
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards a theoretical understanding of excitonic properties of phthalocyanine thin films. I. Low-temperature exciton absorption spectra
Sanghita
Sengupta
 *ag,
Zheng
Pei
ag,
Chance
Lander
a,
Carly R.
Wickizer
a,
Yu
Homma
b,
Hadi
Afshari
c,
Yuezhi
Mao
d,
Pengfei
Huo
e,
Yu
Zhang
f,
Sergei
Tretiak
*f,
Lloyd A.
Bumm
c,
Madalina I.
Furis
*c and
Yihan
Shao
*ag
*ag,
Zheng
Pei
ag,
Chance
Lander
a,
Carly R.
Wickizer
a,
Yu
Homma
b,
Hadi
Afshari
c,
Yuezhi
Mao
d,
Pengfei
Huo
e,
Yu
Zhang
f,
Sergei
Tretiak
*f,
Lloyd A.
Bumm
c,
Madalina I.
Furis
*c and
Yihan
Shao
*ag
aDepartment of Chemistry & Biochemistry, University of Oklahoma, Norman, OK 73019, USA
bResearch Center for Organic Electronics (ROEL), Yamagata University, Jonan 4-3-16, Yonezawa, Yamagata 992-8510, Japan
cHomer L. Dodge Department of Physics, University of Oklahoma, Norman, OK 73019, USA. E-mail: madalina.furis@ou.edu
dDepartment of Chemistry and Biochemistry, San Diego State University, San Diego, CA 92182, USA
eDepartment of Chemistry, University of Rochester, Rochester, NY 14627, USA
fTheoretical Division and Center for Integrated Nanotechnologies, Los Alamos National Laboratory, Los Alamos, NM 87545, USA. E-mail: serg@lanl.gov
gDepartment of Chemistry, Brandeis University, Waltham, MA 02453, USA. E-mail: ssengupta@brandeis.edu; yihanshao@brandeis.edu
First published on 3rd March 2026
Abstract
Phthalocyanine (Pc)-based molecular thin films have emerged in recent years as a promising class of organic semiconductor materials for optoelectronic applications owing to their long exciton coherence length and fast exciton diffusion. However, the dependence of their exciton properties on the dimensionality and thermodynamic conditions, presence of metal ions, and effects of chemical modifications to the PC systems is not yet fully understood. As a first step towards a more comprehensive theoretical understanding of the excitonic properties of Pc thin films, we model their low-temperature exciton absorption spectra by employing the Frenkel Hamiltonian. The latter is derived from quantum-chemical estimates of site energies and exciton–exciton couplings. The predicted exciton absorption spectra of octabutoxy phthalocyanine (H2OBPc) is found to be strongly dependent on the dimensionality of the model as well as the distance cutoff for including the monomer–monomer exciton coupling. We also caution that the widely used dipole–dipole approximation could substantially overestimate the excitonic coupling between different monomers compared to a more accurate evaluation using the respective transition densities.
1 Introduction
Thin films composed of π-conjugated organic molecules are highly versatile materials, providing a robust platform for both fundamental photophysical studies and practical device applications. Their unique optical and electronic properties make them suitable for a range of optoelectronic technologies, including organic light-emitting diodes (OLEDs), organic photovoltaic cells (OPVs), and organic field-effect transistors (OFETs).1–15 A key feature that underscores their applications is their unique ability to support long-distance energy transport via excitons, in addition to charge transport.14,16–20At the microscopic level, the structural and electronic properties of these organic materials are primarily controlled by the nature of the intermolecular interactions.5,9,20 In crystals of uncharged aromatic organic molecules, these interactions are predominantly π–π and van der Waals dispersion.21,22 In contrast, Coulomb interactions between excitons on neighboring molecules enable exciton delocalization across multiple molecules, giving rise to coherent excitonic states that can diffuse in three dimensions.23 As such, a combination of structural adaptability and efficient energy transport underpins the performance of many thin-film-based organic-optoelectronic devices.24 One example is non-peripheral octabutoxy phthalocyanine (H2OBPc) (Fig. 1(a)) which crystallizes in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) triclinic system. It has a two-molecule basis where the two molecules are related by inversion symmetry, Fig. 2 (and SI Section S1 for more details). The molecules are organized into layers of π–π interacting molecules in the ab plane with the Pc rings tilted with respect to the plane. The layers then stack along the c axis. The interaction between the layers is predominantly the van der Waals interactions between the alkoxy chains.
triclinic system. It has a two-molecule basis where the two molecules are related by inversion symmetry, Fig. 2 (and SI Section S1 for more details). The molecules are organized into layers of π–π interacting molecules in the ab plane with the Pc rings tilted with respect to the plane. The layers then stack along the c axis. The interaction between the layers is predominantly the van der Waals interactions between the alkoxy chains.
 | ||
| Fig. 1 (a) Molecular structure of H2OBPc, R = –O(CH2)3CH3. (b) Cross-polarized optical microscope image of an H2OBPc crystalline thin film deposited through the meniscus-guided technique.22 (c) Low-temperature (T = 4 K) absorbance (black line) recorded from the largest crystalline grain in (b). Contributions from several excitonic states are identified through a fitting procedure.26 The contributions of the four near-bandgap excitonic states are highlighted in color. | ||
 | ||
| Fig. 2 Three-dimensional perspective of the molecular arrangement in the H2OBPc crystal structure given by parallel-view stereo pairs. (A) A view of a 3 × 3 × 2 tiling of unit cells. The molecules are arranged in layers of π–π interacting molecules (ab plane). The layers are stacked in the c axis interacting by the alkyl chain van der Waals interactions and longer-range π–π interactions. The unit cell from the literature is shown in green,39 which includes the lower half of the upper layer and the upper half of the lower layer. Here the a axis is aligned with the Cartesian x axis and the b axis is in the xy Cartesian plane. The triclinic P unit cell contains two molecules related by inversion symmetry. These two molecules are distinguished by color. For clarity, the Pc rings are rendered in bold red and blue. The associated alkoxy chains are magenta and cyan, respectively. The upper layer is rendered with reduced opacity for clarity. (B) A view of a single layer in the ab plane tiled 3 × 3 × 1 showing the arrangement of the Pc rings in the distorted hexagonal lattice. The lower ab face of the unit cell from (A) is shown. Note that the [10] lattice direction (the short ab face diagonal) lies in the plane of the Pc rings. The [111] direction (long body diagonal) is perpendicular to the plane of the Pc rings. The hexagonal lattice connecting the centers of the Pc rings is shown in thick gray lines. | ||
Despite their remarkable features, organic molecular thin films remain poorly understood when it comes to the factors governing their static and dynamic properties. This limited understanding has hindered their widespread applications as optoelectronic materials, where precise control over molecular packing, exciton dynamics, and charge transport is crucial for optimal device performance. In the case of oriented mol thin films—whose crystallinity is illustrated in Fig. 1(b)—recent experimental advancements in the fabrication of macroscopically ordered crystalline films have unveiled clear signatures of delocalized, Wannier-like excitonic states formed at low temperatures.22,25–27 Most notably, these excitonic states are characterized by low binding energies (∼30–50 meV), a blue shift of excitonic energy with increasing temperature, and very intense sub-nanosecond radiative recombination at T = 4 K.22,25–27 From a static perspective, it remains unclear how peripheral functionalization of the organic molecules modulates the crystal lattice structure and, in turn, how such crystal packing variations affect both the dimensionality and degree of delocalization of coherent excitonic states. The extent of this delocalization is often characterized by the exciton coherence length (Lcoh), which quantifies the spatial extent of individual molecular units participating in a coherent exciton state. On the other hand, it is equally unclear how dynamical properties [e.g. exciton diffusion coefficient (![[scr D, script letter D]](https://www.rsc.org/images/entities/char_e523.gif) ), exciton lifetime (τ), and ultimately the exciton diffusion length
), exciton lifetime (τ), and ultimately the exciton diffusion length  ] are impacted by local (molecular) and global (lattice) thermal fluctuations.
] are impacted by local (molecular) and global (lattice) thermal fluctuations.
Complementary to experimental techniques, such as X-ray crystallography and ultrafast spectroscopy, the aforementioned two major challenges have been addressed with theoretical and computational modeling. The coherent exciton states in organic molecular thin films are known to be well described by the Frenkel–Davydov exciton model.28 The effects of dynamic disorders in such systems have been modeled primarily through the Frenkel–Davydov–Holstein–Peierls model, which factors in how phonon modes modulate the exciton energies of individual molecules (Holstein coupling) and the exciton coupling between neighboring molecules (Peierls coupling).29–31 Alternatively, dynamic disorders have been captured through atomistic molecular dynamics simulations at certain thermodynamic conditions.32
Inspired by these theoretical advances, especially (a) the 1D modeling of H2OBPc thin films by Fornari, Aragó, and Troisi,29 and (b) recent reports on the significance of model dimensionality and longer-range Coulomb interactions on the excitonic properties of other organic semiconductor systems,30,31 here we undertake a comprehensive re-examination of the excitonic properties of the H2OBPc thin films through a series of theoretical and computational modeling efforts. In the long term, our goal is to help unravel chemical-composition → crystalline-structure → exciton-properties relationships, thereby providing guidance for the development of phthalocyanine (Pc) analogs that maximize the exciton coherence length and energy diffusion in organic thin films. As the first step in this direction, this work focuses on predicting the low-temperature exciton absorption spectra of H2OBPc thin films with the aim of answering three simple but fundamental questions: (a) how does the dimensionality of the modeling (1D vs. 2D vs. 3D) affect the predicted excitonic properties? (b) does long-range Coulomb coupling between H2OBPc molecules (beyond nearest neighbor approximation) contribute substantially to the exciton properties? and (c) how reliable is the dipole–dipole approximation for computing the Coulomb coupling compared to a more accurate approach based on exciton transition densities?
