Precise 3D structure determination of Cu single atoms on an α-Al2O3(0001) surface by polarization-dependent total reflection fluorescence X-ray absorption fine structure and first-principles calculations
Received
22nd September 2025
, Accepted 31st January 2026
First published on 2nd February 2026
Abstract
The valence state and three-dimensional (3D) structure of vacuum-deposited Cu species on an Al-terminated (1 × 1) α-Al2O3(0001) surface have been studied using polarization-dependent total reflection fluorescence X-ray absorption fine structure (PTRF-XAFS) and density functional theory (DFT) calculations. The Cu species were atomically dispersed on the α-Al2O3(0001) surface in its low-coverage region (0.07 ML on Cu/O basis) with a Cu–O distance of 0.194 ± 0.003 nm and were present in the form of Cu(I). The adsorption site for the Cu single atoms was determined to be a three-fold hollow oxygen site corresponding to the imaginary Al position in the next Al layer over the α-Al2O3(0001) surface, accompanied by an upward shift of the nearest neighbor oxygen atoms and expansion of the oxygen–oxygen distance. A similar local structure and surface relaxation mechanism have been reported in our previous PTRF-EXAFS study on Ni single atoms deposited on the α-Al2O3(0001) surface (K. Ijima et al., Chem. Phys. Lett., 2004, 384, 134–138), indicating important implications for stabilizing single metal atoms on the Al2O3 surface.
1. Introduction
Single-atom catalysts (SACs), which consist of isolated single metal atoms dispersed on a powdery oxide support, have attracted much attention due to their high catalytic activity and cost-savings on precious metal atoms, compared with those of conventional nanoparticle catalysts.1–3 For instance, the Cu single-atom sites anchored to γ-Al2O3 oxides are highly active and stable in continuous conversion of methane-steam to methanol at 200 °C, comparable to most of the state-of-the-art Cu–zeolite catalysts.4 The Cu single atoms immobilized on TiO2 also show high activity and excellent stability for photocatalytic hydrogen evolution.5 Since the catalytic performances of such SACs significantly depend on the coordination environment around the active metal site, i.e., metal–oxygen bond distance, metal–oxygen coordination number, coordination symmetry, and valence state of the active metal atoms, etc., unraveling their precise 3D structures is essential to understanding the stabilization mechanism of the single metal atoms and their structure–activity relationship for the development of more active SACs.
X-ray absorption fine structure (XAFS) is one of the most powerful techniques to investigate the structure of the SACs because it provides information about the active metal atom on the valence state through X-ray absorption near edge structure (XANES) analysis and the local structure through extended X-ray absorption fine structure (EXAFS) analysis. However, it is difficult to determine their precise 3D structure by using conventional XAFS techniques because the bonding information is averaged over all directions and provides a 1D radial distribution around the X-ray absorbing atom. Moreover, the powdery oxide-support surfaces are generally ill-defined, exposing various defects and crystal faces, and this often causes formation of non-uniform single metal species, making it difficult to accurately determine their structures. On the other hand, when a well-defined single-crystal oxide surface is used as a support, detailed information about the 3D structure and metal–support interaction for uniform single-metal species can be obtained using the polarization dependence shown in eqn (1),6
| | 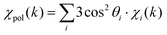 | (1) |
where
χpol(
k) is the polarization-dependent EXAFS signal as a function of wavenumber (
k),
θi is the angle between the electric vector of the incident X-rays and the
i-th bond direction, and
χi(
k) is the EXAFS oscillation originating from the
i-th bond. The problems of a low concentration of surface metal species can be overcome by applying the surface-sensitive polarization-dependent total reflection fluorescence (PTRF)-XAFS technique,
6–8 which has been used to determine the 3D structures of single metal atoms
9–14 and metal nanoclusters
15–18 on TiO
2(110) and α-Al
2O
3(0001) surfaces, based on EXAFS simulation of real-space model structures using the FEFF code.
19 For instance, the Cu single atoms were formed on a TiO
2(110) surface premodified with
o-mercaptobenzoic acid (
o-MBA),
via forming bonds with the sulfur atom of the
o-MBA molecule and the oxygen atom in the TiO
2 lattice, and their 3D structure was precisely determined at less than 0.003 nm resolution in the Cu–S and Cu–O bond distances (0.219 and 0.185 nm, respectively) and at less than 3° precision in the Cu–S and Cu–O bond angles to the surface normal (45° and 43°, respectively).
12 Our previous polarization-dependent total reflection fluorescence (PTRF)-EXAFS studies on single metal atoms deposited on bare oxide single-crystal surfaces have given an important concept for atomic dispersion: a single metal atom is stabilized on an oxide surface through metal–oxygen interactions, not through metal–cation bonding, because no EXAFS oscillation corresponding to a metal–cation bond with a bond distance less than 0.27 nm has been observed.
