe2+) coordinated by two tridentate 2,6-di(pyrazol-1-yl)pyridine (1-bpp) ligands functionalized with ethyl ester groups (–COOC2H5). The resulting cationic complex is charge-balanced by two tetrafluoroborate anions (BF4−) and solvated in acetonitrile (CH3CN), indicative of its crystallization state (molecular and solvent/anion structures in Fig. 1). Taking the tilting of the ethyl ester group into account, the molecule has a symmetry of C2v. As reported in Kuppusamy et al. (2019),18 the complex exhibits a distorted geometry in the HS state, with a trans-N(pyridine)-Fe-N(pyridine) angle of approximately 158.83° (LS: 178.91°), deviating from idealized octahedral symmetry.2.2 Temperature-dependent Raman spectroscopy
Raman spectroscopy was performed using a WITec alpha300RA confocal Raman microscope with a Linkam THMS350EV vacuum stage. To minimize thermal artifacts, samples were maintained in a nitrogen atmosphere and cryogenically cooled with liquid nitrogen. Raman spectra were taken at various temperatures with a 532 nm laser at 0.5 mW for excitation, and a ZeissLS Epiplan-Neofluar 50× objective (NA 0.55) for laser focusing and optical image acquisition. Ultralow frequency modes (down to 10 cm−1) were resolved using a RayShield filter. Spectral peak positions were determined by fitting the data with Gaussian functions. Mode identification in this paper between experiment and simulation was based primarily on frequency shifts and the selection of the modes is based on the relative intensity variations in the experimental measurement. Raman intensities calculated within the non-resonant approximation (not presented here) showed limited correlation with experimental results and are therefore not further discussed. To elucidate switching dynamics and timescales, video recordings were acquired during Raman measurements. These recordings enabled analysis of dynamic features while from temperature-dependent Raman spectroscopy we can identify spectral features for the HS and LS state, seen Fig. 3.2.3 Investigation of the phase boundary movements
Video recordings were analyzed using self-written Python scripts to identify the phase transition region, extract propagation times, and estimate the phase transition boundary velocity (see Table 3). The boundary was determined using an edge detection algorithm, combined with gradient color intensity analysis and Haar-like feature detection (Histogram of Oriented Gradients, HOG42). Variations in video focus occasionally complicated velocity determination; these were mitigated through gradient interpolation, enabling error estimation. This approach facilitated the study of intermolecular coupling dynamics during heating and cooling (see Fig. 7). Various heating and cooling rates and temperature ranges were investigated. However, morphological changes (the “self-grinding” phenomenon35), along with stress-induced strain that introduced cracks, grains, and sometimes caused parts to chip off, significantly complicating rate determination during cooling, necessitating interpolation and dynamic boundary identification to estimate the cooling velocity. Despite these complications, the boundary velocity remained largely unchanged, although the material's history was important for determining the state and the transitions.2.4 Computational method
Density functional theory (DFT) with the finite displacement method was used to compute molecular vibrational spectra. Given the computational complexity associated with the large number of vibrational modes (exceeding 201), the Perdew–Burke–Ernzerhof (PBE) functional43 was selected, based on its demonstrated efficacy in similar investigations.44 DFT calculations were performed using the FHI-aims code,45 an all-electron code with numerically defined atom-centered basis functions, employing the tight basis set (tier 2), van der Waals correction,46 and the atomic zero-order regular approximation (ZORA).47 This method is known for its efficiency in DFT and Raman mode calculations, albeit with substantial computational requirements. For the calculation, we used the standard parameters of a full geometry relaxation with the BFGS algorithm with and without periodic boundary conditions and a threshold on the forces on atoms of 5 × 10−4 eV Å−1 while in the self-consistency cycle forces were converged to 2 × 10−6 eV Å−1 and electronic density changes were less than 2 × 10−7 ea0−3. Subsequent to the converged non-periodic calculation, we used the given core script get_vibrations.py of FHI-aims to calculate (set δ = 0.0025 Å) the vibrational modes and checked for any unstable modes. Raman frequencies were determined for both spin states. Initial geometries from X-ray diffraction data published in the literature18 were geometrically optimized for both HS and LS complexes, considering periodic and free-standing boundary conditions. The free-standing approach provided sufficient accuracy for simulating Raman modes and identifying modes associated with iron-nitrogen or (1-bpp-COOC2H5)2 unit interactions during spin-state switching, compared to periodic calculations.48Calculations were performed on both the full (including counter-anions and lattice solvent) and the minimal (charged Fe(1-bpp-COOC2H5)2+ complex, excluding the smaller molecules) molecular configurations. Comparative analysis showed that including counter-anions and lattice solvent had a negligible effect (0.2% difference) on the calculated bond lengths (Fe–N bonds to pyridine and pyrazole 1.88 Å and 1.96 Å, respectively) and bond angles (179° and 161°) in the LS state, consistent with findings in a different material by Lawson Daku.49
