
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spectral and temporal differentiation between integral and contaminant chlorophyll a in the cytochrome b6f complex
Adrien A. P.
Chauvet†
 *a,
Rachna
Agarwal
bc and
William A.
Cramer
b
*a,
Rachna
Agarwal
bc and
William A.
Cramer
b
aLaboratoire de Spectroscopie Ultrarapide (LSU), ISIC, Faculté des Sciences de Base and Lausanne Centre for Ultrafast Science (LACUS), Ecole Polytechnique Fédérale de Lausanne (EPFL), Station 6, 1015 Lausanne, Switzerland. E-mail: a.chauvet@sheffield.ac.uk
bDepartment of Biological Sciences, Purdue University, West Lafayette, Indiana 47907, USA
cMolecular Biology Division, Bhabha Atomic Research Centre, Mumbai 400 085, India
First published on 2nd December 2025
Abstract
Purification of photosynthetic protein complexes in detergent often results in residual contaminant chlorophyll (Chl) that is dependent on the preparation process. In the case of the cytochrome b6f complex, which contains one molecule of bound Chl a per 130 kDa monomer in the dimeric hetero-oligomeric complex, both complex-bound and contaminant Chl a are present and spectrally indistinguishable in the steady-state absorbance spectra commonly employed to assay photosynthetic protein complexes. We, however, demonstrate that the signals from photo-excited cyt b6f-bound and contaminant Chl a have distinct temporal and spectral signatures, as revealed by ultrafast optical transient spectroscopy. The difference in signals is further amplified by using a non-anisotropic pump–probe scheme to enhance the detection of the contaminant Chl a stimulated emission. Such sharp differences further exemplify the impact of the environment on the photodynamic properties of molecules.
Introduction
The study of interacting pigments, especially in the case of photosynthetic protein complexes, provides valuable information about quantum coherence, energy transfer, and electronic conformation of the proteins involved.1,2 However, ultrafast spectral analyses are highly sensitive to the sample quality, as any contaminant affects the signal and its resolution. In the case of the multi-heme cytochrome (cyt) b6f complex,3 solubilized chlorophyll (Chl) a often remains in the sample even after isolation and purification in detergent. Unfortunately, this Chl a contaminant is spectrally indistinguishable in static measurements compared to the single native embedded Chl a molecule (Fig. 1).4,5 The Chl a purity of these samples is therefore challenging to assess using standard steady state techniques. Although high-quality cyt b6f has been obtained, leading to the crystallographic resolution of the protein complex,6 the extent of Chl contamination in the purified complex remains a problem, and an efficient and quantitative assay would facilitate structural studies as well as studies of the dynamics of the cytochrome complex.6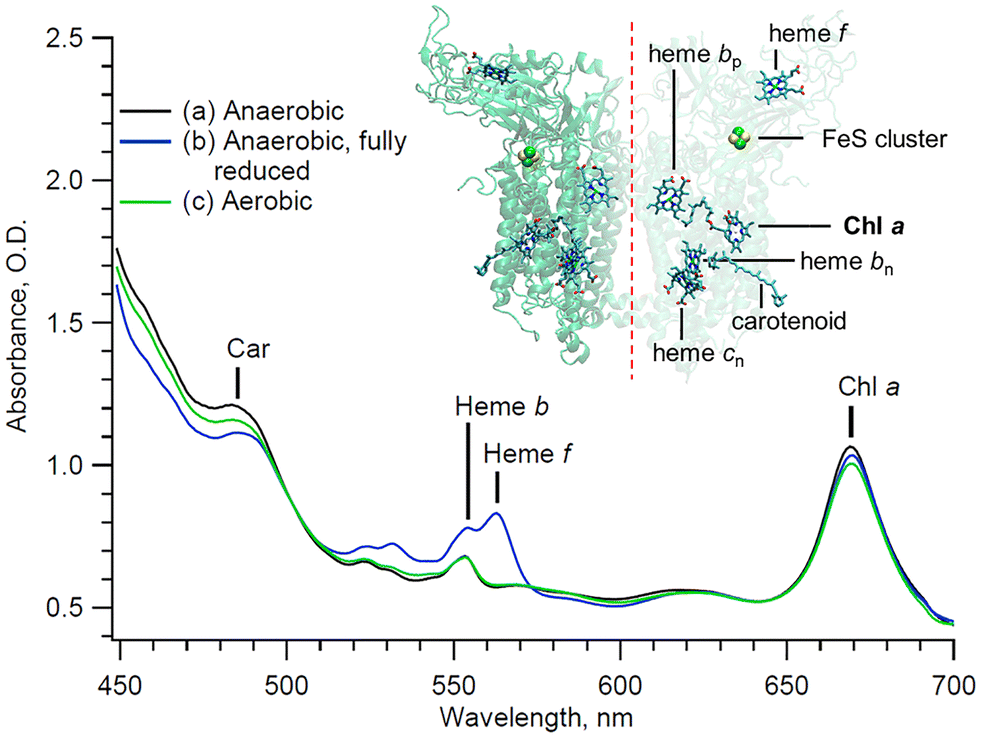 | ||
| Fig. 1 Steady state of the cytochrome b6f complex at different redox states and under different conditions (see text). Inset: Backbone structure and prosthetic groups of the cytochrome b6f complex. The crystallographic data are taken from the 2E74 pdb file.7 | ||
Current optical methods for assessing the Chl a/cyt f ratio are based on the evaluation of the intensity of the sodium dodecyl sulfate (SDS) gel marker or on the absorbance ratio of cyt f to Chl a, which depends on the extinction coefficient used.8,9 Alternatively, Savikhin et al. showed that impure cyt b6f preparations were marked by an additional short ∼ps lifetime, which was then ascribed to contaminant Chl a.4 This work pushes the disambiguation one step further and demonstrates that it is possible to also spectrally distinguish between non-essential contaminant and integral Chl a using ultrafast transient analysis. We show that each Chl a type has distinct spectral and temporal signatures.