7
In this work, we have investigated the valence state and 3D structure of vacuum-deposited Cu on an α-Al2O3(0001) surface in its low-coverage region by using the PTRF-XAFS technique and density functional theory (DFT) calculations, to unveil the metal–oxide interface structure and formation of single-atom species. The Cu/α-Al2O3(0001) system has been extensively studied using X-ray photoelectron spectroscopy (XPS),20–22 scanning force microscopy (SFM),23 and first-principles calculations.24–29 The XPS studies indicated formation of Cu(I) at a Cu coverage of less than ∼0.35 ML (on a Cu/O basis) due to interaction of oxygen atoms of the α-Al2O3(0001) surface.22 First-principles calculations predicted that the Cu atoms are adsorbed preferentially on the three-fold hollow oxygen sites.25–27,29 However, the precise 3D structure of the Cu species has not yet been directly determined experimentally. We carried out EXAFS simulations using real-space model structures obtained by the DFT calculations and determined the precise 3D structure of the Cu species by modifying the DFT-optimized structures so that the simulated EXAFS spectra reproduced the observed EXAFS spectra.
2. Experimental
2.1 Sample preparation
As-received α-Al2O3(0001) single crystals (15 × 15 × 0.5 mm3, Shinkosha Co., Ltd) were annealed in air at 1323 K for 3 h to obtain an atomically flat α-Al2O3(0001) surface.30 The sample was then introduced into an ultra-high vacuum (UHV) chamber and heated at 673 K for 1 h. Afterwards the α-Al2O3(0001) surface was exposed to oxygen plasma (10 W, 10 min) for further surface cleaning, generated by a compact electron cyclotron resonance (ECR) plasma source (Aura-Wave, SAIREM). We have found that the O2 plasma treatment efficiently removed the residual carbon species and gave a sharper low-energy electron diffraction (LEED) pattern with a hexagonal (1 × 1) periodicity, as shown in Fig. S2. Cu was then vacuum-deposited on the α-Al2O3(0001) surface at room temperature via resistive heating of a Cu wire (99.999% purity, Nilaco Co., Japan) in the UHV chamber. The Cu coverage was estimated to be 0.07 ML from the XPS peak intensity ratio of Cu 2p3/2 to O 1s, where 1 ML was defined as 1.5 × 1015 cm−2 with correspondence to the density of the surface O atoms of the α-Al2O3(0001) substrate. The Cu/α-Al2O3(0001) sample was then transferred to the compact UHV cell (operando PTRF-XAFS cell)18 for the PTRF-XAFS measurements.
2.2 PTRF-XAFS measurements
PTRF-XAFS measurements were conducted at the BL-9A beamline of the photon factory at the Institute of Materials Structure Science (KEK-IMSS-PF, Tsukuba, Japan) after the PTRF-XAFS cell was fixed on a 6-axis goniometer placed in the hutch of the beamline. The storage ring energy and ring current were 2.5 GeV and 450 mA, respectively. X-rays were monochromatized using Si(111) double crystals and focused onto the sample with double-bent conical mirrors. The beam size on the sample was regulated via a molybdenum pinhole with a diameter of 0.4 mm, which was attached in front of the ion chamber upstream (I0), to reduce undesirable irradiation outside of the sample surface. The total reflection conditions were adjusted using the 6-axis goniometer. The PTRF-XAFS measurements were carried out at room temperature under the UHV conditions for two different orientations of the sample surface relative to the electric vector of the incident X-rays, i.e., parallel to the surface (s-polarization) and perpendicular to the surface (p-polarization). The Cu Kα fluorescence was detected by a 7-element silicon drift detector (XSDD50-07, TechnoAP, Japan). EXAFS analysis was performed using the REX 2000 (Rigaku Co., Japan) package.31 The EXAFS oscillations were extracted using a spline smoothing method and normalized with respect to the edge height with the energy dependence expressed in a Victoreen equation. The 3D structure was determined via an iterative method using the FEFF8.04 code19 with a Hedin–Lundqvist potential based on a real-space model structure. The goodness of fit between the observed (χobs(k)) and calculated (χcal(k)) EXAFS oscillations was evaluated using eqn (2),| | 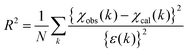 | (2) |
where N and ε(k) are the number of data points and the error of the observed spectrum, respectively. The structure model was adopted when R2 values were lower than 1 for all orientations (s- and p-polarizations). Details of the PTRF-EXAFS analysis are provided in the SI.
