
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photomolecular rotor dynamics of the oxindole-based photoprotective bacterial pigment violacein
Giovanni
Bressan
 a,
Khalid M.
Siddiqui
a,
Eleanor K.
Ashworth
a,
Pratip
Chakraborty
a,
Dipanjan
Banerjee
a,
Erico M.
Braun
ab,
Stephen R.
Meech
a and
James N.
Bull
*a
a,
Khalid M.
Siddiqui
a,
Eleanor K.
Ashworth
a,
Pratip
Chakraborty
a,
Dipanjan
Banerjee
a,
Erico M.
Braun
ab,
Stephen R.
Meech
a and
James N.
Bull
*a
aChemistry, Faculty of Science, University of East Anglia, Norwich NR4 7TJ, UK. E-mail: james.bull@uea.ac.uk
bInstituto de Física, Universidade Federal do Rio Grande do Sul (UFRGS), Avenida Bento Gonçalves, 9500 Porto Alegre, Brazil
First published on 11th December 2025
Abstract
Conjugated molecules incorporating the oxindole motif offer a versatile scaffold for designing high-performance, light-responsive materials that exploit photomolecular rotor motion. Violacein, a purple coloured oxindole-based pigment produced by bacterial strains in superglacial Antarctic ice melts, serves as a natural photoprotectant. Using ultrafast spectroscopy combined with explicitly-solvated potential energy surfaces, we characterise the excited-state relaxation mechanism in violacein to involve initial relaxation to planar locally excited (LE) state(s), followed by passage through a twisted charge-transfer (CT) state along the molecular rotor coordinate to a nearby isomerising-like conical intersection seam. Propagation over the excited-state barrier connecting the LE and CT states along the photomolecular rotor axis leads to a strong viscosity dependence of the excited state lifetimes (ca. 4 ps in acetonitrile and 100 ps in ethylene glycol). These dynamics, leading to efficient electronic-to-vibrational energy conversion, coupled with a fast rate for thermal conversion of the possible Z photoisomer back to the starting E isomer, confer violacein with desirable photoprotectant properties.
1 Introduction
Many organisms rely on photoresponsive molecules to protect them against harmful visible and ultraviolet (UV) radiation.1–6 Well-known examples include the negative phototactic response of the Halorhodospira halophila bacterium (driven by the chromophore in photoactive yellow protein),7,8 and the biosynthesis of UV-absorbing photoprotectants such as mycosporine-like amino acids and scytonemin in cyanobacteria, as well as related compounds in macroalgae and phytoplankton.9–13 Photoprotection is particularly important for organisms inhabiting environments with high UV fluence, including tropical seas9 and polar ecosystems.11 Antarctica, in particular, has experienced elevated UV exposure during the austral summer over recent decades due to stratospheric ozone depletion,14 leading to higher levels of UV-B (280–315 nm) and UV-C (100–280 nm) radiation.15,16 These conditions impose environmental stresses on organisms, with those possessing robust photoprotective mechanisms having enhanced survivability.Over the past few decades, several genera of bacteria that produce photoprotective pigments have been isolated from Antarctic environments.17 Strains that biosynthesise the oxindole-based pigment violacein (Fig. 1) include Janthinobacterium lividum, Chromobacterium violaceum, and Pseudoalteromonas luteoviolacea.18–20 In these microorganisms, violacein (and oxidised derivatives)21 provides resistance towards environmental stresses, including near-UV radiation and potentially low temperatures.19,22 More broadly, violacein-producing bacteria have been identified in diverse habitats spanning marine, freshwater, and soil environments.20 Beyond its photoprotectant function, violacein exhibits antibacterial, antifungal, antiprotozoal, and promising anticancer activity,23–25 which has spurred extensive pharmacological studies.26 The oxindole motif also features prominently in synthetic photoactive materials, including when coupled with further oxindole or conjugated aromatic groups to provide high-isomerisation yield molecular photoswitches that absorb biologically-desirable red light. N,N′-Disubstituted indigos, for example, provide a scaffold for tuning of the thermal half-lives of Z isomers independently while maintaining red-shifted absorption spectra.27 In this vein, oxindole-based photoswitches have served as a stator in Feringa-type photomolecular motors and other light-responsive materials.28–31 While materials applications typically require high photoisomerisation quantum yields and bistable switching, selected applications including photoprotectants and fast phototriggers for in vivo use32,33 benefit from rapid thermal reversion.
 | ||
| Fig. 1 Molecular structure of the bacterial pigment violacein, responsible for purple colouration of Janthinobacterium lividum. The three constituent units, 5-hydroxyindole, 2-pyrrolidone, and oxindole are indicated. The φ and ϕ coordinates are shown, with φ representing the photomolecular rotor axis. Violacein is not soluble in non-polar solvents or water. | ||
Violacein has a deep purple colour, leading to strong absorption both in the visible (molar absorptivity, ε = 3400–3700 mol−1 cm−1 at 570 nm in alcohols), and in the UV (200–300 nm).34 Two rotatable bonds (C–C and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) connecting its ring units have been proposed to mediate excited-state deactivation pathways via passage through an isomerising conical intersection seam,34,35 that converts electronic excitation energy into nuclear motion (vibrational excitation on the S0 electronic state) that is readily dissipated to the surrounding medium. Indeed, preliminary ultrafast spectroscopy revealed a strong dependence of both fluorescence quantum yield and excited state lifetime (by monitoring ground state recovery) with viscosity,34 ranging from ≈3 ps in methanol, to ≈90 ps in ethylene glycol and ≈140 ps glycerol, consistent with such photomolecular rotor motion, although the underlying mechanism remains uncharacterised. This work also demonstrated that UV excitation at 265 nm led to the same recovery lifetimes as measurements directly exciting the S1 state at 600 nm, concluding that near-UV excitation relaxes rapidly to the S1 state and it is the S1 state dynamics that drive the photoprotection mechanism.