Methods
The cytochrome b6f sample was prepared as follows: active dimeric cytochrome b6f complexes were isolated from the leaves of Spinacia as previously described.10 The subunit composition of the b6f preparation was assessed by using SDS polyacrylamide gel electrophoresis (PAGE), clear-native PAGE, and the redox difference spectra using standard procedures.11 All assays were performed in 30 mM tris(hydroxymethyl)aminomethane (Tris)-HCl (pH 7.5), 50 mM NaCl, 0.2 mM ethylenediaminetetraacetic acid (EDTA), and 0.04% undecyl maltoside (UDM). The electron transport activity of the dimeric complex, 150–200 electrons per cyt f s, was assessed using decyl-plastoquinol as an electron donor and Chlamydomonas plastocyanin as an electron acceptor.11 All free-floating Chl a contaminant molecules were effectively washed out during the isolation and purification process. Only Chl a molecules that are integral to the protein complex, as well as Chl a molecules that are non-selectively adsorbed onto the protein complex, remain. The sample, consisting of about 300 µL, was housed in a microfluidic flow-cell.12 The cyt b6f complex was studied under a controlled atmosphere using a bespoke miniature anaerobic chamber (1L).13 The integrity of the complexes over the course of the experiment is directly monitored by recording their steady-state absorbance through the white light continuum of the probe beam. From the steady state absorbance difference spectra, it can be inferred that the preparation contains a Chl a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :cyt f ratio of approximately 1.3:1. The concentration of the sample is about 50 µM for the Cyt b6f dimer.
:cyt f ratio of approximately 1.3:1. The concentration of the sample is about 50 µM for the Cyt b6f dimer.
Transient absorption signals were recorded as follows: the 800-nm output of a 1 kHz regenerative amplifier is used to pump a home-made visible non-collinear optical parametric amplifier (NOPA, see ref. 14 for a detailed description) producing ∼40 fs, 515 nm pump pulses with a full-width-half-maximum of 15 nm. By centering the pump at 515 nm, the goal is to minimize exciting carotenoid (Car) and the various hemes, while avoiding the chlorophyll (Chl) a Qx−y regions, as depicted in Fig. S1 (see the SI). The undisturbed 600–650 region is thus solely characterized by the signal from Car, which can then be modelled. We take advantage of the lack of sharp Car features in the Chl a Qy region to estimate the Car signal as a first order polynomial and subtract it for each time delay and extract the sole Chl a signal, as discussed later. A small fraction of the regenerative amplifier output is focused onto a 5-mm thick CaF2 crystal to provide a broad visible probe. The resulting pump–probe cross-correlation signal is about 150 fs. The relative polarization between the pump and probe beam is either set to the magic angle (54.7°) or parallel in order to enhance the detection of the Chl a signals,15 as discussed in the SI. The probe beam is then focused onto a spectrometer, resulting in a probe window extending from 350 nm to 750 nm with a spectral resolution of 1.3 nm.16 The average excitation power was kept low, within the linear excitation regime (checked via power dependence), at 0.2 mW and focused on a spot size of 100 × 160 µm. Considering 40 fs-long pulses at 1 kHz, we estimate the energy to be about 200 nJ per pulse, 10% of which is lost on passing through the anaerobic chamber's windows. We thus expect the excitation energy to be about 180 nJ per pulse at the sample stage. The estimated proportion of the excited molecules is estimated to be less than 2% based on the direct comparison between the sample's absorbance (∼0.6 OD for Chl a Qy absorption) and the maximum transient signal amplitude (∼0.01 OD for Chl a Qy signal).