3 Results
3.1 Temperature-dependent Raman spectra
The range for temperatures of interest is determined from taking a thermal hysteresis curve, as shown in Fig. 2. The temperature-dependent Raman spectra acquired during such a thermal cycle are shown in Fig. 3, encompassing heating from 300 K to 340 K and subsequent cooling to 220 K, at discrete temperature points (300 K, 318 K, 340 K, 300 K, 270 K, and 220 K), as marked in the hysteresis plot in Fig. 2. Concurrent optical observations, documented as images and for the domain investigation as video, are presented alongside the corresponding Raman spectra (temporally resolved Raman spectra recording was not feasible). | ||
| Fig. 2 Temperature-dependent thermal hysteresis in the temperature regime of 220 to 350 K with a broad hysteresis of 37 K. The symbols mark the temperatures of the Raman measurements shown in the Fig. 3. | ||
 | ||
| Fig. 3 Raman spectra measured at different temperatures with the corresponding optical microscopy images: The left panel of Fig. 3 displays six Raman spectra recorded at 300 K, 318 K, 340 K, 300 K, 270 K, and 220 K, spanning a Raman shift range from 0 to 2000 cm−1. Spectral key features appear and disappear depending on the temperature, particularly in the regions between 300–550 cm−1 (colored blue), 1200–1500 cm−1 (colored orange), and most prominently around 1017 cm−1 (dashed red). These features reflect changes in the vibrational modes of the sample (see Section 3.1). The right panel of Fig. 3 shows the corresponding optical microscope images, revealing a reversible color change associated with the spin state of the sample. At lower temperatures, the material exhibits a grayish-brown hue, corresponding to the low-spin (LS) state. Upon heating, the sample transitions into the high-spin (HS) state, accompanied by an orange-yellow coloration. Cooling the sample leads to a return to the LS state and its original grayish-brown appearance, consistent with the behavior observed in the videos. Additionally, in the intermediate temperature range between 318 K and 270 K, a faint luminescence was observed, resulting in a slight glooming effect in the microscope images and a broad background in the Raman measurements. | ||
Analysis of the Raman spectra reveals several noteworthy features. Adjacent to the Rayleigh line (0 cm−1), a low-intensity feature is observed, which exhibits a significant increase in intensity in the HS state. These low modes will be further discussed in the context of vibrations with a substantial iron contribution. Bands around 50 cm−1 up to 500 cm−1 are anticipated to be crucial for understanding the coupling of the iron between the ligands, as will be discussed later.
A prominent spectral feature is observed around 1000 cm−1, displaying a marked change in intensity and appearing distinctly in the HS state while diminishing upon cooling. This mode is identified as pyridine ring breathing, coupled from one ligand to the other by the iron center (cf.Fig. 6 top row). A neighboring peak exhibits an inverse trend, decreasing in intensity as the 1000 cm−1 peak emerges. In the spectral region between 1100 and 1300 cm−1, several features present in the LS state show a reduction in intensity in the HS state related to the structural change of the complex.
The broad spectral range between 1350 and 1700 cm−1 exhibits both an increase in overall intensity and a shift in the frequencies of individual peaks upon the transition to the HS state. Conversely, the Raman band observed around 1750 cm−1 remains largely unaffected by the spin state switching, suggesting its origin in a functional group of the molecule that is not directly involved in the electronic transition, which we can see in the mode assignment and in the Tables 1 and 2. For a further investigation, we need to assign the modes in a detailed comparison like in the Fig. 4 to understand the iron/bond interaction in the different spin states.
| No. | Vibrational modes in [cm−1] | Deg. | Sym. | Description | |||
|---|---|---|---|---|---|---|---|
| LSexp | LScalc | HSexp | HScalc | ||||
| 1 | 30 | 27.9 | 27 | 24.1 | Deg. (2) | E | Bending of the ligands |
| 2 | 99 | 104.9 | 95 | 104.4 | Deg. (2) | E | Tilting pyridine, pyrazole rings |
| 3 | — | 162.9 | 147 | 139.0 | Non-Deg. | B2 | Iron mode, tilting pyrazole rings |
| 4 | 205 | 210.6 | — | 233.7 | Non-Deg. | A1 | Tilting pyridine, pyrazole rings, iron mode |
| 5 | — | 262.6, 262.9 | 257 | 261.9, 262.3 | Deg. (2) | E | Ethyl group tilting |
| 6 | 310 | 315.7, 308.7, 315.8 | 296 | 279.8, 266.4, 288.02 | Deg. (3) | — | Iron mode, bending C–N bond |