C) connecting its ring units have been proposed to mediate excited-state deactivation pathways via passage through an isomerising conical intersection seam,34,35 that converts electronic excitation energy into nuclear motion (vibrational excitation on the S0 electronic state) that is readily dissipated to the surrounding medium. Indeed, preliminary ultrafast spectroscopy revealed a strong dependence of both fluorescence quantum yield and excited state lifetime (by monitoring ground state recovery) with viscosity,34 ranging from ≈3 ps in methanol, to ≈90 ps in ethylene glycol and ≈140 ps glycerol, consistent with such photomolecular rotor motion, although the underlying mechanism remains uncharacterised. This work also demonstrated that UV excitation at 265 nm led to the same recovery lifetimes as measurements directly exciting the S1 state at 600 nm, concluding that near-UV excitation relaxes rapidly to the S1 state and it is the S1 state dynamics that drive the photoprotection mechanism.
Building on the earlier ultrafast study, we resolve the photomolecular rotor dynamics of violacein in acetonitrile and more viscous (protic) ethylene glycol using ultrafast transient absorption (TA) spectroscopy with a visible-IR probe, with TA bands interpreted using explicitly-solvated potential energy surface calculations within a mixed references spin–flip time-dependent density functional theory (MRSF-TDDDFT) framework. The dynamics involve planar locally excited (LE) state(s), followed by passage through a twisted charge-transfer (CT) state along the molecular rotor coordinate to a nearby isomerising-like conical intersection seam. The potential energy surfaces highlight the importance of including explicit solvation in the calculations in order to stabilise the highly-polarised CT state region.
2 Methods
Samples of violacein were obtained from Merck (>98%) following a Janthinobacterium lividum culture bioextraction. Samples were shipped on dry ice and stored in a −20 °C freezer in order to avoid degradation. Purity was checked using HPLC on shipping and LC-MS on arrival. In dry acetonitrile (Merck, >99%), violacein exhibited good stability over the period of more than 30 days at neutral pH (laboratory temperature regulated at 294 K) and stored under dark conditions, consistent with earlier reports on long-term stability.36 Acetonitrile was chosen as the main solvent because it has non-hydrogen bonding character and has reduced polarity and viscosity compared with methanol, allowing for simpler inclusion of explicit solvent treatment in the calculations. Ethylene glycol (Merck, >99%) was considered to assess the viscosity effect on the excited-state lifetimes.2.1 Steady-state absorption and fluorescence
Steady-state absorption spectra of violacein in solution were measured using a PerkinElmer Lambda XLS spectrometer at T = 294 K in a temperature-controlled laboratory. Fluorescence excitation and emission spectra were recorded using an Edinburgh Instruments FS5 spectrofluorometer. Excitation/emission spectra were monitored/excited at 700/560 nm.2.2 Ultrafast transient absorption
Ultrafast TA data were acquired using a previously reported home-built spectrometer,37–39 using samples in static 1 mm optical path fused-silica cell (Starna). The concentration of violacein in acetonitrile or ethylene glycol was adjusted to achieve a peak OD of ≈0.5 at the maximum in the absorption spectrum. Briefly, ≈450 µJ pulses from the output of a Ti:Sapp regenerative amplifier (Spitfire Ace, Spectra-Physics) operating at 800 nm and 1 kHz repetition rate pumped a commercial non-colinear optical parametric amplifier (NOPA, TOPAS White, Light Conversion) to generate broadband pump pulses centred at 540 nm. Downstream, a commercial acousto-optical programmable dispersive filter (AOPDF, Dazzler, Fastlite) served as a 500 Hz chopper, compensating for second, third, and fourth order spectral phases to achieve nearly transform limited pulses at sample position. Each TA spectrum was averaged over 9000 laser shots. The pump–probe time delay was introduced with a retroreflector in the pump arm mounted on a mechanical linear translation stage (Physik Instrumente). Pump scatter and fluorescence contributions were removed by subtracting a TA spectrum acquired at negative pump–probe delays. The visible to near-IR continuum (500–1200 nm) was generated by seeding the sapphire plate with ≈1 mW of the 1250 nm signal from a commercial OPA (TOPAS Prime, Light Conversion) seeded by 800 µJ of the 800 nm, 1 kHz fundamental. The white-light continuum was split before the sample using a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 beamsplitter as to provide probe and reference beams. The probe was crossed at a 4° angle with the pump beam at the sample position. Pump and probe spot sizes were 80 and 160 µm, respectively. Pump pulse energy and duration at sample position were ≈300 nJ and 30 fs, respectively. The signal was recollimated after the sample and the signal and reference beams were focussed into a home-built dual channel prism-based spectrometer and recorded shot-to-shot by a pair of 1024 pixels CCD detectors (Stresing) synchronised to the AOPDF. The instrument response function (≈45 fs) was measured by spectrally resolving the instantaneous non-resonant solvent response of toluene (see SI). All measurements were conducted at the magic angle to avoid contributions from rotational diffusive motion to the measured signal decay.
:50 beamsplitter as to provide probe and reference beams. The probe was crossed at a 4° angle with the pump beam at the sample position. Pump and probe spot sizes were 80 and 160 µm, respectively. Pump pulse energy and duration at sample position were ≈300 nJ and 30 fs, respectively. The signal was recollimated after the sample and the signal and reference beams were focussed into a home-built dual channel prism-based spectrometer and recorded shot-to-shot by a pair of 1024 pixels CCD detectors (Stresing) synchronised to the AOPDF. The instrument response function (≈45 fs) was measured by spectrally resolving the instantaneous non-resonant solvent response of toluene (see SI). All measurements were conducted at the magic angle to avoid contributions from rotational diffusive motion to the measured signal decay.