Results and discussion
With the pump beam centered at 515 nm, both Chl a and β-carotene (Car) are directly excited. Accordingly, the transient optical signal is dominated by the Car and Chl a responses in the 570 to 730 nm region (Fig. 2). The choice of parallel polarization between the pump and the probe significantly enhances the SE signal detection over other transient absorption signals, and without affecting the motored dynamics of Chl a. Indeed, the anisotropy of the cyt b6f-bound Chl a, measured in the Qy-band, was found to be close to the theoretical maximum of 0.4 and nearly constant over the first 200 ps.4 The transient signal of solubilized Chl a was also shown to be constant at least during the first 5 ps after excitation.17,18 A constant anisotropy indicates that the parallel and perpendicular polarization signals have the same dynamics as the isotropic signal monitored at the magic angle. The signal monitored here can therefore be directly compared to usual isotropic studies found in the literature, at least within these timescales. Furthermore, the Chl a Qy signal, in the 650–710 nm region, is assumed to be free of any spectral contribution from the hemes f and b, which have their α-absorbance bands at 554–563 nm (Fig. 1). The carotenoid signal, on the other hand, has a ‘red tail’ that extends from 600 to 750 nm and beyond. This signal is characteristic of the optically forbidden S1 state of Car, which is populated via vibrational relaxation of the excited S2 at 515 nm.19 | ||
| Fig. 2 (A) Time-wavelength surface and (B) spectra at given time delays for Cyt b6f under anaerobic conditions and parallel pump–probe polarization. | ||
In order to disentangle the Chl a signal from the extended red-tail signal from Car, we take advantage of the lack of sharp features of the latter in the Chl a Qy region and model the Car signal as a first-order polynomial. This background is then evaluated and subtracted for each time delay, thus enabling us to extract the sole Chl a signal shown in Fig. 3 heat map.
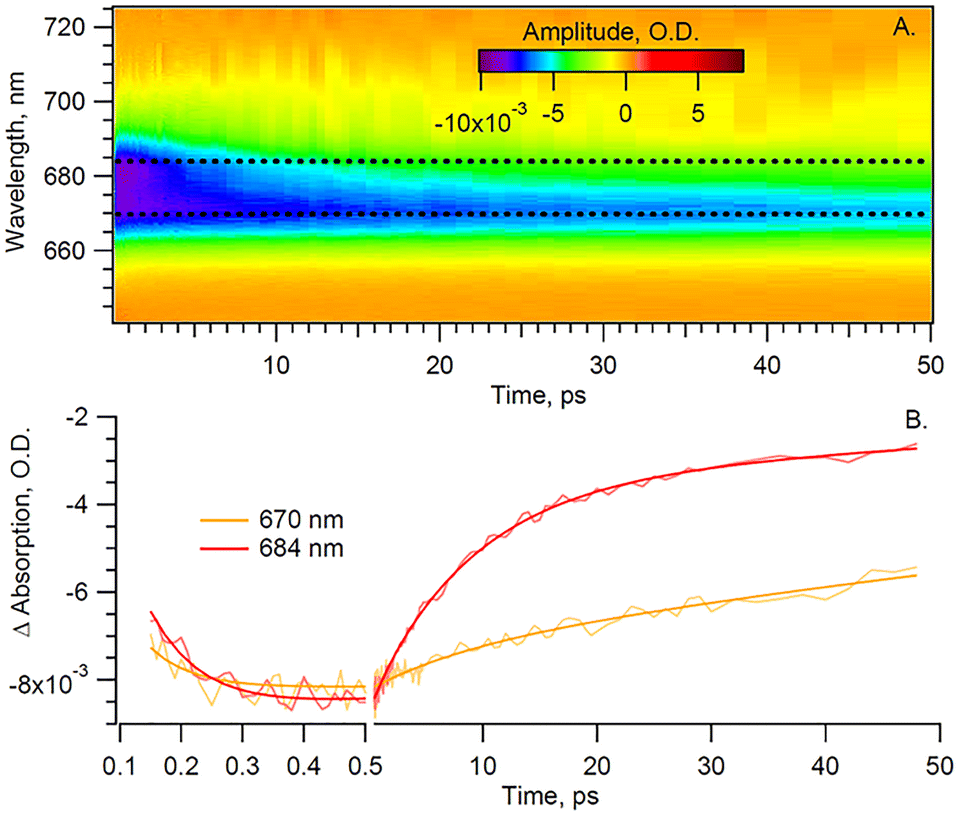 | ||
| Fig. 3 (A) The retrieved Chl a signal and (B) kinetics of the sole Chl a signal from Cyt b6f under anaerobic conditions and parallel pump–probe polarization. The smoothed lines represent fits of the data. | ||
The extracted Chl a signal, shown in Fig. 3A, shows two bands peaking at 670 and 684 nm. We take as a representative of each of the two bands the kinetics at 670 and 684 nm (Fig. 3B). The extracted Chl a signal is analyzed by means of global fit (GF), to estimate the number of exponential components, and singular value decomposition (SVD), to refine the decay times. The resulting decay-associated spectrum (DAS) is shown in Fig. 4.