Our paper is organized as follows. In Section 2, we introduce the Davydov–Frenkel–Holstein–Peierls Hamiltonian, which is subsequently reduced to the Davydov–Frenkel Hamiltonian, suitable for modeling low-temperature behavior in the absence of phonon excitations. Section 3 outlines the computational framework used to extract interaction parameters via first-principles time-dependent density functional theory (TD-DFT) simulations and describes the tight-binding approach for constructing the Hamiltonian of molecular aggregates with varying dimensionalities and distance cutoffs. We also simulate low-temperature absorption spectra using a Voigt profile. The experimental procedure for acquiring absorption spectra in Fig. 1(c), along with the spectral peak fitting, is detailed in Section 3.4. Results from TD-DFT calculations are summarized in Section 4. A comparison of simulated and experimental absorption spectra across different aggregate models is discussed in Section 5. Finally, Section 6 summarizes our key findings and suggests possible extensions to incorporate finite-temperature effects.
2 Theoretical framework: model Hamiltonian for excitons
We consider a model organic semiconductor consisting of N sites (molecules), each having K excitonic states and coupled to a single phonon mode. The excitonic behavior of this system can be well described by the Frenkel–Davydov–Holstein Hamiltonian,28,33–38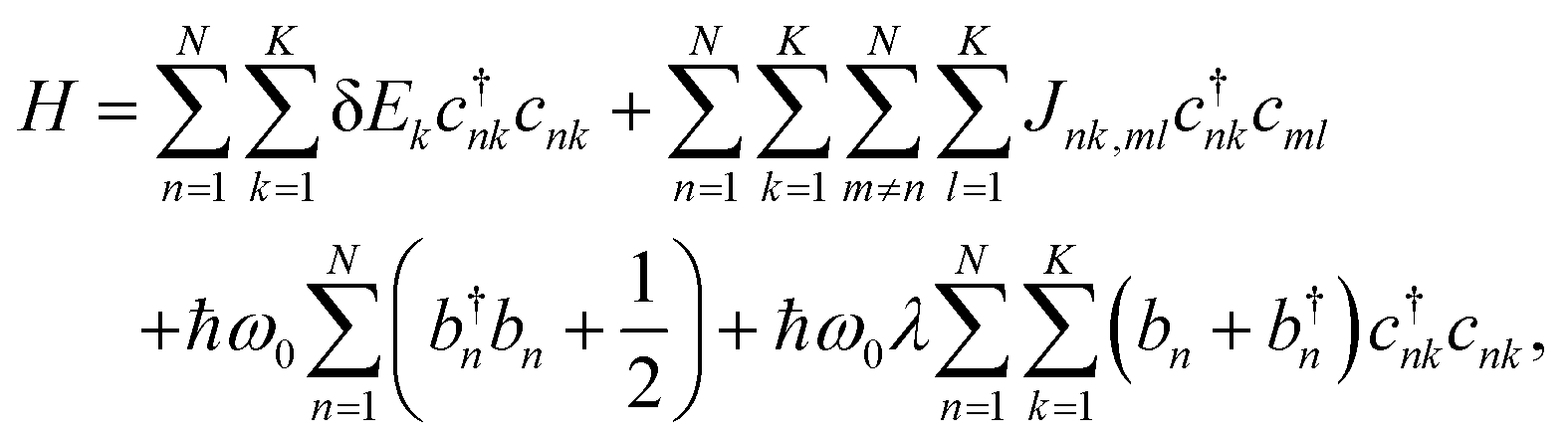 | (1) |
This study employs the low-temperature approximation described in Section 1, where the phonon-related terms are neglected. As a result, eqn (1) reduces to the Frenkel exciton Hamiltonian,
 | (2) |
Here parameters such as excitation energies (δEk) and coupling constants (Jnk,ml) can be directly obtained from first-principle calculations (Section 4), and subsequently incorporated into eqn (2). Diagonalization of the Hamiltonian matrix yields eigenvalues, Ei, and eigenvectors, vnk,i, which are then used to simulate the exciton absorption spectra.
3 Computational & experimental methodology
Our computational methodology includes two main steps, (i) the extraction of excitation energies and other relevant parameters from TD-DFT calculations, and (ii) implementation of a tight-binding formalism to solve the Hamiltonian given in eqn (2). The following subsections detail each step.3.1 Excited state calculations of molecular monomers and dimers
Excited state calculations for H2OBPc monomer and dimers were performed using the TD-DFT formalism40,41 within the Q-Chem software package.42,43 Several standard functionals, including PBE,44 PBE0,45,46 LRC-ωPBE,47 and ωB97X-D,48 were employed with the 6-31G(d,p) basis set;49–51 however, only the results from PBE0 were presented in the following main text for the sake of clarity whereas the rest were provided in SI. The basis set dependence of the on-site energies, transition dipole moments, and Coulomb couplings were benchmarked with PBE0 and provided in Tables S1 and S2. The transition densities were computed using the libwfa package52 as implemented in Q-Chem.To obtain structures of molecular monomer and dimers used for TD-DFT calculations, we first extracted unit cell parameters39 and atomic coordinates from a standard crystallographic information file (CIF) of H2OBPc as shown within a 3D representation in Fig. 2 (and SI Section S1 for more details), including two molecules in a unit cell with inversion symmetry. The relevant dimers were identified based on inter-monomer distance (Dd) between the centers of mass of each monomer as shown in Fig. 3(b). The obtained structures were then subjected to TD-DFT calculations to evaluate the following key parameters: excitation energies for the monomer's Qx and Qy states, representing nearly degenerate excited states oriented along orthogonal directions within the molecular plane, see Fig. 1(a) and Fig. 4; the corresponding transition dipole moment vectors; and excitonic coupling terms for various dimers in Fig. 3(b). These intermolecular coupling terms could be calculated using either of the two approaches below. In the point dipole approximation, the interaction can be modeled as occurring between two transition dipole moments located at the centers of mass of each molecule in the dimer. This method is computationally efficient but assumes a simplified interaction that may neglect important spatial and electronic details. Alternatively, a more rigorous approach that involves evaluating the electrostatic Coulomb interaction between the transition densities associated with the excited states of each molecule in the pair can be employed. This method enables a more accurate and physically realistic assessment of exciton–exciton interactions, particularly for closely spaced molecules where the point dipole approximation may fail. The Q-Chem package includes an implementation of this methodology, dubbed as the direct coupling scheme,53 which we employed in this study. Note that the Coulomb coupling can be further approximated by representing the transition densities as transition density cubes54 or atomic transition charges.32,55,56
 | ||
Fig. 3 Analysis of the molecular arrangement in the H2OBPc crystal. (a) Top view of a 2D layer of the crystal, which corresponds to the xy-plane in Fig. 2(b) and shows three cutoff distances. (b) In the distorted hexagonal lattice, there are three types of nearest neighbors: A (7.43 Å, magenta line), B (9.741 Å, green line), and C (9.903 Å, blue line) within a cutoff distance of 10 Å; three types of second-nearest neighbors (shown in black solid lines): ![[scr Z, script letter Z]](https://www.rsc.org/images/entities/char_e14b.gif) 1 (14.013 Å), 2 (14.309 Å), and 3 (16.936 Å), all within a cutoff distance of 17 Å. Also shown are nine third-nearest neighbors (gray solid line): Ω1 (15.873 Å), Ω2 (18.181 Å), Ω3 (18.807 Å), Ω4 (20.131 Å), Ω5 (20.62 Å), Ω6 (23.1 Å), Ω7 (23.323 Å), Ω8 (25.795 Å) and Ω9 (25.98 Å) and one fourth-nearest neighbor η1 (22.703 Å, red dotted line) which fall within 26 Å. (c) Top, side, and tilted views of two layers of the distorted hexagonal lattice of H2OBPc with the two shortest interlayer distances: Σ1 (16.907 Å, pink line) and Σ2 (14.517 Å, red line). For clarity, only the center-of-mass of each molecule is shown. (d)–(g) Four different 1D models (highlighted with arrows) of H2OBPc: zig-zag orientations of the 1D-AB, 1D-AC, 1D-BC models; 1D-BACA arm-chair model. 1 (14.013 Å), 2 (14.309 Å), and 3 (16.936 Å), all within a cutoff distance of 17 Å. Also shown are nine third-nearest neighbors (gray solid line): Ω1 (15.873 Å), Ω2 (18.181 Å), Ω3 (18.807 Å), Ω4 (20.131 Å), Ω5 (20.62 Å), Ω6 (23.1 Å), Ω7 (23.323 Å), Ω8 (25.795 Å) and Ω9 (25.98 Å) and one fourth-nearest neighbor η1 (22.703 Å, red dotted line) which fall within 26 Å. (c) Top, side, and tilted views of two layers of the distorted hexagonal lattice of H2OBPc with the two shortest interlayer distances: Σ1 (16.907 Å, pink line) and Σ2 (14.517 Å, red line). For clarity, only the center-of-mass of each molecule is shown. (d)–(g) Four different 1D models (highlighted with arrows) of H2OBPc: zig-zag orientations of the 1D-AB, 1D-AC, 1D-BC models; 1D-BACA arm-chair model. | ||
 | ||
| Fig. 4 Frontier molecular orbitals (HOMO, LUMO, and LUMO+1) and transition densities of the Qx and Qy excited states of H2OBPc, obtained from DFT and TD-DFT calculations, respectively, at the crystal monomer geometry using the PBE0 functional and 6-31G(d,p) basis. The percentages indicate the weights of the dominant single-particle excitations (HOMO → LUMO or HOMO → LUMO+1) to the Qx and Qy states. The isosurface values for molecular orbitals and transition densities are set to 0.02 and 0.001, respectively. | ||
3.2 Tight-binding formalism for molecular thin films
In the tight-binding Hamiltonian matrix representation of a system with N H2OBPc molecules, the diagonal elements of the resulting 2N × 2N matrix correspond to the on-site excitation energies, where each molecule contributes two excitonic states, Qx and Qy. The off-diagonal elements capture the excitonic couplings, J, between pairs of molecules that lie within a predefined distance cutoff. These quantities were obtained from TD-DFT calculations and the couplings might correspond to either approximate dipole–dipole interactions or the more accurate direct Coulomb couplings. Eqn (3) exemplifies the structure of the Hamiltonian matrix for a system consisting of 3 or more molecules, | (3) |
The tight-binding models were further constructed to explore different dimensionalities (1D, 2D, and 3D) by applying three distance cutoffs, rcut = 10, 17, 26 Å, labeled as “1NN-10 Å”, “2NN-17 Å”, and “3NN-26 Å”, respectively. In Fig. 3(b), all nearest neighbors (dimers A, B, and C) were located within a center-to-center distance of 10 Å. The 17 Å cutoff extended the interaction range to include all second-nearest neighbors (1, 2, 3) as well as one third-nearest neighbor (Ω1). Expanding the cutoff further to 26 Å incorporated additional third-nearest neighbor interactions (Ω2 through Ω9) and one fourth-nearest neighbor coupling η1. This hierarchical inclusion of longer-range interactions enables a more complete modeling of exciton delocalization and collective optical response across increasing dimensionality and coupling patterns within the molecular crystal.