2.3 Computational
Electronic structure calculations were performed with the OpenQP,40 GAMESS-US (July 2024 R2 release),41 ORCA 6.1.042 and Gaussian 1643 software packages. Geometry optimisations, vibrational frequencies, and MECPs (minimum energy crossing points) were optimised at the MRSF-TDDFT (BH&HLYP/6-31G*)44,45 level of theory.46,47 The potential energy surfaces were constructed by minimum energy pathway searches in OpenQP. Solvation of violacein the was treated with the DOCKER algorithm,48 where explicit acetonitrile molecules were sequentially added with the XTB trajectories at T = 298 K.49 32 (acetonitrile) explicit solvent molecules were chosen as a balance between computational cost and ensuring the chromophore was sufficiently surrounded by solvent molecules. For the solvated S1 state, because the DOCKER algorithm considered S0 state charge distributions, the calculation was polarised by placing partial charges on each of the two ring systems equivalent to charge density found for isolated S1 state from MRSF-TDDFT – this should approximately account for prompt solvent reorientation due to a change in charge distribution;49 it was not computationally feasible to optimise the complete solvated cluster geometry on the S1 state. The solute–solvent cluster geometries were used to simulate the expected TA spectra from the LE, LE2, and CT states with MRSF-TDDFT stick spectra convoluted with Gaussian functions (FWHM = 0.2 eV). Solvated cluster geometries with ethylene glycol were not feasible due to convergence difficulties.3 Results and discussion
3.1 Ultrafast spectroscopy
The steady-state electronic spectra of violacein in acetonitrile (η = 0.36 cP at T = 294 K)50 are shown in Fig. 2a. The main S1 ← S0 absorption band spans 400–680 nm with maximum at 548 nm. The fluorescence maximum is at 673 nm, giving a Stokes shift of ≈3390 cm−1, indicating a degree of nuclear relaxation to reach the fluorescent state. Selected TA traces are shown in Fig. 2b, with ground state bleach (GSB) and stimulated emission (SE) features assigned through comparison to the steady-state spectra in Fig. 2a. Two positive bands are observed (900–1200 nm & ≈620 nm), labelled as near-IR excited state absorption (ESA) and band B. The SE band shows reshaping over 850–1000 nm on the sub-picosecond timescale, which overlaps with the blue end of the near-IR ESA band. Band B also blueshifts (by ≈30 nm) within the first few hundred femtoseconds. The GSB band is narrower than the corresponding steady-state absorption because the intensity of the probe is negligible for wavelengths shorter than ≈500 nm. | ||
Fig. 2 Steady-state spectroscopy and transient absorption (TA) spectroscopy of violacein at T = 294 K: (a) steady-state absorption (solid, purple), fluorescence excitation (dotted, purple), and fluorescence emission (solid, maroon) in acetonitrile. (b) TA spectra in acetonitrile, with assignments: GSB – ground state bleach; SE – stimulated emission; ESA – near-IR excited state absorption; B – band B due to excited-state absorption from LE states. (c) Evolution associated decay spectra (EADS) from a global fit to data in (b) with the sequential kinetic model 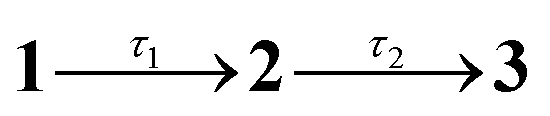 , where 3 is a constant. (d) Representative TA kinetics in acetonitrile at selected wavelengths over GSB, ESA and B band (points) with global model fit (lines). Inset (i) shows a magnified view of the short-lived component over the near-IR ESA band, and inset (ii) displays the data up to 3 ps. (e) Steady-state absorption (purple) and fluorescence emission (maroon) in ethylene glycol. (f) TA spectra in ethylene glycol at selected time delays. (g) EADS from a global fit to data in (f) with the sequential kinetic model , where 3 is a constant. (d) Representative TA kinetics in acetonitrile at selected wavelengths over GSB, ESA and B band (points) with global model fit (lines). Inset (i) shows a magnified view of the short-lived component over the near-IR ESA band, and inset (ii) displays the data up to 3 ps. (e) Steady-state absorption (purple) and fluorescence emission (maroon) in ethylene glycol. (f) TA spectra in ethylene glycol at selected time delays. (g) EADS from a global fit to data in (f) with the sequential kinetic model 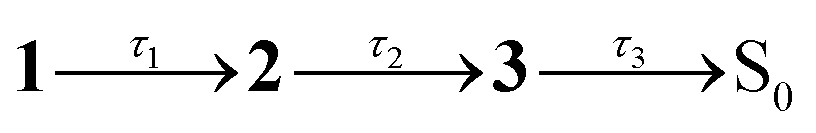 . (h) Representative TA kinetics in ethylene glycol at selected wavelengths. The * in (b), (c), (f), and (g) is an artefact due to subtraction of scattered pump light. . (h) Representative TA kinetics in ethylene glycol at selected wavelengths. The * in (b), (c), (f), and (g) is an artefact due to subtraction of scattered pump light. | ||