2.3 Density functional theory (DFT) calculations
DFT calculations were performed using VASP 5.432–35 with the projector augmented wave (PAW) and plane-wave basis set methods.36 The kinetic energy cutoff for plane-wave expansion was set at 500 eV, and the exchange–correlation interaction between electrons was described by the Perdew–Burke–Ernzerhof functional (PBE).37,38 The calculated lattice constants for the bulk α-Al2O3 (using 4 × 4 × 2 k-points, a = b = 4.778 Å and c = 13.034 Å) are found to be in good agreement with the experimental values (a = b = 4.758 Å and c = 12.990 Å).39 The supercell was obtained by truncating the bulk α-Al2O3 structure, leaving one Al layer exposed with an oxygen layer right beneath it. This surface is known as the single Al layer terminated surface and is accepted to be the most stable unreconstructed (1 × 1) α-Al2O3(0001) surface observed in the UHV conditions.40–42 The formation of this surface was confirmed in this study by the hexagonal (1 × 1) periodicity of the LEED pattern (Fig. S2). The Al-terminated (1 × 1) surface was modeled as a twelve-Al-layer slab with a (3 × 3) supercell, which consists of 108 Al atoms and 162 O atoms. The vacuum layer was set at 20 Å. The bottom three Al layers and two O layers were fixed throughout the calculations, while the upper nine Al layers, four O layers, and the Cu atom were relaxed using a conjugate gradient technique until the residual forces on the atoms were less than 0.005 eV Å−1. The k-point mesh was set as 3 × 3 × 1. The effects of van der Waals forces were considered by the vdw-D3 method.43 The dipole correction along the [0001] direction was included. The adsorption energy (Ead) of single Cu atoms on the α-Al2O3(0001) was calculated as| | | Ead = EAl2O3+Cu − EAl2O3 − ECu | (3) |
where EAl2O3, ECu, and EAl2O3+Cu are the total electron energies of α-Al2O3(0001), gaseous Cu atom, and α-Al2O3(0001) after Cu atom adsorption, respectively.
3. Results and discussion
3.1 Valence state of the Cu species on α-Al2O3(0001)
Fig. 1a and b show the PTRF-XANES spectra of the vacuum-deposited Cu species on the α-Al2O3(0001) surface and the XANES spectra of Cu foil, Cu2O, and CuO as the references, respectively. The XANES spectra of the Cu/α-Al2O3(0001) showed different features from that of Cu foil, which suggested formation of non-metallic Cu species. The mid-edge features observed at approximately 8983 eV for the s- and p-polarizations could be assigned to the 1s to 4pπ* transition, as determined from previous XANES results and ab initio calculations.44 The inflection points of the edge appeared at around 8980.4 eV for both polarization directions. Considering that the inflection points of Cu foil, Cu2O, and CuO were at 8979.5, 8980.6, and 8984.7 eV, respectively, the oxidation state of the Cu species on the α-Al2O3(0001) was monovalent (Cu(I)). Kelber et al. demonstrated that Cu was oxidized to Cu(I) when Cu was sputter-deposited on the α-Al2O3(0001) surface at a coverage of less than ∼0.35 ML (on a Cu/O basis), as evidenced by the Cu(LMM) line shape data obtained via X-ray photoelectron spectroscopy (XPS) measurements.22 A previous first-principles study also indicated that Cu(I) was formed at coverages less than 1/3 (on a Cu/O basis) by interacting with surface oxygen atoms of the α-Al2O3(0001).25
 |
| | Fig. 1 (a) Cu K-edge PTRF-XANES spectra of Cu/α-Al2O3(0001) in s- and p-polarization directions. (b) Cu K-edge XANES spectra of reference compounds. | |
3.2 Three-dimensional (3D) structure of the Cu species on α-Al2O3(0001)
Fig. 2a and b show the PTRF-EXAFS spectra (χs-pol(k) and χp-pol(k)) of the Cu/α-Al2O3(0001) and the EXAFS spectra of the Cu reference compounds, respectively. In Fig. 2a, a slight polarization dependence was found around 50 nm−1 between the two PTRF-EXAFS spectra. The envelopes of the PTRF-EXAFS oscillations are quickly damped in the higher k region, compared to that for Cu foil, which indicates that the nearest neighbor atom of Cu is not Cu, but a lighter atom such as O and Al. The curve fitting results in Table S1 showed no contribution from Cu–Cu interaction, which suggests that no Cu aggregation occurred, while Cu–O interaction (0.194 ± 0.003 nm) was dominant. The coordination number for the Cu–O interaction in the average PTRF-EXAFS spectrum χavg(k), which was calculated as (2 × χs-pol(k) + χp-pol(k))/3, was estimated to be 2.5 ± 0.8, which means that the Cu single atoms form bonds with two or three oxygen atoms on the Al2O3(0001) surface.