| 7 | 361 | 350.12 | 364 | 289.0 | Non-Deg. | B2 | Antisym. N–Fe bond stretching |
| 8 | 387 | 375.9, 377.2 | — | 336.0, 336.3 | Deg. (2) | E | Antisym. and sym. N–Fe bond stretching |
| 9 | — | 523.1, 539.2 | 505 | 507.7, 509.1 | Deg. (2) | E | Tilting pyridine ring |
| 10 | 539 | 521.0, 521.3 | — | 585.1, 585.2 | Deg. (2) | E | Tilting pyridine ring |
| 11 | 734 | 735.2 | — | 751.84 | Deg. (2) | E | Wagging C–H bond at the ethyl group |
| 12 | 965 | 957.2, 957.4 | 966 | 961.8, 963.7 | Deg. (2) | E | Antisym. pyrazole ring breathing |
| 13 | 1017 | 1018.6 | 1016 | 991.1 | Deg. (2) | E | Sym. pyridine ring breathing |
| 14 | 1045 | 1030.6 | 1045 | 996.4 | Non-Deg. | B2 | Antisym. pyridine ring breathing |
| 15 | 1057 | 1057.5 | 1057 | 1058.1 | Non-Deg. | A1 | Sym. pyridine C–C stretching breathing |
| 16 | — | 1072.8, 1072.9 | 1074 | 1076.1, 1076.8 | Deg. (2) | E | Antisym. pyrazole ring breathing |
| 17 | 1140 | 1140.4, 1140.5 | 1145 | 1149.5, 1149.6 | Deg. (2) | E | Antisym. and sym. pyridine ring breathing |
| 18 | 1160 | 1194.9 | — | 1183.4 | Non-Deg. | A1 | Sym. pyridine ring breathing |
| 19 | — | 1205.3, 1205.4 | 1215 | 1198.1, 1198.2 | Deg. (2) | E | Antisym. and sym. pyrazole tilting, H–C rocking |
| No. | Vibrational modes in [cm−1] | Deg. | Sym. | Description | |||
|---|---|---|---|---|---|---|---|
| LSexp | LScalc | HSexp | HScalc | ||||
| 20 | 1223 | 1222.1, 1223.6 | 1223 | 1220.9, 1227.9 | Deg (2) | E | Ethyl group rocking, C–C stretching near the ethyl group |
| 21 | 1262 | 1258.0 | 1262 | 1262.7 | Non-Deg | A1 | Breathing N–N pyrazole mode, sym. C–N stretching |
| 22 | 1287 | 1258.9 | 1286 | 1264.3 | Non-Deg | B2 | N–N pyrazole ring breathing, antisym. C–N stretching |
| 23 | 1303 | 1322.6 | 1301 | 1317.9, 1318.2 | Deg (2) | E | Antisym. and sym. pyrazole ring breathing |
| 24 | 1338 | 1336.8 | — | 1340.6, 1342.4 | Deg (2) | E | Tilting pyridine ring |
| 25 | 1378 | 1373.2, 1373.3 | 1379 | 1373.0, 1373.2 | Deg (2) | E | Wagging H–C bond at the ethyl group |
| 26 | — | 1409.9 | — | 1404.8 | Non-Deg | A2 | Pyridine tilting |
| 27 | 1412 | 1410.0, 1410.4, 1413.0, 1415.5 | 1404 | 1407.1, 1409.3, 1411.5, 1415.1 | Deg (4) | — | Pyridine ring breathing |
| 28 | 1442 | 1441.6, 1457.6 | 1440 | 1441.4, 1441.5, 1457.3, 1457.4 | Deg (4) | — | Tilting functional group |
| 29 | 1472 | 1465.6 | 1462 | 1467.0 | Deg (3) | — | Light pyridine ring breathing mode |
| 30 | 1502 | 1504.8 | 1500 | 1466.3, 1467.0 | Deg (2) | E | Sym. pyridine C–C bending |
| 31 | 1526 | 1547.2, 1547.3 | 1528 | 1554.2, 1555.9 | Non-Deg | A1 | Pyridine ring stretching |
| 32 | 1576 | 1599.6 | 1584 | 1589.0, 1589.3 | Deg (2) | E | Antisym. and sym. C–C pyridine stretching |
| 33 | 1631 | 1602.8, 1627.1 | 1631 | 1626.2, 1630.0 | Deg (2) | E | Antisym. and sym. C–C pyridine stretching |
| 34 | 1730 | 1720.4, 1720.6 | 1737 | 1720.8, 1721.3 | Deg (4) | — | Antisym. and sym. O–C stretching |
| Temperature rate [K min−1] | Average velocity (heating) [µm s−1] | Average velocity (cooling) [µm s−1] |
|---|---|---|
| 5 | 35.60 ± 0.8 | 18.26 ± 8.88 |
| 10 | — | 59.36 ± 12.68 |
| 15 | 110.00 ± 31.22 | 45.65 ± 21.67 |
 | ||
| Fig. 4 Detailed Raman spectra at two different temperatures: 220 K (blue) and 340 K (red). The dashed lines label the vibrational modes listed in the Tables 1 and 2. | ||
3.2 Vibrational mode assignment
For the present research, we are guided by the question of how the spin state of Fe is reflected in the pertaining spectra, in other words, whether there are 'fingerprints' in the spectra that allow one to conclude on the spin state of an individual Fe complex in the crystal. The Raman spectrometer used enables us to examine the low-frequency spectral range (<600 cm−1), where modes involving significant Fe motion are expected. While high-frequency modes are not expected to involve Fe displacements due to the large atomic mass of Fe, even small Fe motions can mediate vibrational coupling between modes with displacements in both ligands.The classification of the Raman-active vibrational modes starts from the experimentally identified peaks in the highly resolved Raman spectra shown in Fig. 4. From the overall 201 calculated vibrational modes, only those are reported in the Tables 1 and 2 for which a corresponding experimental mode could be identified. To establish a one-to-one correspondence between vibrational modes in the LS and HS states, a combined methodology was implemented, encompassing classification, symmetry classification, and density-based spatial clustering of applications with noise (DBSCAN)50 on atomic displacements. The grouping of the modes was accomplished through a symmetry-based approach, wherein modes were assigned by visual inspection according to their closest possible symmetry. If we restrict ourselves to the Fe atoms and its six N neighbors, the complex has an approximate symmetry of D2d (see Fig. 5). It contains a two-fold rotational axis C2 and  and two combined mirror-plus-rotation symmetry operations. In the following, the characterization as “antisymmetric” or “symmetric” refers to the parity of the vibrational displacements with respect to the latter two operations.