In the 1D models, molecules are aligned in a chain-like structure highlighted as arrows in Fig. 3(d)–(g). For example, the 1D-AB model in Fig. 3(d) incorporates only the nearest neighbor interactions for A and B dimers with the “1NN-10 Å” cutoff, while the second-nearest neighbor interactions for 2 dimers are added to the “2NN-17 Å” cutoff, and the “3NN-26 Å” cutoff includes dimers Ω5 and Ω7. Similarly, the “2NN-17 Å” cutoff adds 1 dimers to the 1D-AC model (Fig. 3(e)), 3 dimers to the 1D-BC model (Fig. 3(f)), and 1, 2, and Ω1 dimers to the 1D-BACA model (Fig. 3(g)), in addition to the corresponding nearest neighbors.
In 2D systems, molecules are arranged in a planar grid within a single layer, with intermolecular distances defined in Fig. 3(b). This planar layout supports strong in-plane excitonic interactions, capturing the characteristics of two-dimensional molecular aggregates. In contrast, 3D systems incorporate multiple layers, which are stacked on top of each other as shown in Fig. 3(c). This stacking results in a three-dimensional arrangement, where interactions occur both within individual layers and between adjacent layers (through the Σ1- and Σ2-dimer interactions illustrated in Fig. 3(c)), offering a comprehensive representation of the bulk molecular systems.
3.3 Simulation of absorption spectra
Our modeling of the absorption spectra relies on the computation of the spectral weight A(λ) as a function of wavelength λ. To capture realistic spectral features, the absorption lines are broadened using a Voigt profile, representing a convolution of two broadening mechanisms, namely a Gaussian component typically associated with Doppler broadening due to thermal fluctuations or other inhomogeneous effects, as well as a Lorentzian component accounting for finite excited state lifetimes. The Voigt profile of a center λi is computed using the Faddeeva function ω(z) = e−z2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) erfc(−iz), given as
erfc(−iz), given as | (4) |
 , where γi corresponds to the width in Lorentzian function
, where γi corresponds to the width in Lorentzian function ![[script L]](https://www.rsc.org/images/entities/char_e144.gif) i(λ) ∝ γi/π[(λ − λi)2 + γi2], and σ comes from the Gaussian distribution
i(λ) ∝ γi/π[(λ − λi)2 + γi2], and σ comes from the Gaussian distribution ![[capital G, script]](https://www.rsc.org/images/entities/char_e112.gif) i(λ) ∝ exp(−(λ − λi)2/2σ2) that captures inhomogeneous effects.
i(λ) ∝ exp(−(λ − λi)2/2σ2) that captures inhomogeneous effects.
It is well established that TD-DFT simulations based on different density functional approximations can exhibit systematic errors in excitation energies, often underestimating or overestimating absolute values depending on the choice of functional. Consequently, we introduce an energy shift correction ΔEshift, in order to align the computational results with the experimental peak positions and spectral tails more accurately. Accordingly, the transition wavelength of i-th excitonic state is modified as,
 | (5) |
We also use the oscillator strength fi to weigh the Voigt profile that scales the contribution of each electronic transition to the overall absorption spectrum, which is calculated as,
 | (6) |
![[T with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0054_20d1.gif) k and l represent the transition dipole moment vector of the k- and l-th excited state of each monomer, respectively.
k and l represent the transition dipole moment vector of the k- and l-th excited state of each monomer, respectively.
Finally, we sum over all transitions to get the total absorption spectra,
 | (7) |
3.4 Absorption measurement and spectral fitting
The simulated absorption spectra were compared with the experimental absorption measurements performed at T = 4 K of H2OBPc crystalline thin film fabricated using a meniscus-guided, solution processing deposition technique known as pen-writing.57–60 Briefly, a glass capillary, loaded with a 0.5% H2OBPc solution in toluene, is translated at a speed of 20 µm s−1 onto a dielectric substrate (e.g. glass, indium tin oxide (ITO), or sapphire). At this speed, nucleation takes place exclusively at the line of contact between the solution meniscus and the substrate, resulting in a film morphology that features long macroscopic crystalline grains, as evidenced by the cross-polarized optical microscope image in Fig. 1(b). The contrast between grains observed in this figure originates from the different orientation of crystalline axes and excitonic dipoles in neighboring grains. Extensive grazing incidence X-ray diffraction, absorbance, and linear dichroism studies established that the films are crystalline with the c-axis oriented out of the plane of the substrate.22,25,27Low temperature absorbance is measured at normal incidence using a quasi-monochromatic tunable light source composed of a tungsten halogen lamp and a grating monochromator. Light is focused onto the sample to a diameter less than 100 µm using a short focal length achromat lens, and positioned inside the largest grain using an in situ sample viewing telescope. For low-temperature measurements the sample is placed inside a closed cycle refrigerator. The transmitted light is detected using an amplified Si detector coupled with a locking amplifier. A typical 4 K absorption spectrum is presented in Fig. 1(c) (black line).
A peak-fitting procedure following the protocol reported by Liang et al. in ref. 27 reveals eight distinct contributions to the absorption, among which four near-bandgap excitations originate from the Qx and Qy molecular excitations involving HOMO → LUMO and HOMO → LUMO+1 transitions. The remaining four higher-energy excitonic states are associated with HOMO → LUMO+n (n > 1) transitions. The present work exclusively focuses on the former four excitonic states, highlighted in color in Fig. 1(c). Previous study27 has isolated the contributions of the bandgap excitons using low-temperature magnetic circular dichroism (MCD) and linear dichroism (LD) measurements (see SI of ref. 27 for more details). The same work has also reported that all four transition excitonic dipoles are linearly polarized in the plane of the substrate. Our peak fitting routine considers the same energies for the near-bandgap excitons as those reported in ref. 27. Among the four states in question, the lowest-energy excitonic transition is of particular interest due to its distinct evolution with increasing temperature, associated with the formation of a delocalized state.