 |
| | Fig. 2 (a) Cu K-edge PTRF-EXAFS spectra (χs-pol(k) and χp-pol(k)) of the Cu species on the α-Al2O3(0001) surface. (b) Cu K-edge EXAFS spectra of reference compounds. | |
DFT calculations were conducted to elucidate possible adsorption sites of the Cu single atoms on the α-Al2O3(0001) surface. Fig. 3 shows the optimized structure of the Al-terminated α-Al2O3(0001) surface. As illustrated in Fig. 3b, the outmost Al atoms (Al1) largely relax inward by 88%, compared with those in the bulk-terminated surface, and are practically co-planar with surface oxygen atoms (O1), which agrees with previous calculation results.45,46 Based on the curve fitting results showing that the Cu–O coordination number in the average PTRF-EXAFS spectrum was 2.5 ± 0.8, we considered the bridge and hollow oxygen sites on the α-Al2O3(0001) surface as candidates of adsorption sites for the Cu single atoms. The α-Al2O3(0001) surface has two types of oxygen hollow sites (H1 and H2) and two types of oxygen bridge sites (B1 and B2), as illustrated in Fig. 3a. The O1–O1 distances of the equilateral oxygen triangles consisting of H1 and H2 sites were 0.261 and 0.274 nm, respectively. B1 and B2 are the sites bridging the two oxygen atoms of the H1 and H2 sites, respectively. We put a Cu atom on each of the corresponding positions and calculated the adsorption energies after the structure optimization, which are listed in Table 1. The Cu atoms on the B1 and B2 sites moved to the H1 site after the structure optimization, and we found that the H1 and H2 sites were energetically stable. The adsorption energies at the H1 and H2 sites were calculated to be −2.54 and −2.14 eV, indicating that the H1 site is preferred. The H1 site corresponds to the imaginary Al position in the next Al layer over the α-Al2O3(0001) surface. Previous DFT calculations have also indicated that the H1 site is most stable for adsorption of the Cu single atoms.25–27Fig. 4a and b show the optimized structures for the Cu single atoms adsorbed on the H1 and H2 sites, respectively.
 |
| | Fig. 3 Portion of the Al terminated α-Al2O3(0001) surface showing the adsorption sites. The atomic positions are determined by DFT calculation. (a) top view and (b) side view. Dark blue, blue and light blue spheres represent surface Al atoms (Al1), Al atoms in the 2nd and 3rd Al layer (Al2 and Al3), respectively. Red and orange spheres represent surface O atoms (O1) and O atoms in the 2nd O layer (O2), respectively. | |
Table 1 Adsorption energy of Cu single atoms on the α-Al2O3(0001) surface
|
|
H1 |
H2 |
B1 |
B2 |
| Adsorption energy/eV |
−2.54 |
−2.14 |
Relaxed to H1 |
Relaxed to H1 |
 |
| | Fig. 4 DFT-optimized structures for single Cu atoms on (a) H1 site and (b) H2 site of the α-Al2O3(0001) surface. Brown spheres: Cu atoms. The dark blue, blue and light blue spheres represent the surface Al atoms (Al1), the Al atoms in the 2nd and 3rd Al layer (Al2 and Al3), respectively. The red and orange spheres represent the surface O atoms (O1) and the O atoms in the 2nd O layer (O2), respectively. | |
Since the atomic coordinates of Cu, O, and Al atoms were obtained from the DFT-optimized structures in Fig. 4, we conducted theoretical Cu K-edge EXAFS simulations using the FEFF code to confirm if the simulations reproduced the observed EXAFS spectra. In the simulations, we considered the surrounding atoms around Cu which were located within 0.3 nm because the contribution from the more distant atoms is small and negligible in the EXAFS oscillations. Fig. 5a and b show the simulated EXAFS spectra obtained for the Cu single atoms on the H1 and H2 sites, respectively, and their comparisons with the observed EXAFS spectra. Three O1 atoms and one Al2 (or Al3) atom were included in the EXAFS simulations, and their atomic coordinates are listed in Table S2. The R2 values for the s- and p-polarizations in the H1 site model were 1.6 and 10.3, respectively, and those in the H2 site model were 2.6 and 17.5, respectively, showing that the H1 site provides a better fit although the corresponding R2 values are larger than 1. The primary distinctions between the H1 and H2 site models pertain to the Cu–Al and Cu–O bond distances, which are 0.267 and 0.249 nm for the Cu–Al bond, and 0.207 and 0.214 nm for the Cu–O bond, respectively. The shorter Cu–Al bond distance in the H2 site model made a larger contribution to the total EXAFS oscillations, resulting in lowering of the degree of the fit between the observed and simulated spectra, as indicated by the curve fitting results in Table S1 showing that the Cu–O oscillations are dominant and contribution from the Al atoms should be small in the observed spectra. Moreover, the longer