A global fit of the TA data to a sequential decay model, 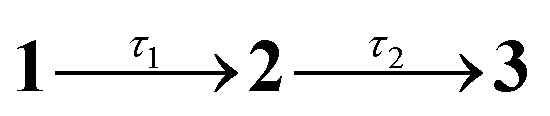 (where 3 is a persistent component), yielded the evolution-associated decay spectra (EADS) shown in Fig. 2c with selected kinetic traces in Fig. 2d. The spectral changes between successive EADS reflect evolution and relaxation of the underlying excited-state population under the assumption of a series of first-order kinetic decay processes. The first EADS (blue trace in Fig. 2c), assigned to the Franck–Condon (FC) state, evolves with τ1 = 0.6 ps into the intermediate EADS (black trace), and is characterised by loss of the near-IR ESA band, partial decay and red-edge broadening of the SE band, and a ≈30 nm blue-shift and growth of band B. This intermediate EADS decays over τ2 = 3.7 ps to the third EADS (red trace in Fig. 2c), characterised by nearly complete recovery of the transient signal except for a weak residual GSB near 580 nm attributed to a photoproduct. The photoproduct, which is presumably the Z isomer, has a lifetime far exceeding the 500 ps measurement window and was therefore treated as constant in the kinetic fit. A slightly red shifted S1(ππ*) absorption band, e.g. by a few to tens of nanometers, is consistent with that expected for Z geometric isomers of stilbenes and oxindole-based photoswitches.30
(where 3 is a persistent component), yielded the evolution-associated decay spectra (EADS) shown in Fig. 2c with selected kinetic traces in Fig. 2d. The spectral changes between successive EADS reflect evolution and relaxation of the underlying excited-state population under the assumption of a series of first-order kinetic decay processes. The first EADS (blue trace in Fig. 2c), assigned to the Franck–Condon (FC) state, evolves with τ1 = 0.6 ps into the intermediate EADS (black trace), and is characterised by loss of the near-IR ESA band, partial decay and red-edge broadening of the SE band, and a ≈30 nm blue-shift and growth of band B. This intermediate EADS decays over τ2 = 3.7 ps to the third EADS (red trace in Fig. 2c), characterised by nearly complete recovery of the transient signal except for a weak residual GSB near 580 nm attributed to a photoproduct. The photoproduct, which is presumably the Z isomer, has a lifetime far exceeding the 500 ps measurement window and was therefore treated as constant in the kinetic fit. A slightly red shifted S1(ππ*) absorption band, e.g. by a few to tens of nanometers, is consistent with that expected for Z geometric isomers of stilbenes and oxindole-based photoswitches.30
The effect of solvent viscosity is illustrated by TA measurements in ethylene glycol (η = 21 cP at T = 294 K).50 The steady-state spectra (Fig. 2e) resemble those in acetonitrile (Fig. 2a) but with a modest red shift in both absorption and fluorescence (spectral features are summarised in Table S1 in the SI). The TA spectra (Fig. 2f) display the same characteristic features observed in acetonitrile (Fig. 2b), with all bands shifted to longer wavelengths. Band B, located between the GSB and SE bands, is less distinct presumably because of the relative shifts of the GSB and SE bands compared with acetonitrile. A global kinetic fit (Fig. 2g), which similarly assumes first-order kinetic decays, used the sequential scheme 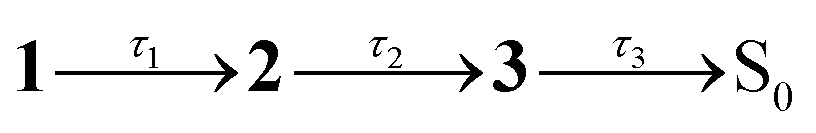 . The first EADS (τ1 = 0.6 ps) due to the FC state is similar in appearance to that obtained in acetonitrile (no apparent viscosity dependence), while the second EADS (τ2 = 27 ps) corresponds to spectral reshaping in the B-band region (with recovery of the overlapping GSB), a slight increase in SE band intensity, but now with a small extent of the near-IR ESA remaining. The third, and longest-lived EADS (τ3 = 80 ps), no longer exhibits near-IR ESA; instead, this EADS appears to be a continuation of the growth of band B and decay of the SE (now extending to ≈1200 nm), suggesting that the dynamics in ethylene glycol are not only much slower than in acetonitrile, but also have more dispersive kinetics, perhaps reflecting the more complex dynamics in a hydrogen-bonding viscous solvent.
. The first EADS (τ1 = 0.6 ps) due to the FC state is similar in appearance to that obtained in acetonitrile (no apparent viscosity dependence), while the second EADS (τ2 = 27 ps) corresponds to spectral reshaping in the B-band region (with recovery of the overlapping GSB), a slight increase in SE band intensity, but now with a small extent of the near-IR ESA remaining. The third, and longest-lived EADS (τ3 = 80 ps), no longer exhibits near-IR ESA; instead, this EADS appears to be a continuation of the growth of band B and decay of the SE (now extending to ≈1200 nm), suggesting that the dynamics in ethylene glycol are not only much slower than in acetonitrile, but also have more dispersive kinetics, perhaps reflecting the more complex dynamics in a hydrogen-bonding viscous solvent.
The pronounced viscosity dependence of the τ2 and τ3 excited-state lifetimes between acetonitrile and ethylene glycol is in accord with the earlier ultrafast study on violacein in methanol, acetone, elthylene glycol, and glycerol, where ground state recovery and loss of stimulated emission were monitored at selected probe wavelengths,34 with the present lifetime (ca. 100 ps) for ethylene glycol consistent with their value of 90 ps for ground state recovery. Ultimately, these viscosity trends are consistent with photomolecular-rotor-type dynamics known for conjugated dyes,51,52 such as cyanines,53 where intramolecular torsion is strongly hindered in viscous environments. Compared with the earlier ultrafast study,34 the present TA spectroscopy has identified two transient absorption bands (band B and near-IR ESA), allowing the detailed photomolecular rotor mechanism to be elucidated using potential energy surfaces described next.