4 Electronic structure of H2OBPc monomer and dimers
In this section, we analyze the TD-DFT results of H2OBPc monomer and dimers, which provided the on-site excitation energies and the exciton–exciton couplings necessary for constructing the model Hamiltonian in eqn (3) for systems of varying dimensionalities.4.1 Molecular orbitals and transition densities
There is a critical interplay between orbital symmetries and electronic excitations, which defines the optoelectronic properties of the H2OBPc system. Fig. 4 displays the frontier molecular orbitals (MOs) and transition densities of the near-degenerate Qx and Qy states (from PBE0 functional as a representative with an energy difference of 73 meV). Across all the four functionals examined (Fig. S2), several consistent trends emerge: (i) the HOMO shows delocalized π-electron character over the central macrocycle and serves as the primary donor orbital in the two low-lying electronic excitations; (ii) the LUMO and LUMO+1 orbitals display complementary delocalization patterns and their symmetries (B2g and B3g if the monomer had a perfect D2h symmetry) differ from that of the HOMO (Au), thus making them orthogonal to the HOMO; (iii) transition density for the Qx state (i.e. the first excited state) is dominated by the HOMO → LUMO transition and is mostly localized on the central macrocycle and extended to the periphery in one direction; and (iv) transition density for the Qy state (i.e. the second excited state) involves the main contribution from the HOMO → LUMO+1 transition with its electronic density distribution extending to the peripheral substituents in another direction. These orbital trends are in agreement with Gouterman's four-orbital model,61,62 which predicts the perpendicular transition dipole moments and near degenerate LUMO and LUMO+1 orbitals. While the LRC-ωPBE functional yields similar frontier orbital shapes as shown in Fig. S2(d), the resulting Qx and Qy states are less dominated by a single orbital pair excitation and the leading contributions are reversed. Furthermore, functional dependence of the excitation energies of Qx and Qy states of H2OBPc and the transition dipole moments (Tx, Ty) are shown in Fig. S3, where the Qx and Qy states display the largest differences in the excitation energies with the PBE functional, a smaller difference with the PBE0 functional, and become nearly degenerate with the ωB97X-D and LRC-ωPBE functionals.As far as the basis set dependence is concerned, Table S1 shows slightly lower on-site Qx and Qy excitation energies (up to 40 meV) with larger basis sets and the same functional (PBE0). However, the energy difference between these two states and their transition dipole moments (Table S1) and Coulomb couplings (Table S2) are all insensitive to the choice of basis set.
4.2 Exciton couplings from TD-DFT calculations at varying inter-molecular distances
In this section, we will focus on the key Frenkel Hamiltonian parameters, the excitonic coupling evaluated using two distinct approaches described above, namely, Coulomb coupling JC and point-dipole approximation JD. The results presented here use the PBE0/6-31G(d,p) level of theory for various dimer distances Dd. Additional results based on other density functionals are compiled in Fig. S5 of the SI.Fig. 5 summarizes the computed Coulomb coupling constants between the two local excited-states in dimers, JC,xx, JC,xy, JC,yx and JC,yy, at varying dimer distances (Dd). For the A-dimer, the JC,xx and JC,yy values (about 26 meV) are much stronger than JC,xy and JC,yx (less than 9 meV). For dimers B and C, on the other hand, the four couplings are more comparable in the range of 13–19 meV. As such, B- and C-dimers display weaker JC,xx and JC,yy couplings compared to the A-dimer but stronger JC,xy and JC,yx couplings. Considering that B- and C-dimers have slightly longer monomer–monomer distances compared to that in the A-dimer (9.74/9.90 Å vs. 7.44 Å), this reflects that the coupling strengths depend not only on the inter-monomer distances but also on the relative orientation of the monomer transition dipole moments.
 | ||
| Fig. 5 Absolute values of calculated Coulomb couplings, JC,xx, JC,xy, JC,yx and JC,yy, at various distances between the molecular pairs (Dd) using the TD-DFT transition densities of Qx and Qy states within PBE0/6-31G(d,p) level of theory at the dimer structures extracted from crystal geometry. The plotted values correspond to the first-nearest neighbor (1NN, denoted by A, B, C), second-nearest neighbor (2NN, denoted by 's) and third-nearest neighbor (3NN, denoted by Ω's) interactions, with η1 being the sole fourth-nearest neighbor interaction within distance <26 Å. Two of the shortest interlayer interactions are shown as Σ2 and Σ1. The remaining unlabeled dimer distances correspond to other interlayer interactions. | ||
In general, the coupling values shown in Fig. 5 for the second-nearest neighbors (dimers 1, 2, and 3) are weaker than those for the nearest neighbors (A, B, and C), reflecting an expected decay of electrostatic interactions as the intermolecular distance increases. However, a simple d−3 decay is not observed. Specifically, JC,xx coupling of 2-dimer is approximately 50% of those for the B- and C-dimers. For JC,xy and JC,yy, both 1 and 3-dimers display rather strong couplings. For JC,yx, 3-dimer coupling appears to be strong as well. The Ω1-dimer, the only third-nearest neighbor within a 17 Å distance cutoff, shows a notably strong JC,yy value. Between the layers, the Σ2-dimer exhibits large JC,xy and JC,yx values (∼12 meV) that are actually comparable to J-values of the A-, B-, and C-dimers. The Σ1-dimer also shows a strong JC,yy coupling value while its other three couplings remain weak. These observations strongly suggest that the second-nearest neighbor and interlayer interactions might substantially affect the excitonic properties of H2OBPc. In addition, many couplings of the third-neighboring dimers (Ω's) are non-negligible in the 17–26 Å inter-molecular distance; for example, dimers Ω2 and Ω3 come with some coupling values close to 10 meV.
Regarding the functional dependence, the couplings generally follow the trend: PBE < PBE0 < ωB97X-D < LRC-ωPBE, as shown in Fig. S5 in SI. This can be attributed to an increased fraction of the Hartree–Fock exchange along the functional sequence, which typically enhances the magnitudes of the transition dipoles (Fig. S3(b)). However, there are noticeable deviations, especially with the LRC-ωPBE functional, which produces relatively small off-diagonal coupling values for the A-dimer, a smaller JC,xy for the 1- and Σ2-dimers, smaller JC,yx value for the Σ2-dimer, and smaller JC,yy for the 3- and Σ1-dimers. On the other hand, this functional also results in larger values for JC,xx for the 3-dimer, JC,xy for the Σ1-dimer, JC,yx for the 2- and Σ1-dimers, and JC,yy for the Σ2-dimer. JC,yx for the 2- and Σ1-dimers, and a small JC,yx value for the Σ2-dimer.
5 Exciton absorption spectra simulated across different dimensionalities
The aforementioned TD-DFT-derived parameters for various dimers were adopted into the Frenkel exciton Hamiltonian with eqn (3) to calculate the low-temperature absorption spectra following the steps outlined in Section 2. Specifically, we modeled exciton absorption spectra for multiple cases including the monomer and the three first-nearest neighboring dimers (shown in Fig. S7 and S8 with Coulomb couplings and dipole–dipole approximation, respectively), as well as 1D, 2D, and 3D models of the H2OBPc thin film (in Fig. 6, 7, S9–S12, S14, and S15). To benchmark the various assumptions in our calculations, we compared our simulated spectra with experimental absorption spectra for polycrystalline, pen-written thin films of H2OBPc from ref. 27. In our comparison of simulated and experimental spectra, an additional energy shift (ΔEshift) wads employed to better mimic the experimental results. The physical origin of ΔEshift could be related to the gas-to-crystal shifts, effects of vinyl stretching mode ω033,37 (see eqn (1)), as well as systematic errors in the TD-DFT methods used for excited state description.63 We optimized the energy shift parameter ΔEshift to minimize the difference between theoretical and experimental spectral areas. While an objective function optimization would be ideal, the shift ΔEshift was manually adjusted here, which should not introduce any qualitative difference. | ||
| Fig. 6 Simulated exciton normalized absorption spectra for various 1D models in Fig. 3(d)–(g) of 600 H2OBPc molecules using Coulomb couplings at (a) 10, (b) 17, and (c) 26 Å distance cutoffs from TD-DFT calculations within PBE0/6-31G(d,p) level of theory, against to the experimental spectra from ref. 27. (a) With a 10 Å distance cutoff, only the nearest-neighbor couplings (A, B, C in Fig. 3) are included. (b) When the cutoff is increased to 17 Å, all second-nearest neighbors (1, 2, 3) and the third-nearest neighbor (Ω1, only in the case of 1D-BACA) are also added to the Frenkel Hamiltonian. ΔEshift corresponds to an additional shift (in nm) to better match the two experimental peaks. Gaussian broadening with σ = 25 nm and Lorentzian broadening with γ = 20 nm are applied in eqn (4). | ||
 | ||
| Fig. 7 Simulated exciton normalized absorption spectra of 600 H2OBPc molecules from 1D-AB, 2D, and 3D models using Coulomb coupling (JC) from TD-DFT PBE0/6-31G(d,p) calculations with broadening parameters σ = 25 nm and γ = 20 nm. Molecular arrangements considered: 1D chain with 600 molecules along the x-axis; 2D lattice with 60 molecules along x and 10 along y; and 3D lattice with 30 molecules along x, 10 along y, and 2 layers along z, yielding 600 molecules in each case. Panels (a), (b), and (c) show the evolution of the absorption spectra with increasing system dimensionality for distance cutoffs of 10, 17, and 26 Å, respectively. The 10 Å cutoff includes only nearest-neighbor couplings (A, B, and C); the 17 Å cutoff incorporates all second-nearest-neighbor interactions (1, 2, and 3); and the 26 Å cutoff captures all third-nearest-neighbor couplings, along with one fourth-nearest-neighbor interaction, as illustrated in Fig. 3(b). | ||
5.1 Exciton absorption spectra for molecular aggregates arranged in 1-dimension
The absorption spectra for four different 1D models—1D-AB, 1D-AC, 1D-BC, and 1D-BACA introduced in Fig. 3(d)–(g)—are shown in Fig. 6 and compared to the experimental spectra of the H2OBPc thin film at 4 K.27 For a comparison of absorption spectra across different functionals, see Section S8 and Fig. S9.When only the nearest-neighbor interactions are considered (1NN-10 Å), the 1D-AB, 1D-AC and 1D-BACA models produce similar absorption spectra, while the 1D-BC arrangement results in a noticeably different absorption spectra, see Fig. 6(a). This difference arises because the B- and C-dimers share similar dimer distances (9.74 and 9.90 Å, respectively) and are both comprised of monomers with inverted symmetries. This is confirmed by the dimer spectra in Fig. S7 or S8, in which the B- and C-dimers give nearly identical absorption profiles with PBE0, ωB97X-D, and LRC-ωPBE functionals. Given these similarities, if one approximates the B-dimer to be equivalent to the C-dimer, then 1D-AC and 1D-BACA are reduced to 1D-AB, and the three models would yield similar absorption spectra. Conversely, since 1D-BC does not contain an A-dimer, the 1D-BC adsorption spectra is expected to differ from that of 1D-AB. Nevertheless, these 1D models all generate single-peak spectra, contradicting to the experimentally two absorption peaks.