Cu–O bond distance in the H2 site model contributes to the further reduced degree of the fit, as evidenced by the larger discrepancy with the experimentally determined Cu–O bond distance (0.194 ± 0.003 nm). Considering the EXAFS simulation results and the adsorption energies in Table 1, we concluded that the H1 site model is most plausible. However, the simulation based on the H1 model did not adequately reproduce the observed spectra, as indicated by the substantial R2 values. One reason for this discrepancy is the Cu–O1 bond distance (0.207 nm), which is slightly longer than the 0.194 ± 0.003 nm obtained by the curve fitting results. A secondary rationale pertains to the position of the Al3 atom, which is located less than 0.27 nm from the Cu atom. This is in contrast to our previous PTRF-EXAFS results on single metal atoms on an oxide surface showing no observable EXAFS oscillation corresponding to metal–cation bond with a bond distance less than 0.27 nm, as already mentioned. And it is imperative to optimize the Cu–O1 bond angle to the surface normal (in the [0001] direction) to accurately reproduce the amplitudes of the EXAFS oscillation in both the s- and p-polarizations, as demonstrated in eqn (1). Then we allowed the positions of the Cu, O1, and Al3 atoms to move from the original positions by keeping the three-fold symmetry and fixing the Cu–O1 bond at 0.194 nm. We systematically changed the distances of the Cu and Al3 atoms from the O1 triangle plane, which are defined as variables d1 and d2, respectively, and simulated the EXAFS oscillations (see Fig. S4). Then we calculated the R2 values for s- and p-polarization EXAFS spectra as functions of d1 and d2. The acceptable ranges of d1 and d2 were determined as those of the region where the R2 values for both the s- and p-polarizations were less than 1. Consequently, that region was successfully found, where d1 lay within the range of 0.116 nm ± 0.001 nm, and d2 lay within the range of 0.156 nm ± 0.002 nm. Thus, we concluded that the H1 site is the most reasonable both from the EXAFS simulations and DFT calculations. A previous PTRF-EXAFS study on vacuum-deposited Ni atoms on α-Al2O3(0001) surface showed that the Ni atoms were atomically dispersed and stabilized on the H1 site, accompanied that the O1 triangle shifted upwards and was expanded.9 We then tested a similar model structure as shown in Fig. 6a, where the Cu single atom on the H1 site is located at d1 = 0.116 nm and d2 = 0.156 nm. The Cu–O1 and Cu–Al3 bond distances are 0.194 nm and 0.272 nm, respectively. This model structure well reproduced the observed EXAFS oscillations for both the s- and p-polarizations, as indicated in Fig. 6b. For Fig. 6a, we assumed that the surface atoms such as Cu and three-fold O1 atoms moved from their original positions while the Al3 atom in the bulk kept its position (see Fig. 4a for the original structure). Three-fold O1 atoms moved upward by 0.032 nm and the O1–O1 distance was expanded from 0.261 nm to 0.269 nm. The Cu–Al3 distance was extended from 0.268 nm to 0.272 nm. The upward shift of the oxygen triangle can be understood as “surface lifting”, which is the surface structural change induced by adsorption of atoms or molecules, particularly the upward displacement of surface atoms. A previous surface X-ray diffraction (SXRD) study of the Cu overlayer on TiO2(110) suggested a significant upward displacement (0.065 nm) of the topmost oxygen atoms after vacuum deposition of Cu.47 In our case, the chemical bond formation between Cu and the triangle oxygen atoms of the α-Al2O3(0001) surface induced the upward displacement (0.032 nm) of the oxygen atoms. The lateral expansion of the oxygen triangle may be attributed to the propensity of three-coordinate Cu(I) complexes to adopt a trigonal planar structure rather than a trigonal pyramidal one, as evidenced by formation of the trigonal planar Cu(I) complexes such as [Cu(CN)3]2−,48 [Cu(C6H7N)3]+,49 and [Cu(etu)3]+ (tris(ethylenethiourea)copper(I)).50 It is worth considering the reasons why the structure obtained from the DFT calculations (the H1 site model in Fig. 5(a)) had to be modified to reproduce the observed PTRF-EXAFS spectra using the theoretical EXAFS simulation. The discrepancy in the local structure around the Cu single atoms, as determined by zero-temperature DFT calculations and room-temperature PTRF-EXAFS measurements, may be attributed to the presence of large, anharmonic vibrations in the Cu–O and Cu–Al bonds.51,52 Given that such vibrations were not taken into account in our zero-temperature DFT calculations, the local surface relaxation observed in our study at room temperature could not be accurately predicted.