 | ||
| Fig. 4 DAS of the retrieved Chl a signals under anaerobic and aerobic conditions. | ||
The extracted anaerobic Chl a signal is satisfactorily fit with a minimum of three exponential decay components: (1) a ∼80 fs DAS component corresponding to the rise of a band maximizing at 684 nm. This component corresponds to the growth of a negative transient absorption signal and is assigned to either the photobleaching (PB) or the SE signal from a pool of Chl a molecules. However, since no specific band is seen near 684 nm in the cyt b6f static spectrum, the signal is solely attributed to SE. (2) A ∼9 ps DAS component, which mirrors the previous ∼80 fs DAS. Accordingly this ∼9 ps component is assigned to the decay of the state from which the SE originates; in agreement with a previous report on solubilized Chl a.20 (3) A ∼0.2 ns component, which has a maximum at 670 nm and coincides with the maximum of the static Chl a Qy absorbance band. This long-lived component is therefore assigned to the ground state recovery of excited Chl a, in accordance with previous studies on the cyt b6f complex.4 This time component coincides with the previously reported 194 ps mono-exponential component from dissolved diffraction grade cyt b6f crystals (i.e. ultra-pure cyt b6f preparation).4 The same referred study showed that an extra 6.5 ps component was found to be characteristic of the conventional preparations (i.e. with Chl a/cyt f ratio > 1) and was assigned to the Chl a contaminant. Accordingly, we assign our 684 nm band signals (Fig. 4) to be exclusively representative of contaminant Chl a. On the other hand, we assign our 670 nm band signal exclusively to the cyt b6f-bound Chl a. The work by Dashdorj et al. also indicates the presence of a small 5.5 ns component (∼10%), which is potentially assigned to both integral and contaminant Chl a. This long-lived signal, if present, would, however, be incorporated in our 0.2 ns component, as our temporal window of 50 ps would not allow us to distinguish between them. It is important to note that this reported 5.5 ns component, because it is assigned to both integral and contaminant Chl a, cannot be used to unambiguously distinguish between the two Chl a types. However, the 684 nm band signals, because they are exclusively assigned to contaminant Chl a, are distinct markers.