When the second- and third-nearest neighbor couplings are added (2NN-17 Å and 3NN-26 Å), shown in Fig. 6(b) and (c), the absorption spectra from the 1D models have no significant differences, except for 1D-BC (dashed black lines). In particular, the spectra of 1D-BC model show two distinct peaks and become closer to the experimental curves and a clear double-peaked profile is produced after all the third-neighbor interactions are included in panel (c). This clearly indicates the importance of including longer-range couplings in the exciton modeling.
5.2 Exciton absorption spectra across different dimensionalities simulated using direct Coulomb coupling
Adopting the formalism given in Section 3.2 and calculated Frenkel exciton Hamiltonian parameters (i.e., JC constants) analyzed in Section 4.2, we next explore the effect of dimensionality on the absorption spectra across different models (1D-AB, 2D, and 3D) encompassing aggregates of 600 H2OBPc molecules. Among of the 1D arrangements studied, we focus on the 1D-AB model, at it was previously employed in an earlier study by Troisi and coworkers.29 For the 3D model, the 600 molecules fall into two layers, each with 300 molecules. As shown in Fig. S14, the simulated adsorption spectra do not change substantially with more molecules for all three models and more layers for the 3D model (see panels (j)–(l) of Fig. S14). From the predicted absorption spectra in Fig. 7 and S10, several key observations can be made regarding the effect of dimensionality, density functional choice, and cutoff distance.(i) Dimensionality: 1D aggregates (red curves in Fig. 7) consistently exhibit narrow spectral shape with 1NN-10 Å, 2NN-17 Å, and 3NN-26 Å models, as the excitonic couplings are confined to a single 1D chain of molecules. As the dimensionality increases from 1D to 2D (yellow curves), the spectra with PBE0 functional broaden substantially with 2NN-17 Å and 3NN-26 Å models, indicating the effect of including exciton couplings across the 2D plane. When the dimensionality increases further to 3D (green), there is a noticeable but much smaller change in the absorption spectra with the 1NN-10 Å and 2NN-17 Å models. This suggests that interlayer exciton couplings through Σ1 and Σ2 dimers play a relatively marginal role in the context of predicting low-temperature absorption spectra with these cutoff distances. For the 3NN-26 Å model, however, the 3D absorption spectra are appreciably different from the 2D case.
(ii) Functional dependence: while PBE and PBE0 calculations often lead to qualitatively similar spectral shapes (Fig. S10 and Fig. 7), PBE produces more pronounced spectral peaks compared to PBE0. This is attributed to differences in the excitation energies and transition dipole moments Tx/y between the PBE and PBE0 approximations as seen in Fig. S3. On the other hand, the predicted absorption spectra of the 2D or 3D models with 17 and 26 Å distance cutoffs also look similar between the two long-range-corrected functionals (LRC-ωPBE and ωB97X-D) as shown in Fig. S10.
(iii) Cutoff radius: when the cutoff radius is increased from 10 to 17 Å, second-nearest neighbors are also considered for the exciton coupling. With the PBE0 functional, there is a noticeable broadening of the overall absorption spectra and, where visible, a larger splitting of the two predicted absorption peaks. This is expected and attributable to the inclusion of longer-range interactions. For other functionals, the dependence of absorption spectra on the cutoff radius is shown in Fig. S10.
(iv) Polarization-dependent absorption spectra: polarization dependence for the lowest absorption peaks for the 1D-AB, 2D, and 3D models using the Coulomb coupling considering different components of the transition dipole moment vector , viz. x, y, or z are shown in Fig. S15. Appreciable changes in the spectra are observed for the different components of the transition dipole moments in the case of the 2D, 3NN-26 Å model.
(v) Comparison with experimental observation: at first glance, our results show the experimental double peak spectrum is better reproduced by the 1D-BC and 2D simulations (Fig. 6(c) and 7(c)), if longer-range interactions (3NN-26 Å) are included in the model and if using broadening parameters of σ = 25 nm and γ = 20 nm. It is, however, premature for us to state which combination of model dimensionality and density functional performs the best, as all predicted absorption spectra are subjected to energy shifts and line broadening empirical parameters. Fig. S16, for instance, illustrates the effects of Gaussian (σ) and Lorentzian (γ) broadening across all dimensionalities, including the 1D-AB, 1D-BC, 2D, and 3D models. Across these spectra, σ and γ tune two complementary broadening mechanisms with distinct visual signatures. Increasing γ (10–30 nm) enhances Lorentzian broadening, causing individual features to become wider and smoother and closely spaced peaks to merge. Increasing σ (5–25 nm) strengthens Gaussian broadening, leading to an overall spreading of the spectrum in which sharp differences between peaks are progressively reduced, while asymmetry and shoulder-like features persist and evolve. In addition to the aforementioned 1D-BC and 2D models, two distinct peaks also arise in Fig. S16(a)–(c) from 3D models with smaller σ and/or γ broadening values.
Intensity-wise, the experimental absorption spectrum shows peaks at 849 and 891 nm, where the latter is slightly higher in its intensity. In benchmarking studies,64 the predicted peak intensities (including the relative strength between different peaks) were reported to be strongly dependent on the choice of the density functional. While such a functional dependence was indeed observed in our study, we found the predicted spectral shape to be also dependent on the dimensionality of the model and the broadening parameters. With our default choice of Gaussian (σ = 25 nm) and Lorentzian (γ = 20 nm) broadening, the absorption profiles from both 1D-BC model in Fig. 6(c) and 2D model in Fig. 7(c) show two peaks of nearly equal intensity with PBE0 functional and at 26 Å distance cutoff. An equal peak intensity can also be found in Fig. S16 with other broadenings, except for panel f (σ = 15 nm and γ = 30 nm) where the shorter-wavefunction peak has a noticeably lower intensity in the spectrum of 1D-BC model. The 3D modeling spectra with PBE0 functional in panels (a) and (b) of Fig. S16, on the other hand, do show the shorter-wavelength peak with a lower intensity. In Fig. S10(a)–(c), the 3D-model also leads to two peaks with the use of PBE functional, but the longer-wavelength peak has substantially lower intensity.
5.3 Exciton absorption spectra across different dimensionalities using dipole–dipole coupling
Fig. S12 shows the absorption spectra across different dimensionalities, functionals, and distance cutoffs, albeit simulated using the dipole–dipole coupling parameters JD summarized in Fig. S6.Overall, JD values are larger compared to their more accurate counterparts, JC. As such, stronger JD couplings lead to spectra with more pronounced peaks across all functionals, highlighting the long-range nature of dipole–dipole interactions, as shown in Fig. S5 and S6. Dimensional differences (1D-AB vs. 2D vs. 3D) are also more pronounced with JD. In general, when the PBE functional is used, our absorption spectra in Fig. S12 modeled with the dipole–dipole couplings tend to overestimate the peak splittings relative to the experimental observations. [In addition, a subtle issue with the dipole–dipole approximation is that it wrongly predicts symmetric excited-state couplings (JD,xy = JD,yx) between the same molecules from different unit cells, whose transition densities should couple in a nonsymmetric way (JC,xy ≠ JC,yx).] Thus, the point-dipole approximation for computing excitonic couplings may not be reliable for modeling absorption spectra in these thin films, especially when employing 2D and 3D models.
6 Discussion & outlook
In this work, we implemented a tight-binding Frenkel exciton Hamiltonian with on-site energies and exciton coupling parameters obtained from TD-DFT calculations to model the low-temperature exciton absorption spectra of organic molecular thin films of H2OBPc molecules. Our model allows us to systematically investigate the effect of different dimensionalities and exciton coupling at varied cutoff distances, thus providing a robust benchmark in the computational study of organic thin films.A key advancement of our work lies in adapting and extending the theoretical formalism beyond the commonly explored 1D models,29,65 which typically include one or two electronic states per molecule,37 to handle 2D and 3D representations of the thin films. For our system, H2OBPc crystalline thin films with a triclinic unit cell,39 the molecules are found to form distorted hexagonal layers. Molecular thin films in two dimensions, most notably pentacene,66,67 have been extensively characterized for their charge-transport properties, whereas theoretical treatments of the charge and exciton properties and their dynamics in phthalocyanine, remain scarce. As such, our framework opens new avenues for probing exciton dynamics and charge conduction in layered organic materials.