 |
| | Fig. 5 The simulated EXAFS spectra (red dotted lines) obtained by assuming (a) H1 site and (b) H2 site for the Cu single atoms and their comparison with the observed EXAFS spectra (black solid lines). The brown spheres are the Cu atoms. The light blue and blue spheres represent surface the Al atoms in the 3rd and 2nd Al layer (Al3 and Al2), respectively. The red spheres show the surface O atoms (O1). | |
 |
| | Fig. 6 (a) Proposed structure for the Cu single atom at H1 site with the in-plane expansion and upward shift of the oxygen atoms. Circles with broken lines indicate the oxygen atom positions before such relaxation, shown in Fig. 4a. (b) Comparison between the observed and simulated PTRF-EXAFS spectra for the Cu/α-Al2O3(0001) (black solid and red dotted lines, respectively) using the structure model shown in (a). The brown spheres are the Cu atoms. The dark blue, blue and light blue spheres represent the surface Al atoms (Al1), the Al atoms in the 2nd and 3rd Al layer (Al2 and Al3), respectively. The red and orange spheres show the surface O atoms (O1) and the O atoms in the 2nd O layer (O2), respectively. | |
The coverage of the H1 adsorption sites of the α-Al2O3(0001) surface that can stabilize the Cu single atoms is 0.33 ML, which may represent the maximum coverage of the Cu single atoms. However, as the Cu loading increases, the limited availability of these specific sites may result in aggregation of vacuum-deposited Cu atoms through their diffusion and collision even at a lower coverage than 0.33 ML. Therefore, the conclusions of the present study are strictly applicable to the low-coverage regime.
3.3 Effect of surface hydroxyl groups on the Cu structure
It has been reported that the hydroxyl groups on the α-Al2O3(0001) surface affect the local structure and energetic stability of the deposited Cu atoms.20–22 The analysis of the XPS O 1s peak of the α-Al2O3(0001) surface suggested that the OH coverage was 0.11 ML (see Fig. S5), which may not be negligible. We then performed additional DFT calculations to investigate the effect of the surface hydroxyl groups on the structure and stability of the Cu single atoms. The origin of the hydroxyl groups may come from dissociative adsorption of residual water molecules in the UHV chamber since additional water exposure results in increase in their coverage.20,21 Therefore, we considered the water-dissociated α-Al2O3(0001) surface, where dissociated OH and H are absorbed at Al1 and the nearest O1 atoms, respectively (the OH coverage is 0.07 ML). Three energetically stable sites for the Cu atoms were found, which are the H1 site, the T1 site consisting of H1 and O of Al1–OH, and the B3 site bridging O1 and O of Al1–OH. The corresponding optimized structures and adsorption energies are shown in Fig. S6. The Cu atoms placed on the T1 and B3 sites were more stable than those on the H1 site probably because they interact with the hydroxyl oxygen. However, they were unlikely to form or would be minor species for the following reasons: (1) the Cu atoms on the T1 sites interact with four oxygen atoms, which may contradict the curve-fitting results in Table S1 showing that that the Cu–O coordination number is 2.5 ± 0.8. (2) The Cu–Al bond distance in the B3 site model (0.260 nm) is shorter than that in the H1 site model (0.267 nm). This is in contrast to the results of the present study, which indicate that the Cu–O oscillations are dominant and the contribution from the Al atoms should be small. Therefore, we concluded that the Cu single atoms on the H1 sites were the dominant Cu species under the applied experimental conditions. In our experiments, the coverages of the OH groups on the α-Al2O3(0001) surface and of the evaporated Cu atoms were low at 0.11 ML and 0.07 ML, respectively. Since diffusion of the evaporated Cu atoms on the α-Al2O3(0001) surface was limited, the probability of them encountering OH groups was very low. Consequently, most of the Cu single atoms might be stabilized on the H1 sites.
4. Conclusions
This work experimentally investigated the valence state and 3D structure of the low-coverage Cu species deposited on α-Al2O3(0001) at room temperature under the UHV conditions. The valence state of the Cu species was determined to be monovalent (Cu(I)) according to the PTRF-XANES spectra. The Cu species was found to be atomically dispersed on the α-Al2O3(0001) surface, as determined by the absence of Cu–Cu interaction in the curve fitting analysis of the PTRF-XAFS spectra. DFT calculations showed that the three-fold hollow oxygen site above the third layer Al atoms was energetically stable for adsorption of the Cu single atom. Local structure modification around the Cu single atom in the DFT-optimized structure was conducted so that the simulated EXAFS spectra reproduce the observed spectra well in the two polarization directions. We proposed local surface relaxation around the Cu atom, where the three-fold oxygen triangle moved upward by 0.032 nm and the oxygen–oxygen distance in the oxygen triangle expanded from 0.261 nm to 0.269 nm. Such relaxation suggests a strong metal–support interaction that can effectively anchor the Cu single atom, thereby suppressing thermal diffusion and aggregation even under reaction conditions. In addition, the modified Cu–O coordination geometry may influence the local electronic structure of the Cu center, which is expected to be relevant for elementary steps involving charge transfer or bond activation in catalytic reactions. Although catalytic measurements are beyond the scope of the present work, the identified adsorption site and relaxation mechanism provide an atomistic basis for understanding the stability and functionality of Cu single-atom catalysts on oxide surfaces.