3.2 Photomolecular rotor mechanism
The ultrafast dynamics and photomolecular rotor mechanism active in violacein are understood with the potential energy surfaces summarised in Fig. 3. We first consider the isloated (gas phase) surface since it provides a straightforward means to compute pathways between critical points. In turn, the impact of solvation at the gas-phase critical points is considered and discussed against the experimental data. | ||
| Fig. 3 Potential energy surface calculations for violacein illustrating the photomolecular rotor mechanism: (a) the planar S0 state is excited to the S1 Franck–Condon (FC) state, with a nearby relaxed planar minimum (S1,P). An excited state barrier (S‡1) of 64 meV along the double-bond twisting coordinate (φ) separates S1,P from a twisted minimum, S1,T, with φ = 89° & ϕ ≈ 180°. A MECP, also with a twisted geometry (φ = 87°), allows for isomerising-like internal conversion. A single-bond twisting coordinate (ϕ) leads to a third excited state minimum, S1,S (ϕ = 0°). An alternative MECP involving pyramidalisation (PYR) of the carbonyl-carbon atom on the 2-pyrrolidone ring (angle θPYR = 35°), which is directly accessible from S1,P, lies much higher in energy and is considered inaccessible. Computed PES using linear interpolation in internal coordinates (LIIC) between the MECP and closest minima are shown inset. (b) Upper –relaxed potential energy surface (rPES) between S1,P and S1,T, separated by S‡1. Middle – fluorescence emission oscillator strength along the rPES, showing that the S1,T state is non-fluorescent in the equilibrium geometry. Lower – Löwdin charges on each ring along the S1 state rPES. (c) Natural orbitals for transition to the S1,P (φ = 180°) and S1,T (φ = 89°) geometries. When planar, the transition has locally excited (LE) ππ* character over the double bond connecting the oxindole and 2-pyrrolidone moieties. When twisted, there is charge-transfer (CT) character. (d) Emission oscillator strength along a rPES for the ϕ coordinate, consistent with S1,S (LE2) being fluorescent. (e) Illustration of explicitly solvated LE state geometry (32 acetonitrile molecules). (f) Solvated surface and calculated absorption spectra for the LE, LE2, and CT states. Solvation with acetonitrile stabilises all critical points, but particularly the MECP along the φ coordinate. Predicted absorption bands for each minimum indicate the LE state consistent with the TA near-IR ESA band and both LE and LE2 are consistent with TA band B. | ||
The gas-phase potential energy surfaces (Fig. 3a and b) show that violacein adopts a planar ground-state (S0) geometry (φ = 180° & ϕ = 180°). A planar excited-state minimum (S1,P) lies near the FC region and is separated from a twisted excited-state minimum (S1,T, φ = 89°) by a small barrier (S‡1) of 64 meV (516 cm−1) shown in Fig. 3b. Passage along this coordinate corresponds to torsion of the alkene bond connecting the oxindole and 2-pyrrolidone units (Fig. 1), forming the photomolecular rotor axis. The fluorescence emission oscillator strength (fems), and therefore SE efficiency, decreases with increasing photomolecular rotor torsion angle φ, with the S1,T state being dark at the equilibrium geometry. Computed charge distributions (Z) across the three ring systems in violacein show increasing charge separation from planar to twisted geometries, in line with natural transition orbitals that reveal locally excited (LE) and charge-transfer (CT) characters along the photomolecular rotor coordinate (Fig. 3c). More formally, the S1,T state can be classified as a twisted intramolecular charge-transfer (TICT) state.54
Further critical points can be identified with twisting of the single bond (ϕ coordinate) connecting the 5-hydroxyindole and 2-pyrrolidone and groups. Namely, this single-bond twist leads over a higher barrier (S‡1,S) to a third excited-state minimum, denoted S1,S, with LE character and is also fluorescent (Fig. 3d). Hereafter, we refer to the S1,P minimum near the FC geometry as the LE state (i.e., φ = 180° & ϕ = 180°), the S1,S minimum as the LE2 state (i.e., φ = 180° & ϕ = 0°), and the S1,T minimum as the CT state. In principle, the LE2 state can lead on to a CT2 state via twisting along the φ coordinate. However, this CT2 state and particularly the associated excited-state barrier (3.02 eV), lie at higher energy than the presented LE → CT coordinate due to increased steric interaction – these will be further discussed with explicit solvation below.
Two S1/S0 conical intersection seams were identified, with MECPs at 2.63 and 3.85 eV. The lower-energy MECP corresponds to an alkene-like E–Z photoisomerisation pathway,35 consistent with the strong viscosity dependence of the excited-state lifetimes.34 The higher-energy intersection (denoted PYR in Fig. 3a) involves partial torsion along the photomolecular rotor coordinate and pronounced pyramidalisation of the carbonyl carbon in the 2-pyrrolidone ring. Although similar to that reported in a semiempirical study,55 the PYR-MECP lies too high in energy to be accessed from the FC region and is inconsistent with the observed viscosity-dependent excited state lifetimes.34 A conical intersection search along the ϕ coordinate or around S‡1,S without φ twisting failed to locate any low energy seam, although a similar MECP2 critical point with ϕ = 0° and φ ≈ 90° exists at higher energy (≈2.80 eV) than the shown MECP due to increased steric interaction.
Incorporating explicit acetonitrile solvation (Fig. 3e) significantly altered the potential energy surface (Fig. 3f). The computed vertical excitation energy (2.32 eV, 534 nm) agrees with the experimental absorption maximum (λmax = 548 nm). Compared with the gas-phase surface in Fig. 3a, critical points S1,T, S‡1,S, and particularly the MECP are stabilised relative to the LE state. Because the MECP now lies close in energy to the CT state with the pathway between the two requiring minimal twisting motion, population accessing the CT state is expected to quickly pass through the conical intersection to recover the ground electronic state. The ϕ twisting coordinate critical points are also stabilised, such that the barriers along φ (S‡1) and ϕ (S‡1,S) relative to the LE state differ by only 30 meV. Because access to the CT2 state from the LE2 state involves a higher energy barrier (critical point situated at 2.55 eV compared with 2.48 eV for S‡1,S) due to steric effects, the LE2 state will likely act as “trap”, requiring LE2 → LE → CT decay rather than decays through CT2 and MECP2 (not shown). Furthermore, the excited-state barriers S‡1 and S‡1,S may depend on solvent identity or hydrogen-bonding character, such that S‡1,S could lie slightly below S‡1 in some environments.
Simulated absorption spectra for the LE, LE2, and CT states in explicitly solvated acetonitrile predict that the LE state should display a near-IR absorption band and a visible absorption band at ≈590 nm, with the LE2 state showing a diminished near-IR absorption band and enhanced absorption band at ≈565 nm. In contrast, the CT state has no significant visible or near-IR absorption features. These calculations enable assignment of the TA bands in Fig. 2b: the near-IR ESA band corresponds to the planar LE state, while band B is consistent with both the LE and LE2 states (but slightly blue-shifted for LE2).
So far we have considered photoexcitation from a single conformation with φ = 180° and ϕ = 180°, which is the lowest energy gas-phase structure. However, with explicit solvation the alternative φ = 180° and ϕ = 0° conformation lies only ≈4 kJ mol−1 higher in energy, suggesting it should have a population of ≈20% at T = 294 K. Excitation of this conformation leads directly to the LE2 state. We acknowledge that a series of sequential first-order kinetic decays, as supplied by EADS analysis, might not capture any complex parallel dynamics in the instance of early bifurcation of excited-state population (e.g. some population undergoing LE2 state trapping) or inhomogeneous ground-state conformations, therefore our assignments focus on the TA bands rather than individual EADS in detail.