While 1D nearest neighbor models of H2OBPc feature sharp absorption peaks, the extension to 2D/3D models and an incorporation of exciton couplings beyond the nearest neighbors lead to a broadening of the predicted spectra (especially when PBE0 functional is used to acquire Frenkel Hamiltonian elements) and a substantially larger splitting between the two main absorption peaks with the PBE0/3NN-26 Å model, which is in better agreement with experimental data. Our findings thus show the importance of adopting the proper dimensionality and including longer-range exciton coupling in modeling the spectral features of molecular aggregates. We also found that the point-dipole approximation for evaluating couplings between monomeric excitons generally exaggerates their interactions and leads to overestimated peak splitting with pure functionals (such as PBE), thus this approximation should be used with caution.
Going beyond the Coulomb coupling, our direct coupling calculations53 also produced Dexter exchange integrals between the transition density matrices, which were found for dimers A, B and C to be at least 100 times smaller than Coulomb coupling. So the Dexter contributions to the exciton absorption spectra are expected to be not substantial. As for charge transfer excitations, these excitations can occur between any pairs of molecules in a molecular crystal. Within the ALMO-TDA model,68–70 we found that these charge transfer excitations have nearly zero oscillator strength even for A-, B- and C-dimers due to non-overlapping electron and hole densities on the two monomers. Furthermore, TD-DFT calculations on these dimers show that the charge transfer excitations do not mix with the local excitations considered so far in our work within the Frenkel–Davydov exciton model. Therefore, these excitations do not contribute to the exciton absorption spectra, either.
So far, our investigations have focused on the low-temperature regime. Work is underway to extend the theoretical models developed here to finite-temperature regimes. This will enable treatment exciton–phonon couplings, including effects of both local/static (Holstein)30,71 and non-local/dynamic (Peierls) disorders30,32,72 on the coherent exciton transport. This development is expected to provide critical insights into the energy-transfer mechanisms that control exciton transport in this class of organic materials.17,30,73–78 For instance, while the simulated adsorption spectra do not change substantially upon the adding of more layers to the 3D model (see Fig. S14(j)–(l)), the exciton transport properties could be much more sensitive.
The H2OBPc crystal is treated in this work using 1D, 2D, and 3D cluster models, where the on-site energies and exciton couplings are computed from TDDFT calculations on isolated molecules and direct coupling calculations on molecular dimers, respectively. Work is underway to use high-level wavefunction methods, such as complete active state self-consistent field (CASSCF) theory79 and complete active space second-order perturbation theory (CASPT2),80,81 to check the accuracy of these on-site energies, transition densities, as well as Coulomb couplings.82 We also note that the absorption spectra and other properties of molecular crystals have also been modeled with electrostatic embedding under the periodic boundary condition (PBC).83 A future systematic comparison of the cluster and PBC approaches would be desirable.
Author contributions
SS, LB, MF, YS: conceptualization; SS, ZP, CL: software; SS, ZP, CL, CRW, YM: data curation and analysis; SS, CL, ST, LB, MF, YS: original draft preparation; all: manuscript editing; YS, PH, YZ, ST, MF: funding acquisition, resources, supervision.Conflicts of interest
The authors declare that they have no conflicts of interest.Data availability
Supplementary information: stereo pairs of the H2OBPc crystal structure. Frontier molecular orbitals, transition densities, on-site energies, and transition dipole moments of H2OBPc monomer computed with different functionals. Coulomb and dipole-dipole coupling for different H2OBPC dimers. Predicted exciton absorption spectra for molecular aggregates of different dimensionalities using various functionals, distance cutoffs, coupling and broadening schemes. See DOI: https://doi.org/10.1039/d5cp04079e.Data for this article, including Q-CHEM input and output files for all functionals in this work are available at https://doi.org/10.5281/zenodo.17237574. The code for exciton absorption spectra can be found at GitHub Repository: https://github.com/cc-ats/exc_absorption.
Acknowledgements
This material is based on work supported by National Science Foundation under grants NSF-OISE 2230706, NSF-Career DMR 1056589 and NSF OAC-2311442. Specifically, YS and PH were supported by NSF grant OAC-2311442 for part of this study, specifically the code development for modeling exciton absorption spectra. MF and HA were supported for the experimental data acquisition and analyses by NSF-OISE 2230706 and NSF-Career DMR 1056589. CL and YZ thank the support from the US DOE, Office of Science, Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division under Triad National Security, LLC (“Triad”) contract Grant 89233218CNA000001 (FWP: LANLECF7). This work was performed in part at the Center for Integrated Nanotechnology (CINT) at Los Alamos National Laboratory (LANL), a U.S. DOE and Office of Basic Energy Sciences user facility. LANL is operated by Triad National Security, LLC, for the National Nuclear Security Administration of the US Department of Energy (Contract No. 89233218CNA000001). YH was supported by JSPS Partnership for International research and Education (PIRE) Grant Number JPJSJRP20221201. The JSPS and NSF grants represent complementary funding in support of “US-Japan Partnership in Excitonic Soft Materials for Clean Energy”. YM acknowledges the support from the San Diego State University Startup Fund and the SDSU Seed Grant. YS also acknowledges the support from Brandeis University through the startup fund.Notes and references
- J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burns and A. B. Holmes, Light-emitting diodes based on conjugated polymers, Nature, 1990, 347, 539–541 CrossRef.
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Highly efficient phosphorescent emission from organic electroluminescent devices, Nature, 1998, 395, 151–154 CrossRef.
- S. R. Forrest, Ultrathin Organic Films Grown by Organic Molecular Beam Deposition and Related Techniques, Chem. Rev., 1997, 97, 1793–1896 CrossRef CAS PubMed.
- R. H. Friend, R. W. Gymer, A. B. Holmes, J. H. Burroughes, R. N. Marks, C. Taliani, D. D. C. Bradley, D. A. dos Santos, J.-L. Brédas, M. Lögdlund and W. R. Salaneck, Electroluminescence in conjugated polymers, Nature, 1999, 397, 121–128 CrossRef CAS.
- M. C. J. M. Vissenberg and M. Matters, Theory of the field-effect mobility in amorphous organic transistors, Phys. Rev. B:Condens. Matter Mater. Phys., 1998, 57, 12964–12967 CrossRef CAS.
- H. Koezuka, A. Tsumura and T. Ando, Field-effect transistor with polythiophene thin film, Synth. Met., 1987, 18, 699–704 CrossRef CAS.
- B. S. Ong, Y. Wu, P. Liu and S. Gardner, High-Performance Semiconducting Polythiophenes for Organic Thin-Film Transistors, J. Am. Chem. Soc., 2004, 126, 3378–3379 CrossRef CAS PubMed.
- R. P. Xu, Y. Q. Li and J. X. Tang, Recent advances in flexible organic light-emitting diodes, J. Mater. Chem. C, 2016, 4, 9116–9142 RSC.
- L. Dou, Y. Liu, Z. Hong, G. Li and Y. Yang, Low-Bandgap Near-IR Conjugated Polymers/Molecules for Organic Electronics, Chem. Rev., 2015, 115, 12633–12665 CrossRef CAS PubMed.
- C. D. Sheraw, T. N. Jackson, D. L. Eaton and J. E. Anthony, Functionalized pentacene active layer organic thin-film transistors, Adv. Mater., 2003, 15, 2009–2011 CrossRef CAS.
- K. Kim, K. Nam, X. Li, D. Y. Lee and S. H. Kim, Programmed Design of Highly Crystalline Organic Semiconductor Patterns with Uniaxial Alignment via Blade Coating for High-Performance Organic Field-Effect Transistors, ACS Appl. Mater. Interfaces, 2019, 11, 42403–42411 CrossRef CAS.
- Y. Xiao, W. Deng, J. Hong, X. Ren, X. Zhang, J. Shi, F. Sheng, X. Zhang and J. Jie, Water-Surface-Mediated Precise Patterning of Organic Single-Crystalline Films via Double-Blade Coating for High-Performance Organic Transistors, Adv. Funct. Mater., 2023, 33, 2213788 CrossRef CAS.
- H. Matsui, Y. Takeda and S. Tokito, Flexible and printed organic transistors: From materials to integrated circuits, Org. Electron., 2019, 75, 105432 CrossRef CAS.
- A. Khasbaatar, Z. Xu, J. H. Lee, G. Campillo-Alvarado, C. Hwang, B. N. Onusaitis and Y. Diao, From Solution to Thin Film: Molecular Assembly of π-Conjugated Systems and Impact on (Opto)electronic Properties, Chem. Rev., 2023, 123, 8395–8487 CrossRef PubMed.
- G. Horowitz, Organic thin film transistors: From theory to real devices, J. Mater. Res., 2004, 19, 1946–1962 CrossRef.
- V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey and J. L. Brédas, Charge Transport in Organic Semiconductors, Chem. Rev., 2007, 107, 926–952 CrossRef.
- J. L. Brédas, D. Beljonne, V. Coropceanu and J. Cornil, Charge-Transfer and Energy-Transfer Processes in π-Conjugated Oligomers and Polymers: A Molecular Picture, Chem. Rev., 2004, 104, 4971–5004 CrossRef.
- J. Zaumseil and H. Sirringhaus, Electron and Ambipolar Transport in Organic Field-Effect Transistors, Chem. Rev., 2007, 107, 1296–1323 Search PubMed.
- O. Ostroverkhova, Organic Optoelectronic Materials: Mechanisms and Applications, Chem. Rev., 2016, 116, 13279–13412 CrossRef PubMed.
- M. Schwoerer and H. C. Wolf, Organic Molecular Solids, Wiley-VCH, Weinheim, 2008 Search PubMed.