Conflicts of interest
There are no conflicts to declare.
Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5cp03666f.
Acknowledgements
The XAFS measurements have been carried out under the approval of PF-PAC (PF advisory committee, PAC Proposal No. 2020G643 and 2022G576). This study was financially supported by the CREST Project (No. JPMJCR19R3) of the Japan Science and Technology Agency (JST), Kakenhi Grants-in-Aid (No. 21H01802, 24H00445, and 24KJ0008), and a Grant-in-Aid for Transformative Research Areas (A) (No. 22H05109) from the Japan Society for the Promotion of Science (JSPS). The computational work was partially supported by JSPS KAKENHI Grant Number 24H02219 (M. G.) in Transformative Research Areas (A) 24A202 Integrated Science of Synthesis by Chemical Structure Reprogramming (SReP). The computational results in this study were achieved through the MANABIYA (ACADEMIC) program conducted by the Institute for Chemical Reaction Design and Discovery (ICReDD), Hokkaido University, which was established by the World Premier International Research Initiative (WPI), MEXT, Japan.
References
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- S. K. Kaiser, Z. Chen, D. Faust Akl, S. Mitchell and J. Pérez-Ramírez, Chem. Rev., 2020, 120, 11703–11809 CrossRef CAS PubMed.
- H. Zhang, X. Zhang, X. Shi, X. Liu, J. Liang, S. Li, S. Ma, X. Xiao, J. Guo, L. Zhang and M. Tan, Appl. Catal., B, 2025, 361, 124648 CrossRef CAS.
- Y. Zhang, J. Zhao, H. Wang, B. Xiao, W. Zhang, X. Zhao, T. Lv, M. Thangamuthu, J. Zhang, Y. Guo, J. Ma, L. Lin, J. Tang, R. Huang and Q. Liu, Nat. Commun., 2022, 13, 58 CrossRef CAS PubMed.
-
K. Asakura, Polarization-dependent total reflection fluorescence extended X-ray absorption fine structure and its application to supported catalysis, in Catalysis, RSC Publishing, 2012, pp. 281–322 Search PubMed.
- S. Takakusagi, W.-J. Chun, H. Uehara, K. Asakura and Y. Iwasawa, Top. Catal., 2013, 56, 1477–1487 CrossRef CAS.
- S. Takakusagi, Y. Iwasawa and K. Asakura, Chem. Rec., 2019, 19, 1244–1255 CrossRef CAS PubMed.
- K. Ijima, Y. Koike, W.-J. Chun, Y. Saito, Y. Tanizawa, T. Shido, Y. Iwasawa, M. Nomura and K. Asakura, Chem. Phys. Lett., 2004, 384, 134–138 CrossRef CAS.
- Y. Koike, K. Ijima, W.-J. Chun, H. Ashima, T. Yamamoto, K. Fujikawa, S. Suzuki, Y. Iwasawa, M. Nomura and K. Asakura, Chem. Phys. Lett., 2006, 421, 27–30 CrossRef CAS.
- K. Asakura, S. Takakusagi, H. Ariga, W.-J. Chun, S. Suzuki, Y. Koike, H. Uehara, K. Miyazaki and Y. Iwasawa, Faraday Discuss., 2013, 162, 165–177 RSC.
- S. Takakusagi, H. Nojima, H. Ariga, H. Uehara, K. Miyazaki, W.-J. Chun, Y. Iwasawa and K. Asakura, Phys. Chem. Chem. Phys., 2013, 15, 14080–14088 RSC.
- C. R. Kim, H. Maeda, B. Lu, Y. Nakamura, Y. Lin, Y. Wakisaka, D. Kido, K. Asakura and S. Takakusagi, Chem. Lett., 2024, 53, upad045 CrossRef CAS.
- Y. Lin, K. Oshiro, J.-Y. Hasegawa, S. Takakusagi, W.-J. Chun, M. Tabuchi and K. Asakura, J. Phys. Chem. C, 2025, 129, 8193–8205 CrossRef CAS.
- Y. Tanizawa, T. Shido, W.-J. Chun, K. Asakura, M. Nomura and Y. Iwasawa, J. Phys. Chem. B, 2003, 107, 12917–12929 CrossRef CAS.
- Y. Koike, K. Fujikawa, S. Suzuki, W.-J. Chun, K. Ijima, M. Nomura, Y. Iwasawa and K. Asakura, J. Phys. Chem. C, 2008, 112, 4667–4675 CrossRef CAS.