Increasing the solvent viscosity with ethylene glycol resulted in three lifetimes associated with the excited states. Although explicitly solvated PESs were not computed for ethylene glycol, the three EADS display the SE band, indicating they are from the LE states. The first EADS (τ1 = 0.6 ps and will be convoluted with the first solvation relaxation lifetime in each solvent56) is due to relaxation from the FC state to the LE minimum, a process that does not require large-amplitude twisting (no viscosity dependence), consistent with the second EADS still showing some weak near-IR ESA. From this minimum, the excited-state population may evolve along either the φ or ϕ coordinate, with the latter acting as a trap from which decay proceeds via the LE2 → LE → CT sequence. The second (τ2 = 27 ps) and third (τ3 = 80 ps) EADS could correspond to these two relaxation pathways. Alternatively, there could be a different initial conformer distribution due to differing stabilisation influence of ethylene glycol (hydrogen bonding) and acetonitrile (non-hydrogen bonding), with a mixture of LE and LE2 states in the FC envelope. Regardless of the precise assignment of the EADS for violacein in ethylene glycol, the assignment of LE-character states is consistent with the observed near-IR ESA band, band B, and SE band across the TA spectra. Because the TA spectra always showed the SE band alongside GSB, the CT state must be very short lived due to proximity with the MECP.
Our measurements focussed on the S1 ← S0 transition in the visible region. Violacein, like most aromatic and highly conjugated organic molecules, also has strong absorption in the near-UV (200–400 nm) – see Fig. 2a inset. Previous 265 nm pump–probe experiments in methanol reported the same GSB recovery lifetime (τ ≈ 5.6 ps) as measurements directly exciting the S1 state at 600 nm.34 We therefore conclude that near-UV excitation relaxes rapidly to the S1 state, without substantial nuclear rearrangement. In turn, the photomolecular-rotor dynamics, as characterised in this work, operate on the S1 state.
The photoprotectant properties of violacein are due to efficient electronic-to-vibrational energy transfer, facilitated by photomolecular rotor motion. Understanding these dynamics with viscosity is relevant biologically. Specifically, in Chromobacterium violaceum, violacein accumulates in bacterial cell membranes.18 Membrane microviscosities (10–100 cP),57,58 with the lower end of the scale corresponding to near-headgroup region of membrane bilayers, overlap with the ethylene-glycol regime used here. Thus, the excited-state dynamics of violacein in ethylene glycol mimic those for violacein in that cellular environment.
3.3 Recyclability of the photoprotectant
An ideal photoprotectant must be efficiently recycled so that the protective mechanism can operate repeatedly without degradation. If a persistent photoproduct was formed in our TA measurements, the GSB band in the TA spectra would fail to recover fully. The nearly complete GSB recovery in acetonitrile indicates a very low photoisomerisation quantum yield and minimal accumulation of a long-lived photoproduct. In principle, the photoisomerisation quantum yield is governed by the topography of the conical intersection seam, specifically whether the intersection has a sloped or peaked topology with respect to the branching-plane coordinates.59 However, rigorous determination of CI topology requires evaluation of both the energy-gap gradient (g) and nonadiabatic coupling (h) vectors, which is beyond the present scope. Experimentally, irradiating a violacein-acetonitrile solution with a high-powered 590 nm LED for 2 minutes to generate a photostationary state, followed by measurement of an absorption spectrum several seconds later produced an identical spectrum to the initial violacein solution (shielded from light) and also a violacein solution left in a dark cupboard for two weeks. We therefore concluded that any Z photoisomer undergoes rapid, sub-second thermal reversion, efficiently regenerating the initial violacein chromophore. This conclusion is consistent with a transition state search, finding a low (gas phase) Z → E thermal isomerisation barrier of ≈65 kJ mol−1, although this barrier will be modified by solvation.Similar self-recovering photoprotectant behaviour has been reported for other natural pigments such as mycosporine-like amino acids and scytonemin, which dissipate absorbed UV energy non-destructively and revert to their ground-state configurations without degradation.9,11,60,61 The analogous photophysical recyclability in violacein is consistent with its biological effectiveness as a robust, reusable molecular photoprotectant.
4 Conclusions
The photoprotectant molecule violacein relaxes through excited-state photomolecular rotor dynamics, in which initially planar locally excited state(s) formed on a sub-picosecond timescale from the Franck–Condon geometry decays over a low barrier to a twisted charge-transfer state that is rapidly lost by internal conversion through a nearby conical intersection seam. Efficient electronic-to-vibrational energy conversion during the photomolecular rotor process and rapid thermal recovery of any transient Z isomer render violacein as a recyclable molecular photoprotectant.62 Such behaviour illustrates how natural photoprotectants achieve efficient, non-destructive energy dissipation through ultrafast excited-state dynamics, helping to offer a guiding principle for designing synthetic light-responsive molecules with inherent self-recovery and photostability. The study highlights how explicit solvation modifies excited-state potential energy surface critical points, particularly in stabilising twisted geometry potential energy barriers, which are often associated with the rate-limiting steps ultrafast dynamics.Beyond its biological role, understanding the mechanistic details of the excited-state pathways in violacein provides a valuable framework for the rational design of synthetic photoprotectants, photoswitches, and other optoelectronic materials that must combine high photostability, efficient internal conversion, and recyclability. Future studies integrating time-resolved spectroscopy with non-adiabatic molecular dynamics simulations could reveal how subtle structural modifications or environmental interactions, such as deprotonation, modify the non-radiative decay channels in violacein and similar oxindole-based chromophores. Indeed, deprotonation of a hydroxy-substituted oxindole-based motors has been proposed to modify charge-transfer character and enhance access to conical intersection seams.63
Conflicts of interest
There are no conflicts to declare.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: steady-state spectroscopy of the violacein; TA spectroscopy IRF characterisation; Cartesian coordinates of potential energy surface critical points. See DOI: https://doi.org/10.1039/d5cp04151a.