- X. Xu, Z. Lou, S. Cheng, P. C. Y. Chow, N. Koch and H. M. Cheng, Understanding and Engineering the Interfacial Interaction in Organic/Inorganic van der Waals Heterostructures for (Opto)electronics, Chem, 2021, 7, 2989–3026 Search PubMed.
- N. Rawat, Z. Pan, L. W. Manning, C. J. Lamarche, I. Cour, R. L. Headrick, R. Waterman, A. R. Woll and M. I. Furis, Macroscopic Molecular Ordering and Exciton Delocalization in Crystalline Phthalocyanine Thin Films, J. Phys. Chem. Lett., 2015, 6, 1834–1840 CrossRef PubMed.
- S. C. J. Meskers, The Exciton Model for Molecular Materials: Past, Present and Future?, ChemPhysChem, 2023, 24, e202300666 Search PubMed.
- H. Najafov, B. Lee, Q. Zhou, L. C. Feldman and V. Podzorov, Observation of long-range exciton diffusion in highly ordered organic semiconductors, Nat. Mater., 2010, 9, 938–943 CrossRef PubMed.
- L. W. Manning, N. Rawat, C. Lamarche, R. Waterman, R. L. Headrick and M. Furis, Exciton Delocalization in H2OBPc1−xMOBPcx (M = Co, Cu, Ni, Mn) Crystalline Thin-Film Organic Alloys, J. Phys. Chem. C, 2016, 120, 11966–11976 CrossRef.
- L. Liang, K. Czar and M. I. Furis, Strain-Enhanced Formation of Delocalized Exciton States in Phthalocyanine Crystalline Thin Films, J. Phys. Chem. C, 2022, 126, 8889–8896 CrossRef.
- L. Liang, K. Burrill and M. I. Furis, Transition Dipole Moments of One-Dimensional Excitons in Soluble Phthalocyanine Thin Films, J. Phys. Chem. C, 2021, 125, 27966–27974 CrossRef.
- A. S. Davydov, Theory of Molecular Excitons, Springer, New York, 1971 Search PubMed.
- R. P. Fornari, J. Aragó and A. Troisi, Exciton Dynamics in Phthalocyanine Molecular Crystals, J. Phys. Chem. C, 2016, 120, 7987–7996 CrossRef.
- A. J. Sneyd, T. Fukui, D. Paleček, S. Prodhan, I. Wagner, Y. Zhang, J. Sung, S. M. Collins, T. J. A. Slater, Z. Andaji-Garmaroudi, L. R. MacFarlane, J. D. Garcia-Hernandez, L. Wang, G. R. Whittell, J. M. Hodgkiss, K. Chen, D. Beljonne, I. Manners, R. H. Friend and A. Rao, Efficient energy transport in an organic semiconductor mediated by transient exciton delocalization, Sci. Adv., 2021, 7, eabh4232 CrossRef PubMed.
- S. Prodhan, S. Giannini, L. Wang and D. Beljonne, Long-Range Interactions Boost Singlet Exciton Diffusion in Nanofibers of π-Extended Polymer Chains, J. Phys. Chem. Lett., 2021, 12, 8188–8193 CrossRef PubMed.
- S. Giannini, W. T. Peng, L. Cupellini, D. Padula, A. Carof and J. Blumberger, Exciton transport in molecular organic semiconductors boosted by transient quantum delocalization, Nat. Commun., 2022, 13, 2755 CrossRef.
- F. C. Spano, The Spectral Signatures of Frenkel Polarons in H- and J-Aggregates, Acc. Chem. Res., 2010, 43, 429–439 Search PubMed.
- F. C. Spano, S. C. J. Meskers, E. Hennebicq and D. Beljonne, Probing Excitation Delocalization in Supramolecular Chiral Stacks by Means of Circularly Polarized Light: Experiment and Modeling, J. Am. Chem. Soc., 2007, 129, 7044–7054 Search PubMed.
- M. Kasha, Energy Transfer Mechanisms and the Molecular Exciton Model for Molecular Aggregates, Radiat. Res., 1963, 20, 55–70 Search PubMed.
- F. C. Spano and H. Yamagata, Vibronic Coupling in J-Aggregates and Beyond: A Direct Means of Determining the Exciton Coherence Length from the Photoluminescence Spectrum, J. Phys. Chem. B, 2011, 115, 5133–5143 Search PubMed.
- N. J. Hestand and F. C. Spano, Expanded Theory of H- and J-Molecular Aggregates: The Effects of Vibronic Coupling and Intermolecular Charge Transfer, Chem. Rev., 2018, 118, 7069–7163 Search PubMed.
- F. C. Spano, J. Clark, C. Silva and R. H. Friend, Determining exciton coherence from the photoluminescence spectral line shape in poly(3-hexylthiophene) thin films, J. Chem. Phys., 2009, 130, 074904 Search PubMed.
- Y. Gao, Y. Chen, R. Li, Y. Bian, X. Li and J. Jiang, Nonperipherally Octa(butyloxy)-Substituted Phthalocyanine Derivatives with Good Crystallinity: Effects of Metal–Ligand Coordination on the Molecular Structure, Internal Structure, and Dimensions of Self-Assembled Nanostructures, Chem. – Eur. J., 2009, 15, 13241–13252 Search PubMed.
- M. E. Casida, Recent Advances in Density Functional Methods Part I., World Scientific, 1995, p. 155 Search PubMed.
- M. E. Casida and M. Huix-Rotllant, Progress in Time-Dependent Density-Functional Theory, Annu. Rev. Phys. Chem., 2012, 63, 287–323 CrossRef PubMed.
- S. Hirata and M. Head-Gordon, Time-Dependent Density Functional Theory for Radicals, Chem. Phys. Lett., 1999, 302, 375–382 CrossRef.
- E. Epifanovsky, A. T. B. Gilbert, X. Feng, J. Lee, Y. Mao, N. Mardirossian, P. Pokhilko, A. F. White, M. P. Coons, A. L. Dempwolff, Z. Gan, D. Hait, P. R. Horn, L. D. Jacobson, I. Kaliman, J. Kussmann, A. W. Lange, K. U. Lao, D. S. Levine, J. Liu, S. C. McKenzie, A. F. Morrison, K. D. Nanda, F. Plasser, D. R. Rehn, M. L. Vidal, Z.-Q. You, Y. Zhu, B. Alam, B. J. Albrecht, A. Aldossary, E. Alguire, J. H. Andersen, V. Athavale, D. Barton, K. Begam, A. Behn, N. Bellonzi, Y. A. Bernard, E. J. Berquist, H. G. A. Burton, A. Carreras, K. Carter-Fenk, R. Chakraborty, A. D. Chien, K. D. Closser, V. Cofer-Shabica, S. Dasgupta, M. de Wergifosse, J. Deng, M. Diedenhofen, H. Do, S. Ehlert, P.-T. Fang, S. Fatehi, Q. Feng, T. Friedhoff, J. Gayvert, Q. Ge, G. Gidofalvi, M. Goldey, J. Gomes, C. E. González-Espinoza, S. Gulania, A. O. Gunina, M. W. D. Hanson-Heine, P. H. P. Harbach, A. Hauser, M. F. Herbst, M. Hernández Vera, M. Hodecker, Z. C. Holden, S. Houck, X. Huang, K. Hui, B. C. Huynh, M. Ivanov, Á. Jász, H. Ji, H. Jiang, B. Kaduk, S. Köhler, K. Khistyaev, J. Kim, G. Kis, P. Klunzinger, Z. Koczor-Benda, J. H. Koh, D. Kosenkov, L. Koulias, T. Kowalczyk, C. M. Krauter, K. Kue, A. Kunitsa, T. Kus, I. Ladjánszki, A. Landau, K. V. Lawler, D. Lefrancois, S. Lehtola, R. R. Li, Y.-P. Li, J. Liang, M. Liebenthal, H.-H. Lin, Y.-S. Lin, F. Liu, K.-Y. Liu, M. Loipersberger, A. Luenser, A. Manjanath, P. Manohar, E. Mansoor, S. F. Manzer, S.-P. Mao, A. V. Marenich, T. Markovich, S. Mason, S. A. Maurer, P. F. McLaughlin, M. F. S. J. Menger, J.-M. Mewes, S. A. Mewes, P. Morgante, J. W. Mullinax, K. J. Oosterbaan, G. Paran, A. C. Paul, S. K. Paul, F. Pavošević, Z. Pei, S. Prager, E. I. Proynov, Á. Rák, E. Ramos-Cordoba, B. Rana, A. E. Rask, A. Rettig, R. M. Richard, F. Rob, E. Rossomme, T. Scheele, M. Scheurer, M. Schneider, N. Sergueev, S. M. Sharada, W. Skomorowski, D. W. Small, C. J. Stein, Y.-C. Su, E. J. Sundstrom, Z. Tao, J. Thirman, G. J. Tornai, T. Tsuchimochi, N. M. Tubman, S. P. Veccham, O. Vydrov, J. Wenzel, J. Witte, A. Yamada, K. Yao, S. Yeganeh, S. R. Yost, A. Zech, I. Y. Zhang, X. Zhang, Y. Zhang, D. Zuev, A. Aspuru-Guzik, A. T. Bell, N. A. Besley, K. B. Bravaya, B. R. Brooks, D. Casanova, J.-D. Chai, S. Coriani, C. J. Cramer, G. Cserey, A. E. DePrince, R. A. DiStasio, A. Dreuw, B. D. Dunietz, T. R. Furlani, W. A. Goddard, S. Hammes-Schiffer, T. Head-Gordon, W. J. Hehre, C.-P. Hsu, T.-C. Jagau, Y. Jung, A. Klamt, J. Kong, D. S. Lambrecht, W. Liang, N. J. Mayhall, C. W. McCurdy, J. B. Neaton, C. Ochsenfeld, J. A. Parkhill, R. Peverati, V. A. Rassolov, Y. Shao, L. V. Slipchenko, T. Stauch, R. P. Steele, J. E. Subotnik, A. J. W. Thom, A. Tkatchenko, D. G. Truhlar, T. Van Voorhis, T. A. Wesolowski, K. B. Whaley, H. L. Woodcock, P. M. Zimmerman, S. Faraji, P. M. W. Gill, M. Head-Gordon, J. M. Herbert and A. I. Krylov, Software for the frontiers of quantum chemistry: An overview of developments in the Q-Chem 5 package, J. Chem. Phys., 2021, 155, 084801 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef PubMed.