and two combined mirror-plus-rotation symmetry operations. In the following, the characterization as “antisymmetric” or “symmetric” refers to the parity of the vibrational displacements with respect to the latter two operations.
 | ||
Fig. 5 Schematic illustrating the D2d symmetry of the complex formed by the Fe atom (blue sphere) and its attached N atoms (brown spheres). Two-fold rotation axes are marked by C2 and 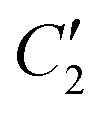 . Mirroring by one of the indicated planes followed by a 90° rotation also maps the complex onto itself. . Mirroring by one of the indicated planes followed by a 90° rotation also maps the complex onto itself. | ||
We note that vibrational modes can be assigned to general categories, such as tilting of the ligands and pyridine/pyrazole breathing modes, and to molecule-specific categories, such as iron modes (e.g., Fe–N stretching, Fe-ligand bending), special rocking modes of the ligands, ethyl group vibrations (e.g., CH3 rocking, CH2 wagging), and carbonyl (CO) stretching.
Inspecting the data in Table 1, we identify a group of modes, labeled 6, centered around 310 cm−1 in the LS state and around 296 cm−1 in the HS state. These modes correspond to Fe motion within the nearly octahedral cage formed by the N atoms. While an ideal octahedron would exhibit three-fold degeneracy, the calculations find a splitting of 10 cm−1 in LS and more than 20 cm−1 in the HS state, indicating a stronger deformation of the octahedral bonding environment in the HS state. Moreover, these vibrational modes are softer in the HS state, consistent with a more spacious cage and elongated Fe–N bonds. We note that other low-lying modes, particularly mode 4, also contain smaller contributions of Fe displacement. This can be understood from the requirement for a conserved center of mass of the whole complex, which necessitates Fe motion when the ligands have different displacement amplitudes. From the experimental perspective, it is important to realize that modes 4 and 6, involving Fe motion, appear as only relatively small peaks. The weak change in dynamic dipole moment for these modes results in weak Raman activity, limiting their usefulness as a fingerprint of the spin state.
The vibrational spectra of the HS and LS states exhibit distinct features of spin crossover-induced structural and electronic changes. Modes involving direct iron-ligand interactions show significant shifts, such as mode 7 (asymmetric N–Fe stretching), which redshifts by 72 cm−1 (LS: 361 cm−1 → HS: 289 cm−1), reflecting stronger N–Fe bonds in the LS state. Similarly, mode 8 (N–Fe stretching) shifts from 387 cm−1 (LS) to 336.0 cm−1 (HS), consistent with bond softening. In contrast, ligand-dominated vibrations, like mode 15 (symmetrical pyridine C–C stretching), remain nearly unchanged (LS: 1057 cm−1 → HS: 1058.1 cm−1), indicating their insensitivity to spin state. The gaps in the spectra are also indicative of spin state: mode 4 (iron-linked tilting) is unobserved in HS, suggesting HS rigidity, while mode 9 (pyridine tilting) is not detected in the LS data, likely due to symmetry or intensity shifts.
 | ||
| Fig. 6 Visualization of selected Raman modes, top row: antisymmetric and symmetric pyridine breathing (13/14); middle row: antisymmetric and symmetric pyrazole ring breathing (12); bottom row: ethyl group modes. | ||
 | ||
| Fig. 7 Optical image (size W: 220 µm × H: 130 µm) of the crystal taken by an optical microscope after heating with a heat rate of 5 K min−1 from 300 K to 318 K, the area or averaged linearized frontier of the transition is shown in gray boxes and with a black line. The direction of movement of the front is also indicated by arrows. | ||
We note that pairs of modes that would be degenerate in an idealized D2d-symmetric complex may show detectable splitting depending on structural distortions. While such splitting remains below the detection limit in mode 19 (pyrazole tilting), mode 20 (ethyl rocking) shifts frequencies (LS: 1223 cm−1 → HS: 1220.9–1227.9 cm−1) and, according to the calculations, increases its splitting in the HS state. Notably, Raman activity depends on mode symmetry. Non-degenerate A1 or B2 modes (e.g., modes 14 or 15) are usually Raman-active while modes with symmetries like A2 (e.g., mode 26) can be Raman-inactive according to molecule symmetry. This explains why some modes are experimentally more intense or less intense in Raman spectra.