In order to verify these assignments, the sample was exposed to the air. Exposing solubilized and unprotected Chl a to oxygen is expected to lead to the formation of singlet oxygens and to the subsequent degradation of the nearby molecules, including the unprotected Chl a molecule itself. Indeed, it has been shown that solubilized Chl a molecules are about 130 times less stable than those embedded in cyt b6f.4 The higher stability of integrant Chl a is expected to be provided by the nearby carotene molecules in cyt b6f. Therefore, if degradation occurs, it is expected to affect primarily the contaminant Chl a molecules. The experiment and data processing are thus repeated on the same cyt b6f preparation, but under aerobic conditions. The sample is left under aerobic conditions for 20 minutes before the start of the experiment. During this short time, we assume that only contaminant Chl a molecules are affected by the presence of oxygen, and that the cyt b6f complexes remain otherwise intact, as indicated by the absorption spectrum in Fig. 1. The GF (not shown) and DAS (Fig. 4) result in a minimum of three exponential decay components that are virtually identical in shape and in timings (but not amplitude, as discussed subsequently), within the error margin of the analysis, to those of the anaerobic case. The presence of dissolved oxygen is indeed not expected to affect the ultrafast dynamics of the complex (i.e. same spectral shape and decay components). However, under aerobic conditions, the amplitude of both the ∼90 fs and ∼8.9 ps components, both peaking at 684 nm, lowers by 12% compared to the anaerobic case, while the ∼0.2 ns component peaking at 670 nm remains identical. Furthermore, it can be seen in the static absorbance spectra (Fig. 1) that the aerobic Chl a Qy band is also reduced compared to the initial anaerobic spectrum. This slight decrease in static absorption reflects the degradation of some Chl a molecules. This degradation directly coincides with the decrease in the 684 nm transient signals (∼90 fs and ∼8.9 ps components). This correspondence confirms that degradation solely affects the 684 nm signal, which is then exclusively assigned to contaminant Chl a. A longer exposure is expected to lead to the degradation of the Chl a contaminant to a greater extent. However, nothing guarantees that only contaminant Chl a would be affected. And without an objective measure of the impact of oxygen on cyt b6f, the interpretation of the data would become even more complex. Here, we assume that cyt b6f remains intact during this short exposure time.
Concerning the nature of the 684 nm band, it could coincide with the SE and the fluorescence signal from Chl a, which are expected to be red-shifted with respect to the static absorbance spectrum.20,21 A similar signal at 684 nm can actually be inferred from the transient isotropic pump–probe Chl a data (data not shown) as well as from the fluorescence spectra from a previous cyt b6f report.15 But in these studies, the 684-nm signal is only present as a faint shoulder to the main 670 nm band, which makes it hardly distinguishable. The present use of parallel polarization fs-spectroscopy greatly improves the resolution of this 684-nm signal and enables its unobstructed spectral and temporal monitoring.
Interestingly, we do not monitor any significant Chl a triplet state signal. While Chl a molecules integral to cyt b6f are not expected to undergo intersystem crossing,4 photo-excited Chl a monomers are expected to form a long-lived triplet state with an efficiency of ∼64%.22,23 The triplet state signal from contaminant Chl a could then be expected to contribute to the 670 nm band bleach in Fig. 4. However, the similarity between the 0.2 ns DAS anaerobic (intact) and aerobic (after oxygen-mediated degradation of contaminant Chl a), shown in Fig. 4, indicates that contaminant Chl a molecules do not significantly contribute to that signal. In light of these conclusions, we offer a tentative Jablonsky diagram in Fig. 5. Because we do not monitor any phosphorescence, contaminant Chl a must therefore have a triplet formation yield much lower compared to solvated Chl a monomers. Unbound Chl a monomers are indeed expected to be washed away during the sample purification process. Hence, we do not expect any unbound Chl a monomers. It is however expected that contaminant Chl a molecules adsorb to the surface of the cyt b6f complexes. It is therefore assumed that adsorption provides a highly effective deactivation mechanism, which hinders triplet formation. This deactivation route does not prohibit entirely the formation of triplet states (given the long-term degradation monitored), but it is effective enough for the triplet state to not be discernable in our transient experiments.