Acknowledgements
This work was funded by an EPSRC New Investigator Award (EP/W018691 to JNB) and an EPSRC Grant (EP/X011410/1 to SRM). GB is grateful to the Leverhulme Trust for funding him through an Early Career Fellowship (grant no. ECF-2023-195). EMB thanks the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) for his studentship (SWE 200581/2025-2). Dr Tony Kao and Mr Alex Cook are thanked for providing initial spectroscopic characterisations.Notes and references
- R. P. Rastogi, Richa, R. P. Sinha, S. P. Singh and D.-P. Häder, J. Ind. Microbiol. Biotechnol., 2010, 37, 537–558 CrossRef CAS PubMed.
- R. Croce and H. van Amerongen, Nat. Chem. Biol., 2014, 10, 492–501 CrossRef CAS PubMed.
- J. M. Woolley, M. Staniforth, M. D. Horbury, G. W. Richings, M. Wills and V. G. Stavros, J. Phys. Chem. Lett., 2018, 9, 3043–3048 CrossRef CAS PubMed.
- N. C. M. Magdaong and R. E. Blankenship, J. Biol. Chem., 2018, 293, 5018–5025 CrossRef CAS PubMed.
- L. Núñez-Pons, C. Avila, G. Romano, C. Verde and D. Giordano, Mar. Drugs, 2018, 16, 336 CrossRef PubMed.
- S. Krishnan P, A. Salian, S. Dutta and S. Mandal, Mat. Chem. Front., 2021, 5, 7696–7723 RSC.
- W. W. Sprenger, W. D. Hoff, J. P. Armitage and K. J. Hellingwerf, J. Bacteriol., 1993, 175, 3096–3104 CrossRef CAS PubMed.
- L. J. G. W. van Wilderen, M. A. van der Horst, I. H. M. van Stokkum, K. J. Hellingwerf, R. van Grondelle and M. L. Groot, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15050–15055 CrossRef CAS PubMed.
- W. C. Dunlap and J. M. Shick, J. Phycol., 1998, 34, 418–430 CrossRef.
- R. P. Sinha, S. P. Singh and D.-P. Häder, J. Photochem. Photobiol., B, 2007, 89, 29–35 CrossRef CAS PubMed.
- R. P. Sinha and D.-P. Häder, Plant Sci., 2008, 174, 278–289 CrossRef CAS.
- J. I. Carreto, S. Roy, K. Whitehead, C. A. Llewellyn and M. O. Carignan, in UV-Absorbing ‘Pigments’: Mycosporine-like Amino Acids, ed. S. Roy, C. A. Llewellyn, E. S. Egeland and G. Johnsen, Cambridge University Press, 2011, pp. 412–442 Search PubMed.
- A. Ekebergh, P. Sandin and J. Mårtensson, Photochem. Photobiol. Sci., 2015, 14, 2179–2186 CrossRef CAS PubMed.
- J. Staehelin, N. R. P. Harris, C. Appenzeller and J. Eberhard, Rev. Geophys., 2001, 39, 231–290 CrossRef CAS.
- J. A. Nienow, C. P. McKay and E. I. Friedmann, Microb. Ecol., 1988, 16, 271–289 CrossRef PubMed.
- J. Rozema, L. O. Björn, J. F. Bornman, A. Gaberščik, D.-P. Häder, T. Trošt, M. Germ, M. Klisch, A. Gröniger, R. P. Sinha, M. Lebert, Y.-Y. He, R. Buffoni-Hall, N. V. J. de Bakker, J. van de Staaij and B. B. Meijkamp, J. Photochem. Photobiol., B, 2002, 66, 2–12 CrossRef CAS PubMed.
- H. J. Smith, C. M. Foreman, T. Akiyama, M. J. Franklin, N. P. Devitt and T. Ramaraj, Genome Announce., 2016, 4, e01468-15 CrossRef PubMed.
- M. Konzen, D. De Marco, C. A. Cordova, T. O. Vieira, R. V. Antônio and T. B. Creczynski-Pasa, Bioorg. Med. Chem., 2006, 14, 8307–8313 CrossRef CAS PubMed.
- N. Mojib, A. Farhoomand, D. T. Andersen and A. K. Bej, Extremophiles, 2013, 17, 367–378 CrossRef CAS PubMed.
- S. Y. Choi, K.-H. Yoon, J. I. Lee and R. J. Mitchell, BioMed Res. Int., 2015, 2015, 1–8 CAS.
- T. Hoshino, Appl. Microbiol. Biotechnol., 2011, 91, 1463–1475 CrossRef CAS PubMed.
- F. Pantanella, F. Berlutti, C. Passariello, S. Sarli, C. Morea and S. Schippa, J. Appl. Microbiol., 2006, 102, 992–999 Search PubMed.
- R. M. Brucker, R. N. Harris, C. R. Schwantes, T. N. Gallaher, D. C. Flaherty, B. A. Lam and K. P. C. Minbiole, J. Chem. Ecol., 2008, 34, 1422–1429 CrossRef CAS PubMed.
- N. R. Myeong, H. J. Seong, H.-J. Kim and W. J. Sul, J. Biotechnol., 2016, 223, 36–37 CrossRef CAS PubMed.
- N. Durán, G. Nakazato, M. Durán, I. R. Berti, G. R. Castro, D. Stanisic, M. Brocchi, W. J. Fáaro, C. V. Ferreira-Halder, G. Z. Justo and L. Tasic, World J. Microbiol. Biotechnol., 2021, 37, 151 CrossRef PubMed.
- Y. Kanade, R. Patwardhan and P. Abhyankar, Properties of Violacein: A Promising Natural Pharmaceutical Secondary Metabolite from Marine Environment with Emphasis on Its Anticancer Activity, Springer Nature, Singapore, 2023, pp. 197–230 Search PubMed.