These trends show that LS strengthens metal–ligand bonds while imposing steric constraints on the ligands (e.g., the ethyl group swing in mode 11 shifts from 735 cm−1 to 751.9 cm−1). Persistent pyridine ring vibrations (e.g., modes 13–15) confirm aromatic rigidity, whereas pyrazole modes are more spin-sensitive due to their aromaticity (see ref. 52). The interplay between symmetry-governed Raman activity and spin-state-dependent frequency shifts provides a dual criterion for distinguishing HS from LS configurations. The data offer a mechanistic framework for spin-state transitions, linking vibrational signatures to structural rearrangements critical for functional spin-crossover materials.
Key observations include the prevalence of pyridine-dominated modes at higher frequencies (>1338 cm−1) indicating stronger C–C bonds in the six-membered ring and the agreement between experimental and calculated frequencies for stretching-dominated vibrations like mode 31 (1526 cm−1 experimental vs. 1547.2 cm−1 calculated), validating our computational method for these motions. Overall, the vibrational signatures reflect the molecular structure and symmetry, highlighting the conformational complexity arising from the coupling of pyridine/pyrazole motions and ethyl group dynamics.
3.3 Motion of the phase boundaries
Next, we turn to the dynamical aspects of the LS-HS transition in polycrystalline samples. The dynamic spin-state transition within the SCO crystal was meticulously examined through in situ optical microscopy and subsequent video analysis, under controlled thermal heat flow. This investigation aimed to elucidate the mechanisms governing the hysteretic spin-state switching, as evidenced by observed morphological and chromatic alterations and the quantified propagation velocity of the transition front. Upon heating from 300 K to 318 K at a rate of 5 K min−1, the SCO crystal underwent a transformation from a grayish-brown to an orange hue, concurrent with the initiation of micro-fracturing. Further elevation of temperature to 340 K intensified this chromatic transition.Conversely, cooling back to 300 K yielded no discernible changes, confirming the material's inherent hysteretic behavior. Subsequent cooling to 220 K induced further micro-fracturing and the grayish-brown coloration returned, signifying the recovery of the LS state. Comparative analysis of the initial LS state at 300 K with the post-thermal cycle LS state at 200 K revealed substantial modifications in crystal morphology, consistent with the previously documented “self-grinding” phenomenon.35
Video microscopy, coupled with linear interpolation of the transition front, allowed for the quantification of the HS to LS transition propagation velocity. During heating from 300 K to 318 K at 5 K min−1, the propagation velocity was determined to be 35.6 ± 0.8 µm s−1 (cf.Fig. 3) equivalent to (36.5 ± 0.8) × 103 molecular diameters per second, assuming a homogeneous molecular volume of 992.83 Å3 per complex. This implies that a row of switched complexes gets attached to an existing domain within 27.3 µs on average.
This observed velocity exceeds previously reported values53–62 even by one order of magnitude, see e.g. ref. 59,62. This is tentatively attributed to intrinsic material properties, such as the broad thermal hysteresis and extrinsic factors, including the high heating rate. Increased heating rates further amplified the propagation velocity. Isothermal analysis at 318 K post-heating yielded similar velocities (31 ± 4 µm s−1). The reverse process, passive cooling, resulted in a propagation velocity of (18 ± 9 µm s−1), corresponding to a molecular interaction velocity of (18 ± 9) × 103 molecular diameters per second.
Increased cooling rates also resulted in higher propagation velocities. The observed disparity in propagation velocities between heating and cooling is attributed to structural changes, variations in Fe–N bond lengths, and micro-fractures, which introduce strain and impede transition front propagation. Micro-fractures and cracks were observed to impede the transition front, leading to pinning effects, which were accounted for in velocity determinations. However, we could not confirm a systematic reduction of the velocity from one heating cycle to the next. The high propagation velocities observed during heating suggest a rapid spin-state transition, influenced by the broad thermal hysteresis and heating rate.
The observed colorimetric variations stem from changes of the electronic structure of the complexes induced by spin-state transitions. Specifically, the observed color can be explained as combined effects of d–d electronic transitions, charge transfer transitions, and alterations in ligand field strength. As has been shown for a different complex,22 the core mechanism hinges on the change in the energy difference (10 Dq) between the Fe(II) ion's 3d orbitals during spin-state transitions, which directly affects d–d electronic transitions and subsequently the absorption spectrum, leading to visible color changes.