 | ||
| Fig. 5 Jablonsky diagram for the integral and contaminant Chl a. After Soret-band activation (hν), the excited Chl a molecules rapidly relax to the lowest Qy state within 80 fs. While the integral Chl a decay back to the ground state (GS) in 200 ps and are marked by a 670-nm signal, the contaminant Chl a decay in 9.1 ps and are marked by a 684-nm signal. Note that no evidence is given for any triplet state formation. | ||
Conclusions
In summary, we demonstrate that, although it is impossible to spectrally disentangle the contribution from cyt b6f-bound to contaminant Chl a using steady state spectroscopy, time resolved spectroscopy enables such distinction. On the one hand, the excited cyt-embedded Chl a is characterized by a single 670 nm band signal that decays mono-exponentially with a decay time of ∼0.2 ns. On the other hand, contaminant Chl a molecules give rise to an extra 684 nm band that appears and decays mono-exponentially in ∼85 fs and ∼9 ps, respectively. The assignment of the latter signals to unbound (thus unprotected) Chl a is further confirmed by the signal's sensitivity to oxygen. The unambiguous monitoring of contaminant Chl a is here made possible by the careful use of parallel polarization between the pump and the probe. This polarization scheme enhances the 684-nm signal, which, based on its lifetime, is taken as a marker of unbound Chl a molecules in cyt b6f preparations.Conflicts of interest
The authors declare no competing financial interests.Abbreviations
| Car | β-Carotene |
| Chl a | Chlorophyll a |
| cyt | Cytochrome |
| DAS | Decay associated spectrum |
| GF | Global fit |
| PAGE | Polyacrylamide gel electrophoresis |
| SE | Stimulated emission |
| SVD | Singular value decomposition |
| SDS | Sodium dodecyl sulfate |
| UDM | Undecyl maltoside |
Data availability
The spectroscopic data (transient UV-vis) pertaining to our work entitled “Spectral and temporal differentiation between integral and contaminant chlorophyll a in the cytochrome b6f complex” is classified as nonspecific and suitable for general or institutional repository. It is currently stored in local university owned servers, as per the project's data management plan. More explicitly, the data consists of static spectra (vector – wavelength) and transient spectra (matrices – time X wavelength) in MATLAB data files, which will be shared directly upon request.The data supporting this article have been included as part of the supplementary information (SI). The SI includes a deconvolution of the b6f complex static absorption spectra in function of its Chl a and Car contributions. The SI also includes calculation of the expected enhancement of signal monitoring due to the use of parallel polarization between pump and probe. See DOI: https://doi.org/10.1039/d5cp03433g.
Acknowledgements
This research was supported by NCCR MUST, funded by the Swiss National Science Foundation (AAPC), by the FP7 Marie Curie COFUND (AAPC), by the EPSRC (EP/R042802/1, EP/R045305/1, AAPC) and by the US NIHGMS-038323 (WAC).References
- M. Cho, Coherent Two-Dimensional Optical Spectroscopy, Chem. Rev., 2008, 108, 1331–1418 CrossRef CAS PubMed.
- S. Mukamel, Multidimensional Femtosecond Correlation Spectroscopies of Electronic and Vibrational Excitons, Annu. Rev. Phys. Chem., 2000, 51, 691–729 CrossRef CAS PubMed.
- W. A. Cramer, H. Zhang, J. Yan, G. Kurisu and J. L. Smith, Transmembrane Traffic in the Cytochrome b6f Complex, Annu. Rev. Biochem., 2006, 75, 769–790 CrossRef CAS PubMed.
- N. Dashdorj, H. Zhang, H. Kim, J. Yan, W. A. Cramer and S. Savikhin, The Single Chlorophyll a Molecule in the Cytochrome b6f Complex: Unusual Optical Properties Protect the Complex against Singlet Oxygen, Biophys. J., 2005, 88, 4178–4187 CrossRef CAS PubMed.
- D. Huang, R. M. Everly and R. H. Cheng, et al., Characterization of the Chloroplast Cytochrome b6f Complex as a Structural and Functional Dimer, Biochemistry, 1994, 12, 4401–4409 CrossRef PubMed.
- S. S. Hasan and W. A. Cramer, Internal Lipid Architecture of the Hetero-Oligomeric Cytochrome b6f Complex, Structure, 2014, 22, 1008–1015 CrossRef CAS PubMed.
- E. Yamashita, H. Zhang and W. A. Cramer, Structure of the cytochrome b6f complex: quinone analogue inhibitors as ligands of heme cn, J. Mol. Biol., 2007, 370, 39–52 CrossRef CAS PubMed.
- Y. Pierre, C. Breyton, Y. Lemoine, B. Robert, C. Vernotte and J.-L. Popot, On the Presence and Role of a Molecule of Chlorophyll a in the Cytochrome b6f Complex, J. Biol. Chem., 1997, 272, 21901–21908 CrossRef CAS PubMed.