- V. Budzák, J. Jovaišaité, C. Huang, P. Baronas, K. Tulaité, S. Juršénas, D. Jacquemin and S. Hecht, Chem. – Eur. J., 2022, 28, e202200496 CrossRef PubMed.
- D. Roke, M. Sen, W. Danowski, S. J. Wezenberg and B. L. Feringa, J. Am. Chem. Soc., 2019, 141, 7622–7627 CrossRef CAS PubMed.
- D. R. S. Pooler, R. Pierron, S. Crespi, R. Costil, L. Pfeifer, J. Léonard, M. Olivucci and B. L. Feringa, Chem. Sci., 2021, 12, 7486–7497 RSC.
- D. Doellerer, D. R. S. Pooler, A. Guinart, S. Crespi and B. L. Feringa, Chem. – Eur. J., 2023, 29, e202301634 CrossRef CAS PubMed.
- M. Ovalle, D. Doellerer and B. L. Feringa, Angew. Chem., Int. Ed., 2025, 64, e202501872 CrossRef CAS PubMed.
- M. Dong, A. Babalhavaeji, S. Samanta, A. A. Beharry and G. A. Woolley, Acc. Chem. Res., 2015, 48, 2662–2670 CrossRef CAS PubMed.
- P. Kobauri, F. J. Dekker, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2023, 62, e202300681 CrossRef CAS PubMed.
- A. A. Beckstead, Y. Zhang, J. K. Hilmer, H. J. Smith, E. Bermel, C. M. Foreman and B. Kohler, J. Phys. Chem. B, 2017, 121, 7855–7861 CrossRef CAS PubMed.
- B. G. Levine and T. J. Martínez, Annu. Rev. Phys. Chem., 2007, 58, 613–634 CrossRef CAS PubMed.
- C. A. Aruldass, R. Rubiyatno, C. K. Venil and W. A. Ahmad, RSC Adv., 2015, 5, 51524–51536 RSC.
- C. R. Hall, J. Conyard, I. A. Heisler, G. Jones, J. Frost, W. R. Browne, B. L. Feringa and S. R. Meech, J. Am. Chem. Soc., 2017, 139, 7408–7414 CrossRef CAS PubMed.
- G. Bressan, I. A. Heisler, G. M. Greetham, A. Edmeades and S. R. Meech, Opt. Express, 2023, 31, 42687 CrossRef CAS PubMed.
- P. Roy, W. R. Browne, B. L. Feringa and S. R. Meech, Nat. Commun., 2023, 14, 1253 CrossRef CAS PubMed.
- V. Mironov, K. Komarov, J. Li, I. Gerasimov, H. Nakata, M. Mazaherifar, K. Ishimura, W. Park, A. Lashkaripour, M. Oh, M. Huix-Rotllant, S. Lee and C. H. Choi, J. Chem. Theory Comput., 2024, 20, 9464–9477 CrossRef CAS PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.:Comput. Mol. Sci., 2011, 2, 73–78 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. M. Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision B.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- S. Lee, E. E. Kim, H. Nakata, S. Lee and C. H. Choi, J. Chem. Phys., 2019, 150, 184111 CrossRef PubMed.
- W. Park, K. Komarov, S. Lee and C. H. Choi, J. Phys. Chem. Lett., 2023, 14, 8896–8908 CrossRef CAS PubMed.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- A. Fatima, E. K. Ashworth, I. Chambrier, A. N. Cammidge, G. Bressan, S. R. Meech and J. N. Bull, Phys. Chem. Chem. Phys., 2025, 27, 9407–9416 RSC.
- CRC handbook of chemistry and physics, ed. J. R. Rumble, T. J. Bruno, M. J. Doa and D. R. Burgess, CRC Press, Taylor & Francis Group, Boca Raton, 105th edn, 2024 Search PubMed.
- A. Vyšniauskas, M. Qurashi, N. Gallop, M. Balaz, H. L. Anderson and M. K. Kuimova, Chem. Sci., 2015, 6, 5773–5778 RSC.
- Y. Wu, M. Štefl, A. Olzyńska, M. Hof, G. Yahioglu, P. Yip, D. R. Casey, O. Ces, J. Humpolíčková and M. K. Kuimova, Phys. Chem. Chem. Phys., 2013, 15, 14986 RSC.
- S. Murphy, B. Sauerwein, H. G. Drickamer and G. B. Schuster, J. Phys. Chem., 1994, 98, 13476–13480 CrossRef CAS.
- S. McConnell, R. H. McKenzie and S. Olsen, J. Chem. Phys., 2015, 142, 084502 CrossRef PubMed.
- X. Chen, W. Wang, J. Han and S.-H. Xia, Chem. Phys., 2024, 581, 112251 CrossRef CAS.
- M. L. Horng, J. A. Gardecki, A. Papazyan and M. Maroncelli, J. Phys. Chem., 1995, 99, 17311–17337 CrossRef CAS.
- M. K. Kuimova, Phys. Chem. Chem. Phys., 2012, 14, 12671 RSC.
- M. Paez-Perez and M. K. Kuimova, Angew. Chem., Int. Ed., 2023, 63, e202311233 CrossRef PubMed.
- A. Migani, M. A. Robb and M. Olivucci, J. Am. Chem. Soc., 2003, 125, 2804–2808 CrossRef CAS PubMed.
- R. P. Sinha and D.-P. Häder, Photochem. Photobiol. Sci., 2002, 1, 225–236 CrossRef CAS PubMed.
- J. M. Shick and W. C. Dunlap, Annu. Rev. Physiol., 2002, 64, 223–262 CrossRef CAS PubMed.
- J.-E. A. Otterstedt, J. Chem. Phys., 1973, 58, 5716–5725 CrossRef CAS.
- M. Mgbukwu, X. Fu, R. Y. Peshkov, D. Doellerer, C. Granados Buitrago, B. L. Feringa, S. Haacke, S. Crespi and J. Léonard, J. Phys. Chem. B, 2025, 129, 3839–3850 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2026 |