Conclusions
This paper investigated the structural and dynamic changes accompanying the spin transition from the low-spin to the high-spin state. Key findings reveal that this transition involves alterations in cellular morphology and a 2% volume increase, exhibiting hysteretic behavior with optically distinguishable phase coexistence. Kinetic analysis of the LS/HS domain boundary velocity within the hysteresis loop provided insights into the switching dynamics. While optical microscopy elucidated macroscopic and mesoscopic strain effects on crystal integrity, Raman spectroscopy probed sub-molecular dynamics and interactions. Distinct Raman bands showed significant intensity and frequency variations correlating with the transition between the low-spin and high-spin states, providing key vibrational signatures of this electronic change. Combined with first-principles calculations, Raman spectroscopy provided a detailed mode assignment, establishing a spectral fingerprint of the spin-state transition. A general downward frequency shift was observed in iron-based modes, revealing bond weakening (softening) during the HS state's approximately 10% bond elongation. Moreover, analysis of video microscopy images enabled the determination of the propagation velocity of phase domain boundaries. Distinct average velocities during heating and cooling, depending on the rate of temperature change, were found. For the present SCO complex, the absolute values were in the range of tens of µm s−1, and thus about an order of magnitude larger than previously reported values for other SCO complexes. In summary, the paper demonstrates characteristic spectral features for the LS and HS states, primarily defined by a general downward frequency shift, with some modes exhibiting dominant intensity variations.Conflicts of interest
There are no conflicts to declare.Data availability
Data for this article, including videos are available at https://uni-duisburg-essen.sciebo.de/s/Ib18Asdp2ewsrUX.Acknowledgements
G. K., L. K., S. S., M. S., H. W., and P. K. gratefully acknowledge the funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project-ID 278162697 – SFB 1242, projects A05, B02, and C05. S. S. and M. S. acknowledge financial support by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project-ID 429784087. G. K. and P. K. gratefully acknowledge the computing time granted by the Center for Computational Sciences and Simulation (CCSS) of the University of Duisburg-Essen and provided on the supercomputer magnitUDE (DFG Grant No. INST 20876/209-1 FUGG and INST 20876/243-1 FUGG) at the Zentrum für Informations- und Mediendienste (ZIM).References
- T. Kitazawa, Crystals, 2019, 9, 382 CrossRef CAS.
- P. Gütlich, Y. Garcia and H. A. Goodwin, Chem. Soc. Rev., 2000, 29, 419–427 RSC.
- C. Lefter, S. Rat, J. S. Costa, M. D. Manrique-Juarez, C. M. Quintero, L. Salmon, I. Seguy, T. Leichle, L. Nicu, P. Demont, A. Rotaru, G. Molnar and A. Bousseksou, Adv. Mater., 2016, 28, 7508–7514 CrossRef CAS PubMed.
- C. Bartual-Murgui, A. Akou, C. Thibault, G. Molnár, C. Vieu, L. Salmon and A. Bousseksou, J. Mater. Chem. C, 2015, 3, 1277–1285 RSC.
- Y. Guo, S. Xue, M. M. Dirtu and Y. Garcia, J. Mater. Chem. C, 2018, 6, 3895–3900 RSC.
- W. Li, A. Rotaru, M. Wolff, S. Demeshko, F. Meyer and Y. Garcia, J. Mater. Chem. C, 2023, 11, 11175–11184 RSC.
- E. Resines-Urien, E. Fernandez-Bartolome, A. Martinez-Martinez, A. Gamonal, L. Pineiro-Lopez and J. S. Costa, Chem. Soc. Rev., 2023, 52, 705–727 RSC.
- O. Kahn, J. Kröber and C. Jay, Adv. Mater., 1992, 4, 718–728 CrossRef CAS.
- S. K. Kuppusamy, L. Spieker, B. Heinrich, H. Wende and M. Ruben, ChemRxiv, 2022, preprint DOI:10.26434/chemrxiv-2022-mnh4n.
- L. Kämmerer, G. Kämmerer, M. Gruber, J. Grunwald, T. Lojewski, L. Mercadier, L. Le Guyader, R. Carley, C. Carinan, N. Gerasimova, D. Hickin, B. E. Van Kuiken, G. Mercurio, M. Teichmann, S. K. Kuppusamy, A. Scherz, M. Ruben, K. Sokolowski-Tinten, A. Eschenlohr, K. Ollefs, C. Schmitz-Antoniak, F. Tuczek, P. Kratzer, U. Bovensiepen and H. Wende, ACS Nano, 2024, 18, 34596–34605 CrossRef PubMed.
- B. Weber, W. Bauer and J. Obel, Angew. Chem., Int. Ed., 2008, 47, 10098–10101 CrossRef CAS PubMed.
- M. Fumanal, F. Jiménez-Gravalos, J. Ribas-Arino and S. Vela, Inorg. Chem., 2017, 56, 4474–4483 CrossRef CAS.
- O. Iasco, E. Riviere, R. Guillot, M. Buron-Le Cointe, J.-F. Meunier, A. Bousseksou and M.-L. Boillot, Inorg. Chem., 2015, 54, 1791–1799 CrossRef CAS PubMed.
- A. Kaiba, H. J. Shepherd, D. Fedaoui, P. Rosa, A. E. Goeta, N. Rebbani, J. F. Letard and P. Guionneau, Dalton Trans., 2010, 39, 2910–2918 RSC.
- K. Takahashi, Inorganics, 2018, 6, 1–4 CrossRef.
- J. G. Park, I.-R. Jeon and T. D. Harris, Inorg. Chem., 2015, 54, 359–369 CrossRef CAS PubMed.
- A. Arroyave, A. Lennartson, A. Dragulescu-Andrasi, K. S. Pedersen, S. Piligkos, S. A. Stoian, S. M. Greer, C. Pak, O. Hietsoi, H. Phan, S. Hill, C. J. McKenzie and M. Shatruk, Inorg. Chem., 2016, 55, 5904–5913 CrossRef CAS PubMed.