- S. U. Metzger, W. A. Cramer and J. Whitmarsh, Critical Analysis of the Extinction Coefficient of Chloroplast Cytochrome f, Biochim. Biophys. Acta., 1997, 1319, 233–241 CrossRef CAS PubMed.
- D. Baniulis, H. Zhang, E. Yamashita, T. Zakharova, S. S. Hasan and W. A. Cramer, Purification and Crystallization of the Cyanobacterial Cytochrome b6f Complex, in Photosynthesis Research Protocols, Methods in Molecular Biology, ed. R. Carpentier, Humana Press Inc., Totowa, NJ, 2011, pp. 65–77 Search PubMed.
- H. Zhang, J. P. Whitelegge and W. A. Cramer, Ferredoxin:NADP+ oxidoreductase is a subunit of the chloroplast cytochrome b6f complex, J. Biol. Chem., 2001, 276, 38159–38165 CrossRef CAS PubMed.
- A. Chauvet, T. Tibiletti, S. Caffarri and M. Chergui, A Microfluidic Flow-Cell for the Study of the Ultrafast Dynamics of Biological Systems, Rev. Sci. Instrum., 2014, 85, 103118 CrossRef PubMed.
- A. A. P. Chauvet, R. Agarwal and W. A. Cramer, Note: Small Anaerobic Chamber for Optical Spectroscopy, Rev. Sci. Instrum., 2015, 86, 106101 CrossRef PubMed.
- J. Helbing, L. Bonacina and R. Pietri, et al., Time-Resolved Visible and Infrared Study of the Cyano Complexes of Myoglobin and of Hemoglobin I from Lucina pectinata, Biophys. J., 2004, 87, 1881–1891 CrossRef CAS PubMed.
- E. J. G. Peterman, S.-O. Wenk and T. Pullerits, et al., Fluorescence and Absorption Spectroscopy of the Weakly Fluorescent Chlorophyll a in Cytochrome b6f of Synechocystis PCC6803, Biophys. J., 1998, 75, 389–398 CrossRef CAS PubMed.
- A. A. P. Chauvet, A. Al Haddad, W.-C. Kao, F. Van Mourik, C. Hunte and M. Chergui, Photo-Induced Dynamics of the Heme Centers in Cytochrome bc1, Phys. Chem. Chem. Phys., 2014, 2143–2151 Search PubMed.
- Y. J. Shiu, Y. Shi, M. Hayashi, C. Su, K. L. Han and S. H. Lin, Femtosecond Spectroscopy Study of Electronically Excited States of Chlorophyll a Molecules in Ethanol, Chem. Phys. Lett., 2003, 378, 202–210 CrossRef CAS.
- P. Martinsson, S. Sundström and E. Akesson, An Ultrafast Time-Resolved Anisotropy Study of Bacteriochlorophyll a in Pyridine, FEBS Lett., 2000, 465, 107–109 CrossRef CAS PubMed.
- P. Zuo, B.-X. Li and X.-H. Zhao, et al., Ultrafast Carotenoid-to-Chlorophyll Singlet Energy Transfer in the Cytochrome b6f Complex from Bryopsis corticulans, Biophys. J., 2006, 90, 4145–4154 CrossRef CAS PubMed.
- Y. Shi, Y. J. Shiu, C. Su, S. H. Lin and K.-L. Han, Transient Absorption of the Chlorophyll a in Ethanol, Chin. J. Chem. Phys., 2006, 19, 6–10 CrossRef CAS.
- D. A. Hartzler, D. M. Niedzwiedzki, D. A. Bryant, R. E. Blankenship, Y. Pushkar and S. Savikhin, Triplet Excited State Energies and Phosphorescence Spectra of (Bacterio)Chlorophylls, J. Phys. Chem. B, 2014, 118, 7221–7232 CrossRef CAS PubMed.
- P. G. Bower and G. Porter, Quantum Yields of Triplet Formation in Solutions of Chlorophyll, Proc. R. Soc. London, Ser. A, 1967, 296, 435–441 Search PubMed.
- G. R. Seely and J. S. Connolly, Fluorescence of Photosynthetic Pigments in Vitro, in Light Emission by Plants and Bacteria, ed. A. J. Govindjee and D. C. Fork, Academic Press, 1986, pp. 99–133 Search PubMed.
Footnote |
| † Present address: School of Mathematic and Physical Sciences, University of Sheffield, Sheffield S3 7HF, UK. |
| This journal is © the Owner Societies 2026 |