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: The PBE0 model, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef.
- C. Adamo, G. E. Scuseria and V. Barone, Accurate excitation energies from time-dependent density functional theory: Assessing the PBE0 model, J. Chem. Phys., 1999, 111, 2889–2899 CrossRef CAS.
- O. A. Vydrov, J. Heyd, A. V. Krukau and G. E. Scuseria, Importance of short-range versus long-range Hartree-Fock exchange for the performance of hybrid density functionals, J. Chem. Phys., 2006, 125, 074106 CrossRef.
- J. D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- R. Ditchfield, W. J. Hehre and J. A. Pople, Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, The influence of polarization functions on molecular orbital hydrogenation energies, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- F. Plasser, A. I. Krylov and A. Dreuw, libwfa: Wavefunction analysis tools for excited and open-shell electronic states, Wiley Interdiscip. Rev.:Comput. Mol. Sci., 2022, 12, e1595 CAS.
- C. P. Hsu, The Electronic Couplings in Electron Transfer and Excitation Energy Transfer, Acc. Chem. Res., 2009, 42, 509–518 CrossRef CAS PubMed.
- B. P. Krueger, G. D. Scholes and G. R. Fleming, Calculation of Couplings and Energy-Transfer Pathways between the Pigments of LH2 by the ab Initio Transition Density Cube Method, J. Phys. Chem. B, 1998, 102, 5378–5386 CrossRef CAS.
- M. E. Madjet, A. Abdurahman and T. Renger, Intermolecular Coulomb Couplings from Ab Initio Electrostatic Potentials: Application to Optical Transitions of Strongly Coupled Pigments in Photosynthetic Antennae and Reaction Centers, J. Phys. Chem. B, 2006, 110, 17268–17281 CrossRef CAS PubMed.
- K. A. Kistler, F. C. Spano and S. Matsika, A Benchmark of Excitonic Couplings Derived from Atomic Transition Charges, J. Phys. Chem. B, 2013, 117, 2032–2044 Search PubMed.
- R. L. Headrick, S. Wo, F. Sansoz and J. E. Anthony, Anisotropic mobility in large grain size solution processed organic semiconductor thin films, Appl. Phys. Lett., 2008, 92, 063302 CrossRef.
- S. Wo, R. L. Headrick and J. E. Anthony, Fabrication and characterization of controllable grain boundary arrays in solution-processed small molecule organic semiconductor films, J. Appl. Phys., 2012, 111, 073716 CrossRef.
- Y. Li, J. Wan, D. M. Smilgies, N. Bouffard, R. Sun and R. L. Headrick, Nucleation and strain-stabilization during organic semiconductor thin film deposition, Sci. Rep., 2016, 6, 32620 CrossRef CAS PubMed.
- Z. Lu, C. Wang, W. Deng, M. T. Achille, J. Jie and X. Zhang, Meniscus-guided coating of organic crystalline thin films for high-performance organic field-effect transistors, J. Mater. Chem. C, 2020, 8, 9133–9146 RSC.
- M. Gouterman, Spectra of porphyrins, J. Mol. Spectrosc., 1961, 6, 138–163 CrossRef CAS.
- C. C. Wamser and A. Ghosh, The Hyperporphyrin Concept: A Contemporary Perspective, JACS Au, 2022, 2, 1543–1560 CrossRef CAS PubMed.
- Y. Shao, Y. Mei, D. Sundholm and V. R. I. Kaila, Benchmarking the Performance of Time-Dependent Density Functional Theory Methods on Biochromophores, J. Chem. Theory Comput., 2020, 16, 587–600 CrossRef.
- M. Caricato, G. W. Trucks, M. J. Frisch and K. B. Wiberg, Oscillator Strength: How Does TDDFT Compare to EOM-CCSD?, J. Chem. Theory Comput., 2011, 7, 456–466 CrossRef CAS.
- A. Troisi and G. Orlandi, Charge-Transport Regime of Crystalline Organic Semiconductors: Diffusion Limited by Thermal Off-Diagonal Electronic Disorder, Phys. Rev. Lett., 2006, 96, 086601 CrossRef PubMed.
- S. Wang, Z. Wei, Y. Yang, X. Zhao, Q. Tang, Y. Tong and Y. Liu, Low surface energy interface-derived low-temperature recrystallization behavior of organic thin films for boosting carrier mobility, J. Mater. Chem. C, 2019, 7, 13778–13785 RSC.
- T. Salzillo, A. Brillante and A. Girlando, Terahertz Raman scattering as a probe for electron–phonon coupling, disorder and correlation length in molecular materials, J. Mater. Chem. C, 2021, 9, 10677–10688 Search PubMed.
- Q. Ge, Y. Mao and M. Head-Gordon, Energy decomposition analysis for exciplexes using absolutely localized molecular orbitals, J. Chem. Phys., 2018, 148, 064105 CrossRef.
- Q. Ge and M. Head-Gordon, Energy Decomposition Analysis for Excimers Using Absolutely Localized Molecular Orbitals within Time-Dependent Density Functional Theory and Configuration Interaction with Single Excitations, J. Chem. Theory Comput., 2018, 14, 5156–5168 CrossRef CAS PubMed.
- X. Ji, Z. Pei, K. N. Huynh, J. Yang, X. Pan, B. Wang, Y. Mao and Y. Shao, On the entanglement of chromophore and solvent orbitals, J. Chem. Phys., 2025, 162, 064107 CrossRef CAS.
- T. Holstein, Studies of polaron motion: Part I. The molecular-crystal model, Ann. Phys., 1959, 8, 325–342 CAS.
- J. H. Fetherolf, D. Golež and T. C. Berkelbach, A Unification of the Holstein Polaron and Dynamic Disorder Pictures of Charge Transport in Organic Crystals, Phys. Rev. X, 2020, 10, 021062 CAS.
- T. Förster, 10th Spiers Memorial Lecture. Transfer mechanisms of electronic excitation, Discuss. Faraday Soc., 1959, 27, 7–17 RSC.
- S. M. Menke and R. J. Holmes, Exciton diffusion in organic photovoltaic cells, Energy Environ. Sci., 2014, 7, 499–512 RSC.
- S. Bai, P. Zhang and D. N. Beratan, Predicting Dexter Energy Transfer Interactions from Molecular Orbital Overlaps, J. Phys. Chem. C, 2020, 124, 18956–18960 CrossRef CAS.
- O. P. Dimitriev, Dynamics of Excitons in Conjugated Molecules and Organic Semiconductor Systems, Chem. Rev., 2022, 122, 8487–8593 CrossRef CAS PubMed.
- S. Cao, A. Rosławska, B. Doppagne, M. Romeo, M. Féron, F. Chérioux, H. Bulou, F. Scheurer and G. Schull, Energy funnelling within multichromophore architectures monitored with subnanometre resolution, Nat. Chem., 2021, 13, 766–770 CrossRef CAS.
- D. L. Andrews, C. Curutchet and G. D. Scholes, Resonance energy transfer: Beyond the limits, Laser Photonics Rev., 2011, 5, 114–123 Search PubMed.
- P. A. Malmqvist and B. O. Roos, The CASSCF state interaction method, Chem. Phys. Lett., 1989, 155, 189–194 Search PubMed.
- J. Finley, P. A. Malmqvist, B. O. Roos and L. Serrano-Andrés, The multi-state CASPT2 method, Chem. Phys. Lett., 1998, 288, 299–306 CrossRef CAS.
- S. Battaglia and R. Lindh, On the role of symmetry in XDW-CASPT2, J. Chem. Phys., 2021, 154, 034102 Search PubMed.
- A. Kaiser, R. E. Daoud, F. Aquilante, O. Kühn, L. De Vico and S. I. Bokarev, A Multiconfigurational Wave Function Implementation of the Frenkel Exciton Model for Molecular Aggregates, J. Chem. Theory Comput., 2023, 19, 2918–2928 CrossRef CAS PubMed.
- M. Rivera, M. Dommett and R. Crespo-Otero, ONIOM(QM:QM) Electrostatic Embedding Schemes for Photochemistry in Molecular Crystals, J. Chem. Theory Comput., 2019, 15, 2504–2516 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2026 |