- S. K. Kuppusamy, A. Mizuno, L. Kammerer, S. Salamon, B. Heinrich, C. Bailly, I. Salitros, H. Wende and M. Ruben, Dalton Trans., 2024, 53, 10851–10865 RSC.
- K. Senthil Kumar, B. Heinrich, S. Vela, E. Moreno-Pineda, C. Bailly and M. Ruben, Dalton Trans., 2019, 48, 3825–3830 RSC.
- H. Hagiwara, T. Masuda, T. Ohno, M. Suzuki, T. Udagawa and K. I. Murai, Cryst. Growth Des., 2017, 17, 6006–6019 CrossRef CAS.
- M. A. Halcrow, Chem. Lett., 2014, 43, 1178–1188 CrossRef CAS.
- C. Lochenie, K. Schötz, F. Panzer, H. Kurz, B. Maier, F. Puchtler, S. Agarwal, A. Köhler and B. Weber, J. Am. Chem. Soc., 2018, 140, 700–709 CrossRef CAS PubMed.
- J.-H. Yang, Y.-X. Zhao, J.-P. Xue, Z.-S. Yao and J. Tao, Inorg. Chem., 2021, 60, 7337–7344 CrossRef CAS PubMed.
- S. Ossinger, L. Kipgen, H. Naggert, M. Bernien, A. J. Britton, F. Nickel, L. M. Arruda, I. Kumberg, T. A. Engesser, E. Golias, C. Nather, F. Tuczek and W. Kuch, J. Phys.: Condens. Matter, 2019, 32, 114003 CrossRef PubMed.
- T. Q. Hung, F. Terki, S. Kamara, M. Dehbaoui, S. Charar, B. Sinha, C. Kim, P. Gandit, I. A. Gural'skiy, G. Molnar, L. Salmon, H. J. Shepherd and A. Bousseksou, Angew. Chem., Int. Ed., 2013, 52, 1185–1188 CrossRef CAS PubMed.
- L. Poggini, M. Gonidec, J. H. Gonzalez-Estefan, G. Pecastaings, B. Gobaut and P. Rosa, Adv. Electron. Mater., 2018, 4, 1800204 CrossRef.
- A. C. Bas, X. Thompson, L. Salmon, C. Thibault, G. Molnár, O. Palamarciuc, L. Routaboul and A. Bousseksou, Magnetochemistry, 2019, 5, 1–14 CrossRef.
- M. Matsuda, K. Kiyoshima, R. Uchida, N. Kinoshita and H. Tajima, Thin Solid Films, 2013, 531, 451–453 CrossRef CAS.
- W. Gawelda, A. Cannizzo, V. T. Pham, F. V. Mourik, C. Bressler and M. Chergui, J. Am. Chem. Soc., 2007, 129, 8199–8206 CrossRef CAS PubMed.
- N. Huse, H. Cho, K. Hong, L. Jamula, F. M. D. Groot, T. K. Kim, J. K. McCusker and R. W. Schoenlein, J. Phys. Chem. Lett., 2011, 2, 880–884 CrossRef CAS PubMed.
- J. E. Monat and J. K. McCusker, J. Am. Chem. Soc., 2000, 122, 4092–4097 CrossRef CAS.
- H. T. Lemke, K. S. Kjær, R. Hartsock, T. B. V. Driel, M. Chollet, J. M. Glownia, S. Song, D. Zhu, E. Pace, S. F. Matar, M. M. Nielsen, M. Benfatto, K. J. Gaffney, E. Collet and M. Cammarata, Nat. Commun., 2017, 8, 15342 CrossRef CAS PubMed.
- M. Cavallini, I. Bergenti, S. Milita, J. C. Kengne, D. Gentili, G. Ruani, I. Salitros, V. Meded and M. Ruben, Langmuir, 2011, 27, 4076–4081 CrossRef CAS.
- N. Suryadevara, A. Mizuno, L. Spieker, S. Salamon, S. Sleziona, A. Maas, E. Pollmann, B. Heinrich, M. Schleberger, H. Wende, S. K. Kuppusamy and M. Ruben, Chem. – A Eur. J., 2021, 28, e202103853 CrossRef PubMed.
- Y. Miyazaki, T. Nakamoto, S. Ikeuchi, K. Saito, A. Inaba, M. Sorai, T. Tojo, T. Atake, G. S. Matouzenko, S. Zein and S. A. Borshch, J. Phys. Chem. B, 2007, 111, 12508–12517 CrossRef CAS PubMed.
- E. J. Devid, P. N. Martinho, M. V. Kamalakar, I. Salitros, U. Prendergast, J.-F. Dayen, V. Meded, T. Lemma, R. González-Prieto, F. Evers, T. E. Keyes, M. Ruben, B. Doudin and S. J. van der Molen, ACS Nano, 2015, 9, 4496–4507 CrossRef CAS PubMed.
- S. Rackwitz, J. A. Wolny, K. Muffler, K. Achterhold, R. Rüffer, Y. Garcia, R. Diller and V. Schünemann, Phys. Chem. Chem. Phys., 2012, 14, 14650–14660 RSC.