- W.-J. Chun, K. Miyazaki, N. Watanabe, Y. Koike, S. Takakusagi, K. Fujikawa, M. Nomura, Y. Iwasawa and K. Asakura, J. Phys. Chem. C, 2013, 117, 252–257 CrossRef CAS.
- B. Lu, D. Kido, Y. Sato, H. Xu, W.-J. Chun, K. Asakura and S. Takakusagi, J. Phys. Chem. C, 2021, 125, 12424–12432 CrossRef CAS.
- A. L. Ankudinov, B. Ravel, J. J. Rehr and S. D. Conradson, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7565–7576 CrossRef CAS.
- Q. Fu, T. Wagner and M. Rühle, Surf. Sci., 2006, 600, 4870–4877 CrossRef CAS.
- C. Niu, K. Shepherd, D. Martini, J. Tong, J. A. Kelber, D. R. Jennison and A. Bogicevic, Surf. Sci., 2000, 465, 163–176 CrossRef CAS.
- J. A. Kelber, C. Niu, K. Shepherd, D. R. Jennison and A. Bogicevic, Surf. Sci., 2000, 446, 76–88 CrossRef CAS.
- M. C. R. Jensen, K. Venkataramani, S. Helveg, B. S. Clausen, M. Reichling, F. Besenbacher and J. V. Lauritsen, J. Phys. Chem. C, 2008, 112, 16953–16960 CrossRef CAS.
- Z. Łodziana and J. K. Nørskov, J. Chem. Phys., 2001, 115, 11261–11267 CrossRef.
- N. C. Hernández and J. F. Sanz, J. Phys. Chem. B, 2002, 106, 11495–11500 CrossRef.
- N. C. Hernández and J. F. Sanz, Appl. Surf. Sci., 2004, 238, 228–232 CrossRef.
- N. C. Hernández, J. Graciani, A. Márquez and J. F. Sanz, Surf. Sci., 2005, 575, 189–196 CrossRef.
- J. F. Sanz and N. C. Hernández, Phys. Rev. Lett., 2005, 94, 016104 CrossRef PubMed.
- H. Zhou, J. Zhu, H. Jin, Y. Li, B. Wang, S. Huang, W. Lin, Y. Li, W. Chen and Y. Zhang, Mol. Catal., 2023, 551, 113646 CAS.
- T. Isono, T. Ikeda, R. Aoki, K. Yamazaki and T. Ogino, Surf. Sci., 2010, 604, 2055–2063 CrossRef CAS.
- T. Taguchi, T. Ozawa and H. Yashiro, Phys. Scr., 2005, 205 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- P. Thompson, D. E. Cox and J. B. Hastings, J. Appl. Crystallogr., 1987, 20, 79–83 CrossRef CAS.
- J. Ahn and J. W. Rabalais, Surf. Sci., 1997, 388, 121–131 CrossRef CAS.
- E. A. Soares, M. A. Van Hove, C. F. Walters and K. F. McCarty, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 195405 CrossRef.
- P. Guénard, G. Renaud, A. Barbier and M. Gautier-Soyer, Surf. Rev. Lett., 1998, 05, 321–324 CrossRef.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- N. Kosugi, H. Kondoh, H. Tajima and H. Kuroda, Chem. Phys., 1989, 135, 149–160 CrossRef.
- C. Rajesh, S. Nigam and C. Majumder, Phys. Chem. Chem. Phys., 2014, 16, 26561–26569 RSC.
- T. Kurita, K. Uchida and A. Oshiyama, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 155319 CrossRef.
- G. Charlton, P. B. Howes, C. A. Muryn, H. Raza, N. Jones, J. S. G. Taylor, C. Norris, R. McGrath, D. Norman, T. S. Turner and G. Thornton, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 16117–16120 CrossRef CAS.
- C. Kappenstein and R. P. Hugel, Inorg. Chem., 1978, 17, 1945–1949 CrossRef CAS.
- A. H. Lewin, R. J. Michl, P. Ganis and U. Lepore, J. Chem. Soc., Chem. Commun., 1972, 661–662 RSC.
- M. S. Weininger, G. W. Hunt and E. L. Amma, J. Chem. Soc., Chem. Commun., 1972, 1140–1141 RSC.
- E. A. Soares, M. A. Van Hove, C. F. Walters and K. F. McCarty, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 195405 CrossRef.
- N. M. Harrison, X.-G. Wang, J. Muscat and M. Scheffler, Faraday Discuss., 1999, 114, 305–312 RSC.
|
| This journal is © the Owner Societies 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Can
Liu
a,
Min
Gao
b,
Haoran
Xu
a,
Daiki
Kido
cd,
Masao
Kimura
*a,
Can
Liu
a,
Min
Gao
b,
Haoran
Xu
a,
Daiki
Kido
cd,
Masao
Kimura