- S. Ossinger, H. Naggert, E. Bill, C. Nather and F. Tuczek, Inorg. Chem., 2019, 58, 12873–12887 CrossRef CAS.
- J. A. Wolny, S. Rackwitz, K. Achterhold, Y. Garcia, K. Muffler, A. D. Naik and V. Schünemann, Phys. Chem. Chem. Phys., 2010, 12, 14782–14788 RSC.
- J. A. Wolny, R. Diller and V. Schünemann, Eur. J. Inorg. Chem., 2012, 2635–2648 CrossRef CAS.
- J.-L. Wang, Q. Liu, X.-J. Lv, R.-L. Wang, C.-Y. Duan and T. Liu, Dalton Trans., 2016, 45, 18552–18558 RSC.
- J. Diaz-Escobar and V. Kober, Applications of Digital Image Processing XXXIX, 2016, p. 99712A Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- D. Gentili, F. Liscio, N. Demitri, B. Schäfer, F. Borgatti, P. Torelli, B. Gobaut, G. Panaccione, G. Rossi, A. D. Esposti, M. Gazzano, S. Milita, I. Bergenti, G. Ruani, I. Šalitroš, M. Ruben and M. Cavallini, Dalton Trans., 2016, 45, 134–143 RSC.
- V. Blum, R. Gehrke, F. Hanke, P. Havu, V. Havu, X. Ren, K. Reuter and M. Scheffler, Comput. Phys. Commun., 2009, 180, 2175–2196 CrossRef CAS.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef PubMed.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783–9792 CrossRef CAS.
- S. Sangeetha, V. A. Danie, N. S. F. Nirmal, T. Alwin and T. F. A. Fen, J. Res. Sci., 2014, 2, 2278–9073 Search PubMed.
- L. M. Lawson Daku, Phys. Chem. Chem. Phys., 2018, 20, 6236–6253 RSC.
- M. Ester, H.-P. Kriegel, J. Sander and X. Xu, Proceedings of the Second International Conference on Knowledge Discovery and Data Mining, 1996, 226–231.
- A. Abhervé, M. J. Recio-Carretero, M. López-Jordá, J. M. Clemente-Juan, J. Canet-Ferrer, A. Cantarero, M. Clemente-León and E. Coronado, Inorg. Chem., 2016, 55, 9361–9367 CrossRef.
- W.-X. Liu, Z. Yang, Z. Qiao, L. Zhang, N. Zhao, S. Luo and J. Xu, Nat. Commun., 2019, 10, 4753 CrossRef PubMed.
- F. Varret, C. Chong, A. Slimani, D. Garrot, Y. Garcia and A. D. Naik, Real-Time Observation of Spin-Transitions by Optical Microscopy, John Wiley & Sons, Ltd, 2013, ch. 16, pp. 425–441 Search PubMed.
- S. Bonnet, G. Molnár, J. S. Costa, M. A. Siegler, A. L. Spek, A. Bousseksou, W. T. Fu, P. Gamez and J. Reedijk, Chem. Mater., 2009, 21, 1123–1136 CrossRef CAS.
- C. Chong, H. Mishra, K. Boukheddaden, S. Denise, G. Bouchez, E. Collet, J. C. Ameline, A. D. Naik, Y. Garcia and F. Varret, J. Phys. Chem. B, 2010, 114, 1975–1984 CrossRef CAS PubMed.
- C. Chong, A. Slimani, F. Varret, K. Boukheddaden, E. Collet, J. C. Ameline, R. Bronisz and A. Hauser, Chem. Phys. Lett., 2011, 504, 29–33 CrossRef CAS.
- M. Sy, D. Garrot, A. Slimani, M. Páez-Espejo, F. Varret and K. Boukheddaden, Angew. Chem., Int. Ed., 2016, 55, 1755–1759 CrossRef CAS PubMed.
- A. Slimani, F. Varret, K. Boukheddaden, C. Chong, H. Mishra, J. Haasnoot and S. Pillet, Phys. Rev. B:Condens. Matter Mater. Phys., 2011, 84, 1–8 CrossRef.
- H. Fourati, E. Milin, A. Slimani, G. Chastanet, Y. Abid, S. Triki and K. Boukheddaden, Phys. Chem. Chem. Phys., 2018, 20, 10142–10154 RSC.
- M. Sy, R. Traiche, H. Fourati, Y. Singh, F. Varret and K. Boukheddaden, J. Phys. Chem. C, 2018, 122, 20952–20962 CrossRef CAS.
- A. Goujon, F. Varret, K. Boukheddaden, C. Chong, J. Jefti, Y. Garcia, A. D. Naik, J. C. Ameline and E. Collet, Inorg. Chim. Acta, 2008, 361, 4055–4064 CrossRef CAS.
- R. Traiche, M. Sy, H. Oubouchou, G. Bouchez, F. Varret and K. Boukheddaden, J. Phys.Chem. C, 2017, 121, 11700–11708 CrossRef CAS.
| This journal is © the Owner Societies 2026 |